15 Cancers of the musculoskeletal system
Primary bone tumours
Aetiology
For osteosarcoma the most important risk factor is prior radiotherapy which accounts for only 3% of cases, with a time interval of 14 years (range 4–40 years). It is the most common secondary malignancy following treatment for childhood cancer. Other factors include chemotherapy with alkylating agents, Paget’s disease, chronic osteomyelitis, and hereditary conditions such as hereditary retinoblastoma and Li–Fraumeni syndrome (p. 53, 55). In hereditary retinoblastoma, the relative risk (RR) of osteosarcoma is 500 for limb osteosarcomas and 2000 for skull osteosarcomas following irradiation for retinoblastoma. Li–Fraumeni syndrome patients have a RR of 15 for osteosarcoma at all sites.
Pathogenesis
Ewing’s tumours consistently have reciprocal chromosomal translocations, usually between the EWS gene on chromosome 22 and FLI-1 on chromosome 11. In 85% of cases this is t(11;22)(q24;q12). Other variants place EWS with other genes such as t(21;22)(q22;q12) EWS-ERG translocation which occurs in up to 10%.
Clinical features
There are some features which are common to many types of bone tumour (Box 15.1).
Staging
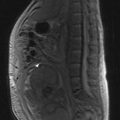
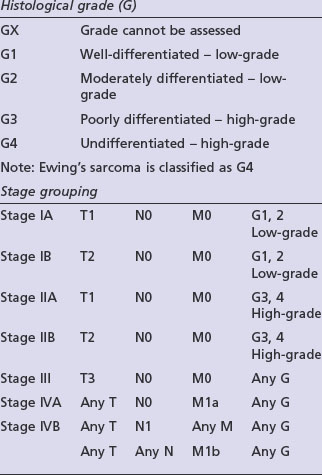
Adequate staging includes optimal imaging of the primary site (MRI) and the entire bone to exclude intra-medullary skip metastases, which occur in osteosarcomas and Ewing’s tumours. A CT scan of the chest should be done to assess for possible pulmonary metastases. For PNETs arising in the lower half of the body a CT of the pelvis and abdomen can be considered as there may be lymph node involvement. A bone scintigram, or in some centres a CT-PET, will assess for bone metastases. A whole body MRI on STIR sequence has shown to be effective in detection of bone metastases which may not be visible with other forms of imaging, particularly in Ewing’s tumours. A bone marrow aspirate should be performed in patients with a Ewing’s tumour to exclude bone marrow involvement. Staging is according to the AJCC bone tumour staging (Table 15.1).

Osteosarcoma
Several subtypes of osteosarcoma exist. Over 90% are high-grade intramedullary tumours, half of which are osteoblastic and the other half are split equally between fibroblastic and chondroblastic osteosarcomas. The remaining 10% are made up of small subgroups including telangiectatic osteosarcoma, extraosseous osteosarcoma, juxtacortical osteosarcoma and malignant fibrous histiocytoma of bone which is treated in the same way as intramedullary osteosarcoma.
Figures 15.1 and 15.2 show imaging features of osteosarcoma. Box 15.2 shows prognostic factors in osteosarcoma. An overview of management is shown in Figure 15.3.
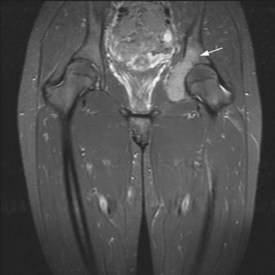
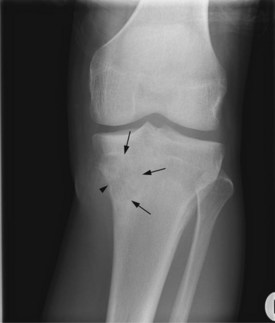
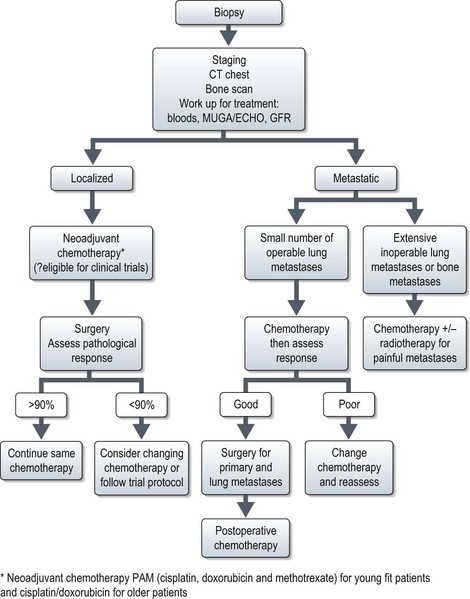
Localized osteosarcoma
Localized osteosarcoma is treated with neoadjuvant chemotherapy followed by surgery (Figure 15.4) and adjuvant chemotherapy.
Chemotherapy
Prior to the 1980s all patients with apparently localized disease had surgery (usually amputation) but only 20–30% survived over 5 years. Most died of disseminated disease suggesting the presence of micrometastases in apparently early disease. A pivotal study showed that giving adjuvant chemotherapy improved survival from 17% to 66%, and neoadjuvant chemotherapy was subsequently implemented. The most frequently used drugs are cisplatin and doxorubicin. Methotrexate at doses up to 12 g/m2 is used in addition to cisplatin and doxorubicin in younger patients and has been found to increase tumour necrosis. The degree of tumour necrosis after neoadjuvant chemotherapy is of prognostic value. Less than 90% necrosis is associated with a poorer prognosis. Whether prognosis can be improved by changing chemotherapy after surgery is one of the roles of the international EURAMOS trial.
Metastatic osteosarcoma
Chemotherapy
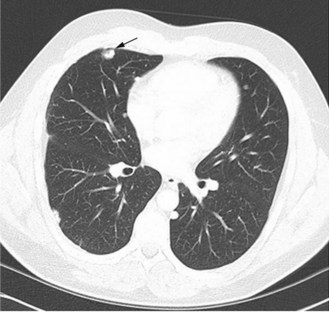
As in the adjuvant setting, the most effective agents are cisplatin, doxorubicin, methotrexate and ifosfamide (Box 15.3). Response rates are between 20–40% although some tumours and metastases may not reduce in size. Apparent calcification of lung metastases can occur (Figure 15.5). If the presentation is with metastases, combination chemotherapy should be the primary treatment.
Box 15.3
Chemotherapy for metastatic osteosarcoma
Ewing’s sarcoma
Presentation
Unlike osteosarcomas which present most often in the epiphyses of long bones, Ewing’s sarcomas occur within the diaphysis and are often associated with a soft tissue mass. The soft tissue reaction may be confused for infection and contribute to delay in diagnosis. The median delay from symptoms (Box 15.1) to diagnosis is 6–9 months. The most common sites are long bones (53%) or the axial skeleton (47%). 25% have a soft tissue primary. Systemic symptoms (fever, anorexia, weight loss, and lethargy) may be present and are associated with advanced disease. 25% patients have metastases at presentation. Subclinical metastases are present in 80–90% with apparently localized disease, necessitating multimodality treatment even in apparently small volume localized disease. Prognostic factors are listed in Box 15.4.
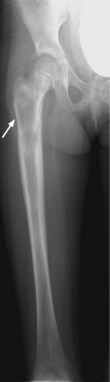
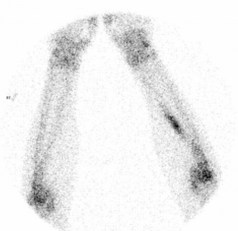
X-ray may show a characteristic destructive lesion within the bone with surrounding periosteal reaction known as an onion skin appearance (Figure 15.6). A soft tissue reaction may also be seen on X-ray and a pathological fracture is seen in 15%. Ewing’s sarcomas, like osteosarcomas, show intense tracer uptake in bone scan (Figure 15.7). Staging is given in Table 15.1 and Figure 15.8 shows an overview of management.
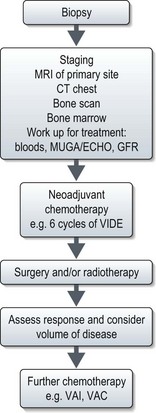
Management of localized disease
Chemotherapy
Stratifying treatment according to risk is under investigation in the EUROEWING99 trial (Figure 15.9). Low-risk patients are randomized between standard treatment and less intense treatment. High-risk/metastatic patients are randomized between standard treatment and high-dose with stem cell rescue.
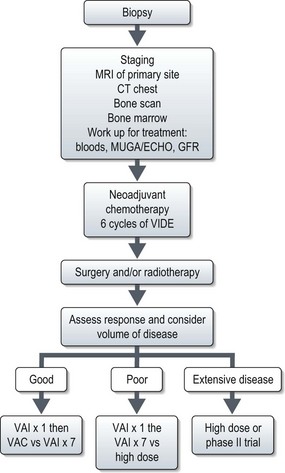
Radiotherapy
Unlike osteosarcomas, Ewing’s tumours are radiosensitive and, radiotherapy used to be the principle method of achieving disease control. Studies have shown radiotherapy without surgery has a worse outcome than surgery for local control but there may be some selection bias. However, radiotherapy still remains a primary method of local control in unresectable tumours when doses of 45–55 Gy have been used (Box 15.5).
Box 15.5
Radiotherapy in sarcomas
Indications
Clinical target volume
Planning target volume – 5–10 mm to CTV depending on immobilization
Management of metastatic disease
Chemotherapy
The same combination chemotherapy agents are used in advanced disease as in localized disease.
Prognosis of Ewing’s sarcoma
Prognosis in localized disease has improved significantly since the introduction of chemotherapy. Poor prognostic features are listed in Box 15.4. With combination chemotherapy and surgery or radiotherapy 5-year survival is 70%. With limited lung metastases and treatment with chemotherapy plus surgery or radiotherapy 5-year survival of 20–40% can be expected. With more extensive disease 5-year survival falls to less than 25%.
Chondrosarcoma
The most common sites are pelvis (Figures 15.10 and 15.11), axial skeleton and proximal limbs. As with other bone tumours axial tumours have a worse prognosis than extremity tumours. It presents with a mass which may be painful or painless or with symptoms due to local disease. Systemic symptoms are rare. Diagnosis may be suggested from plain X-rays but MRI should be undertaken for suspected sarcomas.
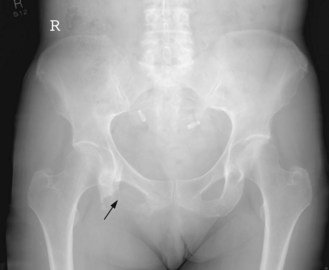
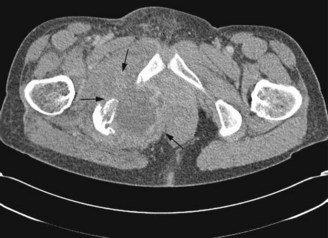
The primary modality of treatment is surgery. Chemotherapy and radiotherapy have limited role except for palliation.
Soft tissue sarcomas
Aetiology
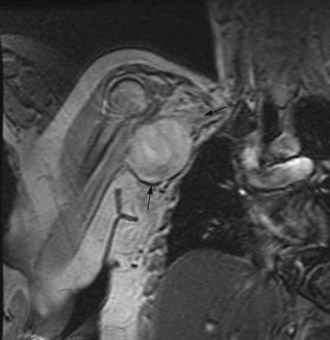
The majority of cases arise de novo. Known predisposing factors include familial cancer syndromes, prior radiotherapy and/or chemotherapy, chronic lymphoedema or infection. Familial cancer syndromes include Li–Fraumeni, hereditary retinoblastoma and familial GIST tumours. Malignant peripheral nerve sheath tumours (MPNST) are more common in neurofibromatosis due to inherited mutations in NF1 associated with a 10% lifetime risk of MPNST (Figure 15.12). Desmoid tumours and aggressive fibromatosis occur in those with APC gene mutations associated with familial adenomatous polyposis.
Pathology
There are over 80 subtypes of soft tissue sarcoma. The most common are leiomyosarcoma, liposarcoma, synovial sarcoma, rhabdomyosarcoma, fibrosarcomas (several subtypes), and malignant peripheral nerve sheath tumour. Many soft tissue sarcomas are characterized and classified according to specific translocations and gene rearrangements (Box 15.6).
Box 15.6
Examples of soft tissue sarcomas which have characteristic chromosomal translocations
Translocations associated with sarcomas
| Translocation | Genes | Type of fusion gene |
|---|---|---|
| Ewing’s family of tumours | ||
| t(11;22)(q24;q12) | EWSR1-FLI1 | Transcription factor |
| t(21;22)(q22;q12) | EWSR1-ERG | Transcription factor |
| t(7;22)(p22;q12) | EWSR1-ETV1 | Transcription factor |
| t(17;22)(q21;q12) | EWSR1-ETV4 | Transcription factor |
| t(2;22)(q33;q12) | EWSR1-FEV | Transcription factor |
| Clear-cell sarcoma | ||
| t(12;22)(q13;q12) | EWS-ATF1 | Transcription factor |
| Desmoplastic small round-cell tumour | ||
| t(11;22)(p13;q12) | EWSR1-WT1 | Transcription factor |
| Myxoid chondrosarcoma | ||
| t(9;22)(q22-31;q11-12) | EWSR1-NR4A3 | Transcription factor |
| Myxoid liposarcoma | ||
| t(12;16)(q13;p11) | FUS-CHOP | Transcription factor |
| t(12;22)(q13;q12) | EWSR1-CHOP | Transcription factor |
| Alveolar rhabdomyosarcoma | ||
| t(2;13)(q35;q14) | PAX3-FKHR | Transcription factor |
| t(1;13)(p36;q14) | PAX7-FKHR | Transcription factor |
| Synovial sarcoma | ||
| t(X;18)(p11;q11) | SYT-SSX | Transcription factor |
Staging
Staging should include evaluation of the primary tumour and a CT of the chest to exclude lung metastases. In certain rare subgroups (rhabdomyosarcoma, synovial sarcoma, clear cell and epithelioid sarcomas) nodal disease may occur so regional CT scanning may be appropriate. The role of bone scintigraphy is less established in soft tissue sarcomas than bone sarcomas. PET has not been fully evaluated (except for GIST) in soft tissue sarcomas. Several staging systems exist, one of the most frequently used is AJCC (Table 15.2) which does not include histology and site, and should not be used for non-extremity sarcomas.
Table 15.2 AJCC staging system for soft tissue sarcomas. This uses four criteria of tumour size, nodal status, grade, and metastasis (TNGM)
| Grade and TNM definitions | |
|---|---|
| Tumour grade (G) | |
| GX | Grade cannot be assessed |
| G1 | Well differentiated |
| G2 | Moderately differentiated |
| G3 | Poorly differentiated |
| G4 | Poorly differentiated or undifferentiated |
| Primary tumour (T) | |
| TX | Primary tumour cannot be assessed |
| T0 | No evidence of primary tumour |
| T1 | Tumour 5 cm or less in greatest dimension: |
General management principles of soft tissue sarcoma
The overall management is shown in Figure 15.13.
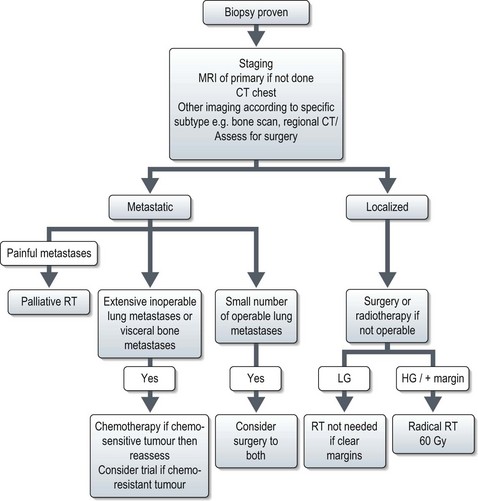
Localized disease
Radiotherapy
Surgery is the primary curative modality for soft tissue sarcomas; radiotherapy has a significant role in the prevention and treatment of local recurrence although there is no evidence that radiotherapy improves overall survival. Many soft tissue sarcomas are radiosensitive. A dose of >60 Gy is needed and the radiotherapy details are shown in Box 15.5.
Preoperative and postoperative radiotherapy
Radiotherapy is usually planned postoperatively in two phases to take into account possible tumour spillage at the edge of the surgical procedure then to focus on the central tumour bed. Planning must take into account preoperative scans to estimate tumour location, but should also encompass the surgical edges. Preoperative radiotherapy allows a single phase approach. An ongoing trial, VORTEX, is examining the possibility of reducing the volume of the postoperative treatment comparing a single phase treatment with a standard 2-phase approach.
Adjuvant chemotherapy
The use of adjuvant chemotherapy in soft tissue sarcomas remains controversial and is compounded by different histological subtypes of sarcoma with varied chemo-sensitivities being included as a group in the majority of studies. There are some sarcomas, which tend to predominate in the paediatric setting, in which adjuvant treatment has been shown to be useful, e.g. rhabdomyosarcomas and the extraskeletal Ewing’s sarcoma. However, for the majority there is no consensus concerning the use of adjuvant chemotherapy (Box 15.7). A meta-analysis showed a statistically significant improvement in local and distant recurrence if adjuvant chemotherapy was given. There was a trend towards an improved overall survival of 4% at 10 years, but this was not statistically significant. A multi-centre international EORTC study randomized 351 patients to standard treatment versus ifosfamide with doxorubicin. A preliminary analysis has shown no statistically significant difference between the two groups in either local recurrence rates or overall survival.
Treatment recommendations
Surgical resection with wide margins remains the mainstay of treatment for soft tissue sarcoma. This should be performed by specialist sarcoma surgeons. Radiotherapy is recommended for the majority of cases except low-grade tumours which have been excised with a wide margin. Radiotherapy is indicated for low grade tumours with involved resection margin when further surgery is not contemplated. The role of adjuvant chemotherapy for most subgroups remains controversial but could be considered within the context of a clinical trial (Box 15.8).
Advanced and metastatic soft tissue sarcomas
Many sarcomas present with locally advanced or metastatic disease. In the majority of cases this presentation carries a very poor prognosis. Prognostic factors are listed in Box 15.9. The median survival with metastatic disease is 12–14 months.
Box 15.9
Prognostic factors in soft tissue sarcomas
Different prognostic factors predict for local and systemic recurrence:
Chemotherapy
Systemic treatment with chemotherapy can be useful but its role is more complex than in other malignancies. There is little evidence that chemotherapy prolongs survival but it can provide significant benefits in symptom control. Response rates as determined by RECIST criteria often do not reflect symptomatic benefit or disease control, especially in response to certain agents such as trabectedin. Different tumour types show different responses to chemotherapy in general and to specific drugs (Box 15.10).
Box 15.10
Specific chemotherapy and histology
Recent studies have shown that some chemotherapy agents are more effective in some tumours:
Doxorubicin is a standard first agent with response rates of 20–25%. Doses below 60 mg/m2 have been shown to be ineffective and above 75 mg/m2 to have increased toxicity without clinical benefit. Ifosfamide is often used as a second line agent and has a response rate of 25% overall. In general combination chemotherapy has been shown to improve response rates up to 46% but with no improvement in PFS or overall survival and with increased toxicity. Combination chemotherapy does have a role in the childhood and adolescent tumours such as rhabdomyosarcoma and extraskeletal Ewing’s sarcoma (Box 15.11).
Surgery
Despite the poor median overall survival with metastatic disease a small group of patients have a prolonged survival most of whom have disease which is amenable to surgery, e.g. lung metastases or isolated intra-abdominal recurrence. No randomized trial has evaluated the benefit of surgical resection of lung metastases but the 5-year survival of patients selected using strict criteria (Box 15.12) is up to 40%.
Isolated limb perfusion
Some patients present with a massive inoperable limb sarcoma, often accompanied by significant symptoms, and too large to be encompassed within a radiotherapy field. For some there may be an option of isolated limb perfusion therapy (ILP) with melphalan and recombinant TNF-alpha within specialist centres.
Specific soft tissue sarcoma subtypes
Liposarcoma
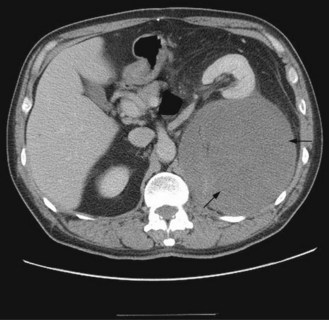
Dedifferentiated liposarcomas usually occur in the retroperitoneum or abdomen (Figure 15.14). They are high-grade, chemoresistant tumours. They have characteristic genetics with abnormalities in CDK4/MDM2.
Uterine sarcomas
The most common histologies are leiomyosarcoma (LMS) and endometrial stromal sarcoma (ESS). Many are found unexpectedly at the time of hysterectomy. A significant proportion of uterine sarcomas express oestrogen (ER) or progesterone (PgR) receptors. Staging is currently according to the FIGO staging system. In ESS pelvic recurrence is more common although distant relapses to the lungs can occur. In LMS pelvic and extra pelvic recurrences can occur. Extra pelvic metastases in LMS can occur in the liver, lung, lymph nodes and bone. Extra pelvic relapse has been shown to be more frequent in those who have had adjuvant radiotherapy.
Retroperitoneal sarcoma
Retroperitoneal sarcomas represent 13% soft tissue sarcomas. The most common histologies at this site are liposarcoma (Figure 15.14) and leiomyosarcoma. Many are found incidentally. When they occur symptoms are due to a large mass (abdominal swelling, leg swelling distal to the mass, pain).
Gastrointestinal stromal tumour (GIST)
The most common sites are stomach (50%) (Figure 15.15) and small bowel (25%) but they can occur in the colon (10%), and other sites. Presentation is commonly with anaemia or abdominal mass but abdominal pain can occur with larger tumours. Patterns of spread include liver metastases (most common) and peritoneal metastases. Bone metastases occur rarely and late, often after years of treatment. Lung metastases are exceptionally rare. Nodal metastases are also uncommon.
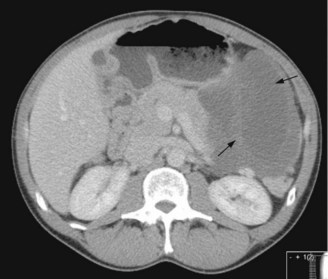
Figure 15.15 Large gastric GIST (shown by arrows) arising from the greater curvature of the stomach.
Pathology and genetics
The malignant potential of GISTs has been shown to be related to size of the tumour, mitotic rate and location of the primary as described in the NIH consensus criteria (Table 15.3 and Box 15.13).
Table 15.3 Risk of aggressive behaviour in GISTs according to NIH consensus criteria
| Risk | Size of tumour | Mitotic count |
|---|---|---|
| Very low risk | <2 cm | <5 per 50 hpf* |
| Low risk | 2–5 cm | <5 per 50 hpf |
| <5 cm | 6–10 per 50 hpf | |
| Intermediate risk | 5–10 cm | <5 per 50 hpf |
| >5 cm | >5 per 50 hpf | |
| High risk | >10 cm | Any mitotic rate |
| Any size | >10 per 50 hpf |
Management
Definitive curative treatment is only possible with resection of the primary tumour in the absence of metastases. High risk (and intermediate risk) tumours are at risk of relapse after a clear resection. Several trials are examining the use of the drug imatinib in the adjuvant setting. Imatinib is an oral tyrosine kinase inhibitor (TKI) which blocks signalling via KIT by binding to the ATP-binding pocket which is essential for phosphorylation and activation of the c-KIT receptor (p. 370 and Box 15.14). Patients with inoperable primary tumours who do not have metastases may be rendered operable by the use of imatinib. Patients with metastatic disease should be treated initially with imatinib 400 mg per day.
Box 15.14
Location of mutations and response to TKIs
At progression dose escalation to 800 mg per day may enable temporary control of disease in 30–35% cases with a median time to progression of 4 months. For patients with an isolated liver metastasis who have responded to imatinib, local hepatic resection or radiofrequency ablation may be considered.
Federman N, Bernthal N, Eilber FC, Tap WD. The multidisciplinary management of osteosarcoma. Curr Treat Options Oncol. 2009;10:82-93.
Ferrari S, Palmerini E. Adjuvant and neoadjuvant combination chemotherapy for osteogenic sarcoma. Curr Opin Oncol. 2007;19:341-346.
Balamuth NJ, Womer RB. Ewing’s sarcoma. Lancet Oncol. 2010;11:184-192.
Riedel RF, Larrier N, Dodd L, et al. The clinical management of chondrosarcoma. Curr Treat Options Oncol. 2009;10:94-106.
Reynoso D, Subbiah V, Trent JC, et al. Neoadjuvant treatment of soft-tissue sarcoma: a multimodality approach. J Surg Oncol. 2010;101:327-333.
Katz SC, Brennan MF. Randomized clinical trials in soft tissue sarcoma. Surg Oncol Clin N Am. 2010;19:1-11.
Verweij J. Soft tissue sarcoma trials: one size no longer fits all. J Clin Oncol. 2009;27:3085-3087.
Judson I. State-of-the-art approach in selective curable tumours: soft tissue sarcoma. Ann Oncol. 2008;19(Suppl 7):vii166-169.
Nam JH, Park JY. Update on treatment of uterine sarcoma. Curr Opin Obstet Gynecol. 2010;22:36-42.
Katz MH, Choi EA, Pollock RE. Current concepts in multimodality therapy for retroperitoneal sarcoma. Expert Rev Anticancer Ther. 2007;7:159-168.
Rubin BP, Heinrich MC, Corless CL. Gastrointestinal stromal tumour. Lancet. 2007;369:1731-1741.