Calmodulin and CaMKII as Ca2+ Switches for Cardiac Ion Channels
Abstract
Changes in intracellular Ca2+ are among the most diverse and important means of cell signaling. In the heart, signaling pathways from excitation-contraction coupling to humoral activation of hypertrophic responses all rely on changes in the concentration of intracellular Ca2+. Ion channels belong to a class of signaling proteins that is particularly sensitive to a change in intracellular Ca2+, which is the final signal of all coordinated ion channel activity. Thus, regulation of ion channel function by Ca2+ provides an essential feedback mechanism for cellular electrical activity. Two intracellular proteins activated by Ca2+, the ubiquitous Ca2+ binding protein calmodulin (CaM), and the Ca2+/CaM-dependent protein kinase CaMKII, dictate most of the actions of intracellular Ca2+ on cardiac ion channels. In this chapter, we highlight key roles for CaM and CaMKII in cardiac myocytes as downstream integrators of Ca2+ signals in the regulation of cardiac ion channels and consequent actions on myocyte excitability.
Ca2+: The Final Signal of Electrical Activity and Regulator of Ion Channels
Unique Features of Ca2+ as a Signaling Ion in Myocytes
Translating Changes in Ca2+ into a Cellular Response: CaM as the Prototypical Cardiac Ca2+ Sensor
CaM Effector Functions on Channels: Direct Binding and Indirect Actions through CaMKII
Calmodulin Regulation of Cardiac Channel Gating
CaMKII Regulation of Cardiac Channels: Indirect Regulation by CaM
Translating Changes in Ca2+ Into a Cellular Response: CaM as the Prototypical Cardiac Ca2+ Sensor
Several Ca2+ binding proteins serve as the Ca2+ sensors to translate changes in intracellular Ca2+ into cellular actions. For example, Ca2+ binding to troponin C causes dissociation of the troponin complex from the active site on actin, allowing myosin interaction and force generation. For regulation of ion channels, the best-characterized Ca2+ sensor is the ubiquitous Ca2+-binding protein CaM, a 16.8 kDa protein that binds four moles of Ca2+ per mole of protein. CaM is highly abundant in cardiac myocytes, but more than 98% CaM is apoCaM (Ca2+-free) sequestered by binding proteins only to be released upon a significant increase in [Ca2+]i.1 The affinity of CaM for Ca2+ varies significantly depending on whether CaM is free in solution or bound in its apo-state to a target protein, such as an ion channel. Ca2+ binding by CaM occurs in the context of 1 to 2 mM intracellular Mg2+, the major competing divalent cation. The structural motif in CaM capable of distinguishing Ca2+ at levels less than 1 : 1000 of Mg2+ is the “EF hand,” a helix-loop-helix domain also found in many other Ca2+-binding proteins, including troponin C, in which it was originally identified. CaM has two EF hands in an N-terminal lobular domain and two more in a C-terminal lobular domain. The two Ca2+-binding domains are connected by an α-helical segment. Upon Ca2+ binding, CaM undergoes significant conformational changes that expose a hydrophobic surface, which can then interact with target proteins in a Ca2+-dependent manner. Within most well-characterized target proteins, such as CaMKII, the CaM interaction domain is an amphipathic helix for which the hydrophobic amino acid side chains become buried within the hydrophobic surface exposed in Ca2+-saturated CaM. In CaMKII, this amphipathic helix blocks access to the kinase’s constitutively active site; CaM binding to this autoinhibitory domain reveals the active site and thereby endows the kinase with a Ca2+-dependent response. In many of the cardiac ion channels to which CaM binds directly, the CaM binding motif has a similar amphipathic pattern. The actions of CaM on the channels, however, are less well understood, and this will be discussed next.
CaM Effector Functions on Channels: Direct Binding and Indirect Actions Through CaMKII
Reactive oxygen species provide an additional means to activate CaMKII. Oxidation of two methionine residues, adjacent to the site of transphosphorylation, endows CaMKII with Ca2+-independent activity in a manner similar to transphosphorylation.2 As with transphosphorylation, Ca2+/CaM binding must occur first, exposing the target methionines to oxidation. Given the association of CaMKII with several adverse cardiac outcomes, including arrhythmias, and an increased redox state with those outcomes, this means of activating CaMKII may have important detrimental effects on cardiac ion channels.
Calmodulin Regulation of Cardiac Channel Gating
CaM Regulation of Ca2+-Dependent Inactivation and Membrane Targeting of CaV1.2 Ca2+ Channels
Ca2+-dependent inactivation (CDI) of Ca2+ channels serves as a classic example of Ca2+/CaM regulation of ion channel function (Figure 19-1). CDI denotes the accelerated channel inactivation seen in experiments in which Ca2+ is used as the charge carrier rather than another permeant divalent cation, such as Ba2+. That the permeant ion regulates channel gating sets CaV1.2 apart from other voltage-gated cardiac channels, in which gating is solely voltage-dependent. CDI of CaV1.2 in myocytes is critical for regulating Ca2+ entry and for controlling the length of the plateau phase of the cardiac action potential. CaM, bound to the “IQ” motif in the C-terminus of the CaV1.2 pore-forming α1C-subunit, serves as the Ca2+ sensor for CDI of CaV1.2. Interaction between CaM and the IQ motif appears essential: homozygous mice with a knock-in mutation in the IQ motif that disrupted CaM interaction died during embryogenesis.3 Adult mice with an IQ motif mutation, obtained with an inducible Cre-recombinase strategy, died within three weeks after inducing the mutant allele.
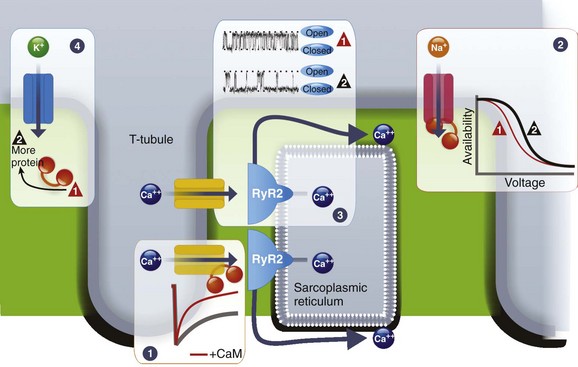
Figure 19-1 CaM regulation of cardiac ion channels. CaM controls Ca2+-dependent inactivation of CaV1.2 L-type Ca2+ channels (1), increases availability of NaV1.5 Na+ channels (2), decreases open probability of RyR2 receptors (3), and increases the abundance of KCNQ1 K+ channels (4). The traces for RyR2 were adapted from Yamaguchi et al.54
An interesting phenotype observed in these mice was a reduction in the number of channels, as indicated by reduced CaV1.2 Ca2+ current density and α1C protein. This finding confirmed previous reports showing that CaM interaction with the α1C IQ motif regulates trafficking of the α1C protein to the plasma membrane.4 Thus, regulation of channel biosynthesis demonstrates another means by which Ca2+, via CaM, can affect cellular electrical activity. Although CaM control of channel biosynthesis is best studied for CaV1.2 Ca2+ channels, CaM appears to play a similar role for other cardiac channels, such as the KCNQ1 K+ channel and the NaV1.5 Na+ channel, as discussed next.
Recent data suggest that CaM interaction with the α1C N-terminus also contributes to CDI.5 The mechanisms by which CaM accelerates channel inactivation, and whether the CaM bound to the α1C C-terminus is the same molecule bound to the N-terminus, are not clear.
Crystal structures that identify putative interaction residues for CaM on the α1C C-terminus6,7 provide a framework for future investigations. These models show an unexpected dimerization of two α1C C-termini through simultaneous binding of multiple CaMs to the two C-termini. In cardiac myocytes, the stoichiometry of CaM with the α1C C-terminus is not known, but experimental evidence from a heterologous expression system suggests that a single CaM can regulate CDI.8 Moreover, these structural models do not account for how any of these CaMs could also bind to the α1C N-terminus. Therefore, these models might not accurately reflect how CaM interacts with α1C within the context of the intact channel. Indeed, the dimerization of α1C C-termini has been suggested to represent a crystallographic artifact.6 On the other hand, recent work presents an intriguing possibility that native α1C subunits can multimerize through their C-termini via a CaM-dependent mechanism.7,9
CaM Regulation of NaV1.5 Na+ Channels
Ca2+ also regulates NaV1.5 Na+ channels (see Figure 19-1). Na+ channels initiate the action potential, and mutations that affect NaV1.5 Na+ channel function have been linked to multiple arrhythmogenic disorders, most prominently long QT syndrome and Brugada syndrome. Elevated Ca2+ increases channel availability,10 but whether CaM affects inactivation of NaV1.5 in myocytes has not yet been demonstrated. Moreover, the mechanism by which Ca2+ affects NaV1.5 Na+ channel function is not clear. As with CaV1.2 Ca2+ channels, CaM binds to an IQ motif in the NaV1.5 Na+ channel’s C-terminus, and thus one candidate Ca2+ sensor is this CaM. Mutations in NaV1.5 designed to disrupt CaM binding to the IQ motif decrease channel availability (hyperpolarize steady-state inactivation) when studied in heterologous systems.10 A recent crystal structure shows how, in the absence of Ca2+, the CaM’s C-lobe wraps around the NaV1.5 Na+ channel’s IQ motif.11 As additional structures with Ca2+/CaM are reported, critical insights into the actions of CaM on NaV1.5 are likely to be revealed. CaM can also bind to a channel’s putative inactivation gate, the III-IV intracellular linker,12 and a Ca2+-dependent complex containing the NaV1.5 C-terminus, CaM, and the III-IV can be formed,13 suggesting a model by which Ca2+/CaM can affect channel inactivation by bridging the channel’s C-terminus and the III-IV linker. Interestingly, an x-ray crystal structure presented evidence for an interaction between Ca2+-loaded CaM C-lobe and the channel’s III-IV intracellular linker.14 In the context of biochemical and structural data showing interaction of the apoCaM C-lobe with the channel’s C-terminus, these data suggest that Ca2+ may induce a movement of the C-lobe from the C-terminus to the III-IV linker and that the CaM N-lobe may instead bind the IQ motif to act as a bridge. Whether the CaM N-lobe interacts with the channel’s C-terminus in the presence of Ca2+ simultaneously with the CaM C-lobe binding to the III-IV linker has not been demonstrated. Understanding how Ca2+/CaM regulates NaV1.5 Na+ channels will likely be greatly aided as more structural information becomes available.
As with CaV1.2 Ca2+ channels, CaM interaction with NaV1.5 may also participate in channel biosynthesis based on analogies from data focused on noncardiac Na+ channels. For the skeletal muscle NaV1.4 and the neuronal NaV1.6 Na+ channels, mutations disrupting the interaction between CaM and the channel’s C-terminus almost completely reduced current amplitude.15 With new structural information, these results should be revisited with a specific focus on NaV1.5 Na+ channels in cardiomyocytes because of recent data showing that fibroblast growth factor homologous factors, which bind to NaV1.5 in the region adjacent to the CaM-binding IQ motif,16 help traffic NaV1.5 to the sarcolemma of cardiomyocytes.17
CaM Regulation of Voltage-gated K+ Channels
Another voltage-gated channel in the heart that contains a CaM-binding IQ motif is the KCNQ1 K+ channel, the most commonly mutated locus in long QT syndrome. KCNQ1 K+ channels control repolarization of the cardiac action potential; loss-of-function mutations hinder repolarization and lengthen the cellular action potential and the electrocardiographic QT interval, providing a basis for life-threatening arrhythmias. As with CaV1.2 Ca2+ channels and NaV1.5 Na+ channels, CaM interaction with the KCNQ1 C-terminus appears to participate in channel biosynthesis (see Figure 19-1). In contrast to CaV1.2 Ca2+ channels and NaV1.5 Na+ channels, CaM interaction with the KCNQ1 C-terminus requires two distinct helices that are separated by approximately 80 amino acids. Biochemical and functional studies implicate that CaM is essential for generating KCNQ1 K+ currents. The significance of these findings is underlined by the association between various long QT syndrome mutations and loss of CaM binding to channel’s C-terminus.18,19 How CaM is deemed necessary was elucidated by studies that implicated a role for CaM in channel trafficking to the plasma membrane rather than in channel assembly.20 Through CaM, Ca2+ also appears to participate in channel gating,18,19 but the molecular mechanisms have not yet been clarified.
CaM Regulation of Ryanodine Receptors
CaM regulates the SR Ca2+ release channel RyR2, the internal source of the Ca2+ that triggers contraction with each heartbeat (see Figure 19-1). How CaM binds to RyR2 and the consequent regulation of channel function are different from the manner described before for CaM interaction with, and regulation of, plasma membrane channels. In vitro experiments show that apoCaM and Ca2+/CaM have a single binding site on the large RyR2 cytoplasmic domain that does not resemble a consensus IQ motif.21
CaM inhibits cardiac muscle RyR2 at greater than 1 µM Ca2+ and either inhibits RyR222 or has no effect23 on RyR2 at less than 1 µM Ca2+. In single-channel measurements, CaM decreased RyR2 channel open probability by decreasing the number of channel events and increasing the duration of close times.24 In permeabilized myocytes, the addition of exogenous CaM decreased Ca2+ spark frequency.25 The physiological importance of CaM binding to RyR2 was shown using mice harboring targeted RyR2 mutations in the CaM binding site. These mutant mice demonstrated an increased ratio of heart weight to body weight and greatly reduced fractional shortening of the left ventricle and lethality at 9 to 16 days of age.26 Interestingly, in a rabbit heart failure model, less CaM coimmunoprecipitated with RyR2, despite unaltered expression.27 Taken together, altered CaM bound to RyR2 may contribute to both the enhanced SR Ca2+ leak in heart failure and to arrhythmogenesis.
CaMKII Regulation of Cardiac Channels: Indirect Regulation by CaM
CaMKII Regulation of CaV1.2 Ca2+ Channels
CaMKII regulation of CaV1.2 Ca2+ channels is a major contributor to the positive force-frequency relationship of cardiac contraction (Figure 19-2). Cardiac output, a product of stroke volume and heart rate, increases during exercise not only because of an increase in heart rate but also because each contraction produces more force. CaMKII, ideally suited to respond to changes in cardiac rhythm, phosphorylates CaV1.2 and thereby increases Ca2+ influx, a process called Ca2+-dependent facilitation. At the single-channel level, CaMKII phosphorylation induces the channel to open frequently and to remain open longer. This increase in so-called “mode 2” gating raises intracellular Ca2+ and initiates a chain of downstream events that increase Ca2+ efflux from internal stores and reduce Ca2+ reuptake in the sarcoplasmic reticulum, providing elevated levels of Ca2+ for the contractile apparatus. Although the end results on cardiac contractility are well established, detailed mechanistic insight into how CaMKII affects Ca2+ influx through CaV1.2 Ca2+ channels, where the process initiates, is lacking. The substrates for CaMKII within CaV1.2 has been proposed as Ser1512 and Ser1570 in the α1C-subunit28 or Thr498 in the auxiliary β2-subunit.29 Recent knock-in data question the role of the phosphorylation sites in the β-subunit.30 The experimental approaches (two Ser to Ala mutations in a knock-in mouse for the α1C sites; adenoviral overexpression of a Thr to Ala mutant for β2 sites) behind each of those conclusions have their limitations, however, and perhaps phosphorylation of both α1C– and β2-subunits are required for CaMKII potentiation.31 Similarly, separate studies have shown that CaMKII can bind directly to the CaV1.2 Ca2+ channel α1C– or β2-subunit.32,33 The advantages of a dedicated Ca2+-frequency detector (CaMKII) at the source of Ca2+ entry (CaV1.2) that is capable of modulating the amount of Ca2+ that enters are obvious, especially in the context of the broad array of CaMKII substrates and the myriad consequences of CaMKII activity in the heart.34 Moreover, mutation of the CaMKII binding site for either α1C or β2 has been shown to ablate Ca2+-dependent facilitation, suggesting that this association is critical. Similar to the debate about which subunit is the “true” CaMKII substrate, the answer of which subunit is the CaMKII scaffold is also not clearly defined; both α1C and β2 may be important for proper channel regulation.
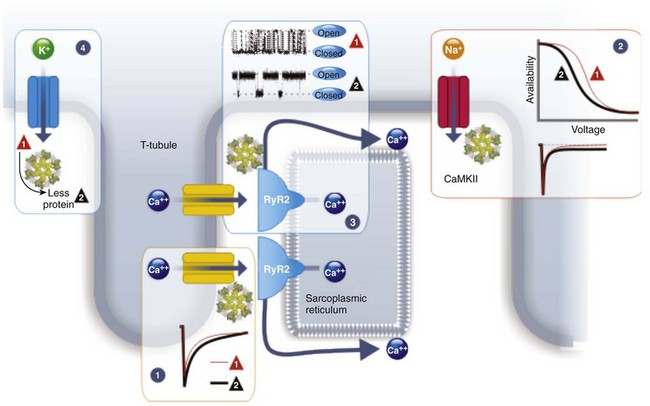
Figure 19-2 CaMKII regulation of cardiac ion channels. CaMKII elicits Ca2+-dependent facilitation of CaV1.2 L-type Ca2+ channels (1), decreases availability of NaV1.5 Na+ channels (2), increases open probability of RyR2 receptors (3), and decreases the abundance of Kv4 and Kir2.1 K+ channels (4). The traces for RyR2 were adapted from Wehrens et al.55
Although CaMKII appears to be essential for Ca2+-dependent facilitation of CaV1.2 Ca2+ channels, the CaM molecule that interacts with the channel’s C-terminus also is required. Mutations that disrupt CaM interaction, and thereby affect CDI, also abolish CaV1.2 Ca2+ channel facilitation in cardiomyocytes.3 How these two separate Ca2+ sensors cooperate to produce facilitation is not known, although it has been speculated that the CaM bound to the channel’s IQ motif may also be able to serve as the CaM that regulates the CaMKII bound to α1C or β2, providing a model that incorporates the binding of CaMKII to both subunits.35 This model is not yet supported by biochemical or structural evidence.
CaMKII Regulation of NaV1.5 Na+ Channels
CaMKII activity decreases NaV1.5 Na+ channel availability and enhances the Na+ “late” or “persistent” current in cardiomyocytes (see Figure 19-2).36 Because CaMKII protein and activity are increased in heart failure, these effects upon NaV1.5 Na+ channels may be particularly relevant for the arrhythmogenesis that accompanies heart failure. In contrast to CaV1.2 Ca2+ channels on which CaMKII binds directly, the CaMKII relevant for NaV1.5 Na+ channel modulation is instead bound to βIV-spectrin,37 an actin-associated protein that has previously been shown to cluster ion channels in neuronal axons. In the heart, βIV-spectrin localizes to the intercalated discs, where a fraction of NaV1.5 Na+ channels is enriched. CaMKII directly phosphorylates NaV1.5, but the specific sites targeted are controversial. Hund et al identified Ser571 within the channel’s I-II intracellular linker as the critical site; mutation of this Ser to Ala abolished the effects of CaMKII on channel inactivation.37 In contrast, Ashpole et al found that the CaMKII targets in the I-II linker were Ser483/Ser484, Ser516, and Thr594, and that Ala-substitution of both Ser516 and Thr594 abolished the CaMKII mediated shift in INa availability to a more negative membrane potential.38 Neither set of experiments was performed in cardiomyocytes, so the relevant sites within their endogenous milieu remain to be determined.
CaMKII Regulation of Kv4 and Kir2.1 K+ Channels
The K+ channels responsible for the resting membrane potential (IK1, mediated by Kir2.1) and for the transient outward current (Ito, which produces the “notch” in the ventricular action potential, mediated by Kv4.2 and Kv4.3) are regulated by CaMKII. Chronic increased CaMKII activity, such as during heart failure, reduced Ito current and reduced Kv4.2 and Kv4.3 protein, suggesting that CaMKII can affect channel biosynthesis (see Figure 19-2). Transcription of Kv4.3 in canine myocytes has been shown to be under the control of CaMKII by a downstream mechanism dependent on the nuclear factor of activated T cells transcription factor.39 In addition to these effects on channel transcription, CaMKII has direct effects on channel function. CaMKII phosphorylation slowed Kv4.3 channel inactivation.40 The current regulating IK was also reduced by chronic CaMKII expression and, similar to Kv4 K+ channels, the effect is through transcriptional regulation.41
CaMKII Regulation of RyR2
CaMKII regulation of RyR2 channel function plays an important role in modulating cardiac contractility and arrhythmogenesis. CaMKII is associated with the RyR2 macromolecular complex42 and phosphorylates the channel in vitro and in vivo.43–45 Although Ser2809 on canine and human RyR2 (Ser2808 in murine RyR2) was originally identified as a protein kinase A and CaMKII phosphorylation site,43,45 site-directed mutagenesis studies and knock-in mice have shown that Ser2809 is exclusively phosphorylated by protein kinase A,46 and Ser2815 (Ser2814 in murine RyR2) is phosphorylated by CaMKII.44,47,48 Other sites for CaMKII phosphorylation on RyR2 may also exist, and the identity of the relevant sites is controversial. CaMKII phosphorylation of RyR2 increases the open probability of RyR2 by sensitizing the channel to cytosolic Ca2+.44,49 CaMKII phosphorylation of RyR2 by endogenous CaMKII increases resting SR Ca2+ release by increasing Ca2+ spark frequency and duration.25 CaMKII phosphorylation of RyR2 plays an important role in mediating the positive force-frequency relationship in the heart. Mice engineered with a RyR2-S2814A mutation have RyR2 channels that cannot be phosphorylated by CaMKII and exhibit a blunted positive force-frequency relationship.47 The positive force-frequency relationship is blunted in heart failure, and in a rat model of heart failure, a rate-dependent increase in CaMKII phosphorylation of RyR2-Ser2814 was not observed.44
Increased activity of CaMKII is believed to promote heart failure progression and arrhythmogenesis, including atrial fibrillation.50,51 Heart failure and arrhythmias are associated with abnormal Ca2+ handling. What is the role of CaMKII phosphorylation of RyR2 in these processes? In patients with nonischemic cardiomyopathy, phosphorylation of RyR2-Ser2814 is increased.48 Mice engineered with a RyR2-S2814A mutation were relatively protected from heart failure development after transverse aortic constriction compared with wild type littermates. These protective effects on cardiac contractility were not observed, however, after myocardial infarction in the S2814A mice.47 The RyR2-S2814A mice are also protected from the development of atrial fibrillation51,52 and pacing-induced arrhythmias after transverse aortic constriction.53 Conversely, mice engineered with a constitutively activated CaMKII phosphorylation site (S2814D) develop sustained ventricular tachycardia and sudden cardiac death with caffeine and epinephrine or programmed electrical stimulation.53 These studies demonstrate that CaMKII phosphorylation of RyR2, although important for the positive force-frequency response, plays a role in the progression of heart failure and the development of atrial and ventricular arrhythmias.
References
1. Wu, X, Bers, DM. Free and bound intracellular calmodulin measurements in cardiac myocytes. Cell Calcium. 2007; 41(4):353–364.
2. Erickson, JR, Joiner, ML, Guan, X, et al. A dynamic pathway for calcium-independent activation of CaMKII by methionine oxidation. Cell. 2008; 133(3):462–474.
3. Poomvanicha, M, Wegener, JW, Blaich, A, et al. Facilitation and Ca2+-dependent inactivation are modified by mutation of the Cav1. 2 channel IQ motif. J Biol Chem. 2011; 286(30):26702–26707.
4. Wang, HG, George, MS, Kim, J, et al. Ca2+/calmodulin regulates trafficking of Ca(V)1. 2 Ca2+ channels in cultured hippocampal neurons. J Neurosci. 2007; 27(34):9086–9093.
5. Dick, IE, Tadross, MR, Liang, H, et al. A modular switch for spatial Ca2+ selectivity in the calmodulin regulation of CaV channels. Nature. 2008; 451(7180):830–834.
6. Kim, EY, Rumpf, CH, Van Petegem, F, et al. Multiple C-terminal tail Ca2+/CaMs regulate CaV1. 2 function but do not mediate channel dimerization. EMBO J. 2010; 29(23):3924–3938.
7. Fallon, JL, Baker, MR, Xiong, L, et al. Crystal structure of dimeric cardiac L-type calcium channel regulatory domains bridged by Ca2+⋅calmodulins. Proc Natl Acad Sci U S A. 2009; 106(13):5135–5140.
8. Mori, MX, Erickson, MG, Yue, DT. Functional stoichiometry and local enrichment of calmodulin interacting with Ca2+ channels. Science. 2004; 304(5669):432–435.
9. Navedo, MF, Cheng, EP, Yuan, C, et al. Increased coupled gating of L-type Ca2+ channels during hypertension and Timothy syndrome. Circ Res. 2010; 106(4):748–756.
10. Biswas, S, DeSilvestre, D, Tian, Y, et al. Calcium-mediated dual-mode regulation of cardiac sodium channel gating. Circ Res. 2009; 104(7):870–878.
11. Wang, C, Chung, BC, Yan, H, et al. Crystal structure of the ternary complex of a NaV C-terminal domain, a fibroblast growth factor homologous factor, and calmodulin. Structure. 2012; 20(7):1167–1176.
12. Potet, F, Chagot, B, Anghelescu, M, et al. Functional interactions between distinct sodium channel cytoplasmic domains through the action of calmodulin. J Biol Chem. 2009; 284(13):8846–8854.
13. Kim, J, Ghosh, S, Liu, H, et al. Calmodulin mediates Ca2+ sensitivity of sodium channels. J Biol Chem. 2004; 279(43):45004–45012.
14. Sarhan, MF, Tung, CC, Van Petegem, F, et al. Crystallographic basis for calcium regulation of sodium channels. Proc Natl Acad Sci. 2012; 109(9):3558–3563.
15. Herzog, RI, Liu, C, Waxman, SG, et al. Calmodulin binds to the C terminus of sodium channels Nav1. 4 and Nav1. 6 and differentially modulates their functional properties. J Neurosci. 2003; 23(23):8261–8270.
16. Wang, C, Wang, C, Hoch, EG, et al. Identification of novel interaction sites that determine specificity between fibroblast growth factor homologous factors and voltage-gated sodium channels. J Biol Chem. 2011; 286(27):24253–24263.
17. Wang, C, Hennessey, JA, Kirkton, RD, et al. Fibroblast growth factor homologous factor 13 regulates Na+ channels and conduction velocity in murine hearts/novelty and significance. Circ Res. 2011; 109(7):775–782.
18. Shamgar, L, Ma, L, Schmitt, N, et al. Calmodulin is essential for cardiac IKS channel gating and assembly: Impaired function in long-QT mutations. Circ Res. 2006; 98(8):1055–1063.
19. Ghosh, S, Nunziato, DA, Pitt, GS. KCNQ1 assembly and function is blocked by long-QT syndrome mutations that disrupt interaction with calmodulin. Circ Res. 2006; 98(8):1048–1054.
20. Wiener, R, Haitin, Y, Shamgar, L, et al. The KCNQ1 (Kv7. 1) COOH terminus, a multitiered scaffold for subunit assembly and protein interaction. J Biol Chem. 2008; 283(9):5815–5830.
21. Balshaw, DM, Xu, L, Yamaguchi, N, et al. Calmodulin binding and inhibition of cardiac muscle calcium release channel (ryanodine receptor). J Biol Chem. 2001; 276(23):20144–20153.
23. Fruen, BR, Bardy, JM, Byrem, TM, et al. Differential Ca(2+) sensitivity of skeletal and cardiac muscle ryanodine receptors in the presence of calmodulin. Am J Physiol Cell physiol. 2000; 279(3):C724–C733.
24. Xu, L, Meissner, G. Mechanism of calmodulin inhibition of cardiac sarcoplasmic reticulum Ca2+ release channel (ryanodine receptor). Biophys J. 2004; 86(2):797–804.
25. Guo, T, Zhang, T, Mestril, R. Bers DM. Ca2+/Calmodulin-dependent protein kinase II phosphorylation of ryanodine receptor does affect calcium sparks in mouse ventricular myocytes. Circ Res. 2006; 99(4):398–406.
26. Yamaguchi, N, Takahashi, N, Xu, L, et al. Early cardiac hypertrophy in mice with impaired calmodulin regulation of cardiac muscle Ca release channel. J Clin Invest. 2007; 117(5):1344–1353.
27. Ai, X, Curran, JW, Shannon, TR, et al. Ca2+/calmodulin-dependent protein kinase modulates cardiac ryanodine receptor phosphorylation and sarcoplasmic reticulum Ca2+ leak in heart failure. Circ Res. 2005; 97(12):1314–1322.
28. Blaich, A, Welling, A, Fischer, S, et al. Facilitation of murine cardiac L-type Cav1. 2 channel is modulated by calmodulin kinase II-dependent phosphorylation of S1512 and S1570. Proc Natl Acad Sci U S A. 2010; 107(22):10285–10289.
29. Grueter, CE, Abiria, SA, Dzhura, I, et al. L-type Ca2+ channel facilitation mediated by phosphorylation of the beta subunit by CaMKII. Mol Cell. 2006; 23(5):641–650.
30. Brandmayr, J, Poomvanicha, M, Domes, K, et al. Deletion of the C-terminal phosphorylation sites in the cardiac beta subunit does not affect the basic β-adrenergic response of the heart and the Cav1. 2 channel. J Biol Chem. 2012; 287(27):22584–22592.
31. Sun, AY, Pitt, GS. Pinning down the CaMKII targets in the L-type Ca2+ channel: An essential step in defining CaMKII regulation. Heart Rhythm. 2011; 8(4):631–633.
32. Koval, OM, Guan, X, Wu, Y, et al. CaV1. 2 beta-subunit coordinates CaMKII-triggered cardiomyocyte death and afterdepolarizations. Proc Natl Acad Sci U S A. 2010; 107(11):4996–5000.
33. Hudmon, A, Schulman, H, Kim, J, et al. CaMKII tethers to L-type Ca2+ channels, establishing a local and dedicated integrator of Ca2+ signals for facilitation. J Cell Biol. 2005; 171(3):537–547.
34. Erickson, JR, He, BJ, Grumbach, IM, et al. CaMKII in the cardiovascular system: Sensing redox states. Physiol Rev. 2011; 91(3):889–915.
35. Pitt, GS. Calmodulin and CaMKII as molecular switches for cardiac ion channels. Cardiovasc Res. 2007; 73(4):641–647.
36. Wagner, S, Dybkova, N, Rasenack, EC, et al. Ca2+/calmodulin-dependent protein kinase II regulates cardiac Na+ channels. J Clin Invest. 2006; 116(12):3127–3138.
37. Hund, TJ, Koval, OM, Li, J, et al. A beta(IV)-spectrin/CaMKII signaling complex is essential for membrane excitability in mice. J Clin Invest. 2010; 120(10):3508–3519.
38. Ashpole, NM, Herren, AW, Ginsburg, KS, et al. Ca2+/calmodulin-dependent protein kinase II (CaMKII) regulates cardiac sodium channel NaV1. 5 gating by multiple phosphorylation sites. J Biol Chem. 2012; 287(24):19856–19869.
39. Xiao, L, Coutu, P, Villeneuve, LR, et al. Mechanisms underlying rate-dependent remodeling of transient putward potassium current in canine ventricular myocytes. Circ Res. 2008; 103(7):733–742.
40. Sergeant, GP, Ohya, S, Reihill, JA, et al. Regulation of Kv4. 3 currents by Ca2+/calmodulin-dependent protein kinase II. Am J Physiol Cell Physiol. 2005; 288(2):C304–C313.
41. Wagner, S, Hacker, E, Grandi, E, et al. Ca/calmodulin kinase II differentially modulates potassium currents. Circ Arrhythm Electrophysiol. 2009; 2(3):285–294.
42. Maier, LS, Zhang, T, Chen, L, et al. Transgenic CaMKIIdeltaC overexpression uniquely alters cardiac myocyte Ca2+ handling: reduced SR Ca2+ load and activated SR Ca2+ release. Circ Res. 2003; 92(8):904–911.
43. Witcher, DR, Kovacs, RJ, Schulman, H, et al. Unique phosphorylation site on the cardiac ryanodine receptor regulates calcium channel activity. J Biol Chem. 1991; 266(17):11144–11152.
44. Wehrens, XH, Lehnart, SE, Reiken, SR, et al. Ca2+/calmodulin-dependent protein kinase II phosphorylation regulates the cardiac ryanodine receptor. Circ Res. 2004; 94(6):e61–e70.
45. Rodriguez, P, Bhogal, MS, Colyer, J. Stoichiometric phosphorylation of cardiac ryanodine receptor on serine 2809 by calmodulin-dependent kinase II and protein kinase A. J Biol Chem. 2003; 278(40):38593–38600.
46. Wehrens, XH, Lehnart, SE, Reiken, S, et al. Ryanodine receptor/calcium release channel PKA phosphorylation: a critical mediator of heart failure progression. Proc Natl Acad Sci U S A. 2006; 103(3):511–518.
47. Kushnir, A, Shan, J, Betzenhauser, MJ, et al. Role of CaMKIIdelta phosphorylation of the cardiac ryanodine receptor in the force frequency relationship and heart failure. Proc Natl Acad Sci U S A. 2010; 107(22):10274–10279.
48. Respress, JL, van Oort, RJ, Li, N, et al. Role of RyR2 phosphorylation at S2814 during heart failure progression. Circ Res. 2012; 110(11):1474–1483.
49. Hain, J, Onoue, H, Mayrleitner, M, et al. Phosphorylation modulates the function of the calcium release channel of sarcoplasmic reticulum from cardiac muscle. J Biol Chem. 1995; 270(5):2074–2081.
50. Anderson, ME, Brown, JH, Bers, DM. CaMKII in myocardial hypertrophy and heart failure. J Mol Cell Cardiol. 2011; 51(4):468–473.
51. Chelu, MG, Sarma, S, Sood, S, et al. Calmodulin kinase II-mediated sarcoplasmic reticulum Ca2+ leak promotes atrial fibrillation in mice. J Clin Invest. 2009; 119(7):1940–1951.
52. Li, N, Wang, T, Wang, W, et al. Inhibition of CaMKII phosphorylation of RyR2 prevents induction of atrial fibrillation in FKBP12. 6 knockout mice. Circ Res. 2012; 110(3):465–470.
53. van Oort, RJ, McCauley, MD, Dixit, SS, et al. Ryanodine receptor phosphorylation by calcium/calmodulin-dependent protein kinase II promotes life-threatening ventricular arrhythmias in mice with heart failure. Circulation. 2010; 122(25):2669–2679.
54. Yamaguchi, N, Takahashi, N, Xu, L, et al. Early cardiac hypertrophy in mice with impaired calmodulin regulation of cardiac muscle Ca2+ release channel. J Clin Invest. 2007; 117(5):1344–1353.
55. Wehrens, XHT, Lehnart, SE, Reiken, SR, et al. Ca2+/calmodulin-dependent protein kinase II phosphorylation regulates the cardiac ryanodine receptor. Circ Res. 2004; 94(6):e61–e70.