Calcium Channels in the Heart
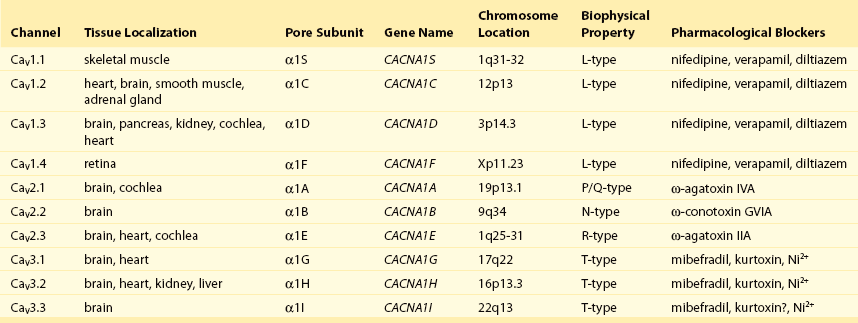
Calcium (Ca2+) permeable ion channels are involved in a number of fundamental processes in the heart, including automaticity in the sinoatrial (SAN) and atrioventricular (AVN) nodes, excitation-contraction coupling in the working myocardium, and regulation of gene expression in hypertrophic signaling.1–4 Several ion channels in the plasma membrane of cardiac myocytes and cardiac fibroblasts are involved in transporting Ca2+ into the cytosol from the extracellular space (Table 2-1). Included among these are two forms of L-type Ca2+ channels (LTCC; CaV1.2 and CaV1.3), T-type Ca2+ channels, and several transient receptor potential (TRP) channels. These different Ca2+ permeable ion channels play distinct roles in different parts of the heart. For example, CaV1.2-mediated L-type Ca2+ current (ICa,L) is present in all cardiomyocytes, whereas CaV1.3-mediated ICa,L is restricted to SAN, AVN, and working atrial myocardium. T-type Ca2+ currents (ICa,T) are mainly expressed in the SAN and AVN in normal conditions, but may be expressed in ventricular myocytes in cardiac hypertrophy. In most cases, TRP channels conduct nonselective cation currents that include Ca2+ influx, which may be important in both cardiac myocytes and cardiac fibroblasts, the latter of which don’t typically express robust voltage-gated Ca2+ channels. The purpose of this chapter is to review the expression patterns, biophysical properties, and structure-function relationships of Ca2+ permeable channels in the heart.
Table 2-1
Nomenclature of the Voltage-Gated Ca2+ Channels Along With Chromosome Location and Biophysical and Pharmacological Properties
L-type Ca2+ Channels
Molecular Composition
LTCCs are multimeric proteins consisting of an α1-subunit that constitutes the pore of the channel and several accessory subunits denoted β, α2-δ, and γ2,5,6 (Figure 2-1). Currently, four α1-subunits for L-type Ca2+ channels are known, and two of these, CaV1.2 (α1C encoded by the CACNA1C gene) and CaV1.3 (α1D encoded by the CACNA1D gene), are expressed in the heart3,5,7 (see Table 2-1). These α1-subunits form the channel pore and contain the voltage sensor that controls channel gating, as well as drug binding sites and regulatory sites targeted by second messengers. Ca2+ channel α1-subunits have a similar structure to voltage-gated Na+ and K+ channels, whereby they are organized into four domains (I through IV), each containing six transmembrane segments (S1 through S6). The voltage sensor is located in the S4 transmembrane segment of each domain, and the pore loops are located between S5 and S6. Alternative splice variants of CaV1.2 have been documented and can impact regulation by second messengers and drug binding. Recently, alternative splice variants of CaV1.3 have also been described.8,9 These variants result in the expression of long and short forms of CaV1.3, which are characterized by differences in biophysical properties such as the activation midpoint; however, not all of the short forms of CaV1.3 are expressed in the heart.8
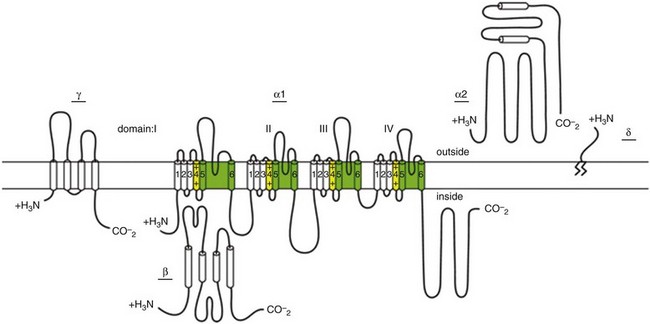
Figure 2-1 Ca2+ channel subunit structure. L-type Ca2+ channels consist of the pore-forming α1-subunit and a series of accessory subunits (β, γ, α2δ). Predicted α-helices are depicted as cylinders. The lengths of the lines correlate with the approximate length of the polypeptide segments. T-type Ca2+ channels consist of the pore-forming α1-subunit; however, whether accessory subunits associate with T-type Ca2+ channels in native cardiomyocytes is less clear. (From Catterall W: Voltage-gated calcium channels. Cold Spring Harb Perspect Biol 3:a003947, 2011.)
Ca2+ channel β-subunits (CaVβ1 through CaVβ4) are encoded by four genes (CACNB1-4) and, like the α1-subunits, can be alternatively spliced to generate a number of isoforms.10,11 Crystal structures of LTCC β-subunits demonstrate that these subunits contain conserved Src homology 3 (SH3) and guanylate kinase domains, as seen in scaffolding proteins in the membrane-associated guanylate kinase (MAGUK) family.12,13 CaVβ-subunits are intracellular proteins (see Figure 2-1) that bind to a single site, called the α interaction domain (AID), on the loop connecting domains I and II on the α1-subunit of the LTCC. CaVβ acts as a chaperone protein that facilitates the trafficking of LTCCs to the plasma membrane and also modulates the biophysical properties of these channels. Specifically, coexpression of CaVβ2 with CaV1.2 increases ICa,L amplitude, accelerates activation and inactivation kinetics, and shifts the steady inactivation curve to more negative membrane potentials. CaVβ also profoundly enhances the affinity of dihydropyridines (blockers of LTCCs) for the α1-subunit by decreasing their dissociation from the channel.14
Four genes (CACNA2D1-4), which can be alternatively spliced, encode the α2-δ-subunit of the LTCC.15,16 The α2-δ1 and α2-δ3 isoforms are expressed in the heart. The protein is cleaved post-translationally and then is relinked via disulfide interactions. This subunit is primarily extracellular, with the δ-subunit portion anchored in the plasma membrane (see Figure 2-1). CaVα2-δ-subunits facilitate the targeting of LTCCs to the plasma membrane and increase ICa,L by shifting the voltage dependence of activation and inactivation to hyperpolarized membrane potentials.
Ca2+ channel γ-subunits are membrane-bound proteins encoded by eight genes (CACNG1-8) with several isoforms, including γ4, γ6, γ7, and γ8, which are present in cardiac muscle. These isoforms associate with CaV1.2 and alter activation and inactivation properties of the channel.17
Biophysical Properties of ICa,L
As their name suggests, LTCCs are highly selective for Ca2+ over monovalent cations. The channel pore is approximately 6 Å in diameter at its narrowest point,18 indicating that size is not the main factor in determining selectivity. Rather, a series of four glutamate residues (EEEE motif) is responsible for conferring ion selectivity through the ability of this motif to bind divalent cations (Ca2+ and Mg2+), which block the passage of monovalent cations.19,20 As additional divalent ions enter the channel, bound Ca2+ ions are displaced from the EEEE motif and are pushed through the pore, which generates ICa,L.
At the single-channel level, ICa,L conductance in ventricular myocytes (i.e., CaV1.2 dependent) has been reported to be approximately 5 pS in 2 mM Ca2+ and approximately 15 pS with Ba2+ as the charge carrier.21 Similarly, single-channel conductance for CaV1.3 has been determined in heterologous expression systems and has been found to be approximately 15 pS with Ba2+ as the charge carrier.8
ICa,L is mediated by CaV1.2 and CaV1.3 in the heart, and these two channel isoforms are distinguished by their unique biophysical properties7,22 (Figure 2-2). CaV1.2-mediated ICa,L, which is expressed throughout the myocardium (atrium, ventricles, and conduction system), has a bell-shaped current-voltage (I-V) relationship in which the current activates at membrane potentials positive to −40 mV and peaks between 0 and +10 mV. The V1/2 of channel activation (V1/2(act) ) is typically between −10 and −15 mV. In contrast, recombinant CaV1.3–dependent ICa,L activates at more negative membrane potentials (i.e., the V1/2(act) is hyperpolarized) and displays slower inactivation kinetics.8,23,24 CaV1.3 is also less sensitive to dihydropyridines than is CaV1.2.
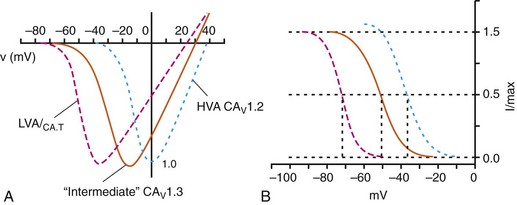
Figure 2-2 Voltage dependence of activation and steady state inactivation of ICa,T, CaV1.3-mediated ICa,L, and CaV1.2-mediated ICa,L from mouse SAN. A, Current-voltage (I-V) relationships illustrating that ICa,T activates most negatively, and CaV1.2-mediated ICa,L activates most positively. CaV1.3-mediated ICa,L has intermediate activation properties. B, Steady state inactivation curves for ICa,T, CaV1.3-mediated ICa,L, and CaV1.2-mediated ICa,L. Dashed lines indicate the voltage at which 50% of channels are inactivated (V1/2(inact) ) for each Ca2+ channel. Dotted lines indicate points of complete current inactivation and full channel availability. (From Mangoni MM, Couette B, Marger L, et al: Voltage-dependent calcium channels and cardiac pacemaker activity: from ionic currents to genes. Prog Biophys Mol Biol 90:38, 2006.)
CaV1.2 is the primary determinant of ICa,L in the ventricular myocardium, where it plays a prominent role in excitation-contraction coupling and the Ca2+ induced–Ca2+ release process.1 Approximately 75% of these LTCCs are located in dyads, in close proximity to the ryanodine receptors located in the junctional sarcoplasmic reticulum, within the t-tubular system (see Figure 2-4). Consistent with the critical role of CaV1.2 in the ventricles, CaV1.2 knockout mice die before birth in association with cardiac failure.25 CaV1.2 is also expressed in the SAN, AVN, and atrial myocardium, along with CaV1.3.22,26 As a result, total ICa,L in these supraventricular tissues is dependent on both α1C and α1D pore-forming subunits and demonstrates biophysical characteristics that are different from those of the ventricles. The generation of CaV1.3 knockout mice has been important in determining the biophysical properties of these channels and in distinguishing them from CaV1.2-mediated ICa,L (and ICa,T).23,27 Specifically, these CaV1.3 knockouts have been used to show that ICa,L in SAN and atrial myocytes activates between −60 and −50 mV in association with a left shift in the V1/2(act) and peaks at membrane potentials around −10 mV. The steady state ICa,L inactivation curve is also shifted to the left when CaV1.3 contributes to total ICa,L. Together, available data show that CaV1.3-dependent ICa,L has intermediate biophysical properties compared with CaV1.2-dependent ICa,L and ICa,T.
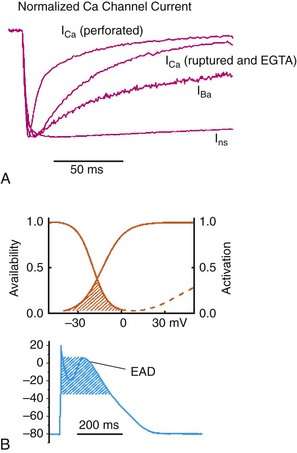
Figure 2-4 Subcellular localization of L-type Ca2+ channels in cardiomyocytes. LTCCs associate with anchoring proteins (AKAPs) and other signaling proteins to create distinct populations of LTCCs in T-tubules and within caveolae. Also, clustering of LTCCs in discrete regions of the plasma membrane results in coupled gating. See text for details. (From Best JM, Kamp TJ: Different subcellular populations of L-type Ca2+ channels exhibit unique regulation and functional roles in cardiomyocytes. J Mol Cell Cardiol 52:376-387, 2012.)
The unique biophysical properties of CaV1.3 enable ICa,L to play a prominent role in pacemaker activity in the SAN and in the electrical conduction in the atrial myocardium. In the SAN, CaV1.3-mediated ICa,L contributes to the diastolic depolarization phase of the action potential and thus is a major determinant of heart rate in vivo. CaV1.2, on the other hand, contributes more prominently to the action potential upstroke in SAN myocytes. Consistent with this, CaV1.3 knockout mice display sinus bradycardia in association with a reduced diastolic depolarization slope.26–28 CaV1.3 knockout mice also show increased susceptibility to atrial fibrillation,24,29 a very common cardiac arrhythmia, confirming that these Ca2+ channels play an important role in atrial conduction.
ICa,L Inactivation
ICa,L undergoes decay during sustained depolarization, which is termed inactivation (Figure 2-3, A). This inactivation process is time, voltage, and calcium dependent.30–32 Voltage-dependent activation is relatively slow, as is indicated by the degree of inactivation with Ba2+ as the charge carrier, or when monovalent cations permeate LTCCs in the absence of divalent cations.33 In the presence of extracellular Ca2+, inactivation is much faster, and this Ca2+-dependent inactivation is further accelerated in the presence of functional SR Ca2+ release (i.e., during normal excitation-contraction coupling).34 This indicates that both the Ca2+ entering the myocytes through the LTCC and the Ca2+ released from the SR during the Ca2+ transient contribute to LTCC inactivation.
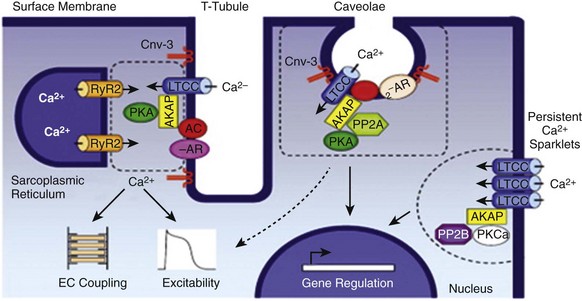
Figure 2-3 A, Ca2+ channel inactivation with Ca2+, Ba2+, or monovalent cations (ns) as the charge carrier. Currents were measured at 0 mV except for Ins at −30 mV to obtain comparable activation, and peak currents were normalized. ICa with SR Ca2+ release (i.e., Ca2+ transients occur) was recorded using the perforated patch-clamp technique and 2 mM external Ca2+. ICa with no SR Ca2+ release (i.e., no Ca2+ transients occur) was recorded in the whole-cell configuration with 10 mM EGTA in the pipette. IBa was recorded in the whole-cell configuration with 2 mM external Ba2+ and 10 mM EGTA in the pipette. Ins was measured in divalent cation-free conditions. The t1/2 of current decline progressively increases from top to bottom, illustrating that ICa inactivation is both Ca2+ and voltage dependent. B, Overlap of steady state activation and inactivation curves for ICa,L illustrates the presence of a window current (dashed lines), which may contribute to the generation of early afterdepolarizations (EADs). ICa,T can also produce window currents at more negative membrane potentials (between −80 and −40 mV). (A, From Bers DM, Perez-Reyes E: Ca channels in cardiac myocytes: structure and function in Ca influx and intracellular Ca release. Cardiovasc Res 42:339-360, 1999. B, From Benitah JP, Alvarez JL, Gomez AM: L-type Ca2+ current in ventricular cardiomyocytes. J Mol Cell Cardiol 48:26-36, 2010.)
Ca2+-dependent inactivation of ICa,L is a calmodulin (CaM) dependent process.35,36 CaM is bound to the C-terminus of the channel via the IQ domain, and when local Ca2+ is elevated, an increase in Ca2+ binding to CaM occurs. This enhances the interaction of CaM with the IQ domain, thereby causing inactivation of the channel. Ca2+-dependent inactivation may serve as a protective negative feedback mechanism to prevent Ca2+ overload in cardiomyocytes.
Steady state inactivation (i.e., channel availability), like voltage-dependent activation, follows a sigmoidal relationship, with a V1/2(inact) of ≈−35 mV for CaV1.2-dependent ICa,L and −45 mV for CaV1.3-dependent ICa,L.22 The activation and inactivation curves for ICa,L can overlap, resulting in a “window current,” which has been postulated to play a role in the generation of arrhythmogenic early afterdepolarizations (EADs; see Figure 2-3, B).37,38
LTCCs are also known to undergo a process called facilitation, whereby a progressive increase in ICa,L amplitude and the time constant of inactivation can occur during increases in pacing frequency.34,39 The facilitation process occurs as the result of a reduction in Ca2+-dependent inactivation and is mediated by calmodulin-dependent kinase II (CaMKII) phosphorylation.40
Organization and Localization of LTCCs
It is now appreciated that LTCC can be organized into distinct subcellular compartments through the actions of scaffolding proteins, including A-kinase anchoring proteins (AKAPs),41,42 within cardiomyocytes43 (Figure 2-4). Examples of subpopulations of LTCCs include those found in dyads within the T-tubules and those in separate plasma membrane domains such as caveolae and lipid rafts.2 This pattern of organization is critical for the precise spatio-temporal modulation and regulation of LTCCs in different physiological functions such as excitation-contraction coupling, transcriptional regulation, and responses to β-adrenergic (β-AR) receptor activation.
Within dyads, LTCCs from a complex with β-ARs, adenylyl cyclase (AC), protein kinase A (PKA), and AKAPs.2,44,45 These proteins area also in close apposition to a complex of proteins associated with the SR, including ryanodine receptors, PKA, protein phosphatase 2A (PP2A), phosphodiesterases, and AKAPs (AKAP5 and AKAP15).41,42 It is the AKAPs that are thought to be responsible for the organization of these signaling complexes. Within caveolae, LTCCs associate with β2-ARs, AC, PKA, PP2A, and caveolin 3, all of which enable LTCCs in caveolae to be locally stimulated by β2-AR activation.46
LTCC also demonstrate a property called coupled gating, which occurs when LTCCs are grouped together into clusters in the plasma membrane, resulting in localized regions with significantly elevated intracellular Ca2+ concentrations. Coupled gating is enhanced by the scaffolding protein AKAP5 and by protein kinase C (PKC), which form the signaling complex with the clusters of LTCCs.43,47
T-type Ca2+ Channels
Molecular Composition and Biophysical Properties
Although L-type Ca2+ channels get the lion’s share of the attention in both cardiac and vascular electrophysiology, low voltage-activated T-type Ca2+ channels also play important roles in normal and diseased hearts. Three T-type Ca2+ channels exist in mammals: Cav3.1 (α1G), Cav3.2 (α1H), and Cav3.3 (α1I), with Cav3.1 and Cav3.2 channels being the major channels in heart48,49 (see Table 2-1). Although L-type Ca2+ channels are complex oligomeric proteins that are heavily regulated by many distinct signal transduction mechanisms, much less is known about the auxiliary subunit structure for T-type Ca2+ channels.50 The electrophysiological properties of T-type α1-subunits expressed alone are very similar to those observed in native channels, suggesting that T-type Ca2+ channels do not require auxiliary subunits, as do L-type Ca2+ channels, for proper function. Consistent with this, antisense depletion of CaVβ-subunits has no effect on ICa,T (in neurons),51,52 and coexpression of cloned CaVβ-subunits has no major effect on ICa,T in heterologous expression systems.53 CaVγ-subunits have been found to have little or no effect on CaV3.3 currents54; however, they have been found to accelerate inactivation and negatively shift steady state inactivation of the CaV3.1 current.55 Coexpression of α2δ-subunits with α1G doubled the size of ICa,T, possibly through increased expression of α1-α2δ complexes at the plasma membrane.53,56 T-type Ca2+ channels have been shown to interact with, and be modulated by, other proteins such as Kelch-like 1 protein57 and caveolin-3.58 In summary, available data indicate that CaV3 α1-subunits can interact with some auxiliary subunits, but additional studies are needed to determine the physiological role for these interactions, particularly in the heart.
The biophysical properties of T-type Ca2+ channels in heart are distinct from those of L-type Ca2+ channels.59 For example, T-type Ca2+ channels activate and inactivate at lower (more negative) membrane potentials with thresholds for activation observed at about −70 mV while inactivation begins to occur at approximately −90 mV50 (see Figure 2-2). T-type Ca2+ channels also show much faster entry and exit from inactivation50,60 that is independent of Ca2+. An analysis of the steady state activation and inactivation curves reveals that T-type Ca2+ channels have relatively large steady state window currents at membrane voltages between −80 and −40 mV,61 which are expected to facilitate depolarization in myocytes of the SAN and the conducting system, thereby promoting spontaneous action potential firing. Although the permeation properties of T-type Ca2+ channels have been challenging to quantify, the single-channel conductance is reported to be 2.5-fold smaller than that of L-type Ca2+ channels (when Ba2+ is the charge carrier).62
T-type Ca2+ channels have distinct pharmacologic properties compared with L-type Ca2+ channels, and these differences have been exploited for dissecting the physiologic role of ICa,T in the myocardium. Relative to L-type channels, T-type channels have a very high sensitivity to block by Ni2+, and this sensitivity varies between different T-type channel isoforms (half maximal inhibitory concentration [IC50] for Cav3.2 is nearly 10-fold lower than that for Cav3.1 channels).50,61,63 Putative T-type channel blockers include pimozide,64 efonidipine,65 and mibefradil,66 although closer scrutiny has revealed that efonidipine and mibefradil also block L-type Ca2+ channels at higher concentrations. The only known specific blocker of T-type Ca2+ currents (generated by the Cav1.3 or Cav3.2 homologues) is kurtoxin67 from scorpions. On the other hand, many putative selective L-type Ca2+ channel blockers (such as dihydropyridines and verapamil) also inhibit T-channels.68 The development of more selective agents will clearly help to improve our understanding of the role of T-type Ca2+ channels in physiological and pathophysiological processes.49
Expression Patterns of T-type Ca2+ Channels
T-type Ca2+ channels show marked developmental and regional differences in expression that have been linked to the unique functions of these channels. For example, T-type Ca2+ currents are present in the fetal heart with the expression of Cav3.1 predominating in mouse and Cav3.2 prevailing in rats.69,70 After birth, ICa,T levels decline progressively in ventricles and become undetectable in adults while remaining at measurable levels in SAN (≈3 pA/pF), conducting system (≈3 pA/pF), and atria.49,71–75 In adult hearts of humans76 and mice,75 Cav3.1 is the more prevalent isoform, and Cav3.2 seems to be more abundant in canine Purkinje fibers,71 although this topic is controversial.77 It is interesting to note that ICa,T appear to vary inversely with body size48,78 in mammals, consistent with the conclusion that these channels influence basal heart rates. Indeed, mice lacking Cav3.1 channels showed bradycardia combined with slowed conduction through the AVN, and no electrical changes were seen in Cav3.2-knockout mice.79–81 A common finding in diseased ventricles is the elevated expression of Cav3.1 and Cav3.2, along with the reappearance of ICa,T,82–85 possibly resulting from fetal gene reactivation. These observations are particularly intriguing because elevations in ICa,T are linked to hypertrophy, disease progression, and arrhythmias.70,78 In addition, ICaT is reported to be preferentially expressed in small mononucleated myocytes of adolescent feline hearts,63 possibly representing nascent myocytes originating from cardiac progenitors. In this regard, T-type Ca2+ channels, along with hyperpolarization activated cyclic nucleotide gated (HCN) channels, are expressed at high levels in embryonic stem cells and undergo a subtype switch during maturation from Cav3.2 to Cav3.1 α-subunits as they differentiate into cardiac myocytes.86 Control of the developmental and regional expression patterns of T-type Ca2+ channels is poorly understood. Tbx3 is reported to be important in SAN development, along with expression of Cav3.1 channels and other channel genes associated with SAN.87,88
T-type Ca2+ Channels in Pacemaking, Cardiac Conduction, and Heart Disease
Unlike L-type Ca2+ channels, the functional effects of T-type Ca2+ channel activity have been more challenging to demonstrate. Pharmacologic and genetic studies support the conclusion that T-type Ca2+ channels contribute to pacemaker activity by providing inward current during the diastolic depolarization,22,62,89 although these effects are relatively modest compared with the contributions of other depolarizing currents in SAN myocytes.70,78 ICa,T also contributes to spontaneous firing and conductance properties of the AVN.22,80,90 Mice with Cav3.1 ablation have reduced heart rates and slowed AVN conduction,79 while Cav3.2-deficient mice are indistinguishable from wild type mice.91 These observations are consistent with the prevalence of Cav3.1 channels, compared with Cav3.2 channels, in the SAN of adult mice, as has been mentioned. A major complication of dissecting the role of T-type Ca2+ channels in pacemaker function is the presence of Cav1.3-dependent L-type Ca2+ channels,23 along with Cav1.2 channels, in SAN and AVN. Cav1.3 channels activate at voltages that partially overlap with T-type Ca2+ channels, and no pharmacologic agent is sufficiently selective to unequivocally dissect the functional contributions of these channels. Moreover, it is conceivable that T-type channels are expressed in distinct regions of the SAN and AVN from Cav1.3 channels, thereby complicating further our understanding of the relative contributions of T-channels to pacemaker activity. A recent report in mice established that Cav1.3 and Cav3.1 contribute synergistically to pacemaker activity in the SAN and conduction in the AVN under baseline conditions and in response to β-adrenergic receptor stimulation.80 These results also confirmed that Cav1.3-mediated ICa,L plays a relatively greater role in pacemaker activity and AVN conduction than Cav3.1-mediated ICa,T. Given that depolarizing current from Na+/Ca2+ exchanger secondary to Ca2+ release from the sarcoplasmic reticulum contributes to diastolic depolarization92,93 (later called the Ca2+ clock94), it remains to be determined to what extent the action of the T-type Ca2+ channel currents on pacemaker activity relies on the activation of RYR2 channels in the SR membrane. In this regard, conditional overexpression of Cav3.1 using the α-MHC promoter suggests that T-type Ca2+ channels are expressed on the surface sarcolemma in regions distinct from the L-type Ca2+ channels and remote from RYR2 channels.95
Many studies have concluded, based on pharmacologic interventions, that re-expression of ICa,T plays a deleterious role in cardiac remodeling and arrhythmogenesis seen in diseased hearts.83,96 For example, treatment with nickel and mibefradil reduced cell proliferation of neonatal cardiomyocytes induced by hyperglycemia.97 Mibefradil also reduced infarct sizes and improved ventricular function in rats subjected to myocardial infarction (MI)98 while protecting dog ventricles from partial coronary artery occlusions99 and pacing-induced arrhythmias.100,101 Both efonidipine and mibefradil were also more effective than nitrendipine in reducing sudden death in mice with heart failure.102 T-type Ca2+ channels have been shown to initiate spontaneous activity in pulmonary veins, thereby potentially inducing paroxysmal atrial fibrillation.103 These findings are consistent with the ability of efonidipine to prevent atrial remodeling in paced dogs.104 By contrast, mibefradil has been shown to cause deleterious effects in animal models,105,106 and treatment of cardiovascular disease in humans with mibefradil has terminated, presumably as a result of off-target effects on the liver.107 The rationale for targeting T-type Ca2+ channels in treating heart disease has come into focus with several recent studies showing that not only do increases in ICaT (via Cav3.1 overexpression in mice) not induce adverse cardiac remodeling,95 they also abrogate cardiac hypertrophy induced by pressure overload, isoproterenol, and exercise.108 Moreover, Cav3.1-deficient mice show enhanced adverse remodeling in these models and also show accelerated functional deterioration and enhanced arrhythmias following MI, in contrast to mice lacking Cav3.2, which responded similarly to wild type mice. This consensus, however, is at odds with a study showing that Cav3.2 ablation, not Cav3.1 deletion, is protective against pressure overload and angiotensin-induced hypertrophy.60
Transient Receptor Potential (TRP) Channels
The TRP channels are a large group of ion channels that were first described in Drosophila based on mutations in trp genes in photoreceptors that resulted in impaired transient responses to light.109 Currently, there are 28 mammalian trp genes, which are divided into six subfamilies denoted as TRPC (canonical), TRPV (vanilloid), TRPM (melastatin), TRPA (ankyrin), TRPP (polycystin), and TRPML (mucolipin).110 All of these channels are cationic in nature; however, there is a high level of diversity in terms of cation selectivity and gating mechanisms. Most TRP channels are nonselective cation channels that are permeable to Na+ and Ca2+ ions (PCa/PNa = 1-10), but this does not apply in all cases. For example, TRPM3α2, TRPV5, and TRPV6 are Ca2+ selective channels (PCa/PNa >100), while TRPM3α 1, TRPM4, and TRPM5 are not Ca2+ permeable (PCa/PNa < 0.05). TRPM6 and TRPM7 are permeable to Mg2+ and Ca2+.110,111
A number of TRP channels have been detected in the heart, at least at the mRNA level, including TRPC1, TRPC3-7, TRPV2, TRPV5, TRPV5, TRPM4, TRPM5, and TRPM7.111–113 Roles for some of these specific TRP channels in the heart have been identified in some instances. For example, TRPC channels have been implicated in cardiac hypertrophy,114 and both TRPC3115 and TRPM7116 have been shown to play a role in cardiac fibroblasts in atrial fibrillation. TRPM7 channels have also been described in ventricular myocytes,117,118 while TRPC channels have been suggested to play a role in pacemaker activity in the SAN.119 In most cases, however, there is still much to be learned about TRP channels in the heart in normal and pathophysiological conditions.
A complete discussion of TRP channels in the heart is beyond the scope of this chapter. Thus, we will provide an overview of the general structure of TRP channels and a brief discussion of the biophysical properties of key members of the TRP family that have been implicated in cardiovascular disease. The reader is directed to several comprehensive reviews of the biology of TRP channels for additional information.110,111,113,120,121
General Structure and Biophysical Properties of TRP Channels
TRP channels are predicted to have a similar membrane topology to other voltage-gated (Na+, Ca2+, K+) ion channels in the heart in which the channel is formed by six transmembrane-spanning domains (S1-S6) with a pore between S5 and S6 (Figure 2-5, A). The N and C termini of these proteins are intracellular and bind a number of regulatory proteins. Similar to the other six transmembrane domain channels, TRP channels are thought to form a tetrameric structure in which each subunit contributes to the selectivity filter and channel pore.110 Most TRP channels, with the exception of TRPA and TRPP, contain a stretch of ≈25 intracellular residues on the C-terminal side of the S6 transmembrane domain (TRP box), from which these channels derive their name.120
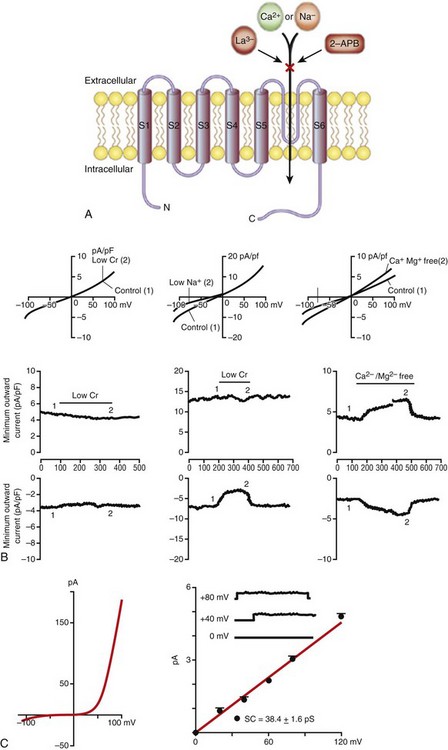
Figure 2-5 Transient receptor potential (TRP) channels in the heart. A, General membrane topology of TRP channels. S1-S6 are the transmembrane segments, and the pore is located between S5 and S6. Most TRP channels are nonselective and permeable to monovalent and divalent cations. Many TRP channels are blocked by lanthanum (La3+) and 2-aminodiethylphenhyl borate (2-APB).
B, Properties of TRPM7-dependent current in human atrial fibroblasts. The I-V relationship (left) elicited by a voltage ramp protocol illustrates that TRPM7 currents strongly outwardly rectify and inward currents are very small in normal conditions. Single-channel conductance (right) of TRPM7-mediated current was 38.4 ± 1.6 pS. C, Properties of TRPC-dependent nonselective cation currents in rat cardiac fibroblasts. Current-voltage (I-V) relations elicited by a voltage ramp protocol (top) and time course of maximum inward and outward currents (bottom) are illustrated. The current is not affected by Cl− replacement with CH3SO3−. Replacing extracellular Na+ with NMDG+ selectively reduces the inward current, while removal of divalent cations linearizes the I-V relationship and increases inward and outward currents. (A, Clapham DE, Runnels LW, Strubig C: The TRP ion channel family. Nat Rev Neurosci 2:387-396, 2001. B, Rose RA, Hatano N, Ohya S, et al: C-type natriuretic peptide activates a non-selective cation current in acutely isolated rat cardiac fibroblasts via natriuretic peptide C receptor-mediated signalling. J Physiol 580:255-274, 2007. C, Du J, Xie J, Zhang Z, et al: TRPM7-mediated Ca2+ signals confer fibrogenesis in human atrial fibrillation. Circ Res 106:992-1003, 2010.)
TRP channels do not contain the series of arginine residues in the S4 transmembrane segment that are typical of other voltage-gated channels, and the location and/or structure of specific gate(s) that control the passage of ions through the channel pore are not well resolved.110 Nevertheless, TRP channel activity is often voltage dependent and characterized by conductance-voltage relationships with relatively shallow slope factors. Furthermore, many TRP channels exhibit the voltage-dependent relaxation seen in tail currents following depolarizing voltage steps.122 TRP channel voltage sensitivity is modulated by a number of factors, including ligand binding, temperature, osmolarity, and mechanical perturbations.
TRPC Channels and Cardiac Hypertrophy
Increases in calcium influx have been implicated in pathological hypertrophic signaling in the heart due at least in part to the activation of calcineurin-nuclear factor of activated T cells (NFAT) signaling.123 Although LTCCs have been shown to contribute to this increase in Ca2+ influx,4,124,125 a number of studies have demonstrated that TRPC channel expression and activity are also upregulated in cardiac hypertrophy and heart failure. For example, pressure overload in rodents results in upregulation of TRPC1 and TRPC3.126,127 Similarly, TRPC6 was upregulated in cardiac hypertrophy and in heart failure.128 TRPC5 has been shown to be elevated in human heart failure patients.126 Consistent with these findings, pro-hypertrophic agents such as endothelin, phenylephrine, and angiotensin II cause the upregulation of TRPC1 and TRPC3 in cultured cardiomyocytes.129,130
Recent studies suggest that Ca2+ influx through TRPC channels initiates signaling pathways associated with hypertrophy and pathological cardiac remodeling.114 For example, activation of TRPC channels leads to activation of fetal genes (including atrial natriuretic peptide and skeletal α-actin), cell enlargement, and apoptosis. These responses have been shown to be dependent on enhanced calcineurin-NFAT signaling. Consistent with a role for TRPC channels in hypertrophic signaling, the TRPC3 blocker Pyr3 (a Bis-trifluoromethyl pyrazole compound) has been shown to prevent cardiac hypertrophy in pressure-overloaded mice,131 and TRPC1 knockout mice are less susceptible to cardiac hypertrophy and heart failure.127 Similar protective responses have been demonstrated in TRPC3-, TRPC4-, and TRPC6-dominant/negative mutant mice.132 Together, these findings suggest an important role for Ca2+ entry through TRP channels in hypertrophy and heart failure.
TRP Channels in Cardiac Fibroblasts
Cardiac fibroblasts account for ≈80% of the nonmyocyte population in the heart and are responsible for the synthesis and secretion of extracellular matrix proteins, including collagen types I and II, as well as matrix metalloproteinases.133,134 In the setting of pathological conditions such as hypertension, cardiac hypertrophy, and heart failure, there is an increase in the proliferation of cardiac fibroblasts, which differentiate into myofibroblasts that contribute to the deposition of extracellular matrix and cardiac fibrosis.133,134 This results in conduction abnormalities, which may be due to both structural remodeling and possible fibroblast-myocyte electrical interactions.135
Several TRP channels are expressed in cardiac fibroblasts. In rat ventricular fibroblasts, mRNA for TRPC2, TRPC3, TRPV2, TRPV6, TRPM4, and TRPM7 has been detected,112 while human atrial fibroblasts were shown to express mRNA for TRPC1, TRPC6, TRPC2, TRPC4, and TRPM7.116 These TRP channels facilitate the influx of Ca2+ into fibroblasts and may serve as a primary source of Ca2+ for these nonexcitable cells, which do not typically express voltage-gated Ca2+ channels.112
Recent data indicate that Ca2+ influx into fibroblasts through TRP channels is a significant contributor to fibroblast proliferation and fibrosis in atrial fibrillation. Du et al116 demonstrated that TRPM7 (Figure 2-5, B) is a major pathway for Ca2+ entry in human atrial fibroblasts, and that this Ca2+ activates transforming growth factor (TGF)-β1, a critical signaling molecule in atrial fibrosis. TRPC channels also constitute a major pathway for Ca2+ entry into cardiac fibroblasts via nonselective cation currents112 (Figure 2-5, C), and a recent study115 suggests that Ca2+ entry in atrial fibroblasts via TRPC3 channels controls fibroblast proliferation and differentiation in an extracellular signal–related kinase (ERK)-dependent fashion in atrial fibrillation. This study further demonstrates a role for microRNA-26 and NFAT in the TRPC3-dependent enhancement of fibroblast proliferation and fibrosis in atrial fibrillation. The pathophysiological role of TRP channels in cardiac arrhythmias is an emerging area of interest, and the aforementioned studies strongly indicate the need to better understand the relationships between these ion channels and conduction disturbances in the heart.136
References
1. Bers, DM. Cardiac excitation-contraction coupling. Nature. 2002; 415:198–205.
2. Best, JM, Kamp, TJ. Different subcellular populations of L-type Ca2+ channels exhibit unique regulation and functional roles in cardiomyocytes. J Mol Cell Cardiol. 2012; 52:376–387.
3. Benitah, JP, Alvarez, JL, Gomez, AM. L-type Ca(2+) current in ventricular cardiomyocytes. J Mol Cell Cardiol. 2010; 48:26–36.
4. Pitt, GS, Dun, W, Boyden, PA. Remodeled cardiac calcium channels. J Mol Cell Cardiol. 2006; 41:373–388.
5. Catterall, WA. Voltage-gated calcium channels. Cold Spring Harb Perspect Biol. 2011; 3:a003947.
6. Catterall, WA. Structure and regulation of voltage-gated Ca2+ channels. Annu Rev Cell Dev Biol. 2000; 16:521–555.
7. Schram, G, Pourrier, M, Melnyk, P, et al. Differential distribution of cardiac ion channel expression as a basis for regional specialization in electrical function. Circ Res. 2002; 90:939–950.
8. Bock, G, Gebhart, M, Scharinger, A, et al. Functional properties of a newly identified C-terminal splice variant of Cav1. 3 L-type Ca2+ channels. J Biol Chem. 2011; 286:42736–42748.
9. Tan, BZ, Jiang, F, Tan, MY, et al. Functional characterization of alternative splicing in the C terminus of L-type CaV1. 3 channels. J Biol Chem. 2011; 286:42725–42735.
10. Foell, JD, Balijepalli, RC, Delisle, BP, et al. Molecular heterogeneity of calcium channel beta-subunits in canine and human heart: evidence for differential subcellular localization. Physiol Genomics. 2004; 17:183–200.
11. Hofmann, F, Biel, M, Flockerzi, V. Molecular basis for Ca2+ channel diversity. Annu Rev Neurosci. 1994; 17:399–418.
12. Chen, YH, Li, MH, Zhang, Y, et al. Structural basis of the alpha1-beta subunit interaction of voltage-gated Ca2+ channels. Nature. 2004; 429:675–680.
13. Van Petegem, F, Clark, KA, Chatelain, FC, et al. Structure of a complex between a voltage-gated calcium channel beta-subunit and an alpha-subunit domain. Nature. 2004; 429:671–675.
14. Mitterdorfer, J, Froschmayr, M, Grabner, M, et al. Calcium channels: The beta-subunit increases the affinity of dihydropyridine and Ca2+ binding sites of the alpha 1-subunit. FEBS Lett. 1994; 352:141–145.
15. Davies, A, Hendrich, J, Van Minh, AT, et al. Functional biology of the alpha(2)delta subunits of voltage-gated calcium channels. Trends Pharmacol Sci. 2007; 28:220–228.
16. Jay, SD, Sharp, AH, Kahl, SD, et al. Structural characterization of the dihydropyridine-sensitive calcium channel alpha 2-subunit and the associated delta peptides. J Biol Chem. 1991; 266:3287–3293.
17. Yang, L, Katchman, A, Morrow, JP, et al. Cardiac L-type calcium channel (Cav1. 2) associates with gamma subunits. FASEB J. 2011; 25:928–936.
18. McCleskey, EW, Almers, W. The Ca channel in skeletal muscle is a large pore. Proc Natl Acad Sci U S A. 1985; 82:7149–7153.
19. Chen, XH, Bezprozvanny, I, Tsien, RW. Molecular basis of proton block of L-type Ca2+ channels. J Gen Physiol. 1996; 108:363–374.
20. Chen, XH, Tsien, RW. Aspartate substitutions establish the concerted action of P-region glutamates in repeats I and III in forming the protonation site of L-type Ca2+ channels. J Biol Chem. 1997; 272:30002–30008.
21. Guia, A, Stern, MD, Lakatta, EG, et al. Ion concentration-dependence of rat cardiac unitary L-type calcium channel conductance. Biophys J. 2001; 80:2742–2750.
22. Mangoni, ME, Couette, B, Marger, L, et al. Voltage-dependent calcium channels and cardiac pacemaker activity: from ionic currents to genes. Prog Biophys Mol Biol. 2006; 90:38–63.
23. Mangoni, ME, Couette, B, Bourinet, E, et al. Functional role of L-type Cav1. 3 Ca2+ channels in cardiac pacemaker activity. Proc Natl Acad Sci U S A. 2003; 100:5543–5548.
24. Zhang, Z, He, Y, Tuteja, D, et al. Functional roles of Cav1. 3(alpha1D) calcium channels in atria: insights gained from gene-targeted null mutant mice. Circulation. 2005; 112:1936–1944.
25. Seisenberger, C, Specht, V, Welling, A, et al. Functional embryonic cardiomyocytes after disruption of the L-type alpha1C (Cav1. 2) calcium channel gene in the mouse. J Biol Chem. 2000; 275:39193–39199.
26. Mangoni, ME, Nargeot, J. Genesis and regulation of the heart automaticity. Physiol Rev. 2008; 88:919–982.
27. Zhang, Z, Xu, Y, Song, H, et al. Functional roles of Ca(v)1. 3 (alpha(1D) ) calcium channel in sinoatrial nodes: insight gained using gene-targeted null mutant mice. Circ Res. 2002; 90:981–987.
28. Platzer, J, Engel, J, Schrott-Fischer, A, et al. Congenital deafness and sinoatrial node dysfunction in mice lacking class D L-type Ca2+ channels. Cell. 2000; 102:89–97.
29. Mancarella, S, Yue, Y, Karnabi, E, et al. Impaired Ca2+ homeostasis is associated with atrial fibrillation in the alpha1D L-type Ca2+ channel KO mouse. Am J Physiol Heart Circ Physiol. 2008; 295:H2017–H2024.
30. Campbell, DL, Giles, WR, Hume, JR, et al. Inactivation of calcium current in bull-frog atrial myocytes. J Physiol. 1988; 403:287–315.
31. Kass, RS, Sanguinetti, MC. Inactivation of calcium channel current in the calf cardiac Purkinje fiber: Evidence for voltage- and calcium-mediated mechanisms. J Gen Physiol. 1984; 84:705–726.
32. Lee, KS, Marban, E, Tsien, RW. Inactivation of calcium channels in mammalian heart cells: Joint dependence on membrane potential and intracellular calcium. J Physiol. 1985; 364:395–411.
33. Hadley, RW, Hume, JR. An intrinsic potential-dependent inactivation mechanism associated with calcium channels in guinea-pig myocytes. J Physiol. 1987; 389:205–222.
34. Richard, S, Perrier, E, Fauconnier, J, et al. ‘Ca(2+)-induced Ca(2+) entry’ or how the L-type Ca(2+) channel remodels its own signalling pathway in cardiac cells. Prog Biophys Mol Biol. 2006; 90:118–135.
35. Peterson, BZ, DeMaria, CD, Adelman, JP, et al. Calmodulin is the Ca2+ sensor for Ca2+ -dependent inactivation of L-type calcium channels. Neuron. 1999; 22:549–558.
36. Qin, N, Olcese, R, Bransby, M, et al. Ca2+-induced inhibition of the cardiac Ca2+ channel depends on calmodulin. Proc Natl Acad Sci U S A. 1999; 96:2435–2438.
37. Bers, DM, Perez-Reyes, E. Ca channels in cardiac myocytes: Structure and function in Ca influx and intracellular Ca release. Cardiovasc Res. 1999; 42:339–360.
38. January, CT, Riddle, JM. Early afterdepolarizations: mechanism of induction and block: A role for L-type Ca2+ current. Circ Res. 1989; 64:977–990.
39. Hryshko, LV, Bers, DM. Ca current facilitation during postrest recovery depends on Ca entry. Am J Physiol. 1990; 259:H951–H961.
40. Zuhlke, RD, Pitt, GS, Deisseroth, K, et al. Calmodulin supports both inactivation and facilitation of L-type calcium channels. Nature. 1999; 399:159–162.
41. Scott, JD, Santana, LF. A-kinase anchoring proteins: Getting to the heart of the matter. Circulation. 2010; 121:1264–1271.
42. Beca, S, Aschars-Sobbi, R, Panama, BK, et al. Regulation of murine cardiac function by phosphodiesterases type 3 and 4. Curr Opin Pharmacol. 2011; 11:714–719.
43. Santana, LF, Navedo, MF. Natural inequalities: Why some L-type Ca2+ channels work harder than others. J Gen Physiol. 2010; 136:143–147.
44. Fuller, MD, Emrick, MA, Sadilek, M, et al. Molecular mechanism of calcium channel regulation in the fight-or-flight response. Sci Signal. 2010; 3:ra70.
45. Hulme, JT, Lin, TW, Westenbroek, RE, et al. Beta-adrenergic regulation requires direct anchoring of PKA to cardiac CaV1. 2 channels via a leucine zipper interaction with A kinase-anchoring protein 15. Proc Natl Acad Sci U S A. 2003; 100:13093–13098.
46. Balijepalli, RC, Foell, JD, Hall, DD, et al. Localization of cardiac L-type Ca(2+) channels to a caveolar macromolecular signaling complex is required for beta(2)-adrenergic regulation. Proc Natl Acad Sci U S A. 2006; 103:7500–7505.
47. Navedo, MF, Cheng, EP, Yuan, C, et al. Increased coupled gating of L-type Ca2+ channels during hypertension and Timothy syndrome. Circ Res. 2010; 106:748–756.
48. Ono, K, Iijima, T. Pathophysiological significance of T-type Ca2+ channels: Properties and functional roles of T-type Ca2+ channels in cardiac pacemaking. J Pharmacol Sci. 2005; 99:197–204.
49. Vassort, G, Talavera, K, Alvarez, JL. Role of T-type Ca2+ channels in the heart. Cell Calcium. 2006; 40:205–220.
50. Perez-Reyes, E. Molecular physiology of low-voltage-activated t-type calcium channels. Physiol Rev. 2003; 83:117–161.
51. Lambert, RC, Maulet, Y, Mouton, J, et al. T-type Ca2+ current properties are not modified by Ca2+ channel beta subunit depletion in nodosus ganglion neurons. J Neurosci. 1997; 17:6621–6628.
52. Leuranguer, V, Bourinet, E, Lory, P, et al. Antisense depletion of beta-subunits fails to affect T-type calcium channels properties in a neuroblastoma cell line. Neuropharmacology. 1998; 37:701–708.
53. Dolphin, AC, Wyatt, CN, Richards, J, et al. The effect of alpha2-delta and other accessory subunits on expression and properties of the calcium channel alpha1G. J Physiol. 1999; 519(Pt 1):35–45.
54. Green, PJ, Warre, R, Hayes, PD, et al. Kinetic modification of the alpha(1I) subunit-mediated T-type Ca(2+) channel by a human neuronal Ca(2+) channel gamma subunit. J Physiol. 2001; 533:467–478.
55. Klugbauer, N, Dai, S, Specht, V, et al. A family of gamma-like calcium channel subunits. FEBS Lett. 2000; 470:189–197.
56. Gao, B, Sekido, Y, Maximov, A, et al. Functional properties of a new voltage-dependent calcium channel alpha(2)delta auxiliary subunit gene (CACNA2D2). J Biol Chem. 2000; 275:12237–12242.
57. Aromolaran, KA, Benzow, KA, Cribbs, LL, et al. T-type current modulation by the actin-binding protein Kelch-like 1. Am J Physiol Cell Physiol. 2010; 298:C1353–1362.
58. Markandeya, YS, Fahey, JM, Pluteanu, F, et al. Caveolin-3 regulates protein kinase A modulation of the Ca(V)3. 2 (alpha1H) T-type Ca2+ channels. J Biol Chem. 2011; 286:2433–2444.
59. Lee, JH, Gomora, JC, Cribbs, LL, et al. Nickel block of three cloned T-type calcium channels: Low concentrations selectively block alpha1H. Biophys J. 1999; 77:3034–3042.
60. Chiang, CS, Huang, CH, Chieng, H, et al. The Ca(v)3. 2 T-type Ca(2+) channel is required for pressure overload-induced cardiac hypertrophy in mice. Circ Res. 2009; 104:522–530.
61. Iftinca, MC, Zamponi, GW. Regulation of neuronal T-type calcium channels. Trends Pharmacol Sci. 2009; 30:32–40.
62. Hagiwara, N, Irisawa, H, Kameyama, M. Contribution of two types of calcium currents to the pacemaker potentials of rabbit sino-atrial node cells. J Physiol. 1988; 395:233–253.
63. Perez-Reyes, E. Molecular characterization of T-type calcium channels. Cell Calcium. 2006; 40:89–96.
64. Xie, X, Van Deusen, AL, Vitko, I, et al. Validation of high throughput screening assays against three subtypes of Ca(v)3 T-type channels using molecular and pharmacologic approaches. Assay Drug Dev Technol. 2007; 5:191–203.
65. Sawada, T, Shinke, T, Shite, J, et al. Impact of cytochrome P450 2C19*2 polymorphism on intra-stent thrombus after drug-eluting stent implantation in Japanese patients receiving clopidogrel. Circ J. 2011; 75:99–105.
66. Tanaka, H, Shigenobu, K. Pathophysiological significance of T-type Ca2+ channels: T-type Ca2+ channels and drug development. J Pharmacol Sci. 2005; 99:214–220.
67. Chuang, RS, Jaffe, H, Cribbs, L, et al. Inhibition of T-type voltage-gated calcium channels by a new scorpion toxin. Nat Neurosci. 1998; 1:668–674.
68. Lacinova, L. T-type calcium channel blockers—new and notable. Gen Physiol Biophys. 2011; 30:403–409.
69. Cribbs, LL, Martin, BL, Schroder, EA, et al. Identification of the t-type calcium channel (Ca(v)3. 1d) in developing mouse heart. Circ Res. 2001; 88:403–407.
70. Cribbs, L. T-type calcium channel expression and function in the diseased heart. Channels (Austin). 2010; 4:447–452.
71. Rosati, B, Dun, W, Hirose, M, et al. Molecular basis of the T- and L-type Ca2+ currents in canine Purkinje fibres. J Physiol. 2007; 579:465–471.
72. Zhou, C, Chen, H, Lu, F, et al. Cav3. 1 (alpha1G) controls von Willebrand factor secretion in rat pulmonary microvascular endothelial cells. Am J Physiol Lung Cell Mol Physiol. 2007; 292:L833–L844.
73. Leuranguer, V, Monteil, A, Bourinet, E, et al. T-type calcium currents in rat cardiomyocytes during postnatal development: contribution to hormone secretion. Am J Physiol Heart Circ Physiol. 2000; 279:H2540–H2548.
74. Mizuta, E, Miake, J, Yano, S, et al. Subtype switching of T-type Ca2+ channels from Cav3. 2 to Cav3. 1 during differentiation of embryonic stem cells to cardiac cell lineage. Circ J. 2005; 69:1284–1289.
75. Niwa, N, Yasui, K, Opthof, T, et al. Cav3. 2 subunit underlies the functional T-type Ca2+ channel in murine hearts during the embryonic period. Am J Physiol Heart Circ Physiol. 2004; 286:H2257–H2263.
76. Chandler, NJ, Greener, ID, Tellez, JO, et al. Molecular architecture of the human sinus node: Insights into the function of the cardiac pacemaker. Circulation. 2009; 119:1562–1575.
77. Han, W, Bao, W, Wang, Z, et al. Comparison of ion-channel subunit expression in canine cardiac Purkinje fibers and ventricular muscle. Circ Res. 2002; 91:790–797.
78. Ono, K, Iijima, T. Cardiac T-type Ca(2+) channels in the heart. J Mol Cell Cardiol. 2010; 48:65–70.
79. Mangoni, ME, Traboulsie, A, Leoni, AL, et al. Bradycardia and slowing of the atrioventricular conduction in mice lacking CaV3. 1/alpha1G T-type calcium channels. Circ Res. 2006; 98:1422–1430.
80. Marger, L, Mesirca, P, Alig, J, et al. Functional roles of Ca(v)1. 3, Ca(v)3. 1 and HCN channels in automaticity of mouse atrioventricular cells: Insights into the atrioventricular pacemaker mechanism. Channels (Austin). 2011; 5:251–261.
81. Le Quang, K, Naud, P, Qi, XY, et al. Role of T-type calcium channel subunits in post-myocardial infarction remodelling probed with genetically engineered mice. Cardiovasc Res. 2011; 91:420–428.
82. Huang, B, Qin, D, Deng, L, et al. Reexpression of T-type Ca2+ channel gene and current in post-infarction remodeled rat left ventricle. Cardiovasc Res. 2000; 46:442–449.
83. Nuss, HB, Houser, SR. T-type Ca2+ current is expressed in hypertrophied adult feline left ventricular myocytes. Circ Res. 1993; 73:777–782.
84. Schmitt, R, Clozel, JP, Iberg, N, et al. Mibefradil prevents neointima formation after vascular injury in rats: Possible role of the blockade of the T-type voltage-operated calcium channel. Arterioscler Thromb Vasc Biol. 1995; 15:1161–1165.
85. Sen, L, Smith, TW. T-type Ca2+ channels are abnormal in genetically determined cardiomyopathic hamster hearts. Circ Res. 1994; 75:149–155.
86. Yanagi, K, Takano, M, Narazaki, G, et al. Hyperpolarization-activated cyclic nucleotide-gated channels and T-type calcium channels confer automaticity of embryonic stem cell-derived cardiomyocytes. Stem Cells. 2007; 25:2712–2719.
87. Wiese, C, Grieskamp, T, Airik, R, et al. Formation of the sinus node head and differentiation of sinus node myocardium are independently regulated by Tbx18 and Tbx3. Circ Res. 2009; 104:388–397.
88. Hoogaars, WM, Engel, A, Brons, JF, et al. Tbx3 controls the sinoatrial node gene program and imposes pacemaker function on the atria. Genes Dev. 2007; 21:1098–1112.
89. Nilius, B, Talavera, K, Verkhratsky, A. T-type calcium channels: The never ending story. Cell Calcium. 2006; 40:81–88.
90. Madle, A, Linhartova, K, Koza, J. Effects of the T-type calcium channel blockade with oral mibefradil on the electrophysiologic properties of the human heart. Med Sci Monit. 2001; 7:74–77.
91. Chen, CC, Lamping, KG, Nuno, DW, et al. Abnormal coronary function in mice deficient in alpha1H T-type Ca2+ channels. Science. 2003; 302:1416–1418.
92. Huser, J, Blatter, LA, Lipsius, SL. Intracellular Ca2+ release contributes to automaticity in cat atrial pacemaker cells. J Physiol. 2000; 524(Pt 2):415–422.
93. Hata, T, Noda, T, Nishimura, M, et al. The role of Ca2+ release from sarcoplasmic reticulum in the regulation of sinoatrial node automaticity. Heart Vessels. 1996; 11:234–241.
94. Lakatta, EG, Maltsev, VA, Vinogradova, TM. A coupled system of intracellular Ca2+ clocks and surface membrane voltage clocks controls the timekeeping mechanism of the heart’s pacemaker. Circ Res. 2010; 106:659–673.
95. Jaleel, N, Nakayama, H, Chen, X, et al. Ca2+ influx through T- and L-type Ca2+ channels have different effects on myocyte contractility and induce unique cardiac phenotypes. Circ Res. 2008; 103:1109–1119.
96. Ertel, SI, Ertel, EA, Clozel, JP. T-type Ca2+ channels and pharmacological blockade: Potential pathophysiological relevance. Cardiovasc Drugs Ther. 1997; 111:723–739.
97. Li, M, Zhang, M, Huang, L, et al. T-type Ca2+ channels are involved in high glucose-induced rat neonatal cardiomyocyte proliferation. Pediatr Res. 2005; 57:550–556.
98. Sandmann, S, Min, JY, Meissner, A, et al. Effects of the calcium channel antagonist mibefradil on haemodynamic parameters and myocardial Ca(2+)-handling in infarct-induced heart failure in rats. Cardiovasc Res. 1999; 44:67–80.
99. Parent de Curzon, O, Ghaleh, B, Hittinger, L, et al. Beneficial effects of the T- and L-type calcium channel antagonist, mibefradil, against exercise-induced myocardial stunning in dogs. J Cardiovasc Pharmacol. 2000; 35:240–248.
100. Fareh, S, Benardeau, A, Nattel, S. Differential efficacy of L- and T-type calcium channel blockers in preventing tachycardia-induced atrial remodeling in dogs. Cardiovasc Res. 2001; 49:762–770.
101. Fareh, S, Benardeau, A, Thibault, B, et al. The T-type Ca(2+) channel blocker mibefradil prevents the development of a substrate for atrial fibrillation by tachycardia-induced atrial remodeling in dogs. Circulation. 1999; 100:2191–2197.
102. Kinoshita, H, Kuwahara, K, Takano, M, et al. T-type Ca2+ channel blockade prevents sudden death in mice with heart failure. Circulation. 2009; 120:743–752.
103. Chen, YC, Chen, SA, Chen, YJ, et al. T-type calcium current in electrical activity of cardiomyocytes isolated from rabbit pulmonary vein. J Cardiovasc Electrophysiol. 2004; 15:567–571.
104. Ohashi, N, Mitamura, H, Tanimoto, K, et al. A comparison between calcium channel blocking drugs with different potencies for T- and L-type channels in preventing atrial electrical remodeling. J Cardiovasc Pharmacol. 2004; 44:386–392.
105. Xia, QG, Reinecke, A, Dorenkamp, M, et al. Comparison of cardioprotective effects of mibefradil and ramipril in stroke-prone spontaneously hypertensive rats. Acta Pharmacol Sin. 2004; 25:763–768.
106. Takebayashi, S, Li, Y, Kaku, T, et al. Remodeling excitation-contraction coupling of hypertrophied ventricular myocytes is dependent on T-type calcium channels expression. Biochem Biophys Res Commun. 2006; 345:766–773.
107. Po, AL, Zhang, WY. What lessons can be learnt from withdrawal of mibefradil from the market? Lancet. 1998; 351:1829–1830.
108. Nakayama, H, Bodi, I, Correll, RN, et al. alpha1G-dependent T-type Ca2+ current antagonizes cardiac hypertrophy through a NOS3-dependent mechanism in mice. J Clin Invest. 2009; 119:3787–3796.
109. Minke, B. Drosophila mutant with a transducer defect. Biophys Struct Mech. 1977; 3:59–64.
110. Ramsey, IS, Delling, M, Clapham, DE. An introduction to TRP channels. Ann Rev Physiol. 2006; 68:619–647.
111. Vennekens, R. Emerging concepts for the role of TRP channels in the cardiovascular system. J Physiol. 2011; 589:1527–1534.
112. Rose, RA, Hatano, N, Ohya, S, et al. C-type natriuretic peptide activates a non-selective cation current in acutely isolated rat cardiac fibroblasts via natriuretic peptide C receptor-mediated signalling. J Physiol. 2007; 580:255–274.
113. Watanabe, H, Murakami, M, Ohba, T, et al. TRP channel and cardiovascular disease. Pharmacol Ther. 2008; 118:337–351.
114. Eder, P, Molkentin, JD. TRPC channels as effectors of cardiac hypertrophy. Circ Res. 2011; 108:265–272.
115. Harada, M, Luo, X, Qi, XY, et al. Transient receptor potential canonical-3 channel-dependent fibroblast regulation in atrial fibrillation. Circulation. 2012; 126:2051–2064.
116. Du, J, Xie, J, Zhang, Z, et al. TRPM7-mediated Ca2+ signals confer fibrogenesis in human atrial fibrillation. Circ Res. 2010; 106:992–1003.
117. Gwanyanya, A, Amuzescu, B, Zakharov, SI, et al. Magnesium-inhibited, TRPM6/7-like channel in cardiac myocytes: Permeation of divalent cations and pH-mediated regulation. J Physiol. 2004; 559:761–776.
118. Gwanyanya, A, Sipido, KR, Vereecke, J, et al. ATP and PIP2 dependence of the magnesium-inhibited, TRPM7-like cation channel in cardiac myocytes. Am J Physiol Cell Physiol. 2006; 291:C627–C635.
119. Ju, YK, Chu, Y, Chaulet, H, et al. Store-operated Ca2+ influx and expression of TRPC genes in mouse sinoatrial node. Circ Res. 2007; 100:1605–1614.
120. Clapham, DE. TRP channels as cellular sensors. Nature. 2003; 426:517–524.
121. Clapham, DE, Runnels, LW, Strubing, C. The TRP ion channel family. Nat Rev Neurosci. 2001; 2:387–396.
122. Nilius, B, Talavera, K, Owsianik, G, et al. Gating of TRP channels: A voltage connection? J Physiol. 2005; 567:35–44.
123. Heineke, J, Molkentin, JD. Regulation of cardiac hypertrophy by intracellular signalling pathways. Nat Rev Mol Cell Biol. 2006; 7:589–600.
124. Richard, S, Leclercq, F, Lemaire, S, et al. Ca2+ currents in compensated hypertrophy and heart failure. Cardiovasc Res. 1998; 37:300–311.
125. Sah, R, Oudit, GY, Nguyen, TT, et al. Inhibition of calcineurin and sarcolemmal Ca2+ influx protects cardiac morphology and ventricular function in K(v)4. 2N transgenic mice. Circulation. 2002; 105:1850–1856.
126. Bush, EW, Hood, DB, Papst, PJ, et al. Canonical transient receptor potential channels promote cardiomyocyte hypertrophy through activation of calcineurin signaling. J Biol Chem. 2006; 281:33487–33496.
127. Seth, M, Zhang, ZS, Mao, L, et al. TRPC1 channels are critical for hypertrophic signaling in the heart. Circ Res. 2009; 105:1023–1030.
128. Niizeki, T, Takeishi, Y, Kitahara, T, et al. Diacylglycerol kinase-epsilon restores cardiac dysfunction under chronic pressure overload: A new specific regulator of Galpha(q) signaling cascade. Am J Physiol Heart Circ Physiol. 2008; 295:H245–H255.
129. Ohba, T, Watanabe, H, Murakami, M, et al. Upregulation of TRPC1 in the development of cardiac hypertrophy. J Mol Cell Cardiol. 2007; 42:498–507.
130. Brenner, JS, Dolmetsch, RE. TrpC3 regulates hypertrophy-associated gene expression without affecting myocyte beating or cell size. PLoS One. 2007; 2:e802.
131. Kiyonaka, S, Kato, K, Nishida, M, et al. Selective and direct inhibition of TRPC3 channels underlies biological activities of a pyrazole compound. Proc Natl Acad Sci U S A. 2009; 106:5400–5405.
132. Wu, X, Eder, P, Chang, B, et al. TRPC channels are necessary mediators of pathologic cardiac hypertrophy. Proc Natl Acad Sci U S A. 2010; 107:7000–7005.
133. Baudino, TA, Carver, W, Giles, W, et al. Cardiac fibroblasts: Friend or foe? Am J Physiol Heart Circ Physiol. 2006; 291:H1015–H1026.
134. Souders, CA, Bowers, SL, Baudino, TA. Cardiac fibroblast: The renaissance cell. Circ Res. 2009; 105:1164–1176.
135. Yue, L, Xie, J, Nattel, S. Molecular determinants of cardiac fibroblast electrical function and therapeutic implications for atrial fibrillation. Cardiovasc Res. 2011; 89:744–753.
136. Rose, RA, Belke, DD, Maleckar, MM, et al. Ca2+ entry through TRP-C channels regulates fibroblast biology in chronic atrial fibrillation. Circulation. 2012; 126:2039–2041.