Chapter 45 Bronchiectasis
Pathology
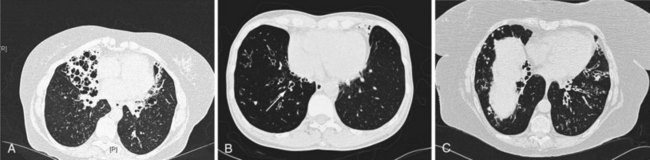
Whitwell classified bronchiectasis into three groups based on pathologic findings—saccular, atelectatic, and follicular—and although these terms may have changed, the descriptions remain the same. Cylindrical bronchiectasis is characterized by bronchi showing a regular outline, with dilated airways only and usually ending abruptly. Varicose bronchiectasis has similarities to the appearance of varicose veins, with dilation that is deformed by areas of relative constriction. These bronchi also have a distorted and bulbous end. Cystic (saccular) bronchiectasis is considered the most severe form of bronchiectasis; its most prominent feature is progressively increasing dilation as the bronchi progress toward the lung periphery and airways, ending in cystlike clusters. These three basic forms of bronchiectasis are demonstrated in Figure 45-1 by high-resolution computed tomography (HRCT) scanning.
Pathogenesis
The cause of non-CF bronchiectasis may only be identifiable in up to 50% of patients. However, a number of recognized conditions and factors are associated with bronchiectasis, and an underlying cause should be assessed in all patients (Box 45-1).
Box 45-1
Underlying Causes of Bronchiectasis
The genetic influence on the development of non-CF bronchiectasis is the subject of ongoing research, often on the role of the cystic fibrosis transmembrane conductance regulator (CFTR) gene. The most common mutation of this gene, ΔF508 (F508del), is associated with severe CF (see Chapter 44). Numerous other mutations have been identified and associated with a milder clinical phenotype. As a consequence, late first presentation of CF has been described in patients who would otherwise have been considered to have idiopathic non-CF bronchiectasis. It is now recognized that mutations of the CFTR gene are more frequently observed in patients with bronchiectasis and a normal sweat chloride test than in the general population. Studies show that the spectrum of CFTR genotypes is associated with a continuum of CFTR dysfunction in the airways. Phenotypes range from patients with bronchiectasis, normal sweat test, and no other features suggestive of CF to those with classic CF. Also, some evidence suggests the CFTR mutations are associated with non-CF bronchiectasis and rheumatoid arthritis. Therefore, CFTR dysfunction can be identified as a cause of bronchiectasis in patients previously diagnosed with idiopathic non-CF bronchiectasis, but without fulfilling the diagnostic criteria for CF. How this affects patient management is currently unknown.
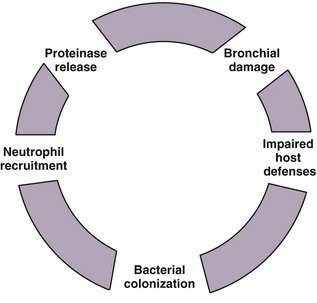
The “vicious cycle” hypothesis of bronchiectasis suggests an initial failure or overwhelmed host defenses, leading to a host-mediated chronic inflammatory response, which in turn causes new or further impairment of mucociliary clearance and defenses, amplifying the problem. This interaction between chronic infection and excessive inflammation, which is predominantly neutrophilic, ensures ongoing damage to the airways and the development and maintenance of the features seen in bronchiectasis. Inflammatory mediators such as the neutrophil chemoattractant interleukin-8 (IL-8) and tumor necrosis factor alpha (TNF-α) are found in bronchial mucosal biopsies and secretions from bronchiectatic airways, in addition to tissue neutrophilia. This initiation of the inflammatory reaction results in the recruitment of phagocytes, dendritic cells, and lymphocytes, which contribute to the adaptive response (Figure 45-2).
Diagnosis and patient assessment
Blood Tests
Inflammatory markers include white blood cell (WBC) count, neutrophil count, erythrocyte sedimentation rate (ESR), and C-reactive protein (CRP) and can be used as nonspecific tests of disease activity and exacerbation severity. These tests can also be used to guide the need for and response to antibiotics in the absence of changes in clinical state or symptomatology of bronchiectatic patients (Box 45-2).
Immunoglobulins
Alpha1-antitrypsin levels should be guided by clinical suspicion of bronchiectasis, as should genetic testing for CF or its variants (Box 45-2).
Sputum Microbiology
An assessment of lower respiratory tract microbiology is imperative in the investigation and management of bronchiectasis. Table 45-1 summarizes the most common organisms isolated from the respiratory tract and their frequency in adults with bronchiectasis.
| Organism | Frequency |
|---|---|
| Haemophilus influenzae | 14%-35% |
| Pseudomonas aeruginosa | 9%-31% |
| Staphylococcus aureus | 0%-14% |
| Streptococcus pneumoniae | 2%-13% |
| Moraxella (Branhamella) catarrhalis | 0%-20% |
| Possibly “nonpathogenic organisms” Corynebacterium spp., Neisseria spp., coagulase-negative Staphylococcus, β-hemolytic Streptococcus | 5%-60% |
exacerbations
Acute Exacerbation
As discussed earlier with microbiologic identification of bronchiectasis exacerbation, presence of bacteria alone does not necessarily indicate an exacerbation requiring antibiotic therapy, in view of the prevalence of colonization in the stable clinical state. A worsening of symptoms from baseline and acute deterioration with a change in bacterial load, however, indicate the need to introduce or add further antibiotics. Box 45-3 lists other symptoms suggestive of an acute bacterial exacerbation.
Recurrent Exacerbation
Furthermore, the clinical problems associated with P. aeruginosa may be further exacerbated by the development of antibodies that reduce bacterial killing. Studies in the 1970s and 1980s of CF patients colonized with P. aeruginosa identified a blocking factor in serum that inhibited killing of Pseudomonas, subsequently shown to be an IgG2 subclass. Recent work supports the role of inhibitory antibodies in the ability of other bacteria to establish infection within the host. This may partly contribute to the failure of the host to control infection, and although the role of this mechanism has yet to be established in bronchiectasis, it is worthy of study. However, this ability of organisms to survive will establish ongoing inflammation as a frustrated attempt by the host to eradicate them, leading to proteolytic damage and perpetuation of the vicious cycle illustrated in Figure 45-2. For these reasons, patients should receive longer-term antibiotic therapy as appropriate, if the cycle can be broken (i.e., sputum purulence disappears).
Antibiotic Therapy
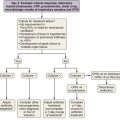
Figure 45-3 provides a decision-making algorithm for antibiotic therapy of the patient with bronchiectasis.
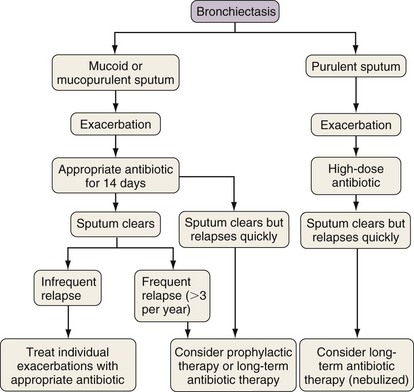
Patient with Acute Exacerbation
Prompt antimicrobial therapy is required once an acute exacerbation is identified, guided by the most up-to-date microbiology results and local guidelines. There are not randomized, placebo controlled trials evaluating the effectiveness of antibiotics in exacerbations in bronchiectasis, but in general high-dose, targeted antibiotics are required for effective treatment. The British Thoracic Society guidelines recommend that many patients with bronchiectasis should receive 14 days of antibiotics rather than the conventional dose. The antibiotics most often used for exacerbations have a broad spectrum of activity. Amoxicillin remains the initial choice for many organisms, especially in the UK, but patterns of resistance need to be recognized and management adapted appropriately. H. influenzae is usually sensitive to amoxicillin, but the production of β-lactamase or changes in penicillin-binding proteins increase resistance, and the presence of resistant strains varies from country to country. Moraxella (Branhamella) catarrhalis also produces β-lactamase. Antibiotic choices based on current UK guidelines are shown in Table 45-2, but local guidelines should be considered.
Table 45-2 Antibiotic Guidelines for Bronchiectasis Exacerbation
| Organism | Antiobiotic/Dose |
|---|---|
| Streptococcus pneumoniae | |
| β-lactamase −ve Haemophilus influenzae |
H. influenzae
Methicillin resistant
Alternative Therapy
Therefore, modulating this process requires further careful investigation, as would abrogation of the other chemoattractants (e.g., LTB4). Neutrophil-derived proteinase activity, particularly that of neutrophil elastase, is also crucial in maintenance of inflammation in chronic lung disease and the ongoing destructive process. This is normally counteracted by the actions of antiproteinases (SLPI, α1-antitrypsin), but overwhelming this central mechanism leads to the ongoing lung destruction. Sputum elastase activity appears to correlate with disease activity, severity, and other inflammatory markers, even in stable-state bronchiectasis. Antielastase therapy may therefore also provide a theoretic disease-modifying therapy for this condition. Indeed, studies of such an approach in CF patients show potential beneficial effects (see Chapter 44).
Management
Surgery
With the advent of the antibiotic era, surgery with the aim of symptomatic relief or cure of bronchiectasis is rare. Current indicators for surgery include severe symptoms unrelieved by usual medical management, lung abscess, or major hemoptysis (see Complications). Removal of a segment or lobe would be considered, especially if a proximal obstruction is leading to distal disease. In principle, however, the bronchiectasis should be localized rather than diffuse, and because many patients tend to have multiple segments of the lung affected, “cure” is not possible by limited resection. Perioperative mortality is reported to be low.
Asad S, Opal S. Bench-to-bedside review: quorum sensing and the role of cell-to-cell communication during invasive bacterial infection. Crit Care. 2008;12:236.
British Thoracic Society Bronchiectasis (non-CF) Guideline Group Guideline for non-CF bronchiectasis. Thorax. 2010;65(Suppl 1). www.brit-thoracic.org.uk.
Cymbala AA, Edmonds LC, Bauer MA, et al. The disease modifying effects of twice weekly azithromycin in patients with bronchiectasis. Treat Respir Med. 2005;4:117–122.
Dupont M, Gacouin A, Lena H, et al. Survival of patients with bronchiectasis after the first ICU stay for respiratory failure. Chest. 2004;125:1815–1820.
Finklea JD, Khan G, Thomas S. Predictors of mortality in hospitalized patients with acute exacerbation of bronchiectasis. Respir Med. 2010;104:816–821.
Hill AT, Campbell EJ, Hill SL, et al. Association between airway bacterial load and markers of airway inflammation in patients with stable chronic bronchitis. Am J Med. 2000;109:288–295.
Hornick DB, Fick RB. The immunoglobulin G subclass composition of immune complexes in cystic fibrosis: implications for the pathogenesis of the Pseudomonas lung lesion. J Clin Invest. 1990;86:185–192.
Jean-Baptiste E. Clinical assessment and management of massive hemoptysis (review). Crit Care Med. 2000;28:1642–1647.
Keistenan T, Saynajakangas O, Tuuponen T, et al. Bronchiectasis: an orphan disease with a poorly understood prognosis. Eur Respir J. 1997;10:2784–2787.
King PT, Holdsworth SR, Farmer M, et al. Phenotypes of adult bronchiectasis: onset of productive cough in childhood and adulthood. J COPD. 2009;6:130–136.
Martinez-Garcia MA, Perpina-Tordera M, Roman-Sanchez P, et al. Quality of life determinants in patients with clinically stable bronchiectasis. Chest. 2005;128:739–745.
Martinez Garcia MA, Soler-Cataluna JJ, Perpino-Tordera M. Factors associated with lung function decline in adult patients with non–cystic fibrosis bronchiectasis. Chest. 2007;132:1565–1572.
Mikami M, Llewellyn-Jones CG, Bayley D, et al. The chemotactic activity of sputum from patients with bronchiectasis. Am J Respir Crit Care Med. 1998;157:723.
Pasteur M, Helliwell SM, Houghton SJ, et al. An investigation into causative factors in patients with bronchiectasis. Am J Respir Crit Care Med. 2001;162:1277–1284.
Tsang KW, Chan K, Ho P, et al. Sputum elastase in steady-state bronchiectasis. Chest. 2000;117:420–426.