FIGURE 105-4 Influence of vismodegib on the hedgehog (Hh) pathway. Normally, one of three Hh ligands (sonic [SHh], Indian, or desert) binds to patched homolog 1 (PTCH1), causing its degradation and release of smoothened homolog (SMO). The downstream events of SMO release are the activation of Gli1, Gli2, and Gli3 through the transcriptional regulator known as SUFU. Gli1 and Gli2 translocate to the nucleus and promote gene transcription. Vismodegib is an SMO antagonist that decreases the interaction between SMO and PTCH1, resulting in decreased Hh pathway signaling, gene transcription, and cell division. The downstream Hh pathway events inhibited by vismodegib are indicated in red.
CLINICAL PRESENTATION
Basal Cell Carcinoma BCC arises from epidermal basal cells. The least invasive of BCC subtypes, superficial BCC, consists of often subtle, erythematous scaling plaques that slowly enlarge and are most commonly seen on the trunk and proximal extremities (Fig. 105-5). This BCC subtype may be confused with benign inflammatory dermatoses, especially nummular eczema and psoriasis. BCC also can present as a small, slowly growing pearly nodule, often with tortuous telangiectatic vessels on its surface, rolled borders, and a central crust (nodular BCC). The occasional presence of melanin in this variant of nodular BCC (pigmented BCC) may lead to confusion with melanoma. Morpheaform (fibrosing), infiltrative, and micronodular BCC, the most invasive and potentially aggressive subtypes, manifest as solitary, flat or slightly depressed, indurated whitish, yellowish, or pink scar-like plaques. Borders are typically indistinct, and lesions can be subtle; thus, delay in treatment is common, and tumors can be more extensive than expected clinically.
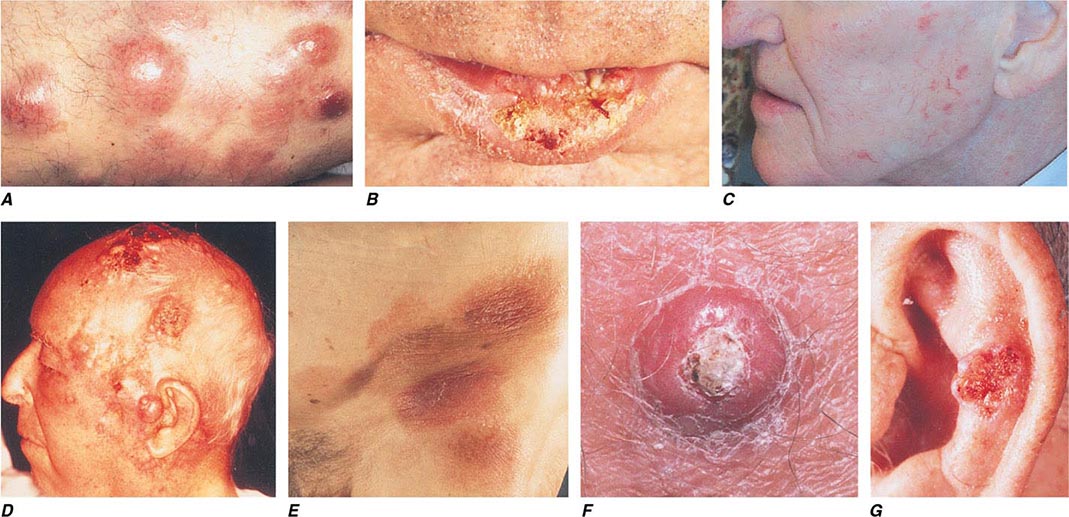
FIGURE 105-5 Cutaneous neoplasms. A. Non-Hodgkin’s lymphoma involves the skin with typical violaceous, “plum-colored” nodules. B. Squamous cell carcinoma is seen here as a hyperkeratotic crusted and somewhat eroded plaque on the lower lip. Sun-exposed skin in areas such as the head, neck, hands, and arms represent other typical sites of involvement. C. Actinic keratoses consist of hyperkeratotic erythematous papules and patches on sun-exposed skin. They arise in middle-aged to older adults and have some potential for malignant transformation. D. Metastatic carcinoma to the skin is characterized by inflammatory, often ulcerated dermal nodules. E. Mycosis fungoides is a cutaneous T cell lymphoma, and plaque-stage lesions are seen in this patient. F. Keratoacanthoma is a low-grade squamous cell carcinoma that presents as an exophytic nodule with central keratinous debris. G. This basal cell carcinoma shows central ulceration and a pearly, rolled telangiectatic tumor border.
Squamous Cell Carcinoma Primary cutaneous SCC is a malignant neoplasm of keratinizing epidermal cells. SCC has a variable clinical course, ranging from indolent to rapid growth kinetics, with the potential for metastasis to regional and distant sites. Commonly, SCC appears as an ulcerated erythematous nodule or superficial erosion on sun-exposed skin of the head, neck, trunk, and extremities (Fig. 105-5). It may also appear as a banal, firm, dome-shaped papule or rough-textured plaque. It is commonly mistaken for a wart or callous when the inflammatory response to the lesion is minimal. Clinically visible overlying telangiectasias are uncommon, although dotted or coiled vessels are a hallmark of SCC when viewed through a dermatoscope. The margins of this tumor may be ill defined, and fixation to underlying structures may occur (“tethering”).
A very rapidly growing but low-grade form of SCC, called keratoacanthoma (KA), typically appears as a large dome-shaped papule with a central keratotic crater. Some KAs regress spontaneously without therapy, but because progression to metastatic SCC has been documented, KAs should be treated in the same manner as other types of cutaneous SCC. KAs are also associated with medications that target BRAF mutations and occur in 15–25% of patients receiving these medications.
Actinic keratoses and cheilitis (actinic keratoses occurring on the lip), both premalignant forms of SCC, present as hyperkeratotic papules on sun-exposed areas. The potential for malignant degeneration in untreated lesions ranges from 0.25 to 20%. SCC in situ, also called Bowen’s disease, is the intraepidermal form of SCC and usually presents as a scaling, erythematous plaque. As with invasive SCC, SCC in situ most commonly arises on sun-damaged skin, but can occur anywhere on the body. Bowen’s disease forming secondary to infection with human papillomavirus (HPV) can arise on skin with minimal or no prior sun exposure, such as the buttock or posterior thigh. Treatment of premalignant and in situ lesions reduces the subsequent risk of invasive disease.
NATURAL HISTORY
Basal Cell Carcinoma The natural history of BCC is that of a slowly enlarging, locally invasive neoplasm. The degree of local destruction and risk of recurrence vary with the size, duration, location, and histologic subtype of the tumor. Location on the central face, ears, or scalp may portend a higher risk. Small nodular, pigmented, cystic, or superficial BCCs respond well to most treatments. Large lesions and micronodular, infiltrative, and morpheaform subtypes may be more aggressive. The metastatic potential of BCC is low (0.0028–0.1% in immunocompetent patients), but the risk of recurrence or a new primary NMSC is about 40% over 5 years.
Squamous Cell Carcinoma The natural history of SCC depends on tumor and host characteristics. Tumors arising on sun-damaged skin have a lower metastatic potential than do those on non-sun-exposed areas. Cutaneous SCC metastasizes in 0.3–5.2% of individuals, most frequently to regional lymph nodes. Tumors occurring on the lower lip and ear develop regional metastases in 13 and 11% of patients, respectively, whereas the metastatic potential of SCC arising in scars, chronic ulcerations, and genital or mucosal surfaces is higher. Recurrent SCC has a much higher potential for metastatic disease, approaching 30%. Large, poorly differentiated, deep tumors with perineural or lymphatic invasion, multifocal tumors, and those arising in immunosuppressed patients often behave aggressively.
PREVENTION
The general principles for prevention are those described for melanoma earlier. Unique strategies for NMSC include active surveillance for patients on immunosuppressive medications or BRAF-targeted therapy. Chemoprophylaxis using synthetic retinoids and immunosuppression reduction when possible may be useful in controlling new lesions and managing patients with multiple tumors.
OTHER NONMELANOMA CUTANEOUS MALIGNANCIES
Neoplasms of cutaneous adnexae and sarcomas of fibrous, mesenchymal, fatty, and vascular tissues make up the remaining 1–2% of NMSCs.
Merkel cell carcinoma (MCC) is a neural crest–derived highly aggressive malignancy with mortality rates approaching 33% at 3 years. An oncogenic Merkel cell polyomavirus is present in 80% of tumors. Many patients have detectable cellular or humoral immune responses to polyoma viral proteins, although this immune response is insufficient to eradicate the malignancy. Survival depends on extent of disease: 90% survive with local disease, 52% with nodal involvement, and only 10% with distant disease at 3 years. MCC incidence tripled over the last 20 years with an estimated 1600 cases per year in the United States. Immunosuppression can increase incidence and diminish prognosis. MCC lesions typically present as an asymptomatic rapidly expanding bluish-red/violaceous tumor on sun-exposed skin of older white patients. Treatment is surgical excision with sentinel lymph node biopsy for accurate staging in patients with localized disease, often followed by adjuvant RT. Patients with extensive disease can be offered systemic chemotherapy; however, there is no convincing survival benefit. Whenever possible a clinical trial should be considered for this rare but aggressive NMSC, especially in light of the potential for new treatments directed at the oncogenic virus that causes this malignancy.
Extramammary Paget’s disease is an uncommon apocrine malignancy arising from stem cells of the epidermis that are characterized histologically by the presence of Paget cells. These tumors present as moist erythematous patches on anogenital or axillary skin of the elderly. Outcomes are generally good with site-directed surgery, and 5-year disease specific survival is approximately 95% with localized disease. Advanced age and extensive disease at presentation are factors that confer diminished prognosis. RT or topical imiquimod can be considered for more extensive disease. Local management may be challenging because these tumors often extend far beyond clinical margins; surgical excision with MMS has the highest cure rates. Similarly, MMS is the treatment of choice in other rare cutaneous tumors with extensive subclinical extension such as dermatofibromasarcoma protuberans.
Kaposi’s sarcoma (KS) is a soft tissue sarcoma of vascular origin that is induced by the human herpesvirus 8. The incidence of KS increased dramatically during the AIDS epidemic, but has now decreased tenfold with the institution of highly active antiretroviral therapy.
ACKNOWLEDGMENT
Carl V. Washington, MD, and Hari Nadiminti, MD, contributed to this chapter in the 18th edition, and material from that chapter is included here. Claudia Taylor, MD, and Steven Kolker, MD, provided valued feedback and suggested many improvements to this chapter.
106 |
Head and Neck Cancer |
Epithelial carcinomas of the head and neck arise from the mucosal surfaces in the head and neck and typically are squamous cell in origin. This category includes tumors of the paranasal sinuses, the oral cavity, and the nasopharynx, oropharynx, hypopharynx, and larynx. Tumors of the salivary glands differ from the more common carcinomas of the head and neck in etiology, histopathology, clinical presentation, and therapy. They are rare and histologically highly heterogeneous. Thyroid malignancies are described in Chap. 405.
INCIDENCE AND EPIDEMIOLOGY
![]() The number of new cases of head and neck cancers (oral cavity, pharynx, and larynx) in the United States was 53,640 in 2013, accounting for about 3% of adult malignancies; 11,520 people died from the disease. The worldwide incidence exceeds half a million cases annually. In North America and Europe, the tumors usually arise from the oral cavity, oropharynx, or larynx. The incidence of oropharyngeal cancers is increasing in recent years. Nasopharyngeal cancer is more commonly seen in the Mediterranean countries and in the Far East, where it is endemic in some areas.
The number of new cases of head and neck cancers (oral cavity, pharynx, and larynx) in the United States was 53,640 in 2013, accounting for about 3% of adult malignancies; 11,520 people died from the disease. The worldwide incidence exceeds half a million cases annually. In North America and Europe, the tumors usually arise from the oral cavity, oropharynx, or larynx. The incidence of oropharyngeal cancers is increasing in recent years. Nasopharyngeal cancer is more commonly seen in the Mediterranean countries and in the Far East, where it is endemic in some areas.
ETIOLOGY AND GENETICS
Alcohol and tobacco use are the most significant risk factors for head and neck cancer, and when used together, they act synergistically. Smokeless tobacco is an etiologic agent for oral cancers. Other potential carcinogens include marijuana and occupational exposures such as nickel refining, exposure to textile fibers, and woodworking.
Some head and neck cancers have a viral etiology. Epstein-Barr virus (EBV) infection is frequently associated with nasopharyngeal cancer, especially in endemic areas of the Mediterranean and Far East. EBV antibody titers can be measured to screen high-risk populations. Nasopharyngeal cancer has also been associated with consumption of salted fish and in-door pollution.
In Western countries, the human papilloma virus (HPV) is associated with a rising incidence of tumors arising from the oropharynx, i.e., the tonsillar bed and base of tongue. Over 50% of oropharyngeal tumors are caused by HPV in the United States. HPV 16 is the dominant viral subtype, although HPV 18 and other oncogenic subtypes are seen as well. Alcohol- and tobacco-related cancers, on the other hand, have decreased in incidence. HPV-related oropharyngeal cancer occurs in a younger patient population and is associated with increased numbers of sexual partners and oral sexual practices. It is associated with a better prognosis, especially for nonsmokers.
Dietary factors may contribute. The incidence of head and neck cancer is higher in people with the lowest consumption of fruits and vegetables. Certain vitamins, including carotenoids, may be protective if included in a balanced diet. Supplements of retinoids, such as cis-retinoic acid, have not been shown to prevent head and neck cancers (or lung cancer) and may increase the risk in active smokers. No specific risk factors or environmental carcinogens have been identified for salivary gland tumors.
HISTOPATHOLOGY, CARCINOGENESIS, AND MOLECULAR BIOLOGY
Squamous cell head and neck cancers are divided into well-differentiated, moderately well-differentiated, and poorly differentiated categories. Poorly differentiated tumors have a worse prognosis than well-differentiated tumors. For nasopharyngeal cancers, the less common differentiated squamous cell carcinoma is distinguished from nonker-atinizing and undifferentiated carcinoma (lymphoepithelioma) that contains infiltrating lymphocytes and is commonly associated with EBV.
Salivary gland tumors can arise from the major (parotid, submandibular, sublingual) or minor salivary glands (located in the submucosa of the upper aerodigestive tract). Most parotid tumors are benign, but half of submandibular and sublingual gland tumors and most minor salivary gland tumors are malignant. Malignant tumors include mucoepidermoid and adenoid cystic carcinomas and adenocarcinomas.
The mucosal surface of the entire pharynx is exposed to alcohol- and tobacco-related carcinogens and is at risk for the development of a premalignant or malignant lesion. Erythroplakia (a red patch) or leukoplakia (a white patch) can be histopathologically classified as hyperplasia, dysplasia, carcinoma in situ, or carcinoma. However, most head and neck cancer patients do not present with a history of premalignant lesions. Multiple synchronous or metachronous cancers can also be observed. In fact, over time, patients with early-stage head and neck cancer are at greater risk of dying from a second malignancy than from a recurrence of the primary disease.
Second head and neck malignancies are usually not therapy-induced; they reflect the exposure of the upper aerodigestive mucosa to the same carcinogens that caused the first cancer. These second primaries develop in the head and neck area, the lung, or the esophagus. Thus, computed tomography (CT) screening for lung cancer in heavy smokers who have already developed a head and neck cancer should be considered. Rarely, patients can develop a radiation therapy–induced sarcoma after having undergone prior radiotherapy for a head and neck cancer.
Much progress has been made in describing the molecular features of head and neck cancer. These features have allowed investigators to describe the genetic and epigenetic alterations and the mutational spectrum of these tumors. Early reports demonstrated frequent overexpression of the epidermal growth factor receptor (EGFR). Overexpression was shown to correlate with poor prognosis. However, it has not proved to be a good predictor of tumor response to EGFR inhibitors, which are successful in only about 10–15% of patients. p53 mutations are also found frequently with other major affected oncogenic driver pathways including the mitotic signaling and Notch pathways and cell cycle regulation. The PI3K pathway is frequently altered, especially in HPV-positive tumors, where it is the only mutated cancer gene identified to date. Overall, these alterations affect mitogenic signaling, genetic stability, cellular proliferation, and differentiation. HPV is known to act through inhibition of the p53 and RB tumor-suppressor genes, thereby initiating the carcinogenic process, and has a mutational spectrum distinct from alcohol- and tobacco-related tumors.
CLINICAL PRESENTATION AND DIFFERENTIAL DIAGNOSIS
Most tobacco-related head and neck cancers occur in patients older than age 60 years. HPV-related malignancies are frequently diagnosed in younger patients, usually in their forties or fifties, whereas EBV-related nasopharyngeal cancer can occur in all ages, including teenagers. The manifestations vary according to the stage and primary site of the tumor. Patients with nonspecific signs and symptoms in the head and neck area should be evaluated with a thorough otolaryngologic exam, particularly if symptoms persist longer than 2–4 weeks. Males are more frequently affected than women by head and neck cancers, including HPV-positive tumors.
Cancer of the nasopharynx typically does not cause early symptoms. However, it may cause unilateral serous otitis media due to obstruction of the eustachian tube, unilateral or bilateral nasal obstruction, or epistaxis. Advanced nasopharyngeal carcinoma causes neuropathies of the cranial nerves due to skull base involvement.
Carcinomas of the oral cavity present as nonhealing ulcers, changes in the fit of dentures, or painful lesions. Tumors of the tongue base or oropharynx can cause decreased tongue mobility and alterations in speech. Cancers of the oropharynx or hypopharynx rarely cause early symptoms, but they may cause sore throat and/or otalgia. HPV-related tumors frequently present with neck lymphadenopathy as the first sign.
Hoarseness may be an early symptom of laryngeal cancer, and persistent hoarseness requires referral to a specialist for indirect laryngoscopy and/or radiographic studies. If a head and neck lesion treated initially with antibiotics does not resolve in a short period, further workup is indicated; to simply continue the antibiotic treatment may be to lose the chance of early diagnosis of a malignancy.
Advanced head and neck cancers in any location can cause severe pain, otalgia, airway obstruction, cranial neuropathies, trismus, odynophagia, dysphagia, decreased tongue mobility, fistulas, skin involvement, and massive cervical lymphadenopathy, which may be unilateral or bilateral. Some patients have enlarged lymph nodes even though no primary lesion can be detected by endoscopy or biopsy; these patients are considered to have carcinoma of unknown primary (Fig. 106-1). If the enlarged nodes are located in the upper neck and the tumor cells are of squamous cell histology, the malignancy probably arose from a mucosal surface in the head or neck. Tumor cells in supraclavicular lymph nodes may also arise from a primary site in the chest or abdomen.
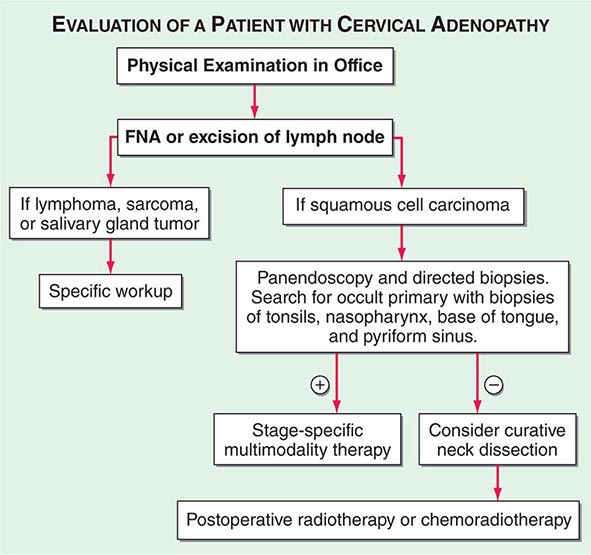
FIGURE 106-1 Evaluation of a patient with cervical adenopathy without a primary mucosal lesion; a diagnostic workup. FNA, fine-needle aspiration.
The physical examination should include inspection of all visible mucosal surfaces and palpation of the floor of the mouth and of the tongue and neck. In addition to tumors themselves, leukoplakia (a white mucosal patch) or erythroplakia (a red mucosal patch) may be observed; these “premalignant” lesions can represent hyperplasia, dysplasia, or carcinoma in situ and require biopsy. Further examination should be performed by a specialist. Additional staging procedures include CT of the head and neck to identify the extent of the disease. Patients with lymph node involvement should have CT scan of the chest and upper abdomen to screen for distant metastases. In heavy smokers, the CT scan of the chest can also serve as a screening tool to rule out a second lung primary tumor. A positron emission tomography (PET) scan may also be administered and can help to identify or exclude distant metastases. The definitive staging procedure is an endoscopic examination under anesthesia, which may include laryngoscopy, esophagoscopy, and bronchoscopy; during this procedure, multiple biopsy samples are obtained to establish a primary diagnosis, define the extent of primary disease, and identify any additional premalignant lesions or second primaries.
Head and neck tumors are classified according to the tumor-node-metastasis (TNM) system of the American Joint Committee on Cancer (Fig. 106-2). This classification varies according to the specific anatomic subsite. In general, primary tumors are classified as T1 to T3 by increasing size, whereas T4 usually represents invasion of another structure such as bone, muscle, or root of tongue. Lymph nodes are staged by size, number, and location (ipsilateral vs contralateral to the primary). Distant metastases are found in <10% of patients at initial diagnosis and are more common in patients with advanced lymph node stage; microscopic involvement of the lungs, bones, or liver is more common, particularly in patients with advanced neck lymph node disease. Modern imaging techniques may increase the number of patients with clinically detectable distant metastases in the future.
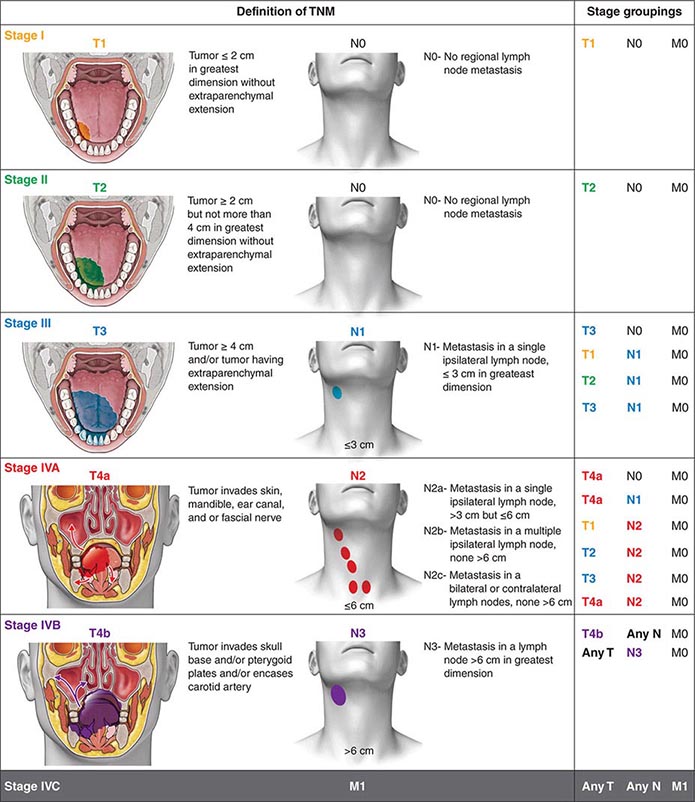
FIGURE 106-2 Tumor-node-metastasis (TNM) staging system.
In patients with lymph node involvement and no visible primary, the diagnosis should be made by lymph node excision (Fig. 106-1). If the results indicate squamous cell carcinoma, a panendoscopy should be performed, with biopsy of all suspicious-appearing areas and directed biopsies of common primary sites, such as the nasopharynx, tonsil, tongue base, and pyriform sinus. HPV-positive tumors especially can have small primary tumors that spread early to locoregional lymph nodes.
SALIVARY GLAND TUMORS
Most benign salivary gland tumors are treated with surgical excision, and patients with invasive salivary gland tumors are treated with surgery and radiation therapy. These tumors may recur regionally; adenoid cystic carcinoma has a tendency to recur along the nerve tracks. Distant metastases may occur as late as 10–20 years after the initial diagnosis. For metastatic disease, therapy is given with palliative intent, usually chemotherapy with doxorubicin and/or cisplatin. Identification of novel agents with activity in these tumors is a high priority.
107 |
Neoplasms of the Lung |
Lung cancer, which was rare prior to 1900 with fewer than 400 cases described in the medical literature, is considered a disease of modern man. By the mid-twentieth century, lung cancer had become epidemic and firmly established as the leading cause of cancer-related death in North America and Europe, killing over three times as many men as prostate cancer and nearly twice as many women as breast cancer. This fact is particularly troubling because lung cancer is one of the most preventable of all of the major malignancies. Tobacco consumption is the primary cause of lung cancer, a reality firmly established in the mid-twentieth century and codified with the release of the U.S. Surgeon General’s 1964 report on the health effects of tobacco smoking. Following the report, cigarette use started to decline in North America and parts of Europe, and with it, so did the incidence of lung cancer. To date, the decline in lung cancer is seen most clearly in men; only recently has the decline become apparent among women in the United States. Unfortunately, in many parts of the world, especially in countries with developing economies, cigarette use continues to increase, and along with it, the incidence of lung cancers is also rising. Although tobacco smoking remains the primary cause of lung cancer worldwide, approximately 60% of new lung cancers in the United States occur in former smokers (smoked ≥100 cigarettes per lifetime, quit ≥1 year), many of whom quit decades ago, or never smokers (smoked <100 cigarettes per lifetime). Moreover, one in five women and one in 12 men diagnosed with lung cancer have never smoked. Given the magnitude of the problem, it is incumbent that every internist has a general knowledge of lung cancer and its management.
EPIDEMIOLOGY
Lung cancer is the most common cause of cancer death among American men and women. More than 225,000 individuals will be diagnosed with lung cancer in the United States in 2013, and over 150,000 individuals will die from the disease. The incidence of lung cancer peaked among men in the late 1980s and has plateaued in women. Lung cancer is rare below age 40, with rates increasing until age 80, after which the rate tapers off. The projected lifetime probability of developing lung cancer is estimated to be approximately 8% among males and approximately 6% among females. The incidence of lung cancer varies by racial and ethnic group, with the highest age-adjusted incidence rates among African Americans. The excess in age-adjusted rates among African Americans occurs only among men, but examinations of age-specific rates show that below age 50, mortality from lung cancer is more than 25% higher among African American than Caucasian women. Incidence and mortality rates among Hispanics and Native and Asian Americans are approximately 40–50% those of whites.
RISK FACTORS
Cigarette smokers have a 10-fold or greater increased risk of developing lung cancer compared to those who have never smoked. A deep sequencing study suggested that one genetic mutation is induced for every 15 cigarettes smoked. The risk of lung cancer is lower among persons who quit smoking than among those who continue smoking; former smokers have a ninefold increased risk of developing lung cancer compared to men who have never smoked versus the 20-fold excess in those who continue to smoke. The size of the risk reduction increases with the length of time the person has quit smoking, although generally even long-term former smokers have higher risks of lung cancer than those who never smoked. Cigarette smoking has been shown to increase the risk of all the major lung cancer cell types. Environmental tobacco smoke (ETS) or second-hand smoke is also an established cause of lung cancer. The risk from ETS is less than from active smoking, with about a 20–30% increase in lung cancer observed among never smokers married for many years to smokers, in comparison to the 2000% increase among continuing active smokers.
Although cigarette smoking is the cause of the majority of lung cancers, several other risk factors have been identified, including occupational exposures to asbestos, arsenic, bischloromethyl ether, hexavalent chromium, mustard gas, nickel (as in certain nickel-refining processes), and polycyclic aromatic hydrocarbons. Occupational observations also have provided insight into possible mechanisms of lung cancer induction. For example, the risk of lung cancer among asbestos-exposed workers is increased primarily among those with underlying asbestosis, raising the possibility that the scarring and inflammation produced by this fibrotic nonmalignant lung disease may in many cases (although likely not in all) be the trigger for asbestos-induced lung cancer. Several other occupational exposures have been associated with increased rates of lung cancer, but the causal nature of the association is not as clear.
The risk of lung cancer appears to be higher among individuals with low fruit and vegetable intake during adulthood. This observation led to hypotheses that specific nutrients, in particular retinoids and carotenoids, might have chemopreventative effects for lung cancer. However, randomized trials failed to validate this hypothesis. In fact, studies found the incidence of lung cancer was increased among smokers with supplementation. Ionizing radiation is also an established lung carcinogen, most convincingly demonstrated from studies showing increased rates of lung cancer among survivors of the atom bombs dropped on Hiroshima and Nagasaki and large excesses among workers exposed to alpha irradiation from radon in underground uranium mining. Prolonged exposure to low-level radon in homes might impart a risk of lung cancer equal or greater than that of ETS. Prior lung diseases such as chronic bronchitis, emphysema, and tuberculosis have been linked to increased risks of lung cancer as well.
Smoking Cessation Given the undeniable link between cigarette smoking and lung cancer (not even addressing other tobacco-related illnesses), physicians must promote tobacco abstinence. Physicians also must help their patients who smoke to stop smoking. Smoking cessation, even well into middle age, can minimize an individual’s subsequent risk of lung cancer. Stopping tobacco use before middle age avoids more than 90% of the lung cancer risk attributable to tobacco. However, there is little health benefit derived from just “cutting back.” Importantly, smoking cessation can even be beneficial in individuals with an established diagnosis of lung cancer, as it is associated with improved survival, fewer side effects from therapy, and an overall improvement in quality of life. Moreover, smoking can alter the metabolism of many chemotherapy drugs, potentially adversely altering the toxicities and therapeutic benefits of the agents. Consequently, it is important to promote smoking cessation even after the diagnosis of lung cancer is established.
Physicians need to understand the essential elements of smoking cessation therapy. The individual must want to stop smoking and must be willing to work hard to achieve the goal of smoking abstinence. Self-help strategies alone only marginally affect quit rates, whereas individual and combined pharmacotherapies in combination with counseling can significantly increase rates of cessation. Therapy with an antidepressant (e.g., bupropion) and nicotine replacement therapy (varenicline, a α4β2 nicotinic acetylcholine receptor partial agonist) are approved by the U.S. Food and Drug Administration (FDA) as first-line treatments for nicotine dependence. However, both drugs have been reported to increase suicidal ideation and must be used with caution. In a randomized trial, varenicline was shown to be more efficacious than bupropion or placebo. Prolonged use of varenicline beyond the initial induction phase proved useful in maintaining smoking abstinence. Clonidine and nortriptyline are recommended as second-line treatments. Of note, reducing cigarettes smoked before quit day and quitting abruptly, with no prior reduction, yield comparable quit rates. Therefore, patients can be given the choice to quit in either of these ways (Chap. 470).
Inherited Predisposition to Lung Cancer Exposure to environmental carcinogens, such as those found in tobacco smoke, induce or facilitate the transformation from bronchoepithelial cells to the malignant phenotype. The contribution of carcinogens on transformation is modulated by polymorphic variations in genes that affect aspects of carcinogen metabolism. Certain genetic polymorphisms of the P450 enzyme system, specifically CYP1A1, and chromosome fragility are associated with the development of lung cancer. These genetic variations occur at relatively high frequency in the population, but their contribution to an individual’s lung cancer risk is generally low. However, because of their population frequency, the overall impact on lung cancer risk could be high. In addition, environmental factors, as modified by inherited modulators, likely affect specific genes by deregulating important pathways to enable the cancer phenotype.
First-degree relatives of lung cancer probands have a two- to threefold excess risk of lung cancer and other cancers, many of which are not smoking-related. These data suggest that specific genes and/or genetic variants may contribute to susceptibility to lung cancer. However, very few such genes have yet been identified. Individuals with inherited mutations in RB (patients with retinoblastoma living to adulthood) and p53 (Li-Fraumeni syndrome) genes may develop lung cancer. Common gene variants involved in lung cancer have been recently identified through large, collaborative, genome-wide association studies. These studies identified three separate loci that are associated with lung cancer (5p15, 6p21, and 15q25) and include genes that regulate acetylcholine nicotinic receptors and telomerase production. A rare germline mutation (T790M) involving the epidermal growth factor receptor (EGFR) maybe be linked to lung cancer susceptibility in never smokers. Likewise, a susceptibility locus on chromosome 6q greatly increases risk lung cancer risk among light and never smokers. Although progress has been made, there is a significant amount of work that remains to be done in identifying heritable risk factors for lung cancer. Currently no molecular criteria are suitable to select patients for more intense screening programs or for specific chemopreventative strategies.
PATHOLOGY
The World Health Organization (WHO) defines lung cancer as tumors arising from the respiratory epithelium (bronchi, bronchioles, and alveoli). The WHO classification system divides epithelial lung cancers into four major cell types: small-cell lung cancer (SCLC), adenocarcinoma, squamous cell carcinoma, and large-cell carcinoma; the latter three types are collectively known as non-small-cell carcinomas (NSCLCs) (Fig. 107-1). Small-cell carcinomas consist of small cells with scant cytoplasm, ill-defined cell borders, finely granular nuclear chromatin, absent or inconspicuous nucleoli, and a high mitotic count. SCLC may be distinguished from NSCLC by the presence of neuroendocrine markers including CD56, neural cell adhesion molecule (NCAM), synaptophysin, and chromogranin. In North America, adenocarcinoma is the most common histologic type of lung cancer. Adenocarcinomas possess glandular differentiation or mucin production and may show acinar, papillary, lepidic, or solid features or a mixture of these patterns. Squamous cell carcinomas of the lung are morphologically identical to extrapulmonary squamous cell carcinomas and cannot be distinguished by immunohistochemistry alone. Squamous cell tumors show keratinization and/or intercellular bridges that arise from bronchial epithelium. The tumor tends to consists of sheets of cells rather than the three-dimensional groups of cells characteristic of adenocarcinomas. Large-cell carcinomas comprise less than 10% of lung carcinomas. These tumors lack the cytologic and architectural features of small-cell carcinoma and glandular or squamous differentiation. Together these four histologic types account for approximately 90% of all epithelial lung cancers.
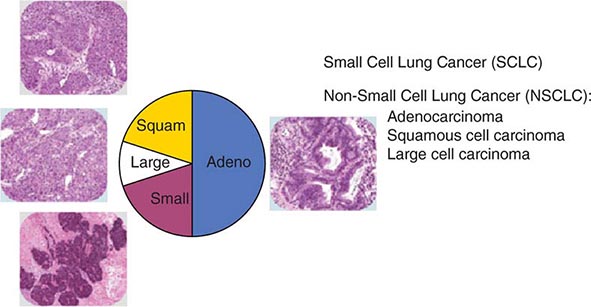
FIGURE 107-1 Traditional histologic view of lung cancer.
All histologic types of lung cancer can develop in current and former smokers, although squamous and small-cell carcinomas are most commonly associated with heavy tobacco use. Through the first half of the twentieth century, squamous carcinoma was the most common subtype of NSCLC diagnosed in the United States. However, with the decline in cigarette consumption over the past four decades, adenocarcinoma has become the most frequent histologic subtype of lung cancer in the United States as both squamous carcinoma and small-cell carcinoma are on the decline. In lifetime never smokers or former light smokers (<10 pack-year history), women, and younger adults (<60 years), adenocarcinoma tends to be the most common form of lung cancer.
Historically, the major pathologic distinction was simply between SCLC and NSCLC, because these tumors have quite different natural histories and therapeutic approaches (see below). Likewise, until fairly recently, there was no apparent need to distinguish among the various subtypes of NSCLC because there were no clear differences in therapeutic outcome based on histology alone. However, this perspective radically changed in 2004 with the recognition that a small percentage of lung adenocarcinomas harbored mutation in EGFR that rendered those tumors exquisitely sensitive to inhibitors of the EGFR tyrosine kinases (e.g., gefitinib and erlotinib). This observation, coupled with the subsequent identification of other “actionable” molecular alterations (Table 107-1) and the recognition that some active chemotherapy agents performed quite differently in squamous carcinomas versus adenocarcinomas, firmly established the need for modifications in the then-existing 2004 WHO lung cancer classification system. The revised 2011 classification system, developed jointly by the International Association for the Study of Lung Cancer, the American Thoracic Society, and the European Respiratory Society, provides an integrated approach to the classification of lung adenocarcinomas that includes clinical, molecular, radiographic, and pathologic information. It also recognizes that most lung cancers present in an advanced stage and are often diagnosed based on small biopsies or cytologic specimens, rendering clear histologic distinctions difficult if not impossible.
|
DRIVER MUTATIONS IN NON-SMALL-CELL LUNG CANCER (NSCLC) |
Previously, in the 2004 classification system, tumors failing to show definite glandular or squamous morphology in a small biopsy or cytologic specimen were simply classified as non-small-cell carcinoma, not otherwise specified. However, because the distinction between adenocarcinoma and squamous carcinoma is now viewed as critical to optimal therapeutic decision making, the modified classification approach recommends these lesions be further characterized using a limited special stain workup. This distinction can be achieved using a single marker for adenocarcinoma (thyroid transcription factor-1 or napsin-A) plus a squamous marker (p40 or p63) and/or mucin stains. The modified classification system also recommends preservation of sufficient specimen material for appropriate molecular testing necessary to help guide therapeutic decision making (see below).
Another significant modification to the WHO classification system is the discontinuation of the terms bronchioloalveolar carcinoma and mixed-subtype adenocarcinoma. The term bronchioloalveolar carcinoma was dropped due to its inconsistent use and because it caused confusion in routine clinical care and research. As formerly used, the term encompassed at least five different entities with diverse clinical and molecular properties. The terms adenocarcinoma in situ and minimally invasive adenocarcinoma are now recommended for small solitary adenocarcinomas (≤3 cm) with either pure lepidic growth (term used to describe single-layered growth of atypical cuboidal cells coating the alveolar walls) or predominant lepidic growth with ≤5 mm invasion. Individuals with these entities experience 100% or near 100% 5-year disease-free survival with complete tumor resection. Invasive adenocarcinomas, representing more than 70–90% of surgically resected lung adenocarcinomas, are now classified by their predominant pattern: lepidic, acinar, papillary, and solid patterns. Lepidic-predominant subtype has a favorable prognosis, acinar and papillary have an intermediate prognosis, and solid-predominant has a poor prognosis. The terms signet ring and clear cell adenocarcinoma have been eliminated from the variants of invasive lung adenocarcinoma, whereas the term micropapillary, a subtype with a particularly poor prognosis, has been added. Although EGFR mutations are encountered most frequently in nonmucinous adenocarcinomas with a lepidic- or papillary-predominant pattern, most adenocarcinoma subtypes can harbor EGFR or KRAS mutations. The same is true of ALK, RET, and ROS1 rearrangements. What was previously termed mucinous bronchioloalveolar carcinoma is now called invasive mucinous adenocarcinoma. These tumors generally lack EGFR mutations and show a strong correlation with KRAS mutations. Overall, the revised WHO reclassification of lung cancer addresses important advances in diagnosis and treatment, most importantly, the critical advances in understanding the specific genes and molecular pathways that initiate and sustain lung tumorigenesis resulting in new “targeted” therapies with improved specificity and better antitumor efficacy.
IMMUNOHISTOCHEMISTRY
The diagnosis of lung cancer most often rests on the morphologic or cytologic features correlated with clinical and radiographic findings. Immunohistochemistry may be used to verify neuroendocrine differentiation within a tumor, with markers such as neuron-specific enolase (NSE), CD56 or NCAM, synaptophysin, chromogranin, and Leu7. Immunohistochemistry is also helpful in differentiating primary from metastatic adenocarcinomas; thyroid transcription factor-1 (TTF-1), identified in tumors of thyroid and pulmonary origin, is positive in over 70% of pulmonary adenocarcinomas and is a reliable indicator of primary lung cancer, provided a thyroid primary has been excluded. A negative TTF-1, however, does not exclude the possibility of a lung primary. TTF-1 is also positive in neuroendocrine tumors of pulmonary and extrapulmonary origin. Napsin-A (Nap-A) is an aspartic protease that plays an important role in maturation of surfactant B7 and is expressed in cytoplasm of type II pneumocytes. In several studies, Nap-A has been reported in >90% of primary lung adenocarcinomas. Notably, a combination of Nap-A and TTF-1 is useful in distinguishing primary lung adenocarcinoma (Nap-A positive, TTF-1 positive) from primary lung squamous cell carcinoma (Nap-A negative, TTF-1 negative) and primary SCLC (Nap-A negative, TTF-1 positive). Cytokeratins 7 and 20 used in combination can help narrow the differential diagnosis; nonsquamous NSCLC, SCLC, and mesothelioma may stain positive for CK7 and negative for CK20, whereas squamous cell lung cancer often will be both CK7 and CK20 negative. p63 is a useful marker for the detection of NSCLCs with squamous differentiation when used in cytologic pulmonary samples. Mesothelioma can be easily identified ultrastructurally, but it has historically been difficult to differentiate from adenocarcinoma through morphology and immunohistochemical staining. Several markers in the last few years have proven to be more helpful including CK5/6, calretinin, and Wilms tumor gene-1 (WT-1), all of which show positivity in mesothelioma.
MOLECULAR PATHOGENESIS
Cancer is a disease involving dynamic changes in the genome. As proposed by Hanahan and Weinberg, virtually all cancer cells acquire six hallmark capabilities: self-sufficiency in growth signals, insensitivity to antigrowth signals, evading apoptosis, limitless replicative potential, sustained angiogenesis, and tissue invasion and metastasis. The order in which these hallmark capabilities are acquired appears quite variable and can differ from tumor to tumor. Events leading to acquisition of these hallmarks can vary widely, although broadly, cancers arise as a result from accumulations of gain-of-function mutations in oncogenes and loss-of-function mutations in tumor-suppressor genes. Further complicating the study of lung cancer, the sequence of events that lead to disease is clearly different for the various histopathologic entities.
The exact cell of origin for lung cancers is not clearly defined. Whether one cell of origin leads to all histologic forms of lung cancer is unclear. However, for lung adenocarcinoma, evidence suggests that type II epithelial cells (or alveolar epithelial cells) have the capacity to give rise to tumors. For SCLC, cells of neuroendocrine origin have been implicated as precursors.
For cancers in general, one theory holds that a small subset of the cells within a tumor (i.e., “stem cells”) are responsible for the full malignant behavior of the tumor. As part of this concept, the large bulk of the cells in a cancer are “offspring” of these cancer stem cells. While clonally related to the cancer stem cell subpopulation, most cells by themselves cannot regenerate the full malignant phenotype. The stem cell concept may explain the failure of standard medical therapies to eradicate lung cancers, even when there is a clinical complete response. Disease recurs because therapies do not eliminate the stem cell component, which may be more resistant to chemotherapy. Precise human lung cancer stem cells have yet to be identified.
Lung cancer cells harbor multiple chromosomal abnormalities, including mutations, amplifications, insertions, deletions, and translocations. One of the earliest sets of oncogenes found to be aberrant was the MYC family of transcription factors (MYC, MYCN, and MYCL). MYC is most frequently activated via gene amplification or transcriptional dysregulation in both SCLC and NSCLC. Currently, there are no MYC-specific drugs.
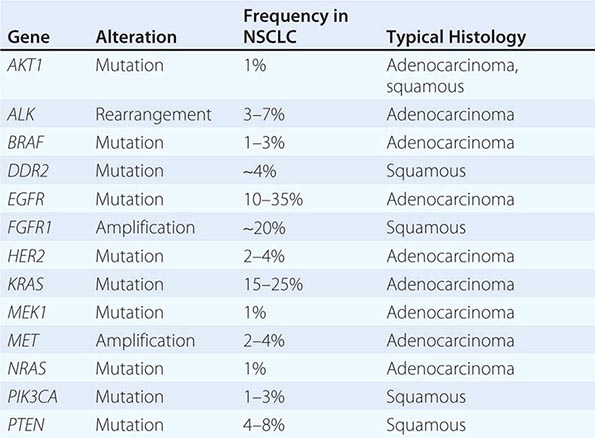
Among lung cancer histologies, adenocarcinomas have been the most extensively catalogued for recurrent genomic gains and losses as well as for somatic mutations (Fig. 107-2). While multiple different kinds of aberrations have been found, a major class involves “driver mutations,” which are mutations that occur in genes encoding signaling proteins that when aberrant, drive initiation and maintenance of tumor cells. Importantly, driver mutations can serve as potential Achilles’ heels for tumors, if their gene products can be targeted appropriately. For example, one set of mutations involves EGFR, which belongs to the ERBB (HER) family of protooncogenes, including EGFR (ERBB1), HER2/neu (ERBB2), HER3 (ERBB3), and HER4 (ERBB4). These genes encode cell-surface receptors consisting of an extracellular ligand-binding domain, a transmembrane structure, and an intracellular tyrosine kinase (TK) domain. The binding of ligand to receptor activates receptor dimerization and TK autophosphorylation, initiating a cascade of intracellular events, and leading to increased cell proliferation, angiogenesis, metastasis, and a decrease in apoptosis. Lung adenocarcinomas can arise when tumors express mutant EGFR. These same tumors display high sensitivity to small-molecule EGFR TK inhibitors (TKIs). Additional examples of driver mutations in lung adenocarcinoma include the GTPase KRAS, the serine-threonine kinase BRAF, and the lipid kinase PIK3CA. More recently, more subsets of lung adenocarcinoma have been identifed as defined by the presence of specific chromsomal rearrangements resulting in the abberant activation of the TKs ALK, ROS1, and RET. Notably, most driver mutations in lung cancer appear to be mutually exclusive, suggesting that acquisition of one of these mutations is sufficient to drive tumorigenesis. Although driver mutations have mostly been found in adenocarinomas, three potential molecular targets recently have been identified in squamous cell lung carcinomas: FGFR1 amplification, DDR2 mutations, and PIK3CA mutations/PTEN loss (Table 107-1). Together, these potentially “actionable” defects occur in up to 50% of squamous carcinomas.
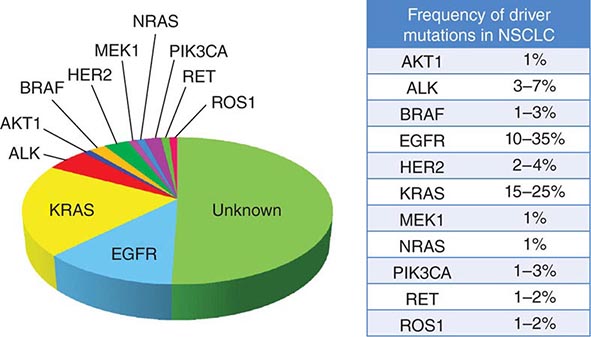
FIGURE 107-2 Driver mutations in adenocarcinomas.
A large number of tumor-suppressor genes have also been identified that are inactivated during the pathogenesis of lung cancer. These include TP53, RB1, RASSF1A, CDKN2A/B, LKB1 (STK11), and FHIT. Nearly 90% of SCLCs harbor mutations in TP53 and RB1. Several tumor-suppressor genes on chromosome 3p appear to be involved in nearly all lung cancers. Allelic loss for this region occurs very early in lung cancer pathogenesis, including in histologically normal smoking-damaged lung epithelium.
EARLY DETECTION AND SCREENING
In lung cancer, clinical outcome is related to the stage at diagnosis, and hence, it is generally assumed that early detection of occult tumors will lead to improved survival. Early detection is a process that involves screening tests, surveillance, diagnosis, and early treatment. Screening refers to the use of simple tests across a healthy population in order to identify individuals who harbor asymptomatic disease. For a screening program to be successful, there must be a high burden of disease within the target population; the test must be sensitive, specific, accessible, and cost effective; and there must be effective treatment that can reduce mortality. With any screening procedure, it is important to consider the possible influence of lead-time bias (detecting the cancer earlier without an effect on survival), length-time bias (indolent cancers are detected on screening and may not affect survival, whereas aggressive cancers are likely to cause symptoms earlier in patients and are less likely to be detected), and overdiagnosis (diagnosing cancers so slow growing that they are unlikely to cause the death of the patient) (Chap. 100).
Because a majority of lung cancer patients present with advanced disease beyond the scope of surgical resection, there is understandable skepticism about the value of screening in this condition. Indeed, randomized controlled trials conducted in the 1960s to 1980s using screening chest x-rays (CXR), with or without sputum cytology, reported no impact on lung cancer–specific mortality in patients characterized as high risk (males age ≥45 years with a smoking history). These studies have been criticized for their design, statistical analyses, and outdated imaging modalities. The results of the more recently conducted Prostate, Lung, Colorectal and Ovarian Cancer Screening Trial (PLCO) are consistent with these earlier reports. Initiated in 1993, participants in the PLCO lung cancer screening trial received annual CXR screening for 4 years, whereas participants in the usual care group received no interventions other than their customary medical care. The diagnostic follow-up of positive screening results was determined by participants and their physicians. The PLCO trial differed from previous lung cancer screening studies in that women and never smokers were eligible. The study was designed to detect a 10% reduction in lung cancer mortality in the interventional group. A total of 154,901 individuals between 55 and 74 years of age were enrolled (77,445 assigned to annual CXR screenings; 77,456 assigned to usual care). Participant demographics and tumor characteristics were well balanced between the two groups. Through 13 years of follow-up, cumulative lung cancer incidence rates (20.1 vs 19.2 per 10,000 person-years; rate ratio [RR], 1.05; 95% confidence interval [CI], 0.98–1.12) and lung cancer mortality (n = 1213 vs n = 1230) were identical between the two groups. The stage and histology of detected cancers in the two groups also were similar. These data corroborate previous recommendations against CXR screening for lung cancer.
In contrast to CXR, low-dose, noncontrast, thin-slice spiral chest computed tomography (LDCT) has emerged as an effective tool to screen for lung cancer. In nonrandomized studies conducted in the 1990s, LDCT scans were shown to detect more lung nodules and cancers than standard CXR in selected high-risk populations (e.g., age ≥60 years and a smoking history of ≥10 pack-years). Notably, up to 85% of the lung cancers discovered in these trials were classified as stage I disease and therefore considered potentially curable with surgical resection.
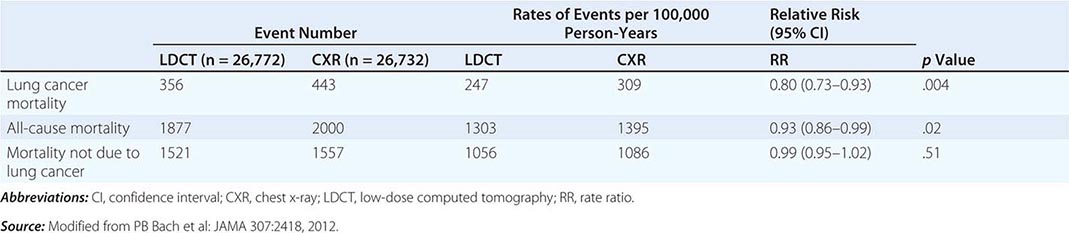
These data prompted the National Cancer Institute (NCI) to initiate the National Lung Screening Trial (NLST), a randomized study designed to determine if LDCT screening could reduce mortality from lung cancer in high-risk populations as compared with standard posterior anterior CXR. High-risk patients were defined as individuals between 55 and 74 years of age, with a ≥30 pack-year history of cigarette smoking; former smokers must have quit within the previous 15 years. Excluded from the trial were individuals with a previous lung cancer diagnosis, a history of hemoptysis, an unexplained weight loss of >15 lb in the preceding year, or a chest CT within 18 months of enrollment. A total of 53,454 persons were enrolled and randomized to annual screening yearly for three years (LDCT screening, n = 26,722; CXR screening, n = 26,732). Any noncalcified nodule measuring ≥4 mm in any diameter found on LDCT and CXR images with any noncalcified nodule or mass were classified as “positive.” Participating radiologists had the option of not calling a final screen positive if a noncalcified nodule had been stable on the three screening exams. Overall, 39.1% of participants in the LDCT group and 16% in the CXR group had at least one positive screening result. Of those who screened positive, the false-positive rate was 96.4% in the LDCT group and 94.5% in the CXR group. This was consistent across all three rounds. In the LDCT group, 1060 cancers were identified compared with 941 cancers in the CXR group (645 vs 572 per 100,000 person-years; RR, 1.13; 95% CI, 1.03 to 1.23). Nearly twice as many early-stage IA cancers were detected in the LDCT group compared with the CXR group (40% vs 21%). The overall rates of lung cancer death were 247 and 309 deaths per 100,000 participants in the LDCT and CXR groups, respectively, representing a 20% reduction in lung cancer mortality in the LDCT-screened population (95% CI, 6.8–26.7%; p = .004). Compared with the CXR group, the rate of death in the LDCT group from any cause was reduced by 6.7% (95% CI, 1.2–13.6; p = .02) (Table 107-2). The number needed to screen (NNTS) to prevent one lung cancer death was calculated to be 320.
|
RESULTS OF NATIONAL LUNG SCREENING TRIAL |
LDCT screening for lung cancer comes with known risks including a high rate of false-positive results, false-negative results, potential for unnecessary follow-up testing, radiation exposure, overdiagnosis, changes in anxiety and quality of life, and substantial financial costs. By far the biggest challenge confronting the use of CT screening is the high false-positive rate. False positives can have a substantial impact on patients through the expense and risk of unneeded further evaluation and emotional stress. The management of these patients usually consists of serial CT scans over time to see if the nodules grow, attempted fine-needle aspirates, or surgical resection. At $300 per scan (NCI estimated cost), the outlay for initial LDCT alone could run into the billions of dollars annually, an expense that only further escalates when factoring in various downstream expenditures an individual might incur in the assessment of positive findings. A formal cost-effectiveness analysis of the NLST is expected soon that should help resolve this crucial concern.
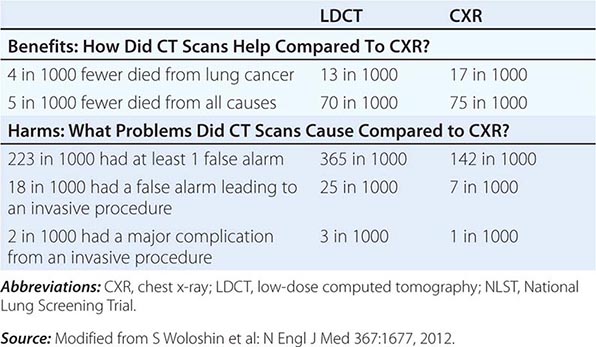
Despite the aforementioned caveats, screening of individuals who meet the NLST criteria for lung cancer risk (or in some cases, modified versions of these criteria) seems warranted, provided comprehensive multidisciplinary coordinated care and follow-up similar to those provided to NLST participants are available. Algorithms to improve candidate selection are under development. When discussing the option of LDCT screening, use of absolute risks rather than relative risks is helpful because studies indicate the public can process absolute terminology more effectively than relative risk projections. A useful guide has been developed by the NCI to help patients and physicians assess the benefits and harms of LDCT screening for lung cancer (Table 107-3). Finally, even a small negative effect of screening on smoking behavior (either lower quit rates or higher recidivism) could easily offset the potential gains in a population. Fortunately no such impact has been reported to date. Nonetheless, smoking cessation must be included as an indispensable component of any screening program.
|
THE BENEFITS AND HARMS OF LDCT SCREENING FOR LUNG CANCER BASED ON NLST DATA |
CLINICAL MANIFESTATIONS
Over half of all patients diagnosed with lung cancer present with locally advanced or metastatic disease at the time of diagnosis. The majority of patients present with signs, symptoms, or laboratory abnormalities that can be attributed to the primary lesion, local tumor growth, invasion or obstruction of adjacent structures, growth at distant metastatic sites, or a paraneoplastic syndrome (Tables 107-4 and 107-5). The prototypical lung cancer patient is a current or former smoker of either sex, usually in the seventh decade of life. A history of chronic cough with or without hemoptysis in a current or former smoker with chronic obstructive pulmonary disease (COPD) age 40 years or older should prompt a thorough investigation for lung cancer even in the face of a normal CXR. A persistent pneumonia without constitutional symptoms and unresponsive to repeated courses of antibiotics also should prompt an evaluation for the underlying cause. Lung cancer arising in a life-time never smoker is more common in women and East Asians. Such patients also tend to be younger than their smoking counterparts at the time of diagnosis. The clinical presentation of lung cancer in never smokers tends to mirror that of current and former smokers.
|
PRESENTING SIGNS AND SYMPTOMS OF LUNG CANCER |
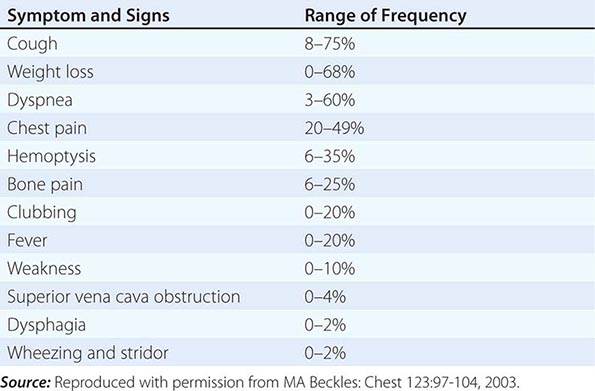
|
CLINICAL FINDINGS SUGGESTIVE OF METASTATIC DISEASE |
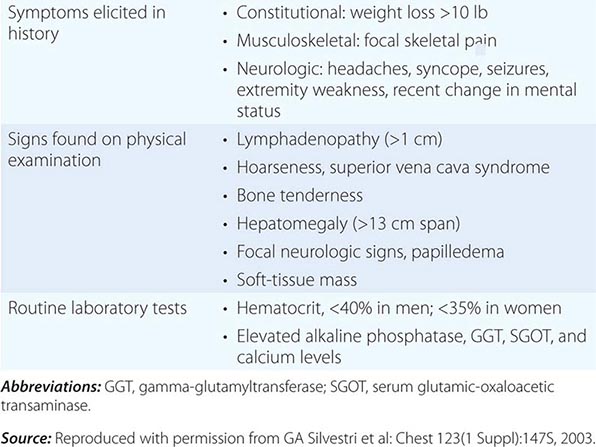
Patients with central or endobronchial growth of the primary tumor may present with cough, hemoptysis, wheeze, stridor, dyspnea, or postobstructive pneumonitis. Peripheral growth of the primary tumor may cause pain from pleural or chest wall involvement, dyspnea on a restrictive basis, and symptoms of a lung abscess resulting from tumor cavitation. Regional spread of tumor in the thorax (by contiguous growth or by metastasis to regional lymph nodes) may cause tracheal obstruction, esophageal compression with dysphagia, recurrent laryngeal paralysis with hoarseness, phrenic nerve palsy with elevation of the hemidiaphragm and dyspnea, and sympathetic nerve paralysis with Horner’s syndrome (enophthalmos, ptosis, miosis, and anhydrosis). Malignant pleural effusions can cause pain, dyspnea, or cough. Pancoast (or superior sulcus tumor) syndromes result from local extension of a tumor growing in the apex of the lung with involvement of the eighth cervical and first and second thoracic nerves, and present with shoulder pain that characteristically radiates in the ulnar distribution of the arm, often with radiologic destruction of the first and second ribs. Often Horner’s syndrome and Pancoast syndrome coexist. Other problems of regional spread include superior vena cava syndrome from vascular obstruction; pericardial and cardiac extension with resultant tamponade, arrhythmia, or cardiac failure; lymphatic obstruction with resultant pleural effusion; and lymphangitic spread through the lungs with hypoxemia and dyspnea. In addition, lung cancer can spread transbronchially, producing tumor growth along multiple alveolar surfaces with impairment of gas exchange, respiratory insufficiency, dyspnea, hypoxemia, and sputum production. Constitutional symptoms may include anorexia, weight loss, weakness, fever, and night sweats. Apart from the brevity of symptom duration, these parameters fail to clearly distinguish SCLC from NSCLC or even from neoplasms metastatic to lungs.
Extrathoracic metastatic disease is found at autopsy in more than 50% of patients with squamous carcinoma, 80% of patients with adenocarcinoma and large-cell carcinoma, and greater than 95% of patients with SCLC. Approximately one-third of patients present with symptoms as a result of distant metastases. Lung cancer metastases may occur in virtually every organ system, and the site of metastatic involvement largely determines other symptoms. Patients with brain metastases may present with headache, nausea and vomiting, seizures, or neurologic deficits. Patients with bone metastases may present with pain, pathologic fractures, or cord compression. The latter may also occur with epidural metastases. Individuals with bone marrow invasion may present with cytopenias or leukoerythroblastosis. Those with liver metastases may present with hepatomegaly, right upper quadrant pain, fever, anorexia, and weight loss. Liver dysfunction and biliary obstructions are rare. Adrenal metastases are common but rarely cause pain or adrenal insufficiency unless they are large.
Paraneoplastic syndromes are common in patients with lung cancer, especially those with SCLC, and may be the presenting finding or the first sign of recurrence. In addition, paraneoplastic syndromes may mimic metastatic disease and, unless detected, lead to inappropriate palliative rather than curative treatment. Often the paraneoplastic syndrome may be relieved with successful treatment of the tumor. In some cases, the pathophysiology of the paraneoplastic syndrome is known, particularly when a hormone with biological activity is secreted by a tumor. However, in many cases, the pathophysiology is unknown. Systemic symptoms of anorexia, cachexia, weight loss (seen in 30% of patients), fever, and suppressed immunity are paraneoplastic syndromes of unknown etiology or at least not well defined. Weight loss greater than 10% of total body weight is considered a bad prognostic sign. Endocrine syndromes are seen in 12% of patients; hypercalcemia resulting from ectopic production of parathyroid hormone (PTH), or more commonly, PTH-related peptide, is the most common life-threatening metabolic complication of malignancy, primarily occurring with squamous cell carcinomas of the lung. Clinical symptoms include nausea, vomiting, abdominal pain, constipation, polyuria, thirst, and altered mental status.
Hyponatremia may be caused by the syndrome of inappropriate secretion of antidiuretic hormone (SIADH) or possibly atrial natriuretic peptide (ANP). SIADH resolves within 1-4 weeks of initiating chemotherapy in the vast majority of cases. During this period, serum sodium can usually be managed and maintained above 128 mEq/L via fluid restriction. Demeclocycline can be a useful adjunctive measure when fluid restriction alone is insufficient. Vasopressin receptor antagonists like tolvaptan also have been used in the management of SIADH. However, there are significant limitations to the use of tolvaptan including liver injury and overly rapid correction of the hyponatremia, which can lead to irreversible neurologic injury. Likewise, the cost of tolvaptan may be prohibitive (as high as $300 per tablet in some areas). Of note, patients with ectopic ANP may have worsening hyponatremia if sodium intake is not concomitantly increased. Accordingly, if hyponatremia fails to improve or worsens after 3–4 days of adequate fluid restriction, plasma levels of ANP should be measured to determine the causative syndrome.
Ectopic secretion of ACTH by SCLC and pulmonary carcinoids usually results in additional electrolyte disturbances, especially hypokalemia, rather than the changes in body habitus that occur in Cushing’s syndrome from a pituitary adenoma. Treatment with standard medications, such as metyrapone and ketoconazole, is largely ineffective due to extremely high cortisol levels. The most effective strategy for management of the Cushing’s syndrome is effective treatment of the underlying SCLC. Bilateral adrenalectomy may be considered in extreme cases.
Skeletal–connective tissue syndromes include clubbing in 30% of cases (usually NSCLCs) and hypertrophic primary osteoarthropathy in 1–10% of cases (usually adenocarcinomas). Patients may develop periostitis, causing pain, tenderness, and swelling over the affected bones and a positive bone scan. Neurologic-myopathic syndromes are seen in only 1% of patients but are dramatic and include the myasthenic Eaton-Lambert syndrome and retinal blindness with SCLC, whereas peripheral neuropathies, subacute cerebellar degeneration, cortical degeneration, and polymyositis are seen with all lung cancer types. Many of these are caused by autoimmune responses such as the development of anti-voltage-gated calcium channel antibodies in Eaton-Lambert syndrome. Patients with this disorder present with proximal muscle weakness, usually in the lower extremities, occasional autonomic dysfunction, and rarely, cranial nerve symptoms or involvement of the bulbar or respiratory muscles. Depressed deep tendon reflexes are frequently present. In contrast to patients with myasthenia gravis, strength improves with serial effort. Some patients who respond to chemotherapy will have resolution of the neurologic abnormalities. Thus, chemotherapy is the initial treatment of choice. Paraneoplastic encephalomyelitis and sensory neuropathies, cerebellar degeneration, limbic encephalitis, and brainstem encephalitis occur in SCLC in association with a variety of antineuronal antibodies such as anti-Hu, anti-CRMP5, and ANNA-3. Paraneoplastic cerebellar degeneration may be associated with anti-Hu, anti-Yo, or P/Q calcium channel autoantibodies. Coagulation or thrombotic or other hematologic manifestations occur in 1–8% of patients and include migratory venous thrombophlebitis (Trousseau’s syndrome), nonbacterial thrombotic (marantic) endocarditis with arterial emboli, and disseminated intravascular coagulation with hemorrhage, anemia, granulocytosis, and leukoerythroblastosis. Thrombotic disease complicating cancer is usually a poor prognostic sign. Cutaneous manifestations such as dermatomyositis and acanthosis nigricans are uncommon (1%), as are the renal manifestations of nephrotic syndrome and glomerulonephritis (≤1%).
DIAGNOSING LUNG CANCER
Tissue sampling is required to confirm a diagnosis in all patients with suspected lung cancer. In patients with suspected metastatic disease, a biopsy of the most distant site of disease is preferred for tissue confirmation. Given the greater emphasis placed on molecular testing for NSCLC patients, a core biopsy is preferred to ensure adequate tissue for analysis. Tumor tissue may be obtained via minimally invasive techniques such as bronchial or transbronchial biopsy during fiberoptic bronchoscopy, by fine-needle aspiration or percutaneous biopsy using image guidance, or via endobronchial ultrasound (EBUS) guided biopsy. Depending on the location, lymph node sampling may occur via transesophageal endoscopic ultrasound-guided biopsy (EUS), EBUS, or blind biopsy. In patients with clinically palpable disease such as a lymph node or skin metastasis, a biopsy may be obtained. In patients with suspected metastatic disease, a diagnosis may be confirmed by percutaneous biopsy of a soft tissue mass, lytic bone lesion, bone marrow, pleural or liver lesion, or an adequate cell block obtained from a malignant pleural effusion. In patients with a suspected malignant pleural effusion, if the initial thoracentesis is negative, a repeat thoracentesis is warranted. Although the majority of pleural effusions are due to malignant disease, particularly if they are exudative or bloody, some may be parapneumonic. In the absence of distant disease, such patients should be considered for possible curative treatment.
The diagnostic yield of any biopsy depends on several factors including location (accessibility) of the tumor, tumor size, tumor type, and technical aspects of the diagnostic procedure including the experience level of the bronchoscopist and pathologist. In general, central lesions such as squamous cell carcinomas, small-cell carcinomas, or endobronchial lesions such as carcinoid tumors are more readily diagnosed by bronchoscopic examination, whereas peripheral lesions such as adenocarcinomas and large-cell carcinomas are more amenable to transthoracic biopsy. Diagnostic accuracy for SCLC versus NSCLC for most specimens is excellent, with lesser accuracy for subtypes of NSCLC.
Bronchoscopic specimens include bronchial brush, bronchial wash, bronchioloalveolar lavage, transbronchial fine-needle aspiration (FNA), and core biopsy. For more accurate histologic classification, mutation analysis, or investigational purposes, reasonable efforts (e.g., a core needle biopsy) should be made to obtain more tissue than what is contained in a routine cytology specimen obtained by FNA. Overall sensitivity for combined use of bronchoscopic methods is approximately 80%, and together with tissue biopsy, the yield increases to 85–90%. Like transbronchial core biopsy specimens, transthoracic core biopsy specimens are also preferred. Sensitivity is highest for larger lesions and peripheral tumors. In general, core biopsy specimens, whether transbronchial, transthoracic, or EUS-guided, are superior to other specimen types. This is primarily due to the higher percentage of tumor cells with fewer confounding factors such as obscuring inflammation and reactive nonneoplastic cells.
Sputum cytology is inexpensive and noninvasive but has a lower yield than other specimen types due to poor preservation of the cells and more variability in acquiring a good-quality specimen. The yield for sputum cytology is highest for larger and centrally located tumors such as squamous cell carcinoma and small-cell carcinoma histology. The specificity for sputum cytology averages close to 100%, although sensitivity is generally <70%. The accuracy of sputum cytology improves with increased numbers of specimens analyzed. Consequently, analysis of at least three sputum specimens is recommended.
STAGING LUNG CANCER
Lung cancer staging consists of two parts: first, a determination of the location of the tumor and possible metastatic sites (anatomic staging), and second, an assessment of a patient’s ability to withstand various antitumor treatments (physiologic staging). All patients with lung cancer should have a complete history and physical examination, with evaluation of all other medical problems, determination of performance status, and history of weight loss. The most significant dividing line is between those patients who are candidates for surgical resection and those who are inoperable but will benefit from chemotherapy, radiation therapy, or both. Staging with regard to a patient’s potential for surgical resection is principally applicable to NSCLC.
ANATOMIC STAGING OF PATIENTS WITH LUNG CANCER
The accurate staging of patients with NSCLC is essential for determining the appropriate treatment in patients with resectable disease and avoiding unnecessary surgical procedures in patients with advanced disease (Fig. 107-3). All patients with NSCLC should undergo initial radiographic imaging with CT scan, positron emission tomography (PET), or preferably CT-PET. PET scanning attempts to identify sites of malignancy based on glucose metabolism by measuring the uptake of 18F-fluorodeoxyglucose (FDG). Rapidly dividing cells, presumably in the lung tumors, will preferentially take up 18F-FDG and appear as a “hot spot.” To date, PET has been mostly used for staging and detection of metastases in lung cancer and in the detection of nodules >15 mm in diameter. Combined 18F-FDG PET-CT imaging has been shown to improve the accuracy of staging in NSCLC compared to visual correlation of PET and CT or either study alone. CT-PET has been found to be superior in identifying pathologically enlarged mediastinal lymph nodes and extrathoracic metastases. A standardized uptake value (SUV) of >2.5 on PET is highly suspicious for malignancy. False negatives can be seen in diabetes, in lesions <8 mm, and in slow-growing tumors (e.g., carcinoid tumors or well-differentiated adenocarcinoma). False positives can be seen in certain infections and granulomatous disease (e.g., tuberculosis). Thus, PET should never be used alone to diagnose lung cancer, mediastinal involvement, or metastases. Confirmation with tissue biopsy is required. For brain metastases, magnetic resonance imaging (MRI) is the most effective method. MRI can also be useful in selected circumstances, such as superior sulcus tumors to rule out brachial plexus involvement, but in general, MRI does not play a major role in NSCLC staging.
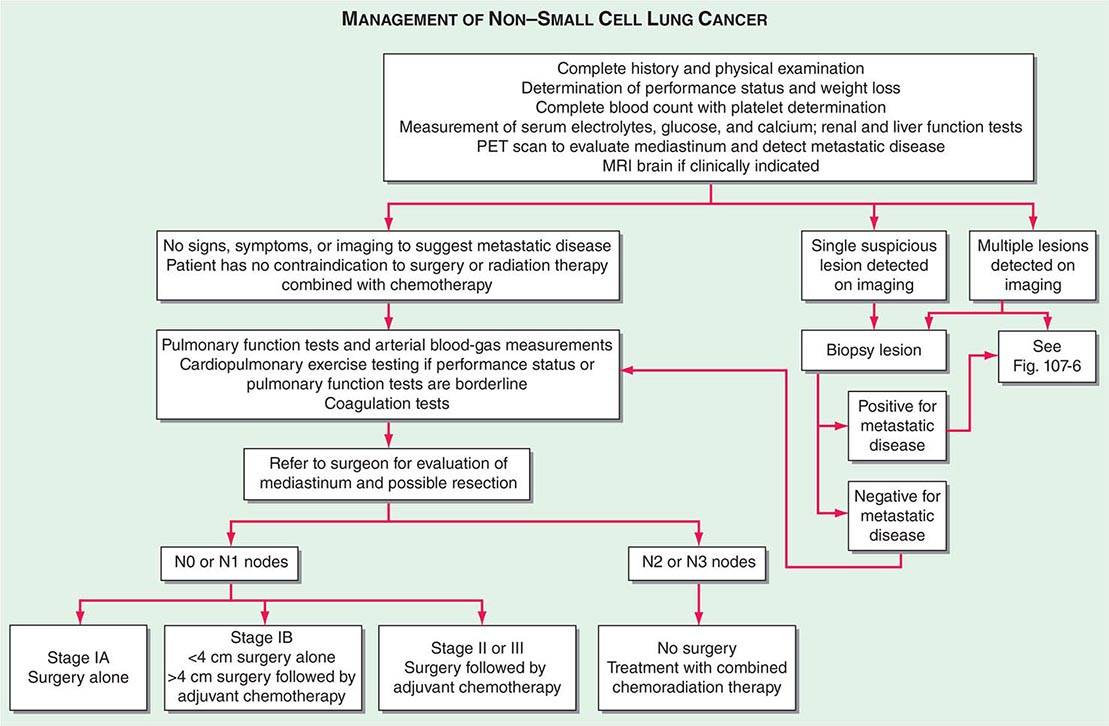
FIGURE 107-3 Algorithm for management of non-small-cell lung cancer. MRI, magnetic resonance imaging; PET, positron emission tomography.
In patients with NSCLC, the following are contraindications to potential curative resection: extrathoracic metastases, superior vena cava syndrome, vocal cord and, in most cases, phrenic nerve paralysis, malignant pleural effusion, cardiac tamponade, tumor within 2 cm of the carina (potentially curable with combined chemoradiotherapy), metastasis to the contralateral lung, metastases to supraclavicular lymph nodes, contralateral mediastinal node metastases (potentially curable with combined chemoradiotherapy), and involvement of the main pulmonary artery. In situations where it will make a difference in treatment, abnormal scan findings require tissue confirmation of malignancy so that patients are not precluded from having potentially curative therapy.
The best predictor of metastatic disease remains a careful history and physical examination. If signs, symptoms, or findings from the physical examination suggest the presence of malignancy, then sequential imaging starting with the most appropriate study should be performed. If the findings from the clinical evaluation are negative, then imaging studies beyond CT-PET are unnecessary and the search for metastatic disease is complete. More controversial is how one should assess patients with known stage III disease. Because these patients are more likely to have asymptomatic occult metastatic disease, current guidelines recommend a more extensive imaging evaluation including imaging of the brain with either CT scan or MRI. In patients in whom distant metastatic disease has been ruled out, lymph node status needs to be assessed via a combination of radiographic imaging and/or minimally invasive techniques such as those mentioned above and/or invasive techniques such as mediastinoscopy, mediastinotomy, thoracoscopy, or thoracotomy. Approximately one-quarter to one-half of patients diagnosed with NSCLC will have mediastinal lymph node metastases at the time of diagnosis. Lymph node sampling is recommended in all patients with enlarged nodes detected by CT or PET scan and in patients with large tumors or tumors occupying the inner third of the lung. The extent of mediastinal lymph node involvement is important in determining the appropriate treatment strategy: surgical resection followed by adjuvant chemotherapy versus combined chemoradiation alone (see below). A standard nomenclature for referring to the location of lymph nodes involved with lung cancer has evolved (Fig. 107-4).
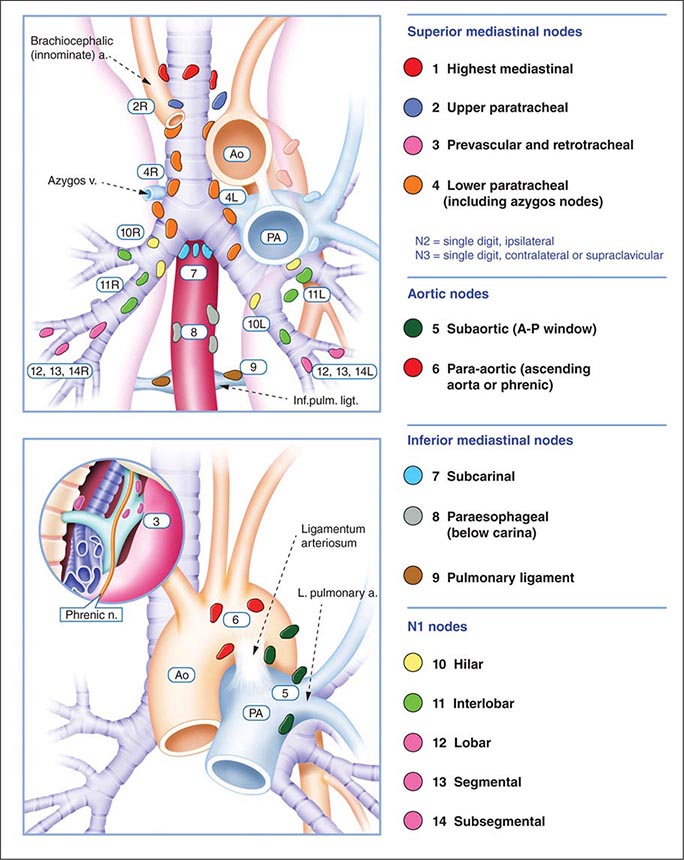
FIGURE 107-4 Lymph node stations in staging non-small-cell lung cancer. The International Association for the Study of Lung Cancer (IASLC) lymph node map, including the proposed grouping of lymph node stations into “zones” for the purposes of prognostic analyses. a., artery; Ao, aorta; Inf. pulm. ligt., inferior pulmonary ligament; n., nerve; PA, pulmonary artery; v., vein.
In SCLC patients, current staging recommendations include a CT scan of the chest and abdomen (because of the high frequency of hepatic and adrenal involvement), MRI of the brain (positive in 10% of asymptomatic patients), and radionuclide bone scan if symptoms or signs suggest disease involvement in these areas (Fig. 107-5). Although there are less data on the use of CT-PET in SCLC, the most recent American College of Chest Physicians Evidence-Based Clinical Practice Guidelines recommend PET scans in patients with clinical stage I SCLC who are being considered for curative intent surgical resection. In addition, invasive mediastinal staging and extrathoracic imaging (head MRI/CT and PET or abdominal CT plus bone scan) is also recommended for patients with clinical stage I SCLC if curative intent surgical resection is contemplated. Some practice guidelines also recommend the use of PET scanning in the staging of SCLC patients who are potential candidates for the addition of thoracic radiotherapy to chemotherapy. Bone marrow biopsies and aspirations are rarely performed now given the low incidence of isolated bone marrow metastases. Confirmation of metastatic disease, ipsilateral or contralateral lung nodules, or metastases beyond the mediastinum may be achieved by the same modalities recommended earlier for patients with NSCLC.
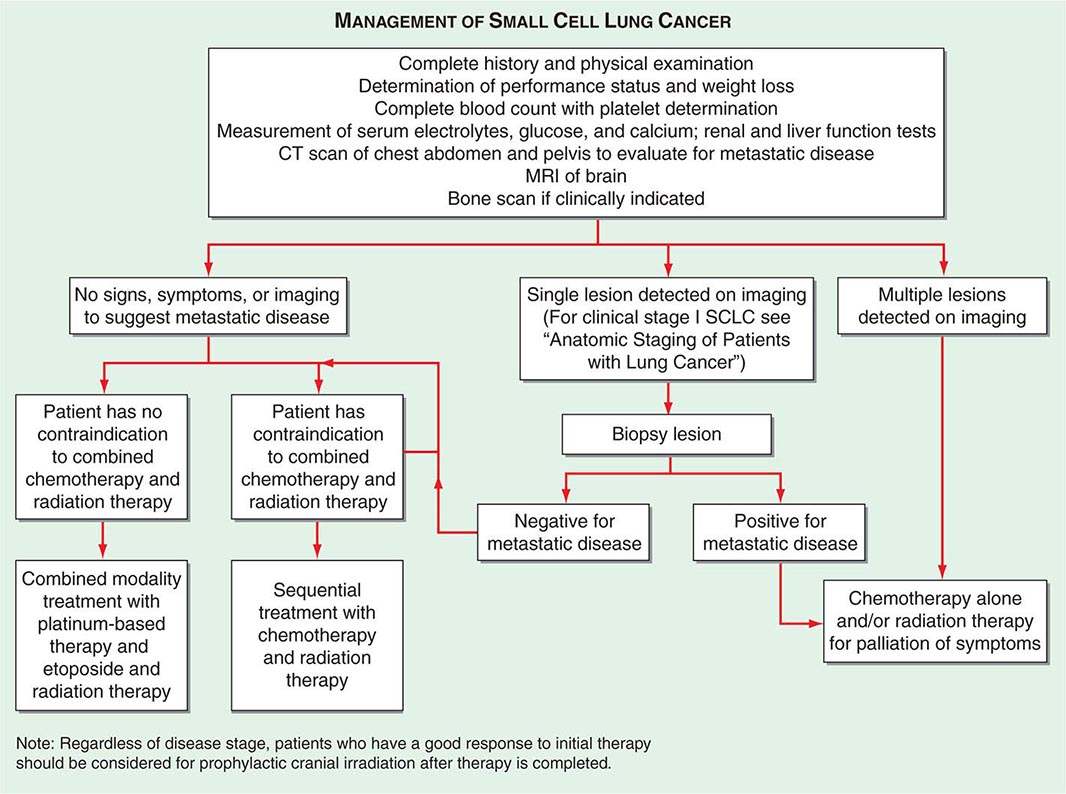
FIGURE 107-5 Algorithm for management of small-cell lung cancer. CT, computed tomography; MRI, magnetic resonance imaging.
If a patient has signs or symptoms of spinal cord compression (pain, weakness, paralysis, urinary retention), a spinal CT or MRI scan and examination of the cerebrospinal fluid cytology should be performed. If metastases are evident on imaging, a neurosurgeon should be consulted for possible palliative surgical resection and/or a radiation oncologist should be consulted for palliative radiotherapy to the site of compression. If signs or symptoms of leptomeningitis develop at any time in a patient with lung cancer, an MRI of the brain and spinal cord should be performed, as well as a spinal tap, for detection of malignant cells. If the spinal tap is negative, a repeat spinal tap should be considered. There is currently no approved therapy for the treatment of leptomeningeal disease.
STAGING SYSTEM FOR NON-SMALL-CELL LUNG CANCER
The tumor-node-metastasis (TNM) international staging system provides useful prognostic information and is used to stage all patients with NSCLC. The various T (tumor size), N (regional node involvement), and M (presence or absence of distant metastasis) are combined to form different stage groups (Tables 107-6 and 107-7). The previous edition of the TNM staging system for lung cancer was developed based on a relatively small database of patients from a single institution. The latest seventh edition of the TNM staging system went into effect in 2010 and developed using a much more robust database of more than 100,000 patients with lung cancer who were treated in multiple countries between 1990 and 2000. Data from 67,725 patients with NSCLC were then used to reevaluate the prognostic value of the TNM descriptors (Table 107-8). The major distinction between the sixth and seventh editions of the international staging systems is within the T classification; T1 tumors are divided into tumors ≤2 cm in size, as these patients were found to have a better prognosis compared to patients with tumors >2 cm but ≤3 cm. T2 tumors are divided into those that are >3 cm but ≤5 cm and those that are >5 cm but ≤7 cm. Tumors that are >7 cm are considered T3 tumors. T3 tumors also include tumors with invasion into local structures such as chest wall and diaphragm and additional nodules in the same lobe. T4 tumors include tumors of any size with invasion into mediastinum, heart, great vessels, trachea, or esophagus or multiple nodules in the ipsilateral lung. No changes have been made to the current classification of lymph node involvement (N). Patients with metastasis may be classified as M1a (malignant pleural or pericardial effusion, pleural nodules, or nodules in the contralateral lung) or M1b (distant metastasis; e.g., bone, liver, adrenal, or brain metastasis). Based on these data, approximately one-third of patients have localized disease that can be treated with curative attempt (surgery or radiotherapy), one-third have local or regional disease that may or may not be amenable to a curative attempt, and one-third have metastatic disease at the time of diagnosis.
|
SEVENTH EDITION TNM STAGING SYSTEMS FOR NON-SMALL-CELL LUNG CANCER |
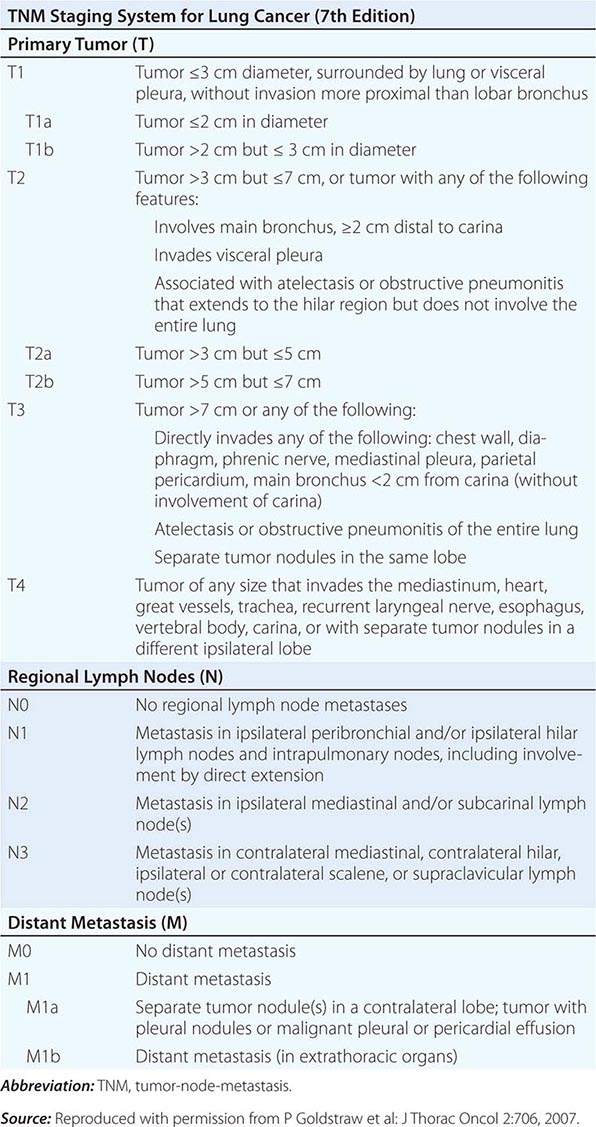
|
SEVENTH EDITION TNM STAGING SYSTEMS FOR NON-SMALL-CELL LUNG CANCER |
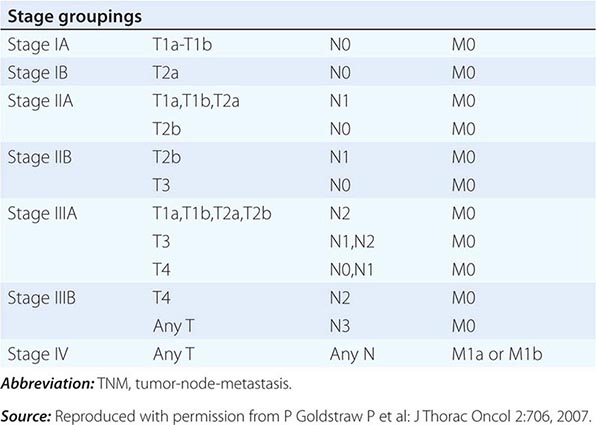
|
FIVE-YEAR SURVIVAL BY STAGE AND TNM CLASSIFICATION OF NON-SMALL-CELL LUNG CANCER (SEVENTH EDITION) |
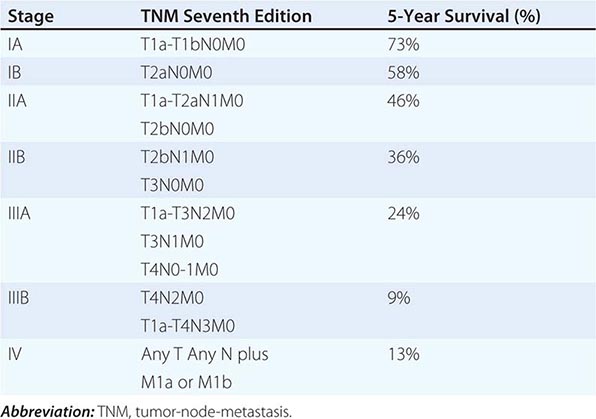
STAGING SYSTEM FOR SMALL-CELL LUNG CANCER
In patients with SCLC, it is now recommended that both the Veterans Administration system and the American Joint Committee on Cancer/International Union Against Cancer seventh edition system (TNM) be used to classify the tumor stage. The Veterans Administration system is a distinct two-stage system dividing patients into those with limited- or extensive-stage disease. Patients with limited-stage disease (LD) have cancer that is confined to the ipsilateral hemithorax and can be encompassed within a tolerable radiation port. Thus, contralateral supraclavicular nodes, recurrent laryngeal nerve involvement, and superior vena caval obstruction can all be part of LD. Patients with extensive-stage disease (ED) have overt metastatic disease by imaging or physical examination. Cardiac tamponade, malignant pleural effusion, and bilateral pulmonary parenchymal involvement generally qualify disease as ED, because the involved organs cannot be encompassed safely or effectively within a single radiation therapy port. Sixty to 70% of patients are diagnosed with ED at presentation. The TNM staging system is preferred in the rare SCLC patient presenting with what appears to be clinical stage I disease (see above).
PHYSIOLOGIC STAGING
Patients with lung cancer often have other comorbid conditions related to smoking including cardiovascular disease and COPD. To improve their preoperative condition, correctable problems (e.g., anemia, electrolyte and fluid disorders, infections, cardiac disease, and arrhythmias) should be addressed, appropriate chest physical therapy should be instituted, and patients should be encouraged to stop smoking. Because it is not always possible to predict whether a lobectomy or pneumonectomy will be required until the time of operation, a conservative approach is to restrict surgical resection to patients who could potentially tolerate a pneumonectomy. Patients with a forced expiratory volume in 1 s (FEV1) of greater than 2 L or greater than 80% of predicted can tolerate a pneumonectomy, and those with an FEV1 greater than 1.5 L have adequate reserve for a lobectomy. In patients with borderline lung function but a resec-table tumor, cardiopulmonary exercise testing could be performed as part of the physiologic evaluation. This test allows an estimate of the maximal oxygen consumption (VO2max). A VO2max <15 mL/(kg⋅min) predicts for a higher risk of postoperative complications. Patients deemed unable to tolerate lobectomy or pneumonectomy from a pulmonary functional standpoint may be candidates for more limited resections, such as wedge or anatomic segmental resection, although such procedures are associated with significantly higher rates of local recurrence and a trend toward decreased overall survival. All patients should be assessed for cardiovascular risk using American College of Cardiology and American Heart Association guidelines. A myocardial infarction within the past 3 months is a contraindication to thoracic surgery because 20% of patients will die of reinfarction. An infarction in the past 6 months is a relative contraindication. Other major contraindications include uncontrolled arrhythmias, an FEV1 of less than 1 L, CO2 retention (resting PCO2 >45 mmHg), DLCO <40%, and severe pulmonary hypertension.
TREATMENT NON-SMALL-CELL LUNG CANCER
The overall treatment approach to patients with NSCLC is shown in Fig. 107-3.
OCCULT AND STAGE 0 CARCINOMAS
Patients with severe atypia on sputum cytology have an increased risk of developing lung cancer compared to those without atypia. In the uncommon circumstance where malignant cells are identified in a sputum or bronchial washing specimen but the chest imaging appears normal (TX tumor stage), the lesion must be localized. More than 90% of tumors can be localized by meticulous examination of the bronchial tree with a fiberoptic bronchoscope under general anesthesia and collection of a series of differential brushings and biopsies. Surgical resection following bronchoscopic localization has been shown to improve survival compared to no treatment. Close follow-up of these patients is indicated because of the high incidence of second primary lung cancers (5% per patient per year).
SOLITARY PULMONARY NODULE AND “GROUND-GLASS” OPACITIES
A solitary pulmonary nodule is defined as an x-ray density completely surrounded by normal aerated lung with circumscribed margins, of any shape, usually 1–6 cm in greatest diameter. The approach to a patient with a solitary pulmonary nodule is based on an estimate of the probability of cancer, determined according to the patient’s smoking history, age, and characteristics on imaging (Table 107-9). Prior CXRs and CT scans should be obtained if available for comparison. A PET scan may be useful if the lesion is greater than 7–8 mm in diameter. If no diagnosis is apparent, Mayo investigators reported that clinical characteristics (age, cigarette smoking status, and prior cancer diagnosis) and three radiologic characteristics (nodule diameter, spiculation, and upper lobe location) were independent predictors of malignancy. At present, only two radiographic criteria are thought to predict the benign nature of a solitary pulmonary nodule: lack of growth over a period >2 years and certain characteristic patterns of calcification. Calcification alone, however, does not exclude malignancy; a dense central nidus, multiple punctuate foci, and “bulls eye” (granuloma) and “popcorn ball” (hamartoma) calcifications are highly suggestive of a benign lesion. In contrast, a relatively large lesion, lack of or asymmetric calcification, chest symptoms, associated atelectasis, pneumonitis, or growth of the lesion revealed by comparison with an old x-ray or CT scan or a positive PET scan may be suggestive of a malignant process and warrant further attempts to establish a histologic diagnosis. An algorithm for assessing these lesions is shown in Fig. 107-6.
|
ASSESSMENT OF RISK OF CANCER IN PATIENTS WITH SOLITARY PULMONARY NODULES |
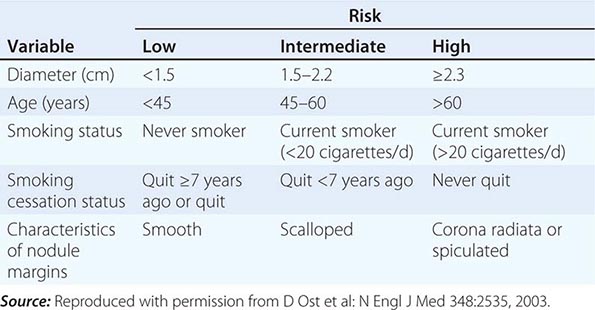
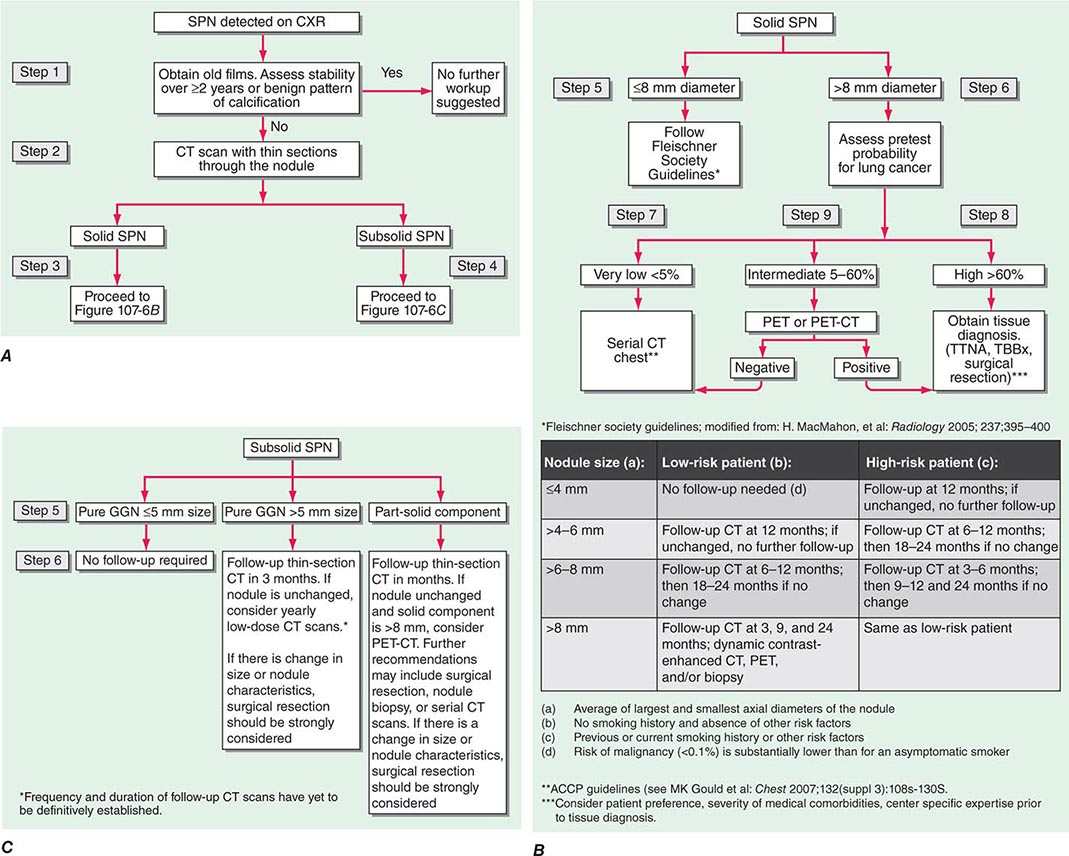
FIGURE 107-6 A. Algorithm for evaluation of solitary pulmonary nodule (SPN). B. Algorithm for evaluation of solid SPN. C. Algorithm for evaluation of semisolid SPN. CT, computed tomography; CXR, chest radiograph; GGN, ground-glass nodule; PET, positron emission tomography; TTBx, transbronchial biopsy; TTNA, transthoracic needle biopsy. (Adapted from VK Patel et al: Chest 143:840, 2013.)
Since the advent of screening CTs, small “ground-glass” opacities (GGOs) have often been observed, particularly as the increased sensitivity of CTs enables detection of smaller lesions. Many of these GGOs, when biopsied, are found to be atypical adenomatous hyperplasia (AAH), adenocarcinoma in situ (AIS), or minimally invasive adenocarcinoma (MIA). AAH is usually a nodule of <5 mm and is minimally hazy, also called nonsolid or ground glass (i.e., hazy slightly increased attenuation, no solid component, and preservation of bronchial and vascular margins). On thin-section CT, AIS is usually a nonsolid nodule and tends to be slightly more opaque than AAH. MIA is mainly solid, usually with a small (<5 mm) central solid component. However, overlap exists among the imaging features of the preinvasive and minimally invasive lesions in the lung adenocarcinoma spectrum. Lepidic adenocarcinomas are usually solid but may be nonsolid. Likewise, the small invasive adenocarcinomas also are usually solid but may exhibit a small nonsolid component.
MANAGEMENT OF STAGES I AND II NSCLC
Surgical Resection of Stage I and II NSCLC Surgical resection, ideally by an experienced thoracic surgeon, is the treatment of choice for patients with clinical stage I and II NSCLC who are able to tolerate the procedure. Operative mortality rates for patients resected by thoracic or cardiothoracic surgeons are lower compared to general surgeons. Moreover, survival rates are higher in patients who undergo resection in facilities with a high surgical volume compared to those performing fewer than 70 procedures per year, even though the higher-volume facilities often serve older and less socioeconomic advantaged populations. The improvement in survival is most evident in the immediate postoperative period. The extent of resection is a matter of surgical judgment based on findings at exploration. In patients with stage IA NSCLC, lobectomy is superior to wedge resection with respect to rates of local recurrence. There is also a trend toward improvement in overall survival. In patients with comorbidities, compromised pulmonary reserve, and small peripheral lesions, a limited resection, wedge resection, and segmentectomy (potentially by video-assisted thoracoscopic surgery) may be reasonable surgical option. Pneumonectomy is reserved for patients with central tumors and should be performed only in patients with excellent pulmonary reserve. The 5-year survival rates are 60–80% for patients with stage I NSCLC and 40–50% for patients with stage II NSCLC.
Accurate pathologic staging requires adequate segmental, hilar, and mediastinal lymph node sampling. Ideally this includes a mediastinal lymph node dissection. On the right side, mediastinal stations 2R, 4R, 7, 8R, and 9R should be dissected; on the left side, stations 5, 6, 7, 8L, and 9L should be dissected. Hilar lymph nodes are typically resected and sent for pathologic review, although it is helpful to specifically dissect and label level 10 lymph nodes when possible. On the left side, level 2 and sometimes level 4 lymph nodes are generally obscured by the aorta. Although the therapeutic benefit of nodal dissection versus nodal sampling is controversial, a pooled analysis of three trials involving patients with stages I to IIIA NSCLC demonstrated a superior 4-year survival in patients undergoing resection and a complete mediastinal lymph node dissection compared with lymph node sampling. Moreover, complete mediastinal lymphadenectomy added little morbidity to a pulmonary resection for lung cancer when carried out by an experienced thoracic surgeon.
Radiation Therapy in Stages I and II NSCLC There is currently no role for postoperative radiation therapy in patients following resection of stage I or II NSCLC. However, patients with stage I and II disease who either refuse or are not suitable candidates for surgery should be considered for radiation therapy with curative intent. Stereotactic body radiation therapy (SBRT) is a relatively new technique used to treat patients with isolated pulmonary nodules (≤5 cm) who are not candidates for or refuse surgical resection. Treatment is typically administered in three to five fractions delivered over 1–2 weeks. In uncontrolled studies, disease control rates are >90%, and 5-year survival rates of up to 60% have been reported with SBRT. By comparison, survival rates typically range from 13 to 39% in patients with stage I or II NSCLC treated with standard external-beam radiotherapy. Cryoablation is another technique occasionally used to treat small, isolated tumors (i.e., ≤3 cm). However, very little data exist on long-term outcomes with this technique.
Chemotherapy in Stages I and II NSCLC Although a landmark meta-analysis of cisplatin-based adjuvant chemotherapy trials in patients with resected stages I to IIIA NSCLC (the Lung Adjuvant Cisplatin Evaluation [LACE] Study) demonstrated a 5.4% improvement in 5-year survival for adjuvant chemotherapy compared to surgery alone, the survival benefit was seemingly confined to patients with stage II or III disease (Table 107-10). By contrast, survival was actually worsened in stage IA patients with the application of adjuvant therapy. In stage IB, there was a modest improvement in survival of questionable clinical significance. Adjuvant chemotherapy was also detrimental in patients with poor performance status (Eastern Cooperative Oncology Group [ECOG] performance status = 2). These data suggest that adjuvant chemotherapy is best applied in patients with resected stage II or III NSCLC. There is no apparent role for adjuvant chemotherapy in patients with resected stage IA or IB NSCLC. A possible exception to the prohibition of adjuvant therapy in this setting is the stage IB patient with a resected lesion ≥4 cm.
|
ADJUVANT CHEMOTHERAPY TRIALS IN NON-SMALL-CELL LUNG CANCER |
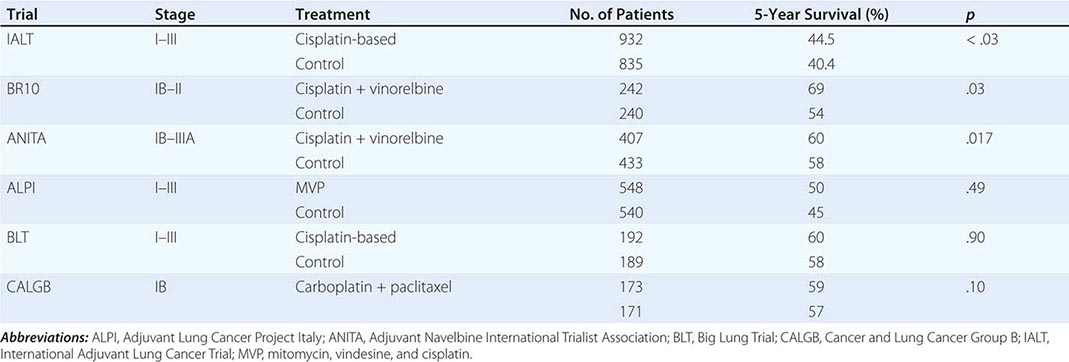
As with any treatment recommendation, the risks and benefits of adjuvant chemotherapy should be considered on an individual patient basis. If a decision is made to proceed with adjuvant chemotherapy, in general, treatment should be initiated 6–12 weeks after surgery, assuming the patient has fully recovered, and should be administered for no more than four cycles. Although a cisplatin-based chemotherapy is the preferred treatment regimen, carboplatin can be substituted for cisplatin in patients who are unlikely to tolerate cisplatin for reasons such as reduced renal function, presence of neuropathy, or hearing impairment. No specific chemotherapy regimen is considered optimal in this setting, although platinum plus vinorelbine is most commonly used.
Neoadjuvant chemotherapy, which is the application of chemotherapy administered before an attempted surgical resection, has been advocated by some experts on the assumption that such an approach will more effectively extinguish occult micrometastases compared to postoperative chemotherapy. In addition, it is thought that preoperative chemotherapy might render an inoperable lesion resectable. With the exception of superior sulcus tumors, however, the role of neoadjuvant chemotherapy in stage I to III disease is not well defined. However, a meta-analysis of 15 randomized controlled trials involving more than 2300 patients with stage I to III NSCLC suggested there may be a modest 5-year survival benefit (i.e., ∼5%) that is virtually identical to the survival benefit achieved with postoperative chemotherapy. Accordingly, neoadjuvant therapy may prove useful in selected cases (see below). A decision to use neoadjuvant chemotherapy should always be made in consultation with an experienced surgeon.
In should be noted that all patients with resected NSCLC are at high risk of recurrence, most of which occurs within 18–24 months of surgery, or developing a second primary lung cancer. Thus, it is reasonable to follow these patients with periodic imaging studies. Given the results of the NLST, periodic CT scans appear to be the most appropriate screening modality. Based on the timing of most recurrences, some guidelines recommend a contrasted chest CT scan every 6 months for the first 3 years after surgery, followed by yearly CT scans of the chest without contrast thereafter.
MANAGEMENT OF STAGE III NSCLC
Management of patients with stage III NSCLC usually requires a combined-modality approach. Patients with stage IIIA disease commonly are stratified into those with “nonbulky” or “bulky” mediastinal lymph node (N2) disease. Although the definition of “bulky” N2 disease varies somewhat in the literature, the usual criteria include the size of a dominant lymph node (i.e., >2–3 cm in short-axis diameter as measured by CT), groupings of multiple smaller lymph nodes, evidence of extracapsular nodal involvement, or involvement of more than two lymph node stations. The distinction between nonbulky and bulky stage IIIA disease is mainly used to select potential candidates for upfront surgical resection or for resection after neoadjuvant therapy. Many aspects of therapy of patients with stage III NSCLC remain controversial, and the optimal treatment strategy has not been clearly defined. Moreover, although there are many potential treatment options, none yields a very high probability of cure. Furthermore, because stage III disease is highly heterogeneous, no single treatment approach can be recommended for all patients. Key factors guiding treatment choices include the particular combination of tumor (T) and nodal (N) disease, the ability to achieve a complete surgical resection if indicated, and the patient’s overall physical condition and preferences. For example, in carefully selected patients with limited stage IIIA disease where involved mediastinal lymph nodes can be completed resected, initial surgery followed by postoperative chemotherapy (with or without radiation therapy) may be indicated. By contrast, for patients with clinically evident bulky mediastinal lymph node involvement, the standard approach to treatment is concurrent chemoradiotherapy. Nevertheless, in some cases, the latter group of patients may be candidates for surgery following chemoradiotherapy.
Absent and Nonbulky Mediastinal (N2, N3) Lymph Node Disease For the subset of stage IIIA patients initially thought to have clinical stage I or II disease (i.e., pathologic involvement of mediastinal [N2] lymph nodes is not detected preoperatively), surgical resection is often the treatment of choice. This is followed by adjuvant chemotherapy in patients with microscopic lymph node involvement in a resection specimen. Postoperative radiation therapy (PORT) may also have a role for those with close or positive surgical margins. Patients with tumors involving the chest wall or proximal airways within 2 cm of the carina with hilar lymph node involvement (but not N2 disease) are classified as having T3N1 stage IIIA disease. They too are best managed with surgical resection, if technically feasible, followed by adjuvant chemotherapy if completely resected. Patients with tumors exceeding 7 cm in size also are now classified as T3 and are consider stage IIIA if tumor has spread to N1 nodes. The appropriate initial management of these patients involves surgical resection when feasible, provided the mediastinal staging is negative, followed by adjuvant chemotherapy for those who achieve complete tumor resection. Patients with T3N0 or T3N1 disease due to the presence of satellite nodules within the same lobe as the primary tumor also are candidates for surgery, as are patients with ipsilateral nodules in another lobe and negative mediastinal nodes (IIIA, T4N0 or T4N1). Although data regarding adjuvant chemotherapy in the latter subsets of patients are limited, it is often recommended.
Patients with T4N0-1 were reclassified as having stage IIIA tumors in the seventh edition of the TNM system. These patients may have involvement of the carina, superior vena cava, or a vertebral body and yet still be candidates for surgical resection in selected circumstances. The decision to proceed with an attempted resection must be made in consultation with an experienced thoracic surgeon often in association with a vascular or cardiac surgeon and an orthopedic surgeon depending on tumor location. However, if an incomplete resection is inevitable or if there is evidence of N2 involvement (stage IIIB), surgery for T4 disease is contraindicated. Most T4 lesions are best treated with chemoradiotherapy.
The role of PORT in patients with completely resected stage III NSCLC is controversial. To a large extent, the use of PORT is dictated by the presence or absence of N2 involvement and, to a lesser degree, by the biases of the treating physician. Using the Surveillance, Epidemiology, and End Results (SEER) database, a recent meta-analysis of PORT identified a significant increase in survival in patients with N2 disease but not in patients with N0 or N1 disease. An earlier analysis by the PORT Meta-analysis Trialist Group using an older database produced similar results.
Known Mediastinal (N2, N3) Lymph Node Disease When pathologic involvement of mediastinal lymph nodes is documented preoperatively, a combined-modality approach is recommended assuming the patient is a candidate for treatment with curative intent. These patients are at high risk for both local and distant recurrence if managed with resection alone. For patients with stage III disease who are not candidates for initial surgical resection, concurrent chemoradiotherapy is most commonly used as the initial treatment. Concurrent chemoradiotherapy has been shown to produce superior survival compared to sequential chemoradiotherapy; however, it also is associated with greater host toxicities (including fatigue, esophagitis, and neutropenia). Therefore, for patients with a good performance status, concurrent chemoradiotherapy is the preferred treatment approach, whereas sequential chemoradiotherapy may be more appropriate for patients with a performance status that is not as good. For patients who are not candidates for a combined-modality treatment approach, typically due to a poor performance status or a comorbidity that makes chemotherapy untenable, radiotherapy alone may provide a modest survival benefit in addition to symptom palliation.
For patients with potentially resectable N2 disease, it remains uncertain whether surgery after neoadjuvant chemoradiotherapy improves survival. In an NCI-sponsored Intergroup randomized trial comparing concurrent chemoradiotherapy alone to concurrent chemoradiotherapy followed by attempted surgical resection, no survival benefit was observed in the trimodality arm compared to the bimodality therapy. In fact, patients subjected to a pneumonectomy had a worse survival outcome. By contrast, those treated with a lobectomy appeared to have a survival advantage based on a retrospective subset analysis. Thus, in carefully selected, otherwise healthy patients with nonbulky mediastinal lymph node involvement, surgery may be a reasonable option if the primary tumor can be fully resected with a lobectomy. This is not the case if a pneumonectomy is required to achieve complete resection.
Superior Sulcus Tumors (Pancoast Tumors) Superior sulcus tumors represent a distinctive subset of stage III disease. These tumors arise in the apex of the lung and may invade the second and third ribs, the brachial plexus, the subclavian vessels, the stellate ganglion, and adjacent vertebral bodies. They also may be associated with Pancoast syndrome, characterized by pain that may arise in the shoulder or chest wall or radiate to the neck. Pain characteristically radiates to the ulnar surface of the hand. Horner’s syndrome (enophthalmos, ptosis, miosis, and anhydrosis) due to invasion of the paravertebral sympathetic chain may be present as well. Patients with these tumors should undergo the same staging procedures as all patients with stage II and III NSCLC. Neoadjuvant chemotherapy or combined chemoradiotherapy followed by surgery is reserved for those without N2 involvement. This approach yields excellent survival outcomes (>50% 5-year survival in patients with an R0 resection). Patients with N2 disease are less likely to benefit from surgery and can be managed with chemoradiotherapy alone. Patients presenting with metastatic disease can be treated with radiation therapy (with or without chemotherapy) for symptom palliation.
MANAGEMENT OF METASTATIC NSCLC
Approximately 40% of NSCLC patients present with advanced, stage IV disease at the time of diagnosis. These patients have a poor median survival (4–6 months) and a 1-year survival of 10% when managed with best supportive care alone. In addition, a significant number of patients who first presented with early-stage NSCLC will eventually relapse with distant disease. Patients who have recurrent disease have a better prognosis than those presenting with metastatic disease at the time of diagnosis. Standard medical management, the judicious use of pain medications, and the appropriate use of radiotherapy and chemotherapy form the cornerstone of management. Chemotherapy palliates symptoms, improves the quality of life, and improves survival in patients with stage IV NSCLC, particularly in patients with good performance status. In addition, economic analysis has found chemotherapy to be cost-effective palliation for stage IV NSCLC. However, the use of chemotherapy for NSCLC requires clinical experience and careful judgment to balance potential benefits and toxicities. Of note, the early application of palliative care in conjunction with chemotherapy is associated with improved survival and a better quality of life.
First-Line Chemotherapy for Metastatic or Recurrent NSCLC A landmark meta-analysis published in 1995 provided the earliest meaningful indication that chemotherapy could provide a survival benefit in metastatic NSCLC as opposed to supportive care alone. However, the survival benefit was seemingly confined to cisplatin-based chemotherapy regimens (hazard ratio 0.73; 27% reduction in the risk of death; 10% improvement in survival at 1 year). These data launched two decades of clinical research aimed at detecting the optimal chemotherapy regimen for advanced NSCLC. For the most part, however, these efforts proved unsuccessful because the overwhelming majority of randomized trials showed no major survival improvement with any one regimen versus another (Table 107-11). On the other hand, differences in progression-free survival, cost, side effects, and schedule were frequently observed. These first-line studies were later extended to elderly patients, where doublet chemotherapy was found to improve overall survival compared to single agents in the “fit” elderly (e.g., elderly patients with no major comorbidities) and in patients with an ECOG performance status of 2. An ongoing debate in the treatment of patients with advanced NSCLC is the appropriate duration of platinum-based chemotherapy. Several large phase III randomized trials have failed to show a meaningful benefit for increasing the duration of platinum-based doublet chemotherapy beyond four to six cycles. In fact, longer duration of chemotherapy has been associated with increased toxicities and impaired quality of life. Therefore, prolonged front-line therapy (beyond four to six cycles) with platinum-based regimens is not recommended. Maintenance therapy following initial platinum-based therapy is discussed below.
|
FIRST-LINE CHEMOTHERAPY TRIALS FOR METASTATIC NON-SMALL-CELL LUNG CANCER |
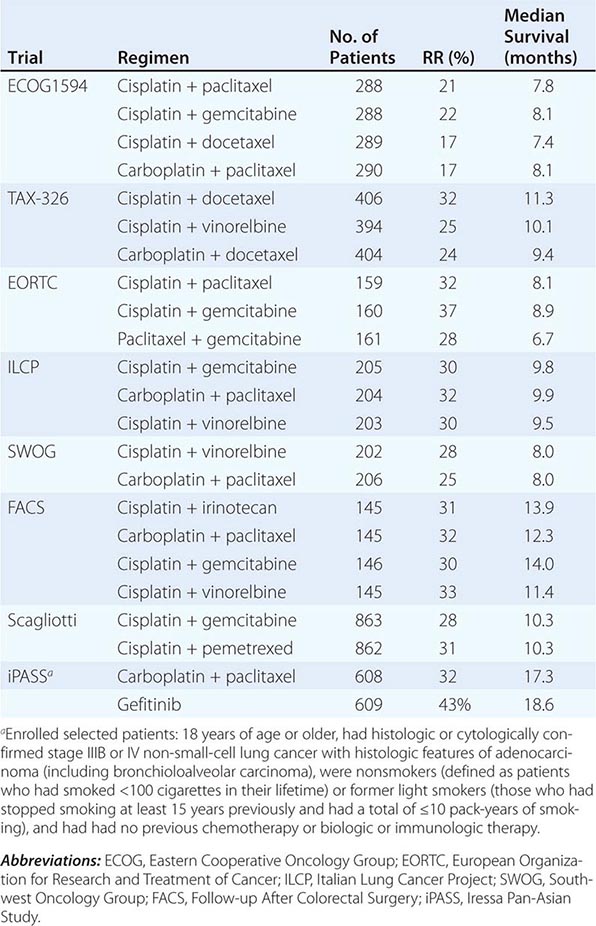
Although specific tumor histology was once considered irrelevant to treatment choice in NSCLC, with the recent recognition that selected chemotherapy agents perform quite differently in squamous versus adenocarcinomas, accurate determination of histology has become essential. Specifically, in a landmark randomized phase III trial, patients with nonsquamous NSCLC were found to have an improved survival when treated with cisplatin and pemetrexed compared to cisplatin and gemcitabine. By contrast, patients with squamous carcinoma had an improved survival when treated with cisplatin and gemcitabine. This survival difference is thought to be related to the differential expression of thymidylate synthase (TS), one of the targets of pemetrexed, between tumor types. Squamous cancers have a much higher expression of TS compared to adenocarcinomas, accounting for their lower responsiveness to pemetrexed. By contrast, the activity of gemcitabine is not impacted by the levels of TS. Bevacizumab, a monoclonal antibody against VEGF, has been shown to improve response rate, progression-free survival, and overall survival in patients with advanced disease when combined with chemotherapy (see below). However, bevacizumab cannot be given to patients with squamous cell histology NSCLC because of their tendency to experience serious hemorrhagic effects.
Agents That Inhibit Angiogenesis Bevacizumab, a monoclonal antibody directed against VEGF, was the first antiangiogenic agent approved for the treatment of patients with advanced NSCLC in the United States. This drug primarily acts by blocking the growth of new blood vessels, which are required for tumor viability. Two randomized phase III trials of chemotherapy with or without bevacizumab had conflicting results. The first trial, conducted in North America, compared carboplatin plus paclitaxel with or without bevacizumab in patients with recurrent or advanced nonsquamous NSCLC and reported a significant improvement in response rate, progression-free survival, and overall survival in patients treated with chemotherapy plus bevacizumab versus chemotherapy alone. Bevacizumab-treated patients had a significantly higher incidence of toxicities. The second trial, conducted in Europe, compared cisplatin/gemcitabine with or without bevacizumab in patients with recurrent or advanced nonsquamous NSCLC and reported a significant improvement in progression-free survival but no improvement in overall survival for bevacizumab-treated patients. A randomized phase III trial compared carboplatin/pemetrexed and bevacizumab to carboplatin/paclitaxel and bevacizumab as first-line therapy in patients with recurrent or advanced nonsquamous NSCLC and reported no significant difference in progression-free survival or overall survival between treatment groups. Therefore, currently carboplatin/paclitaxel and bevacizumab or carboplatin/pemetrexed and bevacizumab are appropriate regimens for first-line treatment for stage IV nonsquamous NSCLC patients. Of note, there are many small-molecule inhibitors of VEGFR; however, these VEGFR TKIs have not proven to be effective in the treatment of NSCLC.
Maintenance Therapy for Metastatic NSCLC Maintenance chemotherapy in nonprogressing patients (patients with a complete response, partial response, or stable disease) is a controversial topic in the treatment of NSCLC. Conceptually, there are two types of maintenance strategies: (1) switch maintenance therapy, where patients receive four to six cycles of platinum-based chemotherapy and are switched to an entirely different regimen; and (2) continuation maintenance therapy, where patients receive four to six cycles of platinum-based chemotherapy and then the platinum agent is discontinued but the agent it is paired with is continued (Table 107-12). Two studies investigated switch maintenance single-agent chemotherapy with docetaxel or pemetrexed in nonprogressing patients following treatment with first-line platinum-based chemotherapy. Both trials randomized patients to immediate single-agent therapy versus observation and reported improvements in progression-free and overall survival. In both trials, a significant portion of patients in the observation arm did not receive therapy with the agent under investigation upon disease progression; 37% of study patients never received docetaxel in the docetaxel study and 81% of patients never received pemetrexed in the pemetrexed study. In the trial of maintenance docetaxel versus observation, survival was identical to the treatment group in the subset of patients who received docetaxel on progression, indicating this is an active agent in NSCLC. These data are not available for the pemetrexed study. Two additional trials evaluated switch maintenance therapy with erlotinib after platinum-based chemotherapy in patients with advanced NSCLC and reported an improvement in progression-free survival and overall survival in the erlotinib treatment group. Currently, maintenance pemetrexed or erlotinib following platinum-based chemotherapy in patients with advanced NSCLC are approved by the U.S. FDA. However, maintenance therapy is not without toxicity and, at this time, should be considered on an individual patient basis.
|
MAINTENANCE THERAPY TRIALS |
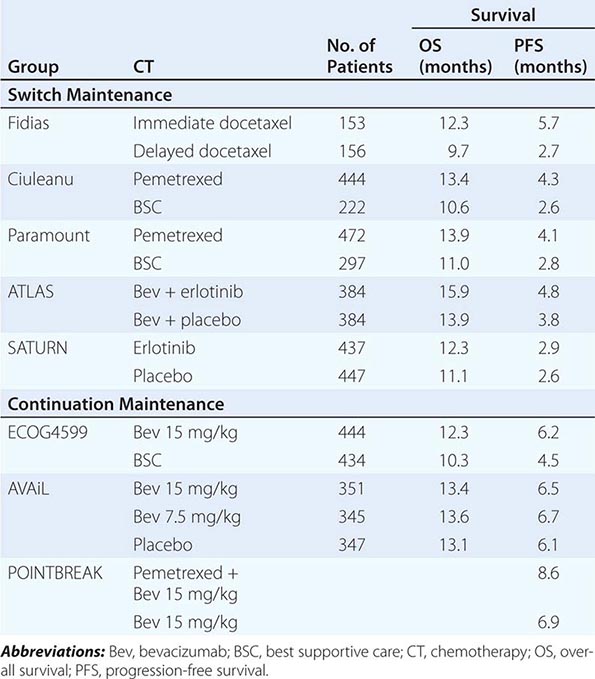
Targeted Therapies for Select Molecular Cohorts of NSCLC As the efficacy of traditional cytotoxic chemotherapeutic agents plateaued in NSCLC, there was a critical need to define novel therapeutic treatment strategies. These novel strategies have largely been based on the identification of somatic driver mutations within the tumor. These driver mutations occur in genes encoding signaling proteins that, when aberrant, drive initiation and maintenance of tumor cells. Importantly, driver mutations can serve as Achilles’ heels for tumors, if their gene products can be targeted therapeutically with small-molecule inhibitors. For example, EGFR mutations have been detected in 10–15% of North American patients diagnosed with NSCLC. EGFR mutations are associated with younger age, light (<10 pack-year) and nonsmokers, and adenocarcinoma histology. Approximately 90% of these mutations are exon 19 deletions or exon 21 L858R point mutations within the EGFR TK domain, resulting in hyperactivation of both EGFR kinase activity and downstream signaling. Lung tumors that harbor activating mutations within the EGFR kinase domain display high sensitivity to small-molecule EGFR TKIs. Erlotinib and afatinib are FDA-approved oral small-molecule TKIs that inhibit EGFR. Outside the United States, gefitinib also is available. Several large, international, phase III studies have demonstrated improved response rates, progression-free survival, and overall survival in patients with EGFR mutation–positive NSCLC patients treated with an EGFR TKI as compared with standard first-line chemotherapy regimens (Table 107-13).
|
RESULTS OF PHASE III TRIALS COMPARING CHEMOTHERAPY AND FIRST-LINE EGFR TKI IN EGFR MUTATION–POSITIVE PATIENTS |
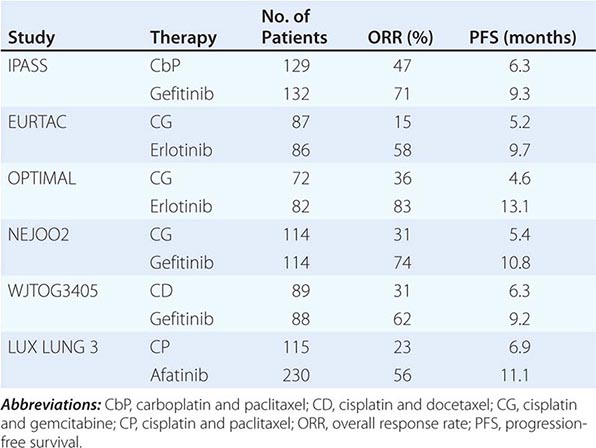
Although response rates with EGFR TKI therapy are clearly superior in patients with lung tumors harboring activating EGFR kinase domain mutations, the EGFR TKI erlotinib is also FDA approved for second- and third-line therapy in patients with advanced NSCLC irrespective of tumor genotype. The reason for this apparent discrepancy is that erlotinib was initially evaluated in lung cancer before the discovery of EGFR activating mutations. In fact, EGFR mutations were initially identified in lung cancer by studying the tumors of patients who had dramatic responses to this agent. With the rapid pace of scientific discovery, additional driver mutations in lung cancer have been identified and targeted therapeutically with impressive clinical results. For example, chromosomal rearrangements involving the anaplastic lymphoma kinase (ALK) gene on chromosome 2 have been found in ∼3–7% of NSCLC. The result of these ALK rearrangements is hyperactivation of the ALK TK domain. Similar to EGFR, ALK rearrangements are typically (but not exclusively) associated with younger age, light (<10 pack-year) and nonsmokers, and adenocarcinoma histology. Remarkably, ALK rearrangements were initially described in lung cancer in 2007, and by 2011, the first ALK inhibitor, crizotinib, received FDA approval for patients with lung tumors harboring ALK rearrangements.
In addition to EGFR and ALK, other driver mutations have been discovered with varying frequencies in NSCLC, including KRAS, BRAF, PIK3CA, NRAS, AKT1, MET, MEK1 (MAP2K1), ROS1, and RET. Mutations within the KRAS GTPase are found in approximately 20% of lung adenocarcinomas. To date, however, no small-molecule inhibitors are available to specifically target mutant KRAS. Each of the other driver mutations occurs in less than 1–3% of lung adenocarcinomas. The great majority of the driver mutations are mutually exclusive, and there are ongoing clinical studies for their specific inhibitors. For example, the BRAF inhibitor vemurafenib and the RET inhibitor cabozantinib have already demonstrated efficacy in patients with lung cancer harboring BRAF mutations or RET gene fusions, respectively. Most of these mutations are present in adenocarcinoma; however, mutations that may be linked to future targeted therapies in squamous cell carcinomas are emerging. In addition, there are active research efforts aimed at defining novel targetable mutations in lung cancer as well as defining mechanisms of acquired resistance to small-molecule inhibitors used in the treatment of patients with NSCLC.
Second-Line Chemotherapy and Beyond Second-line therapy for advanced NSCLC was almost never recommended until a seminal study in 2000 showed that docetaxel improved survival compared to supportive care alone. As first-line chemotherapy regimens improve, a substantial number of patients will maintain a good performance status and a desire for further therapy when they develop recurrent disease. Currently, several agents are FDA approved for second-line use in NSCLC including docetaxel, pemetrexed, erlotinib (approved for second-line therapy regardless of tumor genotype), and crizotinib (for patients with ALK -mutant lung cancer only). Most of the survival benefit for any of these agents is realized in patients who maintain a good performance status.
Immunotherapy For more than 30 years, the investigation of vaccines and immunotherapies in lung cancer has yielded little in the way of meaningful benefit. Recently, however, this perception has changed based on preliminary results of studies using monoclonal antibodies that activate antitumor immunity through blockade of immune checkpoints. For example, ipilimumab, a monoclonal antibody directed at cytotoxic T lymphocyte antigen-4 (CTLA-4), was studied in combination with paclitaxel plus carboplatin in patients with both SCLC and NSCLC. There appeared to be a small but not statistically significant advantage to the combination when ipilimumab was instituted after several cycles of chemotherapy. A randomized phase III trial in SCLC is under way to validate these data. Antibodies to the T cell programmed cell death receptor 1 (PD-1), nivolumab and pembrolizumab, have been shown to produce responses in lung cancer, renal cell cancer, and melanoma. Many of these responses have had very long duration (i.e., >1 year). Monoclonal antibodies to the PD-1 ligand (anti-PDL-1), which may be expressed on the tumor cell, have also been shown to produce responses in patients with melanoma and lung cancer. Preliminary studies in melanoma suggest that the combination of ipilimumab and nivolumab could produce higher response rates compared to either agent alone. A similar strategy is being investigated in SCLC patients. Further evaluation of these agents in both NSCLC and SCLC is ongoing in combination with already approved chemotherapy and targeted agents.
Supportive Care No discussion of the treatment strategies for patients with advanced lung cancer would be complete without a mention of supportive care. Coincident with advances in chemotherapy and targeted therapy was a pivotal study that demonstrated that the early integration of palliative care with standard treatment strategies improved both quality of life and mood for patients with advanced lung cancer. Aggressive pain and symptom control is an important component for optimal treatment of these patients.
TREATMENT SMALL-CELL LUNG CANCER
SURGERY FOR LIMITED-DISEASE SMALL-CELL LUNG CANCER
SCLC is a highly aggressive disease characterized by its rapid doubling time, high growth fraction, early development of disseminated disease, and dramatic response to first-line chemotherapy and radiation. In general, surgical resection is not routinely recommended for patients because even patients with LD-SCLC still have occult micrometastases. However, the most recent American College of Chest Physicians Evidence-Based Clinical Practice Guidelines recommend surgical resection over nonsurgical treatment in SCLC patients with clinical stage I disease after a thorough evaluation for distant metastases and invasive mediastinal stage evaluation (grade 2C). After resection, these patients should receive platinum-based adjuvant chemotherapy (grade 1C). If the histologic diagnosis of SCLC is made in patients on review of a resected surgical specimen, such patients should receive standard SCLC chemotherapy as well.
CHEMOTHERAPY
Chemotherapy significantly prolongs survival in patients with SCLC. Four to six cycles of platinum-based chemotherapy with either cisplatin or carboplatin plus either etoposide or irinotecan has been the mainstay of treatment for nearly three decades and is recommended over other chemotherapy regimens irrespective of initial stage. Cyclophosphamide, doxorubicin (Adriamycin), and vincristine (CAV) may be an alternative for patients who are unable to tolerate a platinum-based regimen. Despite response rates to first-line therapy as high as 80%, the median survival ranges from 12 to 20 months for patients with LD and from 7 to 11 months for patients with ED. Regardless of disease extent, the majority of patients relapse and develop chemotherapy-resistant disease. Only 6–12% of patients with LD-SCLC and 2% of patients with ED-SCLC live beyond 5 years. The prognosis is especially poor for patients who relapse within the first 3 months of therapy; these patients are said to have chemotherapy-resistant disease. Patients are said to have sensitive disease if they relapse more than 3 months after their initial therapy and are thought to have a somewhat better overall survival. These patients also are thought to have the greatest potential benefit from second-line chemotherapy (Fig. 107-7). Topotecan is the only FDA-approved agent for second-line therapy in patients with SCLC. Topotecan has only modest activity and can be given either intravenously or orally. In one randomized trial, 141 patients who were not considered candidates for further IV chemotherapy were randomized to receive either oral topotecan or best supportive care. Although the response rate to oral topotecan was only 7%, overall survival was significantly better in patients receiving chemotherapy (median survival time, 26 weeks vs 14 weeks; p = .01). Moreover, patients given topotecan had a slower decline in quality of life than did those not receiving chemotherapy. Other agents with similar low levels of activity in the second-line setting include irinotecan, paclitaxel, docetaxel, vinorelbine, oral etoposide, and gemcitabine. Clearly novel treatments for this all too common disease are desperately needed.
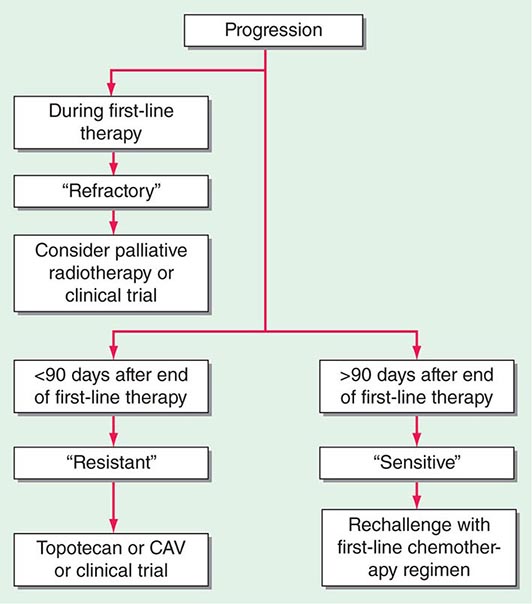
FIGURE 107-7 Management of recurrent small-cell lung cancer (SCLC). CAV, cyclophosphamide, doxorubicin, and vincristine. (Adapted with permission from JP van Meerbeeck et al: Lancet 378:1741, 2011.)
THORACIC RADIATION THERAPY
Thoracic radiation therapy (TRT) is a standard component of induction therapy for good performance status and limited-stage SCLC patients. Meta-analyses indicate that chemotherapy combined with chest irradiation improves 3-year survival by approximately 5% as compared with chemotherapy alone. The 5-year survival rate, however, remains disappointingly low at ∼10–15%. Most commonly, TRT is combined with cisplatin and etoposide chemotherapy due to a superior toxicity profile as compared to anthracycline-containing chemotherapy regimens. As observed in locally advanced NSCLC, concurrent chemoradiotherapy is more effective than sequential chemoradiation but is associated with significantly more esophagitis and hematologic toxicity. Ideally TRT should be administered with the first two cycles of chemotherapy because later application appears slightly less effective. If for reasons of fitness or availability, this regimen cannot be offered, TRT should follow induction chemotherapy. With respect to fractionation of TRT, twice-daily 1.5-Gy fractioned radiation therapy has been shown to improve survival in LD-SCLC patients but is associated with higher rates of grade 3 esophagitis and pulmonary toxicity. Although it is feasible to deliver once-daily radiation therapy doses up to 70 Gy concurrently with cisplatin-based chemotherapy, there are no data to support equivalency of this approach compared with the 45-Gy twice-daily radiotherapy dose. Therefore, the current standard regimen of a 45-Gy dose administered in 1.5-Gy fractions twice daily for 30 days is being compared with higher-dose regimens in two phase III trials, one in the United States and one in Europe. Patients should be carefully selected for concurrent chemoradiation therapy based on good performance status and adequate pulmonary reserve. The role of radiotherapy in ED-SCLC is largely restricted to palliation of tumor-related symptoms such as bone pain and bronchial obstruction.
PROPHYLACTIC CRANIAL IRRADIATION
Prophylactic cranial irradiation (PCI) should be considered in all patients with either LD-SCLC or ED-SCLC who have responded well to initial therapy. A meta-analysis including seven trials and 987 patients with LD-SCLC who had achieved a complete remission after upfront chemotherapy yielded a 5.4% improvement in overall survival for patients treated with PCI. In patients with ED-SCLC who have responded to first-line chemotherapy, a prospective randomized phase III trial showed that PCI reduced the occurrence of symptomatic brain metastases and prolonged disease-free and overall survival compared to no radiation therapy. Long-term toxicities, including deficits in cognition, have been reported after PCI but are difficult to sort out from the effects of chemotherapy or normal aging.
SUMMARY
The management of NSCLC has undergone major change in the past decade. To a lesser extent, the same is true for SCLC. For patients with early-stage disease, advances in radiotherapy and surgical procedures as well as new systemic therapies have greatly improved prognosis in both diseases. For patients with advanced disease, major progress in understanding tumor genetics has led to the development of targeted inhibitors based specifically on the tumor’s molecular profile. Furthermore, increased understanding of how to activate the immune system to drive antitumor immunity is proving to be a promising therapeutic strategy for some patients with advanced lung cancer. In Fig. 107-8, we propose an algorithm of the treatment approach for patient with stage IV NSCLC. However, the reality is that the majority of patients treated with targeted therapies or chemotherapy eventually develop resistance, which provides strong motivation for further research and enrollment of patients onto clinical trials in this rapidly evolving area.
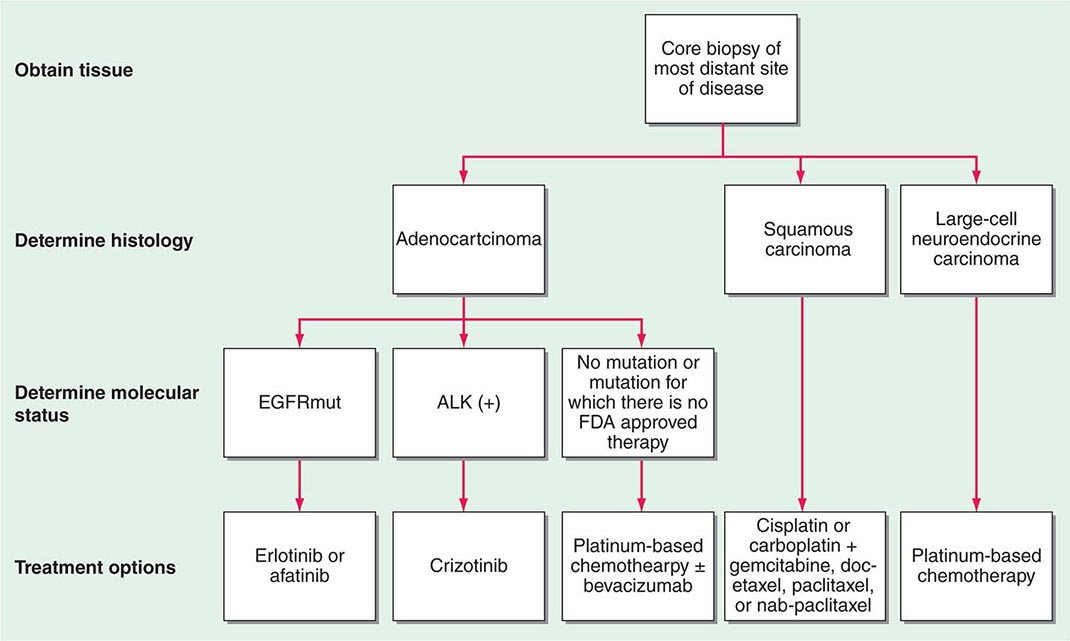
FIGURE 107-8 Approach to first-line therapy in a patient with stage IV non-small-cell lung cancer (NSCLC). EGFRmut, EGFR mutation; FDA, Food and Drug Administration.
108 |
Breast Cancer |
Breast cancer is a malignant proliferation of epithelial cells lining the ducts or lobules of the breast. In the year 2014, about 180,000 cases of invasive breast cancer and 40,000 deaths will occur in the United States. In addition, about 2000 men will be diagnosed with breast cancer. Epithelial malignancies of the breast are the most common cause of cancer in women (excluding skin cancer), accounting for about one-third of all cancer in women. As a result of improved treatment and earlier detection, the mortality rate from breast cancer has begun to decrease very substantially in the United States. This Chapter will not consider rare malignancies presenting in the breast, such as sarcomas and lymphomas, but will focus on the epithelial cancers.
GENETIC CONSIDERATIONS
![]() Human breast cancer is a clonal disease; a single transformed cell—the product of a series of somatic (acquired) or germline mutations—is eventually able to express full malignant potential. Thus, breast cancer may exist for a long period as either a noninvasive disease or an invasive but nonmetastatic disease. These facts have significant clinical ramifications.
Human breast cancer is a clonal disease; a single transformed cell—the product of a series of somatic (acquired) or germline mutations—is eventually able to express full malignant potential. Thus, breast cancer may exist for a long period as either a noninvasive disease or an invasive but nonmetastatic disease. These facts have significant clinical ramifications.
Not more than 10% of human breast cancers can be linked directly to germline mutations. Several genes have been implicated in familial cases. The Li-Fraumeni syndrome is characterized by inherited mutations in the p53 tumor-suppressor gene, which lead to an increased incidence of breast cancer, osteogenic sarcomas, and other malignancies. Inherited mutations in PTEN have also been reported in breast cancer.
Another tumor-suppressor gene, BRCA1, has been identified at the chromosomal locus 17q21; this gene encodes a zinc finger protein, and the protein product functions as a transcription factor and is involved in gene repair. Women who inherit a mutated allele of this gene from either parent have at least a 60–80% lifetime chance of developing breast cancer and about a 33% chance of developing ovarian cancer. The risk is higher among women born after 1940, presumably due to promotional effects of hormonal factors. Men who carry a mutant allele of the gene have an increased incidence of prostate cancer and breast cancer. A fourth gene, termed BRCA2, which has been localized to chromosome 13q12, is also associated with an increased incidence of breast cancer in men and women.
Germline mutations in BRCA1 and BRCA2 can be readily detected; patients with these mutations should be counseled appropriately. All women with strong family histories for breast cancer should be referred to genetic screening programs, particularly women of Ashkenazi Jewish descent who have a high likelihood of a specific founder BRCA1 mutation (substitution of adenine for guanine at position 185).
Even more important than the role these genes play in inherited forms of breast cancer may be their role in sporadic breast cancer. A p53 mutation is present in nearly 40% of human breast cancers as an acquired defect. Acquired mutations in PTEN occur in about 10% of the cases. BRCA1 mutation in sporadic primary breast cancer has not been reported. However, decreased expression of BRCA1 mRNA (possibly via gene methylation) and abnormal cellular location of the BRCA1 protein have been found in some breast cancers. Loss of heterozygosity of BRCA1 and BRCA2 suggests that tumor-suppressor activity may be inactivated in sporadic cases of human breast cancer. Finally, increased expression of a dominant oncogene plays a role in about a quarter of human breast cancer cases. The product of this gene, a member of the epidermal growth factor receptor superfamily, is called erbB2 (HER/2 neu) and is overexpressed in these breast cancers due to gene amplification; this overexpression can contribute to transformation of human breast epithelium and is the target of effective systemic therapy in adjuvant and metastatic disease settings. A series of acquired “driver” mutations have been identified in sporadic breast cancer by major sequencing consortia. Unfortunately, most occur in no more than 5% of cases and generally do not have effective agents to target them, so “personalized medicine” is for now more of a dream than a reality.
EPIDEMIOLOGY
Breast cancer is a hormone-dependent disease. Women without functioning ovaries who never receive estrogen replacement therapy do not develop breast cancer. The female-to-male ratio is about 150:1. For most epithelial malignancies, a log-log plot of incidence versus age shows a single-component straight-line increase with every year of life. A similar plot for breast cancer shows two components: a straight-line increase with age but with a decrease in slope beginning at the age of menopause. The three dates in a woman’s life that have a major impact on breast cancer incidence are age at menarche, age at first full-term pregnancy, and age at menopause. Women who experience menarche at age 16 years have only 50–60% of the breast cancer risk of a woman having menarche at age 12 years; the lower risk persists throughout life. Similarly, menopause occurring 10 years before the median age of menopause (52 years), whether natural or surgically induced, reduces lifetime breast cancer risk by about 35%. Women who have a first full-term pregnancy by age 18 years have a 30–40% lower risk of breast cancer compared with nulliparous women. Thus, length of menstrual life—particularly the fraction occurring before first full-term pregnancy—is a substantial component of the total risk of breast cancer. These three factors (menarche, age of first full-term pregnancy, and menopause) can account for 70–80% of the variation in breast cancer frequency in different countries. Also, duration of maternal nursing correlates with substantial risk reduction independent of either parity or age at first full-term pregnancy.
International variation in incidence has provided some of the most important clues on hormonal carcinogenesis. A woman living to age 80 years in North America has one chance in nine of developing invasive breast cancer. Asian women have one-fifth to one-tenth the risk of breast cancer of women in North America or Western Europe. Asian women have substantially lower concentrations of estrogens and progesterone. These differences cannot be explained on a genetic basis because Asian women living in a Western environment have sex steroid hormone concentrations and risks identical to those of their Western counterparts. These migrant women, and more notably their daughters, also differ markedly in height and weight from Asian women in Asia; height and weight are critical regulators of age of menarche and have substantial effects on plasma concentrations of estrogens.
The role of diet in breast cancer etiology is controversial. While there are associative links between total caloric and fat intake and breast cancer risk, the exact role of fat in the diet is unproven. Increased caloric intake contributes to breast cancer risk in multiple ways: earlier menarche, later age at menopause, and increased postmenopausal estrogen concentrations reflecting enhanced aromatase activities in fatty tissues. On the other hand, central obesity is both a risk factor for occurrence and recurrence of breast cancer. Moderate alcohol intake also increases the risk by an unknown mechanism. Folic acid supplementation appears to modify risk in women who use alcohol but is not additionally protective in abstainers. Recommendations favoring abstinence from alcohol must be weighed against other social pressures and the possible cardioprotective effect of moderate alcohol intake. Chronic low-dose aspirin use is associated with a decreased incidence of breast cancer. Depression is also associated with both occurrence and recurrence of breast cancer.
Understanding the potential role of exogenous hormones in breast cancer is of extraordinary importance because millions of American women regularly use oral contraceptives and postmenopausal hormone replacement therapy. The most credible meta-analyses of oral contraceptive use suggest that these agents cause a small increased risk of breast cancer. By contrast, oral contraceptives offer a substantial protective effect against ovarian epithelial tumors and endometrial cancers. Hormone replacement therapy (HRT) has a powerful effect on breast cancer risk. Data from the Women’s Health Initiative (WHI) trial showed that conjugated equine estrogens plus progestins increased the risk of breast cancer and adverse cardiovascular events but decreased the risk of bone fractures and colorectal cancer. On balance, there were more negative events with HRT; 6–7 years of HRT nearly doubled the risk of breast cancer. A parallel WHI trial with >12,000 women enrolled testing conjugated estrogens alone (estrogen replacement therapy in women who have had hysterectomies) showed no significant increase in breast cancer incidence. Thus, there are serious concerns about long-term HRT use in terms of cardiovascular disease and breast cancer. The WHI trial of conjugated equine estrogen alone demonstrated few adverse effects for women age <70; however, no comparable safety data are available for other more potent forms of estrogen replacement, and they should not be routinely used as substitutes. HRT in women previously diagnosed with breast cancer increases recurrence rates. Rapid decrease in the number of women on HRT has already led to a coincident decrease in breast cancer incidence.
In addition to the other factors, radiation is a risk factor in younger women. Women who have been exposed before age 30 years to radiation in the form of multiple fluoroscopies (200–300 cGy) or treatment for Hodgkin’s disease (>3600 cGy) have a substantial increase in risk of breast cancer, whereas radiation exposure after age 30 years appears to have a minimal carcinogenic effect on the breast.
EVALUATION OF BREAST MASSES IN MEN AND WOMEN
Because the breasts are a common site of potentially fatal malignancy in women, examination of the breast is an essential part of the physical examination. Unfortunately, internists frequently do not examine breasts in men, and in women, they are apt to defer this evaluation to gynecologists. Because of the plausible association between early detection and improved outcome, it is the duty of every physician to identify breast abnormalities at the earliest possible stage and to institute a diagnostic workup. Women should be trained in breast self-examination (BSE). Although breast cancer in men is unusual, unilateral lesions should be evaluated in the same manner as in women, with the recognition that gynecomastia in men can sometimes begin unilaterally and is often asymmetric.
Virtually all breast cancer is diagnosed by biopsy of a nodule detected either on a mammogram or by palpation. Algorithms have been developed to enhance the likelihood of diagnosing breast cancer and reduce the frequency of unnecessary biopsy (Fig. 108-1).
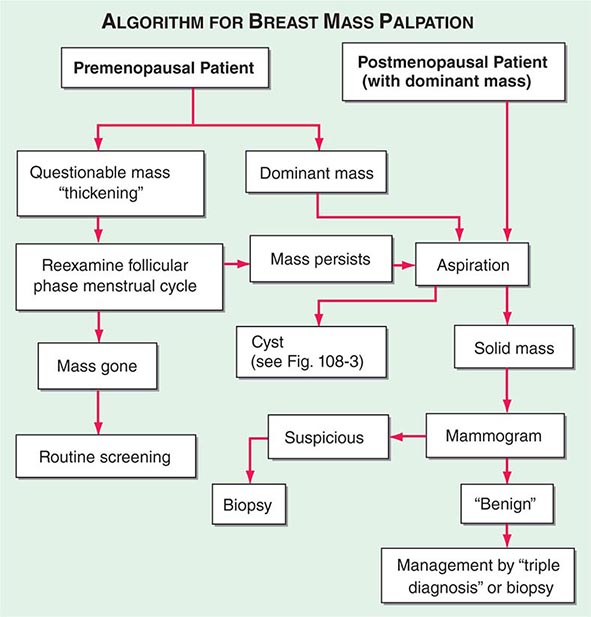
FIGURE 108-1 Approach to a palpable breast mass.
THE PALPABLE BREAST MASS
Women should be strongly encouraged to examine their breasts monthly. A potentially flawed study from China has suggested that BSE does not alter survival, but given its safety, the procedure should still be encouraged. At worst, this practice increases the likelihood of detecting a mass at a smaller size when it can be treated with more limited surgery. Breast examination by the physician should be performed in good light so as to see retractions and other skin changes. The nipple and areolae should be inspected, and an attempt should be made to elicit nipple discharge. All regional lymph node groups should be examined, and any lesions should be measured. Physical examination alone cannot exclude malignancy. Lesions with certain features are more likely to be cancerous (hard, irregular, tethered or fixed, or painless lesions). A negative mammogram in the presence of a persistent lump in the breast does not exclude malignancy. Palpable lesions require additional diagnostic procedures, including biopsy.
In premenopausal women, lesions that are either equivocal or nonsuspicious on physical examination should be reexamined in 2–4 weeks, during the follicular phase of the menstrual cycle. Days 5–7 of the cycle are the best time for breast examination. A dominant mass in a postmenopausal woman or a dominant mass that persists through a menstrual cycle in a premenopausal woman should be aspirated by fine-needle biopsy or referred to a surgeon. If nonbloody fluid is aspirated, the diagnosis (cyst) and therapy have been accomplished together. Solid lesions that are persistent, recurrent, complex, or bloody cysts require mammography and biopsy, although in selected patients the so-called triple diagnostic technique (palpation, mammography, aspiration) can be used to avoid biopsy (Figs. 108-1, 108-2, and 108-3). Ultrasound can be used in place of fine-needle aspiration to distinguish cysts from solid lesions. Not all solid masses are detected by ultrasound; thus, a palpable mass that is not visualized on ultrasound must be presumed to be solid.
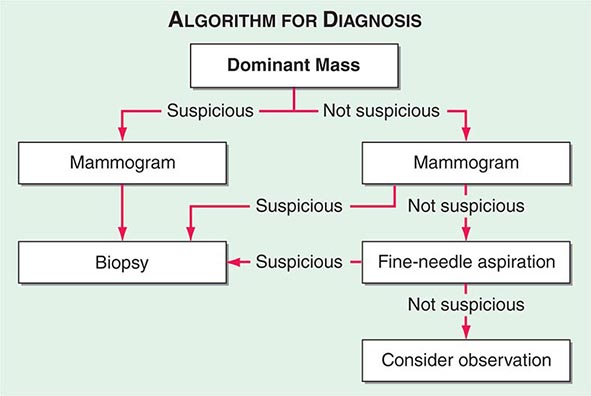
FIGURE 108-2 The “triple diagnosis” technique.
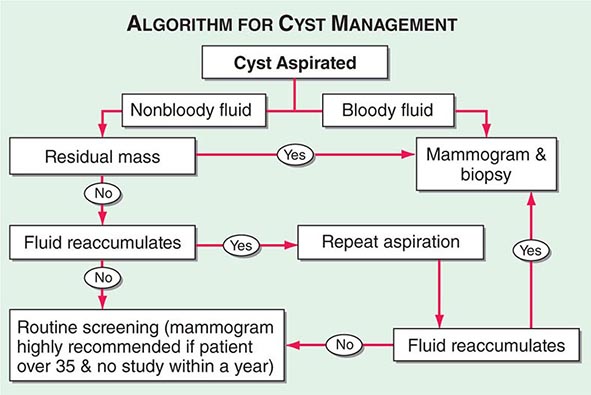
FIGURE 108-3 Management of a breast cyst.
Several points are essential in pursuing these management decision trees. First, risk-factor analysis is not part of the decision structure. No constellation of risk factors, by their presence or absence, can be used to exclude biopsy. Second, fine-needle aspiration should be used only in centers that have proven skill in obtaining such specimens and analyzing them. The likelihood of cancer is low in the setting of a “triple negative” (benign-feeling lump, negative mammogram, and negative fine-needle aspiration), but it is not zero. The patient and physician must be aware of a 1% risk of false negatives. Third, additional technologies such as magnetic resonance imaging (MRI), ultrasound, and sestamibi imaging cannot be used to exclude the need for biopsy, although in unusual circumstances, they may provoke a biopsy.
THE ABNORMAL MAMMOGRAM
Diagnostic mammography should not be confused with screening mammography, which is performed after a palpable abnormality has been detected. Diagnostic mammography is aimed at evaluating the rest of the breast before biopsy is performed or occasionally is part of the triple-test strategy to exclude immediate biopsy.
Subtle abnormalities that are first detected by screening mammography should be evaluated carefully by compression or magnified views. These abnormalities include clustered microcalcifications, densities (especially if spiculated), and new or enlarging architectural distortion. For some nonpalpable lesions, ultrasound may be helpful either to identify cysts or to guide biopsy. If there is no palpable lesion and detailed mammographic studies are unequivocally benign, the patient should have routine follow-up appropriate to the patient’s age. It cannot be stressed too strongly that in the presence of a breast lump a negative mammogram does not rule out cancer.
If a nonpalpable mammographic lesion has a low index of suspicion, mammographic follow-up in 3–6 months is reasonable. Workup of indeterminate and suspicious lesions has been rendered more complex by the advent of stereotactic biopsies. Morrow and colleagues have suggested that these procedures are indicated for lesions that require biopsy but are likely to be benign—that is, for cases in which the procedure probably will eliminate additional surgery. When a lesion is more probably malignant, open biopsy should be performed with a needle localization technique. Others have proposed more widespread use of stereotactic core biopsies for nonpalpable lesions on economic grounds and because diagnosis leads to earlier treatment planning. However, stereotactic diagnosis of a malignant lesion does not eliminate the need for definitive surgical procedures, particularly if breast conservation is attempted. For example, after a breast biopsy with needle localization (i.e., local excision) of a stereotactically diagnosed malignancy, reexcision may still be necessary to achieve negative margins. To some extent, these issues are decided on the basis of referral pattern and the availability of the resources for stereotactic core biopsies. A reasonable approach is shown in Fig. 108-4.
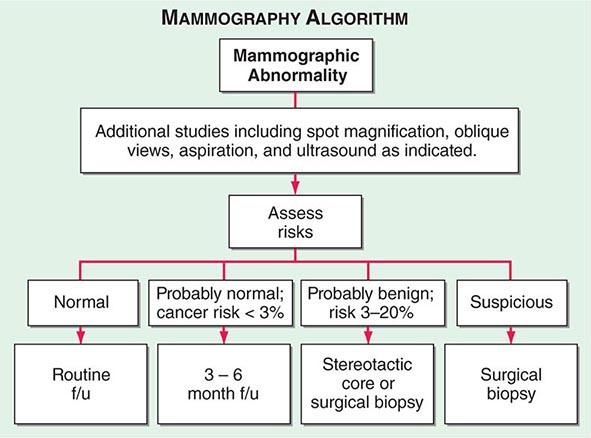
FIGURE 108-4 Approaches to abnormalities detected by mammogram.
BREAST MASSES IN THE PREGNANT OR LACTATING WOMAN
During pregnancy, the breast grows under the influence of estrogen, progesterone, prolactin, and human placental lactogen. Lactation is suppressed by progesterone, which blocks the effects of prolactin. After delivery, lactation is promoted by the fall in progesterone levels, which leaves the effects of prolactin unopposed. The development of a dominant mass during pregnancy or lactation should never be attributed to hormonal changes. A dominant mass must be treated with the same concern in a pregnant woman as any other. Breast cancer develops in 1 in every 3000–4000 pregnancies. Stage for stage, breast cancer in pregnant patients is no different from premenopausal breast cancer in nonpregnant patients. However, pregnant women often have more advanced disease because the significance of a breast mass was not fully considered and/or because of endogenous hormone stimulation. Persistent lumps in the breast of pregnant or lactating women cannot be attributed to benign changes based on physical findings; such patients should be promptly referred for diagnostic evaluation.
BENIGN BREAST MASSES
Only about 1 in every 5–10 breast biopsies leads to a diagnosis of cancer, although the rate of positive biopsies varies in different countries and clinical settings. (These differences may be related to interpretation, medico-legal considerations, and availability of mammograms.) The vast majority of benign breast masses are due to “fibrocystic” disease, a descriptive term for small fluid-filled cysts and modest epithelial cell and fibrous tissue hyperplasia. However, fibrocystic disease is a histologic, not a clinical, diagnosis, and women who have had a biopsy with benign findings are at greater risk of developing breast cancer than those who have not had a biopsy. The subset of women with ductal or lobular cell proliferation (about 30% of patients), particularly the small fraction (3%) with atypical hyperplasia, have a fourfold greater risk of developing breast cancer than those women who have not had a biopsy, and the increase in the risk is about ninefold for women in this category who also have an affected first-degree relative. Thus, careful follow-up of these patients is required. By contrast, patients with a benign biopsy without atypical hyperplasia are at little risk and may be followed routinely.
SCREENING
Breast cancer is virtually unique among the epithelial tumors in adults in that screening (in the form of annual mammography) improves survival. Meta-analysis examining outcomes from every randomized trial of mammography conclusively shows a 25–30% reduction in the chance of dying from breast cancer with annual screening after age 50 years; the data for women between ages 40 and 50 years are almost as positive; however, since the incidence is much lower in younger women, there are more false positives. While controversy continues to surround the assessment of screening mammography, the preponderance of data strongly supports the benefits of screening mammography. New analyses of older randomized studies have occasionally suggested that screening may not work. While the design defects in some older studies cannot be retrospectively corrected, most experts, including panels of the American Society of Clinical Oncology and the American Cancer Society (ACS), continue to believe that screening conveys substantial benefit. Furthermore, the profound drop in breast cancer mortality rate seen over the past decade is unlikely to be solely attributable to improvements in therapy. It seems prudent to recommend annual or biannual mammography for women past the age of 40 years. Although no randomized study of BSE has ever shown any improvement in survival, its major benefit is identification of tumors appropriate for conservative local therapy. Better mammographic technology, including digitized mammography, routine use of magnified views, and greater skill in mammographic interpretation, combined with newer diagnostic techniques (MRI, magnetic resonance spectroscopy, positron emission tomography, etc.) may make it possible to identify breast cancers even more reliably and earlier. Screening by any technique other than mammography is not indicated. However, the ACS suggests that younger women who are BRCA1 or BRCA2 carriers or untested first-degree relatives of women with cancer; women with a history of radiation therapy to the chest between ages 10 and 30 years; women with a lifetime risk of breast cancer of at least 20%; and women with a history of Li-Fraumeni, Cowden, or Bannayan-Riley-Ruvalcaba syndromes may benefit from MRI screening, where the higher sensitivity may outweigh the loss of specificity.
STAGING
Correct staging of breast cancer patients is of extraordinary importance. Not only does it permit an accurate prognosis, but in many cases, therapeutic decision-making is based largely on the TNM (primary tumor, regional nodes, metastasis) classification (Table 108-1). Comparison with historic series should be undertaken with caution, as the staging has changed several times in the past 20 years. The current staging is complex and results in significant changes in outcome by stage as compared with prior staging systems.
|
STAGING OF BREAST CANCER |
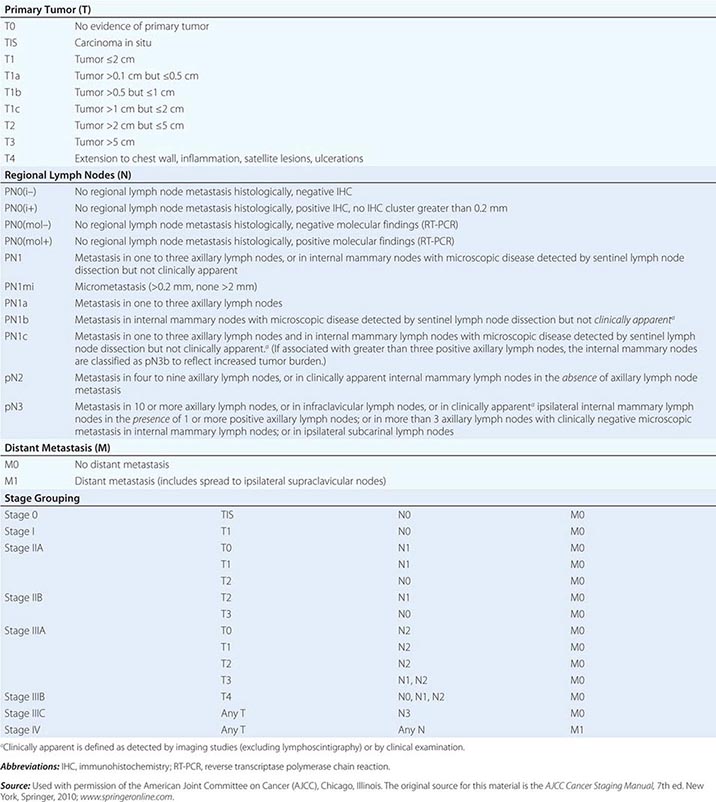
TREATMENT BREAST CANCER
One of the most exciting aspects of breast cancer biology has been its subdivision into at least five subtypes based on gene expression profiling.
1. Luminal A: The luminal tumors express cytokeratins 8 and 18, have the highest levels of estrogen receptor expression, tend to be low grade, are most likely to respond to endocrine therapy, and have a favorable prognosis. They tend to be less responsive to chemotherapy.
2. Luminal B: Tumor cells are also of luminal epithelial origin, but with a gene expression pattern distinct from luminal A. Prognosis is somewhat worse that luminal A.
3. Normal breast–like: These tumors have a gene expression profile reminiscent of nonmalignant “normal” breast epithelium. Prognosis is similar to the luminal B group. This subtype is somewhat controversial and may represent contamination of the sample by normal mammary epithelium.
4. HER2 amplified: These tumors have amplification of the HER2 gene on chromosome 17q and frequently exhibit coamplification and overexpression of other genes adjacent to HER2. Historically the clinical prognosis of such tumors was poor. However, with the advent of trastuzumab and other targeted therapies, the clinical outcome of HER2 -positive patients is markedly improving.
5. Basal: These estrogen receptor/progesterone receptor–negative and HER2-negative tumors (so-called triple negative) are characterized by markers of basal/myoepithelial cells. They tend to be high grade, and express cytokeratins 5/6 and 17 as well as vimentin, p63, CD10, α-smooth muscle actin, and epidermal growth factor receptor (EGFR). Patients with BRCA mutations also fall within this molecular subtype. They also have stem cell characteristics.
PRIMARY BREAST CANCER
Breast-conserving treatments, consisting of the removal of the primary tumor by some form of lumpectomy with or without irradiating the breast, result in a survival that is as good as (or slightly superior to) that after extensive surgical procedures, such as mastectomy or modified radical mastectomy, with or without further irradiation. Postlumpectomy breast irradiation greatly reduces the risk of recurrence in the breast. While breast conservation is associated with a possibility of recurrence in the breast, 10-year survival is at least as good as that after more extensive surgery. Postoperative radiation to regional nodes following mastectomy is also associated with an improvement in survival. Because radiation therapy can also reduce the rate of local or regional recurrence, it should be strongly considered following mastectomy for women with high-risk primary tumors (i.e., T2 in size, positive margins, positive nodes). At present, nearly one-third of women in the United States are managed by lumpectomy. Breast-conserving surgery is not suitable for all patients: it is not generally suitable for tumors >5 cm (or for smaller tumors if the breast is small), for tumors involving the nipple-areola complex, for tumors with extensive intraductal disease involving multiple quadrants of the breast, for women with a history of collagen-vascular disease, and for women who either do not have the motivation for breast conservation or do not have convenient access to radiation therapy. However, these groups probably do not account for more than one-third of patients who are treated with mastectomy. Thus, a great many women still undergo mastectomy who could safely avoid this procedure and probably would if appropriately counseled.
Sentinel lymph node biopsy (SLNB) is generally the standard of care for women with localized breast cancer and clinically negative axilla. If SLNB is negative, more extensive axillary surgery is not required, avoiding much of the risk of lymphedema following more extensive axillary dissections. In the presence of minimal involvement of a sentinel lymph node, further axillary surgery is not required.
An extensive intraductal component is a predictor of recurrence in the breast, and so are several clinical variables. Both axillary lymph node involvement and involvement of vascular or lymphatic channels by metastatic tumor in the breast are associated with a higher risk of relapse in the breast but are not contraindications to breast-conserving treatment. When these patients are excluded, and when lumpectomy with negative tumor margins is achieved, breast conservation is associated with a recurrence rate in the breast of 5% or less. The survival of patients who have recurrence in the breast is somewhat worse than that of women who do not. Thus, recurrence in the breast is a negative prognostic variable for long-term survival. However, recurrence in the breast is not the cause of distant metastasis. If recurrence in the breast caused metastatic disease, then women treated with lumpectomy, who have a higher rate of recurrence in the breast, should have poorer survival than women treated with mastectomy, and they do not. Most patients should consult with a radiation oncologist before making a final decision concerning local therapy. However, a multimodality clinic in which the surgeon, radiation oncologist, medical oncologist, and other caregivers cooperate to evaluate the patient and develop a treatment plan is usually considered a major advantage by patients.
Adjuvant Therapy The use of systemic therapy after local management of breast cancer substantially improves survival. More than half of the women who would otherwise die of metastatic breast cancer remain disease-free when treated with the appropriate systemic regimen. These data have grown more and more impressive with longer follow-up and more effective regimens.
PROGNOSTIC VARIABLES The most important prognostic variables are provided by tumor staging. The size of the tumor and the status of the axillary lymph nodes provide reasonably accurate information on the likelihood of tumor relapse. The relation of pathologic stage to 5-year survival is shown in Table 108-2. For most women, the need for adjuvant therapy can be readily defined on this basis alone. In the absence of lymph node involvement, involvement of microvessels (either capillaries or lymphatic channels) in tumors is nearly equivalent to lymph node involvement. The greatest controversy concerns women with intermediate prognoses. There is rarely justification for adjuvant chemotherapy in most women with tumors <1 cm in size whose axillary lymph nodes are negative. HER2-positive tumors are a potential exception. Detection of breast cancer cells either in the circulation or bone marrow is associated with an increased relapse rate. The most exciting development in this area is the use of gene expression arrays to analyze patterns of tumor gene expression. Several groups have independently defined gene sets that reliably predict disease-free and overall survival far more accurately than any single prognostic variable including the Oncotype DX® analysis of 21 genes. Also, the use of such standardized risk assessment tools such as Adjuvant! Online (www.adjuvantonline.com) is very helpful. These tools are highly recommended in otherwise ambiguous circumstances.
|
5-YEAR SURVIVAL RATE FOR BREAST CANCER BY STAGE |
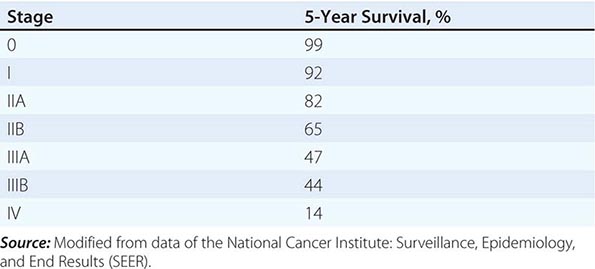
Estrogen receptor status and progesterone receptor status are of prognostic significance. Tumors that lack either or both of these receptors are more likely to recur than tumors that have them.
Several measures of tumor growth rate correlate with early relapse. S-phase analysis using flow cytometry is the most accurate measure. Indirect S-phase assessments using antigens associated with the cell cycle, such as PCNA (Ki67), are also valuable. Tumors with a high proportion (more than the median) of cells in S-phase pose a greater risk of relapse; chemotherapy offers the greatest survival benefit for these tumors. Assessment of DNA content in the form of ploidy is of modest value, with nondiploid tumors having a somewhat worse prognosis.
Histologic classification of the tumor has also been used as a prognostic factor. Tumors with a poor nuclear grade have a higher risk of recurrence than tumors with a good nuclear grade. Semiquantitative measures such as the Elston score improve the reproducibility of this measurement.
Molecular changes in the tumor are also useful. Tumors that overexpress erbB2 (HER2/neu) or have a mutated p53 gene have a worse prognosis. Particular interest has centered on erbB2 overexpression as measured by immunohistochemistry or fluorescence in situ hybridization. Tumors that overexpress erbB2 are more likely to respond to doxorubicin-containing regimens; erbB2 overexpression also predicts those tumors that will respond to HER2/neu antibodies (trastuzumab) (Herceptin) and HER2/neu kinase inhibitors.
Other variables that have also been used to evaluate prognosis include proteins associated with invasiveness, such as type IV collagenase, cathepsin D, plasminogen activator, plasminogen activator receptor, and the metastasis-suppressor gene nm23. None of these has been widely accepted as a prognostic variable for therapeutic decision-making. One problem in interpreting these prognostic variables is that most of them have not been examined in a study using a large cohort of patients.
ADJUVANT REGIMENS Adjuvant therapy is the use of systemic therapies in patients whose known disease has received local therapy but who are at risk of relapse. Selection of appropriate adjuvant chemotherapy or hormone therapy is highly controversial in some situations. Meta-analyses have helped to define broad limits for therapy but do not help in choosing optimal regimens or in choosing a regimen for certain subgroups of patients. A summary of recommendations is shown in Table 108-3. In general, premenopausal women for whom any form of adjuvant systemic therapy is indicated should receive multidrug chemotherapy. Antihormone therapy improves survival in premenopausal patients who are estrogen receptor positive and should be added following completion of chemotherapy. Prophylactic surgical or medically induced castration may also be associated with a substantial survival benefit (primarily in estrogen receptor–positive patients) but is not widely used in this country.
|
SUGGESTED APPROACHES TO ADJUVANT THERAPY |
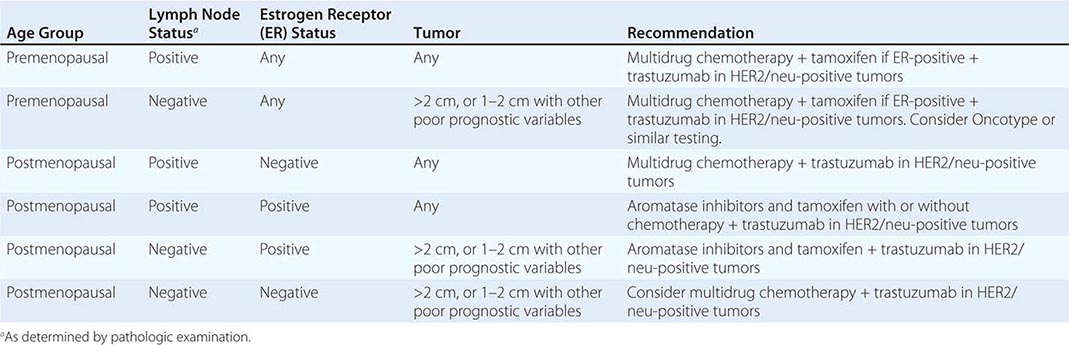
Data on postmenopausal women are also controversial. The impact of adjuvant chemotherapy is quantitatively less clear-cut than in premenopausal patients, particularly in estrogen receptor–positive cases, although survival advantages have been shown. The first decision is whether chemotherapy or endocrine therapy should be used. While adjuvant endocrine therapy (aromatase inhibitors and tamoxifen) improves survival regardless of axillary lymph node status, the improvement in survival is modest for patients in whom multiple lymph nodes are involved. For this reason, it has been usual to give chemotherapy to postmenopausal patients who have no medical contraindications and who have more than one positive lymph node; hormone therapy is commonly given subsequently. For postmenopausal women for whom systemic therapy is warranted but who have a more favorable prognosis (based more commonly on analysis such as the Oncotype DX methodology), hormone therapy may be used alone. Large clinical trials have shown superiority for aromatase inhibitors over tamoxifen alone in the adjuvant setting, although tamoxifen appears essentially equivalent in women who are obese and therefore presumably have higher endogenous concentrations of estrogen. Unfortunately the optimal plan is unclear. Tamoxifen for 5 years followed by an aromatase inhibitor, the reverse strategy, or even switching to an aromatase inhibitor after 2–3 years of tamoxifen has been shown to be better than tamoxifen alone. Continuation of tamoxifen for 10 years yields further benefit and is a reasonable decision for women with less favorable prognoses. Unfortunately, multiple studies have revealed very suboptimal adherence to long-term adjuvant endocrine regimens, and every effort should be made to encourage their continuous use. No valid information currently permits selection among the three clinically approved aromatase inhibitors. Concomitant use of bisphosphonates is almost always warranted; however, it is not finally settled as to whether their prophylactic use increases survival in addition to just decreasing recurrences in bone.
Most comparisons of adjuvant chemotherapy regimens show little difference among them, although small advantages for doxorubicin-containing regimens and “dose-dense” regimens are usually seen.
One approach—so-called neoadjuvant chemotherapy—involves the administration of adjuvant therapy before definitive surgery and radiation therapy. Because the objective response rates of patients with breast cancer to systemic therapy in this setting exceed 75%, many patients will be “downstaged” and may become candidates for breast-conserving therapy. However, overall survival has not been improved using this approach as compared with the same drugs given postoperatively. Patients who achieve a pathologic complete remission after neoadjuvant chemotherapy not unexpectedly have a substantially improved survival. The neoadjuvant setting also provides a wonderful opportunity for the evaluation of new agents. For example, a second HER2 targeting antibody, pertuzumab, has been shown to provide additional benefit when combined with trastuzumab in the neoadjuvant setting.
Other adjuvant treatments under investigation include the use of taxanes, such as paclitaxel and docetaxel, and therapy based on alternative kinetic and biologic models. In such approaches, high doses of single agents are used separately in relatively dose-intensive cycling regimens. Node-positive patients treated with doxorubicin-cyclophosphamide for four cycles followed by four cycles of a taxane have a substantial improvement in survival compared with women receiving doxorubicin-cyclophosphamide alone, particularly in women with estrogen receptor–negative tumors. In addition, administration of the same drug combinations at the same dose but at more frequent intervals (every 2 weeks with cytokine support as compared with the standard every 3 weeks) is even more effective. Among the 25% of women whose tumors overexpress HER2/neu, addition of trastuzumab given concurrently with a taxane and then for a year after chemotherapy produces significant improvement in survival. Although longer follow-up will be important, this is now the standard care for most women with HER2/neu-positive breast cancers. Cardiotoxicity, immediate and long-term, remains a concern, and further efforts to exploit non-anthracycline-containing regimens are being pursued. Very-high-dose therapy with stem cell transplantation in the adjuvant setting has not proved superior to standard-dose therapy and should not be routinely used.
A variety of exciting approaches are close to adoption, and the literature needs to be followed attentively. Tyrosine kinase inhibitors such as lapatinib and additional HER2-targeting antibodies such as pertuzumab are very promising. Finally, as described in the next section, a novel class of agents targeting DNA repair—the so-called poly–ADP ribose polymerase (PARP) inhibitors—is likely to have a major effect on breast cancers either caused by BRCA1 or BRCA2 mutations or sharing similar defects in DNA repair in their etiology.
SYSTEMIC THERAPY OF METASTATIC DISEASE
About one-third of patients treated for apparently localized breast cancer develop metastatic disease. Although a small number of these patients enjoy long remissions when treated with combinations of systemic and local therapy, most eventually succumb to metastatic disease. The median survival for all patients diagnosed with metastatic breast cancer is less than 3 years. Soft tissue, bony, and visceral (lung and liver) metastases each account for approximately one-third of sites of initial relapses. However, by the time of death, most patients will have bony involvement. Recurrences can appear at any time after primary therapy. A very cruel fact about breast cancer recurrences is that at least half of all breast cancer recurrences occur >5 years after initial therapy. It is now clear that a variety of host factors can influence recurrence rates, including depression and central obesity, and these diseases should be managed as aggressively as possible.
Because the diagnosis of metastatic disease alters the outlook for the patient so drastically, it should rarely be made without a confirmatory biopsy. Every oncologist has seen patients with tuberculosis, gallstones, sarcoidosis, or other nonmalignant diseases misdiagnosed and treated as though they had metastatic breast cancer or even second malignancies such as multiple myeloma thought to be recurrent breast cancer. This is a catastrophic mistake and justifies biopsy for virtually every patient at the time of initial suspicion of metastatic disease. Furthermore, there are well-documented changes in hormone receptor status that can occur and substantially alter treatment decisions.
The choice of therapy requires consideration of local therapy needs, the overall medical condition of the patient, and the hormone receptor status of the tumor, as well as clinical judgment. Because therapy of systemic disease is palliative, the potential toxicities of therapies should be balanced against the response rates. Several variables influence the response to systemic therapy. For example, the presence of estrogen and progesterone receptors is a strong indication for endocrine therapy. On the other hand, patients with short disease-free intervals, rapidly progressive visceral disease, lymphangitic pulmonary disease, or intracranial disease are unlikely to respond to endocrine therapy.
In many cases, systemic therapy can be withheld while the patient is managed with appropriate local therapy. Radiation therapy and occasionally surgery are effective at relieving the symptoms of metastatic disease, particularly when bony sites are involved. Many patients with bone-only or bone-dominant disease have a relatively indolent course. Under such circumstances, systemic chemotherapy has a modest effect, whereas radiation therapy may be effective for long periods. Other systemic treatments, such as strontium-89 and/or bisphosphonates, may provide a palliative benefit without inducing objective responses. Most patients with metastatic disease, and certainly all who have bone involvement, should receive concurrent bisphosphonates. Because the goal of therapy is to maintain well-being for as long as possible, emphasis should be placed on avoiding the most hazardous complications of metastatic disease, including pathologic fracture of the axial skeleton and spinal cord compression. New back pain in patients with cancer should be explored aggressively on an emergent basis; to wait for neurologic symptoms is a potentially catastrophic error. Metastatic involvement of endocrine organs can cause profound dysfunction, including adrenal insufficiency and hypopituitarism. Similarly, obstruction of the biliary tree or other impaired organ function may be better managed with a local therapy than with a systemic approach.
Many patients are inappropriately treated with toxic regimens into their last days of life. Often oncologists are unwilling to have the difficult conversations that are required with patients nearing the end of life, and not uncommonly, patients and families can pressure physicians into treatments with very little survival value. Palliative care consultation and realistic assessment of treatment expectations need to be reviewed with patients and families. We urge consideration of palliative care consultations for patients who have received at least two lines of therapy for metastatic disease.
Endocrine Therapy Normal breast tissue is estrogen dependent. Both primary and metastatic breast cancer may retain this phenotype. The best means of ascertaining whether a breast cancer is hormone dependent is through analysis of estrogen and progesterone receptor levels on the tumor. Tumors that are positive for the estrogen receptor and negative for the progesterone receptor have a response rate of ∼30%. Tumors that are positive for both receptors have a response rate approaching 70%. If neither receptor is present, the objective response rates are <5%. Receptor analyses provide information as to the correct ordering of endocrine therapies as opposed to chemotherapy. Because of their lack of toxicity and because some patients whose receptor analyses are reported as negative respond to endocrine therapy, an endocrine treatment should be attempted in virtually every patient with metastatic breast cancer. Potential endocrine therapies are summarized in Table 108-4. The choice of endocrine therapy is usually determined by toxicity profile and availability. In most postmenopausal patients, the initial endocrine therapy should be an aromatase inhibitor rather than tamoxifen. For the subset of postmenopausal women who are estrogen receptor–positive but also HER2/neu-positive, response rates to aromatase inhibitors are substantially higher than to tamoxifen. Aromatase inhibitors are not used in premenopausal women because their hypothalamus can respond to estrogen deprivation by producing gonadotropins that promote estrogen synthesis. Newer “pure” antiestrogens that are free of agonistic effects are also effective. Cases in which tumors shrink in response to tamoxifen withdrawal (as well as withdrawal of pharmacologic doses of estrogens) have been reported. A series of studies with aromatase inhibitors, tamoxifen, and fulvestrant have all shown that the addition of everolimus to the hormonal treatment can lead to significant benefit after progression on the endocrine agent alone. Everolimus (an mTOR inhibitor) in coordination with endocrine agents is now being explored as front-line therapy and in the adjuvant setting. Endogenous estrogen formation may be blocked by analogues of luteinizing hormone–releasing hormone in premenopausal women. Additive endocrine therapies, including treatment with progestogens, estrogens, and androgens, may also be tried in patients who respond to initial endocrine therapy; the mechanism of action of these latter therapies is unknown. Patients who respond to one endocrine therapy have at least a 50% chance of responding to a second endocrine therapy. It is not uncommon for patients to respond to two or three sequential endocrine therapies; however, combination endocrine therapies do not appear to be superior to individual agents, and combinations of chemotherapy with endocrine therapy are not useful. The median survival of patients with metastatic disease is approximately 2 years, although many patients, particularly older persons and those with hormone-dependent disease, may respond to endocrine therapy for 3–5 years or longer.
|
ENDOCRINE THERAPIES FOR BREAST CANCER |
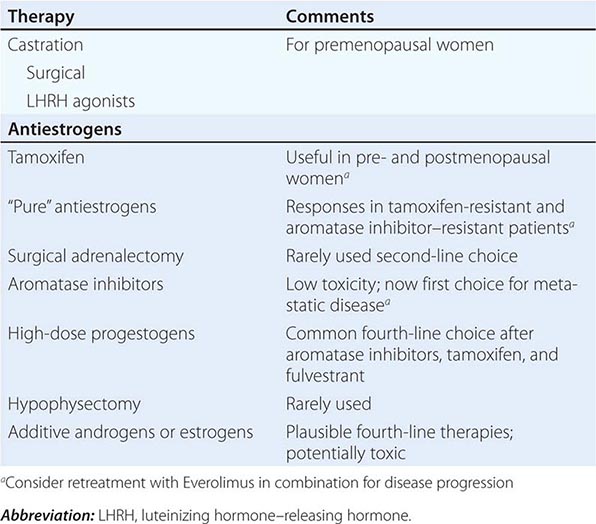
Chemotherapy Unlike many other epithelial malignancies, breast cancer responds to multiple chemotherapeutic agents, including anthracyclines, alkylating agents, taxanes, and antimetabolites. Multiple combinations of these agents have been found to improve response rates somewhat, but they have had little effect on duration of response or survival. The choice among multidrug combinations frequently depends on whether adjuvant chemotherapy was administered and, if so, what type. Although patients treated with adjuvant regimens such as cyclophosphamide, methotrexate, and fluorouracil (CMF regimens) may subsequently respond to the same combination in the metastatic disease setting, most oncologists use drugs to which the patients have not been previously exposed. Once patients have progressed after combination drug therapy, it is most common to treat them with single agents. Given the significant toxicity of most drugs, the use of a single effective agent will minimize toxicity by sparing the patient exposure to drugs that would be of little value. No method to select the drugs most efficacious for a given patient has been demonstrated to be useful.
Most oncologists use either an anthracycline or paclitaxel following failure with the initial regimen. However, the choice has to be balanced with individual needs. One randomized study has suggested that docetaxel may be superior to paclitaxel. A nanoparticle formulation of paclitaxel (Abraxane) is also effective.
The use of a humanized antibody to erbB2 (trastuzumab [Herceptin]) combined with paclitaxel can improve response rate and survival for women whose metastatic tumors overexpress erbB2. A novel antibody conjugate (ADC) that links trastuzumab to a cytotoxic agent has been approved for management of HER2-positive breast cancer. The magnitude of the survival extension is modest in patients with metastatic disease. Similarly, the use of bevacizumab (Avastin) has improved the response rate and response duration to paclitaxel. Objective responses in previously treated patients may also be seen with gemcitabine, vinca alkaloids, capecitabine, vinorelbine, and oral etoposide, as well as a new class of agents, epothilones. There are few comparative trials of one agent versus another in metastatic disease. It is a sad fact that choices are often influenced by aggressive marketing of new very expensive agents that have not been shown to be superior to other generic agents. Platinum-based agents have become far more widely used in both the adjuvant and advanced disease settings for some breast cancers, particularly those of the “triple-negative” subtype.
HIGH-DOSE CHEMOTHERAPY INCLUDING AUTOLOGOUS BONE MARROW TRANSPLANTATION Autologous bone marrow transplantation combined with high doses of single agents can produce objective responses even in heavily pretreated patients. However, such responses are rarely durable and do not alter the clinical course for most patients with advanced metastatic disease.
STAGE III BREAST CANCER
Between 10 and 25% of patients present with so-called locally advanced, or stage III, breast cancer at diagnosis. Many of these cancers are technically operable, whereas others, particularly cancers with chest wall involvement, inflammatory breast cancers, or cancers with large matted axillary lymph nodes, cannot be managed with surgery initially. Although no randomized trials have shown any survival benefit for neoadjuvant regimens as compared to adjuvant therapy, this approach has gained widespread use. More than 90% of patients with locally advanced breast cancer show a partial or better response to multidrug chemotherapy regimens that include an anthracycline. Early administration of this treatment reduces the bulk of the disease and frequently makes the patient a suitable candidate for salvage surgery and/or radiation therapy. These patients should be managed in multimodality clinics to coordinate surgery, radiation therapy, and systemic chemotherapy. Such approaches produce long-term disease-free survival in about 30–50% of patients. The neoadjuvant setting is also an ideal time to evaluate the efficacy of novel treatments because the effect on the tumor can be directly assessed.
BREAST CANCER PREVENTION
Women who have one breast cancer are at risk of developing a contralateral breast cancer at a rate of approximately 0.5% per year. When adjuvant tamoxifen or an aromatase inhibitor is administered to these patients, the rate of development of contralateral breast cancers is reduced. In other tissues of the body, tamoxifen has estrogen-like effects that are beneficial, including preservation of bone mineral density and long-term lowering of cholesterol. However, tamoxifen has estrogen-like effects on the uterus, leading to an increased risk of uterine cancer (0.75% incidence after 5 years on tamoxifen). Tamoxifen also increases the risk of cataract formation. The Breast Cancer Prevention Trial (BCPT) revealed a >49% reduction in breast cancer among women with a risk of at least 1.66% taking the drug for 5 years. Raloxifene has shown similar breast cancer prevention potency but may have different effects on bone and heart. The two agents have been compared in a prospective randomized prevention trial (the Study of Tamoxifen and Raloxifene [STAR] trial). The agents are approximately equivalent in preventing breast cancer with fewer thromboembolic events and endometrial cancers with raloxifene; however, raloxifene did not reduce noninvasive cancers as effectively as tamoxifen, so no clear winner has emerged. A newer selective estrogen receptor modulator (SERM), lasofoxifene, has been shown to reduce cardiovascular events in addition to breast cancer and fractures, and further studies of this agent should be watched with interest. It should be recalled that prevention of contralateral breast cancers in women diagnosed with one cancer is a reasonable surrogate for breast cancer prevention because these are second primaries not recurrences. In this regard, the aromatase inhibitors are all considerably more effective than tamoxifen; however, they are not approved for primary breast cancer prevention. It remains puzzling that agents with the safety profile of raloxifene, which can reduce breast cancer risk by 50% with additional benefits in preventing osteoporotic fracture, are still so infrequently prescribed. They should be far more commonly offered to women than they are.
NONINVASIVE BREAST CANCER
Breast cancer develops as a series of molecular changes in the epithelial cells that lead to ever more malignant behavior. Increased use of mammography has led to more frequent diagnoses of noninvasive breast cancer. These lesions fall into two groups: ductal carcinoma in situ (DCIS) and lobular carcinoma in situ (lobular neoplasia). The management of both entities is controversial.
Ductal Carcinoma In Situ Proliferation of cytologically malignant breast epithelial cells within the ducts is termed DCIS. Atypical hyperplasia may be difficult to differentiate from DCIS. At least one-third of patients with untreated DCIS develop invasive breast cancer within 5 years. However, many low-grade DCIS lesions do not appear to progress over many years; therefore, many patients are overtreated. Unfortunately there is no reliable means of distinguishing patients who require treatment from those who may be safely observed. For many years, the standard treatment for this disease was mastectomy. However, treatment of this condition by lumpectomy and radiation therapy gives survival that is as good as the survival for invasive breast cancer treated by mastectomy. In one randomized trial, the combination of wide excision plus irradiation for DCIS caused a substantial reduction in the local recurrence rate as compared with wide excision alone with negative margins, although survival was identical in the two arms. No studies have compared either of these regimens to mastectomy. Addition of tamoxifen to any DCIS surgical/radiation therapy regimen further improves local control. Data for aromatase inhibitors in this setting are not available.
Several prognostic features may help to identify patients at high risk for local recurrence after either lumpectomy alone or lumpectomy with radiation therapy. These include extensive disease; age <40; and cytologic features such as necrosis, poor nuclear grade, and comedo subtype with overexpression of erbB2. Some data suggest that adequate excision with careful determination of pathologically clear margins is associated with a low recurrence rate. When surgery is combined with radiation therapy, recurrence (which is usually in the same quadrant) occurs with a frequency of ≤10%. Given the fact that half of these recurrences will be invasive, about 5% of the initial cohort will eventually develop invasive breast cancer. A reasonable expectation of mortality for these patients is about 1%, a figure that approximates the mortality rate for DCIS managed by mastectomy. Although this train of reasoning has not formally been proved valid, it is reasonable to recommend that patients who desire breast preservation, and in whom DCIS appears to be reasonably localized, be managed by adequate surgery with meticulous pathologic evaluation, followed by breast irradiation and tamoxifen. For patients with localized DCIS, axillary lymph node dissection is unnecessary. More controversial is the question of what management is optimal when there is any degree of invasion. Because of a significant likelihood (10–15%) of axillary lymph node involvement even when the primary lesion shows only microscopic invasion, it is prudent to do at least a sentinel lymph node sampling for all patients with any degree of invasion. Further management is dictated by the presence of nodal spread.
Lobular Neoplasia Proliferation of cytologically malignant cells within the lobules is termed lobular neoplasia. Nearly 30% of patients who have had adequate local excision of the lesion develop breast cancer (usually infiltrating ductal carcinoma) over the next 15–20 years. Ipsilateral and contralateral cancers are equally common. Therefore, lobular neoplasia may be a premalignant lesion that suggests an elevated risk of subsequent breast cancer, rather than a form of malignancy itself, and aggressive local management seems unreasonable. Most patients should be treated with an SERM or an aromatase inhibitor (for postmenopausal women) for 5 years and followed with careful annual mammography and semiannual physical examinations. Additional molecular analysis of these lesions may make it possible to discriminate between patients who are at risk of further progression and require additional therapy and those in whom simple follow-up is adequate.
MALE BREAST CANCER
Breast cancer is about 1/150th as frequent in men as in women; 1720 men developed breast cancer in 2006. It usually presents as a unilateral lump in the breast and is frequently not diagnosed promptly. Given the small amount of soft tissue and the unexpected nature of the problem, locally advanced presentations are somewhat more common. When male breast cancer is matched to female breast cancer by age and stage, its overall prognosis is identical. Although gynecomastia may initially be unilateral or asymmetric, any unilateral mass in a man older than age 40 years should receive a careful workup including biopsy. On the other hand, bilateral symmetric breast development rarely represents breast cancer and is almost invariably due to endocrine disease or a drug effect. It should be kept in mind, nevertheless, that the risk of cancer is much greater in men with gynecomastia; in such men, gross asymmetry of the breasts should arouse suspicion of cancer. Male breast cancer is best managed by mastectomy and axillary lymph node dissection or SLNB. Patients with locally advanced disease or positive nodes should also be treated with irradiation. Approximately 90% of male breast cancers contain estrogen receptors, and approximately 60% of cases with metastatic disease respond to endocrine therapy. No randomized studies have evaluated adjuvant therapy for male breast cancer. Two historic experiences suggest that the disease responds well to adjuvant systemic therapy, and, if not medically contraindicated, the same criteria for the use of adjuvant therapy in women should be applied to men.
The sites of relapse and spectrum of response to chemotherapeutic drugs are virtually identical for breast cancers in either sex.
FOLLOW-UP OF BREAST CANCER PATIENTS
Despite the availability of sophisticated and expensive imaging techniques and a wide range of serum tumor marker tests, survival is not influenced by early diagnosis of relapse. Surveillance guidelines are given in Table 108-5. Despite pressure from patients and their families, routine computed tomography scans (or other imaging) are not recommended.
|
BREAST CANCER SURVEILLANCE GUIDELINES |
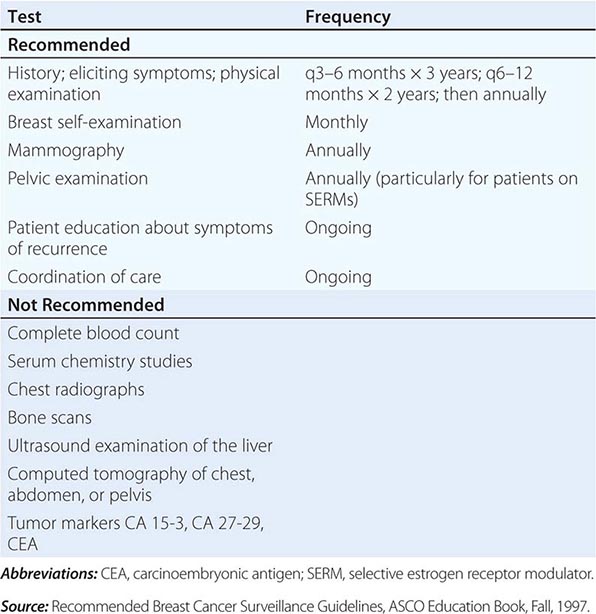
109 |
Upper Gastrointestinal Tract Cancers |
Upper gastrointestinal cancers include malignancies arising in the esophagus, stomach, and small intestine.
ESOPHAGEAL CANCER
INCIDENCE AND ETIOLOGY
![]() Cancer of the esophagus is an increasingly common and extremely lethal malignancy. The diagnosis was made in 18,170 Americans in 2014 and led to 15,450 deaths. Almost all esophageal cancers are either squamous cell carcinomas or adenocarcinomas; the two histologic subtypes have a similar clinical presentation but different causative factors.
Cancer of the esophagus is an increasingly common and extremely lethal malignancy. The diagnosis was made in 18,170 Americans in 2014 and led to 15,450 deaths. Almost all esophageal cancers are either squamous cell carcinomas or adenocarcinomas; the two histologic subtypes have a similar clinical presentation but different causative factors.
Worldwide, squamous cell carcinoma is the more common cell type, having an incidence that rises strikingly in association with geographic location. It occurs frequently within a region extending from the southern shore of the Caspian Sea on the west to northern China on the east, encompassing parts of Iran, central Asia, Afghanistan, Siberia, and Mongolia. Familial increased risk has been observed in regions with high incidence, although gene associations are not yet defined. High-incidence “pockets” of the disease are also present in such disparate locations as Finland, Iceland, Curaçao, southeastern Africa, and northwestern France. In North America and western Europe, the disease is more common in blacks than whites and in males than females; it appears most often after age 50 and seems to be associated with a lower socioeconomic status. Such cancers generally arise in the cervical and thoracic portions of the esophagus.
A variety of causative factors have been implicated in the development of squamous cell cancers of the esophagus (Table 109-1). In the United States, the etiology of such cancers is primarily related to excess alcohol consumption and/or cigarette smoking. The relative risk increases with the amount of tobacco smoked or alcohol consumed, with these factors acting synergistically. The consumption of whiskey is linked to a higher incidence than the consumption of wine or beer. Squamous cell esophageal carcinoma has also been associated with the ingestion of nitrates, smoked opiates, and fungal toxins in pickled vegetables, as well as mucosal damage caused by such physical insults as long-term exposure to extremely hot tea, the ingestion of lye, radiation-induced strictures, and chronic achalasia. The presence of an esophageal web in association with glossitis and iron deficiency (i.e., Plummer-Vinson or Paterson-Kelly syndrome) and congenital hyperkeratosis and pitting of the palms and soles (i.e., tylosis palmaris et plantaris) have each been linked with squamous cell esophageal cancer, as have dietary deficiencies of molybdenum, zinc, selenium, and vitamin A. Patients with head and neck cancer are at increased risk of squamous cell cancer of the esophagus.
|
SOME ETIOLOGIC FACTORS ASSOCIATED WITH SQUAMOUS CELL CANCER OF THE ESOPHAGUS |
For unclear reasons, the incidence of squamous cell esophageal cancer has decreased somewhat in both the black and white populations in the United States over the past 40 years, whereas the rate of adenocarcinoma has risen sevenfold, particularly in white males (male-to-female ratio of 6:1). Whereas squamous cell cancers comprised the vast majority of esophageal cancers in the United States as recently as 40–50 years ago, more than 75% of esophageal tumors are now adenocarcinomas, with the incidence of this histologic subtype continuing to increase rapidly. Understanding the cause for this increase is the focus of current investigation.
Several strong etiologic associations have been observed to account for the development of adenocarcinoma of the esophagus (Table 109-2). Such tumors arise in the distal esophagus in association with chronic gastric reflux, often in the presence of Barrett’s esophagus (replacement of the normal squamous epithelium of the distal esophagus by columnar mucosa), which occurs more commonly in obese individuals. Adenocarcinomas arise within dysplastic columnar epithelium in the distal esophagus. Even before frank neoplasia is detectable, aneuploidy and p53 mutations are found in the dysplastic epithelium. These adenocarcinomas behave clinically like gastric adenocarcinomas, although they are not associated with Helicobacter pylori infections. Approximately 15% of esophageal adenocarcinomas overexpress the HER2/neu gene.
|
SOME ETIOLOGIC FACTORS ASSOCIATED WITH ADENOCARCINOMA OF THE ESOPHAGUS |
CLINICAL FEATURES
About 5% of esophageal cancers occur in the upper third of the esophagus (cervical esophagus), 20% in the middle third, and 75% in the lower third. Squamous cell carcinomas and adenocarcinomas cannot be distinguished radiographically or endoscopically.
Progressive dysphagia and weight loss of short duration are the initial symptoms in the vast majority of patients. Dysphagia initially occurs with solid foods and gradually progresses to include semisolids and liquids. By the time these symptoms develop, the disease is already very advanced, because difficulty in swallowing does not occur until >60% of the esophageal circumference is infiltrated with cancer. Dysphagia may be associated with pain on swallowing (odynophagia), pain radiating to the chest and/or back, regurgitation or vomiting, and aspiration pneumonia. The disease most commonly spreads to adjacent and supraclavicular lymph nodes, liver, lungs, pleura, and bone. Tracheoesophageal fistulas may develop, primarily in patients with upper and mid-esophageal tumors. As with other squamous cell carcinomas, hypercalcemia may occur in the absence of osseous metastases, probably from parathormone-related peptide secreted by tumor cells (Chap. 121).
DIAGNOSIS
Attempts at endoscopic and cytologic screening for carcinoma in patients with Barrett’s esophagus, while effective as a means of detecting high-grade dysplasia, have not yet been shown to reduce the likelihood of death from esophageal adenocarcinoma. Esophagoscopy should be performed in all patients suspected of having an esophageal abnormality, to both visualize and identify a tumor and also to obtain histopathologic confirmation of the diagnosis. Because the population of persons at risk for squamous cell carcinoma of the esophagus (i.e., smokers and drinkers) also has a high rate of cancers of the lung and the head and neck region, endoscopic inspection of the larynx, trachea, and bronchi should also be carried out. A thorough examination of the fundus of the stomach (by retroflexing the endoscope) is imperative as well. The extent of tumor spread to the mediastinum and para-aortic lymph nodes should be assessed by computed tomography (CT) scans of the chest and abdomen and by endoscopic ultrasound. Positron emission tomography scanning provides a useful assessment of the presence of distant metastatic disease, offering accurate information regarding spread to mediastinal lymph nodes, which can be helpful in defining radiation therapy fields. Such scans, when performed sequentially, appear to provide a means of making an early assessment of responsiveness to preoperative chemotherapy.
TUMORS OF THE STOMACH
GASTRIC ADENOCARCINOMA
![]() Incidence and Epidemiology For unclear reasons, the incidence and mortality rates for gastric cancer have decreased in the United States during the past 80 years, although the disease remains the second most frequent cause of worldwide cancer-related death. The mortality rate from gastric cancer in the United States has dropped in men from 28 to 5.8 per 100,000 persons, whereas in women, the rate has decreased from 27 to 2.8 per 100,000. Nonetheless, in 2014, 22,220 new cases of stomach cancer were diagnosed in the United States, and 10,990 Americans died of the disease. Although the incidence of gastric cancer has decreased worldwide, it remains high in such disparate geographic regions as Japan, China, Chile, and Ireland.
Incidence and Epidemiology For unclear reasons, the incidence and mortality rates for gastric cancer have decreased in the United States during the past 80 years, although the disease remains the second most frequent cause of worldwide cancer-related death. The mortality rate from gastric cancer in the United States has dropped in men from 28 to 5.8 per 100,000 persons, whereas in women, the rate has decreased from 27 to 2.8 per 100,000. Nonetheless, in 2014, 22,220 new cases of stomach cancer were diagnosed in the United States, and 10,990 Americans died of the disease. Although the incidence of gastric cancer has decreased worldwide, it remains high in such disparate geographic regions as Japan, China, Chile, and Ireland.
The risk of gastric cancer is greater among lower socioeconomic classes. Migrants from high- to low-incidence nations maintain their susceptibility to gastric cancer, whereas the risk for their offspring approximates that of the new homeland. These findings suggest that an environmental exposure, probably beginning early in life, is related to the development of gastric cancer, with dietary carcinogens considered the most likely factor(s).
Pathology About 85% of stomach cancers are adenocarcinomas, with 15% due to lymphomas, gastrointestinal stromal tumors (GISTs), and leiomyosarcomas. Gastric adenocarcinomas may be subdivided into two categories: a diffuse type, in which cell cohesion is absent, so that individual cells infiltrate and thicken the stomach wall without forming a discrete mass; and an intestinal type, characterized by cohesive neoplastic cells that form glandlike tubular structures. The diffuse carcinomas occur more often in younger patients, develop throughout the stomach (including the cardia), result in a loss of distensibility of the gastric wall (so-called linitis plastica, or “leather bottle” appearance), and carry a poorer prognosis. Diffuse cancers have defective intercellular adhesion, mainly as a consequence of loss of expression of E-cadherin. Intestinal-type lesions are frequently ulcerative, more commonly appear in the antrum and lesser curvature of the stomach, and are often preceded by a prolonged precancerous process, often initiated by H. pylori infection. Although the incidence of diffuse carcinomas is similar in most populations, the intestinal type tends to predominate in the high-risk geographic regions and is less likely to be found in areas where the frequency of gastric cancer is declining. Thus, different etiologic factor(s) are likely involved in these two subtypes. In the United States, ∼30% of gastric cancers originate in the distal stomach, ∼20% arise in the midportion of the stomach, and ∼40% originate in the proximal third of the stomach. The remaining 10% involve the entire stomach.
Etiology The long-term ingestion of high concentrations of nitrates found in dried, smoked, and salted foods appears to be associated with a higher risk. The nitrates are thought to be converted to carcinogenic nitrites by bacteria (Table 109-3). Such bacteria may be introduced exogenously through the ingestion of partially decayed foods, which are consumed in abundance worldwide by the lower socioeconomic classes. Bacteria such as H. pylori may also contribute to this effect by causing chronic inflammatory atrophic gastritis, loss of gastric acidity, and bacterial growth in the stomach. Although the risk for developing gastric cancer is thought to be sixfold higher in people infected with H. pylori, it remains uncertain whether eradicating the bacteria after infection has already occurred actually reduces this risk. Loss of acidity may occur when acid-producing cells of the gastric antrum have been removed surgically to control benign peptic ulcer disease or when achlorhydria, atrophic gastritis, and even pernicious anemia develop in the elderly. Serial endoscopic examinations of the stomach in patients with atrophic gastritis have documented replacement of the usual gastric mucosa by intestinal-type cells. This process of intestinal metaplasia may lead to cellular atypia and eventual neoplasia. Because the declining incidence of gastric cancer in the United States primarily reflects a decline in distal, ulcerating, intestinal-type lesions, it is conceivable that better food preservation and the availability of refrigeration for all socioeconomic classes have decreased the dietary ingestion of exogenous bacteria. H. pylori has not been associated with the diffuse, more proximal form of gastric carcinoma or with cancers arising at the gastroesophageal junction or in the distal esophagus. Approximately 10–15% of adenocarcinomas appearing in the proximal stomach, the gastroesophageal junction, and the distal esophagus overexpress the HER2/neu gene; individuals whose tumors demonstrate this overexpression benefit from treatment directed against this target (i.e., trastuzumab [Herceptin]).
|
NITRATE-CONVERTING BACTERIA AS A FACTOR IN THE CAUSATION OF GASTRIC CARCINOMAa |
aHypothesis: Dietary nitrates are converted to carcinogenic nitrites by bacteria.
Several additional etiologic factors have been associated with gastric carcinoma. Gastric ulcers and adenomatous polyps have occasionally been linked, but data on a cause-and-effect relationship are unconvincing. The inadequate clinical distinction between benign gastric ulcers and small ulcerating carcinomas may, in part, account for this presumed association. The presence of extreme hypertrophy of gastric rugal folds (i.e., Ménétrier’s disease), giving the impression of polypoid lesions, has been associated with a striking frequency of malignant transformation; such hypertrophy, however, does not represent the presence of true adenomatous polyps. Individuals with blood group A have a higher incidence of gastric cancer than persons with blood group O; this observation may be related to differences in the mucous secretion, leading to altered mucosal protection from carcinogens. A germline mutation in the E-cadherin gene (CDH1), inherited in an autosomal dominant pattern and coding for a cell adhesion protein, has been linked to a high incidence of occult diffuse-type gastric cancers in young asymptomatic carriers. Duodenal ulcers are not associated with gastric cancer.
Clinical Features Gastric cancers, when superficial and surgically curable, usually produce no symptoms. As the tumor becomes more extensive, patients may complain of an insidious upper abdominal discomfort varying in intensity from a vague, postprandial fullness to a severe, steady pain. Anorexia, often with slight nausea, is very common but is not the usual presenting complaint. Weight loss may eventually be observed, and nausea and vomiting are particularly prominent in patients whose tumors involve the pylorus; dysphagia and early satiety may be the major symptoms caused by diffuse lesions originating in the cardia. There may be no early physical signs. A palpable abdominal mass indicates long-standing growth and predicts regional extension.
Gastric carcinomas spread by direct extension through the gastric wall to the perigastric tissues, occasionally adhering to adjacent organs such as the pancreas, colon, or liver. The disease also spreads via lymphatics or by seeding of peritoneal surfaces. Metastases to intraabdominal and supraclavicular lymph nodes occur frequently, as do metastatic nodules to the ovary (Krukenberg’s tumor), periumbilical region (“Sister Mary Joseph node”), or peritoneal cul-de-sac (Blumer’s shelf palpable on rectal or vaginal examination); malignant ascites may also develop. The liver is the most common site for hematogenous spread of tumor.
The presence of iron-deficiency anemia in men and of occult blood in the stool in both sexes mandates a search for an occult gastrointestinal tract lesion. A careful assessment is of particular importance in patients with atrophic gastritis or pernicious anemia. Unusual clinical features associated with gastric adenocarcinomas include migratory thrombophlebitis, microangiopathic hemolytic anemia, diffuse seborrheic keratoses (so-called Leser-Trélat sign), and acanthosis nigricans.
Diagnosis The use of double-contrast radiographic examinations has been supplanted by esophagogastroscopy and CT scanning for the evaluation of patients with epigastric complaints.
Gastric ulcers identified at the time of such endoscopic procedure may appear benign but merit biopsy in order to exclude a malignancy. Malignant gastric ulcers must be recognized before they penetrate into surrounding tissues, because the rate of cure of early lesions limited to the mucosa or submucosa is >80%. Because gastric carcinomas are difficult to distinguish clinically or endoscopically from gastric lymphomas, endoscopic biopsies should be made as deeply as possible, due to the submucosal location of lymphoid tumors.
The staging system for gastric carcinoma is shown in Table 109-4.
|
STAGING SYSTEM FOR GASTRIC CARCINOMA |
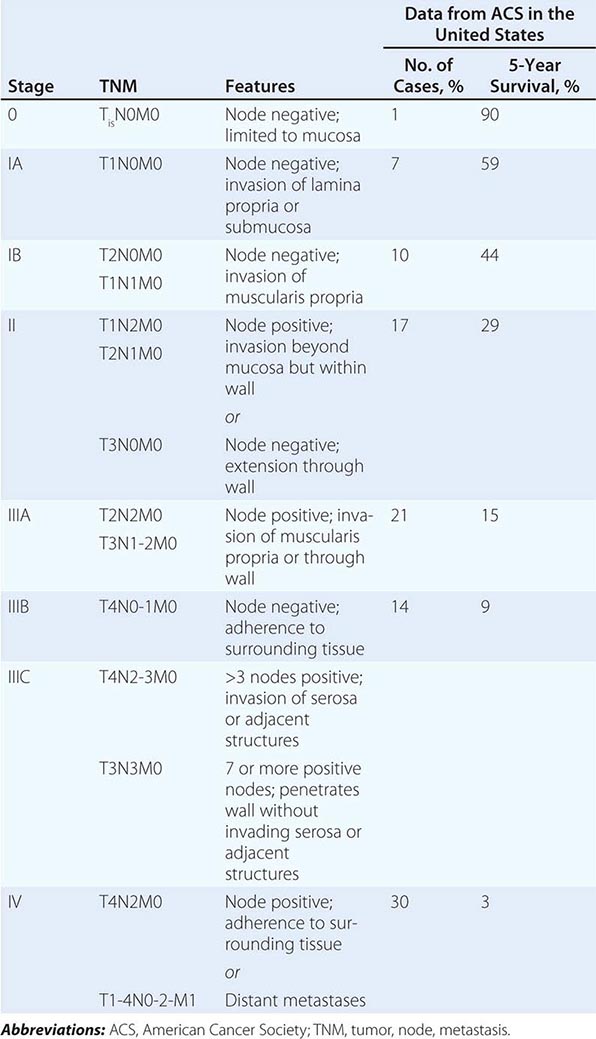
PRIMARY GASTRIC LYMPHOMA
Primary lymphoma of the stomach is relatively uncommon, accounting for <15% of gastric malignancies and ∼2% of all lymphomas. The stomach is, however, the most frequent extranodal site for lymphoma, and gastric lymphoma has increased in frequency during the past 35 years. The disease is difficult to distinguish clinically from gastric adenocarcinoma; both tumors are most often detected during the sixth decade of life; present with epigastric pain, early satiety, and generalized fatigue; and are usually characterized by ulcerations with a ragged, thickened mucosal pattern demonstrated by contrast radiographs or endoscopic appearance. The diagnosis of lymphoma of the stomach may occasionally be made through cytologic brushings of the gastric mucosa but usually requires a biopsy at gastroscopy or laparotomy. Failure of gastroscopic biopsies to detect lymphoma in a given case should not be interpreted as being conclusive, because superficial biopsies may miss the deeper lymphoid infiltrate. The macroscopic pathology of gastric lymphoma may also mimic adenocarcinoma, consisting of either a bulky ulcerated lesion localized in the corpus or antrum or a diffuse process spreading throughout the entire gastric submucosa and even extending into the duodenum. Microscopically, the vast majority of gastric lymphoid tumors are lymphomas of B-cell origin. Histologically, these tumors may range from well-differentiated, superficial processes (mucosa-associated lymphoid tissue [MALT]) to high-grade, large-cell lymphomas. Like gastric adenocarcinoma, infection with H. pylori increases the risk for gastric lymphoma in general and MALT lymphomas in particular. Large-cell lymphomas of the stomach spread initially to regional lymph nodes (often to Waldeyer’s ring) and may then disseminate.
GASTRIC (NONLYMPHOID) SARCOMA
Leiomyosarcomas and GISTs make up 1–3% of gastric neoplasms. They most frequently involve the anterior and posterior walls of the gastric fundus and often ulcerate and bleed. Even those lesions that appear benign on histologic examination may behave in a malignant fashion. These tumors rarely invade adjacent viscera and characteristically do not metastasize to lymph nodes, but they may spread to the liver and lungs. The treatment of choice is surgical resection. Combination chemotherapy should be reserved for patients with metastatic disease. All such tumors should be analyzed for a mutation in the c-kit receptor. GISTs are unresponsive to conventional chemotherapy; yet ∼50% of patients experience objective response and prolonged survival when treated with imatinib mesylate (Gleevec) (400–800 mg PO daily), a selective inhibitor of the c-kit tyrosine kinase. Many patients with GIST whose tumors have become refractory to imatinib subsequently benefit from sunitinib (Sutent) or regorafenib (Stivarga), other inhibitors of the c-kit tyrosine kinase.
TUMORS OF THE SMALL INTESTINE
Small-bowel tumors comprise <3% of gastrointestinal neoplasms. Because of their rarity and inaccessibility, a correct diagnosis is often delayed. Abdominal symptoms are usually vague and poorly defined, and conventional radiographic studies of the upper and lower intestinal tract often appear normal. Small-bowel tumors should be considered in the differential diagnosis in the following situations: (1) recurrent, unexplained episodes of crampy abdominal pain; (2) intermittent bouts of intestinal obstruction, especially in the absence of inflammatory bowel disease (IBD) or prior abdominal surgery; (3) intussusception in the adult; and (4) evidence of chronic intestinal bleeding in the presence of negative conventional and endoscopic examination. A careful small-bowel barium study should be considered in such a circumstance; the diagnostic accuracy may be improved by infusing barium through a nasogastric tube placed into the duodenum (enteroclysis). Alternatively, capsule endoscopic procedures have been used.
BENIGN TUMORS
The histology of benign small-bowel tumors is difficult to predict on clinical and radiologic grounds alone. The symptomatology of benign tumors is not distinctive, with pain, obstruction, and hemorrhage being the most frequent symptoms. These tumors are usually discovered during the fifth and sixth decades of life, more often in the distal rather than the proximal small intestine. The most common benign tumors are adenomas, leiomyomas, lipomas, and angiomas.
Adenomas These tumors include those of the islet cells and Brunner’s glands as well as polypoid adenomas. Islet cell adenomas are occasionally located outside the pancreas; the associated syndromes are discussed in Chap. 113. Brunner’s gland adenomas are not truly neoplastic but represent a hypertrophy or hyperplasia of submucosal duodenal glands. These appear as small nodules in the duodenal mucosa that secrete a highly viscous alkaline mucus. Most often, this is an incidental radiographic finding not associated with any specific clinical disorder.
Polypoid Adenomas About 25% of benign small-bowel tumors are polypoid adenomas (see Table 110-2). They may present as single polypoid lesions or, less commonly, as papillary villous adenomas. As in the colon, the sessile or papillary form of the tumor is sometimes associated with a coexisting carcinoma. Occasionally, patients with Gardner’s syndrome develop premalignant adenomas in the small bowel; such lesions are generally in the duodenum. Multiple polypoid tumors may occur throughout the small bowel (and occasionally the stomach and colorectum) in the Peutz-Jeghers syndrome. The polyps are usually hamartomas (juvenile polyps) having a low potential for malignant degeneration. Mucocutaneous melanin deposits as well as tumors of the ovary, breast, pancreas, and endometrium are also associated with this autosomal dominant condition.
Leiomyomas These neoplasms arise from smooth-muscle components of the intestine and are usually intramural, affecting the overlying mucosa. Ulceration of the mucosa may cause gastrointestinal hemorrhage of varying severity. Cramping or intermittent abdominal pain is frequently encountered.
Lipomas These tumors occur with greatest frequency in the distal ileum and at the ileocecal valve. They have a characteristic radiolucent appearance and are usually intramural and asymptomatic, but on occasion cause bleeding.
Angiomas While not true neoplasms, these lesions are important because they frequently cause intestinal bleeding. They may take the form of telangiectasia or hemangiomas. Multiple intestinal telangiectasias occur in a nonhereditary form confined to the gastrointestinal tract or as part of the hereditary Osler-Rendu-Weber syndrome. Vascular tumors may also take the form of isolated hemangiomas, most commonly in the jejunum. Angiography, especially during bleeding, is the best procedure for evaluating these lesions.
MALIGNANT TUMORS
While rare, small-bowel malignancies occur in patients with long-standing regional enteritis and celiac sprue as well as in individuals with AIDS. Malignant tumors of the small bowel are frequently associated with fever, weight loss, anorexia, bleeding, and a palpable abdominal mass. After ampullary carcinomas (many of which arise from biliary or pancreatic ducts), the most frequently occurring small-bowel malignancies are adenocarcinomas, lymphomas, carcinoid tumors, and leiomyosarcomas.
ADENOCARCINOMAS
The most common primary cancers of the small bowel are adenocarcinomas, accounting for ∼50% of malignant tumors. These cancers occur most often in the distal duodenum and proximal jejunum, where they tend to ulcerate and cause hemorrhage or obstruction. Radiologically, they may be confused with chronic duodenal ulcer disease or with Crohn’s disease if the patient has long-standing regional enteritis. The diagnosis is best made by endoscopy and biopsy under direct vision. Surgical resection is the treatment of choice with suggested postoperative adjuvant chemotherapy options generally following treatment patterns used in the management of colon cancer.
LYMPHOMAS
Lymphoma in the small bowel may be primary or secondary. A diagnosis of a primary intestinal lymphoma requires histologic confirmation in a clinical setting in which palpable adenopathy and hepatosplenomegaly are absent and no evidence of lymphoma is seen on chest radiograph, CT scan, or peripheral blood smear or on bone marrow aspiration and biopsy. Symptoms referable to the small bowel are present, usually accompanied by an anatomically discernible lesion. Secondary lymphoma of the small bowel consists of involvement of the intestine by a lymphoid malignancy extending from involved retroperitoneal or mesenteric lymph nodes (Chap. 134).
Primary intestinal lymphoma accounts for ∼20% of malignancies of the small bowel. These neoplasms are non-Hodgkin’s lymphomas; they usually have a diffuse, large-cell histology and are of T cell origin. Intestinal lymphoma involves the ileum, jejunum, and duodenum, in decreasing frequency—a pattern that mirrors the relative amount of normal lymphoid cells in these anatomic areas. The risk of small-bowel lymphoma is increased in patients with a prior history of malabsorptive conditions (e.g., celiac sprue), regional enteritis, and depressed immune function due to congenital immunodeficiency syndromes, prior organ transplantation, autoimmune disorders, or AIDS.
The development of localized or nodular masses that narrow the lumen results in periumbilical pain (made worse by eating) as well as weight loss, vomiting, and occasional intestinal obstruction. The diagnosis of small-bowel lymphoma may be suspected from the appearance on contrast radiographs of patterns such as infiltration and thickening of mucosal folds, mucosal nodules, areas of irregular ulceration, or stasis of contrast material. The diagnosis can be confirmed by surgical exploration and resection of involved segments. Intestinal lymphoma can occasionally be diagnosed by peroral intestinal mucosal biopsy, but because the disease mainly involves the lamina propria, full-thickness surgical biopsies are usually required.
Resection of the tumor constitutes the initial treatment modality. While postoperative radiation therapy has been given to some patients following a total resection, most authorities favor short-term (three cycles) systemic treatment with combination chemotherapy. The frequent presence of widespread intraabdominal disease at the time of diagnosis and the occasional multicentricity of the tumor often make a total resection impossible. The probability of sustained remission or cure is ∼75% in patients with localized disease but is ∼25% in individuals with unresectable lymphoma. In patients whose tumors are not resected, chemotherapy may lead to bowel perforation.
A unique form of small-bowel lymphoma, diffusely involving the entire intestine, was first described in oriental Jews and Arabs and is referred to as immunoproliferative small intestinal disease (IPSID), Mediterranean lymphoma, or α heavy chain disease. This is a B cell tumor. The typical presentation includes chronic diarrhea and steatorrhea associated with vomiting and abdominal cramps; clubbing of the digits may be observed. A curious feature in many patients with IPSID is the presence in the blood and intestinal secretions of an abnormal IgA that contains a shortened α heavy chain and is devoid of light chains. It is suspected that the abnormal α chains are produced by plasma cells infiltrating the small bowel. The clinical course of patients with IPSID is generally one of exacerbations and remissions, with death frequently resulting from either progressive malnutrition and wasting or the development of an aggressive lymphoma. The use of oral antibiotics such as tetracycline appears to be beneficial in the early phases of the disorder, suggesting a possible infectious etiology. Combination chemotherapy has been administered during later stages of the disease, with variable results. Results are better when antibiotics and chemotherapy are combined.
CARCINOID TUMORS
Carcinoid tumors arise from argentaffin cells of the crypts of Lieberkühn and are found from the distal duodenum to the ascending colon, areas embryologically derived from the midgut. More than 50% of intestinal carcinoids are found in the distal ileum, with most congregating close to the ileocecal valve. Most intestinal carcinoids are asymptomatic and of low malignant potential, but invasion and metastases may occur, leading to the carcinoid syndrome (Chap. 113).
LEIOMYOSARCOMAS
Leiomyosarcomas often are >5 cm in diameter and may be palpable on abdominal examination. Bleeding, obstruction, and perforation are common. Such tumors should be analyzed for the expression of mutant c-kit receptor (defining GIST), and in the presence of metastatic disease, justifying treatment with imatinib mesylate (Gleevec) or, in imatinib-refractory patients, sunitinib (Sutent) or regorafenib (Stivarga).
110 |
Lower Gastrointestinal Cancers |
Lower gastrointestinal cancers include malignant tumors of the colon, rectum, and anus.
COLORECTAL CANCER
INCIDENCE
![]() Cancer of the large bowel is second only to lung cancer as a cause of cancer death in the United States: 136,830 new cases occurred in 2014, and 50,310 deaths were due to colorectal cancer. The incidence rate has decreased significantly during the past 25 years, likely due to enhanced and more compliantly followed screening practices. Similarly, mortality rates in the United States have decreased by approximately 25%, resulting largely from earlier detection and improved treatment.
Cancer of the large bowel is second only to lung cancer as a cause of cancer death in the United States: 136,830 new cases occurred in 2014, and 50,310 deaths were due to colorectal cancer. The incidence rate has decreased significantly during the past 25 years, likely due to enhanced and more compliantly followed screening practices. Similarly, mortality rates in the United States have decreased by approximately 25%, resulting largely from earlier detection and improved treatment.
POLYPS AND MOLECULAR PATHOGENESIS
Most colorectal cancers, regardless of etiology, arise from adenomatous polyps. A polyp is a grossly visible protrusion from the mucosal surface and may be classified pathologically as a nonneoplastic hamartoma (e.g., juvenile polyp), a hyperplastic mucosal proliferation (hyperplastic polyp), or an adenomatous polyp. Only adenomas are clearly premalignant, and only a minority of adenomatous polyps evolve into cancer. Adenomatous polyps may be found in the colons of ∼30% of middle-aged and ∼50% of elderly people; however, <1% of polyps ever become malignant. Most polyps produce no symptoms and remain clinically undetected. Occult blood in the stool is found in <5% of patients with polyps.
A number of molecular changes are noted in adenomatous polyps and colorectal cancers that are thought to reflect a multistep process in the evolution of normal colonic mucosa to life-threatening invasive carcinoma. These developmental steps toward carcinogenesis include, but are not restricted to, point mutations in the K-ras protooncogene; hypomethylation of DNA, leading to gene activation; loss of DNA (allelic loss) at the site of a tumor-suppressor gene (the adenomatous polyposis coli [APC] gene) on the long arm of chromosome 5 (5q21); allelic loss at the site of a tumor-suppressor gene located on chromosome 18q (the deleted in colorectal cancer [DCC] gene); and allelic loss at chromosome 17p, associated with mutations in the p53 tumor-suppressor gene (see Fig. 101e-2). Thus, the altered proliferative pattern of the colonic mucosa, which results in progression to a polyp and then to carcinoma, may involve the mutational activation of an oncogene followed by and coupled with the loss of genes that normally suppress tumorigenesis. It remains uncertain whether the genetic aberrations always occur in a defined order. Based on this model, however, cancer is believed to develop only in those polyps in which most (if not all) of these mutational events take place.




