[level-membership-for-neurology-category]
Chapter 19 Brain Tumors, Metastatic Cancer, and Paraneoplastic Syndromes
Varieties
Primary Brain Tumors
Primary brain tumors arise within the brain or spinal cord tissue (parenchyma) or their coverings (meninges) (Box 19-1). Pathologists have named them after their original cell line. The numerous, mostly small, glial cells, which normally provide the structural, biochemical, and immunologic support for the central nervous system (CNS), give rise to most tumors – gliomas. These tumors arise within the substance of the brain, i.e., in intraparenchymal or intra-axial locations. Meningeal cells give rise to the other large category of brain tumors, meningiomas. Because these tumors arise from the coverings of the CNS, rather than from actual brain or spinal cord tissue, they grow outside the brain, i.e., in extraparenchymal (extra-axial) locations. In contrast, CNS neurons rarely form tumors in adults.
Of the various potential etiologies of primary brain tumors, studies have established that only ionizing radiation, certain neurocutaneous disorders (see Chapter 13), and various genetic mutations constitute risk factors. So far, data have not proven that cellphone use constitutes a risk factor.
Gliomas
Parenchymal tumors, gliomas, include oligodendrogliomas and astrocytomas. Oligodendrocytes, which normally produce the myelin covering that insulates CNS neurons,* may give rise to oligodendrogliomas. These tumors, which occur infrequently and grow slowly, produce similar manifestations to the more commonly occurring astrocytomas (see later).
Glioblastomas, the most malignant variety of astrocytoma, are the most frequently occurring brain tumor. They develop almost exclusively in the cerebrum. The prognosis is grim. These tumors not only grow rapidly and relentlessly, they infiltrate widely. Frontal lobe gliomas frequently cross the corpus callosum to produce the infamous “butterfly glioma” (Figs 19-1, A, 20-8, and 20-20). Contrary to many physicians’ expectations that brain tumors, like most other cancers, arise in the elderly, the age of patients at the time of a glioblastoma diagnosis averages only 54 years.
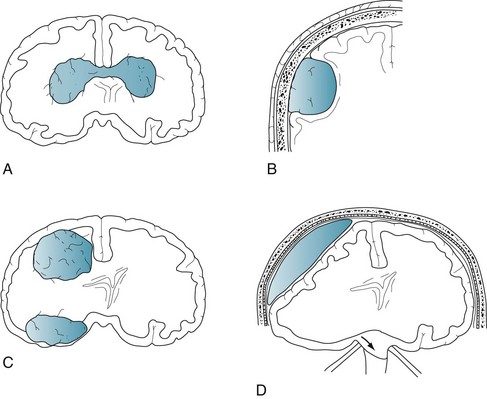
FIGURE 19-1 A, A glioblastoma – the most malignant form of glioma – typically infiltrates along white-matter tracts. Sometimes it spreads through the heavily myelinated corpus callosum in a “butterfly” pattern (see Figs 20-8 and 20-20). B, Meningiomas arise from the meninges overlying the brain or spinal cord and grow slowly (see Fig. 20-10). They compress and irritate, but do not infiltrate, the central nervous system. C, Metastatic tumors, usually multiple and surrounded by edema, destroy large areas of brain and raise intracranial pressure (see Fig. 20-8). D, A subdural hematoma, typically located over one cerebral hemisphere (see Fig. 20-9), compresses the underlying brain and ventricles, and pushes away (shifts) midline structures. Large, acute, rapidly expanding subdural hematomas force the brainstem and ipsilateral oculomotor (third cranial) nerve through the tentorial notch. Such transtentorial herniation, which occurs with epidural as well as subdural hematomas (see Chapter 22), constitutes an immediately life-threatening condition. In contrast, small meningiomas and chronic subdural hematomas cause relatively few symptoms because they are extra-axial and exert little mass effect.
Meningiomas
Arising independently or as an integral part of neurofibromatosis type 1 (see Chapter 13), meningiomas usually create symptoms by compressing the underlying brain or spinal cord (see Figs 19-1, B and 20-10). They grow slowly and predominantly in middle-aged women. Because their expansion is insidious, meningiomas often grow quite large before they produce symptoms.
Primary Cerebral Lymphoma
On CT and MRI, primary cerebral lymphoma resembles cerebral toxoplasmosis (see Figs 20-11 and 20-21). The clinical features of cerebral toxoplasmosis, including focal neurologic deficits and seizures, overlap those of cerebral lymphoma. Although surgery cannot remove lymphomas, steroids often produce dramatic, although temporary, remissions.
Metastatic Tumors
Systemic tumors metastasize to the brain and spinal cord by hematogenous routes. They cannot spread through a lymphatic system because the brain, unlike almost all other organs, does not have one. Metastatic tumors tend to be multiple, surrounded by edema, and rapidly growing. Although individual tumors may each be small, their combined mass constitutes an oppressive intracerebral burden (see Figs 19-1, C and 20-8).
Initial Symptoms
Local Signs
By damaging surrounding tissue, brain tumors usually produce lateralized neurologic deficits, often called “local signs,” such as hemiparesis and dominant- or nondominant-hemisphere neuropsychologic disorders (see Chapter 8). Tumors arising in “eloquent” regions – cerebral cortex areas critical to motor or neuropsychologic function, such as Broca’s or Wernicke’s areas – produce obvious impairments. Tumors that are small, slowly growing, or located in “silent” regions of the brain, such as the right frontal lobe or either of the anterior temporal lobes, notoriously fail to produce symptoms. Tumors that arise from cranial nerves, although rare, almost immediately result in readily recognizable deficits. For example, optic nerve gliomas cause visual loss, and acoustic neuromas cause unilateral progressive hearing loss and tinnitus (see later).
Seizures in individuals older than 60 years are frequently the presenting feature of cerebral tumors. However, because strokes cause seizures nearly as often as tumors, a 60-year-old individual presenting with the first seizure is approximately equally likely to have sustained a stroke as have developed a brain tumor. When the etiology is either a brain tumor or stroke, seizures typically begin as a partial seizure that undergoes secondary generalization (see Chapter 10).
A brain tumor’s tendency to cause seizures also pertains to electroconvulsive therapy (ECT). For example, if a patient harboring a brain tumor were to undergo ECT, the procedure might give rise to multiple, uninterrupted, life-threatening seizures (status epilepticus). However, the benefits of ECT might outweigh the risks if the tumor were small enough not to cause neurologic deficits or surrounding cerebral edema. Large brain tumors, on the other hand, constitute an unequivocal problem regarding ECT. Not only might large brain tumors account for depressive symptoms, but also they might precipitate transtentorial herniation during ECT (Fig. 19-1, D). Thus, before their patients undergo ECT, neurologists order either an MRI or CT.
Signs of Increased Intracranial Pressure
Another sign of increased intracranial pressure, papilledema, occurs as pressure is transmitted along the optic nerve to the optic disks (Fig. 19-2). Although papilledema has a notorious association with brain tumors, it occurs late, if at all, in their course. In fact, among young adults, especially overweight females with menstrual irregularity, idiopathic intracranial hypertension (pseudotumor cerebri) is much more likely than a brain tumor to explain papilledema (see Chapter 9). In general, because only a small proportion of brain tumor patients have papilledema during an initial examination, its absence should not be taken as evidence against the presence of a brain tumor.
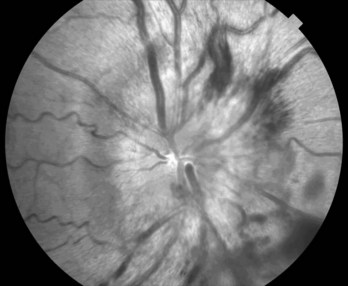
FIGURE 19-2 Papilledema’s main features consist of reddening of the optic disk, which loses its distinct margin, and distension of the retinal veins. In addition, the disk is elevated and hemorrhages appear at its edge. (Compare this disk to the normal optic disk in Figure 4-4.)
In considering manifestations of brain tumors, meningiomas constitute a special category. Unlike gliomas, as discussed previously, small meningiomas are common and usually small. Even large ones may remain asymptomatic. Also, they arise and usually remain entirely in extra-axial locations and produce characteristic syndromes. For example, a meningioma arising from the falx, a parasagittal meningioma, can compress the medial motor cortex and cause spastic paresis of one or both legs. A meningioma arising from the sphenoid wing can damage the adjacent temporal lobe and, because of its proximity to the orbit, cause proptosis and paresis of eye movement. Likewise, an olfactory groove meningioma can compress the adjacent olfactory and optic nerves and the overlying frontal lobe (see Foster–Kennedy syndrome, Chapter 4), causing anosmia, unilateral blindness, and, when large, frontal lobe dysfunction (see Chapter 7).
Initial Mental Symptoms
Direct Effects of Tumors
As a preliminary practical point, most rapidly evolving tumor-related cognitive impairments or personality changes result from a glioblastoma. Another point is that tumors in the frontal lobe produce “frontal lobe personality changes,” consisting of psychomotor retardation, emotional dulling, loss of initiative, poor insight, and reduced capacity to execute complex mental tasks. This clinical picture, like the clinical picture of frontotemporal dementia (see Chapter 7), consists of disturbances in behavior and affect that overshadow cognitive impairments, and those disturbances in turn overshadow physical impairments.
Medication and Other Treatment
In an acute, often debilitating side effect, chemotherapy tends to induce nausea and vomiting (chemotherapy-induced emesis). This problem usually stems from chemotherapy agents triggering the brain’s chemoreceptor zone and its adjacent vomiting center. These zones are located in the area postrema of the medulla, which is one of the few regions of the brain unprotected by the blood–brain barrier. The absence of a blood–brain barrier leaves the chemoreceptor zone freely accessible to any blood-borne substance. Thus, if people inadvertently ingest toxins, such as in poisonous mushrooms, they will immediately vomit. From a medical perspective, morphine, heroin, and high doses of L-dopa, as well as several chemotherapeutic agents, activate the chemoreceptor zone and induce vomiting. On the other hand, both dopamine-blocking agents and 5-HT3 antagonists prevent chemotherapy-induced nausea and vomiting (see Chapter 21).
Depending on the radiation’s total dose and rapidity with which it is administered, radiotherapy sometimes causes inflammatory arteritis and necrosis. Small strokes, which begin to accumulate 6–18 months after a course of radiotherapy, lead to a stepwise progression of cognitive impairments and personality changes resembling vascular cognitive impairment (see Chapter 11). Hemiparesis and dysarthria often accompany neuropsychologic changes. MRIs of patients with radiation-induced cognitive impairments typically reveal white-matter changes (leukoencephalopathy). Overall, radiotherapy induces more cognitive impairment than most chemotherapy agents.
Infections and Organ Failure
Infective agents sometimes invade the CNS, but not other organs. Because cancer patients often cannot respond with fever or leukocytosis to an infection, they may not show the usual markers. In fact, only 5% of cancer patients with meningitis will have the classic triad of fever, nuchal rigidity, and encephalopathy. Also, because of their immunocompromised state, cancer patients are susceptible to opportunistic infections. For example, progressive multifocal leukoencephalopathy (PML) probably results from a papovavirus that attacks CNS myelin. Indeed, CSF analysis in PML cases yields JC virus DNA. Usually complicating the late course of an illness, PML causes dementia and variable physical impairments, but not delirium, fever, or leukocytosis. PML has also complicated AIDS and immunosuppression therapy, including the multiple sclerosis treatment natalizumab (see Chapters 7, 15, and 20).
Paraneoplastic Syndromes
Systemic cancer sometimes causes neurologic syndromes not by invading the nervous system, but by inciting antibody-mediated immune responses directed against the CNS, PNS, or neuromuscular junction. Neurologists previously aptly called these disorders “remote effects of carcinoma,” but now they term them paraneoplastic syndromes. Paraneoplastic syndromes seem to begin with the patient’s synthesizing antibodies against a tumor’s antigens. The antibodies cross-react with neurons’ intracellular components, cell surfaces, or synaptic receptors. Antibodies involved in paraneoplastic syndromes include ones directed against voltage-gated potassium channels (VGKC) and N-methyl-D-aspartate (NMDA) receptors. Of the numerous paraneoplastic syndromes, three are particularly relevant: Cerebellar degeneration, limbic encephalitis, and Lambert–Eaton myasthenic syndrome (LEMS). Their pathophysiology likely represents “molecular mimicry” that is analogous to antistreptococcal antibodies cross-reacting with basal ganglia to cause Sydenham chorea (see Chapter 18).
Lambert–Eaton Myasthenic Syndrome
In LEMS, small cell lung cancer provokes antibodies directed against the presynaptic side of the neuromuscular junction where they impair release of acetylcholine (ACh) (see Chapter 6). The antibody-induced neuromuscular junction dysfunction in LEMS causes proximal limb muscle weakness, which patients partly overcome with repetitive actions. Their weakness has different qualities and different distribution than myasthenia gravis – the well-known disorder of the ACh receptors on the postsynaptic side of the neuromuscular junction. Patients with myasthenia primarily have facial and extraocular muscle weakness that worsens on exertion. Antibody tests and electromyograms readily distinguish these two neuromuscular junction disorders.
Diagnostic Tests for Brain Tumors
Neurologists generally order CT or MRI of the brain, admittedly liberally, for patients who have intellectual decline, those over 50 years who show substantial emotional changes, most adults with headaches not attributable to migraine, cluster, or giant cell arteritis (see Chapter 9), or, as previously discussed, those with excessive concern. Neurologists often also suggest CT or MRI for patients with any new psychiatric illness severe enough to warrant hospitalization or ECT.
CT remains a satisfactory screening procedure in many situations (see Chapter 20). It is sensitive to most tumors and other mass lesions, rapidly performed, relatively inexpensive, permissible for patients with pacemakers, and tolerable for most with claustrophobia. It remains preferable for detecting acute intracranial bleeding, including subarachnoid hemorrhage, subdural hematomas, and intracerebral hemorrhages. CT will also detect fractures and other abnormalities of the skull. On the other hand, its ionizing radiation, at least in children, carries a risk of inducing a malignancy (see Chapter 20).
Neurologists generally do not perform an LP to analyze CSF when they suspect a brain tumor or other intracranial mass lesion because, in such cases, the CSF profile lacks a distinctive profile and rarely reveals malignant cells (see Chapter 20). More important, with large, expanding supratentorial mass lesions, an LP can precipitate transtentorial herniation (Fig. 19-3). Neurologists perform an LP when patients may have either carcinomatous or chronic infectious meningitis. Testing requires large volumes of CSF for neoplastic cells, chemistry studies, fungi, and bacterial and fungal antigens. In an exception to the general rule of not performing an LP in patients with a cerebral lesion, neurologists may perform one to test the CSF for Epstein–Barr virus in an AIDS patient found to have a cerebral tumor because a positive result would indicate a cerebral lymphoma and obviate surgery. Similarly, they may perform one in cases of suspected PML to look for JC virus DNA.
FIGURE 19-3 A patient in transtentorial herniation from a right-sided subdural hematoma (see Fig. 19-1, D) has coma, decerebrate (extensor) posture, Babinski signs, and a dilated right pupil. The right temporal lobe compressing the right-sided (ipsilateral) oculomotor nerve and brainstem through the tentorial notch causes this catastrophe.
Related Conditions
Pituitary Adenomas
Most pituitary adenomas are either prolactinomas, which secrete prolactin, or chromophobe adenomas, which do not. Although prolactinomas usually remain microscopic, they sometimes grow larger than 10 mm, in which case neurologists consider them macroadenomas. Pituitary adenomas of that size may exert pressure on surrounding structures (Fig. 19-4). Their upward pressure on the diaphragm sellae usually causes bitemporal and generalized headache. Compressing the optic chiasm, which is above the diaphragm, causes the visual field deficits. Initially they cause bitemporal superior quadrantanopia and, with further enlargement, bitemporal hemianopsia (see Fig. 12-9).
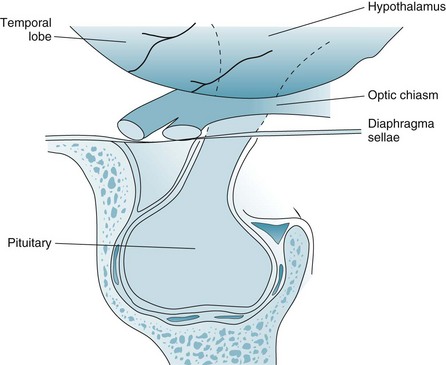
FIGURE 19-4 Pituitary adenomas grow laterally and inferiorly against the walls of the sella turcica and upward against the diaphragm sellae. Large adenomas compress the optic chiasm, which causes a distinctive bitemporal hemianopsia or bitemporal superior quadrantanopia (see Fig. 12-9).
In contrast to these relatively benign pituitary lesions, craniopharyngiomas are large, calcified, cystic, congenital lesion derived from Rathke’s pouch that arise in children as well as in adults. Unlike pituitary adenomas, craniopharyngiomas grow within the hypothalamus, which is located above the diaphragm sellae (see Fig. 19-4). Because craniopharyngiomas cause endocrine deficiency, affected children routinely have delayed or incomplete physical, sexual, and mental maturation; adults tend to have impaired libido, amenorrhea, and apathy; and both children and adults develop diabetes insipidus. When craniopharyngiomas press downward on the optic chiasm, the pressure causes optic atrophy and, as with pituitary tumors, bitemporal hemianopsia. If the tumors completely compress the third ventricle, the obstructive hydrocephalus that ensues leads to papilledema with headache, nausea, and vomiting – classic signs of increased intracranial pressure. Removing them frequently requires extensive surgery and radiotherapy.
Acoustic Neuromas
Most acoustic neuromas grow unilaterally and spontaneously. Bilateral acoustic neuromas, in contrast, characteristically are a manifestation of neurofibromatosis-type 2 (NF2), an autosomal dominant disorder of chromosome 22 (see Chapter 13). Gadolinium-enhanced MRI (see Fig. 20-27), auditory tests, and brainstem auditory-evoked responses (see Chapter 15) detect acoustic neuromas. Stereotactic radiosurgery with a gamma knife or linear accelerator treatments can remove acoustic neuromas while preserving most, if not all, hearing and facial strength.
Spine Metastases
Lung, breast, and other cancers often metastasize to the vertebrae and then grow into the spinal epidural space (Fig. 19-5). Epidural metastases characteristically cause severe pain not only in the affected region (local pain) but also along the path of the affected nerve roots (radicular pain). For example, patients with thoracic spine metastases typically have interscapular spine pain that radiates around the chest in a band-like pattern. Similarly, patients with lumbar spine metastases suffer from lower back pain that, unlike simple musculosketal pain, radiates down the legs.
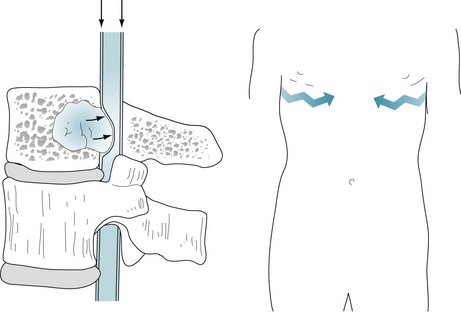
FIGURE 19-5 Left, Vertebral metastases typically grow posteriorly to encroach on the spinal epidural space (arrows). Epidural metastases in the cervical and thoracic regions may cause spinal cord compression, which results in paraplegia or quadriplegia, loss of sensation (hypalgesia) below the level of the lesion, and incontinence. Epidural metastases in the lumbar region may compress the cauda equina (see Fig. 5-11). Right, Patients have “local pain” from destruction of the vertebrae and band-like “radiating pain,” which follows the course of the nerves (jagged arrows). The location of the pain and level of the hypalgesia indicate the site of an epidural metastatic tumor (see Fig. 2-15).
If cervical or thoracic epidural metastases grow large enough, they compress the spinal cord. Metastatic spinal cord compression causes not only local and radicular pain, but also quadriplegia or paraplegia, loss of sensation below the lesion, and urinary and fecal incontinence (see Chapters 2 and 16). In addition, although patients typically retain their cognitive capacity, their pain, physical incapacity, and undeniable progression of their illness often lead to depression.
Other Causes of Limb Weakness in Cancer Patients
Sometimes weakness is iatrogenic. For example, prolonged use of steroids, whether as a medication or an illicit bodybuilding supplement, may result in steroid myopathy or collapse of a hip or vertebrae. Similarly, long-term use of diuretics without potassium supplements results in hypokalemic myopathy. Various chemotherapy agents, such as vincristine, cause polyneuropathy that often includes a disturbing sensory component (see Chapter 5).
In contrast to the generalized weakness of these polyneuropathies, cancer-induced injuries of individual peripheral nerves, mononeuropathies, cause paresis and sensory loss limited to the distribution of those nerves (see Table 5-1). With cancer-induced loss of muscle bulk and subcutaneous fat, mild to moderate pressure – even the patient’s own weight – compresses peripheral nerves deprived of their protective cushions. Compression injuries most often damage the sciatic, peroneal, and radial nerves when bedridden patients remain in one position in bed or secured in a wheelchair for prolonged periods, or are pulled on to stretchers. Sometimes misplaced injections or other accidents injure these nerves.
When lung and breast cancer invades the nearby brachial plexus or pelvic cancer invades the neighboring lumbosacral plexus, patients suffer excruciating pain as well as paresis. Because tumor cells have actually invaded the nerves, routine treatments, such as radiotherapy and opioids, will provide only limited analgesia. Physicians must prescribe treatments for neuropathic pain (see Chapter 14).
Disorders That Resemble Brain Tumors
Subdural hematomas also occur frequently and mimic brain tumors. Head trauma is the most common cause of intracranial bleeding in the potential space between the dura (the thick layer of the meninges) and the underlying brain – the subdural space (see Figs 19-1, D and 20-9). Because subdural bleeding originates from veins, its pressure is low and volume relatively small. After the bleeding forms a hematoma, it accumulates fluid and enlarges. During subsequent weeks, subdural hematomas exert an increasingly greater mass effect that presses broadly against one or both cerebral hemispheres. Subdural hematomas produce headaches, confusion, personality change, dementia, and other nonspecific symptoms of generalized cerebral dysfunction – much more than lateralized signs.
Bacterial abscesses usually result from sepsis originating in intravenous drug abuse, dental procedures, sinusitis, endocarditis, or immunodeficiency. They typically present with signs of a cerebral mass lesion rather than a systemic infection. In their clinical appearance and imaging studies, abscesses resemble brain tumors (see Fig. 20-15).
Patients with neurocysticercosis, the neurologic component of cysticercosis, usually have multiple intracerebral cysts that have originated from a gastrointestinal tapeworm (Taenia solium) infestation. Although each cyst may be small, their total number occupies a considerable space. They also tend to block CSF flow and thus cause obstructive hydrocephalus. Moreover, as they die, their fluid contents leak and provoke an inflammatory response in the surrounding brain. This irritation, as well as their mass effect, often causes seizures. Thus, patients with neurocysticercosis may present with dementia, symptoms and signs of increased intracranial pressure, or, most often, seizures. Almost all patients show cognitive impairment and about 13%, dementia. Because cysticercosis is endemic in Central and South America and the Indian subcontinent, immigrants from these regions often harbor neurocysticercosis. CT and MRI readily detect the cysts (see Fig. 20-12). Whatever the antibiotic regimen, neurologists almost always add AEDs.
As another disorder that mimics brain tumor, idiopathic intracranial hypertension (pseudotumor cerebri) produces headache, papilledema, and, in severe cases, sixth cranial nerve palsies; however, it does not affect cognitive capacity or produce seizures (see before and Chapter 9).
Barry H, Hardiman O, Healy DG, et al. Anti-NMDA receptor encephalitis: an important differential diagnosis in psychosis. Br J Psychiatry. 2012;201:174.
Darnell RB, Posner JB. Paraneoplastic syndromes involving the nervous system. N Engl J Med. 2003;349:1543–1554.
DeAndrade DC, Rodrigues CI, Abraham R. Cognitive impairment and dementia in neurocysticercosis. Neurology. 2010;74:1288–1295.
Duffner PK. Long-term effects of radiation therapy on cognitive and endocrine function in children with leukemia and brain tumors. Neurology. 2004;10:293–310.
Kayser MS, Kohler CG, Dalmau J. Psychiatric manifestations of paraneoplastic disorders. Am J Psychiatry. 2010;16667:1039.
Litofsky NS, Farace E, Anderson F, et al. Depression in patients with high-grade glioma: Results of the Glioma Outcomes Project. Neurosurgery. 2004;54:358–366.
Morris B, Partap S, Yeom K, et al. Cerebrovascular disease in childhood cancer survivors. Neurology. 2009;73:1906–1913.
Pelletier G, Verhoef MJ, Khatri N, et al. Quality of life in brain tumor patients: The relative contributions of depression, fatigue, emotional distress, and existential issues. J Neurooncol. 2002;57:41–49.
Ross L, Johansen C, Dalton SO, et al. Psychiatric hospitalization among survivors of cancer in childhood or adolescence. N Engl J Med. 2003;349:650–657.
Safdieh JE, Mead PA, Sepkowitz KA, et al. Bacterial and fungal meningitis in patients with cancer. Neurology. 2008;70:943–947.
Tess AV, Smetna GW. Medical evaluation of patients undergoing electroconvulsive therapy. N Engl J Med. 2009;360:1437–1444.
Torres IJ, Mundt AJ, Sweeney PJ, et al. A longitudinal neuropsychological study of partial brain radiation in adults with brain tumors. Neurology. 2003;60:1113–1118.
Zaidat OO, Ruff RL. Treatment of spinal epidural metastasis improves patient survival and functional state. Neurology. 2002;58:1360–1366.
Chapter 19Questions and Answers
2. A primary care physician referred a 60-year-old construction worker for evaluation of episodes of impaired consciousness lasting 1–3 minutes. During them, the patient mutters nonsensically and makes kissing movements. He has diabetes and hypertension, and has smoked two packs of cigarettes daily since enlisting in the Navy at 17 years. A neurologist finds that the patient has a left superior quadrantanopia, right dysmetria, and right intention tremor. Electroencephalographic (EEG) monitoring confirms that the episodes represent partial complex seizures originating from the right temporal lobe. Of the following conditions, which is the most likely illness?
5. During the previous 2 years, a 55-year-old woman with multiple café-au-lait spots developed slowly increasing severe paresis of the left leg, which had hyperactive deep tendon reflexes (DTRs) and a Babinski sign. Before undergoing further evaluation, she had a seizure that began with clonic movements of the left foot, then leg, and finally the arm. On examination, she has left hemiparesis, hyperactive DTRs, and a Babinski sign. Of the following, which is the most likely illness?
8. An obese 22-year-old woman has moderately severe, generalized headaches. She has papilledema and paresis of abduction of her right eye but no other neurologic abnormalities. Routine blood and chemistry tests are normal. Computed tomography (CT) shows small ventricles but no mass lesion. What would be the most appropriate next step?
10. A psychiatrist was asked to consult on a 60-year-old retired sanitation truck driver with lung cancer who developed confusion, disorientation, and agitation during his second week of hospitalization. He resisted a full neurologic examination, but a neurologist determined that he had no hemiparesis, papilledema, or nuchal rigidity. A noncontrast head CT was normal. The psychiatrist diagnosed delirium. Which one of the following is its least likely cause?
11. In the above case (Question 10), further testing discovered pneumonia. After a course of antibiotics, his delirium resolved. One month later, his delirium recurred and was more dangerous than the previous time. A CT revealed two ring-shaped lesions with surrounding edema in his cerebral hemispheres. An extensive evaluation found no other potential explanation. His primary care team solicited a psychiatry consultation. Which medication regimen should the psychiatrist recommend?
12. A 65-year-old woman with the onset of dementia over 9 months has no physical or neurologic abnormalities except for frontal release signs and hyperactive DTRs. A complete laboratory and EEG evaluation reveals no specific abnormality. A CT shows atrophy and a small meningioma in the right parietal convexity. Which would be the most appropriate next step?
13. A 75-year-old man, who has had dementia for 6 years, suddenly develops increased irritability and behavioral disturbances. He has no lateralized signs or indication of increased intracranial pressure. He is treated with a major tranquilizer. One week later, he becomes somnolent and has a seizure. He remains comatose with a left hemiparesis. No abnormalities are found on a general medical examination or routine laboratory tests, but a CT shows an extra-axial curved lucency and a shift of midline cerebral structures. Before any treatment can be instituted, the patient dies. An autopsy discloses cerebral atrophy and a large chronic subdural hematoma. Which of the following statements concerning subdural hematomas is false?
19. A 65-year-old man with metastatic prostate carcinoma has been in agony from bone metastases. He is agitated, verbose, and threatening in his demands for morphine. His behavior has become a major management problem, and his family is also becoming disruptive. CT, MRI, and blood tests have shown that the patient has no cerebral metastases, infection, hypercalcemia, or other metabolic aberrations. Which two recommendations should a psychiatry consultant make in response to this situation?
29–31 Match the varieties of hemorrhage with the most likely cause.
29-b. Although trauma and mycotic aneurysms may cause a subarachnoid hemorrhage, the most frequent and important cause is rupture of an aneurysm arising from one of the arteries that comprise the circle of Willis.
30-c. Bleeding from small meningeal veins is the usual cause of subdural hematomas.
31-a. Lacerations of the middle meningeal artery, which often result from a fracture of the temporal bone, are usually the cause of epidural hematomas.
32. A 46-year-old woman with metastatic breast cancer develops a disconcerting numbness in the skin over the right lower chin. She has no weakness of face or jaw muscles and the remainder of her neurologic examination is normal. However, X-rays of her jaw reveal a lytic lesion in the right lower mandible. What is the diagnosis?
33. A family brings a 59-year-old high-school basketball coach for psychiatric consultation because he has become apathetic and markedly forgetful, but intermittently he becomes loud and inexplicably angry. An examination confirms that he has amnesia and personality changes. One month before this consultation, his physicians diagnosed small cell carcinoma of the lung. An MRI with and without gadolinium infusion of the head and three LPs show normal CSF chemistries, absence of cells, and an opening pressure of 150 mmH2O. The results of all his blood tests are also within normal limits. However, an EEG shows focal slowing and spikes over the temporal lobes. Which is the most likely explanation for his memory deficit and other abnormalities?
35. A 59-year-old active professor of English presents with a 3-week history of dulled affect, sleeplessness, and mild morning headaches. Aspirin relieves the headaches. Six years before, following the death of his mother, he had prolonged mourning. On examination, he scores 28 on the Mini-Mental State Examination (MMSE), but he is inattentive and reticent. He has psychomotor impersistence, but no lateralized neurologic deficit. An MRI reveals a single large ring-shaped mass lesion in the right frontal lobe that compresses the anterior horn of the lateral ventricle and shifts midline structures to the left. Which is the most likely nature of the lesion?
36. Eight months after completing whole-brain radiotherapy for metastatic lung cancer, a 47-year-old former newspaper reporter suddenly developed aphasia and right-sided hemiparesis. Her MRI showed an acute infarction in the distribution of a branch of her left middle cerebral artery and small residual metastatic tumors in the right frontal lobe and left cerebellum. Which is the most likely cause of the stroke?
38. The surgical service solicits a psychiatric consultation because the patient, a 79-year-old man, suddenly became “depressed.” The psychiatrist determines that he had been making an uneventful recovery for the preceding 3 weeks following resection of extensive colon cancer. When the psychiatrist examines the patient, she finds that he is intermittently lethargic and often inattentive. When she captures his attention, she finds that he is incompletely oriented and has a poor memory. Although his white count is elevated, he has no fever, asterixis, nuchal rigidity, papilledema, or lateralized neurologic findings. A head CT shows no abnormality. Which is most likely responsible for his change in mental state?
39. A family brings their 74-year-old patriarch, who owns a diner, to the neurologist because he began offending his customers and forgetting their orders. Although he rarely drinks alcohol, he has smoked two packs of cigarettes daily since he came to this country as a teenager. He takes no medications. He has lost 20 lb. His neurologic examination shows pronounced memory impairment, but no lateralized signs or papilledema. An MRI detected no mass lesions, but the medial temporal lobes are smaller than normal. Routine blood tests show mild anemia, but no other abnormality. An EEG contains temporal spikes. Which test should the neurologist now order?
40. A 29-year-old intravenous drug addict, known to have AIDS, comes to the emergency room with uncontrollable involuntary, uncoordinated, large-scale jerky movements of his left arm and leg. He remains alert and free of headache. An MRI shows multiple small ring-enhancing lesions predominantly in the basal ganglia. Which is the most likely etiology of the cerebral lesions?
43. When administered to children, radiation therapy may prevent the spread of acute leukemia to the CNS or obliterate unresectable tumors. However, the treatment has many potential complications. Which of the following is false?
44. A close friend brings a 68-year-old retired nurse for psychiatric evaluation for depression. The patient explained she has lost her liveliness, energy, and interest in other people during the previous 3–6 months. However, she denies memory and other cognitive impairments, headaches, and systemic symptoms. On examination, she scores 28 on the MMSE and has no physical neurologic abnormalities, but she has psychomotor retardation, emotional dulling, and reduced capacity to execute complex mental tasks. During the examination and more so afterwards, she spoke excessively, loudly, and aggressively. With which two conditions is her clinical presentation most consistent?
49. Six months after delivering her third child, a 28-year-old woman seeks a psychiatric consultation for “depression.” She reports loss of interest in her husband and their children. She describes having little appetite or energy, sleeping poorly, and losing interest in sex. Her weight has fallen to below her pre-pregnancy level. Her menses initially returned, but were scant. Then, 2 months before the visit, her menses stopped. Two weeks before the psychiatric consultation, a neurologic evaluation showed no visual field or visual acuity deficits, or other abnormalities. A head MRI, with views of the sella, was normal. Routine blood tests were normal except that the serum prolactin level was slightly elevated. Which is the most likely diagnosis?
50. During a week’s vacation, after a strenuous first semester of college, a 19-year-old woman developed involuntary right arm and leg movements and then unexplained anxiety. While undergoing a medical evaluation, she developed partial complex seizures. Aside from a temperature of 100.0°F, she had no general medical abnormalities. She was confused and disoriented, and had a poor memory, but she had no meningeal or lateralized findings. Her CSF showed 25 lymphocytes and a slightly elevated protein concentration, but no antibodies to herpes simplex virus or any common bacteria. Toxicology studies were consistently negative. A transvaginal ultrasound detected an ovarian lesion. Which is the most likely diagnosis?
[/level-membership-for-neurology-category][not-level-membership-for-neurology-category]
Chapter 19 Brain Tumors, Metastatic Cancer, and Paraneoplastic Syndromes
Varieties
Primary Brain Tumors
Primary brain tumors arise within the brain or spinal cord tissue (parenchyma) or their coverings (meninges) (Box 19-1). Pathologists have named them after their original cell line. The numerous, mostly small, glial cells, which normally provide the structural, biochemical, and immunologic support for the central nervous system (CNS), give rise to most tumors – gliomas. These tumors arise within the substance of the brain, i.e., in intraparenchymal or intra-axial locations. Meningeal cells give rise to the other large category of brain tumors, meningiomas. Because these tumors arise from the coverings of the CNS, rather than from actual brain or spinal cord tissue, they grow outside the brain, i.e., in extraparenchymal (extra-axial) locations. In contrast, CNS neurons rarely form tumors in adults.
Of the various potential etiologies of primary brain tumors, studies have established that only ionizing radiation, certain neurocutaneous disorders (see Chapter 13), and various genetic mutations constitute risk factors. So far, data have not proven that cellphone use constitutes a risk factor.
Gliomas
Parenchymal tumors, gliomas, include oligodendrogliomas and astrocytomas. Oligodendrocytes, which normally produce the myelin covering that insulates CNS neurons,* may give rise to oligodendrogliomas. These tumors, which occur infrequently and grow slowly, produce similar manifestations to the more commonly occurring astrocytomas (see later).
Glioblastomas, the most malignant variety of astrocytoma, are the most frequently occurring brain tumor. They develop almost exclusively in the cerebrum. The prognosis is grim. These tumors not only grow rapidly and relentlessly, they infiltrate widely. Frontal lobe gliomas frequently cross the corpus callosum to produce the infamous “butterfly glioma” (Figs 19-1, A, 20-8, and 20-20). Contrary to many physicians’ expectations that brain tumors, like most other cancers, arise in the elderly, the age of patients at the time of a glioblastoma diagnosis averages only 54 years.
FIGURE 19-1 A, A glioblastoma – the most malignant form of glioma – typically infiltrates along white-matter tracts. Sometimes it spreads through the heavily myelinated corpus callosum in a “butterfly” pattern (see Figs 20-8 and 20-20). B, Meningiomas arise from the meninges overlying the brain or spinal cord and grow slowly (see Fig. 20-10). They compress and irritate, but do not infiltrate, the central nervous system. C, Metastatic tumors, usually multiple and surrounded by edema, destroy large areas of brain and raise intracranial pressure (see Fig. 20-8). D, A subdural hematoma, typically located over one cerebral hemisphere (see Fig. 20-9), compresses the underlying brain and ventricles, and pushes away (shifts) midline structures. Large, acute, rapidly expanding subdural hematomas force the brainstem and ipsilateral oculomotor (third cranial) nerve through the tentorial notch. Such transtentorial herniation, which occurs with epidural as well as subdural hematomas (see Chapter 22), constitutes an immediately life-threatening condition. In contrast, small meningiomas and chronic subdural hematomas cause relatively few symptoms because they are extra-axial and exert little mass effect.
Meningiomas
Arising independently or as an integral part of neurofibromatosis type 1 (see Chapter 13), meningiomas usually create symptoms by compressing the underlying brain or spinal cord (see Figs 19-1, B and 20-10). They grow slowly and predominantly in middle-aged women. Because their expansion is insidious, meningiomas often grow quite large before they produce symptoms.
Primary Cerebral Lymphoma
On CT and MRI, primary cerebral lymphoma resembles cerebral toxoplasmosis (see Figs 20-11 and 20-21). The clinical features of cerebral toxoplasmosis, including focal neurologic deficits and seizures, overlap those of cerebral lymphoma. Although surgery cannot remove lymphomas, steroids often produce dramatic, although temporary, remissions.
Metastatic Tumors
Systemic tumors metastasize to the brain and spinal cord by hematogenous routes. They cannot spread through a lymphatic system because the brain, unlike almost all other organs, does not have one. Metastatic tumors tend to be multiple, surrounded by edema, and rapidly growing. Although individual tumors may each be small, their combined mass constitutes an oppressive intracerebral burden (see Figs 19-1, C and 20-8).
Initial Symptoms
Local Signs
By damaging surrounding tissue, brain tumors usually produce lateralized neurologic deficits, often called “local signs,” such as hemiparesis and dominant- or nondominant-hemisphere neuropsychologic disorders (see Chapter 8). Tumors arising in “eloquent” regions – cerebral cortex areas critical to motor or neuropsychologic function, such as Broca’s or Wernicke’s areas – produce obvious impairments. Tumors that are small, slowly growing, or located in “silent” regions of the brain, such as the right frontal lobe or either of the anterior temporal lobes, notoriously fail to produce symptoms. Tumors that arise from cranial nerves, although rare, almost immediately result in readily recognizable deficits. For example, optic nerve gliomas cause visual loss, and acoustic neuromas cause unilateral progressive hearing loss and tinnitus (see later).
Seizures in individuals older than 60 years are frequently the presenting feature of cerebral tumors. However, because strokes cause seizures nearly as often as tumors, a 60-year-old individual presenting with the first seizure is approximately equally likely to have sustained a stroke as have developed a brain tumor. When the etiology is either a brain tumor or stroke, seizures typically begin as a partial seizure that undergoes secondary generalization (see Chapter 10).
A brain tumor’s tendency to cause seizures also pertains to electroconvulsive therapy (ECT). For example, if a patient harboring a brain tumor were to undergo ECT, the procedure might give rise to multiple, uninterrupted, life-threatening seizures (status epilepticus). However, the benefits of ECT might outweigh the risks if the tumor were small enough not to cause neurologic deficits or surrounding cerebral edema. Large brain tumors, on the other hand, constitute an unequivocal problem regarding ECT. Not only might large brain tumors account for depressive symptoms, but also they might precipitate transtentorial herniation during ECT (Fig. 19-1, D). Thus, before their patients undergo ECT, neurologists order either an MRI or CT.
Signs of Increased Intracranial Pressure
Another sign of increased intracranial pressure, papilledema, occurs as pressure is transmitted along the optic nerve to the optic disks (Fig. 19-2). Although papilledema has a notorious association with brain tumors, it occurs late, if at all, in their course. In fact, among young adults, especially overweight females with menstrual irregularity, idiopathic intracranial hypertension (pseudotumor cerebri) is much more likely than a brain tumor to explain papilledema (see Chapter 9). In general, because only a small proportion of brain tumor patients have papilledema during an initial examination, its absence should not be taken as evidence against the presence of a brain tumor.
FIGURE 19-2 Papilledema’s main features consist of reddening of the optic disk, which loses its distinct margin, and distension of the retinal veins. In addition, the disk is elevated and hemorrhages appear at its edge. (Compare this disk to the normal optic disk in Figure 4-4.)
In considering manifestations of brain tumors, meningiomas constitute a special category. Unlike gliomas, as discussed previously, small meningiomas are common and usually small. Even large ones may remain asymptomatic. Also, they arise and usually remain entirely in extra-axial locations and produce characteristic syndromes. For example, a meningioma arising from the falx, a parasagittal meningioma, can compress the medial motor cortex and cause spastic paresis of one or both legs. A meningioma arising from the sphenoid wing can damage the adjacent temporal lobe and, because of its proximity to the orbit, cause proptosis and paresis of eye movement. Likewise, an olfactory groove meningioma can compress the adjacent olfactory and optic nerves and the overlying frontal lobe (see Foster–Kennedy syndrome, Chapter 4), causing anosmia, unilateral blindness, and, when large, frontal lobe dysfunction (see Chapter 7).
Initial Mental Symptoms
Direct Effects of Tumors
As a preliminary practical point, most rapidly evolving tumor-related cognitive impairments or personality changes result from a glioblastoma. Another point is that tumors in the frontal lobe produce “frontal lobe personality changes,” consisting of psychomotor retardation, emotional dulling, loss of initiative, poor insight, and reduced capacity to execute complex mental tasks. This clinical picture, like the clinical picture of frontotemporal dementia (see Chapter 7), consists of disturbances in behavior and affect that overshadow cognitive impairments, and those disturbances in turn overshadow physical impairments.
Medication and Other Treatment
In an acute, often debilitating side effect, chemotherapy tends to induce nausea and vomiting (chemotherapy-induced emesis). This problem usually stems from chemotherapy agents triggering the brain’s chemoreceptor zone and its adjacent vomiting center. These zones are located in the area postrema of the medulla, which is one of the few regions of the brain unprotected by the blood–brain barrier. The absence of a blood–brain barrier leaves the chemoreceptor zone freely accessible to any blood-borne substance. Thus, if people inadvertently ingest toxins, such as in poisonous mushrooms, they will immediately vomit. From a medical perspective, morphine, heroin, and high doses of L-dopa, as well as several chemotherapeutic agents, activate the chemoreceptor zone and induce vomiting. On the other hand, both dopamine-blocking agents and 5-HT3 antagonists prevent chemotherapy-induced nausea and vomiting (see Chapter 21).
Depending on the radiation’s total dose and rapidity with which it is administered, radiotherapy sometimes causes inflammatory arteritis and necrosis. Small strokes, which begin to accumulate 6–18 months after a course of radiotherapy, lead to a stepwise progression of cognitive impairments and personality changes resembling vascular cognitive impairment (see Chapter 11). Hemiparesis and dysarthria often accompany neuropsychologic changes. MRIs of patients with radiation-induced cognitive impairments typically reveal white-matter changes (leukoencephalopathy). Overall, radiotherapy induces more cognitive impairment than most chemotherapy agents.
Infections and Organ Failure
Infective agents sometimes invade the CNS, but not other organs. Because cancer patients often cannot respond with fever or leukocytosis to an infection, they may not show the usual markers. In fact, only 5% of cancer patients with meningitis will have the classic triad of fever, nuchal rigidity, and encephalopathy. Also, because of their immunocompromised state, cancer patients are susceptible to opportunistic infections. For example, progressive multifocal leukoencephalopathy (PML) probably results from a papovavirus that attacks CNS myelin. Indeed, CSF analysis in PML cases yields JC virus DNA. Usually complicating the late course of an illness, PML causes dementia and variable physical impairments, but not delirium, fever, or leukocytosis. PML has also complicated AIDS and immunosuppression therapy, including the multiple sclerosis treatment natalizumab (see Chapters 7, 15, and 20).
Paraneoplastic Syndromes
Systemic cancer sometimes causes neurologic syndromes not by invading the nervous system, but by inciting antibody-mediated immune responses directed against the CNS, PNS, or neuromuscular junction. Neurologists previously aptly called these disorders “remote effects of carcinoma,” but now they term them paraneoplastic syndromes. Paraneoplastic syndromes seem to begin with the patient’s synthesizing antibodies against a tumor’s antigens. The antibodies cross-react with neurons’ intracellular components, cell surfaces, or synaptic receptors. Antibodies involved in paraneoplastic syndromes include ones directed against voltage-gated potassium channels (VGKC) and N-methyl-D-aspartate (NMDA) receptors. Of the numerous paraneoplastic syndromes, three are particularly relevant: Cerebellar degeneration, limbic encephalitis, and Lambert–Eaton myasthenic syndrome (LEMS). Their pathophysiology likely represents “molecular mimicry” that is analogous to antistreptococcal antibodies cross-reacting with basal ganglia to cause Sydenham chorea (see Chapter 18).
Lambert–Eaton Myasthenic Syndrome
In LEMS, small cell lung cancer provokes antibodies directed against the presynaptic side of the neuromuscular junction where they impair release of acetylcholine (ACh) (see Chapter 6). The antibody-induced neuromuscular junction dysfunction in LEMS causes proximal limb muscle weakness, which patients partly overcome with repetitive actions. Their weakness has different qualities and different distribution than myasthenia gravis – the well-known disorder of the ACh receptors on the postsynaptic side of the neuromuscular junction. Patients with myasthenia primarily have facial and extraocular muscle weakness that worsens on exertion. Antibody tests and electromyograms readily distinguish these two neuromuscular junction disorders.
Diagnostic Tests for Brain Tumors
Neurologists generally order CT or MRI of the brain, admittedly liberally, for patients who have intellectual decline, those over 50 years who show substantial emotional changes, most adults with headaches not attributable to migraine, cluster, or giant cell arteritis (see Chapter 9), or, as previously discussed, those with excessive concern. Neurologists often also suggest CT or MRI for patients with any new psychiatric illness severe enough to warrant hospitalization or ECT.
CT remains a satisfactory screening procedure in many situations (see Chapter 20). It is sensitive to most tumors and other mass lesions, rapidly performed, relatively inexpensive, permissible for patients with pacemakers, and tolerable for most with claustrophobia. It remains preferable for detecting acute intracranial bleeding, including subarachnoid hemorrhage, subdural hematomas, and intracerebral hemorrhages. CT will also detect fractures and other abnormalities of the skull. On the other hand, its ionizing radiation, at least in children, carries a risk of inducing a malignancy (see Chapter 20).
Neurologists generally do not perform an LP to analyze CSF when they suspect a brain tumor or other intracranial mass lesion because, in such cases, the CSF profile lacks a distinctive profile and rarely reveals malignant cells (see Chapter 20). More important, with large, expanding supratentorial mass lesions, an LP can precipitate transtentorial herniation (Fig. 19-3). Neurologists perform an LP when patients may have either carcinomatous or chronic infectious meningitis. Testing requires large volumes of CSF for neoplastic cells, chemistry studies, fungi, and bacterial and fungal antigens. In an exception to the general rule of not performing an LP in patients with a cerebral lesion, neurologists may perform one to test the CSF for Epstein–Barr virus in an AIDS patient found to have a cerebral tumor because a positive result would indicate a cerebral lymphoma and obviate surgery. Similarly, they may perform one in cases of suspected PML to look for JC virus DNA.
FIGURE 19-3 A patient in transtentorial herniation from a right-sided subdural hematoma (see Fig. 19-1, D) has coma, decerebrate (extensor) posture, Babinski signs, and a dilated right pupil. The right temporal lobe compressing the right-sided (ipsilateral) oculomotor nerve and brainstem through the tentorial notch causes this catastrophe.
Related Conditions
Pituitary Adenomas
Most pituitary adenomas are either prolactinomas, which secrete prolactin, or chromophobe adenomas, which do not. Although prolactinomas usually remain microscopic, they sometimes grow larger than 10 mm, in which case neurologists consider them macroadenomas. Pituitary adenomas of that size may exert pressure on surrounding structures (Fig. 19-4). Their upward pressure on the diaphragm sellae usually causes bitemporal and generalized headache. Compressing the optic chiasm, which is above the diaphragm, causes the visual field deficits. Initially they cause bitemporal superior quadrantanopia and, with further enlargement, bitemporal hemianopsia (see Fig. 12-9).
FIGURE 19-4 Pituitary adenomas grow laterally and inferiorly against the walls of the sella turcica and upward against the diaphragm sellae. Large adenomas compress the optic chiasm, which causes a distinctive bitemporal hemianopsia or bitemporal superior quadrantanopia (see Fig. 12-9).
[/not-level-membership-for-neurology-category]
