423 |
Bone and Mineral Metabolism in Health and Disease |
BONE STRUCTURE AND METABOLISM
Bone is a dynamic tissue that is remodeled constantly throughout life. The arrangement of compact and cancellous bone provides strength and density suitable for both mobility and protection. In addition, bone provides a reservoir for calcium, magnesium, phosphorus, sodium, and other ions necessary for homeostatic functions. Bone also hosts and regulates hematopoiesis by providing niches for hematopoietic cell proliferation and differentiation. The skeleton is highly vascular and receives about 10% of the cardiac output. Remodeling of bone is accomplished by two distinct cell types: osteoblasts produce bone matrix, and osteoclasts resorb the matrix.
The extracellular components of bone consist of a solid mineral phase in close association with an organic matrix, of which 90–95% is type I collagen (Chap. 427). The noncollagenous portion of the organic matrix is heterogeneous and contains serum proteins such as albumin as well as many locally produced proteins, whose functions are incompletely understood. Those proteins include cell attachment/signaling proteins such as thrombospondin, osteopontin, and fibronectin; calcium-binding proteins such as matrix gla protein and osteocalcin; and proteoglycans such as biglycan and decorin. Some of the proteins organize collagen fibrils; others influence mineralization and binding of the mineral phase to the matrix.
The mineral phase is made up of calcium and phosphate and is best characterized as a poorly crystalline hydroxyapatite. The mineral phase of bone is deposited initially in intimate relation to the collagen fibrils and is found in specific locations in the “holes” between the collagen fibrils. This architectural arrangement of mineral and matrix results in a two-phase material well suited to withstand mechanical stresses. The organization of collagen influences the amount and type of mineral phase formed in bone. Although the primary structures of type I collagen in skin and bone tissues are similar, there are differences in posttranslational modifications and distribution of intermolecular cross-links. The holes in the packing structure of the collagen are larger in mineralized collagen of bone and dentin than in unmineralized collagens such as those in tendon. Single amino acid substitutions in the helical portion of either the α1 (COL1A1) or α2 (COL1A2) chains of type I collagen disrupt the organization of bone in osteogenesis imperfecta. The severe skeletal fragility associated with this group of disorders highlights the importance of the fibrillar matrix in the structure of bone (Chap. 427).
Osteoblasts synthesize and secrete the organic matrix and regulate its mineralization. They are derived from cells of mesenchymal origin (Fig. 423-1A). Active osteoblasts are found on the surface of newly forming bone. As an osteoblast secretes matrix, which then is mineralized, the cell becomes an osteocyte, still connected with its blood supply through a series of canaliculi. Osteocytes account for the vast majority of the cells in bone. They are thought to be the mechanosensors in bone that communicate signals to surface osteoblasts and their progenitors through the canalicular network and thereby serve as master regulators of bone formation and resorption. Remarkably, osteocytes also secrete fibroblast growth factor 23 (FGF23), a major regulator of phosphate metabolism (see below). Mineralization of the matrix, both in trabecular bone and in osteones of compact cortical bone (Haversian systems), begins soon after the matrix is secreted (primary mineralization) but is not completed for several weeks or even longer (secondary mineralization). Although this mineralization takes advantage of the high concentrations of calcium and phosphate, already near saturation in serum, mineralization is a carefully regulated process that is dependent on the activity of osteoblast-derived alkaline phosphatase, which probably works by hydrolyzing inhibitors of mineralization.
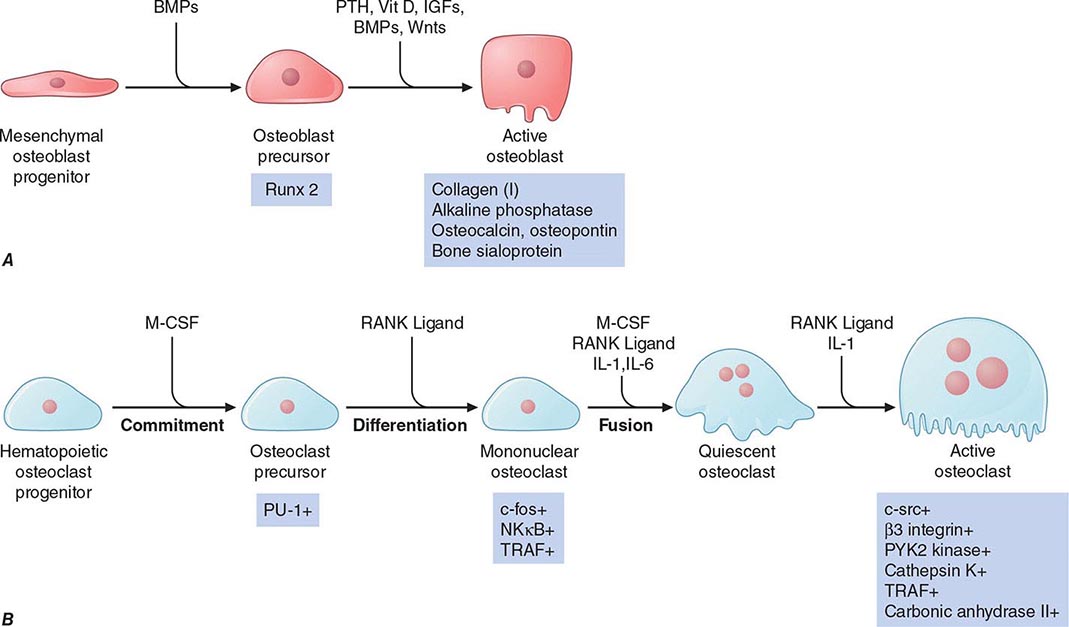
FIGURE 423-1 Pathways regulating development of (A) osteoblasts and (B) osteoclasts. Hormones, cytokines, and growth factors that control cell proliferation and differentiation are shown above the arrows. Transcription factors and other markers specific for various stages of development are depicted below the arrows. BMPs, bone morphogenic proteins; IGFs, insulin-like growth factors; IL-1, interleukin 1; IL-6, interleukin 6; M-CSF, macrophage colony-stimulating factor; NFκB, nuclear factor κB; PTH, parathyroid hormone; PU-1, a monocyte- and B lymphocyte–specific ets family transcription factor; RANK ligand, receptor activator of NFκB ligand; Runx2, Runt-related transcription factor 2; TRAF, tumor necrosis factor receptor–associated factors; Vit D, vitamin D; wnts, wingless-type mouse mammary tumor virus integration site. (Modified from T Suda et al: Endocr Rev 20:345, 1999, with permission.)
Genetic studies in humans and mice have identified several key genes that control osteoblast development. Runx2 is a transcription factor expressed specifically in chondrocyte (cartilage cells) and osteoblast progenitors as well as in hypertrophic chondrocytes and mature osteoblasts. Runx2 regulates the expression of several important osteoblast proteins, including osterix (another transcription factor needed for osteoblast maturation), osteopontin, bone sialoprotein, type I collagen, osteocalcin, and receptor-activator of NFκB (RANK) ligand. Runx2 expression is regulated in part by bone morphogenic proteins (BMPs). Runx2-deficient mice are devoid of osteoblasts, whereas mice with a deletion of only one allele (Runx2 +/–) exhibit a delay in formation of the clavicles and some cranial bones. The latter abnormalities are similar to those in the human disorder cleidocranial dysplasia, which is also caused by heterozygous inactivating mutations in Runx2.
The paracrine signaling molecule, Indian hedgehog (Ihh), also plays a critical role in osteoblast development, as evidenced by Ihh-deficient mice that lack osteoblasts in the type of bone formed on a cartilage mold (endochondral ossification). Signals originating from members of the wnt (wingless-type mouse mammary tumor virus integration site) family of paracrine factors are also important for osteoblast proliferation and differentiation. Numerous other growth-regulatory factors affect osteoblast function, including the three closely related transforming growth factor βs, fibroblast growth factors (FGFs) 2 and 18, platelet-derived growth factor, and insulin-like growth factors (IGFs) I and II. Hormones such as parathyroid hormone (PTH) and 1,25-dihydroxyvitamin D (1,25[OH]2D) activate receptors expressed by osteoblasts to assure mineral homeostasis and influence a variety of bone cell functions.
Resorption of bone is carried out mainly by osteoclasts, multinucleated cells that are formed by fusion of cells derived from the common precursor of macrophages and osteoclasts. Thus, these cells derive from the hematopoietic lineage, quite different from the mesenchymal cells that become osteoblasts. Multiple factors that regulate osteoclast development have been identified (Fig. 423-1B). Factors produced by osteoblasts or marrow stromal cells allow osteoblasts to control osteoclast development and activity. Macrophage colony-stimulating factor (M-CSF) plays a critical role during several steps in the pathway and ultimately leads to fusion of osteoclast progenitor cells to form multinucleated, active osteoclasts. RANK ligand, a member of the tumor necrosis factor (TNF) family, is expressed on the surface of osteoblast progenitors and stromal fibroblasts. In a process involving cell-cell interactions, RANK ligand binds to the RANK receptor on osteoclast progenitors, stimulating osteoclast differentiation and activation. Alternatively, a soluble decoy receptor, referred to as osteoprotegerin, can bind RANK ligand and inhibit osteoclast differentiation. Several growth factors and cytokines (including interleukins 1, 6, and 11; TNF; and interferon γ) modulate osteoclast differentiation and function. Most hormones that influence osteoclast function do not target these cells directly but instead act on cells of the osteoblast lineage to increase production of M-CSF and RANK. Both PTH and 1,25(OH)2D increase osteoclast number and activity by this indirect mechanism. Calcitonin, in contrast, binds to its receptor on the basal surface of osteoclasts and directly inhibits osteoclast function. Estradiol has multiple cellular targets in bone, including osteoclasts, immune cells, and osteoblasts; actions on all these cells serve to decrease osteoclast number and decrease bone resorption.
Osteoclast-mediated resorption of bone takes place in scalloped spaces (Howship’s lacunae) where the osteoclasts are attached through a specific αvβ3 integrin to components of the bone matrix such as osteopontin. The osteoclast forms a tight seal to the underlying matrix and secretes protons, chloride, and proteinases into a confined space that has been likened to an extracellular lysosome. The active osteoclast surface forms a ruffled border that contains a specialized proton pump ATPase that secretes acid and solubilizes the mineral phase. Carbonic anhydrase (type II isoenzyme) within the osteoclast generates the needed protons. The bone matrix is resorbed in the acid environment adjacent to the ruffled border by proteases, such as cathepsin K, that act at low pH.
In the embryo and the growing child, bone develops mostly by remodeling and replacing previously calcified cartilage (endochondral bone formation) or, in a few bones, is formed without a cartilage matrix (intramembranous bone formation). During endochondral bone formation, chondrocytes proliferate, secrete and mineralize a matrix, enlarge (hypertrophy), and then die, enlarging bone and providing the matrix and factors that stimulate endochondral bone formation. This program is regulated by both local factors, such as IGF-I and -II, Ihh, PTH-related peptide (PTHrP), and FGFs, and by systemic hormones, such as growth hormone, glucocorticoids, and estrogen.
New bone, whether formed in infants or in adults during repair, has a relatively high ratio of cells to matrix and is characterized by coarse fiber bundles of collagen that are interlaced and randomly dispersed (woven bone). In adults, the more mature bone is organized with fiber bundles regularly arranged in parallel or concentric sheets (lamellar bone). In long bones, deposition of lamellar bone in a concentric arrangement around blood vessels forms the Haversian systems. Growth in length of bones is dependent on proliferation of cartilage cells and the endochondral sequence at the growth plate. Growth in width and thickness is accomplished by formation of bone at the periosteal surface and by resorption at the endosteal surface, with the rate of formation exceeding that of resorption. In adults, after the growth plates of cartilage close, growth in length and endochondral bone formation cease except for some activity in the cartilage cells beneath the articular surface. Even in adults, however, remodeling of bone (within Haversian systems as well as along the surfaces of trabecular bone) continues throughout life. In adults, ~4% of the surface of trabecular bone (such as iliac crest) is involved in active resorption, whereas 10–15% of trabecular surfaces are covered with osteoid, unmineralized new bone formed by osteoblasts. Radioisotope studies indicate that as much as 18% of the total skeletal calcium is deposited and removed each year. Thus, bone is an active metabolizing tissue that requires an intact blood supply. The cycle of bone resorption and formation is a highly orchestrated process carried out by the basic multicellular unit, which is composed of a group of osteoclasts and osteoblasts (Fig. 423-2).
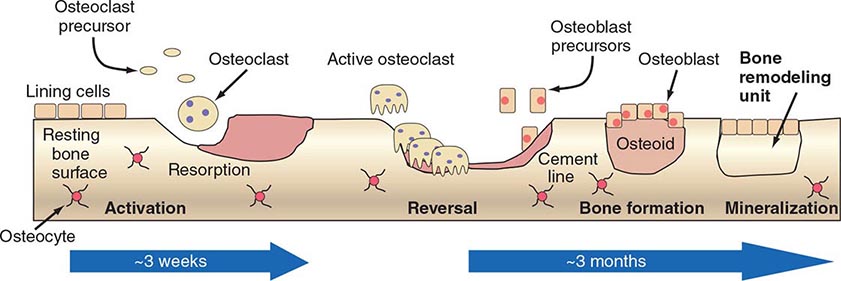
FIGURE 423-2 Schematic representation of bone remodeling. The cycle of bone remodeling is carried out by the basic multicellular unit (BMU), which consists of a group of osteoclasts and osteoblasts. In cortical bone, the BMUs tunnel through the tissue, whereas in cancellous bone, they move across the trabecular surface. The process of bone remodeling is initiated by contraction of the lining cells and the recruitment of osteoclast precursors. These precursors fuse to form multinucleated, active osteoclasts that mediate bone resorption. Osteoclasts adhere to bone and subsequently remove it by acidification and proteolytic digestion. As the BMU advances, osteoclasts leave the resorption site and osteoblasts move in to cover the excavated area and begin the process of new bone formation by secreting osteoid, which eventually is mineralized into new bone. After osteoid mineralization, osteoblasts flatten and form a layer of lining cells over new bone.
The response of bone to fractures, infection, and interruption of blood supply and to expanding lesions is relatively limited. Dead bone must be resorbed, and new bone must be formed, a process carried out in association with growth of new blood vessels into the involved area. In injuries that disrupt the organization of the tissue such as a fracture in which apposition of fragments is poor or when motion exists at the fracture site, progenitor stromal cells recapitulate the endochondral bone formation of early development and form cartilage that is replaced by bone and, variably, fibrous tissue. When there is good apposition with fixation and little motion at the fracture site, repair occurs predominantly by formation of new bone without other mediating tissue.
Remodeling of bone occurs along lines of force generated by mechanical stress. The signals from these mechanical stresses are sensed by osteocytes, which transmit signals to osteoclasts and osteoblasts or their precursors. One such signal made by osteocytes is sclerostin, an inhibitor of wnt signaling. Mechanical forces suppress sclerostin production and thus increase bone formation by osteoblasts. Expanding lesions in bone such as tumors induce resorption at the surface in contact with the tumor by producing ligands such as PTHrP that stimulate osteoclast differentiation and function. Even in a disorder as architecturally disruptive as Paget’s disease, remodeling is dictated by mechanical forces. Thus, bone plasticity reflects the interaction of cells with each other and with the environment.
Measurement of the products of osteoblast and osteoclast activity can assist in the diagnosis and management of bone diseases. Osteoblast activity can be assessed by measuring serum bone-specific alkaline phosphatase. Similarly, osteocalcin, a protein secreted from osteoblasts, is made virtually only by osteoblasts. Osteoclast activity can be assessed by measurement of products of collagen degradation. Collagen molecules are covalently linked to each other in the extracellular matrix through the formation of hydroxypyridinium cross-links (Chap. 427). After digestion by osteoclasts, these cross-linked peptides can be measured both in urine and in blood.
CALCIUM METABOLISM
Over 99% of the 1–2 kg of calcium present normally in the adult human body resides in the skeleton, where it provides mechanical stability and serves as a reservoir sometimes needed to maintain extracellular fluid (ECF) calcium concentration (Fig. 423-3). Skeletal calcium accretion first becomes significant during the third trimester of fetal life, accelerates throughout childhood and adolescence, reaches a peak in early adulthood, and gradually declines thereafter at rates that rarely exceed 1–2% per year. These slow changes in total skeletal calcium content contrast with relatively high daily rates of closely matched fluxes of calcium into and out of bone (~250–500 mg each), a process mediated by coupled osteoblastic and osteoclastic activity. Another 0.5–1% of skeletal calcium is freely exchangeable (e.g., in chemical equilibrium) with that in the ECF.
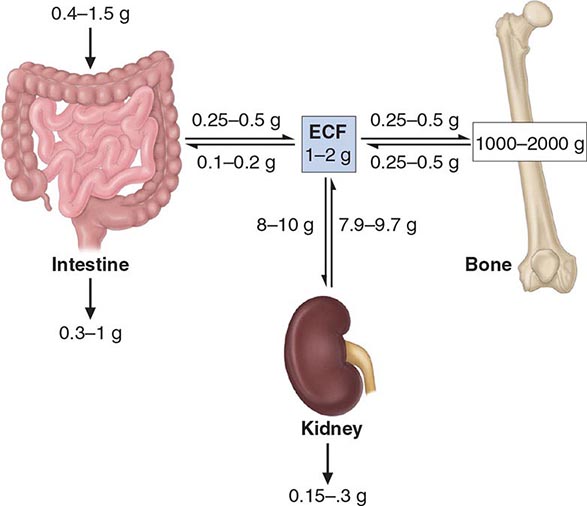
FIGURE 423-3 Calcium homeostasis. Schematic illustration of calcium content of extracellular fluid (ECF) and bone as well as of diet and feces; magnitude of calcium flux per day as calculated by various methods is shown at sites of transport in intestine, kidney, and bone. Ranges of values shown are approximate and were chosen to illustrate certain points discussed in the text. In conditions of calcium balance, rates of calcium release from and uptake into bone are equal.
The concentration of ionized calcium in the ECF must be maintained within a narrow range because of the critical role calcium plays in a wide array of cellular functions, especially those involved in neuromuscular activity, secretion, and signal transduction. Intracellular cytosolic free calcium levels are ~100 nmol/L and are 10,000-fold lower than ionized calcium concentrations in the blood and ECF (1.1–1.3 mmol/L). Cytosolic calcium does not play the structural role played by extracellular calcium; instead, it serves a signaling function. The steep chemical gradient of calcium from outside to inside the cell promotes rapid calcium influx through various membrane calcium channels that can be activated by hormones, metabolites, or neurotransmitters, swiftly changing cellular function. In blood, total calcium concentration is normally 2.2–2.6 mM (8.5–10.5 mg/dL), of which ~50% is ionized. The remainder is bound ionically to negatively charged proteins (predominantly albumin and immunoglobulins) or loosely complexed with phosphate, citrate, sulfate, or other anions. Alterations in serum protein concentrations directly affect the total blood calcium concentration even if the ionized calcium concentration remains normal. An algorithm to correct for protein changes adjusts the total serum calcium (in mg/dL) upward by 0.8 times the deficit in serum albumin (g/dL) or by 0.5 times the deficit in serum immunoglobulin (in g/dL). Such corrections provide only rough approximations of actual free calcium concentrations, however, and may be misleading, particularly during acute illness. Acidosis also alters ionized calcium by reducing its association with proteins. The best practice is to measure blood ionized calcium directly by a method that employs calcium-selective electrodes in acute settings during which calcium abnormalities might occur.
Control of the ionized calcium concentration in the ECF ordinarily is accomplished by adjusting the rates of calcium movement across intestinal and renal epithelia. These adjustments are mediated mainly via changes in blood levels of the hormones, PTH and 1,25(OH)2D. Blood ionized calcium directly suppresses PTH secretion by activating calcium-sensing receptors (CaSRs) in parathyroid cells. Also, ionized calcium indirectly affects PTH secretion by lowering 1,25(OH)2D production. This active vitamin D metabolite inhibits PTH production by an incompletely understood mechanism of negative feedback (Chap. 424).
Normal dietary calcium intake in the United States varies widely, ranging from 10–37 mmol/d (400–1500 mg/d). An Institute of Medicine report recommends a daily allowance of 25–30 mmol (1000–1200 mg) for most adults. Intestinal absorption of ingested calcium involves both active (transcellular) and passive (paracellular) mechanisms. Passive calcium absorption is nonsaturable and approximates 5% of daily calcium intake, whereas active absorption involves apical calcium entry via specific ion channels (TRPV5 and TRPV6), whose expression is controlled principally by 1,25(OH)2D, and normally ranges from 20 to 70%. Active calcium transport occurs mainly in the proximal small bowel (duodenum and proximal jejunum), although some active calcium absorption occurs in most segments of the small intestine. Optimal rates of calcium absorption require gastric acid. This is especially true for weakly dissociable calcium supplements such as calcium carbonate. In fact, large boluses of calcium carbonate are poorly absorbed because of their neutralizing effect on gastric acid. In achlorhydric subjects and for those taking drugs that inhibit gastric acid secretion, supplements should be taken with meals to optimize their absorption. Use of calcium citrate may be preferable in these circumstances. Calcium absorption may also be blunted in disease states such as pancreatic or biliary insufficiency, in which ingested calcium remains bound to unabsorbed fatty acids or other food constituents. At high levels of calcium intake, synthesis of 1,25(OH)2D is reduced; this decreases the rate of active intestinal calcium absorption. The opposite occurs with dietary calcium restriction. Some calcium, ~2.5–5 mmol/d (100–200 mg/d), is excreted as an obligate component of intestinal secretions and is not regulated by calciotropic hormones.
The feedback-controlled hormonal regulation of intestinal absorptive efficiency results in a relatively constant daily net calcium absorption of ~5–7.5 mmol/d (200–400 mg/d) despite large changes in daily dietary calcium intake. This daily load of absorbed calcium is excreted by the kidneys in a manner that is also tightly regulated by the concentration of ionized calcium in the blood. Approximately 8–10 g/d of calcium is filtered by the glomeruli, of which only 2–3% appears in the urine. Most filtered calcium (65%) is reabsorbed in the proximal tubules via a passive, paracellular route that is coupled to concomitant NaCl reabsorption and not specifically regulated. The cortical thick ascending limb of Henle’s loop (cTAL) reabsorbs roughly another 20% of filtered calcium, also via a paracellular mechanism. Calcium reabsorption in the cTAL requires a tight-junctional protein called paracellin-1 and is inhibited by increased blood concentrations of calcium or magnesium, acting via the CaSR, which is highly expressed on basolateral membranes in this nephron segment. Operation of the renal CaSR provides a mechanism, independent of those engaged directly by PTH or 1,25(OH)2D, by which serum ionized calcium can control renal calcium reabsorption. Finally, ~10% of filtered calcium is reabsorbed in the distal convoluted tubules (DCTs) by a transcellular mechanism. Calcium enters the luminal surface of the cell through specific apical calcium channels (TRPV5), whose number is regulated. It then moves across the cell in association with a specific calcium-binding protein (calbindin-D28k) that buffers cytosolic calcium concentrations from the large mass of transported calcium. Ca2+-ATPases and Na+/Ca2+ exchangers actively extrude calcium across the basolateral surface and thereby maintain the transcellular calcium gradient. All these processes are stimulated directly or indirectly by PTH. The DCT is also the site of action of thiazide diuretics, which lower urinary calcium excretion by inducing sodium depletion and thereby augmenting proximal calcium reabsorption. Conversely, dietary sodium loads, or increased distal sodium delivery caused by loop diuretics or saline infusion, induce calciuresis.
The homeostatic mechanisms that normally maintain a constant serum ionized calcium concentration may fail at extremes of calcium intake or when the hormonal systems or organs involved are compromised. Thus, even with maximal activity of the vitamin D–dependent intestinal active transport system, sustained calcium intakes <5 mmol/d (<200 mg/d) cannot provide enough net calcium absorption to replace obligate losses via the intestine, the kidney, sweat, and other secretions. In this case, increased blood levels of PTH and 1,25(OH)2D activate osteoclastic bone resorption to obtain needed calcium from bone, which leads to progressive bone loss and negative calcium balance. Increased PTH and 1,25(OH)2D also enhance renal calcium reabsorption, and 1,25(OH)2D enhances calcium absorption in the gut. At very high calcium intakes (>100 mmol/d [>4 g/d]), passive intestinal absorption continues to deliver calcium into the ECF despite maximally downregulated intestinal active transport and renal tubular calcium reabsorption. This can cause severe hypercalciuria, nephrocalcinosis, progressive renal failure, and hypercalcemia (e.g., “milk-alkali syndrome”). Deficiency or excess of PTH or vitamin D, intestinal disease, and renal failure represent other commonly encountered challenges to normal calcium homeostasis (Chap. 424).
PHOSPHORUS METABOLISM
Although 85% of the ~600 g of body phosphorus is present in bone mineral, phosphorus is also a major intracellular constituent both as the free anion(s) and as a component of numerous organophosphate compounds, including structural proteins, enzymes, transcription factors, carbohydrate and lipid intermediates, high-energy stores (adenosine triphosphate [ATP], creatine phosphate), and nucleic acids. Unlike calcium, phosphorus exists intracellularly at concentrations close to those present in ECF (e.g., 1–2 mmol/L). In cells and in the ECF, phosphorus exists in several forms, predominantly as H2PO4– or NaHPO4–, with perhaps 10% as HPO42–. This mixture of anions will be referred to here as “phosphate.” In serum, about 12% of phosphorus is bound to proteins. Concentrations of phosphates in blood and ECF generally are expressed in terms of elemental phosphorus, with the normal range in adults being 0.75–1.45 mmol/L (2.5–4.5 mg/dL). Because the volume of the intracellular fluid compartment is twice that of the ECF, measurements of ECF phosphate may not accurately reflect phosphate availability within cells that follows even modest shifts of phosphate from one compartment to the other.
Phosphate is widely available in foods and is absorbed efficiently (65%) by the small intestine even in the absence of vitamin D. However, phosphate absorptive efficiency may be enhanced (to 85–90%) via active transport mechanisms that are stimulated by 1,25(OH)2D. These mechanisms involve activation of Na+/PO42– co-transporters that move phosphate into intestinal cells against an unfavorable electrochemical gradient. Daily net intestinal phosphate absorption varies widely with the composition of the diet but is generally in the range of 500–1000 mg/d. Phosphate absorption can be inhibited by large doses of calcium salts or by sevelamer hydrochloride (Renagel), strategies commonly used to control levels of serum phosphate in renal failure. Aluminum hydroxide antacids also reduce phosphate absorption but are used less commonly because of the potential for aluminum toxicity. Low serum phosphate stimulates renal proximal tubular synthesis of 1,25(OH)2D, perhaps by suppressing blood levels of FGF23 (see below).
Serum phosphate levels vary by as much as 50% on a normal day. This reflects the effect of food intake but also an underlying circadian rhythm that produces a nadir between 7:00 and 10:00 A.M. Carbohydrate administration, especially as IV dextrose solutions in fasting subjects, can decrease serum phosphate by >0.7 mmol/L (2 mg/dL) due to rapid uptake into and utilization by cells. A similar response is observed in the treatment of diabetic ketoacidosis and during metabolic or respiratory alkalosis. Because of this wide variation in serum phosphate, it is best to perform measurements in the basal, fasting state.
Control of serum phosphate is determined mainly by the rate of renal tubular reabsorption of the filtered load, which is ~4–6 g/d. Because intestinal phosphate absorption is highly efficient, urinary excretion is not constant but varies directly with dietary intake. The fractional excretion of phosphate (ratio of phosphate to creatinine clearance) is generally in the range of 10–15%. The proximal tubule is the principal site at which renal phosphate reabsorption is regulated. This is accomplished by changes in the levels of apical expression and activity of specific Na+/PO42– co-transporters (NaPi-2a and NaPi-2c) in the proximal tubule. Levels of these transporters at the apical surface of these cells are reduced rapidly by PTH, a major hormonal regulator of renal phosphate excretion. FGF23 can impair phosphate reabsorption dramatically by a similar mechanism. Activating FGF23 mutations cause the rare disorder autosomal dominant hypophosphatemic rickets. In contrast to PTH, FGF23 also leads to reduced synthesis of 1,25(OH)2D, which may worsen the resulting hypophosphatemia by lowering intestinal phosphate absorption. Renal reabsorption of phosphate is responsive to changes in dietary intake such that experimental dietary phosphate restriction leads to a dramatic lowering of urinary phosphate within hours, preceding any decline in serum phosphate (e.g., filtered load). This physiologic renal adaptation to changes in dietary phosphate availability occurs independently of PTH and may be mediated in part by changes in levels of serum FGF23. Findings in FGF23-knockout mice suggest that FGF23 normally acts to lower blood phosphate and 1,25(OH)2D levels. In turn, elevation of blood phosphate increases blood levels of FGF23.
Renal phosphate reabsorption is impaired by hypocalcemia, hypomagnesemia, and severe hypophosphatemia. Phosphate clearance is enhanced by ECF volume expansion and impaired by dehydration. Phosphate retention is an important pathophysiologic feature of renal insufficiency (Chap. 335).
HYPOPHOSPHATEMIA
Causes Hypophosphatemia can occur by one or more of three primary mechanisms: (1) inadequate intestinal phosphate absorption, (2) excessive renal phosphate excretion, and (3) rapid redistribution of phosphate from the ECF into bone or soft tissue (Table 423-1). Because phosphate is so abundant in foods, inadequate intestinal absorption is almost never observed now that aluminum hydroxide antacids, which bind phosphate in the gut, are no longer widely used. Fasting or starvation, however, may result in depletion of body phosphate and predispose to subsequent hypophosphatemia during refeeding, especially if this is accomplished with IV glucose alone.
|
CAUSES OF HYPOPHOSPHATEMIA |
Abbreviations: PTH, parathyroid hormone; PTHrP, parathyroid hormone–related peptide.
Chronic hypophosphatemia usually signifies a persistent renal tubular phosphate-wasting disorder. Excessive activation of PTH/PTHrP receptors in the proximal tubule as a result of primary or secondary hyperparathyroidism or because of the PTHrP-mediated hypercalcemia syndrome in malignancy (Chap. 424) is among the more common causes of renal hypophosphatemia, especially because of the high prevalence of vitamin D deficiency in older Americans. Familial hypocalciuric hypercalcemia and Jansen’s chondrodystrophy are rare examples of genetic disorders in this category (Chap. 424).
Several genetic and acquired diseases cause PTH/PTHrP-independent tubular phosphate wasting with associated rickets and osteomalacia. All these diseases manifest severe hypophosphatemia; renal phosphate wasting, sometimes accompanied by aminoaciduria; inappropriately low blood levels of 1,25(OH)2D; low-normal serum levels of calcium; and evidence of impaired cartilage or bone mineralization. Analysis of these diseases led to the discovery of the hormone FGF23, which is an important physiologic regulator of phosphate metabolism. FGF23 decreases phosphate reabsorption in the proximal tubule and also suppresses the 1α-hydroxylase responsible for synthesis of 1,25(OH)2D. FGF23 is synthesized by cells of the osteoblast lineage, primarily osteocytes. High-phosphate diets increase FGF23 levels, and low-phosphate diets decrease them. Autosomal dominant hypophosphatemic rickets (ADHR) was the first disease linked to abnormalities in FGF23. ADHR results from activating mutations in the gene that encodes FGF23. These mutations alter a cleavage site that ordinarily allows for inactivation of intact FGF23. Several other genetic disorders exhibit elevated FGF23 and hypophosphatemia. The most common of these is X-linked hypophosphatemic rickets (XLH), which results from inactivating mutations in an endopeptidase termed PHEX (phosphate-regulating gene with homologies to endopeptidases on the X chromosome) that is expressed most abundantly on the surface of osteocytes and mature osteoblasts. Patients with XLH usually have high FGF23 levels, and ablation of the FGF23 gene reverses the hypophosphatemia found in the mouse version of XLH. How inactivation of PHEX leads to increased levels of FGF23 has not been determined. Two rare autosomal recessive hypophosphatemic syndromes associated with elevated FGF23 are due to inactivating mutations of dentin matrix protein-1 (DMP1) and ectonucleotide pyrophosphatase/phosphodiesterase 1 (ENPP1), both of which normally are highly expressed in bone and regulate FGF23 production. An unusual hypophosphatemic disorder, tumor-induced osteomalacia (TIO), is an acquired disorder in which tumors, usually of mesenchymal origin and generally histologically benign, secrete FGF23 and/or other molecules that induce renal phosphate wasting. The hypophosphatemic syndrome resolves completely within hours to days after successful resection of the responsible tumor. Such tumors typically express large amounts of FGF23 mRNA, and patients with TIO usually exhibit elevations of FGF23 in their blood.
Dent’s disease is an X-linked recessive disorder caused by inactivating mutations in CLCN5, a chloride transporter expressed in endosomes of the proximal tubule; features include hypercalciuria, hypophosphatemia, and recurrent kidney stones. Renal phosphate wasting is common among poorly controlled diabetic patients and alcoholics, who therefore are at risk for iatrogenic hypophosphatemia when treated with insulin or IV glucose, respectively. Diuretics and certain other drugs and toxins can cause defective renal tubular phosphate reabsorption (Table 423-1).
In hospitalized patients, hypophosphatemia is often attributable to massive redistribution of phosphate from the ECF into cells. Insulin therapy for diabetic ketoacidosis is a paradigm for this phenomenon, in which the severity of the hypophosphatemia is related to the extent of antecedent depletion of phosphate and other electrolytes (Chap. 417). The hypophosphatemia is usually greatest at a point many hours after initiation of insulin therapy and is difficult to predict from baseline measurements of serum phosphate at the time of presentation, when prerenal azotemia can obscure significant phosphate depletion. Other factors that may contribute to such acute redistributive hypophosphatemia include antecedent starvation or malnutrition, administration of IV glucose without other nutrients, elevated blood catecholamines (endogenous or exogenous), respiratory alkalosis, and recovery from metabolic acidosis.
Hypophosphatemia also can occur transiently (over weeks to months) during the phase of accelerated net bone formation that follows parathyroidectomy for severe primary hyperparathyroidism or during treatment of vitamin D deficiency or lytic Paget’s disease. This is usually most prominent in patients who preoperatively have evidence of high bone turnover (e.g., high serum levels of alkaline phosphatase). Osteoblastic metastases can also lead to this syndrome.
Clinical and Laboratory Findings The clinical manifestations of severe hypophosphatemia reflect a generalized defect in cellular energy metabolism because of ATP depletion, a shift from oxidative phosphorylation toward glycolysis, and associated tissue or organ dysfunction. Acute, severe hypophosphatemia occurs mainly or exclusively in hospitalized patients with underlying serious medical or surgical illness and preexisting phosphate depletion due to excessive urinary losses, severe malabsorption, or malnutrition. Chronic hypophosphatemia tends to be less severe, with a clinical presentation dominated by musculoskeletal complaints such as bone pain, osteomalacia, pseudofractures, and proximal muscle weakness or, in children, rickets and short stature.
Neuromuscular manifestations of severe hypophosphatemia are variable but may include muscle weakness, lethargy, confusion, disorientation, hallucinations, dysarthria, dysphagia, oculomotor palsies, anisocoria, nystagmus, ataxia, cerebellar tremor, ballismus, hyporeflexia, impaired sphincter control, distal sensory deficits, paresthesia, hyperesthesia, generalized or Guillain-Barré–like ascending paralysis, seizures, coma, and even death. Serious sequelae such as paralysis, confusion, and seizures are likely only at phosphate concentrations <0.25 mmol/L (<0.8 mg/dL). Rhabdomyolysis may develop during rapidly progressive hypophosphatemia. The diagnosis of hypophosphatemia-induced rhabdomyolysis may be overlooked, as up to 30% of patients with acute hypophosphatemia (<0.7 mM) have creatine phosphokinase elevations that peak 1–2 days after the nadir in serum phosphate, when the release of phosphate from injured myocytes may have led to a near normalization of circulating levels of phosphate.
Respiratory failure and cardiac dysfunction, which are reversible with phosphate treatment, may occur at serum phosphate levels of 0.5–0.8 mmol/L (1.5–2.5 mg/dL). Renal tubular defects, including tubular acidosis, glycosuria, and impaired reabsorption of sodium and calcium, may occur. Hematologic abnormalities correlate with reductions in intracellular ATP and 2,3-diphosphoglycerate and may include erythrocyte microspherocytosis and hemolysis; impaired oxyhemoglobin dissociation; defective leukocyte chemotaxis, phagocytosis, and bacterial killing; and platelet dysfunction with spontaneous gastrointestinal hemorrhage.
|
TREATMENT |
HYPOPHOSPHATEMIA |
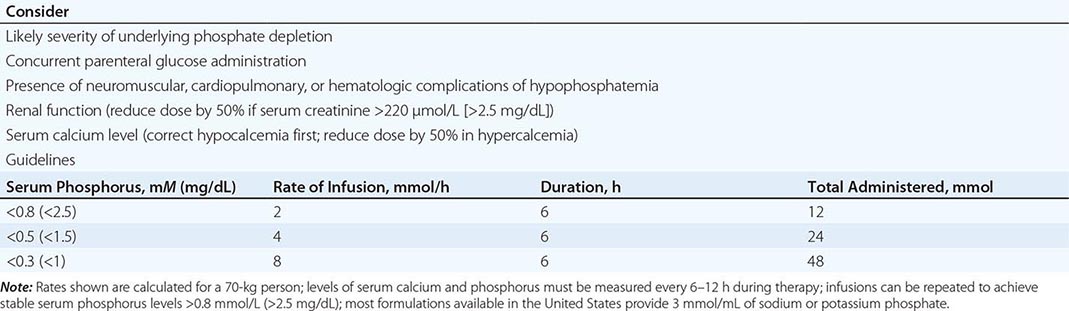
Severe hypophosphatemia (<0.75 mmol/L [<2 mg/dL]), particularly in the setting of underlying phosphate depletion, constitutes a dangerous electrolyte abnormality that should be corrected promptly. Unfortunately, the cumulative deficit in body phosphate cannot be predicted easily from knowledge of the circulating level of phosphate, and therapy must be approached empirically. The threshold for IV phosphate therapy and the dose administered should reflect consideration of renal function, the likely severity and duration of the underlying phosphate depletion, and the presence and severity of symptoms consistent with those of hypophosphatemia. In adults, phosphate may be safely administered IV as neutral mixtures of sodium or potassium phosphate salts at initial doses of 0.2–0.8 mmol/kg of elemental phosphorus over 6 h (e.g., 10–50 mmol over 6 h), with doses >20 mmol/6 h reserved for those who have serum levels <0.5 mmol/L (1.5 mg/dL) and normal renal function. A suggested approach is presented in Table 423-2. Serum levels of phosphate and calcium must be monitored closely (every 6–12 h) throughout treatment. It is necessary to avoid a serum calcium-phosphorus product >50 to reduce the risk of heterotopic calcification. Hypocalcemia, if present, should be corrected before administering IV phosphate. Less severe hypophosphatemia, in the range of 0.5–0.8 mmol/L (1.5–2.5 mg/dL), usually can be treated with oral phosphate in divided doses of 750–2000 mg/d as elemental phosphorus; higher doses can cause bloating and diarrhea.
|
INTRAVENOUS THERAPY FOR HYPOPHOSPHATEMIA |
Management of chronic hypophosphatemia requires knowledge of the cause(s) of the disorder. Hypophosphatemia related to the secondary hyperparathyroidism of vitamin D deficiency usually responds to treatment with vitamin D and calcium alone. XLH, ADHR, TIO, and related renal tubular disorders usually are managed with divided oral doses of phosphate, often with calcium and 1,25(OH)2D supplements to bypass the block in renal 1,25(OH)2D synthesis and prevent secondary hyperparathyroidism caused by suppression of ECF calcium levels. Thiazide diuretics may be used to prevent nephrocalcinosis in patients who are managed this way. Complete normalization of hypophosphatemia is generally not possible in these conditions. Optimal therapy for TIO is extirpation of the responsible tumor, which may be localized by radiographic skeletal survey or bone scan (many are located in bone) or by radionuclide scanning using sestamibi or labeled octreotide. Successful treatment of TIO-induced hypophosphatemia with octreotide has been reported in a small number of patients.
HYPERPHOSPHATEMIA
Causes When the filtered load of phosphate and glomerular filtration rate (GFR) are normal, control of serum phosphate levels is achieved by adjusting the rate at which phosphate is reabsorbed by the proximal tubular NaPi-2 co-transporters. The principal hormonal regulators of NaPi-2 activity are PTH and FGF23. Hyperphosphatemia, defined in adults as a fasting serum phosphate concentration >1.8 mmol/L (5.5 mg/dL), usually results from impaired glomerular filtration, hypoparathyroidism, excessive delivery of phosphate into the ECF (from bone, gut, or parenteral phosphate therapy), or a combination of these factors (Table 423-3). The upper limit of normal serum phosphate concentrations is higher in children and neonates (2.4 mmol/L [7 mg/dL]). It is useful to distinguish hyperphosphatemia caused by impaired renal phosphate excretion from that which results from excessive delivery of phosphate into the ECF (Table 423-3).
|
CAUSES OF HYPERPHOSPHATEMIA |
In chronic renal insufficiency, reduced GFR leads to phosphate retention. Hyperphosphatemia in turn further impairs renal synthesis of 1,25(OH)2D, increases FGF23 levels, and stimulates PTH secretion and hypertrophy both directly and indirectly (by lowering blood ionized calcium levels). Thus, hyperphosphatemia is a major cause of the secondary hyperparathyroidism of renal failure and must be addressed early in the course of the disease (Chaps. 335 and 424).
Hypoparathyroidism leads to hyperphosphatemia via increased expression of NaPi-2 co-transporters in the proximal tubule. Hypoparathyroidism, or parathyroid suppression, has multiple potential causes, including autoimmune disease; developmental, surgical, or radiation-induced absence of functional parathyroid tissue; vitamin D intoxication or other causes of PTH-independent hypercalcemia; cellular PTH resistance (pseudohypoparathyroidism or hypomagnesemia); infiltrative disorders such as Wilson’s disease and hemochromatosis; and impaired PTH secretion caused by hypermagnesemia, severe hypomagnesemia, or activating mutations in the CaSR. Hypocalcemia may also contribute directly to impaired phosphate clearance, as calcium infusion can induce phosphaturia in hypoparathyroid subjects. Increased tubular phosphate reabsorption also occurs in acromegaly, during heparin administration, and in tumoral calcinosis. Tumoral calcinosis is caused by a rare group of genetic disorders in which FGF23 is processed in a way that leads to low levels of active FGF23 in the bloodstream. This may result from mutations in the FGF23 sequence or via inactivating mutations in the GALNT3 gene, which encodes a galactosaminyl transferase that normally adds sugar residues to FGF23 that slow its proteolysis. A similar syndrome results from FGF23 resistance due to inactivating mutations of the FGF23 co-receptor Klotho. These abnormalities cause elevated serum 1,25(OH)2D, parathyroid suppression, increased intestinal calcium absorption, and focal hyperostosis with large, lobulated periarticular heterotopic ossifications (especially at shoulders or hips) and are accompanied by hyperphosphatemia. In some forms of tumoral calcinosis, serum phosphorus levels are normal.
When large amounts of phosphate are delivered rapidly into the ECF, hyperphosphatemia can occur despite normal renal function. Examples include overzealous IV phosphate therapy, oral or rectal administration of large amounts of phosphate-containing laxatives or enemas (especially in children), extensive soft tissue injury or necrosis (crush injuries, rhabdomyolysis, hyperthermia, fulminant hepatitis, cytotoxic chemotherapy), extensive hemolytic anemia, and transcellular phosphate shifts induced by severe metabolic or respiratory acidosis.
Clinical Findings The clinical consequences of acute, severe hyperphosphatemia are due mainly to the formation of widespread calcium phosphate precipitates and resulting hypocalcemia. Thus, tetany, seizures, accelerated nephrocalcinosis (with renal failure, hyperkalemia, hyperuricemia, and metabolic acidosis), and pulmonary or cardiac calcifications (including development of acute heart block) may occur. The severity of these complications relates to the elevation of serum phosphate levels, which can reach concentrations as high as 7 mmol/L (20 mg/dL) in instances of massive soft tissue injury or tumor lysis syndrome.
|
TREATMENT |
HYPERPHOSPHATEMIA |
Therapeutic options for management of severe hyperphosphatemia are limited. Volume expansion may enhance renal phosphate clearance. Aluminum hydroxide antacids or sevelamer may be helpful in chelating and limiting absorption of offending phosphate salts present in the intestine. Hemodialysis is the most effective therapeutic strategy and should be considered early in the course of severe hyperphosphatemia, especially in the setting of renal failure and symptomatic hypocalcemia.
MAGNESIUM METABOLISM
Magnesium is the major intracellular divalent cation. Normal concentrations of extracellular magnesium and calcium are crucial for normal neuromuscular activity. Intracellular magnesium forms a key complex with ATP and is an important cofactor for a wide range of enzymes, transporters, and nucleic acids required for normal cellular function, replication, and energy metabolism. The concentration of magnesium in serum is closely regulated within the range of 0.7–1 mmol/L (1.5–2 meq/L; 1.7–2.4 mg/dL), of which 30% is protein-bound and another 15% is loosely complexed to phosphate and other anions. One-half of the 25 g (1000 mmol) of total body magnesium is located in bone, only one-half of which is insoluble in the mineral phase. Almost all extraskeletal magnesium is present within cells, where the total concentration is 5 mM, 95% of which is bound to proteins and other macromolecules. Because only 1% of body magnesium resides in the ECF, measurements of serum magnesium levels may not accurately reflect the level of total body magnesium stores.
Dietary magnesium content normally ranges from 6 to 15 mmol/d (140–360 mg/d), of which 30–40% is absorbed, mainly in the jejunum and ileum. Intestinal magnesium absorptive efficiency is stimulated by 1,25(OH)2D and can reach 70% during magnesium deprivation. Urinary magnesium excretion normally matches net intestinal absorption and is ~4 mmol/d (100 mg/d). Regulation of serum magnesium concentrations is achieved mainly by control of renal magnesium reabsorption. Only 20% of filtered magnesium is reabsorbed in the proximal tubule, whereas 60% is reclaimed in the cTAL and another 5–10% in the DCT. Magnesium reabsorption in the cTAL occurs via a paracellular route that requires both a lumen-positive potential, created by NaCl reabsorption, and tight-junction proteins encoded by members of the Claudin gene family. Magnesium reabsorption in the cTAL is increased by PTH but inhibited by hypercalcemia or hypermagnesemia, both of which activate the CaSR in this nephron segment.
HYPOMAGNESEMIA
Causes Hypomagnesemia usually signifies substantial depletion of body magnesium stores (0.5–1 mmol/kg). Hypomagnesemia can result from intestinal malabsorption; protracted vomiting, diarrhea, or intestinal drainage; defective renal tubular magnesium reabsorption; or rapid shifts of magnesium from the ECF into cells, bone, or third spaces (Table 423-4). Dietary magnesium deficiency is unlikely except possibly in the setting of alcoholism. A rare genetic disorder that causes selective intestinal magnesium malabsorption has been described (primary infantile hypomagnesemia). Another rare inherited disorder (hypomagnesemia with secondary hypocalcemia) is caused by mutations in the gene encoding TRPM6, a protein that, along with TRPM7, forms a channel important for both intestinal and distal-tubular renal transcellular magnesium transport. Malabsorptive states, often compounded by vitamin D deficiency, can critically limit magnesium absorption and produce hypomagnesemia despite the compensatory effects of secondary hyperparathyroidism and of hypocalcemia and hypomagnesemia to enhance cTAL magnesium reabsorption. Diarrhea or surgical drainage fluid may contain ≥5 mmol/L of magnesium. Proton pump inhibitors (omeprazole and others) may produce hypomagnesemia by an unknown mechanism that does not involve renal wasting of magnesium.
|
CAUSES OF HYPOMAGNESEMIA |
Abbreviations: ATN, acute tubular necrosis; SIADH, syndrome of inappropriate antidiuretic hormone.
Several genetic magnesium-wasting syndromes have been described, including inactivating mutations of genes encoding the DCT NaCl co-transporter (Gitelman’s syndrome), proteins required for cTAL Na-K-2Cl transport (Bartter’s syndrome), claudin 16 or claudin 19 (autosomal recessive renal hypomagnesemia with hypercalciuria), a DCT Na+,K+-ATPase γ-subunit (autosomal dominant renal hypomagnesemia with hypocalciuria), DCT K+ channels (Kv1.1, Kir4.1), and a mitochondrial gene encoding a tRNA. Activating mutations of the CaSR can cause hypomagnesemia as well as hypocalcemia. ECF expansion, hypercalcemia, and severe phosphate depletion may impair magnesium reabsorption, as can various forms of renal injury, including those caused by drugs such as cisplatin, cyclosporine, aminoglycosides, and pentamidine as well as the epidermal growth factor (EGF) receptor inhibitory antibody, cetuximab (EGF action is required for normal DCT apical expression of TRPM6) (Table 423-4). A rising blood concentration of ethanol directly impairs tubular magnesium reabsorption, and persistent glycosuria with osmotic diuresis leads to magnesium wasting and probably contributes to the high frequency of hypomagnesemia in poorly controlled diabetic patients. Magnesium depletion is aggravated by metabolic acidosis, which causes intracellular losses as well.
Hypomagnesemia due to rapid shifts of magnesium from ECF into the intracellular compartment can occur during recovery from diabetic ketoacidosis, starvation, or respiratory acidosis. Less acute shifts may be seen during rapid bone formation after parathyroidectomy, with treatment of vitamin D deficiency, or with osteoblastic metastases. Large amounts of magnesium may be lost with acute pancreatitis, extensive burns, or protracted and severe sweating and during pregnancy and lactation.
Clinical and Laboratory Findings Hypomagnesemia may cause generalized alterations in neuromuscular function, including tetany, tremor, seizures, muscle weakness, ataxia, nystagmus, vertigo, apathy, depression, irritability, delirium, and psychosis. Patients are usually asymptomatic when serum magnesium concentrations are >0.5 mmol/L (1 meq/L; 1.2 mg/dL), although the severity of symptoms may not correlate with serum magnesium levels. Cardiac arrhythmias may occur, including sinus tachycardia, other supraventricular tachycardias, and ventricular arrhythmias. Electrocardiographic abnormalities may include prolonged PR or QT intervals, T-wave flattening or inversion, and ST straightening. Sensitivity to digitalis toxicity may be enhanced.
Other electrolyte abnormalities often seen with hypomagnesemia, including hypocalcemia (with hypocalciuria) and hypokalemia, may not be easily corrected unless magnesium is administered as well. The hypocalcemia may be a result of concurrent vitamin D deficiency, although hypomagnesemia can cause impaired synthesis of 1,25(OH)2D, cellular resistance to PTH, and, at very low serum magnesium (<0.4 mmol/L [0.8 meq/L; <1 mg/dL]), a defect in PTH secretion; these abnormalities are reversible with therapy.
|
TREATMENT |
HYPOMAGNESEMIA |
Mild, asymptomatic hypomagnesemia may be treated with oral magnesium salts (MgCl2, MgO, Mg[OH]2) in divided doses totaling 20–30 mmol/d (40–60 meq/d). Diarrhea may occur with larger doses. More severe hypomagnesemia should be treated parenterally, preferably with IV MgCl2, which can be administered safely as a continuous infusion of 50 mmol/d (100 meq Mg2+/d) if renal function is normal. If GFR is reduced, the infusion rate should be lowered by 50–75%. Use of IM MgSO4 is discouraged; the injections are painful and provide relatively little magnesium (2 mL of 50% MgSO4 supplies only 4 mmol). MgSO4 may be given IV instead of MgCl2, although the sulfate anions may bind calcium in serum and urine and aggravate hypocalcemia. Serum magnesium should be monitored at intervals of 12–24 h during therapy, which may continue for several days because of impaired renal conservation of magnesium (only 50–70% of the daily IV magnesium dose is retained) and delayed repletion of intracellular deficits, which may be as high as 1–1.5 mmol/kg (2–3 meq/kg).
It is important to consider the need for calcium, potassium, and phosphate supplementation in patients with hypomagnesemia. Vitamin D deficiency frequently coexists and should be treated with oral or parenteral vitamin D or 25(OH)D (but not with 1,25[OH]2D, which may impair tubular magnesium reabsorption, possibly via PTH suppression). In severely hypomagnesemic patients with concomitant hypocalcemia and hypophosphatemia, administration of IV magnesium alone may worsen hypophosphatemia, provoking neuromuscular symptoms or rhabdomyolysis, due to rapid stimulation of PTH secretion. This is avoided by administering both calcium and magnesium.
HYPERMAGNESEMIA
Causes Hypermagnesemia is rarely seen in the absence of renal insufficiency, as normal kidneys can excrete large amounts (250 mmol/d) of magnesium. Mild hypermagnesemia due to excessive reabsorption in the cTAL occurs with CaSR mutations in familial hypocalciuric hypercalcemia and has been described in some patients with adrenal insufficiency, hypothyroidism, or hypothermia. Massive exogenous magnesium exposures, usually via the gastrointestinal tract, can overwhelm renal excretory capacity and cause life-threatening hypermagnesemia (Table 423-5). A notable example of this is prolonged retention of even normal amounts of magnesium-containing cathartics in patients with intestinal ileus, obstruction, or perforation. Extensive soft tissue injury or necrosis can also deliver large amounts of magnesium into the ECF in patients who have suffered trauma, shock, sepsis, cardiac arrest, or severe burns.
|
CAUSES OF HYPERMAGNESEMIA |
Clinical and Laboratory Findings The most prominent clinical manifestations of hypermagnesemia are vasodilation and neuromuscular blockade, which may appear at serum magnesium concentrations >2 mmol/L (>4 meq/L; >4.8 mg/dL). Hypotension that is refractory to vasopressors or volume expansion may be an early sign. Nausea, lethargy, and weakness may progress to respiratory failure, paralysis, and coma, with hypoactive tendon reflexes, at serum magnesium levels >4 mmol/L. Other findings may include gastrointestinal hypomotility or ileus; facial flushing; pupillary dilation; paradoxical bradycardia; prolongation of PR, QRS, and QT intervals; heart block; and, at serum magnesium levels approaching 10 mmol/L, asystole.
Hypermagnesemia, acting via the CaSR, causes hypocalcemia and hypercalciuria due to both parathyroid suppression and impaired cTAL calcium reabsorption.
|
TREATMENT |
HYPERMAGNESEMIA |
Successful treatment of hypermagnesemia generally involves identifying and interrupting the source of magnesium and employing measures to increase magnesium clearance from the ECF. Use of magnesium-free cathartics or enemas may be helpful in clearing ingested magnesium from the gastrointestinal tract. Vigorous IV hydration should be attempted, if appropriate. Hemodialysis is effective and may be required in patients with significant renal insufficiency. Calcium, administered IV in doses of 100–200 mg over 1–2 h, has been reported to provide temporary improvement in signs and symptoms of hypermagnesemia.
VITAMIN D
SYNTHESIS AND METABOLISM
1,25-Dihydroxyvitamin D (1,25[OH]2D) is the major steroid hormone involved in mineral ion homeostasis regulation. Vitamin D and its metabolites are hormones and hormone precursors rather than vitamins, since in the proper biologic setting, they can be synthesized endogenously (Fig. 423-4). In response to ultraviolet radiation of the skin, a photochemical cleavage results in the formation of vitamin D from 7-dehydrocholesterol. Cutaneous production of vitamin D is decreased by melanin and high solar protection factor sunblocks, which effectively impair skin penetration by ultraviolet light. The increased use of sunblocks in North America and Western Europe and a reduction in the magnitude of solar exposure of the general population over the last several decades has led to an increased reliance on dietary sources of vitamin D. In the United States and Canada, these sources largely consist of fortified cereals and dairy products, in addition to fish oils and egg yolks. Vitamin D from plant sources is in the form of vitamin D2, whereas that from animal sources is vitamin D3. These two forms have equivalent biologic activity and are activated equally well by the vitamin D hydroxylases in humans. Vitamin D enters the circulation, whether absorbed from the intestine or synthesized cutaneously, bound to vitamin D–binding protein, an α-globulin synthesized in the liver. Vitamin D is subsequently 25-hydroxylated in the liver by cytochrome P450–like enzymes in the mitochondria and microsomes. The activity of this hydroxylase is not tightly regulated, and the resultant metabolite, 25-hydroxyvitamin D (25[OH]D), is the major circulating and storage form of vitamin D. Approximately 88% of 25(OH)D circulates bound to the vitamin D–binding protein, 0.03% is free, and the rest circulates bound to albumin. The half-life of 25(OH)D is approximately 2–3 weeks; however, it is shortened dramatically when vitamin D–binding protein levels are reduced, as can occur with increased urinary losses in the nephrotic syndrome.
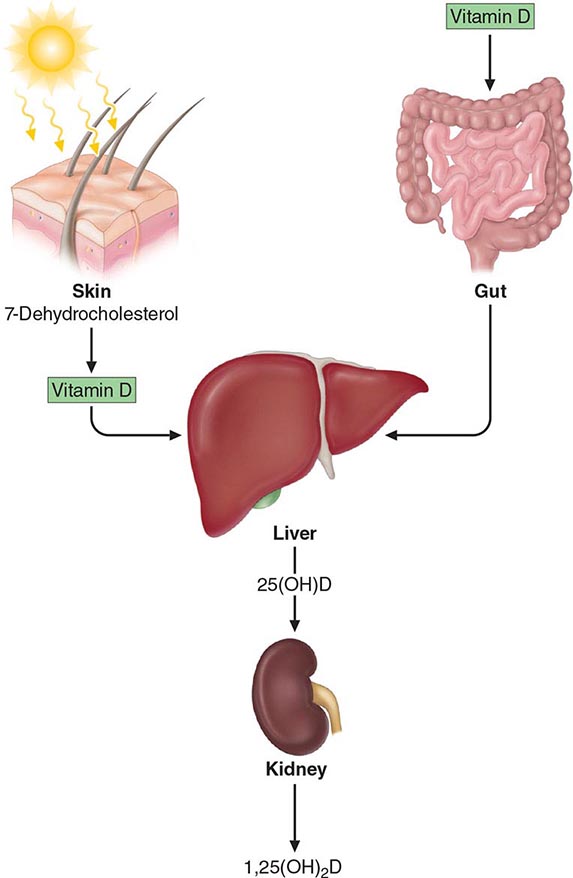
FIGURE 423-4 Vitamin D synthesis and activation. Vitamin D is synthesized in the skin in response to ultraviolet radiation and also is absorbed from the diet. It is then transported to the liver, where it undergoes 25-hydroxylation. This metabolite is the major circulating form of vitamin D. The final step in hormone activation, 1α-hydroxylation, occurs in the kidney.
The second hydroxylation, required for the formation of the mature hormone, occurs in the kidney (Fig. 423-5). The 25-hydroxyvitamin D-1α-hydroxylase is a tightly regulated cytochrome P450–like mixed-function oxidase expressed in the proximal convoluted tubule cells of the kidney. PTH and hypophosphatemia are the major inducers of this microsomal enzyme, whereas calcium, FGF23, and the enzyme’s product, 1,25(OH)2D, repress it. The 25-hydroxyvitamin D-1α-hydroxylase is also present in epidermal keratinocytes, but keratinocyte production of 1,25(OH)2D is not thought to contribute to circulating levels of this hormone. In addition to being present in the trophoblastic layer of the placenta, the 1α-hydroxylase is produced by macrophages associated with granulomas and lymphomas. In these latter pathologic states, the activity of the enzyme is induced by interferon γ and TNF-α but is not regulated by calcium or 1,25(OH)2D; therefore, hypercalcemia, associated with elevated levels of 1,25(OH)2D, may be observed. Treatment of sarcoidosis-associated hypercalcemia with glucocorticoids, ketoconazole, or chloroquine reduces 1,25(OH)2D production and effectively lowers serum calcium. In contrast, chloroquine has not been shown to lower the elevated serum 1,25(OH)2D levels in patients with lymphoma.
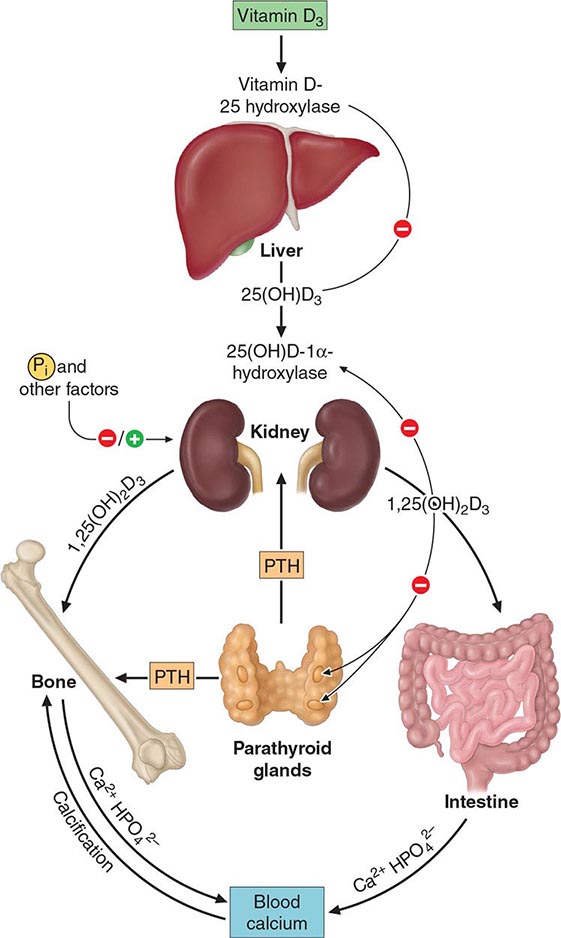
FIGURE 423-5 Schematic representation of the hormonal control loop for vitamin D metabolism and function. A reduction in the serum calcium below ~2.2 mmol/L (8.8 mg/dL) prompts a proportional increase in the secretion of parathyroid hormone (PTH) and so mobilizes additional calcium from the bone. PTH promotes the synthesis of 1,25(OH)2D in the kidney, which in turn stimulates the mobilization of calcium from bone and intestine and regulates the synthesis of PTH by negative feedback.
The major pathway for inactivation of vitamin D metabolites is an additional hydroxylation step by the vitamin D 24-hydroxylase, an enzyme that is expressed in most tissues. 1,25(OH)2D is the major inducer of this enzyme; therefore, this hormone promotes its own inactivation, thereby limiting its biologic effects. Mutations of the gene encoding this enzyme (CYP24A1) can lead to infantile hypercalcemia and, in those less severely affected, long-standing hypercalciuria, nephrocalcinosis, and nephrolithiasis.
Polar metabolites of 1,25(OH)2D are secreted into the bile and reabsorbed via the enterohepatic circulation. Impairment of this recirculation, which is seen with diseases of the terminal ileum, leads to accelerated losses of vitamin D metabolites.
ACTIONS OF 1,25(OH)2D
1,25(OH)2D mediates its biologic effects by binding to a member of the nuclear receptor superfamily, the vitamin D receptor (VDR). This receptor belongs to the subfamily that includes the thyroid hormone receptors, the retinoid receptors, and the peroxisome proliferator–activated receptors; however, in contrast to the other members of this subfamily, only one VDR isoform has been isolated. The VDR binds to target DNA sequences as a heterodimer with the retinoid × receptor, recruiting a series of coactivators that modify chromatin and approximate the VDR to the basal transcriptional apparatus, resulting in the induction of target gene expression. The mechanism of transcriptional repression by the VDR varies with different target genes but has been shown to involve either interference with the action of activating transcription factors or the recruitment of novel proteins to the VDR complex, resulting in transcriptional repression.
The affinity of the VDR for 1,25(OH)2D is approximately three orders of magnitude higher than that for other vitamin D metabolites. In normal physiologic circumstances, these other metabolites are not thought to stimulate receptor-dependent actions. However, in states of vitamin D toxicity, the markedly elevated levels of 25(OH)D may lead to hypercalcemia by interacting directly with the VDR and by displacing 1,25(OH)2D from vitamin D–binding protein, resulting in increased bioavailability of the active hormone.
The VDR is expressed in a wide range of cells and tissues. The molecular actions of 1,25(OH)2D have been studied most extensively in tissues involved in the regulation of mineral ion homeostasis. This hormone is a major inducer of calbindin 9K, a calcium-binding protein expressed in the intestine, which is thought to play an important role in the active transport of calcium across the enterocyte. The two major calcium transporters expressed by intestinal epithelia, TRPV5 and TRPV6 (transient receptor potential vanilloid), are also vitamin D responsive. By inducing the expression of these and other genes in the small intestine, 1,25(OH)2D increases the efficiency of intestinal calcium absorption, and it also has been shown to have several important actions in the skeleton. The VDR is expressed in osteoblasts and regulates the expression of several genes in this cell. These genes include the bone matrix proteins osteocalcin and osteopontin, which are upregulated by 1,25(OH)2D, in addition to type I collagen, which is transcriptionally repressed by 1,25(OH)2D. Both 1,25(OH)2D and PTH induce the expression of RANK ligand, which promotes osteoclast differentiation and increases osteoclast activity, by binding to RANK on osteoclast progenitors and mature osteoclasts. This is the mechanism by which 1,25(OH)2D induces bone resorption. However, the skeletal features associated with VDR-knockout mice (rickets, osteomalacia) are largely corrected by increasing calcium and phosphorus intake, underscoring the importance of vitamin D action in the gut.
The VDR is expressed in the parathyroid gland, and 1,25(OH)2D has been shown to have antiproliferative effects on parathyroid cells and to suppress the transcription of the PTH gene. These effects of 1,25(OH)2D on the parathyroid gland are an important part of the rationale for current therapies directed at preventing and treating hyperparathyroidism associated with renal insufficiency.
The VDR is also expressed in tissues and organs that do not play a role in mineral ion homeostasis. Notable in this respect is the observation that 1,25(OH)2D has an antiproliferative effect on several cell types, including keratinocytes, breast cancer cells, and prostate cancer cells. The effects of 1,25(OH)2D and the VDR on keratinocytes are particularly intriguing. Alopecia is seen in humans and mice with mutant VDRs but is not a feature of vitamin D deficiency; thus, the effects of the VDR on the hair follicle are ligand-independent.
VITAMIN D DEFICIENCY
The mounting concern about the relationship between solar exposure and the development of skin cancer has led to increased reliance on dietary sources of vitamin D. Although the prevalence of vitamin D deficiency varies, the third National Health and Nutrition Examination Survey (NHANES III) revealed that vitamin D deficiency is prevalent throughout the United States. The clinical syndrome of vitamin D deficiency can be a result of deficient production of vitamin D in the skin, lack of dietary intake, accelerated losses of vitamin D, impaired vitamin D activation, or resistance to the biologic effects of 1,25(OH)2D (Table 423-6). The elderly and nursing home residents are particularly at risk for vitamin D deficiency, since both the efficiency of vitamin D synthesis in the skin and the absorption of vitamin D from the intestine decline with age. Similarly, intestinal malabsorption of dietary fats and short bowel syndrome, including that associated with intestinal bypass surgery, can lead to vitamin D deficiency. This is further exacerbated in the presence of terminal ileal disease, which results in impaired enterohepatic circulation of vitamin D metabolites. In addition to intestinal diseases, accelerated inactivation of vitamin D metabolites can be seen with drugs that induce hepatic cytochrome P450 mixed-function oxidases such as barbiturates, phenytoin, and rifampin. Impaired 25-hydroxylation, associated with severe liver disease or isoniazid, is an uncommon cause of vitamin D deficiency. A mutation in the gene responsible for 25-hydroxylation has been identified in one kindred. Impaired 1α-hydroxylation is prevalent in the population with profound renal dysfunction due to an increase in circulating FGF23 levels and a decrease in functional renal mass. Thus, therapeutic interventions should be considered in patients whose creatinine clearance is <0.5 mL/s (30 mL/min). Mutations in the renal 1α-hydroxylase are the basis for the genetic disorder, pseudovitamin D–deficiency rickets. This autosomal recessive disorder presents with the syndrome of vitamin D deficiency in the first year of life. Patients present with growth retardation, rickets, and hypocalcemic seizures. Serum 1,25(OH)2D levels are low despite normal 25(OH)D levels and elevated PTH levels. Treatment with vitamin D metabolites that do not require 1α-hydroxylation results in disease remission, although lifelong therapy is required. A second autosomal recessive disorder, hereditary vitamin D–resistant rickets, a consequence of vitamin D receptor mutations, is a greater therapeutic challenge. These patients present in a similar fashion during the first year of life, but alopecia often accompanies the disorder, demonstrating a functional role of the VDR in postnatal hair regeneration. Serum levels of 1,25(OH)2D are dramatically elevated in these individuals both because of increased production due to stimulation of 1α-hydroxylase activity as a consequence of secondary hyperparathyroidism and because of impaired inactivation, since induction of the 24-hydroxylase by 1,25(OH)2D requires an intact VDR. Because the receptor mutation results in hormone resistance, daily calcium and phosphorus infusions may be required to bypass the defect in intestinal mineral ion absorption.
|
CAUSES OF IMPAIRED VITAMIN D ACTION |
Regardless of the cause, the clinical manifestations of vitamin D deficiency are largely a consequence of impaired intestinal calcium absorption. Mild to moderate vitamin D deficiency is asymptomatic, whereas long-standing vitamin D deficiency results in hypocalcemia accompanied by secondary hyperparathyroidism, impaired mineralization of the skeleton (osteopenia on x-ray or decreased bone mineral density), and proximal myopathy. Vitamin D deficiency also has been shown to be associated with an increase in overall mortality, including cardiovascular causes. In the absence of an intercurrent illness, the hypocalcemia associated with long-standing vitamin D deficiency rarely presents with acute symptoms of hypocalcemia such as numbness, tingling, and seizures. However, the concurrent development of hypomagnesemia, which impairs parathyroid function, or the administration of potent bisphosphonates, which impair bone resorption, can lead to acute symptomatic hypocalcemia in vitamin D–deficient individuals.
Rickets and Osteomalacia In children, before epiphyseal fusion, vitamin D deficiency results in growth retardation associated with an expansion of the growth plate known as rickets. Three layers of chondrocytes are present in the normal growth plate: the reserve zone, the proliferating zone, and the hypertrophic zone. Rickets associated with impaired vitamin D action is characterized by expansion of the hypertrophic chondrocyte layer. The proliferation and differentiation of the chondrocytes in the rachitic growth plate are normal, and the expansion of the growth plate is a consequence of impaired apoptosis of the late hypertrophic chondrocytes, an event that precedes replacement of these cells by osteoblasts during endochondral bone formation. Investigations in murine models demonstrate that hypophosphatemia, which in vitamin D deficiency is a consequence of secondary hyperparathyroidism, is a key etiologic factor in the development of the rachitic growth plate.
The hypocalcemia and hypophosphatemia that accompany vitamin D deficiency result in impaired mineralization of bone matrix proteins, a condition known as osteomalacia. Osteomalacia is also a feature of long-standing hypophosphatemia, which may be a consequence of renal phosphate wasting or chronic use of etidronate or phosphate-binding antacids. This hypomineralized matrix is biomechanically inferior to normal bone; as a result, patients with vitamin D deficiency are prone to bowing of weight-bearing extremities and skeletal fractures. Vitamin D and calcium supplementation have been shown to decrease the incidence of hip fracture among ambulatory nursing home residents in France, suggesting that undermineralization of bone contributes significantly to morbidity in the elderly. Proximal myopathy is a striking feature of severe vitamin D deficiency both in children and in adults. Rapid resolution of the myopathy is observed upon vitamin D treatment.
Although vitamin D deficiency is the most common cause of rickets and osteomalacia, many disorders lead to inadequate mineralization of the growth plate and bone. Calcium deficiency without vitamin D deficiency, the disorders of vitamin D metabolism previously discussed, and hypophosphatemia can all lead to inefficient mineralization. Even in the presence of normal calcium and phosphate levels, chronic acidosis and drugs such as bisphosphonates can lead to osteomalacia. The inorganic calcium/phosphate mineral phase of bone cannot form at low pH, and bisphosphonates bind to and prevent mineral crystal growth. Because alkaline phosphatase is necessary for normal mineral deposition, probably because the enzyme can hydrolyze inhibitors of mineralization such as inorganic pyrophosphate, genetic inactivation of the alkaline phosphatase gene (hereditary hypophosphatasia) also can lead to osteomalacia in the setting of normal calcium and phosphate levels.
Diagnosis of Vitamin D Deficiency, Rickets, and Osteomalacia The most specific screening test for vitamin D deficiency in otherwise healthy individuals is a serum 25(OH)D level. Although the normal ranges vary, levels of 25(OH)D <37 nmol/L (<15 ng/mL) are associated with increasing PTH levels and lower bone density. The Institute of Medicine has defined vitamin D sufficiency as a vitamin D level >50 nmol/L (>20 ng/mL), although higher levels may be required to optimize intestinal calcium absorption in the elderly and those with underlying disease states. Vitamin D deficiency leads to impaired intestinal absorption of calcium, resulting in decreased serum total and ionized calcium values. This hypocalcemia results in secondary hyperparathyroidism, a homeostatic response that initially maintains serum calcium levels at the expense of the skeleton. Due to the PTH-induced increase in bone turnover, alkaline phosphatase levels are often increased. In addition to increasing bone resorption, PTH decreases urinary calcium excretion while promoting phosphaturia. This results in hypophosphatemia, which exacerbates the mineralization defect in the skeleton. With prolonged vitamin D deficiency resulting in osteomalacia, calcium stores in the skeleton become relatively inaccessible, since osteoclasts cannot resorb unmineralized osteoid, and frank hypocalcemia ensues. Because PTH is a major stimulus for the renal 25(OH)D 1α-hydroxylase, there is increased synthesis of the active hormone, 1,25(OH)2D. Paradoxically, levels of this hormone are often normal in severe vitamin D deficiency. Therefore, measurements of 1,25(OH)2D are not accurate reflections of vitamin D stores and should not be used to diagnose vitamin D deficiency in patients with normal renal function.
Radiologic features of vitamin D deficiency in children include a widened, expanded growth plate that is characteristic of rickets. These findings not only are apparent in the long bones but also are present at the costochondral junction, where the expansion of the growth plate leads to swellings known as the “rachitic rosary.” Impairment of intramembranous bone mineralization leads to delayed fusion of the calvarial sutures and a decrease in the radiopacity of cortical bone in the long bones. If vitamin D deficiency occurs after epiphyseal fusion, the main radiologic finding is a decrease in cortical thickness and relative radiolucency of the skeleton. A specific radiologic feature of osteomalacia, whether associated with phosphate wasting or vitamin D deficiency, is pseudofractures, or Looser’s zones. These are radiolucent lines that occur where large arteries are in contact with the underlying skeletal elements; it is thought that the arterial pulsations lead to the radiolucencies. As a result, these pseudofractures are usually a few millimeters wide, are several centimeters long, and are seen particularly in the scapula, the pelvis, and the femoral neck.
|
TREATMENT |
VITAMIN D DEFICIENCY |
Based on the Institute of Medicine 2010 report, the recommended daily intake of vitamin D is 600 IU from 1 to 70 years of age, and 800 IU for those over 70. Based on the observation that 800 IU of vitamin D, with calcium supplementation, decreases the risk of hip fractures in elderly women, this higher dose is thought to be an appropriate daily intake for prevention of vitamin D deficiency in adults. The safety margin for vitamin D is large, and vitamin D toxicity usually is observed only in patients taking doses in the range of 40,000 IU daily. Treatment of vitamin D deficiency should be directed at the underlying disorder, if possible, and also should be tailored to the severity of the condition. Vitamin D should always be repleted in conjunction with calcium supplementation because most of the consequences of vitamin D deficiency are a result of impaired mineral ion homeostasis. In patients in whom 1α-hydroxylation is impaired, metabolites that do not require this activation step are the treatment of choice. They include 1,25(OH)2D3 (calcitriol [Rocaltrol], 0.25–0.5 μg/d) and 1α-hydroxyvitamin D2 (Hectorol, 2.5–5 μg/d). If the pathway required for activation of vitamin D is intact, severe vitamin D deficiency can be treated with pharmacologic repletion initially (50,000 IU weekly for 3–12 weeks), followed by maintenance therapy (800 IU daily). Pharmacologic doses may be required for maintenance therapy in patients who are taking medications, such as barbiturates or phenytoin, that accelerate metabolism of or cause resistance to 1,25(OH)2D. Calcium supplementation should include 1.5–2 g/d of elemental calcium. Normocalcemia is usually observed within 1 week of the institution of therapy, although increases in PTH and alkaline phosphatase levels may persist for 3–6 months. The most efficacious methods to monitor treatment and resolution of vitamin D deficiency are serum and urinary calcium measurements. In patients who are vitamin D replete and are taking adequate calcium supplementation, the 24-h urinary calcium excretion should be in the range of 100–250 mg/24 h. Lower levels suggest problems with adherence to the treatment regimen or with absorption of calcium or vitamin D supplements. Levels >250 mg/24 h predispose to nephrolithiasis and should lead to a reduction in vitamin D dosage and/or calcium supplementation.
424 |
Disorders of the Parathyroid Gland and Calcium Homeostasis |
The four parathyroid glands are located posterior to the thyroid gland. They produce parathyroid hormone (PTH), which is the primary regulator of calcium physiology. PTH acts directly on bone, where it induces calcium release; on the kidney, where it enhances calcium reabsorption in the distal tubules; and in the proximal renal tubules, where it synthesizes 1,25-dihydroxyvitamin D (1,25[OH]2D), a hormone that increases gastrointestinal calcium absorption. Serum PTH levels are tightly regulated by a negative feedback loop. Calcium, acting through the calcium-sensing receptor, and vitamin D, acting through its nuclear receptor, reduce PTH release and synthesis. Additional evidence indicates that fibroblast growth factor 23 (FGF23), a phosphaturic hormone, can suppress PTH secretion. Understanding the hormonal pathways that regulate calcium levels and bone metabolism is essential for effective diagnosis and management of a wide array of hyper- and hypocalcemic disorders.
Hyperparathyroidism, characterized by excess production of PTH, is a common cause of hypercalcemia and is usually the result of autonomously functioning adenomas or hyperplasia. Surgery for this disorder is highly effective and has been shown to reverse some of the deleterious effects of long-standing PTH excess on bone density. Humoral hypercalcemia of malignancy is also common and is usually due to the overproduction of parathyroid hormone–related peptide (PTHrP) by cancer cells. The similarities in the biochemical characteristics of hyperparathyroidism and humoral hypercalcemia of malignancy, first noted by Albright in 1941, are now known to reflect the actions of PTH and PTHrP through the same G protein–coupled PTH/PTHrP receptor.
The genetic basis of multiple endocrine neoplasia (MEN) types 1 and 2, familial hypocalciuric hypercalcemia (FHH), different forms of pseudohypoparathyroidism, Jansen’s syndrome, disorders of vitamin D synthesis and action, and the molecular events associated with parathyroid gland neoplasia have provided new insights into the regulation of calcium homeostasis. PTH and possibly some of its analogues are promising therapeutic agents for the treatment of postmenopausal or senile osteoporosis, and calcimimetic agents, which activate the calcium-sensing receptor, have provided new approaches for PTH suppression.
PARATHYROID HORMONE
PHYSIOLOGY
The primary function of PTH is to maintain the extracellular fluid (ECF) calcium concentration within a narrow normal range. The hormone acts directly on bone and kidney and indirectly on the intestine through its effects on synthesis of 1,25(OH)2D to increase serum calcium concentrations; in turn, PTH production is closely regulated by the concentration of serum ionized calcium. This feedback system is the critical homeostatic mechanism for maintenance of ECF calcium. Any tendency toward hypocalcemia, as might be induced by calcium- or vitamin D–deficient diets, is counteracted by an increased secretion of PTH. This in turn (1) increases the rate of dissolution of bone mineral, thereby increasing the flow of calcium from bone into blood; (2) reduces the renal clearance of calcium, returning more of the calcium and phosphate filtered at the glomerulus into ECF; and (3) increases the efficiency of calcium absorption in the intestine by stimulating the production of 1,25(OH)2D. Immediate control of blood calcium is due to PTH effects on bone and, to a lesser extent, on renal calcium clearance. Maintenance of steady-state calcium balance, on the other hand, probably results from the effects of 1,25(OH)2D on calcium absorption (Chap. 423). The renal actions of the hormone are exerted at multiple sites and include inhibition of phosphate transport (proximal tubule), augmentation of calcium reabsorption (distal tubule), and stimulation of the renal 25(OH)D-1α-hydroxylase. As much as 12 mmol (500 mg) of calcium is transferred between the ECF and bone each day (a large amount in relation to the total ECF calcium pool), and PTH has a major effect on this transfer. The homeostatic role of the hormone can preserve calcium concentration in blood at the cost of bone demineralization.
PTH has multiple actions on bone, some direct and some indirect. PTH-mediated changes in bone calcium release can be seen within minutes. The chronic effects of PTH are to increase the number of bone cells, both osteoblasts and osteoclasts, and to increase the remodeling of bone; these effects are apparent within hours after the hormone is given and persist for hours after PTH is withdrawn. Continuous exposure to elevated PTH (as in hyperparathyroidism or long-term infusions in animals) leads to increased osteoclast-mediated bone resorption. However, the intermittent administration of PTH, elevating hormone levels for 1–2 h each day, leads to a net stimulation of bone formation rather than bone breakdown. Striking increases, especially in trabecular bone in the spine and hip, have been reported with the use of PTH in combination with estrogen. PTH(1–34) as monotherapy caused a highly significant reduction in fracture incidence in a worldwide placebo-controlled trial.
Osteoblasts (or stromal cell precursors), which have PTH/PTHrP receptors, are crucial to this bone-forming effect of PTH; osteoclasts, which mediate bone breakdown, lack such receptors. PTH-mediated stimulation of osteoclasts is indirect, acting in part through cytokines released from osteoblasts to activate osteoclasts; in experimental studies of bone resorption in vitro, osteoblasts must be present for PTH to activate osteoclasts to resorb bone (Chap. 423).
STRUCTURE
PTH is an 84-amino-acid single-chain peptide. The amino-terminal portion, PTH(1–34), is highly conserved and is critical for the biologic actions of the molecule. Modified synthetic fragments of the amino-terminal sequence as small as PTH(1–11) are sufficient to activate the PTH/PTHrP receptor (see below). The carboxyl-terminal region of the full-length PTH(1–84) molecule also can bind to a separate binding protein/receptor (cPTH-R), but this receptor has been incompletely characterized. Fragments shortened at the amino-terminus possibly by binding to cPTH-R can reduce, directly or indirectly, some of the biologic actions of full-length PTH(1–84) and of PTH(1–34).
BIOSYNTHESIS, SECRETION, AND METABOLISM
Synthesis Parathyroid cells have multiple methods of adapting to increased needs for PTH production. Most rapid (within minutes) is secretion of preformed hormone in response to hypocalcemia. Second, within hours, PTH mRNA expression is induced by sustained hypocalcemia. Finally, protracted challenge leads within days to cellular replication to increase parathyroid gland mass.
PTH is initially synthesized as a larger molecule (preproparathyroid hormone, consisting of 115 amino acids). After a first cleavage step to remove the “pre” sequence of 25 amino acid residues, a second cleavage step removes the “pro” sequence of 6 amino acid residues before secretion of the mature peptide comprising 84 residues. Mutations in the preprotein region of the gene can cause hypoparathyroidism by interfering with hormone synthesis, transport, or secretion.
Transcriptional suppression of the PTH gene by calcium is nearly maximal at physiologic calcium concentrations. Hypocalcemia increases transcriptional activity within hours. 1,25(OH)2D strongly suppresses PTH gene transcription. In patients with renal failure, IV administration of supraphysiologic levels of 1,25(OH)2D or analogues of this active metabolite can dramatically suppress PTH overproduction, which is sometimes difficult to control due to severe secondary hyperparathyroidism. Regulation of proteolytic destruction of preformed hormone (posttranslational regulation of hormone production) is an important mechanism for mediating rapid (within minutes) changes in hormone availability. High calcium increases and low calcium inhibit the proteolytic destruction of stored hormone.
Regulation of PTH Secretion PTH secretion increases steeply to a maximum value of about five times the basal rate of secretion as the calcium concentration falls from normal to the range of 1.9–2.0 mmol/L (7.6–8.0 mg/dL; measured as total calcium). However, the ionized fraction of blood calcium is the important determinant of hormone secretion. Severe intracellular magnesium deficiency impairs PTH secretion (see below).
ECF calcium controls PTH secretion by interaction with a calcium-sensing receptor (CaSR), a G protein–coupled receptor (GPCR) for which Ca2+ ions act as the primary ligand (see below). This receptor is a member of a distinctive subgroup of the GPCR superfamily that mediates its actions through the alpha-subunits of two related signaling G proteins, namely Gq and G11, and is characterized by a large extracellular domain suitable for “clamping” the small-molecule ligand. Stimulation of the CaSR by high calcium levels suppresses PTH secretion. The CaSR is present in parathyroid glands and the calcitonin-secreting cells of the thyroid (C cells), as well as in multiple other sites, including brain and kidney. Genetic evidence has revealed a key biologic role for the CaSR in parathyroid gland responsiveness to calcium and in renal calcium clearance. Heterozygous loss-of-function mutations in CaSR cause the syndrome of FHH, in which the blood calcium abnormality resembles that observed in hyperparathyroidism but with hypocalciuria; two more recently defined variants of FHH, FHH2 and FHH3, are caused either by heterozygous mutations in G11, one of the signaling proteins downstream of the CaSR, or by heterozygous mutations in AP2S1. Homozygous loss-of-function mutations in the CaSR are the cause of severe neonatal hyperparathyroidism, a disorder that can be lethal if not treated within the first days of life. On the other hand, heterozygous gain-of-function mutations cause a form of hypocalcemia resembling hypoparathyroidism (see below).
Metabolism The secreted form of PTH is indistinguishable by immunologic criteria and by molecular size from the 84-amino-acid peptide (PTH[1–84]) extracted from glands. However, much of the immunoreactive material found in the circulation is smaller than the extracted or secreted hormone. The principal circulating fragments of immunoreactive hormone lack a portion of the critical amino-terminal sequence required for biologic activity and, hence, are biologically inactive fragments (so-called middle and carboxyl-terminal fragments). Much of the proteolysis of the hormone occurs in the liver and kidney. Peripheral metabolism of PTH does not appear to be regulated by physiologic states (high versus low calcium, etc.); hence, peripheral metabolism of hormone, although responsible for rapid clearance of secreted hormone, appears to be a high-capacity, metabolically invariant catabolic process.
The rate of clearance of the secreted 84-amino-acid peptide from blood is more rapid than the rate of clearance of the biologically inactive fragment(s) corresponding to the middle and carboxyl-terminal regions of PTH. Consequently, the interpretation of results obtained with earlier PTH radioimmunoassays was influenced by the nature of the peptide fragments detected by the antibodies.
Although the problems inherent in PTH measurements have been largely circumvented by use of double-antibody immunometric assays, it is now known that some of these assays detect, besides the intact molecule, large amino-terminally truncated forms of PTH, which are present in normal and uremic individuals in addition to PTH(1–84). The concentration of these fragments relative to that of intact PTH(1–84) is higher with induced hypercalcemia than in eucalcemic or hypocalcemic conditions and is higher in patients with impaired renal function. PTH(7–84) has been identified as a major component of these amino-terminally truncated fragments. Growing evidence suggests that the PTH(7–84) (and probably related amino-terminally truncated fragments) can act, through yet undefined mechanisms, as an inhibitor of PTH action and may be of clinical significance, particularly in patients with chronic kidney disease. In this group of patients, efforts to prevent secondary hyperparathyroidism by a variety of measures (vitamin D analogues, higher calcium intake, higher dialysate calcium, phosphate-lowering strategies, and calcimetic drugs) can lead to oversuppression of the parathyroid glands since some amino-terminally truncated PTH fragments, such as PTH(7–84), react in many immunometric PTH assays (now termed second-generation assays; see below under “Diagnosis”), thus overestimating the levels of biologically active, intact PTH. Such excessive parathyroid gland suppression in chronic kidney disease can lead to adynamic bone disease (see below), which has been associated with further impaired growth in children and increased bone fracture rates in adults, and can furthermore lead to significant hypercalcemia. The measurement of PTH with newer third-generation immunoassays, which use detection antibodies directed against extreme amino-terminal PTH epitopes and thus detect only full-length PTH(1–84), may provide some advantage to prevent bone disease in chronic kidney disease.
PARATHYROID HORMONE–RELATED PROTEIN (PTHrP)
PTHrP is responsible for most instances of humoral hypercalcemia of malignancy (Chap. 121), a syndrome that resembles primary hyperparathyroidism but without elevated PTH levels. Most cell types normally produce PTHrP, including brain, pancreas, heart, lung, mammary tissue, placenta, endothelial cells, and smooth muscle. In fetal animals, PTHrP directs transplacental calcium transfer, and high concentrations of PTHrP are produced in mammary tissue and secreted into milk, but the biologic significance of the very high concentrations of this hormone in breast milk is unknown. PTHrP also plays an essential role in endochondral bone formation and in branching morphogenesis of the breast, and possibly in uterine contraction and other biologic functions.
PTH and PTHrP, although products of different genes, exhibit considerable functional and structural homology (Fig. 424-1) and have evolved from a shared ancestral gene. The structure of the gene encoding human PTHrP, however, is more complex than that of PTH, containing multiple additional exons, which can undergo alternate splicing patterns during formation of the mature mRNA. Protein products of 139, 141, and 173 amino acids are produced, and other molecular forms may result from tissue-specific degradation at accessible internal cleavage sites. The biologic roles of these various molecular species and the nature of the circulating forms of PTHrP are unclear. In fact, it is uncertain whether PTHrP circulates at any significant level in adults. As a paracrine factor, PTHrP may be produced, act, and be destroyed locally within tissues. In adults, PTHrP appears to have little influence on calcium homeostasis, except in disease states, when large tumors, especially of the squamous cell type as well as renal cell carcinomas, lead to massive overproduction of the hormone and hypercalcemia.
FIGURE 424-1 Schematic diagram to illustrate similarities and differences in structure of human parathyroid hormone (PTH) and human PTH-related peptide (PTHrP). Close structural (and functional) homology exists between the first 30 amino acids of hPTH and hPTHrP. The PTHrP sequence may be ≥144 amino acid residues in length. PTH is only 84 residues long; after residue 30, there is little structural homology between the two. Dashed lines in the PTHrP sequence indicate identity; underlined residues, although different from those of PTH, still represent conservative changes (charge or polarity preserved). Ten amino acids are identical, and a total of 20 of 30 are homologues.
PTH AND PTHrP HORMONE ACTION
Both PTH and PTHrP bind to and activate the PTH/PTHrP receptor. The PTH/PTHrP receptor (also known as the PTH-1 receptor, PTH1R) belongs to a subfamily of GPCRs that includes the receptors for calcitonin, glucagon, secretin, vasoactive intestinal peptide, and other peptides. Although both ligands activate the PTH1R, the two peptides induce distinct responses in the receptor, which explains how a single receptor without isoforms can serve two biologic roles. The extracellular regions of the receptor are involved in hormone binding, and the intracellular domains, after hormone activation, bind G protein subunits to transduce hormone signaling into cellular responses through the stimulation of second messenger formation. A second receptor that binds PTH, originally termed the PTH-2 receptor (PTH2R), is primarily expressed in brain, pancreas, and testis. Different mammalian PTH1Rs respond equivalently to PTH and PTHrP, at least when tested with traditional assays, whereas only the human PTH2R responds efficiently to PTH (but not to PTHrP). PTH2Rs from other species show little or no stimulation of second-messenger formation in response to PTH or PTHrP. The endogenous ligand of the PTH2R was shown to be a hypothalamic peptide referred to as tubular infundibular peptide of 39 residues, TIP39, that is distantly related to PTH and PTHrP. The PTH1R and the PTH2R can be traced backward in evolutionary time to fish; in fact, the zebrafish genome contains, in addition to the PTH1R and the PTH2R orthologs, a third receptor, the PTH3R, that is more closely related to the fish PTH1R than to the fish PTH2R. The evolutionary conservation of structure and function suggests important biologic roles for these receptors, even in fish, which lack discrete parathyroid glands but produce two molecules that are closely related to mammalian PTH.
Studies using the cloned PTH1R confirm that it can be coupled to more than one G protein and second-messenger pathway, apparently explaining the multiplicity of pathways stimulated by PTH. Activation of protein kinases (A and C) and calcium transport channels is associated with a variety of hormone-specific tissue responses. These responses include inhibition of phosphate and bicarbonate transport, stimulation of calcium transport, and activation of renal 1α-hydroxylase in the kidney. The responses in bone include effects on collagen synthesis, alkaline phosphatase, ornithine decarboxylase, citrate decarboxylase, and glucose-6-phosphate dehydrogenase activities; phospholipid synthesis; and calcium and phosphate transport. Ultimately, these biochemical events lead to an integrated hormonal response in bone turnover and calcium homeostasis. PTH also activates Na+/Ca2+ exchangers at renal distal tubular sites and stimulates translocation of preformed calcium transport channels, moving them from the interior to the apical surface to increase tubular uptake of calcium. PTH-dependent stimulation of phosphate excretion (reducing reabsorption—the opposite effect from actions on calcium in the kidney) involves the downregulation of two sodium-dependent phosphate co-transporters, NPT2a and NPT2c, and their expression at the apical membrane, thereby reducing phosphate reabsorption in the proximal renal tubules. Similar mechanisms may be involved in other renal tubular transporters that are influenced by PTH. Recent studies reaffirm the critical linkage of blood phosphate lowering to net calcium entry into blood by PTH action and emphasize the participation of bone cells other than osteoclasts in the rapid calcium-elevating actions of PTH.
PTHrP exerts important developmental influences on fetal bone development and in adult physiology. A homozygous ablation of the gene encoding PTHrP (or disruption of the PTH1R gene) in mice causes a lethal phenotype in which animals are born with pronounced acceleration of chondrocyte maturation that resembles a lethal form of chondrodysplasia in humans that is caused by homozygous or compound heterozygous, inactivating PTH1R mutations (Fig. 424-2). Heterozygous PTH1R mutations in humans furthermore can be a cause of delayed tooth eruption, and mice that are heterozygous for ablation of the PTHrP gene display reduced mineral density consistent with osteoporosis. Experiments with these mouse models point to a hitherto unappreciated role of PTHrP as a paracrine/autocrine factor that modulates bone metabolism in adults as well as during bone development.
FIGURE 424-2 Dual role for the actions of the PTH/PTHrP receptor (PTH1R). Parathyroid hormone (PTH; endocrine-calcium homeostasis) and PTH-related peptide (PTHrP; paracrine–multiple tissue actions including growth plate cartilage in developing bone) use the single receptor for their disparate functions mediated by the amino-terminal 34 residues of either peptide. Other regions of both ligands interact with other receptors (not shown).
CALCITONIN
(See also Chap. 408) Calcitonin is a hypocalcemic peptide hormone that in several mammalian species acts as an indirect antagonist to the calcemic actions of PTH. Calcitonin seems to be of limited physiologic significance in humans, at least with regard to calcium homeostasis. It is of medical significance because of its role as a tumor marker in sporadic and hereditary cases of medullary carcinoma and its medical use as an adjunctive treatment in severe hypercalcemia and in Paget’s disease of bone.
The hypocalcemic activity of calcitonin is accounted for primarily by inhibition of osteoclast-mediated bone resorption and secondarily by stimulation of renal calcium clearance. These effects are mediated by receptors on osteoclasts and renal tubular cells. Calcitonin exerts additional effects through receptors present in the brain, the gastrointestinal tract, and the immune system. The hormone, for example, exerts analgesic effects directly on cells in the hypothalamus and related structures, possibly by interacting with receptors for related peptide hormones such as calcitonin gene–related peptide (CGRP) or amylin. Both of these ligands have specific high-affinity receptors that share considerable structural similarity with the PTH1R and can also bind to and activate calcitonin receptors. The calcitonin receptor shares considerable structural similarity with the PTH1R.
The thyroid is the major source of the hormone, and the cells involved in calcitonin synthesis arise from neural crest tissue. During embryogenesis, these cells migrate into the ultimobranchial body, derived from the last branchial pouch. In submammalian vertebrates, the ultimobranchial body constitutes a discrete organ, anatomically separate from the thyroid gland; in mammals, the ultimobranchial gland fuses with and is incorporated into the thyroid gland.
The naturally occurring calcitonins consist of a peptide chain of 32 amino acids. There is considerable sequence variability among species. Calcitonin from salmon, which is used therapeutically, is 10–100 times more potent than mammalian forms in lowering serum calcium.
There are two calcitonin genes, α and β; the transcriptional control of these genes is complex. Two different mRNA molecules are transcribed from the α gene; one is translated into the precursor for calcitonin, and the other message is translated into an alternative product, CGRP. CGRP is synthesized wherever the calcitonin mRNA is expressed (e.g., in medullary carcinoma of the thyroid). The β, or CGRP-2, gene is transcribed into the mRNA for CGRP in the central nervous system (CNS); this gene does not produce calcitonin, however. CGRP has cardiovascular actions and may serve as a neurotransmitter or play a developmental role in the CNS.
The circulating level of calcitonin in humans is lower than that in many other species. In humans, even extreme variations in calcitonin production do not change calcium and phosphate metabolism; no definite effects are attributable to calcitonin deficiency (totally thyroidectomized patients receiving only replacement thyroxine) or excess (patients with medullary carcinoma of the thyroid, a calcitonin-secreting tumor) (Chap. 408). Calcitonin has been a useful pharmacologic agent to suppress bone resorption in Paget’s disease (Chap. 426e) and osteoporosis (Chap. 425) and in the treatment of hypercalcemia of malignancy (see below). However, bisphosphonates are usually more effective, and the physiologic role, if any, of calcitonin in humans is uncertain. On the other hand, ablation of the calcitonin gene (combined because of the close proximity with ablation of the CGRP gene) in mice leads to reduced bone mineral density, suggesting that its biologic role in mammals is still not fully understood.
HYPERCALCEMIA
(See also Chap. 65) Hypercalcemia can be a manifestation of a serious illness such as malignancy or can be detected coincidentally by laboratory testing in a patient with no obvious illness. The number of patients recognized with asymptomatic hypercalcemia, usually hyperparathyroidism, increased in the late twentieth century.
Whenever hypercalcemia is confirmed, a definitive diagnosis must be established. Although hyperparathyroidism, a frequent cause of asymptomatic hypercalcemia, is a chronic disorder in which manifestations, if any, may be expressed only after months or years, hypercalcemia can also be the earliest manifestation of malignancy, the second most common cause of hypercalcemia in the adult. The causes of hypercalcemia are numerous (Table 424-1), but hyperparathyroidism and cancer account for 90% of all cases.
|
CLASSIFICATION OF CAUSES OF HYPERCALCEMIA |
Before undertaking a diagnostic workup, it is essential to be sure that true hypercalcemia, not a false-positive laboratory test, is present. A false-positive diagnosis of hypercalcemia is usually the result of inadvertent hemoconcentration during blood collection or elevation in serum proteins such as albumin. Hypercalcemia is a chronic problem, and it is cost-effective to obtain several serum calcium measurements; these tests need not be in the fasting state.
Clinical features are helpful in differential diagnosis. Hypercalcemia in an adult who is asymptomatic is usually due to primary hyperparathyroidism. In malignancy-associated hypercalcemia, the disease is usually not occult; rather, symptoms of malignancy bring the patient to the physician, and hypercalcemia is discovered during the evaluation. In such patients, the interval between detection of hypercalcemia and death, especially without vigorous treatment, is often <6 months. Accordingly, if an asymptomatic individual has had hypercalcemia or some manifestation of hypercalcemia such as kidney stones for more than 1 or 2 years, it is unlikely that malignancy is the cause. Nevertheless, differentiating primary hyperparathyroidism from occult malignancy can occasionally be difficult, and careful evaluation is required, particularly when the duration of the hypercalcemia is unknown. Hypercalcemia not due to hyperparathyroidism or malignancy can result from excessive vitamin D action, impaired metabolism of 1,25(OH)2D, high bone turnover from any of several causes, or renal failure (Table 424-1). Dietary history and a history of ingestion of vitamins or drugs are often helpful in diagnosing some of the less frequent causes. Immunometric PTH assays serve as the principal laboratory test in establishing the diagnosis.
Hypercalcemia from any cause can result in fatigue, depression, mental confusion, anorexia, nausea, vomiting, constipation, reversible renal tubular defects, increased urine output, a short QT interval in the electrocardiogram, and, in some patients, cardiac arrhythmias. There is a variable relation from one patient to the next between the severity of hypercalcemia and the symptoms. Generally, symptoms are more common at calcium levels >2.9–3.0 mmol/L (11.6–12.0 mg/dL), but some patients, even at this level, are asymptomatic. When the calcium level is >3.2 mmol/L (12.8 mg/dL), calcification in kidneys, skin, vessels, lungs, heart, and stomach occurs and renal insufficiency may develop, particularly if blood phosphate levels are normal or elevated due to impaired renal excretion. Severe hypercalcemia, usually defined as ≥3.7–4.5 mmol/L (14.8–18.0 mg/dL), can be a medical emergency; coma and cardiac arrest can occur.
Acute management of the hypercalcemia is usually successful. The type of treatment is based on the severity of the hypercalcemia and the nature of associated symptoms, as outlined below.
PRIMARY HYPERPARATHYROIDISM
Natural History and Incidence Primary hyperparathyroidism is a generalized disorder of calcium, phosphate, and bone metabolism due to an increased secretion of PTH. The elevation of circulating hormone usually leads to hypercalcemia and hypophosphatemia. There is great variation in the manifestations. Patients may present with multiple signs and symptoms, including recurrent nephrolithiasis, peptic ulcers, mental changes, and, less frequently, extensive bone resorption. However, with greater awareness of the disease and wider use of multiphasic screening tests, including measurements of blood calcium, the diagnosis is frequently made in patients who have no symptoms and minimal, if any, signs of the disease other than hypercalcemia and elevated levels of PTH. The manifestations may be subtle, and the disease may have a benign course for many years or a lifetime. This milder form of the disease is usually termed asymptomatic hyperparathyroidism. Rarely, hyperparathyroidism develops or worsens abruptly and causes severe complications such as marked dehydration and coma, so-called hypercalcemic parathyroid crisis.
The annual incidence of the disease is calculated to be as high as 0.2% in patients >60, with an estimated prevalence, including undiscovered asymptomatic patients, of ≥1%; some reports suggest the incidence may be declining. If confirmed, these changing estimates may reflect less frequent routine testing of serum calcium in recent years, earlier overestimates in incidence, or unknown factors. The disease has a peak incidence between the third and fifth decades but occurs in young children and in the elderly.
Etiology Parathyroid tumors are most often encountered as isolated adenomas without other endocrinopathy. They may also arise in hereditary syndromes such as MEN syndromes. Parathyroid tumors may also arise as secondary to underlying disease (excessive stimulation in secondary hyperparathyroidism, especially chronic renal failure) or after other forms of excessive stimulation such as lithium therapy. These etiologies are discussed below.
SOLITARY ADENOMAS A single abnormal gland is the cause in ~80% of patients; the abnormality in the gland is usually a benign neoplasm or adenoma and rarely a parathyroid carcinoma. Some surgeons and pathologists report that the enlargement of multiple glands is common; double adenomas are reported. In ~15% of patients, all glands are hyperfunctioning; chief cell parathyroid hyperplasia is usually hereditary and frequently associated with other endocrine abnormalities.
HEREDITARY SYNDROMES AND MULTIPLE PARATHYROID TUMORS Hereditary hyperparathyroidism can occur without other endocrine abnormalities but is usually part of a multiple endocrine neoplasia (MEN) syndrome (Chap. 408). MEN 1 (Wermer’s syndrome) consists of hyperparathyroidism and tumors of the pituitary and pancreas, often associated with gastric hypersecretion and peptic ulcer disease (Zollinger-Ellison syndrome). MEN 2A is characterized by pheochromocytoma and medullary carcinoma of the thyroid, as well as hyperparathyroidism; MEN 2B has additional associated features such as multiple neuromas but usually lacks hyperparathyroidism. Each of these MEN syndromes is transmitted in an apparent autosomal dominant manner, although, as noted below, the genetic basis of MEN 1 involves biallelic loss of a tumor suppressor.
The hyperparathyroidism jaw tumor (HPT-JT) syndrome occurs in families with parathyroid tumors (sometimes carcinomas) in association with benign jaw tumors. This disorder is caused by mutations in CDC73 (HRPT2), and mutations in this gene are also observed in parathyroid cancers. Some kindreds exhibit hereditary hyperparathyroidism without other endocrinopathies. This disorder is often termed nonsyndromic familial isolated hyperparathyroidism (FIHP). There is speculation that these families may be examples of variable expression of the other syndromes such as MEN 1, MEN 2, or the HPT-JT syndrome, but they may also have distinctive, still unidentified genetic causes.
Pathology Adenomas are most often located in the inferior parathyroid glands, but in 6–10% of patients, parathyroid adenomas may be located in the thymus, the thyroid, the pericardium, or behind the esophagus. Adenomas are usually 0.5–5 g in size but may be as large as 10–20 g (normal glands weigh 25 mg on average). Chief cells are predominant in both hyperplasia and adenoma. With chief cell hyperplasia, the enlargement may be so asymmetric that some involved glands appear grossly normal. If generalized hyperplasia is present, however, histologic examination reveals a uniform pattern of chief cells and disappearance of fat even in the absence of an increase in gland weight. Thus, microscopic examination of biopsy specimens of several glands is essential to interpret findings at surgery.
Parathyroid carcinoma is often not aggressive. Long-term survival without recurrence is common if at initial surgery the entire gland is removed without rupture of the capsule. Recurrent parathyroid carcinoma is usually slow-growing with local spread in the neck, and surgical correction of recurrent disease may be feasible. Occasionally, however, parathyroid carcinoma is more aggressive, with distant metastases (lung, liver, and bone) found at the time of initial operation. It may be difficult to appreciate initially that a primary tumor is carcinoma; increased numbers of mitotic figures and increased fibrosis of the gland stroma may precede invasion. The diagnosis of carcinoma is often made in retrospect. Hyperparathyroidism from a parathyroid carcinoma may be indistinguishable from other forms of primary hyperparathyroidism but is usually more severe clinically. A potential clue to the diagnosis is offered by the degree of calcium elevation. Calcium values of 3.5–3.7 mmol/L (14–15 mg/dL) are frequent with carcinoma and may alert the surgeon to remove the abnormal gland with care to avoid capsular rupture. Recent findings concerning the genetic basis of parathyroid carcinoma (distinct from that of benign adenomas) indicate the need, in these kindreds, for family screening (see below).
GENETIC DEFECTS ASSOCIATED WITH HYPERPARATHYROIDISM
![]() As in many other types of neoplasia, two fundamental types of genetic defects have been identified in parathyroid gland tumors: (1) overactivity of protooncogenes and (2) loss of function of tumor-suppressor genes. The former, by definition, can lead to uncontrolled cellular growth and function by activation (gain-of-function mutation) of a single allele of the responsible gene, whereas the latter requires loss of function of both allelic copies. Biallelic loss of function of a tumor-suppressor gene is usually characterized by a germline defect (all cells) and an additional somatic deletion/mutation in the tumor (Fig. 424-3).
As in many other types of neoplasia, two fundamental types of genetic defects have been identified in parathyroid gland tumors: (1) overactivity of protooncogenes and (2) loss of function of tumor-suppressor genes. The former, by definition, can lead to uncontrolled cellular growth and function by activation (gain-of-function mutation) of a single allele of the responsible gene, whereas the latter requires loss of function of both allelic copies. Biallelic loss of function of a tumor-suppressor gene is usually characterized by a germline defect (all cells) and an additional somatic deletion/mutation in the tumor (Fig. 424-3).
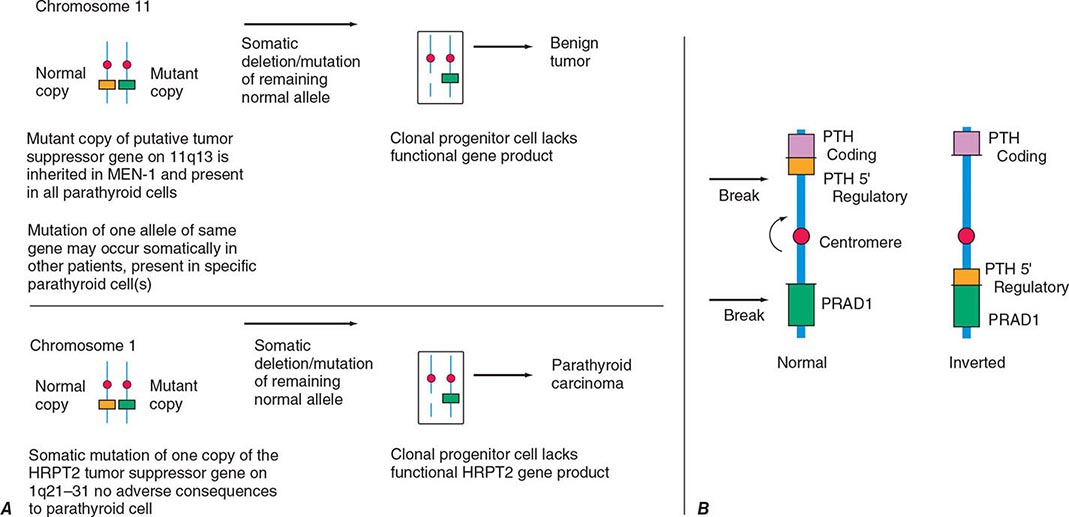
FIGURE 424-3 A. Schematic diagram indicating molecular events in tumor susceptibility. The patient with the hereditary abnormality (multiple endocrine neoplasia [MEN]) is envisioned as having one defective gene inherited from the affected parent on chromosome 11, but one copy of the normal gene is present from the other parent. In the monoclonal tumor (benign tumor), a somatic event, here partial chromosomal deletion, removes the remaining normal gene from a cell. In nonhereditary tumors, two successive somatic mutations must occur, a process that takes a longer time. By either pathway, the cell, deprived of growth-regulating influence from this gene, has unregulated growth and becomes a tumor. A different genetic locus also involving loss of a tumor-suppressor gene termed HRPT2 is involved in the pathogenesis of parathyroid carcinoma. (From A Arnold: J Clin Endocrine Metab 77:1108, 1993. Copyright 1993, The Endocrine Society.) B. Schematic illustration of the mechanism and consequences of gene rearrangement and overexpression of the PRAD1 protooncogene (pericentromeric inversion of chromosome 11) in parathyroid adenomas. The excessive expression of PRAD1 (a cell cycle control protein, cyclin D1) by the highly active parathyroid hormone (PTH) gene promoter in the parathyroid cell contributes to excess cellular proliferation. (From J Habener et al, in L DeGroot, JL Jameson [eds]: Endocrinology, 4th ed. Philadelphia, Saunders, 2001; with permission.)
Mutations in the MEN1 gene locus, encoding the protein MENIN, on chromosome 11q13 are responsible for causing MEN 1; the normal allele of this gene fits the definition of a tumor-suppressor gene. Inheritance of one mutated allele in this hereditary syndrome, followed by loss of the other allele via somatic cell mutation, leads to monoclonal expansion and tumor development. Also, in ~15–20% of sporadic parathyroid adenomas, both alleles of the MEN1 locus on chromosome 11 are somatically deleted, implying that the same defect responsible for MEN 1 can also cause the sporadic disease (Fig. 424-3A). Consistent with the Knudson hypothesis for two-step neoplasia in certain inherited cancer syndromes (Chap. 101e), the earlier onset of hyperparathyroidism in the hereditary syndromes reflects the need for only one mutational event to trigger the monoclonal outgrowth. In sporadic adenomas, typically occurring later in life, two different somatic events must occur before the MEN1 gene is silenced.
Other presumptive anti-oncogenes involved in hyperparathyroidism include a still unidentified gene mapped to chromosome 1p seen in 40% of sporadic parathyroid adenomas and a gene mapped to chromosome Xp11 in patients with secondary hyperparathyroidism and renal failure, who progressed to “tertiary” hyperparathyroidism, now known to reflect monoclonal outgrowths within previously hyperplastic glands.
A more complex pattern, still incompletely resolved, arises with genetic defects and carcinoma of the parathyroids. This appears to be due to biallelic loss of a functioning copy of a gene, HRPT2 (or CDC73), originally identified as the cause of the HPT-JT syndrome. Several inactivating mutations have been identified in HRPT2 (located on chromosome 1q21-31), which encodes a 531-amino-acid protein called parafibromin. The responsible genetic mutations in HRPT2 appear to be necessary, but not sufficient, for parathyroid cancer.
In general, the detection of additional genetic defects in these parathyroid tumor–related syndromes and the variations seen in phenotypic expression/penetrance indicate the multiplicity of the genetic factors responsible. Nonetheless, the ability to detect the presence of the major genetic contributors has greatly aided a more informed management of family members of patients identified in the hereditary syndromes such as MEN 1, MEN 2, and HPT-JT.
An important contribution from studies on the genetic origin of parathyroid carcinoma has been the realization that the mutations involve a different pathway than that involved with the benign gland enlargements. Unlike the pathogenesis of genetic alterations seen in colon cancer, where lesions evolve from benign adenomas to malignant disease by progressive genetic changes, the alterations commonly seen in most parathyroid cancers (HRPT2 mutations) are infrequently seen in sporadic parathyroid adenomas.
Abnormalities at the Rb gene were the first to be noted in parathyroid cancer. The Rb gene, a tumor-suppressor gene located on chromosome 13q14, was initially associated with retinoblastoma but has since been implicated in other neoplasias, including parathyroid carcinoma. Early studies implicated allelic deletions of the Rb gene in many parathyroid carcinomas and decreased or absent expression of the Rb protein. However, because there are often large deletions in chromosome 13 that include many genes in addition to the Rb locus (with similar findings in some pituitary carcinomas), it remains possible that other tumor-suppressor genes on chromosome 13 may be playing a role in parathyroid carcinoma.
Study of the parathyroid cancers found in some patients with the HPT-JT syndrome has led to identification of a much larger role for mutations in the HRPT2 gene in most parathyroid carcinomas, including those that arise sporadically, without apparent association with the HPT-JT syndrome. Mutations in the coding region have been identified in 75–80% of all parathyroid cancers analyzed, leading to the conclusion that, with addition of presumed mutations in the noncoding regions, this genetic defect may be seen in essentially all parathyroid carcinomas. Of special importance was the discovery that, in some sporadic parathyroid cancers, germline mutations have been found; this, in turn, has led to careful investigation of the families of these patients and a new clinical indication for genetic testing in this setting.
Hypercalcemia occurring in family members (who are also found to have the germline mutations) can lead to the finding, at parathyroid surgery, of premalignant parathyroid tumors.
Overall, it seems there are multiple factors in parathyroid cancer, in addition to the HRPT2 and Rb gene, although the HRPT2 gene mutation is the most invariant abnormality. RET encodes a tyrosine kinase type receptor; specific inherited germline mutations lead to a constitutive activation of the receptor, thereby explaining the autosomal dominant mode of transmission and the relatively early onset of neoplasia. In the MEN 2 syndrome, the RET protooncogene may be responsible for the earliest disorder detected, the polyclonal disorder (C cell hyperplasia, which then is transformed into a clonal outgrowth—a medullary carcinoma with the participation of other, still uncharacterized genetic defects).
In some parathyroid adenomas, activation of a protooncogene has been identified (Fig. 424-3B). A reciprocal translocation involving chromosome 11 has been identified that juxtaposes the PTH gene promoter upstream of a gene product termed PRAD-1, encoding a cyclin D protein that plays a key role in normal cell division. This translocation plus other mechanisms that cause an equivalent overexpression of cyclin D1 are found in 20–40% of parathyroid adenomas.
Mouse models have confirmed the role of several of the major identified genetic defects in parathyroid disease and the MEN syndromes. Loss of the MEN1 gene locus or overexpression of the PRAD-1 protooncogene or the mutated RET protooncogene have been analyzed by genetic manipulation in mice, with the expected onset of parathyroid tumors or medullary carcinoma, respectively.
Signs and Symptoms Many patients with hyperparathyroidism are asymptomatic. Manifestations of hyperparathyroidism involve primarily the kidneys and the skeletal system. Kidney involvement, due either to deposition of calcium in the renal parenchyma or to recurrent nephrolithiasis, was present in 60–70% of patients prior to 1970. With earlier detection, renal complications occur in <20% of patients in many large series. Renal stones are usually composed of either calcium oxalate or calcium phosphate. In occasional patients, repeated episodes of nephrolithiasis or the formation of large calculi may lead to urinary tract obstruction, infection, and loss of renal function. Nephrocalcinosis may also cause decreased renal function and phosphate retention.
The distinctive bone manifestation of hyperparathyroidism is osteitis fibrosa cystica, which occurred in 10–25% of patients in series reported 50 years ago. Histologically, the pathognomonic features are an increase in the giant multinucleated osteoclasts in scalloped areas on the surface of the bone (Howship’s lacunae) and a replacement of the normal cellular and marrow elements by fibrous tissue. X-ray changes include resorption of the phalangeal tufts and replacement of the usually sharp cortical outline of the bone in the digits by an irregular outline (subperiosteal resorption). In recent years, osteitis fibrosa cystica is very rare in primary hyperparathyroidism, probably due to the earlier detection of the disease.
Dual-energy x-ray absorptiometry (DEXA) of the spine provides reproducible quantitative estimates (within a few percent) of spinal bone density. Similarly, bone density in the extremities can be quantified by densitometry of the hip or of the distal radius at a site chosen to be primarily cortical. Computed tomography (CT) is a very sensitive technique for estimating spinal bone density, but reproducibility of standard CT is no better than 5%. Newer CT techniques (spiral, “extreme” CT) are more reproducible but are currently available in a limited number of medical centers. Cortical bone density is reduced while cancellous bone density, especially in the spine, is relatively preserved. In symptomatic patients, dysfunctions of the CNS, peripheral nerve and muscle, gastrointestinal tract, and joints also occur. It has been reported that severe neuropsychiatric manifestations may be reversed by parathyroidectomy. When present in symptomatic patients, neuromuscular manifestations may include proximal muscle weakness, easy fatigability, and atrophy of muscles and may be so striking as to suggest a primary neuromuscular disorder. The distinguishing feature is the complete regression of neuromuscular disease after surgical correction of the hyperparathyroidism.
Gastrointestinal manifestations are sometimes subtle and include vague abdominal complaints and disorders of the stomach and pancreas. Again, cause and effect are unclear. In MEN 1 patients with hyperparathyroidism, duodenal ulcer may be the result of associated pancreatic tumors that secrete excessive quantities of gastrin (Zollinger-Ellison syndrome). Pancreatitis has been reported in association with hyperparathyroidism, but the incidence and the mechanism are not established.
Much attention has been paid in recent years to the manifestations of and optimum management strategies for asymptomatic hyperparathyroidism. This is now the most prevalent form of the disease. Asymptomatic primary hyperparathyroidism is defined as biochemically confirmed hyperparathyroidism (elevated or inappropriately normal PTH levels despite hypercalcemia) with the absence of signs and symptoms typically associated with more severe hyperparathyroidism such as features of renal or bone disease.
Three conferences on the topic have been held in the United States over the past two decades, with the most recent in 2008. The published proceedings include discussion of more subtle manifestations of disease, its natural history (without parathyroidectomy), and guidelines both for indications for surgery and medical monitoring in nonoperated patients.
Issues of concern include the potential for cardiovascular deterioration, the presence of subtle neuropsychiatric symptoms, and the longer-term status of skeletal integrity in patients not treated surgically. The current consensus is that medical monitoring rather than surgical correction of hyperparathyroidism may be justified in certain patients. The current recommendation is that patients who show mild disease, as defined by specific criteria (Table 424-2), can be safely followed under management guidelines (Table 424-3). There is, however, growing uncertainty about subtle disease manifestations and whether surgery is therefore indicated in most patients. Among the issues is the evidence of eventual (>8 years) deterioration in bone mineral density after a decade of relative stability. There is concern that this late-onset deterioration in bone density in nonoperated patients could contribute significantly to the well-known age-dependent fracture risk (osteoporosis). One study reported significant and sustained improvements in bone mineral density after successful parathyroidectomy, again raising the issue regarding benefits of surgery. Other randomized studies, however, did not report major gains after surgery.
|
GUIDELINES FOR SURGERY IN ASYMPTOMATIC PRIMARY HYPERPARATHYROIDISMa |
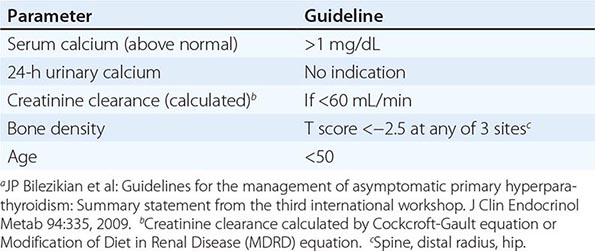
|
GUIDELINES FOR MONITORING IN ASYMPTOMATIC PRIMARY HYPERPARATHYROIDISMa |
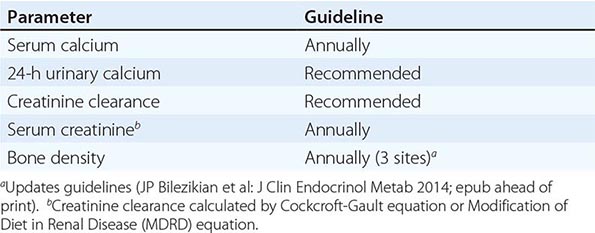
Cardiovascular disease including left ventricular hypertrophy, cardiac functional defects, and endothelial dysfunction have been reported as reversible in European patients with more severe symptomatic disease after surgery, leading to numerous studies of these cardiovascular features in those with milder disease. There are reports of endothelial dysfunction in patients with mild asymptomatic hyperparathyroidism, but the expert panels concluded that more observation is needed, especially regarding whether there is reversibility with surgery.
A topic of considerable interest and some debate is assessment of neuropsychiatric status and health-related quality of life (QOL) status in hyperparathyroid patients both before surgery and in response to parathyroidectomy. Several observational studies suggest considerable improvements in symptom score after surgery. Randomized studies of surgery versus observation, however, have yielded inconclusive results, especially regarding benefits of surgery. Most studies report that hyperparathyroidism is associated with increased neuropsychiatric symptoms, so the issue remains a significant factor in decisions regarding the impact of surgery in this disease.
DIAGNOSIS
The diagnosis is typically made by detecting an elevated immunoreactive PTH level in a patient with asymptomatic hypercalcemia (see “Differential Diagnosis: Special Tests,” below). Serum phosphate is usually low but may be normal, especially if renal failure has developed.
Several modifications in PTH assays have been introduced in efforts to improve their utility in light of information about metabolism of PTH (as discussed above). First-generation assays were based on displacement of radiolabeled PTH from antibodies that reacted with PTH (often also PTH fragments). Double-antibody or immunometric assays (one antibody that is usually directed against the carboxyl-terminal portion of intact PTH to capture the hormone and a second radio- or enzyme-labeled antibody that is usually directed against the amino-terminal portion of intact PTH) greatly improved the diagnostic discrimination of the tests by eliminating interference from circulating biologically inactive fragments, detected by the original first-generation assays. Double-antibody assays are now referred to as second-generation. Such PTH assays have in some centers and testing laboratories been replaced by third-generation assays after it was discovered that large PTH fragments, devoid of only the extreme amino-terminal portion of the PTH molecule, are also present in blood and are detected, incorrectly, as intact PTH. These amino-terminally truncated PTH fragments were prevented from registering in the newer third-generation assays by use of a detection antibody directed against the extreme amino-terminal epitope. These assays may be useful for clinical research studies as in management of chronic renal disease, but the consensus is that either second- or third-generation assays are useful in the diagnosis of primary hyperparathyroidism and for the diagnosis of high-turnover bone disease in chronic kidney disease.
Many tests based on renal responses to excess PTH (renal calcium and phosphate clearance; blood phosphate, chloride, magnesium; urinary or nephrogenous cyclic AMP [cAMP]) were used in earlier decades. These tests have low specificity for hyperparathyroidism and are therefore not cost-effective; they have been replaced by PTH immunometric assays combined with simultaneous blood calcium measurements (Fig. 424-4).
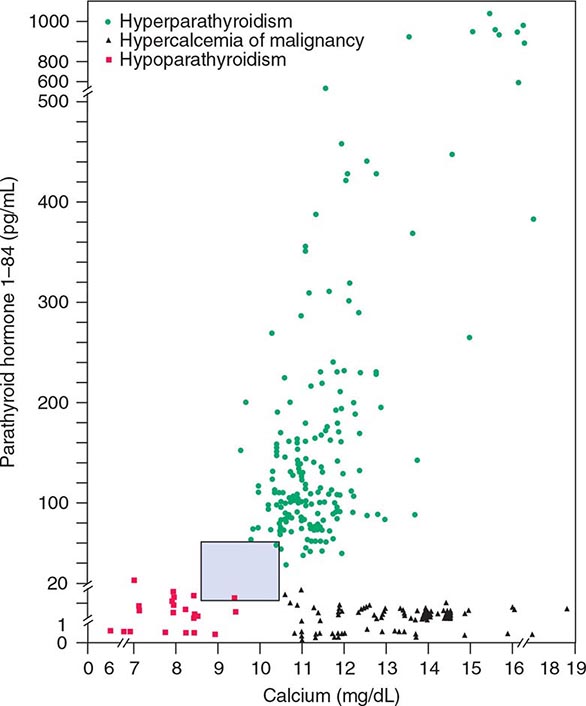
FIGURE 424-4 Levels of immunoreactive parathyroid hormone (PTH) detected in patients with primary hyperparathyroidism, hypercalcemia of malignancy, and hypoparathyroidism. Boxed area represents the upper and normal limits of blood calcium and/or immunoreactive PTH. (From SR Nussbaum, JT Potts, Jr, in L DeGroot, JL Jameson [eds]: Endocrinology, 4th ed. Philadelphia, Saunders, 2001; with permission.)
|
TREATMENT |
HYPERPARATHYROIDISM |
Surgical excision of the abnormal parathyroid tissue is the definitive therapy for this disease. As noted above, medical surveillance without operation for patients with mild, asymptomatic disease is, however, still preferred by some physicians and patients, particularly when the patients are more elderly. Evidence favoring surgery, if medically feasible, is growing because of concerns about skeletal, cardiovascular, and neuropsychiatric disease, even in mild hyperparathyroidism.
Two surgical approaches are generally practiced. The conventional parathyroidectomy procedure was neck exploration with general anesthesia; this procedure is being replaced in many centers, whenever feasible, by an outpatient procedure with local anesthesia, termed minimally invasive parathyroidectomy.
Parathyroid exploration is challenging and should be undertaken by an experienced surgeon. Certain features help in predicting the pathology (e.g., multiple abnormal glands in familial cases). However, some critical decisions regarding management can be made only during the operation.
With conventional surgery, one approach is still based on the view that typically only one gland (the adenoma) is abnormal. If an enlarged gland is found, a normal gland should be sought. In this view, if a biopsy of a normal-sized second gland confirms its histologic (and presumed functional) normality, no further exploration, biopsy, or excision is needed. At the other extreme is the minority viewpoint that all four glands be sought and that most of the total parathyroid tissue mass be removed. The concern with the former approach is that the recurrence rate of hyperparathyroidism may be high if a second abnormal gland is missed; the latter approach could involve unnecessary surgery and an unacceptable rate of hypoparathyroidism. When normal glands are found in association with one enlarged gland, excision of the single adenoma usually leads to cure or at least years free of symptoms. Long-term follow-up studies to establish true rates of recurrence are limited.
Recently, there has been growing experience with new surgical strategies that feature a minimally invasive approach guided by improved preoperative localization and intraoperative monitoring by PTH assays. Preoperative 99mTc sestamibi scans with single-photon emission CT (SPECT) are used to predict the location of an abnormal gland and intraoperative sampling of PTH before and at 5-min intervals after removal of a suspected adenoma to confirm a rapid fall (>50%) to normal levels of PTH. In several centers, a combination of preoperative sestamibi imaging, cervical block anesthesia, minimal surgical incision, and intraoperative PTH measurements has allowed successful outpatient surgical management with a clear-cut cost benefit compared to general anesthesia and more extensive neck surgery. The use of these minimally invasive approaches requires clinical judgment to select patients unlikely to have multiple gland disease (e.g., MEN or secondary hyperparathyroidism). The growing acceptance of the technique and its relative ease for the patient has lowered the threshold for surgery.
Severe hypercalcemia may provide a preoperative clue to the presence of parathyroid carcinoma. In such cases, when neck exploration is undertaken, the tissue should be widely excised; care is taken to avoid rupture of the capsule to prevent local seeding of tumor cells.
Multiple-gland hyperplasia, as predicted in familial cases, poses more difficult questions of surgical management. Once a diagnosis of hyperplasia is established, all the glands must be identified. Two schemes have been proposed for surgical management. One is to totally remove three glands with partial excision of the fourth gland; care is taken to leave a good blood supply for the remaining gland. Other surgeons advocate total parathyroidectomy with immediate transplantation of a portion of a removed, minced parathyroid gland into the muscles of the forearm, with the view that surgical excision is easier from the ectopic site in the arm if there is recurrent hyperfunction.
In a minority of cases, if no abnormal parathyroid glands are found in the neck, the issue of further exploration must be decided. There are documented cases of five or six parathyroid glands and of unusual locations for adenomas such as in the mediastinum.
When a second parathyroid exploration is indicated, the minimally invasive techniques for preoperative localization such as ultrasound, CT scan, and isotope scanning are combined with venous sampling and/or selective digital arteriography in one of the centers specializing in these procedures. Intraoperative monitoring of PTH levels by rapid PTH immunoassays may be useful in guiding the surgery. At one center, long-term cures have been achieved with selective embolization or injection of large amounts of contrast material into the end-arterial circulation feeding the parathyroid tumor.
A decline in serum calcium occurs within 24 h after successful surgery; usually blood calcium falls to low-normal values for 3–5 days until the remaining parathyroid tissue resumes full hormone secretion. Acute postoperative hypocalcemia is likely only if severe bone mineral deficits are present or if injury to all the normal parathyroid glands occurs during surgery. In general, there are few problems encountered in patients with uncomplicated disease such as a single adenoma (the clear majority), who do not have symptomatic bone disease or a large deficit in bone mineral, who are vitamin D and magnesium sufficient, and who have good renal and gastrointestinal function. The extent of postoperative hypocalcemia varies with the surgical approach. If all glands are biopsied, hypocalcemia may be transiently symptomatic and more prolonged. Hypocalcemia is more likely to be symptomatic after second parathyroid explorations, particularly when normal parathyroid tissue was removed at the initial operation and when the manipulation and/or biopsy of the remaining normal glands are more extensive in the search for the missing adenoma.
Patients with hyperparathyroidism have efficient intestinal calcium absorption due to the increased levels of 1,25(OH)2D stimulated by PTH excess. Once hypocalcemia signifies successful surgery, patients can be put on a high-calcium intake or be given oral calcium supplements. Despite mild hypocalcemia, most patients do not require parenteral therapy. If the serum calcium falls to <2 mmol/L (8 mg/dL), and if the phosphate level rises simultaneously, the possibility that surgery has caused hypoparathyroidism must be considered. With unexpected hypocalcemia, coexistent hypomagnesemia should be considered, because it interferes with PTH secretion and causes functional hypoparathyroidism (Chap. 423).
Signs of hypocalcemia include symptoms such as muscle twitching, a general sense of anxiety, and positive Chvostek’s and Trousseau’s signs coupled with serum calcium consistently <2 mmol/L (8 mg/dL). Parenteral calcium replacement at a low level should be instituted when hypocalcemia is symptomatic. The rate and duration of IV therapy are determined by the severity of the symptoms and the response of the serum calcium to treatment. An infusion of 0.5–2 mg/kg per hour or 30–100 mL/h of a 1-mg/mL solution usually suffices to relieve symptoms. Usually, parenteral therapy is required for only a few days. If symptoms worsen or if parenteral calcium is needed for >2–3 days, therapy with a vitamin D analogue and/or oral calcium (2–4 g/d) should be started (see below). It is cost-effective to use calcitriol (doses of 0.5–1 μg/d) because of the rapidity of onset of effect and prompt cessation of action when stopped, in comparison to other forms of vitamin D. A rise in blood calcium after several months of vitamin D replacement may indicate restoration of parathyroid function to normal. It is also appropriate to monitor serum PTH serially to estimate gland function in such patients.
If magnesium deficiency was present, it can complicate the postoperative course since magnesium deficiency impairs the secretion of PTH. Hypomagnesemia should be corrected whenever detected. Magnesium replacement can be effective orally (e.g., MgCl2, MgOH2), but parenteral repletion is usual to ensure postoperative recovery, if magnesium deficiency is suspected due to low blood magnesium levels. Because the depressant effect of magnesium on central and peripheral nerve functions does not occur at levels <2 mmol/L (normal range 0.8–1.2 mmol/L), parenteral replacement can be given rapidly. A cumulative dose as great as 0.5–1 mmol/kg of body weight can be administered if severe hypomagnesemia is present; often, however, total doses of 20–40 mmol are sufficient.
MEDICAL MANAGEMENT
The guidelines for recommending surgical intervention, if feasible (Table 424-2), as well as for monitoring patients with asymptomatic hyperparathyroidism who elect not to undergo parathyroidectomy (Table 424-3), reflect the changes over time since the first conference on the topic in 1990. Medical monitoring rather than corrective surgery is still acceptable, but it is clear that surgical intervention is the more frequently recommended option for the reasons noted above. Tightened guidelines favoring surgery include lowering the recommended level of serum calcium elevation, more careful attention to skeletal integrity through reference to peak skeletal mass at baseline (T scores) rather than age-adjusted bone density (Z scores), as well as the presence of any fragility fracture. The other changes noted in the two guidelines (Tables 424-2 and 424-3) reflect accumulated experience and practical consideration, such as a difficulty in quantity of urine collections. Despite the usefulness of the guidelines, the importance of individual patient and physician judgment and preference is clear in all recommendations.
When surgery is not selected, or not medically feasible, there is interest in the potential value of specific medical therapies. There is no long-term experience regarding specific clinical outcomes such as fracture prevention, but it has been established that bisphosphonates increase bone mineral density significantly without changing serum calcium (as does estrogen, but the latter is not favored because of reported adverse effects in other organ systems). Calcimimetics that lower PTH secretion lower calcium but do not affect bone mineral density.
OTHER PARATHYROID-RELATED CAUSES OF HYPERCALCEMIA
Lithium Therapy Lithium, used in the management of bipolar depression and other psychiatric disorders, causes hypercalcemia in ~10% of treated patients. The hypercalcemia is dependent on continued lithium treatment, remitting and recurring when lithium is stopped and restarted. The parathyroid adenomas reported in some hypercalcemic patients with lithium therapy may reflect the presence of an independently occurring parathyroid tumor; a permanent effect of lithium on parathyroid gland growth need not be implicated as most patients have complete reversal of hypercalcemia when lithium is stopped. However, long-standing stimulation of parathyroid cell replication by lithium may predispose to development of adenomas (as is documented in secondary hyperparathyroidism and renal failure).
At the levels achieved in blood in treated patients, lithium can be shown in vitro to shift the PTH secretion curve to the right in response to calcium; i.e., higher calcium levels are required to lower PTH secretion, probably acting at the calcium sensor (see below). This effect can cause elevated PTH levels and consequent hypercalcemia in otherwise normal individuals. Fortunately, there are usually alternative medications for the underlying psychiatric illness. Parathyroid surgery should not be recommended unless hypercalcemia and elevated PTH levels persist after lithium is discontinued.
GENETIC DISORDERS CAUSING HYPERPARATHYROID-LIKE SYNDROMES
Familial Hypocalciuric Hypercalcemia FHH (also called familial benign hypercalcemia) is inherited as an autosomal dominant trait. Affected individuals are discovered because of asymptomatic hypercalcemia. Most cases of FHH (FHH1) are caused by an inactivating mutation in a single allele of the CaSR (see below), leading to inappropriately normal or even increased secretion of PTH, whereas another hypercalcemic disorder, namely the exceedingly rare Jansen’s disease, is caused by a constitutively active PTH/PTHrP receptor in target tissues. Neither FHH1 nor Jansen’s disease, however, is a growth disorder of the parathyroids. Other forms of FHH are caused either by heterozygous mutations in GNA11 (encoding G11), one of the signaling proteins downstream of the CaSR (FHH2), or by mutations in AP2S1 (FHH3).
The pathophysiology of FHH1 is now understood. The primary defect is abnormal sensing of the blood calcium by the parathyroid gland and renal tubule, causing inappropriate secretion of PTH and excessive reabsorption of calcium in the distal renal tubules. The CaSR is a member of the third family of GPCRs (type C or type III). The receptor responds to increased ECF calcium concentration by suppressing PTH secretion through second-messenger signaling involving the G protein alpha-subunits G11 and Gq, thereby providing negative-feedback regulation of PTH secretion. Many different inactivating CaSR mutations have been identified in patients with FHH1. These mutations lower the capacity of the sensor to bind calcium, and the mutant receptors function as though blood calcium levels were low; excessive secretion of PTH occurs from an otherwise normal gland. Approximately two-thirds of patients with FHH have mutations within the protein-coding region of the CaSR gene. The remaining one-third of kindreds may have mutations in the promoter of the CaSR gene or are caused by mutations in other genes.
Even before elucidation of the pathophysiology of FHH, abundant clinical evidence served to separate the disorder from primary hyperparathyroidism; these clinical features are still useful in differential diagnosis. Patients with primary hyperparathyroidism have <99% renal calcium reabsorption, whereas most patients with FHH have >99% reabsorption. The hypercalcemia in FHH is often detectable in affected members of the kindreds in the first decade of life, whereas hypercalcemia rarely occurs in patients with primary hyperparathyroidism or the MEN syndromes who are age <10 years. PTH may be elevated in the different forms of FHH, but the values are usually normal or lower for the same degree of calcium elevation than is observed in patients with primary hyperparathyroidism. Parathyroid surgery performed in a few patients with FHH before the nature of the syndrome was understood led to permanent hypoparathyroidism; nevertheless, hypocalciuria persisted, establishing that hypocalciuria is not PTH-dependent (now known to be due to the abnormal CaSR in the kidney).
Few clinical signs or symptoms are present in patients with FHH, whereas other endocrine abnormalities are not. Most patients are detected as a result of family screening after hypercalcemia is detected in a proband. In those patients inadvertently operated upon for primary hyperparathyroidism, the parathyroids appeared normal or moderately hyperplastic. Parathyroid surgery is not appropriate, nor, in view of the lack of symptoms, does medical treatment seem needed to lower the calcium. One striking exception to the rule against parathyroid surgery in this syndrome is the occurrence, usually in consanguineous marriages (due to the rarity of the gene mutation), of a homozygous or compound heterozygote state, resulting in severe impairment of CaSR function. In this condition, neonatal severe hypercalcemia, total parathyroidectomy is mandatory, but calcimetics have been used as a temporary measure. Rare but well-documented cases of acquired hypocalciuric hypercalcemia are reported due to antibodies against the CaSR. They appear to be a complication of an underlying autoimmune disorder and respond to therapies directed against the underlying disorder.
Jansen’s Disease Activating mutations in the PTH/PTHrP receptor (PTH1R) have been identified as the cause of this rare autosomal dominant syndrome. Because the mutations lead to constitutive activation of receptor function, one abnormal copy of the mutant receptor is sufficient to cause the disease, thereby accounting for its dominant mode of transmission. The disorder leads to short-limbed dwarfism due to abnormal regulation of chondrocyte maturation in the growth plates of the bone that are formed through the endochondral process. In adult life, there are numerous abnormalities in bone, including multiple cystic resorptive areas resembling those seen in severe hyperparathyroidism. Hypercalcemia and hypophosphatemia with undetectable or low PTH levels are typically observed. The pathogenesis of the growth plate abnormalities in Jansen’s disease has been confirmed by transgenic experiments in which targeted expression of the mutant PTH/PTHrP receptor to the proliferating chondrocyte layer of growth plate emulated several features of the human disorder. Some of these genetic mutations in the parathyroid gland or PTH target cells that affect Ca2+ metabolism are illustrated in Fig. 424-5.
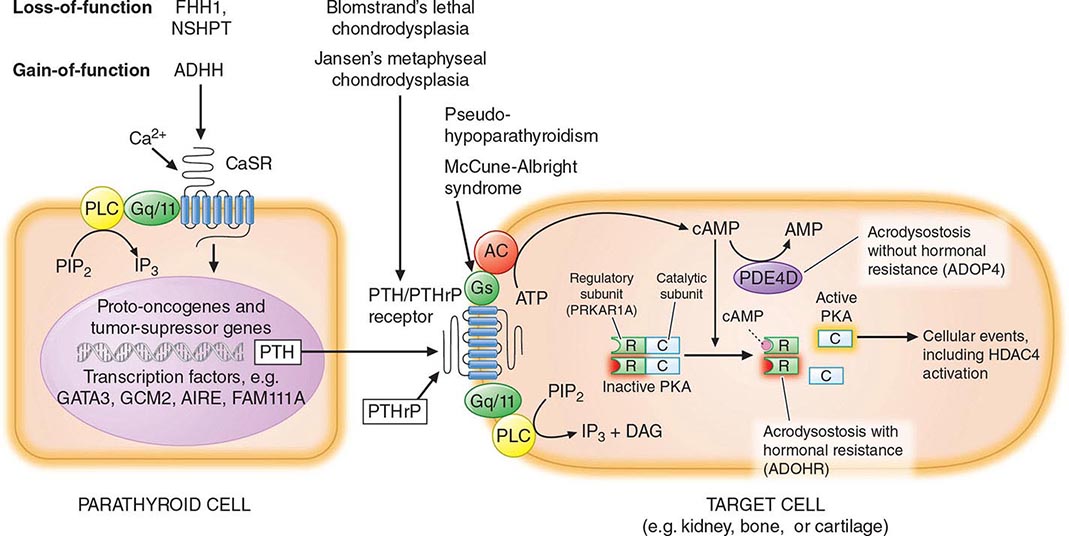
FIGURE 424-5 Illustration of some genetic mutations that alter calcium metabolism by effects on the parathyroid cell or target cells of parathyroid hormone (PTH) action. Alterations in PTH production by the parathyroid cell can be caused by changes in the response to extracellular fluid calcium (Ca2+) that are detected by the calcium-sensing receptor (CaSR). Furthermore, PTH (or PTH-related peptide [PTHrP]) can show altered efficacy in target cells such as in proximal tubular cells, by altered function of its receptor (PTH/PTHrP receptor) or the signal transduction proteins, G proteins such as Gsα, which is linked to adenylate cyclase (AC), the enzyme responsible for producing cyclic AMP (cAMP) (also illustrated are Gq/11, which activate an alternate pathway of receptor signal transmission involving the generation of inositol triphosphate [IP3] or diacylglycerol [DAG]). Heterozygous loss-of-function mutations in the CaSR cause familial benign hypocalciuric hypercalcemia (FBHH), homozygous mutations (both alleles mutated), and severe neonatal hyperparathyroidism (NSHPT); heterozygous gain-of-function causes autosomal dominant hypercalciuric hypocalcemia (ADHH). Other defects in parathyroid cell function that occur at the level of gene regulation (oncogenes or tumor-suppressor genes) or transcription factors are discussed in the text. Blomstrand’s lethal chondrodysplasia is due to homozygous or compound heterozygous loss-of-function mutations in the PTH/PTHrP receptor, a neonatally lethal disorder, while pseudohypoparathyroidism involves inactivation at the level of the G proteins, specifically mutations that eliminate or reduce Gsα activity in the kidney (see text for details). Acrodysostosis can occur with (acrodysostosis with hormonal resistance [ADOHR]; mutant regulatory subunit of PKA) or without hormonal resistance (ADOP4; mutant PDE4D). Jansen’s metaphyseal chondrodysplasia and McCune-Albright syndrome represent gain-of-function mutations in the PTH/PTHrP receptor and Gsα protein, respectively.
MALIGNANCY-RELATED HYPERCALCEMIA
Clinical Syndromes and Mechanisms of Hypercalcemia Hypercalcemia due to malignancy is common (occurring in as many as 20% of cancer patients, especially with certain types of tumor such as lung carcinoma), often severe and difficult to manage, and, on rare occasions, difficult to distinguish from primary hyperparathyroidism. Although malignancy is often clinically obvious or readily detectable by medical history, hypercalcemia can occasionally be due to an occult tumor. Previously, hypercalcemia associated with malignancy was thought to be due to local invasion and destruction of bone by tumor cells; many cases are now known to result from the elaboration by the malignant cells of humoral mediators of hypercalcemia. PTHrP is the responsible humoral agent in most solid tumors that cause hypercalcemia.
The histologic character of the tumor is more important than the extent of skeletal metastases in predicting hypercalcemia. Small-cell carcinoma (oat cell) and adenocarcinoma of the lung, although the most common lung tumors associated with skeletal metastases, rarely cause hypercalcemia. By contrast, many patients with squamous cell carcinoma of the lung develop hypercalcemia. Histologic studies of bone in patients with squamous cell or epidermoid carcinoma of the lung, in sites invaded by tumor as well as areas remote from tumor invasion, reveal increased bone resorption.
Two main mechanisms of hypercalcemia are operative in cancer hypercalcemia. Many solid tumors associated with hypercalcemia, particularly squamous cell and renal tumors, produce and secrete PTHrP that causes increased bone resorption and mediate the hypercalcemia through systemic actions on the skeleton. Alternatively, direct bone marrow invasion occurs with hematologic malignancies such as leukemia, lymphoma, and multiple myeloma. Lymphokines and cytokines (including PTHrP) produced by cells involved in the marrow response to the tumors promote resorption of bone through local destruction. Several hormones, hormone analogues, cytokines, and growth factors have been implicated as the result of clinical assays, in vitro tests, or chemical isolation. The etiologic factor produced by activated normal lymphocytes and by myeloma and lymphoma cells, originally termed osteoclast activation factor, now appears to represent the biologic action of several different cytokines, probably interleukin 1 and lymphotoxin or tumor necrosis factor (TNF). In some lymphomas, there is a third mechanism, caused by an increased blood level of 1,25(OH)2D, produced by the abnormal lymphocytes.
In the more common mechanism, usually termed humoral hypercalcemia of malignancy, solid tumors (cancers of the lung and kidney, in particular), in which bone metastases are absent, minimal, or not detectable clinically, secrete PTHrP measurable by immunoassay. Secretion by the tumors of the PTH-like factor, PTHrP, activates the PTH1R, resulting in a pathophysiology closely resembling hyperparathyroidism, but with normal or suppressed PTH levels. The clinical picture resembles primary hyperparathyroidism (hypophosphatemia accompanies hypercalcemia), and elimination or regression of the primary tumor leads to disappearance of the hypercalcemia.
As in hyperparathyroidism, patients with the humoral hypercalcemia of malignancy have elevated urinary nephrogenous cAMP excretion, hypophosphatemia, and increased urinary phosphate clearance. However, in humoral hypercalcemia of malignancy, immunoreactive PTH is undetectable or suppressed, making the differential diagnosis easier. Other features of the disorder differ from those of true hyperparathyroidism. Although the biologic actions of PTH and PTHrP are exerted through the same receptor, subtle differences in receptor activation by the two ligands must account for some of the discordance in pathophysiology, when an excess of one or the other peptide occurs. Other cytokines elaborated by the malignancy may contribute to the variations from hyperparathyroidism in these patients as well. Patients with humoral hypercalcemia of malignancy may have low to normal levels of 1,25(OH)2D instead of elevated levels as in true hyperparathyroidism. In some patients with the humoral hypercalcemia of malignancy, osteoclastic resorption is unaccompanied by an osteoblastic or bone-forming response, implying inhibition of the normal coupling of bone formation and resorption.
Several different assays (single- or double-antibody, different epitopes) have been developed to detect PTHrP. Most data indicate that circulating PTHrP levels are undetectable (or low) in normal individuals except perhaps in pregnancy (high in human milk) and elevated in most cancer patients with the humoral syndrome. The etiologic mechanisms in cancer hypercalcemia may be multiple in the same patient. For example, in breast carcinoma (metastatic to bone) and in a distinctive type of T cell lymphoma/leukemia initiated by human T cell lymphotropic virus I, hypercalcemia is caused by direct local lysis of bone as well as by a humoral mechanism involving excess production of PTHrP. Hyperparathyroidism has been reported to coexist with the humoral cancer syndrome, and rarely, ectopic hyperparathyroidism due to tumor elaboration of true PTH is reported.
Diagnostic Issues Levels of PTH measured by the double-antibody technique are undetectable or extremely low in tumor hypercalcemia, as would be expected with the mediation of the hypercalcemia by a factor other than PTH (the hypercalcemia suppresses the normal parathyroid glands). In a patient with minimal symptoms referred for hypercalcemia, low or undetectable PTH levels would focus attention on a possible occult malignancy (except for very rare cases of ectopic hyperparathyroidism).
Ordinarily, the diagnosis of cancer hypercalcemia is not difficult because tumor symptoms are prominent when hypercalcemia is detected. Indeed, hypercalcemia may be noted incidentally during the workup of a patient with known or suspected malignancy. Clinical suspicion that malignancy is the cause of the hypercalcemia is heightened when there are other signs or symptoms of a paraneoplastic process such as weight loss, fatigue, muscle weakness, or unexplained skin rash, or when symptoms specific for a particular tumor are present. Squamous cell tumors are most frequently associated with hypercalcemia, particularly tumors of the lung, kidney, head and neck, and urogenital tract. Radiologic examinations can focus on these areas when clinical evidence is unclear. Bone scans with technetium-labeled bisphosphonate are useful for detection of osteolytic metastases; the sensitivity is high, but specificity is low; results must be confirmed by conventional x-rays to be certain that areas of increased uptake are due to osteolytic metastases per se. Bone marrow biopsies are helpful in patients with anemia or abnormal peripheral blood smears.
|
TREATMENT |
MALIGNANCY-RELATED HYPERCALCEMIA |
Treatment of the hypercalcemia of malignancy is first directed to control of tumor; reduction of tumor mass usually corrects hypercalcemia. If a patient has severe hypercalcemia yet has a good chance for effective tumor therapy, treatment of the hypercalcemia should be vigorous while awaiting the results of definitive therapy. If hypercalcemia occurs in the late stages of a tumor that is resistant to antitumor therapy, the treatment of the hypercalcemia should be judicious as high calcium levels can have a mild sedating effect. Standard therapies for hypercalcemia (discussed below) are applicable to patients with malignancy.
VITAMIN D–RELATED HYPERCALCEMIA
Hypercalcemia caused by vitamin D can be due to excessive ingestion or abnormal metabolism of the vitamin. Abnormal metabolism of the vitamin is usually acquired in association with a widespread granulomatous disorder. Vitamin D metabolism is carefully regulated, particularly the activity of renal 1α-hydroxylase, the enzyme responsible for the production of 1,25(OH)2D (Chap. 423). The regulation of 1α-hydroxylase and the normal feedback suppression by 1,25(OH)2D seem to work less well in infants than in adults and to operate poorly, if at all, in sites other than the renal tubule; these phenomena may explain the occurrence of hypercalcemia secondary to excessive 1,25(OH)2D production in infants with Williams’ syndrome (see below) and in adults with sarcoidosis or lymphoma.
Vitamin D Intoxication Chronic ingestion of 40–100 times the normal physiologic requirement of vitamin D (amounts >40,000–100,000 U/d) is usually required to produce significant hypercalcemia in otherwise healthy individuals. The stated upper limit of safe dietary intake is 2000 U/d (50 μg/d) in adults because of concerns about potential toxic effects of cumulative supraphysiologic doses. These recommendations are now regarded as too restrictive, because some estimates are that in elderly individuals in northern latitudes, 2000 U/d or more may be necessary to avoid vitamin D insufficiency.
Hypercalcemia in vitamin D intoxication is due to an excessive biologic action of the vitamin, perhaps the consequence of increased levels of 25(OH)D rather than merely increased levels of the active metabolite 1,25(OH)2D (the latter may not be elevated in vitamin D intoxication). 25(OH)D has definite, if low, biologic activity in the intestine and bone. The production of 25(OH)D is less tightly regulated than is the production of 1,25(OH)2D. Hence concentrations of 25(OH)D are elevated several-fold in patients with excess vitamin D intake.
The diagnosis is substantiated by documenting elevated levels of 25(OH)D >100 mg/mL. Hypercalcemia is usually controlled by restriction of dietary calcium intake and appropriate attention to hydration. These measures, plus discontinuation of vitamin D, usually lead to resolution of hypercalcemia. However, vitamin D stores in fat may be substantial, and vitamin D intoxication may persist for weeks after vitamin D ingestion is terminated. Such patients are responsive to glucocorticoids, which in doses of 100 mg/d of hydrocortisone or its equivalent usually return serum calcium levels to normal over several days; severe intoxication may require intensive therapy.
Sarcoidosis and Other Granulomatous Diseases In patients with sarcoidosis and other granulomatous diseases, such as tuberculosis and fungal infections, excess 1,25(OH)2D is synthesized in macrophages or other cells in the granulomas. Indeed, increased 1,25(OH)2D levels have been reported in anephric patients with sarcoidosis and hypercalcemia. Macrophages obtained from granulomatous tissue convert 25(OH)D to 1,25(OH)2D at an increased rate. There is a positive correlation in patients with sarcoidosis between 25(OH)D levels (reflecting vitamin D intake) and the circulating concentrations of 1,25(OH)2D, whereas normally there is no increase in 1,25(OH)2D with increasing 25(OH)D levels due to multiple feedback controls on renal 1α-hydroxylase (Chap. 423). The usual regulation of active metabolite production by calcium and phosphate or by PTH does not operate in these patients. Clearance of 1,25(OH)2D from blood may be decreased in sarcoidosis as well. PTH levels are usually low and 1,25(OH)2D levels are elevated, but primary hyperparathyroidism and sarcoidosis may coexist in some patients.
Management of the hypercalcemia can often be accomplished by avoiding excessive sunlight exposure and limiting vitamin D and calcium intake. Presumably, however, the abnormal sensitivity to vitamin D and abnormal regulation of 1,25(OH)2D synthesis will persist as long as the disease is active. Alternatively, glucocorticoids in the equivalent of 100 mg/d of hydrocortisone or equivalent doses of glucocorticoids may help control hypercalcemia. Glucocorticoids appear to act by blocking excessive production of 1,25(OH)2D, as well as the response to it in target organs.
Idiopathic Hypercalcemia of Infancy This rare disorder, usually referred to as Williams’ syndrome, is an autosomal dominant disorder characterized by multiple congenital development defects, including supravalvular aortic stenosis, mental retardation, and an elfin facies, in association with hypercalcemia due to abnormal sensitivity to vitamin D. The hypercalcemia associated with the syndrome was first recognized in England after fortification of milk with vitamin D. The cardiac and developmental abnormalities were independently described, but the connection between these defects and hypercalcemia were not described until later. Levels of 1,25(OH)2D can be elevated, ranging from 46 to 120 nmol/L (150–500 pg/mL). The mechanism of the abnormal sensitivity to vitamin D and of the increased circulating levels of 1,25(OH)2D is still unclear. Studies suggest that genetic mutations involving microdeletions at the elastin locus and perhaps other genes on chromosome 7 may play a role in the pathogenesis. Another cause of hypercalcemia in infants and young children is a 24-hydroxylase deficiency that impairs metabolism of 1,25(OH)2D.
HYPERCALCEMIA ASSOCIATED WITH HIGH BONE TURNOVER
Hyperthyroidism As many as 20% of hyperthyroid patients have high-normal or mildly elevated serum calcium concentrations; hypercalciuria is even more common. The hypercalcemia is due to increased bone turnover, with bone resorption exceeding bone formation. Severe calcium elevations are not typical, and the presence of such suggests a concomitant disease such as hyperparathyroidism. Usually, the diagnosis is obvious, but signs of hyperthyroidism may occasionally be occult, particularly in the elderly (Chap. 405). Hypercalcemia is managed by treatment of the hyperthyroidism. Reports that thyroid-stimulating hormone (TSH) itself normally has a bone-protective effect suggest that suppressed TSH levels also play a role in hypercalcemia.
Immobilization Immobilization is a rare cause of hypercalcemia in adults in the absence of an associated disease but may cause hypercalcemia in children and adolescents, particularly after spinal cord injury and paraplegia or quadriplegia. With resumption of ambulation, the hypercalcemia in children usually returns to normal.
The mechanism appears to involve a disproportion between bone formation and bone resorption; the former decreased and the latter increased. Hypercalciuria and increased mobilization of skeletal calcium can develop in normal volunteers subjected to extensive bed rest, although hypercalcemia is unusual. Immobilization of an adult with a disease associated with high bone turnover, however, such as Paget’s disease, may cause hypercalcemia.
Thiazides Administration of benzothiadiazines (thiazides) can cause hypercalcemia in patients with high rates of bone turnover. Traditionally, thiazides are associated with aggravation of hypercalcemia in primary hyperparathyroidism, but this effect can be seen in other high-bone-turnover states as well. The mechanism of thiazide action is complex. Chronic thiazide administration leads to reduction in urinary calcium; the hypocalciuric effect appears to reflect the enhancement of proximal tubular resorption of sodium and calcium in response to sodium depletion. Some of this renal effect is due to augmentation of PTH action and is more pronounced in individuals with intact PTH secretion. However, thiazides cause hypocalciuria in hypoparathyroid patients on high-dose vitamin D and oral calcium replacement if sodium intake is restricted. This finding is the rationale for the use of thiazides as an adjunct to therapy in hypoparathyroid patients, as discussed below. Thiazide administration to normal individuals causes a transient increase in blood calcium (usually within the high-normal range) that reverts to preexisting levels after a week or more of continued administration. If hormonal function and calcium and bone metabolism are normal, homeostatic controls are reset to counteract the calcium-elevating effect of the thiazides. In the presence of hyperparathyroidism or increased bone turnover from another cause, homeostatic mechanisms are ineffective. The abnormal effects of the thiazide on calcium metabolism disappear within days of cessation of the drug.
Vitamin A Intoxication Vitamin A intoxication is a rare cause of hypercalcemia and is most commonly a side effect of dietary faddism (Chap. 96e). Calcium levels can be elevated into the 3–3.5-mmol/L (12–14 mg/dL) range after the ingestion of 50,000–100,000 units of vitamin A daily (10–20 times the minimum daily requirement). Typical features of severe hypercalcemia include fatigue, anorexia, and, in some, severe muscle and bone pain. Excess vitamin A intake is presumed to increase bone resorption.
The diagnosis can be established by history and by measurement of vitamin A levels in serum. Occasionally, skeletal x-rays reveal periosteal calcifications, particularly in the hands. Withdrawal of the vitamin is usually associated with prompt disappearance of the hypercalcemia and reversal of the skeletal changes. As in vitamin D intoxication, administration of 100 mg/d of hydrocortisone or its equivalent leads to a rapid return of the serum calcium to normal.
HYPERCALCEMIA ASSOCIATED WITH RENAL FAILURE
Severe Secondary Hyperparathyroidism The pathogenesis of secondary hyperparathyroidism in chronic kidney disease is incompletely understood. Resistance to the normal level of PTH is a major factor contributing to the development of hypocalcemia, which, in turn, is a stimulus to parathyroid gland enlargement. However, recent findings have indicated that an increase of FGF23 production by osteocytes (and possibly osteoblasts) in bone occurs well before an elevation in PTH is detected. FGF23 is a potent inhibitor of the renal 1-alpha hydroxylase, and the FGF23-dependent reduction in 1,25(OH)2 vitamin D seems to be an important stimulus for the development of secondary hyperparathyroidism.
Secondary hyperparathyroidism occurs not only in patients with renal failure but also in those with osteomalacia due to multiple causes (Chap. 423), including deficiency of vitamin D action and pseudohypoparathyroidism (deficient response to PTH downstream of PTHR1). For both disorders, hypocalcemia seems to be the common denominator in initiating the development of secondary hyperparathyroidism. Primary (1°) and secondary (2°) hyperparathyroidism can be distinguished conceptually by the autonomous growth of the parathyroid glands in primary hyperparathyroidism (presumably irreversible) and the adaptive response of the parathyroids in secondary hyperparathyroidism (typically reversible). In fact, reversal over weeks from an abnormal pattern of secretion, presumably accompanied by involution of parathyroid gland mass to normal, occurs in patients with osteomalacia who have been treated effectively with calcium and vitamin D. However, it is now recognized that a true clonal outgrowth (irreversible) can arise in long-standing, inadequately treated chronic kidney disease (e.g., tertiary [3°] hyperparathyroidism; see below).
Patients with secondary hyperparathyroidism may develop bone pain, ectopic calcification, and pruritus. The bone disease seen in patients with secondary hyperparathyroidism and chronic kidney disease is termed renal osteodystrophy and affects primarily bone turnover. However, osteomalacia is frequently encountered as well and may be related to the circulating levels of FGF23.
Two other skeletal disorders have been frequently associated in the past with chronic kidney disease (CKD) patients treated by long-term dialysis, who received aluminum-containing phosphate binders. Aluminum deposition in bone (see below) leads to an osteomalacia-like picture. The other entity is a low-turnover bone disease termed “aplastic” or “adynamic” bone disease; PTH levels are lower than typically observed in CKD patients with secondary hyperparathyroidism. It is believed that the condition is caused, at least in part, by excessive PTH suppression, which may be even greater than previously appreciated in light of evidence that some of the immunoreactive PTH detected by most commercially available PTH assays is not the full-length biologically active molecule (as discussed above) but may consist of amino-terminally truncated fragments that do not activate the PTH1R.
|
TREATMENT |
SECONDARY HYPERPARATHYROIDISM |
Medical therapy to reverse secondary hyperparathyroidism in CKD includes reduction of excessive blood phosphate by restriction of dietary phosphate, the use of nonabsorbable phosphate binders, and careful, selective addition of calcitriol (0.25–2 μg/d) or related analogues. Calcium carbonate became preferred over aluminum-containing antacids to prevent aluminum-induced bone disease. However, synthetic gels that also bind phosphate (such as sevelamer; Chap. 335) are now widely used, with the advantage of avoiding not only aluminum retention, but also excess calcium loading, which may contribute to cardiovascular calcifications. Intravenous calcitriol (or related analogues), administered as several pulses each week, helps control secondary hyperparathyroidism. Aggressive but carefully administered medical therapy can often, but not always, reverse hyperparathyroidism and its symptoms and manifestations.
Occasional patients develop severe manifestations of secondary hyperparathyroidism, including hypercalcemia, pruritus, extraskeletal calcifications, and painful bones, despite aggressive medical efforts to suppress the hyperparathyroidism. PTH hypersecretion no longer responsive to medical therapy, a state of severe hyperparathyroidism in patients with CKD that requires surgery, has been referred to as tertiary hyperparathyroidism. Parathyroid surgery is necessary to control this condition. Based on genetic evidence from examination of tumor samples in these patients, the emergence of autonomous parathyroid function is due to a monoclonal outgrowth of one or more previously hyperplastic parathyroid glands. The adaptive response has become an independent contributor to disease; this finding seems to emphasize the importance of optimal medical management to reduce the proliferative response of the parathyroid cells that enables the irreversible genetic change.
Aluminum Intoxication Aluminum intoxication (and often hypercalcemia as a complication of medical treatment) in the past occurred in patients on chronic dialysis; manifestations included acute dementia and unresponsive and severe osteomalacia. Bone pain, multiple nonhealing fractures, particularly of the ribs and pelvis, and a proximal myopathy occur. Hypercalcemia develops when these patients are treated with vitamin D or calcitriol because of impaired skeletal responsiveness. Aluminum is present at the site of osteoid mineralization, osteoblastic activity is minimal, and calcium incorporation into the skeleton is impaired. The disorder is now rare because of the avoidance of aluminum-containing antacids or aluminum excess in the dialysis regimen (Chap. 429).
Milk-Alkali Syndrome The milk-alkali syndrome is due to excessive ingestion of calcium and absorbable antacids such as milk or calcium carbonate. It is much less frequent since proton pump inhibitors and other treatments became available for peptic ulcer disease. For a time, the increased use of calcium carbonate in the management of secondary hyperparathyroidism led to reappearance of the syndrome. Several clinical presentations—acute, subacute, and chronic—have been described, all of which feature hypercalcemia, alkalosis, and renal failure. The chronic form of the disease, termed Burnett’s syndrome, is associated with irreversible renal damage. The acute syndromes reverse if the excess calcium and absorbable alkali are stopped.
Individual susceptibility is important in the pathogenesis, because some patients are treated with calcium carbonate and alkali regimens without developing the syndrome. One variable is the fractional calcium absorption as a function of calcium intake. Some individuals absorb a high fraction of calcium, even with intakes ≥2 g of elemental calcium per day, instead of reducing calcium absorption with high intake, as occurs in most normal individuals. Resultant mild hypercalcemia after meals in such patients is postulated to contribute to the generation of alkalosis. Development of hypercalcemia causes increased sodium excretion and some depletion of total-body water. These phenomena and perhaps some suppression of endogenous PTH secretion due to mild hypercalcemia lead to increased bicarbonate resorption and to alkalosis in the face of continued calcium carbonate ingestion. Alkalosis per se selectively enhances calcium resorption in the distal nephron, thus aggravating the hypercalcemia. The cycle of mild hypercalcemia → bicarbonate retention → alkalosis → renal calcium retention → severe hypercalcemia perpetuates and aggravates hypercalcemia and alkalosis as long as calcium and absorbable alkali are ingested.
DIFFERENTIAL DIAGNOSIS: SPECIAL TESTS
Differential diagnosis of hypercalcemia is best achieved by using clinical criteria, but immunometric assays to measure PTH are especially useful in distinguishing among major causes (Fig. 424-6). The clinical features that deserve emphasis are the presence or absence of symptoms or signs of disease and evidence of chronicity. If one discounts fatigue or depression, >90% of patients with primary hyperparathyroidism have asymptomatic hypercalcemia; symptoms of malignancy are usually present in cancer-associated hypercalcemia. Disorders other than hyperparathyroidism and malignancy cause <10% of cases of hypercalcemia, and some of the nonparathyroid causes are associated with clear-cut manifestations such as renal failure.
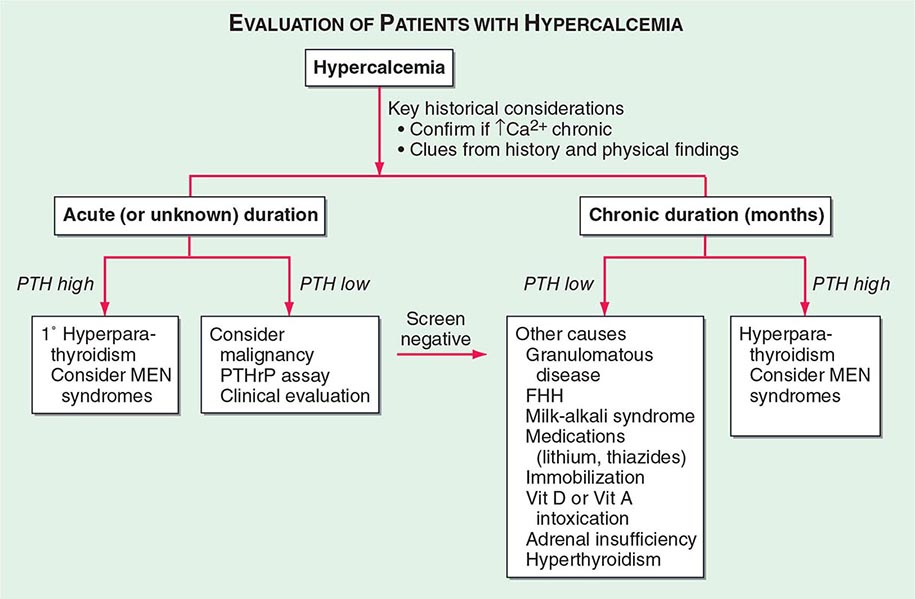
FIGURE 424-6 Algorithm for the evaluation of patients with hypercalcemia. See text for details. FHH, familial hypocalciuric hypercalcemia; MEN, multiple endocrine neoplasia; PTH, parathyroid hormone; PTHrP, parathyroid hormone–related peptide.
Hyperparathyroidism is the likely diagnosis in patients with chronic hypercalcemia. If hypercalcemia has been manifest for >1 year, malignancy can usually be excluded as the cause. A striking feature of malignancy-associated hypercalcemia is the rapidity of the course, whereby signs and symptoms of the underlying malignancy are evident within months of the detection of hypercalcemia. Although clinical considerations are helpful in arriving at the correct diagnosis of the cause of hypercalcemia, appropriate laboratory testing is essential for definitive diagnosis. The immunoassay for PTH usually separates hyperparathyroidism from all other causes of hypercalcemia (exceptions are very rare reports of ectopic production of excess PTH by nonparathyroid tumors). Patients with hyperparathyroidism have elevated PTH levels despite hypercalcemia, whereas patients with malignancy and the other causes of hypercalcemia (except for disorders mediated by PTH such as lithium-induced hypercalcemia) have levels of hormone below normal or undetectable levels. Assays based on the double-antibody method for PTH exhibit very high sensitivity (especially if serum calcium is simultaneously evaluated) and specificity for the diagnosis of primary hyperparathyroidism (Fig. 424-4).
In summary, PTH values are elevated in >90% of parathyroid-related causes of hypercalcemia, undetectable or low in malignancy-related hypercalcemia, and undetectable or normal in vitamin D–related and high-bone-turnover causes of hypercalcemia. In view of the specificity of the PTH immunoassay and the high frequency of hyperparathyroidism in hypercalcemic patients, it is cost-effective to measure the PTH level in all hypercalcemic patients unless malignancy or a specific nonparathyroid disease is obvious. False-positive PTH assay results are rare. Immunoassays for PTHrP are helpful in diagnosing certain types of malignancy-associated hypercalcemia. Although FHH is parathyroid-related, the disease should be managed distinctively from hyperparathyroidism. Clinical features and the low urinary calcium excretion can help make the distinction. Because the incidence of malignancy and hyperparathyroidism both increase with age, they can coexist as two independent causes of hypercalcemia.
1,25(OH)2D levels are elevated in many (but not all) patients with primary hyperparathyroidism. In other disorders associated with hypercalcemia, concentrations of 1,25(OH)2D are low or, at the most, normal. However, this test is of low specificity and is not cost-effective, as not all patients with hyperparathyroidism have elevated 1,25(OH)2D levels and not all nonparathyroid hypercalcemic patients have suppressed 1,25(OH)2D. Measurement of 1,25(OH)2D is, however, critically valuable in establishing the cause of hypercalcemia in sarcoidosis and certain lymphomas.
A useful general approach is outlined in Fig. 424-6. If the patient is asymptomatic and there is evidence of chronicity to the hypercalcemia, hyperparathyroidism is almost certainly the cause. If PTH levels (usually measured at least twice) are elevated, the clinical impression is confirmed and little additional evaluation is necessary. If there is only a short history or no data as to the duration of the hypercalcemia, occult malignancy must be considered; if the PTH levels are not elevated, then a thorough workup must be undertaken for malignancy, including chest x-ray, CT of chest and abdomen, and bone scan. Immunoassays for PTHrP may be especially useful in such situations. Attention should also be paid to clues for underlying hematologic disorders such as anemia, increased plasma globulin, and abnormal serum immunoelectrophoresis; bone scans can be negative in some patients with metastases such as in multiple myeloma. Finally, if a patient with chronic hypercalcemia is asymptomatic and malignancy therefore seems unlikely on clinical grounds, but PTH values are not elevated, it is useful to search for other chronic causes of hypercalcemia such as occult sarcoidosis. A careful history of dietary supplements and drug use may suggest intoxication with vitamin D or vitamin A or the use of thiazides.
|
TREATMENT |
HYPERCALCEMIC STATES |
The approach to medical treatment of hypercalcemia varies with its severity (Table 424-4). Mild hypercalcemia, <3 mmol/L (12 mg/dL), can be managed by hydration. More severe hypercalcemia (levels of 3.2–3.7 mmol/L [13–15 mg/dL]) must be managed aggressively; above that level, hypercalcemia can be life-threatening and requires emergency measures. By using a combination of approaches in severe hypercalcemia, the serum calcium concentration can be decreased by 0.7–2.2 mmol/L (3–9 mg/dL) within 24–48 h in most patients, enough to relieve acute symptoms, prevent death from hypercalcemic crisis, and permit diagnostic evaluation. Therapy can then be directed at the underlying disorder—the second priority.
|
THERAPIES FOR SEVERE HYPERCALCEMIA |
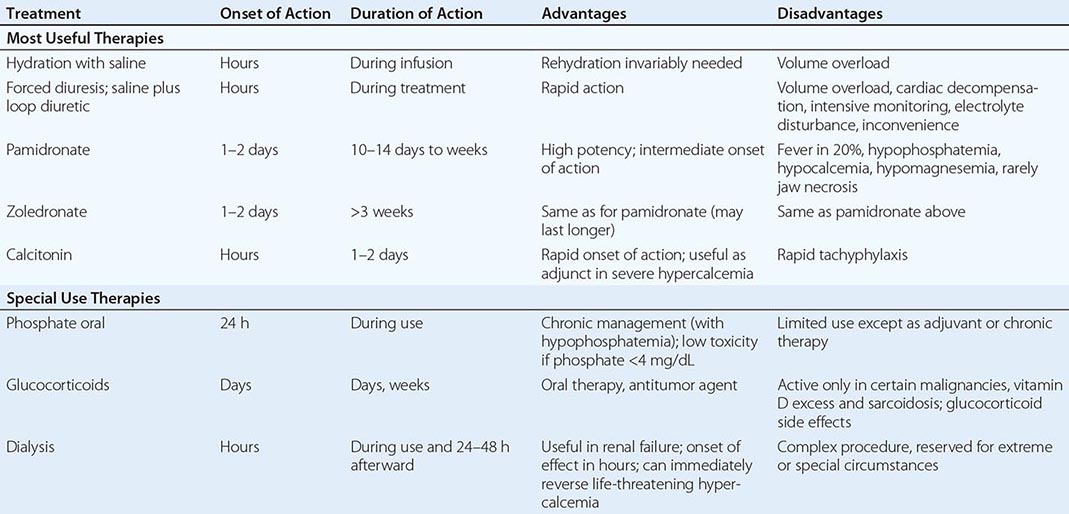
Hypercalcemia develops because of excessive skeletal calcium release, increased intestinal calcium absorption, or inadequate renal calcium excretion. Understanding the particular pathogenesis helps guide therapy. For example, hypercalcemia in patients with malignancy is primarily due to excessive skeletal calcium release and is, therefore, minimally improved by restriction of dietary calcium. On the other hand, patients with vitamin D hypersensitivity or vitamin D intoxication have excessive intestinal calcium absorption, and restriction of dietary calcium is beneficial. Decreased renal function or ECF depletion decreases urinary calcium excretion. In such situations, rehydration may rapidly reduce or reverse the hypercalcemia, even though increased bone resorption persists. As outlined below, the more severe the hypercalcemia, the greater the number of combined therapies that should be used. Rapid-acting (hours) approaches—rehydration, forced diuresis, and calcitonin—can be used with the most effective antiresorptive agents such as bisphosphonates (since severe hypercalcemia usually involves excessive bone resorption).
HYDRATION, INCREASED SALT INTAKE, MILD AND FORCED DIURESIS
The first principle of treatment is to restore normal hydration. Many hypercalcemic patients are dehydrated because of vomiting, inanition, and/or hypercalcemia-induced defects in urinary concentrating ability. The resultant drop in glomerular filtration rate is accompanied by an additional decrease in renal tubular sodium and calcium clearance. Restoring a normal ECF volume corrects these abnormalities and increases urine calcium excretion by 2.5–7.5 mmol/d (100–300 mg/d). Increasing urinary sodium excretion to 400–500 mmol/d increases urinary calcium excretion even further than simple rehydration. After rehydration has been achieved, saline can be administered, or furosemide or ethacrynic acid can be given twice daily to depress the tubular reabsorptive mechanism for calcium (care must be taken to prevent dehydration). The combined use of these therapies can increase urinary calcium excretion to ≥12.5 mmol/d (500 mg/d) in most hypercalcemic patients. Because this is a substantial percentage of the exchangeable calcium pool, the serum calcium concentration usually falls 0.25–0.75 mmol/L (1–3 mg/dL) within 24 h. Precautions should be taken to prevent potassium and magnesium depletion; calcium-containing renal calculi are a potential complication.
Under life-threatening circumstances, the preceding approach can be pursued more aggressively, but the availability of effective agents to block bone resorption (such as bisphosphonates) has reduced the need for extreme diuresis regimens (Table 424-4). Depletion of potassium and magnesium is inevitable unless replacements are given; pulmonary edema can be precipitated. The potential complications can be reduced by careful monitoring of central venous pressure and plasma or urine electrolytes; catheterization of the bladder may be necessary. Dialysis treatment may be needed when renal function is compromised.
BISPHOSPHONATES
The bisphosphonates are analogues of pyrophosphate, with high affinity for bone, especially in areas of increased bone turnover, where they are powerful inhibitors of bone resorption. These bone-seeking compounds are stable in vivo because phosphatase enzymes cannot hydrolyze the central carbon-phosphorus-carbon bond. The bisphosphonates are concentrated in areas of high bone turnover and are taken up by and inhibit osteoclast action; the mechanism of action is complex. The bisphosphonate molecules that contain amino groups in the side chain structure (see below) interfere with prenylation of proteins and can lead to cellular apoptosis. The highly active nonamino group–containing bisphosphonates are also metabolized to cytotoxic products.
The initial bisphosphonate widely used in clinical practice, etidronate, was effective but had several disadvantages, including the capacity to inhibit bone formation as well as blocking resorption. Subsequently, a number of second- or third-generation compounds have become the mainstays of antiresorptive therapy for treatment of hypercalcemia and osteoporosis. The newer bisphosphonates have a highly favorable ratio of blocking resorption versus inhibiting bone formation; they inhibit osteoclast-mediated skeletal resorption yet do not cause mineralization defects at ordinary doses. Although the bisphosphonates have similar structures, the routes of administration, efficacy, toxicity, and side effects vary. The potency of the compounds for inhibition of bone resorption varies more than 10,000-fold, increasing in the order of etidronate, tiludronate, pamidronate, alendronate, risedronate, and zoledronate. The IV use of pamidronate and zoledronate is approved for the treatment of hypercalcemia; between 30 and 90 mg pamidronate, given as a single IV dose over a few hours, returns serum calcium to normal within 24–48 h with an effect that lasts for weeks in 80–100% of patients. Zoledronate given in doses of 4 or 8 mg/5-min infusion has a more rapid and more sustained effect than pamidronate in direct comparison.
These drugs are used extensively in cancer patients. Absolute survival improvements are noted with pamidronate and zoledronate in multiple myeloma, for example. However, although still rare, there are increasing reports of jaw necrosis, especially after dental surgery, mainly in cancer patients treated with multiple doses of the more potent bisphosphonates.
CALCITONIN
Calcitonin acts within a few hours of its administration, principally through receptors on osteoclasts, to block bone resorption. Calcitonin, after 24 h of use, is no longer effective in lowering calcium. Tachyphylaxis, a known phenomenon with this drug, seems to explain the results, since the drug is often effective in the first 24 h of use. Therefore, in life-threatening hypercalcemia, calcitonin can be used effectively within the first 24 h in combination with rehydration and saline diuresis while waiting for more sustained effects from a simultaneously administered bisphosphonate such as pamidronate. Usual doses of calcitonin are 2–8 U/kg of body weight IV, SC, or IM every 6–12 h.
OTHER THERAPIES
Denosumab, an antibody that blocks the RANK ligand (RANKL) and dramatically reduces osteoclast number and function, is approved for therapy of osteoporosis. It also appears to be an effective treatment to reverse hypercalcemia of malignancy, but is not yet approved for this indication. Plicamycin (formerly mithramycin), which inhibits bone resorption, and gallium nitrate, which exerts a hypocalcemic action also by inhibiting bone resorption, are no longer used because of superior alternatives such as bisphosphonates.
Glucocorticoids have utility, especially in hypercalcemia complicating certain malignancies. They increase urinary calcium excretion and decrease intestinal calcium absorption when given in pharmacologic doses, but they also cause negative skeletal calcium balance. In normal individuals and in patients with primary hyperparathyroidism, glucocorticoids neither increase nor decrease the serum calcium concentration. In patients with hypercalcemia due to certain osteolytic malignancies, however, glucocorticoids may be effective as a result of antitumor effects. The malignancies in which hypercalcemia responds to glucocorticoids include multiple myeloma, leukemia, Hodgkin’s disease, other lymphomas, and carcinoma of the breast, at least early in the course of the disease. Glucocorticoids are also effective in treating hypercalcemia due to vitamin D intoxication and sarcoidosis. Glucocorticoids are also useful in the rare form of hypercalcemia, now recognized in certain autoimmune disorders in which inactivating antibodies against the receptor imitate FHH. Elevated PTH and calcium levels are effectively lowered by the glucocorticoids. In all the preceding situations, the hypocalcemic effect develops over several days, and the usual glucocorticoid dosage is 40–100 mg prednisone (or its equivalent) daily in four divided doses. The side effects of chronic glucocorticoid therapy may be acceptable in some circumstances.
Dialysis is often the treatment of choice for severe hypercalcemia complicated by renal failure, which is difficult to manage medically. Peritoneal dialysis with calcium-free dialysis fluid can remove 5–12.5 mmol (200–500 mg) of calcium in 24–48 h and lower the serum calcium concentration by 0.7–3 mmol/L (3–12 mg/dL). Large quantities of phosphate are lost during dialysis, and serum inorganic phosphate concentration usually falls, potentially aggravating hypercalcemia. Therefore, the serum inorganic phosphate concentration should be measured after dialysis, and phosphate supplements should be added to the diet or to dialysis fluids if necessary.
Phosphate therapy, PO or IV, has a limited role in certain circumstances (Chap. 423). Correcting hypophosphatemia lowers the serum calcium concentration by several mechanisms, including bone/calcium exchange. The usual oral treatment is 1–1.5 g of phosphorus per day for several days, given in divided doses. It is generally believed, but not established, that toxicity does not occur if therapy is limited to restoring serum inorganic phosphate concentrations to normal.
Raising the serum inorganic phosphate concentration above normal decreases serum calcium levels, sometimes strikingly. Intravenous phosphate is one of the most dramatically effective treatments available for severe hypercalcemia but is toxic and even dangerous (fatal hypocalcemia). For these reasons, it is used rarely and only in severely hypercalcemic patients with cardiac or renal failure where dialysis, the preferable alternative, is not feasible or is unavailable.
SUMMARY
The various therapies for hypercalcemia are listed in Table 424-4. The choice depends on the underlying disease, the severity of the hypercalcemia, the serum inorganic phosphate level, and the renal, hepatic, and bone marrow function. Mild hypercalcemia (≤3 mmol/L [12 mg/dL]) can usually be managed by hydration. Severe hypercalcemia (≥3.7 mmol/L [15 mg/dL]) requires rapid correction. Calcitonin should be given for its rapid, albeit short-lived, blockade of bone resorption, and IV pamidronate or zoledronate should be administered, although its onset of action is delayed for 1–2 days. In addition, for the first 24–48 h, aggressive sodium-calcium diuresis with IV saline should be given and, following rehydration, large doses of furosemide or ethacrynic acid, but only if appropriate monitoring is available and cardiac and renal function are adequate. Intermediate degrees of hypercalcemia between 3 and 3.7 mmol/L (12 and 15 mg/dL) should be approached with vigorous hydration and then the most appropriate selection for the patient of the combinations used with severe hypercalcemia.
HYPOCALCEMIA
(See also Chap. 65)
PATHOPHYSIOLOGY OF HYPOCALCEMIA: CLASSIFICATION BASED ON MECHANISM
Chronic hypocalcemia is less common than hypercalcemia; causes include chronic renal failure, hereditary and acquired hypoparathyroidism, vitamin D deficiency, pseudohypoparathyroidism, and hypomagnesemia (Table 424-5).
|
FUNCTIONAL CLASSIFICATION OF HYPOCALCEMIA (EXCLUDING NEONATAL CONDITIONS) |
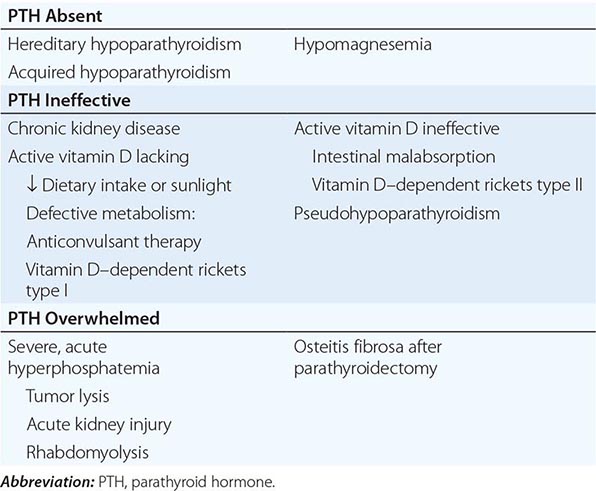
Acute rather than chronic hypocalcemia is seen in critically ill patients or as a consequence of certain medications and often does not require specific treatment. Transient hypocalcemia is seen with severe sepsis, burns, acute kidney injury, and extensive transfusions with citrated blood. Although as many as one-half of patients in an intensive care setting are reported to have calcium concentrations of <2.1 mmol/L (8.5 mg/dL), most do not have a reduction in ionized calcium. Patients with severe sepsis may have a decrease in ionized calcium (true hypocalcemia), but in other severely ill individuals, hypoalbuminemia is the primary cause of the reduced total calcium concentration. Alkalosis increases calcium binding to proteins, and in this setting, direct measurements of ionized calcium should be made.
Medications such as protamine, heparin, and glucagon may cause transient hypocalcemia. These forms of hypocalcemia are usually not associated with tetany and resolve with improvement in the overall medical condition. The hypocalcemia after repeated transfusions of citrated blood usually resolves quickly.
Patients with acute pancreatitis have hypocalcemia that persists during the acute inflammation and varies in degree with disease severity. The cause of hypocalcemia remains unclear. PTH values are reported to be low, normal, or elevated, and both resistance to PTH and impaired PTH secretion have been postulated. Occasionally, a chronic low total calcium and low ionized calcium concentration are detected in an elderly patient without obvious cause and with a paucity of symptoms; the pathogenesis is unclear.
Chronic hypocalcemia, however, is usually symptomatic and requires treatment. Neuromuscular and neurologic manifestations of chronic hypocalcemia include muscle spasms, carpopedal spasm, facial grimacing, and, in extreme cases, laryngeal spasm and convulsions. Respiratory arrest may occur. Increased intracranial pressure occurs in some patients with long-standing hypocalcemia, often in association with papilledema. Mental changes include irritability, depression, and psychosis. The QT interval on the electrocardiogram is prolonged, in contrast to its shortening with hypercalcemia. Arrhythmias occur, and digitalis effectiveness may be reduced. Intestinal cramps and chronic malabsorption may occur. Chvostek’s or Trousseau’s sign can be used to confirm latent tetany.
The classification of hypocalcemia shown in Table 424-5 is based on an organizationally useful premise that PTH is responsible for minute-to-minute regulation of plasma calcium concentration and, therefore, that the occurrence of hypocalcemia must mean a failure of the homeostatic action of PTH. Failure of the PTH response can occur if there is hereditary or acquired parathyroid gland failure, if PTH is ineffective in target organs, or if the action of the hormone is overwhelmed by the loss of calcium from the ECF at a rate faster than it can be replaced.
PTH ABSENT
Whether hereditary or acquired, hypoparathyroidism has a number of common components. Symptoms of untreated hypocalcemia are shared by both types of hypoparathyroidism, although the onset of hereditary hypoparathyroidism can be more gradual and associated with other developmental defects. Basal ganglia calcification and extrapyramidal syndromes are more common and earlier in onset in hereditary hypoparathyroidism. In previous decades, acquired hypoparathyroidism secondary to surgery in the neck was more common than hereditary hypoparathyroidism, but the frequency of surgically induced parathyroid failure has diminished as a result of improved surgical techniques that spare the parathyroid glands and increased use of nonsurgical therapy for hyperthyroidism. Pseudohypoparathyroidism, an example of ineffective PTH action rather than a failure of parathyroid gland production, may share several features with hypoparathyroidism, including extraosseous calcification and extrapyramidal manifestations such as choreoathetotic movements and dystonia.
Papilledema and raised intracranial pressure may occur in both hereditary and acquired hypoparathyroidism, as do chronic changes in fingernails and hair and lenticular cataracts, the latter usually reversible with treatment of hypocalcemia. Certain skin manifestations, including alopecia and candidiasis, are characteristic of hereditary hypoparathyroidism associated with autoimmune polyglandular failure (Chap. 408).
Hypocalcemia associated with hypomagnesemia is associated with both deficient PTH release and impaired responsiveness to the hormone. Patients with hypocalcemia secondary to hypomagnesemia have absent or low levels of circulating PTH, indicative of diminished hormone release despite a maximum physiologic stimulus by hypocalcemia. Plasma PTH levels return to normal with correction of the hypomagnesemia. Thus hypoparathyroidism with low levels of PTH in blood can be due to hereditary gland failure, acquired gland failure, or acute but reversible gland dysfunction (hypomagnesemia).
Genetic Abnormalities and Hereditary Hypoparathyroidism Hereditary hypoparathyroidism can occur as an isolated entity without other endocrine or dermatologic manifestations. More typically, it occurs in association with other abnormalities such as defective development of the thymus or failure of other endocrine organs such as the adrenal, thyroid, or ovary (Chap. 408). Hereditary hypoparathyroidism is often manifest within the first decade but may appear later.
Genetic defects associated with hypoparathyroidism serve to illuminate the complexity of organ development, hormonal biosynthesis and secretion, and tissue-specific patterns of endocrine effector function (Fig. 424-5). Often, hypoparathyroidism is isolated, signifying a highly specific functional disturbance. When hypoparathyroidism is associated with other developmental or organ defects, treatment of the hypocalcemia can still be effective.
A form of hypoparathyroidism associated with defective development of both the thymus and the parathyroid glands is termed the DiGeorge syndrome, or the velocardiofacial syndrome. Congenital cardiovascular, facial, and other developmental defects are present, and patients may die in early childhood with severe infections, hypocalcemia and seizures, or cardiovascular complications. Patients can survive into adulthood, and milder, incomplete forms occur. Most cases are sporadic, but an autosomal dominant form involving microdeletions of chromosome 22q11.2 has been described. Smaller deletions in chromosome 22 are seen in incomplete forms of the DiGeorge syndrome, appearing in childhood or adolescence, that are manifest primarily by parathyroid gland failure. The chromosome 22 defect is now termed DSG1; more recently, a defect in chromosome 10p is also recognized—now called DSG2. The phenotypes seem similar. Studies on the chromosome 22 defect have pinpointed a transcription factor, TBX1. Deletions of the orthologous mouse gene show a phenotype similar to the human syndrome.
Another autosomal dominant developmental defect, featuring hypoparathyroidism, deafness, and renal dysplasia (HDR), has been studied at the genetic level. Cytogenetic abnormalities in some, but not all kindreds, point to translocation defects on chromosome 10, as in DiGeorge syndrome. However, the lack of immunodeficiency and heart defects distinguishes the two syndromes. Mouse models, as well as deletional analysis in some HDR patients, has identified the transcription factor GATA3, which is important in embryonic development and is expressed in developing kidney, ear structures, and the parathyroids.
Another pair of linked developmental disorders involving the parathyroids is recognized. Kenney-Caffey syndrome type I features hypoparathyroidism, short stature, osteosclerosis, and thick cortical bones. A defect seen in Middle Eastern patients, particularly in Saudi Arabia, termed Sanjad-Sakati syndrome, also exhibits growth failure and other dysmorphic features. This syndrome, which is clearly autosomal recessive, involves a gene on chromosome 1q42-q43. Both syndromes apparently involve a chaperone protein, called TBCE, relevant to tubulin function. Recently, a defect in FAM111A was identified as the cause of Kenney-Caffey syndrome type 2.
Hypoparathyroidism can occur in association with a complex hereditary autoimmune syndrome involving failure of the adrenals, the ovaries, the immune system, and the parathyroids in association with recurrent mucocutaneous candidiasis, alopecia, vitiligo, and pernicious anemia (Chap. 408). The responsible gene on chromosome 21q22.3 has been identified. The protein product, which resembles a transcription factor, has been termed the autoimmune regulator, or AIRE. A stop codon mutation occurs in many Finnish families with the disorder, commonly referred to as polyglandular autoimmune type 1 deficiency, whereas another AIRE mutation (Y85C) is typically observed in Jews of Iraqi and Iranian descent.
Hypoparathyroidism is seen in two disorders associated with mitochondrial dysfunction and myopathy, one termed the Kearns-Sayre syndrome (KSS), with ophthalmoplegia and pigmentary retinopathy, and the other termed the MELAS syndrome (mitochondrial encephalopathy, lactic acidosis, and stroke-like episodes). Mutations or deletions in mitochondrial genes have been identified.
Several forms of hypoparathyroidism, each rare in frequency, are seen as isolated defects; the genetic mechanisms are varied. The inheritance includes autosomal dominant, autosomal recessive, and X-linked modes. Three separate autosomal defects involving the parathyroid gene have been recognized: one is dominant and the other two are recessive. The dominant form has a point mutation in the signal sequence, a critical region involved in intracellular transport of the hormone precursor. An Arg for Cys mutation interferes with processing of the precursor and is believed to trigger an apoptotic cellular response, hence acting as a dominant negative. The other two forms are recessive. One point mutation also blocks cleavage of the PTH precursor but requires both alleles to cause hypoparathyroidism. The third involves a single-nucleotide base change that results in an exon splicing defect; the lost exon contains the promoter—hence, the gene is silenced. An X-linked recessive form of hypoparathyroidism has been described in males, and the defect has been localized to chromosome Xq26-q27, perhaps involving the SOX3 gene.
Abnormalities in the CaSR are detected in three distinctive hypocalcemic disorders. All are rare, but more than 10 different gain-of-function mutations have been found in one form of hypocalcemia termed autosomal dominant hypocalcemic hypercalciuria (ADHH). The receptor senses the ambient calcium level as excessive and suppresses PTH secretion, leading to hypocalcemia. The hypocalcemia is aggravated by constitutive receptor activity in the renal tubule causing excretion of inappropriate amounts of calcium. Recognition of the syndrome is important because efforts to treat the hypocalcemia with vitamin D analogues and increased oral calcium exacerbate the already excessive urinary calcium excretion (several grams or more per 24 h), leading to irreversible renal damage from stones and ectopic calcification.
Other causes of isolated hypoparathyroidism include homozygous, inactivating mutations in the parathyroid-specific transcription factor GCM2, which lead to an autosomal recessive form of the disease, or heterozygous point mutations in GCM2, which have a dominant negative effect on the wild-type protein and thus lead to an autosomal dominant form of hypoparathyroidism. Furthermore, heterozygous mutations in G11, one of the two signaling proteins downstream of the CaSR, have been identified as a cause of autosomal dominant hypoparathyroidism.
Bartter’s syndrome is a group of disorders associated with disturbances in electrolyte and acid/base balance, sometimes with nephrocalcinosis and other features. Several types of ion channels or transporters are involved. Curiously, Bartter’s syndrome type V has the electrolyte and pH disturbances seen in the other syndromes but appears to be due to a gain of function in the CaSR. The defect may be more severe than in ADHH and explains the additional features seen beyond hypocalcemia and hypercalciuria.
As with autoimmune disorders that block the CaSR (discussed above under hypercalcemic conditions), there are autoantibodies that at least transiently activate the CaSR, leading to suppressed PTH secretion and hypocalcemia. This disorder may wax and wane.
Acquired Hypoparathyroidism Acquired chronic hypoparathyroidism is usually the result of inadvertent surgical removal of all the parathyroid glands; in some instances, not all the tissue is removed, but the remainder undergoes vascular supply compromise secondary to fibrotic changes in the neck after surgery. In the past, the most frequent cause of acquired hypoparathyroidism was surgery for hyperthyroidism. Hypoparathyroidism now usually occurs after surgery for hyperparathyroidism when the surgeon, facing the dilemma of removing too little tissue and thus not curing the hyperparathyroidism, removes too much. Parathyroid function may not be totally absent in all patients with postoperative hypoparathyroidism.
Rare causes of acquired chronic hypoparathyroidism include radiation-induced damage subsequent to radioiodine therapy of hyperthyroidism and glandular damage in patients with hemochromatosis or hemosiderosis after repeated blood transfusions. Infection may involve one or more of the parathyroids but usually does not cause hypoparathyroidism because all four glands are rarely involved.
Transient hypoparathyroidism is frequent following surgery for hyperparathyroidism. After a variable period of hypoparathyroidism, normal parathyroid function may return due to hyperplasia or recovery of remaining tissue. Occasionally, recovery occurs months after surgery.
|
TREATMENT |
ACQUIRED AND HEREDITARY HYPOPARATHYROIDISM |
Treatment involves replacement with vitamin D or 1,25(OH)2D (calcitriol) combined with a high oral calcium intake. In most patients, blood calcium and phosphate levels are satisfactorily regulated, but some patients show resistance and a brittleness, with a tendency to alternate between hypocalcemia and hypercalcemia. For many patients, vitamin D in doses of 40,000–120,000 U/d (1–3 mg/d) combined with ≥1 g elemental calcium is satisfactory. The wide dosage range reflects the variation encountered from patient to patient; precise regulation of each patient is required. Compared to typical daily requirements in euparathyroid patients of 200 U/d (or in older patients as high as 800 U/d), the high dose of vitamin D (as much as 100-fold higher) reflects the reduced conversion of vitamin D to 1,25(OH)2D. Many physicians now use 0.5–1 μg of calcitriol in management of such patients, especially if they are difficult to control. Because of its storage in fat, when vitamin D is withdrawn, weeks are required for the disappearance of the biologic effects, compared with a few days for calcitriol, which has a rapid turnover.
Oral calcium and vitamin D restore the overall calcium-phosphate balance but do not reverse the lowered urinary calcium reabsorption typical of hypoparathyroidism. Therefore, care must be taken to avoid excessive urinary calcium excretion after vitamin D and calcium replacement therapy; otherwise, nephrocalcinosis and kidney stones can develop, and the risk of CKD is increased. Thiazide diuretics lower urine calcium by as much as 100 mg/d in hypoparathyroid patients on vitamin D, provided they are maintained on a low-sodium diet. Use of thiazides seems to be of benefit in mitigating hypercalciuria and easing the daily management of these patients.
There are now trials of parenterally administered PTH (either PTH[1–34] or PTH[1–84]) in patients with hypoparathyroidism providing greater ease of maintaining serum calcium and reducing urinary calcium excretion (desirable to protect any renal damage). However, PTH therapy for the treatment of hypoparathyroidism is not approved as of yet.
Hypomagnesemia Severe hypomagnesemia (<0.4 mmol/L; <0.8 meq/L) is associated with hypocalcemia (Chap. 423). Restoration of the total-body magnesium deficit leads to rapid reversal of hypocalcemia. There are at least two causes of the hypocalcemia—impaired PTH secretion and reduced responsiveness to PTH. For further discussion of causes and treatment of hypomagnesemia, see Chap. 423.
The effects of magnesium on PTH secretion are similar to those of calcium; hypermagnesemia suppresses and hypomagnesemia stimulates PTH secretion. The effects of magnesium on PTH secretion are normally of little significance, however, because the calcium effects dominate. Greater change in magnesium than in calcium is needed to influence hormone secretion. Nonetheless, hypomagnesemia might be expected to increase hormone secretion. It is therefore surprising to find that severe hypomagnesemia is associated with blunted secretion of PTH. The explanation for the paradox is that severe, chronic hypomagnesemia leads to intracellular magnesium deficiency, which interferes with secretion and peripheral responses to PTH. The mechanism of the cellular abnormalities caused by hypomagnesemia is unknown, although effects on adenylate cyclase (for which magnesium is a cofactor) have been proposed.
PTH levels are undetectable or inappropriately low in severe hypomagnesemia despite the stimulus of severe hypocalcemia, and acute repletion of magnesium leads to a rapid increase in PTH level. Serum phosphate levels are often not elevated, in contrast to the situation with acquired or idiopathic hypoparathyroidism, probably because phosphate deficiency is often seen in hypomagnesmia (Chap. 393).
Diminished peripheral responsiveness to PTH also occurs in some patients, as documented by subnormal response in urinary phosphorus and urinary cAMP excretion after administration of exogenous PTH to patients who are hypocalcemic and hypomagnesemic. Both blunted PTH secretion and lack of renal response to administered PTH can occur in the same patient. When acute magnesium repletion is undertaken, the restoration of PTH levels to normal or supranormal may precede restoration of normal serum calcium by several days.
|
TREATMENT |
HYPOMAGNESEMIA |
Repletion of magnesium cures the condition. Repletion should be parenteral. Attention must be given to restoring the intracellular deficit, which may be considerable. After IV magnesium administration, serum magnesium may return transiently to the normal range, but unless replacement therapy is adequate, serum magnesium will again fall. If the cause of the hypomagnesemia is renal magnesium wasting, magnesium may have to be given long-term to prevent recurrence (Chap. 423).
PTH INEFFECTIVE
PTH is not sufficiently active to fully prevent hypocalcemia (although retaining phosphaturic activity, for example). This problem occurs when the PTH1R–signaling protein complex is defective (as in the different forms of pseudohypoparathyroidism [PHP], discussed below); when PTH action to promote calcium absorption from the diet via the synthesis of 1,25(OH)2D is insufficient because of vitamin D deficiency or because vitamin D is ineffective (defects in vitamin D receptor or vitamin D synthesis); or in CKD in which the calcium-elevating action of PTH is impaired.
Typically, hypophosphatemia is more severe than hypocalcemia in vitamin D deficiency states because of the increased secretion of PTH, which, although only partly effective in elevating blood calcium, is readily capable of promoting urinary phosphate excretion.
PHP, on the other hand, has a pathophysiology that is different from the other disorders of ineffective PTH action. PHP resembles hypoparathyroidism (in which PTH synthesis is deficient) and is manifested by hypocalcemia and hyperphosphatemia, yet elevated PTH levels. The cause of the disorder is defective PTH-dependent activation of the stimulatory G protein complex or the downstream effector protein kinase A, resulting in failure of PTH to increase intracellular cAMP or to respond to elevated cAMP levels (see below).




