BLOOD SUPPLY TO THE HEART
Anatomy of the arterial supply and venous drainage of the Heart
The term ‘coronary’ was first conceived to describe a crown-like arrangement of the arterial blood vessels supplying the heart muscle. If the heart is considered as an upside-down cone with the flat base placed at the level of the atrioventricular groove, then the coronary arteries may be visualized as a ring at the base of the cone with branches which pass towards the tip (Fig. 5.1). This network of coronary arteries arises from two main origins in adjacent aortic sinuses. The anterosuperior sinus gives rise to the right coronary artery (RCA). This passes down the anterior atrioventricular (AV) groove. It gives rise to marginal branches which supply the anterior free wall of the right ventricle (RV). At the junction between the anterior and inferior aspects of the RV it gives off a significant branch—the acute marginal artery. It then passes inferiorly and in 70% of subjects terminates in a right posterior descending branch which supplies the inferior aspects of the RV, left ventricle (LV) and interventricular septum.

The sinoatrial node is supplied by a branch of the RCA in 60% of cases and from the circumflex artery in 40%. The AV node is supplied by the RCA in 90% of cases. Damage to vessels supplying portions of the conducting system may lead to specific defects. In the case of damage to the sinus nodal artery it may lead to ‘sick sinus syndrome’ in which the frequency of generation of cardiac action potentials becomes randomly variable and inappropriate (tachy-brady syndrome). Damage to the supply to the AV node or bundle of His may lead to complete heart block (see Chapter 3) and is particularly associated with inferior myocardial infarction.
An imbalance between the oxygen demands of the heart and amount that can be supplied by the coronary blood supply leads to the development of an hypoxic pain originating in the heart which is called angina (see p. 55). An outline of a clinical case history is shown in Case 5.1:1.
Regulation of coronary blood flow
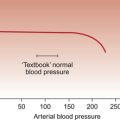
At rest the myocardium receives about 5% of cardiac output. In the normal ‘textbook’ person there is a potential for cardiac output to increase about fivefold during exercise (see Chapter 13). This is roughly paralleled by changes in coronary blood flow and the necessity for this is largely dictated by the high oxygen extraction rate of cardiac muscle.
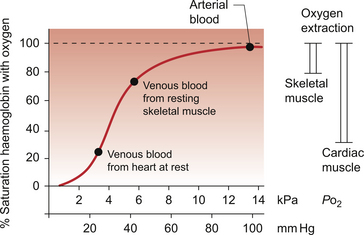
In skeletal muscle, under resting conditions, only of the order of 25–30% of the oxygen carried in arterial blood is extracted for use in the muscle (Fig. 5.2). The saturation of haemoglobin with oxygen in skeletal muscle therefore decreases from about 97–98% (arterial blood) to about 70% (venous blood). Even under resting conditions the venous drainage from cardiac muscle is only 25% saturated, meaning that of the order of 75% of the oxygen in arterial blood has been extracted and used metabolically. During exercise, increased oxygen delivery to contracting skeletal muscle can be provided by a combination of increased blood flow (see Chapter 13) but also by increased (up to 80–90%) extraction of oxygen from haemoglobin. In the heart as oxygen extraction at rest is already about 75% there is limited scope for increasing oxygen delivery by this route. Studies with 11C-acetate positron emission tomography (PET) scanning suggest that, in the heart, oxygen extraction from arterial blood can rise to 90% during exercise but even this is a limited way of increasing oxygen delivery. The bottom line is that if the heart needs increased oxygen supply it must be mainly provided by increased coronary blood flow. The corollary of this is that pathological mechanisms which impair coronary blood flow must limit cardiac performance.
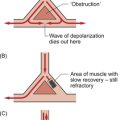
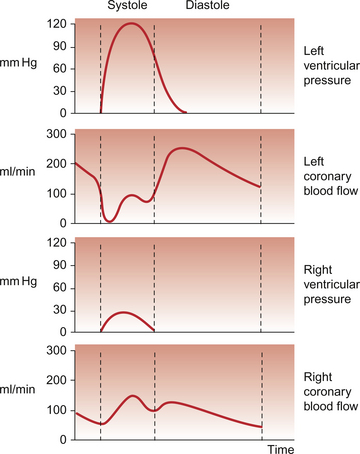
Coronary blood flow, particularly to the left ventricle, is particularly affected by the contraction of the myocardium which crushes coronary vessels (Fig. 5.3). This mainly affects blood vessels in the subendocardial layers of the heart muscle and the blood vessels on or close to the surface of the heart are relatively unaffected. The subendocardial layers are therefore more prone to ischaemic damage. In the left ventricle, because of the high pressures developed in the contracting ventricle, coronary blood flow is much higher during diastole than during systole. In the right side of the heart intraventricular pressures are lower and so the effect of ventricular systole on coronary blood flow is less marked. When heart rate increases during exercise the duration of diastole is shortened more markedly than the duration of systole. This imposes a limitation on increases in coronary blood flow and is probably the limiting factor on maximum exercise ability in normal individuals.
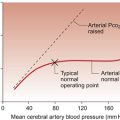
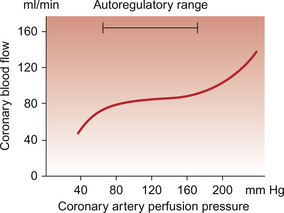
Coronary blood flow is autoregulated (Fig. 5.4). This means that over a range of mean arterial pressures, probably in humans from about 50 to 120 mm Hg, coronary blood flow is relatively independent of arterial pressure. This is thought to result especially from responses of arterioles which are less than 150 μm diameter. Thus, as the arterial pressure increases through the autoregulatory range the smooth muscle in the wall of these arterioles contracts to maintain flow constant.
The major regulatory factor determining coronary blood flow is myocardial oxygen demand coupled to the production of vasodilator metabolites. These metabolites particularly affect blood vessels in the 150–170 μm size range. The vascular smooth muscle is thought to be particularly sensitive to changes in [adenosine], [K+], [H+] and to local changes leading to an increase in interstitial osmolarity (see Chapter 9). A major part of this vasodilator action is mediated by the opening of ATP-sensitive K+ channels. This leads to hyperpolarization and consequently to relaxation of the smooth muscle.
The source of the vasodilator adenosine has been a subject of conjecture. Berne (1980) proposed that it was produced under hypoxic conditions by the complete dephosphorylation of ATP. An alternative pathway was put forward by Deussen (1989), in which adenosine was formed from ATP via the intermediate formation of S-adenosyl methionine and S-adenosyl homocysteine. Both pathways are thought to contribute to interstitial [adenosine]. The following points seem to be relevant to understanding these events. ATP, ADP and AMP are all polar molecules as a result of ionization of their phosphate groups and will not easily cross cell membranes. Adenosine is non-polar and can leave the myocardial cell once it has been formed. Adenosine in the interstitium has a very short half-life (of the order of 10 s) and so must be continuously generated. [ATP] inside cells is about 5 mmol/L but interstitial [adenosine] is about 10 nmol/L, a 500 000-fold difference in concentration. Therefore, only a very small proportion of the intracellular ATP would need to be metabolized to provide relatively big changes in interstitial [adenosine] (Fig. 5.5).
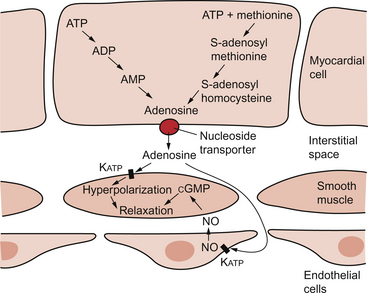
Endothelial influences on blood vessel diameter are described elsewhere (see Chapter 9). Increased shear stress on the endothelium leads to production of nitric oxide and thence to vasodilatation. This occurs particularly in large coronary arteries but it does not appear to contribute to the mechanism of autoregulation. Endothelial dysfunction leading to impaired nitric oxide release is a characteristic of a number of pathological conditions which will affect the coronary blood vessels including hypercholesterolaemia, atherosclerosis and hypertension. Assessment, prevention and treatment of endothelial dysfunction is emerging as an important area of clinical medicine, especially in relation to the coronary circulation.
Modulation of coronary blood flow via the sympathetic nervous system primarily acts through α1-adrenoceptors on relatively large vessels. Vessels less than 100 μm diameter predominantly have α2-adrenoceptors but α1-receptors are also present. Activation of either of these populations of α-receptors leads to vasoconstriction and this is the dominant sympathetically mediated response. In the past there has sometimes been confusion about the role of β-receptor-mediated vasodilatation. Although such receptors do exist in limited numbers on coronary vessels, the vasodilator response which follows β-agonist infusion is mainly a result of increased metabolite (e.g. adenosine) generation following an increased force of ventricular muscle contraction (i.e. an inotropic response). Coronary vasodilatation directly as a result of β-adrenoceptor activation is a very minor component of coronary vascular control.
Ischaemic heart disease
The most frequent cause of obstruction in a main coronary artery is atherosclerosis. As this is not confined to the coronary circuit but may develop in any major vessels in the high-pressure arterial side of the circulation the details of the pathogenesis of atherosclerotic lesions are described in Chapter 8.
Occlusion of the coronary arteries may become critical in various ways:
• Progressive narrowing of vessels by atheromatous plaques.
• The plaque and the associated reduction in blood flow velocity may provide a focus for thrombus formation with consequent further reduction in blood flow. The possibility also exists of embolization of the thrombus.
• With time, a plaque becomes a rigid structure but is still surrounded by pliable blood vessel wall. There is therefore a risk of rupture (fissuring) of the plaque which provides another site for thrombus formation. The processes involved in thrombus formation are described below.
Thrombosis
Change in blood flow is also an important factor. Slow flow, as occurs with incompetent venous valves and dilated veins, can lead to pooling of blood. On the arterial side of the circulation turbulent blood flow near atherosclerotic plaques or aneurysms can lead to damage to the endothelium (see Chapter 8). Both scenarios will lead to increased platelet–vessel wall interaction.
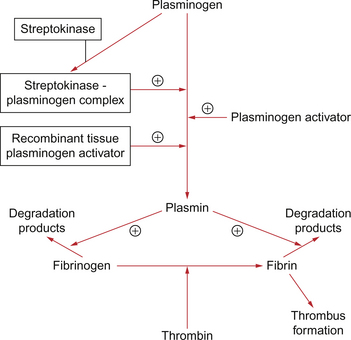
As soon as the platelet plug has formed, fibrinolytic systems are activated which prevent propagation of the thrombus (Fig. 5.6). However, the presence of one or more of Virchow’s triad will tip the scale towards thrombus formation.
Once a thrombus has formed, a number of ‘events’ may occur:
Lysis/dissolution
The fibrinolytic systems (Fig. 5.6) remove the thrombus and the vessel returns to normal.
Embolization
It is not just thrombus that can embolize. Fragments of cholesterol debris can split off from an atheromatous plaque, as for example in carotid artery atherosclerosis leading to cholesterol emboli which pass into the circulation of the eye or brain. Air, fat (especially after trauma or a major operation such as a hip replacement), amniotic fluid, tumour cells and foreign bodies can all embolize (Table 5.1).
Table 5.1
Types of embolus and their characteristics
| Embolic material | Characteristics |
| Thrombus (90% of major emboli) | Venous thrombosis usually from deep veins of legs (95% of cases) becomes pulmonary embolus |
| Thrombus forming over an atheromatous plaque or myocardial infarct or in a fibrillating atrium can give rise to systemic embolus. This leads to infarction, e.g. brain, kidneys, gut and limbs | |
| Atheromatous plaque debris | Frequent cause of problems in lower limbs |
| Infective emboli | Particularly from vegetations on heart valves produced by infective endocarditis |
| Fat | Generated during long bone trauma and in severe burns. Emboli travel to lungs, brain and kidney particularly |
| Gas | May occur during surgery (air embolus) or during rapid decompression of divers (nitrogen) |
| Amniotic fluid | Occurs via damaged uterine blood vessels at childbirth |
| Tumour tissue | Route for tumour metastasis |
| Foreign bodies | Small amounts of material produce a granulomatous reaction where they lodge. At-risk patients include intravenous drug users |
Anti-thrombotic therapy
In hospitalized patients the methods used to try to avoid the formation of thrombus depend on the risk involved. For moderate-risk patients mechanical methods such as elasticated stockings can be used. For higher-risk patients this may be supplemented with low-dose heparin. This is a sulphated mucopolysaccharide (glycosaminoglycan) molecule derived commercially from pig intestinal mucosa or the lungs of cattle. It forms a complex with the clotting cascade protein antithrombin which results in the activation of antithrombin. Thrombin promotes the last stage of the clotting mechanism, conversion of fibrinogen to fibrin (Fig. 5.6). Heparin also inhibits several other stages in the clotting cascade. It is inactive given orally and so must be administered by the intravenous or subcutaneous routes.
Thrombolytic therapy
There are a number of thrombolytic drugs available but the most commonly used are streptokinase and genetically engineered recombinant tissue plasminogen activator (rt-PA). All of the drugs activate plasminogen to form plasmin, an enzyme which promotes the breakdown of fibrin and fibrinogen into degradation products (Fig. 5.6). This leads to lysis of a thrombus and may result in some restoration of blood flow. How effective thrombolytic therapy is depends on factors such as the age of thrombus and the access of the drug to the thrombus.
Angina
Angina is pain which arises from areas of cardiac muscle which are underperfused and lack adequate supplies of oxygen. Typically it is a central, crushing chest pain, of variable severity. Classically it radiates to the left arm or into the neck. Its key feature is that it occurs on exertion and is relieved with rest. In some individuals the pain may be less apparent and the sensation is more of chest tightness and breathlessness (dyspnoea). Relief of the pain by the use of short-acting nitrates such as sublingual glyceryl trinitrate (GTN) (see below) is considered a useful diagnostic feature.
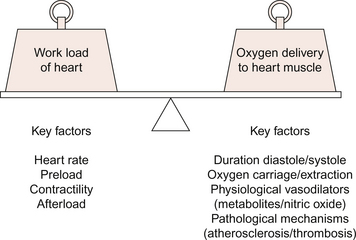
The hypoxia results from a discrepancy between demand for myocardial oxygen and maximum coronary blood flow capacity (Fig. 5.7). The key difference between angina and myocardial infarction is that with angina the myocardial hypoperfusion is reversible and does not cause permanent myocardial damage. A theory is that the pain is elicited by the interstitial accumulation of adenosine which activates unmyelinated nerve fibres. This is based on the observation that angina pain can be mimicked in normal individuals in a dose-related way by coronary artery infusion of adenosine. The lack of associated ECG changes shows that actual hypoxia is not occurring as a result of the adenosine infusion.
‘Unstable angina’ occurs at rest. It is presumably brought about by coronary vasospasm at resting levels of demand and may be difficult to distinguish from infarction, though the pain of infarction is usually more severe and prolonged. ECG changes will reflect myocardial ischaemia, i.e. ST segment depression rather than the elevation associated with infarction (see Chapter 7). Treatment should be supportive using vasodilators and anticoagulants in order to prevent progression to full infarction.
Drugs used in the management of angina
Strategies for the treatment of angina can be targeted at either increasing the coronary blood flow or decreasing the work done by the heart. In the latter case oxygen demand is decreased. This can be achieved by reducing the force of cardiac muscle contraction either by reducing preload on the heart or by reducing cardiac contractility (see Chapter 4). Other changes in workload of the heart can be achieved by reducing heart rate or arterial blood pressure (decreased afterload).
Four major classes of drugs are used:
The organic nitrates are exogenous sources of the natural vasodilator nitric oxide (see Chapter 9). The most widely used drug in this category is glyceryl trinitrate (GTN). There is an increasing number of other drugs including isosorbide dinitrate and isosorbide mononitrate. A major site of action of organic nitrates is on the venous capacitance vessels. Relaxing smooth muscle here leads to a reduced preload on the heart and thus reduces cardiac output (see Chapter 4). Vasodilatation on the arterial side of the circulation, particularly in this case of large arteries, leads to a reduction in blood pressure and reduced afterload on the heart. Effects of nitrates on coronary blood vessels are often minimal. Vessels may already be maximally dilated under the influence of local metabolites (Chapter 9) which have accumulated in the hypoxic cardiac tissue. Nitrates may help to improve flow through collateral vessels and also relieve coronary artery spasm when that is a cause of angina.
β-adrenoceptor blockers (beta-blockers) reduce the force of cardiac contraction (reduce contractility) and lower blood pressure (reduce afterload) (see Chapter 4). These effects reduce oxygen demand by the heart and limit exercise performance. There is also a reduction in heart rate and, with the consequent lengthening of diastole, the phase of the cardiac cycle when most of the coronary perfusion occurs is prolonged. Some β-adrenoceptor blockers, such as atenolol, are referred to as ‘cardioselective’ as they are relatively selective for the β1-subtype of receptors, the main type found in the heart. The first β-adrenoceptor blocker developed, propranolol, is non-selective and has approximately equivalent actions on β1 and β2 receptors. This drug is still widely used but is contraindicated in asthmatics as the β2 blockade may lead to bronchospasm.
The main site of action of calcium channel blockers is on voltage-gated L-type (long-acting) slow Ca++ channels. Voltage-gated T-type (transient) channels are also blocked in pacemaker tissue of the sinoatrial and atrioventricular nodes (see Chapter 4). Beneficial effects of calcium channel blockers in the relief of angina include systemic arteriolar vasodilatation (reduced arterial blood pressure and thus reduced afterload), coronary artery dilatation (in the case of vasospasm), a reduction in heart rate and reduced cardiac contractility (reduced workload). It should be stressed that the different drugs in this broad category have a wide spectrum of activity and they are not identical. For example, verapamil and diltiazem both reduce heart rate but the dihydropyridine drugs nifedipine and amlodipine do not. Amlodipine also does not significantly reduce cardiac contractility but, like nifedipine, it has marked effects leading to arteriolar vasodilatation.
Myocardial infarction
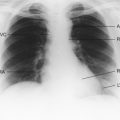
Myocardial infarction (MI) results when there is complete interruption of blood flow to an area of myocardium. It involves necrosis of cardiac muscle followed by inflammatory cell infiltration and, because cardiac myocytes cannot regenerate, eventual fibrous repair. The subendocardial tissue in the left ventricle, which as described earlier is most prone to ischaemia, is most at risk. Figure 5.8 shows an infarcted area of tissue. The classical model for infarction is rupture of an atherosclerotic plaque (see Chapter 8) with thrombosis (see p. 53) and vasospasm completely occluding the lumen of a critical blood vessel. Frequently this is one of the major epicardial blood vessels described at the start of this chapter and shown in Figure 5.1. The infarction occurs downstream from the occluded blood vessel. As already noted, there is considerable variation in the anatomy and distribution of the main coronary arteries. However, some generalizations can be made regarding common sites of obstruction.

Fig. 5.8 Infarcted area of tissue. Myocardial infarct (I) in the lateral wall of the left ventricle. Source: Stevens A, Lowe J 2000.
• Left anterior descending artery obstruction accounts for about 50% of cases. It produces infarcts in the anterior wall of the left ventricle with characteristic ECG changes in the anterior chest leads (V1–V3). Occlusion of this artery is sometimes a cause of sudden death.
• Right coronary artery obstruction occurs in about 30% of cases and leads to inferior wall infarction and sometimes posterior septum infarcts. The ECG changes are seen in leads II, III and aVF.
• Circumflex artery obstruction occurs in about 20% of cases and leads to lateral wall infarction with ECG changes in leads I, aVL and the lateral chest leads (V4–V6).
Investigation of myocardial infarction
MI is typically identified with characteristic ECG changes and increases in the serum level of proteins released from the disrupted myocardial cells. In the past plasma or serum measurements of total creatine kinase (CK), aspartate aminotransferase (also known as serum glutamic oxaloacetic transaminase, SGOT) and total lactate dehydrogenase (LDH) have been used as indicators of cardiac necrosis. However these enzymes are widely distributed in the body and lack specificity to cardiac tissue. More recently the use of other markers has increased. The MB isoenzyme of CK is found in the heart and levels in blood do not start to rise until 4 hours after infarction. CK-MB levels fall again within 72 hours. The CK-MB test is frequently used to provide early confirmation of a diagnosis of MI. The more specific markers troponins T and I are now the gold standard for myocardial cell necrosis as these structural proteins are found solely in myocardial cells. Their physiological function is in the coupling of a rise in intracellular [Ca++] to cross-bridge formation as part of the contraction of cardiac muscle (see Chapter 2). Troponin T and I levels may be modestly raised following the cardiac hypoxia associated with unstable angina. After an acute MI troponin levels are increased within 3–6 hours, reach a peak within 14–20 hours and return to normal after 5–7 days.
Since thrombosis appears to be an important component of the process of infarction the use of thrombolytic therapy, e.g. streptokinase or recombinant tissue-plasminogen activators such as alteplase or reteplase, has become a central component of care in suspected MI. These compounds, rather than being simply anticoagulant, positively promote activation of the fibrinolytic system thereby helping to break down the clot (see p. 54).
Chilian, W. M., Gutterman, D. D. Prologue: new insights into the regulation of the coronary microcirculation. Am. J. Physiol.. 2000; 48:H2585–H2586. [[This paper provides an introduction to a series of specialist reviews on different aspects of coronary blood vessels. ]].
Cohen, M. V., Baines, C. P., Downey, J. M. Ischaemic preconditioning: from adenosine receptor to KATP channel. Annu. Rev. Physiol.. 2000; 62:79–109.
Di Carli, M. F., Tobes, M. C., Mangner, T., et al. Effects of cardiac sympathetic innervation on coronary blood flow. New Engl. J. Med.. 1997; 336:1208–1216.
Foreman, R. D. Mechanisms of cardiac pain. Annu. Rev. Physiol.. 1999; 61:143–167.
Gallagher, P. J. Cardiovascular system. In Underwood J. C. E., Cross S. S., eds. : General and Systematic Pathology, fifth ed., Edinburgh: Churchill Livingstone, 2009.
Jones, J. H., Kuo, L., David, M. J., Chilian, W. M. Regulation of coronary blood flow: co-ordination of heterogeneous control mechanisms in vascular microdomains. Cardiovasc. Res.. 1995; 29:585–596.
Stevens, A., Lowe, J. Pathology, second ed. Edinburgh: Mosby; 2000.
Waller, D. G., Renwick, A. G., Hillier, K. Medical Pharmacology and Therapeutics, third ed. Edinburgh: WB Saunders; 2009.
Widlansky, M. E., Gokce, N., Keaney, J. F., Vita, J. A. The clinical implications of endothelial dysfunction. J. Am. Coll. Cardiol.. 2003; 42:1149–1160.
Wolfe, J. H. N. ABC of Vascular Diseases. London: BMJ Books; 1992.