Chapter 5 Blood cell morphology in health and disease
Examination of blood films
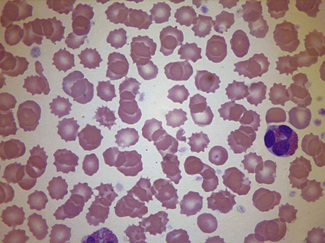
Having selected a suitable area, a ×40 or ×50 objective or ×60 oil-immersion objective should then be used. A much better appreciation of variation in red cell size, shape and staining can be obtained with one of these objectives than with the ×100 oil-immersion lens. It should be possible to detect features such as toxic granulation or the presence of Howell–Jolly bodies or Pappenheimer bodies. The major part of the assessment of a blood film is usually done at this power. The ×100 objective in combination with ×6 or ×10 eyepieces should be used only for the final examination of unusual cells and for looking at fine details such as basophilic stippling (punctate basophilia) or Auer rods. Whether it is necessary to examine a film with a ×100 objective depends on the clinical features, the blood count and the nature of any morphologic abnormality detected at lower power (Fig. 5.1).
Red cell morphology
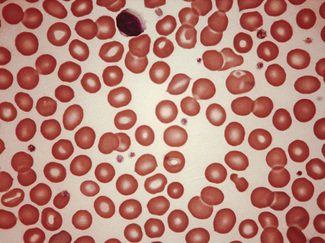
In health, the red blood cells vary relatively little in size and shape (Fig. 5.1). In well-spread, dried and stained films the great majority of cells have round, smooth contours and diameters within the comparatively narrow range of 6.0–8.5 μm. As a rough guide, normal red cell size appears to be about the same as that of the nucleus of a small lymphocyte on the dried film (Fig. 5.1). The red cells stain quite deeply with the eosin component of Romanowsky dyes, particularly at the periphery of the cell as a result of the cell’s normal biconcavity. A small but variable proportion of cells in well-made films (usually <10%) are definitely oval rather than round, and a very small percentage may be contracted and have an irregular contour or appear to have lost part of their substance as the result of fragmentation (schistocytes). According to Marsh, the percentage of ‘pyknocytes’ (irregularly contracted cells) and schistocytes in blood from healthy adults does not exceed 0.1% and the proportion is usually considerably less than this, whereas in normal, full-term infants the proportion is higher, 0.3–1.9%, and in premature infants it is still higher, up to 5.6%.1
Normal and pathological red cells are subject to considerable distortion in the spreading of a film and, as already mentioned, it is imperative to scan films carefully to find an area where the red cells are least distorted before attempting to examine the cells in detail. Such an area can usually be found toward the tail of the film, although not actually at the tail. Rouleaux often form rapidly in blood after withdrawal from the body and may be conspicuous even in films made at a patient’s bedside. They are particularly noticeable in the thicker parts of a film that have dried more slowly. Ideally, red cells should be examined in an area in which there are no rouleaux and the red cells are touching but with little overlap. The film in the chosen area must not be so thin as to cause red cell distortion; if the tail of the film is examined, a false impression of spherocytosis may be gained. The varying appearances of different areas of the same blood film are illustrated in Figures 5.2–5.4. The area illustrated in Figure 5.2 would clearly be the best for looking at red cells critically.
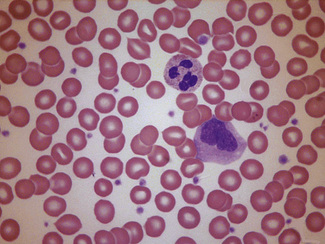
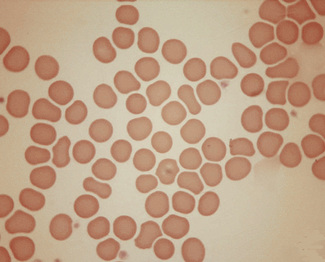
Figure 5.3 Photomicrograph of a blood film showing an area that is too thin for examination (same film as Fig. 5.2).
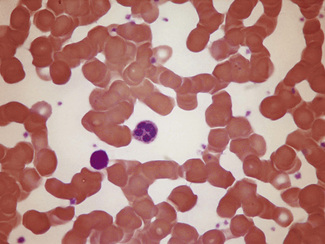
Figure 5.4 Photomicrograph of a blood film showing an area that is too thick for examination (same film as Fig. 5.2).
The advantages and disadvantages of examining red cells suspended in plasma have been referred to briefly in Chapter 4 (see p. 63). By this means, red cells can be seen in the absence of artefacts produced by drying, and abnormalities in size and shape can be better and more reliably appreciated than in films of blood dried on slides. However, the ease and rapidity with which dried films can be made, and their permanence, give them an overwhelming advantage in routine studies.
In disease, abnormality in the red cell picture stems from four main causes, which lead to characteristic cytological abnormalities (Table 5.1).
Table 5.1 Mechanisms of red cell abnormalities and resultant cytological features
| Cause | Resultant abnormality |
|---|---|
| Attempts by the bone marrow to compensate for anaemia by increased erythropoiesis | Signs of less mature cells in the peripheral blood (polychromasia and erythroblastaemia) |
| Inadequate synthesis of haemoglobin | Reduced or unequal haemoglobin content and concentration (hypochromia, anisochromasia or dimorphism) |
| Abnormal erythropoiesis, which may be effective or ineffective | Increased variation in size (anisocytosis) and shape (poikilocytosis), basophilic stippling, sometimes dimorphism |
| Damage to, or changes affecting, the red cells after leaving the bone marrow, including the effects of reduced or absent splenic function | Spherocytosis, irregular contraction, elliptocytosis, ovalocytosis or fragmentation (schistocytosis); the presence of Pappenheimer bodies, Howell–Jolly bodies and variable numbers of certain specific poikilocytes (target cells, acanthocytes and spherocytes) |
Abnormal erythropoiesis
Anisocytosis (αυισoζ, unequal) and Poikilocytosis (πoικιλoζ, varied)
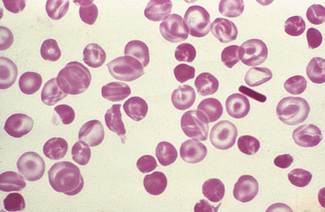
Anisocytosis and poikilocytosis are non-specific features of almost any blood disorder. The terms imply more variation in size or shape than is normally present (Figs 5.5, 5.6). Anisocytosis may be a result of the presence of cells larger than normal (macrocytosis), cells smaller than normal (microcytosis) or both; frequently both macrocytes and microcytes are present (Fig. 5.5).
Poikilocytes are produced in many types of abnormal erythropoiesis, for example, megaloblastic anaemia (Fig. 5.7), iron deficiency anaemia, thalassaemia, myelofibrosis (both idiopathic and secondary) (Fig. 5.8), congenital dyserythropoietic anaemia (Fig. 5.9) and the myelodysplastic syndromes. Elliptocytes and ovalocytes are among the poikilocytes that may be present when there is dyserythropoiesis; they are often present in megaloblastic anaemia (macro-ovalocytes) and in iron deficiency anaemia (‘pencil cells’), but they may also be seen in myelodysplastic syndromes and in primary myelofibrosis (Fig. 5.8). The number of elliptocytes and teardrop poikilocytes has been observed to correlate with the severity of iron deficiency anaemia.2 Poikilocytes are not only characteristic of disordered erythropoiesis but are also seen in various congenital haemolytic anaemias caused by membrane defects and in acquired conditions such as microangiopathic haemolytic anaemia and oxidant damage; in these disorders, the abnormality of shape results from damage to cells after formation and is described later in this chapter.
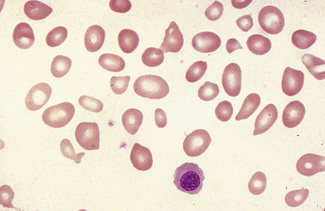
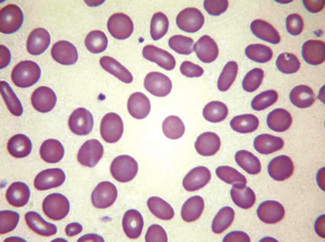
Macrocytes
Classically found in megaloblastic anaemias (Fig. 5.10), but macrocytes are also present in some cases of aplastic anaemia, myelodysplastic syndromes and other dyserythropoietic states. In patients being treated with hydroxycarbamide (previously known as hydroxyurea) the red cells are often macrocytic. A common cause of macrocytosis is excess alcohol intake, and it occurs in alcoholic and other types of chronic liver disease. In these conditions, the red cells tend to be fairly uniform in size and shape and there may also be stomatocytes (Fig. 5.11). In the rare type III form of congenital dyserythropoietic anaemia, some of the macrocytes are exceptionally large. Another rare cause of macrocytosis is benign familial macrocytosis.3 Macrocytosis also occurs whenever there is an increased rate of erythropoiesis, because of the presence of reticulocytes. Their presence is suspected in routinely stained films because of the slight basophilia, giving rise to polychromasia (see p. 86) and is easily confirmed by special stains (e.g. New methylene blue, see p. 33). These polychromatic macrocytes should be distinguished from other macrocytes because the diagnostic significance is quite different.
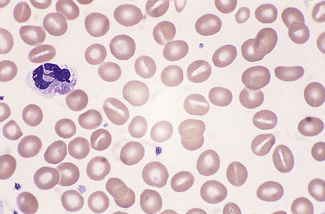
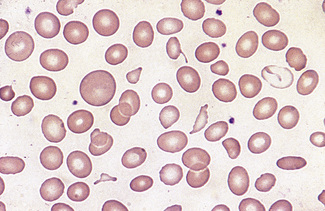
Microcytes
The presence of microcytes usually results from a defect in haemoglobin formation. Microcytosis is characteristic of iron deficiency anaemia (Fig. 5.12), various types of thalassaemia (Fig. 5.13), and severe cases of anaemia of chronic disease. Causes that are rarer include congenital and acquired sideroblastic anaemias. Microcytosis related to a defect in haemoglobin synthesis should be distinguished from red cell fragmentation or schistocytosis (see p. 80). Both abnormalities can lead to a reduction of the mean cell volume (MCV). However, it should be noted that a low MCV is common in association with a defect in haemoglobin synthesis, whereas it is uncommon in fragmentation syndromes because the fragments usually comprise only a small percentage of erythrocytes.
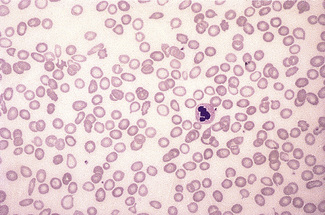
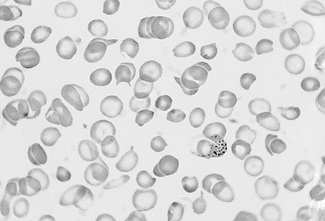
Basophilic Stippling
Basophilic stippling or punctate basophilia means the presence of numerous basophilic granules distributed throughout the cell (Fig. 5.14); in contrast to Pappenheimer bodies (see below), they do not give a positive Perls’ reaction for ionized iron. Punctate basophilia has quite a different significance from diffuse cytoplasmic basophilia. It is indicative of disturbed rather than increased erythropoiesis. It occurs in many blood diseases: thalassaemia, megaloblastic anaemias, infections, liver disease, poisoning by lead and other heavy metals, unstable haemoglobins and pyrimidine-5′-nucleotidase deficiency.4
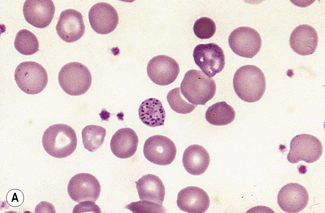
Inadequate haemoglobin formation
Hypochromia (Hypochromasia) (υπoρ, under)
The term hypochromasia, or now, more often, hypochromia, refers to the presence of red cells that stain unusually palely. (In doubtful cases, it is wise to compare the staining of the suspect film with that of a normal film stained at the same time.) There are two possible causes: a lowered haemoglobin concentration and abnormal thinness of the red cells. A lowered haemoglobin concentration results from impaired haemoglobin synthesis. This may stem from failure of haem synthesis – iron deficiency is a very common cause (Fig. 5.15) and sideroblastic anaemia (Fig. 5.16) is a rare cause – or failure of globin synthesis as in the thalassaemias (Fig. 5.17). Haemoglobin synthesis may also be impaired in chronic infections and other inflammatory conditions. Abnormally thin red cells (leptocytes) (see p. 82) can be the result of a defect in haemoglobin synthesis (e.g. in thalassaemias and iron deficiency) but they also occur in liver disease. It cannot be too strongly stressed that a hypochromic blood picture does not necessarily mean iron deficiency, although this is the most common cause. In iron deficiency, the red cells are characteristically hypochromic and microcytic, but the extent of these abnormalities depends on the severity; hypochromia may be minor and may be overlooked if the haemoglobin concentration (Hb) exceeds 100 g/l. In heterozygous or homozygous α+ thalassaemia, heterozygous α° thalassaemia or heterozygous β thalassaemia, hypochromia is often less marked, in relation to the degree of microcytosis, than in iron deficiency. The presence of target cells or basophilic stippling also favours a diagnosis of thalassaemia trait rather than iron deficiency. In homozygous β thalassaemia, the abnormalities are greater than in iron deficiency at the same Hb and nucleated red cells are usually present, whereas they are not a feature of iron deficiency. If the patient is being transfused regularly, normal donor cells will also be present, producing a dimorphic blood film (Fig. 5.18).
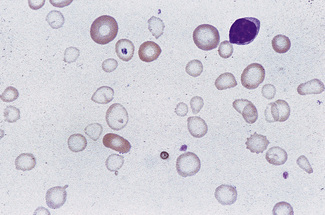
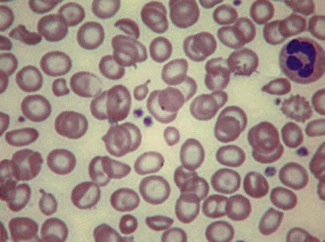
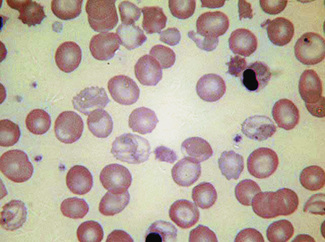
Anisochromasia (αυισoζ, unequal) and Dimorphic Red Cell Population
A distinction should be made between anisochromasia, in which there is abnormal variability in staining of red cells, and a dimorphic picture, in which there are two distinct populations. Anisochromasia, in which some but not all of the red cells stain palely, is characteristic of a changing situation. It can occur during the development or resolution of iron deficiency anaemia (Fig. 5.19) or the anaemia of chronic disease. In thalassaemia trait, in contrast, anisochromasia is much less common. A dimorphic blood film can be seen in several circumstances. It can occur when an iron deficiency anaemia responds to iron therapy, after the transfusion of normal blood to a patient with a hypochromic anaemia (Fig. 5.19), and in sideroblastic anaemia (Fig. 5.20). In acquired sideroblastic anaemia as a feature of a myelodysplastic syndrome, the two populations of cells are usually hypochromic microcytic and normochromic macrocytic, respectively.
Damage to red cells after formation
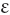 ρ, over)
ρ, over)Spherocytosis (σϕαιρα, a sphere)
Spherocytes are cells that are more spheroidal (i.e. less disc-like) than normal red cells but maintain a regular outline. Their diameter is less and their thickness is greater than normal. Only in extreme instances are they almost spherical in shape. It is useful to draw a distinction between spherocytes of normal size and microspherocytes; the latter result from red cell fragmentation or from removal of a considerable proportion of the red cell membrane by splenic or other macrophages. Spherocytes may result from genetic defects of the red cell membrane as in hereditary spherocytosis (Fig. 5.21); from the interaction between immunoglobulin- or complement-coated red cells and phagocytic cells, as in delayed transfusion reactions, ABO haemolytic disease of the newborn (Fig. 5.22) and autoimmune haemolytic anaemia (Fig. 5.23); and from the action of bacterial toxins (e.g. Clostridium perfringens lecithinase; Fig. 5.24).
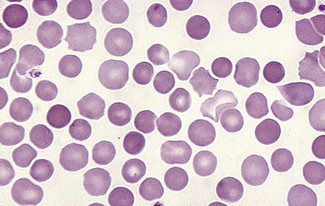
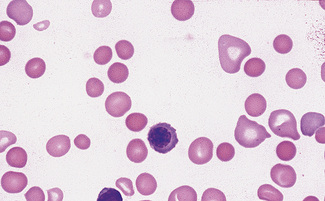
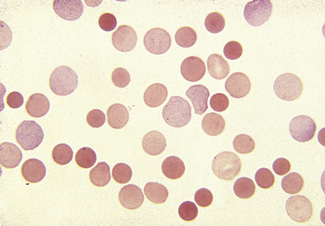
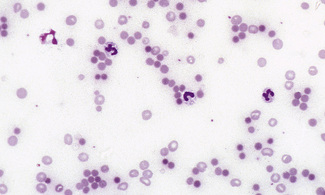
Spherocytes usually appear perfectly round in contour in stained films; they have to be carefully distinguished from both irregularly contracted cells and ‘crenated spheres’ or sphero-echinocytes (Fig. 5.25), which are the end result of crenation (see p. 82). Sphero-echinocytes develop as artefacts, especially in blood that has been allowed to stand before films are spread (Fig. 5.26). The blood film of a patient who has been transfused with stored blood may show a proportion of sphero-echinocytes (Fig. 5.27).
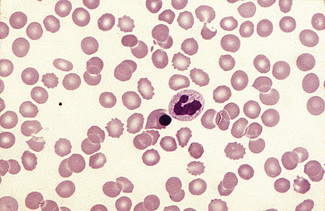
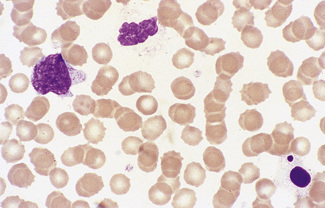
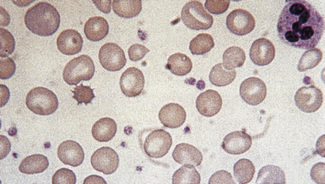
Irregularly Contracted Red Cells
There are a number of causes of irregularly contracted cells. In drug- or chemical-induced haemolytic anaemias, a proportion of the red cells are smaller than normal and unusually densely stained (i.e. they appear contracted) and their margins are slightly or moderately irregular and may be partly concave (Fig. 5.28). These may be cells from which Heinz bodies have been extracted by the spleen. Similar cells may be seen in films of some unstable haemoglobinopathies before splenectomy (e.g. that caused by the presence of Hb Köln or Hb St Mary’s; Fig. 5.29) and in haemoglobin E homozygosity (Fig. 5.30) and, to a lesser extent, haemoglobin E heterozygosity. Heinz bodies are not normally visible in Romanowsky-stained blood films, but they may be seen in such films as pale pink–staining bodies at the cell margin or even protruding from the erythrocytes in severe unstable haemoglobin haemolytic anaemias after splenectomy and in acute oxidant-induced haemolytic anaemia, including that occurring in deficiency of glucose-6-phosphate dehydrogenase. An extreme degree of irregular contraction is characteristic of severe favism or any other very acute haemolytic episode in individuals who are glucose-6-phosphate dehydrogenase deficient. It is typical to see cells in which the haemoglobin appears to have contracted away from the cell membrane, an appearance sometimes referred to as a hemi-ghost (Fig. 5.31); there may also be ghost cells – cells with almost empty membranes containing negligible haemoglobin. Irregularly contracted cells can be seen in small numbers in β thalassaemia trait and in heterozygosity for haemoglobin C. There may be a considerable number in haemoglobin C homozygosity and in this condition haemoglobin C crystals may also be seen (Fig. 5.32).
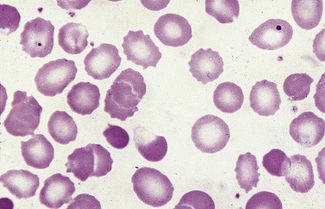
A type of irregular contraction of unknown origin has been described by the term ‘pyknocytosis’.5 The pyknocytes closely resemble chemically damaged red cells. As already mentioned (see p. 70), a small number of pyknocytes may be found in the blood of infants in the first few weeks of life, especially in premature infants. The term ‘infantile pyknocytosis’ refers to a transient haemolytic anaemia, related to glutathione peroxidase and selenium deficiency, affecting infants in whom many pyknocytes are present (Fig. 5.33).5,6
Elliptocytosis and Ovalocytosis
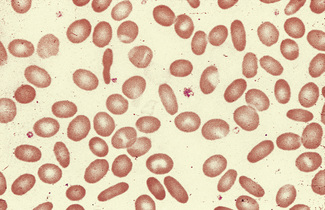
Elliptocytes are often present in large numbers in hereditary elliptocytosis (Fig. 5.34). In hereditary pyropoikilocytosis, elliptocytes are only one of the many types of poikilocyte present (Fig. 5.35). South-east Asian ovalocytosis is characterized by the presence of a variable number of elliptocytes, macro-ovalocytes and stomatocytes (Fig. 5.36). In all these conditions, the reticulocytes are round in contour (i.e. the cell assumes an abnormal shape only in the late stages of maturation). Although the causative condition is inherited, the abnormalities of red cell shape only become apparent as the cells reach maturity.
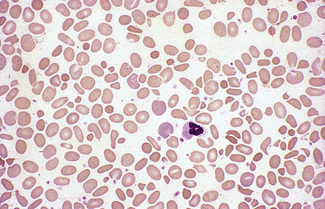
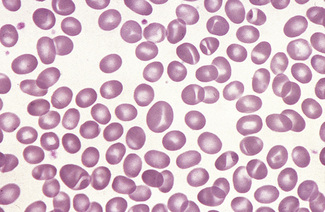
Spiculated cells and red cell fragmentation
The terminology applied to spiculated cells has been confusing because the same terms have been used to designate different types of cells. For this reason the term ‘burr cell’ should be discarded and the terms recommended by Bessis7 should be adopted. On the basis of scanning electron microscopy (discussed later), he distinguished four types of spiculated cell – schistocyte, keratocyte, acanthocyte and echinocyte. The term echinocyte is used for the crenated cell. It is differentiated from the acanthocyte on the basis of the number, shape and disposition of the spicules.
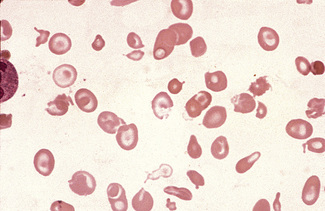
Schistocytosis (Fragmentation) (σχιστoζ, cleft)
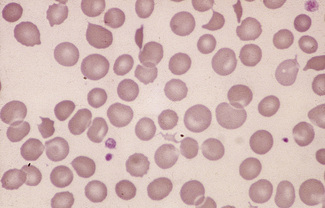
Figure 5.37 Photomicrograph of a blood film. Microangiopathic haemolytic anaemia. Shows angular red cell fragments.
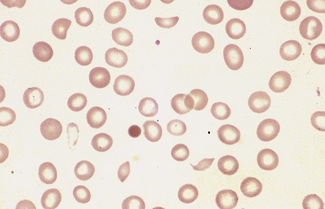
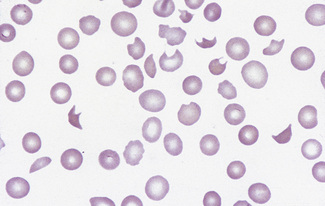
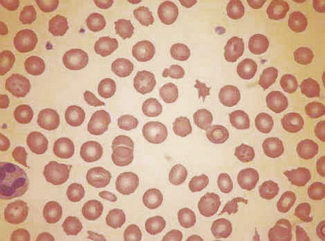
In burns, schistocytes are often rounded, being either microspherocytes or very small disc-shaped fragments. In addition, erythrocytes may be seen to be budding off small rounded blebs of cytoplasm. Not infrequently, as, for instance in the haemolytic-uraemic syndrome in children, the blood picture is made more bizarre by the superimposition of varying degrees of echinocytic change. Schistocytes are also a feature of thrombotic thrombocytopenia purpura.8
Keratocytes (κ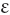 ραζ, horn)
ραζ, horn)
Keratocytes have pairs of spicules, usually either one pair or two pairs. They may be formed either by removal of a Heinz body by the pitting action of the spleen (Fig. 5.42) or by mechanical damage (Figs 5.43, 5.44). The terms ‘helmet cell’ and ‘bite cell’ have sometimes been used to describe keratocytes.
Acanthocytosis (ακανθα, spine)
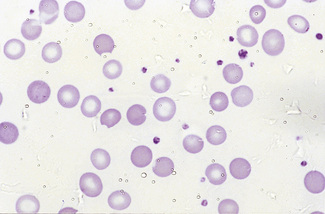
The term acanthocytosis was introduced to describe an abnormality of the red cell in which there are a small number of spicules of inconstant length, thickness and shape, irregularly disposed over the surface of the cell (Fig. 5.45). They are often associated with abnormal phospholipid metabolism9–11 or with inherited abnormalities of red cell membrane proteins, as in the McLeod phenotype, caused by lack of the Kell precursor (Kx).12 They are present in varying numbers following splenectomy and in hyposplenism. A similar cell occurs in severe liver disease (‘spur cell’ anaemia).13
Echinocytosis (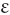 χινoζ, sea-urchin or hedgehog)
χινoζ, sea-urchin or hedgehog)
Echinocytosis or crenation describes the process by which red cells develop numerous short, regular projections from their surface (Figs 5.25, 5.26). First described by Ponder14 as disc-sphere transformation, crenation has many causes. A few crenated cells may be seen in many blood films, even in those from healthy subjects. Crenation regularly develops if blood is allowed to stand overnight at 20°C before films are made (Fig. 5.26). It may be a marked feature, for obscure and probably diverse reasons, in freshly made blood films of patients suffering from a variety of illnesses, especially uraemia. Marked echinocytosis has been reported in premature infants after exchange transfusion or transfusion of normal red cells.15 When crenation is superimposed on an underlying abnormality, the red cells may appear bizarre in the extreme.
Miscellaneous erythrocyte abnormalities
Leptocytosis (λ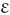 πτoζ, thin)
πτoζ, thin)
The term leptocytosis has been used to describe unusually thin red cells, as in severe iron deficiency or thalassaemia in which the cells may stain as rings of membrane with a little attached haemoglobin with large, almost unstained, central areas (Fig. 5.46). They can also occur when there is an excess of membrane in relation to cytoplasm, as can occur in liver disease.
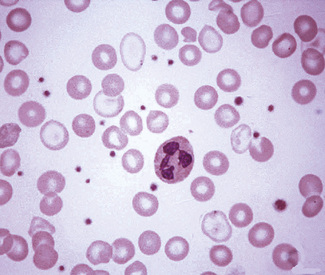
Target Cells
The term target cell refers to a cell in which there is a central round stained area and a peripheral rim of haemoglobinized cytoplasm separated by non-staining or more lightly staining cytoplasm. Target cells result from cells having a surface that is disproportionately large compared with their volume. They may be normal in size, microcytic or macrocytic. They are seen in films in chronic liver diseases in which the cell membrane may be loaded with cholesterol (Fig. 5.47), in hereditary hypo-betalipoproteinaemia,16 and in varying numbers in iron deficiency anaemia and in thalassaemia (Fig. 5.46). They are often conspicuous in certain haemoglobinopathies (e.g. haemoglobin C/β° thalassaemia; Fig. 5.48), haemoglobin C disease (Fig. 5.49), haemoglobin H disease (Fig. 5.50), sickle cell anaemia, sickle cell/haemoglobin C disease (Fig. 5.51), sickle cell/β thalassaemia, and haemoglobin E disease. Smaller numbers are usual in haemoglobin C trait, haemoglobin E trait and postsplenectomy. Splenectomy in thalassaemia may result in an extreme degree of leptocytosis and target cell formation.
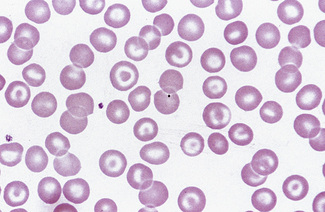
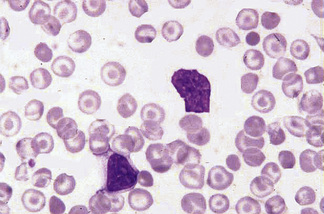
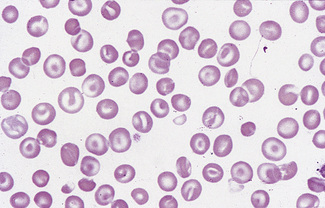
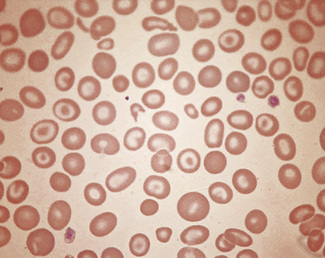
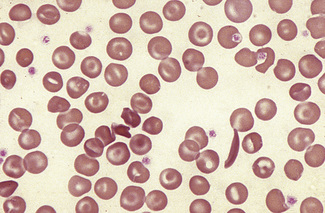
Stomatocytosis (στoμα, mouth)
Stomatocytes are red cells in which the central biconcave area appears slit-like in dried films. In ‘wet’ preparations, the stomatocyte is a cup-shaped red cell. The slit-like appearance of the cell’s concavity, as seen in dried films, is thus to some extent an artefact. The term was first used to describe the appearance of some of the cells in a rare type of haemolytic anaemia, hereditary stomatocytosis.17 They are also a feature of south-east Asian ovalocytosis. They have been described as being particularly frequent in films of Australians of Mediterranean origin.18,19 Subsequently, stomatocytes were recognized in acquired conditions and occasionally they are prominent (Fig. 5.52). They are observed in liver disease, in alcoholism,20 and occasionally in the myelodysplastic syndromes. There is a suspicion that in some films the occurrence of stomatocytosis is an in vitro artefact because it is known that the change can be produced by decreased pH and as the result of exposure to cationic detergent-like compounds and non-penetrating anions.21
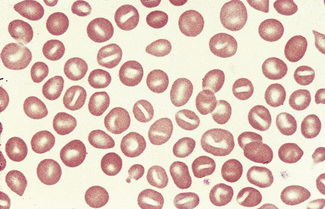
Sickle Cells
The varied film appearances in sickle cell anaemia are illustrated in Figures 5.53–5.55. Sickle cells are almost always present in films of freshly withdrawn blood of adults with homozygosity for haemoglobin S. However, sickle cells are usually absent in neonates and are rare in adult patients with a high haemoglobin F percentage. Sometimes many irreversibly sickled cells are present, and in all cases massive sickling takes place when the blood is subjected to anoxia (see p. 315). In films of fresh blood, the sickled cells vary in shape between boat-shaped forms and sickles. Target cells are also often a feature of blood films from patients with sickle cell anaemia, and Howell–Jolly bodies are found when there is splenic atrophy.
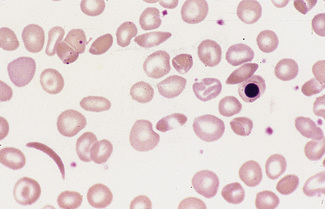
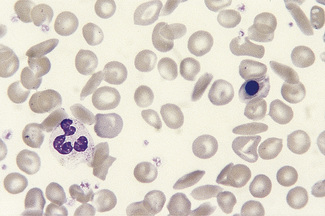
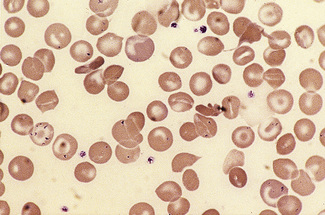
Haemoglobin C Crystals and SC Poikilocytes
In patients with homozygosity for haemoglobin C, target cells and irregularly contracted cells are usually numerous and there may be occasional straight-edged haemoglobin C crystals, either apparently extracellularly (Fig. 5.32) or within the ghost of a red cell. In patients who are compound heterozygotes for both haemoglobin S and haemoglobin C, the film sometimes resembles that of haemoglobin C disease (Fig. 5.32). In other patients, there are elliptical cells, rare sickle cells and sometimes distinctive SC poikilocytes (Fig. 5.56).
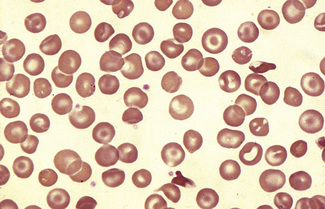
Erythrocyte Inclusions
Howell–Jolly Bodies
Howell–Jolly bodies are nuclear remnants. They are small, round cytoplasmic inclusions that stain purple on a Romanowsky stain. They are regularly present after splenectomy and when there is splenic atrophy (Fig. 5.57). They may be seen in a small percentage of red cells in pernicious anaemia. Usually only a few such inclusions are present, but they may be numerous in cases of coeliac disease and in other conditions in which there is splenic atrophy and megaloblastosis.
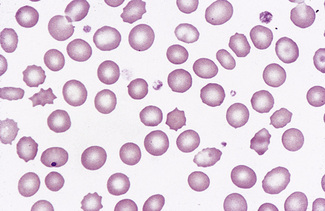
Pappenheimer Bodies
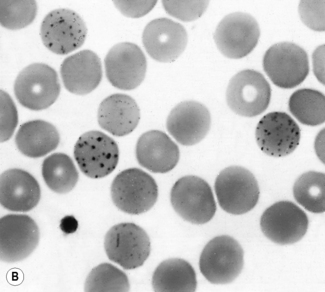
Pappenheimer bodies are small peripherally sited basophilic (almost black) erythrocyte inclusions. They are smaller than Howell–Jolly bodies. Usually only a small number are present in a cell. They are composed of haemosiderin and their presence is related to sideroblastic erythropoiesis and hyposplenism (Figs 5.58, 5.59). Sometimes they are found in the majority of circulating red cells. Their nature can be confirmed by means of a Perls’ stain. They correspond to the siderotic granules of siderocytes and are never distributed in large numbers throughout the cells as in classical punctate basophilia. However, a single cell may show both punctate basophilia and Pappenheimer bodies. With Perls’ stain, the former granules are pink, whereas the latter are blue.
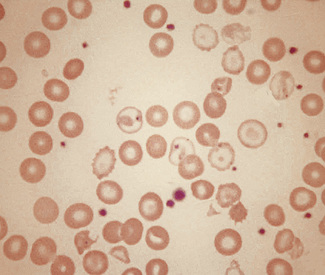
Rouleaux and Autoagglutination
Rouleaux occur to some extent in all films but increased rouleaux formation is significant. The differences between rouleaux and autoagglutination are described on p. 63, and there is usually no difficulty in determining which is which in stained films (Figs 5.60, 5.61). However, in myelomatosis and in other conditions in which there is intense rouleaux formation, the rouleaux may simulate autoagglutination. Even so, if the film, apparently showing autoagglutination, is carefully scanned, an area in which rouleaux can be clearly seen will almost certainly be found, emphasizing the importance of careful selection of the area of film to be examined.
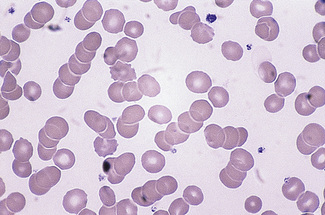
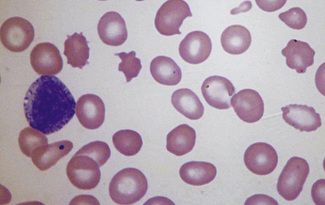
Changes associated with a compensatory increase in erythropoiesis
Polychromasia (πoλθζ, many)
The term polychromasia suggests that the red cells are being stained many colours. In practice, it means that some of the red cells stain shades of bluish grey (Fig. 5.62) – these are the reticulocytes. Cells staining shades of blue, ‘blue polychromasia’, are unusually young reticulocytes. ‘Blue polychromasia’ is most often seen when there is either an intense erythropoietic drive or when there is extramedullary erythropoiesis, as, for instance, in myelofibrosis or carcinomatosis. It should be noted that in certain circumstances the absence of polychromasia is significant; in a patient with severe anaemia it indicates that the bone marrow response is inadequate, e.g. in aplastic anaemia and pure red cell aplasia.
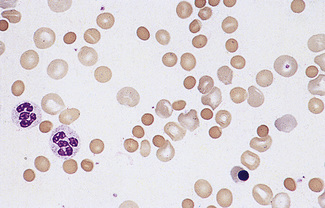
Erythroblastaemia
When large numbers of erythroblasts are present, many of them may be derived from extramedullary foci of erythropoiesis (e.g. in the liver and spleen). This is likely, for instance, in haemolytic disease of the newborn and primary myelofibrosis. In myelofibrosis and carcinomatosis, the number of erythroblasts is often disproportionately high for the degree of anaemia, and a few immature granulocytes are usually also present (designated leucoerythroblastic anaemia) (Fig. 5.63).
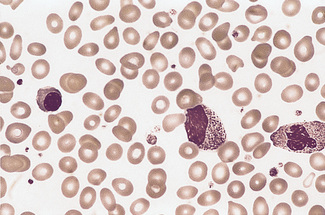
Erythroblasts can usually be found in the peripheral blood after splenectomy, and many may be present in severe anaemia and in the presence of extramedullary erythropoiesis (Fig. 5.64). Large numbers are frequently seen in the blood films of patients with sickle cell anaemia in painful crises. Small numbers of erythroblasts are not uncommon in blood from patients suffering from cyanotic heart failure or septicaemia.
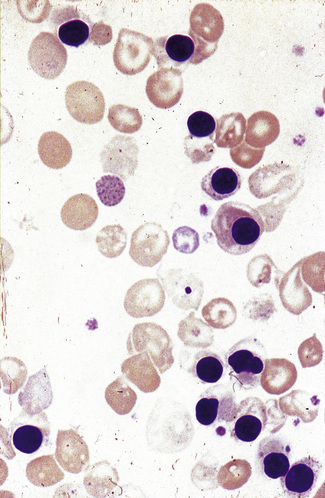
Effects of splenectomy and hyposplenism
Some of the effects of splenectomy and hyposplenism have already been mentioned – namely, the occurrence of target cells, acanthocytes, Howell–Jolly bodies and Pappenheimer bodies (Fig. 5.57). In addition, there may be neutrophilia (early after splenectomy), lymphocytosis, thrombocytosis and giant platelets. In people who are haematologically normal, the blood film features of hyposplenism are variable – sometimes striking and sometimes very minor.
Scanning electron microscopy
The morphology of red cells, as illustrated in this chapter, may be distorted by spreading and drying films in the traditional way. A more authentic portrayal of red cell shape in vivo can be seen by scanning electron microscopy. This provided the means for a critical re-examination of red cell morphology. Bessis and his co-workers published excellent photographs of pathological red cells and, from their appearances, proposed a terminology7,21,22 that has generally been adopted in this chapter. They also discussed the difficult question of the in vivo significance of crenation (echinocytic change) observed in vitro. It seems that neither echinocytosis nor acanthocytosis is necessarily associated with increased haemolysis. It cannot be concluded, either, that crenation is occurring in vivo, when the phenomenon is markedly evident in films made on glass slides. To ensure that cells are crenated in any blood sample as it is withdrawn, Brecher and Bessis recommended that the blood be examined immediately between plastic, instead of glass coverslips or slides, to avoid the known ‘echinocytogenic’ effect of glass surfaces, probably caused by alkalinity.22 Nevertheless, for practical purposes, if a blood film of a freshly drawn blood specimen shows echinocytosis and films from other patients prepared using the same glass slides do not, the abnormality can be accepted as genuine.
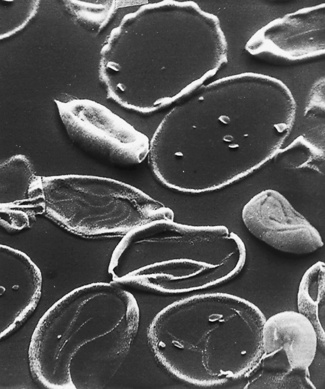
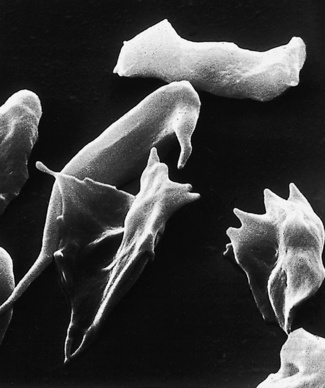
The appearance of various cells by scanning electron microscopy is illustrated in Figures 5.65–5.72.
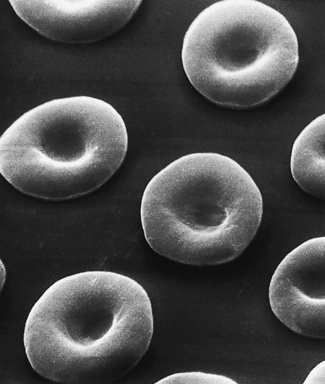
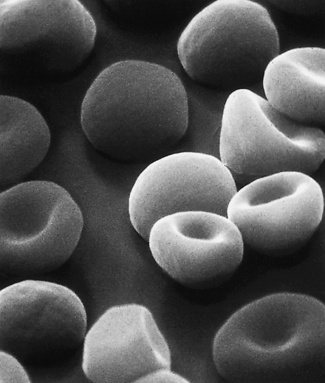
Figure 5.66 Scanning electron microscope photograph. Hereditary spherocytosis. Note the round shape of spherocytes but also that there are cells intermediate in form between spherocytes and normal discocytes. (Compare with Fig. 5.65; also see blood film appearances as shown in Fig. 5.21.)
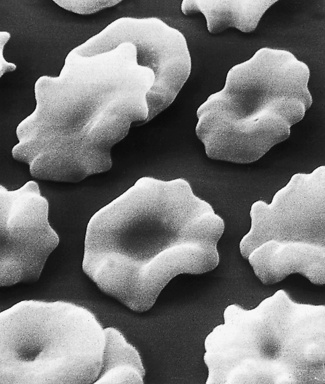
Figure 5.67 Scanning electron microscope photograph. Normal blood after standing overnight. Note crenation.
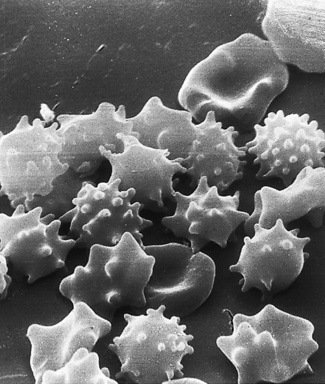
Figure 5.68 Scanning electron microscope photograph. Acanthocytosis. Some cells also show crenation and contraction. (Compare with Fig. 5.67; see also blood film appearances as shown in Fig. 5.45.)
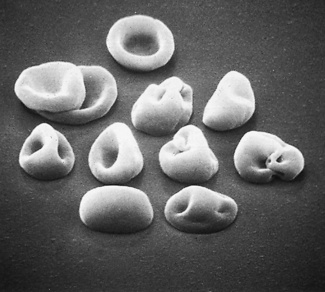
Figure 5.69 Scanning electron microscope photograph. Oxidant drug-induced haemolysis; see blood film appearances shown in Figure 5.43.
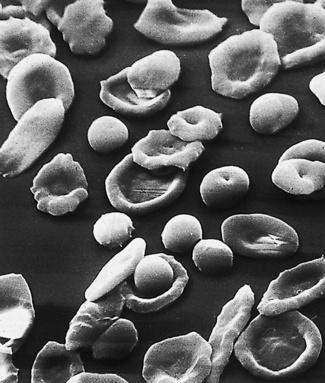
Figure 5.70 Scanning electron microscope photograph. Iron deficiency anaemia. (Compare with Fig. 5.71, and see also Fig. 5.12.)
Morphology of leucocytes
This section will include a description of the normal leucocytes, some congenital anomalies and reactive changes that are commonly encountered. To describe adequately the various changes found in malignant conditions would require a lengthy text and many illustrations that are beyond the scope of this book. They will be referred to briefly here, but for detailed reference readers should consult a specialist test23 or an atlas on blood cells. For classification of the acute leukaemias, see the original description by the FAB (French-American-British) group24 and the subsequent revision.25 There is also a World Health Organization classification,26 which supercedes the FAB classifications when facilities are available for fully characterizing leukaemia and is now in widespread use. However, the FAB classification remains useful as a widely accepted scheme for the initial morphologic description of leukaemias.
Polymorphonuclear neutrophils
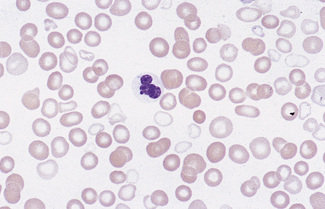
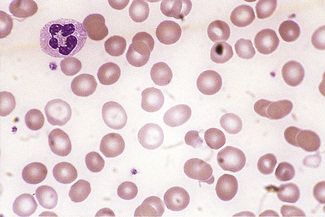
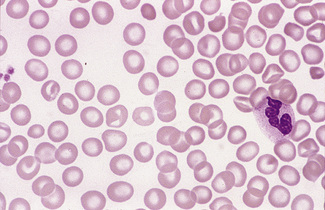
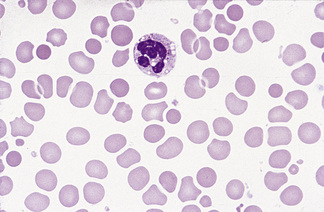
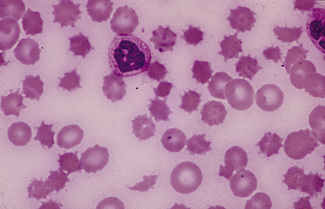
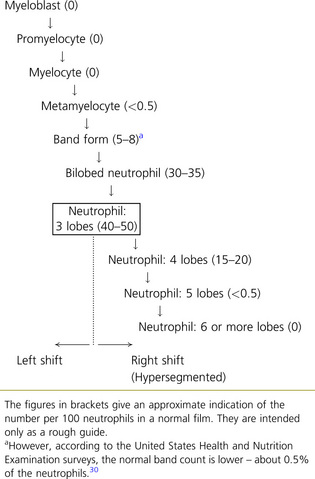
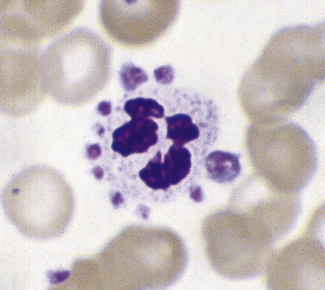
In normal adults, neutrophils account for more than half the circulating leucocytes. They are the main defence of the body against pyogenic bacterial infections. Normal neutrophils are uniform in size, with an apparent diameter of about 13 μm on a film. They have a segmented nucleus and, when stained, pink/orange cytoplasm with fine granulation (Fig. 5.73). The majority of neutrophils have three nuclear segments (lobes) connected by tapering chromatin strands. The chromatin shows clumping and is usually condensed at the nuclear periphery. A small percentage have four lobes, and occasionally five lobes may be seen. Up to 8% of circulating neutrophils are unsegmented or partly segmented (‘band’ forms) (discussed later).
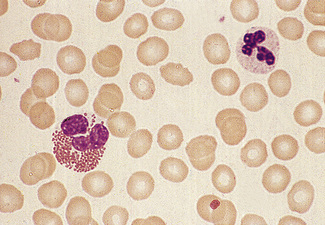
Figure 5.73 Photomicrograph of a blood film. Normal polymorphonuclear neutrophil and normal eosinophil.
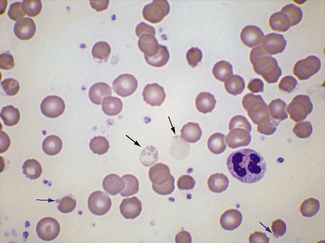
Occasionally, red cells will adhere to neutrophils, forming rosettes. The mechanism is unknown, but it is likely to be immune; usually it appears to be of no clinical significance but occasionally it is seen in an immune haemolytic anaemia. Leucoagglutination also occurs as an in vitro artefact.27 Occasionally neutrophils (and/or monocytes) have phagocytosed erythrocytes (Fig. 5.74). This is particularly common in paroxysmal cold haemoglobinuria.
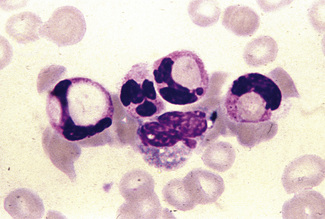
Figure 5.74 Photomicrograph of a blood film. Erythrophagocytosis in a patient with a positive direct antiglobulin test.
It is extremely important to ensure the consistency of staining of the blood films using a standardized Romanowsky method (see Chapter 4) because changes in the staining density, colour and appearance of cytoplasmic granulation, if not artefact, may have diagnostic significance. Common neutrophil abnormalities are described later in this chapter.
Granules
Toxic granulation is the term used to describe an increase in staining density and possibly number of granules that occurs regularly with bacterial infection and often with other causes of inflammation (Fig. 5.75). It can also be a feature of administration of granulocyte colony-stimulating factor (G-CSF) and of aplastic anaemia. Poorly staining (hypogranular) and agranular neutrophils occur in the myelodysplastic syndromes (Fig. 5.76) and in some forms of myeloid leukaemia.
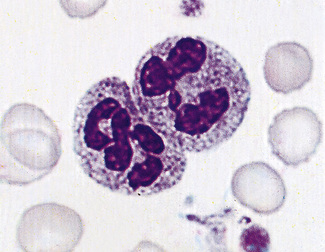
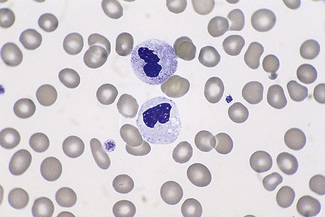
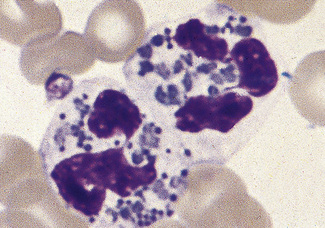
There are rare inherited disorders that are manifest by abnormal neutrophils. In the Alder–Reilly anomaly, the granules are very large, are discrete, stain deep red and may obscure the nucleus (Fig. 5.77). Other leucocytes, including some lymphocytes, also show the abnormal granules. In the Chédiak–Higashi syndrome there are giant but scanty azurophilic granules (Fig. 5.78), and the other leucocyte types may also be affected. Alder–Reilly neutrophils function normally, but in Chédiak–Higashi syndrome there is a functional defect that is manifested by susceptibility to severe infection.
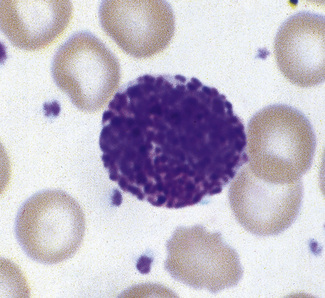
Vacuoles
In blood films spread without delay, the presence of vacuoles in the neutrophils is usually indicative of severe sepsis, when toxic granulation is usually also present. Vacuoles will develop as an artefact with prolonged standing of the blood before films are made (see Chapter 1, Fig. 1.3, see p. 8).
Bacteria
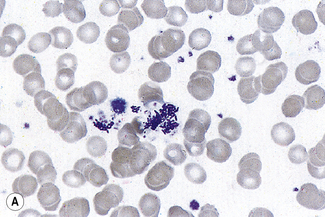
Very rarely, in the presence of overwhelming septicaemia (e.g. meningococcal or pneumococcal), bacteria may be seen within vacuoles or apparently lying free in the cytoplasm of neutrophils. When blood is taken from an infected central line, clumps of bacteria or fungi may be seen scattered in the film as well as in neutrophils in phagocytic vacuoles (Fig. 5.79). In premature infants with staphylococcal septicaemia, the detection of bacteria in neutrophils helps in early diagnosis.28 In endemic areas, detection of bacteria within neutrophils is important in the diagnosis of ehrlichiosis and anaplasmosis.
Nuclei
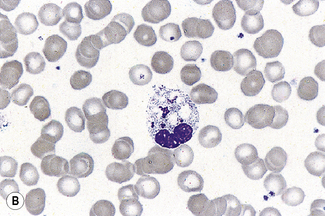
Segmentation of the nucleus of the neutrophil is a normal event as the cell matures from the myelocyte. With the three-lobed neutrophil as a marker, a shift to the left (less mature) or to the right (hypermature) can be recognized (Table 5.2). A left shift with band forms, metamyelocytes and perhaps occasional myelocytes is common in sepsis (Fig. 5.80), when it is usually accompanied by toxic granulation. If promyelocytes and myeloblasts are also present, it is likely to be a feature of a leucoerythroblastic anaemia or leukaemia (Fig. 5.81); occasionally this extreme picture may be seen in very severe infections, when it is called ‘leukaemoid reaction’. A left shift, with a significant number of band forms, occurs normally in pregnancy.
 |
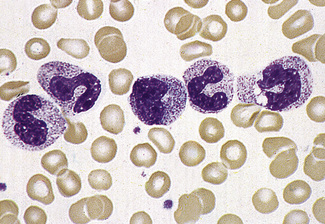
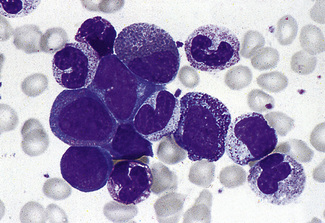
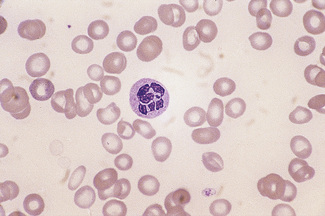
Hypersegmentation
The presence of hypersegmented neutrophils is an important diagnostic feature of megaloblastic anaemias. Neutrophil hypersegmentation can be defined as the presence of neutrophils with six or more lobes or the presence of more than 3% of neutrophils with at least five lobes. In florid megaloblastic states, neutrophils are often enlarged and their nuclei may have six or more segments connected by particularly fine chromatin bridges (Fig. 5.10). A right shift with moderately hypersegmented neutrophils may also be seen in uraemia and not infrequently in iron deficiency.29 Hypersegmentation can be seen after cytotoxic treatment, especially with methotrexate. Patients undergoing treatment with hydroxycarbamide or other drugs that induce megaloblastosis sometimes develop hypersegmented neutrophils (Table 5.2).30
Pelger–Huët Cells
The Pelger–Huët anomaly is a benign inherited condition in which neutrophil nuclei fail to segment properly. The majority of circulating neutrophils have only two discrete equal-sized lobes connected by a thin chromatin bridge (Fig. 5.82). The chromatin is coarsely clumped, and granule content is normal.
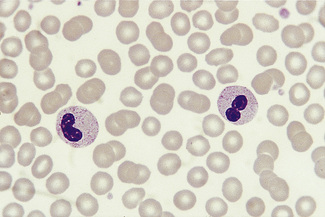
A similar acquired morphologic anomaly, known as pseudo-Pelger cells or the acquired Pelger–Huët anomaly, may be seen in myelodysplastic syndromes, acute myeloid leukaemia with dysplastic maturation, and occasionally in chronic myelogenous leukaemia (during the accelerated phase) (Fig. 5.83). In these conditions, the neutrophils are often hypogranular and they tend to have a markedly irregular nuclear pattern.
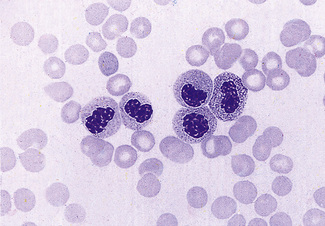
Pyknotic Neutrophils (Apoptosis)
Small numbers of dead or dying cells may normally be found in the blood, especially when there is an infection. They may also develop in normal blood in vitro after standing for 12–18 h, even if kept at 4°C. These cells have round, dense, featureless nuclei and their cytoplasm tends to be dark pink (see p. 8 and Fig. 1.4). It is important not to confuse these cells with erythroblasts.
Eosinophils
Eosinophils are a little larger than neutrophils, 12–17 μm in diameter. They usually have two nuclear lobes or segments, and the cytoplasm is packed with distinctive spherical gold/orange (eosinophilic) granules (Figs 5.73, 5.84). The underlying cytoplasm, which is usually obscured by the granules, is pale blue. Prolonged steroid administration causes eosinopenia. Moderate eosinophilia occurs in allergic conditions; more severe eosinophilia (20–50 × 109/l) may be seen in parasitic infections and even greater numbers may be seen in other reactive eosinophilias, eosinophilic leukaemia and the idiopathic hypereosinophilic syndrome. Reactive eosinophilia with very high counts may be seen in T-cell lymphoma, B-cell lymphoma and acute lymphoblastic leukaemia. Eosinophils are part of the leukaemic population in chronic myelogenous leukaemia, and this is occasionally so in acute myeloid leukaemia. Cytological abnormalities in eosinophils are not very useful in distinguishing between leukaemic and reactive eosinophils.
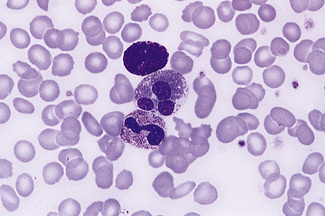
Basophils
Basophils are the rarest (<1%) of the circulating leucocytes. Their nuclear segments tend to fold up on each other, resulting in a compact irregular dense nucleus resembling a closed lotus flower. The distinctive, large, variably sized, dark blue or purple granules of the cytoplasm (Fig. 5.84) often obscure the nucleus; they are rich in histamine, serotonin and heparin. Basophils tend to degranulate, leaving cytoplasmic vacuoles.
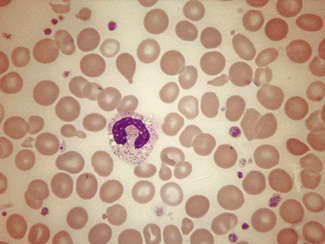
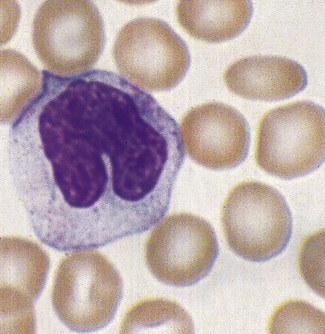
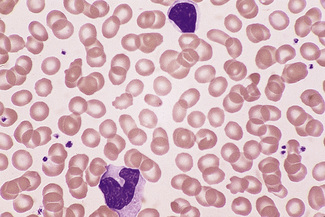
Monocytes
Monocytes are the largest of the circulating leucocytes, 15–18 μm in diameter. They have bluish-grey cytoplasm that contains variable numbers of fine reddish granules. The nucleus is large and curved, often in the shape of a horseshoe, but it may be folded or curled (Figs 5.85, 5.86). It does not undergo segmentation. The chromatin is finer and more evenly distributed than in neutrophil nuclei. An increased number of monocytes occur in some chronic infections and inflammatory conditions such as tuberculosis and Crohn’s disease, in chronic myeloid leukaemias (particularly atypical chronic myeloid leukaemia and chronic myelomonocytic leukaemia) and in acute leukaemias with a monocytic component. In chronic myelomonocytic leukaemia, the mature monocyte count may reach as high as 100 × 109/l. It is occasionally difficult to distinguish abnormal monocytes from the large activated T lymphocytes produced in infectious mononucleosis or from circulating high-grade lymphoma cells.
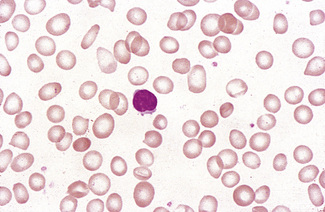
Lymphocytes
The majority of circulating lymphocytes are small cells with a thin rim of cytoplasm, occasionally containing scanty azurophilic granules (Figs 5.1, 5.87). Nuclei are remarkably uniform in size (about 9 μm in diameter). This provides a useful guide for estimating red cell size (normally about 7–8 μm) on the blood film. Some 10% of circulating lymphocytes are larger, with more abundant pale blue cytoplasm containing azurophilic granules (Fig. 5.87). The nuclei of lymphocytes have homogeneous chromatin with some clumping at the nuclear periphery. About 85% of the circulating lymphocytes are T cells or natural killer (NK) cells.
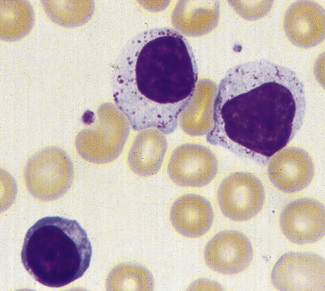
In infections, both bacterial and viral, transformed lymphocytes may be present. These immunoblasts or ‘Türk’ cells are 10–15 μm in diameter, with a round nucleus, often with a large nucleolus, and abundant, deeply basophilic cytoplasm (Fig. 5.88). They may develop into plasmacytoid lymphocytes and plasma cells, and these are occasionally seen in the blood in severe infections. In the absence of infection, multiple myeloma must be excluded. In viral infection, ‘reactive lymphocytes’ appear in the blood. These have slightly larger nuclei with more open chromatin and abundant cytoplasm that may be irregular. The most extreme examples of these cells are usually found in infectious mononucleosis (Fig. 5.89). These ‘glandular fever’ cells have irregular nuclei and abundant cytoplasm that is basophilic at the periphery; they have a tendency to appear, on a blood film, to have flowed around adjacent erythrocytes.
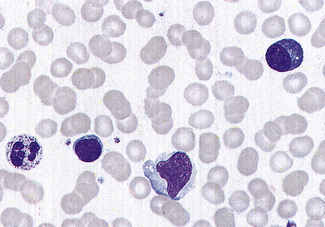
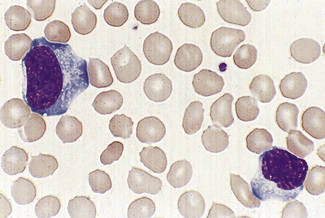
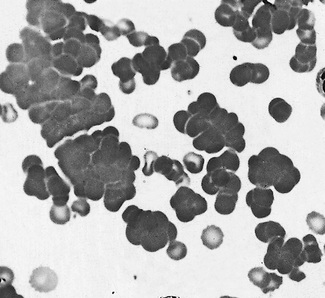
Malignant lymphoid cells vary enormously in their morphology. The commonest malignancy is chronic lymphocytic leukaemia, the leukaemic population being composed almost exclusively of small lymphocytes (Fig. 5.90), sometimes with a few larger nucleolated cells. In prolymphocytic leukaemia, the majority of cells are a little larger than small lymphocytes with more cytoplasm and usually one distinct nucleolus (Fig. 5.91). Lymphoblasts of acute lymphoblastic leukaemia (Figs 5.92, 5.93) vary in size from only slightly larger than lymphocytes to cells of 15–17 μm diameter. The nuclei generally have diffuse chromatin, but there may be some chromatin condensation in the smaller blasts. The cytoplasm varies from weakly to strongly basophilic.
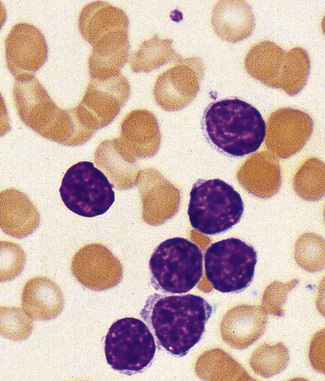
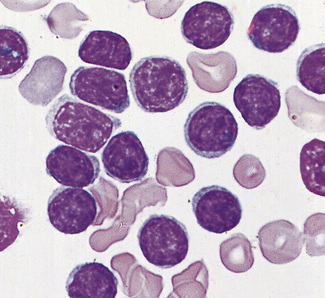
Figure 5.91 Photomicrograph of a blood film. Prolymphocytic leukaemia. There is a uniform population of prolymphocytes.
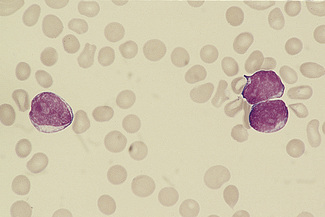
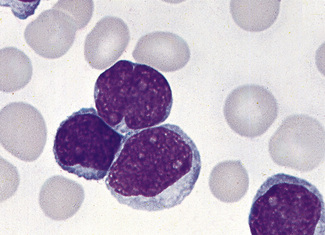
Figure 5.93 Photomicrograph of a blood film. Acute lymphoblastic leukaemia (FAB L1 type). Shows lymphoblasts.
Circulating lymphoma cells vary markedly in size, depending on the type of lymphoma. When there is a lymphocytosis, the lymphocytes are usually far less uniform than in chronic lymphocytic leukaemia, and the lymphoma cells frequently have irregular lobed, indented or cleaved nuclei and relatively scanty agranular cytoplasm that varies in its degree of basophilia. Lobulated lymphocytes are a feature of HTLV-I (human T-lymphotropic virus type I) infection and of adult T-cell leukaemia/lymphoma. However, lymphocytes with definite lobulation are also a common storage artefact in blood kept for 18–24 h at room temperature (see p. 7).
Platelet morphology
Normal platelets are 1–3 μm in diameter. They are irregular in outline with fine red granules that may be scattered or centralized. A small number of larger platelets, up to 5 μm in diameter, may be seen in normal films. Larger platelets are seen in the blood when platelet production is increased (Fig. 5.94) and in hyposplenism (Fig. 5.95). Thus, for example, in severe immune thrombocytopenia, some large platelets will be seen on the film. Very high platelet counts as a feature of a myeloproliferative neoplasm may be associated with extreme platelet anisocytosis, with some platelets being as large as red cells and often with some agranular or hypogranular platelets (Fig. 5.96). The platelet count frequently increases with acute inflammatory stress or bleeding but seldom to more than 1000 × 109/l. More than this, unless the patient is critically ill or hyposplenic, makes a myeloproliferative neoplasm a definite possibility.
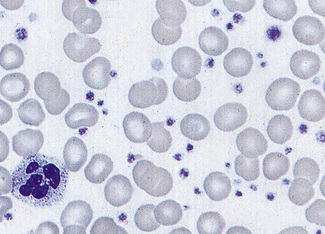
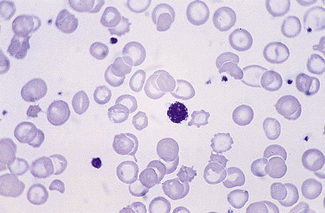
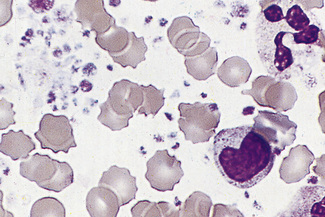
Characteristic morphologic features are seen in various uncommon inherited platelet disorders associated with bleeding. These include the Bernard–Soulier syndrome, in which there are giant platelets with a defective ristocetin response, and the grey platelet syndrome, in which the platelets have decreased granules and have a ghost-like appearance on the stained blood film (Fig. 5.97). Thrombocytopenia with large platelets is also a feature of the May–Hegglin anomaly (see p. 92).
Figure 5.97 Photomicrograph of a blood film. Grey platelet syndrome. Shows severely hypogranular platelets.
In about 1% of individuals, ethylenediaminetetra-acetic acid (EDTA) anticoagulant causes platelet clumping, resulting in pseudothrombocytopenia.31 It is much less likely to occur when the blood is collected into any other anticoagulant.32 This phenomenon may be detected when it gives rise to a ‘flag’ on an automated blood cell counter; it is identifiable on the blood film. It is not associated with any coagulation disturbance and platelet function is normal. When this abnormality is detected it is important that the erroneous platelet count is deleted from the report. If the blood film shows that the platelet count is clearly normal, the statement ‘Platelet count normal’ is sufficient, rather than a repeat sample in an alternative anticoagulant being requested. Occasionally, EDTA inhibits the staining of platelets.33
Occasionally, platelets may be seen adhering to neutrophils (Fig. 5.98).34–37 This has been reported in patients who have demonstrable antiplatelet autoantibodies,38 but it is more commonly seen in apparently healthy individuals. It is not seen in films made directly from blood that has not been anticoagulated. Sometimes the platelets are ingested by neutrophils.39 If a blood film in a patient with platelet satellitism or phagocytosis shows that the automated count is erroneous and the platelet count is in fact normal, the erroneous count should be deleted from the report and an explanation given.
1 Marsh G.W. Abnormal contraction, distortion and fragmentation in human red cells. MD thesis: London University; 1966.
2 Rodgers M.S., Chang C.-C., Kass L. Elliptocytosis and tailed poikilocytes correlate with severity of iron deficiency anemia. Am J Clin Pathol. 1999;111:672-675.
3 Sechi L.A., De Carll S., Catena C., et al. Benign familial macrocytosis. Clin Lab Haematol. 1996;18:41-43.
4 Rees D.C., Dudley J.A., Marinaki A. Review: pyrimidine 5′nucleotidase deficiency. Br J Haematol. 2003;120:375-383.
5 Tuffy P., Brown A.K., Zuelzer W.W. Infantile pyknocytosis: a common erythrocyte abnormality of the first trimester. Am J Dis Child. 1959;98:227-241.
6 Keimowitz R., Desforges J.F. Infantile pyknocytosis. N Engl J Med. 1965;273:1152-1154.
7 Bessis M. Red cell shapes: an illustrated classification and its rationale. Nouv Rev Fr Hematol. 1972;12:721-745.
8 Burns E.R., Lou Y., Pathak A. Morphological diagnosis of thrombotic thrombocytopenic purpura. Am J Hematol. 2004;75:18-21.
9 Estes J.W., Morley T.J., Levine I.M., et al. A new hereditary acanthocytosis syndrome. Am J Med. 1967;42:868-881.
10 Mier M., Schwartz S.O., Boshes B. Acanthrocytosis [sic], pigmented degeneration of the retina and ataxic neuropathy: a genetically determined syndrome with associated metabolic disorder. Blood. 1960;16:1586-1608.
11 Salt H.B., Wolfe O.H., Lloyd J.K., et al. On having no beta-lipoprotein: a syndrome comprising a-beta-lipoprotinaemia, acanthocytosis, and steatorrhoea. Lancet. 1960;ii:325-329.
12 Wimer B.M., Marsh W.L., Taswel H.F., et al. Haematological changes associated with the McLeod phenotype of the Kell blood group system. Br J Haematol. 1977;36:219-224.
13 Silber R., Amorosi E.L., Howe J., et al. Spur-shaped erythrocytes in Laennec’s cirrhosis. N Engl J Med. 1966;275:639-643.
14 Ponder E. Hemolysis and Related Phenomena. New York: Grune & Stratton; 1948.
15 Feo C.J., Tchernia G., Subtu E., et al. Observation of echinocytosis in eight patients: a phase contrast and SEM study. Br J Haematol. 1978;40:519-526.
16 Crook M., Williams W., Schey S. Target cells and stomatocytes in heterozygous familial hypobetalipoproteinaemia. Eur J Haematol. 1998;60:68-69.
17 Lock S.P., Sephton Smith R., Hardisty R.M. Stomatocytosis: a hereditary red cell anomaly associated with haemolytic anaemia. Br J Haematol. 1961;7:303-314.
18 Ducrou W., Kimber R.J. Stomatocytes, haemolytic anaemia and abdominal pain in Mediterranean migrants: some examples of a new syndrome? Med J Aust. 1969;ii:1087-1091.
19 Norman J.G. Stomatocytosis in migrants of Mediterranean origin. Med J Aust. 1969;i:315.
20 Douglass C., Twomey J. Transient stomatocytosis with hemolysis: a previously unrecognized complication of alcoholism. Ann Intern Med. 1970;72:159-164.
21 Weed R.I., Bessis M. The discocyte-stomatocyte equilibrium of normal and pathologic red cells. Blood. 1973;41:471-475.
22 Brecher G., Bessis M. Present status of spiculated red cells and their relationship to the discocyte–echinocyte transformation: a critical review. Blood. 1972;40:333-344.
23 Bain B.J. Leukaemia Diagnosis, 4th ed. Wiley-Blackwell, Oxford, 2010.
24 Bennett J.M., Catovsky D., Daniel M.T., et al. Proposals for the classification of the acute leukaemias (FAB cooperative group). Br J Haematol. 1976;33:451-458.
25 Bennett J.M., Catovsky D., Daniel M.T., et al. Proposed revised criteria for the classification of acute myeloid leukemia. Ann Intern Med. 1985;103:620-625.
26 Swerdlow S.H., Campo E., Harris N.L., et al. World Health Classification of Tumours of Haematopoietic and Lymphoid Tissues. Lyon: IARC; 2008.
27 Deal J., Hernandez A.M., Pierre R.V. Ethylenediamine tetraacetic acid-associated leukoagglutination. Am J Clin Pathol. 1995;103:338-340.
28 Howard M.R., Smith R.A. Early diagnosis of septicaemia in preterm infants from examination of the peripheral blood films. Clin Lab Haematol. 1999;21:365-368.
29 Van Assendelft O.W., McGrath C., Murphy R.S., et al. The differential distribution of leukocytes. In: Koepke J.A., editor. CAP Aspen Conference: Differential Leukocyte Counting. Northfield, IL: College of American Pathologists, 1977.
30 Westerman D.A., Evans D., Metz J. Neutrophil hypersegmentation in iron deficiency anaemia: a case control study. Br J Haematol. 1999;107:512-515.
31 Gowland E., Kay H.E.M., Spillman J.C., et al. Agglutination of platelets by a serum factor in the presence of EDTA. J Clin Pathol. 1969;22:460-1454.
32 Criswell K.A., Breider M.A., Bleavins M.R. EDTA-dependent platelet phagocytosis. A cytochemical, ultrastructural and functional characterization. Am J Clin Pathol. 2001;115:376-384.
33 Stavem P., Berg K. A macromolecular serum component acting on platelets in the presence of EDTA–’platelet stain preventing factor? Scand J Haematol. 1973;10:202-208.
34 Crome P.E., Barkhan P. Platelet adherence to polymorphs. Br Med J. 1963;ii:871.
35 Field E.J., Macleod I. Platelet adherence to polymorphs. Br Med J. 1963;ii:388.
36 Greipp P.R., Gralnick H.R. Platelet to leucocyte adherence phenomena associated with thrombocytopenia. Blood. 1976;47:513-521.
37 Skinnider L.F., Musclow C.E., Kahn W. Platelet satellitism: an ultrastructural study. Am J Hematol. 1978;4:179-185.
38 White L.A.Jr, Brubaker L.H., Aster R.H., et al. Platelet satellitism and phagocytosis by neutrophils: association with antiplatelet antibodies and lymphoma. Am J Hematol. 1978;4:313-323.
39 Campbell V., Fosbury E., Bain B.J. Platelet phagocytosis as a cause of pseudothrombocytopenia. Am J Hematol. 2009;84:362.