[level-membership-for-hematology-oncology-and-palliative-medicine-category]
Chapter 21 Blood cell antigens and antibodies
erythrocytes, platelets and granulocytes
Erythrocytes
Red Cell Antigens
A total of 30 blood group systems have been described (Table 21.1). Each system is a series of red cell antigens, determined either by a single genetic locus or very closely linked loci. In addition to the blood group systems, there are six ‘collections’ of antigens (e.g. Cost), which bring together other genetically, biochemically or serologically related sets of antigens and a separate series of low-frequency (e.g. Rd) and high-frequency (e.g. Vel) antigens, which do not fit into any system or collection. A numeric catalogue of red cell antigens is being maintained by an International Society of Blood Transfusion (ISBT) Working Party.1
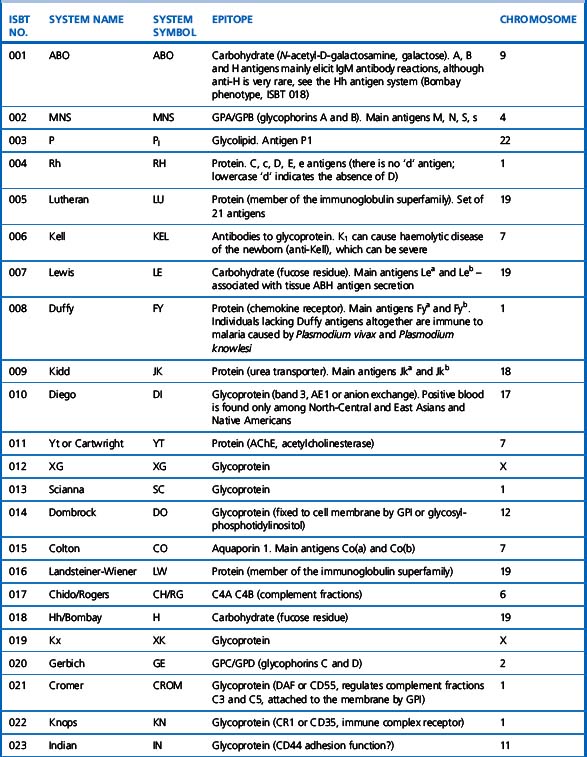
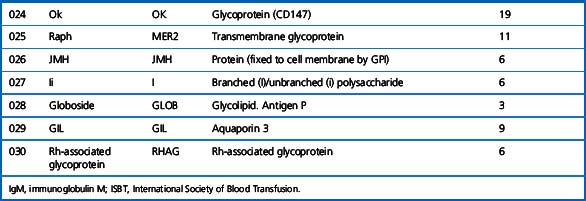
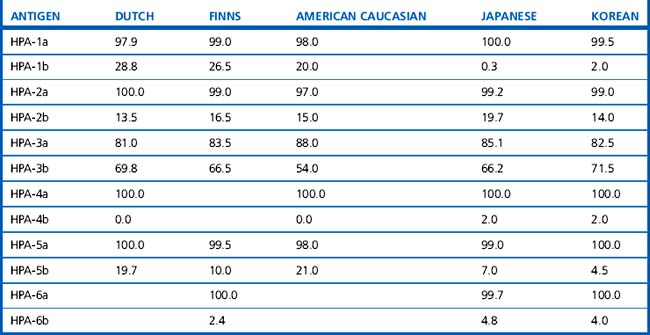
Alternative forms of a gene coding for red cell antigens at a particular locus are called alleles and individuals may inherit identical or non-identical alleles. Most blood group genes have been assigned to specific chromosomes (e.g. ABO system on chromosome 9, Rh system on chromosome 1). The term genotype is used for the sum of the inherited alleles of a particular gene (e.g. AA, AO) and most red cell genes are expressed as codominant antigens (i.e. both genes are expressed in the heterozygote). The phenotype refers to the recognizable product of the alleles and there are many racial differences in the frequencies of red cell phenotypes, as shown in Table 21.2.
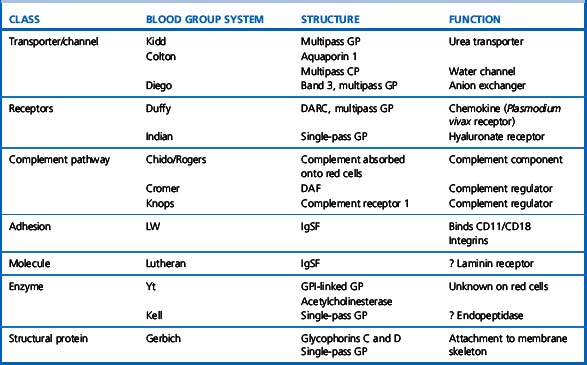
Red cell antigens are determined either by carbohydrate structures or protein structures. Carbohydrate-defined antigens are indirect gene products (e.g. ABO, Lewis, P). The genes code for an intermediate product, usually an enzyme that creates the antigenic specificity by transferring sugar molecules onto the protein or lipid. Protein-defined antigens are direct gene products and the specificity is determined by the inherited amino acid sequence and/or the conformation of the protein. Proteins carrying red cell antigens are inserted into the membrane in one of three ways: single pass, multipass or linked to phosphatidylinositol (GPI-linked). Only a few red cell antigens are erythroid-specific (Rh, LW, Kell and MNSs), the remainder being expressed in many other tissues. The structure and functions of the membrane proteins and glycoproteins carrying blood group antigens have been reviewed by Daniels.2 An illustration of the putative functions of molecules containing blood group antigens is provided in Table 21.3.
ABO System
Discovery of the ABO system by Landsteiner marked the beginning of safe blood transfusion. The ABO antigens, although most important in relation to transfusion, are also expressed on most endothelial and epithelial membranes and are important histocompatibility antigens.3 Transplantation of ABO-incompatible solid organs increases the potential for hyperacute graft rejection, although ABO-incompatible renal transplantation can be successfully carried out with plasmapheresis in addition to immunosuppression of the recipient.4 Major ABO-incompatible stem cell transplants (e.g. group A stem cells into a group O recipient) will provoke haemolysis, unless the donation is depleted of red cells.
ABO Antigens and Encoding Genes
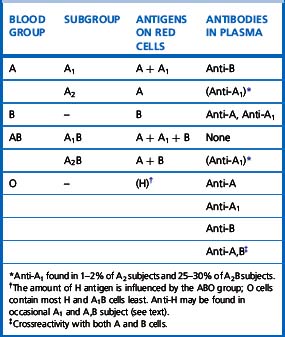
There are four main blood groups: A, B, AB and O (Table 21.4). In the British Caucasian population, the frequency of group A is 42%, B 9%, AB 3% and O 46%, but there is racial variation in these frequencies.5 The epitopes of ABO antigens are determined by carbohydrates (sugars), which are linked either to polypeptides (forming glycoproteins) or to lipids (glycolipids).
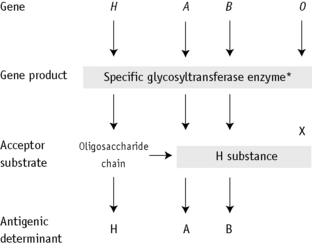
The expression of ABO antigens is controlled by three separate genetic loci: ABO located on chromosome 9 and FUT1 (H) and FUT2 (Se), both of which are located on chromosome 19. The genes from each locus are inherited in pairs as Mendelian dominants. Each gene codes for a different enzyme (glycosyltransferase), which attaches specific monosaccharides onto precursor disaccharide chains (Table 21.5). There are four types of disaccharide chains known to occur on red cells, on other tissues and in secretions. The Type 1 disaccharide chain is found in plasma and secretions and is the substrate for the FUT2 (Se) gene, whereas Types 2, 3 and 4 chains are only found on red cells and are the substrate for the FUT1 (H) gene. It is likely that the O and B genes arose by mutation of the A gene. The O gene does not encode for the production of a functional enzyme; group O individuals commonly have a deletion at nucleotide 261 (the O1 allele), which results in a frame-shift and premature termination of the translated polypeptide and the production of an enzyme with no catalytic activity. The B gene differs from A by consistent nucleotide substitutions.6 The expression of A and B antigens is dependent on the H and Se genes, which both give rise to glycosyltransferases that add L-fucose, producing the H antigen. The presence of an A or B gene (or both) results in the production of further glycosyltransferases, which convert H substance into A and B antigens by the terminal addition of N-acetyl-D-galactosamine and D-galactose, respectively (Fig. 21.1). Because the O gene produces an inactive transferase, H substance persists unchanged as group O. In the extremely rare Oh Bombay phenotype, the individual is homozygous for the h allele of FUT1 and hence cannot form the H precursor of the A and B antigen. Their red cells type as group O, but their plasma contains anti-H, in addition to anti-A, anti-B and anti-A,B, which are all active at 37°C. As a consequence, individuals with an Oh Bombay phenotype can only be safely transfused with other Oh red cells.
Table 21.5 Glycosyltransferases produced by genes encoding antigens within the ABO, H and Lewis blood group systems
| Gene | Allele | Transferase |
|---|---|---|
| FUT1 | H | α-2-L-fucosyltransferase |
| h | None | |
| A | A | α-3-N-acetyl-D-galactosaminyltransferase |
| B | B | α-3-D-galactosyltransferase |
| O | O | None |
| FUT2 | Se | α-2-L-fucosyltransferase |
| se | None | |
| FUT3 | Le | α-3/4-L-fucosyltransferase |
| le | None |
Serologists have defined two common subgroups of the A antigen. Approximately 20% of group A and group AB individuals belong to group A2 and group A2B, respectively, the remainder belonging to group A1 and group A1B. These subgroups arise as a result of inheritance of either the A1 or A2 alleles. The A2 transferase is less efficient in transferring N-acetyl-D-galactosamine to available H antigen sites and cannot utilize Types 3 and 4 disaccharide chains. As a consequence, A2 red cells have fewer A antigen sites than A1 cells and the plasma of group A2 and group A2B individuals may also contain anti-A1. The distinction between these subgroups can be made using the lectin Dolichos biflorus, which only reacts with A1 cells. The H antigen content of red cells depends on the ABO group and, when assessed by agglutination reactions with anti-H, the strength of reaction tends to be graded O > A2 > A2B > B > A1 > A1B. Other subgroups of A are occasionally found (e.g. A3, Ax) that result from mutant forms of the glycosyltransferases produced by the A gene and are less efficient at transferring N-acetyl-D-galactosamine onto H substance.6
Secretors and Non-Secretors
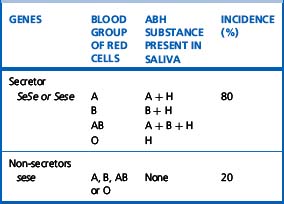
The ability to secrete A, B and H substances in water-soluble form is controlled by FUT2 (dominant allele Se). In a Caucasian population, about 80% are secretors (genotype SeSe or Sese) and 20% are non-secretors (genotype sese) (Table 21.6). Secretors have H substance in the saliva and other body fluids together with A substances, B substances or both, depending on their blood group. Only traces of these substances are present in the secretions of non-secretors, although the antigens are expressed normally on their red cells and other tissues.
An individual’s secretor status can be determined by testing for ABH substance in saliva (see p. 504).
ABO Antigens and Disease
Group A individuals rarely may acquire a B antigen from a bacterial infection that results in the release of a deacetylase enzyme. This converts N-acetyl-D-galactosamine into α-galactosamine, which is similar to galactose, the immunodominant sugar of group B, thereby sometimes causing the red cells to appear to be group AB. In the original reported cases, five out of seven of the patients had carcinoma of the gastrointestinal tract. Case reports attest to the danger of individuals with an acquired B antigen being transfused with AB red cells, resulting in a fatal haemolytic transfusion reaction following the production of hyperimmune anti-B.7
The inheritance of ABH antigens is also known to be weakly associated with predisposition to certain diseases. Group A individuals have 1.2 times the risk of developing carcinoma of the stomach than group O or B; group O individuals have 1.4 times more risk of developing peptic ulcer than non-group O individuals; and non-secretors of ABH have 1.5 times the risk of developing peptic ulcer than secretors.8 The ABO group also affects plasma von Willebrand factor (VWF) and factor VIII levels; group O healthy individuals have levels around 25% lower than those of other ABO groups.9 ABO blood group appears to mediate its effect by accelerating clearance of VWF but the mechanism is not yet clear10 ABH antigens are also frequently more weakly expressed on the red cells of persons with leukaemia.
ABO Antibodies
Anti-A and anti-B
ABO antibodies, in the absence of the corresponding antigens, appear during the first few months after birth, probably as a result of exposure to ABH antigen-like substances in the diet or the environment (i.e. they are ‘naturally occurring’) (Table 21.4). This allows for reverse (serum/plasma) grouping as a means of confirming the red cell phenotype. The antibodies are a potential cause of dangerous haemolytic transfusion reactions if transfusions are given without regard to ABO compatibility. Anti-A and anti-B are always, to some extent, immunoglobulin M (IgM). Although they react best at low temperatures, they are nevertheless potentially lytic at 37°C. Hyperimmune anti-A and anti-B occur less frequently, usually in response to transfusion or pregnancy, but they may also be formed following the injection of some toxoids and vaccines. They are predominantly of IgG class and are usually produced by group O and sometimes by group A2 individuals. Hyperimmune IgG anti-A and/or anti-B from group O or group A2 mothers may cross the placenta and cause haemolytic disease of the newborn (HDN). These antibodies react over a wide thermal range and are more effective haemolysins than the naturally occurring antibodies. Group O donors should always be screened for high-titre anti-A and anti-B antibodies, which may cause haemolysis when group O platelets or plasma are transfused to recipients with A and B phenotypes.
Anti-A1 and anti-H
Anti-H reacts most strongly with group O and A2 red cells and also normally acts as a cold agglutinin. A notable, but rare, exception is the anti-H that occurs in the Oh Bombay phenotype, which is an IgM antibody and causes lysis at 37°C (Table 21.4) so that Oh Bombay phenotype blood would be required for transfusion.
Rh System
Rh Antigens and Encoding Genes
In Caucasian, D-negative individuals, the RHD gene is deleted, whereas in Black races and other populations, single-point mutations, partial deletions or recombinations have been described. In individuals with a weak D antigen (Du), there is a quantitative reduction in D antigen sites, believed to arise from an uncharacterized transcriptional defect. These individuals do not make anti-D antibodies following a D antigen challenge. Partial D individuals lack one or more epitopes of the D antigen, defined using panels of monoclonal reagents. Dvi is perhaps the most important partial D phenotype because such individuals not infrequently make anti-D. Partial D phenotypes arise from DNA exchanges between RHD and RHCE genes and from other rearrangements. Comprehensive reviews of this system have been provided by Avent and Reid11 and Daniels et al.12
The Rh haplotypes are named either by the component antigens (e.g. CDe, cde) or by a single shorthand symbol (e.g. R1 = CDe, r = cde). Thus, a person may inherit CDe (R1) from one parent and cde (r) from the other and have the genotype CDe/cde (R1r). The haplotypes in order of frequency and the corresponding shorthand notation are given in Table 21.7. Although two other nomenclatures are also used to describe the Rh system, namely, Wiener’s Rh-Hr terminology and Rosenfield’s numeric notation, the CDE nomenclature, derived from Fisher’s original theory, is recommended by a World Health Organization Expert Committee13 in the interest of simplicity and uniformity. The Rh antigens are defined by corresponding antisera, with the exception of ‘anti-d’, which does not exist. Consequently, the distinction between homozygous DD and the heterozygous Dd cannot be made by direct serological testing but may be resolved by informative family studies. It is still routine practice to predict the genotype from the phenotype on the basis of probability tables for the various Rh genotypes in the population (Table 21.7). However, in women with immune anti-D and a history of an infant affected by HDN, RH DNA typing is used in prenatal testing for the fetal D status to decide on the clinical management of the pregnancy, e.g. the need for monitoring for fetal anaemia using middle cerebral artery Doppler ultrasound. Suitable sources include amniotic fluid (amniocytes) and trophoblastic cells (chorionic villi) or after 15 weeks’ gestation, maternal blood can be used because it contains fetal DNA.14,15 In practice, multiplex polymerase chain reaction (PCR) is used, with more than two primer sets, to detect the different molecular bases for D-negative phenotypes in non-Caucasians. RH DNA typing also has applications in paternity testing and forensic medicine. There are racial differences in the distribution of Rh antigens, e.g. D negativity is more common in Caucasians (approximately 15%), whereas Ro (cDe) is found in approximately 48% of Black Americans but is uncommon (approximately 2%) in Caucasians. The Rh antigens are present only on red cells and are a structural part of the cell membrane. Complete absence of Rh antigens (Rh-null phenotype) may be associated with a congenital haemolytic anaemia with spherocytes and stomatocytes in the blood film, increased osmotic fragility and increased cation transport. This phenotype arises either as a result of homozygosity for silent alleles at the RH locus (the amorph type) or more commonly by homozygosity for an autosomal suppressor gene (X), genetically independent of the RH locus (the regulator type). Rh antigens are well-developed before birth and can be demonstrated on the red cells of very early fetuses.
Table 21.7 The Rh haplotypes in order of frequency (Fisher nomenclature) in Caucasians and the corresponding short notations
| Fisher | Short Notations | Approximate Frequency (%) |
|---|---|---|
| CDe | R1 | 41 |
| cde | r | 39 |
| cDE | R2 | 14 |
| cDe | R0 | 3 |
| CwDe | R1w | 1 |
| cdE | r″ | 1 |
| Cde | r′ | 1 |
| CDE | RZ | Rare |
| CdE | ry | Rare |
MNSs System
Clinical Significance of Red Cell Alloantibodies
The significance of the alloantibodies described, with respect to the nature of the haemolytic transfusion reaction they produce, is provided in Table 21.8. The majority of haemolytic transfusion reactions, however, are the result of ABO incompatibility.16
Table 21.8 Antibody specificities related to the mechanism of immune haemolytic destruction
| Blood Group System | Intravascular Haemolysis | Extravascular Haemolysis |
|---|---|---|
| ABO,H | A, B, H | |
| Rh | All | |
| Kell | K | K, k, Kpa, Kpb, Jsa, Jsb |
| Kidd | Jka | Jka, Jkb, Jk3 |
| Duffy | Fya, Fyb | |
| MNS | M, S, s, U | |
| Lutheran | Lub | |
| Lewis | Lea | |
| Cartwright | Yta | |
| Colton | Coa, Cob | |
| Dombrock | Doa, Dob |
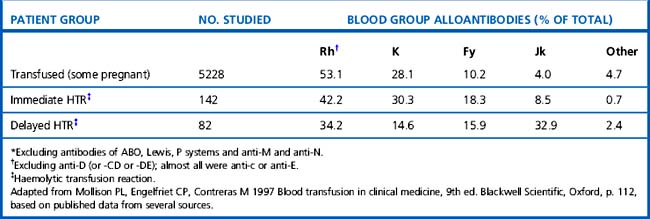
Mollison et al.17 analysed the significance of blood group antigens other than those of the ABO system and D by looking at the prevalence of transfusion-induced red cell alloantibodies, excluding anti-D, -CD and -DE (Table 21.9). Rh antibodies, mainly anti-c or anti-E, accounted for 53% of the total and anti-K and anti-Fya accounted for a further 38%, leaving only about 9% for all other specificities. A similar distribution of the different red cell antibodies was found in a smaller group of patients who experienced immediate haemolytic transfusion reactions (HTR). However, the figures for delayed HTR showed a striking increase in the relative frequency of Jk antibodies, which reflects the outlined characteristics of Jk antibodies.
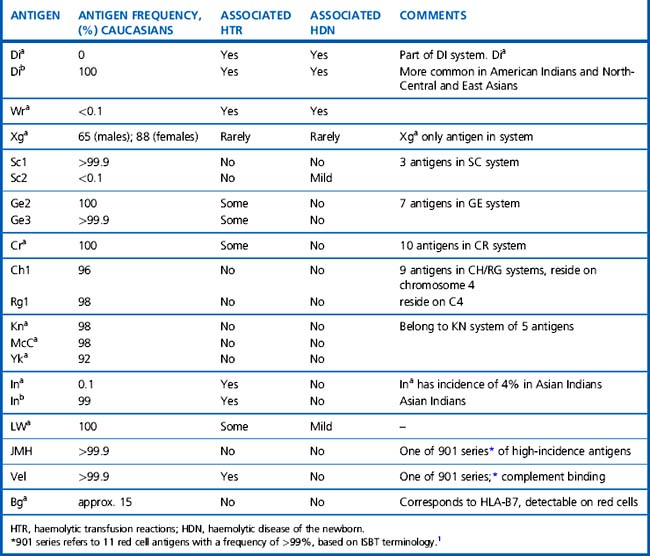
The significance of the many other blood group antigens not referred to in the text is summarized in Table 21.10. However, it should be noted that the antibodies listed are usually wholly or predominantly IgG and would be detectable in routine pretransfusion testing using the indirect anti-globulin test (IAT).
Mechanisms of Immune Destruction of Red Cells
Immune-mediated haemolysis of red cells depends on the following:18
The mechanism of immune haemolysis also determines the site of haemolysis:
Macrophage activity is an important component of cell destruction and further study of cellular interactions at this stage of immune haemolysis may provide an explanation for the differing severity of haemolysis in patients with apparently similar antibodies. In vitro macrophage (monocyte) assays have been used sometimes to supplement conventional serological techniques to assess this aspect of immune haemolysis.20
Interleukin-6 also enhances FcγRII activation and increased activity of the CR1 receptor occurs through the action of T-cell cytokines and through chemotactic agents released in the inflammatory response.22 The increased levels of proinflammatory cytokines and other biological mediators and their effects on the activity of the monocyte phagocytic system have been monitored in patients with systemic inflammatory response syndrome.23 It is therefore possible that release of cytokines during viral and bacterial infections could, at least in part, trigger some episodes of autoimmune cell destruction.
Antigen–Antibody Reactions
Some complement-binding antibodies (especially IgM) may cause lysis in vitro (without noticeable agglutination), which can be enhanced by the addition of fresh serum as a source of complement. However, complement activation may only proceed to the C3 stage; in these circumstances cell-bound C3 can be detected by the antiglobulin test using an appropriate anticomplement reagent (see p. 500).
Quality Assurance within the Laboratory
It has long been appreciated that the test systems used for routine pre-transfusion testing are of the utmost importance because errors can and do lead to patient morbidity and mortality. It is therefore of little surprise that within the European Union all reagents, calibrators and control materials for red cell typing and for determining the presence of ‘irregular anti-erythrocytic antibodies’ have been included under the In-vitro Diagnostics (IVD) Medical Devices Directive24 (see p. 588). This means that all reagents sold within the European Union must display the CE mark to show that they conform to the agreed Common Technical Specifications (CTS). In each European country, a Competent Authority will be able to withdraw or suspend certification of any reagent, depending on the information received from its Notified Body, which will perform batch release approval and monitor the performance of the manufacturer and the product.
The majority of the following points are taken from the British Committee for Standards in Haematology (BCSH) guidelines25 for pre-transfusion compatibility testing:
General Points of Serological Technique
Serum versus Plasma
When plasma is used, complement is inactivated by the ethylenediaminetetra-acetic acid (EDTA) anticoagulant. This is relevant for the detection of some complement-binding antibodies (e.g. of Kidd specificity) that may be missed or give only weak reactions with anti-IgG in the routine antiglobulin test but can be readily detected by anticomplement (see p. 500). It is therefore essential, before using plasma, to optimize the sensitivity of techniques for detecting weak IgG antibodies and to validate the procedure (see p. 500). For example, in antibody screening, increased sensitivity can be achieved by using panel cells with homozygous expression of selected antigens (see p. 528).
Red Cell Suspensions
Normal ionic strength saline
A 2–3% suspension of washed red cells in phosphate buffered saline (PBS), pH 7.0, is generally recommended. Cells suspended in normal ionic strength saline (NISS) are routinely used for antibody titrations, but their use in routine pretransfusion testing has declined over the last decade as observations from external quality assessment exercises have demonstrated that laboratories using NISS have a significantly lower detection rate of antibodies than those using other technologies.32
Low ionic strength saline
It is known that the rate of association of antibodies with red cell antigens is enhanced by lowering the ionic strength of the medium in which the reactions take place. Hence, a major advantage of low ionic strength saline (LISS) is that the incubation period in the IAT (see p. 529) can be shortened while maintaining or increasing sensitivity to the majority of red cell antibodies. The LISS solution can be made up in the laboratory (see p. 620) or purchased commercially.
To avoid false-positive results, the following rules should be followed:
False-positive reactions may still infrequently occur with some sera/plasma. If plasma is used, subsequent serological work may be performed using NISS; if serum is used, anti-IgG should replace the polyspecific antiglobulin reagent.
Reagent Red Cells
Red cells of selected phenotypes are required for ABO and RhD grouping, Rh phenotyping and antibody screening and identification (see Chapter 20). Such cells are available commercially or from blood transfusion centres.
Use of Enzyme-Treated Cells
The most sensitive techniques are those using washed enzyme-pretreated red cells (two-stage), which should match the performance of the spin-tube LISS antiglobulin test (see p. 501) One-stage mixtures and papain inhibitor techniques are relatively insensitive and are not recommended. An ISBT/International Council for Standardization in Haematology (ICSH) protease enzyme standard and an agreed method for its use are available.
Methods for the preparation of papain solution (Low’s method and the two-stage method) and for preparation of bromelin solution have been described.34–36
Agglutination of Red Cells by Antibody: A Basic Method
Agglutination tests are usually carried out in tubes, microtitre plates or column agglutination (gel) technology, using centrifugation or sedimentation. Slide tests are rarely used for emergency ABO and D grouping (see p. 524). For microplate tests, see p. 525.
Tubes
Glass tubes should always be used if the contents are to be heated to 50°C or higher or if organic solvents are being used. Glass tubes, however, are difficult to clean satisfactorily, particularly small-bore tubes and cleaning methods such as those given on p. 623 should be followed carefully.
Reading Results of Tube Tests
Only the strongest complete (C) grade of agglutination seems to be able to withstand a shake procedure without some degree of disruption, which may downgrade the strength of reaction. The BCSH Blood Transfusion Task Force has therefore recommended the following reading procedure.37
Microscopic reading
A scheme of scoring the results is given in Table 21.11.
Table 21.11 Scoring of results in red cell agglutination tests
| Symbol | Agglutination Score* | Description |
|---|---|---|
| 4+ or C (complete) | 12 | Cell button remains in one clump, macroscopically visible |
| 3+ | 10 | Cell button dislodges into several large clumps, macroscopically visible |
| 2+ | 8 | Cell button dislodges into many small clumps, macroscopically visible |
| 1+ | 5 | Cell button dislodges into finely granular clumps, macroscopically just visible |
| (+) or w (weak) | 3 | Cell button dislodges into fine granules, only visible microscopically† |
| − | 0 | Negative result – all cells free and evenly distributed |
* Titration scores are the summation of the agglutination scores at each dilution.
† May be further classified depending on the number of cells in the clumps (e.g. clumps of 12–20 cells [score 3]; 8–10 cells [score 2]; 4–6 cells [score 1]. This is the minimum agglutination that should be considered positive.
Macroscopic reading
A gentle agitation tip-and-roll ‘macroscopic’ method is recommended. It is possible to read agglutination tests macroscopically with the aid of a hand reading glass or concave mirror, but it is then difficult to distinguish reactions weaker than + (microscopic reading) from the normal slight granular appearance of unagglutinated red cells in suspension. Macroscopic reading thus gives lower titration values than does microscopic reading, but the former is recommended. Follow the system of scoring in Table 21.11.
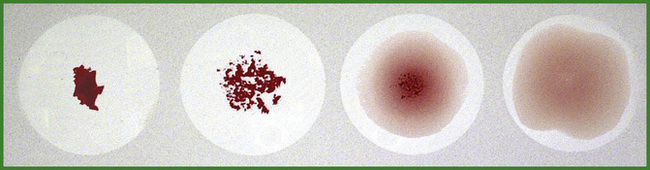
A good idea of the presence or absence of agglutination can often be obtained by inspection of the deposit of sedimented cells: a perfectly smooth round button suggests no agglutination, whereas agglutination is shown by varying degrees of irregularity, ‘graininess’, or dispersion of the deposit (Fig. 21.2).
Demonstration of Lysis
Many blood-group antibodies lyse red cells under suitable conditions in the presence of complement. This is particularly true of anti-A and anti-B, anti-P, anti-Lea and Leb, anti-PP1Pk (anti-Tja) and certain autoantibodies (see p. 284). If it is necessary to add fresh complement, this should be mixed with the serum being tested before the addition of red cells. Otherwise, agglutination occurs and could block complement access. Lysis should be looked for at the end of the incubation period before the tubes are centrifuged, if the cells have sedimented sufficiently; lysis may be scored semiquantitatively after centrifuging the suspensions and comparing the colour of the supernatant with that of the control.
Lysis tests are usually carried out at 37°C, but with cold antibodies a lower temperature (e.g. 20°C or 30°C) would be appropriate, depending on the upper thermal range of activity of the antibody or, in the case of the Donath–Landsteiner antibody, 0°C followed by 37°C (see p. 277).
Antiglobulin Test
The antiglobulin test (Coombs test) was introduced by Coombs and colleagues in 194538 as a method for detecting ‘incomplete’ Rh antibodies (i.e. IgG antibodies capable of sensitizing red cells but incapable of causing agglutination of the same cells suspended in saline), as opposed to ‘complete’ IgM antibodies, which do agglutinate saline-suspended red cells.
The antiglobulin test is probably the most important test in the serologist’s repertoire. The DAT is used to demonstrate in vivo attachment of antibodies to red cells, as in autoimmune haemolytic anaemia (see p. 275), alloimmune HDN (see p. 535) and alloimmune haemolysis following an incompatible transfusion (see p. 542). The IAT has wide application in blood transfusion serology, including antibody screening and identification and crossmatching.
Antiglobulin Reagents
Quality Control of Antiglobulin Reagents
An ISBT/ICSH freeze-dried reference reagent is available for evaluating either polyspecific antihuman globulin reagents or those containing their separate monospecific components.26 The validation of a new antiglobulin reagent should assess the following qualities of the reagent:
Only proceed further if the antiglobulin reagent passes the previously listed tests.
The quality control of Ig class and subclass specific antiglobulin reagents, although following the previously listed general principles, is more complex. Details of the appropriate techniques are beyond the scope of this chapter; the reader should consult the review by Engelfriet et al.40
Recommended Antiglobulin Test Procedure
A spin-tube technique is recommended for the routine antiglobulin test; the procedure described here is based on BCSH Guidelines for Compatibility Testing in Hospital Blood Banks.27,37 Reliable performance depends on the correct procedure at each stage of the test and appropriate quality-control measures.

The production of satisfactory antiglobulin control cells can be achieved by limiting the level of anti-D sensitization to that which gives a negative test in the presence of 1 in 1000 parts serum in saline.37
The suitability of the antiglobulin control cells can be checked as follows:
The test containing 1 in 1000 serum in saline should be negative and the control tube should give at least 2+ reaction. A negative reaction with the control tube suggests a washing deficiency and demands corrective action. If an automated cell-washing centrifuge is used, the washing efficiency should be checked.30,37
Alternative Technology for Antibody Detection by the Antiglobulin Test
Alternative techniques, now commonly in use, have a simpler reading phase than the manually read spin-tube IAT. These are of two main types: solid-phase red cell adherence methods41 and column agglutination techniques. A well-performed spin-tube IAT, as described earlier, is the standard against which any new system should be compared.
With column agglutination techniques very small volumes of serum and cells are mixed in a reservoir at the top of a narrow column that contains either a Dextran gel (DiaMed, AG, Switzerland) or glass beads (Bio Vue, Ortho-Clinical Diagnosis, NJ).42 The columns with the integral reservoirs are supplied in card or cassette form, respectively. After a suitable incubation period, the cards/cassettes containing the tests are spun in a centrifuge in which the axis of the column is strictly in line with the centrifugal force. The red cells, but not the medium in which they are suspended, enter the column. Agglutinated red cells are trapped at the top of the column and unagglutinated red cells form a pellet at the bottom of the column (see Fig. 22.6, see p. 526).
The advantages of column agglutination technology are as follows:
Assessment of Individual Worker Performance
It is recommended that all staff (including ‘on-call’ staff who do not routinely work in the blood bank) should be assessed at regular intervals. A procedure based on ‘blind’ replicate antiglobulin tests may be used for this purpose.31,37
Titration of Antibodies
A method for preparing primary dilutions of serum and subsequent antibody titration is illustrated in Figure 21.3.
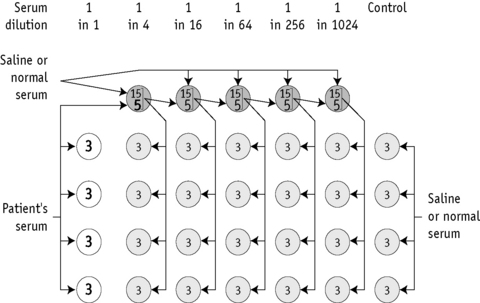
External quality assessment exercises have demonstrated the wide range of titres reported for a single sample, reflecting the differing sensitivities of technologies in use and have also highlighted the lack of reproducibility.44 The following points are taken from an addendum to the BCSH guidelines.45
Preparation of serial dilutions of patient’s or other sera
Red Cell Genotyping
Red cell genotyping is not currently in widespread use, but is emerging as a potential alternative to serological provision of compatible blood for patients in future and is currently used occasionally in situations when serological techniques prove difficult, e.g. phenotyping of a recently transfused patient or in a patient with autoimmune haemolytic anaemia.46
Platelet and Neutrophils
Platelet and Neutrophil Alloantigen Systems
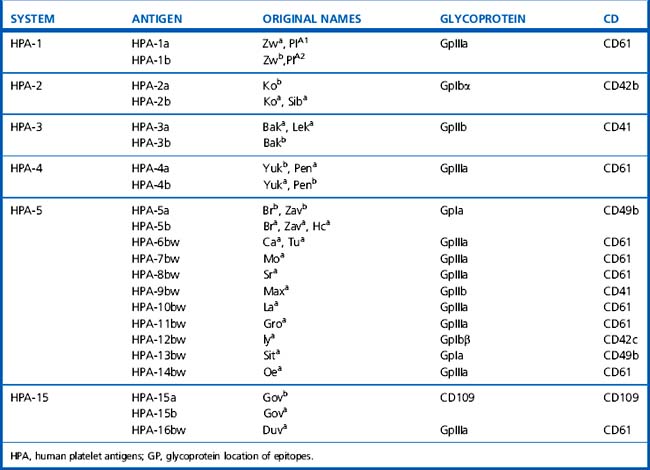
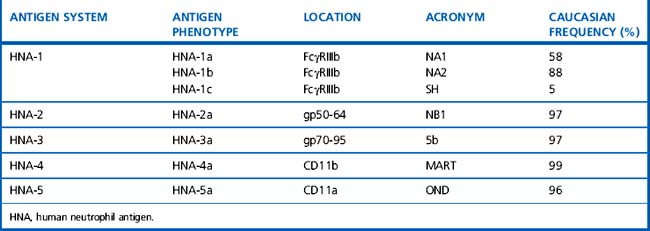
Platelet and neutrophil alloantigens may be exclusive to each cell type (cell-specific) or shared with other cells. The currently recognized human platelet antigens (HPA) and human neutrophil antigens (HNA) are shown in Tables 21.12–21.14.47–49 The historical nomenclature for granulocyte antigens used the letter N to indicate neutrophil specificity and this has been retained, although it is recognized that many studies used granulocytes rather than pure neutrophils and many ‘neutrophil-specific’ antibodies can also target granulocyte precursors. In the HPA nomenclature, HPA-1, -2, -3, -4 and -5 were designated as separate diallelic alloantigen systems. The high-frequency allele of a system was designated with the letter ‘a’ and the low-frequency allele was designated with the letter ‘b’. However, this system is difficult to reconcile with recent molecular genetic knowledge, which suggests that each new base exchange does not constitute a new diallelic alloantigen system but rather defines a single allele that expresses a single new epitope. Currently, nine different GpIIIa alleles have been found in the human gene pool,50 four allelic variants have been described for GpIa and GpIIb and two allelic variants have been found for the GpIbα and GpIbβ subunits50 (a database of human platelet antigens is available at: www.ebi.ac.uk/ipd/hpa).51 Of the shared antigens, the HLA system is the most important clinically; only class 1 antigens (HLA-A, -B and to a lesser extent -C) are expressed on platelets and granulocytes. ABH antigens are also expressed on platelets (in part absorbed from the plasma) but cannot be demonstrated on granulocytes.
Alloantibodies
Alloimmunization to platelet and neutrophil antigens is most commonly a result of transfusion or pregnancy. The associated clinical problems depend on the specificity of the antibody, which determines the target cell involved. Cell-specific alloantibodies are associated with well-defined clinical conditions, which are summarized in Tables 21.15 and 21.16.
Table 21.15 Clinical significance of platelet-specific alloantibodies52
| 1. | Neonatal alloimmune thrombocytopenia |
| 2. | Post-transfusion purpura |
| 3. | Refractoriness to platelet transfusion Usually as a result of human leucocyte antigen antibodies. |
Table 21.16 Clinical significance of neutrophil-specific alloantibodies53–55
| 1. | Neonatal alloimmune neutropenia |
| 2. | Febrile reactions following transfusion (HLA antibodies also involved) |
| 3. | Transfusion-related acute lung injury (TRALI) (transfusion of high-titre antibody) |
| 4. | Poor survival and function of transfused neutrophils (HLA antibodies also involved) |
| 5. | Autoimmune neutropenia – some autoantibodies have allospecificity for HNA system antigens. |
HLA, human leucocyte antigen; HNA, human neutrophil antigen.
Alloimmune fetal and neonatal thrombocytopenia are commonly caused by anti-HPA-1a and less frequently by anti-HPA-5b. The chance of HPA-1a alloimmunization is strongly associated with maternal HLA class-II DRB3*0101 (DR52a) type.56 Partners should be offered HPA genotyping and, if heterozygous, with a severely affected previous child, fetal HPA grouping should be considered in the first trimester of the next pregnancy using amniocyte DNA. Potential strategies for routine antenatal screening and the acceptability and cost-effectiveness of such a programme are discussed in several publications.57,58
Isoantibodies
Rarely, after blood transfusion or pregnancy, patients with type I Glanzmann’s disease make antibodies that react with platelet glycoprotein (Gp) IIb/IIIa not present on their own platelets but present on normal platelets (i.e. isotypic determinants).62–65 Similarly, patients with Bernard–Soulier syndrome may make antibodies against isotypic determinants on GpIbVIX not present on their own platelets.66 This may present a serious clinical problem because no functional compatible donor platelets can be found to treat severe bleeding episodes.
Autoantibodies
Autoimmune thrombocytopenia may be idiopathic or secondary in association with other conditions. Demonstration of a platelet autoantibody is not required; even with the most suitable techniques now available, platelet autoantibodies remain elusive in a variable proportion (10–20%) of patients. The autoreactive antibodies target epitopes on certain glycoproteins. In 30–40% of patients these are directed against epitopes on the αIIbβ3 integrin heterodimer, platelet glycoprotein GpIIbIIIa (CD41) and in 30–40% against the von Willebrand receptor or complex GpIbα/GPIbβ/IX (CD42).67–70
Autoimmune neutropenia may be idiopathic or secondary. Idiopathic autoimmune neutropenia is more common in infants than in adults, in whom it is usually associated with other disorders that have in common a postulated imbalance of the immune system.73 However, it is the least well-studied of the autoimmune cytopenias because it is rare and performing granulocyte assays is difficult, lengthy, labour-intensive and expensive.
Neutrophil autoantibodies (which are usually IgG) are unusual in that they often have well-defined specificity for alloantigens, especially NA1 or NA2.74 These autoantibodies may suppress granulocyte precursors in the bone marrow and cause more severe neutropenia. The investigation of suspected autoimmune neutropenia should, when possible, include granulocyte immunology and studies of colony growth (e.g. CFU-GM) to identify any suppression of bone marrow precursors, as a result of interaction with autoantibodies.
Drug-Induced Antibodies
Drug-induced antibodies may cause selective haemolytic anaemia (see p. 289), thrombocytopenia or neutropenia or various combinations of these in the same patient.75,76
A drug may cause an immune cytopenia by stimulating production of either an autoantibody (which reacts directly with the target cell independently of the drug itself) or a drug-dependent antibody (which destroys the target cell by reacting with a drug–membrane complex on the target cell).77 Laboratory tests may demonstrate both types of antibody in some patients.78
Demonstration of Platelet and Neutrophil Antibodies
Alloantibodies
It is important to combine a sensitive binding assay, such as the platelet immunofluorescence test (PIFT), with an antigen-capture method, such as the monoclonal antibody immobilization of platelet antigens (MAIPA),81 to increase the chance of detecting weak antibodies or those that react with relatively few antigen sites.
An important consideration in platelet serology is the occasional occurrence of antibodies against hidden (cryptic) antigens of the GpIIbIIIa complex, which are exposed by EDTA and paraformaldehyde (PFA) fixation.82 These antibodies, which are only active in vitro, are unpredictable but when suspected can be avoided by using unfixed test platelets from citrated blood.
The detection and identification of granulocyte alloantibodies should be left to experienced reference laboratories, but should follow a similar schedule with the use of monoclonal antibody immobilization of granulocyte antigens (MAIGA)83,84 or adsorption of the sera with pooled platelets to differentiate between granulocyte-specific and HLA antibodies.
Autoantibodies
The detection of autoantibodies and drug-induced antibodies requires special consideration.
A major interest in platelet autoimmunity has been the quantitative measurement of platelet-associated immunoglobulins as an indication of in vivo sensitization. A criticism of these quantitative methods is that they detect not only platelet autoantibody but also Ig nonspecifically trapped or bound to platelets and platelet fragments.85 and are therefore generally nonspecific in the diagnosis of autoimmune thrombocytopenia.86 It is now customary to use the direct PIFT,87,88 using flow cytometry. The patient’s platelets are incubated with isotype-specific fluorescein-isothiocyanate (FITC)-labelled conjugates (anti-IgG, anti-IgM and anti-IgA) and the test is reported as positive when the fluorescence intensity is >mean + 2SD when compared with the results obtained with pooled (10 or more) normal donor platelet suspensions. In a study of 75 patients with idiopathic thrombocytopenic purpura, using microscopy rather than flow cytometry, von dem Borne and colleagues89 found a weak positive (± to +) direct PIFT in 60% of patients and strong reactions (++ to ++++) in only 26% of patients. In the same study, the indirect PIFT was positive with the patient’s serum in 66% of cases who had a positive direct PIFT and it was positive with an ether eluate of the patient’s platelets in 94% of the same cases. Although these results may be a reflection of the relative insensitivity of the method, they may result from a low-affinity antibody that is easily eluted during the assay procedure85 or indicate an alternative immune mechanism for thrombocytopenia in some cases.
The Ig class of platelet autoantibodies is similar in idiopathic and secondary autoimmune thrombocytopenia; mostly it is IgG (92%), but often (also) it is IgM (42%) and sometimes (also) IgA (9%).89 All IgG subclasses occur, but IgG1 and/or IgG3 are the most frequent.
A combination of the granulocyte immunofluorescence test (GIFT)90 and the granulocyte agglutination test (GAT)91 provides the most effective means of granulocyte antibody detection. However, immune complexes and aggregates in a patient’s serum can still cause false-positive results. This can cause a problem for sera from adult patients with secondary autoimmune neutropenia, which should also be investigated for immune complexes (e.g. Clq-enzyme-linked immunosorbent assay). The granulocyte chemiluminescence test (GCLT)92 is relatively insensitive to the presence of immune complexes when inactivated serum is used, but it is unable to detect antibodies of the IgM. Several reviews provide an appraisal of the techniques available for detecting granulocyte-specific antibodies and antigens,93,94 including a recent review of investigations for transfusion-related acute lung injury (TRALI).95
Drug-Induced Antibodies
The serological investigation of drug-induced immune thrombocytopenia (neutropenia) follows the same pattern as for haemolytic anaemia (see p. 289), with the exception that it is not always possible to collect enough cells to test at the nadir of thrombocytopenia or neutropenia. The following blood samples are therefore necessary:
If the patient’s acute-phase serum is tested against normal donor cells, it is essential to take account of positive reactions owing to HLA or cell-specific alloantibody in the patient’s serum. Furthermore, negative results with normal donor cells may be the result of absence of the antigen for the particular drug-dependent antibody (e.g. owing to genetic restriction of the antigen concerned).96
Methods of Demonstrating Antibodies
The Immunofluorescent Antiglobulin Methods
The immunofluorescent antiglobulin methods are based on the conventional antiglobulin technique (see p. 500) and are suitable for platelet,80 granulocyte90 and lymphocyte98 serology. The PIFT and GIFT (Fison Ltd, Loughborough, UK) are described in detail in this chapter.
These tests can either be read by direct examination of a cell suspension using fluorescence microscopy or by flow cytometry. These tests can detect allo-, auto- and drug-induced antibodies and, by using appropriate monospecific antiglobulin reagents, can determine the Ig class and subclass of the antibody and cell-bound complement components. Both tests can be used with chloroquine-treated cells to differentiate cell-specific from HLA antibodies.99
Patient’s and Screening Panel Cells
The best results are obtained with the freshest cell preparations, but some delay is tolerable (see later). Neutrophils are more susceptible to storage damage than platelets; cells should be fixed (see later) on the day of collection, but serology may be delayed to the following day. Platelets are more resilient and an anticoagulated blood sample may be satisfactory for testing for up to 2 days at ambient temperature (c20°C). Once fixed, platelets may be kept for 3–4 days at 4°C before serological testing. For longer storage, platelet-rich plasma may be kept at −40°C for at least 2 months; however, there is some membrane damage after recovery of frozen platelets, which causes increased background fluorescence that may limit the sensitivity of the test.100,101 For longer-term storage a cryoprotectant (e.g. DMSO) may be used.101
Eluate from Patient’s Sensitized Cells
Elution by lowering the pH of the medium, by ether (or DMSO) and by heating to 56°C, has been used.102 For routine platelet serology, ether elution for platelet autoantibodies or heating to 56°C for platelet-specific alloantibodies could be used.
Platelet Preparation
Granulocyte Preparation
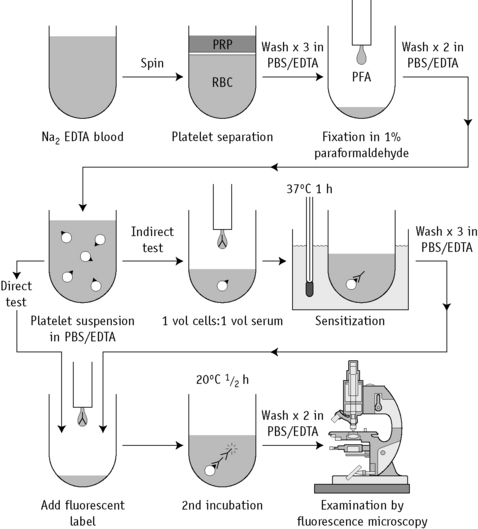
Platelet and Granulocyte Immunofluorescence Tests
The serological methods for testing platelets and granulocytes in the suspension immunofluorescence test are similar, except that platelets are washed throughout in PBS/EDTA buffer and granulocytes are washed in PBS/BSA buffer. A flow diagram of the PIFT is shown in Figure 21.5.
Positive and negative controls (as described earlier) should be included with each batch of tests.
Indirect Test
Scoring Results
Reactions in the PIFT and GIFT may be scored on a scale from negative (−) through graded positives + to + + + +. Although subjective, this method of scoring in experienced hands can produce semiquantitative results in the PIFT.103
Use of Flow Cytometry
With simplification of flow cytometers and improved software, more platelet reference laboratories are using them for primary analysis in PIFT because sensitivity is improved. Nevertheless, platelets are more difficult to work with flow cytometrically than other cells and particular attention has to be paid to prevent aggregation and to ensure single cell suspensions. Presence of platelet particles and debris may also cause confusion. The technical considerations of applying flow cytometry to platelet work are the subject of several reviews.87,88
Chloroquine Treatment of Platelets and Granulocytes
Platelets for chloroquine treatment should be prepared from fresh blood or blood stored overnight at 4°C; granulocytes are suitable only if freshly prepared.52,104 An important consideration is the extent of chloroquine-induced cell membrane damage, which is minimal with fresh cells.
Interpretation of Results with Chloroquine-Treated Cells
Typical results with HLA- and cell-specific antibodies are shown in Table 21.17. If a serum that has been shown to contain HLA antibodies by LCT and/or LIFT gives equal or stronger reactions with chloroquine-treated cells than with untreated cells, then a cell-specific antibody is also present. The Second Canadian Workshop on Platelet Serology100 concluded that a weaker reaction with chloroquine-treated platelets should be interpreted with caution; this could indicate residual HLA reactivity, especially in the presence of high-titre multispecific HLA antibodies. If a platelet-specific antibody is nevertheless still suspected, other methods should be used to confirm this (e.g. MAIPA using appropriate monoclonal antibodies for capture; see later).
Table 21.17 Platelet and granulocyte antibody reactions using cells prepared with and without treatment with chloroquine
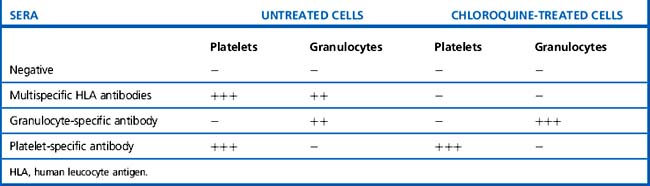
Similar caution should be observed in interpreting the GIFT results with chloroquine-treated cells.
MAIPA Assay
The principle of the MAIPA assay is shown in Figure 21.6. The test is based on the use of monoclonal antibodies, such as anti-IIbIIIa, anti-IbIX, anti-IaIIa and anti-HLA class I, to ‘capture’ specific platelet membrane glycoproteins. The availability of appropriate monoclonal antibodies has led to the wider clinical application of this method.81 The same principle can be used with granulocytes, depending on the availability of appropriate monoclonal antibodies.81,105
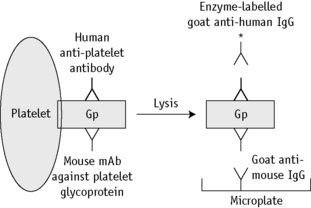
The following assay protocol was developed from the original method described by Kiefel.81,106
Other Methods
Several other methods have been developed for the detection of platelet antibodies.
Solid-phase red cell adherence (SPRCA) techniques (some commercially available) evolved as alternatives to the microscopic reading initially required for the PIFT. These assays combine traditional red cell serology technology with platelet serology. Platelets are captured on microtitre wells; test antibodies are applied; and, after washing and addition of antihuman globulin, platelet or HLA alloantibody binding is detected using tanned sheep red cells107 or anti-D sensitized RhD-positive red cells.108 SPRCA are robust, sensitive tests that lend themselves to automation and the chloroquine treatment of platelets can be used effectively to screen out HLA antibodies.
GTI PakPlus is a platelet antibody kit based on an ELISA principle (Quest Biomedical, Solihull, UK). Microwells coated with platelet glycoproteins or HLA class I antigens are incubated with test serum. After incubation, followed by washing to remove unbound proteins, any antibody bound to the microwell is detected using an alkaline-phosphatase-conjugated antihuman globulin reagent (anti-immunoglobulin or anti-IgG) and the appropriate substrate. Results are considered positive when the ratio of the mean absorbance of the test sample to that of the normal control sera is ≥2.0.109
With respect to testing for granulocyte antibodies when working with the GIFT or GAT, elucidation of the alloantibody requires panels of typed granulocytes, which cannot be preserved for more than a few hours. A technique has been reported that uses extracted granulocyte antigens coated onto U-well Terasaki plates and a micromixed passive haemagglutination test. Patient’s serum and appropriate controls (sera known to contain granulocyte-specific antibodies, monoclonal antibodies, such as anti-CD16 and anti-NA1 and sera from normal donors) are added to the wells and, following incubation and washing, indicator blood cells are added (sheep red blood cells coated with antihuman IgG and antimouse IgG).110
Molecular Genotyping of Platelet Alloantigens
The application of DNA technology for platelet genotyping is based on the knowledge that the platelet antigen systems are the result of single DNA base changes, which lead to single amino acid substitutions in the platelet membrane glycoproteins (Table 21.13).
Molecular genotyping involves amplification of the relevant segments of genomic DNA from any nucleated cell by the PCR in combination with sequence-specific primers111 or by allele-specific restriction enzyme analysis112 or allele-specific oligonucleotide dot blot hybridization.113
1 International Society of Blood Transfusion (ISBT) Working Party. Available at: http://ibgrl.blood.co.uk Accessed 01.06.10
2 Daniels G. Functions of red cell surface proteins. Vox Sang. 2007;93(4):331-340.
3 Eastlund T. The histo-blood group ABO system and tissue transplantation. Transfusion. 1998;38:975-988.
4 Galliford J., Charif R., Chan K.K., et al. ABO incompatible renal transplantation in a steroid sparing protocol. Transplantation. 2008;86(7):901-906.
5 Mourant A.E., Kopec A.C., Domaniewska-Sobczak K. The Distribution of the Human Blood Groups and Other Biochemical Polymorphisms, 2nd ed. Oxford: Oxford University Press; 1976.
6 Yainamoto F. Molecular genetics of the ABO histo-blood group system. Vox Sang. 1995;69:1-7.
7 Garratty G., Arndt P., Co S. Fatal ABO haemolytic transfusion reaction resulting from acquired B antigen only detectable by some monoclonal grouping reagents. Transfusion. 1993;33(suppl):47S.
8 Garratty G. Association of blood groups and disease: do blood group antigens and antibodies have a biological role? Hist Philos Life Sci. 1996;18:321-344.
9 O’Donnell J., Laffan M. The relationship between ABO histo-blood group, factor VIII and von Willebrand factor. Transfus Med. 2001;11:343-351.
10 Jenkins PV, O’Donnell JS. ABO blood group determines plasma von Willebrand factor levels: a biologic function after all? Transfusion. 46(10):1836–1844.
11 Avent N.D., Reid M.E. The Rh blood group system: a review. Blood. 2000;95:375-387.
12 Daniels G., Poole G., Poole J. Partial D and weak D: can they be distinguished? Transfus Med. 2007;17(2):145-146.
13 World Health Organization (WHO). Twenty-eighth Report of WHO Expert Committee on Biological Standardization. Geneva: WHO; 1977. Technical Report Series 610
14 Lo Y.M., Bowell P.J., Selinger M., et al. Prenatal determination of fetal rhesus D status by DNA amplification of peripheral blood of rhesus-negative mothers. Ann N Y Acad Sci. 1994;731:229-236.
15 Lo Y.M., Tein M.S., Lau T.K., et al. Quantitative analysis of fetal DNA in maternal plasma and serum: implications for noninvasive prenatal diagnosis. Am J Hum Genet. 1998;62:768-775.
16 Serious Hazards of Transfusion Annual Report. Available at: www.shotuk.org, 2008. Accessed 01.06.10
17 Mollison P.L., Engelfriet C.P., Contreras M. Blood Transfusion in Clinical Medicine, 10th ed. Blackwell Scientific, Oxford, 1997;89.
18 Engelfriet C.P. The immune destruction of red cells. Transfus Med. 1992;2:1-6.
19 Horsewood P., Kelton J.G. Macrophage-mediated cell destruction. In: Garratty G., editor. Immunobiology of Transfusion Medicine. New York: Marcel Dekker; 1994:434-464.
20 Zupanska B.A. Cellular bioassays and their use in predicting the clinical significance of antibodies. In: Garratty G., editor. Immunobiology of Transfusion Medicine. New York: Marcel Dekker; 1994:465-492.
21 Guyre P.M., Miller R. Recombinant immune interferon increases immunoglobulin G Fc receptors on cultured human mononuclear phagocytes. J Clin Invest. 1983;72:393-397.
22 Griffin J.A., Griffin F.M. Augmentation of macrophage complement receptor function in vitro. J Exp Med. 1979;150:653-675.
23 Volk H.D., Reinke P., Krausch D. Monocyte deactivation-rationale for a new therapeutic strategy in sepsis. Intensive Care Med. 1996;22:S474-S481.
24 EU. In-vitro diagnostics IVD Medical Devices Directive 1998. Official Journal of the European Communities. 1998:1331. (7.12.98)
25 British Committee for Standards in Haematology. Guidelines for compatibility procedures in blood transfusion laboratories. Transfus Med. 2004;14:59-73.
26 Case J., Ford D.S., Chung A. International reference reagents: antihuman globulin. Vox Sang. 1999;77:121-127.
27 Department of Health. Guidelines for the blood transfusion services in the United Kingdom, 4th ed. London: HMSO; 1998.
28 British Committee for Standards in Haematology. Recommendations for the evaluation, validation and implementation of new techniques for blood grouping antibody screening and crossmatching. Transfus Med. 1995;5:145-150.
29 Voak D. Validation of new technology for antibody detection by antiglobulin tests. Transfus Med. 1992;2:177-179.
30 Phillips P.K., Voak D., Whitton C.M., et al. BCSH-NIBSC anti-D reference reagent for antiglobulin tests: the in-house assessment of red cell washing centrifuges and of operator variability in the detection of weak macroscopic agglutination. Transfus Med. 1993;3:143-148.
31 Voak D., Downie D.M., Moore B.P. Replicate tests for the detection and correction of errors in anti-human globulin AHG tests: optimum conditions and quality control. Haematologia (Budap). 1988;2:3-16.
32 Knowles S.M., Milkins C.E., Chapman J.F., et al. The United Kingdom National External Quality Assessment Scheme (Blood Transfusion Laboratory practice): trends in proficiency and practice between 1985 and 2000. Transfus Med. 2002;12:11-23.
33 Voak D., Downie D.M., Haigh T., et al. Improved antiglobulin tests to detect difficult antibodies: detection of anti-Kell by LISS. Med Lab Sci. 1982;39:363-370.
34 Scott M.L., Voak D., Downie D.M. Optimum enzyme activity in blood grouping and a new technique for antibody detection: an explanation for the poor performance of the one-stage mix technique. Med Lab Sci. 1988;45:7-18.
35 Scott M.L., Phillips P.K. A sensitive two-stage papain technique without cell washing. Vox Sang. 1987;52:67-70.
36 Pirofsky B. The use of bromelin in establishing a standard cross-match. Am J Clin Pathol. 1959;32:350-356.
37 British Committee for Standards in Haematology. Roberts B., editor. Standard Haematology Practice. Blackwell Scientific, Oxford, 1991;150-163.
38 Coombs R.R., Mourant A.E., Race R.R. A new test of the detection of weak and ‘incomplete’ Rh agglutinins. Br J Exp Pathol. 1945;26:255-266.
39 Voak D., Downie D.M., Moore P.B., et al. Antihuman globulin reagent specification: the European and ISBT/ICSH view. Biotest Bulletin. 1986;1:7.
40 Engelfriet C.P., Overbeeke M.A., Voak D. The antiglobulin test, Coombs test and the red cell. Cash J.D., editor. Progress in Transfusion Medicine, Vol. 2. Churchill Livingstone, Edinburgh, 1987;74-98.
41 Sinor L.T. Advances in solid-phase red cell adherence methods and transfusion serology. Transfus Med Rev. 1992;6:26-31.
42 Lapierre Y., Rigel D., Adams J., et al. The gel test: a new way to detect red cell antigen–antibody reactions. Transfusion. 1990;30:109-113.
43 Phillips P., Voak D., Downie M. New reference reagent for the quality assurance of anti-D antibody detection. Transfus Med. 1998;8:225-230.
44 O’Hagan J., Milkins C.E., Chapman J.F., et al. Antibody titres – results of a UK NEQAS BGS exercise and accompanying questionnaire. Transfus Med. 7(suppl 1), 1997.
45 British Committee for Standards in Haematology. Addendum for guidelines for blood grouping and red cell antibody testing during pregnancy. Transfus Med. 1999;9:99.
46 Van der Schoot C.E., de Haas M., Engelfriet C.P., et al. Genotyping for red cell polymorphisms. Vox Sang. 2009;96(2):167-179.
47 Santoso S. Human platelet-specific alloantigens: update. Vox Sang. 1998;74(suppl 2):249-253.
48 Bux J., von dem Borne A.E.G., de Haas L. ISBT Working Party on Platelet and Granulocyte Serology, Granulocyte Antigen Working Party: Nomenclature of granulocyte alloantigens. Vox Sang. 1999;77:251.
49 Lucas G.F., Metcalfe P. Platelet and granulocyte polymorphisms. Transfus Med. 2000;10:157-174.
50 Metcalfe P., Watkins N.A., Ouwehand W.H., et al. Nomenclature of human platelet antigens (HPA). Vox Sang. 2003;85:240-245.
51 IPD–HPA Database. Available at: www.ebi.ac.uk/ipd/hpa Accessed 30.07.10
52 Nordhagen R., Flaathen S.T. Chloroquine removal of HLA antigens from platelets for the platelet immunofluorescence test. Vox Sang. 1985;48:156-159.
53 Engelfriet C.P., Tetteroo P.A., van der Veen J.P., et al. Granulocyte-specific antigens and methods for their detection. In: McCullough J., Sandler S.G., Sweeney G.E., editors. Advances in Immunobiology: Blood Cell Antigens and Bone Marrow Transplantation. New York: Liss; 1984:121.
54 McCullough J. The clinical significance of granulocyte antibodies and in vivo studies of the fate of granulocytes. In: Garraty G., editor. Current Concepts in Transfusion Therapy. Arlington, VA, USA: American Association of Blood Banks; 1985:125.
55 Minchinton R.M., Waters A.H. The occurrence and significance of neutrophil antibodies. Br J Haematol. 1984;56:521-528.
56 Decary F., L’Abbe D., Tremblay L., et al. The immune response to the HPA-1a antigen: association with HLA-DRw52a. Transfus Med. 1991;1:55-62.
57 Williamson L.W., Hackett G., Rennie J., et al. The natural history of fetomaternal alloimmunization to the platelet-specific antigen HPA-1a PIAI Zwa as determined by antenatal screening. Blood. 1998;92:2280-2287.
58 Flug F., Karpatkin M., Karpatkin S. Should all pregnant women be tested for their platelet PLA Zw HPA-1 phenotype? Br J Haematol. 1994;86:1-5.
59 Mueller-Eckhardt C., Kroll H., Kiefel V., et al. European PTP Study Group: post-transfusion purpura. In: Kaplan-Gouet C., Schlegel N., Salmon C., et al, editors. Platelet Immunology: Fundamental and Clinical Aspects. INSERM/John Libbey, Eurotext, 1991.
60 Schnaidt M., Northoff H., Wernet D. Frequency and specificity of platelet-specific allo-antibodies in HLA-immunised haematologic-oncologic patients. Transfus Med. 1996;6:111-114.
61 Kurz M., Hildegard G., Hocker P., et al. Specificities of anti-platelet antibodies in multitransfused patients with haemato-oncological disorders. Br J Haematol. 1996;95:564-569.
62 Bierling P., Fromont P., Elbez A., et al. Early immunization against platelet glycoprotein IIIa in a new born Glanzmann type I patient. Vox Sang. 1988;55:109-113.
63 Brown C.H.3rd, Weisberg R.J., Natelson E.A., et al. Glanzmann’s thrombasthenia: assessment of the response to platelet transfusion. Transfusion. 1975;15:124-131.
64 Ribera A., Martin-Vega C., Pico M., et al. Sensitization against platelet antigens in Glanzmann disease. Congress of the International Society of Blood Transfusion in Association with British Blood Transfusion Society (BBTS), Manchester, 1988;240. Abstract XX
65 Van Leeuwen E.F., von dem Borne A.E., von Riesz L.E., et al. Absence of platelet specific alloantigens in Glanzmann’s thrombasthenia. Blood. 1981;57:49-54.
66 Degos L., Tobelem G., Lethielliux P., et al. A molecular defect in platelets of patients with Bernard-Soulier syndrome. Blood. 1977;50:899-903.
67 Van Leeuwen E.F., Helmerhorst F.M., Engelfriet C.P., et al. Specificity of auto-antibodies in autoimmune thrombocytopenia. Blood. 1982;59:23-26.
68 Fujisawa K., Tani P., O’Toole T.E., et al. Different specificities of platelet-associated and plasma auto-antibodies to platelet GPIIb-IIIa in patients with chronic immune thrombocytopenic purpura. Blood. 1992;79:1441-1446.
69 Fujisawa K., Tani P., McMillan R. Platelet-associated antibody to glycoprotein IIb/IIIa from chronic immune thrombocytopenic purpura patients often binds to divalent cation-dependent antigens. Blood. 1993;81:1284-1289.
70 Hou M., Stockelberg D., Kutti J., et al. Glycoprotein IIb/IIIa autoantigenic repertoire in chronic idiopathic thrombocytopenic purpura. Br J Haematol. 1995;91:971-975.
71 Kelton J.G., Sheridan D.P., Santos A.V., et al. Heparin-induced thrombocytopenia: laboratory studies. Blood. 1988;72:925-930.
72 Pegels J.G., Bruynes E.C., Engelfriet C.P., et al. Pseudothrombocytopenia: an immunologic study on platelet antibodies dependent on ethylene diamine tetra-acetate. Blood. 1982;59:157-161.
73 Shastri K.A., Logue G.L. Autoimmune neutropenia. Blood. 1993;81:1984-1995.
74 McCullough J., Clay M.E., Thompson H.W. Autoimmune granulocytopenia. Baillière’s Clinical Immunology and Allergy. 1987;1:303.
75 Bux J., Mueller-Eckhardt C. Autoimmune neutropenia. Semin Hematol. 1992;29:45-53.
76 Mueller-Eckhardt C. Drug-induced immune thrombocytopenia. Baillière’s Clinical Immunology and Allergy. 1987;1:369.
77 Mueller-Eckhardt C., Salama A. Drug-induced immune cytopenias: a unifying pathogenetic concept with special emphasis on the role of drug metabolites. Transfus Med Rev. 1990;4:69-77.
78 Salama A., Schutz B., Kiefel V., et al. Immune-mediated agranulocytosis related to drugs and their metabolites: mode of sensitization and heterogeneity of antibodies. Br J Haematol. 1989;72:127-132.
79 Metcalfe P., Waters A.H. Report on the fourth ISBT/ICSH platelet serology workshop. Vox Sang. 1990;58:170-175.
80 Von dem Borne A.E., Verheught F.W., Oosterhof F., et al. A simple immunofluorescence test for the detection of platelet antibodies. Br J Haematol. 1978;39:195-207.
81 Kiefel V. The MAIPA assay and its applications in immunohaematology. Transfus Med. 1992;2:181-188.
82 von dem Borne A.E., van der Lelie J., Vos J.J., et al. Antibodies against crypt antigens of platelets. Characterization and significance for the serologist. Curr Stud Hematol Blood Transfus. 1986;52:33.
83 Bux J., Kober B., Kiefel V., et al. Analysis of granulocyte-reactive antibodies using an immunoassay based upon monoclonal-antibody-specific immobilization of granulocyte antigens. Transfus Med. 1993;3:157-162.
84 Minchinton R.M., Noonan K., Johnson T.J. Examining technical aspects of the monoclonal antibody immobilization of granulocyte antigen assay. Vox Sang. 1997;73:87-92.
85 Shulman N.R., Leissinger C.A., Hotchkiss A.J., et al. The non-specific nature of platelet associated IgG. Trans Assoc Am Physicians. 1982;95:213-220.
86 Von dem Borne A.E. Autoimmune thrombocytopenia. Baillière’s Clinical Immunology and Allergy. 1987;1:269.
87 Goodall A.H., Macey M.G. Platelet-associated molecules and immunoglobulins. In: Macey M.R., editor. Flow Cytometry – Clinical Applications. London: Blackwell Scientific; 1994:148-191.
88 Ault K.A. Flow cytometric measurement of platelet-associated immunoglobulin. Pathol Immunopathol Res. 1988;7:395-408.
89 Von dem Borne A.E., Vos J.J., van der Lelie J., et al. Clinical significance of positive platelet immunofluorescence test in thrombocytopenia. Br J Haematol. 1986;64:767-776.
90 Verheugt F.W., von dem Borne A.E., Decary F., et al. The detection of granulocyte allo-antibodies with an indirect immunofluorescence test. Br J Haematol. 1977;36:533-544.
91 McCullough J., Clay M.E., Press C., et al. Granulocyte serology. A clinical and laboratory guide. Chicago: ACSP; 1988.
92 Lucas G.F. Prospective evaluation of the chemiluminescence test for the detection of granulocyte antibodies: comparison with the immunofluorescence test. Vox Sang. 1994;66:141-147.
93 Bux J. Challenges in the determination of clinically significant granulocyte antibodies and antigens. Transfus Med Rev. 1996;10:222-232.
94 Stroncek D.F. Granulocyte immunology: is there a need to know? Transfusion. 1997;37:886-888.
95 ISBT Working Party. Recommendations of the ISBT Working Party on granulocyte immunobiology for leukocyte antibody screening in the investigation and prevention of antibody mediated transfusion related acute lung injury. Vox Sang. 2009;96:266-269.
96 Claas F.H., Langerak J., de Beer L.L., et al. Drug-induced antibodies: interaction of the drug with a polymorphic platelet antigen. Tissue Antigens. 1981;17:64-66.
97 Salama A., Mueller-Eckhardt C., Kissel K., et al. Ex vivo antigen preparation for the serological detection of drug-dependent antibodies in immune haemolytic anaemias. Br J Haematol. 1984;58:525-531.
98 Decary F., Vermeulen A., Engelfriet C.P. A look at HLA antisera in the indirect immunofluorescence technique LIFT. Histocompatibility testing. Munksgaard, Copenhagen, 1975;380.
99 Metcalfe P., Minchinton R.M., Murphy M.F., et al. Use of chloroquine-treated granulocytes and platelets in the diagnosis of immune cytopenias. Vox Sang. 1985;49:340-345.
100 Decary F. Report on the second Canadian workshop on platelet serology. Curr Stud Hematol Blood Transfus. 1988;54:1.
101 Helmerhorst F.M., Ten Boerge M.L., van der Plas-van Dalen C., et al. Platelet freezing for serological purposes with and without a cryopreservative. Vox Sang. 1984;46:318-322.
102 Helmerhorst F.M., van Oss C.J., Bruynes E.C.E., et al. Elution of granulocyte and platelet antibodies. Vox Sang. 1982;43:196-204.
103 Vos J.J., Huisman J.G., Winkel I.N., et al. Quantification of platelet-bound allo-antibodies by radioimmunoassay: a study on some variables. Vox Sang. 1987;53:108-116.
104 Minchinton R.M., Waters A.H. Chloroquine stripping of HLA antigens from neutrophils without removal of neutrophil specific antigens. Br J Haematol. 1984;57:703-706.
105 Metcalfe P., Waters A.H. Location of the granulocyte-specific antigen LAN on the Fc-receptor III. Transfus Med. 1992;2:283-287.
106 Kiefel V., Santoso S., Weisheit M., et al. Monoclonal antibody specific immobilization of platelet antigens MAIPA: a new tool for the identification of platelet-reactive antibodies. Blood. 1987;70:1722-1726.
107 Shibata Y., Juji T., Nishizawa Y., et al. Detection of platelet antibodies by a newly developed mixed agglutination with platelets. Vox Sang. 1981;41:25-31.
108 Lown J.A., Ivey J.G. Evaluation of solid phase red cell adherence technique for platelet antibody screening. Transfus Med. 1991;1:163-167.
109 Lucas G.F., Rogers S.E. Evaluation of an enzyme-linked immunosorbent assay kit GTI PakPlus® for the detection of antibodies against human platelet antigens. Transfus Med. 1999;9:63-67.
110 Araki N., Nose Y., Kohsaki M., et al. Antigranulocyte antibody screening with extracted granulocyte antigens by a micro-mixed passive haemagglutination method. Vox Sang. 1999;77:44-51.
111 Metcalfe P., Waters A.H. HPA-1 typing by PCR amplification with sequence-specific primers PCR-SSP: a rapid and simple technique. Br J Haematol. 1993;85:227-229.
112 Simsek S., Faber N.M., Bleeker P.M., et al. Determination of human platelet antigen frequencies in the Dutch population by immunophenotyping and DNA allele-specific restriction enzyme analysis. Blood. 1993;81:835-840.
113 McFarland J.G., Aster R.H., Bussel J.B., et al. Prenatal diagnosis of neonatal alloimmune thrombocytopenia using allele-specific oligonucleotide probes. Blood. 1991;78:2276-2282.
* The microplate is prepared by adding to each well 100 μl of goat antimouse IgG (Sera-Lab, code SBA 1030–01) at 3 μg/ml in carbonate coating buffer, pH 9.6. Leave the plate to stand overnight at 4°C. Next morning, wash the plate four times with TBS wash buffer. Leave the last wash supernatant for 30 min to ‘block’ nonspecific protein adsorption to the plastic and then decant.
[/level-membership-for-hematology-oncology-and-palliative-medicine-category][not-level-membership-for-hematology-oncology-and-palliative-medicine-category]
Chapter 21 Blood cell antigens and antibodies
erythrocytes, platelets and granulocytes
Erythrocytes
Red Cell Antigens
A total of 30 blood group systems have been described (Table 21.1). Each system is a series of red cell antigens, determined either by a single genetic locus or very closely linked loci. In addition to the blood group systems, there are six ‘collections’ of antigens (e.g. Cost), which bring together other genetically, biochemically or serologically related sets of antigens and a separate series of low-frequency (e.g. Rd) and high-frequency (e.g. Vel) antigens, which do not fit into any system or collection. A numeric catalogue of red cell antigens is being maintained by an International Society of Blood Transfusion (ISBT) Working Party.1
Alternative forms of a gene coding for red cell antigens at a particular locus are called alleles and individuals may inherit identical or non-identical alleles. Most blood group genes have been assigned to specific chromosomes (e.g. ABO system on chromosome 9, Rh system on chromosome 1). The term genotype is used for the sum of the inherited alleles of a particular gene (e.g. AA, AO) and most red cell genes are expressed as codominant antigens (i.e. both genes are expressed in the heterozygote). The phenotype refers to the recognizable product of the alleles and there are many racial differences in the frequencies of red cell phenotypes, as shown in Table 21.2.
Red cell antigens are determined either by carbohydrate structures or protein structures. Carbohydrate-defined antigens are indirect gene products (e.g. ABO, Lewis, P). The genes code for an intermediate product, usually an enzyme that creates the antigenic specificity by transferring sugar molecules onto the protein or lipid. Protein-defined antigens are direct gene products and the specificity is determined by the inherited amino acid sequence and/or the conformation of the protein. Proteins carrying red cell antigens are inserted into the membrane in one of three ways: single pass, multipass or linked to phosphatidylinositol (GPI-linked). Only a few red cell antigens are erythroid-specific (Rh, LW, Kell and MNSs), the remainder being expressed in many other tissues. The structure and functions of the membrane proteins and glycoproteins carrying blood group antigens have been reviewed by Daniels.2 An illustration of the putative functions of molecules containing blood group antigens is provided in Table 21.3.
ABO System
Discovery of the ABO system by Landsteiner marked the beginning of safe blood transfusion. The ABO antigens, although most important in relation to transfusion, are also expressed on most endothelial and epithelial membranes and are important histocompatibility antigens.3 Transplantation of ABO-incompatible solid organs increases the potential for hyperacute graft rejection, although ABO-incompatible renal transplantation can be successfully carried out with plasmapheresis in addition to immunosuppression of the recipient.4 Major ABO-incompatible stem cell transplants (e.g. group A stem cells into a group O recipient) will provoke haemolysis, unless the donation is depleted of red cells.
ABO Antigens and Encoding Genes
There are four main blood groups: A, B, AB and O (Table 21.4). In the British Caucasian population, the frequency of group A is 42%, B 9%, AB 3% and O 46%, but there is racial variation in these frequencies.5 The epitopes of ABO antigens are determined by carbohydrates (sugars), which are linked either to polypeptides (forming glycoproteins) or to lipids (glycolipids).
The expression of ABO antigens is controlled by three separate genetic loci: ABO located on chromosome 9 and FUT1 (H) and FUT2 (Se), both of which are located on chromosome 19. The genes from each locus are inherited in pairs as Mendelian dominants. Each gene codes for a different enzyme (glycosyltransferase), which attaches specific monosaccharides onto precursor disaccharide chains (Table 21.5). There are four types of disaccharide chains known to occur on red cells, on other tissues and in secretions. The Type 1 disaccharide chain is found in plasma and secretions and is the substrate for the FUT2 (Se) gene, whereas Types 2, 3 and 4 chains are only found on red cells and are the substrate for the FUT1 (H) gene. It is likely that the O and B genes arose by mutation of the A gene. The O gene does not encode for the production of a functional enzyme; group O individuals commonly have a deletion at nucleotide 261 (the O1 allele), which results in a frame-shift and premature termination of the translated polypeptide and the production of an enzyme with no catalytic activity. The B gene differs from A by consistent nucleotide substitutions.6 The expression of A and B antigens is dependent on the H and Se genes, which both give rise to glycosyltransferases that add L-fucose, producing the H antigen. The presence of an A or B gene (or both) results in the production of further glycosyltransferases, which convert H substance into A and B antigens by the terminal addition of N-acetyl-D-galactosamine and D-galactose, respectively (Fig. 21.1). Because the O gene produces an inactive transferase, H substance persists unchanged as group O. In the extremely rare Oh Bombay phenotype, the individual is homozygous for the h allele of FUT1 and hence cannot form the H precursor of the A and B antigen. Their red cells type as group O, but their plasma contains anti-H, in addition to anti-A, anti-B and anti-A,B, which are all active at 37°C. As a consequence, individuals with an Oh Bombay phenotype can only be safely transfused with other Oh red cells.
Table 21.5 Glycosyltransferases produced by genes encoding antigens within the ABO, H and Lewis blood group systems
| Gene | Allele | Transferase |
|---|---|---|
| FUT1 | H | α-2-L-fucosyltransferase |
| h | None | |
| A | A | α-3-N-acetyl-D-galactosaminyltransferase |
| B | B | α-3-D-galactosyltransferase |
| O | O | None |
| FUT2 | Se | α-2-L-fucosyltransferase |
| se | None | |
| FUT3 | Le | α-3/4-L-fucosyltransferase |
| le | None |
Serologists have defined two common subgroups of the A antigen. Approximately 20% of group A and group AB individuals belong to group A2 and group A2B, respectively, the remainder belonging to group A1 and group A1B. These subgroups arise as a result of inheritance of either the A1 or A2 alleles. The A2 transferase is less efficient in transferring N-acetyl-D-galactosamine to available H antigen sites and cannot utilize Types 3 and 4 disaccharide chains. As a consequence, A2 red cells have fewer A antigen sites than A1 cells and the plasma of group A2 and group A2B individuals may also contain anti-A1. The distinction between these subgroups can be made using the lectin Dolichos biflorus, which only reacts with A1 cells. The H antigen content of red cells depends on the ABO group and, when assessed by agglutination reactions with anti-H, the strength of reaction tends to be graded O > A2 > A2B > B > A1 > A1B. Other subgroups of A are occasionally found (e.g. A3, Ax) that result from mutant forms of the glycosyltransferases produced by the A gene and are less efficient at transferring N-acetyl-D-galactosamine onto H substance.6
Secretors and Non-Secretors
The ability to secrete A, B and H substances in water-soluble form is controlled by FUT2 (dominant allele Se). In a Caucasian population, about 80% are secretors (genotype SeSe or Sese) and 20% are non-secretors (genotype sese) (Table 21.6). Secretors have H substance in the saliva and other body fluids together with A substances, B substances or both, depending on their blood group. Only traces of these substances are present in the secretions of non-secretors, although the antigens are expressed normally on their red cells and other tissues.
An individual’s secretor status can be determined by testing for ABH substance in saliva (see p. 504).
ABO Antigens and Disease
Group A individuals rarely may acquire a B antigen from a bacterial infection that results in the release of a deacetylase enzyme. This converts N-acetyl-D-galactosamine into α-galactosamine, which is similar to galactose, the immunodominant sugar of group B, thereby sometimes causing the red cells to appear to be group AB. In the original reported cases, five out of seven of the patients had carcinoma of the gastrointestinal tract. Case reports attest to the danger of individuals with an acquired B antigen being transfused with AB red cells, resulting in a fatal haemolytic transfusion reaction following the production of hyperimmune anti-B.7
The inheritance of ABH antigens is also known to be weakly associated with predisposition to certain diseases. Group A individuals have 1.2 times the risk of developing carcinoma of the stomach than group O or B; group O individuals have 1.4 times more risk of developing peptic ulcer than non-group O individuals; and non-secretors of ABH have 1.5 times the risk of developing peptic ulcer than secretors.8 The ABO group also affects plasma von Willebrand factor (VWF) and factor VIII levels; group O healthy individuals have levels around 25% lower than those of other ABO groups.9 ABO blood group appears to mediate its effect by accelerating clearance of VWF but the mechanism is not yet clear10 ABH antigens are also frequently more weakly expressed on the red cells of persons with leukaemia.
ABO Antibodies
Anti-A and anti-B
ABO antibodies, in the absence of the corresponding antigens, appear during the first few months after birth, probably as a result of exposure to ABH antigen-like substances in the diet or the environment (i.e. they are ‘naturally occurring’) (Table 21.4). This allows for reverse (serum/plasma) grouping as a means of confirming the red cell phenotype. The antibodies are a potential cause of dangerous haemolytic transfusion reactions if transfusions are given without regard to ABO compatibility. Anti-A and anti-B are always, to some extent, immunoglobulin M (IgM). Although they react best at low temperatures, they are nevertheless potentially lytic at 37°C. Hyperimmune anti-A and anti-B occur less frequently, usually in response to transfusion or pregnancy, but they may also be formed following the injection of some toxoids and vaccines. They are predominantly of IgG class and are usually produced by group O and sometimes by group A2 individuals. Hyperimmune IgG anti-A and/or anti-B from group O or group A2 mothers may cross the placenta and cause haemolytic disease of the newborn (HDN). These antibodies react over a wide thermal range and are more effective haemolysins than the naturally occurring antibodies. Group O donors should always be screened for high-titre anti-A and anti-B antibodies, which may cause haemolysis when group O platelets or plasma are transfused to recipients with A and B phenotypes.
Anti-A1 and anti-H
Anti-H reacts most strongly with group O and A2 red cells and also normally acts as a cold agglutinin. A notable, but rare, exception is the anti-H that occurs in the Oh Bombay phenotype, which is an IgM antibody and causes lysis at 37°C (Table 21.4) so that Oh Bombay phenotype blood would be required for transfusion.
Rh System
Rh Antigens and Encoding Genes
In Caucasian, D-negative individuals, the RHD gene is deleted, whereas in Black races and other populations, single-point mutations, partial deletions or recombinations have been described. In individuals with a weak D antigen (Du), there is a quantitative reduction in D antigen sites, believed to arise from an uncharacterized transcriptional defect. These individuals do not make anti-D antibodies following a D antigen challenge. Partial D individuals lack one or more epitopes of the D antigen, defined using panels of monoclonal reagents. Dvi is perhaps the most important partial D phenotype because such individuals not infrequently make anti-D. Partial D phenotypes arise from DNA exchanges between RHD and RHCE genes and from other rearrangements. Comprehensive reviews of this system have been provided by Avent and Reid11 and Daniels et al.12
The Rh haplotypes are named either by the component antigens (e.g. CDe, cde) or by a single shorthand symbol (e.g. R1 = CDe, r = cde). Thus, a person may inherit CDe (R1) from one parent and cde (r) from the other and have the genotype CDe/cde (R1r). The haplotypes in order of frequency and the corresponding shorthand notation are given in Table 21.7. Although two other nomenclatures are also used to describe the Rh system, namely, Wiener’s Rh-Hr terminology and Rosenfield’s numeric notation, the CDE nomenclature, derived from Fisher’s original theory, is recommended by a World Health Organization Expert Committee13 in the interest of simplicity and uniformity. The Rh antigens are defined by corresponding antisera, with the exception of ‘anti-d’, which does not exist. Consequently, the distinction between homozygous DD and the heterozygous Dd cannot be made by direct serological testing but may be resolved by informative family studies. It is still routine practice to predict the genotype from the phenotype on the basis of probability tables for the various Rh genotypes in the population (Table 21.7). However, in women with immune anti-D and a history of an infant affected by HDN, RH DNA typing is used in prenatal testing for the fetal D status to decide on the clinical management of the pregnancy, e.g. the need for monitoring for fetal anaemia using middle cerebral artery Doppler ultrasound. Suitable sources include amniotic fluid (amniocytes) and trophoblastic cells (chorionic villi) or after 15 weeks’ gestation, maternal blood can be used because it contains fetal DNA.14,15 In practice, multiplex polymerase chain reaction (PCR) is used, with more than two primer sets, to detect the different molecular bases for D-negative phenotypes in non-Caucasians. RH DNA typing also has applications in paternity testing and forensic medicine. There are racial differences in the distribution of Rh antigens, e.g. D negativity is more common in Caucasians (approximately 15%), whereas Ro (cDe) is found in approximately 48% of Black Americans but is uncommon (approximately 2%) in Caucasians. The Rh antigens are present only on red cells and are a structural part of the cell membrane. Complete absence of Rh antigens (Rh-null phenotype) may be associated with a congenital haemolytic anaemia with spherocytes and stomatocytes in the blood film, increased osmotic fragility and increased cation transport. This phenotype arises either as a result of homozygosity for silent alleles at the RH locus (the amorph type) or more commonly by homozygosity for an autosomal suppressor gene (X), genetically independent of the RH locus (the regulator type). Rh antigens are well-developed before birth and can be demonstrated on the red cells of very early fetuses.
Table 21.7 The Rh haplotypes in order of frequency (Fisher nomenclature) in Caucasians and the corresponding short notations
| Fisher | Short Notations | Approximate Frequency (%) |
|---|---|---|
| CDe | R1 | 41 |
| cde | r | 39 |
| cDE | R2 | 14 |
| cDe | R0 | 3 |
| CwDe | R1w | 1 |
| cdE | r″ | 1 |
| Cde | r′ | 1 |
| CDE | RZ | Rare |
| CdE | ry | Rare |
MNSs System
Clinical Significance of Red Cell Alloantibodies
The significance of the alloantibodies described, with respect to the nature of the haemolytic transfusion reaction they produce, is provided in Table 21.8. The majority of haemolytic transfusion reactions, however, are the result of ABO incompatibility.16
Table 21.8 Antibody specificities related to the mechanism of immune haemolytic destruction
| Blood Group System | Intravascular Haemolysis | Extravascular Haemolysis |
|---|---|---|
| ABO,H | A, B, H | |
| Rh | All | |
| Kell | K | K, k, Kpa, Kpb, Jsa, Jsb |
| Kidd | Jka | Jka, Jkb, Jk3 |
| Duffy | Fya, Fyb | |
| MNS | M, S, s, U | |
| Lutheran | Lub | |
| Lewis | Lea | |
| Cartwright | Yta | |
| Colton | Coa, Cob | |
| Dombrock | Doa, Dob |
Mollison et al.17 analysed the significance of blood group antigens other than those of the ABO system and D by looking at the prevalence of transfusion-induced red cell alloantibodies, excluding anti-D, -CD and -DE (Table 21.9). Rh antibodies, mainly anti-c or anti-E, accounted for 53% of the total and anti-K and anti-Fya accounted for a further 38%, leaving only about 9% for all other specificities. A similar distribution of the different red cell antibodies was found in a smaller group of patients who experienced immediate haemolytic transfusion reactions (HTR). However, the figures for delayed HTR showed a striking increase in the relative frequency of Jk antibodies, which reflects the outlined characteristics of Jk antibodies.
The significance of the many other blood group antigens not referred to in the text is summarized in Table 21.10. However, it should be noted that the antibodies listed are usually wholly or predominantly IgG and would be detectable in routine pretransfusion testing using the indirect anti-globulin test (IAT).
Mechanisms of Immune Destruction of Red Cells
Immune-mediated haemolysis of red cells depends on the following:18
The mechanism of immune haemolysis also determines the site of haemolysis:
Macrophage activity is an important component of cell destruction and further study of cellular interactions at this stage of immune haemolysis may provide an explanation for the differing severity of haemolysis in patients with apparently similar antibodies. In vitro macrophage (monocyte) assays have been used sometimes to supplement conventional serological techniques to assess this aspect of immune haemolysis.20
Interleukin-6 also enhances FcγRII activation and increased activity of the CR1 receptor occurs through the action of T-cell cytokines and through chemotactic agents released in the inflammatory response.22 The increased levels of proinflammatory cytokines and other biological mediators and their effects on the activity of the monocyte phagocytic system have been monitored in patients with systemic inflammatory response syndrome.23 It is therefore possible that release of cytokines during viral and bacterial infections could, at least in part, trigger some episodes of autoimmune cell destruction.
Antigen–Antibody Reactions
Some complement-binding antibodies (especially IgM) may cause lysis in vitro (without noticeable agglutination), which can be enhanced by the addition of fresh serum as a source of complement. However, complement activation may only proceed to the C3 stage; in these circumstances cell-bound C3 can be detected by the antiglobulin test using an appropriate anticomplement reagent (see p. 500).
Quality Assurance within the Laboratory
It has long been appreciated that the test systems used for routine pre-transfusion testing are of the utmost importance because errors can and do lead to patient morbidity and mortality. It is therefore of little surprise that within the European Union all reagents, calibrators and control materials for red cell typing and for determining the presence of ‘irregular anti-erythrocytic antibodies’ have been included under the In-vitro Diagnostics (IVD) Medical Devices Directive24 (see p. 588). This means that all reagents sold within the European Union must display the CE mark to show that they conform to the agreed Common Technical Specifications (CTS). In each European country, a Competent Authority will be able to withdraw or suspend certification of any reagent, depending on the information received from its Notified Body, which will perform batch release approval and monitor the performance of the manufacturer and the product.
[/not-level-membership-for-hematology-oncology-and-palliative-medicine-category]
