6 BLOOD AND HEMATOPOIESIS
BLOOD
Plasma
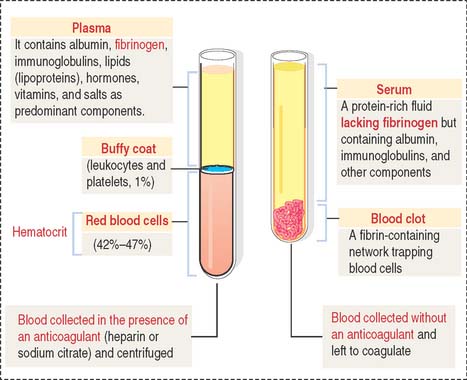
Plasma is the fluid component of blood (Figure 6-1). Plasma contains salts and organic compounds (including amino acids, lipids, vitamins, proteins, and hormones). In the absence of anticoagulants, the cellular elements of blood, together with plasma proteins (mostly fibrinogen), form a clot in the test tube. The fluid portion is called serum, which is essentially fibrinogen-free plasma.
Cellular elements of the blood: Red blood cells (erythrocytes)
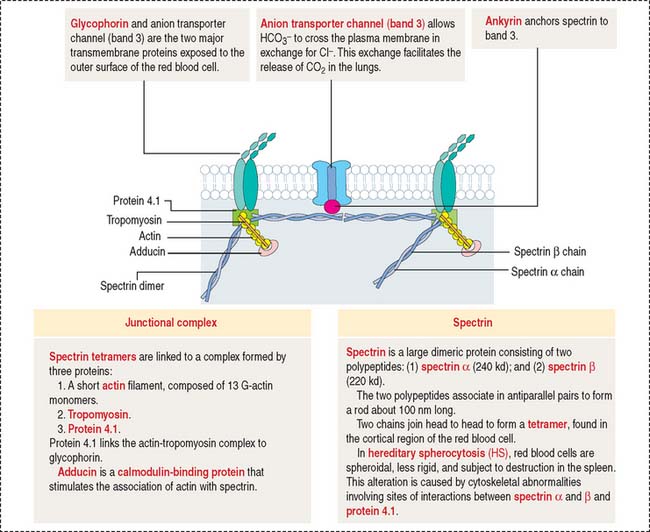
RBCs, also called erythrocytes (Greek erythros, red; kytos, cell), are non-nucleated, biconcave-shaped cells measuring 7.8 μm in diameter (unfixed). RBCs lack organelles and consist only of a plasma membrane, its underlying cytoskeleton (Figure 6-2), hemoglobin, and glycolytic enzymes.
Clinical significance: Cytoskeletal and hemoglobin abnormalities
Elliptocytosis and spherocytosis are alterations in the shape of RBCs caused by defects in the cytoskeleton. Elliptocytosis, an autosomal dominant disorder characterized by the presence of oval-shaped RBCs, is caused by defective self-association of spectrin subunits, defective binding of spectrin to ankyrin, protein 4.1 defects, and abnormal glycophorin (see Figure 6-2). Spherocytosis is also an autosomal dominant condition involving a deficiency in spectrin. The common clinical features of elliptocytosis and spherocytosis are anemia, jaundice, and splenomegaly (enlargement of the spleen). Splenectomy is usually curative, because the spleen is the primary site responsible for the destruction of elliptocytes and spherocytes.
Hemoglobin genetic defects (α2βS2) cause sickle cell anemia and thalassemia (Greek thalassa, sea; observed in populations along the Greek and Italian coasts). Sickle cell anemia results from a point mutation in which glutamic acid is replaced by valine at the sixth position in the β-globin chain. Defective hemoglobin (Hb S) tetramers aggregate and polymerize in deoxygenated RBCs, changing the biconcave disk shape into a rigid and less deformable sickle-shaped cell. Hb S leads to severe chronic hemolytic anemia and obstruction of postcapillary venules (see Spleen in Chapter 10, Immune-Lymphatic System).
Thalassemia syndromes are heritable anemias characterized by defective synthesis of either the α or β chains of the normal hemoglobin tetramer (α2β2). The specific thalassemia syndromes are designated by the affected globin chain: α-thalassemia and β-thalassemia. Thalassemia syndromes are defined by anemia caused by defective synthesis of the hemoglobin molecule and hemolysis.
Clinical significance: Erythroblastosis fetalis
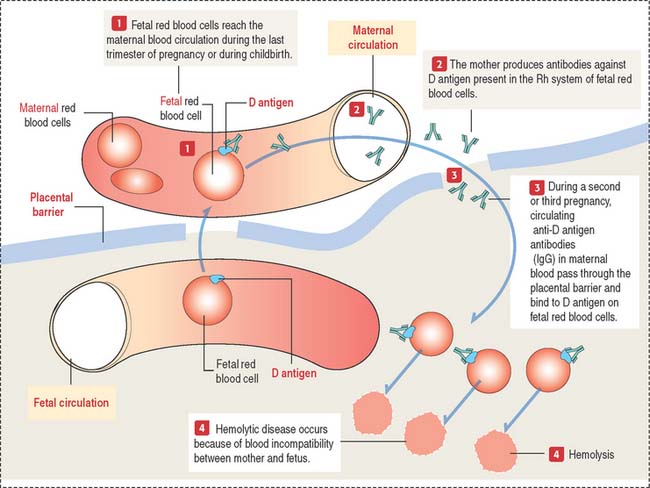
Erythroblastosis fetalis is an antibody-induced hemolytic disease in the newborn that is caused by blood group incompatibility between mother and fetus (Figure 6-3 and Box 6-A). This incompatibility occurs when the fetus inherits RBC antigenic determinants that are foreign to the mother. ABO and Rh blood group antigens are of particular interest.
Box 6-A Hemolysis in erythroblastosis fetalis
Essentially, the mother becomes sensitized to blood group antigens on red blood cells, which can reach maternal circulation during the last trimester of pregnancy (when the cytotrophoblast is no longer present as a barrier, as we discuss in Chapter 23, Fertilization, Placentation, and Lactation) or during childbirth. Within the Rh system, D antigen is the major cause of Rh incompatibility. The initial exposure to the Rh antigen during the first pregnancy does not cause erythroblastosis fetalis because immunoglobulin M (IgM) is produced and IgMs cannot cross the placenta because of their large size.
LEUKOCYTES
Leukocytes (6 to 10 × 103 per mm3; see Box 6-B) are categorized as either granulocytes (containing primary, and specific or secondary cytoplasmic granules, Box 6-C) or agranulocytes (containing only primary granules). In response to an appropriate stimulus, leukocytes may leave the bloodstream (diapedesis) and enter the connective tissue by the homing mechanism (see Figure 6-9).
Box 6-B Blood cells/μL or mm3
| Erythrocytes | 4-6 × 106 | |
| Leukocytes | 6000 to 10,000 | |
| Neutrophils | 5000 | (60% to 70%) |
| Eosinophils | 150 | (2% to 4%) |
| Basophils | 30 | (0.5%) |
| Lymphocytes | 2400 | (28%) |
| Monocytes | 350 | (5%) |
| Platelets | 300,000 | |
| Hematocrit | ~48% for men and ~38% for women | |
Box 6-C Primary and specific granules
Granulocytes
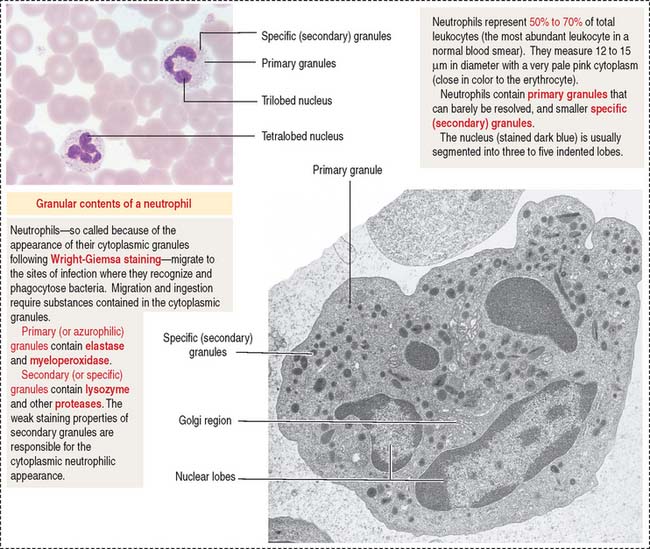
Enzymes contained in the primary granules (elastase and myeloperoxidase) and secondary granules (lysozyme and other proteases), specific receptors for C5a (produced by the complement system pathway, see Chapter 10, Immune-Lymphatic System), and L-selectin, and integrins (with binding affinity to endothelial cell ligands such as intercellular-adhesion molecules 1 and 2 [ICAM-1 and ICAM-2]) enable the antibacterial and homing function of neutrophils (see Figure 6-9).
Agranulocytes
Lymphocytes are either large (3% of lymphocytes; 9 to 12 μm) or small (97% of lymphocytes; 6 to 8 μm (Figure 6-7) cells. In either case, the nucleus is round and may be slightly indented. The cytoplasm is basophilic, often appearing as a thin rim around the nucleus (see Figure 6-7). A few primary granules may be present. Lymphocytes may live for a few days or several years.
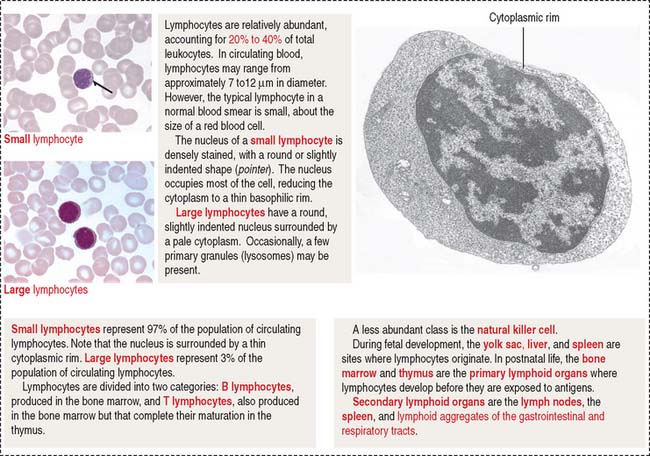
Lymphocytes are divided into two categories: B lymphocytes (also called B cells) are produced and mature in bone marrow. Antigen-stimulated B cells differentiate into antibody-secreting plasma cells. T lymphocytes (also called T cells) are produced in bone marrow but complete their maturation in the thymus. Activated T cells participate in cell-mediated immunity (for additional details, see Chapter 10, Immune-Lymphatic System).
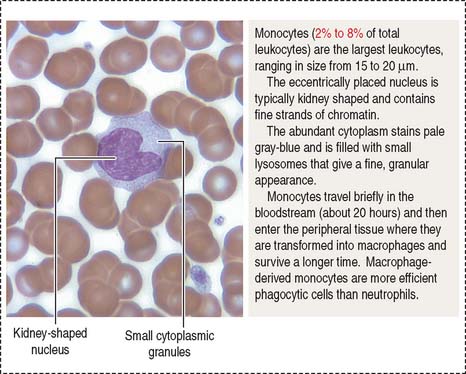
Monocytes (Figure 6-8) can measure 12 to 20 μm in diameter. Their nucleus is kidney shaped or oval. Cytoplasmic granules are small and may not be resolved on light microscopy. Monocytes circulate in blood for 12 to 100 hours and then enter the connective tissue. In the connective tissue, monocytes differentiate into macrophages, which are involved in bacterial phagocytosis, antigen presentation, and clean-up of dead cell debris. In bone, monocytes differentiate into osteoclasts under the control of osteoblasts (see Chapter 4, Connective Tissue).
Clinical significance: Homing and inflammation
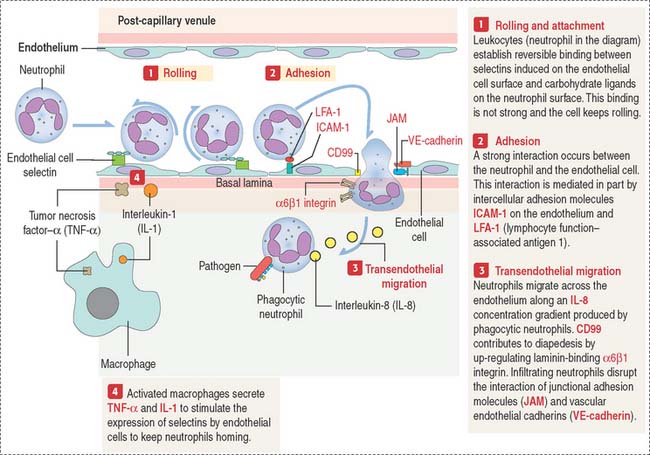
We have studied in Chapter 1, Epithelium (see Figure 1-13), the molecular principles of homing. We will expand the concept of homing by studying the mechanism of migration of phagocytic neutrophils to the site of infection and inflammation (Figure 6-9).
Clinical significance of homing: Leukocyte adhesion deficiencies
Leukocyte adhesion deficiency I is caused by a defect in the β subunit of the integrin molecule. As a consequence, leukocytes are unable to leave blood vessels to enter the tissue by transendothelial migration. In these patients, inflammatory cell infiltrates are devoid of neutrophils.
In leukocyte adhesion deficiency II, the fucosyl-containing ligands for selectins are absent due to a congenital defect of endogenous fucose metabolism. As shown in Figure 6-9, selectin-carbohydrate interactions have a role in the rolling of leukocytes on an endothelial cell surface, a step required for the transendothelial migration of leukocytes into extravascular areas of inflammation.
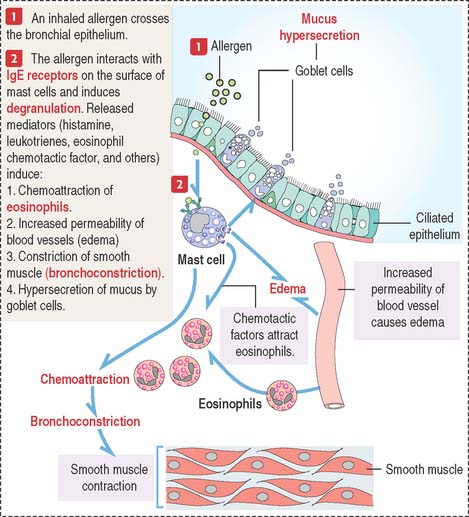
Clinical significance: Mast cell-eosinophil interaction in asthma
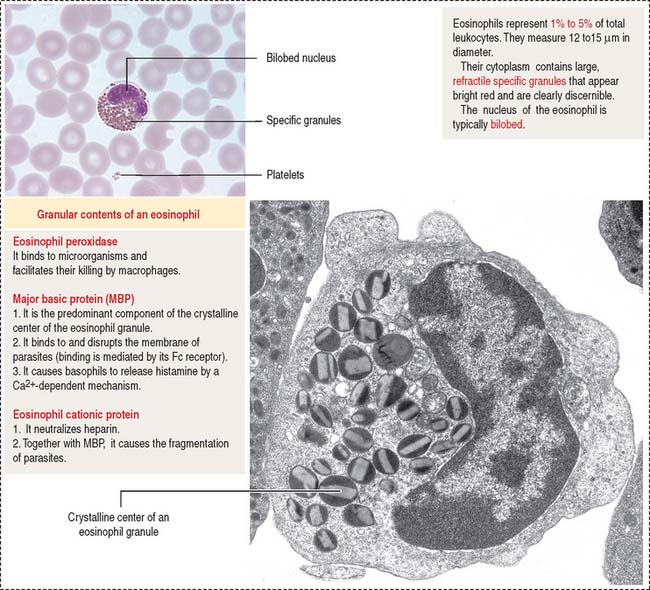
When mast cells degranulate and release chemical mediators, eosinophils and neutrophils are attracted from blood vessels into the connective tissue of the respiratory mucosa. Eosinophils, in turn, release additional mediators (leukotriene B4 and others) to enhance bronchoconstriction and edema. The release of eosinophil cationic protein and major basic protein into the bronchial lumen damages the epithelial cell lining and disturbs mucociliary function (Figure 6-10).
PLATELETS
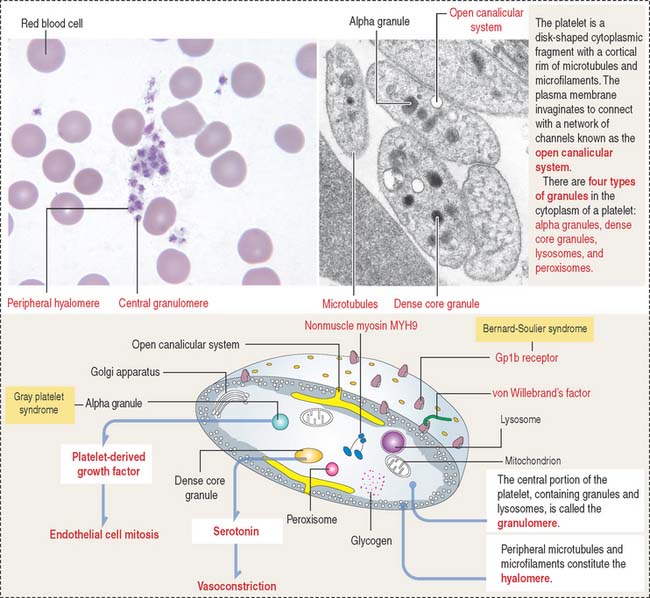
Platelets are small (2 to 4 μm) cytoplasmic fragments derived from the mega-karyocyte (Figure 6-11) under the control of thrombopoietin, a 35- to 70-kd glycoprotein produced in the kidneys and liver. Megakaryocytes develop cytoplasmic projections that become proplatelets, which fragment into platelets. This differentiation process takes 10 to 12 days. Platelets bind and degrade thrombopoietin, a mechanism that regulates platelet production.
Clinical significance: Thrombocytopenia
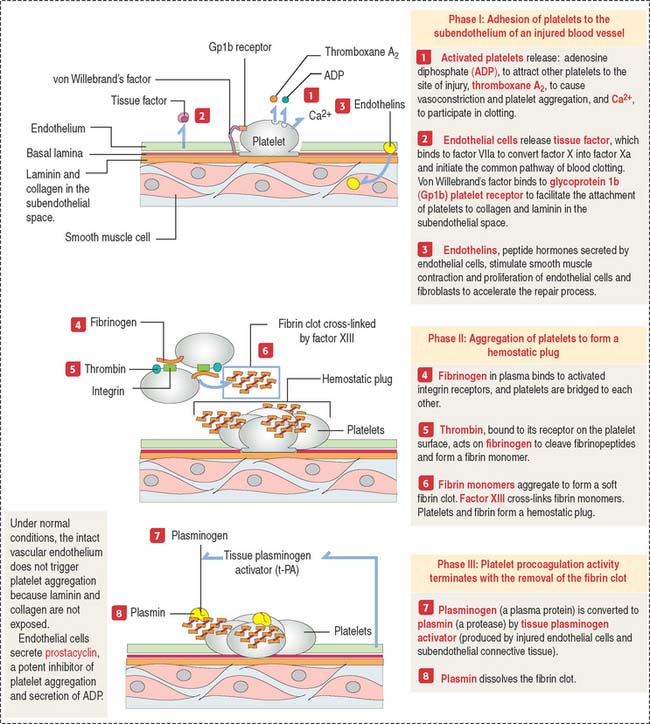
Deficiency of the glycoprotein 1b—factor IX complex, or von Willebrand’s factor, a protein associated with factor VIII, leads to two congenital bleeding disorders, Bernard-Soulier syndrome and von Willebrand’s disease, respectively (see Figures 6-11 to 6-13) (see Box 6-D). These two diseases are characterized by the inability of platelets to attach to vascular subendothelial surfaces. The glycoprotein 1b—factor IX—von Willebrand’s factor complex is relevant for the aggregation of normal platelets when they are exposed to injured subendothelial tissues.
Box 6-D Hemophilia
Clinical significance: Hemostasis and the blood clotting cascade
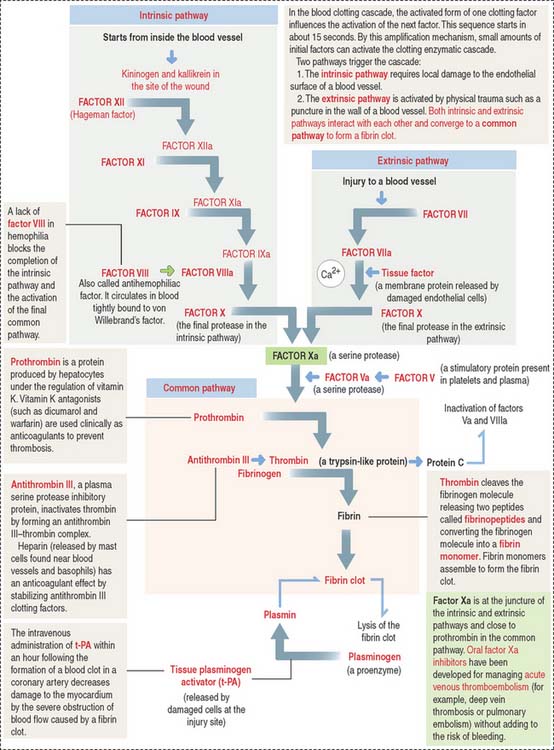
The blood clotting or coagulation cascade depends on the sequential activation of proenzymes to enzymes and the participation of endothelial cells and platelets to achieve hemostasis or arrest of bleeding. Hemostasis occurs when fibrin is formed to reinforce the platelet plug (Figure 6-12).
The blood clotting cascade has the following characteristics:
Extrinsic and intrinsic pathways converge to a crucial step in which fibrinogen is converted to fibrin, which forms mesh that enables platelets to attach. The convergence starts with the activation of factor X to factor Xa, together with activated factor Va, resulting in the cleavage of prothrombin to thrombin. The initial hemostatic plug consists of a platelet scaffold for the conversion of prothrombin to thrombin, which changes fibrinogen into fibrin (see Figure 6-12).
Fibrinogen, produced by hepatocytes, consists of three polypeptide chains, which contain numerous negatively charged amino acids in the amino terminal. These characteristics allow fibrinogen to remain soluble in plasma. After cleavage, the newly formed fibrin molecules aggregate forming a mesh. Fibrin, with the addition of plasma fibronectin, stabilizes the blood clot (Figure 6-13).
HEMATOPOIESIS
In the fetus, hematopoiesis (Greek haima, blood; poiein, to make) starts during the first trimester in islands of hematopoiesis found in the yolk sac. The islands develop from hemangioblasts, the progenitors of both hematopoietic and endothelial cells. Fetal hematopoiesis continues after the second trimester in the liver and then in the spleen. During the seventh month of intrauterine life, the bone marrow becomes the primary site of hematopoiesis, where it remains during adulthood. In the adult, an approximate volume of 1.7 L of marrow contains 1012 hematopoietic cells.
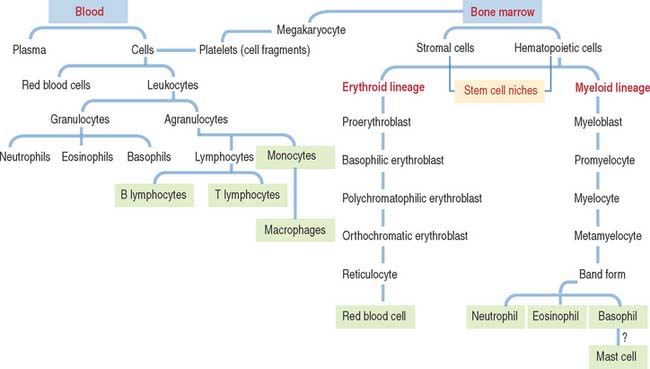
The bone marrow consists of two compartments: (1) the marrow stromal compartment and (2) the hematopoietic cell compartment. The marrow stromal compartment is a framework of adipose cells, fibroblasts, stromal cells, vascular endothelial cells, macrophages, and blood vessels interspersed within trabecular bone (Figures 6-14 to 6-16). The marrow stromal compartment provides niches for the maintenance, self-renewal, and expansion of stem cells of the hematopoietic cell compartment. Hematopoietic stem cell niches are located near bone surfaces, forming the endosteal bone marrow-hematopoietic stem cell niche, or associated with the sinusoidal endothelium as part of the vascular bone marrow-hematopoietic cell niche.
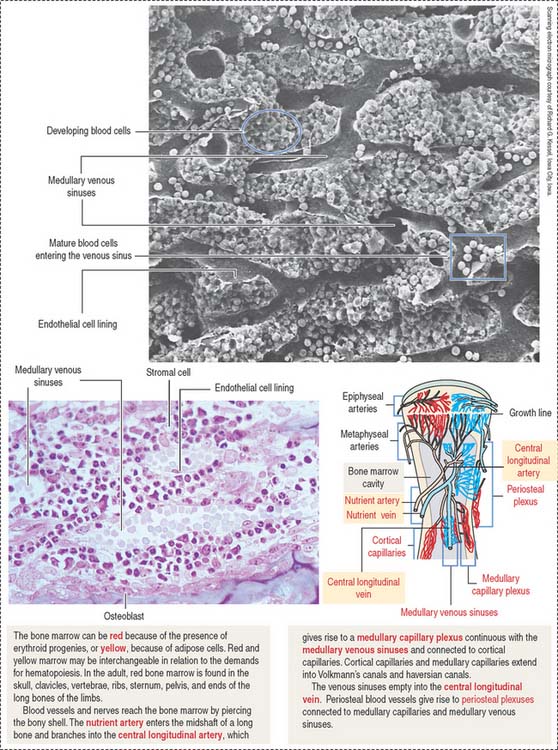
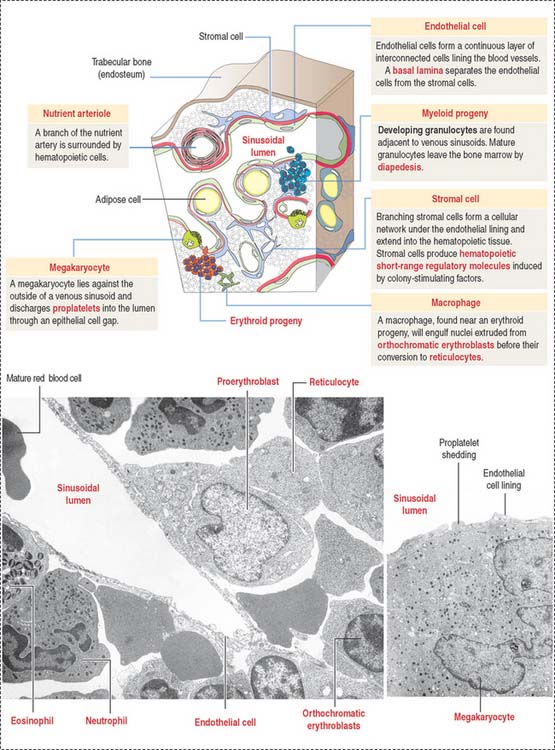
The hematopoietic cell compartment is highly vascularized. It is supplied by the central longitudinal artery, derived from the nutrient artery. Medullary capillary plexuses and periosteal capillary plexuses are interconnected. Medullary sinusoids drain into the central longitudinal vein before leaving through the nutrient vein (see Figure 6-14).
Mature hematopoietic cells translocate from the site of growth through the sinusoid wall by active transendothelial migration across openings into the sinuses (see Figure 6-15) before entering the circulation through the central vein. Immature hematopoietic cells lack the capacity of transendothelial migration and are retained in the extravascular space by the vascular endothelial cells. The sinusoids of the marrow are lined by specialized endothelial cells with significant phagocytic activity and a capacity to produce growth factors that stimulate the proliferation and differentiation of hematopoietic cells.
Hematopoietic cell populations
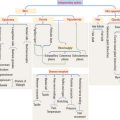
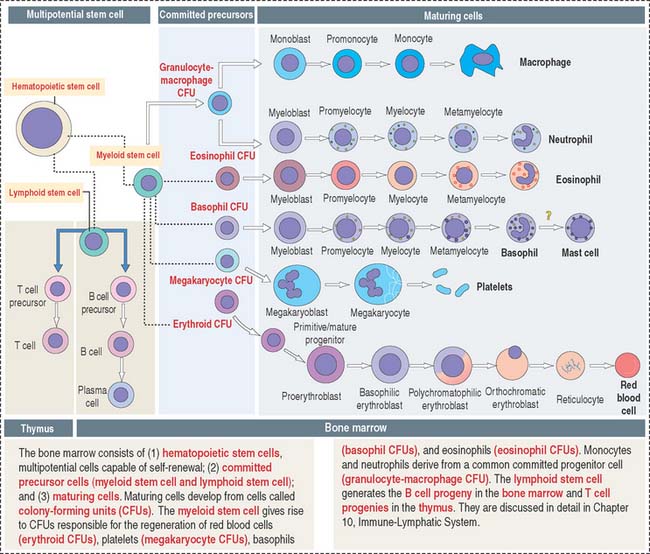
The bone marrow consists of three major populations (see Figure 6-16): (1) the hematopoietic stem cells, capable of self-renewal; (2) committed precursor cells, responsible for the generation of distinct cell lineages; and (3) maturing cells, resulting from the differentiation of the committed precursor cell population.
Myeloid and lymphoid stem cells are multipotential cells (see Figure 6-16). They are committed to the formation of cells of the blood and lymphoid organs.
Five colony-forming units (CFUs) derive from the myeloid stem cell: the erythroid CFU, the megakaryocyte CFU, the basophil CFU, the eosinophil CFU, and the granulocyte-macrophage CFU. The erythroid CFU produces red blood cells. The megakaryocyte CFU generates platelets. The granulocyte-macrophage CFU produces both monocytes and neutrophils. Basophils and eosinophils derive from the basophil and eosinophil CFUs, respectively. The lymphoid stem cell gives rise to T cell and B cell precursors.
Clinical significance: Hematopoietic growth factors
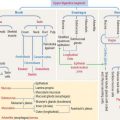
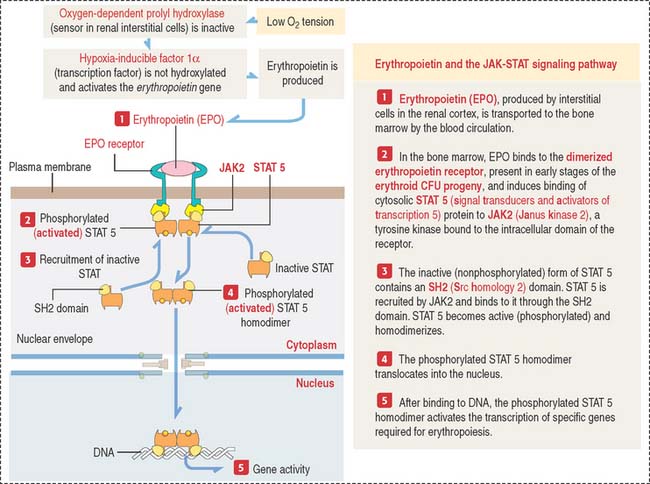
There are three major groups of hematopoietic growth factors: (1) colony-stimulating factors, (2) erythropoietin (Figure 6-17) and thrombopoietin (Greek thrombos, clot; poietin, to make), and (3) cytokines (primarily interleukins).
Colony-stimulating factors are so named because they are able to stimulate committed precursor cells to grow in vitro into cell clusters or colonies. Interleukins are produced by leukocytes (mainly lymphocytes) and affect other leukocytes (paracrine mechanism) or themselves (autocrine mechanism).
Hematopoietic cells express distinct patterns of growth factor receptors as they differentiate. Binding of the ligand to the receptor leads to a conformational change, activation of intracellular kinases, and the final induction of cell proliferation (see Chapter 3, Cell Signaling).
We discuss the roles of specific hematopoietic growth factors when we analyze each cell lineage.
Erythroid lineage
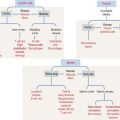
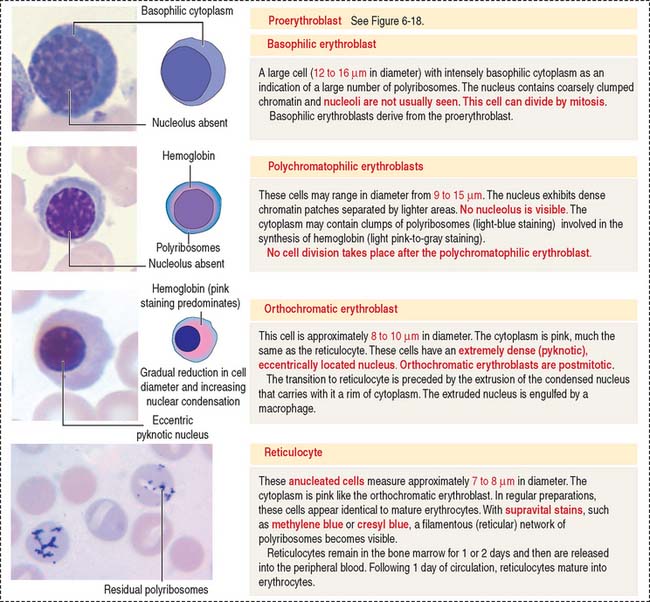
Erythropoiesis includes the following sequence (Figure 6-18): proerythroblast, basophilic erythroblast, polychromatophilic erythroblast, orthochromatic erythroblast, reticulocyte, and erythrocyte.
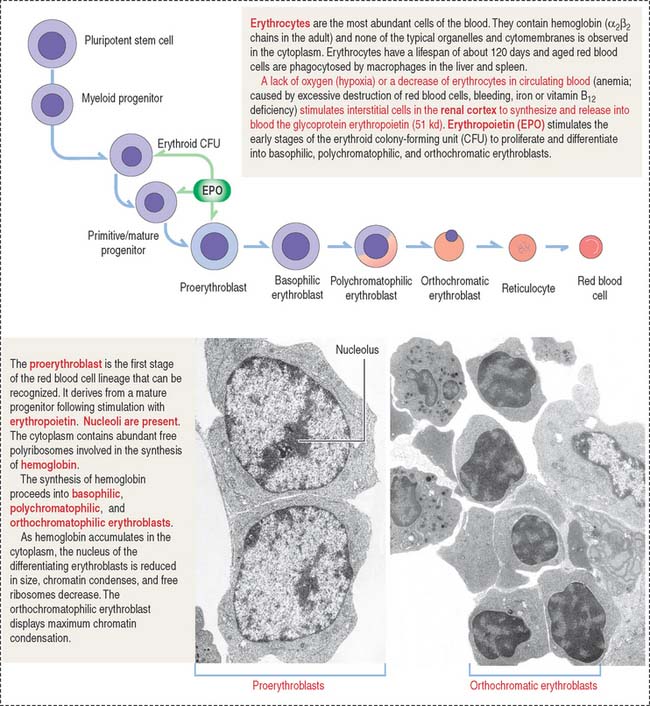
The major regulator of erythropoiesis is erythropoietin (EPO) (see Figure 6-17), a glycoprotein produced primarily (90%) in the kidneys (juxtatubular interstitial cells in the renal cortex) in response to hypoxia (a decrease in oxygen level in inspired air or tissues).
Erythropoietin production in chronic renal diseases is severely impaired. Recombinant erythropoietin can be administered intravenously or subcutaneously for the treatment of anemia caused by a decrease in the production of erythropoietin by the kidneys. The effectiveness of erythropoietin treatment can be monitored by an increase of reticulocytes in circulating blood. Reticulocytes can be identified by the supravital stain of residual polyribosomes forming a reticular network (Figure 6-19).
Leukopoiesis
Leukopoiesis (Greek leukos, white; poietin, to make) results in the formation of cells belonging to the granulocyte and agranulocyte series. The granulocyte lineage (Figure 6-20) includes the myeloblast, promyelocyte, myelocyte, metamyelocyte, band cell, and mature form. The granulocyte-macrophage precursor gives rise to neutrophils and monocytes. The myeloid stem cell generates eosinophil and basophil progenies. Agranulocytes include lymphocytes and monocytes.
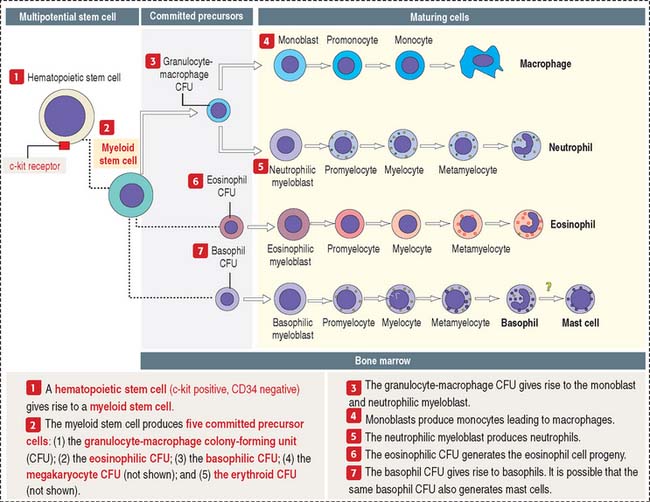
Granulocytes
Neutrophilic and macrophage cell lines share a common precursor cell: the granulocyte-macrophage CFU (see Figure 6-20). Eosinophils and basophils derive from independent eosinophil and basophil CFUs. Neutrophil, eosinophil, and basophil granulocytes follow a similar pattern of proliferation, differentiation, maturation, and storage in the bone marrow. Details of these processes are better recognized for neutrophils, the most abundant granulocyte in the bone marrow and blood. It takes 10 to 14 days for neutrophils to develop from early precursors, but this timing is accelerated in the presence of infections or by treatment with granulocyte colony-stimulating factor (CSF) or granulocyte-macrophage CSF (see below).
Myeloblasts, promyelocytes, and myelocytes are mitotically dividing cells; metamyelocytes and band cells cannot divide but continue to differentiate (see Figure 6-20). A typical feature of the maturation process of granulocytes is the appearance of primary (azurophilic) granules and “specific” or secondary granules in the cytoplasm (Figures 6-21 and 6-22).
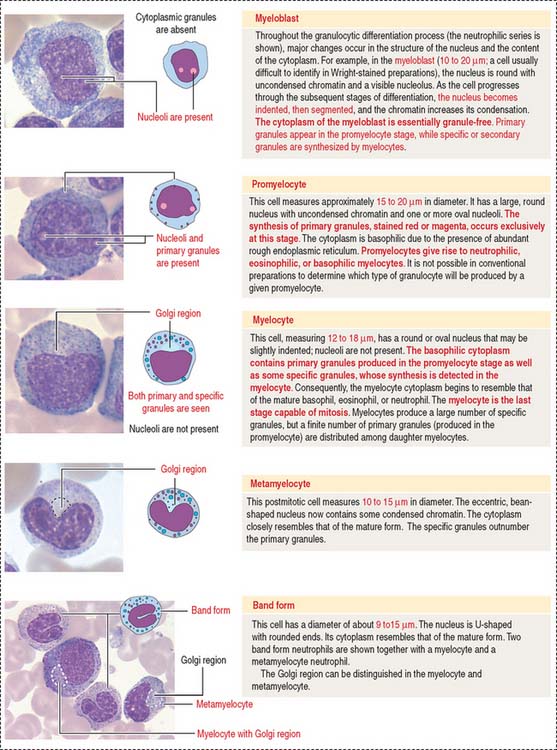
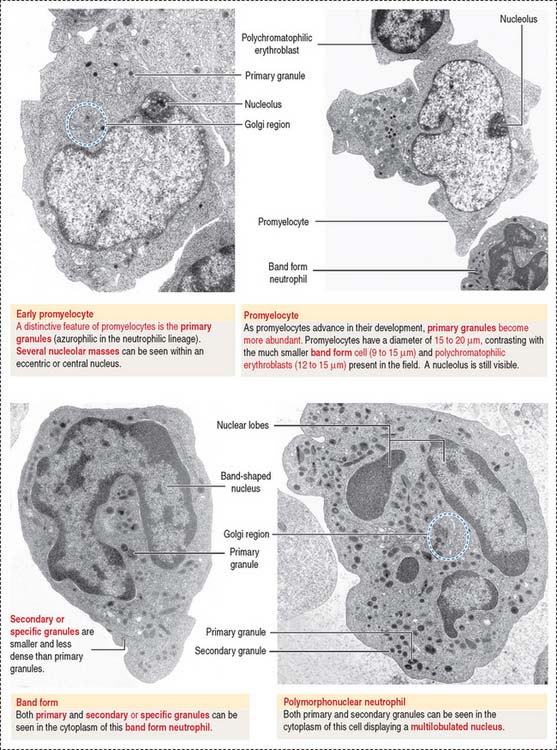
Myeloblasts are undifferentiated cells lacking cytoplasmic granules. Promyelocytes and myelocytes display primary granules in cells of the neutrophil, eosinophil, and basophil series. Secondary granules appear in myelocytes. Primary granules do not transform into specific granules. Primary granules persist as such throughout the cell differentiation sequence (see Figure 6-22).
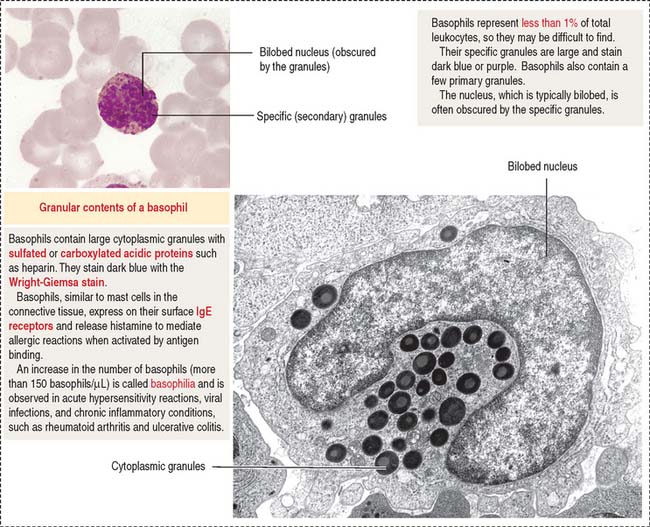
Basophils are distinguished by their large, coarse, and darkly stained granules that fill the cytoplasm and often obscure the nucleus (Figure 6-23). The granules contain peroxidase, heparin, and histamine as well as kallikrein, a substance that attracts eosinophils.
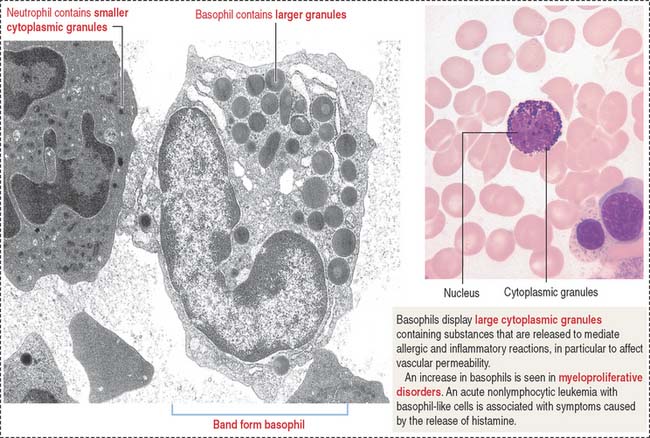
We discuss in Chapter 4, Connective Tissue, that mast cells are structurally similar to basophils. However, mast cells are larger cells and are found in tissues, close to blood vessels. A notable difference is that mast cells contain serotonin and 5-hydroxytryptamine, which basophils do not contain. In addition, mast cells discharge their granules into the extracellular space in contrast with basophils, which usually undergo diffuse internal degranulation.
Agranulocytes: Lymphocytes
The pluripotent stem cell gives rise to all hematopoietic cells, including lymphocytes phocytes of the B and T cell lineage. B cells mature in the bone marrow and then migrate to other lymphoid organs. T cells complete their maturation in the thymus and then migrate to specific lymphoid organs.
A lymphoblast gives rise to a prolymphocyte, an intermediate stage that precedes the mature lymphocyte. B and T lymphocytes are nonphagocytic cells. They are morphologically similar but functionally different, as discussed in Chapter 10, Immune-Lymphatic System.
Monocytes
Monocytes derive from the granulocyte-macrophage CFU. We have already discussed that the granulocyte-macrophage CFU gives rise to the neutrophil lineage and the macrophage lineage. Under the influence of a specific CSF, each precursor cell establishes its own hierarchy: the granulocyte colony-stimulating factor (G-CSF) takes the granulocyte precursor cell into the myeloblast pathway; the granulocyte-macrophage colony-stimulating factor (GM-CSF) guides the monocyte precursor cell into the monoblast pathway, leading to the production of peripheral blood monocytes and tissue macrophages. Receptors for the macrophage-stimulating factor (M-CSF) are restricted to the monocyte lineage (see Osteoclastogenesis in Chapter 5, Osteogenesis).
Monocytes (12 to 20 μm in diameter) in the bone marrow and the blood have a large indented nucleus found in the central portion of the cytoplasm (Figure 6-24). Granules (primary lysosomes) and small vacuoles are typical features. Lysosomes lack peroxidase but contain other proteases and hydrolases. Monocytes are motile in response to chemotactic signals and attach to a surface.
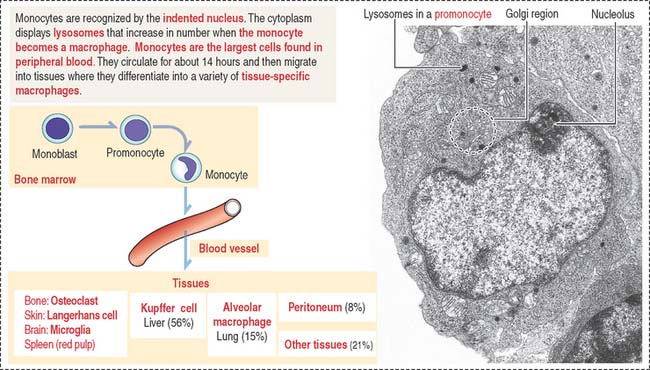
The structural and functional characteristics of tissue macrophages are discussed in Chapter 4, Connective Tissue. In Chapter 11, Integumentary System, we discuss the antigenic reactivity of monocyte-derived Langerhans cells in epidermis. In Chapter 17, Digestive Glands, we explore the important role of Kupffer cells in liver function, and in Chapter 10, Immune-Lymphatic System, we examine the phagocytic properties of macrophages in spleen.
Clinical significance: Colony-stimulating factors and interleukins
GM-CSF is also a glycoprotein produced by endothelial cells, T cells, fibroblasts, and monocytes that stimulates the formation of neutrophils, eosinophils, basophils, monocytes, and dendritic cells (Figure 6-25). However, GM-CSF is less potent than G-CSF in increasing the levels of neutrophils during neutropenia. As is the case with G-CSF, a synthetic form of GM-CSF (sargramostim or molgramostim) is available for the treatment of neutropenia.
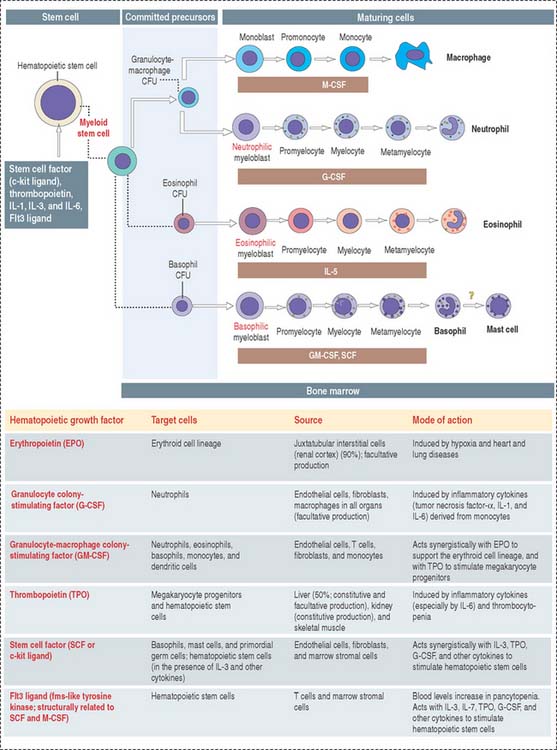
Interleukins have a relevant function in the formation and function of type B and T cells as we discuss in Chapter 10, Immune-Lymphatic System. IL-3 stimulates proliferation of hematopoietic stem cells and acts together with other growth factors, including stem cell factor, thrombopoietin, IL-1, IL-6, and Flt3 (fms-like tyrosine kinase 3) ligand (see Figure 6-25)• IL-5 acts specifically on the eosinophil progeny.
Platelets and megakaryocytes
The precursor cell of the platelet (also called thrombocyte; Greek thrombos, clot) is the megakaryoblast, a cell derived from the megakaryocyte CFU (see Figure 6-16).
The megakaryocyte (35 to 160 μm in diameter; Figure 6-26) contains an irregularly lobed nucleus produced by an endomitotic nuclear division process in which nuclear divisions occur without cell division (polyploid nucleus). Nucleoli are not detected.
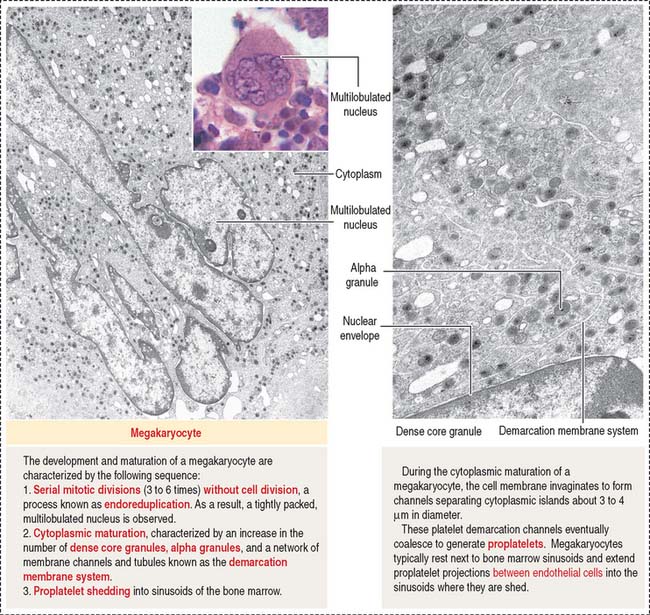
Platelets play important roles in maintaining the integrity of blood vessels (see Figure 6-12). Keep in mind that platelet activation during hemostasis involves sequentially:
Clinical significance: Thrombopoietin
Platelets bind and degrade thrombopoietin, a process that autoregulates platelet production.
Clinical significance: Stem cell factor (also known as c-kit ligand)
Stem cell factor exists in two forms: membrane-associated and soluble forms, the latter generated by proteolytic cleavage of the membrane-associated protein. The c-kit receptor has an extracellular domain consisting of five immunoglobulin motif repeats responsible for stem cell factor binding and dimerization (Figure 6-27). Binding of stem cell factor induces the dimerization of the c-kit receptor, followed by autophosphorylation. Autophosphorylated c-kit receptor is the docking site of specific signaling molecules.
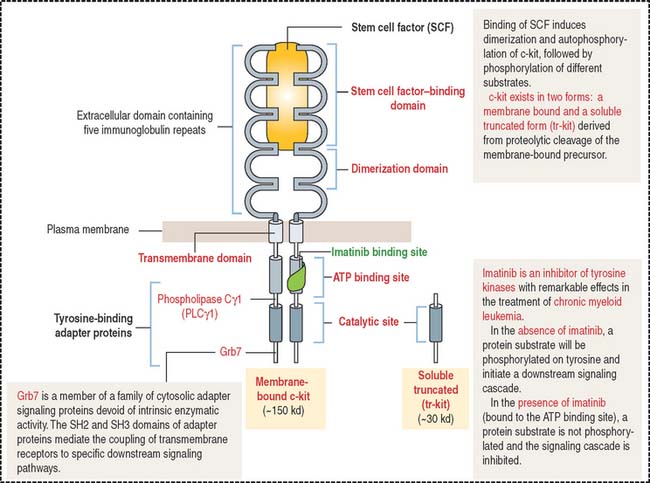
The stem cell factor ligand by itself is a weak stimulator of hematopoiesis but makes hematopoietic stem cells responsive to other cytokines (see Figure 6-25). It does not induce the formation of cell colonies by itself. Flt3 (fms-like tyrosine kinase 3) ligand is closely related to c-kit receptor and stem cell factor. Similar to stem cell factor, Flt3 ligand acts on the pluripotent stem cell in synergy with thrombopoietin, stem cell factor, and interleukins.
The stem cell factor receptor is expressed by the c-kit proto-oncogene. A mutation in genes expressing the components of the stem cell factor receptor–ligand complex causes anemia and affects the development of melanocytes in skin and the survival and proliferation of primordial germinal cells in the developing ovaries and testes (see Chapter 21, Sperm Transport and Maturation). Stem cell factor is potentially useful for the treatment of inherited and acquired disorders of hematopoiesis as well as in bone marrow transplantation.
In Chapter 4, Connective Tissue, we note that mast cells derive from a bone marrow precursor. The storage and release of histamine- and heparin-containing granules from mast cells are affected in mutants lacking stem cell factor.
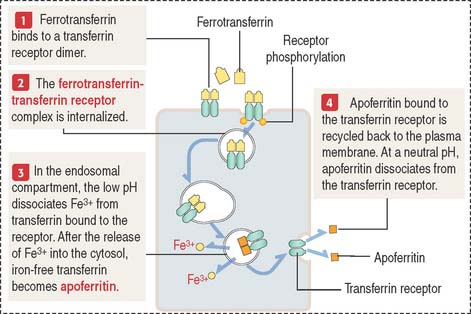
Clinical significance: Transferrin and iron metabolites
Transferrin, a serum protein produced in the liver, and lactoferrin, a protein present in maternal milk, are nonheme proteins involved in the transport of iron (Figure 6-28). Transferrin complexed to two Fe3+ ions is called ferrotransferrin. Transferrin devoid of iron is known as apotransferrin.
Vitamin B12 (known as extrinsic factor) binds to intrinsic factor, a protein produced by the parietal cells in the gastric glands. The vitamin B12 –intrinsic factor complex binds to specific receptor sites in the ileum, transported across enterocytes, and released in blood, where it binds to the transport protein trans-cobalaphilin III.
Vitamin B12 deficiency is rare because the liver stores up to a 6-year supply of vitamin B12. Under deficiency conditions, the maturation of the erythroid cell progeny slows down, causing abnormally large RBCs (megaloblasts) with fragile cell membranes, resulting in the destruction of RBCs (megaloblastic anemia; see Box 6-E).
Box 6-E Anemia
Essential concepts
There are two types of agranulocytes: lymphocytes and monocytes.
The hematopoietic stem cell gives rise to the myeloid stem cell and the lymphoid stem cell.