7 Blood and cancer
Anaemia
Iron deficiency anaemia
As shown in Case 7.1, tiredness, lethargy and listlessness are suggestive of anaemia. Pallor may be harder to detect in darker skin but may be apparent to parents, and will be clearly evident from inspection of mucous membranes. A pale blue tinge to the sclera is characteristic of iron deficiency. The findings of tachycardia with a flow murmur reflect the compensatory increase in cardiac output.
Iron deficiency causes a range of effects:
• Symptoms of anaemia (see above)
• Impaired or abnormal appetite
• Mucosal abnormalities, e.g. angular cheilitis and atrophic glossitis, pruritis vulvae
• Impaired cognitive and motor development in toddlers; impaired short-term memory in older children
• Malabsorption, e.g. coeliac disease (see Chapter 12, p. 172)
• Chronic blood loss from gastroesophageal reflux (see Chapter 13, p. 164) or Meckel’s diverticulum, when ectopic gastric mucosa secretes acid, leading to ulceration and bleeding in the small bowel.
Haemolytic anaemia
Haemolytic disease of the newborn
Haemolytic disease of the newborn is a condition of rapid red cell destruction in the fetus or newborn infant caused by maternal antibodies raised against the infant’s red cells (see also Chapter 17, p. 252). Where a pregnant mother is blood group rhesus negative and the fetus rhesus positive, the leak of even a few fetal cells into the maternal circulation may be sufficient to trigger the production of IgG antibodies. These cross the placenta in the same, or more commonly in a subsequent, pregnancy to cause immune-mediated destruction of antibody-coated red cells. The same process occurs with ABO incompatibility (as shown in Case 7.2), although this usually causes milder haemolysis.
Haemolytic disorders of childhood
Haemolytic anaemias are characterized by shortened red cell lifespan, with a compensatory increase in erythropoiesis, increased synthesis of bilirubin, and anaemia. Presentation may occur at birth with early-onset or severe neonatal jaundice, with inter-current infection provoking haemolysis or aplastic crisis, with symptoms of anaemia, with specific complications such as dactylitis in sickle-cell anaemia (see below), or by chance, following routine investigations (see Case 7.3).
General principles of treatment of haemolytic anaemias
• Folic acid requirements are increased due to high red cell turnover, and supplementation is usually required to optimize erythropoiesis
• Chronic anaemia is usually well-tolerated and transfusion is done as infrequently as symptoms allow
• Transfusion may be required for acute haemolytic crises. These occur secondary to inter-current infection, notably with parvovirus which causes a temporary depression of erythropoiesis – aplastic crisis. In G6PD deficiency (an X-linked condition, therefore very rare in girls) a variety of oxidant drugs will provoke acute haemolysis
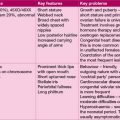
• Blood transfusions ultimately may lead to iron overload, particularly in thalassaemia, requiring use of iron chelation therapy with desferrioxamine administered by overnight subcutaneous infusion 5–6 days per week. Newer oral agents, deferiprone (DFP) and deferasirox (DFX), approved for use in 2006, are available. DFP is less effective than DFO in treating liver disease, and so is usually used in combination with DFO. DFX is as effective as DFO, but long-term efficacy and safety is not yet established, and use is not recommended in patients with cardiac involvement. All chelation agents have significant side-effects, notably agranulocytosis with DFP.
• Symptomatic pigment gallstones require cholecystectomy
• Bone marrow transplantation may be appropriate for thalassaemia and severe sickle-cell disease
Hereditary spherocytosis
Thalassaemia
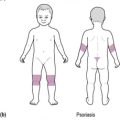
The hallmark of thalassaemia major is hypochromic, microcytic anaemia, which may be severe, with associated hypersplenism due to increased red cell destruction (see Figure 7.1). Beta-thalassaemia also produces skeletal abnormalities, most notably frontal bossing, due to hyperplasic marrow, and growth failure.
Sickle-cell disease
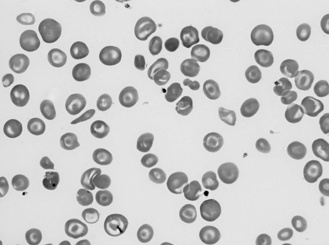
Sickle-cell anaemia results from a glutamate to valine substitution in position 6 of the beta-globin gene. When present in the homozygous form this renders the red cell prone to sickling. Common triggers are hypoxia, dehydration and fever, and the result is less compliant sickle-shaped red cells which occlude small vessels causing tissue ischaemia and end-organ damage. The sickle cells are rapidly removed from the circulation causing anaemia and hypersplenism (as shown in Case 7.4). Ultimately, hyposplenism results from recurrent splenic infarction. Hydroxycarbamide (see below) may help preserve splenic function, thereby reducing infection risk.
There are a number of clinical problems particular to sickle-cell disease:
• Vaso-occlusive crises leading to:
• Sequestration crises where large amounts of blood are sequestered to the spleen or liver, leading to anaemia, shock and severe abdominal pain
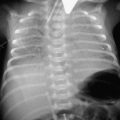
• Sickle chest in which widespread pulmonary infiltrates occur, leading to respiratory failure
• Infection with encapsulated organisms notably Pneumococcus, Haemophilus and Salmonella, due to progressive infarction of the spleen, leading to hyposplenism
• Impaired growth with delayed puberty (special growth charts are available in the UK for children with sickle-cell anaemia).
Bruising and bleeding
Idiopathic thrombocytopenic purpura
The acute-onset, easy bruising in Case 7.5 suggests thrombocytopenia, which was confirmed. The most likely cause is idiopathic thrombocytopenic purpura (ITP). If there is severe bleeding, platelets may be given. Most cases of acute ITP resolve spontaneously over days or a few weeks. Treatment with steroids or intravenous immunoglobulin will accelerate the rate of recovery in most cases. Treatment is considered in children with a platelet count below 30 × 1012/L with significant bleeding, depending on severity. Steroids can disguise aleukaemic leukaemia, which may present with isolated thrombocytopenia, and bone marrow examination is recommended before starting steroids. Treatment may incur significant side-effects and will not reduce the risk of developing chronic ITP, which occurs in 10–30% of cases and most cases can safely be managed conservatively.
Haemophilia
The two main forms are haemophilia A (80–85% of cases) and haemophilia B. Both are inherited conditions with X-linked recessive inheritance and consequently boys are affected, with deficient production of clotting factors and an increased tendency to haemorrhage, either spontaneously or after trauma. In haemophilia A, factor VIII is affected (as in Case 7.6) and in haemophilia B, factor IX levels are reduced; both are involved in the conversion of fibrinogen to fibrin in the clotting cascade.
Bone marrow failure
Aplastic anaemia
Aplastic anaemia results from reduced numbers of all haematopoietic cells – erythroblasts, myeloblasts and megakaryocytes – resulting in pancytopenia (as shown in Case 7.7). The majority of cases (50–75%) are idiopathic, but it may be congenital – Fanconi anaemia, or acquired following viral infection or toxin exposure.
• Immunosuppressive drugs, e.g. ciclosporin, anti-thymocyte globulin
Aplastic anaemia is severe, and without bone marrow transplantation (BMT) the 12-month survival is under 10%. With BMT, survival is 70–90%.
Malignant disease
Risk factors
For the great majority of childhood cancers, there are no identifiable risk factors.
• Radiation. Radiation is a recognized cause of cancer, and children are more susceptible to the effects of radiation than are adults. Most evidence comes from high-dose, relatively short-term radiation such as following nuclear accidents or radiotherapy. The tumour most strongly associated with radiation exposure is papillary thyroid cancer. Risk can be reduced in the aftermath of exposure by taking potassium iodide. There is a lack of knowledge about the long-term risks of low-dose exposure, such as domestic radon, over prolonged periods.
• Genetic factors. Genetic factors may increase the likelihood of cancer through a range of mechanisms.
• Infection. Infection may be a major trigger factor for some forms of cancer, e.g. hepatitis B and hepatocellular carcinoma, AIDS and non-Hodgkin’s lymphoma. Epstein–Barr virus has also been implicated in non-Hodgkin’s lymphoma, particularly Burkitt’s lymphoma.
• Environmental factors. Chemical and drug exposure may be implicated in the development of particular cancers, e.g. antenatal exposure to the barbiturate sodium pentothal (thiopental sodium) has been linked to brain and spinal cord tumours, and there is limited evidence suggesting a link between pesticide exposure and soft-tissue sarcomas.
Symptoms and signs
• Visible or palpable mass. The most obvious presentation of cancer is with a visible enlarging mass, such as a neck, soft-tissue or scrotal swelling, or incidentally recognized abdominal mass.
• Headaches and neurological symptoms and signs. Brain and spinal cord tumours produce elevated intracranial pressure and neurological symptoms or signs depending on their location and speed of growth. Epilepsy is an uncommon manifestation.
• Local effects. Osteosarcomas lead to pain and disability at the site of the tumour. Retinoblastoma causes visual loss. Wilms’ tumour may produce haematuria.
• Distant tumour effects. ‘Dancing-eyes syndrome’ is a characteristic neurological syndrome arising from neuroblastoma. The mechanism is unknown. Tumours may also produce distant effects by impairing endocrine function or by secreting hormones. For example, hypothalamic tumours such as craniopharyngiomas will result in panhypopituitarism with failure of growth and puberty, hypothyroidism, diabetes insipidus and adrenal insufficiency (see Chapter 12). Pituitary tumours, phaechromocytomas and neuroblastomas may all secrete hormones causing distant effects. The most common functional pituitary tumour is a prolactinoma, which produces breast enlargement and galactorrhoea. Catecholamines from neuroblastomas or phaeochromocytomas may produce labile hypertension. These catecholamines may be detected in urine and are useful as tumour markers for diagnosis and monitoring.
• Bone marrow failure (see above). Bone marrow failure is seen most commonly with leukaemia, and results in anaemia with pallor, thrombocytopenia causing easy bruising and bleeding, or leucopenia resulting in severe infection. Bone marrow metastases, most commonly seen in neuroblastoma, may produce a similar picture. Bone pain is common.
Treatment
Treatment of childhood malignancy is a complex area, but the principal elements of management are:
Presentations of principal childhood malignancies
Leukaemia
Around 2–4/100 000 children per year develop acute leukaemia and over 80% of those have acute lymphoblastic leukaemia (ALL) (as in Case 7.8), most of the rest having acute myeloid leukaemia (AML). The peak incidence is between 2 and 5 years, and although there is an increased incidence in children with chromosomal disorders, e.g. Down syndrome, or with immunodeficiency, this condition mostly occurs in previously well children.
Innocent cervical lymphadenopathy
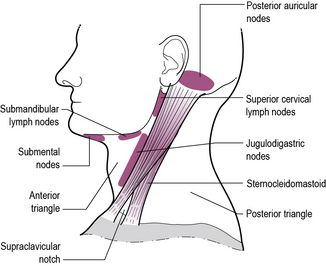
Cervical lymphadenopathy is universal in children. Prevalence studies indicate that up to 45% of otherwise healthy children have cervical lymphadenopathy at any one time. Usually (as in Case 7.9) the lymphadenopathy is bilateral, generalized and transient, in association with an infection, typically of the upper respiratory tract, and predominantly involving glands in the anterior triangle (see Figure 7.2 for an anatomical description of cervical lymph nodes). Unilateral lymphadenopathy signifies staphylococcal or streptococcal infection in 40–80% of cases. Subacute and chronic lymphadenitis occurs with cat scratch disease, toxoplasmosis and occasionally atypical mycobacteria. Persistent enlargement of glands in the supraclavicular region is much more commonly associated with malignancy. Malignant disease or mycobacterial infection is more likely if there are associated constitutional symptoms such as fever, night sweats, malaise and weight loss.
Cervical lymphadenopathy secondary to malignancy is characteristically progressive, firm and frequently associated with constitutional symptoms. In the young child, neuroblastoma and leukaemia are the commonest causes, but in the older child and adolescent, Hodgkin’s disease and non-Hodgkin’s lymphoma are more common (Case 7.10).
Lymphoma
• Hodgkin’s lymphoma. Hodgkin’s lymphoma is distinguished clinically by the contiguous spread of disease from one lymph node group to the next, and pathologically by a unique histological feature, the Reed–Sternberg cell, an atypical histiocyte. About 50% of cases are triggered by Epstein-Barr virus infection, and probably represents an abnormal immune response in a genetically susceptible individual.
• Non-Hodgkin’s lymphoma. Non-Hodgkin’s lymphoma (NHL) arises from B- or T-cell lines, including primitive precursors. T-cell lymphoma is uncommon. Although arising in lymphoid tissue, extra-nodal spread is common. Thirty-five subtypes of non-Hodgkin’s lymphoma are recognized. Some cases are triggered by Epsten-Barr virus, notably Burkitt’s lymphoma. Human T-cell leukaemia/lymphoma virus type 1 (HTLV-1) is predominantly seen in patients of Japanese or Afro-Caribbean origin. Helicobacter pylori infection of the stomach may cause mucosa-associated lymphoid tissue (MALT) lymphoma. Antibiotics are curative in 70-80%.
A cervical lymphoma presents with painless neck swelling, as in Case 7.10. Mediastinal involvement leads to general malaise, shortness of breath and signs of pleural effusions or superior vena caval obstruction. Abdominal lymphoma causes nausea and vomiting, bowel disturbance including obstruction, pain, weight loss, melaena, ascites and fever.
Treatment is with a multi-agent cytotoxic chemotherapy regime similar to that for acute leukaemia.
Brain tumours
Children usually present with symptoms of rising intracranial pressure, particularly headache and vomiting. Features in the history that should elicit concern are a short history of headaches, with progressive symptoms, particularly if associated with functional impairment such as difficulty walking, unsteadiness, etc. (see also Chapter 14, p. 187).
As Case 7.11 shows, manifestations of cerebral tumours are primarily related to anatomical site and pressure effects – tumours may obstruct CSF (cerebrospinal fluid) flow leading to hydrocephalus (see Chapter 14, p. 187). This may lead to a rapid rise in intracranial pressure.
Other presentations of brain tumours include:
• Focal neurological signs such as hemiplegia or cranial nerve palsies
• Rapidly rising head circumference in an infant
• Ataxia due to cerebellar dysfunction
• Neck stiffness (posterior fossa tumours)
• Temporal visual field loss (lesions affecting the optic nerves or chiasm, e.g. hypothalamic tumours)
• Percussion of the skull may reveal a ‘cracked-pot’ percussion note, typical of hydrocephalus.
Medulloblastoma
Thirty per cent of brain tumours are due to embryonal primitive neuro-ectodermal tumours (PNETs), which, in the cerebellum, are referred to as medulloblastomas. Medulloblastomas account for 90% of all PNETs. For a case history of medulloblastoma, see Chapter 14, Case 14.2, p. 187. PNETs are malignant with a propensity to metastasize within the brain and spinal cord. Treatment of medulloblastoma entails surgery, followed in children over 4 years by craniospinal radiotherapy.
Craniopharyngioma
Craniopharyngioma, an embryonal tumour, arises in the suprasellar region, and is a benign tumour, but with a high rate of local recurrence. As in Case 7.12, progressive growth failure, often going back a number of years, is the earliest sign. The tumour contains solid and cystic elements, often with calcification, readily seen on CT scan. Treatment is with surgery and post-operative radiotherapy. Proximity to the pituitary gland and optic chiasm commonly leads to panhypopituitarism and/or blindness.
Neuroblastoma
Neuroblastoma (as in Case 7.13) is a tumour of neural crest cells and arises from the adrenal glands or sympathetic nerve root ganglia. The tumour secretes catecholamines, whose metabolites, vanillylmandelic acid and homovanillic acid, may be measured in urine.
Wilms’ tumour
The differential diagnosis of an abdominal mass includes Wilms’ tumour (Case 7.14), neuroblastoma, hepatoblastoma and non-Hodgkin’s lymphoma.
The majority of patients with Wilms’ tumour do well, with survival rates over 60%.
Osteosarcoma and Ewing’s sarcoma
The majority of osteosarcomas occur in adolescents and young adults, presenting with limb pain, swelling or limp (Case 7.15). The metaphyses of long bones are most commonly affected and metastases may occur in the lung. Disease is confirmed radiologically with X-ray, bone scan and CT scan of the chest. After preliminary chemotherapy, surgical resection is attempted, with limb preservation where possible. Further chemotherapy is usually required.
Supportive and palliative care
• Treatment to protect the child from foreseeable complications, e.g. rehydration, and allopurinol to prevent tumour lysis syndrome (see above), or prophylactic antibiotic and antifungal therapy to protect the neutropenic child after intensive chemotherapy
• Prompt treatment of possible and confirmed intercurrent infection
• Treatment of pain and nausea
• Replacement of blood products
• Nutritional support to maintain weight and growth
• Monitoring for side-effects and complications of treatment.
Late sequelae of cancer and their treatment
Specific problems may occur following chemo- or radiotherapy; for example:
• Cardiomyopathy (anthracyclines), pulmonary fibrosis (bleomycin), renal impairment (ifosfamide), deafness (platinum), etc.
• Cranial radiotherapy also commonly leads to hypopituitarism with growth failure, hypothyroidism and hypogonadism, although precocious puberty may also occur, particularly in girls
• Infertility is very common secondarily to chemo- or radiotherapy and pituitary dysfunction. It is important to address fertility issues, as they are a significant cause of psychological morbidity
• Spinal radiotherapy will impair spinal growth, resulting in relative skeletal disproportion, with short stature.