chapter 22 Blood
INTRODUCTION AND OVERVIEW
In recent years, patients with haematological conditions may have been treated with new therapies, including immunotherapy (e.g. rituximab for non-Hodgkin’s lymphomas) or molecularly targeted therapies (‘designer drugs’ or ‘magic bullets’ such as imatinib mesylate for chronic myeloid leukaemia (CML)).1 The impact of these new therapies and the rapidly changing treatment of blood cancers is discussed.
ANAEMIA AND HAEMATINIC DEFICIENCY STATES
Symptoms that are often present, and may alert you to the presence of anaemia, include:
The history, in a simplified clinical approach, should focus on:
In the patient without obvious cause, anaemia itself is best regarded as a symptom, not a diagnosis.
Microcytic anaemia results mostly from iron deficiency, anaemia of chronic disease or thalassaemia/haemoglobinopathy. The most common causes of macrocytic anaemia are alcohol, pregnancy, and vitamin B12 and folate deficiency (Table 22.1).
TABLE 22.1 Red cell size as a pointer to the likely cause of anaemia
| Microcytic | Normocytic | Macrocytic |
|---|---|---|
| Iron deficiency | Chronic disease* | Pregnancy |
| Chronic disease* | Combined haematinic deficiencies | Reticulocytosis |
| Thalassaemias & haemoglobinopathies | Renal failure |
* Anaemia of chronic disease can cause normocytic or microcytic anaemia.
When the bone marrow responds to an insult such as haemorrhage or haemolysis with a brisk increase in reticulocytes (immature red cell forms), they can increase the MCV. For example, a reticulocytosis of over 10% may bring the MCV up to 105 fL (normal range: approx. 80–100). Rarer causes include hypothyroidism, myeloma and many chemotherapy drugs.
ANAEMIA OF CHRONIC DISEASE
The onset can be within 2 weeks of acute illness. The pathophysiology results in part from suboptimal erythropoietin activity, due to inhibition of gene expression by the inflammatory cytokines interleukin-1 and tumour necrosis factor. However, we now know that the principal mediator is hepcidin, which is synthesised in the liver, and this is upregulated by (other) inflammatory cytokines. High levels of hepcidin reduce iron absorption from the gut and inhibit iron release from macrophages. The net effect is that iron is unavailable for erythropoiesis.2 The resulting anaemia can be normocytic or microcytic (25% of cases). In renal failure, lack of erythropoietin probably plays a larger part, and the anaemia can be profound, even with relatively mild renal insufficiency.
IRON DEFICIENCY ANAEMIA
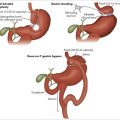
In iron deficiency anaemia (Fig 22.1), the usual aetiology is negative iron balance, either from inadequate dietary intake or from gastrointestinal loss, menorrhagia or, more rarely, haemolysis. Iron deficiency is often symptomatic in the absence of anaemia. It can be associated with impaired mentation (‘thinking through a cloud of cotton wool’), excessive fatigue, restless legs, a mild subjective peripheral neuritis and the rare pica syndrome (the urge to eat bizarre foods such as plaster or ice in excessive amounts). The history should include careful assessment of dietary iron, blood loss with menses, and asking about possible melaena, and haematuria or haemoglobinuria with dark urine. Physical examination may reveal classic stigmata, including glossitis, angular cheilosis and koilonychia, but these may not be present even with severe deficiency.
In the asymptomatic patient, test for faecal blood loss and consider panendoscopy. In men and postmenopausal women, iron deficiency usually signifies gastrointestinal blood loss.3
Occasional patients are seen in whom dietary intake of iron is adequate and no ongoing source of blood or iron loss is identified. Recently described mutations of the TMPRSS6 gene leading to impaired iron absorption may explain why some patients are refractory to oral iron replacement therapy. However, it appears that patients with these mutations are rare.4,5
Oral iron supplements are effective and well tolerated in about 80% of patients.6 Slow-release ferrous sulfate preparations are better tolerated than the iron salt, but rates of discontinuation in randomised trials are the same. Patients should be warned of possible constipation, and increased bloating, wind or flatulence, probably attributable to rapid bacterial overgrowth in the presence of increased available iron. Co-prescribing live culture yoghurts or probiotics may minimise this. Bowel motions will turn black (it is, however, a greenish-black, rather than the reddish-black of melaena). Oral iron should never be taken with tea (because of its tannin content), but rather with orange juice for its vitamin C content. Indeed, some iron formulations incorporate vitamin C. One tablet a day is usually enough; some patients tolerate two tablets daily without problems, but others find even one a day difficult. For these patients, taking the supplement with food may still provide enough iron to raise body stores. You will need to check the elemental iron equivalent in different supplements.
Some 20% of patients are genuinely intolerant of oral iron.6 It can lead to pseudo bowel obstruction and even surgical abdominal crises. For these patients, intravenous iron infusions are greatly preferable to intramuscular iron. The latter is poorly bio-available, and persists under the skin, leaving scarring and pigmentation. Whereas earlier IV preparations (iron dextran) were associated with cardiac arrhythmias, modern preparations (e.g. iron polymaltose, iron sucrose) have a far lower incidence of anaphylaxis and can be given rapidly without requiring electrocardiographic monitoring. This is best done in a specialised unit. Medical supervision should be on hand for the rare instance of anaphylaxis. Some patients experience arthralgia for 48 hours and this may require paracetamol. The response in many is gratifying, although it can take 2–6 weeks to become maximal. Thereafter, patients can be monitored with iron studies every 3 months, as many, but not all, will need further infusions in the future.
MEGALOBLASTIC ANAEMIA
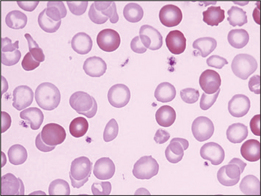
A megaloblast is a morphologically abnormal red cell precursor with features of delayed nuclear maturation (Fig 22.2). The common causes are dietary vitamin B12 or folate deficiency, some antimetabolite drugs and, infrequently, myelodysplasia with ‘megaloblastoid’ changes. In the history, check diet, drugs and past history of abdominal surgery or autoimmune disease. The physical examination may reveal evidence of dementia or loss of ankle reflexes and proprioception in the lower limbs. Vitamin B12 deficiency anaemia can occur without the neurological deficits (subacute combined degeneration of the spinal cord, dementia) and vice versa.
Folate deficiency is mostly seen with poor diet and alcoholism.
The combination of folate and iron deficiency is highly suggestive of coeliac disease.
Some medications inhibit dihydrofolate reductase and antagonise folate metabolism. Examples are trimethoprim, primethamine, phenytoin and methotrexate. Because both vitamin B12 and folate are not injurious, therapeutic trials are a reasonable manoeuvre in cases where megaloblastic anaemia has been identified.
IMMUNE-MEDIATED CYTOPENIA
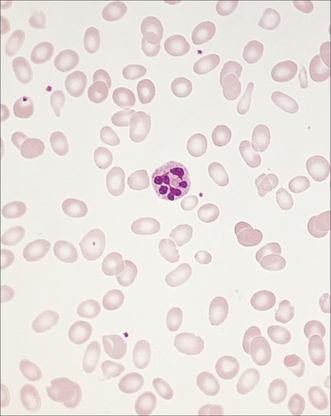
Whereas white cells (Fig 22.3) mostly leave the circulation to perform their various functions, red blood cells (Fig 22.4) and platelets are recycled. This recycling is one of the two functions of the spleen. The other function of the spleen is to act as a lymph gland for the bloodstream. Splenectomised patients are therefore at increased risk of septicaemia, with well-known encapsulated organisms including Neisseria meningitidis, Streptococcus pneumoniae and Haemophilus influenzae. Galen believed that the spleen was the ‘seat of the soul’. In evolutionary terms, the spleen was also the site of haematopoiesis, so it made sense to recycle blood cell components there.
It follows that treatment of ITP and AIHA is aimed at:
IMMUNE-MEDIATED THROMBOCYTOPENIA
Therapeutics
Splenectomy is effective in about 50–75% of cases of chronic refractory ITP or AIHA. Other indications include diagnostic splenectomy (rare, for suspected primary splenic lymphoma), symptomatic hereditary spherocytosis, Gaucher’s disease, Felty’s syndrome, and after traumatic or spontaneous splenic rupture. Electively, it can be done laparoscopically, which is far less invasive and traumatic for the patient, who can often go home by the second postoperative day. (Much smaller incisions are made; the splenic pedicle is clamped and the spleen enclosed in a plastic bag; the organ is then mulched to a deformable state and passed out through one of the small incisions.) Recent guidelines for the management of splenectomised patients are provided in Box 22.1.7
COAGULATION DISORDERS
The modern understanding of haemostasis describes a primary phase and a secondary phase. The actions of blood vessels and platelets constitute the primary phase. In response to injury, blood vessels constrict. Platelets, in response to newly exposed collagen, ADP and adrenaline, undergo the shape change reaction, rather like inflating an inverted rubber glove. Thus, previously hidden, or cryptic, molecules are exposed on the platelet surface, allowing interaction principally with von Willebrand factor (vWf). Platelets can now bind to injured endothelium (adhesion) and each other (aggregation), leading to a ‘stacks on the mill’ aggregation of platelets, and thrombus formation.
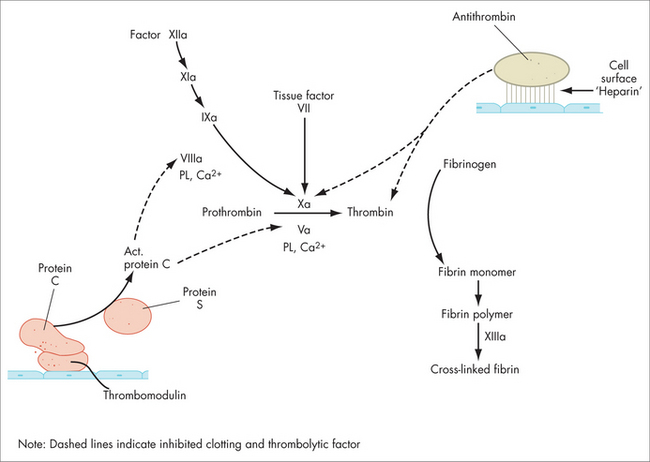
The coagulation protein reactions constitute the second phase of haemostasis. This generates fibrin in high local concentration at the site of injury or thrombosis, in effect ‘throwing a net’ over the platelet aggregate, anchoring it in place. These protein reactions occur as a sequence of macromolecular aggregates (not a ‘cascade’, Fig 22.6), mediated on the platelet surface, at the site of injury. In the current view, tissue factor has a crucial role as the initiator of haemostasis, down both the intrinsic and the extrinsic pathways (see below). The last step involves the conversion of fibrinogen to fibrin (factors I and Ia respectively) by thrombin (also called factor IIa). Fibrinogen’s shape becomes linear and it spontaneously polymerises (side-to-side and end-to-end, making D-dimers) to create the fibrin net. Finally, this fibrin mesh is stabilised by cross-linkage with factor XIII.
The endothelial cell has a key role in locally regulating pro-coagulant and anti-coagulant proteins, to allow a thrombus to form where needed, but inhibit propagation too widely. There are complex local mechanisms controlling levels of required proteins, cofactors and substrates in these reactions. Ex vivo testing of plasma cannot accurately reflect these events in the body.
Causes of a bleeding diathesis (referring here to mild or asymptomatic) are listed in Table 22.2. Congenital causes include mild phenotypes of the classic bleeding disorders, haemophilia A (factor VIII deficiency), haemophilia B (factor IX deficiency) and von Willebrand’s disease, and rarer deficiencies of other clotting factors.
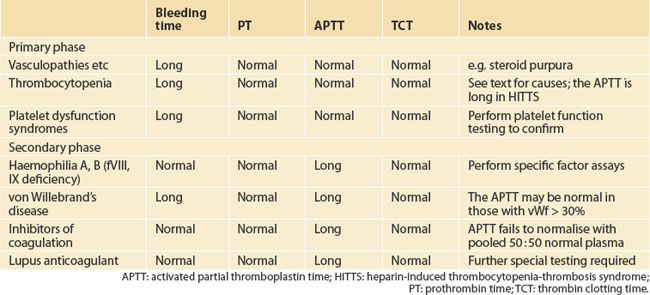
INVESTIGATING THE PATIENT WITH EASY BRUISING
In the examination, the findings may point to a platelet-type bleeding defect or to a coagulation pathway problem with deep tissue bleeding. Look for bruises of differing ages, gum bleeding, haematuria and, possibly, fundoscopy for retinal haemorrhages. Tall stature, cardiac murmurs or loose skin could point to a collagen disorder, such as Marfan’s syndrome.
DISORDERS OF PLATELET NUMBER AND FUNCTION
Referral to a haematologist is urgent, for accurate diagnosis and prompt treatment. This includes stopping heparin and, often, finding an alternative anticoagulant such as lepirudin or danaparoid.8 Re-exposure to heparin may induce an anamnestic response and a much more rapid fall in platelet count. This rapid-onset HITTS occurs mostly when the prior exposure was within the previous 3 months or, more typically, 30 days.
Other drugs causing thrombocytopenia include quinine (or quinidine), even in trace amounts in tonic water or angostura bitters; ranitidine, rifampicin, cotrimoxazole and others are rarer culprits.9 The full list is daunting, and any drug should be suspected.
COAGULATION PROFILE TESTS AND ASSOCIATED DISORDERS
Mild von Willebrand’s disease requires specifically assaying vWf. Various subtypes are recognised. Bleeding may manifest only after an antiplatelet agent is started, including some herbs as above. A distinction is increasingly being drawn between those with low vWf (30–50%) and those with mild von Willebrand’s disease (levels usually 10–30%).10,11
VENOUS THROMBOEMBOLIC DISEASE
CAUSES AND DIAGNOSIS
Hypercoagulable states (Fig 22.7) refer to clinical settings associated with increased thrombosis risk:
TREATMENT
If heparin is used, a baseline APTT should be raised two-fold over baseline within 8–24 hours, to reduce the chance of VTE progression.12,13 Monitoring of the APTT is required.
Low molecular weight heparins have a molecular weight of 4000–7000 Dalton, greater predictability in effect, longer half-life, greater specificity of action (inhibition of activated factor Xa and IIa, i.e. thrombin), and better correlation of effect with body weight. A Cochrane review concluded that once-daily treatment for deep vein thrombosis (DVT) and sub-massive PE at home with LMWH is safer than with heparin.14 Levels may accumulate in renal failure or be low in the very obese. Reversibility with protamine is incomplete. Monitoring is not usually needed, but can be achieved by measuring anti-factor Xa activity in, for example, those with renal insufficiency or the very obese.
Five milligrams daily will achieve therapeutic levels in 80% within 4 days.15
The duration of anticoagulation for a DVT should be at least 3 months.16 A meta-analysis found that recurrence is 40% less with 12–24 weeks of anticoagulation after unprovoked DVT, than with 3–6 weeks, without increased risk of major bleeding.17 For ‘provoked’ (e.g. associated with a venous catheter, or injury) and calf vein DVT, some say a shorter course can be given. With the latter, a repeat ultrasound at 10 days to exclude more proximal extension and higher risk of PE may be worthwhile.18 For superficial vein thrombosis and thrombophlebitis, topical hyaluronidase and aspirin are more appropriate therapy. For a PE, anticoagulation is usually for 6–12 months.
For a second confirmed episode of VTE, lifelong anticoagulation should be strongly considered.19 The intensity of anticoagulation with warfarin (target INR) is 2.0–3.0 in all circumstances, save one: mechanical left-sided cardiac valves require anticoagulation to an intensity of INR 3.0–4.5. See Table 22.3 for guidelines on target and duration of therapy, based on published Australian recommendations.20
| Intensity (target INR) | |
|---|---|
| All patients, except … | 2.0–3.0 |
| Mechanical heart valves (caged ball, caged disc) | 3.0–4.5 |
| Duration (risk stratification) | |
|---|---|
| First episode | 3 months |
| First episode, provoked (e.g. venous catheter) or calf vein DVT | 6–12 weeks |
| First episode, permanent risk factors | 3–6 months, consider indefinite |
| Two episodes | Indefinite |
There has been much interest in warfarin pharmacogenomics. CYPC29 polymorphisms are seen in 1% of the population, in whom a lower dose of warfarin may be needed and there is higher risk of bleeding. Conversely, VKORC1 refers to genetic variations of the vitamin K epoxide reductase subunit 1 gene; they are rare and associated with clotting factor deficiencies and warfarin resistance. The value of screening the population is debated.21
Warfarin reversal can be achieved with vitamin K, fresh frozen plasma or Prothrombinex®, depending upon urgency (see Baker et al 200422 for guidelines). Prothrombinex® contains concentrates of factors II, VII and IX; although it is recommended that cryoprecipitate or fresh frozen plasma (FFP) be given as well, to supply factor X, it might not be needed.23
PREVENTION OF VTE
Primary prevention, using either low-dose heparin or LMWH, is recommended by most authorities perioperatively in all patients for some forms of surgery (such as orthopaedics) and in hospitalised or critically ill patients (Table 22.4).
| Indication | Medication | Duration |
|---|---|---|
| Orthopaedic surgery (total hip, total knee, hip fracture) | LMWH + GCS/IPC | 35 days |
| Major abdominal surgery and age > 40 | LMWH + GCS/IPC | Until discharge or 28 days if high risk |
| Acute medical illness requiring immobility, with other risk factors | LMWH | Until ambulant |
| Pregnancy and postpartum with other risk factor | LMWH | During pregnancy and 4–6 weeks post-partum |
| Travel over 8 hours |
GCS: graduated compression stockings; IPC: intermittent pneumatic compression; LMWH: low molecular weight heparin.
Secondary prevention measures should be applied equally to those with a prior episode of VTE, a thrombophilic state (see below) or both.22,24 Long-haul travel (in planes, cars and trains for longer than 8 hours) is associated with a small absolute increase in thrombosis, possibly as little as one per two million arriving passengers over the subsequent 30 days.25 Recommended measures include wearing loose clothing, drinking plenty of fluids, frequent calf muscle contraction and, for those at higher risk (past VTE, thrombophilia), fitted compression stockings and LMWH immediately pre-travel (repeating the dose 24 hours later).
Aspirin is not an effective thromboprophylactic in this setting.24 Travel within 3 weeks of a VTE or high-risk event (e.g. surgery) is best avoided, even for those on warfarin. In cancer patients, VTE is best treated indefinitely with LMWH, not warfarin, as recurrence is halved.26
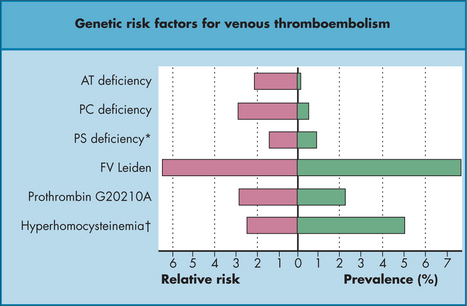
HYPERCOAGULABLE STATES AND THROMBOPHILIA
Hypercoagulable states refer to clinical settings associated with increased thrombosis risk: pregnancy, malignancy, sepsis, oestrogen therapy, postoperative status and prolonged immobility during travel (‘economy class syndrome’). Thrombophilia is a growing list of measurable inherited and acquired disorders that predispose an individual to thrombosis to varying degrees (Table 22.5). There is much debate over who to screen, but probably the indications include younger VTE patients (< 40 years); those with unprovoked ‘atypical’ VTE; those with VTE in an unusual site such as cerebral, renal, hepatic or mesenteric veins; those with a strong family history; and perhaps those in whom elective pro-thrombotic therapy is contemplated. Patients should be assessed individually, as the familial predispositions tend to ‘run true’ in kindreds, and family history is important.
TABLE 22.5 Frequency and effect (increased thrombosis risk) of selected thrombophilias27
| Syndrome | Frequency in Caucasian population | Prothrombotic effect |
|---|---|---|
| Congenital | ||
| Factor V Leiden | 3–5% | × 3–7* |
| Anti-thrombin III deficiency | < 1% | × 10 |
| Protein C deficiency | < 1% | × 10 |
| Protein S deficiency | < 1% | × 10 |
| Prothrombin 20210A mutation | 3% | × 3 |
| Hyperhomocysteinaemia | 5% have level > 18 μmol/L | mild |
| Acquired | ||
| Lupus anticoagulant | Rare | × 10 |
* Homozygotes, however, have a 30-fold increased risk.
Inherited thrombophilias include:
Thrombophilic individuals after one episode of VTE are at only a modestly increased risk of a second DVT, compared with others who have had a VTE episode.28,29 Anticoagulation should be at the usual level and duration. Exceptions include homozygosity for factor V Leiden and those with two thrombophilic states, which confer a much higher thrombosis risk. Such individuals should be reviewed by a haematologist, and lifelong therapy offered.
EXOGENOUS OESTROGEN
Screening all women contemplating HRT for a thrombophilic state is not feasible, and this raises a dilemma. A meta-analysis of 81 studies concluded that screening of all women is impractical and not cost-effective, whereas screening of higher-risk groups based on individual assessment of risk is.30
For thrombophilic or post-VTE women, other forms of contraception should be encouraged. There is no evidence that progesterone alone is pro-thrombotic. Some women find menopausal symptoms so bad that they will take HRT against haematological advice. Some find symptom relief with phyto-oestrogens and other treatments (see Ch 53, Menopause). Transcutaneous oestrogen patches, due to first-pass metabolism, are less thrombogenic.31,32 The French ESTHER study performed a multi-centre case-control study of 271 consecutive cases of VTE among postmenopausal women aged 45–70 years and 610 controls (426 hospital controls, 184 community controls) matched for centre, age and admission date. After adjustment for potential confounding factors, the odds ratios (ORs) for VTE in current users of oral and transdermal oestrogen compared with non-users were 4.2 (95% CI 1.5–11.6) and 0.9 (95% CI 0.4–2.1), respectively.31 These findings suggest that transdermal oestrogen may not be thrombogenic at all.
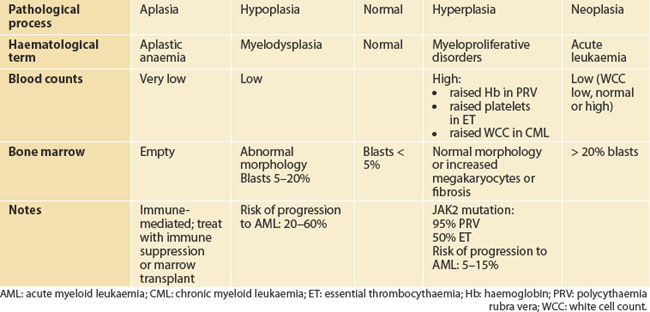
MALIGNANT HAEMATOLOGY
After an infection, the white cell count rises, sometimes with the platelet count, and then falls to the same premorbid level. Thus, marrow haemopoietic tissue, some 5 kg scattered around the long and flat bones of the body, behaves in a coordinated way, as a single organ. Moreover, it is subject to the same pathological processes as other organs. For historical reasons, the terminology is different. Table 22.6 shows how haematological terms correspond conceptually to the more familiar terms aplasia, dysplasia, hyperplasia and neoplasia in the spectrum of primary bone marrow pathologies. On the deficit side, complete loss of marrow or marrow function is called aplastic anaemia (a better term would be aplastic pancytopenia). Hypoplasia of the marrow usually maps to the myelodysplastic syndromes. In contrast, hyperplastic disorders of the marrow are referred to as the myeloproliferative syndromes. Neoplasia of the marrow stem cell is acute leukaemia.
Symptoms in patients with haematological malignancies can be considered negative or positive.
Negative symptoms arise from marrow hypofunction, manifesting with:
Physical examination should include assessment of all peripheral lymph node stations, including visualising the tonsils and palpating the epitrochlear and popliteal lymph nodes; plus a careful check for splenomegaly; and gently testing for bone tenderness over the sternum.
A brief summary of the main haematological cancers now follows.
CHRONIC MYELOID LEUKAEMIA
Chronic myeloid leukaemia (CML) (Fig 22.8) is a misnomer, as it is really a myeloproliferative pre-leukaemia, albeit one that had a 100% risk of progression to acute leukaemia before effective therapies were available. It has a unique triphasic natural history, from chronic phase, to accelerated and, finally, transformed CML. Patients may present late in the chronic phase, with a white cell count as high as 600,000 × 1012/L. Fatigue, moderate or massive splenomegaly, early satiety, night sweats and weight loss are classic symptoms. Increasingly the diagnosis is made in asymptomatic patients as an incidental finding—at a health or insurance check, for example.
Treatment has changed from busulphan in the 1970s, to interferon in the 1980s, to the astonishing success of imatinib mesylate in the early 2000s. This has provided proof-of-concept that the molecular biology revolution can deliver to the clinic. Imatinib is the first widely used drug targeted at a pathological molecule—that is, BCR-ABL, a tyrosine kinase, generated by the famous Philadelphia chromosome (translocation 9;22). The full story has been told recently elsewhere.1 For many with CML, lifelong disease control with tyrosine kinase inhibitors, such as imatinib and its successors, appears probable, after our first 10 years’ experience at least; bone marrow transplantation, although still the only cure, is reserved for those with accelerated or transformed disease, which remains much feared.
CHRONIC LYMPHOCYTIC LEUKAEMIA
Rather than abnormal cell growth, chronic lymphocytic leukaemia (CLL) (Fig 22.9) is characterised by the accumulation over time of lymphocytes in the bloodstream, lymph glands and/or bone marrow, or combinations thereof. Again, increasingly the diagnosis is made incidentally. The diagnostic test is no longer the bone marrow biopsy. Peripheral blood flow cytometry is easier and gives a pathognomic result, although the marrow often shows unexpectedly high numbers of CLL cells, and this may influence clinical decision-making. Treatment is indicated for symptomatic disease, bulky or disfiguring lymphadenopathy, or cytopenia. Most haematologists recommend treatment when the lymphocyte count hits 100 × 1012/L, for at this level, the other triggers for treatment usually soon follow.
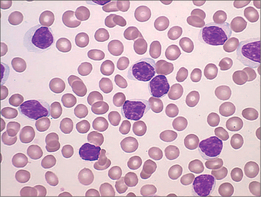
The treatment paradigm has shifted from oral chlorambucil to more aggressive combination chemotherapy, with good evidence that this confers better survival and longer remissions.33
For many newly diagnosed patients, treatment is not immediately indicated. Nevertheless, all patients should be referred for haematological review, even if asymptomatic, as the immediate implications for health include advice to start antibiotics promptly for suspected bacterial infection, to take measures to reduce solar skin damage and to assess for complications. As with any B-cell tumour, hypogammaglobulinaemia may occur (IgG < 2 g/L). Other complications include increased second cancers including skin and other cancers, autoimmune haemolytic anaemia in up to 20% of cases, and Richter’s transformation, a rare progression to aggressive non-Hodgkin’s lymphoma.
ACUTE LEUKAEMIA
When the term leukaemia was coined, in the nineteenth century, it referred to an excess of white cells in the bloodstream. Those with normal-appearing white cells lived longer (hence chronic leukaemia), whereas those with abnormal, or blast, cells, died rapidly, and so this was called acute leukaemia. We now recognise acute leukaemia (Fig 22.10) as a cancer of the bone marrow stem cell. The modern definition is: more than 20% blast cells in the marrow. (Less than 2% blast cells is normal; 5–20% blast cells characterises myelodysplasia.) Acute lymphoblastic leukaemia (ALL) is seven times more common than acute myeloid leukaemia (AML) in children; the ratio is reversed in adults.
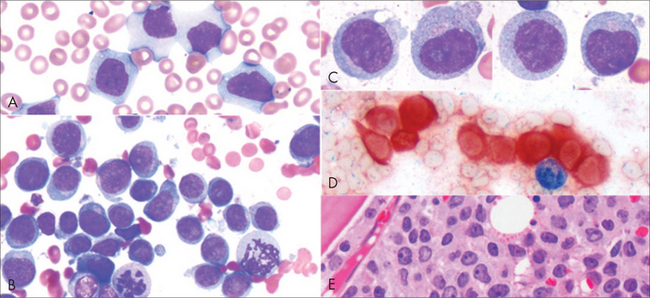
Therapeutics
Curative management of acute leukaemia requires 3 or 4 months of intensive chemotherapy in or near hospital. In ALL, the central nervous system is recognised as a possible sanctuary site for future relapse, and chemotherapy or adjunctive radiotherapy is given accordingly. Some 80% of patients achieve an initial response or remission; the problem then is keeping it. Paediatric ALL has cure rates approaching 90%, a great success story. Present cure rates in adults rest at 40% for AML and 25% for ALL. There are better results with some subtypes, including AML with certain karyotypic abnormalities and APML. Bone marrow transplantation is offered to selected patients in first remission as the best chance of cure, and has become less dangerous with improvements in tissue typing, viral and fungal antibiotics and supportive care for graft versus host disease. For the very elderly, a palliative chemotherapy approach may give surprising longevity (over a year) with good quality of life.
MYELODYSPLASIA
The myelodysplastic syndromes occur mostly in the elderly. Causes include prior chemotherapy and/or radiotherapy. Characteristically there are low peripheral blood counts, leading to negative symptoms of marrow failure. The cells look abnormal in the bone marrow biopsy (Fig 22.11). It is a morphogical diagnosis. Abnormal marrow cytogenetics are found in up 60% of cases, and there is a 20–80% risk of progression to acute leukaemia.
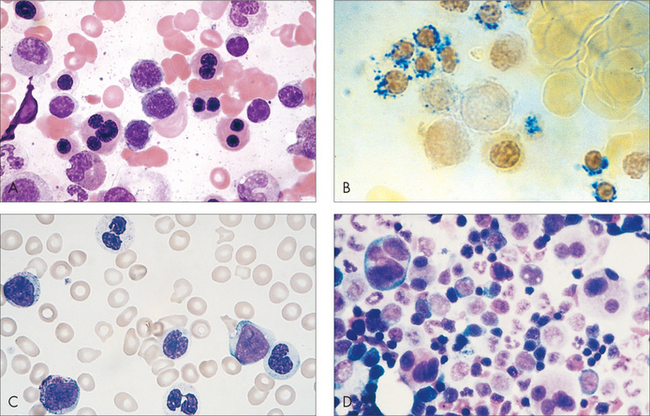
MYELOPROLIFERATIVE DISORDERS
Symptoms common to all these diseases include:
High haemoglobin levels may cause facial plethora, conjunctival injection and gritty eyes. A patient with an elevated haemoglobin may have plasma volume contraction (Gaisbock’s syndrome, associated with diuretic and tobacco use), secondary polycythaemia (to chronic hypoxia from cardiorespiratory disease) or true polycythaemia rubra vera (PRV). Rarer causes of secondary polycythaemia include various erythropoietin-secreting tumours.
The recent description of an acquired mutation of the JAK2 kinase gene in the MPS has revolutionised the diagnosis.34 It is positive in 95% of those with PRV and 50% of those with ET. Previously, nuclear medicine scans and bone marrow biopsy were needed to diagnose PRV and ET respectively. Now, screening for the JAK2 kinase mutation is diagnostic in most cases (now readily available in many countries) and it is an appropriate step for the GP to perform prior to referral.
LYMPHOMA
Lymphoma (Fig 22.12) is cancer of the immune system, and this in part explains the difficulty in classification over many years. Modern understanding of the immune system only began with the identification of B- and T-lymphocytes around 1970. Seeing lymphomas in this way also explains their heterogeneity, as there are many types of lymphocytes, with specialised roles and organ sites. Nevertheless, some broad generalisations can be made.
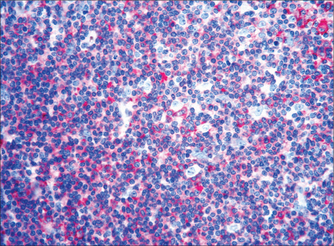
Therapeutics
Lymphoma management is complex and changing.35 Multidisciplinary care, as for many solid tumours, is increasingly used, principally with consolidative radiotherapy. In aggressive NHL, cure rates are high (around 70%). Randomised, controlled clinical trials have generally shown no benefit for more intensive regimens or for up-front bone marrow transplantation for aggressive NHL. Instead, cure rates improved in B-cell lymphomas with the addition of rituximab to the familiar CHOP regimen (R-CHOP). This is a humanised antibody to CD20, a pan-B-cell antigen found on most B-lymphoma cell types; it seems to work essentially by ‘lighting up’ the B-cells for the patient’s own T-cells to kill. Rituximab has rewritten the NHL landscape and represents an advance that proved elusive with intensified chemotherapy. Very high-grade lymphomas are treated essentially as for acute leukaemia.
Cases of follicular, or low-grade, NHL often present beyond stage II and are rarely curable with radiotherapy. Worldwide, with evidence that the addition of rituximab improves survival, the treatment paradigm has shifted to a more aggressive approach, also using R-CHOP, followed by maintenance rituximab, after which fully 80% can expect a remission lasting 4 years.36
Hodgkin’s lymphoma, now recognised as a B-cell tumour although lacking the usual B-cell surface antigens, has a cure rate of around 85–90%, mostly with the relatively non-toxic regimen of ABVD. Nevertheless, using a prognostic scoring system,37 cases at high risk of relapse can be identified and should be treated with a more intensive approach, such as BEACOPP or variants.
INVESTIGATION OF A PATIENT WITH LYMPHADENOPATHY
However, open biopsy will still be necessary to provide tissue for the large battery of tests in modern histopathology to grade a lymphoma. There are often up to 12 different immuno-histochemical stains, plus fluorescence in situ hybridisation, flow cytometry of cell suspensions and molecular work-up of tumour cell DNA. Lymphoma work-up in a larger pathology laboratory with an interest in lymphoma diagnosis will reclassify about 1 in 20 cases, leading to possible changes in treatment.38–40
MULTIPLE MYELOMA
Multiple myeloma (Fig 22.13) is not a tumour of bone, as the name suggests. Rather, it is a tumour of antibody-secreting (into the plasma, hence ‘plasma cells’) immune cells that reside in the marrow. It is an insidious process of unknown cause, and the diagnosis may easily be missed for several years. These cells can erode bone, and crowd out the normal production of blood cells, and the protein they secrete can induce renal failure. Classic presentations are covered by the CRAB acronym: hypercalcaemia, renal failure, anaemia and bone pain or fractures. It may mimic osteoporotic spinal fractures. Rarer presentations include neutropenia or other cytopenia, hyperviscosity syndrome (headaches, blurred vision, dyspnoea and organ dysfunction), pyrexia of unknown origin and splenomegaly. Physical examination has little diagnostic value.
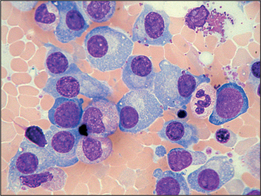
Therapeutics
Treatment is indicated for those with symptoms, high tumour burden or evidence of end-organ damage, such as lytic lesions or fractures, anaemia or renal failure. Components of treatment include chemotherapy, radiotherapy and bisphosphonates. Management of myeloma is also complex and rapidly changing—the past 15 years or so have seen the introduction of autologous stem cell transplantation, thalidomide, bortezomib and lenalidomide, all effective and capable of improving survival.41 Thus, for many, myeloma can be brought into remission for periods lasting several years at a time between lines of therapy and a more optimistic message can and should be given to myeloma patients. Oral or IV bisphosphonate therapy reduces bony adverse events by some 40%; recently, a rare complication, osteonecrosis of the jaw, has been recognised.42
INTEGRATIVE MANAGEMENT OF BLOOD CANCERS
The GP has a responsibility to be well informed about the adjunctive treatments that may be of benefit to patients in alleviating side effects of cancer treatments and enhancing the patient’s ability to cope with the disease and its treatment. There is also a valuable role for adjunctive therapies in rehabilitation after treatment with surgery, chemotherapy or radiotherapy (see Ch 24, Cancer).
Encouraging the patient to develop a personal approach to their illness can be very empowering for them, and can improve compliance with mainstream treatment.43 The patient must be involved in treatment and have the chance to buy into the therapeutic decisions, rather than having assumptions made and choice removed. There is good (level 2) evidence that lifestyle interventions can alleviate or reduce cancer symptoms and treatment side effects. This has recently been reviewed.44 A number of these interventions are now discussed here.
Lifestyle
Exercise
Perhaps the best single lifestyle intervention before, during and after cancer is exercise. It has been shown to have primary cancer preventive effect in the Californian teachers’ study, in which women who exercised (very hard, it must be said) reduced their breast cancer risk by 20%.45 Secondary cancer preventive benefit has been suggested in the setting of colorectal cancer,46,47 breast cancer and prostate cancer. Exercise promotes immune cell function and changes circulating white cell and lymphocyte subsets. Chemotherapy causes muscle catabolism and widespread loss of muscle mass; this may be potentiated by steroid myopathy. Resistance training in various muscle groups is more likely to help retain muscle mass, and reduce fatigue, than is mild aerobic exercise such as walking.48 High-intensity exercise is inappropriate, if not impossible, during chemotherapy, and a rough rule of thumb is: never exercise so hard that you can’t complete a sentence.
Diet
Diet is a focus for many cancer patients. Patients are encouraged to include all the food groups and specifically vitamin B12, folate and iron, in their diet, to aid haemopoietic recovery after each cycle of chemotherapy. (This is specious, as no dietary manipulation has been demonstrated to improve white cell or platelet counts, save that 10% of iron-deficient individuals manifest thrombocytopenia.) The dangers of mono-food and fad diets should be expressly mentioned. Weight gain above the healthy BMI range increases mortality after cancer.43
Antioxidants
Antioxidants clearly have some anti-cancer effects in vitro, and their proponents argue that they may confer cytoprotective effects upon normal cells exposed to chemo-radiotherapy as well. However, human clinical trials have been disappointing. Indeed, a 2008 review cast a shadow over supplemental antioxidants during chemotherapy, raising the possibility of worse outcomes.49 Furthermore, a Cochrane non-cancer-related review that included 67 randomised trials with 232,550 participants found that although vitamin C and selenium appeared safe, vitamin A, beta-carotene and vitamin E may be associated with increased mortality.50 Long-term selenium use has been linked to increased risk of type II diabetes,51 but appeared to reduce radiation-associated diarrhoea seen with treatment of gynaecological cancer.52
High-dose vitamin C may potentiate the activity of some chemotherapy regimens. Higher serum levels are achievable with IV than with oral preparations, and only recently has this been investigated in a more open way.53 Evidence of benefit remains scanty. Although it appears safe, it may precipitate haemolysis in G6PD-deficient individuals and promote urinary stone formation.
These are cautionary notes and, for these reasons, a blended fruit/vegetable drink is recommended as a healthier source of antioxidants, and possibly a multivitamin three times a week, as B-group vitamins may aid neurological recovery after insult (e.g. peripheral neuropathy after vinca alkaloids).
Asking patients what they are taking, and checking with a desktop reference of alternative and herbal preparations,54 shows a preparedness to discuss CAM use, and can help prevent dangerous herb–drug interactions.
Acupuncture
There is Cochrane meta-analysis evidence from 11 trials that acupuncture reduces acute post-chemotherapy vomiting (less so nausea or late vomiting).55
Music therapy
Music therapy can be passive (listening) or active (playing in any form, singing, etc). In an RCT of patients undergoing autologous stem cell transplantation, subjects receiving an individualised program of live music therapy had a significant total improvement in mood.56
Massage
There are many forms of massage, such as therapeutic massage, aromatherapy and reflexology. There is no single report in the literature that massage promotes cancer spread, although it seems prudent not to massage known sites of disease. Massage seems mostly to have an anti-anxiety effect. A recent well-conducted multi-centre RCT of aromatherapy massage showed short-term benefit (6 weeks, but not 10 weeks).57,58
Stress
There is some evidence that major life stresses predispose to cancer, but the small number of papers reporting a positive association are outweighed by many more finding no such link. A study in Sydney failed to show increased breast cancer rates in 239 women with breast carcinoma and 275 women with benign breast disease, after multiple regression analyses of carefully measured psychosocial factors.59
Positive mental attitude: ‘fighting the cancer’
Cancer patients are often encouraged and expected to show the much-quoted ‘positive mental attitude’. Many don’t feel they can achieve this, and feel guilty as a result of not meeting the expectations of those around them. A meta-analysis failed to show any survival benefit of any particular coping style.60 Admittedly, the point here is not survival, but making treatment more tolerable.
Instead, patients should be encouraged to reach a state of calmness about their cancer. How to do this will vary from person to person. Learning about the cancer may vary from acceptance of medical advice for some, to long hours on the internet to others. Patients should satisfy themselves that their mainstream advice is good, and decide how to face their illness in their own way. GPs can have a key role in providing some support to this process. There is growing evidence that some complementary therapies can reduce anxiety, ameliorate pain and fatigue and empower cancer patients.43,61 Positive self-imagery—an image of lying on a beach in Bali after treatment, for example—helps some.
Carers
Carers are part of the equation, and their attitude and capacity to assist a loved one through illness is clearly important. They should be encouraged to touch the patient—touch is a powerful form of human communication. They must respect the patient’s approach to their illness, which may be difficult, and keep aside some time and energy for self-care. Appointing a spokesperson for updates outside the cancer-afflicted household can save much energy, distress and time. Friends can help by providing ready-made meals, transport assistance and acceptance of the path chosen by the patient.
HAEMATOLOGICAL CANCER SURVIVORSHIP
Identified unmet needs in haematological cancer survivors include advice on financial and employment implications, the surveillance plan and information on lifestyle changes and long-term side effects, including infertility and second cancer risk; but the major unmet need by far is the fear of relapse.62
Haematology Education Resource. http://www.haematology.org/.
Haematology Society of Australia and New Zealand. http://www.hsanz.org.au/.
Hoffbrand AV, Moss PAH, Pettit JE. Essential haematology. 5th edn. Oxford: Blackwell, 2006.
Rosenthal DS, Dean-Clower E. Integrative medicine in hematology/oncology: benefits, ethical considerations, and controversies. Hematology. 2005;1:491-497. Online. Available: http://asheducationbook.hematologylibrary.org/cgi/content/full/2005/1/491.
1 Joske DJ. Chronic myeloid leukemia: the evolution of gene-targeted therapy. Med J Aust. 2008;189(5):277-282.
2 Argawal N, Prchal J. Anemia of chronic disease (anaemia of inflammation). Acta Haematologica. 2009;122:103-108.
3 Ioannou GN, Rockey DC, Bryson CL, et al. Iron deficiency and gastrointestinal malignancy: a population cohort study. Am J Med. 2002;113:276-280.
4 Finberg K, Heeney MM, Campagna DR, et al. Mutations in TMPRSS6 cause iron-refractory iron deficiency anemia (IRIDA). Nat Genet. 2008;40(5):569-571.
5 Beutler E, van Geet C, te Loo DM, et al. Polymorphisms and mutations of human TMPRSS6 in iron deficiency anemia. Blood Cell Molec Dis. 2010;44:16-21.
6 McDiarmid T, Johnson ED. Clinical inquiries. Are any oral iron formulations better tolerated than ferrous sulphate? J Fam Prac. 2002;51(6):576.
7 Spelman D, Buttery J, Daley A, et al. Guidelines for the prevention of sepsis in asplenic and hyposplenic patients. Australasian Society for Infectious Diseases. Intern Med J. 2008;38(5):349-356.
8 Ortel T. Heparin-induced thrombocytopenia: when a low platelet count is a mandate for anticoagulation. Hematology. 2009;1:225-232.
9 George J, Aster R. Drug-induced thrombocytopenia: pathogenesis, evaluation and management. Hematology. 2009:153-158.
10 Hayward C. Diagnosis and management of mild bleeding disorders. Hematology. 2005;1:423-428.
11 Leung L. Perioperative evaluation of a bleeding diathesis. Hematology. 2006;1:457-461.
12 Hull RD, Raskob GE, Rosenbloom D, et al. Optimal therapeutic level of heparin therapy in patients with venous thrombosis. Arch Intern Med. 1992;152:1589-1595.
13 Raschke RA, Reilly BM, Guidry JR, et al. The weight-based heparin dosing nomogram compared with a ‘standard care’ nomogram: a randomised controlled trial. Ann Intern Med. 1993;119:874-881.
14 Othieno R, Abu Affan M, Okpo E. Home versus in-patient treatment for deep vein thrombosis. Cochrane Database Syst Rev. 2007;3:CD003076.
15 Crowther MA, Ginsberg JB, Kearon C, et al. A randomized trial comparing 5 mg and 10 mg warfarin loading doses. Arch Intern Med. 1999;159:46-48.
16 Schulman S, Rhedin AS, Lindmaker P, et alDuration of Anticoagulation Study Group. A comparison of six weeks with six months of oral anticoagulant therapy after a first episode of venous thrombo-embolism. N Eng J Med. 1995;332:1661-1665.
17 Pinede L, Duhaut P, Cucherat M, et al. Comparison of long versus short duration of anticoagulant therapy after a first episode of venous thromboembolism a meta-analysis of randomised controlled trials. J Intern Med. 2000;247(5):553-562.
18 Prandoni P, Prins MH, Lensing AW, et al. AESOPUS Investigators. Residual thrombosis on ultrasonography to guide the duration of anticoagulation in patients with deep vein thrombosis: a randomized trial. Ann Int Med. 2009;150(9):577-585.
19 Schulman S, Granqvist S, Holmstrom M, et al. The duration of oral anticoagulant therapy after a second episode of venous thromboembolism. N Engl J Med. 1997;336(6):393-398.
20 Gallus A, Baker R, Chong B, et al. Consensus guidelines for warfarin therapy. Recommendations from the Australasian Society of Thrombosis and Haemostasis. Med J Aust. 2000;172:600-605.
21 Rosove MH, Grody WW. Should we be applying warfarin pharmacogenetics to clinical practice? No, not now. Ann Intern Med. 2009;151:270-273.
22 Baker R, Coughlin PB, Gallus AS, et al. Warfarin reversal: consensus guidelines, on behalf of the Australasian Society of Thrombosis and Haemostasis. Med J Aust. 2004;181:492-497.
23 Crawford JH, Augustson BM. Prothrombinex use for the reversal of warfarin: is fresh frozen plasma needed? Med J Aust. 2006;184:365-366.
24 Geerts WD, Berqvist D, Pineo GF, et al. Prevention of venous thromboembolism: American College of Chest Physicians, evidence-based clinical practice guidelines, 8th edn. Chest. 2008;133(6 Suppl):381S-453S.
25 Kelman CW, Kortt MA, Becker NG, et al. Deep vein thrombosis and air travel: record linkage study. BMJ. 2003;327:1072-1075.
26 Lee AY, Levine MN, Baker RI, et al. Low-molecular-weight heparin versus coumarin for the prevention of recurrent venous thromboembolism in patients with cancer. N Engl J Med. 2003;349(2):146-153.
27 De Stefano V, Finazzi G, Mannucci PM. Inherited thrombophilia: pathogenesis, clinical syndromes, and management. Blood. 1996;87:3531-3544.
28 De Stefano V, Martinelli I, Mannuci PM, et al. The risk of recurrent deep vein thrombosis among heterozygous carriers of both factor V Leiden and the G20210 A prothrombin mutation. N Engl J Med. 1999;341(11):801-806.
29 Ho WK, Hankey GJ, Quinlan DJ, et al. Risk of recurrent venous thromboembolism in patients with common thrombophilia: a systematic review. Arch Intern Med. 2006;166:729-736.
30 Wu O, Robertson L, Twaddle S, et al. Screening for thrombophilia in high-risk situations: systematic review and cost-effectiveness analysis. The Thrombosis; Risk and Economic Assessment of Thrombophilia Screening (TREATS) study. Health Technol Assess. 2006:1-110.
31 Canonico M, Oger E, Plu-Bureau G, et alEstrogen and Thromboembolism Risk (ESTHER) Study Group. Hormone therapy and venous thromboembolism among postmenopausal women: impact of the route of estrogen administration and progestogens: the ESTHER study. Circulation. 2007;115(7):840-845.
32 Canonico M, Plu-Bureau G, Lowe GD, et al. Hormone replacement therapy and risk of venous thromboembolism in postmenopausal women: systematic review and meta-analysis. BMJ. 2008;36(7655):1227-1231.
33 Carney DA, Mulligan SP. Chronic lymphocytic leukemia: current first-line therapy. Intern Med J. 2009;39(1):44-48.
34 Levine RL, Gilliland DG. Myeloproliferative disorders. Blood. 2008;112(6):2190-2198. Review.
35 Young GA, Iland HJ. Clinical perspectives in lymphoma. Intern Med J. 2007;37(7):478-484.
36 van Oers MH, Klasa R, Marcus RE, et al. Rituximab maintenance improves clinical outcomes of relapsed/resistant follicular non-Hodgkin lymphoma in patients both with and without rituximab during induction: results of a prospective randomised phase 3 intergroup trial. Blood. 2006;108(10):3295-3301.
37 Hasenclever D, Diehl V. A prognostic score for advanced Hodgkin’s disease. International Prognostic Factors Project on Advanced Hodgkin’s Disease. N Engl J Med. 1998;339(21):1506-1514.
38 Clarke CA, Undurraga DM, Harasty PJ, et al. Changes in cancer registry coding for lymphoma subtypes: reliability over time and relevance for surveillance and study. Cancer Epidemiology Biomarkers Prev. 2006;15(4):630-638.
39 LaCasce A, Kho M, Friedberg J, et al. Comparison of referring and final pathology for patients with non-Hodgkin’s lymphoma in the National Comprehensive Cancer Network. J Clin Oncol. 2008;26(31):5107-5112.
40 Prescott R, Wells S, Bisset DL, et al. Audit of tumour histopathology reviewed by a regional oncology centre. J Clin Path. 1995;48:245-249.
41 Raab MS, Podar K, Breitkreutz I, et al. Multiple myeloma. Lancet. 2009;374(9686):324-339. Review.
42 Dickinson M, Prince HM, Kirsa S, et al. Osteonecrosis of the jaw complicating bisphosphonate treatment for bone disease in multiple myeloma: an overview with recommendations for prevention and treatment. Intern Med J. 2009;39(5):304-316.
43 Jones LW, Demark-Wahnefried W. Diet, exercise and complementary therapies after primary treatment for cancer. Lancet Oncol. 2006;7:1017-1026.
44 Joske DJ, Rao A, Kristjanson L. Critical review of complementary therapies in haemato-oncology. Intern Med J. 2006;36(9):579-586.
45 Dallal CM, Sullivan-Halley J, Ross RK, et al. Long-term recreational physical activity and risk of invasive in situ breast cancer: the California teachers’ study. Arch Intern Med. 2007;167(4):408-415.
46 Meyerhardt JA, Giovannucci EL, Holmes MD, et al. Physical activity and survival after colorectal cancer diagnosis. J Clin Oncol. 2006;24(22):3527-3534.
47 Meyerhardt JA, Giovannucci EL, Ogino S, et al. Physical activity and male colorectal cancer survival. Arch Intern Med. 2009;169(22):2102-2108.
48 Galvao DA, Newton RU. Review of exercise intervention studies in cancer patients. J Clin Oncol. 2005;23(4):899-909.
49 Lawenda BD, Kelly KM, Ladas EJ, et al. Should supplemental antioxidant administration be avoided during chemotherapy and radiation therapy? J Natl Cancer Inst. 2008;100(11):773-783.
50 Bjelakovic G, Nikolova D, Gluud LL, et al. Antioxidant supplements for prevention of mortality in healthy participants and patients with various diseases. Cochrane Database Syst Rev. 2008;2:CD007176.
51 Stranges S, Marshall JR, Natarajan R, et al. Effects of long-term selenium supplementation on the incidence of type 2 diabetes: a randomised trial. Ann Intern Med. 2007;147(4):217-223.
52 Muecke R, Schomburg L, Glatzel M, et al. Multicentre, phase 3 trial comparing selenium supplementation with observation in gynecologic radiation oncology. Int J Radiat Oncol Biol Phys. 2010; 2 Feb. [Epub ahead of print].
53 Verrax J, Buc Calderon P. The controversial place of vitamin C in cancer treatment. Biochem Pharmacol. 2008;76(12):1644-1652.
54 Ernst E, editor. The desktop guide to complementary and alternative medicine. An evidence-based approach. Edinburgh: Mosby, 2001.
55 Ezzo JM, Richardson MA, Vickers A, et al. Acupuncture point stimulation for chemotherapy induced nausea or vomiting. Cochrane Database Syst Rev. 2006;2:CD002285.
56 Casileth BR, Vickers AJ, Magill LA. Music therapy for mood disturbance during hospitalisation for autologous stem cell transplantation: a randomised controlled trial. Cancer. 2003;98(12):2723-2729.
57 Post-White J, Kinney ME, Savik K, et al. Therapeutic massage and healing touch improve symptoms in cancer. Integr Cancer Ther. 2003;2(4):332-334.
58 Wilkinson SM, Love SB, Westcombe AM, et al. Effectiveness of aromatherapy massage in the management of anxiety and depression in patients with cancer: a multicentre randomised controlled trial. J Clin Oncol. 2007;25(5):532-539.
59 Price MA, Tennant CC, Butow PN, et al. The role of psychosocial factors in the development of breast carcinoma, Part II. Life event stressors, social support, defense style emotional control and their interactions. Cancer. 2001;91(4):686-697.
60 Petticrew M, Bell R, Hunter D. Influence of psychological coping on survival and recurrence in people with cancer: systematic review. BMJ. 2002;325(7372):1066. Review.
61 Diehl V. The bridge between patient and doctor: the shift from CAM to integrative medicine. Hematology. 2009;1:320-325. Review.
62 Lobb E, Joske D, Butow P, et al. When the safety net of treatment has been removed: patients’ unmet needs at the completion of treatment for haematological malignancies. Patient Educ Couns. 2009;77(1):103-108.