[level-membership-for-cardiovascular-category]9
Biophysics of Normal and Abnormal Cardiac Sodium Channel Function
Abstract
Voltage-gated sodium channels (Nav) underlie the activity of many excitable cells. In the heart, Nav channels are responsible for the rapid cardiomyocyte action potential upstroke that promotes rapid conduction of the electrical impulse leading to coordinated mechanical contraction. Central to this function, Nav channels activate (and then inactivate) rapidly in response to a small depolarization of the membrane, resulting in a large influx of Na+ ions and further membrane depolarization. Dysfunction in Nav channel activity results in human diseases and disorders, including epilepsy, ataxia, cardiac arrhythmia, and myotonia.1–3 Variants in SCN5A, the gene encoding the primary cardiac Nav α-subunit Nav1.5, have been linked to human arrhythmia syndromes including long QT type 3 (LQT3), Brugada syndrome, cardiac conduction disease, sinus node disease, and atrial fibrillation.2,4,5 A detailed discussion of fundamental aspects of Nav structure function, gating, and pharmacology can be found in Chapter 1. Here we discuss current understanding regarding regulation of Nav biophysical activity and cellular function in health and disease.
The Nav channel pore-forming α-subunit in vertebrates is a single polypeptide with four homologous transmembrane domains (DI-DIV) comprised of six membrane-spanning α-helices (S1-S6; Figure 9-1). Nav channels share structural similarities with voltage-gated Ca2+ channels from which they may have evolved.6 Ten different Nav channel α-subunits are present in mammals, each with specific biophysical, expression, and regulatory signatures. The primary cardiac isoform, Nav1.5, is derived from 28 exons and undergoes alternative splicing in mammalian heart (including human).4,7–11 Although Nav1.5 is the primary Nav channel expressed in the heart, limited expression of neuronal α-subunits has also been reported and likely introduces important heterogeneity in Nav biophysical activity, localization, and drug sensitivity within the cell.12,13 Although the α-subunit is capable of forming a functional channel by itself, several auxiliary β-subunits have been identified (β1-β4) that modulate channel gating and/or trafficking. Human mutations in both the α and β-subunits have been linked to arrhythmia.14,15 Importantly, Nav1.5 and associated β-subunits reside with large macromolecular complexes comprised of adaptor, accessory, cytoskeletal, and regulatory proteins (Figure 9-2).4,5,16–22 An exciting area of research going forward, and the focus of this and other chapters in this volume (Chapters 18 and 21), is the physiologic function of Nav macromolecular complexes and their role in arrhythmias and mechanical dysfunction in the setting of human disease.
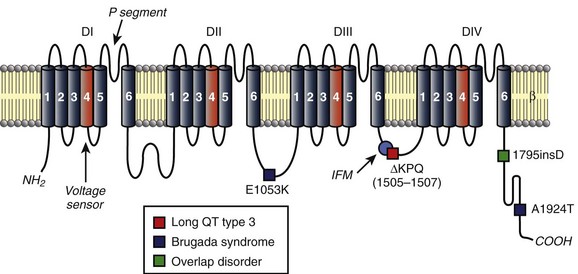
Figure 9-1 Topology of cardiac Nav1.5 channel (SCN5A) and human variants linked to congenital cardiac arrhythmia. Charged S4 segments (orange) in each domain function as voltage sensors. P segments between S5 and S6 form the channel pore. IFM motif in DIII-DIV linker (light blue circle) acts as inactivation particle to facilitate rapid voltage-dependent inactivation. The location of several human variants in SCN5A linked to LQT3 (red square), Brugada syndrome (blue square), or overlap disorder (green square) are indicated.
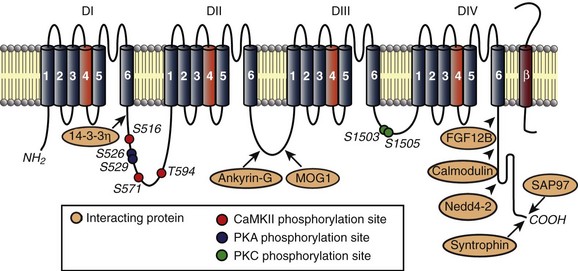
Figure 9-2 Nav1.5 resides in macromolecular complexes comprised of β-subunits (red) and cytoskeletal, adapter, trafficking, and regulatory molecules. Known interacting proteins are shown along with binding regions. Phosphorylation sites identified to date are indicated for CaMKII (red), PKA (blue), and protein kinase C (green).
Nav Channel Activity in Health and Disease
Over the course of the action potential, Na+ channels undergo a remarkable series of conformational changes that are essential for the rapid and controlled influx of Na+ ions that supports the action potential upstroke (detailed in Chapter 1).23 Recent work to crystallize the bacterial Na+ channel NavAb (forms as a tetramer of identical subunits akin to most K+ channels) has added to decades of important research on the complex Nav structure–function relationship governing channel activity.24,25 Briefly, in response to membrane depolarization, channels first activate rapidly as the four charged S4 segments (see Figure 9-1) move outward and open the channel pore.26 Channel activation is followed almost immediately by rapid inactivation caused by further outward movement of the S4 sensors in DIII and DIV and blocking of the channel pore by the DIII-DIV linker that likely depends on the interaction between hydrophobic residues (isoleucine-phenylalanine-methionine, IFM) in the III-IV linker and multiple sites in the linkers between S4 and S5 in DIII and DIV and the cytoplasmic end of S6 in DIV.27–29 Rapid inactivation is followed by transition into several different slow inactivation states that are controlled, in part, by P segments linking S5 and S6 segments that form the inner channel pore (see Figure 9-1). Importantly, disruption of Na+ channel gating at any step during this highly coordinated set of movements may result in inappropriate (elevated, reduced) current and give rise to arrhythmias.
Dysfunction in Nav1.5 activity has been identified as a potential cause of arrhythmia in several forms of congenital and acquired disease (see Figure 9-1). Classic examples of the link between inherited defects in Nav1.5 function and disease may be found in two inherited arrhythmia syndromes closely linked to mutations in SCN5A: Brugada syndrome and LQT3. An overview of molecular basis for these diseases is provided here; both are addressed in detail in later chapters. Brugada syndrome is an autosomal, dominant cardiac arrhythmia syndrome characterized by ST-segment elevation in the right precordial leads (V1-V3) and sudden death in the absence of overt structural disease.30,31 The syndrome is caused by loss-of-function variants in SCN5A (although identified in only about 20% of Brugada cases31) that result in loss of Na+ current early in the action potential because of a loss of functional channel expression/localization or defects in channel gating.20,32,33 The substrate for Brugada syndrome arrhythmia is thought to arise in the epicardium of the right ventricle where high expression of transient outward K+ current Ito may shift the balance of current to favor exaggerated phase 1 repolarization (notch) and even loss of the action potential plateau.34,35 Loss-of-function SCN5A variants have also been associated with isolated cardiac conduction disease characterized by slow conduction throughout the heart (even requiring pacemaker implantation) without the repolarization abnormalities or ventricular tachyarrhythmia observed in Brugada syndrome.36 In general, gene variants that give rise to cardiac conduction disease produce a less severe effect on channel function than those associated with Brugada syndrome, by introducing a secondary gain-of-function change in Nav activity (e.g., depolarizing shift in voltage dependence of inactivation) that partially balances the primary loss-of-function change (e.g., depolarizing shift in voltage dependence of activation).
Although Brugada syndrome is a disease associated with Nav loss-of-function, LQT3 represents the other extreme and illustrates how Nav gain-of-function may also produce arrhythmia. A heterogeneous autosomal dominant genetic disease, LQT3 is associated with abnormal QT-interval prolongation on the electrocardiogram, syncope, polymorphic ventricular tachycardia, and sudden death.14 Variants in SCN5A are the cause of LQT3, which is associated with increased risk of cardiac events during rest and decreased efficacy of β-blocker therapy.14,37,38 At least 50 variants in SCN5A have been identified as causal for LQT3, and although these variants are scattered throughout the cytoplasmic face of the channel (with a cluster in the S4 segments), they primarily produce an increase in Na+-channel current (gain-of-Nav function) by disrupting rapid inactivation, by shifting the voltage-dependence of activation or inactivation, or by altering recovery from inactivation. A prototypical case study of the causal link between SCN5A mutations and LQT3 comes from the first reported LQT3 mutation resulting in three–amino acid deletion in the DIII-DIV linker that is important for rapid channel inactivation (ΔKPQ, see Figure 9-1).38 The variant ΔKPQ allele disrupts rapid inactivation, producing a small (~0.5% of peak) but sustained Na+ current during the action potential plateau that shifts the balance of current to prolong action potential and increase the likelihood of arrhythmogenic afterdepolarizations.38,39 Although LQT3 and Brugada syndrome are often presented as two very different faces of Nav dysfunction, overlap syndromes have been reported, with features of both diseases showing QT prolongation at slow rates coupled with ST elevation during exercise. Notably, the 1795insD mutation (see Figure 9-1) interrupts rapid inactivation to enhance persistent Na+ current and prolong action potential duration, especially at slow pacing rates, but it also enhances an intermediate inactivation state that compromises Nav recovery between stimuli, leading to loss of current at fast rates.40,41 Thus, as discussed before, for cardiac conduction disease, a single molecular defect may alter channel activity at more than one phase (e.g., rapid and slow inactivation processes) in the gating cycle, giving rise to a complex disease phenotype.
Studies of rare congenital conditions such as LQT3 and Brugada syndrome have generated important insight into Nav channel function and have increased our understanding of arrhythmogenesis in the more complex setting of acquired heart disease, where Nav channel dysfunction is a common finding. In heart failure, for example, an increase in persistent (late) Na+ current has been observed both in patients and animal models of human disease.42–46 Similar to LQT3, in heart failure it is likely that increased late Na+ current delays action potential repolarization and promotes pro-arrhythmogenic afterdepolarizations.45 Increased Na+ entry via the late current may also disrupt normal Ca2+ homeostasis and promote mechanical dysfunction and progression of disease.47 The underlying mechanism for changes in Nav function in heart failure is unknown but is most likely not a result of changes in expression of Nav α or β-subunits.42,45 In light of known defects in β-adrenergic and calcium-dependent signaling pathways in heart failure, it is likely that defects in channel posttranslational modification play a role in Nav dysfunction. Regardless of Na+ current mechanism, mounting studies support the late Na+ current as a viable therapeutic target (e.g., inhibition with ranolazine) for preventing arrhythmias and progression of disease in heart failure.48–51
There have also been many studies on the link between defects in Nav function and arrhythmias after myocardial infarction, where reentrant arrhythmias are highly localized to the highly remodeled tissue surrounding the infarct (border zone). In particular, in the canine heart, dramatic electrical and structural remodeling has been identified coupled with anisotropic conduction and reentrant arrhythmias in the border zone region. Conduction through this region is highly irregular, characterized by slow and discontinuous conduction.52 Myocytes isolated from the border zone regions display a characteristic time course of changes in ion channel activity and action potential, including significant decrease in peak Na+ current, altered subcellular localization, decreased channel availability, and slow recovery from inactivation.53–55 Computational studies demonstrate that measured defects in Nav channel activity prolong refractoriness, decrease conduction velocity, and increase susceptibility to initiation of reentrant arrhythmias.53,56–59 Although the precise mechanism underlying abnormal Nav function after myocardial infarction is likely multifactorial and not fully understood, roles for increased oxidative stress and/or increased CaMKII-dependent channel phosphorylation have been identified.57,60 Interestingly, adenoviral overexpression of skeletal muscle Nav channel, with inactivation shifted to more depolarized potentials compared with cardiac isoform, has been shown to improve conduction and suppress arrhythmias in the canine after myocardial infarction.61 It is intriguing to consider the possibility that targeting channel regulatory pathways (e.g., CaMKII) may be a second, and conceivably more practical, method to alter channel availability for therapeutic benefit.
NaV Channel Trafficking and Stabilization in Health and Disease
As outlined before, Nav function is critical for normal heart function, and alteration of Nav activity is a hallmark of many forms of inherited and acquired diseases. Despite major advances, the mechanistic link among specific molecular defects, Nav channel dysfunction, and arrhythmias associated with many human arrhythmia variants and in common disease remains elusive. Mounting evidence, in particular from human arrhythmia variants in genes encoding ion channel accessory proteins (e.g., adapter and scaffolding proteins, channel subunits, chaperones), highlight the importance of proper Nav channel localization within specific cellular domains for normal heart function.62–69
Voltage-gated Nav channels are targeted to specific membrane domains in cardiomyocytes (Figure 9-3). Residing primarily at cell ends in the region of the intercalated disc (ID), Nav channels are also found at secondary sites at lateral and transverse-tubule membranes (see Chapter 18). Although the cardiac isoform Nav1.5 largely comprises the Nav population at the ID, neuronal isoforms (e.g., Nav1.1, Nav1.3, Nav1.6) are found almost exclusively at transverse tubules.12,13,70 Although the functional consequences for differential targeting of Nav isoforms within the cardiomyocyte remain unclear, computational studies demonstrate that concentration of Nav channels at the ID (Nav1.5), where cells are electrically and mechanically coupled, supports electrical impulse propagation.71,72 Likewise, a role for the neuronal isoforms (largely outside the ID) has been explored using low doses of tetrodotoxin (TTX) (insufficient to block TTX-resistant Nav1.5). These experiments show that although neuronal channels are not necessary for conduction under normal conditions, they may play modulatory roles in excitation-contraction coupling and ventricular function.12,13
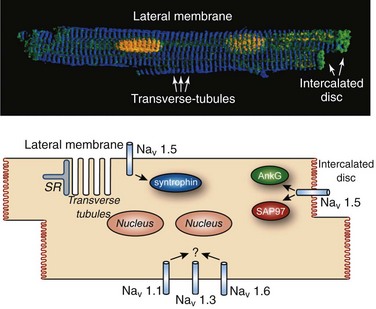
Figure 9-3 Nav channels are differentially localized within cardiomyocyte. A, Three-dimensional rendering of isolated murine ventricular cardiomyocyte stained with markers for the nucleus (yellow), intercalated disc (green), and z-line (blue). B, Schematic indicating Nav localization and interacting partners at specific membrane domains within the cardiomyocyte.
An important outstanding question is how are Nav channels differentially targeted and regulated within the cardiomyocyte? The answer may be found in the growing family of adapter, accessory, cytoskeletal and regulatory proteins—including α1-syntrophin, ankyrin-G, βIV-spectrin, glycerol-3-phosphate dehydrogenase 1–like protein, MOG1, Nedd4-2, SAP97, and calmodulin—that reside in macromolecular complexes with Nav channels (see Figure 9-2).4,5,16–22 Recent studies demonstrate that interactions between Nav and these related proteins: (1) regulate channel function, (2) differ depending on local membrane domain, and (3) are altered in disease.
The Nav1.5 α-subunit associates with one or more β-subunits (β1-β4). These β-subunits are type I integral membrane proteins with an extracellular N-terminus containing an immunoglobulin domain with homology to domains found in cell adhesion molecules, a single transmembrane domain, and a cytoplasmic C-terminal sequence.73 Differential localization of β-subunits has been reported in mouse heart, with co-localization of β1 and β2 found at the intercalated disc and T-tubules, β3 at T-tubules, and β4 at the intercalated disc.12 Beta-subunits are reported to alter Nav1.5 membrane expression and gating. Importantly, human mutations in Nav β-subunits have been linked to diseases and conditions, including Brugada syndrome (SCN1B and SCN3B), long QT (SCN4B), cardiac conduction disease (SCN1B), and atrial fibrillation (SCN1B, SCN2B and SCN3B).4,67,73–76
The ankyrin family of multifunctional adapter proteins (e.g., ankyrin-R, ankyrin-B, and ankyrin-G expressed in the heart) coordinates macromolecular complexes at specific membrane domains in cardiomyocytes. The most studied ankyrin in the heart, ankyrin-B, is involved in targeting of the Na+/Ca2+ exchanger, Na+/K+ adenosine triphosphatase (ATPase), Kir6.2, InsP3 receptor, and protein phosphatase 2A to transverse tubules.77–80 Ankyrin-G, on the other hand, is concentrated at the cardiomyocyte ID, where it associates directly with Nav1.5 through binding to a conserved motif in the DII-DIII linker (see Figures 9-2 and 9-3).19,20,81 Interfering with ankyrin-G/Nav1.5 interaction, either through knockdown of ankyrin-G or expression of mutant Nav1.5 lacking ankyrin-binding activity, disrupts Nav1.5 membrane, targeting resulting in loss of Na+ current.19,20 Recently, ankyrin-G has been identified in a functional macromolecular complex in vivo with Nav1.5, the actin-associated polypeptide βIV-spectrin, and calmodulin kinase II (CaMKII) for targeting and regulation of Nav function and cell membrane excitability.82 Ankyrin-G also has been shown to associate and co-localize with desmosome and gap junction proteins plakophilin-2 and connexin43,83 supporting the notion that ankyrin-G is an important nodal point for coordinating mechanical and electrical signaling at the cardiomyocyte ID. Importantly, defects in ankyrin-based pathways have been identified as the underlying cause for abnormal channel targeting and arrhythmias in congenital and acquired forms of cardiac disease.20,62,84–86 In the case of Nav1.5, a human SCN5A variant linked to Brugada syndrome (E1053K) is located in the ankyrin-binding motif of Nav1.5 and disrupts ankyrin-binding activity in vitro (see Figure 9-1).20
Nav channels display unique biophysical properties based on their specific membrane localization.70 Consistent with these findings, work from several groups has recently shown that Nav1.5 complexes with distinct partners at the ID versus lateral membranes.87 In addition to ankyrin-G, Nav1.5 has been found to associate with synapse-associated protein SAP97, a member of the membrane-associated guanylate kinase family, at the cardiomyocyte ID (see Figure 9-3).87 This interaction depends on the terminal three amino acids of the Nav1.5 C-terminus (see Figure 9-2), and knockdown of SAP97 leads to a decrease in INa and reorganization of Nav1.5 in adult cardiomyocytes. Nav1.5 is also found, albeit to a much lower degree, at the lateral membrane and is likely regulated by interaction with the dystrophin-syntrophin complex through direct binding of Nav1.5 C-terminus with a PDZ domain in syntrophin.88–91 Na+ current and Nav1.5 expression are decreased in mice lacking dystrophin (mdx5cv), and human mutations in SNTA1 (encodes α1-syntrophin) have been identified as the cause of increased Na+ current in congenital long QT syndrome, and sudden infant death syndrome.90–92,87 It will be interesting in the future to determine the precise role of Nav1.5 interacting partners at the intercalated disc and lateral membrane in targeting, retention, and function of the channel.
Nav Channel Posttranslational Regulation in Health and Disease
Tight spatial and temporal control of local signaling domains is essential for proper regulation and activity of Nav channels in cardiomyocytes. Importantly, changes in posttranslational modification of membrane proteins are associated with increased susceptibility to congenital and acquired arrhythmia.90,93–95 In particular, Nav channel activity is heavily regulated by posttranslational modification, including phosphorylation by protein kinase C,96–98 protein kinase A (PKA),99–102 CaMKII,82,103–106 and glycosylation.4,107 Important in the setting of heart disease, β-adrenergic stimulation is known to increase INa through PKA-direct and PKA-indirect modulation.99–101,108 As discussed before, increased INa (late) likely contributes to arrhythmogenesis in heart failure. In parallel, β-adrenergic stimulation is enhanced in failing hearts. Therefore, it is interesting to consider the possibility that defects in Nav1.5 posttranslational modification downstream of β-adrenergic stimulation may underlie INa dysfunction in heart failure. Although PKA directly phosphorylates Nav1.5 at specific residues in the DI-DII linker (see Figure 9-2),102 studies with PKA inhibitors suggest that this accounts for only part of the effect of β-adrenergic stimulation on Nav function.101,109 PKA-independent effects likely involve regulation of Nav1.5 surface expression by the stimulatory G protein α-subunit (Gαs) and caveolin-3 (CAV3).101,109,110 Human gene variants in CAV3 have been linked to increased late (persistent) INa and arrhythmias in long QT syndrome type 9.65 Although the link between mutant CAV3 and changes in Nav function remain unknown, computational studies suggest that disruption of CAV3/Gαs interaction may alter the dynamics of caveolae membrane fusion to give rise to increased late Na+ current.111
Nav channels are also regulated by Ca2+/CaM-dependent pathways, which are known to be dysregulated in disease.112,113 CaM interacts directly with multiple Nav isoforms, including Nav1.5 via a C-terminal IQ motif and a domain in the DIII-DIV loop with variable results.113–119 Although the net effect of CaM on channel activity likely depends on factors such as specific isoform and experimental conditions, binding of CaM to the IQ motif of human Nav1.5 has been shown to enhance slow component of inactivation,113 with other studies showing CaM-dependent effects on steady-state inactivation.105,114,118 Interestingly, the human mutation A1924T in the Nav1.5 IQ motif (see Figure 9-1) results in Brugada syndrome and alters channel activity by interrupting CaM binding to Nav1.5.113–115 More recently, a role for Ca2+/CaM regulation of Nav1.5 via CaMKII-dependent phosphorylation has been described.82,104–106,112 Specifically, initial studies showed that overexpression of CaMKIIδc in rabbit (adenoviral, acute) and mouse (transgenic, chronic) ventricular cardiomyocytes shifted steady-state inactivation in the hyperpolarizing direction (decreased availability), increased intermediate activation, slowed recovery from inactivation, and enhanced late Na+ current.104 At the cellular level, CaMKIIδ overexpression resulted in accumulation of intracellular Na+ and prolongation of APD, which translates to increased QRS (marker for slow intraventricular conduction) and QT intervals on the electrocardiogram, as well as increased susceptibility to ventricular arrhythmias.104 It is now clear that CaMKII phosphorylates the channel at multiple sites in the DI-DII linker (including S516, S571, T594) (see Figure 9-2) to alter steady-state channel availability, recovery from inactivation and persistent current.82,105,106 Furthermore, CaMKII-dependent regulation of Nav1.5 depends on direct interaction with the actin-associated polypeptide βIV-spectrin, which targets CaMKII with Nav1.5 to the cardiomyocyte intercalated disc.82 Loss of spectrin/CaMKIIδ interaction disrupts CaMKII regulation of Nav1.5 and abnormal cell membrane excitability. Future studies should determine the broader role for βIV-spectrin in coordinating Nav macromolecular complexes at the cardiomyocyte intercalated disc and its role in arrhythmogenesis.
References
1. Nattel, S, Maguy, A, Le Bouter, S, et al. Arrhythmogenic ion-channel remodeling in the heart: heart failure, myocardial infarction, and atrial fibrillation. Physiol Rev. 2007; 87:425–456.
2. Wilde, AA, Brugada, R. Phenotypical manifestations of mutations in the genes encoding subunits of the cardiac sodium channel. Circ Res. 2011; 108:884–897.
3. Waxman, SG. Channel, neuronal and clinical function in sodium channelopathies: from genotype to phenotype. Nat Neurosci. 2007; 10:405–409.
4. Rook, MB, Evers, MM, Vos, MA, et al. Biology of cardiac sodium channel Nav1. 5 expression. Cardiovasc Res. 2012; 93:12–23.
5. Abriel, H. Cardiac sodium channel Na(v)1. 5 and interacting proteins: Physiology and pathophysiology. J Mol Cell Cardiol. 2010; 48:2–11.
6. Zakon, HH. Adaptive evolution of voltage-gated sodium channels: The first 800 million years. Proc Natl Acad Sci U S A. 2012; 109(Suppl 1):10619–10625.
7. Wang, Q, Li, Z, Shen, J, et al. Genomic organization of the human SCN5A gene encoding the cardiac sodium channel. Genomics. 1996; 34:9–16.
8. Schroeter, A, Walzik, S, Blechschmidt, S, et al. Structure and function of splice variants of the cardiac voltage-gated sodium channel Na(v)1. 5. J Mol Cell Cardiol. 2010; 49:16–24.
9. Shang, LL, Dudley, SC, Jr. Tandem promoters and developmentally regulated 5’- and 3’-mRNA untranslated regions of the mouse Scn5a cardiac sodium channel. J Biol Chem. 2005; 280:933–940.
10. Shang, LL, Pfahnl, AE, Sanyal, S, et al. Human heart failure is associated with abnormal C-terminal splicing variants in the cardiac sodium channel. Circ Res. 2007; 101:1146–1154.
11. Walzik, S, Schroeter, A, Benndorf, K, et al. Alternative splicing of the cardiac sodium channel creates multiple variants of mutant T1620K channels. PLoS One. 2011; 6:e19188.
12. Maier, SK, Westenbroek, RE, McCormick, KA, et al. Distinct subcellular localization of different sodium channel alpha and beta subunits in single ventricular myocytes from mouse heart. Circulation. 2004; 109:1421–1427.
13. Maier, SK, Westenbroek, RE, Schenkman, KA, et al. An unexpected role for brain-type sodium channels in coupling of cell surface depolarization to contraction in the heart. Proc Natl Acad Sci U S A. 2002; 99:4073–4078.
14. Moss, AJ, Kass, RS. Long QT syndrome: from channels to cardiac arrhythmias. J Clin Invest. 2005; 115:2018–2024.
15. Napolitano, C, Bloise, R, Monteforte, N, et al. Sudden cardiac death and genetic ion channelopathies: long QT, Brugada, short QT, catecholaminergic polymorphic ventricular tachycardia, and idiopathic ventricular fibrillation. Circulation. 2012; 125:2027–2034.
16. Abriel, H, Kass, RS. Regulation of the voltage-gated cardiac sodium channel Nav1. 5 by interacting proteins. Trends Cardiovasc Med. 2005; 15:35–40.
17. Wu, L, Yong, SL, Fan, C, et al. Identification of a new co-factor, MOG1, required for the full function of cardiac sodium channel Nav 1. 5. J Biol Chem. 2008; 283:6968–6978.
18. Lemaillet, G, Walker, B, Lambert, S. Identification of a conserved ankyrin-binding motif in the family of sodium channel alpha subunits. J Biol Chem. 2003; 278:27333–27339.
19. Lowe, JS, Palygin, O, Bhasin, N, et al. Voltage-gated Nav channel targeting in the heart requires an ankyrin-G dependent cellular pathway. J Cell Biol. 2008; 180:173–186.
20. Mohler, PJ, Rivolta, I, Napolitano, C, et al. Nav1. 5 E1053K mutation causing Brugada syndrome blocks binding to ankyrin-G and expression of Nav1. 5 on the surface of cardiomyocytes. Proc Natl Acad Sci U S A. 2004; 101:17533–17538.
21. Abriel, H, Kamynina, E, Horisberger, JD, et al. Regulation of the cardiac voltage-gated Na+ channel (H1) by the ubiquitin-protein ligase Nedd4. FEBS Lett. 2000; 466:377–380.
22. van Bemmelen, MX, Rougier, JS, Gavillet, B, et al. Cardiac voltage-gated sodium channel Nav1. 5 is regulated by Nedd4-2 mediated ubiquitination. Circ Res. 2004; 95:284–291.
23. Catterall, WA. Voltage-gated sodium channels at 60: Structure, function, and pathophysiology. J Physiol. 2012; 590:2557–2589.
24. Payandeh, J, Gamal El-Din, TM, Scheuer, T, et al. Crystal structure of a voltage-gated sodium channel in two potentially inactivated states. Nature. 2012; 486:135–139.
25. Payandeh, J, Scheuer, T, Zheng, N, et al. The crystal structure of a voltage-gated sodium channel. Nature. 2011; 475:353–358.
26. Catterall, WA. Ion channel voltage sensors: structure, function, and pathophysiology. Neuron. 2010; 67:915–928.
27. Vassilev, PM, Scheuer, T, Catterall, WA. Identification of an intracellular peptide segment involved in sodium channel inactivation. Science. 1988; 241:1658–1661.
28. West, JW, Patton, DE, Scheuer, T, et al. A cluster of hydrophobic amino acid residues required for fast Na(+)-channel inactivation. Proc Natl Acad Sci U S A. 1992; 89:10910–10914.
29. Cha, A, Ruben, PC, George, AL, Jr., et al. Voltage sensors in domains III and IV, but not I and II, are immobilized by Na+ channel fast inactivation. Neuron. 1999; 22:73–87.
30. Brugada, P, Brugada, J. Right bundle branch block, persistent ST segment elevation and sudden cardiac death: a distinct clinical and electrocardiographic syndrome. A multicenter report. J Am Coll Cardiol. 1992; 20:1391–1396.
31. Antzelevitch, C, Brugada, P, Brugada, J, et al. Brugada syndrome: a decade of progress. Circ Res. 2002; 91:1114–1118.
32. Chen, Q, Kirsch, GE, Zhang, D, et al. Genetic basis and molecular mechanism for idiopathic ventricular fibrillation. Nature. 1998; 392:293–296.
33. Dumaine, R, Towbin, JA, Brugada, P, et al. Ionic mechanisms responsible for the electrocardiographic phenotype of the Brugada syndrome are temperature dependent. Circ Res. 1999; 85:803–809.
34. Yan, GX, Antzelevitch, C. Cellular basis for the Brugada syndrome and other mechanisms of arrhythmogenesis associated with ST-segment elevation. Circulation. 1999; 100:1660–1666.
35. Gima, K, Rudy, Y. Ionic current basis of electrocardiographic waveforms: a model study. Circ Res. 2002; 90:889–896.
36. Tan, HL, Bink-Boelkens, MT, Bezzina, CR, et al. A sodium-channel mutation causes isolated cardiac conduction disease. Nature. 2001; 409:1043–1047.
37. Wang, Q, Shen, J, Li, Z, et al. Cardiac sodium channel mutations in patients with long QT syndrome, an inherited cardiac arrhythmia. Hum Mol Genet. 1995; 4:1603–1607.
38. Wang, Q, Shen, J, Splawski, I, et al. SCN5A mutations associated with an inherited cardiac arrhythmia, long QT syndrome. Cell. 1995; 80:805–811.
39. Clancy, CE, Rudy, Y. Linking a genetic defect to its cellular phenotype in a cardiac arrhythmia. Nature. 1999; 400:566–569.
40. Bezzina, C, Veldkamp, MW, van Den Berg, MP, et al. A single Na+ channel mutation causing both long-QT and Brugada syndromes. Circ Res. 1999; 85:1206–1213.
41. Clancy, CE, Rudy, Y. Na+ channel mutation that causes both Brugada and long-QT syndrome phenotypes: a simulation study of mechanism. Circulation. 2002; 105:1208–1213.
42. Valdivia, CR, Chu, WW, Pu, J, et al. Increased late sodium current in myocytes from a canine heart failure model and from failing human heart. J Mol Cell Cardiol. 2005; 38:475–483.
43. Jacques, D, Bkaily, G, Jasmin, G, et al. Early fetal like slow Na+ current in heart cells of cardiomyopathic hamster. Mol Cell Biochem. 1997; 176:249–256.
44. Maltsev, VA, Sabbah, HN, Higgins, RS, et al. Novel, ultraslow inactivating sodium current in human ventricular cardiomyocytes. Circulation. 1998; 98:2545–2552.
45. Undrovinas, AI, Maltsev, VA, Sabbah, HN. Repolarization abnormalities in cardiomyocytes of dogs with chronic heart failure: role of sustained inward current. Cell Mol Life Sci. 1999; 55:494–505.
46. Maltsev, VA, Silverman, N, Sabbah, HN, et al. Chronic heart failure slows late sodium current in human and canine ventricular myocytes: implications for repolarization variability. Eur J Heart Fail. 2007; 9:219–227.
47. Bers, DM, Despa, S, Bossuyt, J. Regulation of Ca2+ and Na+ in normal and failing cardiac myocytes. Ann N Y Acad Sci. 2006; 1080:165–177.
48. Undrovinas, NA, Maltsev, VA, Belardinelli, L, et al. Late sodium current contributes to diastolic cell Ca2+ accumulation in chronic heart failure. J Physiol Sci. 2010; 60:245–257.
49. Jacobshagen, C, Belardinelli, L, Hasenfuss, G, et al. Ranolazine for the treatment of heart failure with preserved ejection fraction: background, aims, and design of the RALI-DHF study. Clin Cardiol. 2011; 34:426–432.
50. Scirica, BM, Morrow, DA, Hod, H, et al. Effect of ranolazine, an antianginal agent with novel electrophysiological properties, on the incidence of arrhythmias in patients with non ST-segment elevation acute coronary syndrome: results from the Metabolic Efficiency With Ranolazine for Less Ischemia in Non ST-Elevation Acute Coronary Syndrome Thrombolysis in Myocardial Infarction 36 (MERLIN-TIMI 36) randomized controlled trial. Circulation. 2007; 116:1647–1652.
51. Song, Y, Shryock, JC, Wu, L, et al. Antagonism by ranolazine of the pro-arrhythmic effects of increasing late INa in guinea pig ventricular myocytes. J Cardiovasc Pharmacol. 2004; 44:192–199.
52. El-Sherif, N, Scherlag, BJ, Lazzara, R, et al. Re-entrant ventricular arrhythmias in the late myocardial infarction period. 1. Conduction characteristics in the infarction zone. Circulation. 1977; 55:686–702.
53. Baba, S, Dun, W, Cabo, C, et al. Remodeling in cells from different regions of the reentrant circuit during ventricular tachycardia. Circulation. 2005; 112:2386–2396.
54. Pu, J, Boyden, PA. Alterations of Na+ currents in myocytes from epicardial border zone of the infarcted heart. A possible ionic mechanism for reduced excitability and postrepolarization refractoriness. Circ Res. 1997; 81:110–119.
55. Lue, WM, Boyden, PA. Abnormal electrical properties of myocytes from chronically infarcted canine heart. Alterations in Vmax and the transient outward current. Circulation. 1992; 85:1175–1188.
56. Cabo, C, Boyden, P. Electrical remodeling of the epicardial border zone in the canine infarcted heart: a computational analysis. Am J Physiol Heart Circ Physiol. 2003; 284:H372–H384.
57. Christensen, MD, Dun, W, Boyden, PA, et al. Oxidized calmodulin kinase II regulates conduction following myocardial infarction: A computational analysis. PLoS Comput Biol. 2009; 5:e1000583.
58. Decker, KF, Heijman, J, Silva, JR, et al. Properties and ionic mechanisms of action potential adaptation, restitution and accommodation in canine epicardium. Am J Physiol Heart Circ Physiol. 2009; 296:H1017–1026.
59. Starmer, CF, Colatsky, TJ, Grant, AO. What happens when cardiac Na channels lose their function? 1—numerical studies of the vulnerable period in tissue expressing mutant channels. Cardiovasc Res. 2003; 57:82–91.
60. Fukuda, K, Davies, SS, Nakajima, T, et al. Oxidative mediated lipid peroxidation recapitulates proarrhythmic effects on cardiac sodium channels. Circ Res. 2005; 97:1262–1269.
61. Lau, DH, Clausen, C, Sosunov, EA, et al. Epicardial border zone overexpression of skeletal muscle sodium channel SkM1 normalizes activation, preserves conduction, and suppresses ventricular arrhythmia: an in silico, in vivo, in vitro study. Circulation. 2009; 119:19–27.
62. Mohler, PJ, Schott, JJ, Gramolini, AO, et al. Ankyrin-B mutation causes type 4 long-QT cardiac arrhythmia and sudden cardiac death. Nature. 2003; 421:634–639.
63. Chen, L, Marquardt, ML, Tester, DJ, et al. Mutation of an A-kinase-anchoring protein causes long-QT syndrome. Proc Natl Acad Sci U S A. 2007; 104:20990–20995.
64. London, B, Michalec, M, Mehdi, H, et al. Mutation in glycerol-3-phosphate dehydrogenase 1 like gene (GPD1-L) decreases cardiac Na+ current and causes inherited arrhythmias. Circulation. 2007; 116:2260–2268.
65. Vatta, M, Ackerman, MJ, Ye, B, et al. Mutant caveolin-3 induces persistent late sodium current and is associated with long-QT syndrome. Circulation. 2006; 114:2104–2112.
66. Cronk, LB, Ye, B, Kaku, T, et al. Novel mechanism for sudden infant death syndrome: persistent late sodium current secondary to mutations in caveolin-3. Heart Rhythm. 2007; 4:161–166.
67. Medeiros-Domingo, A, Kaku, T, Tester, DJ, et al. SCN4B-encoded sodium channel beta4 subunit in congenital long-QT syndrome. Circulation. 2007; 116:134–142.
68. Abbott, GW, Sesti, F, Splawski, I, et al. MiRP1 forms IKr potassium channels with HERG and is associated with cardiac arrhythmia. Cell. 1999; 97:175–187.
69. Splawski, I, Tristani-Firouzi, M, Lehmann, M, et al. Mutations in the hminK gene cause long QT syndrome and suppress IKs function. Nat Genet. 1997; 17:338–340.
70. Lin, X, Liu, N, Lu, J, et al. Subcellular heterogeneity of sodium current properties in adult cardiac ventricular myocytes. Heart Rhythm. 2011; 8:1923–1930.
71. Kucera, JP, Rohr, S, Rudy, Y. Localization of sodium channels in intercalated disks modulates cardiac conduction. Circ Res. 2002; 91:1176–1182.
72. Mori, Y, Fishman, GI, Peskin, CS. Ephaptic conduction in a cardiac strand model with 3D electrodiffusion. Proc Natl Acad Sci U S A. 2008; 105:6463–6468.
73. Meadows, LS, Isom, LL. Sodium channels as macromolecular complexes: implications for inherited arrhythmia syndromes. Cardiovasc Res. 2005; 67:448–458.
74. Watanabe, H, Darbar, D, Kaiser, DW, et al. Mutations in sodium channel beta1- and beta2-subunits associated with atrial fibrillation. Circ Arrhythm Electrophysiol. 2009; 2:268–275.
75. Valdivia, CR, Medeiros-Domingo, A, Ye, B, et al. Loss-of-function mutation of the SCN3B-encoded sodium channel β3 subunit associated with a case of idiopathic ventricular fibrillation. Cardiovasc Res. 2010; 86:392–400.
76. Watanabe, H, Koopmann, TT, Le Scouarnec, S, et al. Sodium channel beta1 subunit mutations associated with Brugada syndrome and cardiac conduction disease in humans. J Clin Invest. 2008; 118:2260–2268.
77. Bhasin, N, Cunha, SR, Mduannayake, M, et al. Molecular basis for PP2A regulatory subunit B56 alpha targeting in cardiomyocytes. Am J Physiol Heart Circ Physiol. 2007; 293:H109–H119.
78. Hashemi, SM, Hund, TJ, Mohler, PJ. Cardiac ankyrins in health and disease. J Mol Cell Cardiol. 2009; 47:203–209.
79. Cunha, SR, Bhasin, N, Mohler, PJ. Targeting and stability of Na/Ca exchanger 1 in cardiomyocytes requires direct interaction with the membrane adaptor ankyrin-B. J Biol Chem. 2007; 282:4875–4883.
80. Mohler, PJ, Davis, JQ, Bennett, V. Ankyrin-B coordinates the Na/K ATPase, Na/Ca exchanger, and InsP3 receptor in a cardiac T-tubule/SR microdomain. PLoS Biol. 2005; 3:e423.
81. Garrido, JJ, Giraud, P, Carlier, E, et al. A targeting motif involved in sodium channel clustering at the axonal initial segment. Science. 2003; 300:2091–2094.
82. Hund, TJ, Koval, OM, Li, J, et al. A betaIV spectrin/CaMKII signaling complex is essential for membrane excitability in mice. J Clin Invest. 2010; 120:3508–3519.
83. Sato, PY, Coombs, W, Lin, X, et al. Interactions between ankyrin-g, plakophilin-2, and connexin43 at the cardiac intercalated disc. Circ Res. 2011; 109:193–201.
84. Hund, TJ, Wright, PJ, Dun, W, et al. Regulation of the ankyrin-B-based targeting pathway following myocardial infarction. Cardiovasc Res. 2009; 81:742–749.
85. Le Scouarnec, S, Bhasin, N, Vieyres, C, et al. Dysfunction in ankyrin-B-dependent ion channel and transporter targeting causes human sinus node disease. Proc Natl Acad Sci U S A. 2008; 105:15617–15622.
86. Cunha, SR, Hund, TJ, Hashemi, S, et al. Defects in ankyrin-based membrane protein targeting pathways underlie atrial fibrillation. Circulation. 2011; 124:1212–1222.
87. Petitprez, S, Zmoos, AF, Ogrodnik, J, et al. SAP97 and dystrophin macromolecular complexes determine two pools of cardiac sodium channels Nav1. 5 in cardiomyocytes. Circ Res. 2011; 108:294–304.
88. Gavillet, B, Rougier, JS, Domenighetti, AA, et al. Cardiac sodium channel Nav1. 5 is regulated by a multiprotein complex composed of syntrophins and dystrophin. Circ Res. 2006; 99:407–414.
89. Gee, SH, Madhavan, R, Levinson, SR, et al. Interaction of muscle and brain sodium channels with multiple members of the syntrophin family of dystrophin-associated proteins. J Neurosci. 1998; 18:128–137.
90. Ueda, K, Valdivia, C, Medeiros-Domingo, A, et al. Syntrophin mutation associated with long QT syndrome through activation of the nNOS-SCN5A macromolecular complex. Proc Natl Acad Sci U S A. 2008; 105:9355–9360.
91. Wu, G, Ai, T, Kim, JJ, et al. alpha-1-syntrophin mutation and the long-QT syndrome: a disease of sodium channel disruption. Circ Arrhythm Electrophysiol. 2008; 1:193–201.
92. Cheng, J, Van Norstrand, DW, Medeiros-Domingo, A, et al. Alpha1-syntrophin mutations identified in sudden infant death syndrome cause an increase in late cardiac sodium current. Circ Arrhythm Electrophysiol. 2009; 2:667–676.
93. Liu, N, Ruan, Y, Denegri, M, et al. Calmodulin kinase II inhibition prevents arrhythmias in RyR2(R4496C+/-) mice with catecholaminergic polymorphic ventricular tachycardia. J Mol Cell Cardiol. 2011; 50:214–222.
94. Ai, X, Curran, JW, Shannon, TR, et al. Ca2+/calmodulin-dependent protein kinase modulates cardiac ryanodine receptor phosphorylation and sarcoplasmic reticulum Ca2+ leak in heart failure. Circ Res. 2005; 97:1314–1322.
95. Bers, DM. Altered cardiac myocyte Ca regulation in heart failure. Physiology (Bethesda). 2006; 21:380–387.
96. Qu, Y, Rogers, JC, Tanada, TN, et al. Phosphorylation of S1505 in the cardiac Na+ channel inactivation gate is required for modulation by protein kinase C. J Gen Physiol. 1996; 108:375–379.
97. Qu, Y, Rogers, J, Tanada, T, et al. Modulation of cardiac Na+ channels expressed in a mammalian cell line and in ventricular myocytes by protein kinase C. Proc Natl Acad Sci U S A. 1994; 91:3289–3293.
98. Murray, KT, Hu, NN, Daw, JR, et al. Functional effects of protein kinase C activation on the human cardiac Na+ channel. Circ Res. 1997; 80:370–376.
99. Frohnwieser, B, Chen, LQ, Schreibmayer, W, et al. Modulation of the human cardiac sodium channel alpha-subunit by cAMP-dependent protein kinase and the responsible sequence domain. J Physiol. 1997; 498(Pt 2):309–318.
100. Zhou, J, Yi, J, Hu, N, et al. Activation of protein kinase A modulates trafficking of the human cardiac sodium channel in Xenopus oocytes. Circ Res. 2000; 87:33–38.
101. Lu, T, Lee, HC, Kabat, JA, et al. Modulation of rat cardiac sodium channel by the stimulatory G protein alpha subunit. J Physiol. 1999; 518(Pt 2):371–384.
102. Murphy, BJ, Rogers, J, Perdichizzi, AP, et al. cAMP-dependent phosphorylation of two sites in the alpha subunit of the cardiac sodium channel. J Biol Chem. 1996; 271:28837–28843.
103. Yoon, JY, Ho, WK, Kim, ST, et al. Constitutive CaMKII activity regulates Na+ channel in rat ventricular myocytes. J Mol Cell Cardiol. 2009; 47:475–484.
104. Wagner, S, Dybkova, N, Rasenack, EC, et al. Ca/calmodulin-dependent protein kinase II regulates cardiac Na channels. J Clin Invest. 2006; 116:3127–3138.
105. Aiba, T, Hesketh, GG, Liu, T, et al. Na+ channel regulation by Ca2+/calmodulin and Ca2+/calmodulin-dependent protein kinase II in guinea-pig ventricular myocytes. Cardiovasc Res. 2010; 85:454–463.
106. Ashpole, NM, Herren, AW, Ginsburg, KS, et al. Ca2+/calmodulin-dependent protein kinase II (CaMKII) regulates cardiac sodium channel NaV1. 5 gating by multiple phosphorylation sites. J Biol Chem. 2012; 287:19856–19869.
107. Ufret-Vincenty, CA, Baro, DJ, Lederer, WJ, et al. Role of sodium channel deglycosylation in the genesis of cardiac arrhythmias in heart failure. J Biol Chem. 2001; 276:28197–28203.
108. Schreibmayer, W, Frohnwieser, B, Dascal, N, et al. Beta-adrenergic modulation of currents produced by rat cardiac Na+ channels expressed in Xenopus laevis oocytes. Recept Channels. 1994; 2:339–350.
109. Yarbrough, TL, Lu, T, Lee, HC, et al. Localization of cardiac sodium channels in caveolin-rich membrane domains: regulation of sodium current amplitude. Circ Res. 2002; 90:443–449.
110. Palygin, OA, Pettus, JM, Shibata, EF. Regulation of caveolar cardiac sodium current by a single Gsalpha histidine residue. Am J Physiol Heart Circ Physiol. 2008; 294:H1693–H1699.
111. Besse, IM, Mitchell, CC, Hund, TJ, et al. A computational investigation of cardiac caveolae as a source of persistent sodium current. Front Physiol. 2011; 2:87.
112. Deschenes, I, Neyroud, N, DiSilvestre, D, et al. Isoform-specific modulation of voltage-gated Na+ channels by calmodulin. Circ Res. 2002; 90:E49–57.
113. Tan, HL, Kupershmidt, S, Zhang, R, et al. A calcium sensor in the sodium channel modulates cardiac excitability. Nature. 2002; 415:442–447.
114. Potet, F, Chagot, B, Anghelescu, M, et al. Functional interactions between distinct sodium channel cytoplasmic domains through the action of calmodulin. J Biol Chem. 2009; 284:8846–8854.
115. Shah, VN, Wingo, TL, Weiss, KL, et al. Calcium-dependent regulation of the voltage-gated sodium channel hH1: intrinsic and extrinsic sensors use a common molecular switch. Proc Natl Acad Sci U S A. 2006; 103:3592–3597.
116. Kim, J, Ghosh, S, Liu, H, et al. Calmodulin mediates Ca2+ sensitivity of sodium channels. J Biol Chem. 2004; 279:45004–45012.
117. Young, KA, Caldwell, JH. Modulation of skeletal and cardiac voltage-gated sodium channels by calmodulin. J Physiol. 2005; 565:349–370.
118. Sarhan, MF, Van Petegem, F, Ahern, CA. A double tyrosine motif in the cardiac sodium channel domain III-IV linker couples calcium-dependent calmodulin binding to inactivation gating. J Biol Chem. 2009; 284:33265–33274.
119. Wang, C, Chung, BC, Yan, H, et al. Crystal structure of the ternary complex of a NaV C-terminal domain, a fibroblast growth factor homologous factor, and calmodulin. Structure. 2012; 20:1167–1176.
[/level-membership-for-cardiovascular-category][not-level-membership-for-cardiovascular-category]9
Biophysics of Normal and Abnormal Cardiac Sodium Channel Function
Abstract
Voltage-gated sodium channels (Nav) underlie the activity of many excitable cells. In the heart, Nav channels are responsible for the rapid cardiomyocyte action potential upstroke that promotes rapid conduction of the electrical impulse leading to coordinated mechanical contraction. Central to this function, Nav channels activate (and then inactivate) rapidly in response to a small depolarization of the membrane, resulting in a large influx of Na+ ions and further membrane depolarization. Dysfunction in Nav channel activity results in human diseases and disorders, including epilepsy, ataxia, cardiac arrhythmia, and myotonia.1–3 Variants in SCN5A, the gene encoding the primary cardiac Nav α-subunit Nav1.5, have been linked to human arrhythmia syndromes including long QT type 3 (LQT3), Brugada syndrome, cardiac conduction disease, sinus node disease, and atrial fibrillation.2,4,5 A detailed discussion of fundamental aspects of Nav structure function, gating, and pharmacology can be found in Chapter 1. Here we discuss current understanding regarding regulation of Nav biophysical activity and cellular function in health and disease.
The Nav channel pore-forming α-subunit in vertebrates is a single polypeptide with four homologous transmembrane domains (DI-DIV) comprised of six membrane-spanning α-helices (S1-S6; Figure 9-1). Nav channels share structural similarities with voltage-gated Ca2+ channels from which they may have evolved.6 Ten different Nav channel α-subunits are present in mammals, each with specific biophysical, expression, and regulatory signatures. The primary cardiac isoform, Nav1.5, is derived from 28 exons and undergoes alternative splicing in mammalian heart (including human).4,7–11 Although Nav1.5 is the primary Nav channel expressed in the heart, limited expression of neuronal α-subunits has also been reported and likely introduces important heterogeneity in Nav biophysical activity, localization, and drug sensitivity within the cell.12,13 Although the α-subunit is capable of forming a functional channel by itself, several auxiliary β-subunits have been identified (β1-β4) that modulate channel gating and/or trafficking. Human mutations in both the α and β-subunits have been linked to arrhythmia.14,15 Importantly, Nav1.5 and associated β-subunits reside with large macromolecular complexes comprised of adaptor, accessory, cytoskeletal, and regulatory proteins (Figure 9-2).4,5,16–22 An exciting area of research going forward, and the focus of this and other chapters in this volume (Chapters 18 and 21), is the physiologic function of Nav macromolecular complexes and their role in arrhythmias and mechanical dysfunction in the setting of human disease.
Figure 9-1 Topology of cardiac Nav1.5 channel (SCN5A) and human variants linked to congenital cardiac arrhythmia. Charged S4 segments (orange) in each domain function as voltage sensors. P segments between S5 and S6 form the channel pore. IFM motif in DIII-DIV linker (light blue circle) acts as inactivation particle to facilitate rapid voltage-dependent inactivation. The location of several human variants in SCN5A linked to LQT3 (red square), Brugada syndrome (blue square), or overlap disorder (green square) are indicated.
Figure 9-2 Nav1.5 resides in macromolecular complexes comprised of β-subunits (red) and cytoskeletal, adapter, trafficking, and regulatory molecules. Known interacting proteins are shown along with binding regions. Phosphorylation sites identified to date are indicated for CaMKII (red), PKA (blue), and protein kinase C (green).
Nav Channel Activity in Health and Disease
Over the course of the action potential, Na+ channels undergo a remarkable series of conformational changes that are essential for the rapid and controlled influx of Na+ ions that supports the action potential upstroke (detailed in Chapter 1).23 Recent work to crystallize the bacterial Na+ channel NavAb (forms as a tetramer of identical subunits akin to most K+ channels) has added to decades of important research on the complex Nav structure–function relationship governing channel activity.24,25 Briefly, in response to membrane depolarization, channels first activate rapidly as the four charged S4 segments (see Figure 9-1) move outward and open the channel pore.26 Channel activation is followed almost immediately by rapid inactivation caused by further outward movement of the S4 sensors in DIII and DIV and blocking of the channel pore by the DIII-DIV linker that likely depends on the interaction between hydrophobic residues (isoleucine-phenylalanine-methionine, IFM) in the III-IV linker and multiple sites in the linkers between S4 and S5 in DIII and DIV and the cytoplasmic end of S6 in DIV.27–29 Rapid inactivation is followed by transition into several different slow inactivation states that are controlled, in part, by P segments linking S5 and S6 segments that form the inner channel pore (see Figure 9-1). Importantly, disruption of Na+ channel gating at any step during this highly coordinated set of movements may result in inappropriate (elevated, reduced) current and give rise to arrhythmias.
Dysfunction in Nav1.5 activity has been identified as a potential cause of arrhythmia in several forms of congenital and acquired disease (see Figure 9-1). Classic examples of the link between inherited defects in Nav1.5 function and disease may be found in two inherited arrhythmia syndromes closely linked to mutations in SCN5A: Brugada syndrome and LQT3. An overview of molecular basis for these diseases is provided here; both are addressed in detail in later chapters. Brugada syndrome is an autosomal, dominant cardiac arrhythmia syndrome characterized by ST-segment elevation in the right precordial leads (V1-V3) and sudden death in the absence of overt structural disease.30,31 The syndrome is caused by loss-of-function variants in SCN5A (although identified in only about 20% of Brugada cases31) that result in loss of Na+ current early in the action potential because of a loss of functional channel expression/localization or defects in channel gating.20,32,33 The substrate for Brugada syndrome arrhythmia is thought to arise in the epicardium of the right ventricle where high expression of transient outward K+ current Ito may shift the balance of current to favor exaggerated phase 1 repolarization (notch) and even loss of the action potential plateau.34,35 Loss-of-function SCN5A variants have also been associated with isolated cardiac conduction disease characterized by slow conduction throughout the heart (even requiring pacemaker implantation) without the repolarization abnormalities or ventricular tachyarrhythmia observed in Brugada syndrome.36 In general, gene variants that give rise to cardiac conduction disease produce a less severe effect on channel function than those associated with Brugada syndrome, by introducing a secondary gain-of-function change in Nav activity (e.g., depolarizing shift in voltage dependence of inactivation) that partially balances the primary loss-of-function change (e.g., depolarizing shift in voltage dependence of activation).
Although Brugada syndrome is a disease associated with Nav loss-of-function, LQT3 represents the other extreme and illustrates how Nav gain-of-function may also produce arrhythmia. A heterogeneous autosomal dominant genetic disease, LQT3 is associated with abnormal QT-interval prolongation on the electrocardiogram, syncope, polymorphic ventricular tachycardia, and sudden death.14 Variants in SCN5A are the cause of LQT3, which is associated with increased risk of cardiac events during rest and decreased efficacy of β-blocker therapy.14,37,38 At least 50 variants in SCN5A have been identified as causal for LQT3, and although these variants are scattered throughout the cytoplasmic face of the channel (with a cluster in the S4 segments), they primarily produce an increase in Na+-channel current (gain-of-Nav function) by disrupting rapid inactivation, by shifting the voltage-dependence of activation or inactivation, or by altering recovery from inactivation. A prototypical case study of the causal link between SCN5A mutations and LQT3 comes from the first reported LQT3 mutation resulting in three–amino acid deletion in the DIII-DIV linker that is important for rapid channel inactivation (ΔKPQ, see Figure 9-1).38 The variant ΔKPQ allele disrupts rapid inactivation, producing a small (~0.5% of peak) but sustained Na+ current during the action potential plateau that shifts the balance of current to prolong action potential and increase the likelihood of arrhythmogenic afterdepolarizations.38,39 Although LQT3 and Brugada syndrome are often presented as two very different faces of Nav dysfunction, overlap syndromes have been reported, with features of both diseases showing QT prolongation at slow rates coupled with ST elevation during exercise. Notably, the 1795insD mutation (see Figure 9-1) interrupts rapid inactivation to enhance persistent Na+ current and prolong action potential duration, especially at slow pacing rates, but it also enhances an intermediate inactivation state that compromises Nav recovery between stimuli, leading to loss of current at fast rates.40,41 Thus, as discussed before, for cardiac conduction disease, a single molecular defect may alter channel activity at more than one phase (e.g., rapid and slow inactivation processes) in the gating cycle, giving rise to a complex disease phenotype.
Studies of rare congenital conditions such as LQT3 and Brugada syndrome have generated important insight into Nav channel function and have increased our understanding of arrhythmogenesis in the more complex setting of acquired heart disease, where Nav channel dysfunction is a common finding. In heart failure, for example, an increase in persistent (late) Na+ current has been observed both in patients and animal models of human disease.42–46 Similar to LQT3, in heart failure it is likely that increased late Na+ current delays action potential repolarization and promotes pro-arrhythmogenic afterdepolarizations.45 Increased Na+ entry via the late current may also disrupt normal Ca2+ homeostasis and promote mechanical dysfunction and progression of disease.47 The underlying mechanism for changes in Nav function in heart failure is unknown but is most likely not a result of changes in expression of Nav α or β-subunits.42,45 In light of known defects in β-adrenergic and calcium-dependent signaling pathways in heart failure, it is likely that defects in channel posttranslational modification play a role in Nav dysfunction. Regardless of Na+ current mechanism, mounting studies support the late Na+ current as a viable therapeutic target (e.g., inhibition with ranolazine) for preventing arrhythmias and progression of disease in heart failure.48–51
There have also been many studies on the link between defects in Nav function and arrhythmias after myocardial infarction, where reentrant arrhythmias are highly localized to the highly remodeled tissue surrounding the infarct (border zone). In particular, in the canine heart, dramatic electrical and structural remodeling has been identified coupled with anisotropic conduction and reentrant arrhythmias in the border zone region. Conduction through this region is highly irregular, characterized by slow and discontinuous conduction.52 Myocytes isolated from the border zone regions display a characteristic time course of changes in ion channel activity and action potential, including significant decrease in peak Na+ current, altered subcellular localization, decreased channel availability, and slow recovery from inactivation.53–55 Computational studies demonstrate that measured defects in Nav channel activity prolong refractoriness, decrease conduction velocity, and increase susceptibility to initiation of reentrant arrhythmias.53,56–59 Although the precise mechanism underlying abnormal Nav function after myocardial infarction is likely multifactorial and not fully understood, roles for increased oxidative stress and/or increased CaMKII-dependent channel phosphorylation have been identified.57,60 Interestingly, adenoviral overexpression of skeletal muscle Nav channel, with inactivation shifted to more depolarized potentials compared with cardiac isoform, has been shown to improve conduction and suppress arrhythmias in the canine after myocardial infarction.61 It is intriguing to consider the possibility that targeting channel regulatory pathways (e.g., CaMKII) may be a second, and conceivably more practical, method to alter channel availability for therapeutic benefit.
NaV Channel Trafficking and Stabilization in Health and Disease
As outlined before, Nav function is critical for normal heart function, and alteration of Nav activity is a hallmark of many forms of inherited and acquired diseases. Despite major advances, the mechanistic link among specific molecular defects, Nav channel dysfunction, and arrhythmias associated with many human arrhythmia variants and in common disease remains elusive. Mounting evidence, in particular from human arrhythmia variants in genes encoding ion channel accessory proteins (e.g., adapter and scaffolding proteins, channel subunits, chaperones), highlight the importance of proper Nav channel localization within specific cellular domains for normal heart function.62–69
Voltage-gated Nav channels are targeted to specific membrane domains in cardiomyocytes (Figure 9-3). Residing primarily at cell ends in the region of the intercalated disc (ID), Nav channels are also found at secondary sites at lateral and transverse-tubule membranes (see Chapter 18). Although the cardiac isoform Nav1.5 largely comprises the Nav population at the ID, neuronal isoforms (e.g., Nav1.1, Nav1.3, Nav1.6) are found almost exclusively at transverse tubules.12,13,70 Although the functional consequences for differential targeting of Nav isoforms within the cardiomyocyte remain unclear, computational studies demonstrate that concentration of Nav channels at the ID (Nav1.5), where cells are electrically and mechanically coupled, supports electrical impulse propagation.71,72 Likewise, a role for the neuronal isoforms (largely outside the ID) has been explored using low doses of tetrodotoxin (TTX) (insufficient to block TTX-resistant Nav1.5). These experiments show that although neuronal channels are not necessary for conduction under normal conditions, they may play modulatory roles in excitation-contraction coupling and ventricular function.12,13
Figure 9-3 Nav channels are differentially localized within cardiomyocyte. A, Three-dimensional rendering of isolated murine ventricular cardiomyocyte stained with markers for the nucleus (yellow), intercalated disc (green), and z-line (blue). B, Schematic indicating Nav localization and interacting partners at specific membrane domains within the cardiomyocyte.
An important outstanding question is how are Nav channels differentially targeted and regulated within the cardiomyocyte? The answer may be found in the growing family of adapter, accessory, cytoskeletal and regulatory proteins—including α1-syntrophin, ankyrin-G, βIV-spectrin, glycerol-3-phosphate dehydrogenase 1–like protein, MOG1, Nedd4-2, SAP97, and calmodulin—that reside in macromolecular complexes with Nav channels (see Figure 9-2).4,5,16–22
[/not-level-membership-for-cardiovascular-category]
