[level-membership-for-internal-medicine-category]
462e |
Muscular Dystrophies and Other Muscle Diseases |
Skeletal muscle diseases, or myopathies, are disorders with structural changes or functional impairment of muscle. These conditions can be differentiated from other diseases of the motor unit (e.g., lower motor neuron or neuromuscular junction pathologies) by characteristic clinical and laboratory findings.
Myasthenia gravis and related disorders are discussed in Chap. 461; dermatomyositis, polymyositis, and inclusion body myositis are discussed in Chap. 388.
CLINICAL FEATURES
Most myopathies present with proximal, symmetric limb weakness (arms or legs) with preserved reflexes and sensation. However, asymmetric and predominantly distal weakness can be seen in some myopathies. An associated sensory loss suggests injury to a peripheral nerve or the central nervous system (CNS) rather than myopathy. On occasion, disorders affecting the motor nerve cell bodies in the spinal cord (anterior horn cell disease), the neuromuscular junction, or peripheral nerves can mimic findings of myopathy.
Muscle Weakness Symptoms of muscle weakness can be either intermittent or persistent. Disorders causing intermittent weakness (Fig. 462e-1) include myasthenia gravis, periodic paralyses (hypokalemic, hyperkalemic, and paramyotonia congenita), and metabolic energy deficiencies of glycolysis (especially myophosphorylase deficiency), fatty acid utilization (carnitine palmitoyltransferase deficiency), and some mitochondrial myopathies. The states of energy deficiency cause activity-related muscle breakdown accompanied by myoglobinuria, appearing as light-brown- to dark-brown-colored urine.
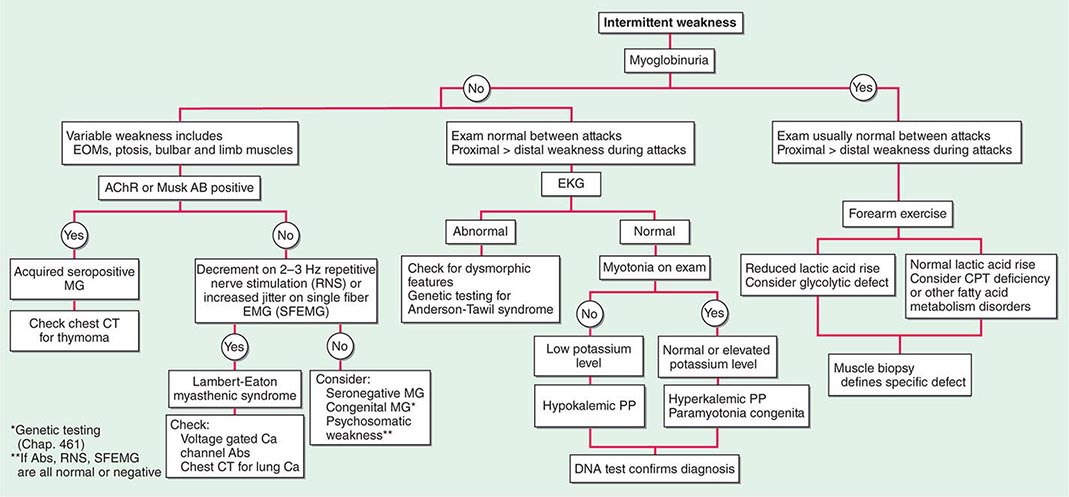
FIGURE 462e-1 Diagnostic evaluation of intermittent weakness. AChR AB, acetylcholine receptor antibody; CPT, carnitine palmitoyltransferase; EOMs, extraocular muscles; MG, myasthenia gravis; PP, periodic paralysis.
Most muscle disorders cause persistent weakness (Fig. 462e-2). In the majority of these, including most types of muscular dystrophy, polymyositis, and dermatomyositis, the proximal muscles are weaker than the distal and are symmetrically affected, and the facial muscles are spared, a pattern referred to as limb-girdle. The differential diagnosis is more restricted for other patterns of weakness. Facial weakness (difficulty with eye closure and impaired smile) and scapular winging (Fig. 462e-3) are characteristic of facioscapulohumeral dystrophy (FSHD). Facial and distal limb weakness associated with hand grip myotonia is virtually diagnostic of myotonic dystrophy type 1. When other cranial nerve muscles are weak, causing ptosis or extraocular muscle weakness, the most important disorders to consider include neuromuscular junction disorders, oculopharyngeal muscular dystrophy, mitochondrial myopathies, or some of the congenital myopathies (Table 462e-1). A pathognomonic pattern characteristic of inclusion body myositis is atrophy and weakness of the flexor forearm (e.g., wrist and finger flexors) and quadriceps muscles that is often asymmetric. Less frequently, but important diagnostically, is the presence of a dropped head syndrome indicative of selective neck extensor muscle weakness. The most important neuromuscular diseases associated with this pattern of weakness include myasthenia gravis, amyotrophic lateral sclerosis, late-onset nemaline myopathy, hyperparathyroidism, focal myositis, and some forms of inclusion body myopathy. A final pattern, recognized because of preferential distal extremity weakness, is typical of a unique category of muscular dystrophy, the distal myopathies.
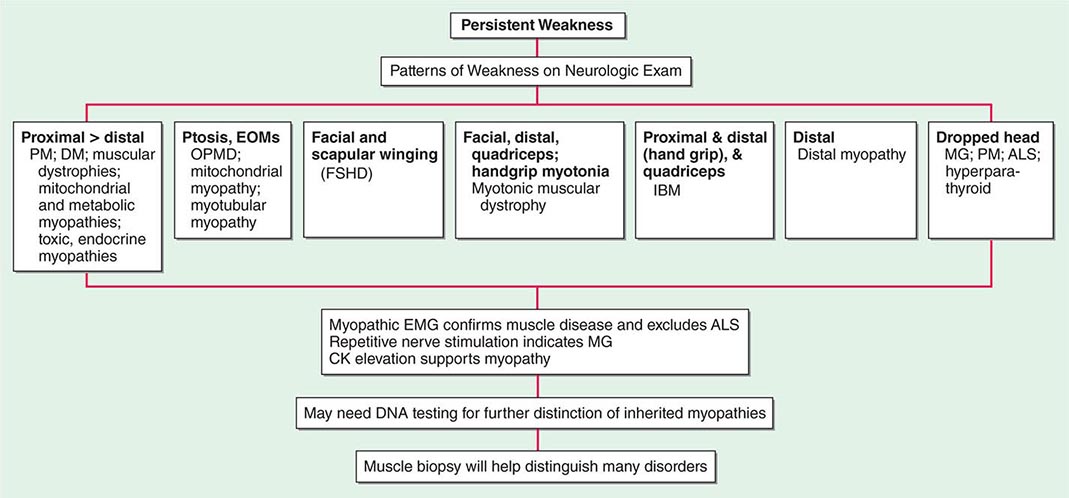
FIGURE 462e-2 Diagnostic evaluation of persistent weakness. Examination reveals one of seven patterns of weakness. The pattern of weakness in combination with the laboratory evaluation leads to a diagnosis. ALS, amyotrophic lateral sclerosis; CK, creatine kinase; DM, dermatomyositis; EMG, electromyography; EOMs, extraocular muscles; FSHD, facioscapulohumeral dystrophy; IBM, inclusion body myositis; MG, myasthenia gravis; OPMD, oculopharyngeal muscular dystrophy; PM, polymyositis.
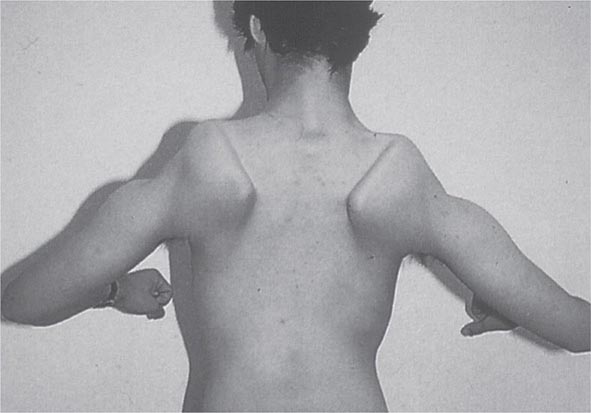
FIGURE 462e-3 Facioscapulohumeral dystrophy with prominent scapular winging.
|
NEUROMUSCULAR CAUSES OF PTOSIS OR OPHTHALMOPLEGIA |
It is important to examine functional capabilities to help disclose certain patterns of weakness (Table 462e-2). The Gowers’ sign (Fig. 462e-4) is particularly useful. Observing the gait of an individual may disclose a lordotic posture caused by combined trunk and hip weakness, frequently exaggerated by toe walking (Fig. 462e-5). A waddling gait is caused by the inability of weak hip muscles to prevent hip drop or hip dip. Hyperextension of the knee (genu recurvatum or back-kneeing) is characteristic of quadriceps muscle weakness; and a steppage gait, due to footdrop, accompanies distal weakness.
|
OBSERVATIONS ON EXAMINATION THAT DISCLOSE MUSCLE WEAKNESS |
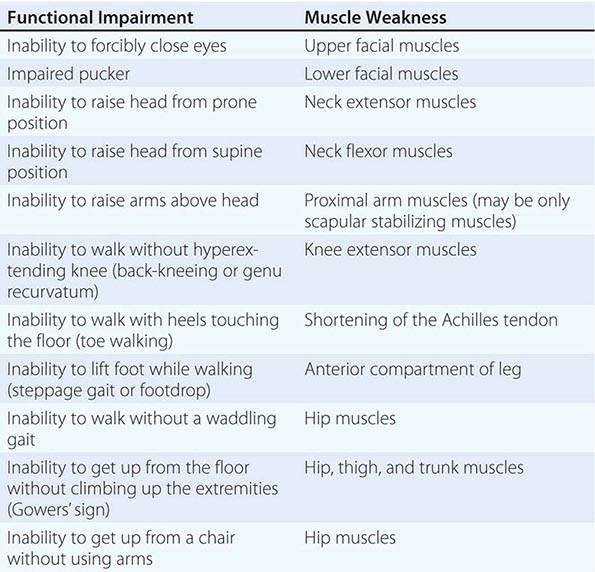
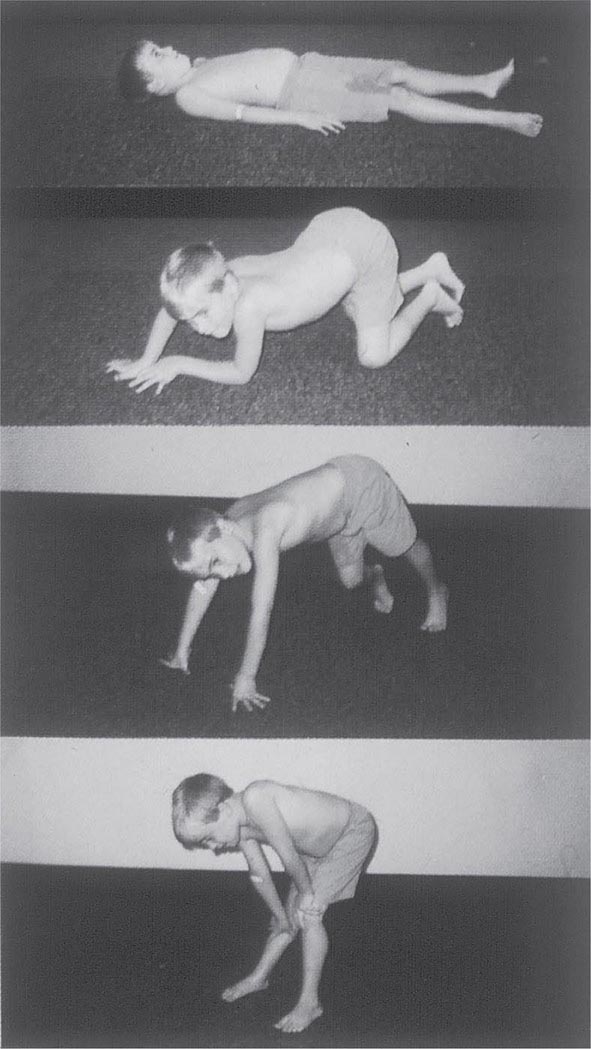
FIGURE 462e-4 Gowers’ sign showing a patient using his arms to climb up the legs in attempting to get up from the floor.
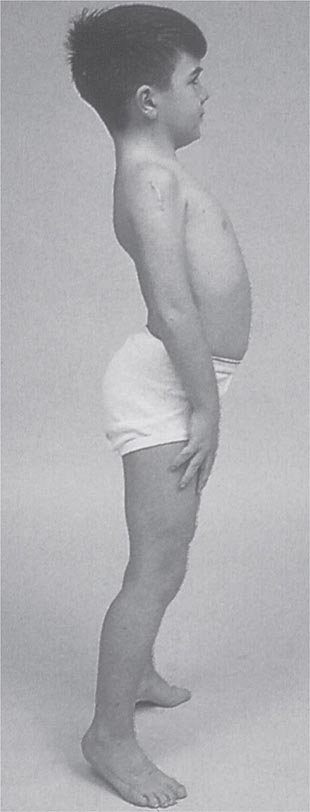
FIGURE 462e-5 Lordotic posture, exaggerated by standing on toes, associated with trunk and hip weakness.
Any disorder causing muscle weakness may be accompanied by fatigue, referring to an inability to maintain or sustain a force (pathologic fatigability). This condition must be differentiated from asthenia, a type of fatigue caused by excess tiredness or lack of energy. Associated symptoms may help differentiate asthenia and pathologic fatigability. Asthenia is often accompanied by a tendency to avoid physical activities, complaints of daytime sleepiness, necessity for frequent naps, and difficulty concentrating on activities such as reading. There may be feelings of overwhelming stress and depression. Thus, asthenia is not a myopathy. In contrast, pathologic fatigability occurs in disorders of neuromuscular transmission and in disorders altering energy production, including defects in glycolysis, lipid metabolism, or mitochondrial energy production. Pathologic fatigability also occurs in chronic myopathies because of difficulty accomplishing a task with less muscle. Pathologic fatigability is accompanied by abnormal clinical or laboratory findings. Fatigue without those supportive features almost never indicates a primary muscle disease.
Muscle Pain (Myalgias), Cramps, and Stiffness Muscle pain can be associated with cramps, spasms, contractures, and stiff or rigid muscles. In distinction, true myalgia (muscle aching), which can be localized or generalized, may be accompanied by weakness, tenderness to palpation, or swelling. Certain drugs cause true myalgia (Table 462e-3).
|
DRUGS THAT CAUSE TRUE MYALGIA |
There are two painful muscle conditions of particular importance, neither of which is associated with muscle weakness. Fibromyalgia is a common, yet poorly understood, type of myofascial pain syndrome. Patients complain of severe muscle pain and tenderness and have specific painful trigger points, sleep disturbances, and easy fatigability. Serum creatine kinase (CK), erythrocyte sedimentation rate (ESR), electromyography (EMG), and muscle biopsy are normal (Chap. 396). Polymyalgia rheumatica occurs mainly in patients >50 years and is characterized by stiffness and pain in the shoulders, lower back, hips, and thighs (Chap. 385). The ESR is elevated, while serum CK, EMG, and muscle biopsy are normal. Temporal arteritis, an inflammatory disorder of medium- and large-sized arteries, usually involving one or more branches of the carotid artery, may accompany polymyalgia rheumatica. Vision is threatened by ischemic optic neuritis. Glucocorticoids can relieve the myalgias and protect against visual loss.
Localized muscle pain is most often traumatic. A common cause of sudden abrupt-onset pain is a ruptured tendon, which leaves the muscle belly appearing rounded and shorter in appearance compared to the normal side. The biceps brachii and Achilles tendons are particularly vulnerable to rupture. Infection or neoplastic infiltration of the muscle is a rare cause of localized muscle pain.
A muscle cramp or spasm is a painful, involuntary, localized, muscle contraction with a visible or palpable hardening of the muscle. Cramps are abrupt in onset, short in duration, and may cause abnormal posturing of the joint. The EMG shows firing of motor units, reflecting an origin from spontaneous neural discharge. Muscle cramps often occur in neurogenic disorders, especially motor neuron disease (Chap. 452), radiculopathies, and polyneuropathies (Chap. 459), but are not a feature of most primary muscle diseases. Duchenne muscular dystrophy is an exception because calf muscle complaints are a common complaint. Muscle cramps are also common during pregnancy.
A muscle contracture is different from a muscle cramp. In both conditions, the muscle becomes hard, but a contracture is associated with energy failure in glycolytic disorders. The muscle is unable to relax after an active muscle contraction. The EMG shows electrical silence. Confusion is created because contracture also refers to a muscle that cannot be passively stretched to its proper length (fixed contracture) because of fibrosis. In some muscle disorders, especially in Emery-Dreifuss muscular dystrophy and Bethlem myopathy, fixed contractures occur early and represent distinctive features of the disease.
Muscle stiffness can refer to different phenomena. Some patients with inflammation of joints and periarticular surfaces feel stiff. This condition is different from the disorders of hyperexcitable motor nerves causing stiff or rigid muscles. In stiff-person syndrome, spontaneous discharges of the motor neurons of the spinal cord cause involuntary muscle contractions mainly involving the axial (trunk) and proximal lower extremity muscles. The gait becomes stiff and labored, with hyperlordosis of the lumbar spine. Superimposed episodic muscle spasms are precipitated by sudden movements, unexpected noises, and emotional upset. The muscles relax during sleep. Serum antibodies against glutamic acid decarboxylase are present in approximately two-thirds of cases. In neuromyotonia (Isaacs’ syndrome), there is hyperexcitability of the peripheral nerves manifesting as continuous muscle fiber activity. Myokymia (groups of fasciculations associated with continuous undulations of muscle) and impaired muscle relaxation are the result. Muscles of the leg are stiff, and the constant contractions of the muscle cause increased sweating of the extremities. This peripheral nerve hyperexcitability is mediated by antibodies that target voltage-gated potassium channels. The site of origin of the spontaneous nerve discharges is principally in the distal portion of the motor nerves.
Myotonia is a condition of prolonged muscle contraction followed by slow muscle relaxation. It always follows muscle activation (action myotonia), usually voluntary, but may be elicited by mechanical stimulation (percussion myotonia) of the muscle. Myotonia typically causes difficulty in releasing objects after a firm grasp. In myotonic muscular dystrophy type 1 (DM1), distal weakness usually accompanies myotonia, whereas in DM2, proximal muscles are more affected; thus the related term proximal myotonic myopathy (PROMM) is used to describe this condition. Myotonia also occurs with myotonia congenita (a chloride channel disorder), but in this condition muscle weakness is not prominent. Myotonia may also be seen in individuals with sodium channel mutations (hyperkalemic periodic paralysis or potassium-sensitive myotonia). Another sodium channelopathy, paramyotonia congenita, also is associated with muscle stiffness. In contrast to other disorders associated with myotonia in which the myotonia is eased by repetitive activity, paramyotonia congenita is named for a paradoxical phenomenon whereby the myotonia worsens with repetitive activity.
Muscle Enlargement and Atrophy In most myopathies muscle tissue is replaced by fat and connective tissue, but the size of the muscle is usually not affected. However, in many limb-girdle muscular dystrophies (and particularly the dystrophinopathies), enlarged calf muscles are typical. The enlargement represents true muscle hypertrophy; thus the term pseudohypertrophy should be avoided when referring to these patients. The calf muscles remain very strong even late in the course of these disorders. Muscle enlargement can also result from infiltration by sarcoid granulomas, amyloid deposits, bacterial and parasitic infections, and focal myositis. In contrast, muscle atrophy is characteristic of other myopathies. In dysferlinopathies (LGMD2B) and anoctaminopathies (LGMD2L), there is a predilection for early atrophy of the gastrocnemius muscles, particularly the medial aspect. Atrophy of the humeral muscles is characteristic of FSHD.
LABORATORY EVALUATION
A limited battery of tests can be used to evaluate a suspected myopathy. Nearly all patients require serum enzyme level measurements and electrodiagnostic studies as screening tools to differentiate muscle disorders from other motor unit diseases. The other tests described—DNA studies, the forearm exercise test, and muscle biopsy—are used to diagnose specific types of myopathies.
Serum Enzymes CK is the preferred muscle enzyme to measure in the evaluation of myopathies. Damage to muscle causes the CK to leak from the muscle fiber to the serum. The MM isoenzyme predominates in skeletal muscle, whereas creatine kinase-myocardial bound (CK-MB) is the marker for cardiac muscle. Serum CK can be elevated in normal individuals without provocation, presumably on a genetic basis or after strenuous activity, minor trauma (including the EMG needle), a prolonged muscle cramp, or a generalized seizure. Aspartate aminotransferase (AST), alanine aminotransferase (ALT), aldolase, and lactic dehydrogenase (LDH) are enzymes sharing an origin in both muscle and liver. Problems arise when the levels of these enzymes are found to be elevated in a routine screening battery, leading to the erroneous assumption that liver disease is present when in fact muscle could be the cause. An elevated γ-glutamyl transferase (GGT) helps to establish a liver origin because this enzyme is not found in muscle.
Electrodiagnostic Studies EMG, repetitive nerve stimulation, and nerve conduction studies (Chap. 442e) are essential methods for evaluation of the patient with suspected muscle disease. In combination, they provide the information necessary to differentiate myopathies from neuropathies and neuromuscular junction diseases. Routine nerve conduction studies are typically normal in myopathies but reduced amplitudes of compound muscle action potentials may be seen in atrophied muscles. The needle EMG may reveal irritability on needle placement suggestive of a necrotizing myopathy (inflammatory myopathies, dystrophies, toxic myopathies, myotonic myopathies), whereas a lack of irritability is characteristic of long-standing myopathic disorders (muscular dystrophies, endocrine myopathies, disuse atrophy, and many of the metabolic myopathies). In addition, the EMG may demonstrate myotonic discharges that will narrow the differential diagnosis (Table 462e-4). Another important EMG finding is the presence of short-duration, small-amplitude, polyphasic motor unit action potentials (MUAPs). Such MUAPs can be seen in both myopathic and neuropathic disorders; however, the recruitment or firing pattern is different. In myopathies, the MUAPs fire early but at a normal rate to compensate for the loss of individual muscle fibers, whereas in neurogenic disorders the MUAPs fire faster. The EMG is usually normal in steroid or disuse myopathy, both of which are associated with type 2 fiber atrophy; this is because the EMG preferentially assesses the physiologic function of type 1 fibers. The EMG can also be invaluable in helping to choose an appropriately affected muscle to sample for biopsy.
|
MYOTONIC DISORDERS |
aAssociated with myotonic discharges on electromyography but no clinical myotonia.
DNA Analysis This serves as an important tool for the definitive diagnosis of many muscle disorders. Nevertheless, there are a number of limitations in currently available molecular diagnostics. For example, in Duchenne and Becker dystrophies, two-thirds of patients have deletion or duplication mutations in the dystrophin gene that are easy to detect, while the remainder have point mutations that are much more difficult to find. For patients without identifiable gene defects, the muscle biopsy remains the main diagnostic tool.
Forearm Exercise Test In myopathies with intermittent symptoms, and especially those associated with myoglobinuria, there may be a defect in glycolysis. Many variations of the forearm exercise test exist. For safety, the test should not be performed under ischemic conditions to avoid an unnecessary insult to the muscle, causing rhabdomyolysis. The test is performed by placing a small indwelling catheter into an antecubital vein. A baseline blood sample is obtained for lactic acid and ammonia. The forearm muscles are exercised by asking the patient to vigorously open and close the hand for 1 min. Blood is then obtained at intervals of 1, 2, 4, 6, and 10 min for comparison with the baseline sample. A three- to fourfold rise of lactic acid is typical. The simultaneous measurement of ammonia serves as a control, because it should also rise with exercise. In patients with myophosphorylase deficiency or other glycolytic defects, the lactic acid rise will be absent or below normal, while the rise in ammonia will reach control values. If there is lack of effort, neither lactic acid nor ammonia will rise. Patients with selective failure to increase ammonia may have myoadenylate deaminase deficiency. This condition has been reported to be a cause of myoglobinuria, but deficiency of this enzyme in asymptomatic individuals makes interpretation controversial.
Muscle Biopsy Muscle biopsy is an important step in establishing the diagnosis of a suspected myopathy. The biopsy is usually obtained from a quadriceps or biceps brachii muscle, less commonly from a deltoid muscle. Evaluation includes a combination of techniques—light microscopy, histochemistry, immunocytochemistry with a battery of antibodies, and electron microscopy. Not all techniques are needed for every case. A specific diagnosis can be established in many disorders. Endomysial inflammatory cells surrounding and invading muscle fibers are seen in polymyositis; similar endomysial infiltrates associated with muscle fibers containing rimmed vacuoles and amyloid deposits consisting of SMI-31-, p62-, and TDP-43-positive inclusions within fibers are characteristic of inclusion body myositis; and perivascular, perimysial inflammation associated with perifascicular atrophy is a feature of dermatomyositis. In addition, the congenital myopathies have distinctive light and electron microscopy features essential for diagnosis. Mitochondrial and metabolic (e.g., glycogen and lipid storage diseases) myopathies also demonstrate distinctive histochemical and electron-microscopic profiles. Biopsied muscle tissue can be sent for metabolic enzyme or mitochondrial DNA analyses. A battery of antibodies is available for the identification of abnormal proteins to help diagnose specific types of muscular dystrophies. Western blot analysis on muscle specimens can be performed to determine whether specific muscle proteins are reduced in quantity or are of abnormal size.
HEREDITARY MYOPATHIES
Muscular dystrophy refers to a group of hereditary progressive diseases each with unique phenotypic and genetic features (Tables 462e-5, 462e-6, and 462e-7).
|
PROGRESSIVE MUSCULAR DYSTROPHIES |
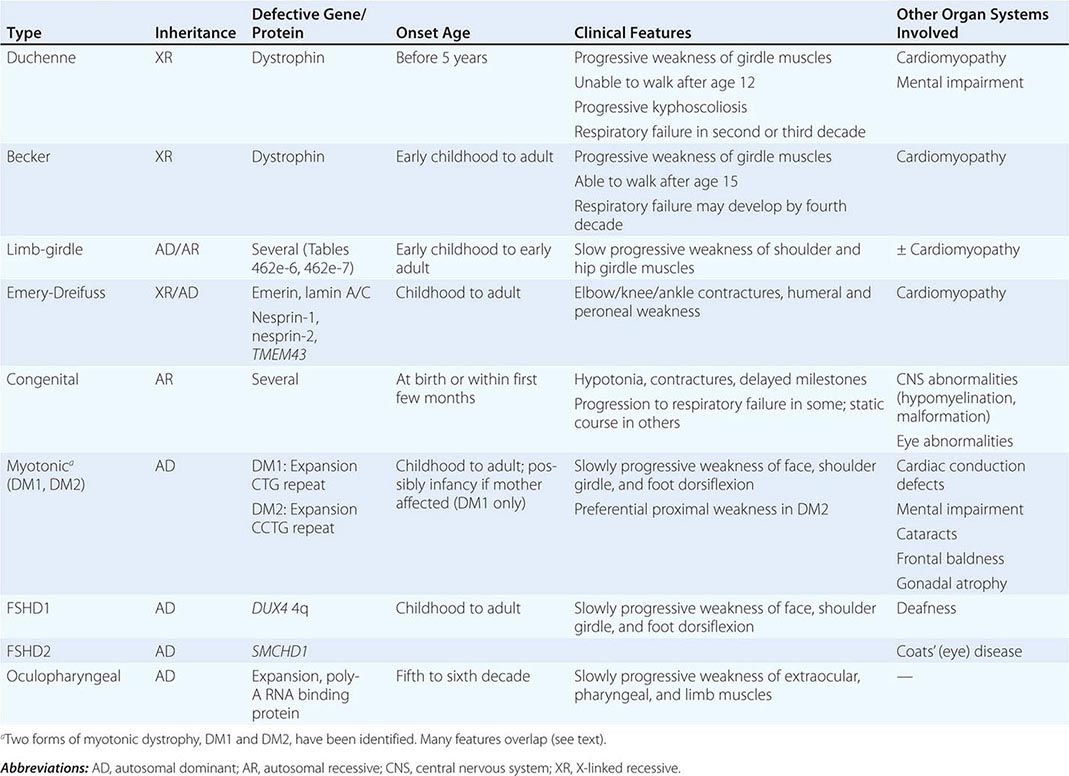
|
AUTOSOMAL DOMINANT LIMB-GIRDLE MUSCULAR DYSTROPHIES (LGMDs) |
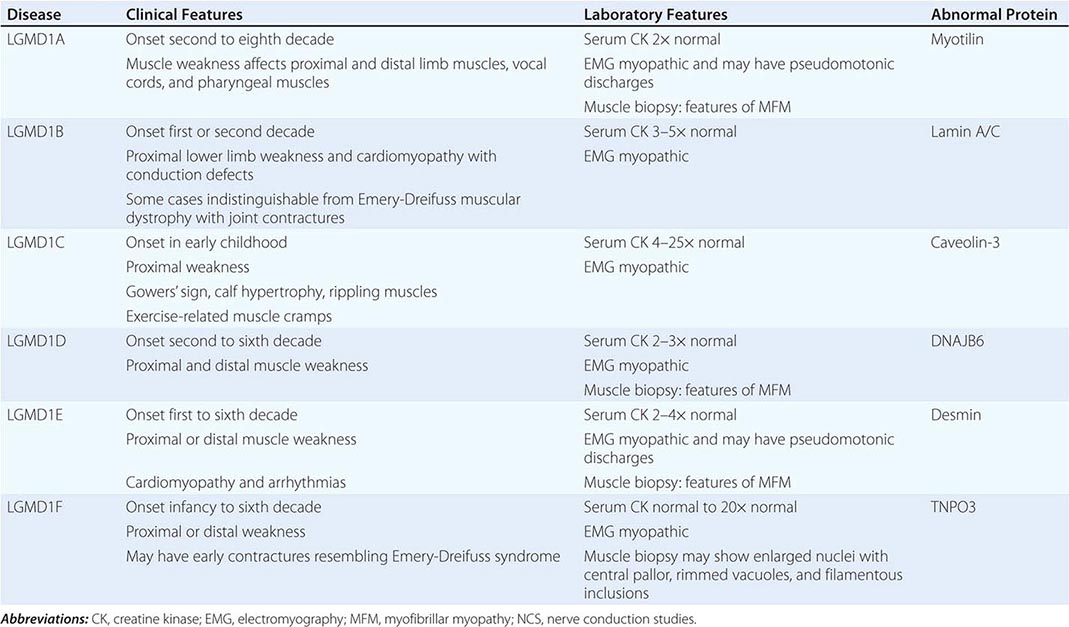
|
AUTOSOMAL RECESSIVE LIMB-GIRDLE MUSCULAR DYSTROPHIES (LGMDs) |
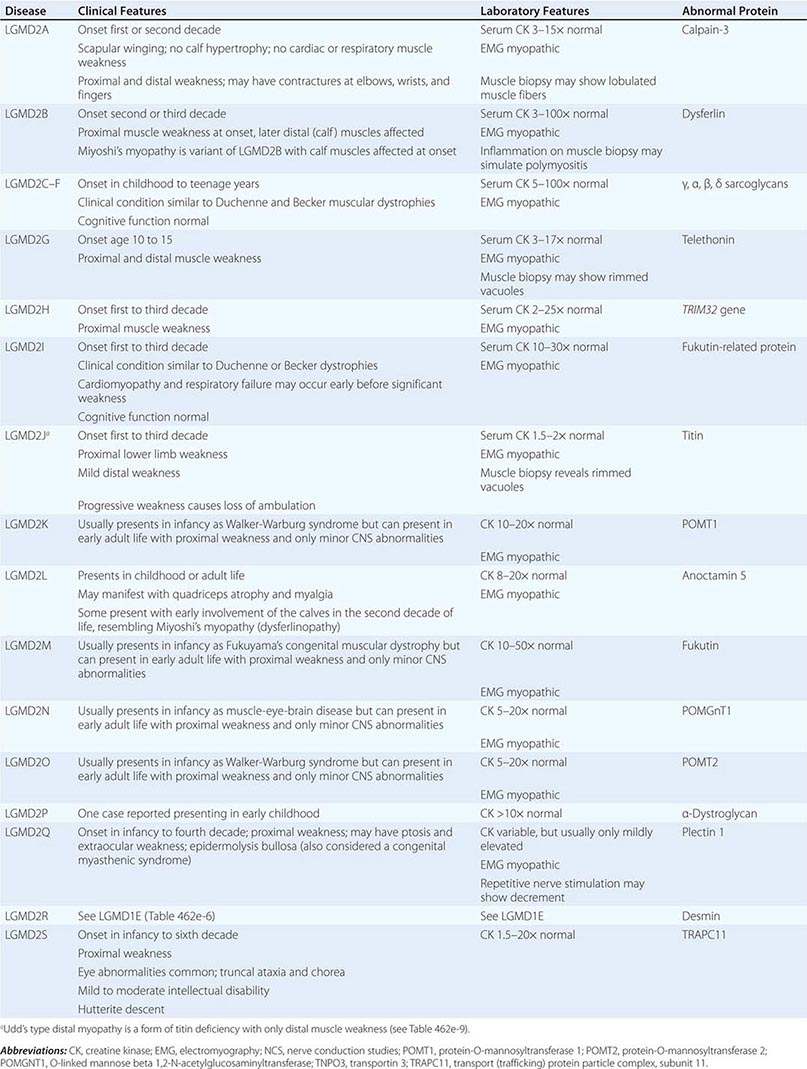
DUCHENNE MUSCULAR DYSTROPHY
This X-linked recessive disorder, sometimes also called pseudohypertrophic muscular dystrophy, has an incidence of ~1 per 5200 live-born males.
Clinical Features Duchenne dystrophy is present at birth, but the disorder usually becomes apparent between ages 3 and 5 years. The boys fall frequently and have difficulty keeping up with friends when playing. Running, jumping, and hopping are invariably abnormal. By age 5 years, muscle weakness is obvious by muscle testing. On getting up from the floor, the patient uses his hands to climb up himself (Gowers’ maneuver [Fig. 462e-4]). Contractures of the heel cords and iliotibial bands become apparent by age 6 years, when toe walking is associated with a lordotic posture. Loss of muscle strength is progressive, with predilection for proximal limb muscles and the neck flexors; leg involvement is more severe than arm involvement. Between ages 8 and 10 years, walking may require the use of braces; joint contractures and limitations of hip flexion, knee, elbow, and wrist extension are made worse by prolonged sitting. Prior to the use of glucocorticoids, most boys became wheelchair dependent by 12 years of age. Contractures become fixed, and a progressive scoliosis often develops that may be associated with pain. The chest deformity with scoliosis impairs pulmonary function, which is already diminished by muscle weakness. By age 16–18 years, patients are predisposed to serious, sometimes fatal pulmonary infections. Other causes of death include aspiration of food and acute gastric dilation.
A cardiac cause of death is uncommon despite the presence of a cardiomyopathy in almost all patients. Congestive heart failure seldom occurs except with severe stress such as pneumonia. Cardiac arrhythmias are rare. The typical electrocardiogram (ECG) shows an increased net RS in lead V1; deep, narrow Q waves in the precordial leads; and tall right precordial R waves in V1. Intellectual impairment in Duchenne dystrophy is common; the average intelligence quotient (IQ) is ~1 standard deviation (SD) below the mean. Impairment of intellectual function appears to be nonprogressive and affects verbal ability more than performance.
Laboratory Features Serum CK levels are invariably elevated to between 20 and 100 times normal. The levels are abnormal at birth but decline late in the disease because of inactivity and loss of muscle mass. EMG demonstrates features typical of myopathy. The muscle biopsy shows muscle fibers of varying size as well as small groups of necrotic and regenerating fibers. Connective tissue and fat replace lost muscle fibers. A definitive diagnosis of Duchenne dystrophy can be established on the basis of dystrophin deficiency in a biopsy of muscle tissue or mutation analysis on peripheral blood leukocytes, as discussed below.
Duchenne dystrophy is caused by a mutation of the gene that encodes dystrophin, a 427-kDa protein localized to the inner surface of the sarcolemma of the muscle fiber. The dystrophin gene is >2000 kb in size and thus is one of the largest identified human genes. It is localized to the short arm of the × chromosome at Xp21. The most common gene mutation is a deletion. The size varies but does not correlate with disease severity. Deletions are not uniformly distributed over the gene but rather are most common near the beginning (5′ end) and middle of the gene. Less often, Duchenne dystrophy is caused by a gene duplication or point mutation. Identification of a specific mutation allows for an unequivocal diagnosis, makes possible accurate testing of potential carriers, and is useful for prenatal diagnosis.
A diagnosis of Duchenne dystrophy can also be made by Western blot analysis of muscle biopsy specimens, revealing abnormalities on the quantity and molecular weight of dystrophin protein. In addition, immunocytochemical staining of muscle with dystrophin antibodies can be used to demonstrate absence or deficiency of dystrophin localizing to the sarcolemmal membrane. Carriers of the disease may demonstrate a mosaic pattern, but dystrophin analysis of muscle biopsy specimens for carrier detection is not reliable.
Pathogenesis Dystrophin is part of a large complex of sarcolemmal proteins and glycoproteins (Fig. 462e-6). Dystrophin binds to F-actin at its amino terminus and to β-dystroglycan at the carboxyl terminus. β-Dystroglycan complexes to α-dystroglycan, which binds to laminin in the extracellular matrix (ECM). Laminin has a heterotrimeric molecular structure arranged in the shape of a cross with one heavy chain and two light chains, β1 and γ1. The laminin heavy chain of skeletal muscle is designated laminin α2. Collagen proteins IV and VI are also found in the ECM. Like β-dystroglycan, the transmembrane sarcoglycan proteins also bind to dystrophin; these five proteins (designated α- through ε-sarcoglycan) complex tightly with each other. More recently, other membrane proteins implicated in muscular dystrophy have been found to be loosely affiliated with constituents of the dystrophin complex. These include caveolin-3, α7 integrin, and collagen VI.
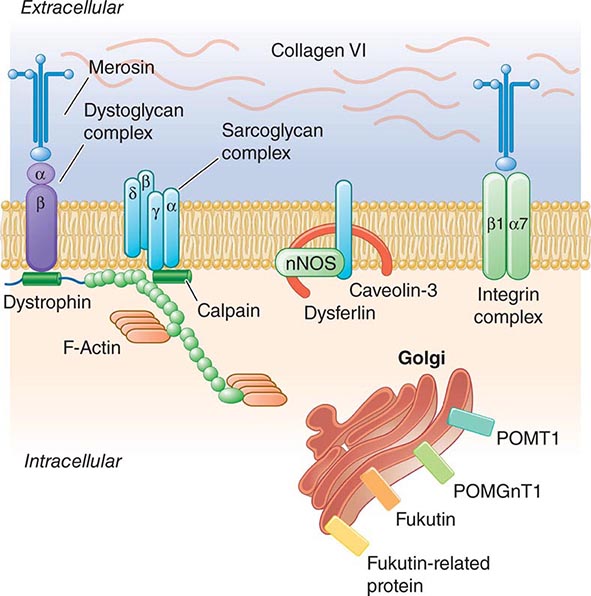
FIGURE 462e-6 Selected muscular dystrophy–associated proteins in the cell membrane and Golgi complex.
Dystrophin localizes to the cytoplasmic face of the muscle cell membrane. It complexes with two transmembrane protein complexes, the dystroglycans and the sarcoglycans. The dystroglycans bind to the extracellular matrix protein merosin, which is also complexed with β1 and α7 integrins (Tables 462e-5, 462e-6, and 462e-7). Dysferlin complexes with caveolin-3 (which binds to neuronal nitric oxide synthase, or nNOS) but not with the dystrophin-associated proteins or the integrins. In some of the congenital dystrophies and limb-girdle muscular dystrophies (LGMDs), there is loss of function of different enzymes that glycosylate α-dystroglycan, which thereby inhibits proper binding to merosin: POMT1, POMT2, POMGnT1, Fukutin, Fukutin-related protein, and LARGE.
The dystrophin-glycoprotein complex appears to confer stability to the sarcolemma, although the function of each individual component of the complex is incompletely understood. Deficiency of one member of the complex may cause abnormalities in other components. For example, a primary deficiency of dystrophin (Duchenne dystrophy) may lead to secondary loss of the sarcoglycans and dystroglycan. The primary loss of a single sarcoglycan (see “Limb-Girdle Muscular Dystrophy,” below) results in a secondary loss of other sarcoglycans in the membrane without uniformly affecting dystrophin. In either instance, disruption of the dystrophin-glycoprotein complexes weakens the sarcolemma, causing membrane tears and a cascade of events leading to muscle fiber necrosis. This sequence of events occurs repeatedly during the life of a patient with muscular dystrophy.
BECKER MUSCULAR DYSTROPHY
This less severe form of X-linked recessive muscular dystrophy results from allelic defects of the same gene responsible for Duchenne dystrophy. Becker muscular dystrophy is ~10 times less frequent than Duchenne.
Clinical Features The pattern of muscle wasting in Becker muscular dystrophy closely resembles that seen in Duchenne. Proximal muscles, especially of the lower extremities, are prominently involved. As the disease progresses, weakness becomes more generalized. Significant facial muscle weakness is not a feature. Hypertrophy of muscles, particularly in the calves, is an early and prominent finding.
Most patients with Becker dystrophy first experience difficulties between ages 5 and 15 years, although onset in the third or fourth decade or even later can occur. By definition, patients with Becker dystrophy walk beyond age 15, whereas patients with Duchenne dystrophy are typically in a wheelchair by the age of 12. Patients with Becker dystrophy have a reduced life expectancy, but most survive into the fourth or fifth decade.
Mental retardation may occur in Becker dystrophy, but it is not as common as in Duchenne. Cardiac involvement occurs in Becker dystrophy and may result in heart failure; some patients manifest with only heart failure. Other less common presentations are asymptomatic hyper-CK-emia, myalgias without weakness, and myoglobinuria.
Laboratory Features Serum CK levels, results of EMG, and muscle biopsy findings closely resemble those in Duchenne dystrophy. The diagnosis of Becker muscular dystrophy requires Western blot analysis of muscle biopsy samples, demonstrating a reduced amount or abnormal size of dystrophin or mutation analysis of DNA from peripheral blood leukocytes. Genetic testing reveals deletions or duplications of the dystrophin gene in 65% of patients with Becker dystrophy, approximately the same percentage as in Duchenne dystrophy. In both Becker and Duchenne dystrophies, the size of the DNA deletion does not predict clinical severity; however, in ~95% of patients with Becker dystrophy, the DNA deletion does not alter the translational reading frame of messenger RNA. These “in-frame” mutations allow for production of some dystrophin, which accounts for the presence of altered rather than absent dystrophin on Western blot analysis.
LIMB-GIRDLE MUSCULAR DYSTROPHY
The syndrome of LGMD represents more than one disorder. Both males and females are affected, with onset ranging from late in the first decade to the fourth decade. The LGMDs typically manifest with progressive weakness of pelvic and shoulder girdle musculature. Respiratory insufficiency from weakness of the diaphragm may occur, as may cardiomyopathy.
A systematic classification of LGMD is based on autosomal dominant (LGMD1) and autosomal recessive (LGMD2) inheritance. Superimposed on the backbone of LGMD1 and LGMD2, the classification uses a sequential alphabetical lettering system (LGMD1A, LGMD2A, etc.). Disorders receive letters in the order in which they are found to have chromosomal linkage. This results in an ever-expanding list of conditions summarized in Tables 462e-6 and 462e-7. None of the conditions is as common as the dystrophinopathies; however, prevalence data for the LGMDs have not been systematically gathered for any large heterogeneous population. In referral-based clinical populations, Fukutin-related protein (FKRP) deficiency (LGMD2I), calpainopathy (LGMD2A), anoctaminopathy (LGMD2L), and to a lesser extent dysferlinopathy (LGMD2B) have emerged as the most common disorders.
EMERY-DREIFUSS MUSCULAR DYSTROPHY
There are at least five genetically distinct forms of Emery-Dreifuss muscular dystrophy (EDMD). Emerin mutations are the most common cause of X-linked EDMD, although mutations in FHL1 may also be associated with a similar phenotype, which is X-linked as well. Mutations involving the gene for lamin A/C are the most common cause of autosomal dominant EDMD (also known as LGMD1B) and are also a common cause of hereditary cardiomyopathy. Less commonly, autosomal dominant EDMD has been reported with mutations in nesprin-1, nesprin-2, and TMEM43.
Clinical Features Prominent contractures can be recognized in early childhood and teenage years, often preceding muscle weakness. The contractures persist throughout the course of the disease and are present at the elbows, ankles, and neck. Muscle weakness affects humeral and peroneal muscles at first and later spreads to a limb-girdle distribution. The cardiomyopathy is potentially life threatening and may result in sudden death. A spectrum of atrial rhythm and conduction defects includes atrial fibrillation and paralysis and atrioventricular heart block. Some patients have a dilated cardiomyopathy. Female carriers of the X-linked variant may have cardiac manifestations that become clinically significant.
Laboratory Features Serum CK may be elevated two- to tenfold. EMG is myopathic. Muscle biopsy usually shows nonspecific dystrophic features, although cases associated with FHL1 mutations have features of myofibrillar myopathy. Immunohistochemistry reveals absent emerin staining of myonuclei in X-linked EDMD due to emerin mutations. ECGs demonstrate atrial and atrioventricular rhythm disturbances.
X-linked EDMD usually arises from defects in the emerin gene encoding a nuclear envelope protein. FHL1 mutations are also a cause of X-linked scapuloperoneal dystrophy, but can also present with an X-linked form of EDMD. The autosomal dominant disease can be caused by mutations in the LMNA gene encoding lamin A and C; in the synaptic nuclear envelope protein 1 (SYNE1) or 2 (SYNE2) encoding nesprin-1 and nesprin-2, respectively; and most recently in TMEM43 encoding LUMA. These proteins are essential components of the filamentous network underlying the inner nuclear membrane. Loss of structural integrity of the nuclear envelope from defects in emerin, lamin A/C, nesprin-1, nesprin-2, and LUMA accounts for overlapping phenotypes (Fig. 462e-7).
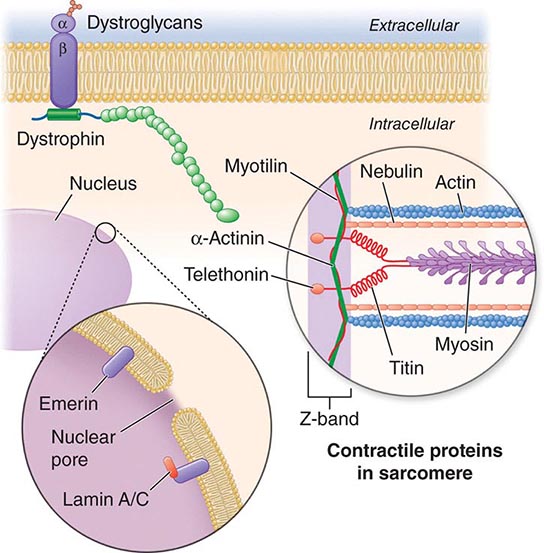
FIGURE 462e-7 Selected muscular dystrophy–associated proteins in the nuclear membrane and sarcomere. As shown in the exploded view, emerin and lamin A/C are constituents of the inner nuclear membrane. Several dystrophy-associated proteins are represented in the sarcomere including titin, nebulin, calpain, telethonin, actinin, and myotilin. The position of the dystrophin-dystroglycan complex is also illustrated.
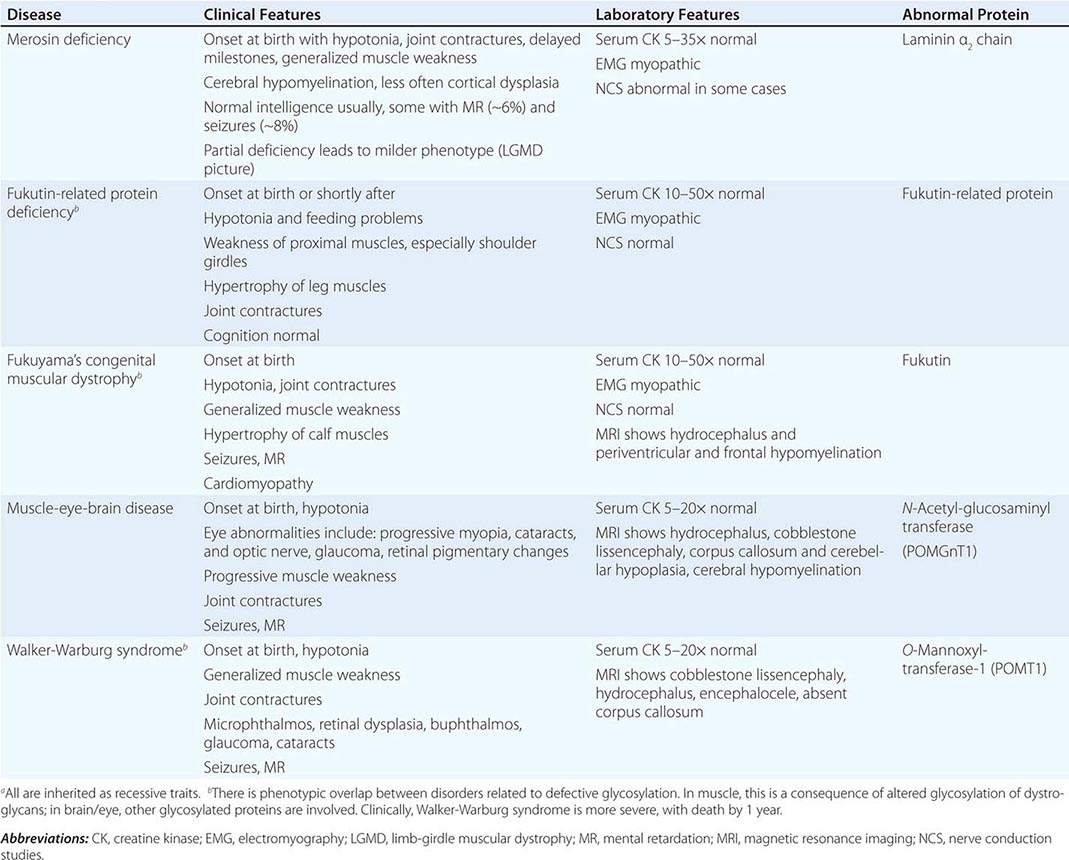
CONGENITAL MUSCULAR DYSTROPHY (CMD)
This is not one entity but rather a group of disorders with varying degrees of muscle weakness, CNS impairment, and eye abnormalities.
Clinical Features As a group, CMDs present at birth or in the first few months of life with hypotonia and proximal or generalized muscle weakness. Calf muscle hypertrophy is seen in some patients. Facial muscles may be weak, but other cranial nerve–innervated muscles are spared (e.g., extraocular muscles are normal). Most patients have joint contractures of varying degrees at elbows, hips, knees, and ankles. Contractures present at birth are referred to as arthrogryposis. Respiratory failure may be seen in some cases.
The CNS is affected in some forms of CMD. In merosin and FKRP deficiency, cerebral hypomyelination may be seen by magnetic resonance imaging (MRI), although only a small number of patients have mental retardation and seizures. Three forms of congenital muscular dystrophy have severe brain impairment. These include Fukuyama’s congenital muscular dystrophy (FCMD), muscle-eye-brain (MEB) disease, and Walker-Warburg syndrome (WWS). Patients are severely disabled in all three of these conditions. In MEB disease and WWS, but not in FCMD, ocular abnormalities impair vision. WWS is the most severe congenital muscular dystrophy, causing death by 1 year of age.
Laboratory Features Serum CK is markedly elevated in all of these conditions. The EMG is myopathic and muscle biopsies show nonspecific dystrophic features. Merosin, or laminin α2 chain (a basal lamina protein), is deficient in surrounding muscle fibers in merosin deficiency. Skin biopsies can also demonstrate defects in laminin α2 chain. In the other disorders (FKRP deficiency, FCMD, MEB disease, WWS), there is abnormal α-dystroglycan staining in muscle. In merosin deficiency, cerebral hypomyelination is common, and a host of brain malformations are seen in FCMD, MEB disease, and WWS.
All forms of CMD are inherited as autosomal recessive disorders. Chromosomal linkage and specific gene defects are presented in Table 462e-8. With the exception of merosin, the other gene defects affect posttranslational glycosylation of α-dystroglycan. This abnormality is thought to impair binding with merosin and leads to weakening of the dystrophin-glycoprotein complex, instability of the muscle membrane, and/or abnormalities in muscle contraction. CMDs with brain and eye phenotypes probably involve defective glycosylation of additional proteins, accounting for the more extensive phenotypes.
|
CONGENITAL MUSCULAR DYSTROPHIESa |
MYOTONIC DYSTROPHY
Myotonic dystrophy is also known as dystrophia myotonica (DM). The condition is composed of at least two clinical disorders with overlapping phenotypes and distinct molecular genetic defects: myotonic dystrophy type 1 (DM1), the classic disease originally described by Steinert, and myotonic dystrophy type 2 (DM2), also called proximal myotonic myopathy (PROMM).
Clinical Features The clinical expression of DM1 varies widely and involves many systems other than muscle. Affected patients have a typical “hatchet-faced” appearance due to temporalis, masseter, and facial muscle atrophy and weakness. Frontal baldness is also characteristic of the disease. Neck muscles, including flexors and sternocleidomastoids, and distal limb muscles are involved early. Weakness of wrist extensors, finger extensors, and intrinsic hand muscles impairs function. Ankle dorsiflexor weakness may cause footdrop. Proximal muscles remain stronger throughout the course, although preferential atrophy and weakness of quadriceps muscles occur in many patients. Palatal, pharyngeal, and tongue involvement produce a dysarthric speech, nasal voice, and swallowing problems. Some patients have diaphragm and intercostal muscle weakness, resulting in respiratory insufficiency.
Myotonia, which usually appears by age 5 years, is demonstrable by percussion of the thenar eminence, the tongue, and wrist extensor muscles. Myotonia causes a slow relaxation of hand grip after a forced voluntary closure. Advanced muscle wasting makes myotonia more difficult to detect.
Cardiac disturbances occur commonly in patients with DM1. ECG abnormalities include first-degree heart block and more extensive conduction system involvement. Complete heart block and sudden death can occur. Congestive heart failure occurs infrequently but may result from cor pulmonale secondary to respiratory failure. Mitral valve prolapse also occurs commonly. Other associated features include intellectual impairment, hypersomnia, posterior subcapsular cataracts, gonadal atrophy, insulin resistance, and decreased esophageal and colonic motility.
Congenital myotonic dystrophy is a more severe form of DM1 and occurs in ~25% of infants of affected mothers. It is characterized by severe facial and bulbar weakness, transient neonatal respiratory insufficiency, and mental retardation.
DM2, or PROMM, has a distinct pattern of muscle weakness affecting mainly proximal muscles. Other features of the disease overlap with DM1, including cataracts, testicular atrophy, insulin resistance, constipation, hypersomnia, and cognitive defects. Cardiac conduction defects occur but are less common, and the hatchet face and frontal baldness are less consistent features. A very striking difference is the failure to clearly identify a congenital form of DM2.
Laboratory Features The diagnosis of myotonic dystrophy can usually be made on the basis of clinical findings. Serum CK levels may be normal or mildly elevated. EMG evidence of myotonia is present in most cases of DM1 but may be more patchy in DM2. Muscle biopsy shows muscle atrophy, which selectively involves type 1 fibers in 50% of cases, and ringed fibers in DM1 but not in DM2. Typically, numerous internalized nuclei can be seen in individual muscle fibers as well as atrophic fibers with pyknotic nuclear clumps in both DM1 and DM2. Necrosis of muscle fibers and increased connective tissue, common in other muscular dystrophies, are less apparent in myotonic dystrophy.
DM1 and DM2 are both autosomal dominant disorders. New mutations do not appear to contribute to the pool of affected individuals. DM1 is transmitted by an intronic mutation consisting of an unstable expansion of a CTG trinucleotide repeat in a serine-threonine protein kinase gene (named DMPK) on chromosome 19q13.3. An increase in the severity of the disease phenotype in successive generations (genetic anticipation) is accompanied by an increase in the number of trinucleotide repeats. A similar type of mutation has been identified in fragile × syndrome (Chap. 451e). The unstable triplet repeat in myotonic dystrophy can be used for prenatal diagnosis. Congenital disease occurs almost exclusively in infants born to affected mothers; it is possible that sperm with greatly expanded triplet repeats do not function well.
DM2 is caused by a DNA expansion mutation consisting of a CCTG repeat in intron 1 of the ZNF9 gene located at chromosome 3q13.3-q24. The gene is believed to encode an RNA-binding protein expressed in many different tissues, including skeletal and cardiac muscle.
The DNA expansions in DM1 and DM2 almost certainly impair muscle function by a toxic gain of function of the mutant mRNA. In both DM1 and DM2, the mutant RNA appears to form intranuclear inclusions composed of aberrant RNA. These RNA inclusions sequester RNA-binding proteins essential for proper splicing of a variety of other mRNAs. This leads to abnormal transcription of multiple proteins in a variety of tissues/organ systems, in turn causing the systemic manifestations of DM1 and DM2.
FACIOSCAPULOHUMERAL (FSH) MUSCULAR DYSTROPHY
This form of muscular dystrophy has a prevalence of ~1 in 20,000. There are two forms of FSHD that have similar pathogenesis, as will be discussed. Most patients have FSHD type 1 (95%), whereas approximately 5% have FSHD2. FSHD1 and FSHD2 are clinically and histopathologically identical. FSHD is not to be confused with the genetically distinct scapuloperoneal dystrophies.
Clinical Features The condition typically has an onset in childhood or young adulthood. In most cases, facial weakness is the initial manifestation, appearing as an inability to smile, whistle, or fully close the eyes. Weakness of the shoulder girdles, rather than the facial muscles, usually brings the patient to medical attention. Loss of scapular stabilizer muscles makes arm elevation difficult. Scapular winging (Fig. 462e-3) becomes apparent with attempts at abduction and forward movement of the arms. Biceps and triceps muscles may be severely affected, with relative sparing of the deltoid muscles. Weakness is invariably worse for wrist extension than for wrist flexion, and weakness of the anterior compartment muscles of the legs may lead to footdrop.
In most patients, the weakness remains restricted to facial, upper extremity, and distal lower extremity muscles. In 20% of patients, weakness progresses to involve the pelvic girdle muscles, and severe functional impairment and possible wheelchair dependency result.
Characteristically, patients with FSHD do not have involvement of other organ systems, although labile hypertension is common, and there is an increased incidence of nerve deafness. Coats’ disease, a disorder consisting of telangiectasia, exudation, and retinal detachment, also occurs.
Laboratory Features The serum CK level may be normal or mildly elevated. EMG usually indicates a myopathic pattern. The muscle biopsy shows nonspecific features of a myopathy. A prominent inflammatory infiltrate, which is often multifocal in distribution, is present in some biopsy samples. The cause or significance of this finding is unknown.
An autosomal dominant inheritance pattern with almost complete penetrance has been established, but each family member should be examined for the presence of the disease, since ~30% of those affected are unaware of involvement. FSHD1 is associated with deletions of tandem 3.3-kb repeats at 4q35. The deletion reduces the number of repeats to a fragment of <35 kb in most patients. Within these repeats lies the DUX4 gene, which usually is not expressed. In patients with FSHD1 these deletions in the setting of a specific polymorphism leads to hypomethylation of the region and toxic expression of the DUX4 gene. In patients with FSHD2, there is no deletion, but a mutation in SMCHD1. Interestingly, in the setting of the same polymorphism, there again is seen hypomethylation of the region and the permissive expression of the DUX4 gene. In both FSHD1 and FSHD2, there is overexpression of the DUX4 transcript.
OCULOPHARYNGEAL DYSTROPHY
This form of muscular dystrophy represents one of several disorders characterized by progressive external ophthalmoplegia, which consists of slowly progressive ptosis and limitation of eye movements with sparing of pupillary reactions for light and accommodation. Patients usually do not complain of diplopia, in contrast to patients having conditions with a more acute onset of ocular muscle weakness (e.g., myasthenia gravis).
Clinical Features Oculopharyngeal muscular dystrophy has a late onset; it usually presents in the fourth to sixth decade with ptosis and/or dysphagia. The extraocular muscle impairment is less prominent in the early phase but may be severe later. The swallowing problem may become debilitating and result in pooling of secretions and repeated episodes of aspiration. Mild weakness of the neck and extremities also occurs.
Laboratory Features The serum CK level may be two to three times normal. Myopathic EMG findings are typical. On biopsy, muscle fibers are found to contain rimmed vacuoles, which by electron microscopy are shown to contain membranous whorls, accumulation of glycogen, and other nonspecific debris related to lysosomes. A distinct feature of oculopharyngeal dystrophy is the presence of tubular filaments, 8.5 nm in diameter, in muscle cell nuclei.
Oculopharyngeal dystrophy has an autosomal dominant inheritance pattern with complete penetrance. The incidence is high in French-Canadians and in Spanish-American families of the southwestern United States. Large kindreds of Italian and of eastern European Jewish descent have been reported. The molecular defect in oculopharyngeal muscular dystrophy is a subtle expansion of a modest polyalanine repeat tract in a poly-RNA-binding protein (PABP2) in muscle.
DISTAL MYOPATHIES
A group of muscle diseases, the distal myopathies, are notable for their preferential distal distribution of muscle weakness in contrast to most muscle conditions associated with proximal weakness. The major distal myopathies are summarized in Table 462e-9.
|
DISTAL MYOPATHIES |
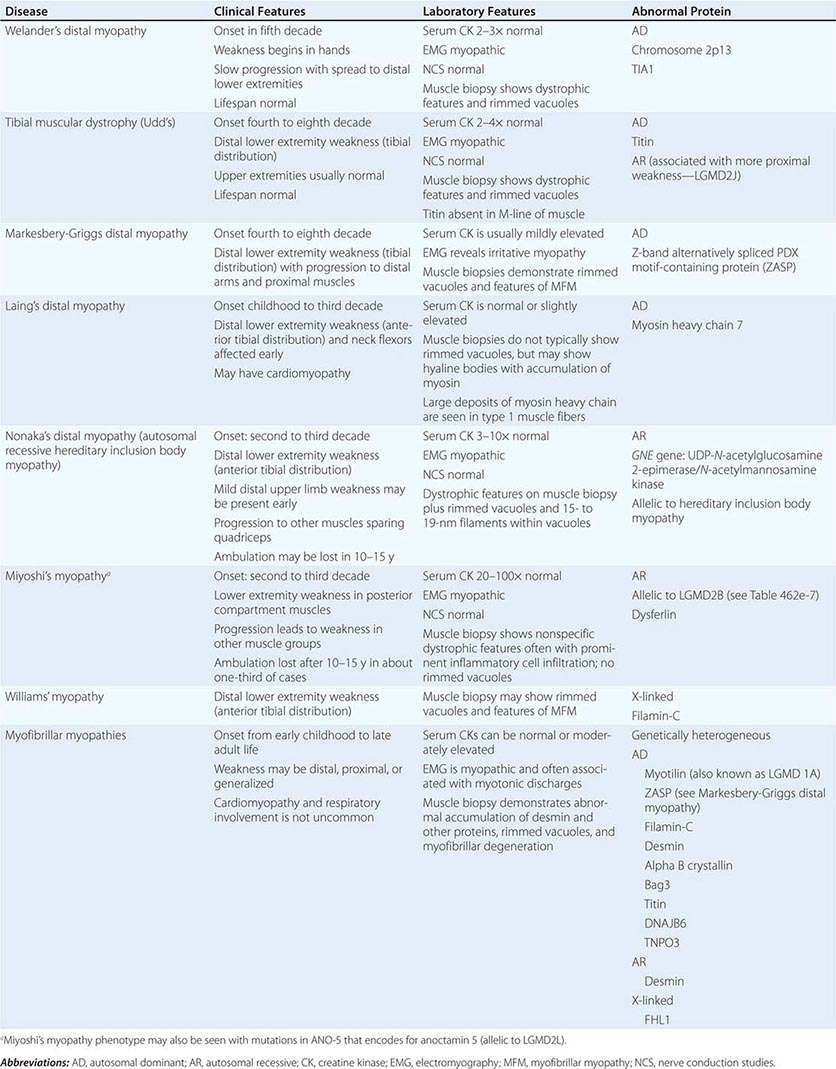
Clinical Features Welander’s, Udd’s, and Markesbery-Griggs type distal myopathies are all late-onset, dominantly inherited disorders of distal limb muscles, usually beginning after age 40 years. Welander’s distal myopathy preferentially involves the wrist and finger extensors, whereas the others are associated with anterior tibial weakness leading to progressive footdrop. Laing’s distal myopathy is also a dominantly inherited disorder heralded by tibial weakness; however, it is distinguished by onset in childhood or early adult life. Nonaka’s distal myopathy and Miyoshi’s myopathy are distinguished by autosomal recessive inheritance and onset in the late teens or twenties. Nonaka’s and Williams’ myopathy entails anterior tibial weakness, whereas Miyoshi’s myopathy is unique in that gastrocnemius muscles are preferentially affected at onset. Finally, the myofibrillar myopathies (MFMs) are a clinically and genetically heterogeneous group of disorders that can be associated with prominent distal weakness; they can be inherited in an autosomal dominant or recessive pattern. Of note, Markesbery-Griggs myopathy (caused by mutations in ZASP) and LGMD1B (caused by mutations in myotilin) are in fact subtypes of myofibrillar myopathy.
Laboratory Features Serum CK level is particularly helpful in diagnosing Miyoshi’s myopathy because it is very elevated. In the other conditions, serum CK is only slightly increased. EMGs are myopathic. In the MFMs, myotonic or pseudomyotonic discharges are common. Muscle biopsy shows nonspecific dystrophic features and, with the exception of Laing’s and Miyoshi’s myopathies, often shows rimmed vacuoles. MFM is associated with the accumulation of dense inclusions, as well as amorphous material best seen on Gomori trichrome and myofibrillar disruption on electron microscopy. Immune staining sometimes demonstrates accumulation of desmin and other proteins in MFM, large deposits of myosin heavy chain in the subsarcolemmal region of type 1 muscle fibers in Laing’s myopathy, and reduced or absent dysferlin in Miyoshi’s myopathy.
The affected genes and their gene products are listed in Table 462e-9.
CONGENITAL MYOPATHIES
These rare disorders are distinguished from muscular dystrophies by the presence of specific histochemical and structural abnormalities in muscle. Although primarily disorders of infancy or childhood, three forms that may present in adulthood are described here: central core disease, nemaline (rod) myopathy, and centronuclear (myotubular) myopathy. Sarcotubular myopathy is caused by mutations in TRIM-32 and is identical to LGMD2H. Other types, such as minicore myopathy (multi-minicore disease), fingerprint body myopathy, and cap myopathy, are not discussed.
CENTRAL CORE DISEASE
Patients with central core disease may have decreased fetal movements and breech presentation. Hypotonia and delay in motor milestones, particularly in walking, are common. Later in childhood, patients develop problems with stair climbing, running, and getting up from the floor. On examination, there is mild facial, neck-flexor, and proximal-extremity muscle weakness. Legs are more affected than arms. Skeletal abnormalities include congenital hip dislocation, scoliosis, and pes cavus; clubbed feet also occur. Most cases are nonprogressive, but exceptions are well documented. Susceptibility to malignant hyperthermia must be considered as a potential risk factor for patients with central core disease. Recent series have demonstrated that many cases of late-onset axial myopathy in which patients manifest with bent spine (camptocormia) or neck extensor weakness (neck extensor myopathy) are caused by mutations in the ryanodine receptor gene (RYR1). This illustrates the interesting spectrum of RYR1 mutations.
The serum CK level is usually normal. Needle EMG demonstrates a myopathic pattern. Muscle biopsy shows fibers with single or multiple central or eccentric discrete zones (cores) devoid of oxidative enzymes. Cores occur preferentially in type 1 fibers and represent poorly aligned sarcomeres associated with Z disk streaming.
Autosomal dominant inheritance is characteristic; sporadic cases also occur. As alluded above, this myopathy is caused by point mutations of RYR1, encoding the calcium-release channel of the sarcoplasmic reticulum of skeletal muscle; mutations of this gene also account for some cases of inherited malignant hyperthermia (Chap. 23). Malignant hyperthermia is an allelic condition; C-terminal mutations of the RYR1 gene predispose to this complication.
Specific treatment is not required, but establishing a diagnosis of central core disease is extremely important because these patients have a known predisposition to malignant hyperthermia during anesthesia.
NEMALINE MYOPATHY
The term nemaline refers to the distinctive presence in muscle fibers of rods or threadlike structures (Greek nema, “thread”). Nemaline myopathy is clinically heterogeneous. A severe neonatal form presents with hypotonia and feeding and respiratory difficulties, leading to early death. Nemaline myopathy usually presents in infancy or childhood with delayed motor milestones. The course is nonprogressive or slowly progressive. The physical appearance is striking because of the long, narrow facies, high-arched palate, and open-mouthed appearance due to a prognathous jaw. Other skeletal abnormalities include pectus excavatum, kyphoscoliosis, pes cavus, and clubfoot deformities. Facial and generalized muscle weakness, including respiratory muscle weakness, is common. An adult-onset disorder with progressive proximal or distal weakness may be seen. Myocardial involvement is occasionally present in both the childhood and adult-onset forms. The serum CK level is usually normal or slightly elevated. The EMG demonstrates a myopathic pattern. Muscle biopsy shows clusters of small rods (nemaline bodies), which occur preferentially, but not exclusively, in the sarcoplasm of type 1 muscle fibers. Occasionally, the rods are also apparent in myonuclei. The muscle often shows type 1 muscle fiber predominance. Rods originate from the Z disk material of the muscle fiber.
Six genes have been associated with nemaline myopathy. Five of these code for thin filament–associated proteins, suggesting disturbed assembly or interplay of these structures as a pivotal mechanism. Mutations of the nebulin (NEB) gene account for most cases, including both severe neonatal and early childhood forms, inherited as autosomal recessive disorders. Neonatal and childhood cases, inherited as predominantly autosomal dominant disorders, are caused by mutations of the skeletal muscle a-actinin (ACTA1) gene. In milder forms of the disease with autosomal dominant inheritance, mutations have been identified in both the slow a-tropomyosin (TPM3) and β;-tropomyosin (TPM2) genes accounting for <3% of cases. Muscle troponin T (TNNT1) gene mutations appear to be limited to the Amish population in North America. Mutations may also be seen in NEM6 that encodes a putative BTB/Kelch protein. No specific treatment is available.
CENTRONUCLEAR (MYOTUBULAR) MYOPATHY
Three distinct variants of centronuclear myopathy occur. A neonatal form, also known as myotubular myopathy, presents with severe hypotonia and weakness at birth. The late infancy–early childhood form presents with delayed motor milestones. Later, difficulty with running and stair climbing becomes apparent. A marfanoid, slender body habitus, long narrow face, and high-arched palate are typical. Scoliosis and clubbed feet may be present. Most patients exhibit progressive weakness, some requiring wheelchairs. Progressive external ophthalmoplegia with ptosis and varying degrees of extraocular muscle impairment are characteristic of both the neonatal and the late-infantile forms. A third variant, the late childhood–adult form, has an onset in the second or third decade. Patients have full extraocular muscle movements and rarely exhibit ptosis. There is mild, slowly progressive limb weakness that may be distally predominant (some of these patients have been classified as having Charcot-Marie-Tooth disease type 2 [CMT2; Chap. 459]).
Normal or slightly elevated CK levels occur in each of the forms. Nerve conduction studies may reveal reduced amplitudes of distal compound muscle action potentials, in particular in adult-onset cases that resemble CMT2. EMG studies often give distinctive results, showing positive sharp waves and fibrillation potentials, complex and repetitive discharges, and rarely myotonic discharges. Muscle biopsy specimens in longitudinal section demonstrate rows of central nuclei, often surrounded by a halo. In transverse sections, central nuclei are found in 25–80% of muscle fibers.
A gene for the neonatal form of centronuclear myopathy has been localized to Xq28; this gene encodes myotubularin, a protein tyrosine phosphatase. Missense, frameshift, and splice-site mutations predict loss of myotubularin function in affected individuals. Carrier identification and prenatal diagnosis are possible. Autosomal recessive forms are caused by mutations in BIB1 that encodes for amphyphysin-2, whereas some autosomal dominant cases, which are allelic to a form of CMT2, are associated with mutations in the gene that encodes dynamin-2. No specific medical treatments are available at this time.
DISORDERS OF MUSCLE ENERGY METABOLISM
There are two principal sources of energy for skeletal muscle—fatty acids and glucose. Abnormalities in either glucose or lipid utilization can be associated with distinct clinical presentations that can range from an acute, painful syndrome with rhabdomyolysis and myoglobinuria to a chronic, progressive muscle weakness simulating muscular dystrophy.
GLYCOGEN STORAGE AND GLYCOLYTIC DEFECTS
Disorders of Glycogen Storage Causing Progressive Weakness • α-GLUCOSIDASE, OR ACID MALTASE, DEFICIENCY (POMPE’S DISEASE) Three clinical forms of α-glucosidase, or acid maltase, deficiency (type II glycogenosis) can be distinguished. The infantile form is the most common, with onset of symptoms in the first 3 months of life. Infants develop severe muscle weakness, cardiomegaly, hepatomegaly, and respiratory insufficiency. Glycogen accumulation in motor neurons of the spinal cord and brainstem contributes to muscle weakness. Death usually occurs by 1 year of age. In the childhood form, the picture resembles muscular dystrophy. Delayed motor milestones result from proximal limb muscle weakness and involvement of respiratory muscles. The heart may be involved, but the liver and brain are unaffected. The adult form usually begins in the third or fourth decade but can present as late as the seventh decade. Respiratory failure and diaphragmatic weakness are often initial manifestations, heralding progressive proximal muscle weakness. The heart and liver are not involved.
The serum CK level is 2–10 times normal in infantile or childhood-onset Pompe’s disease but can be normal in adult-onset cases. EMG examination demonstrates a myopathic pattern, but other features are especially distinctive, including myotonic discharges, trains of fibrillation and positive waves, and complex repetitive discharges. EMG discharges are very prominent in the paraspinal muscles. The muscle biopsy in infants typically reveals vacuoles containing glycogen and the lysosomal enzyme acid phosphatase. Electron microscopy reveals membrane-bound and free tissue glycogen. However, muscle biopsies in late-onset Pompe’s disease may demonstrate only nonspecific abnormalities. Enzyme analysis of dried blood spots is a sensitive technique to screen for Pompe’s disease. A definitive diagnosis is established by enzyme assay in muscle or cultured fibroblasts or by genetic testing.
Pompe’s disease is inherited as an autosomal recessive disorder caused by mutations of the α-glucosidase gene. Enzyme replacement therapy (ERT) with IV recombinant human α-glucosidase has been shown to be beneficial in infantile-onset Pompe’s disease. Clinical benefits in the infantile disease include reduced heart size, improved muscle function, reduced need for ventilatory support, and longer life. In late-onset cases, ERT has not been associated with the dramatic response that can be seen in classic infantile Pompe’s disease, yet it appears to stabilize the disease process.
OTHER GLYCOGEN STORAGE DISEASES WITH PROGRESSIVE WEAKNESS In de-branching enzyme deficiency (type III glycogenosis), a slowly progressive form of muscle weakness can develop after puberty. Rarely, myoglobinuria may be seen. Patients are usually diagnosed in infancy, however, because of hypotonia and delayed motor milestones, hepatomegaly, growth retardation, and hypoglycemia. Branching enzyme deficiency (type IV glycogenosis) is a rare and fatal glycogen storage disease characterized by failure to thrive and hepatomegaly. Hypotonia and muscle wasting may be present, but the skeletal muscle manifestations are minor compared to liver failure.
Disorders of Glycolysis Causing Exercise Intolerance Several glycolytic defects are associated with recurrent myoglobinuria: myophosphorylase deficiency (type V glycogenosis), phosphofructokinase deficiency (type VII glycogenosis), phosphoglycerate kinase deficiency, phosphorylase kinase deficiency (type IX glycogenosis), phosphoglycerate mutase deficiency (type × glycogenosis), lactate dehydrogenase deficiency (glycogenosis type XI), and β-enolase deficiency. Myophosphorylase deficiency, also known as McArdle’s disease, is by far the most common of the glycolytic defects associated with exercise intolerance. These glycolytic defects result in a common failure to support energy production at the initiation of exercise, although the exact site of energy failure remains controversial.
Clinical muscle manifestations in these conditions usually begin in adolescence. Symptoms are precipitated by brief bursts of high-intensity exercise such as running or lifting heavy objects. A history of myalgia and muscle stiffness usually precedes the intensely painful muscle contractures, which may be followed by myoglobinuria. Acute renal failure accompanies significant pigmenturia.
Certain features help distinguish some enzyme defects. In McArdle’s disease, exercise tolerance can be enhanced by a slow induction phase (warm-up) or brief periods of rest, allowing for the start of the “second-wind” phenomenon (switching to utilization of fatty acids). Varying degrees of hemolytic anemia accompany deficiencies of both phosphofructokinase (mild) and phosphoglycerate kinase (severe). In phosphoglycerate kinase deficiency, the usual clinical presentation is a seizure disorder associated with mental retardation; exercise intolerance is an infrequent manifestation.
In all of these conditions, the serum CK levels fluctuate widely and may be elevated even during symptom-free periods. CK levels >100 times normal are expected, accompanying myoglobinuria. All patients with suspected glycolytic defects leading to exercise intolerance should undergo a forearm exercise test. An impaired rise in venous lactate is highly indicative of a glycolytic defect. In lactate dehydrogenase deficiency, venous levels of lactate do not increase, but pyruvate rises to normal. A definitive diagnosis of glycolytic disease is made by muscle biopsy and subsequent enzyme analysis or by genetic testing.
Myophosphorylase deficiency, phosphofructokinase deficiency, and phosphoglycerate mutase deficiency are inherited as autosomal recessive disorders. Phosphoglycerate kinase deficiency is X-linked recessive. Mutations can be found in the respective genes encoding the abnormal proteins in each of these disorders.
Training may enhance exercise tolerance, perhaps by increasing perfusion to muscle. Dietary intake of free glucose or fructose prior to activity may improve function but care must be taken to avoid obesity from ingesting too many calories.
LIPID AS AN ENERGY SOURCE AND ASSOCIATED DEFECTS
Lipid is an important muscle energy source during rest and during prolonged, submaximal exercise. Fatty acids are derived from circulating very-low-density lipoprotein (VLDL) in the blood or from triglycerides stored in muscle fibers. Oxidation of fatty acids occurs in the mitochondria. To enter the mitochondria, a fatty acid must first be converted to an “activated fatty acid,” acyl-CoA. The acyl-CoA must be linked with carnitine by the enzyme carnitine palmitoyltransferase (CPT) I for transport into the mitochondria. CPT I is present on the inner side of the outer mitochondrial membrane. Carnitine is removed by CPT II, an enzyme attached to the inside of the inner mitochondrial membrane, allowing transport of acyl-CoA into the mitochondrial matrix for β-oxidation.
Carnitine Palmitoyltransferase Deficiency CPT II deficiency is the most common recognizable cause of recurrent myoglobinuria, more common than the glycolytic defects. Onset is usually in the teenage years or early twenties. Muscle pain and myoglobinuria typically occur after prolonged exercise but can also be precipitated by fasting or infections; up to 20% of patients do not exhibit myoglobinuria, however. Strength is normal between attacks. In contrast to disorders caused by defects in glycolysis, in which muscle cramps follow short, intense bursts of exercise, the muscle pain in CPT II deficiency does not occur until the limits of utilization have been exceeded and muscle breakdown has already begun. Episodes of rhabdomyolysis may produce severe weakness. In young children and newborns, CPT II deficiency can present with a very severe clinical picture including hypoketotic hypoglycemia, cardiomyopathy, liver failure, and sudden death.
Serum CK levels and EMG findings are both usually normal between episodes. A normal rise of venous lactate during forearm exercise distinguishes this condition from glycolytic defects, especially myophosphorylase deficiency. Muscle biopsy does not show lipid accumulation and is usually normal between attacks. The diagnosis requires direct measurement of muscle CPT or genetic testing.
CPT II deficiency is much more common in men than women (5:1); nevertheless, all evidence indicates autosomal recessive inheritance. A mutation in the gene for CPT II (chromosome 1p36) causes the disease in some individuals. Attempts to improve exercise tolerance with frequent meals and a low-fat, high-carbohydrate diet, or by substituting medium-chain triglycerides in the diet, have not proven to be beneficial.
Myoadenylate Deaminase Deficiency The muscle enzyme myoadenylate deaminase converts adenosine-5′-monophosphate (5′-AMP) to inosine monophosphate (IMP) with liberation of ammonia. Myoadenylate deaminase may play a role in regulating adenosine triphosphate (ATP) levels in muscles. Most individuals with myoadenylate deaminase deficiency have no symptoms. There have been a few reports of patients with this disorder who have exercise-exacerbated myalgia and myoglobinuria. Many questions have been raised about the clinical effects of myoadenylate deaminase deficiency, and, specifically, its relationship to exertional myalgia and fatigability, but there is no consensus.
MITOCHONDRIAL MYOPATHIES
In 1972, Olson and colleagues recognized that muscle fibers with significant numbers of abnormal mitochondria could be highlighted with the modified trichrome stain; the term ragged red fibers was coined. By electron microscopy, the mitochondria in ragged red fibers are enlarged and often bizarrely shaped and have crystalline inclusions. Since that seminal observation, the understanding of these disorders of muscle and other tissues has expanded (Chap. 82).
Mitochondria play a key role in energy production. Oxidation of the major nutrients derived from carbohydrate, fat, and protein leads to the generation of reducing equivalents. The latter are transported through the respiratory chain in the process known as oxidative phosphorylation. The energy generated by the oxidation-reduction reactions of the respiratory chain is stored in an electrochemical gradient coupled to ATP synthesis.
A novel feature of mitochondria is their genetic composition. Each mitochondrion possesses a DNA genome that is distinct from that of the nuclear DNA. Human mitochondrial DNA (mtDNA) consists of a double-strand, circular molecule comprising 16,569 base pairs. It codes for 22 transfer RNAs, 2 ribosomal RNAs, and 13 polypeptides of the respiratory chain enzymes. The genetics of mitochondrial diseases differ from the genetics of chromosomal disorders. The DNA of mitochondria is directly inherited from the cytoplasm of the gametes, mainly from the oocyte. The sperm contributes very little of its mitochondria to the offspring at the time of fertilization. Thus, mitochondrial genes are derived almost exclusively from the mother, accounting for maternal inheritance of some mitochondrial disorders.
Patients with mitochondrial myopathies have clinical manifestations that usually fall into three groups: chronic progressive external ophthalmoplegia (CPEO), skeletal muscle–CNS syndromes, and pure myopathy simulating muscular dystrophy or metabolic myopathy.
PROGRESSIVE EXTERNAL OPHTHALMOPLEGIA SYNDROMES WITH RAGGED RED FIBERS
The single most common sign of a mitochondrial myopathy is CPEO, occurring in >50% of all mitochondrial myopathies. Varying degrees of ptosis and weakness of extraocular muscles are seen, usually in the absence of diplopia, a point of distinction from disorders with fluctuating eye weakness (e.g., myasthenia gravis).
KEARNS-SAYRE SYNDROME (KSS)
KSS is a widespread multiorgan system disorder with a defined triad of clinical findings: onset before age 20, CPEO, and pigmentary retinopathy, plus one or more of the following features: complete heart block, cerebrospinal fluid (CSF) protein >1 g/L (100 mg/dL), or cerebellar ataxia. Some patients with CPEO and ragged red fibers may not fulfill all of the criteria for KSS. The cardiac disease includes syncopal attacks and cardiac arrest related to the abnormalities in the cardiac conduction system: prolonged intraventricular conduction time, bundle branch block, and complete atrioventricular block. Death attributed to heart block occurs in ~20% of the patients. Varying degrees of progressive limb muscle weakness and easy fatigability affect activities of daily living. Endocrine abnormalities are common, including gonadal dysfunction in both sexes with delayed puberty, short stature, and infertility. Diabetes mellitus is a cardinal sign of mitochondrial disorders and is estimated to occur in 13% of KSS patients. Other less common endocrine disorders include thyroid disease, hyperaldosteronism, Addison’s disease, and hypoparathyroidism. Both mental retardation and dementia are common accompaniments to this disorder. Serum CK levels are normal or slightly elevated. Serum lactate and pyruvate levels may be elevated. EMG is myopathic. Nerve conduction studies may be abnormal related to an associated neuropathy. Muscle biopsies reveal ragged red fibers, highlighted in oxidative enzyme stains, many showing defects in cytochrome oxidase. By electron microscopy, there are increased numbers of mitochondria that often appear enlarged and contain paracrystalline inclusions.
KSS is a sporadic disorder. The disease is caused by single mtDNA deletions presumed to arise spontaneously in the ovum or zygote. The most common deletion, occurring in about one-third of patients, removes 4977 bp of contiguous mtDNA. Monitoring for cardiac conduction defects is critical. Prophylactic pacemaker implantation is indicated when ECGs demonstrate a bifascicular block. In KSS, no benefit has been shown for supplementary therapies, including multivitamins or coenzyme Q10. Of all the proposed options, exercise might be the most applicable but must be approached cautiously because of defects in the cardiac conduction system.
PROGRESSIVE EXTERNAL OPHTHALMOPLEGIA (PEO)
This condition is caused by nuclear DNA mutations affecting mtDNA copy number and integrity and is thus inherited in a Mendelian fashion. Onset is usually after puberty. Fatigue, exercise intolerance, and complaints of muscle weakness are typical. Some patients notice swallowing problems. The neurologic examination confirms the ptosis and ophthalmoplegia, usually asymmetric in distribution. A sensorineural hearing loss may be encountered. Mild facial, neck flexor, and proximal weakness are typical. Rarely, respiratory muscles may be progressively affected and may be the direct cause of death. Serum CK is normal or mildly elevated. The resting lactate level is normal or slightly elevated but may rise excessively after exercise. CSF protein is normal. The EMG is myopathic, and nerve conduction studies are usually normal. Ragged red fibers are prominently displayed in the muscle biopsy. Southern blots of muscle reveal a normal mtDNA band at 16.6 kb and several additional mtDNA deletion bands with genomes varying from 0.5 to 10 kb.
This autosomal dominant form of CPEO has been linked to loci on three chromosomes: 4q35, 10q24, and 15q22–26. In the chromosome 4q-related form of disease, mutations of the gene encoding the heart and skeletal muscle–specific isoform of the adenine nucleotide translocator 1 (ANT1) gene are found. This highly abundant mitochondrial protein forms a homodimeric inner mitochondrial channel through which adenosine diphosphate (ADP) enters and ATP leaves the mitochondrial matrix. In the chromosome 10q–related disorder, mutations of the gene C10orf2 are found. Its gene product, twinkle, co-localizes with the mtDNA and is named for its punctate, starlike staining properties. The function of twinkle is presumed to be critical for lifetime maintenance of mitochondrial integrity. In the cases mapped to chromosome 15q, a mutation affects the gene encoding mtDNA polymerase (POLG), an enzyme important in mtDNA replication. Autosomal recessive PEO has also been described with mutations in the POLG gene. Point mutations have been identified within various mitochondrial tRNA (Leu, Ile, Asn, Trp) genes in families with maternal inheritance of PEO.
Exercise may improve function but will depend on the patient’s ability to participate.
MITOCHONDRIAL DNA SKELETAL MUSCLE–CENTRAL NERVOUS SYSTEM SYNDROMES
Myoclonic Epilepsy with Ragged Red Fibers (MERRF) The onset of MERRF is variable, ranging from late childhood to middle adult life. Characteristic features include myoclonic epilepsy, cerebellar ataxia, and progressive muscle weakness. The seizure disorder is an integral part of the disease and may be the initial symptom. Cerebellar ataxia precedes or accompanies epilepsy. It is slowly progressive and generalized. The third major feature of the disease is muscle weakness in a limb-girdle distribution. Other more variable features include dementia, peripheral neuropathy, optic atrophy, hearing loss, and diabetes mellitus.
Serum CK levels are normal or slightly increased. The serum lactate may be elevated. EMG is myopathic, and in some patients nerve conduction studies show a neuropathy. The electroencephalogram is abnormal, corroborating clinical findings of epilepsy. Typical ragged red fibers are seen on muscle biopsy. MERRF is caused by maternally inherited point mutations of mitochondrial tRNA genes. The most common mutation found in 80% of MERRF patients is an A to G substitution at nucleotide 8344 of tRNA lysine (A8344G tRNAlys). Other tRNAlys mutations include base-pair substitutions T8356C and G8363A. Only supportive treatment is possible, with special attention to epilepsy.
Mitochondrial Myopathy, Encephalopathy, Lactic Acidosis, and Strokelike Episodes (MELAS) MELAS is the most common mitochondrial encephalomyopathy. The term strokelike is appropriate because the cerebral lesions do not conform to a strictly vascular distribution. The onset in the majority of patients is before age 20. Seizures, usually partial motor or generalized, are common and may represent the first clearly recognizable sign of disease. The cerebral insults that resemble strokes cause hemiparesis, hemianopia, and cortical blindness. A presumptive stroke occurring before age 40 should place this mitochondrial encephalomyopathy high in the differential diagnosis. Associated conditions include hearing loss, diabetes mellitus, hypothalamic pituitary dysfunction causing growth hormone deficiency, hypothyroidism, and absence of secondary sexual characteristics. In its full expression, MELAS leads to dementia, a bedridden state, and a fatal outcome. Serum lactic acid is typically elevated. The CSF protein is also increased but is usually ≤1 g/L (100 mg/dL). Muscle biopsies show ragged red fibers. Neuroimaging demonstrates basal ganglia calcification in a high percentage of cases. Focal lesions that mimic infarction are present predominantly in the occipital and parietal lobes. Strict vascular territories are not respected, and cerebral angiography fails to demonstrate lesions of the major cerebral blood vessels.
MELAS is caused by maternally inherited point mutations of mitochondrial tRNA genes. Most of the tRNA mutations are lethal, accounting for the paucity of multigeneration families with this syndrome. The A3243G point mutation in tRNALeu(UUR) is the most common, occurring in ~80% of MELAS cases. About 10% of MELAS patients have other mutations of the tRNALeu(UUR) gene, including 3252G, 3256T, 3271C, and 3291C. Other tRNA gene mutations have also been reported in MELAS, including G583A tRNAPhe, G1642A tRNAVal, G4332A tRNAGlu, and T8316C tRNALys. Mutations have also been reported in mtDNA polypeptide-coding genes. Two mutations were found in the ND5 subunit of complex I of the respiratory chain. A missense mutation has been reported at mtDNA position 9957 in the gene for subunit III of cytochrome C oxidase. No specific treatment is available. Supportive treatment is essential for the strokelike episodes, seizures, and endocrinopathies.
PURE MYOPATHY SYNDROMES
Muscle weakness and fatigue can be the predominant manifestations of mtDNA mutations. When the condition affects exclusively muscle (pure myopathy), the disorder becomes difficult to recognize. Occasionally, mitochondrial myopathies can present with recurrent myoglobinuria without fixed weakness and thus resemble a glycogen storage disorder or CPT deficiency.
Mitochondrial DNA Depletion Syndromes Mitochondrial DNA depletion syndrome (MDS) is a heterogeneous group of disorders that are inherited in an autosomal recessive fashion and can present in infancy or adults. MDS can be caused by mutations in genes (TK2, DGUOK, RRM2B, TYMP, SUCLA1, and SUCLA2) that lead to depletion of pools of mitochondrial deoxyribonucleotide (dNTP) pools necessary for mtDNA replication The other major cause of MDS is a set of mutations in genes essential for mtDNA replication (e.g., POLG1 and C10orf2). The clinical phenotypes associated with MDS vary. Patients may develop a severe encephalopathy (e.g., Leigh’s syndrome), PEO, an isolated myopathy, myo-neuro-gastrointestinal-encephalopathy (MNGIE), and a sensory neuropathy with ataxia.
DISORDERS OF MUSCLE MEMBRANE EXCITABILITY
Muscle membrane excitability is affected in a group of disorders referred to as channelopathies. The heart may also be involved, resulting in life-threatening complications (Table 462e-10).
|
CLINICAL FEATURES OF PERIODIC PARALYSIS AND NONDYSTROPHIC MYOTONIAS |
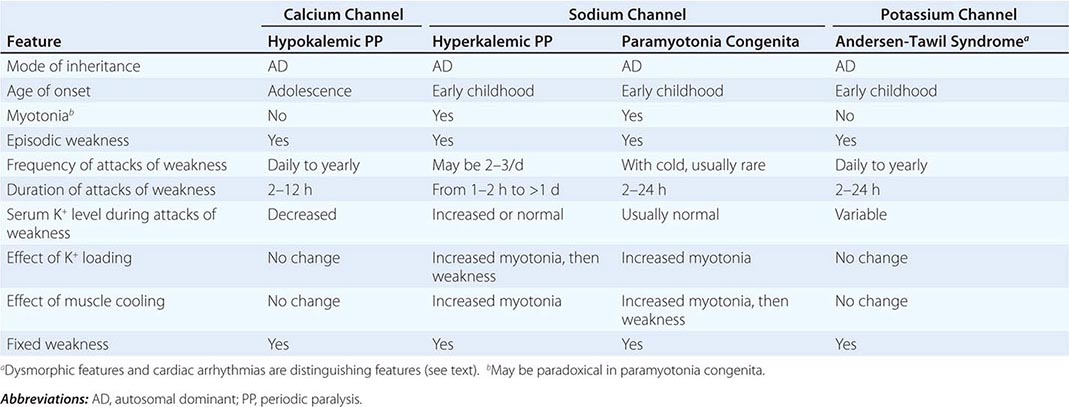
CALCIUM CHANNEL DISORDERS OF MUSCLE
Hypokalemic Periodic Paralysis (HypoKPP) Onset occurs at adolescence. Men are more often affected because of decreased penetrance in women. Episodic weakness with onset after age 25 is almost never due to periodic paralyses, with the exception of thyrotoxic periodic paralysis (see below). Attacks are often provoked by meals high in carbohydrates or sodium and may accompany rest following prolonged exercise. Weakness usually affects proximal limb muscles more than distal. Ocular and bulbar muscles are less likely to be affected. Respiratory muscles are usually spared, but when they are involved, the condition may prove fatal. Weakness may take as long as 24 h to resolve. Life-threatening cardiac arrhythmias related to hypokalemia may occur during attacks. As a late complication, patients commonly develop severe, disabling proximal lower extremity weakness.
Attacks of thyrotoxic periodic paralysis resemble those of primary HypoKPP. Despite a higher incidence of thyrotoxicosis in women, men, particularly those of Asian descent, are more likely to manifest this complication. Attacks abate with treatment of the underlying thyroid condition.
A low serum potassium level during an attack, excluding secondary causes, establishes the diagnosis. Interattack muscle biopsies show the presence of single or multiple centrally placed vacuoles or tubular aggregates. Provocative tests with glucose and insulin to establish a diagnosis are usually not necessary and are potentially hazardous.
In the midst of an attack of weakness, motor conduction studies may demonstrate reduced amplitudes, whereas EMG may show electrical silence in severely weak muscles. In between attacks, the EMG and routine nerve conduction studies are normal. However, a long exercise test may demonstrate a decrementing amplitude, and myopathic MUAPs may be seen on EMG in patients with fixed weakness.
HypoKPP is caused by mutations in either of two genes. HypoKPP type 1, the most common form, is inherited as an autosomal dominant disorder with incomplete penetrance. These patients have mutations in the voltage-sensitive, skeletal muscle calcium channel gene, CALCL1A3 (Fig. 462e-8). Approximately 10% of cases are HypoKPP type 2, arising from mutations in the voltage-sensitive sodium channel gene (SCN4A). In either instance, the mutations lead to an abnormal gating pore current that predisposes the muscle cell to depolarize when potassium levels are low. It is also now recognized that some cases of thyrotoxic HypoKPP are caused by genetic variants in a potassium channel (Kir 2.6), whose expression is regulated by thyroid hormone.
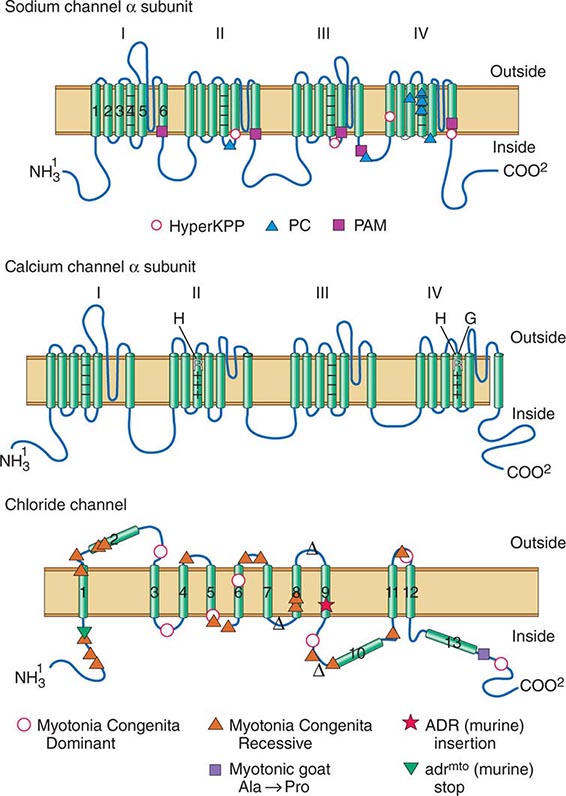
FIGURE 462e-8 The sodium and calcium channels are depicted here as containing four homologous domains, each with six membrane-spanning segments. The fourth segment of each domain bears positive charges and acts as the “voltage sensor” for the channel. The association of the four domains is thought to form a pore through which ions pass. Sodium channel mutations are shown along with the phenotype that they confer. HyperKPP, hyperkalemic periodic paralysis; PC, paramyotonia congenita; PAM, potassium-aggravated myotonia. See text for details.
The chloride channel is envisioned to have 10 membrane-spanning domains. The positions of mutations causing dominantly and recessively inherited myotonia congenita are indicated, along with mutations that cause this disease in mice and goats.
SODIUM CHANNEL DISORDERS OF MUSCLE
Hyperkalemic Periodic Paralysis (HyperKPP) The term hyperkalemic is misleading because patients are often normokalemic during attacks. The fact that attacks are precipitated by potassium administration best defines the disease. The onset is in the first decade; males and females are affected equally. Attacks are brief and mild, usually lasting 30 min to 4 ho. Weakness affects proximal muscles, sparing bulbar muscles. Attacks are precipitated by rest following exercise and fasting. In a variant of this disorder, the predominant symptom is myotonia without weakness (potassium-aggravated myotonia). The symptoms are aggravated by cold, and myotonia makes the muscles stiff and painful. This disorder can be confused with paramyotonia congenita, myotonia congenita, and proximal myotonic myopathy (DM2).
Potassium may be slightly elevated but may also be normal during an attack. As in HypoKPP, nerve conduction studies in HyperKPP muscle may demonstrate reduced motor amplitudes and the EMG may be silent in very weak muscles. In between attacks of weakness, the conduction studies are normal. The EMG will often demonstrate myotonic discharges during and between attacks.
The muscle biopsy shows vacuoles that are smaller, less numerous, and more peripheral compared to the hypokalemic form or tubular aggregates. Provocative tests by administration of potassium can induce weakness but are usually not necessary to establish the diagnosis. HyperKPP and potassium-aggravated myotonia are inherited as autosomal dominant disorders. Mutations of the voltage-gated sodium channel SCN4A gene (Fig. 462e-8) cause these conditions. For patients with frequent attacks, acetazolamide (125–1000 mg/d) is helpful. We have found mexiletine to be helpful in patients with significant myotonia.
Paramyotonia Congenita In paramyotonia congenita (PC), the attacks of weakness are cold-induced or occur spontaneously and are mild. Myotonia is a prominent feature but worsens with muscle activity (paradoxical myotonia). This is in contrast to classic myotonia in which exercise alleviates the condition. Attacks of weakness are seldom severe enough to require emergency room treatment. Over time patients develop interattack weakness as they do in other forms of periodic paralysis. PC is usually associated with normokalemia or hyperkalemia.
Serum CK is usually mildly elevated. Routine sensory and motor nerve conduction studies are normal. Short exercise test may be abnormal however, and cooling of the muscle often dramatically reduces the amplitude of the compound muscle action potentials. EMG reveals diffuse myotonic potentials in PC. Upon local cooling of the muscle, the myotonic discharges disappear as the patient becomes unable to activate MUAPs.
PC is inherited as an autosomal dominant condition; voltage-gated sodium channel mutations (Fig. 462e-8) are responsible, and thus this disorder is allelic with HyperKPP and potassium-aggravated myotonia. Patients with PC seldom seek treatment during attacks. Oral administration of glucose or other carbohydrates hastens recovery. Because interattack weakness may develop after repeated episodes, prophylactic treatment is usually indicated. Thiazide diuretics (e.g., chlorothiazide, 250–1000 mg/d) and mexiletine (slowly increase dose from 450 mg/d) are reported to be helpful. Patients should be advised to increase carbohydrates in their diet.
POTASSIUM CHANNEL DISORDERS
Andersen-Tawil Syndrome This rare disease is characterized by episodic weakness, cardiac arrhythmias, and dysmorphic features (short stature, scoliosis, clinodactyly, hypertelorism, small or prominent low-set ears, micrognathia, and broad forehead). The cardiac arrhythmias are potentially serious and life threatening. They include long QT, ventricular ectopy, bidirectional ventricular arrhythmias, and tachycardia. For many years, the classification of this disorder was uncertain because episodes of weakness are associated with elevated, normal, or reduced levels of potassium during an attack. In addition, the potassium levels differ among kindreds but are consistent within a family. Inheritance is autosomal dominant, with incomplete penetrance and variable expressivity. The disease is caused by mutations of the inwardly rectifying potassium channel (Kir 2.1) gene that heighten muscle cell excitability. The treatment is similar to that for other forms of periodic paralysis and must include cardiac monitoring. The episodes of weakness may differ between patients because of potassium variability. Acetazolamide may decrease the attack frequency and severity.
CHLORIDE CHANNEL DISORDERS
Two forms of this disorder, autosomal dominant (Thomsen’s disease) and autosomal recessive (Becker disease), are related to the same gene abnormality. Symptoms are noted in infancy and early childhood. The severity lessens in the third to fourth decade. Myotonia is worsened by cold and improved by activity. The gait may appear slow and labored at first but improves with walking. In Thomsen’s disease, muscle strength is normal, but in Becker disease, which is usually more severe, there may be muscle weakness. Muscle hypertrophy is usually present. Myotonic discharges are prominently displayed by EMG recordings.
Serum CK is normal or mildly elevated. The muscle biopsy shows hypertrophied fibers. The disease is inherited as dominant or recessive and is caused by mutations of the chloride channel gene (Fig. 462e-8) that increase muscle cell excitability. Many patients will not require treatment and learn that the symptoms improve with activity. Medications that can be used to decrease myotonia include quinine, phenytoin, and mexiletine.
ENDOCRINE AND METABOLIC MYOPATHIES
Many endocrine disorders cause weakness. Muscle fatigue is more common than true weakness. The cause of weakness in these disorders is not well defined. It is not even clear that weakness results from disease of muscle as opposed to another part of the motor unit, since the serum CK level is often normal (except in hypothyroidism) and the muscle histology is characterized by atrophy rather than destruction of muscle fibers. Nearly all endocrine myopathies respond to treatment.
THYROID DISORDERS
(See also Chap. 405) Abnormalities of thyroid function can cause a wide array of muscle disorders. These conditions relate to the important role of thyroid hormones in regulating the metabolism of carbohydrates and lipids as well as the rate of protein synthesis and enzyme production. Thyroid hormones also stimulate calorigenesis in muscle, increase muscle demand for vitamins, and enhance muscle sensitivity to circulating catecholamines.
Hypothyroidism Patients with hypothyroidism have frequent muscle complaints, and proximal muscle weakness occurs in about one-third of them. Muscle cramps, pain, and stiffness are common. Some patients have enlarged muscles. Features of slow muscle contraction and relaxation occur in 25% of patients; the relaxation phase of muscle stretch reflexes is characteristically prolonged and best observed at the ankle or biceps brachii reflexes. The serum CK level is often elevated (up to 10 times normal), even when there is minimal clinical evidence of muscle disease. EMG is typically normal. The cause of muscle enlargement has not been determined, and muscle biopsy shows no distinctive morphologic abnormalities.
Hyperthyroidism Patients who are thyrotoxic commonly have proximal muscle weakness and atrophy on examination, but they rarely complain of myopathic symptoms. Activity of deep tendon reflexes may be enhanced. Bulbar, respiratory, and even esophageal muscles may occasionally be affected, causing dysphagia, dysphonia, and aspiration. When bulbar involvement occurs, it is usually accompanied by chronic proximal limb weakness, but occasionally it presents in the absence of generalized thyrotoxic myopathy. Fasciculations may be apparent and, when coupled with increased muscle stretch reflexes, may lead to an erroneous diagnosis of amyotrophic lateral sclerosis. A form hypokalemic periodic paralysis can occur in patients who are thyrotoxic. Recently, mutations in the KCNJ18 gene that encodes for the inwardly rectifying potassium channel, Kir 2.6, have been discovered in up to a third of cases. Other neuromuscular disorders that occur in association with hyperthyroidism include myasthenia gravis (Chap. 461) and a progressive ocular myopathy associated with proptosis (Graves’ ophthalmopathy). Serum CK levels are not elevated in thyrotoxic myopathy, the EMG is normal, and muscle histology usually shows only atrophy of muscle fibers.
PARATHYROID DISORDERS
(See also Chap. 424)
Hyperparathyroidism Muscle weakness is an integral part of primary and secondary hyperparathyroidism. Proximal muscle weakness, muscle wasting, and brisk muscle stretch reflexes are the main features of this endocrinopathy. Some patients develop neck extensor weakness (part of the dropped head syndrome). Serum CK levels are usually normal or slightly elevated. Serum parathyroid hormone levels are elevated. Serum calcium and phosphorus levels show no correlation with the clinical neuromuscular manifestations. Muscle biopsies show only varying degrees of atrophy without muscle fiber degeneration.
Hypoparathyroidism An overt myopathy due to hypocalcemia rarely occurs. Neuromuscular symptoms are usually related to localized or generalized tetany. Serum CK levels may be increased secondary to muscle damage from sustained tetany. Hyporeflexia or areflexia is usually present and contrasts with the hyperreflexia in hyperparathyroidism.
ADRENAL DISORDERS
(See also Chap. 406) Conditions associated with glucocorticoid excess cause a myopathy; in fact, steroid myopathy is the most commonly diagnosed endocrine muscle disease. Glucocorticoid excess, either endogenous or exogenous (see “Drug-Induced Myopathies,” below), produces various degrees of proximal limb weakness. Muscle wasting may be striking. A cushingoid appearance usually accompanies clinical signs of myopathy. Histologic sections demonstrate muscle fiber atrophy, preferentially affecting type 2b fibers, rather than degeneration or necrosis of muscle fibers. Adrenal insufficiency commonly causes muscle fatigue. The degree of weakness may be difficult to assess but is typically mild. In primary hyperaldosteronism (Conn’s syndrome), neuromuscular complications are due to potassium depletion. The clinical picture is one of persistent muscle weakness. Long-standing hyperaldosteronism may lead to proximal limb weakness and wasting. Serum CK levels may be elevated, and a muscle biopsy may demonstrate degenerating fibers, some with vacuoles. These changes relate to hypokalemia and are not a direct effect of aldosterone on skeletal muscle.
PITUITARY DISORDERS
(See also Chap. 403) Patients with acromegaly usually have mild proximal weakness without muscle atrophy. Muscles often appear enlarged but exhibit decreased force generation. The duration of acromegaly, rather than the serum growth hormone levels, correlates with the degree of myopathy.
DIABETES MELLITUS
(See also Chap. 417) Neuromuscular complications of diabetes mellitus are most often related to neuropathy, with cranial and peripheral nerve palsies or distal sensorimotor polyneuropathy. Diabetic amyotrophy is a clumsy term because the condition represents a neuropathy affecting the proximal major nerve trunks and lumbosacral plexus. More appropriate terms for this disorder include diabetic proximal neuropathy and lumbosacral radiculoplexus neuropathy.
The only notable myopathy of diabetes mellitus is ischemic infarction of leg muscles, usually involving one of the thigh muscles but on occasion affecting the distal leg. This condition occurs in patients with poorly controlled diabetes and presents with abrupt onset of pain, tenderness, and edema of one thigh. The area of muscle infarction is hard and indurated. The muscles most often affected include the vastus lateralis, thigh adductors, and biceps femoris. Computed tomography (CT) or MRI can demonstrate focal abnormalities in the affected muscle. Diagnosis by imaging is preferable to muscle biopsy, if possible, as hemorrhage into the biopsy site can occur.
VITAMIN DEFICIENCY
Vitamin D deficiency (Chap. 96e) due to decreased intake, decreased absorption, or impaired vitamin D metabolism (as occurs in renal disease) may lead to chronic muscle weakness. Pain reflects the underlying bone disease (osteomalacia). Vitamin E deficiency may result from malabsorption. Clinical manifestations include ataxic neuropathy due to loss of proprioception and myopathy with proximal weakness. Progressive external ophthalmoplegia is a distinctive finding. It has not been established that deficiency of other vitamins causes a myopathy.
MYOPATHIES OF SYSTEMIC ILLNESS
Systemic illnesses such as chronic respiratory, cardiac, or hepatic failure are frequently associated with severe muscle wasting and complaints of weakness. Fatigue is usually a more significant problem than weakness, which is typically mild.
Myopathy may be a manifestation of chronic renal failure (CRF), independent of the better known uremic polyneuropathy. Abnormalities of calcium and phosphorus homeostasis and bone metabolism in chronic renal failure result from a reduction in 1,25-dihydroxyvitamin D, leading to decreased intestinal absorption of calcium. Hypocalcemia, further accentuated by hyperphosphatemia due to decreased renal phosphate clearance, leads to secondary hyperparathyroidism. Renal osteodystrophy results from the compensatory hyperparathyroidism, which leads to osteomalacia from reduced calcium availability and to osteitis fibrosa from the parathyroid hormone excess. The clinical picture of the myopathy of CRF is identical to that of primary hyperparathyroidism and osteomalacia. There is proximal limb weakness with bone pain.
Gangrenous calcification represents a separate, rare, and sometimes fatal complication of CRF. In this condition, widespread arterial calcification occurs and results in ischemia. Extensive skin necrosis may occur, along with painful myopathy and even myoglobinuria.
DRUG-INDUCED MYOPATHIES
Drug-induced myopathies are relatively uncommon in clinical practice with the exception of those caused by the cholesterol-lowering agents and glucocorticoids. Others impact practice to a lesser degree but are important to consider in specific situations. Table 462e-11 provides a comprehensive list of drug-induced myopathies with their distinguishing features.
|
DRUG-INDUCED MYOPATHIES |

MYOPATHY FROM LIPID-LOWERING AGENTS
All classes of lipid-lowering agents have been implicated in muscle toxicity, including fibrates (clofibrate, gemfibrozil), HMG-CoA reductase inhibitors (referred to as statins), niacin (nicotinic acid), and ezetimibe. Myalgia, malaise, and muscle tenderness are the most common manifestations. Muscle pain may be related to exercise. Patients may exhibit proximal weakness. Varying degrees of muscle necrosis are seen, and in severe reactions rhabdomyolysis and myoglobinuria occur. Concomitant use of statins with fibrates and cyclosporine is more likely to cause adverse reactions than use of one agent alone. Elevated serum CK is an important indication of toxicity. Muscle weakness is accompanied by a myopathic EMG, and muscle necrosis is observed by muscle biopsy. Severe myalgias, muscle weakness, significant elevations in serum CK (>three times baseline), and myoglobinuria are indications for stopping the drug. Patients usually improve with drug cessation, although this may take several weeks. Rare cases continue to progress after the offending agent is discontinued. It is possible that in such cases the statin may have triggered an immune-mediated necrotizing myopathy, as these individuals require aggressive immunotherapy (e.g., prednisone and sometimes other agents) to improve and often relapse when these therapies are discontinued. Interestingly, antibodies directed against the 100-kDa HMG-CoA reductase receptor on muscle fibers have been identified in many of these cases.
GLUCOCORTICOID-RELATED MYOPATHIES
Glucocorticoid myopathy occurs with chronic treatment or as “acute quadriplegic” myopathy secondary to high-dose IV glucocorticoid use. Chronic administration produces proximal weakness accompanied by cushingoid manifestations, which can be quite debilitating; the chronic use of prednisone at a daily dose of ≥30 mg/d is most often associated with toxicity. Patients taking fluorinated glucocorticoids (triamcinolone, betamethasone, dexamethasone) appear to be at especially high risk for myopathy. In chronic steroid myopathy, the serum CK is usually normal. Serum potassium may be low. The muscle biopsy in chronic cases shows preferential type 2 muscle fiber atrophy; this is not reflected in the EMG, which is usually normal.
Patients receiving high-dose IV glucocorticoids for status asthmaticus, chronic obstructive pulmonary disease, organ transplantation, or other indications may develop severe generalized weakness (critical illness myopathy). This myopathy, also known as acute quadriplegic myopathy, can also occur in the setting of sepsis. Involvement of the diaphragm and intercostal muscles causes respiratory failure and requires ventilatory support. In these settings, the use of glucocorticoids in combination with nondepolarizing neuromuscular blocking agents potentiates this complication. In critical illness myopathy, the muscle biopsy is abnormal, showing a distinctive loss of thick filaments (myosin) by electron microscopy. By light microscopy, there is focal loss of ATPase staining in central or paracentral areas of the muscle fiber. Calpain stains show diffusely reactive atrophic fibers. Withdrawal of glucocorticoids will improve the chronic myopathy. In acute quadriplegic myopathy, recovery is slow. Patients require supportive care and rehabilitation.
DRUG-INDUCED MITOCHONDRIAL MYOPATHY
Zidovudine, used in the treatment of HIV infection, is a thymidine analogue that inhibits viral replication by interrupting reverse transcriptase. Myopathy is a well-established complication of this agent. Patients present with myalgias, muscle weakness, and atrophy affecting the thigh and calf muscles. The complication occurs in about 17% of patients treated with doses of 1200 mg/d for 6 months. The introduction of protease inhibitors for treatment of HIV infection has led to lower doses of zidovudine therapy and a decreased incidence of myopathy. Serum CK is elevated and EMG is myopathic. Muscle biopsy shows ragged red fibers with minimal inflammation; the lack of inflammation serves to distinguish zidovudine toxicity from HIV-related myopathy. If the myopathy is thought to be drug related, the medication should be stopped or the dosage reduced.
DRUGS OF ABUSE AND RELATED MYOPATHIES
Myotoxicity is a potential consequence of addiction to alcohol and illicit drugs. Ethanol is one of the most commonly abused substances with potential to damage muscle. Other potential toxins include cocaine, heroin, and amphetamines. The most deleterious reactions occur from overdosing leading to coma and seizures, causing rhabdomyolysis, myoglobinuria, and renal failure. Direct toxicity can occur from cocaine, heroin, and amphetamines causing muscle breakdown and varying degrees of weakness. The effects of alcohol are more controversial. Direct muscle damage is less certain, since toxicity usually occurs in the setting of poor nutrition and possible contributing factors such as hypokalemia and hypophosphatemia. Alcoholics are also prone to neuropathy (Chap. 467).
Focal myopathies from self-administration of meperidine, heroin, and pentazocine can cause pain, swelling, muscle necrosis, and hemorrhage. The cause is multifactorial; needle trauma, direct toxicity of the drug or vehicle, and infection may all play a role. When severe, there may be overlying skin induration and contractures with replacement of muscle by connective tissue. Elevated serum CK and myopathic EMG are characteristic of these reactions. The muscle biopsy shows widespread or focal areas of necrosis. In conditions leading to rhabdomyolysis, patients need adequate hydration to reduce serum myoglobin and protect renal function. In all of these conditions, counseling is essential to limit drug abuse.
DRUG-INDUCED AUTOIMMUNE MYOPATHIES
As mentioned previously, an autoimmune necrotizing myopathy associated with autoantibodies directed against HMG-CoA rarely occurs in the setting of statin use. An inflammatory myopathy also may occur with D-penicillamine, sometimes used in the treatment of Wilson’s disease scleroderma, rheumatoid arthritis, and primary biliary cirrhosis. The incidence of this inflammatory muscle disease is about 1%. Myasthenia gravis is also induced by D-penicillamine, with a higher incidence estimated at 7%. These disorders resolve with drug withdrawal, although immunosuppressive therapy may be warranted in severe cases.
Scattered reports of other drugs causing an inflammatory myopathy are rare and include a heterogeneous group of agents: cimetidine, phenytoin, procainamide, and propylthiouracil. In most cases, a cause-and-effect relationship is uncertain. A complication of interest was related to L-tryptophan. In 1989 an epidemic of eosinophilia-myalgia syndrome (EMS) in the United States was caused by a contaminant in the product from one manufacturer. The product was withdrawn, and incidence of EMS diminished abruptly following this action.
OTHER DRUG-INDUCED MYOPATHIES
Certain drugs produce painless, largely proximal, muscle weakness. These drugs include the amphophilic cationic drugs (amiodarone, chloroquine, hydroxychloroquine) and antimicrotubular drugs (colchicine) (Table 462e-11). Muscle biopsy can be useful in the identification of toxicity because autophagic vacuoles are prominent pathologic features of these toxins.
463e |
Special Issues in Inpatient Neurologic Consultation |
Inpatient neurologic consultations usually involve questions regarding specific disease processes or prognostication after various cerebral injuries. Common reasons for neurologic consultation include stroke (Chap. 446), seizures (Chap. 445), altered mental status (Chap. 34), headache (Chap. 21), and management of coma and other neurocritical care conditions (Chaps. 328 and 330). This chapter focuses on additional common reasons for consultation that are not addressed elsewhere in the text.
CONSULTATIONS REGARDING CENTRAL NERVOUS SYSTEM DYSFUNCTION
HYPERPERFUSION STATES LEADING TO POSTERIOR REVERSIBLE ENCEPHALOPATHY SYNDROME
A group of neurologic disorders shares the common feature of hyperperfusion, probably related to endothelial dysfunction, playing a key role in pathogenesis. These seemingly diverse syndromes include hypertensive encephalopathy, eclampsia, postcarotid endarterectomy syndrome, and toxicity from calcineurin-inhibitor and other medications. Modern imaging techniques and experimental models suggest that vasogenic edema is typically the primary process leading to neurologic dysfunction; therefore, prompt recognition and management of this condition should allow for clinical recovery as long as superimposed hemorrhage or infarction has not occurred.
The brain’s autoregulatory capability successfully maintains a fairly stable cerebral blood flow in adults despite alterations in systemic mean arterial pressure (MAP) ranging from 50 to 150 mmHg (Chap. 330). In patients with chronic hypertension, this cerebral autoregulation curve is shifted, resulting in autoregulation working over a much higher range of pressures (e.g., 70–175 mmHg). In these hypertensive patients, cerebral blood flow is kept steady at higher MAP, but a rapid lowering of pressure can lead to ischemia on the lower end of the autoregulatory curve, even at values typically thought of as normotensive. This autoregulatory phenomenon is achieved through both myogenic and neurogenic influences causing small arterioles to contract and dilate. When the systemic blood pressure exceeds the limits of this mechanism, breakthrough of autoregulation occurs, resulting in hyperperfusion via increased cerebral blood flow, capillary leakage into the interstitium, and resulting edema. The predilection of all of the hyperperfusion disorders to affect the posterior rather than anterior portions of the brain may be due to a lower threshold for autoregulatory breakthrough in the posterior circulation or a vasculopathy that is more common in these blood vessels.
Although elevated or relatively elevated blood pressure is common in many of these disorders, some hyperperfusion states such as calcineurin-inhibitor toxicity occur with no apparent pressure rise. In these cases, vasogenic edema is likely due primarily to dysfunction of the capillary endothelium itself, leading to breakdown of the blood-brain barrier. It is useful to separate disorders of hyperperfusion into those caused primarily by increased pressure and those due mostly to endothelial dysfunction from a toxic or autoimmune etiology (Table 463e-1). In reality, both of these pathophysiologic processes likely play some role in each of these disorders.
|
SOME COMMON ETIOLOGIES OF HYPERPERFUSION SYNDROME |
The clinical presentation of all of the hyperperfusion syndromes is similar with prominent headaches, seizures, or focal neurologic deficits. Headaches have no specific characteristics, range from mild to severe, and may be accompanied by alterations in consciousness ranging from confusion to coma. Seizures may be present, and these can be of multiple types depending on the severity and location of the edema. Nonconvulsive seizures have been described in hyperperfusion states; therefore, a low threshold for obtaining an electroencephalogram (EEG) in these patients should be maintained. The typical focal deficit in hyperperfusion states is cortical visual loss, given the tendency of the process to involve the occipital lobes. However, any focal deficit can occur depending on the area affected, as evidenced by patients who, after carotid endarterectomy, exhibit neurologic dysfunction referable to the ipsilateral newly reperfused hemisphere. In conditions where increased cerebral blood flow plays a role, examination of the inpatient vital signs record will usually reveal a systemic blood pressure that is increased above the patient’s baseline. It appears as if the rapidity of rise, rather than the absolute value of pressure, is the most important risk factor.
The diagnosis in all of these conditions is clinical. The symptoms of these disorders are common and nonspecific, so a long differential diagnosis should be entertained, including consideration of other causes of confusion, focal neurologic deficits, headache, and seizures. Magnetic resonance imaging (MRI) has improved the ability of clinicians to diagnose hyperperfusion syndromes, although cases have been reported with normal imaging. Patients classically exhibit the high T2 signal of edema primarily in the posterior occipital lobes, not respecting any single vascular territory (Fig. 463e-1). Diffusion-weighted images are typically normal, emphasizing the vasogenic rather than cytotoxic nature of this edema. Imaging with computed tomography (CT) is less sensitive but may show a pattern of patchy hypodensity in the involved territory. Previously this classic radiographic appearance had been termed reversible posterior leukoencephalopathy (RPLE). However, this term has fallen out of favor because none of its elements are completely accurate. The radiographic and clinical changes are not always reversible; the territory involved is not uniquely posterior; and gray matter may be affected as well, rather than purely white matter as the term “leukoencephalopathy” intimates. The now more commonly used radiologic term posterior reversible encephalopathy syndrome (PRES) suffers from many of these same limitations. Vessel imaging may demonstrate narrowing of the cerebral vasculature, especially in the posterior circulation; whether this noninflammatory vasculopathy is a primary cause of the edema or occurs as a secondary phenomenon remains unclear. Other ancillary studies such as cerebrospinal fluid (CSF) analysis often yield nonspecific results. It should be noted that many of the substances that have been implicated, such as cyclosporine, can cause this syndrome even at low doses or after years of treatment. Therefore, normal serum levels of these medications do not exclude them as inciting agents.
FIGURE 463e-1 Axial fluid-attenuated inversion recovery (FLAIR) magnetic resonance imaging (MRI) of the brain in a patient taking cyclosporine after liver transplantation, who presented with seizures, headache, and cortical blindness. Increased signal is seen bilaterally in the occipital lobes predominantly involving the white matter, consistent with a hyperperfusion state secondary to calcineurin-inhibitor exposure.
In cases of hyperperfusion syndromes, treatment should commence urgently once the diagnosis is considered. Hypertension plays a key role commonly, and judicious lowering of the blood pressure with IV agents such as labetalol or nicardipine is advised along with continuous cardiac and blood pressure monitoring, often through an arterial line. It is reasonable to lower MAP by ~20% initially, as further lowering of the pressure may cause secondary ischemia and possibly infarction as pressure drops below the lower range of the patient’s autoregulatory capability. In cases where there is an identified cause of the syndrome, these etiologies should be treated promptly, including discontinuation of offending substances such as calcineurin inhibitors in toxic processes, treatment of immune-mediated disorders such as thrombotic thrombocytopenic purpura (TTP), and prompt delivery of the fetus in eclampsia. Seizures must be identified and controlled, often necessitating continuous EEG monitoring. Anticonvulsants are effective when seizure activity is identified, but in the special case of eclampsia, there is evidence to support the use of magnesium sulfate for seizure control.
POST-CARDIAC BYPASS BRAIN INJURY
Central nervous system (CNS) injuries following open heart or coronary artery bypass grafting (CABG) surgery are common and include acute encephalopathy, stroke, and a chronic syndrome of cognitive impairment. Hypoperfusion and embolic disease are frequently involved in the pathogenesis of these syndromes, although multiple mechanisms may be involved in these critically ill patients who are at risk for various metabolic and polypharmaceutical complications.
The frequency of hypoxic injury secondary to inadequate blood flow intraoperatively has been markedly decreased by the use of modern surgical and anesthetic techniques. Despite these advances, some patients still experience neurologic complications from cerebral hypoperfusion or may suffer focal ischemia from carotid or focal intracranial stenoses in the setting of regional hypoperfusion. Postoperative infarcts in the border zones between vascular territories commonly are blamed on systemic hypotension, although these infarcts can also result from embolic disease (Fig. 463e-2).
FIGURE 463e-2 Coronal fluid-attenuated inversion recovery (FLAIR) magnetic resonance imaging (MRI) of the brain in a patient presenting with altered mental status after an episode of hypotension during coronary artery bypass grafting (CABG). Increased signal is seen in the border zones bilaterally between the middle cerebral artery and anterior cerebral artery territories. Diffusion-weighted MRI sequences demonstrated restricted diffusion in these same locations, suggesting acute infarction.
Embolic disease is likely the predominant mechanism of cerebral injury during cardiac surgery as evidenced by diffusion-weighted MRI and intraoperative transcranial Doppler studies. It should be noted that some of the emboli that are found histologically in these patients are too small to be detected by standard imaging sequences; therefore, a negative MRI after surgery does not exclude the diagnosis of emboli-related complications. Thrombus in the heart itself as well as atheromas in the aortic arch can become dislodged during cardiac surgeries, releasing a shower of particulate matter into the cerebral circulation. Cross-clamping of the aorta, manipulation of the heart, extracorporeal circulation techniques (“bypass”), arrhythmias such as atrial fibrillation, and introduction of air through suctioning have all been implicated as potential sources of emboli. Histologic studies indicate that literally millions of tiny emboli may be released, even using modern surgical techniques.
This shower of microemboli results in a number of clinical syndromes. Occasionally, a single large embolus leads to an isolated large-vessel stroke that presents with obvious clinical focal deficits. More commonly, the emboli released are multiple and smaller. When there is a high burden of these small emboli, an acute encephalopathy can occur postoperatively, presenting as either a hyperactive or hypoactive confusional state, the latter of which is frequently and incorrectly ascribed to depression or a sedative-induced delirium. When the burden of microemboli is lower, no acute syndrome is recognized, but the patient may suffer a chronic cognitive deficit. Cardiac surgery can be viewed, like delirium, as a “stress test for the brain.” Some patients with a low cerebral reserve due to underlying cerebrovascular disease or an early neurodegenerative process will develop a chronic, cognitive deficit, whereas others with higher reserves may remain asymptomatic despite a similar dose of microemboli. In this manner, cardiac surgery may serve to unmask the early manifestations of neurodegenerative disorders such as Alzheimer’s disease.
Since modern techniques have successfully minimized hypoperfusion complications during these surgeries, much attention is now focused on reducing this inevitable shower of microemboli. Off-pump CABG surgeries have the advantages of reducing length of stay and perioperative complications; however, off-pump CABG probably does not preserve cognitive function compared with on-pump CABG. Filters placed in the aortic arch may have some promise in capturing these emboli, although convincing evidence is lacking. Development of successful endovascular operative approaches may provide a reasonable alternative to conventional CABG procedures, especially for patients at high risk of developing cognitive dysfunction after surgery due to advanced age, previous stroke, underlying neurodegenerative disorders, or severe atheromatous disease of the carotid arteries or aortic arch.
POST-SOLID ORGAN TRANSPLANT BRAIN INJURY
Patients who have undergone solid organ transplantation are at risk for neurologic injury in the postoperative period and for months to years thereafter. Neurologic consultants should view these patients as a special population at risk for both unique neurologic complications as well as for the usual disorders found in any critically ill inpatient.
Immunosuppressive medications are administered in high doses to patients after solid organ transplant, and many of these compounds have well-described neurologic complications. In patients with headache, seizures, or focal neurologic deficits taking calcineurin inhibitors, the diagnosis of hyperperfusion syndrome should be considered, as discussed above. This neurotoxicity occurs mainly with cyclosporine and tacrolimus and can present even in the setting of normal serum drug levels. Treatment primarily involves lowering the drug dosage or discontinuing the drug. Sirolimus has very few recorded cases of neurotoxicity and may be a reasonable alternative for some patients. Other examples of immunosuppressive medications and their neurologic complications include OKT3-associated akinetic mutism and the leukoencephalopathy seen with methotrexate, especially when it is administered intrathecally or with concurrent radiotherapy. In any solid organ transplant patient with neurologic complaints, a careful examination of the medication list is required to search for these possible drug effects.
Cerebrovascular complications of solid organ transplant are often first recognized in the immediate postoperative period. Border zone territory infarctions can occur, especially in the setting of systemic hypotension during cardiac transplant surgery. Embolic infarctions classically complicate cardiac transplantation, but all solid organ transplant procedures place patients at risk for systemic emboli. When cerebral embolization accompanies renal or liver transplantation surgery, a careful search for right-to-left shunting should include evaluation of the heart with agitated saline echocardiography (i.e., “bubble study”), as well as looking for intrapulmonary shunting. Renal and some cardiac transplant patients often have advanced atherosclerosis, providing yet another mechanism for stroke. Imaging with CT or MRI with diffusion is advised when cerebrovascular complications are suspected to confirm the diagnosis and to exclude intracerebral hemorrhage, which most often occurs in the setting of coagulopathy secondary to liver failure or after cardiac bypass procedures.
Given that patients with solid organ transplants are chronically immunosuppressed, infections are a common concern (Chap. 169). In any transplant patient with new CNS signs or symptoms such as seizure, confusion, or focal deficit, the diagnosis of a CNS infection should be considered and evaluated through imaging (usually MRI) and possibly lumbar puncture. The most common pathogens responsible for CNS infections in these patients vary based on time since transplant. In the first month posttransplant, common pathogens include the usual bacterial organisms associated with surgical procedures and indwelling catheters. Starting in the second month posttransplant, opportunistic infections of the CNS become more common, including Nocardia and Toxoplasma species as well as fungal infections such as aspergillosis. Viral infections that can affect the brain of the immunosuppressed patient, such as herpes simplex virus, cytomegalovirus, human herpesvirus type 6 (HHV-6), and varicella, also become more common after the first month posttransplant. After 6 months posttransplant, immunosuppressed patients still remain at risk for these opportunistic bacterial, fungal, and viral infections but can also suffer late CNS infectious complications such as progressive multifocal leukoencephalopathy (PML) associated with JC virus and Epstein-Barr virus–driven clonal expansions of B cells resulting in posttransplant lymphoproliferative disorder or CNS lymphoma.
COMMON NEUROLOGIC COMPLICATIONS OF ELECTROLYTE DISTURBANCES
A wide variety of neurologic conditions can result from abnormalities in serum electrolytes, and consideration of electrolyte disturbances should be part of any inpatient neurologic consultation. A complete general discussion of fluid and electrolyte imbalance and homeostasis can be found in Chap. 63.
HYPERNATREMIA AND HYPEROSMOLALITY
The normal range of serum osmolality is around 275–295 mOsm/kg, but neurologic manifestations are usually seen only at levels >325 mOsm/kg. Hyperosmolality is usually due to hypernatremia, hyperglycemia, azotemia, or the addition of extrinsic osmoles such as mannitol, which is commonly used in critically ill neurologic patients. Hyperosmolality itself can lead to a generalized encephalopathy that is nonspecific and without focal findings; however, an underlying lesion such as a mass can become symptomatic under the metabolic stress of a hyperosmolar state, producing focal neurologic signs. Some patients with hyperosmolality from severe hyperglycemia can present, for unclear reasons, with generalized seizures or unilateral movement disorders, which usually respond to lowering of the serum glucose. The treatment of all forms of hyperosmolality involves calculation of apparent water losses and slow replacement so that the serum sodium declines no faster than 2 mmol/L (2 meq/L) per hour.
Hypernatremia leads to the loss of intracellular water, leading to cell shrinkage. In the cells of the brain, solutes such as glutamine and urea are generated under these conditions in order to minimize this shrinkage. Despite this corrective mechanism, when hypernatremia is severe (serum sodium >160 mmol/L [>160 meq/L]) or occurs rapidly, cellular metabolic processes fail and encephalopathy will result. There are many etiologies of hypernatremia including, most commonly, renal and extrarenal losses of water. Causes of neurologic relevance include central diabetes insipidus, where hyperosmolality is accompanied by submaximal urinary concentration due to inadequate release of arginine vasopressin (AVP) from the posterior pituitary, resulting often from pituitary injury in the setting of surgery, hemorrhage, infiltrative processes, or cerebral herniation.
HYPONATREMIA
Hyponatremia is commonly defined as a serum sodium <135 mmol/L (<135 meq/L). Neurologic symptoms occur at different levels of low sodium, depending not only on the absolute value but also on the rate of fall. In patients with hyponatremia that develops over hours, life-threatening seizures and cerebral edema may occur at values as high as 125 mmol/L. In contrast, some patients with more chronic hyponatremia that has slowly developed over months to years may be asymptomatic even with serum levels <110 mmol/L. Correction of hyponatremia, especially when chronic, must take place slowly in order to avoid additional neurologic complications. Cells in the brain swell in hypotonic hyponatremic states but may compensate over time by excreting solute into the extracellular space, leading to restoration of cell volume when water follows the solute out of the cells. If treatment of hyponatremia results in a rapid rise in serum sodium, cells in the brain may quickly shrink, leading to osmotic demyelination, a process that previously was thought to be limited exclusively to the brainstem (central pontine myelinolysis; see Fig. 330-6), but now has been described elsewhere in the CNS.
Treatment of hyponatremia is dependent on the cause. Hypertonic hyponatremia treatment focuses on correcting the underlying condition, such as hyperglycemia. Isovolemic hyponatremia (syndrome of inappropriate antidiuretic hormone [SIADH]) is managed with water restriction or administration of AVP antagonists. The management of choice for patients with hypervolemic hypotonic hyponatremia is free-water restriction and treatment of the underlying edematous disorder, such as hepatic failure, nephrotic syndrome, or congestive heart failure. Finally, in hypovolemic hypotonic hyponatremia, volume is replaced with isotonic saline while underlying conditions of the kidneys, adrenals, and gastrointestinal tract are addressed.
One neurologic cause of hypovolemic hypotonic hyponatremia is the cerebral salt-wasting syndrome that accompanies subarachnoid hemorrhage and, less commonly, other cerebral processes such as meningitis, stroke, or traumatic brain injury. In these cases, the degree of renal sodium excretion can be remarkable, and large amounts of saline, hypertonic saline, or oral sodium may need to be given in a judicious fashion in order to avoid complications from cerebral edema.
HYPOKALEMIA
Hypokalemia, defined as a serum potassium level < 3.5 mmol/L (<3.5 meq/L), occurs either because of excessive potassium losses (from the kidneys or gut) or due to an abnormal potassium distribution between the intracellular and extracellular spaces. At very low levels (<1.5 mmol/L), hypokalemia may be life threatening due to the risk of cardiac arrhythmia and may present neurologically with severe muscle weakness and paralysis. Hypokalemic periodic paralysis is a rare disorder caused by excessive intracellular potassium uptake in the setting of a calcium or sodium channel mutation. Treatment of hypokalemia is dependent on the etiology but usually includes replacement of potassium through oral or IV routes as well as correcting the cause of potassium balance problems (e.g., eliminating β2-adrenergic agonist medications or treating the underlying cause of severe diarrhea).
HYPERKALEMIA
Hyperkalemia is defined as a serum potassium level >5.5 mmol/L (>5.5 meq/L) and can neurologically present as muscle weakness with or without paresthesias. Hyperkalemia becomes life threatening when it produces electrocardiographic abnormalities such as peaked T waves or a widened QRS complex. In these cases, prompt treatment is essential and consists of strategies that protect the heart against arrhythmias (calcium gluconate administration); promote potassium redistribution into cells (with glucose, insulin, and β2-agonist medications); and increase potassium removal (through sodium polystyrene sulfonate, loop diuretics, or hemodialysis).
CALCIUM DISTURBANCES
Hypercalcemia usually occurs in the setting of either hyperparathyroidism or systemic malignancy. Neurologic manifestations include encephalopathy as well as muscle weakness due to reduced neuromuscular excitability. Seizures can occur but are more common in states of low calcium.
Hypocalcemia in adults often follows surgical treatment of the thyroid or parathyroid. Seizures and altered mental status dominate the neurologic picture and usually resolve with calcium repletion. Tetany is due to spontaneous, repetitive action potentials in peripheral nerves and remains the classic sign of symptomatic hypocalcemia.
MAGNESIUM DISTURBANCES
Disorders of magnesium are difficult to correlate with serum levels because a very small amount of total-body magnesium is located in the extracellular space. Hypomagnesemia presents neurologically with seizures, tremor, and myoclonus. When intractable seizures occur in the setting of hypomagnesemia, only administration of magnesium will lead to their resolution.
High levels of magnesium, in contrast, lead to CNS depression. Hypermagnesemia usually only occurs in the setting of renal failure or magnesium administration and can lead to confusion and muscular paralysis when severe.
CONSULTATIONS REGARDING PERIPHERAL NERVOUS SYSTEM DYSFUNCTION
ENTRAPMENT NEUROPATHIES
Polyneuropathy is a common cause of outpatient neurologic consultation (Chap. 459). In the inpatient setting, however, mononeuropathies are more frequent, especially the entrapment neuropathies that complicate many surgical procedures and medical conditions. Median neuropathy at the wrist (carpal tunnel syndrome) is the most frequent entrapment neuropathy by far, but it is rarely a cause for inpatient consultation. Mechanisms for perioperative mononeuropathy include traction, compression, and ischemia of the nerve. Imaging with MRI, including neurography, may allow these causes to be distinguished. In all cases of mononeuropathy, the diagnosis can be made through the clinical examination and then confirmed with electrodiagnostic studies in the subacute period, if necessary. Treatment consists mainly of avoidance of repetitive trauma but may also include surgical approaches to relieve pressure or repair the nerve.
RADIAL NEUROPATHY
Radial nerve injury classically presents with weakness of extension of the wrist and fingers (“wrist drop”) with or without more proximal weakness of extensor muscles of the upper extremity, depending on the site of injury. Sensory loss is in the distribution of the radial nerve, which includes the dorsum of the hand (Fig. 463e-3A). Compression at the level of the axilla, e.g., resulting from use of crutches, leads to weakness of the triceps, brachioradialis, and supinator muscles in addition to wrist drop. A more common site of compression occurs in the spiral groove of the upper arm in the setting of a humerus fracture or from sleeping with the arm draped over a bench or chair (“Saturday night palsy”). Sparing of the triceps is the rule when the nerve is injured in this location. Because extensors of the upper extremity are injured preferentially in radial nerve injury, these lesions may be mistaken for the pyramidal distribution of weakness that accompanies upper motor neuron lesions from brain or spinal cord processes, prompting neuroimaging to exclude acute stroke or mass lesion.
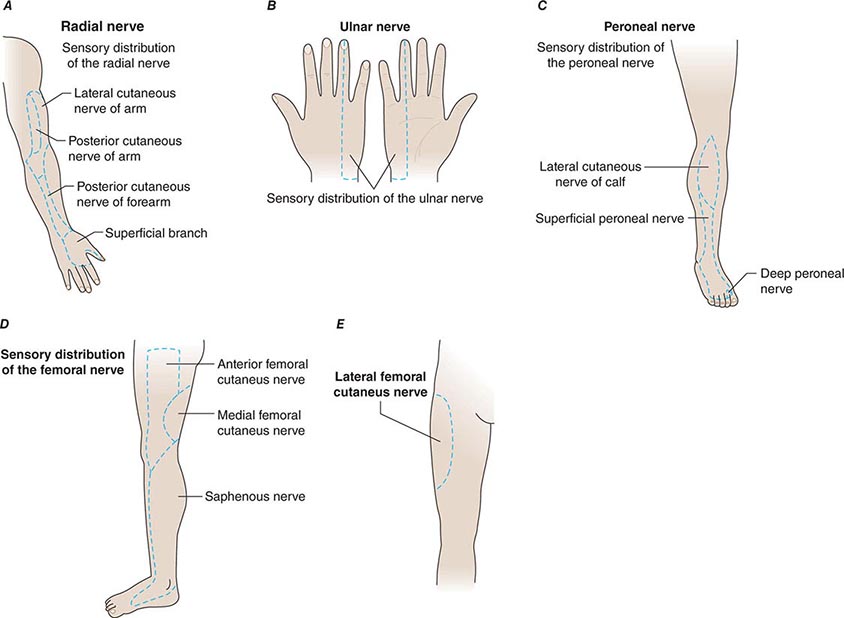
FIGURE 463e-3 Sensory distribution of peripheral nerves commonly affected by entrapment neuropathies. A. Radial nerve. B. Ulnar nerve. C. Peroneal nerve. D. Femoral nerve. E. Lateral femoral cutaneous nerve.
ULNAR NEUROPATHY
Compression of the ulnar nerve is the second most common entrapment neuropathy after carpal tunnel syndrome. The most frequent site of compression is at the elbow where the nerve passes superficially in the ulnar groove. Symptoms usually begin with tingling in the ulnar distribution, including the fourth and fifth digits of the hand (Fig. 463e-3B). Sensory symptoms may be worsened by elbow flexion due to increased pressure on the nerve, hence the tendency of patients to complain of increasing paresthesias at night when the arm is flexed at the elbow during sleep. Motor dysfunction can be disabling and involves most of the intrinsic hand muscles, limiting dexterity and strength of grasp and pinch. Etiologies of ulnar entrapment include trauma to the nerve (hitting the “funny bone”), malpositioning during anesthesia for surgical procedures, and chronic arthritis of the elbow. When a perioperative ulnar nerve injury is considered, stretch injury or trauma to the lower trunk of the brachial plexus should be entertained as well since its symptoms can mimic those of an ulnar neuropathy. If the clinical examination is equivocal, electrodiagnostic studies can definitively distinguish between plexus and ulnar nerve lesions a few weeks after the injury. Conservative methods of treatment are often the first step including bracing of the elbow to prevent flexion while sleeping. A variety of surgical approaches may also be effective in refractory or recurrent cases, including anterior ulnar nerve transposition and release of the flexor carpi ulnaris aponeurosis.
PERONEAL NEUROPATHY
The peroneal (also known as the fibular) nerve winds around the head of the fibula in the leg below the lateral aspect of the knee, and its superficial location at this site makes it vulnerable to trauma. Patients present with weakness of foot dorsiflexion (“foot drop”) as well as with weakness in eversion but not inversion at the ankle. Sparing of inversion, which is a function of muscles innervated by the tibial nerve, helps to distinguish peroneal neuropathies from L5 radiculopathies. Sensory loss involves the lateral aspect of the leg as well as the dorsum of the foot (Fig. 463e-3C). Fractures of the fibular head may be responsible for peroneal neuropathies, but in the perioperative setting, poorly applied braces exerting pressure on the nerve while the patient is unconscious, often in lithotomy position, are more often responsible. Tight-fitting stockings or casts of the upper leg can also cause a peroneal neuropathy, and thin individuals and those with recent weight loss are at increased risk.
PROXIMAL FEMORAL NEUROPATHY
Lesions of the proximal femoral nerve are relatively uncommon but may present dramatically with weakness of hip flexion, quadriceps atrophy, weakness of knee extension (often manifesting with leg-buckling falls), and an absent patellar reflex. Adduction of the thigh is spared as these muscles are supplied by the obturator nerve, thereby distinguishing a femoral neuropathy from a more proximal lumbosacral plexus lesion. The sensory loss found is in the distribution of the femoral nerve sensory branches including the anterior part of the thigh (Fig. 463e-3D). Compressive lesions from retroperitoneal hematomas or masses are common, and a CT of the pelvis should be obtained in all cases of femoral neuropathy to exclude these conditions. Bleeding into the pelvis resulting in hematoma can occur spontaneously, following trauma, or after intrapelvic surgeries such as renal transplantation. In intoxicated or comatose patients, stretch injuries to the femoral nerve are seen following prolonged, extreme hip flexion or extension. Rarely, attempts at femoral vein or arterial puncture can be complicated by injury to this nerve.
LATERAL FEMORAL CUTANEOUS NERVE
The symptoms of lateral femoral cutaneous nerve entrapment, commonly known as “meralgia paresthetica,” include sensory loss, pain, and dysesthesia in part of the area supplied by the nerve (Fig. 463e-3E). There is no motor component to the nerve, and therefore weakness is not a part of this syndrome. Symptoms often are worsened by standing or walking. Compression of the nerve occurs where it enters the leg near the inguinal ligament, usually in the setting of tight-fitting belts, pants, corsets, or recent weight gain, including that of pregnancy. The differential diagnosis of these symptoms includes hip problems such as trochanteric bursitis.
OBSTETRIC NEUROPATHIES
Pregnancy and delivery place women at special risk for a variety of nerve injuries. Radiculopathy due to a herniated lumbar disc is not common during pregnancy, but compressive injuries of the lumbosacral plexus do occur secondary to either the fetal head passing through the pelvis or the use of forceps during delivery. These plexus injuries are more frequent with cephalopelvic disproportion and often present with a painless unilateral foot drop which must be distinguished from a peroneal neuropathy caused by pressure on the nerve while in lithotomy position during delivery. Other compressive mononeuropathies of pregnancy include meralgia paresthetica, carpal tunnel syndrome, femoral neuropathy when the thigh is abducted severely in an effort to facilitate delivery of the fetal shoulder, and isolated obturator neuropathy during lithotomy positioning. The latter presents with medial thigh pain that may be accompanied by weakness of thigh adduction. There is also a clear association between pregnancy and an increased frequency of idiopathic facial palsy (Bell’s palsy).
SECTION 4 |
CHRONIC FATIGUE SYNDROME |
464e |
Chronic Fatigue Syndrome |
DEFINITION
Chronic fatigue syndrome (CFS) is a disorder characterized by persistent and unexplained fatigue resulting in severe impairment in daily functioning. Besides intense fatigue, most patients with CFS report concomitant symptoms such as pain, cognitive dysfunction, and unrefreshing sleep. Additional symptoms can include headache, sore throat, tender lymph nodes, muscle aches, joint aches, feverishness, difficulty sleeping, psychiatric problems, allergies, and abdominal cramps. Criteria for the diagnosis of CFS have been developed by the U.S. Centers for Disease Control and Prevention (Table 464e-1).
|
DIAGNOSTIC CRITERIA FOR CHRONIC FATIGUE SYNDROME |
EPIDEMIOLOGY
 CFS is seen worldwide, with adult prevalence rates varying between 0.2% and 0.4%. In the United States, the prevalence is higher among women (∼75% of cases), members of minority groups (African and Native Americans), and individuals with lower levels of education and occupational status. The mean age of onset is between 29 and 35 years. Many patients probably go undiagnosed and/or do not seek help.
CFS is seen worldwide, with adult prevalence rates varying between 0.2% and 0.4%. In the United States, the prevalence is higher among women (∼75% of cases), members of minority groups (African and Native Americans), and individuals with lower levels of education and occupational status. The mean age of onset is between 29 and 35 years. Many patients probably go undiagnosed and/or do not seek help.
ETIOLOGY
There are numerous hypotheses about the etiology of CFS; there is no definitively identified cause. Distinguishing between predisposing, precipitating, and perpetuating factors in CFS helps to provide a framework for understanding this complex condition (Table 464e-2).
|
PREDISPOSING, PRECIPITATING, AND PERPETUATING FACTORS IN CHRONIC FATIGUE SYNDROME |
Predisposing Factors Physical inactivity and trauma in childhood tend to increase the risk of CFS in adults. Neuroendocrine dysfunction may be associated with childhood trauma, reflecting a biological correlate of vulnerability. Psychiatric illness and physical hyperactivity in adulthood raise the risk of CFS in later life. Twin studies suggest a familial predisposition to CFS, but no causative genes have been identified.
Precipitating Factors Physical or psychological stress may elicit the onset of CFS. Most patients report an infection (usually a flulike illness or infectious mononucleosis) as the trigger of their fatigue. Relatively high percentages of CFS cases follow Q fever and Lyme disease. However, no differences in Epstein-Barr virus load and immunologic reactivity were found between individuals who developed CFS and those who did not. While antecedent infections are associated with CFS, a direct microbial causality is unproven and unlikely. One study identified a murine leukemia virus–related retrovirus (XMRV); however, several subsequent studies have established this virus as a laboratory artifact. Patients also often report other precipitating somatic events such as serious injury, surgery, pregnancy, or childbirth. Serious life events, such as the loss of a loved one or a job, military combat, and other stressful situations, may also precipitate CFS. One-third of all patients cannot recall a trigger.
Perpetuating Factors Once CFS has developed, numerous factors may impede recovery. Physicians may contribute to chronicity by ordering unnecessary diagnostic procedures, by persistently suggesting psychological causes, and by not acknowledging CFS as a diagnosis.
A patient’s focus on illness and avoidance of activities may perpetuate symptoms. A firm belief in a physical cause, a strong focus on bodily sensations, and a poor sense of control over symptoms may also prolong or exacerbate the fatigue and functional impairment. In most patients, inactivity is caused by negative illness perceptions rather than by poor physical fitness. Solicitous behavior of others may reinforce a patient’s illness-related perceptions and behavior. A lack of social support is another known perpetuating factor.
PATHOPHYSIOLOGY
The pathophysiology of CFS is unclear. Neuroimaging studies have found that CFS is associated with reduced gray matter volume, which in turn is associated with a decline in physical activity; these changes have been partially reversed following cognitive behavioral therapy (CBT). In addition, functional MRI data have suggested that abnormal patterns of activation correlate with self-reported problems with information processing. Neurophysiologic studies have shown altered CNS activation patterns during muscle contraction.
Evidence for immunologic dysfunction is inconsistent. Modest elevations in titers of antinuclear antibodies, reductions in immunoglobulin subclasses, deficiencies in mitogen-driven lymphocyte proliferation, reductions in natural killer cell activity, disturbances in cytokine production, and shifts in lymphocyte subsets have been described. None of these immune findings appears in most patients, nor does any correlate with the severity of CFS. In theory, symptoms of CFS could result from excessive production of a cytokine, such as interleukin 1, that induces asthenia and other flulike symptoms; however, compelling data in support of this hypothesis are lacking. There is some evidence that CFS patients have mild hypocortisolism, the degree of which is associated with a poorer response to CBT. Discrepancies in perceived and actual cognitive performance are consistent findings in patients with CFS.
DIAGNOSIS
In addition to a thorough history, a systematic physical examination is warranted to exclude disorders causing fatigue (e.g., endocrine disorders, neoplasms, heart failure). The heart rate of CFS patients is often slightly above normal. Laboratory tests serve primarily to exclude other diagnoses; no test can diagnose CFS. The following laboratory screen usually suffices: complete blood count; erythrocyte sedimentation rate; C-reactive protein; serum creatinine, electrolytes, calcium, and iron; blood glucose; creatine kinase; liver function tests; thyroid-stimulating hormone; anti-gliadin antibodies; and urinalysis. Serology for viral or bacterial infections usually is not helpful. No specific abnormalities have been identified on MRI or CT scans. The decrease in gray matter volume observed at a population level by MRI is not useful for diagnosis in the individual patient. Extensive, unfocused, and expensive testing in a search for the “hidden” cause of the fatigue is not productive. CFS is a constellation of symptoms with no pathognomonic features and remains a diagnosis of exclusion.
Bipolar disorders, schizophrenia, and substance abuse exclude a diagnosis of CFS, as do eating disorders, unless these issues have been resolved ≥5 years before symptom onset. In addition, CFS is excluded if the chronic fatigue developed immediately after a depressive episode. Depression developing in the course of the fatigue, however, does not preclude CFS. Concurrent psychiatric disorders, especially anxiety and mood disorders, are present in 30–60% of cases.
INITIAL MANAGEMENT
In cases of suspected CFS, the clinician should acknowledge the impact of the patient’s symptoms on daily functioning. Disbelief or denial can provoke an exacerbation of genuine symptoms, which in turn strengthens the clinician’s disbelief, leading to an unfortunate cycle of miscommunication. The possibility of CFS should be considered if a patient fulfils all criteria (Table 464e-1) and if other diagnoses have been excluded.
The patient should be asked to describe the symptoms (fatigue and accompanying symptoms) and their duration as well as their consequences (reduction in daily activities). To assess symptom severity and the extent of daily-life impairment, the patient should describe a typical day, from waking to retiring, and, for comparison, an average day prior to symptom onset. Next, potential fatigue-precipitating factors are sought. The severity of fatigue is commonly difficult to assess quantitatively; a brief questionnaire is often helpful (Fig. 464e-1).

FIGURE 464e-1 Shortened fatigue questionnaire.
The patient should be informed of the current understanding of precipitating and perpetuating factors and effective treatments and should be offered general advice about disease management. If CBT for CFS is not available as an initial option (see below) and depression and anxiety are present, these symptoms should be treated. For patients with headache, diffuse pain, and feverishness, nonsteroidal anti-inflammatory drugs may be helpful. Even modest improvements in symptoms can make an important difference in the patient’s degree of self-sufficiency and ability to appreciate life’s pleasures.
Controlled therapeutic trials have established that acyclovir, fludrocortisone, galantamine, modafinil, and IV immunoglobulin, among other agents, offer no significant benefit in CFS. Countless anecdotes circulate regarding other traditional and nontraditional therapies. It is important to guide patients away from those therapeutic modalities that are toxic, expensive, or unreasonable.
The patient should be encouraged to maintain regular sleep patterns, to remain as active as possible, and to gradually return to previous levels of exercise and other activity (work).
PROGNOSIS
Full recovery from untreated CFS is rare: the median annual recovery rate is 5% (range, 0–31%), and the median improvement rate is 39% (range, 8–63%). Patients with an underlying psychiatric disorder and those who continue to attribute their symptoms to an undiagnosed medical condition have poorer outcomes.
SECTION 5 |
PSYCHIATRIC AND ADDICTION DISORDERS |
465e |
Biology of Psychiatric Disorders |
Psychiatric disorders are central nervous system diseases characterized by disturbances in emotion, cognition, motivation, and socialization. They are highly heritable, with genetic risk comprising 20–90% of disease vulnerability. As a result of their prevalence, early onset, and persistence, they contribute substantially to the burden of illness worldwide. All psychiatric disorders are broad heterogeneous syndromes that currently lack well-defined neuropathology and bona fide biologic markers. Therefore, diagnoses continue to be made solely from clinical observations using criteria in the Diagnostic and Statistical Manual of Mental Disorders (DSM) of the American Psychiatric Association, which is in its fifth edition as of 2013.
There is increasing agreement that the classification of psychiatric illnesses in DSM does not accurately reflect the underlying biology of these disorders. Uncertainties in diagnosis make it extremely difficult to study the neurobiologic and genetic basis of mental illness. This has led to the development of an alternative diagnostic scheme, termed Research Domain Criteria (RDoCs), which classifies mental illness on the basis of core abnormalities—e.g., psychosis (loss of reality) or anhedonia (decreased ability to experience pleasure), which are common symptoms of several illnesses—with the idea that such classifications will assist in defining the biologic basis of at least key symptoms. Other factors that have impeded progress in understanding mental illness include the lack of access to pathologic brain tissue except upon death and the inherent limitations of animal models for disorders defined largely by behavioral abnormalities (e.g., hallucinations, delusions, guilt, suicidality) that are inaccessible in animals.
Despite these limitations, the past decade has seen significant advances. Neuroimaging methods are beginning to provide evidence of brain pathology, genome-wide association studies and high-throughput sequencing are at last revealing genes that confer risk for severe forms of mental illness, and investigations using better validated animal models are offering new insight into the molecular, cellular, and circuit mechanisms of disease pathogenesis. There is also excitement in the potential utility of neuron-like cells induced in vitro from a patient’s peripheral tissues (e.g., fibroblasts) providing novel ways of studying disease pathophysiology and screening for new treatments. There is consequently justified optimism that the field of psychiatry will transition from behaviorally defined syndromes to true biologic disease entities and that such advances will drive the development of improved treatments and eventually cures and preventive measures. This chapter describes several examples of recent discoveries in basic neuroscience that have informed our current understanding of disease mechanisms in psychiatry.
NEUROGENETICS
Because the human brain can only be examined indirectly during life, genome analyses have been extremely important for obtaining molecular clues about the pathogenesis of psychiatric disorders. A wealth of new information has been made possible by recent technological developments that have permitted affordable, large-scale genome-wide association studies and fine-scale sequencing. As an example, significant progress has been made in the genetics of autism spectrum disorders (ASDs), which are a heterogeneous group of neurodevelopmental diseases that share clinical features of impaired reciprocal social communication and interaction and restricted, repetitive patterns of behavior. ASDs are highly heritable; concordance rates in monozygotic twins (~60–90%) are roughly 10-fold higher than in dizygotic twins and siblings, whereas first-degree relatives show about 50-fold increased risk compared with the general population. ASDs are also genetically heterogeneous. More than 100 known mutations account for up to 20% of cases, although none individually accounts for more than 1% (Table 465e-1). It appears that most cases result from complex genetic mechanisms, including inheritance of multiple genetic risk variants and epigenetic modifications. For example, up to 10% of patients with autism have large (>500 kb) de novo copy number variations scattered across the genome, suggesting that hundreds of different genes can influence autism risk.
|
EXAMPLES OF GENES IMPLICATED IN AUTISM |
Amid this genetic heterogeneity, however, some common themes have emerged that inform pathogenesis of ASDs. For instance, many identified mutations are in genes that encode proteins involved in synaptic function and early transcriptional regulation (Table 465e-1) and have a clear relationship to activity-dependent neural responses that can affect the development of neural systems underlying cognition and social behaviors. Mutations in these genes may be detrimental by altering the balance of excitatory versus inhibitory synaptic signaling in local and extended circuits and by altering mechanisms that control brain growth. Some mutations affect genes (e.g., PTEN, TSC1, and TSC2) that negatively regulate signaling from several types of extracellular stimuli, including those transduced by receptor tyrosine kinases. Their dysregulation can alter neuronal growth as well as synaptic development and function.
With further understanding of pathogenesis and the definition of specific ASD subtypes, there is reason to believe that effective therapies will be identified. Work in mouse models has already demonstrated that some autism-like behaviors can be reversed, even in fully developed adult animals, by modifying the underlying pathology; these results encourage hope for many affected individuals. Treatments that target excitation-inhibition imbalance or altered mRNA translation appear to offer early promise. For example, the genes TSC1, TSC2, and PTEN are negative regulators of signaling through the target of rapamycin complex 1 (TORC1), which regulates protein synthesis. Rapamycin, a selective inhibitor of TORC1, can reverse several behavioral and synaptic defects in mice carrying null mutations in these genes. Another example is fragile × syndrome, which is the leading cause of inherited autism and mental disability and is due to mutations in FMR1 that result in loss of the encoded fragile × mental retardation protein (FMRP). FMRP is a polyribosome-associated mRNA-binding protein that represses the translation of a subset (~5%) of all mRNAs, several of which encode proteins that comprise the postsynaptic density, including the metabotropic glutamate receptor 5 (mGluR5). Treatment of Fmr1 knockout mice with mGluR5 antagonists reduces several behavioral and morphologic abnormalities in these mice; these promising preclinical results have led to ongoing trials of mGluR5 antagonists in humans with fragile × syndrome.
The ability to catalog common genetic variants and assay them on array-based platforms and, more recently, to carry out whole-exome sequencing has allowed investigators to collect sample sizes sufficient to detect genetic risk loci for schizophrenia with genome-wide significance. Several of the identified genes are parts of molecular complexes, such as voltage-gated calcium channels (in particular, CACNA1C and CACNB2) and the postsynaptic density of excitatory synapses. As in ASDs, copy number variants, single-nucleotide polymorphisms, and small insertions and deletions are common in schizophrenia. Genes that promote risk for drug addiction have also begun to emerge from large family and population studies. The best-established susceptibility loci are regions on chromosomes 4 and 5 containing GABAA receptor gene clusters linked to alcoholism and the CHRNA5-A3-B4 nicotinic acetylcholine receptor gene cluster on chromosome 15 associated with nicotine and alcohol addiction.
A recurrent theme that has emerged from genetic studies of psychiatric disorders is pleiotropy, namely, that many genes are associated with multiple psychiatric syndromes. For example, mutations in MECP2, FMR1, and TSC1 and TSC2 (see Table 465e-1 for abbreviations) can cause mental retardation without ASD, others in MECP2 can cause obsessive-compulsive and attention-deficit hyperactivity disorders, some alleles of NRXN1 are associated with symptoms of both ASD and schizophrenia, and common polymorphisms in CACNA1C are strongly associated with both schizophrenia and bipolar disorder. Likewise, duplication of chromosome 16p is associated with both schizophrenia and autism, whereas DiGeorge’s (velocardiofacial) syndrome region deletions and the DISC1 (disrupted in schizophrenia 1) locus on chromosome 22 are associated with schizophrenia, autism, and bipolar disorder. The association of genes with multiple syndromes attests to the complexity of psychiatric disorders and the influence of additional factors that combine to specify the ultimate phenotype, including regulatory variants that determine cell-type specificity and timing of gene expression, protective variants, and epigenetic effects.
SIGNAL TRANSDUCTION
Studies of signal transduction have revealed numerous intracellular signaling pathways that are perturbed in psychiatric disorders, and such research has provided insight into development of new therapeutic agents. For example, lithium is a highly effective drug for bipolar disorder and competes with magnesium to inhibit numerous magnesium-dependent enzymes, including the enzyme GSK3β and several enzymes involved in phosphoinositide signaling that lead to activation of protein kinase C. These findings have led to discovery programs focused on developing GSK3β or protein kinase C inhibitors as potential novel treatments for mood disorders.
The observations that tricyclic antidepressants (e.g., imipramine) inhibit serotonin and/or norepinephrine reuptake and that monoamine oxidase inhibitors (e.g., tranylcypromine) are effective antidepressants initially led to the view that depression is caused by a deficiency of these monoamines. However, this hypothesis has not been substantiated. A cardinal feature of these drugs is that long-term administration is needed for their antidepressant effects. This means that their short-term actions, namely promotion of serotonin or norepinephrine function, are not per se antidepressant but rather induce a cascade of adaptations in the brain that underlie their clinical effects. The nature of these therapeutic drug-induced adaptations has not been identified with certainty. One theory holds that, in a subset of depressed patients who display upregulation of the hypothalamic-pituitary-adrenal (HPA) axis characterized by increased secretion of corticotropin-releasing factor (CRF) and glucocorticoids, excessive glucocorticoids cause atrophy of hippocampal neurons, which is associated with reduced hippocampal volumes seen clinically. Chronic antidepressant administration might reverse this atrophy by increasing brain-derived neurotrophic factor (BDNF) in hippocampus. A role for stress-induced decreases in the generation of newly born hippocampal granule cell neurons, and its reversal by antidepressants through BDNF and other growth factors, has also been suggested.
A major advance in recent years has been the identification of several rapidly acting antidepressants with non–monoamine-based mechanisms of action. The best established is ketamine, a noncompetitive antagonist of N-methyl-D-aspartate (NMDA) glutamate receptors, which exerts rapid (hours) and robust antidepressant effects in severely depressed patients who have not responded to other treatments. Ketamine, which at higher doses is psychotomimetic and anesthetic, exerts these antidepressant effects at low doses with minimal side effects. However, the response to ketamine is transient, which has led to several approaches to maintain treatment response, such as repeated ketamine delivery. The mechanism underlying ketamine’s antidepressant action is not known, but its striking clinical efficacy has stimulated animal research on the role of glutamate neurotransmission and synaptic plasticity in key limbic regions. Recent evidence supports a role for TORC1 activation because administration of rapamycin blocks the antidepressant-like effects of ketamine in animal models. Mechanisms by which ketamine activates TORC1 are currently an active area of investigation.
A major goal in the field of drug abuse has been to identify neuroadaptive mechanisms that lead from recreational use to addiction. Such research has determined that repeated intake of abused drugs induces specific changes in cellular signal transduction, leading to changes in synaptic strength (long-term potentiation or depression) and neuronal structure (altered dendritic branching or cell soma size) within the brain’s reward circuitry. These modifications are mediated in part by changes in gene expression, achieved by drug regulation of transcription factors (e.g., CREB [cAMP response element-binding protein] and ΔFosB [a Fos family protein]) and their target genes. Such alterations in gene expression are associated with lasting alterations in epigenetic modifications, including histone acetylation and methylation and DNA methylation. These adaptations provide opportunities for developing treatments targeted to drug-addicted individuals. The fact that the spectrum of these adaptations partly differs depending on the particular addictive substance used creates opportunities for treatments that are specific for different classes of addictive drugs and that may, therefore, be less likely to disturb basic mechanisms that govern normal motivation and reward.
Increasingly, causal relationships are being established between individual molecular and cellular adaptations and specific behavioral abnormalities that characterize the addicted state. For example, acute activation of μ-opioid receptors by morphine or other opiates activates Gi/o proteins, leading to inhibition of adenylyl cyclase, resulting in reduced cyclic AMP (cAMP) production, protein kinase A (PKA) activation, and activation of the transcription factor CREB. Repeated administration of these drugs (Fig. 465e-1) evokes a homeostatic response involving upregulation of adenylyl cyclases and PKA and increased activation of CREB. Such upregulation of cAMP-CREB signaling has been identified in the locus coeruleus, periaqueductal gray, ventral tegmental area (VTA), nucleus accumbens, and several other CNS regions, and contributes to opiate craving and signs of opiate withdrawal. The fact that endogenous opioid peptides do not produce tolerance and dependence, while morphine and heroin do, may relate to the observation that, unlike endogenous opioids, morphine and heroin are weak inducers of μ-opioid receptor desensitization and endocytosis. Therefore, these drugs cause prolonged receptor activation and inhibition of adenylyl cyclases, which provides a powerful stimulus for the upregulation of cAMP-CREB signaling that characterizes the opiate-dependent state.
FIGURE 465e-1 Opiate action in the locus coeruleus (LC). Binding of opiate agonists to μ-opioid receptors catalyzes nucleotide exchange on Gi and Go proteins, leading to inhibition of adenylyl cyclase, neuronal hyperpolarization via activation of K+ channels, and inhibition of neurotransmitter release via inhibition of Ca2+ channels. Inhibition of adenylyl cyclase (AC) reduces protein kinase A (PKA) activity and phosphorylation of several PKA substrate proteins, thereby altering their function. For example, opiates reduce phosphorylation of the cAMP response element-binding protein (CREB), which appears to initiate longer term changes in neuronal function. Chronic administration of opiates increases levels of AC isoforms, PKA catalytic (C) and regulatory (R) subunits, and the phosphorylation of several proteins, including CREB (indicated by red arrows). These changes contribute to the altered phenotype of the drug-addicted state. For example, the excitability of LC neurons is increased by enhanced cAMP signaling, although the ionic basis of this effect remains unknown. Activation of CREB causes upregulation of AC isoforms and tyrosine hydroxylase, the rate-limiting enzyme in catecholamine biosynthesis.
SYSTEMS NEUROSCIENCE
The study of interconnected brain circuits that drive behavior has been greatly advanced through newer methods in brain imagining that have documented abnormalities in neural function and connectivity in psychiatric disorders. The past decade has also witnessed the development of revolutionary new techniques—optogenetics and designer receptors and ligands—that provide unprecedented temporal and spatial control of neural circuits and permit detection of neural activity in real time in awake, behaving animals.
Positron emission tomography (PET), diffusion tensor imaging (DTI), and functional magnetic resonance imaging (fMRI) have identified neural circuits that contribute to psychiatric disorders, for example, defining the neural circuitry of mood within the brain’s limbic system (Fig. 465e-2). Integral to this system are the nucleus accumbens (important also for brain reward—see below), amygdala, hippocampus, and regions of prefrontal cortex. Recent optogenetic research in animals, where the activity of specific types of neurons in defined circuits can be controlled with light, has confirmed the importance of this limbic circuitry in controlling depression-related behavioral abnormalities. Given that many symptoms of depression (so-called neurovegetative symptoms) involve physiologic functions, a key role for the hypothalamus is also presumed. A subset of depressed individuals shows a small reduction in hippocampal size, as noted above. In addition, brain imaging investigations have revealed increased activation of the amygdala by negative stimuli and reduced activation of the nucleus accumbens by rewarding stimuli. There is also evidence for altered activity in prefrontal cortex, such as hyperactivity of subgenual area 25 in anterior cingulate cortex. Such findings have led to trials of deep brain stimulation (DBS) of either the nucleus accumbens or subgenual area 25, which appears to be therapeutic in some severely depressed individuals.
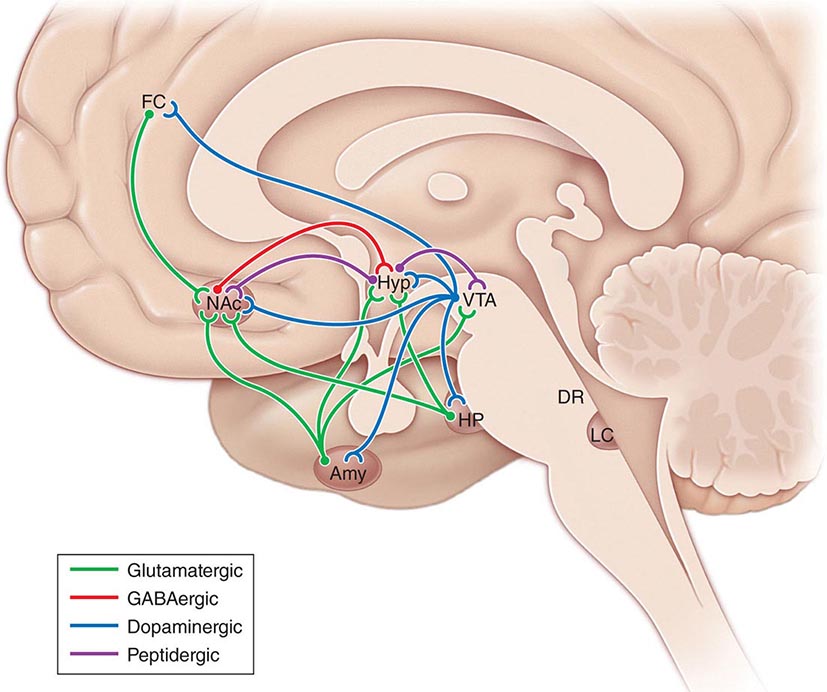
FIGURE 465e-2 Neural circuitry of depression and addiction. The figure shows a simplified summary of a series of limbic circuits in brain that regulate mood and motivation and are implicated in depression and addiction. Shown in the figure are the hippocampus (HP) and amygdala (Amy) in the temporal lobe, regions of prefrontal cortex, nucleus accumbens (NAc), and hypothalamus (Hyp). Only a subset of the known interconnections among these brain regions is shown. Also shown is the innervation of several of these brain regions by monoaminergic neurons. The ventral tegmental area (VTA) provides dopaminergic input to each of the limbic structures. Norepinephrine (from the locus coeruleus [LC]) and serotonin (from the dorsal raphe [DR] and other raphe nuclei) innervate all of the regions shown. In addition, there are strong connections between the hypothalamus and the VTA-NAc pathway. Important peptidergic projections from the hypothalamus include those from the arcuate nucleus that release β-endorphin and melanocortin and from the lateral hypothalamus that release orexin.
In schizophrenia, structural and functional imaging studies have identified a 3% loss of brain volume, most of which is in gray matter. This loss is progressive, and cortical gray matter appears to be particularly affected over time. The temporal lobes, particularly the left superior temporal gyrus, Heschl gyrus, and planum temporale, are often the most severely affected. The rate of loss in these regions as well as in frontal and parietal lobes appears to be greatest early in the course of the disease. Functional imaging studies provide evidence of reduced metabolic (presumably neural) activity in the dorsolateral prefrontal cortex at rest and when performing tests of executive function, including working memory. There is also evidence for impaired structural and task-related functional connectivity, mainly in frontal and temporal lobes.
These neuroimaging findings in schizophrenia have been confirmed in pathologic studies that show enlargement of the ventricular system and reduction of cortical and subcortical gray matter in frontal and temporal lobes and in the limbic system. The reduction in cortical thickness is associated with increased cell packing density and reduced neuropil (defined as axons, dendrites, and glial cell processes) without an apparent change in neuronal cell number. Specific classes of interneurons in prefrontal cortex consistently show reduced expression of the gene encoding the enzyme glutamic acid decarboxylase 1 (GAD1), which synthesizes γ-aminobutyric acid (GABA), the principal inhibitory neurotransmitter in the brain. Neuregulin 1 (NRG1), a member of the epidermal growth factor (EGF) family of growth factors, and its receptor ERBB4, have been implicated in schizophrenia, and they serve important roles in the maturation of GABAergic interneurons in cerebral cortex; loss of NRG1-ERBB4 in mice leads to a reduced neuropil, thus phenocopying a pathologic finding in schizophrenia. These findings are consistent with one working hypothesis of schizophrenia as a developmental neurodegenerative disorder due in part to loss of cortical interneurons in frontal and temporal lobes.
Work in rodent and nonhuman primate models of addiction has established the brain’s reward regions as key neural substrates for the acute actions of drugs of abuse and for addiction induced by repeated drug administration (Fig. 465e-2). Midbrain dopamine neurons in the VTA function normally as rheostats of reward: they are activated by natural rewards (food, sex, social interaction) or even by the expectation of such rewards, and many are suppressed by the absence of an expected reward or by aversive stimuli. These neurons thereby transmit crucial survival signals to the rest of the limbic brain to promote reward-related behavior, including motor responses to seek and obtain the rewards (nucleus accumbens), memories of reward-related cues (amygdala, hippocampus), and executive control of obtaining rewards (prefrontal cortex).
Drugs of abuse alter neurotransmission through initial actions at different classes of ion channels, neurotransmitter receptors, or neurotransmitter transporters (Table 465e-2). Studies in animal models have demonstrated that although the initial targets differ, the actions of these drugs converge on the brain’s reward circuitry by promoting dopamine neurotransmission in the nucleus accumbens and other limbic targets of the VTA. In addition, some drugs promote activation of opioid and cannabinoid receptors, which modulate this reward circuitry. By these mechanisms, drugs of abuse produce powerful rewarding signals, which, after repeated drug administration, corrupt a vulnerable brain’s reward circuitry in ways that promote addiction. Three major pathologic adaptations have been described. First, drugs produce tolerance and dependence in reward circuits, which promote escalating drug intake and a negative emotional state during drug withdrawal that promotes relapse. Second, sensitization to the rewarding effects of the drugs and associated cues is seen during prolonged abstinence and also triggers relapse. Third, executive function is impaired in such a way as to increase impulsivity and compulsivity, both of which promote relapse.
|
INITIAL ACTIONS OF DRUGS OF ABUSE |
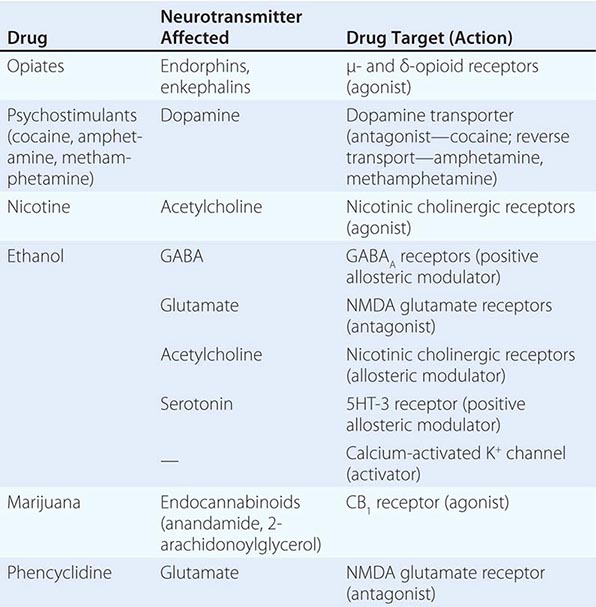
Imaging studies in humans confirm that addictive drugs, as well as craving for them, activate the brain’s reward circuitry. In addition, patients who abuse alcohol or psychostimulants show reduced gray matter in the prefrontal cortex as well as reduced activity in anterior cingulate and orbitofrontal cortex during tasks of attention and inhibitory control. It is thought that damage to these cortical areas contributes to addiction by impairing decision-making and increasing impulsivity.
NEUROINFLAMMATION
There is increasing evidence for the involvement of inflammatory mechanisms in a subset of depressed patients. These individuals display elevated blood levels of interleukin 6 (IL-6), tumor necrosis factor α (TNF-α), and other cytokines. Moreover, rodents exposed to chronic stress exhibit similar increases in peripheral cytokines, and peripheral or central delivery of those cytokines to normal rodents increases their susceptibility to chronic stress. These findings have led to the novel idea of using peripheral cytokines as biomarkers of a subtype of depression and the potential utility of developing new antidepressants that oppose cytokine action.
Recent evidence has also linked proinflammatory signaling in the brain to addiction, particularly to alcohol. Human alcoholism is associated with impaired innate immunity, increases in circulating proinflammatory cytokines, and an increase in brain levels of the cytokine monocyte chemotactic protein-1 (MCP-1, also referred to as CCL2). Many of these cytokines are produced by astrocytes and microglia, and by neurons under certain pathologic conditions, where they play important roles in modifying neuronal function and plasticity. For example MCP-1 modulates the release of certain neurotransmitters and, when administered into the VTA, increases neuronal excitability, promotes dopamine release, and increases locomotor activity. Recent gene expression array studies of alcohol drinking in mice have identified a network of regulated cytokines in brain, and a role in regulation of alcohol consumption has been recently validated for several of them, including IL-6. A major focus of current research is to define the site and mechanism by which proinflammatory cytokines impair brain function to elicit a depressive episode or promote drug abuse.
CONCLUSIONS
This brief narrative illustrates the substantial progress that is being made in understanding the genetic and neurobiologic basis of mental illness. It is anticipated that biologic measures will be used increasingly to diagnosis and subtype a psychiatric disorder and that targeted therapeutics will become available to treat them.
466 |
Mental Disorders |
Mental disorders are common in medical practice and may present either as a primary disorder or as a comorbid condition. The prevalence of mental or substance use disorders in the United States is approximately 30%, but only one-third of affected individuals are currently receiving treatment. Global burden of disease statistics indicate that 4 of the 10 most important causes of morbidity and attendant health care costs worldwide are psychiatric in origin.
Changes in health care delivery underscore the need for primary care physicians to assume responsibility for the initial diagnosis and treatment of the most common mental disorders. Prompt diagnosis is essential to ensure that patients have access to appropriate medical services and to maximize the clinical outcome. Validated patient-based questionnaires have been developed that systematically probe for signs and symptoms associated with the most prevalent psychiatric diagnoses and guide the clinician into targeted assessment. The Primary Care Evaluation of Mental Disorders (PRIME-MD; and a self-report form, the Patient Health Questionnaire) and the Symptom-Driven Diagnostic System for Primary Care (SDDS-PC) are inventories that require only 10 min to complete and link patient responses to the formal diagnostic criteria of anxiety, mood, somatoform, and eating disorders and to alcohol abuse or dependence.
A physician who refers patients to a psychiatrist should know not only when doing so is appropriate but also how to refer, because societal misconceptions and the stigma of mental illness impede the process. Primary care physicians should base referrals to a psychiatrist on the presence of signs and symptoms of a mental disorder and not simply on the absence of a physical explanation for a patient’s complaint. The physician should discuss with the patient the reasons for requesting the referral or consultation and provide reassurance that he or she will continue to provide medical care and work collaboratively with the mental health professional. Consultation with a psychiatrist or transfer of care is appropriate when physicians encounter evidence of psychotic symptoms, mania, severe depression, or anxiety; symptoms of posttraumatic stress disorder (PTSD); suicidal or homicidal preoccupation; or a failure to respond to first-order treatment. This chapter reviews the clinical assessment and treatment of some of the most common mental disorders presenting in primary care and is based on the Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition (DSM-5), the framework for categorizing psychiatric illness used in the United States. Eating disorders are discussed later in this chapter, and the biology of psychiatric and addictive disorders is discussed in Chap. 465e.
GLOBAL CONSIDERATIONS
![]() The DSM-5 and the tenth revision of the International Classification of Diseases (ICD-10), which is used more commonly worldwide, have taken somewhat differing approaches to the diagnosis of mental illness, but considerable effort has been expended to provide an operational translation between the two nosologies. Both systems are in essence purely descriptive and emphasize clinical pragmatism, in distinction to the Research Domain Criteria (RDOC) proposed by National Institute of Mental Health, which aspires to provide a causal framework for classification of behavioral disturbance. None of these diagnostic systems has as yet achieved adequate validation. The Global Burden of Disease Study 2010, using available epidemiologic data, nevertheless has reinforced the conclusion that, regardless of nosologic differences, mental and substance abuse disorders are the major cause of life-years lost to disability among all medical illnesses. There is general agreement that high-income countries will need to build capacity in professional training in low- and middle-income countries in order to provide an adequate balanced care model for the delivery of evidence-based therapies for mental disorders. Recent surveys that indicate a dramatic increase in mental disorder prevalence in rapidly developing countries, such as China, may reflect both an increased recognition of the issue, but also the consequence of social turmoil, stigma, and historically inadequate resources. The need for improved prevention strategies and for more definitive and effective interventional treatments remains a global concern.
The DSM-5 and the tenth revision of the International Classification of Diseases (ICD-10), which is used more commonly worldwide, have taken somewhat differing approaches to the diagnosis of mental illness, but considerable effort has been expended to provide an operational translation between the two nosologies. Both systems are in essence purely descriptive and emphasize clinical pragmatism, in distinction to the Research Domain Criteria (RDOC) proposed by National Institute of Mental Health, which aspires to provide a causal framework for classification of behavioral disturbance. None of these diagnostic systems has as yet achieved adequate validation. The Global Burden of Disease Study 2010, using available epidemiologic data, nevertheless has reinforced the conclusion that, regardless of nosologic differences, mental and substance abuse disorders are the major cause of life-years lost to disability among all medical illnesses. There is general agreement that high-income countries will need to build capacity in professional training in low- and middle-income countries in order to provide an adequate balanced care model for the delivery of evidence-based therapies for mental disorders. Recent surveys that indicate a dramatic increase in mental disorder prevalence in rapidly developing countries, such as China, may reflect both an increased recognition of the issue, but also the consequence of social turmoil, stigma, and historically inadequate resources. The need for improved prevention strategies and for more definitive and effective interventional treatments remains a global concern.
ANXIETY DISORDERS
Anxiety disorders, the most prevalent psychiatric illnesses in the general community, are present in 15–20% of medical clinic patients. Anxiety, defined as a subjective sense of unease, dread, or foreboding, can indicate a primary psychiatric condition or can be a component of, or reaction to, a primary medical disease. The primary anxiety disorders are classified according to their duration and course and the existence and nature of precipitants.
When evaluating the anxious patient, the clinician must first determine whether the anxiety antedates or postdates a medical illness or is due to a medication side effect. Approximately one-third of patients presenting with anxiety have a medical etiology for their psychiatric symptoms, but an anxiety disorder can also present with somatic symptoms in the absence of a diagnosable medical condition.
PANIC DISORDER
Clinical Manifestations Panic disorder is defined by the presence of recurrent and unpredictable panic attacks, which are distinct episodes of intense fear and discomfort associated with a variety of physical symptoms, including palpitations, sweating, trembling, shortness of breath, chest pain, dizziness, and a fear of impending doom or death. Paresthesias, gastrointestinal distress, and feelings of unreality are also common. Diagnostic criteria require at least 1 month of concern or worry about the attacks or a change in behavior related to them. The lifetime prevalence of panic disorder is 2–3%. Panic attacks have a sudden onset, developing within 10 min and usually resolving over the course of an hour, and they occur in an unexpected fashion. Some may occur when waking from sleep. The frequency and severity of panic attacks vary, ranging from once a week to clusters of attacks separated by months of well-being. The first attack is usually outside the home, and onset is typically in late adolescence to early adulthood. In some individuals, anticipatory anxiety develops over time and results in a generalized fear and a progressive avoidance of places or situations in which a panic attack might recur. Agoraphobia, which occurs commonly in patients with panic disorder, is an acquired irrational fear of being in places where one might feel trapped or unable to escape. It may, however, be diagnosed even if panic disorder is not present. Typically, it leads the patient into a progressive restriction in lifestyle and, in a literal sense, in geography. Frequently, patients are embarrassed that they are housebound and dependent on the company of others to go out into the world and do not volunteer this information; thus, physicians will fail to recognize the syndrome if direct questioning is not pursued.
Differential Diagnosis A diagnosis of panic disorder is made after a medical etiology for the panic attacks has been ruled out. A variety of cardiovascular, respiratory, endocrine, and neurologic conditions can present with anxiety as the chief complaint. Patients with true panic disorder will often focus on one specific feature to the exclusion of others. For example, 20% of patients who present with syncope as a primary medical complaint have a primary diagnosis of a mood, anxiety, or substance abuse disorder, the most common being panic disorder. The differential diagnosis of panic disorder is complicated by a high rate of comorbidity with other psychiatric conditions, especially alcohol and benzodiazepine abuse, which patients initially use in an attempt at self-medication. Some 75% of panic disorder patients will also satisfy criteria for major depression at some point in their illness.
When the history is nonspecific, physical examination and focused laboratory testing must be used to rule out anxiety states resulting from medical disorders such as pheochromocytoma, thyrotoxicosis, or hypoglycemia. Electrocardiogram (ECG) and echocardiogram may detect some cardiovascular conditions associated with panic such as paroxysmal atrial tachycardia and mitral valve prolapse. In two studies, panic disorder was the primary diagnosis in 43% of patients with chest pain who had normal coronary angiograms and was present in 9% of all outpatients referred for cardiac evaluation. Panic disorder has also been diagnosed in many patients referred for pulmonary function testing or with symptoms of irritable bowel syndrome.
Etiology and Pathophysiology The etiology of panic disorder is unknown but appears to involve a genetic predisposition, altered autonomic responsivity, and social learning. Panic disorder shows familial aggregation; the disorder is concordant in 30–45% of monozygotic twins, and genome-wide screens have identified suggestive risk loci. Acute panic attacks appear to be associated with increased noradrenergic discharges in the locus coeruleus. Intravenous infusion of sodium lactate evokes an attack in two-thirds of panic disorder patients, as do the α2-adrenergic antagonist yohimbine, cholecystokinin tetrapeptide (CCK-4), and carbon dioxide inhalation. It is hypothesized that each of these stimuli activates a pathway involving noradrenergic neurons in the locus coeruleus and serotonergic neurons in the dorsal raphe. Agents that block serotonin reuptake can prevent attacks. Patients with panic disorder have a heightened sensitivity to somatic symptoms, which triggers increasing arousal, setting off the panic attack; accordingly, therapeutic intervention involves altering the patient’s cognitive interpretation of anxiety-producing experiences as well as preventing the attack itself.
|
TREATMENT |
PANIC DISORDER |
Achievable goals of treatment are to decrease the frequency of panic attacks and to reduce their intensity. The cornerstone of drug therapy is antidepressant medication (Tables 466-1 through 466-3). Selective serotonin reuptake inhibitors (SSRIs) benefit the majority of panic disorder patients and do not have the adverse effects of tricyclic antidepressants (TCAs). Fluoxetine, paroxetine, sertraline, and the selective serotonin-norepinephrine reuptake inhibitor (SNRI) venlafaxine have received approval from the U.S. Food and Drug Administration (FDA) for this indication. These drugs should be started at one-third to one-half of their usual antidepressant dose (e.g., 5–10 mg fluoxetine, 25–50 mg sertraline, 10 mg paroxetine, venlafaxine 37.5 mg). Monoamine oxidase inhibitors (MAOIs) are also effective and may specifically benefit patients who have comorbid features of atypical depression (i.e., hypersomnia and weight gain). Insomnia, orthostatic hypotension, and the need to maintain a low-tyramine diet (avoidance of cheese and wine) have limited their use, however. Antidepressants typically take 2–6 weeks to become effective, and doses may need to be adjusted based on the clinical response.
|
ANTIDEPRESSANTS |
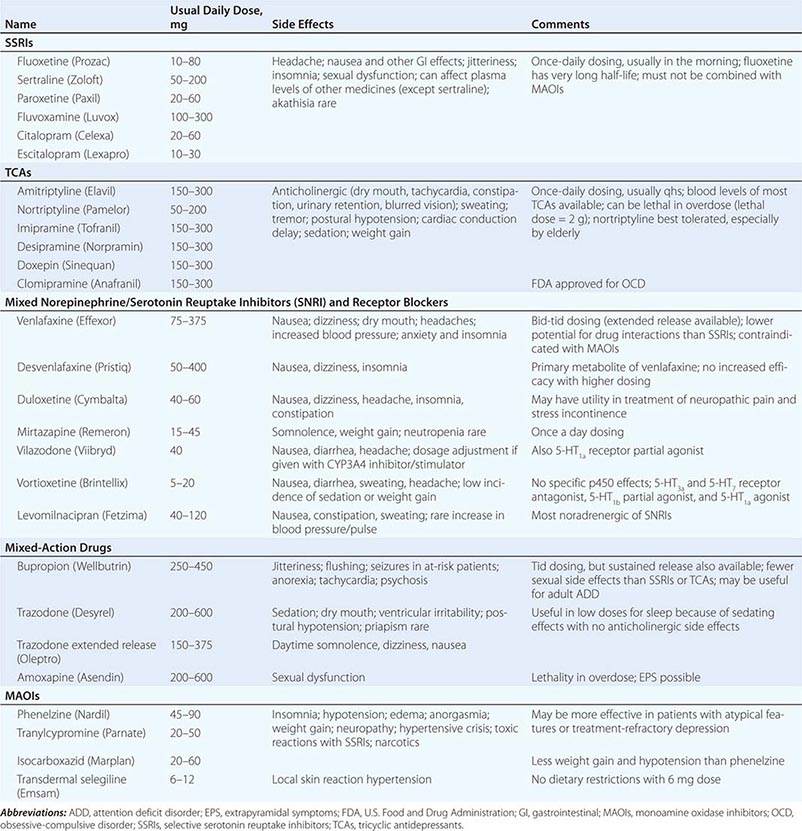
|
MANAGEMENT OF ANTIDEPRESSANT SIDE EFFECTS |
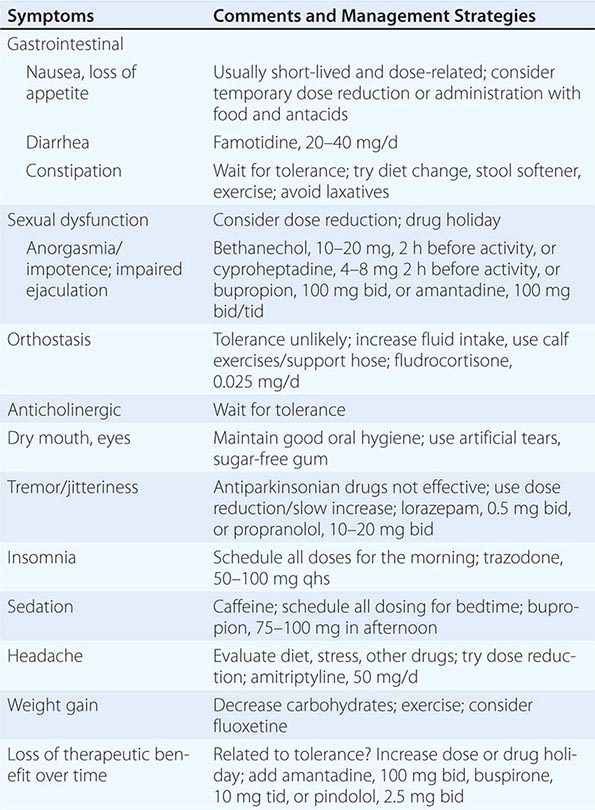
|
POSSIBLE DRUG INTERACTIONS WITH SELECTIVE SEROTONIN REUPTAKE INHIBITORS |

Because of anticipatory anxiety and the need for immediate relief of panic symptoms, benzodiazepines are useful early in the course of treatment and sporadically thereafter (Table 466-4). For example, alprazolam, starting at 0.5 mg qid and increasing to 4 mg/d in divided doses, is effective, but patients must be monitored closely, as some develop dependence and begin to escalate the dose of this medication. Clonazepam, at a final maintenance dose of 2–4 mg/d, is also helpful; its longer half-life permits twice-daily dosing, and patients appear less likely to develop dependence on this agent.
|
ANXIOLYTICS |
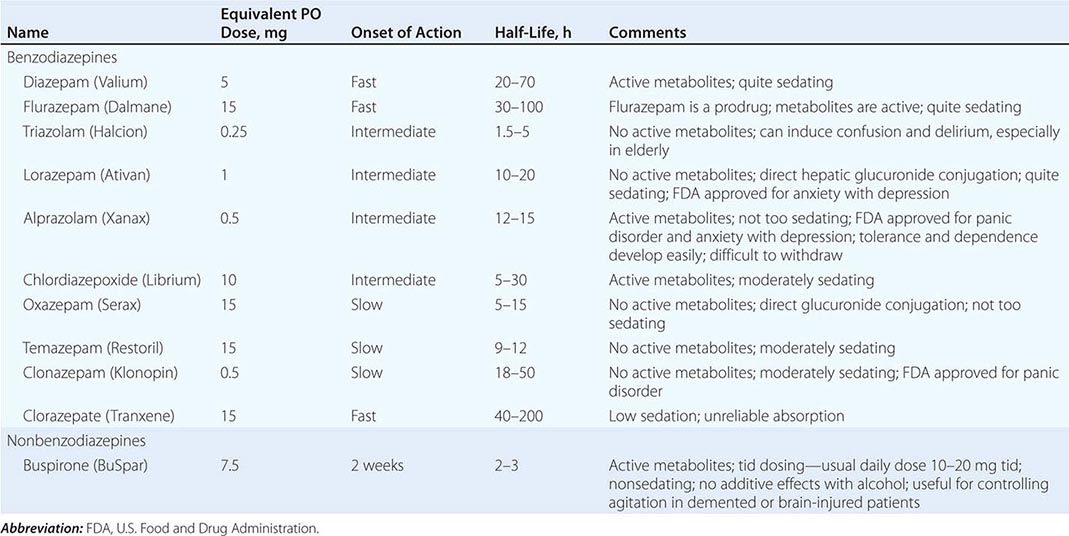
Early psychotherapeutic intervention and education aimed at symptom control enhance the effectiveness of drug treatment. Patients can be taught breathing techniques, be educated about physiologic changes that occur with panic, and learn to expose themselves voluntarily to precipitating events in a treatment program spanning 12–15 sessions. Homework assignments and monitored compliance are important components of successful treatment. Once patients have achieved a satisfactory response, drug treatment should be maintained for 1–2 years to prevent relapse. Controlled trials indicate a success rate of 75–85%, although the likelihood of complete remission is somewhat lower.
[/level-membership-for-internal-medicine-category][not-level-membership-for-internal-medicine-category]
462e |
Muscular Dystrophies and Other Muscle Diseases |
Skeletal muscle diseases, or myopathies, are disorders with structural changes or functional impairment of muscle. These conditions can be differentiated from other diseases of the motor unit (e.g., lower motor neuron or neuromuscular junction pathologies) by characteristic clinical and laboratory findings.
Myasthenia gravis and related disorders are discussed in Chap. 461; dermatomyositis, polymyositis, and inclusion body myositis are discussed in Chap. 388.
CLINICAL FEATURES
Most myopathies present with proximal, symmetric limb weakness (arms or legs) with preserved reflexes and sensation. However, asymmetric and predominantly distal weakness can be seen in some myopathies. An associated sensory loss suggests injury to a peripheral nerve or the central nervous system (CNS) rather than myopathy. On occasion, disorders affecting the motor nerve cell bodies in the spinal cord (anterior horn cell disease), the neuromuscular junction, or peripheral nerves can mimic findings of myopathy.
Muscle Weakness Symptoms of muscle weakness can be either intermittent or persistent. Disorders causing intermittent weakness (Fig. 462e-1) include myasthenia gravis, periodic paralyses (hypokalemic, hyperkalemic, and paramyotonia congenita), and metabolic energy deficiencies of glycolysis (especially myophosphorylase deficiency), fatty acid utilization (carnitine palmitoyltransferase deficiency), and some mitochondrial myopathies. The states of energy deficiency cause activity-related muscle breakdown accompanied by myoglobinuria, appearing as light-brown- to dark-brown-colored urine.
FIGURE 462e-1 Diagnostic evaluation of intermittent weakness. AChR AB, acetylcholine receptor antibody; CPT, carnitine palmitoyltransferase; EOMs, extraocular muscles; MG, myasthenia gravis; PP, periodic paralysis.
Most muscle disorders cause persistent weakness (Fig. 462e-2). In the majority of these, including most types of muscular dystrophy, polymyositis, and dermatomyositis, the proximal muscles are weaker than the distal and are symmetrically affected, and the facial muscles are spared, a pattern referred to as limb-girdle. The differential diagnosis is more restricted for other patterns of weakness. Facial weakness (difficulty with eye closure and impaired smile) and scapular winging (Fig. 462e-3) are characteristic of facioscapulohumeral dystrophy (FSHD). Facial and distal limb weakness associated with hand grip myotonia is virtually diagnostic of myotonic dystrophy type 1. When other cranial nerve muscles are weak, causing ptosis or extraocular muscle weakness, the most important disorders to consider include neuromuscular junction disorders, oculopharyngeal muscular dystrophy, mitochondrial myopathies, or some of the congenital myopathies (Table 462e-1). A pathognomonic pattern characteristic of inclusion body myositis is atrophy and weakness of the flexor forearm (e.g., wrist and finger flexors) and quadriceps muscles that is often asymmetric. Less frequently, but important diagnostically, is the presence of a dropped head syndrome indicative of selective neck extensor muscle weakness. The most important neuromuscular diseases associated with this pattern of weakness include myasthenia gravis, amyotrophic lateral sclerosis, late-onset nemaline myopathy, hyperparathyroidism, focal myositis, and some forms of inclusion body myopathy. A final pattern, recognized because of preferential distal extremity weakness, is typical of a unique category of muscular dystrophy, the distal myopathies.
FIGURE 462e-2 Diagnostic evaluation of persistent weakness. Examination reveals one of seven patterns of weakness. The pattern of weakness in combination with the laboratory evaluation leads to a diagnosis. ALS, amyotrophic lateral sclerosis; CK, creatine kinase; DM, dermatomyositis; EMG, electromyography; EOMs, extraocular muscles; FSHD, facioscapulohumeral dystrophy; IBM, inclusion body myositis; MG, myasthenia gravis; OPMD, oculopharyngeal muscular dystrophy; PM, polymyositis.
FIGURE 462e-3 Facioscapulohumeral dystrophy with prominent scapular winging.
|
NEUROMUSCULAR CAUSES OF PTOSIS OR OPHTHALMOPLEGIA |
It is important to examine functional capabilities to help disclose certain patterns of weakness (Table 462e-2). The Gowers’ sign (Fig. 462e-4) is particularly useful. Observing the gait of an individual may disclose a lordotic posture caused by combined trunk and hip weakness, frequently exaggerated by toe walking (Fig. 462e-5). A waddling gait is caused by the inability of weak hip muscles to prevent hip drop or hip dip. Hyperextension of the knee (genu recurvatum or back-kneeing) is characteristic of quadriceps muscle weakness; and a steppage gait, due to footdrop, accompanies distal weakness.
|
OBSERVATIONS ON EXAMINATION THAT DISCLOSE MUSCLE WEAKNESS |
FIGURE 462e-4 Gowers’ sign showing a patient using his arms to climb up the legs in attempting to get up from the floor.
FIGURE 462e-5 Lordotic posture, exaggerated by standing on toes, associated with trunk and hip weakness.
Any disorder causing muscle weakness may be accompanied by fatigue, referring to an inability to maintain or sustain a force (pathologic fatigability). This condition must be differentiated from asthenia, a type of fatigue caused by excess tiredness or lack of energy. Associated symptoms may help differentiate asthenia and pathologic fatigability. Asthenia is often accompanied by a tendency to avoid physical activities, complaints of daytime sleepiness, necessity for frequent naps, and difficulty concentrating on activities such as reading. There may be feelings of overwhelming stress and depression. Thus, asthenia is not a myopathy. In contrast, pathologic fatigability occurs in disorders of neuromuscular transmission and in disorders altering energy production, including defects in glycolysis, lipid metabolism, or mitochondrial energy production. Pathologic fatigability also occurs in chronic myopathies because of difficulty accomplishing a task with less muscle. Pathologic fatigability is accompanied by abnormal clinical or laboratory findings. Fatigue without those supportive features almost never indicates a primary muscle disease.
Muscle Pain (Myalgias), Cramps, and Stiffness Muscle pain can be associated with cramps, spasms, contractures, and stiff or rigid muscles. In distinction, true myalgia (muscle aching), which can be localized or generalized, may be accompanied by weakness, tenderness to palpation, or swelling. Certain drugs cause true myalgia (Table 462e-3).
|
DRUGS THAT CAUSE TRUE MYALGIA |
There are two painful muscle conditions of particular importance, neither of which is associated with muscle weakness. Fibromyalgia is a common, yet poorly understood, type of myofascial pain syndrome. Patients complain of severe muscle pain and tenderness and have specific painful trigger points, sleep disturbances, and easy fatigability. Serum creatine kinase (CK), erythrocyte sedimentation rate (ESR), electromyography (EMG), and muscle biopsy are normal (Chap. 396). Polymyalgia rheumatica occurs mainly in patients >50 years and is characterized by stiffness and pain in the shoulders, lower back, hips, and thighs (Chap. 385). The ESR is elevated, while serum CK, EMG, and muscle biopsy are normal. Temporal arteritis, an inflammatory disorder of medium- and large-sized arteries, usually involving one or more branches of the carotid artery, may accompany polymyalgia rheumatica. Vision is threatened by ischemic optic neuritis. Glucocorticoids can relieve the myalgias and protect against visual loss.
Localized muscle pain is most often traumatic. A common cause of sudden abrupt-onset pain is a ruptured tendon, which leaves the muscle belly appearing rounded and shorter in appearance compared to the normal side. The biceps brachii and Achilles tendons are particularly vulnerable to rupture. Infection or neoplastic infiltration of the muscle is a rare cause of localized muscle pain.
A muscle cramp or spasm is a painful, involuntary, localized, muscle contraction with a visible or palpable hardening of the muscle. Cramps are abrupt in onset, short in duration, and may cause abnormal posturing of the joint. The EMG shows firing of motor units, reflecting an origin from spontaneous neural discharge. Muscle cramps often occur in neurogenic disorders, especially motor neuron disease (Chap. 452), radiculopathies, and polyneuropathies (Chap. 459), but are not a feature of most primary muscle diseases. Duchenne muscular dystrophy is an exception because calf muscle complaints are a common complaint. Muscle cramps are also common during pregnancy.
A muscle contracture is different from a muscle cramp. In both conditions, the muscle becomes hard, but a contracture is associated with energy failure in glycolytic disorders. The muscle is unable to relax after an active muscle contraction. The EMG shows electrical silence. Confusion is created because contracture also refers to a muscle that cannot be passively stretched to its proper length (fixed contracture) because of fibrosis. In some muscle disorders, especially in Emery-Dreifuss muscular dystrophy and Bethlem myopathy, fixed contractures occur early and represent distinctive features of the disease.
Muscle stiffness can refer to different phenomena. Some patients with inflammation of joints and periarticular surfaces feel stiff. This condition is different from the disorders of hyperexcitable motor nerves causing stiff or rigid muscles. In stiff-person syndrome, spontaneous discharges of the motor neurons of the spinal cord cause involuntary muscle contractions mainly involving the axial (trunk) and proximal lower extremity muscles. The gait becomes stiff and labored, with hyperlordosis of the lumbar spine. Superimposed episodic muscle spasms are precipitated by sudden movements, unexpected noises, and emotional upset. The muscles relax during sleep. Serum antibodies against glutamic acid decarboxylase are present in approximately two-thirds of cases. In neuromyotonia (Isaacs’ syndrome), there is hyperexcitability of the peripheral nerves manifesting as continuous muscle fiber activity. Myokymia (groups of fasciculations associated with continuous undulations of muscle) and impaired muscle relaxation are the result. Muscles of the leg are stiff, and the constant contractions of the muscle cause increased sweating of the extremities. This peripheral nerve hyperexcitability is mediated by antibodies that target voltage-gated potassium channels. The site of origin of the spontaneous nerve discharges is principally in the distal portion of the motor nerves.
Myotonia is a condition of prolonged muscle contraction followed by slow muscle relaxation. It always follows muscle activation (action myotonia), usually voluntary, but may be elicited by mechanical stimulation (percussion myotonia) of the muscle. Myotonia typically causes difficulty in releasing objects after a firm grasp. In myotonic muscular dystrophy type 1 (DM1), distal weakness usually accompanies myotonia, whereas in DM2, proximal muscles are more affected; thus the related term proximal myotonic myopathy (PROMM) is used to describe this condition. Myotonia also occurs with myotonia congenita (a chloride channel disorder), but in this condition muscle weakness is not prominent. Myotonia may also be seen in individuals with sodium channel mutations (hyperkalemic periodic paralysis or potassium-sensitive myotonia). Another sodium channelopathy, paramyotonia congenita, also is associated with muscle stiffness. In contrast to other disorders associated with myotonia in which the myotonia is eased by repetitive activity, paramyotonia congenita is named for a paradoxical phenomenon whereby the myotonia worsens with repetitive activity.
Muscle Enlargement and Atrophy In most myopathies muscle tissue is replaced by fat and connective tissue, but the size of the muscle is usually not affected. However, in many limb-girdle muscular dystrophies (and particularly the dystrophinopathies), enlarged calf muscles are typical. The enlargement represents true muscle hypertrophy; thus the term pseudohypertrophy should be avoided when referring to these patients. The calf muscles remain very strong even late in the course of these disorders. Muscle enlargement can also result from infiltration by sarcoid granulomas, amyloid deposits, bacterial and parasitic infections, and focal myositis. In contrast, muscle atrophy is characteristic of other myopathies. In dysferlinopathies (LGMD2B) and anoctaminopathies (LGMD2L), there is a predilection for early atrophy of the gastrocnemius muscles, particularly the medial aspect. Atrophy of the humeral muscles is characteristic of FSHD.
LABORATORY EVALUATION
A limited battery of tests can be used to evaluate a suspected myopathy. Nearly all patients require serum enzyme level measurements and electrodiagnostic studies as screening tools to differentiate muscle disorders from other motor unit diseases. The other tests described—DNA studies, the forearm exercise test, and muscle biopsy—are used to diagnose specific types of myopathies.
Serum Enzymes CK is the preferred muscle enzyme to measure in the evaluation of myopathies. Damage to muscle causes the CK to leak from the muscle fiber to the serum. The MM isoenzyme predominates in skeletal muscle, whereas creatine kinase-myocardial bound (CK-MB) is the marker for cardiac muscle. Serum CK can be elevated in normal individuals without provocation, presumably on a genetic basis or after strenuous activity, minor trauma (including the EMG needle), a prolonged muscle cramp, or a generalized seizure. Aspartate aminotransferase (AST), alanine aminotransferase (ALT), aldolase, and lactic dehydrogenase (LDH) are enzymes sharing an origin in both muscle and liver. Problems arise when the levels of these enzymes are found to be elevated in a routine screening battery, leading to the erroneous assumption that liver disease is present when in fact muscle could be the cause. An elevated γ-glutamyl transferase (GGT) helps to establish a liver origin because this enzyme is not found in muscle.
Electrodiagnostic Studies EMG, repetitive nerve stimulation, and nerve conduction studies (Chap. 442e) are essential methods for evaluation of the patient with suspected muscle disease. In combination, they provide the information necessary to differentiate myopathies from neuropathies and neuromuscular junction diseases. Routine nerve conduction studies are typically normal in myopathies but reduced amplitudes of compound muscle action potentials may be seen in atrophied muscles. The needle EMG may reveal irritability on needle placement suggestive of a necrotizing myopathy (inflammatory myopathies, dystrophies, toxic myopathies, myotonic myopathies), whereas a lack of irritability is characteristic of long-standing myopathic disorders (muscular dystrophies, endocrine myopathies, disuse atrophy, and many of the metabolic myopathies). In addition, the EMG may demonstrate myotonic discharges that will narrow the differential diagnosis (Table 462e-4). Another important EMG finding is the presence of short-duration, small-amplitude, polyphasic motor unit action potentials (MUAPs). Such MUAPs can be seen in both myopathic and neuropathic disorders; however, the recruitment or firing pattern is different. In myopathies, the MUAPs fire early but at a normal rate to compensate for the loss of individual muscle fibers, whereas in neurogenic disorders the MUAPs fire faster. The EMG is usually normal in steroid or disuse myopathy, both of which are associated with type 2 fiber atrophy; this is because the EMG preferentially assesses the physiologic function of type 1 fibers. The EMG can also be invaluable in helping to choose an appropriately affected muscle to sample for biopsy.
|
MYOTONIC DISORDERS |
aAssociated with myotonic discharges on electromyography but no clinical myotonia.
DNA Analysis This serves as an important tool for the definitive diagnosis of many muscle disorders. Nevertheless, there are a number of limitations in currently available molecular diagnostics. For example, in Duchenne and Becker dystrophies, two-thirds of patients have deletion or duplication mutations in the dystrophin gene that are easy to detect, while the remainder have point mutations that are much more difficult to find. For patients without identifiable gene defects, the muscle biopsy remains the main diagnostic tool.
Forearm Exercise Test In myopathies with intermittent symptoms, and especially those associated with myoglobinuria, there may be a defect in glycolysis. Many variations of the forearm exercise test exist. For safety, the test should not be performed under ischemic conditions to avoid an unnecessary insult to the muscle, causing rhabdomyolysis. The test is performed by placing a small indwelling catheter into an antecubital vein. A baseline blood sample is obtained for lactic acid and ammonia. The forearm muscles are exercised by asking the patient to vigorously open and close the hand for 1 min. Blood is then obtained at intervals of 1, 2, 4, 6, and 10 min for comparison with the baseline sample. A three- to fourfold rise of lactic acid is typical. The simultaneous measurement of ammonia serves as a control, because it should also rise with exercise. In patients with myophosphorylase deficiency or other glycolytic defects, the lactic acid rise will be absent or below normal, while the rise in ammonia will reach control values. If there is lack of effort, neither lactic acid nor ammonia will rise. Patients with selective failure to increase ammonia may have myoadenylate deaminase deficiency. This condition has been reported to be a cause of myoglobinuria, but deficiency of this enzyme in asymptomatic individuals makes interpretation controversial.
Muscle Biopsy Muscle biopsy is an important step in establishing the diagnosis of a suspected myopathy. The biopsy is usually obtained from a quadriceps or biceps brachii muscle, less commonly from a deltoid muscle. Evaluation includes a combination of techniques—light microscopy, histochemistry, immunocytochemistry with a battery of antibodies, and electron microscopy. Not all techniques are needed for every case. A specific diagnosis can be established in many disorders. Endomysial inflammatory cells surrounding and invading muscle fibers are seen in polymyositis; similar endomysial infiltrates associated with muscle fibers containing rimmed vacuoles and amyloid deposits consisting of SMI-31-, p62-, and TDP-43-positive inclusions within fibers are characteristic of inclusion body myositis; and perivascular, perimysial inflammation associated with perifascicular atrophy is a feature of dermatomyositis. In addition, the congenital myopathies have distinctive light and electron microscopy features essential for diagnosis. Mitochondrial and metabolic (e.g., glycogen and lipid storage diseases) myopathies also demonstrate distinctive histochemical and electron-microscopic profiles. Biopsied muscle tissue can be sent for metabolic enzyme or mitochondrial DNA analyses. A battery of antibodies is available for the identification of abnormal proteins to help diagnose specific types of muscular dystrophies. Western blot analysis on muscle specimens can be performed to determine whether specific muscle proteins are reduced in quantity or are of abnormal size.
HEREDITARY MYOPATHIES
Muscular dystrophy refers to a group of hereditary progressive diseases each with unique phenotypic and genetic features (Tables 462e-5, 462e-6, and 462e-7).
|
PROGRESSIVE MUSCULAR DYSTROPHIES |
|
AUTOSOMAL DOMINANT LIMB-GIRDLE MUSCULAR DYSTROPHIES (LGMDs) |
|
AUTOSOMAL RECESSIVE LIMB-GIRDLE MUSCULAR DYSTROPHIES (LGMDs) |
DUCHENNE MUSCULAR DYSTROPHY
This X-linked recessive disorder, sometimes also called pseudohypertrophic muscular dystrophy, has an incidence of ~1 per 5200 live-born males.
Clinical Features Duchenne dystrophy is present at birth, but the disorder usually becomes apparent between ages 3 and 5 years. The boys fall frequently and have difficulty keeping up with friends when playing. Running, jumping, and hopping are invariably abnormal. By age 5 years, muscle weakness is obvious by muscle testing. On getting up from the floor, the patient uses his hands to climb up himself (Gowers’ maneuver [Fig. 462e-4]). Contractures of the heel cords and iliotibial bands become apparent by age 6 years, when toe walking is associated with a lordotic posture. Loss of muscle strength is progressive, with predilection for proximal limb muscles and the neck flexors; leg involvement is more severe than arm involvement. Between ages 8 and 10 years, walking may require the use of braces; joint contractures and limitations of hip flexion, knee, elbow, and wrist extension are made worse by prolonged sitting. Prior to the use of glucocorticoids, most boys became wheelchair dependent by 12 years of age. Contractures become fixed, and a progressive scoliosis often develops that may be associated with pain. The chest deformity with scoliosis impairs pulmonary function, which is already diminished by muscle weakness. By age 16–18 years, patients are predisposed to serious, sometimes fatal pulmonary infections. Other causes of death include aspiration of food and acute gastric dilation.
A cardiac cause of death is uncommon despite the presence of a cardiomyopathy in almost all patients. Congestive heart failure seldom occurs except with severe stress such as pneumonia. Cardiac arrhythmias are rare. The typical electrocardiogram (ECG) shows an increased net RS in lead V1; deep, narrow Q waves in the precordial leads; and tall right precordial R waves in V1. Intellectual impairment in Duchenne dystrophy is common; the average intelligence quotient (IQ) is ~1 standard deviation (SD) below the mean. Impairment of intellectual function appears to be nonprogressive and affects verbal ability more than performance.
Laboratory Features Serum CK levels are invariably elevated to between 20 and 100 times normal. The levels are abnormal at birth but decline late in the disease because of inactivity and loss of muscle mass. EMG demonstrates features typical of myopathy. The muscle biopsy shows muscle fibers of varying size as well as small groups of necrotic and regenerating fibers. Connective tissue and fat replace lost muscle fibers. A definitive diagnosis of Duchenne dystrophy can be established on the basis of dystrophin deficiency in a biopsy of muscle tissue or mutation analysis on peripheral blood leukocytes, as discussed below.
Duchenne dystrophy is caused by a mutation of the gene that encodes dystrophin, a 427-kDa protein localized to the inner surface of the sarcolemma of the muscle fiber. The dystrophin gene is >2000 kb in size and thus is one of the largest identified human genes. It is localized to the short arm of the × chromosome at Xp21. The most common gene mutation is a deletion. The size varies but does not correlate with disease severity. Deletions are not uniformly distributed over the gene but rather are most common near the beginning (5′ end) and middle of the gene. Less often, Duchenne dystrophy is caused by a gene duplication or point mutation. Identification of a specific mutation allows for an unequivocal diagnosis, makes possible accurate testing of potential carriers, and is useful for prenatal diagnosis.
A diagnosis of Duchenne dystrophy can also be made by Western blot analysis of muscle biopsy specimens, revealing abnormalities on the quantity and molecular weight of dystrophin protein. In addition, immunocytochemical staining of muscle with dystrophin antibodies can be used to demonstrate absence or deficiency of dystrophin localizing to the sarcolemmal membrane. Carriers of the disease may demonstrate a mosaic pattern, but dystrophin analysis of muscle biopsy specimens for carrier detection is not reliable.
Pathogenesis Dystrophin is part of a large complex of sarcolemmal proteins and glycoproteins (Fig. 462e-6). Dystrophin binds to F-actin at its amino terminus and to β-dystroglycan at the carboxyl terminus. β-Dystroglycan complexes to α-dystroglycan, which binds to laminin in the extracellular matrix (ECM). Laminin has a heterotrimeric molecular structure arranged in the shape of a cross with one heavy chain and two light chains, β1 and γ1. The laminin heavy chain of skeletal muscle is designated laminin α2. Collagen proteins IV and VI are also found in the ECM. Like β-dystroglycan, the transmembrane sarcoglycan proteins also bind to dystrophin; these five proteins (designated α- through ε-sarcoglycan) complex tightly with each other. More recently, other membrane proteins implicated in muscular dystrophy have been found to be loosely affiliated with constituents of the dystrophin complex. These include caveolin-3, α7 integrin, and collagen VI.
FIGURE 462e-6 Selected muscular dystrophy–associated proteins in the cell membrane and Golgi complex.
Dystrophin localizes to the cytoplasmic face of the muscle cell membrane. It complexes with two transmembrane protein complexes, the dystroglycans and the sarcoglycans. The dystroglycans bind to the extracellular matrix protein merosin, which is also complexed with β1 and α7 integrins (Tables 462e-5, 462e-6, and 462e-7). Dysferlin complexes with caveolin-3 (which binds to neuronal nitric oxide synthase, or nNOS) but not with the dystrophin-associated proteins or the integrins. In some of the congenital dystrophies and limb-girdle muscular dystrophies (LGMDs), there is loss of function of different enzymes that glycosylate α-dystroglycan, which thereby inhibits proper binding to merosin: POMT1, POMT2, POMGnT1, Fukutin, Fukutin-related protein, and LARGE.
The dystrophin-glycoprotein complex appears to confer stability to the sarcolemma, although the function of each individual component of the complex is incompletely understood. Deficiency of one member of the complex may cause abnormalities in other components. For example, a primary deficiency of dystrophin (Duchenne dystrophy) may lead to secondary loss of the sarcoglycans and dystroglycan. The primary loss of a single sarcoglycan (see “Limb-Girdle Muscular Dystrophy,” below) results in a secondary loss of other sarcoglycans in the membrane without uniformly affecting dystrophin. In either instance, disruption of the dystrophin-glycoprotein complexes weakens the sarcolemma, causing membrane tears and a cascade of events leading to muscle fiber necrosis. This sequence of events occurs repeatedly during the life of a patient with muscular dystrophy.
BECKER MUSCULAR DYSTROPHY
This less severe form of X-linked recessive muscular dystrophy results from allelic defects of the same gene responsible for Duchenne dystrophy. Becker muscular dystrophy is ~10 times less frequent than Duchenne.
Clinical Features The pattern of muscle wasting in Becker muscular dystrophy closely resembles that seen in Duchenne. Proximal muscles, especially of the lower extremities, are prominently involved. As the disease progresses, weakness becomes more generalized. Significant facial muscle weakness is not a feature. Hypertrophy of muscles, particularly in the calves, is an early and prominent finding.
Most patients with Becker dystrophy first experience difficulties between ages 5 and 15 years, although onset in the third or fourth decade or even later can occur. By definition, patients with Becker dystrophy walk beyond age 15, whereas patients with Duchenne dystrophy are typically in a wheelchair by the age of 12. Patients with Becker dystrophy have a reduced life expectancy, but most survive into the fourth or fifth decade.
Mental retardation may occur in Becker dystrophy, but it is not as common as in Duchenne. Cardiac involvement occurs in Becker dystrophy and may result in heart failure; some patients manifest with only heart failure. Other less common presentations are asymptomatic hyper-CK-emia, myalgias without weakness, and myoglobinuria.
Laboratory Features Serum CK levels, results of EMG, and muscle biopsy findings closely resemble those in Duchenne dystrophy. The diagnosis of Becker muscular dystrophy requires Western blot analysis of muscle biopsy samples, demonstrating a reduced amount or abnormal size of dystrophin or mutation analysis of DNA from peripheral blood leukocytes. Genetic testing reveals deletions or duplications of the dystrophin gene in 65% of patients with Becker dystrophy, approximately the same percentage as in Duchenne dystrophy. In both Becker and Duchenne dystrophies, the size of the DNA deletion does not predict clinical severity; however, in ~95% of patients with Becker dystrophy, the DNA deletion does not alter the translational reading frame of messenger RNA. These “in-frame” mutations allow for production of some dystrophin, which accounts for the presence of altered rather than absent dystrophin on Western blot analysis.
LIMB-GIRDLE MUSCULAR DYSTROPHY
The syndrome of LGMD represents more than one disorder. Both males and females are affected, with onset ranging from late in the first decade to the fourth decade. The LGMDs typically manifest with progressive weakness of pelvic and shoulder girdle musculature. Respiratory insufficiency from weakness of the diaphragm may occur, as may cardiomyopathy.
A systematic classification of LGMD is based on autosomal dominant (LGMD1) and autosomal recessive (LGMD2) inheritance. Superimposed on the backbone of LGMD1 and LGMD2, the classification uses a sequential alphabetical lettering system (LGMD1A, LGMD2A, etc.). Disorders receive letters in the order in which they are found to have chromosomal linkage. This results in an ever-expanding list of conditions summarized in Tables 462e-6 and 462e-7. None of the conditions is as common as the dystrophinopathies; however, prevalence data for the LGMDs have not been systematically gathered for any large heterogeneous population. In referral-based clinical populations, Fukutin-related protein (FKRP) deficiency (LGMD2I), calpainopathy (LGMD2A), anoctaminopathy (LGMD2L), and to a lesser extent dysferlinopathy (LGMD2B) have emerged as the most common disorders.
EMERY-DREIFUSS MUSCULAR DYSTROPHY
There are at least five genetically distinct forms of Emery-Dreifuss muscular dystrophy (EDMD). Emerin mutations are the most common cause of X-linked EDMD, although mutations in FHL1 may also be associated with a similar phenotype, which is X-linked as well. Mutations involving the gene for lamin A/C are the most common cause of autosomal dominant EDMD (also known as LGMD1B) and are also a common cause of hereditary cardiomyopathy. Less commonly, autosomal dominant EDMD has been reported with mutations in nesprin-1, nesprin-2, and TMEM43.
Clinical Features Prominent contractures can be recognized in early childhood and teenage years, often preceding muscle weakness. The contractures persist throughout the course of the disease and are present at the elbows, ankles, and neck. Muscle weakness affects humeral and peroneal muscles at first and later spreads to a limb-girdle distribution. The cardiomyopathy is potentially life threatening and may result in sudden death. A spectrum of atrial rhythm and conduction defects includes atrial fibrillation and paralysis and atrioventricular heart block. Some patients have a dilated cardiomyopathy. Female carriers of the X-linked variant may have cardiac manifestations that become clinically significant.
Laboratory Features Serum CK may be elevated two- to tenfold. EMG is myopathic. Muscle biopsy usually shows nonspecific dystrophic features, although cases associated with FHL1 mutations have features of myofibrillar myopathy. Immunohistochemistry reveals absent emerin staining of myonuclei in X-linked EDMD due to emerin mutations. ECGs demonstrate atrial and atrioventricular rhythm disturbances.
X-linked EDMD usually arises from defects in the emerin gene encoding a nuclear envelope protein. FHL1 mutations are also a cause of X-linked scapuloperoneal dystrophy, but can also present with an X-linked form of EDMD. The autosomal dominant disease can be caused by mutations in the LMNA gene encoding lamin A and C; in the synaptic nuclear envelope protein 1 (SYNE1) or 2 (SYNE2) encoding nesprin-1 and nesprin-2, respectively; and most recently in TMEM43 encoding LUMA. These proteins are essential components of the filamentous network underlying the inner nuclear membrane. Loss of structural integrity of the nuclear envelope from defects in emerin, lamin A/C, nesprin-1, nesprin-2, and LUMA accounts for overlapping phenotypes (Fig. 462e-7).
FIGURE 462e-7 Selected muscular dystrophy–associated proteins in the nuclear membrane and sarcomere. As shown in the exploded view, emerin and lamin A/C are constituents of the inner nuclear membrane. Several dystrophy-associated proteins are represented in the sarcomere including titin, nebulin, calpain, telethonin, actinin, and myotilin. The position of the dystrophin-dystroglycan complex is also illustrated.
CONGENITAL MUSCULAR DYSTROPHY (CMD)
This is not one entity but rather a group of disorders with varying degrees of muscle weakness, CNS impairment, and eye abnormalities.
Clinical Features As a group, CMDs present at birth or in the first few months of life with hypotonia and proximal or generalized muscle weakness. Calf muscle hypertrophy is seen in some patients. Facial muscles may be weak, but other cranial nerve–innervated muscles are spared (e.g., extraocular muscles are normal). Most patients have joint contractures of varying degrees at elbows, hips, knees, and ankles. Contractures present at birth are referred to as arthrogryposis. Respiratory failure may be seen in some cases.
The CNS is affected in some forms of CMD. In merosin and FKRP deficiency, cerebral hypomyelination may be seen by magnetic resonance imaging (MRI), although only a small number of patients have mental retardation and seizures. Three forms of congenital muscular dystrophy have severe brain impairment. These include Fukuyama’s congenital muscular dystrophy (FCMD), muscle-eye-brain (MEB) disease, and Walker-Warburg syndrome (WWS). Patients are severely disabled in all three of these conditions. In MEB disease and WWS, but not in FCMD, ocular abnormalities impair vision. WWS is the most severe congenital muscular dystrophy, causing death by 1 year of age.
Laboratory Features Serum CK is markedly elevated in all of these conditions. The EMG is myopathic and muscle biopsies show nonspecific dystrophic features. Merosin, or laminin α2 chain (a basal lamina protein), is deficient in surrounding muscle fibers in merosin deficiency. Skin biopsies can also demonstrate defects in laminin α2 chain. In the other disorders (FKRP deficiency, FCMD, MEB disease, WWS), there is abnormal α-dystroglycan staining in muscle. In merosin deficiency, cerebral hypomyelination is common, and a host of brain malformations are seen in FCMD, MEB disease, and WWS.
All forms of CMD are inherited as autosomal recessive disorders. Chromosomal linkage and specific gene defects are presented in Table 462e-8. With the exception of merosin, the other gene defects affect posttranslational glycosylation of α-dystroglycan. This abnormality is thought to impair binding with merosin and leads to weakening of the dystrophin-glycoprotein complex, instability of the muscle membrane, and/or abnormalities in muscle contraction. CMDs with brain and eye phenotypes probably involve defective glycosylation of additional proteins, accounting for the more extensive phenotypes.
|
CONGENITAL MUSCULAR DYSTROPHIESa |
MYOTONIC DYSTROPHY
Myotonic dystrophy is also known as dystrophia myotonica (DM). The condition is composed of at least two clinical disorders with overlapping phenotypes and distinct molecular genetic defects: myotonic dystrophy type 1 (DM1), the classic disease originally described by Steinert, and myotonic dystrophy type 2 (DM2), also called proximal myotonic myopathy (PROMM).
Clinical Features The clinical expression of DM1 varies widely and involves many systems other than muscle. Affected patients have a typical “hatchet-faced” appearance due to temporalis, masseter, and facial muscle atrophy and weakness. Frontal baldness is also characteristic of the disease. Neck muscles, including flexors and sternocleidomastoids, and distal limb muscles are involved early. Weakness of wrist extensors, finger extensors, and intrinsic hand muscles impairs function. Ankle dorsiflexor weakness may cause footdrop. Proximal muscles remain stronger throughout the course, although preferential atrophy and weakness of quadriceps muscles occur in many patients. Palatal, pharyngeal, and tongue involvement produce a dysarthric speech, nasal voice, and swallowing problems. Some patients have diaphragm and intercostal muscle weakness, resulting in respiratory insufficiency.
Myotonia, which usually appears by age 5 years, is demonstrable by percussion of the thenar eminence, the tongue, and wrist extensor muscles. Myotonia causes a slow relaxation of hand grip after a forced voluntary closure. Advanced muscle wasting makes myotonia more difficult to detect.
Cardiac disturbances occur commonly in patients with DM1. ECG abnormalities include first-degree heart block and more extensive conduction system involvement. Complete heart block and sudden death can occur. Congestive heart failure occurs infrequently but may result from cor pulmonale secondary to respiratory failure. Mitral valve prolapse also occurs commonly. Other associated features include intellectual impairment, hypersomnia, posterior subcapsular cataracts, gonadal atrophy, insulin resistance, and decreased esophageal and colonic motility.
Congenital myotonic dystrophy is a more severe form of DM1 and occurs in ~25% of infants of affected mothers. It is characterized by severe facial and bulbar weakness, transient neonatal respiratory insufficiency, and mental retardation.
DM2, or PROMM, has a distinct pattern of muscle weakness affecting mainly proximal muscles. Other features of the disease overlap with DM1, including cataracts, testicular atrophy, insulin resistance, constipation, hypersomnia, and cognitive defects. Cardiac conduction defects occur but are less common, and the hatchet face and frontal baldness are less consistent features. A very striking difference is the failure to clearly identify a congenital form of DM2.
Laboratory Features The diagnosis of myotonic dystrophy can usually be made on the basis of clinical findings. Serum CK levels may be normal or mildly elevated. EMG evidence of myotonia is present in most cases of DM1 but may be more patchy in DM2. Muscle biopsy shows muscle atrophy, which selectively involves type 1 fibers in 50% of cases, and ringed fibers in DM1 but not in DM2. Typically, numerous internalized nuclei can be seen in individual muscle fibers as well as atrophic fibers with pyknotic nuclear clumps in both DM1 and DM2. Necrosis of muscle fibers and increased connective tissue, common in other muscular dystrophies, are less apparent in myotonic dystrophy.
DM1 and DM2 are both autosomal dominant disorders. New mutations do not appear to contribute to the pool of affected individuals. DM1 is transmitted by an intronic mutation consisting of an unstable expansion of a CTG trinucleotide repeat in a serine-threonine protein kinase gene (named DMPK) on chromosome 19q13.3. An increase in the severity of the disease phenotype in successive generations (genetic anticipation) is accompanied by an increase in the number of trinucleotide repeats. A similar type of mutation has been identified in fragile × syndrome (Chap. 451e). The unstable triplet repeat in myotonic dystrophy can be used for prenatal diagnosis. Congenital disease occurs almost exclusively in infants born to affected mothers; it is possible that sperm with greatly expanded triplet repeats do not function well.
DM2 is caused by a DNA expansion mutation consisting of a CCTG repeat in intron 1 of the ZNF9 gene located at chromosome 3q13.3-q24. The gene is believed to encode an RNA-binding protein expressed in many different tissues, including skeletal and cardiac muscle.
The DNA expansions in DM1 and DM2 almost certainly impair muscle function by a toxic gain of function of the mutant mRNA. In both DM1 and DM2, the mutant RNA appears to form intranuclear inclusions composed of aberrant RNA. These RNA inclusions sequester RNA-binding proteins essential for proper splicing of a variety of other mRNAs. This leads to abnormal transcription of multiple proteins in a variety of tissues/organ systems, in turn causing the systemic manifestations of DM1 and DM2.
FACIOSCAPULOHUMERAL (FSH) MUSCULAR DYSTROPHY
This form of muscular dystrophy has a prevalence of ~1 in 20,000. There are two forms of FSHD that have similar pathogenesis, as will be discussed. Most patients have FSHD type 1 (95%), whereas approximately 5% have FSHD2. FSHD1 and FSHD2 are clinically and histopathologically identical. FSHD is not to be confused with the genetically distinct scapuloperoneal dystrophies.
Clinical Features The condition typically has an onset in childhood or young adulthood. In most cases, facial weakness is the initial manifestation, appearing as an inability to smile, whistle, or fully close the eyes. Weakness of the shoulder girdles, rather than the facial muscles, usually brings the patient to medical attention. Loss of scapular stabilizer muscles makes arm elevation difficult. Scapular winging (Fig. 462e-3) becomes apparent with attempts at abduction and forward movement of the arms. Biceps and triceps muscles may be severely affected, with relative sparing of the deltoid muscles. Weakness is invariably worse for wrist extension than for wrist flexion, and weakness of the anterior compartment muscles of the legs may lead to footdrop.
In most patients, the weakness remains restricted to facial, upper extremity, and distal lower extremity muscles. In 20% of patients, weakness progresses to involve the pelvic girdle muscles, and severe functional impairment and possible wheelchair dependency result.
Characteristically, patients with FSHD do not have involvement of other organ systems, although labile hypertension is common, and there is an increased incidence of nerve deafness. Coats’ disease, a disorder consisting of telangiectasia, exudation, and retinal detachment, also occurs.
Laboratory Features The serum CK level may be normal or mildly elevated. EMG usually indicates a myopathic pattern. The muscle biopsy shows nonspecific features of a myopathy. A prominent inflammatory infiltrate, which is often multifocal in distribution, is present in some biopsy samples. The cause or significance of this finding is unknown.
An autosomal dominant inheritance pattern with almost complete penetrance has been established, but each family member should be examined for the presence of the disease, since ~30% of those affected are unaware of involvement. FSHD1 is associated with deletions of tandem 3.3-kb repeats at 4q35. The deletion reduces the number of repeats to a fragment of <35 kb in most patients. Within these repeats lies the DUX4 gene, which usually is not expressed. In patients with FSHD1 these deletions in the setting of a specific polymorphism leads to hypomethylation of the region and toxic expression of the DUX4 gene. In patients with FSHD2, there is no deletion, but a mutation in SMCHD1. Interestingly, in the setting of the same polymorphism, there again is seen hypomethylation of the region and the permissive expression of the DUX4 gene. In both FSHD1 and FSHD2, there is overexpression of the DUX4 transcript.
OCULOPHARYNGEAL DYSTROPHY
This form of muscular dystrophy represents one of several disorders characterized by progressive external ophthalmoplegia, which consists of slowly progressive ptosis and limitation of eye movements with sparing of pupillary reactions for light and accommodation. Patients usually do not complain of diplopia, in contrast to patients having conditions with a more acute onset of ocular muscle weakness (e.g., myasthenia gravis).
Clinical Features Oculopharyngeal muscular dystrophy has a late onset; it usually presents in the fourth to sixth decade with ptosis and/or dysphagia. The extraocular muscle impairment is less prominent in the early phase but may be severe later. The swallowing problem may become debilitating and result in pooling of secretions and repeated episodes of aspiration. Mild weakness of the neck and extremities also occurs.
Laboratory Features The serum CK level may be two to three times normal. Myopathic EMG findings are typical. On biopsy, muscle fibers are found to contain rimmed vacuoles, which by electron microscopy are shown to contain membranous whorls, accumulation of glycogen, and other nonspecific debris related to lysosomes. A distinct feature of oculopharyngeal dystrophy is the presence of tubular filaments, 8.5 nm in diameter, in muscle cell nuclei.
Oculopharyngeal dystrophy has an autosomal dominant inheritance pattern with complete penetrance. The incidence is high in French-Canadians and in Spanish-American families of the southwestern United States. Large kindreds of Italian and of eastern European Jewish descent have been reported. The molecular defect in oculopharyngeal muscular dystrophy is a subtle expansion of a modest polyalanine repeat tract in a poly-RNA-binding protein (PABP2) in muscle.
DISTAL MYOPATHIES
A group of muscle diseases, the distal myopathies, are notable for their preferential distal distribution of muscle weakness in contrast to most muscle conditions associated with proximal weakness. The major distal myopathies are summarized in Table 462e-9.
|
DISTAL MYOPATHIES |
Clinical Features Welander’s, Udd’s, and Markesbery-Griggs type distal myopathies are all late-onset, dominantly inherited disorders of distal limb muscles, usually beginning after age 40 years. Welander’s distal myopathy preferentially involves the wrist and finger extensors, whereas the others are associated with anterior tibial weakness leading to progressive footdrop. Laing’s distal myopathy is also a dominantly inherited disorder heralded by tibial weakness; however, it is distinguished by onset in childhood or early adult life. Nonaka’s distal myopathy and Miyoshi’s myopathy are distinguished by autosomal recessive inheritance and onset in the late teens or twenties. Nonaka’s and Williams’ myopathy entails anterior tibial weakness, whereas Miyoshi’s myopathy is unique in that gastrocnemius muscles are preferentially affected at onset. Finally, the myofibrillar myopathies (MFMs) are a clinically and genetically heterogeneous group of disorders that can be associated with prominent distal weakness; they can be inherited in an autosomal dominant or recessive pattern. Of note, Markesbery-Griggs myopathy (caused by mutations in ZASP) and LGMD1B (caused by mutations in myotilin) are in fact subtypes of myofibrillar myopathy.
Laboratory Features Serum CK level is particularly helpful in diagnosing Miyoshi’s myopathy because it is very elevated. In the other conditions, serum CK is only slightly increased. EMGs are myopathic. In the MFMs, myotonic or pseudomyotonic discharges are common. Muscle biopsy shows nonspecific dystrophic features and, with the exception of Laing’s and Miyoshi’s myopathies, often shows rimmed vacuoles. MFM is associated with the accumulation of dense inclusions, as well as amorphous material best seen on Gomori trichrome and myofibrillar disruption on electron microscopy. Immune staining sometimes demonstrates accumulation of desmin and other proteins in MFM, large deposits of myosin heavy chain in the subsarcolemmal region of type 1 muscle fibers in Laing’s myopathy, and reduced or absent dysferlin in Miyoshi’s myopathy.
The affected genes and their gene products are listed in Table 462e-9.
CONGENITAL MYOPATHIES
These rare disorders are distinguished from muscular dystrophies by the presence of specific histochemical and structural abnormalities in muscle. Although primarily disorders of infancy or childhood, three forms that may present in adulthood are described here: central core disease, nemaline (rod) myopathy, and centronuclear (myotubular) myopathy. Sarcotubular myopathy is caused by mutations in TRIM-32 and is identical to LGMD2H. Other types, such as minicore myopathy (multi-minicore disease), fingerprint body myopathy, and cap myopathy, are not discussed.
CENTRAL CORE DISEASE
Patients with central core disease may have decreased fetal movements and breech presentation. Hypotonia and delay in motor milestones, particularly in walking, are common. Later in childhood, patients develop problems with stair climbing, running, and getting up from the floor. On examination, there is mild facial, neck-flexor, and proximal-extremity muscle weakness. Legs are more affected than arms. Skeletal abnormalities include congenital hip dislocation, scoliosis, and pes cavus; clubbed feet also occur. Most cases are nonprogressive, but exceptions are well documented. Susceptibility to malignant hyperthermia must be considered as a potential risk factor for patients with central core disease. Recent series have demonstrated that many cases of late-onset axial myopathy in which patients manifest with bent spine (camptocormia) or neck extensor weakness (neck extensor myopathy) are caused by mutations in the ryanodine receptor gene (RYR1). This illustrates the interesting spectrum of RYR1 mutations.
The serum CK level is usually normal. Needle EMG demonstrates a myopathic pattern. Muscle biopsy shows fibers with single or multiple central or eccentric discrete zones (cores) devoid of oxidative enzymes. Cores occur preferentially in type 1 fibers and represent poorly aligned sarcomeres associated with Z disk streaming.
Autosomal dominant inheritance is characteristic; sporadic cases also occur. As alluded above, this myopathy is caused by point mutations of RYR1, encoding the calcium-release channel of the sarcoplasmic reticulum of skeletal muscle; mutations of this gene also account for some cases of inherited malignant hyperthermia (Chap. 23). Malignant hyperthermia is an allelic condition; C-terminal mutations of the RYR1 gene predispose to this complication.
Specific treatment is not required, but establishing a diagnosis of central core disease is extremely important because these patients have a known predisposition to malignant hyperthermia during anesthesia.
NEMALINE MYOPATHY
The term nemaline refers to the distinctive presence in muscle fibers of rods or threadlike structures (Greek nema, “thread”). Nemaline myopathy is clinically heterogeneous. A severe neonatal form presents with hypotonia and feeding and respiratory difficulties, leading to early death. Nemaline myopathy usually presents in infancy or childhood with delayed motor milestones. The course is nonprogressive or slowly progressive. The physical appearance is striking because of the long, narrow facies, high-arched palate, and open-mouthed appearance due to a prognathous jaw. Other skeletal abnormalities include pectus excavatum, kyphoscoliosis, pes cavus, and clubfoot deformities. Facial and generalized muscle weakness, including respiratory muscle weakness, is common. An adult-onset disorder with progressive proximal or distal weakness may be seen. Myocardial involvement is occasionally present in both the childhood and adult-onset forms. The serum CK level is usually normal or slightly elevated. The EMG demonstrates a myopathic pattern. Muscle biopsy shows clusters of small rods (nemaline bodies), which occur preferentially, but not exclusively, in the sarcoplasm of type 1 muscle fibers. Occasionally, the rods are also apparent in myonuclei. The muscle often shows type 1 muscle fiber predominance. Rods originate from the Z disk material of the muscle fiber.
Six genes have been associated with nemaline myopathy. Five of these code for thin filament–associated proteins, suggesting disturbed assembly or interplay of these structures as a pivotal mechanism. Mutations of the nebulin (NEB) gene account for most cases, including both severe neonatal and early childhood forms, inherited as autosomal recessive disorders. Neonatal and childhood cases, inherited as predominantly autosomal dominant disorders, are caused by mutations of the skeletal muscle a-actinin (ACTA1) gene. In milder forms of the disease with autosomal dominant inheritance, mutations have been identified in both the slow a-tropomyosin (TPM3) and β;-tropomyosin (TPM2) genes accounting for <3% of cases. Muscle troponin T (TNNT1) gene mutations appear to be limited to the Amish population in North America. Mutations may also be seen in NEM6 that encodes a putative BTB/Kelch protein. No specific treatment is available.
CENTRONUCLEAR (MYOTUBULAR) MYOPATHY
Three distinct variants of centronuclear myopathy occur. A neonatal form, also known as myotubular myopathy, presents with severe hypotonia and weakness at birth. The late infancy–early childhood form presents with delayed motor milestones. Later, difficulty with running and stair climbing becomes apparent. A marfanoid, slender body habitus, long narrow face, and high-arched palate are typical. Scoliosis and clubbed feet may be present. Most patients exhibit progressive weakness, some requiring wheelchairs. Progressive external ophthalmoplegia with ptosis and varying degrees of extraocular muscle impairment are characteristic of both the neonatal and the late-infantile forms. A third variant, the late childhood–adult form, has an onset in the second or third decade. Patients have full extraocular muscle movements and rarely exhibit ptosis. There is mild, slowly progressive limb weakness that may be distally predominant (some of these patients have been classified as having Charcot-Marie-Tooth disease type 2 [CMT2; Chap. 459]).
[/not-level-membership-for-internal-medicine-category]
