Biological Pacing
These studies were supported by USPHS-NHLBI grant HL-28958.
The Natural Pacemaker
Like any biological system, the sinoatrial node is affected by aging and pathology in ways that can lead to dysfunction. For much of human history, this dysfunction, expressed as sinoatrial arrest or block or atrioventricular (AV) block, was accompanied by syncope, a marginal quality of life, and—over varying time spans—death.1,2 The major therapy until the mid-20th century was a stopgap: ephedrine or sublingual isoproterenol, administered every 2 hours.
In the 1960s, electronic pacing became available as devices that could be implanted transvenously and with little risk.3,4 Early units were relatively massive—rather like hockey pucks in appearance—but they were lifesaving. Technology improved and the size of units diminished over the second half of the 20th century; improvement and innovation continue in the current era.
The Normal Cardiac Pacemaker
The mammalian sinoatrial node was identified structurally in 1907 by Keith and Flack,5 and its function as the primary source of the cardiac impulse was demonstrated electrophysiologically by Lewis et al in 1910.6 These investigators recognized that they were dealing with a heterogeneous structure that appeared to serve as the dominant site of origin of the normal heartbeat. Yet the mechanisms determining sinoatrial node function remained a mystery for decades. One school of thought held that the node—and indeed the entire cardiac conduction system—consisted of specialized neural fibers, with a neural impulse initiating the heartbeat.7 That sinus rhythm persisted after cardiac denervation8 argued against central neural origin, although a role for local neuronal activity as the cause of the heartbeat was a continuing belief.7
The modern era of molecular biophysics has determined the why and the wherefore of the origin of cardiac impulses, although the origin of the sinoatrial node cells themselves remains a subject of active investigation.9 As is shown in Figure 26-1, A, multiple currents contribute to the sinoatrial node action potential.10–13 That the resting membrane potentials of normal sinoatrial node cells are more depolarized than those of most other cardiac myocytes is the result of a small inward rectifier current, IK1. Action potentials initiated at these voltages have calcium rather than sodium as the inward charge carrier. The result is a slowly propagating impulse similar to that in the normal AV node, the AV valves, and the coronary sinus.
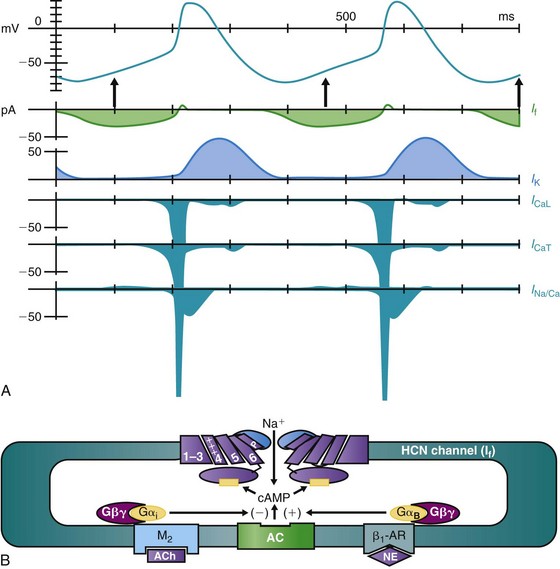
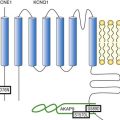
Figure 26-1 A, The sinoatrial node action potential and the currents contributing to impulse generation. B, The HCN channel. See text for discussion. (Reproduced and modified with permission from DiFrancesco D, Camm AJ: Heart rate lowering by specific and selective lf current inhibition with ivabradine: A new therapeutic perspective in cardiovascular disease. Drugs 64:1757–1765, 2004; and Biel M, Schneider A, Wahl C: Cardiac HCN channels: Structure, function, and modulation. Trends Cardiovasc Med 12:202–216, 2002.)
As sinoatrial node cells repolarize to their maximum diastolic potentials, their hyperpolarization permits the opening of HCN (for Hyperpolarization-activated Cyclic Nucleotide gated) channels, through which an inward sodium current, referred to as If, flows and begins to depolarize the membrane (Figure 26-1, A, B).10–12 A major contributor to pacemaker activity is the calcium clock mechanism, which depends on calcium cycling between intracellular uptake and release sites, as well as calcium entry via L- and T-type Ca channels.13,14 This affects the activity of the Na/Ca exchanger, which directly modulates phase 4 depolarization.13,14 All this activity occurs against the backdrop of outward (repolarizing) potassium current, such that any increase in inward current and/or decrease in outward current will increase the slope of phase 4 depolarization and the pacemaker rate.
The sympathetic and parasympathetic nervous systems are the principal modulators of phase 4 depolarization. Their respective neurohumors, norepinephrine and acetylcholine, act via β-adrenergic or muscarinic receptor-G protein-linked pathways to increase (β-adrenergic) or decrease (muscarinic) cyclic adenosine monophosphate (cAMP) synthesis (see Figure 26-1, B). Cyclic AMP binding to a site near the carboxy terminus of the HCN channel shifts channel activation positively, resulting in an increase in phase 4 depolarization.12
Whereas all the currents described contribute to phase 4 depolarization and to pacemaker rate, the initiator of the process appears to be If.10,11 That all pacemaker activity is not attributable to If has been demonstrated in experiments using If-blocking drugs. In both experimental models and the clinic, the result is significant slowing of sinus rate but not termination of the rhythm.15,16 Indeed, the inability to suppress sinus node function using any one blocker highlights the redundancy within the system such that it continues to function even under challenge.
The HCN channels that initiate pacemaker function have 6 transmembrane-spanning domains, are ubiquitous in the heart, and are found elsewhere in the body as well (see Figure 26-1, B). Four channel isoforms, HCN1-4, are present, with 4 and 1 in the sinoatrial node, 2 in much of the conducting system and the myocardium, and 3 largely in neural tissues.12 Each has differing activation and deactivation kinetics and differing cAMP sensitivity. The critical aspects of these channels with regard to pacemaker activity include their activation on hyperpolarization and their modulation by autonomic neurohumors.
Pathologies Influencing Normal Pacemaker Function and a Brief History of Therapies
In 1827 and 1846, respectively, Adams1 and Stokes2 noted the clinical characteristics of an event that had likely afflicted mankind for centuries: They described patients who became pale, whose pulse became slow or was absent, and who then collapsed. We now recognize that the pathology was a high-degree heart block. In the 20th century, the availability of synthetic catecholamines and later of β-agonists afforded one form of treatment: sublingual isoproterenol every 2 hours.17 Although this therapy increased the rate of idioventricular pacemakers to a more physiological range in many patients, it also could and did cause ventricular tachycardia. Moreover, this stopgap therapy could not ensure any long-term survival.
The answer to this dilemma faced by patients and physicians was the electronic pacemaker. Discovery of this device is traced to the 1889 report of McWilliam, who described a means of delivering shocks at about 60 to 70 times per minute to a patient in whom the heartbeat had failed.18 In 1928, Lidwell and Booth used an electrical power source attached to a needle plunged into the heart to revive “a stillborn infant.”19 The term cardiac pacemaker is attributed to Hyman, who used a hand-cranked device to generate electrical shocks in 1932.20 However, the modern era in pacing commenced in the early 1950s, when Hopps21 in Canada and then Zoll22 in the United States reported devices that delivered transcutaneous shocks. Pacing via shocks delivered through an intramyocardial needle in the setting of postsurgical complete heart block was reported by Weirich et al in 195723—the same year in which Bakken fabricated the first portable external pacemaker.24
Initially, pacemaker implantation in the human heart required a thoracotomy for intramyocardial electrode placement.4 The first report of temporary transvenous approaches was published by Furman and Schwedel in 1959.3 The early 1960s then saw the rapid adoption of transvenous pacing with implanted units. Early pacemakers had no sensing function, paced at a fixed rate, and, although internally implanted, were cumbersome and required frequent power pack changes because of their use of mercury batteries. Moreover, their failure to sense often led to competition between implanted and idioventricular pacemakers and, in far too many instances, arrhythmogenesis. But the 1960s and the 1970s saw rapid improvement in batteries, electrodes, and software, such that pacemakers could be placed in a demand mode, sensing spontaneously occurring heartbeats and re-setting appropriately.4,24,25
Innovation continues through the present time. For example, units are being tested that vary heart rate according to the body’s physiological needs,26 and leadless electrodes are being developed that hold the promise of stimulating the heart without incorporating a catheter.27 So the history of electronic pacing has been meteoric, and its future is bright.
Rationale for Developing Biological Pacemakers
Given the previous description of electronic pacemakers, why bother with biological pacing—or indeed with any other approach? Simply because the electronic pacemaker, although a wonderful treatment, is a panacea, not a cure. Table 26-1 summarizes the reasons why biological pacemakers are being investigated to normalize cardiac rhythm in a way that more nearly approximates the physiological state when compared with electronic pacemakers. If biological pacing succeeds, the two technologies would likely be used in tandem for some time to maximize patient safety. But the eventual goal would be to see the phasing out of electronic pacing. Therefore, biological pacing represents a disruptive technology.
Table 26-1
Limitations of Cardiac Pacemakers
• Not responsive to the autonomic nervous system—to the demands of exercise and emotion
• Require monitoring and maintenance, including at times battery and/or electrode replacement
• Not optimal for pediatric patients
• Problems with infection, interference from other devices
• Often cannot implant at sites that optimize contraction in individual patients
Strategies, Successes, and Failures in Building Biological Pacemakers
In the late 1980s, biological pacing was a pipe dream occasionally discussed around coffee tables among investigators working on mechanisms of impulse initiation. These discussions were likely stimulated by Dario DiFrancesco’s discovery of the pacemaker current If,10,11 and usually began with statements like, “Wouldn’t it be wonderful if we could put pacemaker currents into diseased sinus nodes or ventricles and create a site of normal impulse initiation?” Surely, this was the stuff of science fiction, but as we’ve learned time and again, science fiction often explores desirable outcomes before the needed technology has been developed; the works of Jules Verne are a prime example.
By the late 1990s, advances in genetic manipulation and stem cell science brought the concept of biological pacing to the realm of the possible, as we and others ventured to state in lectures and in print rather than simply at the coffee table. Several templates for exploration of biological pacing are suggested by Figure 26-1. For example, β-adrenergic stimulation can modulate If to increase pacemaker rate, and an initial strategy for biological pacing was to overexpress β2-adrenergic receptors in murine, then pig, hearts.28,29 Injecting a naked plasmid while incorporating the β2 receptor into pig atria led to increased basal rate and catecholamine response. Although overexpressing β-adrenergic receptors augmented sinus node function and increased the rate of normal rhythm, the following concerns arose: (1) In a diseased heart in which the sinus node is not functioning well and/or AV block is present, β-receptor overexpression can be arrhythmogenic; (2) upregulating β receptors implies that pacemaker cells already in the heart will respond normally to increased catecholaminergic effect—which may or may not be the case; and (3) transfection using naked plasmids has marginal success in the heart in situ.
Viral Vector-Based Delivery of Constructs
As can be seen in Figure 26-1, A, the initial ion channel-based approach reported transfection of guinea pig ventricles with an adenoviral vector incorporating a dominant negative Kir2.1 construct to reduce the repolarizing current, IK1.30 As is shown on electrocardiogram (ECG) in the intact heart and with microelectrode and ion current recordings, an ectopic ventricular rhythm was expressed that was associated with phase 4 depolarization and reduced IK1 in myocytes. A shortcoming was that reducing repolarizing current increases action potential duration, and a subsequent publication showed that the repolarization characteristics of Anderson’s syndrome were created by this approach to biological pacing,31 such that its continued development was undesirable.
Our group has worked primarily with means to modify and magnify the pacemaker current, If, as originally reported by DiFrancesco.10,11 We first demonstrated in neonatal rat myocytes in cell culture that using an adenoviral vector to overexpress the HCN2 channel isoform resulted in a robust and rapid pacemaker that far exceeded in rate and stability the rhythm normally recorded in non-transfected cultures.32,33 For this reason, we adopted the strategy of using HCN genes to build biological pacemakers and centered the approach on HCN2.34 An additional attraction of working with HCN channels was that their inward current flows only during diastole; hence, although we were increasing an inward current, this would not prolong action potential duration, thereby avoiding the problem that had afflicted IK1 downregulation.
Our initial in vivo study involved injecting adenoviral HCN2 and green fluorescent protein (GFP) constructs into canine left atria, and several days later using vagal stimulation to suppress sinus rhythm.34 The result consisted of escape beats originating from the implant site. Cells disaggregated from this site showed If (actually IHCN2) about 100 times greater in magnitude than that in native atrial myocytes. Additional vagal stimulation terminated firing of this pacemaker locus, consistent with parasympathetic control.
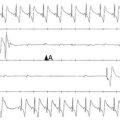
A follow-up study used a custom-designed electrode catheter to locate the left bundle branch system in dogs and to inject the same adenoviral-HCN2 complex into the bundle branch.35 When the vagi were stimulated to terminate sinus rhythm and induce AV block, a rhythm emerged from the injection site. Microelectrode studies demonstrated spontaneous and rapid impulse initiation at these bundle branch sites. Subsequently, we found that in dogs with radiofrequency-induced complete heart block, a biological pacemaker inserted into the left bundle branch system drove the heart regularly and stably.36 These rhythms were mapped to their bundle branch origins (Figure 26-2).
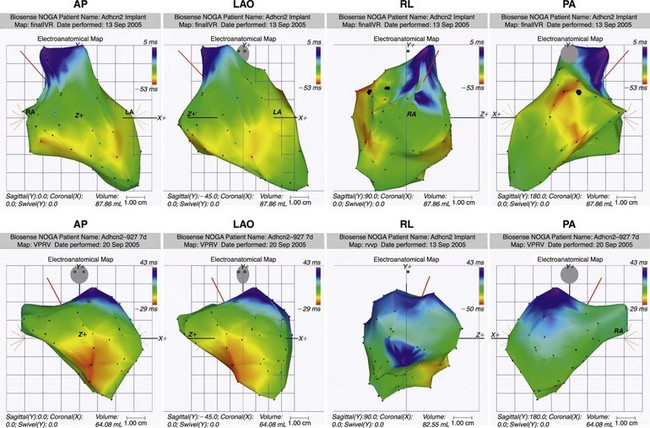
Figure 26-2 Noncontact mapping of left ventricle (LV) with CARTO system in dog with complete heart block, an electronic right ventricular (RV) apical endocardial pacemaker, and an HCN2 adenoviral construct administered into the proximal left bundle branch. Panels demonstrate four projections, showing early (red) through late (blue) activation. Upper panels show an impulse activating LV endocardium at several sites simultaneously, reflecting arrival of an impulse initiated in the left bundle branch system. Lower panels show early activation of LV septum via electronic pacemaker.
For the 2 weeks during which stable HCN2 expression occurred, about 70% of beats originated from the injection site. Basal rates averaged 50 to 60 beats/minute (bpm).36 The remaining 30% of beats were provided by the demand electronic pacemaker, set at an escape rate of 45 bpm. A control group of saline-injected animals with electronic pacemakers, also set at 45 bpm, had 90% of beats electrically stimulated, with the remainder arising from various idioventricular sites. Finally, the biological pacemaker manifested an approximate 50% rate increase in response to catecholamine injection. Taking this together with our earlier report,34 we concluded that both vagal and sympathetic (or at least β-adrenergic) modulation of biological pacing was possible.
Optimizing Pacemaker Gene Constructs
Attempts are ongoing to replicate or improve on the function of HCN2 using designer-based K channel genes,37 mutant or chimeric HCN channels,36,38,39 and combinations of two channels. We first studied a mutant HCN2 (E324A) that showed some enhanced catecholamine responsiveness, but only modest improvement over wild type HCN2.36 We then designed a chimera, HCN212, whose transmembrane portion included the pore of HCN1 (which has more positive activation than HCN2) and the amino- and carboxy-termini (the latter incorporating the cAMP-binding site) of HCN2.39 We hypothesized that this channel would generate faster basal pacemaker rates than HCN2 while exhibiting greater autonomic responsiveness than HCN1 alone. However, HCN212 resulted in an overshoot, such that ventricular tachycardias having rates in excess of 200 bpm occurred. Although this result was unfortunate with regard to construct design, it permitted us to test whether If blockade might be useful in suppressing arrhythmias generated by biological pacemakers. The If blocker ivabradine was highly effective here.39
Another mutant gene was reported by Kashiwakura et al,37 who used a K channel engineered to carry inward sodium current that had many of the properties of If. The shortcoming was the absence of a cAMP-binding site, without which there is no autonomic modulation. Although these and other constructs so far developed have not offered advantages over the wild type, it is anticipated that continued efforts at discovery will lead to robust alternatives.
In contrast to this, combinations of two genes have been promising. One such approach uses the dominant negative Kir2.1 construct previously described.40 This was tested in an adenoviral vector together with an HCN gene to increase inward current. The intent was to decrease hyperpolarizing current and increase inward current simultaneously in a small region of the ventricle. This was done successfully in pigs40 and is being explored by investigators as a potential treatment in settings where electronic pacemakers have become infected, necessitating hardware extraction from the heart. The concern here is that the adenoviral vector, which itself can cause inflammation, may contribute to the inflammatory process in the heart. Therefore, it is likely that this approach will be tested in a large animal model of endocarditis before the therapy is advanced.
Another approach uses HCN2 together with the gene for the skeletal muscle sodium channel, SkM1.41,42 SkM1 not only improves propagation of the cardiac impulse at depolarized membrane potentials,43,44 it also moves threshold potential to more negative voltages.42 The net result is that impulse initiation occurs earlier in phase 4, speeding pacemaker rate. When tested in a canine model of complete heart block, the outcome has been basal rates of approximately 80 bpm, with responses to catecholamine or exercise of up to 140 to 150 bpm. In addition, in this setting, there has been no need for the backup electronic pacemaker to fire.41
A different attempt to improve function uses the Ca2+-stimulated adenylyl cyclase AC1 gene expressed alone or in combination with HCN2.45,46 Basal beating rates were excessively high—around 140 bpm with HCN2/AC1, and only in the 50 to 65-bpm range with AC1 alone. Escape times in both groups were within the 1- to 2-second range following overdrive pacing, and the percent of electronic beats was in the 2% to 7% range. Instantaneous and long-term heart rate variability and circadian rhythm on 24-hour recordings showed greater sensitivity to parasympathetic modulation in animals injected with AC1 and a high degree of sympathetic modulation in those injected with HCN2/AC1. In vitro and in vivo data suggested that enhancement not only of If but of other, Ca-based pacemaker mechanisms occurs with AC1, and this is a matter of concern.46 But given the robust autonomic response to this intervention, its continued exploration is warranted, although better control of basal rates will be required.
Cell Therapies
Two differing philosophies have provided the framework for using cell therapies in biological pacing. The first uses cells that have a normal complement of ion channel genes to develop a biological pacemaker. The most advanced therapy reported uses human embryonic stem cells that have been “forced” along a cardiogenic lineage.47 Within this lineage a subset of cells manifest typical pacemaker characteristics, including action potentials that show prominent phase 4 depolarization.47 The underlying populations of ion channels include a very weak IK1 and a robust If, which together would tend to generate depolarization during phase 4.48 Unlike in a sinoatrial node, however, the major contributor of inward current is reportedly sodium, rather than L-type Ca current.48
Clusters of these cells in culture have been shown to originate and propagate spontaneous rhythms, and, when implanted into the ventricles of pigs in complete heart block, they have initiated stable spontaneous rhythms that persist for up to 3 months.47 Major issues regarding this approach have been and remain the need to use immunosuppression and questions regarding the eventual fate of the cells (e.g., Will they persist as pacemaker cells? Will they mature into nonbeating ventricular myocytes or into other inimical cell types?).
Other attempts to work with cells containing full ion channel complements have included the grafting of fetal49 or neonatal50 pacemaker cells. These research directions have not yet been published in sufficient detail to permit objective evaluation of the possibilities they offer. An alternative cell therapy approach has involved the generation of induced pluripotent stem cells (IPSCs), initially from dermal fibroblasts51 and more recently from keratinocytes.52 At the cellular level, good pacemaker function has been demonstrated. The advantage of IPSCs is that they permit pacemaker cells to be developed that have the full complement of pacemaker genes, such as the embryonic stem cell. An advantage over the embryonic stem cell is that IPSCs can be used autologously, thereby creating no immune response. Questions remain regarding the possibility of tumor development, although the oncogenes originally used to create IPSCs no longer need be used.53,54 In addition, questions continue regarding whether autologous stem cells developed from tissues of aging populations (such as most patients requiring pacemakers) will be as robust as those from young individuals. Animal studies with biological pacemakers developed from IPSCs are awaited.
The second broad approach to cell therapy uses cell types that do not have the proper ion channel population to generate pacemaker function but have other characteristics that make them candidates for platform therapy, that is, they can be loaded with a gene or genes of interest that result in delivery of a pacemaker signal to adjacent cells. Although some work has been reported using fibroblasts as platforms and fusing them with ventricular myocytes,55 the more completely reported approach to date has been our own—using adult human mesenchymal stem cells (hMSCs).56–58 Gene chip analysis of hMSCs has shown that they have no appreciable signal for the pacemaker genes, HCN1-4, but they do have a robust signal for the cardiac connexins, Cx40 and Cx43.57 In deciding to work with hMSCs as biological pacemakers, we hypothesized that if we could load them with a pacemaker gene, they might couple adequately to myocytes such that the depolarizing signal generated by If would be transmitted to adjacent myocytes, causing them to depolarize (Figure 26-3).59–61 Using both dye transfer and current injection experiments in cell pairs, we showed that hMSCs could indeed communicate with myocytes.57 This prompted us to load them with HCN2 via electroporation (avoiding any viral vector) (Figure 26-4, A-C) and to implant them in canine left ventricle.56,58 In an initial study, we used vagal stimulation to suppress sinoatrial node impulse initiation and AV conduction. Here, an escape rhythm that pace-mapped to the injection site was generated by the hMSC-based pacemakers (Figure 26-4, D). We also demonstrated connexins at interstices between hMSCs and cardiac myocytes (Figure 26-4, E).
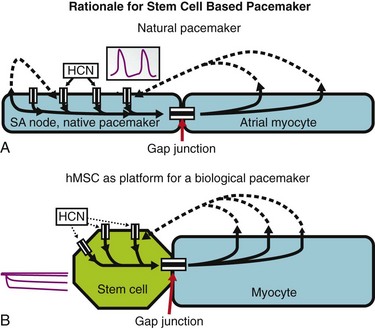
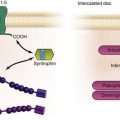
Figure 26-3 Initiation of spontaneous rhythms by wild type pacemaker cells and by genetically engineered stem cell pacemakers. A, In a native pacemaker cell (or in a myocyte engineered to incorporate pacemaker current via gene transfer), action potentials (inset) are initiated via inward current flowing through transmembrane HCN channels. These open when the membrane repolarizes to its maximum diastolic potential and close when the membrane has depolarized during the action potential. Current flowing via gap junctions to adjacent myocytes results in their excitation and the propagation of impulses through the conducting system. B, A stem cell has been engineered to incorporate HCN channels in its membrane. These channels can open, and current can flow through them (inset) only when the membrane is hyperpolarized; such hyperpolarization can be delivered only if an adjacent myocyte is tightly coupled to the stem cell via gap junctions. In the presence of such coupling and opening of the HCN channels to induce local current flow, the adjacent myocyte will be excited and will initiate an action potential that then propagates through the conducting system. Depolarization of the action potential will result in closing of the HCN channels until the next repolarization restores a high negative membrane potential. In summary, wild type and genetically engineered pacemaker cells incorporate in each cell all the machinery needed to initiate and propagate action potentials. In contrast, in the stem cell–myocyte pairing, two cells together work as a single functional unit whose operation is critically dependent on the gap junctions that form between the two disparate cell types. (Reproduced with permission from Rosen MR, Brink PR, Cohen IS, et al: Genes, stem cells and biological pacemakers. Cardiovasc Res 64:12–23, 2004.)
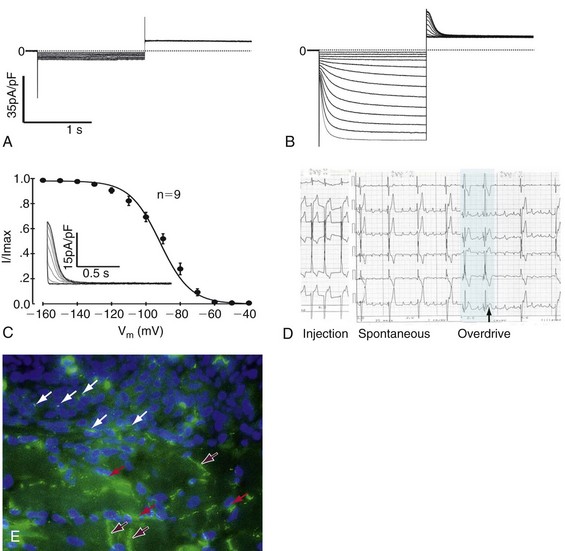
Figure 26-4 Example of loading human mesenchymal stem cell (hMSC) with a gene of interest, studying it biophysically, and determining its effect on the heart. A, Pacemaker current, If, is not seen in a non-electroporated hMSC. B, Functional expression of If in hMSC transfected with the mHCN2 gene. C, Fit by the Boltzmann equation to the normalized tail currents of If gives a midpoint of −91.8 ± 0.9 mV and a slope of 8.8 ± 0.5 mV (n=9). If is fully activated around −140 mV with an activation threshold of −60 mV. Inset shows representative tail currents used for activation curves. Voltage protocol: Hold at −30 mV, hyperpolarize × 1.5 s to −40 to −160 mV in 10-mV increments, followed by 1.5 s voltage step to +20 mV. D, Pacemaker function in canine heart in situ. Top to bottom, Electrocardiogram (ECG) leads I, II, III, AVR, AVL, and AVF. Left, Pacing from hMSC injection site showing ECG configuration. Middle, Spontaneous rhythm pace-maps to site of injection, suggesting that it is initiated at the injection site; Right, Last two beats of overdrive pacing (black bar) at 80 bpm followed by escape rhythm. Escape time = 1.3 sec. E, Immunostaining for cardiac connexin (Cx)43 in a region of interface between an injection site and myocardium. DAPI staining reveals nuclei. Purple arrows are intercalated discs; white arrows show Cx43 staining between hMSCs; red arrows show Cx43 staining between hMSCs and myocytes. (Modified with permission from Potapova I, Plotnikov A, Lu Z, et al: Human mesenchymal stem cell as a gene delivery system to create cardiac pacemakers. Circ Res 94:841–959, 2004.)
In a subsequent study of dogs in complete heart block, hMSCs generated pacemaker function for at least 6 weeks when 700,000 or more cells, approximately 50% of which were loaded with HCN2, were implanted at a single left ventricular site.58 Longer-term follow-up has revealed an issue of major concern, that is, by 8 to 10 weeks, most of the hMSCs have migrated from the site of administration. For this reason, attempts are now being made to encapsulate them in nanofabrics that will maintain them at the site of administration while still permitting gap junction formation and transmission of signals across the fabric.
Critical questions regarding these—and other—stem cells involve not only the possibility of evolution into inimical or ineffective cell types, but also the possibility of rejection or apoptosis.60,61 In a 6-week trial of hMSCs xenotransplanted into canine hearts, no rejection was reported (Figure 26-5).58 This finding is consistent with the hypothesis that hMSCs are immunoprotected, most likely by one of several humoral factors that they release.62–66 However, we have seen robust rejection of occasional lots of hMSCs by canine hearts, and this is not unexpected with xenotransplantation. As for the eventual human application of hMSCs, studies of their allogeneic administration to human subjects for myocardial repair have reported no rejection response, but a high degree of cell loss,62,63 mirroring our own experience in the dog. Clearly, issues remain regarding how to best bring this therapy to human patients.
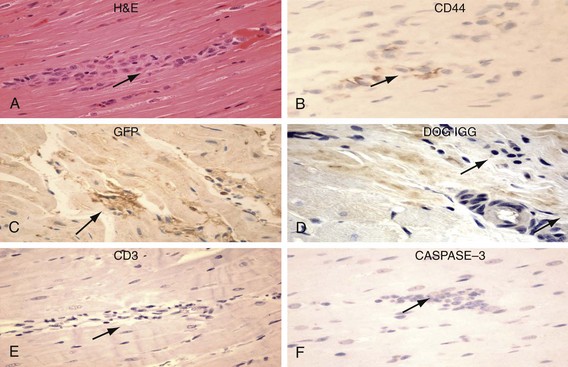
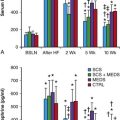
Figure 26-5 Human mesenchymal stem cells (hMSCs) 6 weeks after left ventricular (LV) anterior wall intramyocardial injection. Hematoxylin and eosin (H&E) identifies basophilic cells (upper left) that are CD44 positive (upper right) and GFP positive (peroxidase stain, middle left). hMSCs do not display labeling/binding of dog-IGG to their surface (middle right)—evidence against humoral rejection. CD3-positive T lymphocytes are rarely noted in association with clusters of hMSCs (lower left)—evidence against cellular rejection. Staining is negative for activated CASPASE 3 (lower right)—evidence against apoptosis. (Original magnification, ×400.) (Reproduced with permission from Plotnikov AP, Shlapakova I, Szaboks MJ, et al: Xenografted adult human mesenchymal stem cells provide a platform for sustained biological pacemaker function in canine heart. Circulation 116:706–713, 2007.)
Challenges
Some of the challenges we will discuss are unique to biological pacing; others hold for a variety of other approaches to gene and cell therapy. A major challenge is to identify the optimal construct as the groups working in this area continue attempts at optimizing mutants and chimeras. HCN2 alone manifests properties that likely are adequate for first-generation biological pacing. However, it might be preferable to have a construct that maintains basal rates in the 60 to 70-bpm range and reaches peaks of 120 to 140 bpm on catecholamine stimulation, as has been reported for the HCN2/SkM1 combination.41,42
Although we have demonstrated that vagal and β-adrenergic responsiveness is a characteristic of HCN2-based biological pacemakers, further understanding of autonomic control is required. Use of HCN2 in both viral and hMSC constructs has shown that vagal control depends on the site at which the pacemaker is implanted,67 that is, in vagally innervated regions, Poincaré plots of heart rate variability are consistent with vago-sympathetic input. In contrast, on the lateral free wall of the left ventricle, where little to no vagal innervation has been noted, no evidence of vagal input has been found, and only sympathetic responsiveness is seen.
Preclinical and clinical testing of biological pacing is also a major challenge. At the preclinical level, questions that arise relate largely to safety, efficacy, and duration of action. Another challenge involves effective utilization of the relationship between biological and electronic pacing. When and if biological pacing is ready for clinical trials, it cannot simply be administered to human subjects in need of support of their ventricular rate. Rather, it will have to be used together with electronic pacemakers, because these represent the current state of the art. For this reason, we have tested integration of biological and electronic pacemakers in tandem operation.36 We found that tandem biological-electronic pacemaker therapy manifests (1) a seamless interface between biological and electronic components, maintaining heart rate greater than 45 bpm; (2) conservation of total electronic beats delivered (which should prolong battery life); (3) the possibility of using the memory function of the electronic pacemaker to track the function of the biological component, and (4) a more physiological and catecholamine responsive heart rate than is provided by electronic pacemakers alone.
The need to reclaim normal cardiac activation in the setting of normal sinus rhythm and complete heart block has led investigators to engineer bypass tracts such that sinus impulses can gain access to the ventricles. This design would facilitate AV conduction and would potentially rival the function of AV sequential electronic pacing. However, this work is still in its infancy as it relates to biological pacemaking. Through one innovative approach, rat skeletal myoblasts were placed in an artificial matrix and were inserted into the AV groove of rats whose adjacent atrium and ventricle had been denuded of epicardium. AV conduction and survival of transplanted cells were then documented.68 Replication of results and data regarding long-term success of this approach are awaited.
A variety of methods are available for tracking cells (Table 26-2). Ideally, tracking should use markers that stay with the cell forever and cannot pass from cell to cell, and whose signal can be read from the body surface via imaging techniques. To date, no method has been adequate. Variations on magnetic resonance imaging (MRI) have been used most frequently in in vivo studies to track iron or ferritin particles loaded into stem cells. The shortcoming is that if a cell or cells die, these particles can be taken up by or can reside in other cells and/or can reside extracellularly. Upon being read on MRI, they may be correctly identified as a persistent signal, thus giving rise to a false-positive conclusion of the continued presence of stem cells.
Table 26-2
| Agent | Limitations |
| Reporter Proteins | |
| LacZ/b-gal | Require secondary detection |
| GFP | Unstable, may be weaker than autofluorescence |
| Ex Vivo Staining | |
| FISH | Expensive, false-positives |
| Surface markers | Lost/unexpressed over time; require secondary detection |
| Inorganic | |
| Radiometals | Toxicity, uniformity of loading |
| Fluorescent microspheres | Significant aggregation, unstable signal |
| Nanoparticles (i.e., quantum dots) | New technology, many unknowns |
FISH, Fluorescent in situ hybridization; GFP, green fluorescent protein.
We have worked with quantum dots as a tracking method (Figure 26-6).69 These nanoparticles offer the advantages of passive loading into cells, inability to pass through gap junctions, uptake and removal by the reticuloendothelial system if host cells die, and a strong emission signal that can be readily detected. Although they are readily identified in tissue sections, tracing the fate of cells loaded with quantum dots using standard imaging techniques in vivo remains a major challenge.
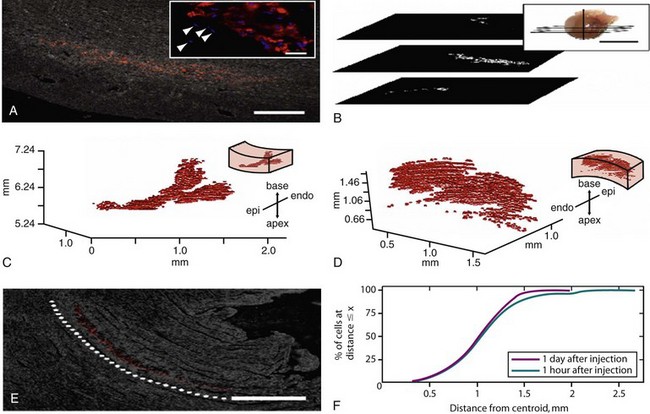
Figure 26-6 Quantum dots (QD) are used to identify single human mesenchymal stem cells (hMSCs) after injection into the rat heart and are further used to reconstruct the three-dimensional (3D) distribution of all delivered cells. Rat hearts were injected with QD-hMSCs. Fixed, frozen sections were cut transversely (plane shown in B, inset) at 10 µm and were mounted onto glass slides. Sections were imaged for QD fluorescence emission (655 nm) with phase overlay for visualization of tissue borders. QD-hMSCs can be visualized at (A) low power, and (A, inset) high power (Hoechst 33342 dye used to stain nuclei blue). In (A, inset), endogenous nuclei can be seen adjacent to the delivered cells in the mid-myocardium (arrowheads). Serial low-power images were registered with respect to one another, and (B) binary masks were generated, where white pixels depict QD-positive zones in the images. The vertical line in (B, inset) represents the z-axis, which has a zero value at the apex of the heart. The binary masks for all QD-positive sections of the heart were compiled and were used to generate the 3D reconstruction of delivered cells in the tissue. QD-hMSCs remaining in the tissue adhesive on the epicardial surface (not within the cardiac syncytium) were excluded from the reconstruction. (C) QD-hMSC reconstruction in an animal that was terminated 1 hour after injection. (D) Reconstruction from an animal euthanized 1 day after injection, with orientation noted in the inset. Our reconstructions in (C) and (D) do not account for all of the approximately 100,000 hMSCs delivered through the needle. Some of these cells undoubtedly leaked out of the needle track, and others may not have survived the injection protocol. Views of both reconstructions—(C) and (D)—are oriented for optimal static visualization (and have different scales and are situated at different positions along the z-axis, depending on the distance of the injection site from the apex of the heart). (E) One day after injection into the heart, the pattern of QD-hMSCs is well organized and appears to mimic the endogenous myocardial orientation (dotted white line highlights myofibril alignment). Complete representations of the spatial localization of QD-hMSCs in the heart permit further quantitative analyses. (F) One parameter that can be computed is the distance of individual cells from the centroid of the total cell mass. The plots show the percentage of cells at a distance less than or equal to x for both 1-hour and 1-day rats. At both time points, most of the cells are within 1.5 mm of the centroid.
500 µm, inset = 20 µm Scale bar on B, inset Scale bar on a = 1 cm Scale bar on e = 500 µm (Reproduced with permission from Rosen AB, Kelly DJ, Schuldt AJ, et al: Finding fluorescent needles in the cardiac haystack: Tracking human mesenchymal stem cells labeled with quantum dots for quantitative in vivo 3-D fluorescence analysis. Stem Cells 25:2128–2138, 2007.)
Cell systems must be optimized and their persistence documented. Autologous cells elicit no immune response when re-administered to the donor. However, they would make biological pacing into a designer therapy whose expense and assurance of quality control from patient to patient would be nightmarish in comparison with electronic pacing. It has been shown that hMSCs can be administered allogeneically to human subjects with no adverse reactions, although long-term persistence remains an issue.61 These cells may ultimately offer a readily standardized cell type, but more research is needed to test this hypothesis. Human ESCs have required the use of immunosuppression in experiments to date.47 Whether this will be the case in human experimentation remains to be seen, but a lifetime of electronic pacing appears far preferable to a lifetime of immunosuppression.
The choice of promoter and the regulation of nucleic acids are additional issues. To date, many of us have worked with a cytomegalovirus (CMV) promoter. However, temporal changes in the expression of promoters may be noted,70 such that CMV may not be the best long-term choice. Moreover, because of safety concerns, the idea of a cardiac-specific (and maybe even a cardiac region–specific) promoter would be of importance. This would seem especially relevant with regard to viral delivery.
One issue on which some progress has been made is the design of preclinical and clinical trials of biological pacemakers. One could not envisage a standard phase 1 trial in which healthy volunteers are sought. Rather, a reasonable trial design in human subjects would enroll patients in chronic atrial fibrillation and with complete heart block or significant bradycardia, in whom demand ventricular pacing is needed. The canine equivalent of this trial would include animals in ablation-induced complete heart block. Either setting would incorporate implantation of a right ventricular apical endocardial demand pacemaker, as well as implantation of the biological pacemaker. The latter would be introduced at a site that optimizes cardiac output, such as the high interventricular septum. The trial would proceed in such a way that the electronic pacemaker would provide a safety net should temporary or permanent failure of the biological occur. In addition, the electronic pacemaker can be used to track and record the function of the biological. The biological unit, in turn, would likely conserve battery power in the electronic unit—our work to date has shown that tandem biological-electronic operation sees the electronic unit firing only 30% of the time, which would indeed prolong the life of the battery.36 In addition, the biological component would be the heart’s primary pacemaker, would allow autonomic responsiveness, and would provide cardiac activation and output characteristics superior to those provided by the electronic unit. As reviewed previously, proof of concept experiments of tandem pacing have been promising and have shown no competition between biological and electronic units.36
References
1. Adams, R. Cases of diseases of the heart, accompanied with pathological observations. Dublin Hospital Reports. 1827; 4:353–453.
2. Stokes, W. Observations on some cases of permanently slow pulse. Dublin Quarterly Journal of Medical Science. 1846; 2:73–85.
3. Furman, S, Schwedel, JB. An intracardiac pacemaker for Stokes-Adams seizures. N Engl J Med. 1959; 261:943–948.
4. Jeffrey, K. The invention and reinvention of cardiac pacing. Cardiol Clin. 1992; 10:561–571.
5. Keith, A, Flack, MW. The form and nature of the muscular connections between the primary divisions of the vertebrate heart. J Anat Physiol. 1907; 41:172–189.
6. Lewis, T, Oppenheimer, BS, Oppenheimer, A. The site of origin of the mammalian heart-beat; the pacemaker in the dog. Heart. 1910; 2:147–169.
7. Fye, WB. The origin of the heart beat: a tale of frogs, jellyfish, and turtles. Circulation. 1987; 76:493–500.
8. Geison, G. The Royal Institution Lectures of 1869. In: Michael Foster and the Cambridge School of Physiology. Princeton, NJ: Princeton University Press; 1978:200.
9. Mommersteeg, MT, Hoogaars, WM, Prall, OW, et al. Molecular pathway for the localized formation of the sinoatrial node. Circ Res. 2007; 100:354–362.
10. DiFrancesco, D. A study of the ionic nature of the pacemaker current in calf Purkinje fibres. J Physiol. 1981; 314:377–393.
11. DiFrancesco, D. Block and activation of the pacemaker channel in calf Purkinje fibres: Effects of potassium, caesium and rubidium. J Physiol. 1982; 222:329–347.
12. Biel, M, Schneider, A, Wahl, C. Cardiac HCN channels: Structure, function, and modulation. Trends Cardiovasc Med. 2002; 12:202–216.
13. Lakatta, EG, Maltsev, VA, Vinogradova, TM. A coupled SYSTEM of intracellular Ca2+ clocks and surface membrane voltage clocks controls the timekeeping mechanism of the heart’s pacemaker. Circ Res. 2010; 106:659–673.
14. Lakatta, EG, DiFrancesco, D. What keeps us ticking: A funny current, a calcium clock, or both? J Mol Cell Cardiol. 2009; 47:157–170.
15. Thollon, C, Bedut, S, Villeneuve, N, et al. Use-dependent inhibition of hHCN4 by ivabradine and relationship with reduction in pacemaker activity. Br J Pharmacol. 2007; 150:37–46.
16. Borer, JS, Fox, K, Jaillon, P, et al. for the Ivabradine Investigators Group: Antianginal and antiischemic effects of ivabradine, an If inhibitor, in stable angina: A randomized, double-blind, multicentered, placebo-controlled trial. Circulation. 2003; 107:817–823.
17. Scherf, D, Schott, A. Extrasystoles and Allied Arrhythmias, ed 2. Chicago: Year Book Medical Publishers; 1973.
18. McWilliam, JA. Electrical stimulation of the heart in man. BMJ. 1889; 1:348–350.
19. Lidwell MC: Cardiac disease in relation to anaesthesia. In Transactions of the Third Session, Australasian Medical Congress, September 2-7, 1929, Sydney, Australia, p 160.
20. Furman, S, Szarka, G, Layvand, D. Reconstruction of Hyman’s second pacemaker. Pacing Clin Electrophysiol. 2005; 28:446–453.
21. Bigelow, WG, Callaghan, JC, Hopps, JA. General hypothermia for experimental intracardiac surgery: The use of electrophrenic respirations, an artificial pacemaker for cardiac standstill, and radio-frequency rewarming in general hypothermia. Trans Meet Am Surg Assoc Am Surg Assoc. 1950; 68:211–219.
22. Zoll, PM. Resuscitation of the heart in ventricular standstill by external electric stimulation. N Engl J Med. 1952; 247:768–771.
23. Weirich, W, Gott, V, Lillehei, C. The treatment of complete heart block by the combined use of a myocardial electrode and an artificial pacemaker. Surg Forum. 1957; 8:360–363.
24. Nelson, G. A brief history of cardiac pacing. Texas Heart Inst J. 1993; 20:12–18.
25. Zivin, A, Mehra, R, Bardy, GH. Cardiac pacemakers. In: Spooner PM, Rosen MR, eds. Foundations of Cardiac Arrhythmias. New York: Marcel Dekker Inc; 2001:571–598.
26. Alt, E, Matula, M, Theres, H, et al. The basis for activity controlled rate variable cardiac pacemakers: An analysis of mechanical forces on the human body induced by exercise and environment. Pac Clin Electrophysiol. 1989; 12:1667–1680.
27. Lee, KL. In the wireless era: Leadless pacing. Expert Rev Cardiovasc Ther. 2010; 8:171–174.
28. Edelberg, JM, Aird, WC, Rosenberg, RD. Enhancement of murine cardiac chronotropy by the molecular transfer of the human β2-adrenergic receptor cDNA. J Clin Invest. 1998; 101:337–343.
29. Edelberg, JM, Huang, DT, Josephson, ME, et al. Molecular enhancement of porcine cardiac chronotropy. Heart. 2001; 86:559–562.
30. Miake, J, Marbán, E, Nuss, HB. Gene therapy: Biological pacemaker created by gene transfer. Nature. 2002; 419:132–133.
31. Miake, J, Marban, E, Nuss, HB. Functional role of inward rectifier current in heart probed by Kir2. 1 overexpression and dominant-negative-suppression. J Clin Invest. 2003; 111:1529–1536.
32. Yu, H, Wu, J, Potapova, I, et al. MinK-related peptide 1: A β subunit for the HCN ion channel subunit family enhances expression and speeds activation. Circ Res. 2001; 88:E84–E87.
33. Qu, J, Barbuti, A, Protas, L, et al. HNC2 overexpression in newborn and adult ventricular myocytes: Distinct effects on gating and excitability. Circ Res. 2001; 89:E8–E14.
34. Qu, J, Plotnikov, AN, Danilo, P, Jr., et al. Expression and function of a biological pacemaker in canine heart. Circulation. 2003; 107:1106–1109.
35. Plotnikov, AN, Sosunov, EA, Qu, J, et al. A biological pacemaker implanted in the canine left bundle branch provides ventricular escape rhythms having physiologically acceptable rates. Circulation. 2004; 109:506–512.
36. Bucchi, A, Plotnikov, AN, Shlapakova, I, et al. Wild-type and mutant HCN channels in a tandem biological-electronic cardiac pacemaker. Circulation. 2006; 114:992–999.
37. Kashiwakura, Y, Cho, HC, Barth, AS, et al. Gene transfer of a synthetic pacemaker channel into the heart: A novel strategy for biological pacing. Circulation. 2006; 114:1682–1686.
38. Tse, HF, Xue, T, Lau, CP, et al. Bioartificial sinus node constructed via in vivo gene transfer of an engineered pacemaker HCN channel reduces the dependence on electronic pacemaker in a sick-sinus syndrome model. Circulation. 2006; 114:1000–1011.
39. Plotnikov, AN, Bucchi, A, Shlapakova, I, et al. HCN212-channel biological pacemakers manifesting ventricular tachyarrhythmias are responsive to treatment with If blockade. Heart Rhythm. 2008; 5:282–288.
40. Cingolani, E, Yee, K, Shehata, M, et al. Biological pacemaker created by percutaneous gene delivery via venous catheters in a porcine model of complete heart block. Heart Rhythm. 2011; 8:S112.
41. Boink, GJJ, Kryukova, Y, Lau, DH, et al. HCN2/SkM1 gene transfer into the canine left bundle branch induces highly stable biological pacing at physiological beating rates. Heart Rhythm. 2011; 8:S54.
42. Boink, GJJ, Sosunov, EA, Shlapakova, IN, et al. SkM1 gene transfer into the canine ventricular myocardium decreases the threshold potential for action potential initiation. Circulation. 2011; 123:A4013.
43. Protas, L, Dun, W, Lu, J, et al. Expression of skeletal but not cardiac Na+ channel isoform preserves normal conduction in a depolarized cardiac syncytium. Cardiovasc Res. 2009; 81:528–535.
44. Lau, DH, Clausen, C, Sosunov, EA, et al. Epicardial border zone overexpression of skeletal muscle sodium channel, SkM1, normalizes activation, preserves conduction and suppresses ventricular arrhythmia: An in silico, in vivo, in vitro study. Circulation. 2009; 119:19.
45. Kryukova, Y, Protas, L, Robinson, RB. Ca2+-activated adenylyl cyclase 1 introduces Ca2+-dependence to beta-adrenergic stimulation of HCN2 current. J Mol Cell Cardiol. 2012; 52:1233–1239.
46. Boink, GJJ, Kryukova, Y, Lau, DH, et al. Introducing the Ca2+-stimulated adenylyl cyclase AC1 into HCN2-based biological pacemakers enhances their function. Circulation. 2010; 122:A16415.
47. Kehat, I, Khimovich, L, Caspi, O, et al. Electromechanical integration of cardiomyocytes derived from human embryonic stem cells. Nat Biotechnol. 2004; 22:1282–1289.
48. Satin, J, Kehat, I, Caspi, O, et al. Mechanism of spontaneous excitability in human embryonic stem cell derived cardiomyocytes. J Physiol. 2004; 559:479–496.
49. Lin, G, Cai, J, Jiang, H, et al. Biological pacemaker created by fetal cardiomyocyte transplantation. J Biomed Sci. 2005; 12:513–519.
50. Cai, J, Lin, G, Jiang, H, et al. Transplanted neonatal cardiomyocytes as a potential biological pacemaker in pigs with complete atrioventricular block. Transplantation. 2006; 81:1022–1026.
51. Zhang, J, Wilson, GF, Soerens, AG, et al. Functional cardiomyocytes derived from human induced pluripotent stem cells. Circ Res. 2009; 104:e30–e41.
52. Novak, A, Shtrichman, R, Germanguz, I, et al. Enhanced reprogramming and cardiac differentiation of human keratinocytes derived from plucked hair follicles, using a single excisable lentivirus. Cell Reprogram. 2010; 12:665–678.
53. Efe, JA, Hilcove, S, Kim, J, et al. Conversion of mouse fibroblasts into cardiomyocytes using a direct reprogramming strategy. Nat Cell Biol. 2011; 13:215–222.
54. Warren, L, Manos, PD, Ahfeldt, T, et al. Highly efficient reprogramming to pluripotency and directed differentiation of human cells with synthetic modified mRNA. Cell Stem Cell. 2010; 7:618–630.
55. Cho, HC, Kashiwakura, Y, Marban, E. Creation of a biological pacemaker by cell fusion. Circ Res. 2007; 100:1112–1115.
56. Potapova, I, Plotnikov, A, Lu, Z, et al. Human mesenchymal stem cell as a gene delivery system to create cardiac pacemakers. Circ Res. 2004; 94:841–959.
57. Valiunas, V, Doronin, S, Valiuniene, L, et al. Human mesenchymal stem cells make cardiac connexins and form functional gap junctions. J Physiol. 2004; 555:617–626.
58. Plotnikov, AP, Shlapakova, I, Szabolcs, MJ, et al. Xenografted adult human mesenchymal stem cells provide a platform for sustained biological pacemaker function in canine heart. Circulation. 2007; 116:706–713.
59. Rosen, MR, Brink, PR, Cohen, IS, et al. Genes, stem cells and biological pacemakers. Cardiovasc Res. 2004; 64:12–23.
60. Rosen, M. Biological pacemaking: In our lifetime? Heart Rhythm. 2005; 2:418–428.
61. Rosen, MR, Robinson, RB, Brink, PR, et al. The road to biological pacing. Nat Rev Cardiol. 2011; 8:656–666.
62. Zimmett, JM, Hare, JM. Emerging role for bone marrow derived mesenchymal stem cells in myocardial regenerative therapy. Basic Res Cardiol. 2005; 100:471–481.
63. Rosen, MR. Are stem cells drugs? The regulation of stem cell research and development. Circulation. 2006; 114:1992–2000.
64. Groh, ME, Maitra, B, Szekely, E, et al. Human mesenchymal stem cells require monocyte-mediated activation to suppress alloreactive T cells. Exp Hematol. 2005; 33:928–934.
65. Di Nicola, M, Carlo-Stella, C, Magni, M, et al. Human bone marrow stromal cells suppress T lymphocyte proliferation induced by cellular or nonspecific mitogenic stimuli. Blood. 2002; 99:3838–3843.
66. Tse, WT, Pendleton, JD, Beyer, WM, et al. Suppression of allogeneic T-cell proliferation by human marrow stromal cells: Implications in transplantation. Transplantation. 2003; 75:389–397.
67. Shlapakova, IN, Nearing, BD, Lau, DH, et al. Biological pacemakers in canines exhibit positive chronotropic response to emotional arousal. Heart Rhythm. 2010; 7:1835–1840.
68. Choi, YH, Stamm, C, Hammer, PE, et al. Cardiac conduction through engineered tissue. Am J Pathol. 2006; 169:72–85.
69. Rosen, AB, Kelly, DJ, Schuldt, AJ, et al. Finding fluorescent needles in the cardiac haystack: Tracking human mesenchymal stem cells labeled with quantum dots for quantitative in vivo 3-D fluorescence analysis. Stem Cells. 2007; 25:2128–2138.
70. Reinhard, E, Nedivi, E, Wegner, J, et al. Neural selective activation and temporal regulation of a mammalian GAP-43 promoter in zebrafish. Development. 1994; 120:1767–1775.