29 Biochemical, Cellular, and Molecular Mechanisms of Neuronal Death and Secondary Brain Injury in Critical Care
In this chapter, we provide a general discussion of the biochemical, cellular, and molecular mechanisms of neuronal death and secondary brain injury that are germane to the central nervous system (CNS) insults that require neurointensive care, highlighting the important shared mechanisms in these conditions. In Chapter 30, Dr. Kofke builds upon the biochemical and molecular mechanisms to address general pathophysiologic principles in neurointensive care, focusing on intracranial dynamics and the cerebral circulation. Chapters 31 through 42 of Part 2 address other important facets of neurointensive care, such as monitoring and coma, along with the specific pathophysiology and treatment of the key disease processes central to neurointensive care in both adults and children. This includes traumatic brain injury (TBI), cardiopulmonary arrest, stroke, subarachnoid hemorrhage (SAH), and seizures, among other insults.
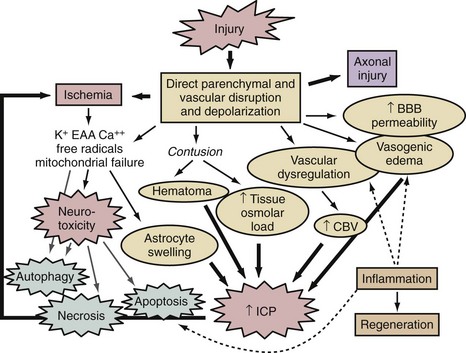
A thumbnail sketch of the most important mechanisms of secondary injury involved in the brain after a traumatic or ischemic insult is provided in Figure 29-1. Central to all brain insults relevant to neurointensive care is the occurrence of cerebral ischemia and/or cerebral energy failure. The principal consequence of ischemic injury and/or energy failure is neuronal death. The two principal forms of ischemia in neurointensive care are global and focal, as seen in cases of cardiopulmonary arrest and stroke, respectively.
 Global Cerebral Ischemia
Global Cerebral Ischemia
In patients with global cerebral ischemia, insults are dense and often square-wave in nature.1 The classic example of a global cerebral ischemic insult in neurointensive care is ventricular fibrillation cardiopulmonary arrest (see Chapter 33). Using conventional approaches, patients can be successfully resuscitated from these insults only if they are brief—that is, circulation must be restored in 5 to 12 minutes, although the maximal duration compatible with intact neurologic outcome can depend on a variety of factors, such as temperature. In cases of complete global cerebral ischemia, adenosine triphosphate (ATP) and phosphocreatine levels in brain are depleted in less than 2 minutes.2,3 Membrane failure ensues, with loss of ion homeostasis that includes cellular release of K+ and uptake of Ca++, Na+, and Cl−.2,3 Upon reperfusion, a complex sequence of events is set into motion that depends on the duration of the insult. Disturbances in lipid metabolism such as free fatty acid release and DNA damage result, along with a series of deleterious cascades including oxidative and nitrosative stress, excitotoxicity, poly-ADP-ribose polymerase (PARP) activation, mitochondrial and endoplasmic reticulum (ER) dysfunction, and a host of cell-signaling abnormalities. A number of endogenous neuroprotectant responses are also initiated. The specific biochemical, cellular, and molecular events are discussed later. The aforementioned increases in intracellular calcium level are believed to play a critical role in initiating many of these events. In situations where the patient is potentially salvageable, such as with threshold insults, reperfusion results in transient hyperemia (minutes) followed by delayed hypoperfusion (hours).1,4,5 The pattern of neuronal damage seen after global cerebral ischemia is classically termed selective vulnerability. This is often delayed and primarily neuronal in nature, and it is believed to result from complex biological cascades involving some features of programmed cell death (discussed later).
A number of brain regions are specifically vulnerable to ischemia, including the CA1 region of the hippocampus, cortical layers 3 and 5, portions of the amygdaloid nucleus, and cerebellar Purkinje cells, among others.2,3,5 Global ischemic insults from cardiopulmonary arrest from which there is some potential for recovery are generally believed to be devoid of important increases in intracranial pressure, since, based on studies in animal models, it has been shown that the threshold for producing poor outcome in patients with global ischemic insults is less than that needed to generate clinically significant intracranial hypertension.6 Thus, brain edema and vascular injury are not believed to represent important therapeutic targets after global cerebral ischemia. Two relevant but atypical global insults in neurointensive care are asphyxial cardiopulmonary arrest (particularly important in children and discussed in Chapter 42), and near-hanging episodes. In the latter, obstruction of cerebral venous drainage during the asphyxial insult compounds the ischemic insult.
 Focal Cerebral Ischemia
Focal Cerebral Ischemia
Focal ischemic insults in neurointensive care are produced by thrombotic or embolic events and generally produce a dense ischemic focus that is surrounded by a periischemic penumbral region with intermediate cerebral blood flow (CBF) values.2 The ischemic focus is generally believed to be unsalvageable unless reperfused almost immediately. In contrast, the ischemic penumbra is a region with some collateral flow and represents a therapeutic target for reperfusion with thrombolytics and/or pharmacologic therapy. In cases of focal cerebral ischemia, a hierarchy of CBF thresholds has been demonstrated in experimental studies, with inhibition of protein synthesis being the most sensitive to CBF reductions, followed by loss of electrical activity (evoked potentials and electroencephalogram), and eventually membrane failure.7–8 Unlike the selective vulnerability seen in global ischemic insults, focal cerebral ischemia produces pan-necrosis of the vasculature and astrocytes, resulting in infarction. However, cell death in the penumbra can demonstrate necrotic, apoptotic, and mixed phenotypes. Again, however, classic apoptosis is not seen. Astrocyte swelling and blood-brain barrier injury with focal cerebral edema can play important roles. In the penumbra, spreading depression waves resulting in depolarization can enhance excitotoxic damage with expansion of the lesion core. Reperfusion can occur spontaneously or with the administration of thrombolytics and can produce a microcosm of the aforementioned oxidative and nitrosative stress, mitochondrial and ER damage, and cell signaling abnormalities seen in global cerebral ischemia. In patients with focal cerebral ischemia with large infarcts, brain swelling can be substantial enough that secondary ischemia can result from intracranial hypertension. Dr. Kofke discusses these concepts in greater detail in Chapter 30. Focal cerebral ischemia from delayed vasospasm is also the most common critical complication of SAH and is discussed in Chapter 35.
 Traumatic Brain Injury
Traumatic Brain Injury
In cases of severe TBI, the biochemical and molecular mechanisms involved depend on the specific type of injury. In cases of focal contusion, direct disruption of parenchyma with local necrosis and hemorrhage results in superimposed vascular disruption, blood-brain barrier permeability, and local ischemia. This sets the stage for excitotoxicity and necrotizing cascades in the contusion penumbra, including oxidative and nitrosative stress, and calpain-mediated proteolysis, among other mechanisms.9,10 Local axonal injury is also seen in patients with contusions. Focal contusions are commonly complicated by marked local swelling and often by intracranial hypertension, with the potential for secondary focal or global ischemic insults or herniation syndromes. In contrast in diffuse injury, a constellation of diffuse axonal and vascular disruption can be seen, with characteristic findings of petechial hemorrhages in the white matter.11 This insult can be devastating even in the absence of intracranial hypertension.12 The biochemical and molecular events involved in axonal injury are discussed later. In cases of severe TBI, combined insults that include both multiple contusions and diffuse injury are also common. Finally, in addition to secondary compression ischemia from refractory intracranial hypertension, secondary extracerebral insults such as hypotension and hypoxemia can also negatively affect outcome and, importantly, complicate the biochemical and molecular response to severe TBI, markedly enhancing delayed neuronal death in brain regions that might otherwise have recovered.13,14
 Key Biochemical and Molecular Mechanisms of Neuronal Secondary Damage
Key Biochemical and Molecular Mechanisms of Neuronal Secondary Damage
Excitotoxicity
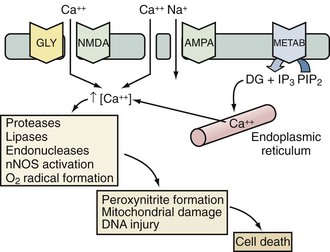
Excitotoxicity describes the process by which glutamate and other excitatory amino acids cause neuronal damage. Lucas and Newhouse15 first described the toxicity of glutamate. Olney16 subsequently reported that intraperitoneal administration of glutamate produces brain injury in experimental animals. Although glutamate is the most abundant neurotransmitter in the brain, exposure to toxic levels produces neuronal death.17 Glutamate exposure produces neuronal injury in two phases. Minutes after exposure, sodium-dependent neuronal swelling occurs.18 This is followed by delayed calcium-dependent degeneration. These effects are mediated through both ionophore-linked receptors, labeled according to specific agonists (N-methyl-D-aspartate [NMDA], kainite, and α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid [AMPA]), and receptors linked to second messenger systems, called metabotropic receptors. Activation of these receptors leads to calcium influx through receptor-gated or voltage-gated channels, or through the release of intracellular calcium stores. Increased intracellular calcium concentration is the trigger for a number of processes that can lead to cellular injury or death (Figure 29-2). One mechanism involves activation of neuronal nitric oxide synthase (nNOS), leading to nitric oxide (NO) production, peroxynitrite formation, and resultant DNA damage. PARP is an enzyme normally operative in DNA repair. In the face of overwhelming DNA damage, PARP overactivation leads to depletion of NAD+ and ATP, metabolic failure, and cell death.19–21 PARP may also impair ATP production directly via posttranslational modification of electron transport chain proteins.22 This may be important, since PARP knockout mice exhibit improved outcome versus controls after experimental stroke or TBI.20,23
There is considerable evidence in experimental laboratory models supporting an important contribution of excitotoxicity to the evolution of secondary damage in cases of global and focal cerebral ischemia, severe TBI, SAH, and status epilepticus.24–31 Evidence supporting an important role for excitotoxicity in humans has similarly been provided in cases of severe TBI, stroke, and SAH. Persson and Hillered32 reported increases in brain interstitial levels of glutamate in a patient with SAH as early as 1992. Palmer et al.33 first demonstrated increased concentrations of excitatory amino acids in ventricular cerebrospinal fluid (CSF) from adult patients with TBI. Glutamate concentrations were about fivefold greater than in control patients (up to 7 µM)—levels sufficient to cause neuronal death in cell culture.34 Bullock et al.35 characterized patterns of glutamate release by measuring excitatory amino acids by microdialysis in patients after TBI. Patients with a normal head computed tomography (CT) scan and no secondary ischemic events had interstitial concentrations of glutamate that were increased early in their course then returned to normal. In contrast, patients with a progressively rising level of glutamate died. Similarly, in cases of human stroke, Bullock et al.36 also reported massive increases in the excitatory amino acids, glutamate and aspartate, in a patient who required decompressive craniectomy to prevent brainstem herniation.
Despite these and many other clinical reports, clinical trials with anti-excitotoxic therapies have been unsuccessful in patients with either stroke or TBI. This may be due to problems with patient selection, side effects of the anti-excitotoxic agents that were tested, and the likelihood that treatment was initiated too late.37 Inhibition of plasticity by anti-excitotoxic therapies may also limit their efficacy, especially at the interface between the acute and subacute periods after injury.38
Programmed Cell Death Cascades
It is now increasingly clear from experimental models and human data that cells dying after global or focal cerebral ischemia or TBI can be categorized on a morphologic continuum ranging from necrosis to apoptosis.39,40 Recently, additional phenotypic definitions have been included within this continuum: those of autophagic degeneration, programmed necrosis, and “parthanatos.”41–43 Apoptosis is a morphologic description of cell death defined by cell shrinkage and nuclear condensation, internucleosomal DNA fragmentation, and the formation of apoptotic bodies.44 In contrast, cells dying of necrosis display cellular and nuclear swelling with dissolution of membranes. Apoptosis requires a cascade of intracellular events for completion of cell death; thus, the term apoptosis was previously used synonymously with programmed cell death.45 Because other types of cell death have now been characterized that can also be considered “programmed,” apoptosis now refers primarily to the phenotypic definition as classically defined by Kerr et al.44 In diseases with complex and multiple mechanisms, such as stroke and TBI, it is typically the rule rather than the exception to detect dying cells with many or all currently defined cell-death phenotypes.46 For example, some cells may display DNA fragmentation and activation of proteases involved in apoptosis, despite having nuclear and cellular swelling. Dying cells with mixed phenotypes may represent particularly difficult therapeutic targets.
Biochemical Pathways in Delayed Neuronal Death
Apoptosis is an evolutionarily conserved process required for selective cell elimination during development, and it occurs in all tissues, including brain. Execution of apoptosis requires novel gene expression and protein synthesis.47–49 Apoptosis is an intricate and critical mechanism for balancing cell proliferation, remodeling of tissues during development, and maintenance of tissues with a high rate of cell turnover. Apoptosis can be thought of as “molecular débridement,” delicately eliminating unwanted cells, with minimal disturbance of neighboring cells. Apoptosis is cybernetic and may occur via multiple pathways that can be independent (Figure 29-3); however, cross-talk between these (and other nonapoptotic) pathways also may occur.50,51 At present, neuronal apoptosis can be segregated into two pathways, one involving the activation of a family of cysteine proteases termed caspases, and one that is caspase independent.52
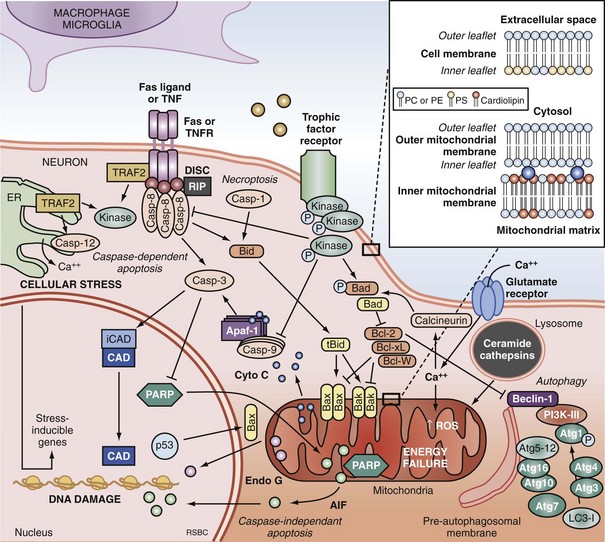
Caspase family proteases include 14 currently identified members that are synthesized as proenzymes, which for the most part are proteolytically activated.50 Initiator caspases, including caspase 8, 9, and 10, are activated by autocleavage and aggregation. Executioner caspases, including caspase 3, 6, and 7, are cleaved and activated by initiator caspases. The proteolytic cleavage of caspase substrates produces the phenotypic changes characteristic of apoptosis, including cytoskeletal disintegration, DNA fragmentation, and disruption of cellular and DNA repair processes (Figure 29-4). Cytoskeletal caspase targets include spectrin and nuclear lamin53; in addition, caspase 3 activates the enzyme, gelsolin, which cleaves actin.54 Active caspase 3 can also cleave the inhibitor of caspase-dependent deoxyribonuclease, permitting caspase-dependent deoxyribonuclease to digest DNA into small oligonucleosomal fragments.55 These small DNA fragments (multiples of approximately 180 base pairs) can be seen on a DNA gel as a ladder and are a hallmark of caspase-dependent apoptosis. Caspase 3 also inhibits DNA repair by proteolytically inactivating many DNA repair proteins, including PARP.56–58 This combination of features—silencing of the genome and incapacitation of DNA repair processes, and destruction of key cytoskeletal components, all with surgical-like precision and ultimately leading to cell death—illustrates why apoptosis has been referred to as “cell suicide.”
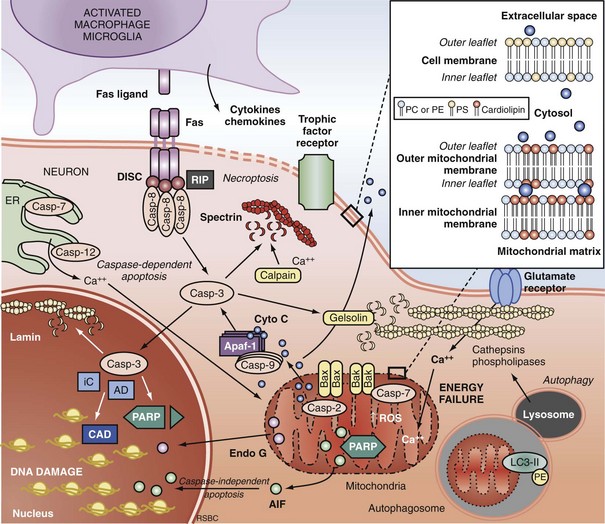
Extrinsic Pathways of Apoptosis and Programmed Necrosis
Programmed cell death can be initiated by extrinsic or intrinsic signals. Extrinsic signals include cell surface death receptor-ligand interactions and cell signaling pathways. The most prominent cell death receptor family is the tumor necrosis factor (TNF) receptor superfamily, which includes TNF-α and Fas.59 The coupling of cell surface TNF or Fas receptors with extracellular TNF-α or Fas ligand induces trimerization of the receptors that leads to the formation of submembrane complexes with intracellular death domain–signaling molecules. This death-inducing signaling complex then activates caspase 860 or 10.61 Caspase 3 is then cleaved and activated, perpetuating the cascade. The extrinsic pathway can also be regulated by multiple intracellular signal transduction pathways that are initiated by G-protein coupled cell surface receptors, which can be either activated by neurotransmitters (e.g., cyclic nucleotides) or inactivated by interruption of trophic factors (e.g., nerve growth factor) after injury.62 Perturbations in neurotransmitters and trophic factors controlling these pathways occur after ischemia and TBI. Multiple interrelated pro-death or pro-survival kinase pathways have been identified, including those involving mitogen-activated protein kinases, and protein kinase B and protein kinase C.63,64 Caspase 8 and 3 cleavage consistent with activation has been demonstrated in humans after TBI.39,65
More recently, programmed cell death with phenotypic characteristics of necrosis (programmed necrosis) or shared characteristics of apoptosis and necrosis (necroptosis) have been described.66 Programmed necrosis occurs through TNF receptor signaling involving receptor interacting protein 1 (RIP-1) and TNF receptor-associated factors (TRAFs) and regulation by protein ubiquitination and phosphorylation.66 Effector mechanisms of programmed necrosis are thought to involve caspase 8 but may also occur via direct effects on mitochondrial permeability transition. Thus, caspase 8 activation may reflect either extrinsic apoptosis or programmed necrosis, or both.
Intrinsic Pathways of Apoptosis
The intrinsic pathway of apoptosis is triggered by stress on cellular organelles, notably mitochondria and ER. Mitochondrial stress can lead to caspase-dependent apoptosis via mitochondrial release of cytochrome C induced upon mitochondrial membrane depolarization. Egress of cytochrome C into the cytosol enables interaction with apoptotic protease activating factor-1 (Apaf-1), dATP, and procaspase 9 to form a complex termed an apoptosome. Apaf-1 activates caspase 9 and subsequently caspase 3.67 Several mitochondrial proteins are capable of inducing apoptosis without direct activation of the caspase cascade, thus exemplifying pathways that are caspase independent. Apoptosis-inducing factor (AIF) within the mitochondria serves as an antioxidant68; however, upon mitochondrial membrane depolarization, it can translocate from the mitochondria to the nucleus, where it is sufficient to induce apoptosis.69 Translocation of AIF into the nuclei induces the formation of large-scale DNA fragmentation (>50 kilobase pairs), in contrast to cytochrome C-mediated, caspase-dependent apoptosis, which leads to oligonucleosomal DNA fragmentation (180-1200 base pairs). AIF-mediated apoptosis occurs in neurons under conditions of experimental TBI52 and cerebral ischemia.70 It is now accepted that PARP-1 overactivation mediates AIF-translocation and subsequent cell death.19,71 As noted earlier, “parthanatos” was recently coined to describe poly(ADP-ribose)-related cell death (from the Greek thanatos, referring to the personification of death).42
Other mitochondrial proteins related to programmed cell death include endonuclease G,72 Htr2A/Omi,73 and Smac/Diablo74; however, their roles in neuronal death after brain injury remain unexplored. Disruption of ER calcium homeostasis and/or accumulation of excess proteins can lead to ER stress, which in turn can trigger programmed cell death via activation of ER-localized caspase 12, an upstream initiator caspase. ER stress-related activation of caspase 12 has been detected in experimental models of cerebral ischemia75 and TBI.76
Autophagic Neurodegeneration
Autophagy is a homeostatic physiologic process important for recycling amino acids by digestion of proteins and organelles. Literally meaning “eating oneself,” this is an important response to nutrient deprivation in every organism. Like apoptosis, disrupted autophagy results in disease, in this case resulting in accumulation of intracellular proteins and aged organelles.77 Possibly like apoptosis, too much autophagy may also contribute to disease, depending upon the insult, organ, and cell type involved. For example, even under conditions of starvation, inhibition of autophagy protects neurons, whereas it exacerbates cell death in fibroblasts.78 Although there is considerable controversy regarding its role, increased autophagy has been demonstrated in models of cerebral ischemia79 and TBI80 and in brain tissue from humans with critical illness.81 The controversy arises in terms of whether or not inhibition or promotion of autophagy is beneficial after brain injury, insofar as both of these divergent strategies have been shown to be protective in various experimental models.80,82–84 There is also cross-talk between autophagy and apoptosis, perhaps at the level of the Bcl-2 protein family.51,85
Regulation of Programmed Cell Death by the Bcl-2 Protein Family
Caspase-dependent and caspase-independent apoptosis, as well as autophagy, are regulated by the B-cell lymphoma-2 (Bcl-2) family of proteins. The Bcl-2 family contains both pro-death and pro-survival members.86 Bcl-2 family proteins regulate changes in permeability of the mitochondrial outer membrane independent of permeability transition pore formation. Bcl-2 family proteins contain highly conserved Bcl-2 homology domains (BH1-BH4) essential for homo- and hetero-complex formation.87 Complexes formed between proteins containing BH3 domains such as Bax, truncated Bid, and Bad can facilitate mitochondrial cytochrome C release.88,89 The antiapoptotic members Bcl-2, Bcl-xL, and Mcl-1L prevent the release of mitochondrial proteins by inhibiting the pore formation.90 Bax expression is associated with neuronal cell death after cardiac arrest in dogs.91 Transgenic mice overexpressing Bcl-2 are partially protected from the neuropathologic sequelae of TBI versus wild-type mice,92 and overexpression of Bcl-xL also inhibits neuronal cell death after focal cerebral ischemia.93 The Bcl-2 interacting partner Beclin 1 contains a BH3-only domain and is required for autophagy.94 It is postulated that binding of Beclin 1 to Bcl-2 or Bcl-xL via the BH3 domain is how cross-talk occurs between apoptosis and autophagy.85
Programmed Cell Death in Human Brain Injury
Phenotypic descriptions of programmed cell death occurring after brain injury in humans date back to the 1940s.95,96 However, biochemical evidence of programmed cell death after brain injury in humans has been reported only within the last decade and has now been reported after TBI,39,65,81,97,98 stroke,99 and epilepsy.100 Brain tissue samples from TBI patients requiring decompressive craniectomy for the treatment of life-threatening intracranial hypertension were found to have evidence of DNA fragmentation by terminal deoxynucleotidyl transferase–mediated nick-end labeling (TUNEL) and cleavage of caspase 1 and 3, suggesting activation of the apoptotic cascade.39 The up-regulation of caspase-8 in human brain after TBI at both transcriptional and translational levels has also been reported.65 Caspase 8 was found predominantly in neurons and was associated with relative levels of the death receptor Fas, providing evidence of the extrinsic apoptotic pathway within neurons. Increases in Fas and Fas ligand have also been reported in CSF from TBI patients, with Fas levels correlating with intracranial pressure.101,102 Activation of the intrinsic apoptotic pathway also occurs after TBI. Alteration of Bcl-2-family proteins has been reported in human brain from adults and in CSF from infants and children after TBI.39,97,103 In pediatric patients, lower concentrations of Bcl-2 were detected in patients who died than in those who survived, supporting a pro-survival role for Bcl-2.97 After TBI in adults, the presence of pro-death Bcl-2 family protein Bax in patients in whom Bcl-2 was also detectable represented a more favorable outcome as compared with patients in whom Bax but not Bcl-2 was detectable.104 In contrast to TBI patients, patients after stroke demonstrate reductions in soluble Bcl-2 and soluble Fas within CSF,99 suggesting dysregulation of apoptosis after stroke. In adolescents and young adults with refractory seizures, increases in Bcl-2 and Bcl-xL, as well as increases in expression and proteolysis of caspase 1 and 3, occur in resected temporal lobe.100 These patients have had medically refractory seizures for several years, implying both protracted and acute apoptosis within the brain. Protracted programmed cell death after TBI also occurs. Cells with apoptotic morphologies and DNA damage detected by TUNEL have been reported in autopsy specimens from patients dying up to 12 months after injury,105 perhaps implying that a relatively wide therapeutic window exists for the administration of treatments aimed at reducing programmed cell death.
Many of these clinical observational studies suggest potential sex differences in cell death mechanisms operative after brain injury. For example, CSF levels of cytochrome C are associated with female gender after TBI in children,106 and CSF levels of the biochemical footprint of PARP activation are associated with male gender after TBI in both children107 and adults.108 These studies are strikingly consistent with experimental studies of neuronal death in vitro109 and in vivo.110,111
Several notes of caution are in order. First, it is unclear what the quantitative contribution of programmed cell death, particularly apoptosis, is in clinical cases of cerebral ischemia or TBI. It is likely that dying cells demonstrate some biochemical and phenotypic features of programmed cell death, but that the actual deathblow to the cell is not dependent on an active process.112 Even if programmed cell death mechanisms do play a key role, it is unclear whether inhibiting neuronal death after injury is entirely beneficial, since apoptosis is a vital mechanism for biological systems to eliminate abnormal or aging cells, and autophagy is important for protein and organelle turnover. In other words, quiet elimination via “cell suicide” of damaged or dysfunctional cells and/or organelles may lead to overall benefit to the patient, in essence “molecular débridement.” Only clinical trials of novel therapies targeting individual programmed cell death cascades will be able to determine whether these mechanisms, alone or in combination, represent important targets in neurointensive care. Recent studies of the efficacy of mild hypothermia after experimental and clinical cardiopulmonary arrest, however, suggest that the success of this intervention may be derived from its effects on programmed cell death.113–115
Axonal Injury
White matter damage is important in infarction that results from stroke but probably plays only a limited role in the pathology of reversible global cerebral ischemia. In contrast, axonal injury is of paramount importance in patients with TBI. This has been demonstrated both clinically116,117 and in experimental models.118–120 The extent and distribution of traumatic axonal injury depends on injury severity and category (focal versus diffuse).121 The classic view that traumatic axonal injury occurs because of immediate physical shearing is represented primarily in cases of severe injury in which frank axonal tears occur.116,122,123 However, recent experimental studies suggest that axonal damage predominantly occurs by a delayed process termed secondary axotomy.118,124,125 Two hypothetical sequences have attempted to explain secondary axotomy, one attributing axolemmal permeability and calcium influx as the initiating event (Figure 29-5), and the other a direct cytoskeletal abnormality impairing axoplasmic flow.118,125,126 It has been posited that both forms of reactive axonal swelling take place but in different proportions depending on the severity of injury. Superimposed on these theories is the finding that hypoxic/ischemic insults can also produce axonal swelling.127 As a result, differing as well as unifying theories for axonal injuries in patients with brain injury have been proposed.128 Common mechanistic features include focal ion flux, calcium dysregulation, and mitochondrial and cytoskeletal dysfunction.
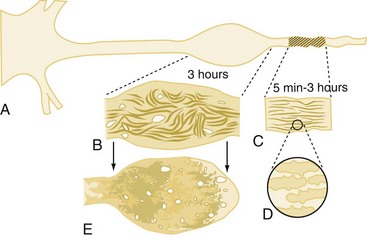
Traumatic axonal injury contributes to morbidity and mortality after TBI.118,121,122 Until recently, the contributions of axonal injury to morbidity have remained speculative, since traumatic axonal injury has remained refractory to treatment even in the laboratory. However, recent studies in experimental TBI models have shown that hypothermia or cyclosporin-A can both reduce white matter damage.129,130 These therapeutic advances should help determine more definitively the contributions of traumatic axonal injury to secondary damage. Recent application of magnetic resonance imaging (MRI) to the study of traumatic axonal injury and axonal connectivity may improve our understanding of both this injury mechanism and axonal regeneration.131,132
Cerebral Swelling
In addition to cascades of neuronal death and axonal damage, brain swelling is a hallmark finding in cases of focal cerebral ischemia, severe TBI, and severe global cerebral ischemia from prolonged cardiopulmonary arrest. Brain swelling often results in the development of intracranial hypertension. Cerebral swelling and accompanying intracranial hypertension contribute to secondary damage in two ways. Intracranial hypertension can compromise cerebral perfusion, leading to secondary ischemia. It can also produce the devastating consequences of brain deformation and vascular compression through herniation syndromes. Intracranial hypertension results from increases in intracranial volume from a variety of sources, outlined in Figure 29-1. In some cases of TBI or spontaneous intracranial hemorrhage, such as with epidural, subdural, or parenchymal hematoma formation, an extraaxial or parenchymal blood collection is the key culprit and can be addressed by surgical evacuation.133 However, there are several important mechanisms more uniformly involved in the development of intracranial hypertension. These are related to either brain swelling from vasogenic edema, astrocyte swelling, and an increase in tissue osmolar load, or vascular dysregulation with swelling secondary to an increase in cerebral blood volume (CBV).
Most of the mechanistic work in this area has come from studies in the field of TBI. Recent data suggest that brain swelling after severe TBI results from edema rather than increased CBV. Marmarou and colleagues134 measured both CBV and brain water in adults with TBI. Using a dye indicator technique (coupled to CT) to measure CBV and MRI to quantify brain water, increases in brain water were commonly observed but were generally associated with reduced (not increased) CBV (Figure 29-6).
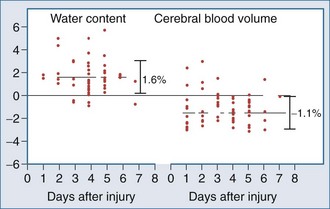
Thus, edema rather than increased CBV appears to be the predominant contributor to cerebral swelling after TBI. Both cytotoxic and vasogenic edema may play important roles in cerebral swelling, but the biochemical and molecular pathways involved in our traditional concept of cytotoxic and vasogenic edema are evolving. There appear to be four putative mechanisms for edema formation in the injured brain. First, vasogenic edema may form in the extracellular space as a result of disruption of the blood-brain barrier. Second, cellular swelling can be produced in two ways. Astrocyte swelling can occur as part of the homeostatic uptake of substances such as glutamate. Glutamate uptake is coupled to glucose utilization via a sodium/potassium ATPase, with sodium and water accumulation in astrocytes. Astrocyte swelling appears to be importantly linked to water movement through the aquaporin-4 channel found in the astrocyte foot processes near capillaries.135–137 Studies have demonstrated reduced cerebral edema in mice genetically deficient in this channel.138 Swelling of both neurons and other cells in the neuropil can also result from ischemia- or trauma-induced ionic pump failure. This can be important in the penumbral regions of focal cerebral ischemia and around cerebral contusions. Finally, osmolar swelling may also contribute to edema formation in the extracellular space, particularly in maturing cerebral contusions. Osmolar swelling, however, is actually dependent on an intact blood-brain barrier or an alternative solute barrier.
In both cerebral ischemia and TBI, cellular swelling may be of greatest importance. Using a model of diffuse TBI in rats, Barzo and colleagues139 applied diffusion-weighted MRI to localize the increase in brain water. A decrease in the apparent diffuse coefficient after injury suggested predominantly cellular swelling rather than vasogenic edema in the development of intracranial hypertension. Cellular swelling may be of even greater importance in the setting of TBI with a secondary hypoxemic-ischemic insult.140 Katayama et al.141 also suggested that the role of the blood-brain barrier in the development of posttraumatic edema might have been overstated, even in the setting of cerebral contusion. One intriguing possibility is that as macromolecules are degraded within injured brain regions, the osmolar load in the contused tissue or infarcts increases. As the blood-brain barrier reconstitutes (or as other osmolar barriers are formed), a considerable osmolar driving force for the local accumulation of water develops, resulting in the marked swelling so often seen in and around cerebral contusions (Figure 29-7). This has been supported by recent clinical studies of human cerebral contusion.142
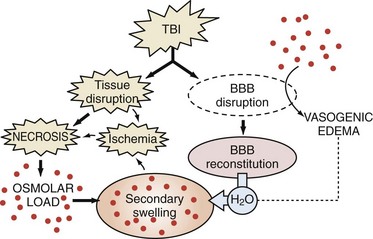
Figure 29-7 Schematic based on hypothesis of Katayama et al.,141 suggesting that as osmolar load increases (breakdown of macromolecules in the region of contusion necrosis), a considerable driving force develops for the accumulation of water, resulting in the secondary swelling so often seen in and around cerebral contusions.
In some cases, increases in CBV can be seen after TBI and contribute to intracranial hypertension. When an increase in CBV is seen, it may result from local increases in cerebral glycolysis, “hyperglycolysis” as described by Bergsneider and colleagues.143 In regions with increases in glutamate levels, such as in contusions, increases in glycolysis are observed because astrocyte uptake of glutamate is coupled to glycolysis rather than oxidative metabolism. Recall that oxidative metabolism is generally depressed by approximately 50% in comatose victims of severe TBI in the intensive care unit.144 Hyperglycolysis results in a marked local increase in cerebral glucose utilization, with a coupled increase in CBF and CBV and resultant local brain swelling. That said, the contribution of hyperglycolysis to the pathogenesis of TBI remains unclear, and there have been few recent reports focusing on hyperglycolysis after brain injury.
A detailed discussion of this topic is beyond the scope of this chapter, but an expanded discussion of intracranial dynamics and vascular dysregulation in neurointensive care is provided in the next chapter. As MRI and magnetic resonance spectroscopic methods continue to develop and become applied to critically ill patients,132 our knowledge of the mechanisms involved in cerebral swelling should greatly advance. It must be remembered that although neuronal and axonal injury are key downstream events in the evolution of damage after severe TBI, brain swelling and resultant intracranial hypertension is still the principal target for titration of therapy in the intensive care unit.
Inflammation and Regeneration
There appear to be both acute detrimental and subacute/chronic beneficial aspects of inflammation in cerebral ischemia and TBI. Inflammatory mechanisms in the evolution of secondary injury and repair have the greatest support in stroke and TBI, although some support for a role of inflammation in the regulation of neuronal death has been suggested even in cases of transient global ischemic insults.145–149 There is robust acute inflammation after stroke and TBI in both experimental models146,150,151 and in patients.152–155 Nuclear factor-κB,156 TNF-α,99,157–160 interleukin (IL)-1β,161,162 eicosanoids,163 neutrophils,164,165 and macrophages166,167 contribute to both secondary damage and repair.
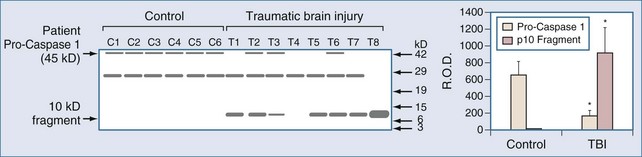
Markers of inflammation after TBI have been assessed in humans using two general strategies, (1) examination of inflammation in contused brain tissue or cerebral infarcts resected from patients with refractory intracranial hypertension, and (2) study of mediator levels in CSF. Consistent with a role for IL-1β in the evolution of tissue damage in cases of human TBI, Clark et al.39 performed western blot analysis of brain samples resected from adults with refractory intracranial hypertension secondary to severe contusion. Interleukin-1-converting enzyme (ICE), also known as caspase 1, was activated, as evidenced by specific cleavage in patients with TBI. ICE activation is critical to the production of IL-1β. ICE activation was not detected in patients who died of non-CNS causes (Figure 29-8). This supports the production of IL-1β, a pivotal proinflammatory mediator, in the traumatically injured brain in humans. Similar support for increases in a variety of inflammatory mediators exists in human stroke.99,155,159,160,164
Studies of CSF further support a role for inflammation in TBI. Marion and associates154 demonstrated increases in IL-1β in CSF after severe TBI in adults. These increases were attenuated by the use of moderate therapeutic hypothermia. Satchell et al.106 demonstrated increases in ICE that were followed by a reduction in pro-IL-1β and an increase in IL-1β in CSF after severe TBI in children. Similarly, there are increases of a number of cytokines in CSF after severe TBI and stroke, including IL-6 and IL-8.159,168 Contusion and local tissue necrosis appear to be important to trigger neutrophil influx, with resultant secondary tissue damage.169 Neutrophil influx is accompanied by increases in inducible nitric oxide synthase (iNOS) in brain155,170 and is followed by macrophage infiltration, which peaks between 24 and 72 hours after injury.171 Macrophage infiltration and the differentiation of endogenous microglia into resident macrophages may signal the link between inflammation and regeneration with elaboration of a number of trophic factors (i.e., nerve growth factor [NGF], nitrosothiols, vascular endothelial growth factor).161,168,172,173 Kossmann et al.168 reported a link between IL-6 production and the production of neurotrophins such as NGF in human head injury. Cultured astrocytes treated with either IL-6 or IL-8 in CSF from brain-injured adults produced NGF. Cytokine production after cerebral ischemia and TBI may be important to neuronal plasticity and repair, as discussed later.
Studies in models of TBI suggest early detrimental effects of a number of inflammatory mediators but beneficial effects of inflammation on long-term outcome.157,174 Mice deficient in TNF-α exhibit improved functional outcome (versus wild-type) early after TBI. However, the long-term consequences of TNF-α deficiency on outcome are detrimental.157 Similarly, despite a detrimental role for iNOS in the initial 72 hours after trauma,175 iNOS-deficient mice demonstrated impaired long-term outcome versus controls176; iNOS is important in wound healing, and iNOS-derived nitrosylation of proteins may play a role.172 Regeneration and plasticity play important roles in mediating beneficial long-term effects on recovery, and these responses are linked to inflammation. Analogs of these beneficial consequences of inflammation are anticipated in humans but remain to be demonstrated.
The contribution of the inflammatory response to cerebral ischemia and TBI remains to be determined. Although there are a few promising reports in models of the use of antiinflammatory therapies in TBI and ischemia (targeting IL-1β, ICE/caspase 1, and TNF-α), it is unclear whether antiinflammatory therapies will improve outcome after stroke or TBI in humans. Initial trials have not been promising.164 Finally, the consequences of antiinflammatory therapies on the incidence of sepsis or secondary infectious complications must also be considered.177 Similarly, the potential CNS consequences of novel immunostimulatory therapies (such as GCSF or GMCSF) for the treatment of sepsis and multiple organ failure must also be carefully considered when these agents are used in patients with multisystem disease that includes CNS injury.177
Endogenous Neuroprotectants
Ischemia, excitotoxicity, or their combination, are key facets of secondary injury. These mechanisms are linked to calcium overload, oxidative stress, and mitochondrial failure. There is, however, a coupled endogenous retaliatory response to these ischemic and excitotoxic insults. Two important components of this cascade are adenosine and heat shock protein 70 (Hsp70). Adenosine is an endogenous neuroprotectant produced in response to both ischemia and excitotoxicity. It antagonizes a number of events thought to mediate neuronal death.178 Breakdown of ATP leads to formation of adenosine, a purine nucleoside that decreases neuronal metabolism and increases CBF among other mechanisms. Adenosine binding to A1 receptors decreases metabolism by increasing K+ and Cl− and decreasing Ca++ conductances in the neuronal membrane. A1 receptors bind adenosine with high affinity and are located on neurons in brain regions that are susceptible to injury and are spatially associated with NMDA receptors.179 Thus, locally released adenosine minimizes excitotoxicity. Binding of adenosine to lower-affinity A2 receptors (on cerebrovascular smooth muscle) causes vasodilation, although binding to A2a receptors on neurons may be detrimental. Brain interstitial levels of adenosine are increased 50- to 100-fold early after experimental cerebral ischemia or TBI.180–183
In clinical studies, marked increases in brain interstitial levels of adenosine in adults with TBI were seen during episodes of jugular venous desaturation (secondary insults), supporting a role of adenosine as a “retaliatory” defense metabolite.184 Surprisingly, increases in CSF levels of the commonly consumed adenosine receptor antagonist, caffeine, were associated with favorable outcome after severe TBI in humans, a finding that may be explained by up-regulation of A1 receptors by chronic caffeine exposure.185,186 Another endogenous neuroprotectant that plays a role after cerebral ischemia, severe TBI, and SAH is Hsp70. Hsp70 optimizes protein folding as a molecular chaperone. It also inhibits proinflammatory signaling.187 Hsp70 is induced as part of the preconditioning response in brain and has been shown to be increased in both CSF and brain tissue after severe TBI in humans.103,188,189 Thus, the brain mounts an important endogenous defense response to TBI. Therapies designed to augment these pathways have not been examined adequately.
 Summary
SummaryKey Points
Barone FC, Feuerstein GZ. Inflammatory mediators and stroke: new opportunities for novel therapeutics. J Cereb Blood Flow Metab. 1999;19(8):819-834.
Bullock R, Zauner A, Woodward JJ, et al. Factors affecting excitatory amino acid release following severe human head injury. J Neurosurg. 1998;89(4):507-518.
Clark RS, Kochanek PM, Chen M, et al. Increases in Bcl-2 and cleavage of caspase-1 and caspase-3 in human brain after head injury. FASEB J. 1999;13(8):813-821.
Povlishock JT. Traumatically induced axonal injury: pathogenesis and pathobiological implications. Brain Pathol. 1992;2(1):1-12.
Siesjo BK. Cell damage in the brain: a speculative synthesis. J Cereb Blood Flow Metab. 1981;1(2):155-185.
Siesjö BK, Katsura K, Zhao Q, et al. Mechanisms of secondary brain damage in global and focal ischemia: a speculative synthesis. J Neurotrauma. 1995;12(5):943-956.
Snyder JV, Nemoto EM, Carroll RG, Safar P. Global ischemia in dogs: intracranial pressures, brain blood flow and metabolism. Stroke. 1975;6(1):21-27.
1 Snyder JV, Nemoto EM, Carroll RG, et al. Global ischemia in dogs: intracranial pressures, brain blood flow and metabolism. Stroke. 1975;6:21-27.
2 Siesjo BK. Cell damage in the brain: A speculative synthesis. J Cereb Blood Flow Metab. 1981;1:155.
3 Siesjo BK, Katsura K-I, Zhao Q, et al. Mechanisms of secondary brain damage in global and focal ischemia: A speculative synthesis. J Neurotrauma. 1995;12:943-956.
4 Beckstead JE, Tweed WA, Lee J, et al. Cerebral blood flow and metabolism in man following cardiac arrest. Stroke. 1978;9:569-573.
5 Bromont C, Marie C, Bralet J. Increased lipid peroxidation in vulnerable brain regions after transient forebrain ischemia in rats. Stroke. 1989;20:918-924.
6 Safar P, Grenvik A, Safar P, editors. Clinics in Critical Care Medicine: Brain Failure and Resuscitation. New York: Churchill Livingstone; 1981:155-184.
7 Hermann DM, Kilic E, Hata R, et al. Relationship between metabolic dysfunctions, gene responses and delayed cell death after mild focal cerebral ischemia in mice. Neuroscience. 2001;104:947-955.
8 Hossmann KA. Viability thresholds and the penumbra of focal ischemia. Ann Neurol. 1994;36:557-565.
9 Kochanek PM, Clark RS, Ruppel RA, et al. Biochemical, cellular, and molecular mechanisms in the evolution of secondary damage after severe traumatic brain injury in infants and children: Lessons learned from the bedside. Pediatr Crit Care Med. 2000;1:4-19.
10 McIntosh TK. Novel pharmacologic therapies in the treatment of experimental traumatic brain injury: A review. J Neurotrauma. 1993;10:215-261.
11 Pittella JE, Gusmao SN. Diffuse vascular injury in fatal road traffic accident victims: its relationship to diffuse axonal injury. J Forensic Sci. 2003;48:626-630.
12 Lee TT, Galarza M, Villanueva PA. Diffuse axonal injury (DAI) is not associated with elevated intracranial pressure (ICP). Acta Neurochir. 1998;140:41-46.
13 Chestnut RM, Marshall LF, Klauber MR, et al. The role of secondary brain injury in determining outcome from severe head injury. J Trauma. 1993;34:216-222.
14 Clark RSB, Kochanek PM, Dixon CE, et al. Early neuropathologic effects of mild or moderate hypoxemia after controlled cortical impact injury in rats. J Neurotrauma. 1997;14:179-189.
15 Lucas DR, Newhouse JP. The toxic effect of sodium L-glutamate on the inner layers of the retina. AMA Arch Ophthalmol. 1957;58:193-201.
16 Olney JW. Brain lesions, obesity, and other disturbances in mice treated with monosodium glutamate. Science. 1969;164:719-721.
17 Choi DW, Maulucci-Gedde M, Kriegstein AR. Glutamate neurotoxicity in cortical cell culture. J Neurosci. 1987;7:357-368.
18 Choi DW. Ionic dependence of glutamate neurotoxicity. J Neurosci. 1987;7:369-379.
19 Du L, Zhang X, Han YY, et al. Intra-mitochondrial poly-ADP-ribosylation contributes to NAD+ depletion and cell death induced by oxidative stress. J Biol Chem. 2003;278:18426-18433.
20 Whalen MJ, Clark RS, Dixon CE, et al. Reduction of cognitive and motor deficits after traumatic brain injury in mice deficient in poly(ADP-ribose) polymerase. J Cereb Blood Flow Metab. 1999;19:835-842.
21 Zhang J, Dawson VL, Dawson TM, et al. Nitric oxide activation of poly(ADP-ribose) synthetase in neurotoxicity. Science. 1994;263:687-689.
22 Lai Y, Chen Y, Watkins SC, et al. Identification of poly-ADP-ribosylated mitochondrial proteins after traumatic brain injury. J Neurochem. 2008;104:1700-1711.
23 Eliasson MJ, Sampei K, Mandir AS, et al. Poly(ADP-ribose) polymerase gene disruption renders mice resistant to cerebral ischemia. Nat Med. 1997;3:1089-1095.
24 Benveniste H, Harper AM, Macrae IM, editors. Cerebrovascular Brain Metabolism Reviews. New York: Raven Press; 1991:213-245.
25 Faden AI, Demediuk P, Panter SS, et al. The role of excitatory amino acids and NMDA receptors in traumatic brain injury. Science. 1989;244:798-800.
26 Goodman JC, Valadka AB, Gopinpath SP, et al. Lactate and excitatory amino acids measured by microdialysis are decreased by pentobarbital coma in head-injured patients. J Neurotrauma. 1996;13:549-556.
27 Hayes RL, Jenkins LW, Lyeth BG, et al. Pretreatment with phencyclidine, an N-methyl-D-aspartate antagonist, attenuates long-term behavioral deficits in the rat produced by traumatic brain injury. J Neurotrauma. 1988;5:259-274.
28 Huettner JE. Indole-2-carboxylic acid: a competitive antagonist of potentiation by glycine at the NMDA receptor. Science. 1989;243:1611-1613.
29 McIntosh TK, Vink R, Soares H, et al. Effect of noncompetitive blockade of N-methyl-D-aspartate receptors on the neurochemical sequelae of experimental brain injury. J Neurochem. 1990;55:1170-1179.
30 Smith DH, Okiyama K, Gennarelli TA, et al. Magnesium and ketamine attenuate cognitive dysfunction following experimental brain injury. Neurosci Lett. 1993;157:211-214.
31 Suehiro E, Fujisawa H, Ito H, et al. Brain temperature modifies glutamate neurotoxicity in vivo. J Neurotrauma. 1999;16:285-297.
32 Persson L, Hillered L. Chemical monitoring of neurosurgical intensive care patients using intracerebral microdialysis. J Neurosurg. 1992;76:72-80.
33 Palmer AM, Marion DW, Botscheller ML, et al. Increased transmitter amino acid concentration in human ventricular CSF after brain injury. Neuroreport. 1994;6:153-156.
34 Meldrum B, Garthwaite J. Excitatory amino acid neurotoxicity and neurodegenerative disease. Trends Pharmacol Sci. 1990;11:379-387.
35 Bullock R, Zauner A, Woodward JJ, et al. Factors affecting excitatory amino acid release following severe human head injury. J Neurosurg. 1998;89:507-518.
36 Bullock R, Zauner A, Woodward J, et al. Massive persistent release of excitatory amino acids following human occlusive stroke. Stroke. 1995;26:2187-2189.
37 Doppenberg EM, Choi SC, Bullock R. Clinical trials in traumatic brain injury. What can we learn from previous studies? Ann N Y Acad Sci. 1997;825:305-322.
38 Hendricson AW, Thomas MP, Lippmann MJ, et al. Suppression of L-type voltage-gated calcium channel-dependent synaptic plasticity by ethanol: analysis of miniature synaptic currents and dendritic calcium transients. J Pharmacol Exp Ther. 2003;307:550-558.
39 Clark RS, Kochanek PM, Chen M, et al. Increases in Bcl-2 and cleavage of caspase-1 and caspase-3 in human brain after head injury. FASEB J. 1999;13:813-821.
40 Rink A, Fung K-M, Trojanowski JQ, et al. Evidence of apoptotic cell death after experimental traumatic brain injury in the rat. Am J Pathol. 1995;147:1575-1583.
41 Chu CT, Plowey ED, Dagda RK, et al. Autophagy in neurite injury and neurodegeneration: in vitro and in vivo models. Methods Enzymol. 2009;453:217-249.
42 David KK, Andrabi SA, Dawson TM, et al. Parthanatos, a messenger of death. Front Biosci. 2009;14:1116-1128.
43 Degterev A, Huang Z, Boyce M, et al. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol. 2005;1:112-119.
44 Kerr JF, Wyllie AH, Currie AR. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer. 1972;26:239-257.
45 Steller H. Mechanisms and genes of cellular suicide. Science. 1995;267:1445-1449.
46 Portera-Cailliau C, Price DL, Martin LJ. Excitotoxic neuronal death in the immature brain is an apoptosis-necrosis morphological continuum. J Comp Neurol. 1997;378:70-87.
47 Lockshin RA. Programmed cell death. Activation of lysis by a mechanism involving the synthesis of protein. J Insect Physiol. 1969;15:1505-1516.
48 Marovitz WF, Shugar JM, Khan KM. The role of cellular degeneration in the normal development of (rat) otocyst. Laryngoscope. 1976;86:1413-1425.
49 Webster DA, Gross J. Studies on possible mechanisms of programmed cell death in the chick embryo. Dev Biol. 1970;22:157-184.
50 Liou AK, Clark RS, Henshall DC, et al. To die or not to die for neurons in ischemia, traumatic brain injury and epilepsy: a review on the stress-activated signaling pathways and apoptotic pathways. Prog Neurobiol. 2003;69:103-142.
51 Maiuri MC, Zalckvar E, Kimchi A, et al. Self-eating and self-killing: crosstalk between autophagy and apoptosis. Nat Rev Mol Cell Biol. 2007;8:741-752.
52 Zhang X, Chen J, Graham SH, et al. Intranuclear localization of apoptosis-inducing factor (AIF) and large scale DNA fragmentation after traumatic brain injury in rats and in neuronal cultures exposed to peroxynitrite. J Neurochem. 2002;82:181-191.
53 Eldadah BA, Faden AI. Caspase pathways, neuronal apoptosis, and CNS injury. J Neurotrauma. 2000;17:811-829.
54 Kothakota S, Azuma T, Reinhard C, et al. Caspase-3-generated fragment of gelsolin: effector of morphological change in apoptosis. Science. 1997;278:294-298.
55 Cao G, Pei W, Lan J, et al. Caspase-activated DNase/DFF40 mediates apoptotic DNA fragmentation in transient cerebral ischemia and in neuronal cultures. J Neurosci. 2001. in press
56 Clark RSB, Kochanek PM, Watkins SC, et al. Caspase-3 mediated neuronal death after traumatic brain injury in rats. J Neurochem. 2000;74:740-753.
57 Nicholson DW, Ali A, Thornberry NA, et al. Identification and inhibition of the ICE/CED-3 protease necessary for mammalian apoptosis [see comments]. Nature. 1995;376:37-43.
58 Shackelford DA, Tobaru T, Zhang S, et al. Changes in expression of the DNA repair protein complex DNA-dependent protein kinase after ischemia and reperfusion. J Neurosci. 1999;19:4727-4738.
59 Ashkenazi A, Dixit VM. Death receptors: signaling and modulation. Science. 1998;281:1305-1308.
60 Qiu J, Whalen MJ, Lowenstein P, et al. Upregulation of the Fas receptor death-inducing signaling complex after traumatic brain injury in mice and humans. J Neurosci. 2002;22:3504-3511.
61 Kischkel FC, Lawrence DA, Tinel A, et al. Death receptor recruitment of endogenous caspase-10 and apoptosis initiation in the absence of caspase-8. J Biol Chem. 2001;276:46639-46646.
62 Jenkins LW, Dixon CE, Peters G, et al. Clark RS, Kochanek PM, editors. Brain Injury. vol. 2. Boston: Kluwer Academic Publishers; 2001:163-180.
63 Mori T, Wang X, Jung JC, et al. Mitogen-activated protein kinase inhibition in traumatic brain injury: in vitro and in vivo effects. J Cereb Blood Flow Metab. 2002;22:444-452.
64 Otani N, Nawashiro H, Fukui S, et al. Differential activation of mitogen-activated protein kinase pathways after traumatic brain injury in the rat hippocampus. J Cereb Blood Flow Metab. 2002;22:327-334.
65 Zhang X, Graham SH, Kochanek PM, et al. Caspase-8 expression and proteolysis in human brain after severe head injury. FASEB J. 2003;17:1367-1369.
66 Moquin D, Chan FK. The molecular regulation of programmed necrotic cell injury. Trends Biochem Sci. 2010.
67 Zou H, Henzel WJ, Liu X, et al. Apaf-1, a human protein homologous to C. elegans CED-4, participates in cytochrome c-dependent activation of caspase-3. Cell. 1997;90:405-413.
68 Klein JA, Longo-Guess CM, Rossmann MP, et al. The harlequin mouse mutation downregulates apoptosis-inducing factor. Nature. 2002;419:367-374.
69 Susin SA, Lorenzo HK, Zamzami N, et al. Molecular characterization of mitochondrial apoptosis-inducing factor. Nature. 1999;397:441-446.
70 Cao G, Clark RS, Pei W, et al. Translocation of apoptosis-inducing factor in vulnerable neurons after transient cerebral ischemia and in neuronal cultures after oxygen-glucose deprivation. J Cereb Blood Flow Metab. 2003;23:1137-1150.
71 Yu SW, Wang H, Poitras MF, et al. Mediation of poly(ADP-ribose) polymerase-1-dependent cell death by apoptosis-inducing factor. Science. 2002;297:259-263.
72 Li LY, Luo X, Wang X. Endonuclease G is an apoptotic DNase when released from mitochondria. Nature. 2001;412:95-99.
73 Suzuki Y, Imai Y, Nakayama H, et al. A serine protease, HtrA2, is released from the mitochondria and interacts with XIAP, inducing cell death. Mol Cell. 2001;8:613-621.
74 Chai J, Du C, Wu JW, et al. Structural and biochemical basis of apoptotic activation by Smac/DIABLO. Nature. 2000;406:855-862.
75 Shibata M, Hattori H, Sasaki T, et al. Activation of caspase-12 by endoplasmic reticulum stress induced by transient middle cerebral artery occlusion in mice. Neuroscience. 2003;118:491-499.
76 Larner SF, Hayes RL, McKinsey DM, et al. Increased expression and processing of caspase-12 after traumatic brain injury in rats. J Neurochem. 2004;88:78-90.
77 Komatsu M, Waguri S, Chiba T, et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature. 2006;441:880-884.
78 Du L, Hickey RW, Bayir H, et al. Starving neurons show sex difference in autophagy. J Biol Chem. 2009;284:2383-2396.
79 Koike M, Shibata M, Tadakoshi M, et al. Inhibition of autophagy prevents hippocampal pyramidal neuron death after hypoxic-ischemic injury. Am J Pathol. 2008;172:454-469.
80 Lai Y, Hickey RW, Chen Y, et al. Autophagy is increased after traumatic brain injury in mice and is partially inhibited by the antioxidant gamma-glutamylcysteinyl ethyl ester. J Cereb Blood Flow Metab. 2008;28:540-550.
81 Clark RS, Bayir H, Chu CT, et al. Autophagy is increased in mice after traumatic brain injury and is detectable in human brain after trauma and critical illness. Autophagy. 2008;4:88-90.
82 Carloni S, Buonocore G, Balduini W. Protective role of autophagy in neonatal hypoxia-ischemia induced brain injury. Neurobiol Dis. 2008.
83 Koike M, Shibata M, Waguri S, et al. Participation of autophagy in storage of lysosomes in neurons from mouse models of neuronal ceroid-lipofuscinoses (Batten disease). Am J Pathol. 2005;167:1713-1728.
84 Puyal J, Vaslin A, Mottier V, et al. Postischemic treatment of neonatal cerebral ischemia should target autophagy. Ann Neurol. 2009.
85 Djavaheri-Mergny M, Maiuri MC, Kroemer G. Cross talk between apoptosis and autophagy by caspase-mediated cleavage of Beclin 1. Oncogene. 2010;29:1717-1719.
86 Graham SH, Chen J, Clark RS. Bcl-2 family gene products in cerebral ischemia and traumatic brain injury. J Neurotrauma. 2000;17:831-841.
87 Yin X-M, Oltvai ZN, Korsmeyer SJ. BH1 and BH2 domains of bcl-2 are required for inhibition of apoptosis and heterodimerization with bax. Nature. 1994;369:321-323.
88 Henshall DC, Bonislawski DP, Skradski SL, et al. Cleavage of bid may amplify caspase-8-induced neuronal death following focally evoked limbic seizures. Neurobiol Dis. 2001;8:568-580.
89 Oltvai ZN, Milliman CL, Korsmeyer SJ. Bcl-2 heterodimerizes in vivo with a conserved homolog, bax, that accelerates programmed cell death. Cell. 1993;74:609-619.
90 Antonsson B, Conti F, Ciavatta AM, et al. Inhibition of Bax Channel-forming activity by Bcl-2. Science. 1997;277:370-372.
91 Krajewski S, Mai JK, Krajewska M, et al. Upregulation of Bax protein levels in neurons following cerebral ischemia. J Neurosci. 1995;15:6364-6376.
92 Raghupathi R, Fernandez SC, Murai H, et al. BCL-2 overexpression attenuates cortical cell loss after traumatic brain injury in transgenic mice. J Cereb Blood Flow Metab. 1998;18:1259-1269.
93 Cao G, Pei W, Ge H, et al. In vivo delivery of a Bcl-xL fusion protein containing the TAT protein transduction domain protects against ischemic brain injury and neuronal apoptosis. J Neurosci. 2002;22:5423-5431.
94 Yue Z, Jin S, Yang C, et al. Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc Natl Acad Sci U S A. 2003;100:15077-15082.
95 Evans JP, Scheinker IM. Histologic studies of the brain following head trauma. IV. Late changes: Atrophic sclerosis of the white matter. J Neurosurg. 1944;1:306-320.
96 Evans JP, Scheinker IM. Histologic studies of the brain following head trauma: I. Post-traumatic cerebral swelling and edema. J Neurosurg. 1945;2:306-314.
97 Clark RS, Kochanek PM, Adelson PD, et al. Increases in bcl-2 protein in cerebrospinal fluid and evidence for programmed cell death in infants and children after severe traumatic brain injury. J Pediatr. 2000;137:197-204.
98 Zhang X, Alber S, Watkins SC, et al. Proteolysis consistent with activation of caspase-7 after severe traumatic brain injury in humans. J Neurotrauma. 2006. in press
99 Tarkowski E, Rosengren L, Blomstrand C, et al. Intrathecal expression of proteins regulating apoptosis in acute stroke. Stroke. 1999;30:321-327.
100 Henshall DC, Clark RS, Adelson PD, et al. Alterations in bcl-2 and caspase gene family protein expression in human temporal lobe epilepsy. Neurology. 2000;55:250-257.
101 Ertel W, Keel M, Stocker R, et al. Detectable concentrations of Fas ligand in cerebrospinal fluid after severe head injury. J Neuroimmunol. 1997;80:93-96.
102 Lenzlinger PM, Marx A, Trentz O, et al. Prolonged intrathecal release of soluble Fas following severe traumatic brain injury in humans. J Neuroimmunol. 2002;122:167-174.
103 Seidberg N, Clark RS, Zhang X, et al. Alterations in inducible 72 kilodalton heat shock protein and the chaperone cofactor BAG-1 in human brain after head injury. J Neurochem. 2003;84:514-521.
104 Ng I, Yeo TT, Tang WY, et al. Apoptosis occurs after cerebral contusions in humans. Neurosurgery. 2000;46:949-956.
105 Williams S, Raghupathi R, MacKinnon MA, et al. In situ DNA fragmentation occurs in white matter up to 12 months after head injury in man. Acta Neuropathologica. 2001;102:581-590.
106 Satchell MA, Lai Y, Kochanek PM, et al. Cytochrome c, a biomarker of apoptosis, is increased in cerebrospinal fluid from infants with inflicted brain injury from child abuse. J Cereb Blood Flow Metab. 2005;25:919-927.
107 Fink EL, Lai Y, Zhang X, et al. Quantification of poly(ADP-ribose)-modified proteins in cerebrospinal fluid from infants and children after traumatic brain injury. J Cereb Blood Flow Metab. 2008.
108 Sarnaik AA, Conley YP, Okonkwo DO, et al. Influence of PARP-1 polymorphisms in patients after traumatic brain injury. J Neurotrauma. 2010;27:465-471.
109 Du L, Bayir H, Lai Y, et al. Innate gender-based proclivity in response to cytotoxicity and programmed cell death pathway. J Biol Chem. 2004;279:38563-38570.
110 Hagberg H, Wilson MA, Matsushita H, et al. PARP-1 gene disruption in mice preferentially protects males from perinatal brain injury. J Neurochem. 2004;90:1068-1075.
111 Renolleau S, Fau S, Goyenvalle C, et al. Specific caspase inhibitor Q-VD-OPh prevents neonatal stroke in P7 rat: a role for gender. J Neurochem. 2007;100:1062-1071.
112 Sloviter RS. Apoptosis: a guide for the perplexed. Trends Pharmacol Sci. 2002;23:19-24.
113 Mild therapeutic hypothermia to improve the neurologic outcome after cardiac arrest. N Engl J Med. 2002;346:549-556.
114 Bernard SA, Gray TW, Buist MD, et al. Treatment of comatose survivors of out-of-hospital cardiac arrest with induced hypothermia. N Engl J Med. 2002;346:557-563.
115 Safar PJ, Kochanek PM. Therapeutic hypothermia after cardiac arrest. N Engl J Med. 2002;346:612-613.
116 Adams JH, Graham DI, Murray LS, et al. Diffuse axonal injury due to nonmissile head injury in humans: an analysis of 45 cases. Ann Neurol. 1982;12:557-563.
117 Genneralli TA, Thibault LF, Adams TH, et al. Diffuse axonal injury and traumatic coma in the primate. Ann Neurol. 1982;12:564-568.
118 Fitzpatrick MO, Maxwell WL, Graham DI. The role of the axolemma in the initiation of traumatically induced axonal injury. J Neurol Neurosurg Psychiatry. 1998;64:285-287.
119 Povlishock JT. Traumatically induced axonal injury: pathogenesis and pathobiological implications. Brain Pathol. 1992;2:1-12.
120 Smith DH, Chen XH, Xu BN, et al. Characterization of diffuse axonal pathology and selective hippocampal damage following inertial brain trauma in the pig. J Neuropathol Exp Neurol. 1997;56:822-834.
121 Gennarelli TA. Mechanisms of brain injury. J Emerg Med. 1993;11(Suppl 1):5-11.
122 Graham DI, Ford I, Adams JH, et al. Fatal head injury in children. J Clin Pathol. 1989;42:18-22.
123 Graham DI, Lawrence AE, Adams JH, et al. Brain damage in fatal non-missile head injury without high intracranial pressure. J Clin Pathol. 1988;41:34-37.
124 Povlishock JT, Buki A, Koiziumi H, et al. Initiating mechanisms involved in the pathobiology of traumatically induced axonal injury and interventions targeted at blunting their progression. Acta Neurochir Suppl. 1999;73:15-20.
125 Povlishock JT, Jenkins LW. Are the pathobiological changes evoked by traumatic brain injury immediate and irreversible? Brain Pathol. 1995;5:415-426.
126 Maxwell WL, Povlishock JT, Graham DL. A mechanistic analysis of nondisruptive axonal injury: A review. J Neurotrauma. 1997;14:419-440.
127 Stys PK. Anoxic and ischemic injury of myelinated axons in CNS white matter: from mechanistic concepts to therapeutics. J Cereb Blood Flow Metab. 1998;18:2-25.
128 Kampfl A, Posmantur RM, Zhao X, et al. Mechanisms of calpain proteolysis following traumatic brain injury: implications for pathology and therapy: implications for pathology and therapy: a review and update. J Neurotrauma. 1997;14:121-134.
129 Buki A, Koizumi H, Povlishock JT. Moderate posttraumatic hypothermia decreases early calpain-mediated proteolysis and concomitant cytoskeletal compromise in traumatic axonal injury. Exp Neurol. 1999;159:319-328.
130 Buki A, Okonkwo DO, Povlishock JT. Postinjury cyclosporin A administration limits axonal damage and disconnection in traumatic brain injury. J Neurotrauma. 1999;16:511-521.
131 Singh M, Jeong J, Hwang D, et al. Novel diffusion tensor imaging methodology to detect and quantify injured regions and affected brain pathways in traumatic brain injury. Magn Reson Imaging. 2010;28:22-40.
132 Tshibanda L, Vanhaudenhuyse A, Galanaud D, et al. Magnetic resonance spectroscopy and diffusion tensor imaging in coma survivors: promises and pitfalls. Prog Brain Res. 2009;177:215-229.
133 Seelig JM, Becker DP, Miller JD, et al. Traumatic acute subdural hematoma: major mortality reduction in comatose patients treated within four hours. N Engl J Med. 1981;304:1511-1518.
134 Marmarou A, Barzo P, Fatouros P, et al. Traumatic brain swelling in head injured patients: brain edema or vascular engorgement? Acta Neurochir Suppl. 1997;70:68-70.
135 Nag S, Manias JL, Stewart DJ. Pathology and new players in the pathogenesis of brain edema. Acta Neuropathol. 2009;118:197-217.
136 Nicchia GP, Frigeri A, Liuzzi GM, et al. Inhibition of aquaporin-4 expression in astrocytes by RNAi determines alteration in cell morphology, growth, and water transport and induces changes in ischemia-related genes. FASEB J. 2003;17:1508-1510.
137 Sun MC, Honey CR, Berk C, et al. Regulation of aquaporin-4 in a traumatic brain injury model in rats. J Neurosurg. 2003;98:565-569.
138 Griesdale DE, Honey CR. Aquaporins and brain edema. Surg Neurol. 2004;61:418-421.
139 Barzo P, Marmarou A, Fatouros P, et al. Contribution of vasogenic and cellular edema to traumatic brain swelling measured by diffusion-weighted imaging. J Neurosurg. 1997;87:900-907.
140 Barzo P, Marmarou A, Fatouros P, et al. Magnetic resonance imaging-monitored acute blood-brain barrier changes in experimental traumatic brain injury. J Neurosurg. 1996;85:1113-1121.
141 Katayama Y, Mori T, Maeda T, et al. Pathogenesis of the mass effect of cerebral contusions: rapid increase in osmolality within the contusion necrosis. Acta Neurochir Suppl. 1998;71:289-292.
142 Katayama Y, Kawamata T. Edema fluid accumulation within necrotic brain tissue as a cause of the mass effect of cerebral contusion in head trauma patients. Acta Neurochir Suppl. 2003;86:323-327.
143 Bergsneider M, Hovda DA, Shalmon E, et al. Cerebral hyperglycolysis following severe traumatic brain injury in humans: a positron emission tomography study. J Neurosurg. 1997;86:241-251.
144 Obrist WD, Langfitt TW, Jaggi JL, et al. Cerebral blood flow and metabolism in comatose patients with acute head injury. Relationship to intracranial hypertension. J Neurosurg. 1984;61:241-253.
145 Clemens JA, Stephenson DT, Smalstig EB, et al. Global ischemia activates nuclear factor-kappa B in forebrain neurons of rats. Stroke. 1997;28:1073-1080. discussion 80-1
146 Kochanek P, Hallenbeck J. Polymorphonuclear leukocytes and monocytes/macrophages in the pathogenesis of cerebral ischemia and stroke. Stroke. 1992;23:1367-1379.
147 Mizushima H, Zhou CJ, Dohi K, et al. Reduced postischemic apoptosis in the hippocampus of mice deficient in interleukin-1. J Comp Neurol. 2002;448:203-216.
148 Saito K, Suyama K, Nishida K, et al. Early increases in TNF-alpha, IL-6 and IL-1 beta levels following transient cerebral ischemia in gerbil brain. Neurosci Lett. 1996;206:149-152.
149 Tang M, Alexander H, Clark RS, et al. Minocycline reduces neuronal death and attenuates microglial response after pediatric asphyxial cardiac arrest. J Cereb Blood Flow Metab. 2009.
150 Barone FC, Feuerstein GZ. Inflammatory mediators and stroke: new opportunities for novel therapeutics. J Cereb Blood Flow Metab. 1999;19:819-834.
151 Rosomoff HL, Clasen RA, Hartstock R, et al. Brain reaction to experimental injury after hypothermia. Arch Neurol. 1965;13:337-345.
152 Kossmann T, Hans VHJ, Imhof H-G, et al. Intrathecal and serum interleukin-6 and the acute-phase response in patients with severe traumatic brain injuries. Shock. 1995;4:311-317.
153 Kossmann T, Stahel PF, Lenzlinger PM, et al. Interleukin-8 released into the cerebrospinal fluid after brain injury is associated with blood-brain barrier dysfunction and nerve growth factor production. J Cereb Blood Flow Metab. 1997;17:280-289.
154 Marion DW, Penrod LE, Kelsey SF, et al. Treatment of traumatic brain injury with moderate hypothermia. N Engl J Med. 1997;336:540-546.
155 Forster C, Clark HB, Ross ME, et al. Inducible nitric oxide synthase expression in human cerebral infarcts. Acta Neuropathol. 1999;97:215-220.
156 Bethea JR, Castro M, Keane RW, et al. Traumatic spinal cord injury induces nuclear factor-kappaB activation. J Neurosci. 1998;18:3251-3260.
157 Scherbel U, Raghupathi R, Nakamura M, et al. Differential acute and chronic responses of tumor necrosis factor-deficient mice to experimental brain injury. Proc Natl Acad Sci U S A. 1999;96:8721-8726.
158 Shohami E, Bass R, Wallach D, et al. Inhibition of tumor necrosis factor alpha (TNFa) activity in rat brain is associated with cerebroprotection after closed head injury. J Cereb Blood Flow Metab. 1996;16:378-384.
159 Vila N, Castillo J, Davalos A, et al. Proinflammatory cytokines and early neurological worsening in ischemic stroke. Stroke. 2000;31:2325-2329.
160 Zaremba J, Losy J. Early TNF-alpha levels correlate with ischaemic stroke severity. Acta Neurol Scand. 2001;104:288-295.
161 DeKosky ST, Styren SD, O’Malley ME, et al. Interleukin-1 receptor antagonist suppresses neurotrophin response in injured rat brain. Ann Neurol. 1996;39:123-127.
162 Toulmond S, Rothwell NJ. Interleukin-1 receptor antagonist inhibits neuronal damage caused by fluid percussion injury in the rat. Brain Res. 1995;671:261-266.
163 Shapira Y, Artru AA, Yadid G, et al. Methylprednisolone does not decrease eicosanoid concentrations or edema in brain tissue or improve neurologic outcome after head trauma in rats. Anesth Analg. 1992;75:238-244.
164 Use of anti-ICAM-1 therapy in ischemic stroke: results of the Enlimomab Acute Stroke Trial. Neurology. 2001;57:1428-1434.
165 Uhl MW, Biagas KV, Grundl PD, et al. Effects of neutropenia on edema, histology, and cerebral blood flow after traumatic brain injury in rats. J Neurotrauma. 1994;11:303-315.
166 Blight AR. Effects of silica on the outcome from experimental spinal cord injury: implication of macrophages in secondary tissue damage. Neuroscience. 1994;60:263-273.
167 Popovich PG, Guan Z, Wei P, et al. Depletion of hematogenous macrophages promotes partial hindlimb recovery and neuroanatomical repair after experimental spinal cord injury. Exp Neurol. 1999;158:351-365.
168 Kossmann T, Hans V, Imhof H-G, et al. Interleukin-6 released in human cerebrospinal fluid following traumatic brain injury may trigger nerve growth factor production in astrocytes. Brain Res. 1996;713:143-152.
169 Schoettle RJ, Kochanek PM, Magargee MJ, et al. Early polymorphonuclear leukocyte accumulation correlates with the development of posttraumatic cerebral edema in rats. J Neurotrauma. 1990;7:207-217.
170 Clark RSB, Schiding JK, Kaczorowski SL, et al. Neutrophil accumulation after traumatic brain injury in rats: Comparison of weight-drop and controlled cortical impact models. J Neurotrauma. 1994;11:499-506.
171 Sinz EH, Kochanek PM, Heyes MP, et al. Quinolinic acid is increased in CSF and associated with mortality after traumatic brain injury in humans. J Cereb Blood Flow Metab. 1998;18:610-615.
172 Bayir H, Kochanek PM, Liu SX, et al. Increased S-nitrosothiols and S-nitrosoalbumin in cerebrospinal fluid after severe traumatic brain injury in infants and children: indirect association with intracranial pressure. J Cereb Blood Flow Metab. 2003;23:51-61.
173 Shore PM, Jackson EK, Wisniewski SR, et al. Vascular endothelial growth factor is increased in cerebrospinal fluid after traumatic brain injury in infants and children. Neurosurgery. 2004;54:605-611. discussion 11-2
174 Sinz EH, Kochanek PM, Dixon CE, et al. Inducible nitric oxide synthase is an endogenous neuroprotectant after traumatic brain injury in rats and mice. J Clin Invest. 1999;104:647-656.
175 Yamasaki K, Edington HD, McClosky C, et al. Reversal of impaired wound repair in iNOS-deficient mice by topical adenoviral-mediated iNOS gene transfer. J Clin Invest. 1998;101:967-971.
176 Wada K, Chatzipanteli K, Kraydieh S, et al. Inducible nitric oxide synthase expression after traumatic brain injury and neuroprotection with aminoguanidine treatment in rats. Neurosurgery. 1998;43:1427-1436.
177 Heard SO, Fink MP, Gamelli RL, et al. Effect of prophylactic administration of recombinant human granulocyte colony-stimulating factor (filgrastim) on the frequency of nosocomial infections in patients with acute traumatic brain injury or cerebral hemorrhage. Crit Care Med. 1998;26:748-754.
178 Rudolphi KA, Schubert P, Parkinson FE, et al. Neuroprotective role of adenosine in cerebral ischaemia. Trends Pharmacol Sci. 1992;13:439-445.
179 Deckert J, Jorgensen MB. Evidence for pre- and postsynpatic localization of adenosine A1 receptors in the CA1 region of rat hippocampus: a quantitative autoradiographic study. Brain Res. 1988;446:161-164.
180 Bell MJ, Kochanek PM, Carcillo JA, et al. Interstitial adenosine, inosine, and hypoxanthine are increased after experimental traumatic brain injury in the rat. J Neurotrauma. 1998;15:163-170.
181 Hagberg H, Andersson P, Lacarewicz J, et al. Extracellular adenosine, inosine, hypoxanthine, and xanthine in relation to tissue nucleotides and purines in rat striatum during transient ischemia. J Neurochem. 1987;49:227-231.
182 Headrick JP, Bendall MR, Faden AI, et al. Dissociation of adenosine levels from bioenergetic state in experimental brain trauma: Potential role in secondary injury. J Cereb Blood Flow Metab. 1994;14:853-861.
183 Nilsson P, Hillered L, Ponten U, et al. Changes in cortical extracellular levels of energy-related metabolites and amino acids following concussive brain injury in rats. J Cereb Blood Flow Metab. 1990;10:631-637.
184 Bell MJ, Robertson CS, Kochanek PM, et al. Interstitial brain adenosine and xanthine increase during jugular venous oxygen desaturations in humans after traumatic brain injury. Crit Care Med. 2001;29:399-404.
185 Sachse KT, Jackson EK, Wisniewski SR, et al. Increases in cerebrospinal fluid caffeine concentration are associated with favorable outcome after severe traumatic brain injury in humans. J Cereb Blood Flow Metab. 2008;28:395-401.
186 Rudolphi KA, Keil M, Fastbom J, et al. Ischaemic damage in gerbil hippocampus is reduced following upregulation of adenosine (A1) receptors by caffeine treatment. Neurosci Lett. 1989;103:275-280.
187 Simon MM, Reikerstorfer A, Schwarz A, et al. Heat shock protein 70 overexpression affects the response to ultraviolet light in murine fibroblasts. Evidence for increased cell viability and suppression of cytokine release. J Clin Invest. 1995;95:926-933.
188 Dutcher SA, Underwood BD, Michael DB, et al. Heat-shock protein 72 expression in excitotoxic versus penetrating injuries of the rodent cerebral cortex. J Neurotrauma. 1998;15:421-432.
189 Lai Y, Kochanek PM, Adelson PD, et al. Induction of the stress response after inflicted and non-inflicted traumatic brain injury in infants and children. J Neurotrauma. 2004;21:229-238.
