Biliary Atresia
Biliary atresia is a relatively rare obstructive condition of the bile ducts causing neonatal jaundice. There is a variable incidence around the world (eg, Europe, 1 in 18,000 live births; France, 1 in 19,500 live births; the UK and Ireland, 1 in 16,700 live births; Sweden, 1 in 14,000 live births; and Japan, 1 in 9640 live births),1–6 with the highest recorded incidence in French Polynesia (1 in 3124 live births).7 There is a slight female predominance.
Biliary atresia first appeared as a distinct entity in the Edinburgh Medical Journal in 1891.8 The concept of ‘correctable’ and ‘noncorrectable’ forms was introduced in 1916.9 Despite the first successful surgical treatment for the correctable type being reported in 1928, there were only a few long-term survivors over the next three decades, all with the correctable type.10–12
In the 1950s and 60s, a variety of procedures for ‘noncorrectable’ disease were developed, but none provided consistent biliary decompression.13–17 Also, the timing of operative intervention was controversial, with reports of ‘spontaneous’ cures, and second-look explorations for the rather mystical belief that a totally fibrotic extrahepatic ductal system might subsequently become patent.18–22
In 1959, the now common Kasai hepatic portoenterostomy procedure was first described and ended a long, hopeless era for patients with the noncorrectable-type disease.23 Kasai’s original report was in Japanese and received little attention until it was published in English in 1968.24 Although effective bile drainage could be achieved after portoenterostomy in about 50% of patients, early repair was crucial and needed to be performed before the age of 2 months. Conversely, effective bile drainage was observed in only 7% of patients if correction was performed after the age of 4 months.25 Kasai’s portoenterostomy procedure gradually gained popularity in the USA, and by the 1990s, more than 90% of infants with biliary atresia had undergone this procedure.26
Recently, the authors had an opportunity to review an original video of Professor Kasai performing his own portoenterostomy.27 Interestingly, his original portal dissection was actually quite shallow and limited, resulting in a narrow portoenterostomy anastomosis, with sutures placed shallowly at 2 and 10 o’clock where the native right and left bile ducts would have been, probably to minimize microscopic bile duct injury. Although not supported by research, the original technique as performed by Kasai may result in superior outcomes as it focuses specifically on the physiologic and anatomic characteristics of the liver in biliary atresia. The authors currently perform a modified version of Kasai’s original portoenterostomy (KOPE) using the minimally invasive approach.
Pathogenesis
There are syndromic and nonsyndromic forms of biliary atresia.28 Syndromic biliary atresia (also known as the embryonic type) is associated with other congenital anomalies, including interrupted inferior vena cava, preduodenal portal vein, intestinal malrotation, situs inversus, cardiac defects, and polysplenia.29 Syndromic biliary atresia accounts for 10–20% of all cases, and is likely to be due to a developmental insult occurring during differentiation of the hepatic diverticulum from the foregut of the embryo. A possible relation between syndromic biliary atresia and maternal diabetes has been reported.28 Nonsyndromic biliary atresia (also known as the perinatal type) may have its origins later in gestation, and may have a different clinical course, with biliary obstruction being progressive.
Reovirus type 3 infection, rotavirus, cytomegalovirus (CMV), papillomavirus, and Epstein–Barr virus have all been proposed as possible etiologic agents, but conclusive evidence is lacking. In one report, CMV infection was found in four of ten patients with biliary atresia,30 and reovirus infection has been found in the livers of up to 55% of biliary atresia patients versus 10–20% in a control group.31 The identification of viruses in children with biliary atresia is inconsistent in the literature, and several viruses have been used to create animal models that may be valuable for assessing the pathogenesis and treatment of biliary atresia.
Generally, biliary atresia is not considered an inherited disorder. However, genetic mutations that result in defective morphogenesis may be important in syndromic biliary atresia. Transgenic mice with a recessive deletion of the inversin gene have situs inversus and an interrupted extrahepatic biliary tree.32 Mutations of the CFC1 gene, which is involved in left–right axis determination in humans, have been identified in a few patients with syndromic biliary atresia.33 The importance of the macrophage migration inhibitory factor gene, which is a pleiotropic lymphocyte and macrophage cytokine in biliary atresia pathogenesis, has also been reported.34 Other studies have identified abnormalities in laterality genes in a small number of patients with biliary atresia, including the transcription factor ZIC3.35 A high incidence of polymorphic variants in the jagged-1, keratin-8, and keratin-18 genes have also been described in a series of 18 children with biliary atresia.36,37 Taken together, the increased incidence of nonhepatic anomalies in children with biliary atresia and genetic mutations reported in subsets of patients with laterality defects suggest that multiple genes are involved, each affecting a small number of patients.
Intrahepatic bile ducts are derived from primitive hepatocytes that form a sleeve (the ductal plate) around the intrahepatic portal vein branches and associated mesenchyme early in gestation. Remodeling of the ductal plate in fetal life results in the formation of the intrahepatic biliary system. This is supported by similarities in cytokeratin immunostaining between biliary ductules in biliary atresia and normal first-trimester fetal bile ducts.38 These findings suggest that nonsyndromic biliary atresia might be caused by a failure of bile duct remodeling at the hepatic hilum, with persistence of fetal bile ducts poorly supported by mesenchyme.
Several studies have investigated whether bile duct epithelial cells are susceptible to an immune/inflammatory attack because of abnormal expression of human leukocyte antigen (HLA) antigens or intracellular adhesion molecules on their surfaces.39,40 A greater than threefold increase in HLA-B12 antigen has been found in babies with biliary atresia compared with controls, particularly in those with no associated malformations.41 Aberrant expression of class II HLA-DR antigens on biliary epithelial cells and damaged hepatocytes in patients with biliary atresia may render these tissues more susceptible to immune-mediated damage by cytotoxic T-cells or locally released cytokines.42 Increased expression of intercellular adhesion molecule-1 (ICAM-1) has been noted on bile duct epithelium in patients with biliary atresia, a finding that may play a role in immune-mediated damage.40 Strong expression of ICAM-1 also has been found on proliferating bile ductules, endothelial cells, and hepatocytes in biliary atresia. A direct relationship exists between the degree of ductal expression of ICAM-1 and disease severity, suggesting that ICAM-1 might be important in the development of cirrhosis.43
Interest has also focused on co-stimulatory molecules. Two processes are involved in the activation of T lymphocytes by antigen-presenting cells (APC). One relates to the expression of major histocompatibility complex class II molecules, which interact directly with T-cell receptors. The other depends on the expression of B7 antigens on APC, and provides the second (co-stimulatory) signal to T-lymphocytes through CD28.44 In postoperative biliary atresia patients with good liver function, co-stimulatory antigens (B7-1, B7-2, and CD40) are expressed only on bile duct epithelial cells, whereas in patients with failing livers these markers are found on the surfaces of Kupffer cells, dendritic cells, and sinusoidal endothelial cells and in the cytoplasm of hepatocytes.45 This suggests that the biliary epithelium and hepatocytes in biliary atresia are susceptible to immune recognition and destruction. Agents that block or prevent co-stimulatory pathways might offer a new therapeutic approach for reducing liver damage.
Two studies have involved comprehensive molecular and cellular surveys of liver biopsies and found a proinflammatory gene expression signature, with increased activation of interferon-γ, osteopontin, tumor necrosis factor-α, and other inflammatory mediators.46,47 These studies may prove to be helpful in delineating the molecular networks responsible for the proinflammatory response and autoimmunity thought to be involved in the pathogenesis of biliary atresia. However, none of these mechanisms appears to be mutually exclusive. Moreover, it is not clear which signs and symptoms are primary and which are secondary.
In summary, the etiology of nonsyndromic biliary atresia is hypothesized to involve a viral or other toxic insult to the bile duct epithelium that induces the expression of new antigens on the surfaces of biliary epithelial cells.48 Coupled with a genetically predetermined susceptibility that is mediated via histocompatibility antigens, these neoantigens are recognized by circulating T-lymphocytes, resulting in an immune cell mediated, fibrosclerosing response.
Classification
Biliary atresia can be classified by using macroscopic appearance and cholangiography findings into three main categories: atresia of the common bile duct (CBD) (type I), atresia of the common hepatic duct (type IIa) or atresia of the CBD and the common hepatic duct (type IIb), and atresia of all extrahepatic bile ducts up to the porta hepatis (type III) (Fig. 43-1). Most patients have type III. In patients with a patent CBD and cystic duct (correctable type) (5% of cases), the gallbladder can be anastomosed to the porta hepatis, (gallbladder Kasai). In more than 90% of cases, however, no patent extrahepatic ductal structures are found at the porta hepatis (i.e., ‘noncorrectable’ type).49
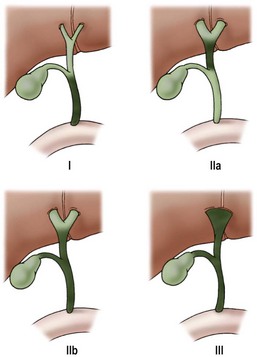
FIGURE 43-1 Morphologic classification of biliary atresia based on macroscopic and cholangiographic findings. Type I, occlusion of common bile duct; type IIa, obliteration of common hepatic duct; type IIb, obliteration of common bile duct, hepatic and cystic ducts, with cystic dilatation of ducts at the porta hepatis, and no gallbladder involvement; type III, obliteration of common, hepatic, and cystic ducts without anastomosable ducts at porta hepatis. (From Lefkowitch JH. Biliary atresia. Mayo Clin Proc 1998;73:90–5.)
Histopathology
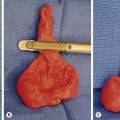
Early in the course of biliary atresia, the liver becomes enlarged, firm, and green. The gallbladder may be small and filled with white mucus, or it may be completely atretic (Fig. 43-2). Microscopically, the biliary tracts contain inflammatory and fibrous cells surrounding miniscule ducts, which are probably remnants of the original embryonic duct system. The liver parenchyma is fibrotic and shows signs of cholestasis. Proliferation of biliary neoductules is seen (Fig. 43-3). This process develops into end-stage cirrhosis if adequate biliary drainage cannot be achieved. These early changes are often nonspecific and may be confused with neonatal hepatitis and metabolic diseases.
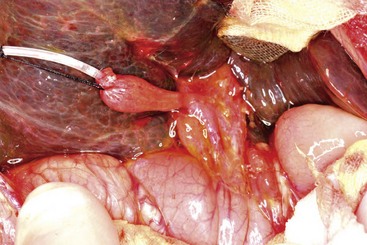
FIGURE 43-2 Type III biliary atresia with an enlarged, firm, green liver and hypoplastic small gallbladder was found in this infant.
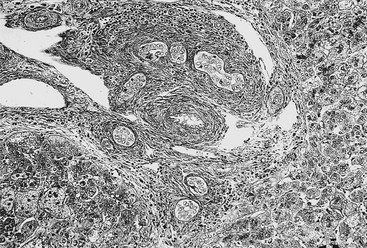
FIGURE 43-3 Photomicrograph of the portal tract of the liver in a 60-day-old infant with biliary atresia. Ductal plate malformation can be seen in the center of the portal space with portal fibrosis. Note ductal metaplasia of hepatocytes (Azan stain, ×100).
It is generally accepted that the pathologic changes seen in biliary atresia are panductal, affecting the intrahepatic biliary tree as well as the extrahepatic bile duct system. Moreover, the intrahepatic bile ducts can be narrowed, distorted in configuration, or irregular in shape.50–52 However, some authors believe that secondary damage occurs only to the extrahepatic biliary system as a result of obliteration of extrahepatic bile ducts during liver formation.53 This theory is strongly supported by the fact that outcome is better if the portoenterostomy is performed early.
The intrahepatic biliary tree is important not only pathologically, but also clinically. The degree of damage that has already occurred in the intrahepatic biliary system is actually responsible for much of the morbidity after hepatic portoenterostomy. Intrahepatic bile duct proliferation likely results from disturbances in formation of the ductal plate as well as ductular metaplasia of hepatocytes.54,55
Certain substances can act as prognostic factors in biliary atresia. Serum levels of interleukin (IL)-6, IL-1ra, insulin-like growth factor-1 (IGF-1), vascular cell adhesion molecule-1 (VCAM-1), and ICAM-1 correlate with liver dysfunction in postoperative biliary atresia patients.43,56,57 Immunohistochemically, a reduction in the expression of CD68 and ICAM-1 at the time of portoenterostomy is associated with a better prognosis.58 The presence of ductal plate malformation in the liver predicts poor bile flow after hepatoportoenterostomy in infants with biliary atresia.59 Growth failure and poor mean weight z-scores three months after hepatoportoenterostomy are also associated with a poor clinical outcome.60
Diagnosis
The cardinal signs and symptoms of biliary atresia are jaundice, clay-colored stools, and hepatomegaly. However, meconium staining is normal in most patients. In the neonatal period, feces are yellow in more than half of patients.61 The newborn’s urine becomes dark brown. Although the neonates are active, and their growth is usually normal for the first few months, anemia, malnutrition, and growth retardation develop gradually because of malabsorption of fat-soluble vitamins. Jaundice that persists beyond two weeks should no longer be considered physiologic, particularly if the elevation in bilirubin is mainly in the direct fraction. Neonatal hepatitis and interlobular biliary hypoplasia are most likely to be confused with biliary atresia. Conventional liver function tests alone are useless for establishing a definitive diagnosis of biliary atresia.
Although a number of diagnostic protocols have been published, the emphasis must always be on early diagnosis (Box 43-1).62,63 The definitive diagnosis of biliary atresia requires further investigations, including special biochemical studies, tests to confirm the patency of the extrahepatic bile ducts, and needle biopsy of the liver. Several authors consider liver biopsy to be the most reliable test for establishing the diagnosis.64,65 Serum lipoprotein-X is positive in all patients with biliary atresia, although it also is positive in 20–40% of patients with neonatal hepatitis. Serum bile acid levels increase in infants with cholestatic disease, but both the total bile acid level and the ratio of chenodeoxycholic acid to cholic acid have no value for differentiating biliary atresia from other cholestatic diseases.66 Hyaluronic acid, which has been considered a serum marker for liver function, has also been reported to be a biochemical marker for evaluating infants with biliary atresia.67 Duodenal aspiration is an easy, noninvasive, and rapid test because biliary atresia can be excluded if bilirubin-stained fluid is aspirated.68 Hepatobiliary scintigraphy with technetium-labeled agents is widely used for differentiating biliary atresia from other cholestatic diseases. In biliary atresia, nucleotide uptake by hepatocytes is rapid, but excretion into the bowel is absent, even on delayed images. In hepatocellular jaundice, isotope uptake is delayed owing to parenchymal disease. Excretion into the intestine may or may not be seen.
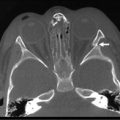
Ultrasonography (US) should be performed on all jaundiced infants. Hepatobiliary ultrasound will exclude other surgical causes of jaundice such as choledochal cyst and inspissated bile syndrome. In biliary atresia, the intrahepatic ducts are not dilated on ultrasound because they are affected by the inflammatory process. Various sonographic features have been targeted in an attempt to distinguish biliary atresia from other causes of conjugated hyperbilirubinemia in infants.69–73 In biliary atresia, the gallbladder is small, shrunken, and noncontractile, and there is increased echogenicity of the liver. The presence of other associated anomalies of the polysplenia syndrome is pathognomonic of biliary atresia.74 Differentiation from choledochal cyst and type I biliary atresia also is rapid and simple with ultrasound.75 Irrespective of interobserver variation, failure to visualize the CBD is not diagnostic of biliary atresia because a patent distal CBD can be found in up to 20% of biliary atresia cases. However, an absent gallbladder or one with an irregular outline is suggestive of biliary atresia.73 In some cases, a well-defined triangular area of high reflectivity is seen at the porta hepatis, corresponding to fibrotic ductal remnants (the ‘triangular cord’ sign) (Fig. 43-4).70,71
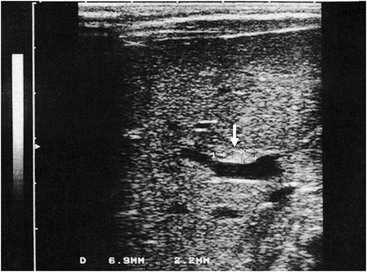
FIGURE 43-4 Ultrasonography shows a well-defined triangular area of high echogenicity (arrow) at the porta hepatis, corresponding to fibrotic ductal remnants (the ‘triangular cord’ sign).
Nonvisualization of the fetal gallbladder may indicate abnormalities ranging from gallbladder agenesis to biliary atresia.76 Amniotic fluid digestive enzymes, which are synthesized by the biliary epithelium, gradually decrease until 24 weeks of gestation. As it is no longer possible to differentiate between abnormally low and physiologically low levels of the enzymes after 24 weeks of gestation, the prenatal diagnosis of biliary atresia is difficult.77
Most patients with biliary atresia can be correctly diagnosed by using an appropriate combination of the investigations just outlined. However, to accurately differentiate biliary atresia, biliary hypoplasia, and severe neonatal hepatitis, cholangiography is usually required.
Recently, laparoscopy-assisted cholangiography has been used to display the anatomic structure of the biliary tree.78 Also, percutaneous cholecystocholangiography may be a useful option to prevent unnecessary laparotomy in infants whose cholestasis is caused by diseases other than biliary atresia.79
Operative Management
Open Surgical Technique
The portoenterostomy procedure for biliary atresia has been repeatedly modified to achieve better rates of jaundice clearance and survival, and hardly resembles Kasai’s original portoenterostomy (Fig. 43-5B). The current favored technique involves an extended lateral dissection around the porta hepatis with a very wide anastomosis (extended portoenterostomy, or EPE) (Fig. 43-5C).80,81
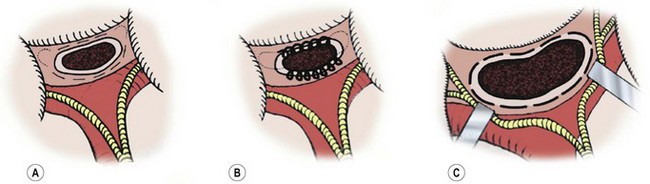
FIGURE 43-5 Salient features of three portoenterostomy procedures are depicted. (A) Modified Kasai portoenterostomy: Interrupted shallow sutures (thin broken lines) are placed in the liver parenchyma around the transected biliary remnant, except at the 2 and 10 o’clock positions, where the right and left bile ducts should be. If sutures are necessary at the 2 and 10 o’clock positions to prevent an anastomotic leak, shallow interrupted sutures (thin dotted lines) are placed only in the connective tissue near the right and left hepatic arteries or the hepatoduodenal ligament at the porta hepatis. (B) Kasai’s original portoenterostomy:23,25 A continuous suture (looped line) is placed in the side of the transected biliary remnant, except at the 2 and 10 o’clock positions, where sutures (dotted lines) are placed in the connective tissue. (C) Extended portoenterostomy: Deep interrupted sutures (bold broken lines) are placed in the liver parenchyma, even at the 2 and 10 o’clock positions. (From Nakamura H, Koga H, Wada M, et al. Reappraising the portoenterostomy procedure according to sound physiological/anatomic principles enhances postoperative jaundice clearance in biliary atresia. Pediatr Surg 2012;28:205; and From Yamataka A, Lane GJ, Cazares J. Laparoscopic surgery for biliary atresia and choledochal cyst. Sem Pediatr Surg 2012;21:201.)
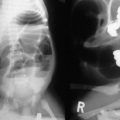
EPE is the most widely used open approach for treating biliary atresia. The patient is placed supine on an operating table with the capability for intraoperative cholangiography. An extended right subcostal incision, dividing the muscle layers, is used to expose the inferior margin of the liver. After division of the falciform and triangular ligaments, the liver is delivered from the abdominal cavity. This maneuver provides an excellent operative field for dissection of the porta hepatis. Cholangiography is recommended to identify the anatomy (Fig. 43-6). The fundus of the gallbladder is mobilized from the liver bed, and a 4–6 French feeding tube is passed into the gallbladder through a small cholecystotomy incision. If bile is detected on aspiration from the gallbladder, a small amount of contrast material is injected. However, in most patients with biliary atresia, the lumen of the atrophic gall bladder is already obstructed, and it is impossible to insert even a 4 French catheter. Thus, cholangiography cannot be performed. Unless normal anatomy of the intrahepatic biliary system can be confirmed, hepatic portoenterostomy is performed.24,82
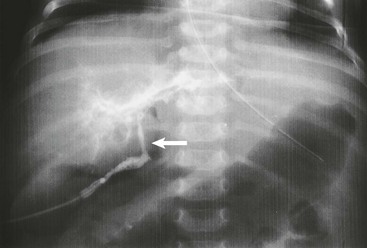
FIGURE 43-6 Intraoperative cholangiogram, type III biliary atresia. Note the almost atretic common bile duct (arrow).
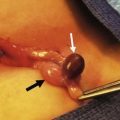
The cystic artery is ligated and divided and the gallbladder is dissected from the liver bed. The mobilized gallbladder is used as a guide for locating the fibrous remnant of the CBD. After the caudal end of the CBD is ligated and divided at the upper border of the duodenum, the cephalad portion of the CBD with the gallbladder attached is dissected above the bifurcation of the portal vein. The portal vein and the hepatic arteries are exposed along their entire course. For better portal dissection, the right and left hepatic arteries and the right and left portal branches are individually encircled with vessel loops (Fig. 43-7A). The hepatic ducts usually form a cone-shaped fibrous mass anterior to the bifurcation of the portal vein. Several small vessels connecting the portal vein to the fibrous cone are divided after being ligated. The posterior aspect of the fibrous cone is freed completely. The anterior aspect of the fibrous cone and the quadrate lobe of the liver are then exposed. The fibrous biliary plate should be dissected as far as the entrance of the anterior branch of the right hepatic artery on the right side and as far left as the entrance of the obliterated umbilical vein into the left portal vein. The fibrous cone is sharply transected at the level of the posterior surface of the portal vein with scissors or a scalpel, and is removed. The fibrous cone should have an extensive transected surface that allows a wide anastomosis. In other words, in EPE, dissection of the porta hepatis is not confined just to the area around the base of the fibrous ductal plate.83,84 As much of the transected hilar surface as possible, including all potentially usable remnants of the intrahepatic ducts in the area between and beneath the branches of the right and left portal veins, is incorporated in the anastomosis. Also, the right and left portal veins and hepatic arteries must be retracted to allow extensive resection of the biliary remnant and a wide portoenterostomy.
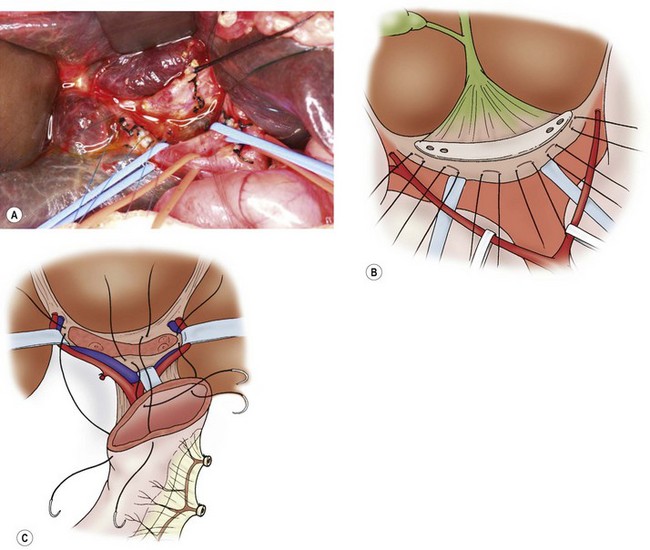
FIGURE 43-7 The current favored technique for a portoenterostomy involves an extended lateral dissection around the porta hepatis with a very wide anastomosis. (A) Photograph of the initial mobilization of the gallbladder and atretic bile ducts and dissection/exposure of the porta hepatis. After the common bile duct remnants are severed from the duodenal side, the dissection proceeds cephalad and the portal bile duct remnants are freed from the underlying structures. The portal vein and hepatic artery have been encircled with vessel loops. Several small vessel branches between the portal vein to the fibrous remnant can be identified and divided between ligatures. (B) The portal bile duct remnants must be dissected 5 or 6 mm proximal to the anterior branch of the right hepatic artery on the right side and as far left as the entrance of the obliterated umbilical vein into the left portal vein. The fibrous cone is sharply transected at the level of the posterior surface with scissors or a scalpel, and is removed. The fibrous cone should have an extensive transected surface, which allows a wide anastomosis. (C) The end of the Roux-en-Y limb is anastamosed around the transected end of the fibrous remnant. Sutures should not be placed into the transected surface of the bile duct remnant, because minute bile ducts may be present. As much of the transected hilar surface as possible, including all potentially usable remnants of the intrahepatic ducts in the area between and beneath the branches of the right and left portal veins, is incorporated in the anastomosis. It is important to retract the right and left portal veins and hepatic arteries to allow extensive reception of the biliary remnant and a wide portoenterostomy.
During the dissection, 5-0 or 6-0 absorbable monofilament sutures are usually placed in the liver surface posterior and caudal to the fibrous plate before transecting it (Fig. 43-7B). It is important to have adequate distance between these sutures and the remnant fibrous plate because it will be difficult to transect the fibrous plate if the sutures are too close to it. Bleeding points are controlled by packing with gauze. Diathermy should not be used as it can cause damage to any microscopic patent bile ducts. A liver biopsy should be performed on the right lobe to obtain histopathologic data for prognostic purposes. Also, if intraoperative histology of the transected portal fibrous plate does not identify microscopic (110–150 µm) patent bile duct structures at the transected surface, further cephalad dissection and transection of the portal fibrous plate is needed.85–87
A Roux loop of 30–40 cm is prepared by dividing the jejunum approximately 15 cm downstream from the ligament of Treitz. The distal end may be oversewn or left open, and is passed in a retrocolic position to the hepatic hilum. Small bowel continuity is re-established with an end-to-side enteroenterostomy. The hepatic portoenterostomy is performed in an end-to-side or end-to-end fashion using interrupted 5-0 or 6-0 sutures. Using the 5-0 or 6-0 sutures that were previously placed posterior to the fibrous plate, the posterior aspect of the portoenterostomy is performed first (Fig. 44-7C). Next, the anterior edge of the jejunum is anastomosed to the liver parenchyma anterior to the transected fibrous hilar remnant with interrupted 5-0 or 6-0 monofilament absorbable sutures, including the 2 and 10 o’clock positions. There should be adequate space between the anterior margin and the remnant fibrous plate as well. A small drain is positioned in the foramen of Winslow through a separate stab incision in the right abdominal wall and the incision is closed.
Modified Kasai Original Portoenterostomy
In contrast to the EPE technique just described, in both Kasai’s original portoenterostomy (KOPE) and our preferred modified KOPE (M-KOPE), dissection of the porta hepatis is confined to the area around the base of the fibrous biliary plate which is not transected very deep.27,88,89 As a result, the portoenterostomy anastomosis is not as wide as in the EPE technique. We place interrupted sutures in the liver parenchyma around the outer edge of the transected biliary remnant except at the 2 and 10 o’clock positions. At the 2 and 10 o’clock positions, where the right and left hepatic ducts should be, sutures are placed very shallow in the connective tissue near the right and left hepatic arteries or the hepatoduodenal ligament at the porta hepatis, and not to the side of the transected biliary remnant.27,89 These sutures are placed shallow, especially at the 2 and 10 o’clock positions, to minimize microscopic bile duct injury, but deep enough to prevent leakage at the portoenterostomy. See Figure 43-5 for a comparison of M-KOPE (Fig. 43-5A), KOPE (Fig. 43-5B), and EPE (Fig. 43-5C).
Hepaticojejunostomy
Kimura reported that in correctable biliary atresia, although a wide and deep portal dissection is not required, excision of the patent CBD cephalad to the liver hilum is important.90 Any cyst-like structure should be excised and should not be used for anastomosis to the intestine. Failure to remove all abnormal CBD or hepatic duct tissue may result in anastomotic stricture or cholangitis.
Nio reported on 323 biliary atresia patients undergoing operation between 1953 and 2004.91 Fifty patients were classified as type I. In these 50 patients, 28 underwent portoenterostomy and 22 underwent hepaticoenterostomy. The overall survival rate for type I patients who did not require liver transplantation was significantly better than type II/III patients (52% vs 33%). However, the authors also found a higher incidence of cholangitis in type I patients, but this difference was not statistically significant.
Recently, in a review of 200 patients from 1963–2008 at one institution, the authors reported excellent long-term outcomes in 12 patients who had a hilar cyst and underwent hepaticojejunostomy (type I cyst: 9 cases and a subtype of type II: 3 cases).92 The overall survival with the patient’s native liver was 83.3%.
Minimally Invasive Kasai Operation
The first laparoscopic Kasai (lap-Kasai) procedure was described in 2002.93 Since that report, there have been few others performed, probably because the procedure itself is technically complicated. Moreover, it may be associated with an increased incidence of postoperative complications and a worse early clinical outcome.94 It has been suggested that the pneumoperitoneum compromises liver perfusion with subsequent liver cell damage.95 In a rat model, it was shown that elevated intra-abdominal pressure can decrease hepatocyte proliferation and induce liver cell apoptosis.96 One report found there was no measurable benefit of laparoscopy compared with open portoenterostomy regarding the amount of scarring and adhesions at the time of liver transplantation.97 On the other hand, one recent study found relatively good outcomes at a median follow-up of 72 months (range: 33–89 months) after lap-Kasai, with 50% (eight cases) of the patients being jaundice-free with normal bilirubin levels.98 Also, recently, a prospective study compared lap-Kasai to conventional Kasai and found that lap-Kasai was technically feasible.99 However, the study also found worse survival after 24 months in the 12 infants who had lap-Kasai compared with infants who had conventional surgery.
The authors began performing the M-KOPE via laparoscopy in 2009.100 During lap-Kasai, the Ligasure is used to divide portal vein branches draining into the caudate lobe at the porta hepatis. The Ligasure generates much less heat laterally when compared to monopolar hook diathermy that can cause thermal injury to the microscopic bile ductules in the fibrous plate during dissection.101,102 The Roux limb is created by exteriorizing the jejunum through the umbilical port. The length of the Roux limb is customized by measuring it to reach just above the xiphoid process (Fig. 43-8). The Roux limb tends to be shorter than with the open approach. The jejuno-jejunostomy is performed extracorporeally. The portoenterostomy is then performed as described using the M-KOPE technique.
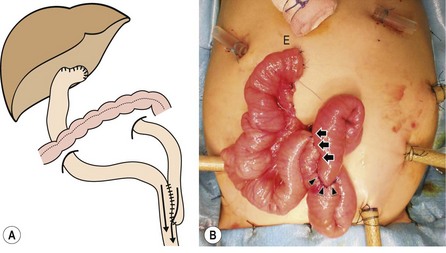
FIGURE 43-8 The length of the Roux-en-Y (RY) limb is determined by exteriorizing the jejunal loop through the umbilicus and measuring the distal end (E) of the limb to be 3 cm above the xiphoid process. The jejunojejunostomy (arrowheads) will then fit naturally into the splenic flexure after anastomosis. Arrows show where the RY limb has been approximated to the native jejunum for 8 cm cranially to streamline flow into the distal jejunum and eliminate reflux into and stasis in the RY limb. (From Yamataka A, Lane GJ, Cazares J. Laparoscopic surgery for biliary atresia and choledochal cyst. Sem Pediatr Surg 2012;21:201.)
Repeated Hepatic Portoenterostomy
Bile drainage after reoperation is significant only in patients with good bile excretion after the initial surgery.103,104 As liver transplantation is a good option, repeated hepatic portoenterostomy should only be considered for selected patients in whom good bile flow suddenly ceases, or in those patients who might benefit from a delay in transplantation.105
Postoperative Management
Most centers have standardized protocols for postoperative corticosteroid, antibiotic, and choleretic usage. Corticosteroids are used routinely both for their choleretic effect and to decrease scarring at the anastomosis site.106,107 Ursodeoxycholic acid may be useful in augmenting bile flow, but only in the presence of patent bile ductules. Fat-soluble vitamins (A, D, E, and K) and formula feeding enriched with medium-chain triglycerides are also used.
The authors’ postoperative management protocol includes decreasing the dose of intravenous prednisolone once the C-reactive protein level falls below 1.0 mg/dL.88 Each dose is given for three days, commencing with an initial dose of 4 mg/kg/day, then 3, 2, 1 mg/kg/day, and finishing with 0.5 mg/kg/day. This 15 day cycle can be repeated up to four to five times if jaundice persists (total bilirubin >1.2 mg/dL) and if there is evidence of clinical benefit (ie, lower serum bilirubin or improvement in stool color). However, if jaundice persists without clinical improvement, then only three cycles are administered and the patient is considered for liver transplantation. Also, if stools begin to turn pale, the cycle is either restarted from the beginning, or the previous dose is re-administered. Double agent antibiotic therapy, usually a cephalosporin and an aminoglycoside, is routine and stopped once the C-reactive protein is less than 0.3 mg/dL. An intravenous cholagog (usually dehydrocholic acid) is begun on day 2 after portoenterostomy and continued until jaundice clears. Oral choleretics such as ursodeoxycholic acid or aminoethylsulfonic acid are administered once oral feeding is started and continued thereafter.
Complications
Cholangitis
Approximately 40% of infants develop cholangitis. Patients present initially with fever, decreased quantity and quality of bile excretion, elevation in serum bilirubin, and signs of infection. Prompt treatment is necessary because recurring attacks cause progressive liver damage. After initial blood cultures, broad-spectrum antibiotics with good Gram-negative coverage are started, and a favorable response usually results. If stools become acholic, a pulse of corticosteroids is given. To decrease the risk for cholangitis, Roux-en-Y biliary reconstruction has been modified by various maneuvers, including lengthening the Roux-en-Y limb from 50 cm to 70 cm, total diversion of the biliary conduit, creation of an intestinal valve, and the use of a physiologic intestinal valve.108–112 The antireflux intussusception valve may be the best modification, and may result in a reduced rate of cholangitis. A gallbladder conduit is not recommended when the lumen of the patent duct is narrow or when pancreaticobiliary anomalies are demonstrated on cholangiography. Stomas complicate liver transplantation, which may be required later.
Cessation of Bile Flow
Loss of bile pigment in the stool in a patient with an apparent well-functioning portoenterostomy is an ominous sign, and parents should be encouraged to report changes in stool color or signs of cholangitis immediately. Prompt re-establishment of bile flow is imperative to avoid liver damage. If bile flow ceases, a corticosteroid pulse is given as corticosteroids both augment bile flow and reduce inflammation.106,107 If bile flow is re-established, then the corticosteroid dose is reduced. If bile flow is not re-established, the corticosteroids are stopped. If bile flow was initially good, it is reasonable to consider reoperation in selected cases. However, multiple attempts at reoperation generally increase the technical difficulty of subsequent transplantation.
Portal Hypertension
Portal hypertension is common after portoenterostomy, even in infants with good bile flow. The basic inflammatory process affecting the extrahepatic ducts also damages the intrahepatic branches. Persistent hepatic fibrosis has been found in some children despite successful portoenterostomy.113 Clinical manifestations of portal hypertension include esophageal varices, hypersplenism, and ascites (Fig. 43-9). Over time, the susceptibility to complications from portal hypertension seems to decrease, resulting in reduced frequency and severity of variceal bleeding. This observation is difficult to explain and may be related to improvement in hepatic histology or the development of spontaneous portosystemic shunts. This observation justifies a nonsurgical approach to the portal hypertension as long as hepatic function is preserved (i.e., the patient remains anicteric with no coagulopathy and normal serum albumin level). However, in the presence of poor hepatic function, complications from portal hypertension are an indication for liver transplantation.
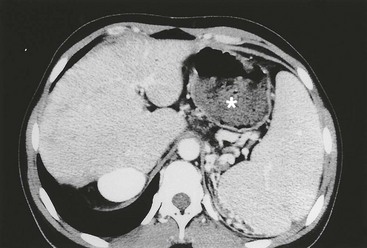
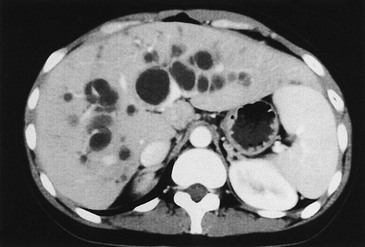
Intrahepatic Cysts
Biliary cysts, or ‘lakes,’ can develop within the livers of long-term survivors, and cause recurring attacks of cholangitis (Fig. 43-10).114 Prolonged antibiotics and ursodeoxycholic acid may be helpful in preventing cholangitis, but unremitting infection is an indication for liver transplantation.
Hepatic Malignancy
Rarely, malignant changes (hepatocellular carcinoma or cholangiocarcinoma) complicate long- standing biliary cirrhosis after portoenterostomy. A case of hepatocellular carcinoma has been reported in a 19-year-old post-Kasai male biliary atresia patient, indicating the need for a high index of suspicion for the development of carcinoma, even in young patients.115
Other Complications
Due to the presence of residual hepatic disease, metabolic problems associated with malabsorption of fat, protein, vitamins, and trace minerals can occur postoperatively because of impairment of bile flow to the gut.116,117 Weight gain after portoenterostomy may be retarded if hepatic dysfunction persists. Essential fatty acid deficiencies and rickets are common problems related to metabolic derangements.118 Long-term monitoring of clinical symptoms and adequate nutritional supplementation are needed. Ectopic intestinal variceal bleeding and pulmonary arteriovenous fistulas are sometimes seen in long-term survivors with incomplete relief of impaired liver function.119,120
As more postoperative biliary atresia patients are now reaching adulthood, the issue of pregnancy in females is becoming more common. In nontransplanted corrected biliary atresia patients, preterm cesarean delivery at around the 34th week appears to be reasonable because this is a high risk pregnancy in a patient with poor hepatic reserve. Conversely, delivery at full term may be possible for selected mothers with good liver function.121
Results and Prognosis
Without question, the Kasai hepatic portoenterostomy has greatly improved outcomes of infants with biliary atresia, and the results of surgical treatment have improved steadily over the past 30 years. Classically, the major determinants of satisfactory outcome after portoenterostomy are (1) age at initial operation; (2) successful achievement of postoperative bile flow; (3) presence of microscopic ductal structures at the hilum; (4) the degree of parenchymal disease at diagnosis; and (5) technical factors at the anastomosis. Age at portoenterostomy is the single most widely quoted prognostic variable. A favorable outcome is expected if the procedure is performed before 60 days of age, because cirrhosis will develop by 3 to 4 months of age. However, a wide discrepancy exists in reported long-term results. One study from Japan found postoperative outcome to be excellent, with a ten-year survival rate of more than 70%, if the portoenterostomy was performed before 60 days of age.122 However, a nationwide survey of the Surgical Section of the American Academy of Pediatrics found that long-term survival was only 25%.26 Other reported 10-year survival rates include 40–50% from the UK and 68% from France.1,123 Outcomes are considerably worse if the infant is older than 100 days at the time of portoenterostomy because obliterative cholangiopathy and hepatic fibrosis have already developed.123,124
The impact of age at portoenterostomy on long-term outcome, especially in patients with type III biliary atresia, was recently evaluated.125 Age at portoenterostomy was found to have a significant impact on jaundice clearance, but not on long-term survival in type III biliary atresia, suggesting that age at portoenterostomy might be less significant as a prognostic factor over time. In another study from Hong Kong, portoenterostomies performed beyond the age of 60 days was not associated with worse outcome, and a high percentage of patients could still achieve good bile flow and a normal bilirubin postoperatively.126 Of particular interest is a report by Kuroda et al, who reviewed postoperative biliary atresia patients in relation to pregnancy and found that age at portoenterostomy may not necessarily influence long-term outcome.127 However, liver function at puberty may be a useful indicator and may be predictive for the safety of pregnancy. Also, management strategies may need to be revised accordingly after puberty.
The presence of microscopic ducts at the hilum is somewhat controversial.128,129 Some authors have suggested that duct size is important, but there is not universal agreement. Types I and II biliary atresia generally have a good prognosis if treated early. In the more typical type III disease, the presence of larger bile ductules at the porta hepatis (>150 µm in diameter) is associated with a better prognosis. The subgroup of infants with syndromic biliary atresia have worse outcomes in terms of both clearance of jaundice and survival.1,28 The latter is related to associated malformations (particularly congenital heart disease), a predisposition to developing hepatopulmonary syndrome, and possible immune compromise from functional hyposplenism. Personal experience suggests that infants with concomitant CMV infection fare less well after a portoenterostomy.
The importance of surgeon experience was shown in a British survey in which patients who underwent portoenterostomy at centers treating one case per year had significantly worse outcomes than patients who were treated at centers performing more than five cases per year.130 Since 1999, the management of biliary atresia has been centralized to three centers in England and Wales that are able to offer both portoenterostomy and transplantation. In 2011, the results of this policy change were reported, and it was found that outcomes were better than previously reported.131 These improved outcomes may be due to centralization of surgical and medical resources.
Recently, outcomes in infants enrolled in the prospective Childhood Liver Disease Research and Education Network who underwent portoenterostomy were reported. Liver anatomy, splenic malformation, presence of ascites, liver nodularity at portoenterostomy, and early postoperative clearance of jaundice were found to be significant predictors of transplant-free survival.132
Irrespective of age and other factors related to the timing of portoenterostomy, a significant decrease in serum bilirubin and signs of good bile excretion in the stool may be predictive of good long-term outcome.133 Due to the possibility of sudden hepatic deterioration and the constant concern for cholangitis and portal hypertension, recent reports about long-term outcomes in biliary atresia patients consistently emphasize lifelong follow-up.134,135
Liver Transplantation
A high hepatic artery resistance index measured on Doppler ultrasound is an indication for relatively urgent transplantation.136 Deterioration in hepatic status may be precipitated by adolescence or pregnancy. However, as many as 20% of patients undergoing portoenterostomy will remain healthy and reach adulthood with good liver function.
The dramatic improvement in survival with the use of cyclosporine and tacrolimus immunosuppression after liver transplantation raises the question of transplantation becoming a more conventional form of treatment for biliary atresia. The donor supply is always a problem, alleviated to some extent by reduced-size liver transplantation. Five-year survival after liver transplantation for biliary atresia is currently 80–90%, and techniques such as split-liver grafting and living-related liver transplantation have decreased waiting times.137 Furthermore, long-term studies of post-transplant biliary atresia patients have shown that survivors have an acceptable to good quality of life.138,139
A recent study summarized the largest series (n = 464) of postportoenterostomy patients who had undergone living-related liver transplantation.140 The outcome of living-related liver transplantation in adults with biliary atresia was significantly worse than in infants and children. The overall five- and ten-year survival rates were 70% and 56% in adults versus 87% and 81% in pediatric patients, respectively. On the other hand, there is a report concluding that living-related liver transplantation after portoenterostomy can be performed safely in adults with a long-term survival rate equivalent to that for pediatric patients.141 Longer immunosuppression might ultimately lead to increased morbidity, including higher rates of cancer, infection, and metabolic diseases later in life. In addition, in living-related liver transplantation, the risk to the donor is always a concern.142 The optimal timing of transplantation in postportoenterostomy patients has yet to be established.
References
1. Chardot, C, Carton, M, Spire-Bendelac, N, et al. Prognosis of biliary atresia in the era of liver transplantation: French national study from 1986 to 1996. Hepatology. 1999; 30:606–611.
2. McKiernan, PJ, Baker, AJ, Kelly, DA. The frequency and outcome of biliary atresia in the UK and Ireland. Lancet. 2000; 355:25–29.
3. Yoon, PW, Bresee, JS, Olney, RS, et al. Epidemiology of biliary atresia: A population-based study. Pediatrics. 1997; 99:376–382.
4. Fischler, B, Haglund, B, Hjern, A. A population-based study on the incidence and possible pre- and perinatal etiologic risk factors of biliary atresia. J Pediatr. 2002; 141:217–222.
5. Petersen, C, Harder, D, Abola, Z, et al. European biliary atresia registries: Summary of a symposium. Eur J Pediatr Surg. 2008; 18:111–116.
6. Nio, M, Ohi, R, Miyano, T, et al. Five- and 10-year survival rates after surgery for biliary atresia: A report from Japanese biliary atresia registry. J Pediatr Surg. 2003; 38:997–1000.
7. Vic, P, Gestas, P, Mallet, EC, et al. Biliary atresia in French Polynesia: Retrospective study of 10 years. Arch Pediatr. 1994; 1:646–651.
8. Thomson, J. On congenital obliteration of the bile ducts. Edinburgh Med J. 1891; 37:523–531.
9. Holmes, JB. Congenital obliteration of the bile ducts: Diagnosis and suggestions for treatment. Am J Dis Child. 1916; 11:405–431.
10. Ladd, WE. Congenital atresia and stenosis of the bile ducts. JAMA. 1928; 91:1082–1085.
11. Bill, AH. Biliary atresia. World J Surg. 1987; 2:557–559.
12. Gross, RE. The Surgery of Infancy and Children. Philadelphia: WB Saunders; 1953.
13. Sterling, JA. Experiences with Congenital Biliary Atresia. Springfield, IL: Charles C Thomas; 1960.
14. Potts, WJ. The Surgeons and the Child. Philadelphia: WB Saunders; 1959.
15. Longmire, WP, Sanford, MC. Intrahepatic cholangiojejunostomy with partial hepatectomy for biliary obstruction. Surgery. 1948; 24:264–276.
16. Fonkalsrud, EW, Kitagawa, S, Longmire, WP. Hepatic drainage to the jejunum for congenital biliary atresia. Am J Surg. 1966; 112:188–194.
17. Williams, LF, Dooling, JA. Thoracic duct-esophagus anastomosis for relief of congenital biliary atresia. Surg Forum. 1963; 14:189–191.
18. Swenson, O, Fisher, JH. Utilization of cholangiogram during exploration for biliary atresia. N Engl J Med. 1952; 249:247–248.
19. Thaler, MM, Gellis, SS. Studies in neonatal hepatitis and biliary atresia: II. The effect of diagnostic laparotomy on long-term prognosis of neonatal hepatitis. Am J Dis Child. 1968; 116:262–270.
20. Kanof, A, Donovan, EJ, Berner, H. Congenital atresia of the biliary system: Delayed development of correctability. Am J Dis Child. 1953; 86:780–787.
21. Kravetz, LJ. Congenital biliary atresia. Surgery. 1960; 47:453–467.
22. Carlson, E. Salvage of the ‘non-correctable’ case of congenital extrahepatic biliary atresia. Arch Surg. 1960; 81:893–898.
23. Kasai, M, Suzuki, S. A new operation for non-correctable biliary atresia: Hepatic portoenterostomy. Shujutu. 1959; 13:733–739.
24. Kasai, M, Kimura, S, Asakura, Y, et al. Surgical treatment of biliary atresia. J Pediatr Surg. 1968; 3:665–675.
25. Kasai, M. Treatment of biliary atresia with special reference to hepatic portoenterostomy and its modification. Progr Pediatr Surg. 1974; 6:5–52.
26. Karrer, FM, Lilly, JR, Stewart, BA, et al. Biliary atresia registry, 1976–1989. J Pediatr Surg. 1990; 25:1076–1081.
27. Kasai M. Surgery for biliary atresia, Japan Surgical Society Video library: No.78-07.
28. Perlmutter, DH, Shepherd, RW. Extrahepatic biliary atresia: A disease or a phenotype? Hepatology. 2002; 35:1297–1304.
29. Davenport, M, Savage, M, Mowat, AP, et al. The biliary atresia splenic malformation syndrome. Surgery. 1993; 113:662–668.
30. Morecki, R, Glaser, JH, Cho, S, et al. Biliary atresia and reovirus type 3 infection. N Engl J Med. 1984; 310:1610.
31. Tyler, KL, Sokol, RJ, Oberhaus, SM, et al. Detection of reovirus RNA in hepatobiliary tissues from patients with extrahepatic biliary atresia and choledochal cysts. Hepatology. 1998; 27:1475–1482.
32. Mazziotti, MV, Willis, LK, Heuckeroth, RO, et al. Anomalous development of the hepatobiliary system in the Inv mouse. Hepatology. 1999; 30:372–378.
33. Jacquemin, E, Cresteil, D, Raynaud, N, et al. CFC1 gene mutation and biliary atresia with polysplenia syndrome. J Pediatr Gastroenterol Nutr. 2002; 34:326–327.
34. Arikan, C, Berdeli, A, Ozgenc, F, et al. Positive association of macrophage migration inhibitory factor gene-173G/C polymorphism with biliary atresia. J Pediatr Gastroenterol Nutr. 2006; 42:77–82.
35. Ware, SM, Peng, J, Zhu, L, et al. Identification and functional analysis of ZIC3 mutations in heterotaxy and related congenital heart defects. Am J Hum Genet. 2004; 74:93–105.
36. Kohsaka, T, Yuan, ZR, Guo, SX, et al. The significance of human jagged 1 mutations detected in severe cases of extrahepatic biliary atresia. Hepatology. 2002; 36:904–912.
37. Ku, NO, Darling, JM, Krams, SM, et al. Keratin 8 and 18 mutations are risk factors for developing liver disease of multiple etiologies. Proc Natl Acad Sci U S A. 2003; 13:6063–6068.
38. Tan, CEL, Driver, M, Howard, ER, et al. Extrahepatic biliary atresia: A first-trimester event? Clues from light microscopy and immunohistochemistry. J Pediatr Surg. 1994; 29:808–814.
39. Seidman, SL, Duquesnoy, RJ, Zeevi, A, et al. Recognition of major histocompatibility complex antigens on cultured human biliary epithelial cells by alloreactive lymphocytes. Hepatology. 1991; 13:239–246.
40. Dillon, P, Belchis, D, Tracy, T, et al. Increased expression of intercellular adhesion molecules in biliary atresia. Am J Pathol. 1994; 145:263–267.
41. Silveira, TR, Salzano, FM, Donaldson, PT, et al. Association between HLA and extrahepatic biliary atresia. J Pediatr Gastroenterol Nutr. 1993; 16:114–117.
42. Kobayashi, H, Puri, P, O’Brian, DS, et al. Hepatic overexpression of MHC class II antigens and macrophage-associated antigen (CD68) in patients with biliary atresia of poor prognosis. J Pediatr Surg. 1997; 32:590–593.
43. Kobayashi, H, Horikoshi, K, Li, L, et al. Serum concentration of adhesion molecules in postoperative biliary atresia patients: Relationship to disease activity and cirrhosis. J Pediatr Surg. 2001; 36:1297–1301.
44. Allison, JP. CD28-B7 interactions in T-cell activation. Curr Opin Immunol. 1994; 6:414–419.
45. Kobayashi, H, Li, Z, Yamataka, A, et al. Role of immunologic co-stimulatory factors in the pathogenesis of biliary atresia. J Pediatr Surg. 2003; 38:892–896.
46. Bezerra, JA, Tiao, G, Ryckman, FC, et al. Genetic induction of proinflammatory immunity in children with biliary atresia. Lancet. 2002; 23:1653–1659.
47. Mack, CL, Tucker, RM, Sokol, RJ, et al. Biliary atresia is associated with CD4+ Th1 cell-mediated portal tract inflammation. Pediatr Res. 2004; 56:79–87.
48. Sokol, RJ, Mack, C. Etiopathogenesis of biliary atresia. Semin Liver Dis. 2001; 21:517–524.
49. O’Neill, JA, Rowe, MI, Grosfeld, JL, et al. Pediatric Surgery, 5th ed. St. Louis: Mosby–Year Book; 1998.
50. Ito, T, Horisawa, M, Ando, H. Intrahepatic bile ducts in biliary atresia: A possible factor determining the prognosis. J Pediatr Surg. 1983; 18:124–130.
51. Raweily, EA, Gibson, AAM, Burt, AD. Abnormalities of intrahepatic bile ducts in extrahepatic biliary atresia. Histopathology. 1990; 17:521–527.
52. Lilly, JR, Altman, RP. Hepatic portoenterostomy (the Kasai operation) for biliary atresia. Surgery. 1975; 78:76–86.
53. Ohi, R, Chiba, T, Endo, N. Morphologic studies of the liver and bile ducts in biliary atresia. Acta Paediatr Jpn. 1987; 29:584–589.
54. Desmet, VJ. Intrahepatic bile ducts under the lens. J Hepatol. 1987; 1:545–559.
55. Sherlock, S. The syndrome of disappearing intrahepatic bile ducts. Lancet. 1987; 2:493–496.
56. Phavichitr, N, Theamboonlers, A, Poovorawan, Y. Insulin-like growth factor-1 (IGF-1) in children with postoperative biliary atresia: A cross-sectional study. Asian Pac J Allergy Immunol. 2008; 26:57–61.
57. Kobayashi, H, Yamataka, A, Lane, GJ, et al. Levels of circulating anti-inflammatory cytokine interleukin-1 receptor antagonist and proinflammatory cytokines at different stages of biliary atresia. J Pediatr Surg. 2002; 37:1038–1041.
58. Davenport, M, Gonde, C, Redkar, R, et al. Immunohistochemistry of the liver and biliary tree in extrahepatic biliary atresia. J Pediatr Surg. 2001; 36:1017–1025.
59. Shimadera, S, Iwai, N, Deguchi, E, et al. Significance of ductal plate malformation in the postoperative clinical course of biliary atresia. J Pediatr Surg. 2008; 43:304–307.
60. DeRusso, PA, Ye, W, Shepherd, R, et al. Growth failure and outcomes in infants with biliary atresia: A report from the Biliary Atresia Research Consortium. Hepatology. 2007; 46:1632–1638.
61. Chiba, T, Ohi, R, Kamiyama, T, Nio, M, Ibrahim, M. Japanese biliary atresia registry: Biliary atresia. Tokyo: Icom Assoc; 1991.
62. Altman, RP, Levy, J. Biliary atresia. Pediatr Ann. 1985; 14:481–485.
63. Okazaki, T, Kobayashi, H, Yamataka, A, et al. Long-term post surgical outcome of biliary atresia. J Pediatr Surg. 1998; 34:312–315.
64. Balistreri, WF. Neonatal cholestasis. J Pediatr. 1985; 106:171–184.
65. Brough, H, Houssin, D. Conjugated hyperbilirubinemia in early infancy: A reassessment of liver biopsy. Hum Pathol. 1974; 5:507–516.
66. Javitt, NB, Keating, JP, Grand, RJ, et al. Serum bile acid patterns in neonatal hepatitis and extrahepatic biliary atresia. J Pediatr. 1977; 90:736–739.
67. Ukarapol, N, Wongsawasdi, L, Ong-Chai, S, et al. Hyaluronic acid: Additional biochemical marker in the diagnosis of biliary atresia. Pediatr Int. 2007; 49:608–611.
68. Faweya, AG, Akinyinka, OO, Sodeinde, O. Duodenal intubation and aspiration test: Utility in the differential diagnosis of infantile cholestasis. J Pediatr Gastroenterol Nutr. 1991; 13:290–292.
69. Azuma T, Nakamura T, Moriuchi T, et al. Preoperative ultrasonographic diagnosis of biliary atresia with reference to the presence or absence of the extrahepatic bile duct. Paper presented at the 38th Annual Congress of the Japanese Society of Pediatric Surgeons. Tokyo, Japan, June 2001.
70. Park, WH, Choi, SO, Lee, HJ. The ultrasonographic ‘triangular cord’ coupled with gallbladder images in the diagnostic prediction of biliary atresia from infantile intrahepatic cholestasis. J Pediatr Surg. 1999; 34:1706–1710.
71. Kotb, MA, Kotb, A, Sheba, MF, et al. Evaluation of the triangular cord sign in the diagnosis of biliary atresia. Pediatrics. 2001; 108:416–420.
72. Tan Kendrick, AP, Ooi, BC, Tan, CE. Biliary atresia: Making the diagnosis by the gallbladder ghost triad. Pediatr Radiol. 2003; 33:311–315.
73. Farrant, P, Meire, HB, Mieli-Vergani, G. Ultrasound features of the gallbladder in infants presenting with conjugated hyperbilirubinaemia. Br J Radiol. 2000; 73:1154–1158.
74. Abramson, SJ, Berdon, WE, Altman, RP, et al. Biliary atresia and noncardiac polysplenia syndrome: Ultrasound and surgical consideration. Radiology. 1987; 163:377–379.
75. Han, BK, Babcock, DS, Gelfand, MM. Choledochal cyst with bile duct dilatation: Sonographic and 99mTc-IDA cholescintigraphy. AJR Am J Roentgenol. 1981; 136:1075–1079.
76. Ochshorn, Y, Rosner, G, Barel, D, et al. Clinical evaluation of isolated nonvisualized fetal gallbladder. Prenat Diagn. 2007; 27:699–703.
77. Boughanim, M, Benachi, A, Dreux, S, et al. Nonvisualization of the fetal gallbladder by second-trimester ultrasound scan: Strategy of clinical management based on four examples. Prenat Diagn. 2008; 28:46–48.
78. Okazaki, T, Miyano, G, Yamataka, A, et al. Diagnostic laparoscopy-assisted cholangiography in infants with prolonged jaundice. Pediatr Surg Int. 2006; 22:140–143.
79. Nwomeh, BC, Caniano, DA, Hogan, M. Definitive exclusion of biliary atresia in infants with cholestatic jaundice: The role of percutaneous cholecysto-cholangiography. Pediatr Surg Int. 2007; 23:845–849.
80. Nio, M, Ohi, R. Biliary atresia. Semin Pediatr Surg. 2000; 9:177–186.
81. Davenport, M. Surgery for biliary atresia. In: Spitz L, Coran AG, eds. Operative pediatric surgery. New York: Hodder Arnold; 2006:661–672.
82. Miyano, T, Fujimoto, T, Ohya, T, et al. Current concept of the treatment of biliary atresia. World J Surg. 1993; 17:332–336.
83. Kobayashi, H, Yamataka, A, Urao, M, et al. Innovative modification of the hepatic portoenterostomy. Our experience of treating biliary atresia. J Pediatr Surg. 2006; 41:19–22.
84. Miyano, T, Ohya, T, Kimura, K, et al. Current state of the treatment of congenital biliary atresia (in Japanese). J Jpn Surg Soc. 1989; 90:1343–1347.
85. Kobayashi, H, Horikoshi, K, Yamataka, A, et al. Alpha-glutathione-S-transferase as a new sensitive marker of hepatocellular damage in biliary atresia. Pediatr Surg Int. 2000; 16:302–305.
86. Kobayashi, H, Horikoshi, K, Yamataka, A, et al. Hyaluronic acid: A specific prognostic indicator of hepatic damage in biliary atresia. J Pediatr Surg. 1999; 34:1791–1794.
87. Miyano, T, Suruga, K, Tsuchiya, H, et al. A histopathological study of the remnant of extrahepatic bile duct in so-called uncorrectable biliary atresia. J Pediatr Surg. 1977; 12:19–25.
88. Nakamura, H, Koga, H, Wada, M, et al. Reappraising the portoenterostomy procedure according to sound physiological/anatomic principles enhances postoperative jaundice clearance in biliary atresia. Pediatr Surg Int. 2012; 28:205–209.
89. Kasai, M. Treatment of biliary atresia with special reference to hepatic porto-enterostomy and its modifications. Prog Pediatr Surg. 1974; 6:5–52.
90. Kimura, K, Tsugawa, C, Matsumoto, T, et al. The surgical management of the unusual forms of biliary atresia. J Pediatr Surg. 1979; 14:653–660.
91. Nio, M, Sano, N, Ishii, T, et al. Long-term outcome in type I biliary atresia. J Pediatr Surg. 2006; 41:1973–1975.
92. Takahashi, Y, Matsuura, T, Saeki, I, et al. Excellent long-term outcome of hepaticojejunostomy for biliary atresia with a hilar cyst. J Pediatr Surg. 2009; 44:231–2315.
93. Esteves, E, Clemente Neto, E, Ottaiano Neto, M, et al. Laparoscopic Kasai portoenterostomy for biliary atresia. Pediatr Surg Int. 2002; 18:737–740.
94. Wong, KK, Chung, PH, Chan, KL, et al. Should open Kasai portoenterostomy be performed for biliary atresia in the era of laparoscopy? Pediatr Surg Int. 2008; 24:931–933.
95. Kuebler, JF, Kos, M, Jesch, NK, et al. Carbon dioxide suppresses macrophage superoxide anion production independent of extracellular pH and mitochondrial activity. J Pediatr Surg. 2007; 42:244–248.
96. Mogilner, JG, Bitterman, H, Hayari, L, et al. Effect of elevated intraabdominal pressure and hyperoxia on portal vein blood flow, hepatocyte proliferation and apoptosis in a rat model. Eur J Pediatr Surg. 2008; 18:380–386.
97. Von Sochaczewski, OC, Petersen, C, Ure, BM, et al. Laparoscopic versus conventional Kasai portoenterostomy does not facilitate subsequent liver transplantation in infants with biliary atresia. J Laparoendosc Adv Surg Tech. 2012; 22:408–411.
98. Chan, KWE, Lee, KH, Mou, JWC, et al. The outcome of laparoscopic portoenterostomy for biliary atresia in children. Pediatr Surg Int. 2011; 27:671–674.
99. Ure, BM, Kueblaer, JF, Schukfeh, N, et al. Survival with the native liver after laparoscopic versus conventional Kasai portoenterostomy in infants with biliary atresia. A prospective trial. Ann Surg. 2011; 253:826–830.
100. Koga, H, Miyano, G, Takahashi, T, et al. Laparoscopic portoenterostomy for uncorrectable biliary atresia using Kasai’s original technique. J Laparoendosc Adv Surg Tech A. 2011; 21:291–294.
101. Yamataka, A, Lane, GJ, Cazares, J. Laparoscopic surgery for biliary atresia and choledochal cyst. Semi Pediatr Surg. 2012; 21:201–210.
102. Davenport, M, Yamataka, A. Surgery for biliary atresia. In: Spitz L, Goran AG, eds. Operative Pediatric Surgery. 7th ed. Florida: CRC Press; 2013:655–666.
103. Freitas, L, Gauthier, F, Valayer, J. Second operation for repair of biliary atresia. J Pediatr Surg. 1987; 22:857–860.
104. Ibrahim, M, Ohi, R, Chiba, T, et al. Indication and Results of Reoperation for Biliary Atresia. Tokyo: Icom Association; 1991.
105. Bondoc, AJ, Taylor, JA, Alonso, MH, et al. The beneficial impact of revision of Kasai portoenterostomy for biliary atresia. Ann Surg. 2012; 255:570–576.
106. Muraji, T, Higashimoto, Y. The improved outlook for biliary atresia with corticosteroid therapy. J Pediatr Surg. 1997; 32:1103–1107.
107. Karrer, FM, Lilly, JR. Corticosteroid therapy in biliary atresia. J Pediatr Surg. 1985; 20:693–695.
108. Suruga, K, Miyano, T, Kimura, K, et al. Reoperation in the treatment of biliary atresia. J Pediatr Surg. 1982; 17:1–6.
109. Sawaguchi, S., Y. Akiyama, M. Saeki, Y. Ohta. The treatment of congenital biliary atresia with special reference to hepatic portoenteroanastomosis. Paper presented at the fifth annual meeting of the Pacific Association of Pediatric Surgeons, Tokyo, 1972.
110. Nakajo, T, Hashizume, K, Saeki, M, et al. Intussusception-type antireflux valve in Roux-en-Y loop to prevent ascending cholangitis after hepatic portojejunostomy. J Pediatr Surg. 1990; 25:311–314.
111. Tanaka, K, Shirahase, I, Utsunomiya, H, et al. A valved hepatic portoduodenal intestinal conduit for biliary atresia. Ann Surg. 1990; 213:230–235.
112. Endo, M, Katsumata, K, Yokoyama, J, et al. Extended dissection of the porta hepatis and creation of an intussuscepted ileocolic conduit for biliary atresia. J Pediatr Surg. 1983; 12:784–793.
113. Altman, RP, Chandra, R, Lilly, JR. Ongoing cirrhosis after successful portoenterostomy with biliary atresia. J Pediatr Surg. 1975; 10:685–691.
114. Bu, LN, Chen, HL, Ni, YH, et al. Multiple intrahepatic biliary cysts in children with biliary atresia. J Pediatr Surg. 2002; 37:1183–1187.
115. Hol, L, van den Bos, IC, Hussain, SM, et al. Hepatocellular carcinoma complicating biliary atresia after Kasai portoenterostomy. Eur J Gastroenterol Hepatol. 2008; 20:227–231.
116. Andrews, WS, Pau, CM, Chase, HP, et al. Fat soluble vitamin deficiency in biliary atresia. J Pediatr Surg. 1981; 16:284–290.
117. Greene, HL, Helinek, GL, Moran, R, et al. A diagnostic approach to prolonged obstructive jaundice by 24-hour collection of duodenal fluid. J Pediatr Surg. 1979; 95:412–414.
118. Barkin, RM, Lilly, JR. Biliary atresia and the Kasai operation: Continuing care. J Pediatr Surg. 1980; 96:1015–1019.
119. Raffensperger, JG. A long-term follow-up of three patients with biliary atresia. J Pediatr Surg. 1991; 26:176–177.
120. Agarwal, GS, Saxena, A, Bhatnagar, V. The development of intrapulmonary arteriovenous shunts as a poor prognostic factor following surgery for biliary atresia. Trop Gastroenterol. 2009; 30:110–112.
121. Kuroda, T, Saeki, M, Morikawa, N, et al. Biliary atresia and pregnancy: Puberty may be an important point for predicting the outcome. J Pediatr Surg. 2005; 40:1852–1855.
122. Kasai, M, Mochizuki, I, Ohkohchi, N, et al. Surgical limitation for biliary atresia: Indication for liver transplantation. J Pediatr Surg. 1989; 24:851–854.
123. Davenport, M, Kerkar, N, Mieli-Vergani, G, et al. Biliary atresia: The King’s College Hospital experience (1974–1995). J Pediatr Surg. 1997; 32:479–485.
124. Chardot, C, Carton, M, Spire-Bendelac, N, et al. Is the Kasai operation still indicated in children older than 3 months diagnosed with biliary atresia? J Pediatr Surg. 2001; 138:224–228.
125. Nio, M, Sasaki, H, Wada, M, et al. Impact of age at Kasai operation on short- and long-term outcomes of type III biliary atresia at a single institution. J Pediatr Surg. 2010; 45:2361–2363.
126. Wong, KKY, Chung, PHY, Chan, IHY, et al. Performing Kasai portoenterostomy beyond 60 days of life is not necessarily associated with a worse outcome. J Pediatr Gastroenterol Nutr. 2010; 51:631–634.
127. Kuroda, T, Saeki, M, Morikawa, N, et al. Management of adult biliary atresia patients: Should hard work and pregnancy be discouraged? J Pediatr Surg. 2007; 42:2106–2109.
128. Chandra, RS, Altman, RP. Ductal remnants in extrahepatic biliary atresia: A histopathologic study with clinical correlation. J Pediatr Surg. 1978; 93:196–200.
129. Tan, EL, Davenport, M, Driver, M, et al. Does the morphology of the extrahepatic biliary remnants in biliary atresia influence survival? A review of 205 cases. J Pediatr Surg. 1994; 29:1459–1464.
130. McClement, JW, Howard, ER, Mowat, AP. Results of surgical treatment for extrahepatic biliary atresia in the United Kingdom, 1980–1982. BMJ. 1985; 290:345–347.
131. Davenport, M, Ong, E, Sharif, K, et al. Biliary atresia in England and Wales: Results of centralization and new benchmark. J Pediatr Surg. 2011; 46:1689–1694.
132. Superina, RS, Magee, JC, Brandt, ML, et al. The anatomic pattern of biliary atresia identified at time of Kasai hepatoportoenterostomy and early postoperative clearance of jaundice are significant predictors of transplant-free survival. Ann Surg. 2011; 254:577–585.
133. Ohhama, Y, Shinkai, M, Fujita, S, et al. Early prediction of long-term survival and the timing of liver transplantation after the Kasai operation. J Pediatr Surg. 2000; 35:1031–1034.
134. Kumagi, T, Drenth, JPH, Guttman, O, et al. Biliary atresia and survival into adulthood without transplantation: A collaborative multicenter clinic review. Liver Int. 2011. [DOI:10.1111/j.1478-3231].
135. Shinkai, M, Ohhama, Y, Take, H, et al. Long-term outcome of children with biliary atresia who were not transplanted after the Kasai operation: > 20-year experience at a children’s hospital. J Pediatr Gastroenterol Nut. 2009; 48:443–450.
136. Broide, E, Farrant, P, Reid, F, et al. Hepatic artery resistance index can predict early death in children with biliary atresia. Liver Transplant Surg. 1997; 3:604–610.
137. Tanaka, K, Uemoto, S, Tokunaga, Y, et al. Surgical techniques and innovations in living related liver transplantation. Ann Surg. 1993; 217:82–91.
138. Howard, ER, MacClean, G, Nio, M, et al. Biliary atresia: Survival patterns after portoenterostomy and comparison of a Japanese with a UK cohort of long-term survivors. J Pediatr Surg. 2001; 36:892–897.
139. Bucuvalas, JC, Britto, M, Krug, S, et al. Health-related quality of life in pediatric liver transplant recipients: A single-center study. Liver Transplant. 2003; 9:62–71.
140. Uchida, Y, Kasahara, M, Egawa, H, et al. Long-term outcome of adult-to-adult living donor liver transplantation for post-Kasai biliary atresia. Am J Transplant. 2006; 6:2443–2448.
141. Kyoden, Y, Tamura, S, Sugawara, Y, et al. Outcome of living donor liver transplantation for post-Kasai biliary atresia in adults. Liver Transpl. 2008; 14:186–192.
142. Trotter, JF, Adam, R, Lo, CM, et al. Documented deaths of hepatic lobe donors for living donor liver transplantation. Liver Transpl. 2006; 12:1485–1488.