[level-membership-for-gastroenterology-and-hepatology-category]
CHAPTER 64 Bile Secretion and the Enterohepatic Circulation
Bile formation is essential for intestinal lipid digestion and absorption, cholesterol homeostasis, and hepatic excretion of lipid-soluble xenobiotics, drug metabolites, and heavy metals. The process of bile formation depends on hepatic synthesis and canalicular secretion of bile acids, the predominant organic anions in bile, and maintenance of hepatic bile formation is essential for normal liver function. Most of the bile acids secreted by the hepatocyte have been secreted previously into the small intestine and have undergone enterohepatic cycling. As a result, disturbances in bile acid synthesis, biliary secretion, and intestinal absorption have profound effects on hepatic and gastrointestinal physiology. Identification of the hepatic, biliary, and intestinal bile acid transporters has advanced our understanding of genetic and acquired disorders of bile formation and secretion. Transporter gene mutations have been identified in disorders such as progressive familial intrahepatic cholestasis (PFIC) types 1 to 3, intrahepatic cholestasis of pregnancy (ICP), low phospholipid-associated cholelithiasis (LPAC), Dubin-Johnson syndrome, and primary bile acid malabsorption (PBAM).1 In addition, developments in the nuclear and G-protein–coupled receptor fields have provided new insights into the regulation of bile acid synthesis, secretion, and enterohepatic cycling.2 This chapter reviews the current knowledge of the hepatic synthesis, biliary secretion, and enterohepatic circulation of bile acids.
Bile is a complex, lipid-rich micellar solution that is isosmotic with plasma and composed primarily of water, inorganic electrolytes, and organic solutes such as bile acids, phospholipids (mostly phosphatidylcholine), cholesterol, and bile pigments (Table 64-1). The relative proportions of the major organic solutes in bile is illustrated in Figure 64-1.The volume of hepatic bile secreted is estimated to range from 500 to 600 mL per day, and bile acids are the dominant organic components. Actively secreted across the canalicular membrane, bile acids induce the secretion of other biliary constituents. In healthy humans, canalicular secretion is efficient and remarkably concentrative; the intracellular monomeric concentration of bile acid is less than 10 mmol/L in the hepatocyte and more than 1000 mmol/L in canalicular bile. Bile acids travel down the biliary tree and are stored in the gallbladder. After a meal the gallbladder contracts and empties its contents into the duodenum, where bile acids facilitate absorption of cholesterol and fat-soluble vitamins. Bile acids are poorly absorbed in the proximal small intestine but are absorbed almost completely by the terminal ileum. The bile acids are returned to the liver in the portal circulation, actively absorbed at the hepatocyte sinusoidal membrane, and re-secreted into bile.3
Table 64-1 Composition of Hepatic Bile
| COMPONENT | CONCENTRATION |
|---|---|
| Electrolytes and minerals (mmol/L): | |
| Sodium | 140-160 |
| Potassium | 3-8 |
| Chloride | 70-120 |
| Bicarbonate | 20-50 |
| Calcium | 1-5 |
| Phosphate | 0-1.2 |
| Magnesium | 1-3 |
| Metals (µmol/L): | |
| Iron | 2-72 |
| Copper | 12-21 |
| Organic constituents (mmol/L): | |
| Bile acids | 5-50 |
| Bilirubin (total) | 1-2 |
| Phospholipid (lecithin) | 0.5-20.0 |
| Cholesterol | 0.5-1.0 |
| Glutathione | 3-5 |
| Glucose | 0.2-1.0 |
| Urea | 2.2-6.5 |
| Protein (g/dL) | 0.2-3.0 |
Values obtained from measurements of human bile are drawn from Albers CJ, Huizenga JR, Krom RA, et al. Composition of human hepatic bile. Ann Clin Biochem 1985; 22:129-32; Keulemans YC, Mok KS, de Wit LT, et al. Hepatic bile versus gallbladder bile: A comparison of protein and lipid concentration and composition in cholesterol gallstone patients. Hepatology 1998; 28:11-16; and Ho KJ. Biliary electrolytes and enzymes in patients with and without gallstones. Dig Dis Sci 1996; 41:2409-16.
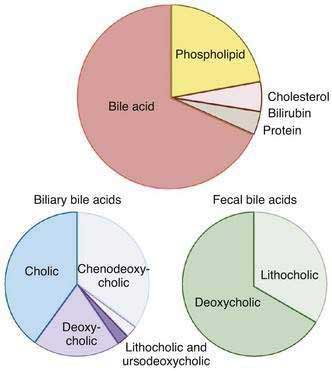
Figure 64-1. Top, Typical solute composition of hepatic and gallbladder bile in healthy humans. Bile acids are the primary solute in bile, constituting approximately 67% of bile. Phospholipid accounts for approximately 22%, cholesterol 4%, bilirubin 0.3%, and protein 4.5% of bile. Biliary bile acid composition is shown in the bottom left. Cholic acid, chenodeoxycholic acid, and deoxycholic acid constitute more than 95% of the biliary bile acids, and virtually all of the biliary bile acids are in conjugated form. The proportion of biliary lithocholic acid and ursodeoxycholic acid varies but rarely exceeds 5%. The majority of lithocholic acid in bile is present in sulfated form. When administered in therapeutic doses, the proportion of ursodeoxycholic acid in bile may rise to as much as 40%. Fecal bile acid composition is shown in the bottom right. Fecal bile acids are almost entirely unconjugated as a result of bacterial deconjugating enzymes in the colon and consist primarily of the dehydroxylated bile acids, deoxycholic acid, and lithocholic acid (see Figure 64-2).
The functions of bile acids in the liver and gastrointestinal tract are multiple.3,4 First, bile acids induce bile flow and hepatic secretion of biliary lipids (phospholipid and cholesterol). The vectorial movement of bile acids from blood into the bile canaliculus generates an osmotic water flow and is a major determinant of bile formation. Second, bile acids play an important role in the digestion of dietary fats and are essential for the intestinal absorption of cholesterol and fat-soluble vitamins. Bile acids promote intestinal absorption by solubilizing dietary lipids and lipid digestion products as mixed micelles to facilitate their aqueous diffusion across the intestinal mucosa. Fat-soluble vitamins (A, D, E, and K) are poorly absorbed in the absence of bile acid micelles, and disturbances in the secretion or enterohepatic cycling of bile acids lead to fat-soluble vitamin deficiency. Along with their major role in promoting dietary lipid absorption, bile acids may facilitate intestinal absorption of protein by accelerating protein hydrolysis by pancreatic proteases.5 Third, bile acids play a complex role in maintaining cholesterol homeostasis. On one hand, bile acids promote cholesterol intake by facilitating the intestinal absorption of biliary and dietary cholesterol. On the other hand, bile acids function through several mechanisms to promote cholesterol elimination from the body. Bile acids are water-soluble end products of cholesterol catabolism and a major route for the elimination of cholesterol via fecal excretion. Bile acids also promote hepatic secretion of cholesterol into bile by inducing bile flow and solubilizing biliary cholesterol, thereby enabling cholesterol to move from the hepatocyte to the intestinal lumen for elimination. Fourth, bile acids contribute to the antimicrobial defense mechanisms of the intestine through direct bacteriostatic actions of bile acid–fatty acid mixed micelles in the proximal small intestine and by inducing expression of antimicrobial genes in the distal small intestine.6,7 Fifth, bile acids act to prevent the formation of calcium gallstones and oxalate kidney stones.8 Sixth, bile acids can function as hormones to regulate the enterohepatic circulation as well as fat, glucose, and energy homeostasis by signaling through nuclear and G-protein–coupled receptors.2
BILE ACID SYNTHESIS AND METABOLISM
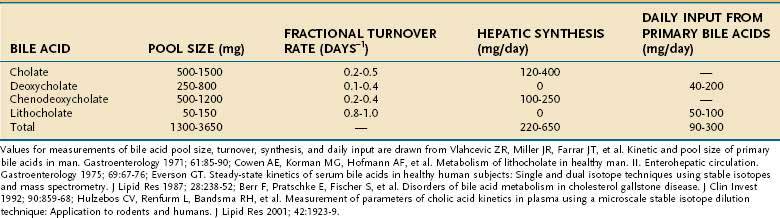
Bile acids are synthesized from cholesterol in pericentral hepatocytes of the hepatic acini. In this process, cholesterol, a lipophilic compound, is converted into a water-soluble product. In humans the newly synthesized (primary) bile acids are cholic acid (CA), a trihydroxy-bile acid with hydroxy groups at the C-3, C-7, and C-12 positions, and chenodeoxycholic acid (CDCA), a dihydroxy-bile acid with hydroxy groups at the C-3 and C-7 positions (Fig. 64-2). The kinetics for bile acid synthesis and turnover in humans are summarized in Table 64-2. Hepatic bile acid synthesis involves two major pathways, the “classical” neutral pathway (cholesterol 7α-hydroxylase pathway) that favors CA biosynthesis and the “alternative” acidic pathway (oxysterol 7α-hydroxylase pathway) that favors CDCA biosynthesis.9 In the classical pathway, the enzyme cholesterol 7α-hydroxylase (cytochrome P-450 7A1 [CYP7A1]) converts cholesterol directly into 7α-hydroxycholesterol. In the alternative pathway, cholesterol must first be converted by C-24, C-25, or C-27 sterol hydroxylases into an oxysterol, the major species being 27-hydroxycholesterol, before conversion by the oxysterol 7α-hydroxylase (CYP7B1).
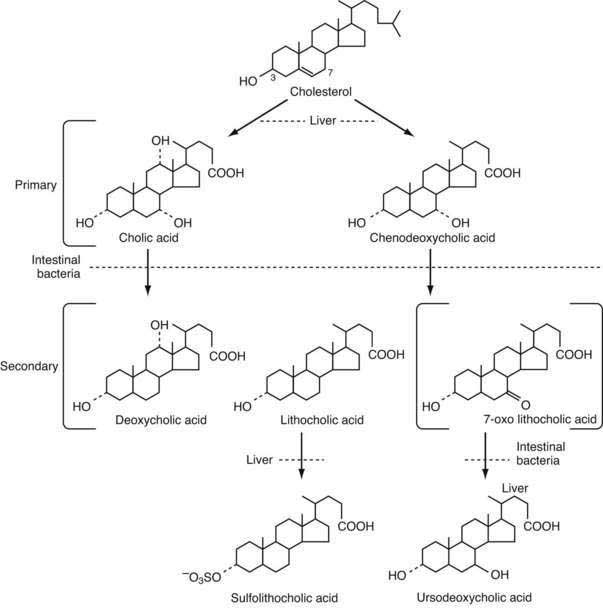
Table 64-3 Proteins Involved in the Regulation of Bile Acid Synthesis and Enterohepatic Cycling
| PROTEIN (GENE) | DESCRIPTION AND FUNCTION OF PROTEIN IN BILE ACID METABOLISM |
|---|---|
| FXR (NR1H4) | Bile acid–activated nuclear receptor; regulation of bile acid synthesis, transport, and metabolism |
| HNF4α (NR2A1) | Nuclear receptor; positive regulator of cytochrome P-450 7a1 (CYP7a1) expression and hepatic bile acid synthesis |
| SHP (NR0B2) | Nuclear receptor; negative feedback regulation of hepatic bile acid synthesis by antagonizing HNF4α, LRH-1; regulation of bile acid transport and metabolism |
| LRH-1 (NR5A2) | Nuclear receptor; positive regulator of CYP7a1 expression and hepatic bile acid synthesis |
| PXR (NR1I2) | Bile acid and xenobiotic-activated nuclear receptor involved in detoxification of secondary bile acids |
| VDR (NR1I1) | Vitamin D and bile acid–activated nuclear receptor; involved in detoxification of LCA |
| FGFR4 (FGFR4) | Membrane receptor; negative feedback regulation of CYP7a1 and hepatic bile acid synthesis |
| β-klotho (KLB) | Membrane co-receptor associated with FGFR4; confers liver specificity to FGFR4-FGF19 pathway; negative feedback regulation of CYP7a1 and hepatic bile acid synthesis |
| FGF19 (FGF19) | Protein growth factor; secreted by intestine in response to bile acids; regulates hepatic bile acid synthesis via FGFR4:β-klotho |
FXR, farnesoid X-receptor; FGF19, fibroblast growth factor 19; FGFR4, fibroblast growth factor receptor 4; HNF4α, hepatocyte nuclear factor 4 alpha; LCA, lithocolic acid; LRH-1, liver receptor homolog 1; PXR, pregnane X-receptor; SHP, small heterodimer partner; VDR, vitamin D receptor.
The overall process of bile acid biosynthesis is complex, involving 17 different enzymes divided into two broad groups.9 The first group performs modifications to the sterol ring structure, whereas the second group modifies the sterol side chain. In the classical pathway, sterol ring modifications precede side chain changes, whereas in the alternative pathway the side chain modifications occur before or during changes to the sterol ring structure. Of the two major biosynthetic pathways, the classical (CYP7A1) pathway is quantitatively more important in humans. This conclusion is supported by the finding that bile acid production is decreased by almost 90% in an adult patient with an inherited mutation in the CYP7A1 gene.10 In contrast to adults, the alternative pathway may be dominant in neonates, as evidenced by the low expression of CYP7A1 in newborns and the finding of severe cholestatic liver disease in an infant with an inherited CYP7B1 mutation.11
The rate-limiting step for the classical pathway is the enzyme CYP7A1. Bile acid feedback inhibition of CYP7A1 is well established experimentally; bile acid synthesis is decreased after administration of hydrophobic bile acids and increased by interruption of the enterohepatic circulation following ileal resection or administration of bile acid sequestrants.12 In contrast to the classical (CYP7A1) pathway, the alternative pathway for bile acid synthesis is not subject to regulation by bile acids but is controlled by cholesterol delivery to the mitochondria, the site of 27-hydroxylation.13 The molecular mechanisms responsible for the negative feedback regulation of the CYP7A1 pathway involve the liver and small intestine and have been elucidated. For the major pathway, bile acids act as ligands for the farnesoid X receptor (FXR) in ileal enterocytes to induce synthesis of an endocrine hormone, the fibroblast growth factor-19 (FGF19). FGF19 is secreted into the portal circulation and acts on hepatocytes through its cell surface receptor, a complex of the β-klotho protein and fibroblast growth factor-4, to repress CYP7A1 expression and bile acid synthesis.14–16 In a second pathway, excess bile acids in the hepatocyte activate FXR and indirectly suppress expression of CYP7A1 through a complex mechanism that involves the orphan nuclear receptor designated small heterodimer partner (SHP). SHP interferes with the activity of several nuclear receptors including hepatocyte nuclear factor 4α (HNF4α) and liver receptor homolog 1 (LRH-1), which are required for expression of the CYP7A1 gene.12,17 Finally, bile acids also inhibit CYP7A1 gene transcription in a SHP and FGFR4-independent fashion through activation of the c-Jun NH2-terminal kinase (JNK) pathway.18,19 These complex molecular titrations link bile acid synthesis to changes in intestinal as well as hepatic bile acid levels. The receptors and protein factors that participate in the regulation of bile acid synthesis and the enterohepatic circulation of bile acids are summarized in Table 64-3.
Before secretion into the bile canaliculus, both CA and CDCA are N-acyl amidated with glycine or taurine, a process commonly termed conjugation. This conjugation enhances the hydrophilicity of the bile acid and the acidic strength of the side chain, in essence converting a weak acid (pKa ≈ 5.0 for the unconjugated bile acid) to a strong acid (pKa ≈ 3.9 for the glycine conjugate; pKa ≈ 2.0 for the taurine conjugate). The major function of conjugation is to decrease the passive diffusion of bile acids across cell membranes during their transit through the enterohepatic circulation. As a result, conjugated bile acids are absorbed only if a specific membrane carrier is present. Compared with unconjugated bile acids, conjugated bile acids are also more soluble at acidic pH and more resistant to precipitation in the presence of high concentrations of calcium. The net effect of conjugation is to maintain high intraluminal concentrations of bile acids down the length of the small intestine to facilitate lipid digestion and absorption. The importance of bile acid conjugation is illustrated by the finding that inherited defects in bile acid conjugation present with the clinical phenotype of fat-soluble vitamin malabsorption and steatorrhea.11,20
Most of the conjugated bile acids secreted into the small intestine are efficiently absorbed intact; however, bile acids are also metabolized during their passage down the intestine, by the endogenous bacterial flora. In the distal small intestine, the bacterial flora deconjugates a small fraction, ≈15%, of the bile acids. The unconjugated bile acids are absorbed passively or actively and returned to the liver, where they are efficiently reconjugated and mix with newly synthesized bile acids to be re-secreted into bile. This process of intestinal deconjugation and hepatic reconjugation is a normal part of bile acid metabolism. An additional bacterial modification is the epimerization of the 3α-hydroxy or 7α-hydroxy groups to their corresponding 3β- or 7β-hydroxy forms.21,22 The 7α-hydroxy group of CDCA is epimerized to form the 3α,7β-dihydroxy bile acid, ursodeoxycholic acid (UDCA). After being absorbed from the intestine, UDCA is conjugated to taurine or glycine in the liver and circulates as a minor component, normally less than 5%, of the bile acid pool. In addition to endogenous formation, UDCA is used as a therapeutic agent in various forms of cholestatic liver disease (see Chapters 76 and 89).23
A small fraction of bile acids secreted into the small intestine escapes absorption and passes into the large intestine, where deconjugation is almost complete. In the colon, the action of bacterial 7α-dehydroxylase converts CA to deoxycholic acid (DCA), a dihydroxy bile acid with hydroxy groups at the C-3 and C-12 positions, and converts CDCA to lithocholic acid (LCA), a monohydroxy bile acid with a hydroxy group at position C-3 (see Fig. 64-2).21 The colon absorbs about 50% of the DCA formed as well as a small fraction of the LCA. After returning to the liver, DCA is reconjugated and circulates with the primary bile acids. Hepatic conjugation of the circulating bile acids is extremely efficient, so virtually all the biliary bile acids (primarily CA, CDCA, DCA, and UDCA) are in conjugated form. Bacterial deconjugation and dehydroxylation in the colon are also efficient, so the feces contain mainly the unconjugated secondary bile acids DCA and LCA (see Fig. 64-1).
The secondary bile acids can be metabolized further by various pathways, including hepatic re-epimerization of 3β-hydroxy bile acids, hepatic reduction of 7-oxo-lithocholate to CDCA or UDCA, and sulfation of bile acids by the liver or kidney (see Fig. 64-2). Additional minor pathways include hydroxylation and glucuronidation of bile acids. The conjugation of LCA with sulfate or glucuronide blocks intestinal absorption, and the modified LCA is rapidly lost from the circulating pool of bile acids. In humans, sulfation of LCA plays an important protective role, because unmodified LCA is intrinsically hepatoxic.24 The molecular mechanisms responsible for inducing these hepatoprotective modifications are being elucidated and involve induction of cytosolic sulfotransferases and cytochrome P-450 enzymes by the nuclear receptors pregnane X receptor (PXR) and constitutive androsterone receptor (CAR).25 For example, LCA, as well as other xenobiotic inducers such as rifampin, bind and directly activate PXR, thereby stimulating important mechanisms for hepatic detoxification and transport of bile acids.26
THE ENTEROHEPATIC CIRCULATION
The anatomic components of the enterohepatic circulation are the liver, biliary tract, gallbladder, small intestine, portal venous circulation, and, to a lesser extent, colon, systemic circulation, and kidneys (Fig. 64-3). At a fundamental level, the enterohepatic circulation of bile acids can be considered to consist of a series of storage chambers (gallbladder, small intestine), valves (sphincter of Oddi, ileocecal valve), mechanical pumps (bile canaliculi, biliary tract, small intestine), and chemical pumps (hepatocyte, cholangiocyte, and ileocyte).
Efficient intestinal reabsorption and hepatic extraction of bile acids permit an effective recycling and conservation mechanism that largely restricts bile acids to the intestinal and hepatobiliary compartments. During fasting, bile acids move down the biliary tract and are concentrated approximately 10-fold in the gallbladder. After an overnight fast, most of the bile acids are sequestered in the gallbladder, resulting in low levels of bile acids in the small intestine, portal vein, systemic circulation, and liver. In response to a meal, cholecystokinin is released from the intestinal mucosa and acts on the biliary tree to relax the sphincter of Oddi and stimulate gallbladder contraction. A concentrated solution of mixed micelles (bile acids, phospholipids, and cholesterol) is emptied from the gallbladder via the bile duct into the small intestine, where the micelles facilitate fat absorption by stimulating the action of pancreatic lipase on triglyceride, solubilizing the hydrolytic products, and delivering lipids to the mucosal surface. During the digestion of a large meal, the gallbladder remains contracted, and bile acids secreted by the liver bypass the gallbladder and empty directly into the duodenum. During this period, the bile acid concentration in the small intestine is approximately 5 to 10 mmol/L; micelle formation requires that the intraluminal bile acid concentration be greater than 1.5 mmol/L. During the interdigestive period, the sphincter of Oddi contracts and the gallbladder relaxes, causing a larger fraction of the secreted bile acid to enter the gallbladder for storage. This gallbladder relaxation is mediated in part by the ileal release of fibroblast growth factor 19 (FGF19).27 Therefore, the enterohepatic cycling of bile acids accelerates during digestion and slows between meals and during overnight fasting. This rhythm of bile acid secretion is maintained even after cholecystectomy.28 When the gallbladder is absent, bile acids are stored in the proximal small intestine. After ingestion of a meal, small intestinal contractions propel the stored bile acids to the distal ileum, where they are actively reabsorbed.
The enterohepatic cycling of bile acids is extremely efficient; less than 10% of the intestinal bile acids escape reabsorption and are eliminated in the feces. Bile acids are absorbed from the small intestine predominantly by an active transport system restricted to the terminal ileum and, to a lesser extent, by passive absorption down the length of the intestine. Of all the conjugated anions secreted into bile, only bile acids are actively absorbed in conjugated form and undergo an enterohepatic circulation. In adult humans, the enterohepatic circulation maintains a bile acid pool size of 50 to 60 mmol per kg body weight, corresponding to approximately 2 to 4 g. The bile acid pool cycles two to three times per meal, resulting in 6 to 10 cycles per day, and the intestine may reabsorb between 10 and 30 g of bile acid per day. Approximately 0.2 to 0.6 g of bile acids escapes reabsorption and is eliminated in the stool each day. Hepatic conversion of cholesterol to bile acid balances fecal excretion, and this process represents an important route for elimination of cholesterol from the body. The kinetics of bile acid turnover in humans are summarized in Table 64-2.
HEPATIC BILE ACID TRANSPORT AND BILE SECRETION
Hepatic adenosine triphosphate (ATP)–dependent carriers actively secrete bile acids into the canalicular lumen, from which they are too large to diffuse back across the paracellular junctions that line the canaliculi. Solutes such as the conjugated bile acids that are actively pumped across the canalicular membrane generate bile flow and are termed primary solutes. The list of primary solutes includes conjugated bilirubin, glutathione, heavy metals, and conjugates of various metabolites and xenobiotics. Water, plasma electrolytes, calcium, glucose, amino acids, bicarbonate, and other low-molecular-weight solutes that flow into the canaliculus in response to the osmotic gradient are termed secondary solutes. The choleretic activity of each primary solute is defined as the amount of bile flow induced per amount of solute secreted. The choleretic activity varies for different bile acid species and ranges from 8 to 30 microliters of bile flow induced per micromole of bile acid secreted. In humans most canalicular bile flow is generated by bile acid secretion; however, the secretion of other primary solutes by the hepatocyte and biliary epithelium also contributes to bile formation. Newly secreted hepatic canalicular bile is modified during its transit in the biliary tract via the action of ductule epithelial cells (cholangiocytes), and this ductular secretion may account for up to 40% of the volume of bile. The ductular modifications to hepatic bile include the absorption of solutes such as glucose, amino acids, and bile acids; the secretion of solutes such as bicarbonate and chloride; and the movement of water through specific channels (aquaporins) and paracellularly.29,30
BILE ACID–INDEPENDENT BILE FLOW
Canalicular bile flow is generated by active secretion of primary solutes in addition to bile acids. Reduced glutathione (GSH) and bicarbonate (HCO3−) constitute the major components of the bile acid–independent fraction of bile flow. The ATP-dependent canalicular secretion of GSH and GSH conjugates via the multidrug resistance–associated protein 2 (MRP2) plays a particularly important role. In addition to being secreted at high concentrations into bile, intraluminal catabolism of GSH by gamma glutamyl transpeptidase (GGTP) further raises the solute concentration and contributes to the osmotic driving force for canalicular bile formation. Besides the ATP-dependent secretion of organic anions into bile, hepatic and biliary ATP-independent secretion of bicarbonate via the HCO3−/Cl− anion exchanger AE2 contributes to the bile acid–independent bile flow.31 The majority of this HCO3− secretion occurs at the level of the bile duct epithelial cells in response to stimulation by a variety of hormones and neuropeptides, such as secretin and vasoactive intestinal peptide.32
CHOLEHEPATIC SHUNT PATHWAY
The term cholehepatic shunt was coined to describe the cycle whereby unconjugated dihydroxy bile acids secreted into bile are absorbed passively by cholangiocytes, returned to the hepatocyte via the periductular capillary plexus, and resecreted into bile. Absorption of the protonated unconjugated bile acid molecule generates a bicarbonate anion, resulting in a bicarbonate-rich choleresis. Premature absorption and resecretion of the bile acid also increases bile acid-dependent bile flow. This cycle explains the hypercholeresis observed for unconjugated C-24 dihydroxy bile acids such as UDCA, unconjugated C-23 bile acid analogs such as norursodeoxycholate (norUDCA), and certain drugs such as the nonsteroidal anti-inflammatory drug sulindac.33 The physiologic significance of the cholehepatic shunt pathway for hepatic bile secretion is unclear, because as originally proposed, the cholehepatic shunt has only a passive absorption component. Unlike exogenously administered UDCA or norUDCA, which may be conjugated incompletely, endogenous bile acids are conjugated efficiently to taurine or glycine before their biliary secretion. As such, the majority of the biliary bile acid pool is ionized and unable to diffuse passively across the biliary epithelium.
The finding that the biliary epithelium expresses the apical sodium-dependent bile acid transporter (ASBT; gene symbol SLC10A2) has provided a physiologic mechanism for cholehepatic shunting of conjugated bile acids.34 ASBT activity in cholangiocytes and cholehepatic shunting contribute to the choleresis and increase in biliary lipid secretion and bile flow associated with bile acid feeding.35 Because bile acids ultimately empty from the biliary tract into the small intestine, the quantitative contribution of this pathway to bile production under physiologic or pathophysiologic conditions remains to be determined. Indeed, the major function of the ASBT in biliary epithelium may be to permit cholangiocytes to sample biliary bile acid concentrations to activate cellular signaling pathways rather than to transport significant quantities of bile acids.36
HEPATIC BILE ACID TRANSPORT
Approximately 95% of the bile acids secreted into bile are derived from the recirculating pool. To maintain this process, liver parenchymal cells must transport bile acids efficiently from the portal blood into bile. This vectorial trans-hepatocellular movement of bile acids is a concentrative transport process that is driven by a distinct set of primary (ATP-dependent), secondary (Na+ gradient dependent), and tertiary (OH−– or HCO3−-dependent anion exchange) transport systems at the sinusoidal and canalicular plasma membranes.37,38 Bile acid flux through the liver and the number of participating hepatocytes vary. In the fasting state, bile acids are taken up predominantly by the periportal hepatocytes (the hepatocytes closest to the portal venules in the liver acinus), whereas during feeding, more hepatocytes in the liver acinus participate in bile acid uptake. In periportal hepatocytes, bile acid synthesis is repressed, but in pericentral (perivenous) hepatocytes, bile acids are actively synthesized. Therefore, periportal hepatocytes primarily absorb and secrete recirculating bile acids; pericentral cells predominantly secrete newly synthesized bile acids; and the transport of recirculating bile acids through periportal hepatocytes drives the majority of bile flow.39
The concentration of bile acids in the portal blood of healthy humans is 20 to 50 µmol/L. Uptake by the liver is typically expressed as fractional extraction, or first-pass extraction, and represents the percentage of bile acids removed during a single passage through the hepatic acinus. The fractional extraction of bile acids from sinusoidal blood ranges from 50% to 90% and remains constant irrespective of systemic bile acid concentrations. The hepatic fractional extraction is related to bile acid structure and albumin binding and is largest (80% to 90%) for conjugated hydrophilic bile acids such as conjugated CA and smallest (50% to 60%) for unconjugated hydrophobic protein-bound bile acids such as CDCA. The concentration of total bile acids in the systemic circulation reflects this efficient hepatic extraction, averaging 2 to 5 µmol/L and 5 to 15 µmol/L in the fasting and fed states, respectively.40
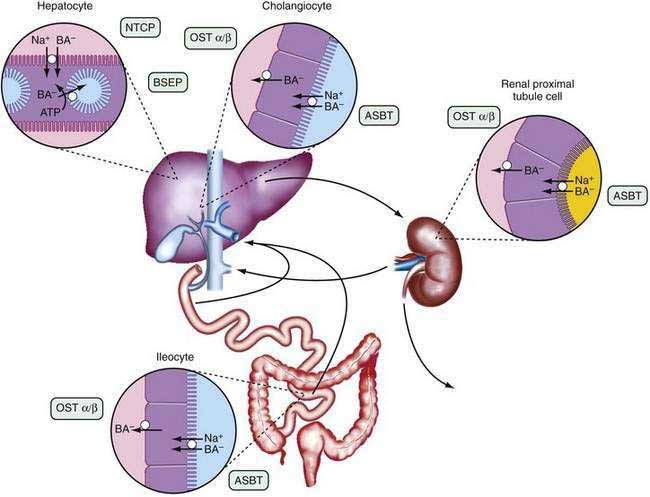
Remarkable progress has been made in identifying the transport proteins that function to maintain the enterohepatic circulation of bile acids.37,38 The major bile acid carriers in the human hepatocyte, cholangiocyte, ileal enterocyte, and renal proximal tubule cell are shown in Figures 64-3 and 64-4, and the general properties of these carriers are listed in Table 64-4. Because of their importance for bile secretion, the bile acid transporters are highlighted; however, the hepatocyte sinusoidal and canalicular membranes also express specialized transport proteins for a wide spectrum of endogenous and exogenous compounds besides bile acids.41,42
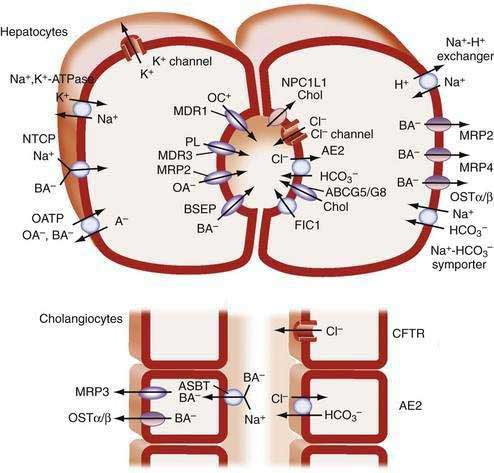
Table 64-4 Function of Transport Proteins Involved in Bile Formation and the Enterohepatic Circulation of Bile Acids
| TRANSPORTER (GENE SYMBOL) | LOCATION | FUNCTION |
|---|---|---|
| Hepatocyte | Bile Acid–Dependent Bile Flow | |
| NTCP (SLC10A1) | Basolateral membrane | Na+-dependent bile acid and xenobiotic uptake |
| OATP1B1 (SLCO1B1) | Basolateral membrane | Na+-independent bile acid and xenobiotic uptake |
| OATP1B3 (SLCO1B3) | Basolateral membrane | Na+-independent bile acid and xenobiotic uptake |
| Na+,K+-ATPase | Basolateral membrane | Secretion of 2 Na+ in exchange for 3 K+ |
| BSEP (ABCB11) | Canalicular membrane | ATP-dependent bile acid export |
| MDR3 (ABCB4) | Canalicular membrane | ATP-dependent phosphatidylcholine export |
| ABCG5 | Canalicular membrane | ATP-dependent sterol export |
| ABCG8 | Canalicular membrane | ATP-dependent sterol export |
| NPC1L1 | Canalicular membrane | Sterol import |
| FIC1 (ATP8B1) | Canalicular membrane | ATP-dependent aminophospholipid flipping |
| Bile Acid–Independent Bile Flow | ||
| MRP2 (ABCC2) | Canalicular membrane | ATP-dependent transport of glucuronide, glutathione, and sulfate conjugates |
| OATP (1B1, 1B3, 2B1) | Basolateral membrane | Na+-independent transport of organic anions, cations, and neutral steroids |
| Sinusoidal Bile Acid Export | ||
| MRP3 (ABCC3) | Basolateral membrane | ATP-dependent export of bile acids and glucuronide conjugates |
| MRP4 (ABCC4) | Basolateral membrane | ATP-dependent export of glutathione and bile acids |
| OSTα-OSTβ | Basolateral membrane | Bile acid export |
| Cholangiocyte | Ductular Secretion | |
| Aquaporin 1 (AQP1) | Apical membrane | Water transport |
| Aquaporin 4 (AQP4) | Basolateral membrane | Water transport |
| AE2 (SLC4A2) | Apical membrane | HCO3− secretion in exchange for Cl− |
| CFTR (ABCC7) | Apical membrane | Cl− secretion |
| ASBT (SLC10A2) | Apical membrane | Bile acid uptake (cholehepatic shunt) |
| Ileal Enterocyte | ||
| ASBT (SLC10A2) | Apical membrane | Na+-dependent bile acid uptake |
| NPC1L1 | Apical membrane | Sterol import |
| OSTα-OSTβ | Basolateral membrane | Bile acid export |
| MRP3 (ABCC3) | Basolateral membrane | Bile acid export |
ABC, ATP-binding cassette; AE2, chloride-bicarbonate anion exchanger isoform 2; ASBT, apical Na+ bile acid transporter; BSEP, bile salt export pump; CFTR, cystic fibrosis transmembrane regulator; FIC1, P-type ATPase mutated in progressive familial intrahepatic cholestasis type 1; MDR, multidrug resistance protein; MRP, multidrug resistance–associated protein; NPC1L1, Niemann-Pick C1 Like 1; NTCP, Na+-taurocholate cotransporting polypeptide; OATP, organic anion transporting polypeptide; OST, organic solute transporter; SLC, solute carrier.
HEPATIC SINUSOIDAL Na+-DEPENDENT BILE ACID UPTAKE
Hepatocellular uptake of bile acids occurs against an unfavorable electrochemical ion gradient, resulting in a 5- to 10-fold concentration gradient between the portal blood and hepatocyte cytosol. The uptake of conjugated bile acids at the sinusoidal (basolateral) membrane is mediated predominantly (>80%) by a secondary active Na+-dependent transport system. The driving force for the Na+-dependent uptake is generated by the basolateral Na+,K+-ATPase that maintains the prevailing out-to-in Na+ gradient. In contrast to conjugated bile acids, Na+-dependent uptake accounts for less than half of the uptake of unconjugated bile acids such as CA and UDCA. The Na+-taurocholate cotransporting polypeptide (NTCP; gene symbol SLC10A1) is responsible for the hepatocytic Na+-coupled uptake of all the major species of bile acids as well as certain drugs such as rosuvastatin.43,44 The presence of additional sinusoidal Na+-dependent bile acid transporters has been suggested, but support for the existence of such transport systems is equivocal. No inherited defect in NTCP has been described, but polymorphisms that are specific to different ethnic groups and that interfere with bile acid transport in vitro have been reported for the human NTCP gene.45 Unfortunately, clinical data for these subjects were not reported. In addition, although inherited disorders characterized by hypercholanemia have been described, mutations in genes other than NTCP, such as the tight junction proteins ZO-2 and claudin-1, were found to be responsible for these diseases.20,46 These findings leave open the possibility that an isolated NTCP gene defect may be asymptomatic because the liver also expresses Na+-independent bile acid transporters.
HEPATIC SINUSOIDAL Na+-INDEPENDENT BILE ACID UPTAKE
Unconjugated bile acids such as cholate are taken up predominantly by hepatic Na+-independent transport systems. Identification of Na+-independent hepatic transporters was originally confounded by the broad substrate specificity of these transporters, and the multiplicity of substrates could not be reconciled easily with the properties of known hepatic transport systems. The list of substrates that share apparently common transport pathways included conjugated and unconjugated bile acids, sulfobromphthalein (BSP), cardiac glycosides and other neutral steroids, linear and cyclic peptides, and numerous drugs. This conundrum was resolved with the discovery of the organic anion-transporting polypeptide (OATP) gene family and the finding that multiple OATP family members contribute to the hepatic uptake of bile acids, organic anions, and drugs.47 The OATP-type transporters include more than 39 members identified in human, mouse, and rat tissues. The original Human Genome Organization (HUGO) Gene Nomenclature Committee designation for the OATP solute carriers was SLC21A, but confusion related to species differences led to the adoption and widespread acceptance of a new species-independent classification and nomenclature system designated OATP/SLCO.47 In contrast to well-understood Na+-cotransporters such as the NTCP, the driving force for OATP-mediated solute uptake remains poorly understood and may vary among individual OATP family members. Potential transport mechanisms include facilitative diffusion and electroneutral exchange that couple solute uptake to GSH efflux.48,49
The human OATP family members, OATP1B1 (gene symbol SLCO1B1; original protein name OATP-C) and OATP1B3 (gene symbol SLCO1B3; original protein name OATP8) are expressed primarily in liver, exhibit partially overlapping substrate specificities, and account for the majority of hepatic Na+-independent bile acid clearance. OATP2B1 (gene symbol SLCO2B1; original protein name OATP-B) is expressed in numerous human tissues including liver. OATP2B1 does not transport bile acids but rather functions along with OATP1B1 and OATP1B3 to transport organic anions and solutes such as BSP, bilirubin glucuronides, steroid metabolites (estradiol-17β-glucuronide, dehydroepiandrosterone-3-sulfate, and estrone-3-sulfate), arachidonic acid products (prostaglandin E2, thromboxane B2, and leukotriene C4), and a wide variety of drugs (such as rifampin, pravastatin, rosuvastatin, digoxin, and fexofenadine).50 Because the majority of hepatic bile acid uptake is sodium dependent, the major physiologic role of these broad-specificity solute carriers may be hepatic clearance of non–bile acid metabolites and xenobiotics. Indeed, inherited polymorphisms in the OATP genes are now recognized to influence drug metabolism and contribute to some forms of drug toxicity.51
HEPATIC SINUSOIDAL BILE ACID EFFLUX
At the basolateral membrane of hepatocytes, bile acid efflux is an important protective mechanism to reduce bile acid overload under cholestatic conditions.52 Hepatic sinusoidal bile acid export is mediated by the heteromeric organic solute transporter, OSTα-OSTβ, and by members of the MRP family including MRP3 (gene symbol ABCC3) and MRP4 (gene symbol ABCC4).53 This is an important part of the adaptive response to conditions of bile acid overload that also includes down-regulation of the major liver bile acid uptake transporters, NTCP, and members of the OATP family.52
CANALICULAR BILE ACID TRANSPORT
Canalicular secretion is the rate-limiting step in hepatic transport of bile acids from blood into bile. Whereas bile acid concentrations within the hepatocyte are in the micromolar range, canalicular bile acid concentrations are more than 1000-fold higher, necessitating active transport across the canalicular membrane. Functional evidence of ATP-dependent bile acid transport by the canalicular membrane existed since the early 1990s, and functional expression and characterization studies revealed that a novel ABC transporter closely related to the multidrug resistance protein (MDR1)/P-glycoprotein gene is the canalicular bile acid transporter. This 160-kd protein (originally named “sister of P-glycoprotein” [Spgp]) was shown to transport conjugated bile acids efficiently and was subsequently renamed the bile salt export pump (BSEP; gene symbol ABCB11).54 The role of BSEP as the major canalicular bile acid efflux pump was confirmed by the identification of ABCB11 mutations in patients with PFIC type 2, a hepatic disorder characterized by biliary bile acid concentrations less than 1% of normal (see Chapter 76).55,56
INTESTINAL AND RENAL BILE ACID TRANSPORT
MOLECULAR MECHANISMS OF ILEAL AND RENAL BILE ACID TRANSPORT
Bile acids are transported actively across the ileal brush-border membrane by the well-characterized apical sodium bile acid transporter (ASBT; gene symbol SLC10A2). The relationship between the hepatic, biliary, ileal, and renal Na+–bile acid cotransport systems was resolved with the cloning of the bile acid carriers from those tissues. The liver and ileum express distinct, but related, Na+–bile acid cotransporters, NTCP and ASBT, whereas the ileal enterocyte, renal proximal tubule cell, and cholangiocyte all express the same Na+–bile acid cotransporter (ASBT).38 The inwardly directed Na+ gradient maintained by the basolateral Na+,K+-ATPase as well as the negative intracellular potential provide the driving force for ASBT-mediated bile acid uptake. The ASBT transports all the major species of bile acids but does not appear to transport any non–bile acid–related metabolites or drugs. The finding that ASBT gene mutations are responsible for PBAM, a disorder associated with intestinal bile acid malabsorption and steatorrhea, demonstrated that most intestinal bile acid absorption in humans is mediated by the ASBT.57
Little is known about the intracellular transport of bile acids in the ileal enterocyte. The ileal lipid-binding protein (ILBP, also called the ileal bile acid binding protein [IBABP]; gene symbol FABP6) is a member of the fatty acid binding protein family and a major ileal enterocyte cytosolic protein. ILBP is believed to be involved in the transcellular transport of bile acids; however, a mouse model lacking appreciable ILBP expression did not exhibit impaired intestinal bile acid absorption, indicating that ILBP is not essential for this process.58 The proteins responsible for bile acid export across the basolateral membrane of the ileal enterocyte, cholangiocytes, and renal proximal tubule cell have been identified. The heteromeric organic solute transporter, OSTα-OSTβ, is expressed on the basolateral membrane of ileal enterocytes, hepatocytes, cholangiocytes, and renal proximal tubule cells and transports bile acids as well as a variety of organic anions.59 Although no inherited defects have been reported for the OSTα or OSTβ genes in humans, targeted inactivation of the OSTα gene in mice resulted in impaired intestinal bile acid absorption and altered bile acid metabolism, indicating that OSTα-OSTβ is a major basolateral bile acid transporter.60,61
DISORDERS OF THE ENTEROHEPATIC CIRCULATION
The measurement of serum bile acid levels has only a limited diagnostic role for disorders of the enterohepatic circulation. Specific diagnosis of the rare inherited bile acid synthesis defects is made by analysis of body fluids (bile, blood, and urine) using fast atom bombardment–mass spectroscopy (FAB-MS) and gas chromatography–mass spectroscopy (GC-MS).11,62 These disorders present with markedly reduced or complete absence of CA and CDCA levels and greatly elevated concentrations of atypical bile acids in bile, serum, and urine. Measurement of serum bile acid concentrations, however, offers little, if any, benefit over conventional liver biochemical tests in the diagnosis or management of most forms of liver disease or bile acid malabsorption.
BILE ACID SYNTHESIS
Continuous bile acid synthesis from cholesterol is required to maintain the bile acid pool in the enterohepatic circulation. The maximal rate of bile acid synthesis is on the order of 4 to 6 g/day. Although inherited defects in biosynthesis are extremely rare, these disorders serve to illustrate the importance of bile acid synthesis for normal hepatic function. The effects of a cessation in bile acid synthesis include depletion of the bile acid pool by fecal excretion, loss of bile acid–dependent bile flow, decreased biliary excretion of cholesterol and xenobiotics, reduced intestinal absorption of cholesterol and fat-soluble vitamins, and accumulation of cytotoxic bile acid biosynthetic intermediates. Inherited defects in eight of the bile acid biosynthetic enzymes have been reported; because the side chain modification steps in bile acid biosynthesis occur in the peroxisome, disorders that disrupt peroxisome biogenesis such as Zellwegner syndrome also affect bile acid synthesis (see Chapter 76). The list of inborn errors includes cholesterol 7α-hydroxylase (CYP7A1), sterol 27-hydroxylase (CYP27A1), oxysterol 7α-hydroxylase (CYP7B1), 3β-hydroxy-Δ5-C27-steroid oxidoreductase (HSD3B7), Δ4-3-oxosteroid 5β-reductase (AKR1D1), 2-methylacyl-coenzyme A racemase (AMACR), D-bifunctional protein (HSD17B4), and the bile acid coenzyme A:amino acid N-acyl-transferase (BAAT).11,62
In general, genetic defects that affect early steps in the bile acid biosynthetic pathway cause disease in newborns, whereas the consequences of defects in later steps are varied. In some cases, a single enzyme defect is not sufficient to eliminate production of all bile acids, because multiple biosynthetic pathways exist.9 For example, cerebrotendinous xanthomatosis (CTX) is a rare, inherited disease caused by mutations in the mitochondrial enzyme sterol 27-hydroxylase (CYP27A1). In CTX, the alternative pathway of bile acid synthesis is blocked, and bile acid synthesis is diminished but not eliminated. Liver disease does not occur in patients with CTX, which is characterized by progressive neurologic disturbances, premature atherosclerosis, cataracts, and tendinous xanthomas. Bile acid therapy with hydrophobic bile acids such as CDCA has been reported to suppress the biochemical abnormalities in CTX and to slow progression of the disease.
The most commonly reported defect in bile acid synthesis is 3β-hydroxy-C27-steroid oxidoreductase (HSD3B7) deficiency, and this enzyme defect affects both the classical and alternative pathways for bile acid biosynthesis. The disease is characterized by progressive intrahepatic cholestasis and accumulation of abnormal bile acids. These unusual conjugates of dihydroxy- and trihydroxy-5-cholenoic acid are poorly transported by the BSEP and interfere with the canalicular secretion of other bile acids. Clinical manifestations include unconjugated bilirubinemia, jaundice, serum aminotransferase elevations, steatorrhea, pruritus, and poor growth. Disease progression varies but ultimately results in cirrhosis and hepatic failure in a high proportion of affected patients. Treatment with exogenous primary bile acids such as UDCA reverses the biochemical abnormalities and may be life-saving.11
MEMBRANE TRANSPORT OF BILE ACIDS AND BILIARY LIPIDS
A growing number of disorders have been found to be associated with mutations in the bile acid transporter or biliary organic solute transporter genes.1 The list of these disorders includes PFIC types 1 to 3, benign recurrent intrahepatic cholestasis (BRIC) types 1 and 2, LPAC, ICP, Dubin-Johnson syndrome, sitosterolemia, and PBAM and is summarized in Table 64-5.
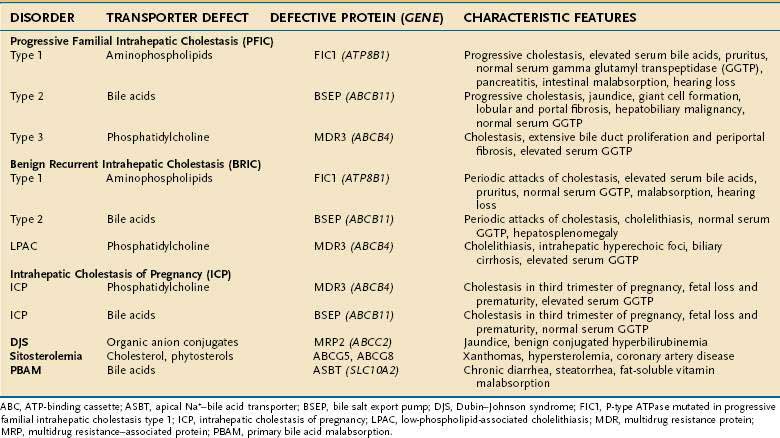
PFIC type 1 (PFIC1; formerly called Byler’s disease) manifests primarily as chronic intrahepatic cholestasis and coarse granular bile in patients with normal serum GGTP levels. The gene defect in patients with PFIC1 had been mapped to chromosome 18 in the same region where a similar but milder disease phenotype, BRIC, has been localized. A combined search for the two disease loci identified a P-type ATPase, designated FIC1 (gene symbol ATP8B1), as the defective gene for both PFIC1 and BRIC type 1. P-type ATPases are distinct from ABC transporters and constitute a large family that includes ion pumps such as the Na+,K+-ATPase, Ca2+-ATPase, and the copper-transporting Wilson disease gene product (see Chapter 75). An analysis of the spectrum of ATP8B1 mutations in patients with PFIC1 and BRIC revealed that mutation type and location in the gene generally correlate with clinical severity. More innocuous ATP8B1 missense mutations are common in BRIC type 1 (BRIC1), whereas nonsense, frame-shifting, and large-deletion mutations are more common in PFIC1.63 FIC1 functions as an aminophospholipid (phosphatidylserine) flippase, and activity requires the coexpression of CDC50 proteins that promote the trafficking of FIC1 to the plasma membrane.64 In humans, the FIC1 protein is expressed in many tissues, including the pancreas, small intestine, urinary bladder, stomach, and prostate. The expression and activity of FIC1 in many extrahepatic tissues may help explain the high frequency of diarrhea, pancreatitis, respiratory symptoms, and hearing loss in patients with PFIC1 disease. Whereas the mechanism responsible for cholestasis associated with FIC1 deficiency remains to be fully elucidated, results from the analysis of an ATP8B1 mutant mouse indicate that loss of aminophospholipid flippase activity makes the canalicular membrane more susceptible to damage from hydrophobic bile acids.65 In addition, defective bile acid signaling pathways may contribute to the pathophysiology of PFIC1.66 Treatment options for PFIC1 and BRIC1 include supplementation with fat-soluble vitamins, partial external biliary diversion, and, for patients with PFIC1, liver transplantation.67
PFIC2 (also called BSEP disease) is associated with low bile acid secretion, progressive cholestasis, normal serum GGTP levels, lobular and portal fibrosis, hepatic giant cell transformation, and the lack of bile duct proliferation on examination of liver histology. The disease was mapped to chromosome 2q24, and the defective gene has been shown to encode the canalicular BSEP protein (ABCB11). BSEP protein could not be detected on the canalicular membrane in liver biopsy specimens from patients with PFIC2, therefore suggesting that mutations in ABCB11 impair the synthesis, cellular trafficking, or stability of BSEP protein.56 In addition to a progressive intrahepatic cholestatic phenotype, ABCB11 missense mutations were found in patients with a milder disease similar to BRIC.68 Unlike the form of BRIC that is associated with ATP8B1 mutations, patients with BRIC caused by ABCB11 mutations lack extrahepatic symptoms such as pancreatitis and are more likely to form gallstones. On the basis of these findings and consistent with the genetic classification of PFIC into subtypes, BRIC is classified as types 1 and 2, corresponding to ATP8B1- and ABCB11-mutation associated forms, respectively. The ABCB11 mutations associated with milder disease may also confer an increased risk for the development of ICP and drug-induced cholestasis.69
Patients with PFIC2 present as infants with high serum bile acid levels, intractable pruritus, intestinal malabsorption, failure to thrive, and cholestasis, ultimately developing fibrosis and end-stage liver disease before adulthood.67 In addition to cholestasis, outcome data have confirmed that severe BSEP deficiency significantly increases the risk for hepatobiliary malignancy, including hepatocellular carcinoma and cholangiocarcinoma.56,70 The treatment of PFIC2 is similar to that for PFIC1, with liver transplantation as the ultimate therapy for patients with severe forms of the disease.
PFIC3 is quite different from the other PFIC subtypes. Serum GGTP levels are markedly elevated in these patients, and liver histologic examination shows extensive bile duct proliferation and portal and periportal fibrosis. The most common clinical presentations include jaundice, pale stools, pruritus, and hepatomegaly. The defect in PFIC3 lies in MDR3 (gene symbol ABCB4), a canalicular phosphatidylcholine (PC) transporter that belongs to the ABC transporter superfamily.71 In PFIC3, hepatic bile acid secretion is unimpaired, but PC transport is greatly diminished. In bile, PC normally forms mixed micelles with bile acids and acts to buffer the cytotoxic detergent properties of the bile acids. In the absence of biliary phospholipid, the bile acids are highly toxic and cause cholestatic liver damage. In addition to PFIC3, mutations in MDR3 have been associated with LPAC and ICP.1 PFIC3 has been treated with UDCA, with variable results, and with liver transplantation.67
BILE ACID BIOTRANSFORMATION (DECONJUGATION AND DEHYDROXYLATION)
Bile acid deconjugation normally begins in the distal small intestine and is mediated by spilling of colonic bacteria across the ileocecal valve. In patients with small intestinal stasis and bacterial overgrowth, deconjugation of bile acids also occurs in the proximal intestine. The unconjugated bile acids are absorbed passively, and extensive bacterial deconjugation can reduce the intraluminal concentration of bile acids and impair formation of micelles in the small intestine. Increased bacterial deconjugation can be detected indirectly by measuring levels of unconjugated bile acids in the systemic venous plasma. This test is rarely used, however, because bacterial overgrowth is detected more easily with a hydrogen breath test after ingestion of a glucose-containing meal (see Chapter 102).
In the colon bile acids are first deconjugated and then 7α-dehydroxylated.22 In healthy humans, colonic bile acids eventually undergo nearly complete 7α-dehydroxylation to yield the secondary bile acids DCA and LCA.21 The conversion of CA to DCA and subsequent colonic absorption of DCA is increased in subjects with an elevated proportion of DCA in their bile acid pool. The underlying mechanism is related to prolonged colonic transit time that results in increases in the concentration of bacteria, 7α-dehydroxylase activity, and colonic pH and DCA solubility.72 Because accumulation of DCA in bile can contribute indirectly to cholesterol cholelithiasis, strategies to accelerate colonic transit or acidify the colonic luminal contents may be useful in preventing cholesterol gallstones in these subjects.
BILE ACID CIRCULATION
Biliary Obstruction and Biliary Fistula
Biliary obstruction caused, for example, by a stone that obstructs the bile duct leads to retention of hepatic bile acids and, ultimately, hepatocyte necrosis or apoptosis. A portion of the retained bile acids is modified by sulfation, and both sulfated and unsulfated bile acids are regurgitated from hepatocytes back into the systemic circulation. Despite increased urinary excretion of bile acids, plasma concentrations of bile acids rise as much as 20-fold. When biliary obstruction is incomplete, bile acids continue to be secreted into the small intestine, efficiently absorbed by the ileum, and returned to the liver via the portal circulation. In this clinical condition, administration of bile acid sequestrants decreases the intestinal absorption of cytotoxic bile acids and may slow the progression of liver damage.73 With complete bile duct obstruction, bile acids are not secreted into the small intestine, and intestinal malabsorption of fat-soluble vitamins and steatorrhea results. Secondary bile acids are not formed, and fecal bile acid output diminishes.
Cholecystectomy
Despite removal of a major storage pool of bile acids, the overall effect of cholecystectomy on biliary secretion is small, and daily bile acid secretion is not altered substantially.28 In the absence of a gallbladder, the bile acid pool is stored in the small intestine during the fasting state. After ingestion of a meal, the bile acid pool moves to the terminal ileum, where it is actively absorbed and returned to the liver via the portal circulation. A change in the composition of the bile acid pool has been reported and is characterized by increased dehydroxylation of CA to DCA. In the small subset of patients with postcholecystectomy diarrhea, the movement of the bile acid pool to the small intestine may overwhelm the ileal transport system, thereby leading to bile acid malabsorption. The diarrhea is usually only transitory, and affected patients generally respond to administration of a bile acid sequestrant.
Ileal Resection
Resection of the terminal ileum causes intestinal bile acid malabsorption. If the resection is short (<100 cm), the effect on bile acid metabolism is minimal because greater biosynthesis balances increased fecal loss. With longer resections, hepatic bile acid synthesis rises more dramatically to compensate for the increased loss. The unabsorbed bile acids enter the colon in greater amounts and act to inhibit water absorption or induce secretion, thereby resulting in mild, watery diarrhea. Symptomatic response is obtained with administration of a bile acid sequestrant. When more than 100 cm of ileum is resected, including the ileocecal valve, bile acid secretion diminishes because the bile acid biosynthetic potential is well below the normal hepatic secretion rate. The bile acid pool becomes progressively depleted during the day, and intestinal fat malabsorption appears because of the lack of micelles and loss of intestinal mucosal surface. The increased fatty acid flux through the colon inhibits water absorption and results in severe diarrhea that responds poorly to bile acid sequestrants. If the diarrhea is of sufficiently large volume and accompanied by malabsorption of other nutrients, the patient may be diagnosed as having short bowel syndrome. Therapy is complex and has only limited success. In some patients, fecal weight and frequency are reduced by elimination of fat from the diet. Other therapies include bile acid replacement and administration of glutamine and growth factors (see Chapter 103).
Bile Acid Malabsorption and Diarrhea
The enterohepatic circulation conserves bile acids efficiently, thereby maintaining bile flow and adequate intraluminal bile acid concentrations for micellar solubilization and absorption of lipids. Impaired intestinal absorption of bile acids may contribute to the pathogenesis of a number of gastrointestinal disorders, including idiopathic chronic diarrhea, chronic ileitis, gallstone disease, postcholecystectomy diarrhea, Crohn’s disease, and irritable bowel syndrome. Symptomatic bile acid malabsorption results from failure of the active intestinal transport component and is most commonly caused by ileal resection, ileal disease such as Crohn’s disease, ileal bypass, and radiation enteritis. Bile acid malabsorption is associated also with conditions such as peptic ulcer surgery, chronic pancreatitis, celiac disease, diabetes mellitus, cystic fibrosis, and the use nonsteroidal anti-inflammatory drugs (which may cause intestinal ulcers and strictures), colchicine, olsalazine, and antineoplastic agents. Primary and idiopathic forms of bile acid malabsorption are rare disorders. Mutations in the ASBT gene cause congenital or primary bile acid malabsorption but are not responsible for a more common condition termed idiopathic bile acid malabsorption that describes patients with adult-onset intestinal bile acid malabsorption, chronic diarrhea, and a morphologically normal ileum.57
Bile acid malabsorption permits higher concentrations of dihydroxy bile acids to reach the colon, where they alter water and electrolyte movement and lead to diarrhea. Water transport in the colon is critical for the regulation of intestinal fluid and electrolyte balance and is the ultimate determinant of diarrhea (see Chapter 15). Bile acids play a role in colonic water transport apparently by increasing chloride secretion in the perfused colon by an inositol 1,4,5-triphosphate and calcium–dependent mechanism. Trihydroxy bile acids have no effect, but dihydroxy bile acids induce net fluid secretion at high concentrations and block absorption of fluid and water at low concentrations. To induce net secretion, bile acids must (1) have an appropriate structure (hydrophobic dihydroxy bile acids, DCA or CDCA), (2) be present in high concentrations (>1.5 mM) in the aqueous phase, and (3) exist in an environment with the appropriate alkaline pH (fecal pH > 6.8).
An additional consequence of bile acid malabsorption and steatorrhea is increased renal oxalate excretion. Under conditions of intestinal bile acid malabsorption, the concentration of long-chain fatty acids is elevated in the colon, where the fatty acids form insoluble calcium soaps. Consequently, less free calcium is available to precipitate dietary oxalate. The higher soluble oxalate concentrations in the lumen and increased colonic permeability promote hyperabsorption of oxalate. The enteric hyperoxaluria contributes to the formation of kidney stones in affected patients, such as those with Crohn’s disease or short bowel syndrome. Therapeutic approaches to the fat malabsorption and diarrhea associated with bile acid malabsorption are discussed in Chapters 15 and 101.
BILE ACID THERAPY, SEQUESTRANTS, AND TRANSPORT INHIBITORS
BILE ACID THERAPY
Bile acid therapy traditionally has been divided into two types: displacement and replacement. The goal of displacement therapy is to alter the composition of the bile acid pool to reduce either the cytotoxicity of endogenous bile acids or biliary cholesterol secretion, whereas the aim of replacement therapy is to correct a bile acid deficiency. More recently, the recognition that bile acids signal through nuclear and G-protein–coupled receptors to induce hepatoprotective mechanisms and modulate fat, glucose, and energy homeostasis has renewed interest in therapeutic uses for bile acids or bile acid derivatives.2 With the exception of guggul lipid, however, an FXR modulator used in traditional Ayurvedic medicine and sold in the West as a food supplement, these agents are still in preclinical or clinical development. Because of its safety and lack of hepatotoxicity, UDCA is the most widely used form of bile acid therapy.23 After oral administration, UDCA accumulates and ultimately constitutes up to 40% of the circulating bile acid pool, displacing the more hydrophobic endogenous bile acids.23 UDCA was originally administered and approved by the U.S. Food and Drug Administration (FDA) for gallstone dissolution but is not commonly used today for that purpose because of the success of laparoscopic cholecystectomy. The FDA has also approved bile acid therapy for treatment of primary biliary cirrhosis, in which UDCA may delay the progression of liver fibrosis and improve survival (see Chapter 89). UDCA therapy may also have favorable effects in other conditions, such as ICP, viral hepatitis, and parenteral nutrition–associated cholestasis.23 A chemical analog of UDCA, norUDCA, has one less carbon atom in the side chain, is poorly amidated, and undergoes significant cholehepatic shunting. Therapy with norUDCA is effective in treating peribiliary fibrosis in the MDR2 knockout mouse, an animal model of primary sclerosis cholangitis, and is in preclinical development as a potentially new agent for cholestatic liver disease.74
Bile acid replacement therapy is used to treat rare inborn errors of bile acid biosynthesis. In affected patients, administration of a mixture of UDCA and CA suppresses the synthesis of cytotoxic bile acid precursors and restores the input of primary bile acids into the enterohepatic circulation.11 Bile acid replacement therapy is also indicated in patients with severe bile acid malabsorption or short bowel syndrome, in which a deficiency of bile acids in the proximal small intestine leads to impairment of micellar solubilization and intestinal fat malabsorption.75
BILE ACID SEQUESTRANTS
Bile acid sequestrants are positively charged polymeric resins that bind bile acids in the intestinal lumen to decrease the aqueous concentration and the efficiency of intestinal conservation of bile acids. In patients with mild bile acid malabsorption, bile acid sequestrants reduce diarrhea by lowering the concentration of free bile acids in the colon. Bile acid sequestrants also are used to decrease pruritus in patients with cholestasis, presumably by reducing the concentration of bile acids (or other anionic biliary constituents) in the systemic circulation.67 Efficacy is only moderate because of the weak bile acid–binding efficiency of the resins and poor patient adherence to therapy. In addition to the older preparations, cholestyramine and colestipol, more potent sequestrants with superior bile acid-binding properties have been developed. Colesevelam is a newer bile acid sequestrant and has been approved by the FDA for the treatment of hypercholesterolemia and for the improvement of glycemic control in adults with type 2 diabetes mellitus.
BILE ACID TRANSPORT INHIBITORS
An alternative to luminal sequestration of bile acids with binding resins is direct inhibition of the ileal ASBT. Several potent ASBT inhibitors have been developed and evaluated in animal models. Although these agents are being targeted primarily for the treatment of hypercholesterolemia, they may also prove useful for blocking the inappropriate ileal conservation of bile acids that contributes to hepatocellular damage and pruritus in cholestatic liver disease.73
Ballatori N, Christian WV, Lee JY, et al. OSTalpha-OSTbeta: A major basolateral bile acid and steroid transporter in human intestinal, renal, and biliary epithelia. Hepatology. 2005;42:1270-9. (Ref 59.)
Fickert P, Wagner M, Marschall HU, et al. 24-norUrsodeoxycholic acid is superior to ursodeoxycholic acid in the treatment of sclerosing cholangitis in Mdr2 (Abcb4) knockout mice. Gastroenterology. 2006;130:465-81. (Ref 74.)
Hagenbuch B, Meier PJ. Organic anion transporting polypeptides of the OATP/SLC21 family: Phylogenetic classification as OATP/ SLCO superfamily, new nomenclature and molecular/functional properties. Pflugers Arch. 2004;447:653-65. (Ref 47.)
Hamilton JP, Xie G, Raufman JP, et al. Human cecal bile acids: Concentration and spectrum. Am J Physiol Gastrointest Liver Physiol. 2007;293:G256-63. (Ref 21.)
Hofmann AF, Hagey LR. Bile acids: Chemistry, pathochemistry, biology, pathobiology, and therapeutics. Cell Mol Life Sci. 2008;65:2461-83. (Ref 3.)
Inagaki T, Choi M, Moschetta A, et al. Fibroblast growth factor 15 functions as an enterohepatic signal to regulate bile acid homeostasis. Cell Metab. 2005;2:217-25. (Ref 14.)
Inagaki T, Moschetta A, Lee YK, et al. Regulation of antibacterial defense in the small intestine by the nuclear bile acid receptor. Proc Natl Acad Sci U S A. 2006;103:3920-5. (Ref 7.)
Klomp LW, Vargas JC, van Mil SW, et al. Characterization of mutations in ATP8B1 associated with hereditary cholestasis. Hepatology. 2004;40:27-38. (Ref 63.)
Knisely AS, Strautnieks SS, Meier Y, et al. Hepatocellular carcinoma in ten children under five years of age with bile salt export pump deficiency. Hepatology. 2006;44:478-86. (Ref 70.)
Lundasen T, Galman C, Angelin B, Rudling M. Circulating intestinal fibroblast growth factor 19 has a pronounced diurnal variation and modulates hepatic bile acid synthesis in man. J Intern Med. 2006;260:530-6. (Ref 15.)
Marschall HU, Wagner M, Zollner G, et al. Complementary stimulation of hepatobiliary transport and detoxification systems by rifampicin and ursodeoxycholic acid in humans. Gastroenterology. 2005;129:476-85. (Ref 26.)
Oude Elferink RP, Paulusma CC, Groen AK. Hepatocanalicular transport defects: Pathophysiologic mechanisms of rare diseases. Gastroenterology. 2006;130:908-25. (Ref 1.)
Paulusma CC, Groen A, Kunne C, et al. Atp8b1 deficiency in mice reduces resistance of the canalicular membrane to hydrophobic bile salts and impairs bile salt transport. Hepatology. 2006;44:195-204. (Ref 65.)
Strautnieks SS, Byrne JA, Pawlikowska L, et al. Severe bile salt export pump deficiency: 82 different ABCB11 mutations in 109 families. Gastroenterology. 2008;134:1203-14. (Ref 56.)
Thomas C, Pellicciari R, Pruzanski M, et al. Targeting bile-acid signalling for metabolic diseases. Nat Rev Drug Discov. 2008;7:678-93. (Ref 2.)
1. Oude Elferink RP, Paulusma CC, Groen AK. Hepatocanalicular transport defects: Pathophysiologic mechanisms of rare diseases. Gastroenterology. 2006;130:908-25.
2. Thomas C, Pellicciari R, Pruzanski M, et al. Targeting bile-acid signalling for metabolic diseases. Nat Rev Drug Discov. 2008;7:678-93.
3. Hofmann AF, Hagey LR. Bile acids: Chemistry, pathochemistry, biology, pathobiology, and therapeutics. Cell Mol Life Sci. 2008;65:2461-83.
4. Hofmann AF. Biliary secretion and excretion in health and disease: Current concepts. Ann Hepatol. 2007;6:15-27.
5. Gass J, Vora H, Hofmann AF, Gray GM, et al. Enhancement of dietary protein digestion by conjugated bile acids. Gastroenterology. 2007;133:16-23.
6. Lorenzo-Zúñiga V, Bartoli R, Planas R, et al. Oral bile acids reduce bacterial overgrowth, bacterial translocation, and endotoxemia in cirrhotic rats. Hepatology. 2003;37:551-7.
7. Inagaki T, Moschetta A, Lee YK, et al. Regulation of antibacterial defense in the small intestine by the nuclear bile acid receptor. Proc Natl Acad Sci U S A. 2006;103:3920-5.
8. Emmett M, Guirl MJ, Santa Ana CA, et al. Conjugated bile acid replacement therapy reduces urinary oxalate excretion in short bowel syndrome. Am J Kidney Dis. 2003;41:230-7.
9. Russell DW. The enzymes, regulation, and genetics of bile acid synthesis. Annu Rev Biochem. 2003;72:137-74.
10. Pullinger CR, Eng C, Salen G, et al. Human cholesterol 7alpha-hydroxylase (CYP7A1) deficiency has a hypercholesterolemic phenotype. J Clin Invest. 2002;110:109-17.
11. Sundaram SS, Bove KE, Lovell MA, Sokol RJ. Mechanisms of disease: Inborn errors of bile acid synthesis. Nat Clin Pract Gastroenterol Hepatol. 2008;5:456-68.
12. Gilardi F, Mitro N, Godio C, et al. The pharmacological exploitation of cholesterol 7alpha-hydroxylase, the key enzyme in bile acid synthesis: From binding resins to chromatin remodelling to reduce plasma cholesterol. Pharmacol Ther. 2007;116:449-72.
13. Ren S, Hylemon PB, Marques D, et al. Overexpression of cholesterol transporter StAR increases in vivo rates of bile acid synthesis in the rat and mouse. Hepatology. 2004;40:910-17.
14. Inagaki T, Choi M, Moschetta A, et al. Fibroblast growth factor 15 functions as an enterohepatic signal to regulate bile acid homeostasis. Cell Metab. 2005;2:217-25.
15. Lundasen T, Galman C, Angelin B, Rudling M. Circulating intestinal fibroblast growth factor 19 has a pronounced diurnal variation and modulates hepatic bile acid synthesis in man. J Intern Med. 2006;260:530-6.
16. Kurosu H, Choi M, Ogawa Y, et al. Tissue-specific expression of betaKlotho and fibroblast growth factor (FGF) receptor isoforms determines metabolic activity of FGF19 and FGF21. J Biol Chem. 2007;282:26687-95.
17. Li T, Chiang JY. A novel role of transforming growth factor beta1 in transcriptional repression of human cholesterol 7alpha-hydroxylase gene. Gastroenterology. 2007;133:1660-9.
18. Xu Z, Tavares-Sanchez OL, Li Q, et al. Activation of bile acid biosynthesis by the p38 mitogen-activated protein kinase (MAPK): Hepatocyte nuclear factor-4alpha phosphorylation by the p38 MAPK is required for cholesterol 7alpha-hydroxylase expression. J Biol Chem. 2007;282:24607-14.
19. Yu C, Wang F, Jin C, et al. Independent repression of bile acid synthesis and activation of c-Jun N-terminal kinase (JNK) by activated hepatocyte fibroblast growth factor receptor 4 (FGFR4) and bile acids. J Biol Chem. 2005;280:17707-14.
20. Carlton VE, Harris BZ, Puffenberger EG, et al. Complex inheritance of familial hypercholanemia with associated mutations in TJP2 and BAAT. Nat Genet. 2003;34:91-6.
21. Hamilton JP, Xie G, Raufman JP, et al. Human cecal bile acids: Concentration and spectrum. Am J Physiol Gastrointest Liver Physiol. 2007;293:G256-63.
22. Ridlon JM, Kang DJ, Hylemon PB. Bile salt biotransformations by human intestinal bacteria. J Lipid Res. 2006;47:241-59.
23. Beuers U. Drug insight: Mechanisms and sites of action of ursodeoxycholic acid in cholestasis. Nat Clin Pract Gastroenterol Hepatol. 2006;3:318-28.
24. Hofmann AF. Detoxification of lithocholic acid, a toxic bile acid: Relevance to drug hepatotoxicity. Drug Metab Rev. 2004;36:703-22.
25. Uppal H, Toma D, Saini SP, et al. Combined loss of orphan receptors PXR and CAR heightens sensitivity to toxic bile acids in mice. Hepatology. 2005;41:168-76.
26. Marschall HU, Wagner M, Zollner G, et al. Complementary stimulation of hepatobiliary transport and detoxification systems by rifampicin and ursodeoxycholic acid in humans. Gastroenterology. 2005;129:476-85.
27. Choi M, Moschetta A, Bookout AL, et al. Identification of a hormonal basis for gallbladder filling. Nat Med. 2006;12:1253-5.
28. Kullak-Ublick GA, Paumgartner G, Berr F. Long-term effects of cholecystectomy on bile acid metabolism. Hepatology. 1995;21:41-5.
29. Alpini G, McGill JM, Larusso NF. The pathobiology of biliary epithelia. Hepatology. 2002;5:1256-68.
30. Feranchak AP, Fitz JG. Thinking outside the cell: The role of extracellular adenosine triphosphate in bile formation. Gastroenterology. 2007;133:1726-8.
31. Salas JT, Banales JM, Sarvide S, et al. Ae2a,b-deficient mice develop antimitochondrial antibodies and other features resembling primary biliary cirrhosis. Gastroenterology. 2008;134:1482-93.
32. Kanno N, LeSage G, Glaser S, Alpini G. Regulation of cholangiocyte bicarbonate secretion. Am J Physiol Gastrointest Liver Physiol. 2001;281:G612-25.
33. Hofmann AF, Zakko SF, Lira M, et al. Novel biotransformation and physiological properties of norursodeoxycholic acid in humans. Hepatology. 2005;42:1391-8.
34. Xia X, Francis H, Glaser S, et al. Bile acid interactions with cholangiocytes. World J Gastroenterol. 2006;12:3553-63.
35. Alpini G, Glaser S, Baiocchi L, et al. Secretin activation of the apical Na+-dependent bile acid transporter is associated with cholehepatic shunting in rats. Hepatology. 2005;41:1037-45.
36. Alpini G, Glaser S, Alvaro D, et al. Bile acid depletion and repletion regulate cholangiocyte growth and secretion by a phosphatidylinositol 3-kinase-dependent pathway in rats. Gastroenterology. 2002;123:1226-37.
37. Trauner M, Boyer JL. Bile salt transporters: Molecular characterization, function, and regulation. Physiol Rev. 2003;83:633-71.
38. Alrefai WA, Gill RK. Bile acid transporters: structure, function, regulation and pathophysiological implications. Pharm Res. 2007;24:1803-23.
39. Roberts MS, Magnusson BM, Burczynski FJ, Weiss M. Enterohepatic circulation: Physiological, pharmacokinetic and clinical implications. Clin Pharmacokinet. 2002;41:751-90.
40. Galman C, Angelin B, Rudling M. Bile acid synthesis in humans has a rapid diurnal variation that is asynchronous with cholesterol synthesis. Gastroenterology. 2005;129:1445-53.
41. Zamek-Gliszczynski MJ, Hoffmaster KA, Nezasa K, et al. Integration of hepatic drug transporters and phase II metabolizing enzymes: Mechanisms of hepatic excretion of sulfate, glucuronide, and glutathione metabolites. Eur J Pharm Sci. 2006;27:447-86.
42. Geier A, Wagner M, Dietrich CG, Trauner M. Principles of hepatic organic anion transporter regulation during cholestasis, inflammation and liver regeneration. Biochim Biophys Acta. 2007;1773:283-308.
43. Hagenbuch B, Dawson P. The sodium bile salt cotransport family SLC10. Pflugers Arch. 2004;447:566-70.
44. Ho RH, Tirona RG, Leake BF, et al. Drug and bile acid transporters in rosuvastatin hepatic uptake: Function, expression, and pharmacogenetics. Gastroenterology. 2006;130:1793-806.
45. Ho RH, Leake BF, Roberts RL, et al. Ethnicity-dependent polymorphism in Na+-taurocholate cotransporting polypeptide (SLC10A1) reveals a domain critical for bile acid substrate recognition. J Biol Chem. 2004;279:7213-22.
46. Hadj-Rabia S, Baala L, Vabres P, et al. Claudin-1 gene mutations in neonatal sclerosing cholangitis associated with ichthyosis: A tight junction disease. Gastroenterology. 2004;127:1386-90.
47. Hagenbuch B, Meier PJ. Organic anion transporting polypeptides of the OATP/SLC21 family: Phylogenetic classification as OATP/SLCO superfamily, new nomenclature and molecular/functional properties. Pflugers Arch. 2004;447:653-65.
48. Ballatori N, Hammond CL, Cunningham JB, et al. Molecular mechanisms of reduced glutathione transport: Role of the MRP/CFTR/ABCC and OATP/SLC21A families of membrane proteins. Toxicol Appl Pharmacol. 2005;204:238-55.
49. Mahagita C, Grassl SM, Piyachaturawat P, Ballatori N. Human organic anion transporter 1B1 and 1B3 function as bidirectional carriers and do not mediate GSH-bile acid cotransport. Am J Physiol Gastrointest Liver Physiol. 2007;293:G271-8.
50. Konig J, Seithel A, Gradhand U, Fromm MF. Pharmacogenomics of human OATP transporters. Naunyn Schmiedebergs Arch Pharmacol. 2006;372:432-43.
51. Link E, Parish S, Armitage J, et al. SLCO1B1 variants and statin-induced myopathy—A genomewide study. N Engl J Med. 2008;359:789-99.
52. Zollner G, Marschall HU, Wagner M, Trauner M. Role of nuclear receptors in the adaptive response to bile acids and cholestasis: Pathogenetic and therapeutic considerations. Mol Pharm. 2006;3:231-51.
53. Mennone A, Soroka CJ, Cai SY, et al. Mrp4-/- mice have an impaired cytoprotective response in obstructive cholestasis. Hepatology. 2006;43:1013-21.
54. Stieger B, Meier Y, Meier PJ. The bile salt export pump. Pflugers Arch. 2007;453:611-20.
55. Strautnieks SS, Bull LN, Knisely AS, et al. A gene encoding a liver-specific ABC transporter is mutated in progressive familial intrahepatic cholestasis. Nat Genet. 1998;20:233-8.
56. Strautnieks SS, Byrne JA, Pawlikowska L, et al. Severe bile salt export pump deficiency: 82 different ABCB11 mutations in 109 families. Gastroenterology. 2008;134:1203-14.
57. Oelkers P, Kirby LC, Heubi JE, Dawson PA. Primary bile acid malabsorption caused by mutations in the ileal sodium-dependent bile acid transporter gene (SLC10A2). J Clin Invest. 1997;99:1880-7.
58. Kok T, Hulzebos CV, Wolters H, et al. Enterohepatic circulation of bile salts in farnesoid X receptor-deficient mice: Efficient intestinal bile salt absorption in the absence of ileal bile acid-binding protein. J Biol Chem. 2003;278:41930-7.
59. Ballatori N, Christian WV, Lee JY, et al. OSTalpha-OSTbeta: A major basolateral bile acid and steroid transporter in human intestinal, renal, and biliary epithelia. Hepatology. 2005;42:1270-9.
60. Rao A, Haywood J, Craddock AL, et al. The organic solute transporter alpha-beta, Ostalpha-Ostbeta, is essential for intestinal bile acid transport and homeostasis. Proc Natl Acad Sci U S A. 2008;105:3891-6.
61. Ballatori N, Fang F, Christian WV, et al. Ostalpha-Ostbeta is required for bile acid and conjugated steroid disposition in the intestine, kidney, and liver. Am J Physiol Gastrointest Liver Physiol. 2008;295:G179-G86.
62. Heubi JE, Setchell KD, Bove KE. Inborn errors of bile acid metabolism. Semin Liver Dis. 2007;27:282-94.
63. Klomp LW, Vargas JC, van Mil SW, et al. Characterization of mutations in ATP8B1 associated with hereditary cholestasis. Hepatology. 2004;40:27-38.
64. Paulusma CC, Folmer DE, Ho-Mok KS, et al. ATP8B1 requires an accessory protein for endoplasmic reticulum exit and plasma membrane lipid flippase activity. Hepatology. 2008;47:268-78.
65. Paulusma CC, Groen A, Kunne C, et al. Atp8b1 deficiency in mice reduces resistance of the canalicular membrane to hydrophobic bile salts and impairs bile salt transport. Hepatology. 2006;44:195-204.
66. Frankenberg T, Miloh T, Chen FY, et al. The membrane protein ATPase class I type 8B member 1 signals through protein kinase C zeta to activate the farnesoid X receptor. Hepatology. 2008. [Epub ahead of print]
67. Alissa FT, Jaffe R, Shneider BL. Update on progressive familial intrahepatic cholestasis. J Pediatr Gastroenterol Nutr. 2008;46:241-52.
68. van Mil SW, van der Woerd WL, van der Brugge G, et al. Benign recurrent intrahepatic cholestasis type 2 is caused by mutations in ABCB11. Gastroenterology. 2004;127:379-84.
69. Pauli-Magnus C, Meier PJ. Hepatobiliary transporters and drug-induced cholestasis. Hepatology. 2006;44:778-87.
70. Knisely AS, Strautnieks SS, Meier Y, et al. Hepatocellular carcinoma in ten children under five years of age with bile salt export pump deficiency. Hepatology. 2006;44:478-86.
71. Jacquemin E, De Vree JM, Cresteil D, et al. The wide spectrum of multidrug resistance 3 deficiency: from neonatal cholestasis to cirrhosis of adulthood. Gastroenterology. 2001;120:1448-58.
72. Thomas LA, Veysey MJ, Murphy GM, et al. Octreotide induced prolongation of colonic transit increases faecal anaerobic bacteria, bile acid metabolising enzymes, and serum deoxycholic acid in patients with acromegaly. Gut. 2005;54:630-5.
73. Hofmann AF. Inappropriate ileal conservation of bile acids in cholestatic liver disease: homeostasis gone awry. Gut. 2003;52:1239-41.
74. Fickert P, Wagner M, Marschall HU, et al. 24-norUrsodeoxycholic acid is superior to ursodeoxycholic acid in the treatment of sclerosing cholangitis in Mdr2 (Abcb4) knockout mice. Gastroenterology. 2006;130:465-81.
75. Kapral C, Wewalka F, Praxmarer V, et al. Conjugated bile acid replacement therapy in short bowel syndrome patients with a residual colon. Z Gastroenterol. 2004;42:583-9.
[/level-membership-for-gastroenterology-and-hepatology-category][not-level-membership-for-gastroenterology-and-hepatology-category]
CHAPTER 64 Bile Secretion and the Enterohepatic Circulation
Bile formation is essential for intestinal lipid digestion and absorption, cholesterol homeostasis, and hepatic excretion of lipid-soluble xenobiotics, drug metabolites, and heavy metals. The process of bile formation depends on hepatic synthesis and canalicular secretion of bile acids, the predominant organic anions in bile, and maintenance of hepatic bile formation is essential for normal liver function. Most of the bile acids secreted by the hepatocyte have been secreted previously into the small intestine and have undergone enterohepatic cycling. As a result, disturbances in bile acid synthesis, biliary secretion, and intestinal absorption have profound effects on hepatic and gastrointestinal physiology. Identification of the hepatic, biliary, and intestinal bile acid transporters has advanced our understanding of genetic and acquired disorders of bile formation and secretion. Transporter gene mutations have been identified in disorders such as progressive familial intrahepatic cholestasis (PFIC) types 1 to 3, intrahepatic cholestasis of pregnancy (ICP), low phospholipid-associated cholelithiasis (LPAC), Dubin-Johnson syndrome, and primary bile acid malabsorption (PBAM).1 In addition, developments in the nuclear and G-protein–coupled receptor fields have provided new insights into the regulation of bile acid synthesis, secretion, and enterohepatic cycling.2 This chapter reviews the current knowledge of the hepatic synthesis, biliary secretion, and enterohepatic circulation of bile acids.
Bile is a complex, lipid-rich micellar solution that is isosmotic with plasma and composed primarily of water, inorganic electrolytes, and organic solutes such as bile acids, phospholipids (mostly phosphatidylcholine), cholesterol, and bile pigments (Table 64-1). The relative proportions of the major organic solutes in bile is illustrated in Figure 64-1.The volume of hepatic bile secreted is estimated to range from 500 to 600 mL per day, and bile acids are the dominant organic components. Actively secreted across the canalicular membrane, bile acids induce the secretion of other biliary constituents. In healthy humans, canalicular secretion is efficient and remarkably concentrative; the intracellular monomeric concentration of bile acid is less than 10 mmol/L in the hepatocyte and more than 1000 mmol/L in canalicular bile. Bile acids travel down the biliary tree and are stored in the gallbladder. After a meal the gallbladder contracts and empties its contents into the duodenum, where bile acids facilitate absorption of cholesterol and fat-soluble vitamins. Bile acids are poorly absorbed in the proximal small intestine but are absorbed almost completely by the terminal ileum. The bile acids are returned to the liver in the portal circulation, actively absorbed at the hepatocyte sinusoidal membrane, and re-secreted into bile.3
Table 64-1 Composition of Hepatic Bile
| COMPONENT | CONCENTRATION |
|---|---|
| Electrolytes and minerals (mmol/L): | |
| Sodium | 140-160 |
| Potassium | 3-8 |
| Chloride | 70-120 |
| Bicarbonate | 20-50 |
| Calcium | 1-5 |
| Phosphate | 0-1.2 |
| Magnesium | 1-3 |
| Metals (µmol/L): | |
| Iron | 2-72 |
| Copper | 12-21 |
| Organic constituents (mmol/L): | |
| Bile acids | 5-50 |
| Bilirubin (total) | 1-2 |
| Phospholipid (lecithin) | 0.5-20.0 |
| Cholesterol | 0.5-1.0 |
| Glutathione | 3-5 |
| Glucose | 0.2-1.0 |
| Urea | 2.2-6.5 |
| Protein (g/dL) | 0.2-3.0 |
Values obtained from measurements of human bile are drawn from Albers CJ, Huizenga JR, Krom RA, et al. Composition of human hepatic bile. Ann Clin Biochem 1985; 22:129-32; Keulemans YC, Mok KS, de Wit LT, et al. Hepatic bile versus gallbladder bile: A comparison of protein and lipid concentration and composition in cholesterol gallstone patients. Hepatology 1998; 28:11-16; and Ho KJ. Biliary electrolytes and enzymes in patients with and without gallstones. Dig Dis Sci 1996; 41:2409-16.
Figure 64-1. Top, Typical solute composition of hepatic and gallbladder bile in healthy humans. Bile acids are the primary solute in bile, constituting approximately 67% of bile. Phospholipid accounts for approximately 22%, cholesterol 4%, bilirubin 0.3%, and protein 4.5% of bile. Biliary bile acid composition is shown in the bottom left. Cholic acid, chenodeoxycholic acid, and deoxycholic acid constitute more than 95% of the biliary bile acids, and virtually all of the biliary bile acids are in conjugated form. The proportion of biliary lithocholic acid and ursodeoxycholic acid varies but rarely exceeds 5%. The majority of lithocholic acid in bile is present in sulfated form. When administered in therapeutic doses, the proportion of ursodeoxycholic acid in bile may rise to as much as 40%. Fecal bile acid composition is shown in the bottom right. Fecal bile acids are almost entirely unconjugated as a result of bacterial deconjugating enzymes in the colon and consist primarily of the dehydroxylated bile acids, deoxycholic acid, and lithocholic acid (see Figure 64-2).
The functions of bile acids in the liver and gastrointestinal tract are multiple.3,4 First, bile acids induce bile flow and hepatic secretion of biliary lipids (phospholipid and cholesterol). The vectorial movement of bile acids from blood into the bile canaliculus generates an osmotic water flow and is a major determinant of bile formation. Second, bile acids play an important role in the digestion of dietary fats and are essential for the intestinal absorption of cholesterol and fat-soluble vitamins. Bile acids promote intestinal absorption by solubilizing dietary lipids and lipid digestion products as mixed micelles to facilitate their aqueous diffusion across the intestinal mucosa. Fat-soluble vitamins (A, D, E, and K) are poorly absorbed in the absence of bile acid micelles, and disturbances in the secretion or enterohepatic cycling of bile acids lead to fat-soluble vitamin deficiency. Along with their major role in promoting dietary lipid absorption, bile acids may facilitate intestinal absorption of protein by accelerating protein hydrolysis by pancreatic proteases.5 Third, bile acids play a complex role in maintaining cholesterol homeostasis. On one hand, bile acids promote cholesterol intake by facilitating the intestinal absorption of biliary and dietary cholesterol. On the other hand, bile acids function through several mechanisms to promote cholesterol elimination from the body. Bile acids are water-soluble end products of cholesterol catabolism and a major route for the elimination of cholesterol via fecal excretion. Bile acids also promote hepatic secretion of cholesterol into bile by inducing bile flow and solubilizing biliary cholesterol, thereby enabling cholesterol to move from the hepatocyte to the intestinal lumen for elimination. Fourth, bile acids contribute to the antimicrobial defense mechanisms of the intestine through direct bacteriostatic actions of bile acid–fatty acid mixed micelles in the proximal small intestine and by inducing expression of antimicrobial genes in the distal small intestine.6,7 Fifth, bile acids act to prevent the formation of calcium gallstones and oxalate kidney stones.8 Sixth, bile acids can function as hormones to regulate the enterohepatic circulation as well as fat, glucose, and energy homeostasis by signaling through nuclear and G-protein–coupled receptors.2
BILE ACID SYNTHESIS AND METABOLISM
Bile acids are synthesized from cholesterol in pericentral hepatocytes of the hepatic acini. In this process, cholesterol, a lipophilic compound, is converted into a water-soluble product. In humans the newly synthesized (primary) bile acids are cholic acid (CA), a trihydroxy-bile acid with hydroxy groups at the C-3, C-7, and C-12 positions, and chenodeoxycholic acid (CDCA), a dihydroxy-bile acid with hydroxy groups at the C-3 and C-7 positions (Fig. 64-2). The kinetics for bile acid synthesis and turnover in humans are summarized in Table 64-2. Hepatic bile acid synthesis involves two major pathways, the “classical” neutral pathway (cholesterol 7α-hydroxylase pathway) that favors CA biosynthesis and the “alternative” acidic pathway (oxysterol 7α-hydroxylase pathway) that favors CDCA biosynthesis.9 In the classical pathway, the enzyme cholesterol 7α-hydroxylase (cytochrome P-450 7A1 [CYP7A1]) converts cholesterol directly into 7α-hydroxycholesterol. In the alternative pathway, cholesterol must first be converted by C-24, C-25, or C-27 sterol hydroxylases into an oxysterol, the major species being 27-hydroxycholesterol, before conversion by the oxysterol 7α-hydroxylase (CYP7B1).
Table 64-3 Proteins Involved in the Regulation of Bile Acid Synthesis and Enterohepatic Cycling
| PROTEIN (GENE) | DESCRIPTION AND FUNCTION OF PROTEIN IN BILE ACID METABOLISM |
|---|---|
| FXR (NR1H4) | Bile acid–activated nuclear receptor; regulation of bile acid synthesis, transport, and metabolism |
| HNF4α (NR2A1) | Nuclear receptor; positive regulator of cytochrome P-450 7a1 (CYP7a1) expression and hepatic bile acid synthesis |
| SHP (NR0B2) | Nuclear receptor; negative feedback regulation of hepatic bile acid synthesis by antagonizing HNF4α, LRH-1; regulation of bile acid transport and metabolism |
| LRH-1 (NR5A2) | Nuclear receptor; positive regulator of CYP7a1 expression and hepatic bile acid synthesis |
| PXR (NR1I2) | Bile acid and xenobiotic-activated nuclear receptor involved in detoxification of secondary bile acids |
| VDR (NR1I1) | Vitamin D and bile acid–activated nuclear receptor; involved in detoxification of LCA |
| FGFR4 (FGFR4) | Membrane receptor; negative feedback regulation of CYP7a1 and hepatic bile acid synthesis |
| β-klotho (KLB) | Membrane co-receptor associated with FGFR4; confers liver specificity to FGFR4-FGF19 pathway; negative feedback regulation of CYP7a1 and hepatic bile acid synthesis |
| FGF19 (FGF19) | Protein growth factor; secreted by intestine in response to bile acids; regulates hepatic bile acid synthesis via FGFR4:β-klotho |
FXR, farnesoid X-receptor; FGF19, fibroblast growth factor 19; FGFR4, fibroblast growth factor receptor 4; HNF4α, hepatocyte nuclear factor 4 alpha; LCA, lithocolic acid; LRH-1, liver receptor homolog 1; PXR, pregnane X-receptor; SHP, small heterodimer partner; VDR, vitamin D receptor.
The overall process of bile acid biosynthesis is complex, involving 17 different enzymes divided into two broad groups.9 The first group performs modifications to the sterol ring structure, whereas the second group modifies the sterol side chain. In the classical pathway, sterol ring modifications precede side chain changes, whereas in the alternative pathway the side chain modifications occur before or during changes to the sterol ring structure. Of the two major biosynthetic pathways, the classical (CYP7A1) pathway is quantitatively more important in humans. This conclusion is supported by the finding that bile acid production is decreased by almost 90% in an adult patient with an inherited mutation in the CYP7A1 gene.10 In contrast to adults, the alternative pathway may be dominant in neonates, as evidenced by the low expression of CYP7A1 in newborns and the finding of severe cholestatic liver disease in an infant with an inherited CYP7B1 mutation.11
The rate-limiting step for the classical pathway is the enzyme CYP7A1. Bile acid feedback inhibition of CYP7A1 is well established experimentally; bile acid synthesis is decreased after administration of hydrophobic bile acids and increased by interruption of the enterohepatic circulation following ileal resection or administration of bile acid sequestrants.12 In contrast to the classical (CYP7A1) pathway, the alternative pathway for bile acid synthesis is not subject to regulation by bile acids but is controlled by cholesterol delivery to the mitochondria, the site of 27-hydroxylation.13 The molecular mechanisms responsible for the negative feedback regulation of the CYP7A1 pathway involve the liver and small intestine and have been elucidated. For the major pathway, bile acids act as ligands for the farnesoid X receptor (FXR) in ileal enterocytes to induce synthesis of an endocrine hormone, the fibroblast growth factor-19 (FGF19). FGF19 is secreted into the portal circulation and acts on hepatocytes through its cell surface receptor, a complex of the β-klotho protein and fibroblast growth factor-4, to repress CYP7A1 expression and bile acid synthesis.14–16 In a second pathway, excess bile acids in the hepatocyte activate FXR and indirectly suppress expression of CYP7A1 through a complex mechanism that involves the orphan nuclear receptor designated small heterodimer partner (SHP). SHP interferes with the activity of several nuclear receptors including hepatocyte nuclear factor 4α (HNF4α) and liver receptor homolog 1 (LRH-1), which are required for expression of the CYP7A1 gene.12,17 Finally, bile acids also inhibit CYP7A1 gene transcription in a SHP and FGFR4-independent fashion through activation of the c-Jun NH2-terminal kinase (JNK) pathway.18,19 These complex molecular titrations link bile acid synthesis to changes in intestinal as well as hepatic bile acid levels. The receptors and protein factors that participate in the regulation of bile acid synthesis and the enterohepatic circulation of bile acids are summarized in Table 64-3.
Before secretion into the bile canaliculus, both CA and CDCA are N-acyl amidated with glycine or taurine, a process commonly termed conjugation. This conjugation enhances the hydrophilicity of the bile acid and the acidic strength of the side chain, in essence converting a weak acid (pKa ≈ 5.0 for the unconjugated bile acid) to a strong acid (pKa ≈ 3.9 for the glycine conjugate; pKa ≈ 2.0 for the taurine conjugate). The major function of conjugation is to decrease the passive diffusion of bile acids across cell membranes during their transit through the enterohepatic circulation. As a result, conjugated bile acids are absorbed only if a specific membrane carrier is present. Compared with unconjugated bile acids, conjugated bile acids are also more soluble at acidic pH and more resistant to precipitation in the presence of high concentrations of calcium. The net effect of conjugation is to maintain high intraluminal concentrations of bile acids down the length of the small intestine to facilitate lipid digestion and absorption. The importance of bile acid conjugation is illustrated by the finding that inherited defects in bile acid conjugation present with the clinical phenotype of fat-soluble vitamin malabsorption and steatorrhea.11,20
Most of the conjugated bile acids secreted into the small intestine are efficiently absorbed intact; however, bile acids are also metabolized during their passage down the intestine, by the endogenous bacterial flora. In the distal small intestine, the bacterial flora deconjugates a small fraction, ≈15%, of the bile acids. The unconjugated bile acids are absorbed passively or actively and returned to the liver, where they are efficiently reconjugated and mix with newly synthesized bile acids to be re-secreted into bile. This process of intestinal deconjugation and hepatic reconjugation is a normal part of bile acid metabolism. An additional bacterial modification is the epimerization of the 3α-hydroxy or 7α-hydroxy groups to their corresponding 3β- or 7β-hydroxy forms.21,22 The 7α-hydroxy group of CDCA is epimerized to form the 3α,7β-dihydroxy bile acid, ursodeoxycholic acid (UDCA). After being absorbed from the intestine, UDCA is conjugated to taurine or glycine in the liver and circulates as a minor component, normally less than 5%, of the bile acid pool. In addition to endogenous formation, UDCA is used as a therapeutic agent in various forms of cholestatic liver disease (see Chapters 76 and 89).23
A small fraction of bile acids secreted into the small intestine escapes absorption and passes into the large intestine, where deconjugation is almost complete. In the colon, the action of bacterial 7α-dehydroxylase converts CA to deoxycholic acid (DCA), a dihydroxy bile acid with hydroxy groups at the C-3 and C-12 positions, and converts CDCA to lithocholic acid (LCA), a monohydroxy bile acid with a hydroxy group at position C-3 (see Fig. 64-2).21 The colon absorbs about 50% of the DCA formed as well as a small fraction of the LCA. After returning to the liver, DCA is reconjugated and circulates with the primary bile acids. Hepatic conjugation of the circulating bile acids is extremely efficient, so virtually all the biliary bile acids (primarily CA, CDCA, DCA, and UDCA) are in conjugated form. Bacterial deconjugation and dehydroxylation in the colon are also efficient, so the feces contain mainly the unconjugated secondary bile acids DCA and LCA (see Fig. 64-1).
The secondary bile acids can be metabolized further by various pathways, including hepatic re-epimerization of 3β-hydroxy bile acids, hepatic reduction of 7-oxo-lithocholate to CDCA or UDCA, and sulfation of bile acids by the liver or kidney (see Fig. 64-2). Additional minor pathways include hydroxylation and glucuronidation of bile acids. The conjugation of LCA with sulfate or glucuronide blocks intestinal absorption, and the modified LCA is rapidly lost from the circulating pool of bile acids. In humans, sulfation of LCA plays an important protective role, because unmodified LCA is intrinsically hepatoxic.24 The molecular mechanisms responsible for inducing these hepatoprotective modifications are being elucidated and involve induction of cytosolic sulfotransferases and cytochrome P-450 enzymes by the nuclear receptors pregnane X receptor (PXR) and constitutive androsterone receptor (CAR).25 For example, LCA, as well as other xenobiotic inducers such as rifampin, bind and directly activate PXR, thereby stimulating important mechanisms for hepatic detoxification and transport of bile acids.26
THE ENTEROHEPATIC CIRCULATION
The anatomic components of the enterohepatic circulation are the liver, biliary tract, gallbladder, small intestine, portal venous circulation, and, to a lesser extent, colon, systemic circulation, and kidneys (Fig. 64-3). At a fundamental level, the enterohepatic circulation of bile acids can be considered to consist of a series of storage chambers (gallbladder, small intestine), valves (sphincter of Oddi, ileocecal valve), mechanical pumps (bile canaliculi, biliary tract, small intestine), and chemical pumps (hepatocyte, cholangiocyte, and ileocyte).
Efficient intestinal reabsorption and hepatic extraction of bile acids permit an effective recycling and conservation mechanism that largely restricts bile acids to the intestinal and hepatobiliary compartments. During fasting, bile acids move down the biliary tract and are concentrated approximately 10-fold in the gallbladder. After an overnight fast, most of the bile acids are sequestered in the gallbladder, resulting in low levels of bile acids in the small intestine, portal vein, systemic circulation, and liver. In response to a meal, cholecystokinin is released from the intestinal mucosa and acts on the biliary tree to relax the sphincter of Oddi and stimulate gallbladder contraction. A concentrated solution of mixed micelles (bile acids, phospholipids, and cholesterol) is emptied from the gallbladder via the bile duct into the small intestine, where the micelles facilitate fat absorption by stimulating the action of pancreatic lipase on triglyceride, solubilizing the hydrolytic products, and delivering lipids to the mucosal surface. During the digestion of a large meal, the gallbladder remains contracted, and bile acids secreted by the liver bypass the gallbladder and empty directly into the duodenum. During this period, the bile acid concentration in the small intestine is approximately 5 to 10 mmol/L; micelle formation requires that the intraluminal bile acid concentration be greater than 1.5 mmol/L. During the interdigestive period, the sphincter of Oddi contracts and the gallbladder relaxes, causing a larger fraction of the secreted bile acid to enter the gallbladder for storage. This gallbladder relaxation is mediated in part by the ileal release of fibroblast growth factor 19 (FGF19).27 Therefore, the enterohepatic cycling of bile acids accelerates during digestion and slows between meals and during overnight fasting. This rhythm of bile acid secretion is maintained even after cholecystectomy.28 When the gallbladder is absent, bile acids are stored in the proximal small intestine. After ingestion of a meal, small intestinal contractions propel the stored bile acids to the distal ileum, where they are actively reabsorbed.
The enterohepatic cycling of bile acids is extremely efficient; less than 10% of the intestinal bile acids escape reabsorption and are eliminated in the feces. Bile acids are absorbed from the small intestine predominantly by an active transport system restricted to the terminal ileum and, to a lesser extent, by passive absorption down the length of the intestine. Of all the conjugated anions secreted into bile, only bile acids are actively absorbed in conjugated form and undergo an enterohepatic circulation. In adult humans, the enterohepatic circulation maintains a bile acid pool size of 50 to 60 mmol per kg body weight, corresponding to approximately 2 to 4 g. The bile acid pool cycles two to three times per meal, resulting in 6 to 10 cycles per day, and the intestine may reabsorb between 10 and 30 g of bile acid per day. Approximately 0.2 to 0.6 g of bile acids escapes reabsorption and is eliminated in the stool each day. Hepatic conversion of cholesterol to bile acid balances fecal excretion, and this process represents an important route for elimination of cholesterol from the body. The kinetics of bile acid turnover in humans are summarized in Table 64-2.
HEPATIC BILE ACID TRANSPORT AND BILE SECRETION
Hepatic adenosine triphosphate (ATP)–dependent carriers actively secrete bile acids into the canalicular lumen, from which they are too large to diffuse back across the paracellular junctions that line the canaliculi. Solutes such as the conjugated bile acids that are actively pumped across the canalicular membrane generate bile flow and are termed primary solutes. The list of primary solutes includes conjugated bilirubin, glutathione, heavy metals, and conjugates of various metabolites and xenobiotics. Water, plasma electrolytes, calcium, glucose, amino acids, bicarbonate, and other low-molecular-weight solutes that flow into the canaliculus in response to the osmotic gradient are termed secondary solutes. The choleretic activity of each primary solute is defined as the amount of bile flow induced per amount of solute secreted. The choleretic activity varies for different bile acid species and ranges from 8 to 30 microliters of bile flow induced per micromole of bile acid secreted. In humans most canalicular bile flow is generated by bile acid secretion; however, the secretion of other primary solutes by the hepatocyte and biliary epithelium also contributes to bile formation. Newly secreted hepatic canalicular bile is modified during its transit in the biliary tract via the action of ductule epithelial cells (cholangiocytes), and this ductular secretion may account for up to 40% of the volume of bile. The ductular modifications to hepatic bile include the absorption of solutes such as glucose, amino acids, and bile acids; the secretion of solutes such as bicarbonate and chloride; and the movement of water through specific channels (aquaporins) and paracellularly.29,30
BILE ACID–INDEPENDENT BILE FLOW
Canalicular bile flow is generated by active secretion of primary solutes in addition to bile acids. Reduced glutathione (GSH) and bicarbonate (HCO3−) constitute the major components of the bile acid–independent fraction of bile flow. The ATP-dependent canalicular secretion of GSH and GSH conjugates via the multidrug resistance–associated protein 2 (MRP2) plays a particularly important role. In addition to being secreted at high concentrations into bile, intraluminal catabolism of GSH by gamma glutamyl transpeptidase (GGTP) further raises the solute concentration and contributes to the osmotic driving force for canalicular bile formation. Besides the ATP-dependent secretion of organic anions into bile, hepatic and biliary ATP-independent secretion of bicarbonate via the HCO3−/Cl− anion exchanger AE2 contributes to the bile acid–independent bile flow.31 The majority of this HCO3− secretion occurs at the level of the bile duct epithelial cells in response to stimulation by a variety of hormones and neuropeptides, such as secretin and vasoactive intestinal peptide.32
CHOLEHEPATIC SHUNT PATHWAY
The term cholehepatic shunt was coined to describe the cycle whereby unconjugated dihydroxy bile acids secreted into bile are absorbed passively by cholangiocytes, returned to the hepatocyte via the periductular capillary plexus, and resecreted into bile. Absorption of the protonated unconjugated bile acid molecule generates a bicarbonate anion, resulting in a bicarbonate-rich choleresis. Premature absorption and resecretion of the bile acid also increases bile acid-dependent bile flow. This cycle explains the hypercholeresis observed for unconjugated C-24 dihydroxy bile acids such as UDCA, unconjugated C-23 bile acid analogs such as norursodeoxycholate (norUDCA), and certain drugs such as the nonsteroidal anti-inflammatory drug sulindac.33
[/not-level-membership-for-gastroenterology-and-hepatology-category]
