14. Beta Thalassemia
Definition
Beta thalassemia is an inherited blood disease of abnormal hemoglobin production in which both beta-globin subunit components of normal hemoglobin are absent.
Incidence
The estimated incidence rates of beta thalassemia vary by population and it is more commonly found in areas around the Mediterranean Sea; in Italian, Sicilian, and Greek populations, the incidence is approximately 10%; in African populations, it is approximately 1.5%; in the African American population, the incidence is approximately 1.5%; and in Southeastern Asian populations, it is approximately 5%.
Etiology
The manufacture of beta-globin molecules has been traced to chromosome 11. More than 200 mutations of the beta-globin genes on chromosome 11 that can result in thalassemia have been documented. The result of the particular mutation is either the absence of the beta-globin, called beta(0)thalassemia, or diminished beta-globin, called beta(+) thalassemia.
Signs and Symptoms
• Abdominal swelling
• Amenorrhea
• Cardiac failure
• Cardiomegaly
• Dark urine
• Dysrhythmias
• Facial deformities
• Failure to grow/thrive
• Frontal bossing
• Gallstones
• Infection
• Irritability
• Jaundice
• Leg ulcers
• Mongoloid facies
• Pallor
• Progressive hepatosplenomegaly
• Severe anemia
• Sexual development retardation
• Skeletal deformities
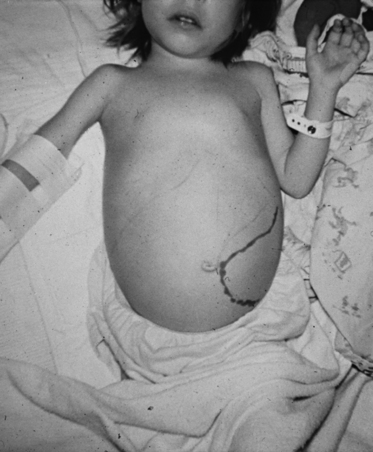 |
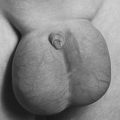
| Beta Thalassemia. A child with beta thalassemia major who has severe splenomegaly. |
Medical Management
The primary goal of treatment is to achieve and maintain correction of the anemia. Anemia is evident by 6 months of age as the neonate’s physiology converts from fetal hemoglobin to adult hemoglobin. The patient becomes dependent on transfusions very early in life and remains transfusion-dependent for his or her entire life. The patient should be placed on a regular schedule of transfusion. The transfusion protocols are classified as conservative, hypertransfusion, and supertransfusion (see box on p. 57). The hypertransfusion regimen has the secondary benefit of reducing the patient’s ineffective erythropoiesis.
Transfusion Protocols for the Patient with Beta Thalassemia
• Conservative: Transfuse when the hemoglobin level is >6 to 8 g/dL or the patient becomes symptomatic.
• Hypertransfusion: Transfuse frequently enough to ensure the hemoglobin level is maintained at >10 g/dL and the hematocrit is >30%. Proper hemoglobin level maintenance also suppresses endogenous erythroid activity, prevents bone marrow expansion, and reduces absorption of dietary iron. This method is associated with a high incidence of hemosiderosis.
• Supertransfusion: Transfuse frequently enough to achieve and maintain a hematocrit level >35%; usually two or three units are transfused every 2 to 4 weeks. This method is also associated with a high incidence of hemosiderosis.
The patient with beta thalassemia should receive broad-spectrum antibiotics after febrile illnesses, based on the results of urine and/or blood cultures. The patient who has undergone splenectomy frequently receives prophylactic penicillin therapy.
The only treatment option currently available that is considered to be potentially curative is bone marrow transplantation. Other therapies being investigated and evaluated include augmentation of fetal hemoglobin synthesis, bisphosphonate therapies, and gene therapy.
Surgical interventions related to beta thalassemia are limited. Typical surgical interventions are placement of permanent, indwelling central venous catheter(s) and splenectomy. Splenectomy is frequently avoidable if the child is maintained on an aggressive transfusion protocol and should be delayed or deferred until the patient is older than 6 years of age to minimize the potential of overwhelming infection.
Complications
• Aplastic and megaloblastic crises
• Cholelithiasis
• Chronic hemolysis
• Death
• Delayed/absent puberty
• Heart failure
• Hemolytic anemia
• Hepatic siderosis
• Impaired growth rate
• Iron overload
• Pathologic fractures
• Septicemia
• Severe anemia
• Splenomegaly
Anesthesia Implications
The patient with beta thalassemia may present to the operating room with significant anemia, depending on where he or she is in the transfusion protocol. The degree of anemia dictates this patient’s O 2-carrying capacity. The anesthetist should strive to optimize the patient’s O 2-carrying capabilities by performing preoperative and/or perioperative transfusion(s) and providing as close to 100% oxygen as possible for the anesthetic. Preoperative complete blood count and basic metabolic panel are imperative laboratory studies for the patient with beta thalassemia.
The anesthetist must be very discerning with regard to the liver function test results. The patient with beta thalassemia is prone to develop hemochromatosis secondary to the multiple transfusions received throughout his or her lifetime. As a result of the induced liver dysfunction, metabolism may be prolonged for a host of anesthesia-related drugs, from opiates to hypnotics to benzodiazepines to muscle relaxants. The anesthetist may need to reduce the doses of these drugs. If the liver dysfunction becomes severe enough, the patient may experience coagulation irregularities. The anesthetist may need to administer coagulation adjuncts, such as vitamin K or various coagulation factors or fresh frozen plasma. As a result, the anesthetist should anticipate greater than expected blood volume losses for a given surgical intervention.
Beta thalassemia causes bone marrow hyperplasia and accelerated hematopoiesis. The hematopoiesis acceleration produces distortion of some of the bones, most notably distortion of the craniofacial bones. The patient with beta thalassemia is characterized by the “chipmunk” face that results from the craniofacial anatomy distortion. Cranial radiographs demonstrate what is known as “hair-on-end” patterns. The craniofacial distortions produced by beta thalassemia may make direct laryngoscopy more difficult than would otherwise be anticipated.
The patient with beta thalassemia is prone to pathologic fractures. As a result, the anesthetist must take great care during the process of positioning the patient and ensure adequate padding to bony prominences. Because of their greater ability to disperse pressure, gelatin-filled pads are superior to foam padding.