120 Beta-Lactam Drugs
The β-lactam antibiotics are the most commonly prescribed antibiotics in the critical care setting. Their individual microbiological spectra and relative safety have made them first-line therapy for prophylaxis and treatment of infection. From the oldest (penicillin) to the newest (doripenem) agents, β-lactams continue to be useful for the myriad infectious complications of critical illness. Table 120-1 lists the parenteral β-lactam antibiotics commonly used in the intensive care unit (ICU).
TABLE 120-1 Beta-Lactam Antibiotics
| Natural Penicillins |
| Penicillin GK |
| Penicillinase-Resistant Penicillins |
| Aminopenicillins |
| Anti-Pseudomonal Penicillins |
| Cephalosporins |
| Carbapenems |
| Monobactams |
| Aztreonam |
* This drug is not yet approved by the FDA and has not been designated a fourth-generation agent.
 Mechanism of Action
Mechanism of Action
β-Lactam antibiotics are similar in that each contains a β-lactam ring in addition to other pharmacologically active side chains stemming from this central structure. Side chain manipulation is largely responsible for both spectrum of activity and stability against enzymatic degradation, pharmacokinetics, and adverse effects. β-Lactam antibiotics inhibit bacterial wall synthesis by binding to penicillin-binding proteins (PBPs). These PBPs are transpeptidases, carboxypeptidases, and endopeptidases involved in the structure and function of the cell wall.1,2 The cell wall is made up of a peptidoglycan consisting of long polysaccharide chains of N-acetylglucosamine and N-acetylmuramic acid cross-linked by shorter peptide chains.3 There are three stages to peptidoglycan formation, including accumulation of peptidoglycan precursors in the cytoplasm, linkage of precursor products in a long polymer, followed by cross-linking by transpeptidation. β-Lactams inhibit this final transpeptidation step.
Transpeptidation cross-links adjacent sugar chains via their pentapeptides. Peptidoglycan transglycosylase and D-alanyl-D-alanine transpeptidase are responsible for this activity. β-Lactams inhibit D-alanyl-D-alanine transpeptidase activity by acetylation, forming stable esters with the open lactam ring attached to the enzyme’s active site. The propensity of D-alanyl-D-alanine trans- and carboxypeptidase to form stable bonds with β-lactams provides these enzymes with their collective name of penicillin-binding proteins (PBPs).3 PBPs lie on the outer side of the cytoplasmic membrane in gram-positive bacteria and are shielded only by the peptidoglycan and outer capsule. In gram-negative bacteria, most β-lactams must cross the outer membrane via porin channels to reach PBPs. Entry through the porin channels is determined by size, charge, and hydrophobicity.
Bacterial killing and clinical efficacy for β-lactam antibiotics is associated with the percent of time during the dosing interval that the drug concentration is above the minimum inhibitory concentration (MIC). Maximal killing occurs when the antibiotic concentration is maintained at 4 to 5 times the MIC. Carbapenems have faster killing rates than penicillins; cephalosporins have the slowest killing rates of the β-lactam class.4 Therefore, percentages for time above the MIC required for bacterial killing are highest for the cephalosporins and lowest for the carbapenems.4 Near-maximal bactericidal effect is typically observed when the free drug serum concentration exceeds the MIC for 60% to 70% of the dosing interval for cephalosporins, 50% for penicillins, and 40% for carbapenems. In vitro data in an experimental Pseudomonas aeruginosa aortic endocarditis model in rabbits suggested that bacterial resistance to β-lactams may develop if the antibiotic concentration falls below the MIC for more than half the dosing interval.4
All intravenous (IV) β-lactam antibiotics are recommended to be given in several daily intervals. Administering a β-lactam agent as an infusion for longer than the conventional 30- to 60-minute infusion produces a lower peak concentration of the drug while maintaining a serum drug concentration in excess of the pathogen MIC for a longer period of time. Continuous infusion of these agents is also an attractive administration method to maintain serum drug concentrations above the MIC. Several clinical trials have validated the use of extended infusion and continuous infusion β-lactams in the critically ill, and institutions may institute these administration methods to improve outcomes and reduce daily drug costs.5–10
 Mechanisms of Resistance
Mechanisms of Resistance
Bacteria resist the cytotoxic activity of the β-lactams by modifying the normal PBPs, bypassing the normal PBPs, reducing the permeability of drug through the outer membrane (gram-negative bacteria), actively removing drug from the cell through the efflux pump mechanism, and producing β-lactamases. PBP modification and bypassing of normal PBPs are the most important mechanisms of resistance in gram-positive cocci, but β-lactamases are important mechanisms of antibiotic resistance in gram-negative bacteria.11
Alteration of PBPs, including decreased expression of PBPs and structural modifications to the PBPs to decrease antibiotic binding affinity, are seen in both gram-positive and gram-negative bacteria.11 In gram-positive bacteria, altered PBPs occur commonly in Streptococcus pneumoniae, Enterococcus faecium, and Staphylococcus aureus. Genes encoding these PBP changes in S. pneumoniae contain segments from several different organisms, including the viridans streptococci.12 In S. aureus and E. faecium, novel PBPs may be inducible through exposure to certain antibiotics.13,14 These novel PBPs have a low affinity for β-lactam antibiotics. PBP alterations are best illustrated in methicillin-resistant S. aureus (MRSA). Methicillin resistance occurs through the actions of the mecA gene that encodes PBP2′ (PBP2a). MRSA produces PBP2′ as a fifth PBP in addition to the four PBPs found in all S. aureus strains.15 β-Lactam antibiotics have very low affinity for PBP2′, so the enzyme’s function continues even in the presence of β-lactams.
Gram-negative bacteria, including Neisseria meningitides, Haemophilus influenzae, and Escherichia coli, also produce altered PBPs.11,16–19 Imipenem resistance due to altered PBPs has been reported in P. aeruginosa, Acinetobacter baumannii, and Proteus mirabilis, although this PBP alteration is not the primary mechanism responsible for most imipenem resistance.20–22
β-Lactamase production is largely responsible for β-lactam antibiotic resistance among gram-negative bacteria in the critical care setting. β-Lactamase hydrolyzes the β-lactam ring structure within the antibiotic molecule, rendering the drug inactive. Most β-lactamases function by a serine ester hydrolysis mechanism, but a few use a zinc ion to attack the β-lactam ring.11 β-Lactamase can be chromosomal (inherent within the chromosome of the organism) or can be encoded by plasmids or transposons, which are mobile genetic elements that can carry genes for resistance mechanisms. β-Lactamase production may be constitutive or inducible, and β-lactam antibiotics vary in their ability to induce β-lactamase production.23,24 Penicillin G, ampicillin, cefoxitin, imipenem, clavulanate, and first-generation cephalosporins are strong β-lactamase inducers.24 Third-generation cephalosporins, ureidopenicillins, aztreonam, and semisynthetic penicillinase-stable penicillins are weak β-lactamase inducers.24
Some measure of β-lactamase stability can be achieved through addition to the β-lactam ring of a substituent that hinders hydrolysis.25 For example, the semisynthetic penicillinase-stable drugs such as oxacillin and nafcillin remain active against methicillin-susceptible S. aureus because of this ring structure manipulation. β-Lactamase stability has been difficult to achieve in compounds with activity against gram-negative bacteria and may be due to the periplasmic location of β-lactamase in the gram-negative cell structure.11 Antibiotics including the β-lactams have difficulty accessing the gram-negative cell wall owing to the presence of an outer membrane. Porins within the membrane permit limited access through to the peptidoglycan layer of the cell, but the periplasmic space between the membrane and peptidoglycan layer allows β-lactamase to overwhelm the limited concentrations of drug that enter.
Third-generation cephalosporins have activity against β-lactamase-producing Enterobacteriaceae because they do not induce enzyme synthesis. However, these drugs may select spontaneous “derepressed” mutants that constitutively produce β-lactamase.11 Emergence of derepressed mutants of Enterobacter spp. during third-generation cephalosporin therapy may be significant, particularly in pneumonia and bacteremia.26 Through this selective pressure, organisms have developed that overproduce their chromosomal AmpC (class C) β-lactamase.27 This type of β-lactamase is broad spectrum and inactivates most cephalosporins and aztreonam. AmpC resistance has been demonstrated in many clinically important gram-negative bacteria, including Acinetobacter spp., Citrobacter freundii, Enterobacter spp., E. coli, Morganella morganii, P. aeruginosa, and Serratia marcescens.26,27 AmpC β-lactamase is not inhibited by β-lactamase inhibitors such as clavulanic acid, sulbactam, or tazobactam.27 Unfortunately, these chromosomal AmpC β-lactamases have been found on plasmids worldwide, suggesting that this broad-spectrum class of enzymes may be spread much more readily in clinical settings.27
Enterobacter spp. are intrinsically resistant to aminopenicillins, cefazolin, and cefoxitin due to production of constitutive chromosomal AmpC β-lactamases, which hydrolyze third-generation or expanded spectrum cephalosporins, and are resistant to inhibition by clavulanate or other β-lactamase inhibitors.26 β-Lactam antibiotic exposure drives AmpC-mediated resistance, leading to development of resistance to third-generation cephalosporins and mutations that may result in permanent enzyme hyperproduction. Exposure of Enterobacter organisms to third-generation cephalosporins may select for mutant strains associated with hyperproduction of AmpC β-lactamase.26
Other plasmid-mediated β-lactamases with more limited hydrolytic capacity have been found in Klebsiella pneumoniae, E. coli, Enterobacter spp., and other common Enterobacteriaceae. These so-called extended-spectrum β-lactamases (ESBL) are active against the oxyiminocephalosporins and aztreonam but not 7-α-methoxycephalosporins (cefoxitin, cefotetan) and are blocked by clavulanic acid, sulbactam, and tazobactam.28 There are numerous reports of outbreaks of ESBL-producing Klebsiella and Enterobacter infections in ICUs.28,29–35 Most organisms producing AmpC and ESBL enzymes remain susceptible to carbapenems such as imipenem. However, β-lactamase that uses zinc as an active site for β-lactam hydrolysis is able to hydrolyze carbapenems along with every other β-lactam presently available.36 Carbapenemases found in Enterobacteriaceae can be either metallo-β-lactamases, expanded-spectrum oxacillinases, or clavulanic acid–inhibited β-lactamases. The most concerning carbapenemases prevalent worldwide today are the K. pneumoniae carbapenemase (KPC) enzymes, a group of mostly plasmid-encoded enzymes from K. pneumoniae. Klebsiella pneumoniae carbapenemase enzymes hydrolyze all β-lactam antibiotics including penicillins, cephalosporins, and aztreonam, although cephamycins and ceftazidime are weakly hydrolyzed.37 The KPC enzymes may be mistaken for extended-spectrum β-lactamases (ESBLs), since they also hydrolyze expanded-spectrum cephalosporins, but unlike extended-spectrum β-lactamases, they also weakly hydrolyze carbapenems. The hydrolytic activity of KPC enzymes is not sufficient to produce resistance against carbapenems, but increases in MICs can occur.
 Penicillins
Penicillins
The microbiological activity of the penicillins is shown in Tables 120-2 to 120-4. Natural penicillins are most active against non-β-lactamase-producing gram-positive aerobic and anaerobic bacteria as well as selected gram-negative cocci such as Neisseria spp. Penicillin G is effectively the only natural penicillin used in the critical care setting. Gram-positive bacteria inhibited by natural penicillins are generally more susceptible to these penicillins than to semisynthetic penicillins. Penicillin and ampicillin remain the drugs of choice for enterococcal infections, but resistance to ampicillin among enterococcal isolates in North America is nearly 20%.60 Semisynthetic penicillins (oxacillin, nafcillin) are the agents of choice for penicillin-resistant S. aureus and Staphylococcus epidermidis, because penicillins exhibit faster bactericidal activity and improved clinical outcomes when compared with vancomycin.61,62 Semisynthetic penicillins should be reserved for staphylococcal infections, even though they are active against streptococci. Methicillin is seldom used because of an associated higher incidence of interstitial nephritis than oxacillin or nafcillin. Nafcillin and oxacillin have similar antistaphylococcal activity and can be used interchangeably for this indication.
TABLE 120-2 Microbiological Activity of Beta-Lactam Antibiotics Against Aerobic Gram-Positive Bacteria*
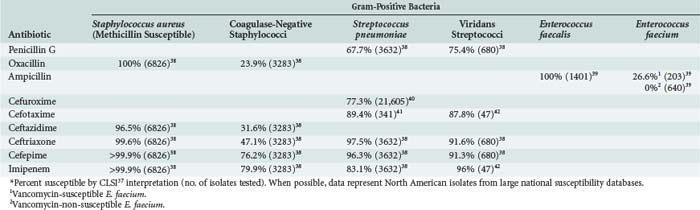
TABLE 120-3 Microbiological Activity of Beta-Lactam Antibiotics Against Aerobic Gram-Negative Bacteria*
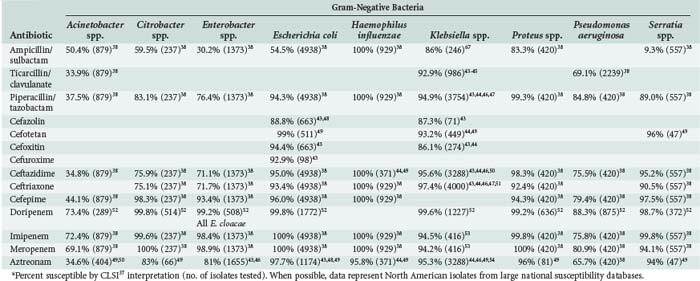
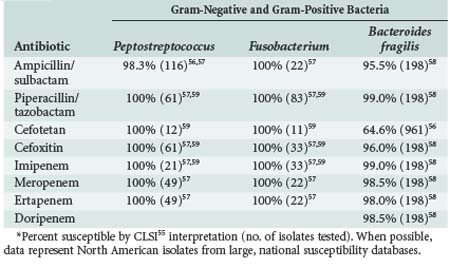
TABLE 120-4 Microbiological Activity of Beta-Lactam Antibiotics Against Anaerobic Gram-Positive and Gram-Negative Bacteria*
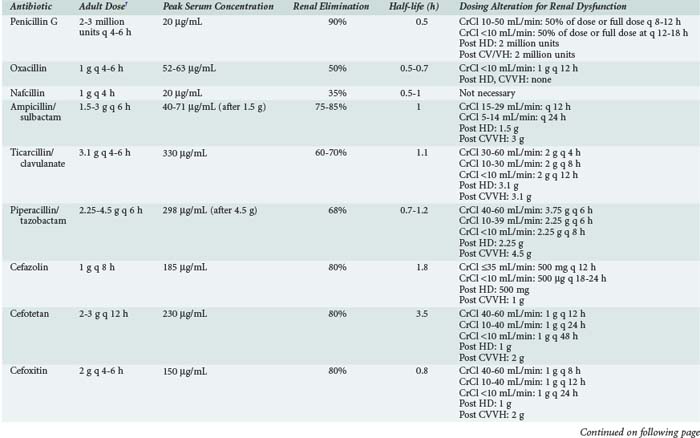
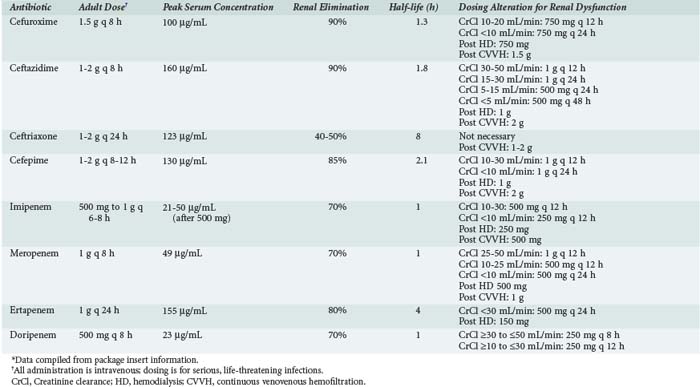
The pharmacokinetics of the penicillins and their dosing guidelines and administration are shown in Table 120-5. The pharmacokinetics of these agents has not been well investigated in critically ill patients, so extrapolation from healthy volunteers and less acutely ill patients is required. When penicillin was first available in the 1940s, the drug was administered as a continuous infusion to treat bacterial endocarditis. Nearly 70 years later, there is a resurgence of interest in using a continuous infusion or extended infusion of a β-lactam to improve bacterial killing activity and reduce development of resistance.
Piperacillin has been well studied (with and without tazobactam) as a continuous infusion. Several studies have demonstrated improved clinical cure rates and reduced overall drug exposure and drug costs compared to traditional intermittent 30- to 60-minute infusions.63 Patients with ventilator-associated pneumonia (VAP) caused by gram-negative pathogens with MICs of 8 to 16 µg/mL demonstrated higher probability of clinical cure when piperacillin-tazobactam was administered by continuous compared with intermittent infusion.64
In a study of 194 seriously ill patients with P. aeruginosa infection, the use of piperacillin/tazobactam in an extended infusion period (4-hour infusion with doses administered every 8 hours) demonstrated reduced 14-day mortality for patients with high Acute Physiology and Chronic Health Evaluation [APACHE] II scores (>17) when compared to conventional 30-minute infusions (12.2% versus 31.6%; P < 0.04).65 Extended infusion may offer some advantages over continuous infusion. A continuous infusion requires a dedicated IV line or lumen of a catheter. This is not always practical in the critically ill, especially for patients who have limited IV access, patients who require multiple daily infusions, or in situations where drug compatibility concerns may occur. An extended infusion provides a period of time in which the IV line is available. For either administration method, intensive nursing attention is required to make sure the drug is delivered properly.
The most common adverse event with the penicillins is hypersensitivity reaction. The most frequent clinical presentations of such hypersensitivity reactions are maculopapular or urticarial rashes and angioedema, but severe reactions such as anaphylaxis can also occur. The avoidance of β-lactams based only on the clinical history may exclude the use of several effective and cost-effective agents. Clinical data in over 500 cases suggest that patients who report penicillin allergies are unlikely to experience hypersensitivity reactions if penicillin skin testing is negative.66 Although some critically ill patients may be anergic, Arroliga and associates demonstrated that 106 of 117 ICU patients with a history of nonanaphylactic penicillin allergy responded to histamine control as part of a penicillin skin testing protocol, and 105 (90%) tested negative for penicillin reaction.67,68 Cross-reactivity between penicillins and cephalosporins has been reported at anywhere between 0.1% and 10%.69 The true rate is probably closer to 1%.69 In most allergic reactions to cephalosporins, the side chain on the β-lactam ring is responsible for the hypersensitivity response.70 Because older cephalosporins have side chains similar to penicillin and may have contained penicillin contaminants, the rates of cross-reactivity reported with early use of the cephalosporins were high.70 Cross-reactivity between penicillin and carbapenems is low, but this may reflect the low underlying rate of reaction with rechallenge of penicillin rather than a lack of cross-reactivity. Aztreonam is unlikely to elicit a reaction in penicillin-allergic individuals, owing to the unique side chain structure on the monobactam chemical.
Robinson and associates have published an approach to the treatment of patients with a possible or probable β-lactam allergy.70 For patients with a history suggestive of hypersensitivity to β-lactams, such as urticarial rash, pruritus, angioedema, hyperperistalsis, bronchospasm, hypotension, or arrhythmia, a penicillin skin test should be performed before initiating therapy. If the test is negative, β-lactam therapy can be started. Patients who react to the test should avoid β-lactams or undergo desensitization.
Penicillin is rarely used in the ICU except for treatment of meningococcal meningitis, tertiary syphilis, streptococcal endocarditis, or streptococcal necrotizing fasciitis. The semisynthetic penicillins are indicated for nonurinary methicillin-susceptible staphylococcal infections. Ampicillin/sulbactam is useful for a variety of infections in the critically ill, including urinary tract infections, community-acquired respiratory tract infections, meningitis, endocarditis, biliary infections, skin and skin structure infections, and intraabdominal infections. Piperacillin/tazobactam and ticarcillin/clavulanate are workhorse agents for many infections that arise in the critically ill, including pneumonia, bacteremia, urinary and biliary tract infections, intraabdominal infections, and skin and skin structure infections. These agents can be used alone, but growing resistance problems dictate that empirical combination therapy with aminoglycosides or fluoroquinolones may be optimal until culture data are available. Piperacillin/tazobactam has maintained activity against P. aeruginosa in recent years, but trends toward slightly decreased susceptibility have been noted across the United States.71
 Cephalosporins
Cephalosporins
The microbiological activity of the cephalosporins is shown in Tables 120-2 to 120-4. Only parenteral cephalosporins are useful in the critical care setting, because higher serum and tissue concentrations are required for serious infections, and these can be achieved only through parenteral administration. The cephalosporins can be divided into generations based on their microbiological activity. Cefazolin is effectively the only parenteral first-generation cephalosporin in general use, although cephapirin and cephradine are also marketed in North America. Cefazolin has activity against methicillin-susceptible S. aureus and coagulase-negative staphylococci but may be susceptible to staphylococcal β-lactamase. Cefazolin is also active against most streptococci, but all cephalosporins lack clinically useful activity against the enterococci. Cefazolin activity against gram-negative bacteria is limited to Moraxella catarrhalis, E. coli, P. mirabilis, K. pneumoniae, Salmonella spp., and Shigella spp.
The second-generation cephalosporins may be divided by their anaerobic activity, with cefoxitin and cefotetan (cephamycins) active against most gram-negative anaerobic organisms, including Prevotella spp., Fusobacterium spp., and B. fragilis. Cephamycins have less gram-positive potency than the first-generation cephalosporins but increased activity against Enterobacteriaceae, such as M. morganii, Proteus vulgaris, Providencia spp., and S. marcescens. Cefoxitin is a potent inducer of chromosomally mediated β-lactamases.72 True parenteral second-generation cephalosporins include cefonicid and cefuroxime. Cefuroxime is stable to most β-lactamases produced by gram-negative bacilli and is more active against methicillin-susceptible staphylococci and streptococci than is cefazolin. Cefuroxime has good potency against H. influenzae and is effective against most typical community-acquired respiratory tract pathogens.
Eradication of ESBL-producing organisms with standard cefepime regimens (1-2 g every 12 hours) is low due to the higher MICs of these bacteria. Cefepime pharmacodynamic exposure (time above MIC [T>MIC]) was determined for 18 patients with ESBL and non-ESBL infections using a published population pharmacokinetic model.73 Eradication was 80% when T>MIC was 50%, compared with 0% when T>MIC was less than 50% (P < 0.05), regardless of ESBL-production. Since median cefepime MICs for ESBL-producing isolates are generally several-fold higher than non-ESBL-producing isolates, higher doses of 4 to 6 grams administered IV as a continuous infusion, or 2 grams IV every 6 to 8 hours with a 4-hour infusion, are required to optimize therapy against ESBL-producing organisms that still retain cefepime susceptibility. It is important to note that critically ill patients, such as after trauma, may have an increased glomerular filtration rate or increased apparent volume of distribution, making the ability to optimize the time exceeding the MIC less likely.
Ceftobiprole is an investigational agent and the first of a new generation of cephalosporins with activity against MRSA. Potential applications for compounds such as ceftobiprole may include many infections commonly observed in critically ill patients. Ceftobiprole is an extended-spectrum β-lactam with in vitro activity against many gram-positive, gram-negative, and anaerobic bacteria.74,75 Ceftobiprole’s MRSA activity is due to its strong affinity for PBP2a and PBP2x, which are responsible for resistance in staphylococci and streptococci, respectively.74 Its activity against gram-positive bacteria includes S. aureus (methicillin-resistant, vancomycin-intermediate, and resistant strains), methicillin-resistant S. epidermidis, penicillin-susceptible and -resistant S. pneumoniae, and Enterococcus (ampicillin-susceptible E. faecalis and E. faecium as well as vancomycin-resistant E. faecalis).74–76 Studies have reported that ceftobiprole has a MIC for methicillin-susceptible S. aureus ranging from less than 0.12 to 1 µg/mL (MIC required to kill 90% of organisms [MIC90], 0.5 µg/mL) and for MRSA ranging from 0.25 to 4 µg/mL (MIC90, 1 µg/mL).77
The pharmacokinetics of the cephalosporins and their dosing guidelines and administration are shown in Table 120-5. Most cephalosporins have short half-lives and undergo extensive renal elimination. Cefoperazone and ceftriaxone, with significant biliary excretion, do not require dosing adjustments in renal dysfunction. The half-life of cefotaxime is not significantly increased in patients with renal failure; however, its active metabolite, desacetylcefotaxime, accumulates significantly, and thus dosing adjustments are required.
Pharmacokinetics in the critically ill have been studied for ceftazidime, ceftriaxone, and cefepime. Ceftazidime volume of distribution (VD) and terminal half-life were increased in critically ill patients without renal dysfunction.78,79 Ceftazidime area under the concentration time curve (AUC) was increased 1.8-fold, clearance was increased 1.3-fold, VD was increased 4.1-fold, and half-life was increased from 1.8 hours to 4.75 hours.80 This expansion of the VD may lead to inadequate serum concentrations throughout the dosing interval with intermittent bolus dosing.80 Continuous infusion of ceftazidime, 60 mg/kg/d, in trauma patients was shown to maintain serum concentrations at well above the MIC90 for most ICU pathogens.5 Continuous infusion of ceftazidime, 3 g/d, has been effective in treating nosocomial pneumonia, and the dose of drug administered is typically less than that required for intermittent bolus dosing.78
Ceftriaxone clearance in critically ill patients correlates to the degree of glomerular filtration function and is typically halved, even in patients with normal renal function.54 VD is also increased by up to 90% in the critically ill, possibly resulting in suboptimal serum concentrations with daily dosing of 2 g.54 Cefepime VD is also expanded in the critically ill, with a delay in renal clearance resulting in serum trough concentrations below the MIC50 for many P. aeruginosa isolates with 2-g, every-12-hour dosing.81 Pharmacokinetic modeling suggests that shorter dosing intervals, such as 1 g every 4 hours, extended infusions (over 3-4 hours), or continuous infusion could be used to improve serum trough concentrations.81
Continuous infusion of cefuroxime has also been studied in critically ill patients after coronary artery bypass grafting.82 A continuous infusion of 3 g over 24 hours provided serum concentrations above the MIC for common ICU pathogens throughout the 24-hour dosing interval and prevented sternal wound infection in the 54 patients studied.82
Cefepime has been studied in adult critical care patients with ventilator-associated pneumonia. Thirty two patients treated with high-dose cefepime (2 g every 8 hours [3-h infusion] or a renal function-adjusted equivalent dose) were studied. The likelihood of 2 g every 8 hours (3-hour infusion) achieving free drug concentrations above the MIC for 50% of the dosing interval were 91.8%, 78.1%, and 50.3% for MICs of 8, 16, and 32 µg/mL, respectively.83 A recent study suggested that maintaining a T>MIC of 100% for cefepime or ceftazidime is associated with significantly greater clinical cure (82% versus 33%; P <0.002) and bacteriologic eradication (97% versus 44%; P <0.001) in patients with severe infections.84
 Carbapenems and Monobactams
Carbapenems and Monobactams
The carbapenems have a broad antibacterial spectrum of activity, including most aerobic and anaerobic gram-positive and gram-negative bacteria. They are useful for the treatment of infection due to gram-negative bacteria resistant to other antibiotics or to streamline complex polypharmacy. The microbiological activity of the carbapenems is shown in Tables 120-2 to 120-4.
Aztreonam, a monobactam, has broad aerobic gram-negative activity but lacks gram-positive activity or efficacy against anaerobes. Carbapenems are not active against MRSA. Imipenem and doripenem have clinically useful potency against most enterococci, but meropenem and ertapenem have significantly less activity against these bacteria. Doripenem is active against MRSA, but breakpoints have not been established against this organism. Against 22,389 oxacillin-susceptible S. aureus isolates, doripenem inhibited 100% of all strains at ≤4 mcg/ml.85 Against 16,515 MRSA isolates, doripenem inhibited 59.3% at ≤4 µg/mL, and 69.3% at ≤8 µg/mL.85
Meropenem and ertapenem are less active against gram-positive aerobic bacteria than are imipenem and doripenem. Ertapenem, meropenem, and doripenem are more active than imipenem against Enterobacteriaceae. Doripenem has more potent activity against P. aeruginosa than any other carbapenem. Imipenem and meropenem have similar activity against P. aeruginosa and Acinetobacter spp., but ertapenem has no activity against important nonfermenting gram-negative rods, including P. aeruginosa, S. maltophilia, and Acinetobacter spp. Aztreonam generally has activity against P. aeruginosa, but ceftazidime is usually twice as active.86 The carbapenems have potent activity against gram-positive and gram-negative anaerobic bacteria (see Table 120-4).
Carbapenems are generally stable against most β-lactamases; however, metalloenzymes that can hydrolyze the carbapenem ring are increasing in the ICU setting.87 The most concerning carbapenemases prevalent worldwide today are the KPC enzymes, a group of mostly plasmid-encoded enzymes from K. pneumoniae. Klebsiella pneumoniae carbapenemase enzymes hydrolyze all β-lactam antibiotics including penicillins, cephalosporins, and aztreonam, although cephamycins and ceftazidime are weakly hydrolyzed. The KPC enzymes may be mistaken for ESBLs, since they also hydrolyze expanded-spectrum cephalosporins, but unlike extended spectrum β-lactamases, they also weakly hydrolyze carbapenems. The hydrolytic activity of KPC enzymes is not sufficient enough to produce resistance against carbapenems, but increases in MICs can occur. To achieve full resistance to carbapenems, organisms must also exhibit impaired outer membrane permeability.
Carbapenems are stable to ESBLs, but aztreonam is not. Carbapenems are also affected by multidrug efflux pumps and porin channel changes, particularly in P. aeruginosa. Imipenem is not affected by the common efflux pump mediated by MexA-MexB-OprM.88 However, imipenem readily selects resistant mutants of P. aeruginosa that lack a crucial porin channel (OprD) necessary for bacterial permeability to carbapenems but not other β-lactamase drugs.88 Loss of this porin channel produces imipenem MICs for P. aeruginosa of 8 to 32 mg/mL, conferring clinical resistance.88 Meropenem is recognized and ejected by the Mex-B-mediated efflux pump, as well as being affected by the loss of the OprD porin channel.88 Although either mechanism produces a threefold rise in meropenem MICs, neither mutation alone produces clinical resistance to meropenem. Rather, the combination of resistance mechanisms is required to preclude meropenem’s clinical effectiveness.
The pharmacokinetics of the carbapenems and aztreonam are shown in Table 120-5. Ertapenem is highly protein bound and has a 4-hour half-life, compared with 1 hour for doripenem, imipenem, and meropenem. Consequently, it is administered once daily. In critically ill patients, imipenem, meropenem, and doripenem have an expanded VD and prolonged half-life.89–91 Similar changes were observed for critically ill patients receiving aztreonam.89 These data suggest that trough concentrations of carbapenems and aztreonam may be low in critically ill patients, and aggressive dosing may be warranted to minimize treatment failure and drug resistance. For carbapenems, extended infusions (2-6 hours) of intermittent dosing may also be advantageous. When MICs are low, either intermittent or extended-infusion dosing strategies may be effective. However, with higher MICs, as are often observed in ICU-related infections, extended infusion dosing may be advantageous. A recent large clinical trial supported this concept, with clinical cure rates for doripenem administered as a 4-hour infusion greater than for imipenem administered as a 30-minute infusion in patients with P. aeruginosa infections (16/20 [80%] versus 6/14 [42.9%], respectively).92
Imipenem is metabolized extensively by renal dehydropeptidase-1 (DHP-1), producing nephrotoxic metabolites that can produce proximal tubular necrosis. Cilastatin is a competitive inhibitor of DHP-1 that results in protection against the toxic metabolites of imipenem and increases the imipenem urine delivery to approximately 70%.93 Doripenem, meropenem, and ertapenem do not require cilastatin coadministration. Imipenem has proconvulsive activity when administered in higher doses (4 g/d) or to patients with significant renal dysfunction in whom the drug may accumulate.
Key Points
Lodise TP, Lomaestro BM, Drusano GL. Application of antimicrobial pharmacodynamic concepts into clinical practice: focus on beta-lactam antibiotics: insights from the Society of Infectious Diseases Pharmacists. Pharmacotherapy. 2006;26:1320-1332.
Ramphal R, Ambrose PG. Extended-spectrum beta-lactamases and clinical outcomes: current data. Clin Infect Dis. 2006;42:S164-S172.
Roberts JA, Webb S, Paterson D, Ho KM, Lipman J. A systematic review on clinical benefits of continuous administration of beta-lactam antibiotics. Crit Care Med. 2009;37:2071-2078.
Livermore DM. Of Pseudomonas, porins, pumps and carbapenems. J Antimicrob Chemother. 2001;47:247-250.
Robinson JL, Hameed T, Carr S. Practical aspects of choosing an antibiotic for patients with a reported allergy to an antibiotic. Clin Infect Dis. 2002;35:26-31.
1 Yocum R, Waxman D, Strominger J. The mechanism of action of penicillin. J Biol Chem. 1980;255:3977-3986.
2 Koch AL. Penicillin binding proteins, beta-lactams, and lactamases: Offensives, attacks, and defensive countermeasures. Crit Rev Microbiol. 2000;26:205-220.
3 Tomasz A. Penicillin-binding proteins and the antibacterial effectiveness of the beta-lactam antibiotics. Rev Infect Dis. 1986;8:S270-S278.
4 Roberts JA, Paratz J, Paratz E, Krueger WA, Lipman J. Continuous infusion of beta-lactam antibiotics in severe infections: a review of its role. Int J Antimicrob Agents. 2007;30:11-18.
5 Nicolau DP, McNabb J, Lacy MK, et al. Continuous versus intermittent administration of ceftazidime in intensive care unit patients with nosocomial pneumonia. Int J Antimicrob Agents. 2001;17:497-504.
6 Patel GW, Patel N, Lat A, Trombley K, Enbawe S, Manor K, et al. Outcomes of extended infusion piperacillin/tazobactam for documented gram-negative infections. Diagn Microbiol Infect Dis. 2009;64:236-240.
7 Roberts JA, Kirkpatrick CM, Roberts MS, Robertson TA, Dalley AJ, Lipman J. Meropenem dosing in critically ill patients with sepsis and without renal dysfunction: intermittent bolus versus continuous administration? Monte Carlo dosing simulations and subcutaneous tissue distribution. J Antimicrob Chemother. 2009;64:142-150.
8 Wang D. Experience with extended-infusion meropenem in the management of ventilator-associated pneumonia due to multidrug-resistant Acinetobacter baumannii. Int J Antimicrob Agents. 2009;33:290-291.
9 Lee SY, Kuti JL, Nicolau DP. Cefepime pharmacodynamics in patients with extended spectrum beta-lactamase (ESBL) and non-ESBL infections. J Infect. 2007;54:463-468.
10 Reese AM, Frei CR, Burgess DS. Pharmacodynamics of intermittent and continuous infusion piperacillin/tazobactam and cefepime against extended-spectrum beta-lactamase-producing organisms. Int J Antimicrob Agents. 2005;26:114-119.
11 Livermore DM. Beta-lactamases- the threat renews. Curr Protein Pept Sci. 2009;10:397-400.
12 Hakenbeck R, Grebe T, Zahner D, Stock JB. Beta-lactam resistance in Streptococcus pneumoniae: penicillin-binding proteins and non–penicillin-binding proteins. Mol Microbiol. 1999;33:673-678.
13 Rossi L, Tonin E, Cheng YR, Fontana R. Regulation of penicillin-binding protein activity: description of a methicillin-inducible penicillin-binding protein in Staphylococcus aureus. Antimicrob Agents Chemother. 1985;27:828-831.
14 Fontana R, Canepari P, Lleo MM, Satta G. Mechanisms of resistance of enterococci to beta-lactam antibiotics. Eur J Clin Microbiol Infect Dis. 1990;9:103-105.
15 Hiramatsu K, Cui L, Kuroda M, Ito T. The emergence and evolution of methicillin-resistant Staphylococcus aureus. Trends Microbiol. 2001;9:486-493.
16 Ferreira RC, Costa SO, Ferreira LC, Trabulsi LR. Penicillin-binding proteins of pathogenic Escherichia coli strains. FEMS Microbiol Lett. 1998;168:313-317.
17 Hakenbeck R. Target-mediated resistance to beta-lactam antibiotics. Biochem Pharmacol. 1995;50:1121-1127.
18 Mendelman PM, Chaffin DO, Kalaitzoglou G. Penicillin-binding proteins and ampicillin resistance in Haemophilus influenzae. J Antimicrob Chemother. 1990;25:525-534.
19 Saez-Nieto JA, Lujan R, Berron S, et al. Epidemiology and molecular basis of penicillin-resistant Neisseria meningitidis in Spain: A 5-year history (1985-1989). Clin Infect Dis. 1992;14:394-402.
20 Bellido F, Veuthey C, Blaser J, et al. Novel resistance to imipenem associated with an altered PBP-4 in a Pseudomonas aeruginosa clinical isolate. J Antimicrob Chemother. 1990;25:57-68.
21 Gehrlein M, Leying H, Cullmann W, et al. Imipenem resistance in Acinetobacter baumannii is due to altered penicillin-binding proteins. Chemotherapy. 1991;37:405-412.
22 Neuwirth C, Siebor E, Duez JM, et al. Imipenem resistance in clinical isolates of Proteus mirabilis associated with alterations in penicillin-binding proteins. J Antimicrob Chemother. 1995;36:335-342.
23 Danziger LH, Pendland SL. Bacterial resistance to beta-lactam antibiotics. Am J Health Syst Pharm. 1995;52:S3-S8.
24 McManus MC. Mechanisms of bacterial resistance to antimicrobial agents. Am J Health Syst Pharm. 1997;54:1420-1433. quiz 1444-6
25 Frere JM. Beta-lactamases and bacterial resistance to antibiotics. Mol Microbiol. 1995;16:385-395.
26 Paterson DL. Resistance in gram-negative bacteria: Enterobacteriaceae. Am J Infect Control. 2006;34:S20-S28.
27 Philippon A, Arlet G, Jacoby GA. Plasmid-determined AmpC-type beta-lactamases. Antimicrob Agents Chemother. 2002;46:1-11.
28 Paterson DL, Bonomo RA. Extended-spectrum beta-lactamases: a clinical update. Clin Microbiol Rev. 2005;18:657-686. 30
29 Paterson DL. Serious infections caused by enteric gram-negative bacilli—mechanisms of antibiotic resistance and implications for therapy of gram-negative sepsis in the transplanted patient. Semin Respir Infect. 2002;17:260-264.
30 Vogelaers D, De Bels D, Forêt F, Cran S, Gilbert E, Schoonheydt K, Blot S. Patterns of antimicrobial therapy in severe nosocomial infections: empiric choices, proportion of appropriate therapy, and adaptation rates–a multicentre, observational survey in critically ill patients. Int J Antimicrob Agents. 2010;35:375-8131.
31 Cohen MJ, Anshelevich O, Raveh D, Broide E, Rudensky B, Yinnon AM. Acquisition of multidrug-resistant organisms among hospital patients hospitalized in beds adjacent to critically ill patients. Infect Control Hosp Epidemiol. 2006;27:675-681.
32 Gupta A. Hospital-acquired infections in the neonatal intensive care unit—Klebsiella pneumoniae. Semin Perinatol. 2002;26:340-345.
33 Kocazeybek BS. Antimicrobial resistance surveillance of gram-negative bacteria isolated from intensive care units of four different hospitals in Turkey: Evaluation of the prevalence of extended-spectrum and inducible beta-lactamases using different E-test strips and direct induction methods. Chemotherapy. 2001;47:396-408.
34 Pena C, Pujol M, Ardanuy C, et al. An outbreak of hospital-acquired Klebsiella pneumoniae bacteraemia, including strains producing extended-spectrum beta-lactamase. J Hosp Infect. 2001;47:53-59.
35 Rebuck JA, Olsen KM, Fey PD, et al. Characterization of an outbreak due to extended-spectrum beta-lactamase–producing Klebsiella pneumoniae in a pediatric intensive care unit transplant population. Clin Infect Dis. 2000;31:1368-1372.
36 Nordmann P, Cuzon G, Naas T. The real threat of Klebsiella pneumoniae carbapenemase producing bacteria. Lancet Infect Dis. 2009;9:228-236.
37 Clinical and Laboratory Standards Institute. M100-S17, Performance Standards for Antimicrobial Susceptibility Testing, 17th Informational Supplement. Wayne, PA: CLSI; 2007.
38 Fritsche TR, Sader HS, Jones RN. Antimicrobial activity of ceftobiprole, a novel anti-methicillin-resistant Staphylococcus aureus cephalosporin, tested against contemporary pathogens: results from the SENTRY Antimicrobial Surveillance Program (2005-2006). Diagn Microbiol Infect Dis. 2008;61:86-95.
39 Sader HS, Jones RN. Antimicrobial susceptibility of gram-positive bacteria isolated from US medical centers; results of the Daptomycin Surveillance Program (2007-2008). Diagn Microbiol Infect Dis. 2009;65:158-162.
40 Pottumarthy S, Fritsche TR, Jones RN. Comparative activity of oral and parenteral cephalosporins tested against multidrug-resistant Streptococcus pneumoniae: report from the SENTRY Antimicrobial Surveillance Program (1997-2003). Diagn Microbiol Infect Dis. 2005;51:147-150.
41 Pfaller MA, Jones RN, Doern GV, et al. Survey of blood stream infections attributable to gram-positive cocci: Frequency of occurrence and antimicrobial susceptibility of isolates collected in 1997 in the United States, Canada, and Latin America from the SENTRY Antimicrobial Surveillance Program. SENTRY Participants Group. Diagn Microbiol Infect Dis. 1999;33:283-297.
42 Kuriyama T, Karasawa T, Nakagawa K, et al. Bacteriology and antimicrobial susceptibility of gram-positive cocci isolated from pus specimens of orofacial odontogenic infections. Oral Microbiol Immunol. 2002;17:132-135.
43 Rennie RP, Jones RN, Mutnick AH. Occurrence and antimicrobial susceptibility patterns of pathogens isolated from skin and soft tissue infections: Report from the SENTRY Antimicrobial Surveillance Program (United States and Canada, 2000). Diagn Microbiol Infect Dis. 2003;45:287-293.
44 Hoban DJ, Biedenbach DJ, Mutnick AH, Jones RN. Pathogen of occurrence and susceptibility patterns associated with pneumonia in hospitalized patients in North America: Results of the SENTRY Antimicrobial Surveillance Study (2000). Diagn Microbiol Infect Dis. 2003;45:279-285.
45 Gordon KA, Beach ML, Biedenbach DJ, et al. Antimicrobial susceptibility patterns of beta-hemolytic and viridans group streptococci: Report from the SENTRY Antimicrobial Surveillance Program (1997-2000). Diagn Microbiol Infect Dis. 2002;43:157-162.
46 Jones RN, Biedenbach DJ, Gales AC. Sustained activity and spectrum of selected extended-spectrum beta-lactams (carbapenems and cefepime) against Enterobacter spp. and ESBL-producing Klebsiella spp.: Report from the SENTRY antimicrobial surveillance program (USA, 1997-2000). Int J Antimicrob Agents. 2003;21:1-7.
47 Jones RN, Varnam DJ. Antimicrobial activity of broad-spectrum agents tested against gram-negative bacilli resistant to ceftazidime: report from the SENTRY Antimicrobial Surveillance Program (North America, 2001). Diagn Microbiol Infect Dis. 2002;44:379-382.
48 Diekema DJ, Pfaller MA, Jones RN, et al. Trends in antimicrobial susceptibility of bacterial pathogens isolated from patients with bloodstream infections in the USA, Canada and Latin America. SENTRY Participants Group. Int J Antimicrob Agents. 2000;13:257-271.
49 Friedland I, Stinson L, Ikaiddi M, et al. Phenotypic antimicrobial resistance patterns in Pseudomonas aeruginosa and Acinetobacter: results of a multicenter intensive care unit surveillance study, 1995-2000. Diagn Microbiol Infect Dis. 2003;45:245-250.
50 Blondeau JM, Vaughan D. In vitro activity of 19 antimicrobial agents against 3513 nosocomial pathogens collected from 48 Canadian medical centres. The Canadian Antimicrobial Study Group. Int J Antimicrob Agents. 2000;15:213-219.
51 Jones RN, Biedenbach DJ. Comparative activity of garenoxacin (BMS 284756), a novel desfluoroquinolone, tested against 8,331 isolates from community-acquired respiratory tract infections: North American results from the SENTRY Antimicrobial Surveillance Program (1999-2001). Diagn Microbiol Infect Dis. 2003;45:273-278.
52 Pillar CM, Torres MK, Brown NP, Shah D, Sahm DF. In vitro activity of doripenem, a carbapenem for the treatment of challenging infections caused by Gram-negative bacteria, against recent clinical isolates from the United States. Antimicrob Agents Chemother. 2008;52:4388-4399.
53 Rhomberg PR, Jones RN. Summary trends for the Meropenem Yearly Susceptibility Test Information Collection Program: a 10-year experience in the United States (1999-2008). Diagn Microbiol Infect Dis. 2009;65:414-426.
54 Joynt GM, Lipman J, Gomersall CD, et al. The pharmacokinetics of once-daily dosing of ceftriaxone in critically ill patients. J Antimicrob Chemother. 2001;47:421-429.
55 Clinical and Standards Institute (CLSI). Methods for antimicrobial susceptibility testing of anaerobic bacteria; approved standard-seventh edition. M11-A7. Wayne PA: CLSI; 2007.
56 Snydman DR, Jacobus NV, McDermott LA, et al. Multicenter study of in vitro susceptibility of the Bacteroides fragilis group, 1995 to 1996, with comparison of resistance trends from 1990 to 1996. Antimicrob Agents Chemother. 1999;43:2417-2422.
57 Aldridge KE. Ertapenem (MK-0826), a new carbapenem: Comparative in vitro activity against clinically significant anaerobes. Diagn Microbiol Infect Dis. 2002;44:181-186.
58 Snydman DR, Jacobus NV, McDermott LA. In vitro activity od doripenem, a new broad-spectrum carbapenem against recently collected clinical anaerobic isolates, with emphasis on the Bacteroides fragilis group. Antimicrob Agents Chemother. 2008;52:4492-4496.
59 Citron DM, Appleman MD. Comparative in vitro activities of trovafloxacin (CP-99,219) against 221 aerobic and 217 anaerobic bacteria isolated from patients with intra-abdominal infections. Antimicrob Agents Chemother. 1997;41:2312-2316.
60 Mutnick AH, Biedenbach DJ, Jones RN. Geographic variations and trends in antimicrobial resistance among Enterococcus faecalis and Enterococcus faecium in the SENTRY Antimicrobial Surveillance Program (1997-2000). Diagn Microbiol Infect Dis. 2003;46:63-68.
61 Ackerman BH, Vannier AM, Eudy EB. Analysis of vancomycin time-kill studies with Staphylococcus species by using a curve stripping program to describe the relationship between concentration and pharmacodynamic response. Antimicrob Agents Chemother. 1992;36:1766-1769.
62 Gonzalez C, Rubio M, Romero-Vivas J, et al. Bacteremic pneumonia due to Staphylococcus aureus: A comparison of disease caused by methicillin-resistant and methicillin-susceptible organisms. Clin Infect Dis. 1999;29:1171-1177.
63 Patel GW, Patel N, Lat A, et al. Outcomes of extended infusion piperacillin/tazobactam for documented gram-negative infections. Diagn Micro Infect Dis. 2009;64:236-240.
64 Lorente L, Jiménez A, Martín MM, Iribarren JL, Jiménez JJ, Mora ML. Clinical cure of ventilator-associated pneumonia treated with piperacillin/tazobactam administered by continuous or intermittent infusion. Int J Antimicrob Agents. 2009;33:464-468.
65 Lodise TP, Lomaestro B, Drusano GL. Piperacillin-Tazobactam for Pseudomonas aeruginosa Infection: Clinical Implications of an Extended-Infusion Dosing Strategy. Clin Infect Dis. 2007;44:357-363.
66 Macy E, Mangat R, Burchette RJ. Penicillin skin testing in advance of need: Multiyear follow-up in 568 test result-negative subjects exposed to oral penicillins. J Allergy Clin Immunol. 2003;111:1111-1115.
67 Arroliga ME, Wagner W, Bobek MB, et al. A pilot study of penicillin skin testing in patients with a history of penicillin allergy admitted to a medical ICU. Chest. 2000;118:1106-1108.
68 Arroliga ME, Radojicic C, Gordon SM, et al. A prospective observational study of the effect of penicillin skin testing on antibiotic use in the intensive care unit. Infect Control Hosp Epidemiol. 2003;24:347-350.
69 Leviton I. Separating fact from fiction: the data behind allergies and side effects caused by penicillins, cephalosporins, and carbapenem antibiotics. Curr Pharm Des. 2003;9:983-988.
70 Robinson JL, Hameed T, Carr S. Practical aspects of choosing an antibiotic for patients with a reported allergy to an antibiotic. Clin Infect Dis. 2002;35:26-31.
71 Jones RN, Stilwell MG, Rhomberg PR, Sader HS. Antipseudomonal activity of piperacillin/tazobactam: more than a decade of experience from the SENTRY Antimicrobial Surveillance Program (1997-2007). Diagn Microbiol Infect Dis. 2009;65:331-334.
72 Moritz VA, Carson PB. Cefoxitin sensitivity as a marker for inducible beta-lactamases. J Med Microbiol. 1986;21:203-207.
73 Lee SY, Kuti JL, Nicolau DP. Cefepime pharmacodynamics in patients with extended spectrum b-lactamase (ESBL) and non-ESBL infections. J Infect. 2007;54:463-468.
74 Murthy B, Schmitt-Hoffmann A. Pharmacokinetics and pharmacodynamics of ceftobiprole, an anti-MRSA cephalosporin with broad-spectrum activity. Clin Pharmacokinet. 2008;47:21-33.
75 Anderson SD, Gums JG. Ceftobiprole: an extended spectrum anti-methicillin-resistant Staphylococcus aureus cephalosporin. Ann Pharmacother. 2008;42:806-816.
76 Fritsche TR, Sader HS, Jones RN. Antimicrobial activity of ceftobiprole, a novel anti-methicillin-resistant Staphylococcus aureus cephalosporin, tested against contemporary pathogens: results from the SENTRY Antimicrobial Surveillance Program (2005-2006). Diagn Microbiol Infect Dis. 2008;61:86-95.
77 Deresinski SC. Ceftobiprole: breaking therapeutic dogmas of the beta-lactam class. Diagn Microbiol Infect Dis. 2008;61:82-85.
78 Hanes SD, Wood GC, Herring V, et al. Intermittent and continuous ceftazidime infusion for critically ill trauma patients. Am J Surg. 2000;179:436-440.
79 Gomez CM, Cordingly JJ, Palazzo MG. Altered pharmacokinetics of ceftazidime in critically ill patients. Antimicrob Agents Chemother. 1999;43:1798-1802.
80 Young RJ, Lipman J, Gin T, et al. Intermittent bolus dosing of ceftazidime in critically ill patients. J Antimicrob Chemother. 1997;40:269-273.
81 Lipman J, Wallis SC, Rickard C. Low plasma cefepime levels in critically ill septic patients: pharmacokinetic modeling indicates improved troughs with revised dosing. Antimicrob Agents Chemother. 1999;43:2559-2561.
82 Pass SE, Miyagawa CI, Healy DP, Ivey TD. Serum concentrations of cefuroxime after continuous infusion in coronary bypass graft patients. Ann Pharmacother. 2001;35:409-413.
83 Nicasio AM, Ariano RE, Zelenitsky SA, Kim A, Crandon JL, Kuti JL, et al. Population pharmacokinetics of high-dose, prolonged-infusion cefepime in adult critically ill patients with ventilator-associated pneumonia. Antimicrob Agents Chemother. 2009;53:1476-1481.
84 McKinnon PS, Paladino JA, Schentag JJ. Evaluation of area under the inhibitory curve (AUIC) and time above the minimum inhibitory concentration (T>MIC) as predictors of outcome for cefepime and ceftazidime in serious bacterial infections. Int J Antimicrob Agents. 2008;31:345-351.
85 Fritsche TR, Sader HS, Stillwell MG, Jones RN. Antimicrobial activity of doripenem tested against prevalent Gram-positive pathogens: results from a global surveillance study (2003-2007). Diagn Micro Infect Dis. 2009;6:440-446.
86 Kucers ACS, Grayson ML, Hoy JF. Aztreonam. In: Kucers ACS, Grayson ML, Hoy JF, editors. The use of antibiotics; a clinical review of antibacterial, antifungal, and antiviral drugs. Avon, England: The Bath Press; 1997:385-394.
87 Miriagou V, Cornaglia G, Edelstein M, et al. Acquired carbapenemases in gram-negative bacterial pathogens: detection and surveillance issues. Clin Microbiol Infect. 2010;16:112-122.
88 Livermore DM. Of Pseudomonas, porins, pumps and carbapenems. J Antimicrob Chemother. 2001;47:247-250.
89 Thalhammer F, Traunmuller F, El Menyawi I, et al. Continuous infusion versus intermittent administration of meropenem in critically ill patients. J Antimicrob Chemother. 1999;43:523-527.
90 McKindley DS, Boucher BA, Hess MM, et al. Pharmacokinetics of aztreonam and imipenem in critically ill patients with pneumonia. Pharmacotherapy. 1996;16:924-931.
91 Van Wart SA, Andes DR, Ambrose PG, Bhavnani SM. Pharmacokinetic-pharmacodynamic modeling to support doripenem dose regimen optimization for critically ill patients. Diagn Microbiol Infect Dis. 2009;63:409-414.
92 Chastre J, Wunderink R, Prokocimer P, Lee M, Kaniga K, Friedland I. Efficacy and safety of intravenous infusion of doripenem versus imipenem in ventilator-associated pneumonia: a multicenter, randomized study. Crit Care Med. 2008;36:1089-1096.
93 Mouton JW, Touzw DJ, Horrevorts AM, Vinks AA. Comparative pharmacokinetics of the carbapenems: clinical implications. Clin Pharmacokinet. 2000;39:185-201.
