CHAPTER 49 Benign Tumors of the Sinonasal Tract
Significant refinements in imaging techniques and the widespread application of endoscopic surgery have led to revived interest in the management of benign tumors of the sinonasal tract. Not unexpectedly, this anatomic region may be involved by a large variety of different histopathologic entities that, according to the World Health Organization (WHO) classification, include epithelial tumors (papillomas and salivary gland–type adenomas), soft tissue tumors (myxoma, leiomyoma, hemangioma, Schwannoma, neurofibroma, meningioma), and tumors of bone and cartilage (giant cell lesion, giant cell tumor, chondroma, osteoma, chondroblastoma, chondromyxoid fibroma, osteochondroma, osteoid osteoma, osteoblastoma, ameloblastoma, nasal chondromesenchymal hamartoma).1 Although originating from the pterygomaxillary fossa, juvenile angiofibroma is habitually included in the group of sinonasal tract tumors because of its common presentation as a nasal or nasopharyngeal mass.
This chapter discusses in detail inverted papilloma, osteoma, and juvenile angiofibroma, which are the most common histologic types seen in our series of 635 benign tumors of the sinonasal tract encountered over 15 years at two University Hospitals (Table 49-1). We also provide information on other benign tumors of less common observation.
Inverted Papilloma
Inverted papilloma (or Schneiderian papilloma, inverted type), which is included in the group of sinonasal papillomas together with the oncocytic and exophytic variants, is the second most common benign tumor of the sinonasal tract after osteoma, even though it represents the most common surgical indication for a benign sinonasal tumor. The lesion is estimated to represent 0.4% to 4.7% of all surgically removed nasal tumors, with an incidence ranging from 0.6 to 1.5 cases per 100,000 inhabitants per year.2,3 Men are more commonly affected than women, and the lesion is prevalently observed in the fifth and sixth decades of life.
With regard to the histologic appearance, inverted papilloma is composed exclusively or almost exclusively of hyperplastic ribbons of basement membrane–enclosed epithelium that grow endophytically into the underlying stroma. The epithelium is multilayered and formed of squamous or ciliated columnar cells mixed with mucocytes.4
The association of inverted papilloma with squamous cell carcinoma has been overemphasized, with reported frequencies as high as 56%.5 Recent data clearly show that the prevalence varies between 3.4%6 and 9.7%7 and that synchronous occurrence is more common than metachronous.
Inverted papilloma is suspected to have a viral etiology. Human papillomavirus DNA has been demonstrated by in situ hybridization or the polymerase chain reaction in papillomas, with a prevalence of serotypes 6, 11, 16, and 18.8,9 The last two serotypes have been specifically found to be associated with inverted papillomas that show histologic signs of malignant transformation. Not unexpectedly, in this setting an increase in levels of epidermal growth factor receptor and tumor growth factor-α has been demonstrated.10
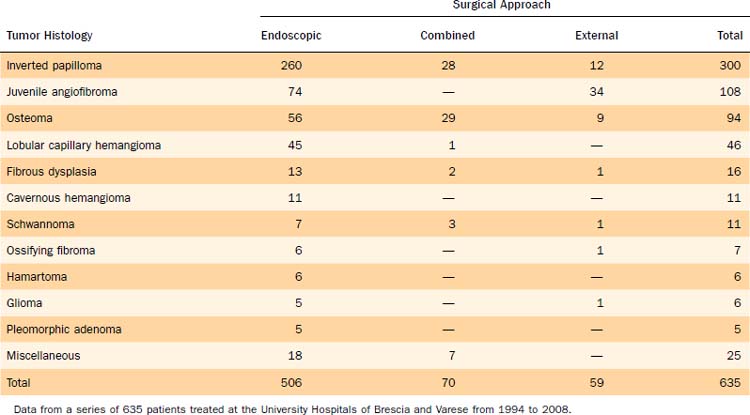
Unilateral nasal obstruction with watery rhinorrhea is the most common symptom prompting the patient to seek otolaryngologic consultation, whereas epiphora, proptosis, diplopia, and headache may be associated with advanced lesions involving the orbit or the skull base. Endoscopy of the nose, usually showing a pale, polypoid lesion with a papillary appearance protruding from the middle meatus (Fig. 49-1), may easily suggest the diagnosis, which is sometimes made less obvious by the concomitant presence of inflammatory polyps. A biopsy performed under endoscopic guidance is indicated to establish the histologic diagnosis.
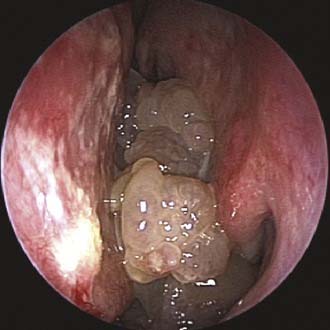
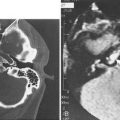
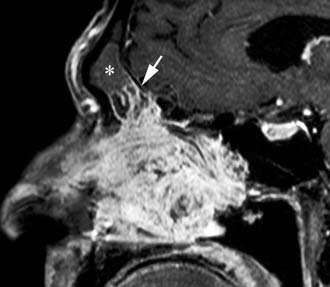
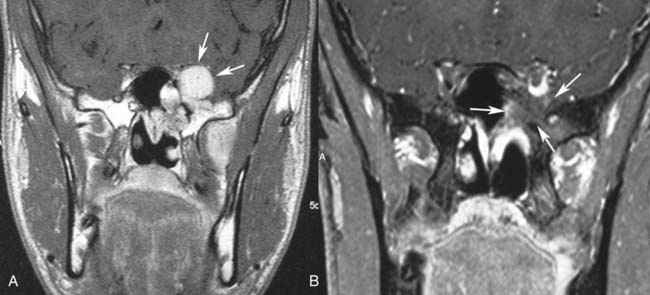
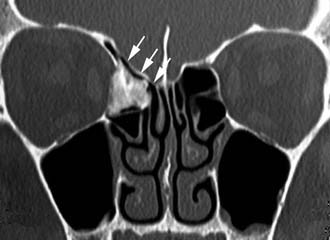
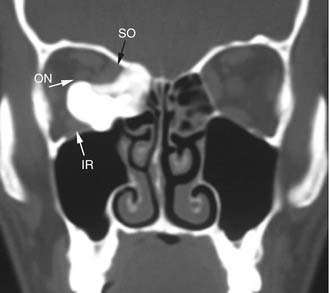
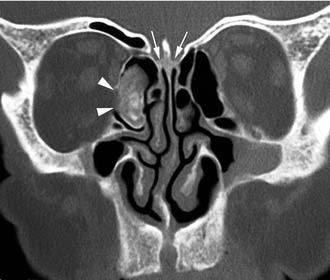
Imaging studies are required to assess the extent and three-dimensional configuration of the lesion and to disclose its relationship with surrounding structures (i.e., orbit, skull base, optic nerve, internal carotid artery). In our experience, these goals are best achieved by MRI with gadolinium enhancement, which also has the advantage over CT of better differentiating tumor from inflammatory mucosal changes and to disclose the so-called cerebriform-columnar pattern (Fig. 49-2). This reflects the histologic arrangement of inverted papilloma characterized by the alternation of regular parallel folds made of a highly cellular metaplastic epithelium and of an underlying less cellular stroma. It is therefore highly predictive for inverted papilloma diagnosis.11 However, even MRI has limitations, especially in lesions completely filling the maxillary, sphenoid, or frontal sinus, in differentiating inverted papillomas growing inside the sinus but arising from a small area of insertion from those extensively involving the mucosa. Recent studies indicate that focal hyperostosis12 and osteitic changes13 upon CT may be considered reliable predictors of tumor origin. These findings may also be identified by MRI (Fig. 49-3).
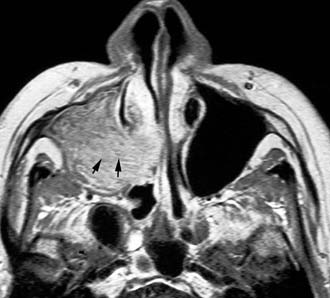
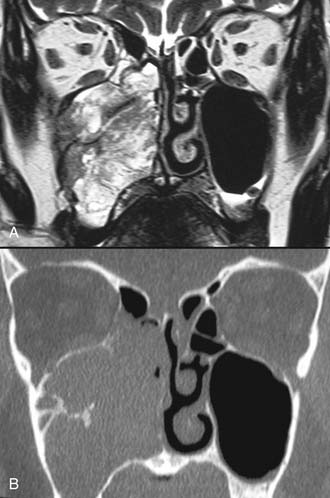
As clearly shown by a 2006 meta-analysis14 and by our experience based on 300 inverted papillomas, endoscopic surgery is a reliable alternative to traditional external techniques for the vast majority of lesions. An exclusive endoscopic approach may be contraindicated in the following situations: (1) massive involvement of the mucosa of the frontal sinus and/or of a supraorbital cell, (2) intradural extension or transorbital extension, both of which are very uncommon situations usually found in patients who have already undergone one or more surgical procedures, (3) concomitant presence of a malignancy involving critical areas, and (4) presence of abundant scar tissue from previous surgery.
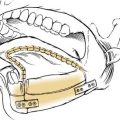
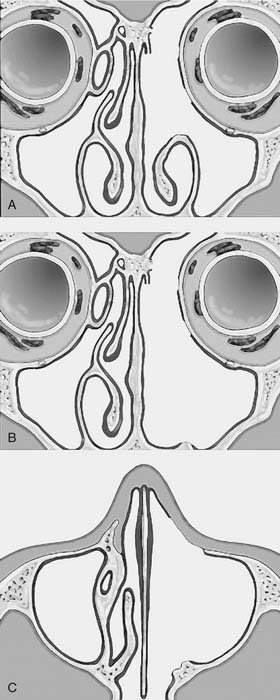
One criticism of endoscopic approaches is the impossibility in always obtaining an en bloc resection. However, it is not the concept of en bloc resection per se that has to be fulfilled to achieve complete removal; the key point is to dissect the involved mucosa along the subperiosteal plane and to drill the underlying bone whenever required by imaging and/or intraoperative findings.15 The extent of the operation is dictated by the site of the lesion and the area of mucosa involved by the lesion. Apart from cases with a clearly identifiable small attachment of the lesion, which can be managed with a very conservative approach,16 there are basically three different types of endoscopic resections available, according to our classification.17 Type I resection (Fig. 49-4A) is indicated for inverted papillomas involving the middle meatus, ethmoid, superior meatus, sphenoid sinus, or a combination of these structures; even lesions protruding into the maxillary sinus without direct involvement of the mucosa are amenable to this approach. Type II resection (Fig. 49-4B), which corresponds to an endoscopic medial maxillectomy, is indicated for tumors originating within the nasoethmoidal complex and secondarily extending into the maxillary sinus or for primary maxillary lesions not involving the anterior and lateral walls of the sinus itself. The nasolacrimal duct can be included in the specimen to increase the exposure of the anterior part of the maxillary sinus. Type III resection (Fig. 49-4C), also known as the Sturman-Canfield operation or endonasal Denker operation,18 entails removal of the medial portion of the anterior wall of the maxillary sinus to enable access to all the antrum walls. It is therefore recommended for inverted papillomas extensively involving the anterior compartment of the maxillary sinus.
Frontal sinus involvement can be observed in different scenarios, ranging from a limited lesion marginally growing from the ethmoid into the frontal recess (Fig. 49-5) to very complex cases in which most or all of the sinus mucosa is diseased. Because the key point for ultimate success is removal of the lesion along the subperiosteal plane with the possibility to drill out the underlying bone, the surgical choice may involve a Draf IIB or Draf III endoscopic sinusotomy or a combination of an endoscopic approach with external sinusotomy through an osteoplastic flap depending on the extent of frontal sinus mucosa involvement, a finding that can usually be definitively assessed only at the time of surgery. Even the involvement of an extensively pneumatized supraorbital cell is frequently challenging. Exposure through the nose may be increased by coagulation and sectioning of the anterior ethmoid artery and by displacement of the orbit after drilling of the upper portion of the lamina papyracea. However, whenever the lesion involves a cell that extends far posteriorly and/or laterally over the orbit and complete resection cannot be achieved transnasally, the surgeon should resort to a frontal osteoplastic flap.
In the era when transnasal resection without endoscopic or microscopic assistance was the most commonly used technique, the rate of recurrences ranged from 40%19 to 78%.20 These extremely high values indicate that these “recurrences,” mostly occurring at the site of primary resection, should have been more appropriately regarded as residual lesions. They reflected the inadequacy of transnasal surgery in affording a radical excision of the lesion. In view of such limitations, medial maxillectomy through lateral rhinotomy was established in the 1970s and 1980s as the gold standard for treatment of inverted papilloma. Although the frequency of recurrences decreased, reportedly from 0%21 to 29%,22 this technique was associated with potential aesthetic sequelae. Therefore, midfacial degloving became the most popular approach for the management of inverted papilloma. The routine use of available staging systems specific for inverted papilloma23–27 would facilitate the comparison of results among different institutions.
Juvenile Angiofibroma
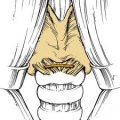
Juvenile angiofibroma is a benign lesion histologically characterized by vascular endothelium–lined spaces embedded in a fibrous stroma that typically affects young male adolescents. Immunohistochemical and electron microscopy studies suggested that the lesion could be considered a vascular malformation (or hamartoma) rather than a tumor.28 These observations led Schick and colleagues29 to postulate that juvenile angiofibroma might develop from incomplete regression of a branchial artery, which arises in embryogenesis between day 22 and 24 and forms a temporary connection between the ventral aorta and dorsal aorta. This artery commonly regresses and forms a vascular plexus that either involutes or may leave remnants, potentially leading to development of juvenile angiofibroma (Fig. 49-6). This theory is supported by the finding that juvenile angiofibroma vessels express laminin α2, which is considered a marker for early angiogenesis.30
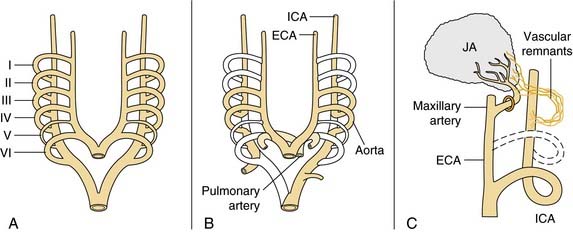
Figure 49-6. Schematic drawing summarizing the theory postulated by Schick and colleagues29 on the origin of juvenile angiofibroma (JA) from remnants of the branchial artery. A, During embryogenesis the sixth branchial arch arteries temporarily connect the ventral and dorsal aortas. B, Physiologic regression of several vascular structures (white vessels) subsequently occurs, leading to definitive configuration of the vascular system. C, Regression of the first branchial arch artery takes place via the formation of a vascular plexus, which is usually complete at birth. Incomplete regression of this structure can explain the typical blood supply for juvenile angiofibromas, from the maxillary and sphenopalatine arteries, with persistent vascular connections to the internal carotid pathway. I through VI, six branchial arteries; ECA, external carotid artery; ICA, internal carotid artery.
The lesion has a pathognomonic epicenter of origin at the level of the pterygopalatine fossa and subsequently grows through different pathways of spread that typically follow foramina and fissures of the skull base. The bone may be involved basically with two mechanisms, resorption by pressure coming from subperiosteal growth or invasion of the cancellous component initially at the level of the root of the pterygoid process, with subsequent expansion within the greater wing and erosion of the floor of the middle cranial fossa. In its early phase, juvenile angiofibroma extends through the sphenopalatine foramen into the nasopharynx and the nasal cavity and along the vidian nerve into the floor of the sphenoid sinus. Lateral extension through the pterygomaxillary fissure leads to invasion of the infratemporal fossa, which in advanced lesions may be completely filled. When the lesion expands anteriorly, the posterior wall of the maxillary sinus is progressively pushed forward. Although benign, juvenile angiofibroma may extend intracranially through the orbit via the inferior and superior orbital fissure or along the maxillary nerve to the parasellar region. Encroachment upon the anterior skull base through the ethmoid is less commonly observed. Regardless of the site and pattern of intracranial involvement, transdural growth of the lesion is very rare.31
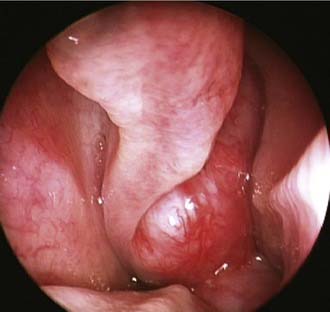
Unilateral nasal obstruction and epistaxis are the most common heralding symptoms of small to intermediate-size juvenile angiofibromas. In advanced lesions, swelling of the cheek, proptosis, or headache may be present, indicating an involvement of the infratemporal fossa, the orbit, or the cranial fossa, respectively. The endoscopic finding of a smooth, hypervascularized lesion originating behind the middle turbinate, which is usually laterally displaced against the lateral wall (Fig. 49-7), in a teenage boy strongly suggests a diagnosis of juvenile angiofibroma, which is usually confirmed by CT and MRI. Therefore, resorting to a biopsy, which is associated with a high risk of hemorrhage, is rarely if ever justified.
On CT and MRI the diagnosis of juvenile angiofibroma is based on the following three features: the area of origin invariably located at the level of pterygopalatine fossa, its hypervascular appearance after contrast enhancement, and its pattern of growth.32 On MRI, the presence on both T1- and T2-weighted sequences of several signal voids within the lesion, indicating major intralesional vessels, further corroborates a diagnosis of juvenile angiofibroma. Sometimes differentiation of this lesion from lobular capillary hemangioma, hemangiopericytoma, and schwannoma can be difficult in view of a similar pattern of enhancement. However, these other lesions do not commonly involve the pterygopalatine fossa and occur in a different age group.
Intraoperative bleeding has always been considered one of the most challenging issues in the management of juvenile angiofibroma, leading in the past to a high rate of persistent disease and to a notable morbidity. The introduction in the early 1970s of preoperative embolization,33 which is commonly performed 48 hours before surgery, has revolutionized the treatment of this lesion by dramatically decreasing intraoperative bleeding and therefore making the assessment of tumor borders at dissection more accurate. Nowadays, the availability of intra-arterial digital subtraction angiography, microcatheters, and embolic agents such as polyvinyl-alcohol particles makes superselective embolization of feeding vessels even easier.34 Furthermore, angiography provides a detailed map of the lesion’s vascular supply, showing connections with the internal carotid artery, the vertebral artery, and branches of the contralateral carotid system. Although the currently available methods of embolization provide excellent devascularization of feeding vessels from the internal maxillary artery and its branches as well as from the ascending pharyngeal artery, control of major vascularization from the internal carotid artery is demanding. Devascularization by direct tumor puncture and embolization is associated with an unacceptable risk of major neurologic complications35 and has therefore been abandoned. In case of encasement of the internal artery, which is indeed a very rare event, a balloon occlusion test and sacrifice of the internal carotid artery or, as a less invasive procedure, stenting of the intratemporal carotid artery36 may be considered.
Not all authorities concur about routinely performing preoperative embolization. In fact, the modifications induced by this procedure at the tumor periphery have been shown to increase the likelihood of leaving residual tissue behind.37
Several classification systems for juvenile angiofibroma are available in the literature.38–41 They should be used with the intent to stratify cases according to lesion extent and to make comparison among different series easier.
Surgery is considered the mainstay in the management of juvenile angiofibroma. Several approaches are currently available, ranging from microendoscopic techniques to midfacial degloving and infratemporal fossa resection. Independent of the technique selected, the key steps to minimizing bleeding and achieving radical resection are the dissection of the lesion in the subperiosteal plane with the help of bipolar coagulation and an extensive drilling of the basisphenoid where the tumor is growing with digitations that are difficult to identify even under magnification. The importance of this latter surgical step has been emphasized by Howard and associates,42 who have observed a dramatic reduction of recurrences in their series with the systematic removal of the cancellous bone of the sphenoid involved by the lesion.
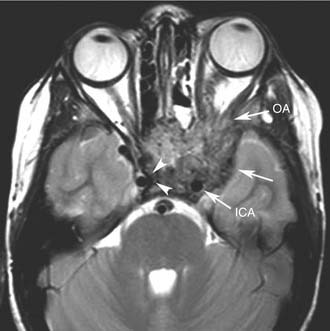
Endoscopic surgery has also emerged as a viable alternative to traditional external approaches in the management of small to intermediate size juvenile angiofibroma.43–45 Involvement of the infratemporal fossa, orbit, and parasellar region cannot be considered contraindications to endoscopic surgery (Fig. 49-8), but advanced lesions with extensive involvement of the cavernous sinus or encasement of the internal carotid artery (Fig. 49-9) are better treated through a combined endonasal-external approach. Midfacial degloving or an infratemporal fossa approach may be selected according to the location of the intracranial component and the surgeon’s preference. For lesions with a large intracranial extension, another option is resecting the extracranial component and the intracranial component in two sessions. For residual lesions involving critical areas, the possibility of switching to a combined procedure should always be discussed with the patient, in view of possible scarring that makes dissection through an exclusively endonasal approach problematic. One should also keep in mind that radiotherapy at a low dose (30-36 Gy) has been demonstrated to be effective in cases of advanced or recurrent lesions deemed not amenable to complete resection with acceptable morbidity.46 The availability of new techniques such as Gamma knife radiosurgery47 and the Cyberknife system48 might be alternatives with a low morbidity when used for lesions of limited size.
Postoperative surveillance is based on periodic endoscopic and imaging examination. However, because most of the residual lesions tend to grow submucosally, contrast-enhanced CT or MRI plays a key role in their early detection. Imaging studies performed in the immediate postoperative period would identify residual juvenile angiofibroma tissue more easily, because of the absence of any inflammatory changes.49 The actual frequency of residual lesions, although difficult to assess, ranges from 6% to 39%.47 Even though there is a great variability in postoperative surveillance with respect to the timing and imaging techniques used by different investigators, most residual lesions are diagnosed within 1 year after surgery. Independent of the surgical approach used (external vs. endoscopic), invasion of the sphenoid or pterygoid process, involvement of the infratemporal fossa, foramen lacerum, and/or cavernous sinus in more advanced lesions have a negative impact on local control.41 Endoscopic surgery, when appropriately planned, is associated with a low rate of residual lesions, which are reported in 6.6%43 to 17%41 of patients.
Osteoma
Osteoma, a benign, slow-growing osteoblastic lesion, is the most common benign tumor of the sinonasal tract. It is found in 1% of subjects undergoing plain sinus radiographs and in 3% of those undergoing CT for sinus symptoms.50 Osteomas are generally diagnosed between the second and fifth decades of life, with a slight male preponderance commonly reported in the literature. The frontal sinus is the most frequently involved anatomic site (approximately 80% of cases), followed by the ethmoid, the maxillary sinus, and, more rarely, the sphenoid sinus. Osteomas can be observed in conjunction with Gardner’s syndrome, a genetic disorder characterized by multiple polyps of the colon in association with osteomas of the skull and multiple soft tissue tumors.
According to the Fu and Perzin51 classification, osteomas are histologically divided into the following three categories:
There is no general agreement on the mechanisms leading to the development of osteomas. Three main theories have been proposed. The embryologic theory, which proposes that osteoma develops at the junction between the embryonic cartilaginous ethmoid and the membranous frontal bone, does not explain the occurrence of osteomas distant from the area where the frontal bone and the ethmoid labyrinth join together. The traumatic theory correlates the development of osteoma with a previous trauma, whereas the infective theory is based on the belief that local inflammation may alter adjacent bone metabolism by activating osteogenesis.32 One additional possibility is that the etiology of osteomas is variable and the lesions should be more properly considered slow-growing osseous hamartomas arising in childhood.52
Endoscopic examination of the nasal cavity is usually normal, because the lesion is deeply located inside a paranasal cavity. Only in very rare instances in which the osteoma is growing toward the nasal cavity can it be visualized as a firm mass covered by normal or atrophic mucosa.32
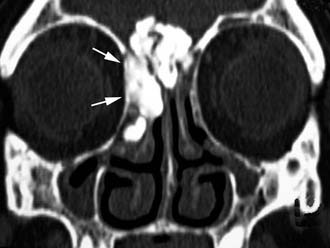
Imaging assessment of osteomas is currently based on CT. According to the amount of mineralized bone within the lesion, osteomas may exhibit a very high density, resembling cortical bone (Fig. 49-10), or a gradually decreasing density to a ground-glass pattern (Figs. 49-11 and 49-12). Contrast enhancement is usually unnecessary to reach a diagnosis.32 Multiplanar reconstructions may help identify the sinus wall of origin of the lesion, which is a relevant finding for selecting the best surgical approach. Whenever the lesion is massively eroding the bony interface with the anterior skull base or the orbit, MRI may better delineate the relationship between the osteoma and adjacent soft tissues (i.e., dura, brain, orbital content).
Different external surgical approaches (i.e., frontoethmoidectomy through a Lynch-Howarth incision, midfacial degloving, lateral rhinotomy, Caldwell-Luc procedure, and osteoplastic frontal sinusotomy via a coronal incision) have all been extensively used in the past according to the site and size of osteomas. The introduction and subsequent refinement of microendoscopic techniques led to a revolution in the surgical management of these lesions, and most are now treated through a transnasal approach (video clip available on website). A surgical trick that helps resect even large osteomas through the nose is cavitation, which consists of drilling the core of the lesion with a diamond bur, thus leaving a very thin shell of bone that can be easily fractured and dissected from the adjacent tissues.53 The possibility of using a device inducing ultrasound bone emulsification has also been proposed.54 Cerebrospinal fluid leak can be expected during these maneuvers if the lesion is in contact with dura. Consequently, the patient should be informed about this possibility and the need to repair the defect with a multilayer technique.55
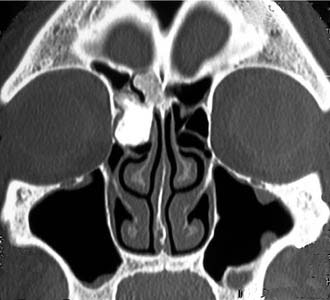
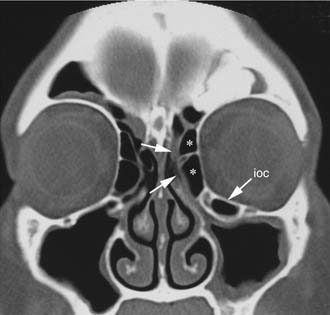
Schick and colleagues56 have precisely established the indications for and limits of endoscopic removal of osteomas. Lesions involving the ethmoid, the sphenoid, the medial wall of the maxillary sinus, and, in some selected cases, even the inferior and medial wall of the orbit can be resected transnasally (Fig. 49-13). Frontal osteomas are amenable to endoscopic surgery if they are located medial to a virtual sagittal plane prolonged upward from the lamina papyracea, if they originate from the inferior portion of the posterior frontal sinus wall, and if the anteroposterior diameter of the frontal sinus is at least 10 mm. Performing a Draf III procedure may sometimes help resect the lateral part of an osteoma of the frontal infundibulum extending over the orbit. However, for lesions extending far laterally in a well-pneumatized frontal sinus, radical removal cannot be achieved with a purely endoscopic procedure (Fig. 49-14). Such a lesion can be completely exposed and drilled out through the combination of an endoscopic approach with an image-guided frontal trephination57 or, in more extensive lesions, with osteoplastic flap sinusotomy. In our experience, obliteration of the frontal sinus is contraindicated whenever a large frontal sinusotomy, which can maintain satisfactory sinus drainage, is performed. On the other hand, when a frontal osteoma is located laterally and does not involve the frontal infundibulum, resection is better achieved through an exclusive osteoplastic flap without disturbing the frontal sinus outflow. In the case of a lesion that is massively eroding the anterior wall of the frontal sinus, reconstruction is required to maintain a regular frontal contour. This can be accomplished through the harvest of a split calvarial graft from the parietal bone, which is fixed in place by microplates.
Lobular Capillary Hemangioma
Lobular capillary hemangioma is a rapidly growing lesion characterized by a proliferation of capillaries arranged in lobules and separated by a loose connective tissue stroma, often infiltrated by inflammatory cells.58 The tumor has been commonly reported in the literature as “pyogenic granuloma,” which is currently considered inadequate in view of the fact that the lesion is neither the result of a bacterial infection nor a true granuloma.
Most mucosal lobular capillary hemangiomas of the head and neck arise in the oral cavity, and fewer in the nasal cavity. In a series of 40 patients reported by Puxeddu and colleagues,59 age ranged from 10 months to 72 years, with a mean age of 42 years and a peak incidence in the fifth decade of life. No gender preponderance was observed.
In its typical presentation, lobular capillary hemangioma appears at endoscopy as a red to purple mass, not larger than 1 cm, associated with epistaxis.59 However, in more rare instances the lesion reaches a considerable size, filling the nasal cavity entirely and leading to a complaint of unilateral nasal obstruction. In this setting, differential diagnosis from other hypervascularized lesions, such as angiofibroma, angiomatous polyp, hemangioma, hemangiopericytoma, paraganglioma, angiosarcoma, and metastases from highly vascularized tumors (i.e., kidney or thyroid carcinoma), can be problematic and requires appropriate imaging studies as a first step. On CT, the lesion appears as a unilateral mass with soft tissue density, whereas the MRI pattern consists of T2 hyperintensity and spontaneous T1 hypointensity; on both sequences, vivid enhancement can be observed after administration of paramagnetic contrast medium.32 Once angiofibroma is excluded, because of its peculiar location and epidemiologic profile, only histologic examination may establish a definitive diagnosis.
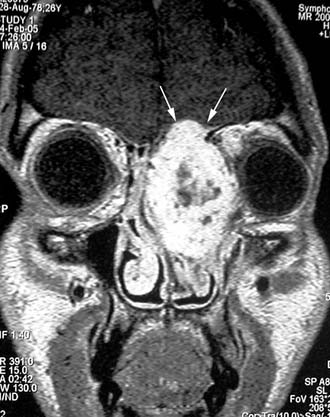
Surgery is the treatment of choice for lobular capillary hemangioma, and radical resection can be performed through an endoscopic approach even in large lesions.32 In our experience, only in one case was formal anterior craniofacial resection necessary; in that case, the lesion extensively involved the orbit and anterior skull base and was associated with intravascular papillary endothelial hyperplasia (Fig. 49-15).60
Ossifying Fibroma and Fibrous Dysplasia
In the past, ossifying fibroma and fibrous dysplasia were traditionally grouped together because of their histologic similarities. In 1963, however, Reed61 suggested that they should be regarded as two distinct entities. Ossifying fibroma is a true benign neoplasm, whereas fibrous dysplasia is a genetically based developmental anomaly of the bone-forming mesenchyme with a defect in osteoblastic differentiation and maturation that leads to a replacement of normal bony tissue by fibrous tissue of variable cellularity and immature woven bone. The main histologic differences between the two lesions lie in the absence of a capsule and the presence of more immature bone without osteoblastic activity in fibrous dysplasia.62 Psammomatoid ossifying fibroma is a variant of ossifying fibroma characterized by the presence of numerous small ossicles in stroma that resemble psammoma bodies found in extracranial meningiomas, which are indeed acellular and test positive for epithelial membrane antigens.63
The CT appearance of fibrous dysplasia is strictly related to the degree of mineralization of the tissue. In the early phases, associated with a high density of fibrous tissue, the lesion displays a radiolucent or lytic appearance, which is difficult to differentiate from a simple bone cyst. As the amount of bony tissue increases, a ground-glass (Fig. 49-16) or sclerotic appearance is seen. On MRI, the signal is reported as rather variable on T2-weighted sequences, whereas the T1 signal is more commonly hypointense. Nonhomogeneous enhancement may be obtained after gadolinium administration.32
Although differentiation on imaging between fibrous dysplasia and ossifying fibroma may be problematic, the latter more commonly appears on CT as a well-defined, multiloculated lesion, bordered by a peripheral eggshell-like dense rim.64 On MRI, ossifying fibroma displays a hyperintense signal on T2-weighted sequences, but on T1-weighted sequences, the signal is intermediately intense to hyperintense in the central part of the lesion and hypointense in the outer shell.32
The objective of surgical treatment is different for the two lesions. Ossifying fibroma requires radical resection, in view of both the high rate of relapses, accounting for 44% of lesions localized in the ethmoids,65 and the aggressive behavior of recurring tumors, with local destruction and potential invasion of adjacent vital structures.66 Successful removal of ossifying fibroma via an endoscopic approach has been reported in the literature.67–69 For fibrous dysplasia, surgery is intended to relieve symptoms such as visual impairment due to compression of the optic nerve or to correct aesthetic deformities, but not to remove the entire lesion. Optic nerve decompression has been traditionally achieved through external transfacial, neurosurgical, or combined approaches, although most cases can nowadays be managed with endonasal endoscopic surgery.67,70–73 Radiotherapy is contraindicated because of the risk of malignant transformation and the possible side effects on facial skeleton growth in young patients.
Medical treatment of fibrous dysplasia based on the use of bisphosphonates (i.e., pamidronate), which inhibit osteoclastic activity, has been used with success in patients with extensive lesions associated with significant disfigurement and pain.74,75
Schwannoma
Schwannoma is a neurogenic tumor arising from the Schwann cells of the sheath of myelinated nerves. This is a rare neoplasm that can be found in any part of the body; in 25% to 45% of cases, it is localized at the level of the head and neck. Only 4% of the lesions involve the sinonasal tract, where the ethmoid, maxillary sinus, nasal fossa, and sphenoid are involved (listed in order of decreasing frequency). According to a review of 160 cases,76 the age distribution ranges from 6 to 78 years, with most patients between 25 and 55 years of age, and no gender preference. The lesion has been only rarely observed in association with von Recklinghausen’s disease.
Macroscopically, the tumor appears as a well-delineated but nonencapsulated globular, firm to rubbery yellow-tan mass.77 Histologically, schwannomas are composed of cellular Antoni A areas with Verocay bodies and hypocellular myxoid Antoni B areas. Tumor cells are strongly and diffusely immunoreactive for S-100 protein. The pathologist should differentiate schwannoma from neurofibroma, solitary fibrous tumor, leiomyoma, fibrous histiocytoma, and fibrosarcoma.
At endoscopy, the appearance of a sinonasal schwannoma is quite nonspecific; the presence of a network of capillaries on the surface may sometimes suggest a diagnosis of a hypervascular lesion (Fig. 49-17). CT findings are not usually diagnostic, but MRI reflects the histologic features of the lesion (Fig. 49-18). Specifically, lesions with a prevalent Antoni A component have an intermediate signal on both T1- and T2-weighted images, whereas in those with a predominant Antoni B pattern, which is related to a loose myxoid stroma, hyperintensity is observed on T2-weighted images.78,79
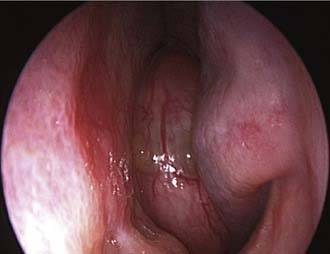
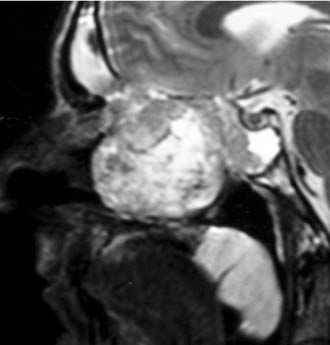
Radical surgery is the treatment of choice for sinonasal schwannoma. Before the advent of endoscopic surgery, a large variety of external approaches were employed in relation to the site and size of the lesion. After the initial report by Klossek and coworkers80 on the endoscopic management of sinonasal schwannomas, other cases with successful outcome have appeared in the literature,81–83 thus supporting the idea that even for this tumor, endoscopic surgery can be considered a viable alternative to traditional techniques.
Barnes L, Eveson JW, Reichart P, et al. Pathology and Genetics of Head and Neck Tumours. (World Health Organization Classification of Tumours.). Lyon: IARC Press; 2005.
Bignami M, Dallan I, Terranova P, et al. Frontal sinus osteomas: the window of endonasal endoscopic approach. Rhinology. 2007;45:315-320.
Brors D, Draf W. The treatment of inverted papilloma. Curr Opin Otolaryngol Head Neck Surg. 1999;7:33-38.
Busquets JM, Hwang PH. Endoscopic resection of sinonasal inverted papilloma: a meta-analysis. Otolaryngol Head Neck Surg. 2006;134:476-482.
Coutinho-Camillo CM, Brentani MM, Nagai MA. Genetic alterations in juvenile nasopharyngeal angiofibromas. Head Neck. 2008;30:390-400.
Danesi G, Panciera DT, Harvey RJ, et al. Juvenile nasopharyngeal angiofibroma: evaluation and management of advanced disease. Otolaryngol Head Neck Surg. 2008;138:581-586.
Hofmann T, Bernal-Sprekelsen M, Koele W, et al. Endoscopic resection of juvenile angiofibromas—long term results. Rhinology. 2005;43:282-289.
Howard DJ, Lloyd G, Lund V. Recurrence and its avoidance in juvenile angiofibroma. Laryngoscope. 2001;111:1509-1511.
Hyams VJ. Papillomas of the nasal cavity and paranasal sinuses: a clinicopathological study of 315 cases. Ann Otol Rhinol Laryngol. 1971;80:192-206.
Kamel RH. Transnasal endoscopic medial maxillectomy in inverted papilloma. Laryngoscope. 1995;105:847-853.
Kamel R, Kbaled A, Kandil T. Inverted papilloma: new classification and guidelines for endoscopic surgery. Am J Rhinol. 2005;19:358-364.
Kania RE, Sauvaget E, Guichard JP, et al. Early postoperative CT scanning for juvenile nasopharyngeal angiofibroma: detection of residual disease. AJNR Am J Neuroradiol. 2005;26:82-88.
Krouse JH. Development of a staging system for inverted papilloma. Laryngoscope. 2000;110:965-968.
Lee DK, Chung SK, Dhong HJ, et al. Focal hyperostosis on CT of sinonasal inverted papilloma as a predictor of tumor origin. AJNR Am J Neuroradiol. 2007;28:618-621.
Maroldi R, Farina D, Palvarini L, et al. Magnetic resonance imaging findings of inverted papilloma: differential diagnosis with malignant sinonasal tumors. Am J Rhinol. 2004;18:305-310.
Marshall AH, Bradley PJ. Management dilemmas in the treatment and follow-up of advanced juvenile nasopharyngeal angiofibroma. ORL J Otorhinolaryngol Relat Spec. 2005;68:273-278.
McCary WS, Gross CW, Reibel JF, et al. Preliminary report: endoscopic versus external surgery in the management of inverting papilloma. Laryngoscope. 1994;104:415-419.
Nicolai P, Berlucchi M, Tomenzoli D, et al. Endoscopic surgery for juvenile angiofibroma: when and how. Laryngoscope. 2003;113:775-782.
Onerci M, Ogretmenoglu O, Yücel T. Juvenile nasopharyngeal angiofibroma: a revised staging system. Rhinology. 2006;44:39-45.
Puxeddu R, Berlucchi M, Ledda GP, et al. Lobular capillary hemangioma of the nasal cavity: a retrospective study on 40 patients. Am J Rhinol. 2006;20:480-484.
Schick B, Steigerwald C, El Tahan AER, et al. The role of endonasal surgery in the management of frontoethmoidal osteomas. Rhinology. 2001;39:66-70.
Starlinger V, Wendler O, Gramann M, et al. Laminin expression in juvenile angiofibroma indicates vessel’s early developmental stage. Acta Otolaryngol. 2007;127:1310-1315.
Tanna N, Edwards JD, Aghdam H, et al. Transnasal endoscopic medial maxillectomy as the initial oncologic approach to sinonasal neoplasms: the anatomic basis. Arch Otolaryngol Head Neck Surg. 2007;133:1139-1142.
Woodworth BA, Bhargave GA, Palmer JN, et al. Clinical outcomes of endoscopic and endoscopic-assisted resection of inverted papillomas: a 15-year experience. Am J Rhinol. 2007;21:591-600.
Wormald PJ, van Hesselt A. Endoscopic removal of juvenile angiofibroma. Otolaryngol Head Neck Surg. 2003;129:684-691.
1. Barnes L, Eveson JW, Reichart P, et al. Pathology and Genetics of Head and Neck Tumours. (World Health Organization Classification of Tumours.). Lyon: IARC Press; 2005.
2. Buchwald C, Franzmann MB, Tos S. Sinonasal papillomas: a report of 82 cases in Copenhagen County, including a longitudinal epidemiological and clinical study. Laryngoscope. 1995;105:72-79.
3. Outzen KE, Grontveld A, Jorgensen K, et al. Inverted papilloma: incidence and late results of surgical treatment. Rhinology. 1996;34:114-118.
4. Barnes L, Tse LLY, Hunt JL. Schneiderian papillomas. In: Barnes L, Eveson JW, Reichart P, Sidransky D, editors. Pathology and Genetics of Head and Neck Tumours. Lyon: IARC Press, 2005.
5. Yamaguchi KT, Shapshay SN, Incze GS, et al. Inverted papilloma and squamous cell carcinoma. J Otolaryngol. 1979;8:171-178.
6. Pasquini E, Sciarrettta V, Farneti G, et al. Inverted papilloma: report of 89 cases. Am J Otolaryngol. 2004;25:178-185.
7. Yoskovitch AY, Braverman I, Nachtigal D, et al. Sinonasal schneiderian papilloma. J Otolaryngol. 1998;27:122-126.
8. Buchwald C, Franzmann MB, Tos M. Human papillomavirus (HPV) in sinonasal papillomas: a study of 78 cases using in situ hybridization and polymerase chain reaction. Laryngoscope. 1995;105:66-71.
9. Hwang CS, Yang HS, Hong MK. Detection of human papillomavirus (HPV) in sinonasal inverted papillomas using polymerase chain reaction (PCR). Am J Rhinol. 1998;12:363-366.
10. Katori H, Nozawa A, Tsukuda M. Markers of malignant transformation of sinonasal inverted papilloma. Eur J Surg Oncol. 2005;31:905-911.
11. Maroldi R, Farina D, Palvarini L, et al. Magnetic resonance imaging findings of inverted papilloma: differential diagnosis with malignant sinonasal tumors. Am J Rhinol. 2004;18:305-310.
12. Lee DK, Chung SK, Dhong HJ, et al. Focal hyperostosis on CT of sinonasal inverted papilloma as a predictor of tumor origin. Am J Neuroradiol. 2007;28:618-621.
13. Yousuf K, Wright ED. Site of attachment of inverted papilloma predicted by CT findings of osteitis. Am J Rhinol. 2007;21:32-36.
14. Busquets JM, Hwang PH. Endoscopic resection of sinonasal inverted papilloma: a meta-analysis. Otolaryngol Head Neck Surg. 2006;134:476-482.
15. Nicolai P, Tomenzoli D, Lombardi D, et al. Different endoscopic options in the treatment of inverted papilloma. Op Tech Otolaryngol Head Neck Surg. 2006;17:80-86.
16. Landsberg R. Attachment-oriented endoscopic surgical approach for sinonasal inverted papilloma. Op Tech Otolaryngol Head Neck Surg. 2006;17:87-96.
17. Tomenzoli D, Castelnuovo P, Pagella F, et al. Different endoscopic surgical strategies in the management of inverted papilloma of the sinonasal tract: experience with 47 patients. Laryngoscope. 2004;114:193-200.
18. Brors D, Draf W. The treatment of inverted papilloma. Curr Opin Otolaryngol Head Neck Surg. 1999;7:33-38.
19. Oberman HA. Papillomas of the nose and paranasal sinuses. Am J Clin Pathol. 1964;42:254-258.
20. Calcaterra TC, Thompson JW, Paglia DE. Inverting papillomas of the nose and paranasal sinuses. Laryngoscope. 1980;90:53-60.
21. Weissler MC, Montgomery WW, Turner PA, et al. Inverted papilloma. Ann Otol Rhinol Laryngol. 1986;95:215-221.
22. Myers EN, Schramm VL, Burns EL. Management of inverted papilloma of the nose and paranasal sinuses. Laryngoscope. 1981;91:2071-2084.
23. Krouse JH. Endoscopic treatment of inverted papilloma: safety and efficacy. Am J Otolaryngol. 2001;22:87-99.
24. Han JK, Smith TL, Loehrl T, et al. An evolution in the management of sinonasal inverting papilloma. Laryngoscope. 2001;111:1395-1400.
25. Oikawa K, Furuta Y, Oridate N, et al. Preoperative staging of sinonasal inverted papilloma by magnetic resonance imaging. Laryngoscope. 2003;113:1983-1987.
26. Kamel R, Kbaled A, Kandil T. Inverted papilloma: new classification and guidelines for endoscopic surgery. Am J Rhinol. 2005;19:358-364.
27. Kannady SB, Batra PS, Sautter NB, et al. New staging system for sinonasal inverted papilloma in the endoscopic era. Laryngoscope. 2007;117:1283-1287.
28. Beham A, Beham-Scmid C, Regauer S, et al. Nasopharyngeal angiofibroma: true neoplasm or vascular malformation? Adv Anat Pathol. 2000;7:36-46.
29. Schick B, Plinkert PK, Prescher A. Aetiology of angiofibromas: reflection on their specific vascular component [in German]. Laryngorhinootologie. 2002;81:280-284.
30. Starlinger V, Wendler O, Gramann M, et al. Laminin expression in juvenile angiofibroma indicates vessel’s early developmental stage. Acta Otolaryngol. 2007;127:1310-1315.
31. Danesi G, Panciera DT, Harvey RJ, et al. Juvenile nasopharyngeal angiofibroma: evaluation and management of advanced disease. Otolaryngol Head Neck Surg. 2008;138:581-586.
32. Maroldi R, Berlucchi M, Farina D, et al. Benign neoplasms and tumor-like lesions. In: Maroldi R, Nicolai P, editors. Imaging in Treatment Planning for Sinonasal Diseases. Berlin: Springer, 2005.
33. Robertson GH, Biller H, Sessions DG, et al. Presurgical internal maxillary artery embolization in juvenile angiofibroma. Laryngoscope. 1972;82:1524-1532.
34. Valavanis A, Christoforidis G. Applications of interventional neuroradiology in the head and neck. Semin Roentgenol. 2000;35:72-83.
35. Casasco A, Houdart E, Biondi A, et al. Major complications of percutaneous embolization of skull-base tumors. AJNR Am J Neuroradiol. 1999;20:179-181.
36. Sanna M, Khrais T, Menozi R, et al. Surgical removal of jugular paragangliomas after stenting of the intratemporal internal carotid artery: a preliminary report. Laryngoscope. 2006;116:742-746.
37. McCombe A, Lund VJ, Hoeard JJ. Recurrence in juvenile angiofibroma. Rhinology. 1990;109:140-147.
38. Chandler JR, Goulding R, Moskowitz L, et al. Nasopharyngeal angiofibromas: staging and management. Ann Otol Rhinol Laryngol. 1984;93:322-329.
39. Andrews JC, Fish U, Valavanis A, et al. The surgical management of extensive nasopharyngeal angiofibromas with the infratemporal fossa approach. Laryngoscope. 1989;99:429-437.
40. Radkovski D, McGill T, Healy GB, et al. Angiofibroma: changes in staging and treatment. Arch Otolaryngol Head Neck Surg. 1996;122:122-129.
41. Onerci M, Ogretmenoglu O, Yücel T. Juvenile nasopharyngeal angiofibroma: a revised staging system. Rhinology. 2006;44:39-45.
42. Howard DJ, Lloyd G, Lund V. Recurrence and its avoidance in juvenile angiofibroma. Laryngoscope. 2001;111:1509-1511.
43. Nicolai P, Berlucchi M, Tomenzoli D, et al. Endoscopic surgery for juvenile angiofibroma: when and how. Laryngoscope. 2003;113:775-782.
44. Onerci TM, Yucel OT, Ogretmenoglu O. Endoscopic surgery in treatment of juvenile nasopharyngeal angiofibroma. Int J Pediatr Otolaryngol. 2003;67:1219-1225.
45. Wormald PJ, van Hesselt A. Endoscopic removal of juvenile angiofibroma. Otolaryngol Head Neck Surg. 2003;129:684-691.
46. McAfee WJ, Morris CG, Amdur RJ, et al. Definitive radiotherapy for juvenile nasopharyngeal angiofibroma. Am J Clin Oncol. 2006;29:168-170.
47. Roche PH, Paris J, Régis J, et al. Management of invasive juvenile nasopharyngeal angiofibromas: the role of a multimodality approach. Neurosurgery. 2007;61:768-777.
48. Deguchi K, Fukuiwa T, Saito K, et al. Application of Cyberknife for the treatment of juvenile nasopharyngeal angiofibroma: a case report. Auris Nasus Larynx. 2002;29:395-400.
49. Kania RE, Sauvaget E, Guichard JP, et al. Early postoperative CT scanning for juvenile nasopharyngeal angiofibroma: detection of residual disease. AJNR Am J Neuroradiol. 2005;26:82-88.
50. Earwaker J. Paranasal sinus osteomas: a review of 46 cases. Skeletal Radiol. 1993;22:417-423.
51. Fu YS, Perzin KH. Non-epithelial tumors of the nasal cavity, paranasal sinuses, and nasopharynx: a clinicopathologic study. Cancer. 1974;33:1289-1333.
52. Harrison D, Lund VJ. Tumours of the Upper Jaw. Churchill Livingstone: Edinburgh; 1993.
53. Bignami M, Dallan I, Terranova P, et al. Frontal sinus osteomas: the window of endonasal endoscopic approach. Rhinology. 2007;45:315-320.
54. Pagella F, Giourgos G, Matti E, et al. Removal of a fronto-ethmoidal osteoma using the Sonopet Omni Ultrasonic Bone Curette: first impressions. Laryngoscope. 2008;118:307-309.
55. Locatelli D, Rampa F, Acchiardi I, et al. Endoscopic endonasal approaches for repair of cerebrospinal fluid leaks: nine-year experience. Neurosurgery. 2006;58:246-256.
56. Schick B, Steigerwald C, El Tahan AER, et al. The role of endonasal surgery in the management of frontoethmoidal osteomas. Rhinology. 2001;39:66-70.
57. Zacharek MA, Fong KJ, Hwang PH. Image-guided frontal trephination: a minimally invasive approach for hard-to-reach frontal sinus disease. Otolaryngol Head Neck Surg. 2006;135:518-522.
58. Mills SE, Cooper PH, Fechner RE. Lobular capillary hemangioma: the underlying lesion of pyogenic granuloma: a study of 73 cases from the oral and nasal mucosa membranes. Am J Surg Pathol. 1980;4:470-479.
59. Puxeddu R, Berlucchi M, Ledda GP, et al. Lobular capillary hemangioma of the nasal cavity: a retrospective study on 40 patients. Am J Rhinol. 2006;20:480-484.
60. Lombardi D, Galtelli C, Khrais T, et al. Giant hypervascular lesion of the sinonasal tract invading the anterior skull base and orbit: a puzzling case. Ann Otol Rhinol Laryngol. 2008;117:653-658.
61. Reed RJ. Fibrous dysplasia of bone: a review of 25 cases. Arch Pathol. 1963;75:480-495.
62. Hyams VJ, Batsakis JG, Michaels L. Tumors of the Upper Respiratory Tract and Ear. Washington: Armed Forces Institute of Pathology; 1988.
63. Slootveg PJ, El Mofty SK. Ossifying fibroma. In: Barnes L, Eveson JW, Reichart P, Sidransky D, editors. Pathology and Genetics of Head and Neck Tumours. Lyon: IARC Press, 2005.
64. Han MH, Chang KH, Lee CH, et al. Sinonasal psammomatoid ossifying fibroma: CT and MR manifestations. AJNR Am J Neuroradiol. 1991;12:25-30.
65. Redaelli de Zinis LO, Ansarin M, Galli G, et al. Approccio chirurgico transbasale-transfrontale in un caso di fibroma ossificante etmoidale. Gior Pat Chir Cr Fa. 1996;2:42-45.
66. Karkuzhali P, Gnanaguruparan A, Bhattachryya M. Psammomatoid ossifying fibroma of sinonasal tract. Otolaryngol Head Neck Surg. 2006;134:705-707.
67. London SD, Schlosser RJ, Gross CW. Endoscopic management of benign sinonasal tumors: a decade of experience. Am J Rhinol. 2002;16:221-227.
68. Castelnuovo P, Pagella F, Semino L, et al. Endoscopic treatment of the isolated sphenoid sinus lesions. Eur Arch Otorhinolaryngol. 2005;262:142-147.
69. Post G, Kountakis SE. Endoscopic resection of large sinonasal ossifying fibroma. Am J Otolaryngol. 2005;26:54-56.
70. Ikeda K, Suzuki H, Oshima T, et al. Endonasal endoscopic management in fibrous dysplasia of the paranasal sinuses. Am J Otolaryngol. 1997;18:415-418.
71. Brodish BN, Morgan CE, Sillers MJ. Endoscopic resection of fibro-osseous lesions of the paranasal sinuses. Am J Rhinol. 1999;13:111-116.
72. Kutluhan A, Kiroglu AF, Yurttas V, et al. Monostotic fibrous dysplasia originating from ethmoid bone: treatment with endoscopic approach. Ann Otol Rhinol Laryngol. 2004;113:139-141.
73. Pletcher SD, Metson R. Endoscopic optic nerve decompression for nontraumatic optic neuropathy. Arch Otolaryngol Head Neck Surg. 2007;133:780-783.
74. Chapurlat RD, Hugueny P, Delmas PD, et al. Treatment of fibrous dysplasia of bone with intravenous pamidronate: long term effectiveness and evaluation of predictors of response to treatment. Bone. 2004;35:235-242.
75. Mäkitie AA, Törnwall J, Mäkitie O. Bisphosphonate treatment in craniofacial fibrous dysplasia: a case report and review of the literature. Clin Rheumatol. 2008;27:809-812.
76. Higo R, Yamasoba T, Kikuchi S. Nasal neurinoma: case report and review of the literature. Auris Nasus Larynx. 1993;20:297-301.
77. Fanburg-Smith JC, Thompson LDR. Benign soft tissues tumors. In: Barnes L, Eveson JW, Reichart P, Sidransky D, editors. Pathology and Genetics of Head and Neck Tumours. Lyon: IARC Press, 2005.
78. Cakmak O, Yavuz H, Yucel T. Nasal and paranasal sinus schwannomas. Eur Arch Otorhinolaryngol. 2003;260:195-197.
79. Quesada JL, Enrique A, Lorente J, et al. Sinonasal schwannoma treated with endonasal microsurgery. Otolaryngol Head Neck Surg. 2003;129:300-302.
80. Klossek JM, Ferrie JC, Goujon JM, et al. Les schwannomes naso-sinusiens: à propos de deux cas. Intérèt de l’endoscopie nasale pour le diagnostic et le traitement. Ann Otolaryngol Chir Cervicofac. 1993;110:341-345.
81. Pasquini E, Sciarretta V, Farneti G, et al. Endoscopic endonasal approach for the treatment of benign schwannoma of the sinonasal tract and pterygopalatine fossa. Am J Rhinol. 2002;16:113-118.
82. Shinohara K, Hashimoto K, Yamashita M, et al. Schwannoma of the nasal septum removed with endoscopic surgery. Otolaryngol Head Neck Surg. 2005;132:963-964.
83. Park EH, Lee SS, Byun SW. A schwannoma in the nasal septum. Eur Arch Otorhinolaryngol. 2008;265:983-985.