[level-membership-for-internal-medicine-category]
114 |
Bladder and Renal Cell Carcinomas |
BLADDER CANCER
Transitional cell epithelium lines the urinary tract from the renal pelvis to the ureter, urinary bladder, and the proximal two-thirds of the urethra. Cancers can occur at any point: 90% of malignancies develop in the bladder, 8% in the renal pelvis, and 2% in the ureter or urethra. Bladder cancer is the fourth most common cancer in men and the thirteenth in women, with an estimated 72,570 new cases and 15,210 deaths in the United States predicted for the year 2013. The almost 5:1 ratio of incidence to mortality reflects the higher frequency of the less lethal superficial variants compared to the more lethal invasive and metastatic variants. The incidence is roughly four times higher in men than in women and twofold higher in white men than in black men, with a median age of 65 years.
Once diagnosed, urothelial tumors exhibit polychronotropism, which is the tendency to recur over time in new locations in the urothelial tract. As long as urothelium is present, continuous monitoring is required.
EPIDEMIOLOGY
Cigarette smoking is believed to contribute to up to 50% of urothelial cancers in men and nearly 40% in women. The risk of developing a urothelial cancer in male smokers is increased two- to fourfold relative to nonsmokers and continues for 10 years or longer after cessation. Other implicated agents include aniline dyes, the drugs phenacetin and chlornaphazine, and external beam radiation. Chronic cyclophosphamide exposure also increases risk, whereas vitamin A supplements appear to be protective. Exposure to Schistosoma haematobium, a parasite found in many developing countries, is associated with an increase in both squamous and transitional cell carcinomas of the bladder.
PATHOLOGY
Clinical subtypes are grouped into three categories: 75% are superficial, 20% invade muscle, and 5% are metastatic at presentation. Staging of the tumor within the bladder is based on the pattern of growth and depth of invasion. The revised tumor, node, metastasis (TNM) staging system is illustrated in Fig. 114-1. About half of invasive tumors presented originally as superficial lesions that later progressed. Tumors are also rated by grade. Low-grade (highly differentiated) tumors rarely progress to a higher stage, whereas high-grade tumors do.
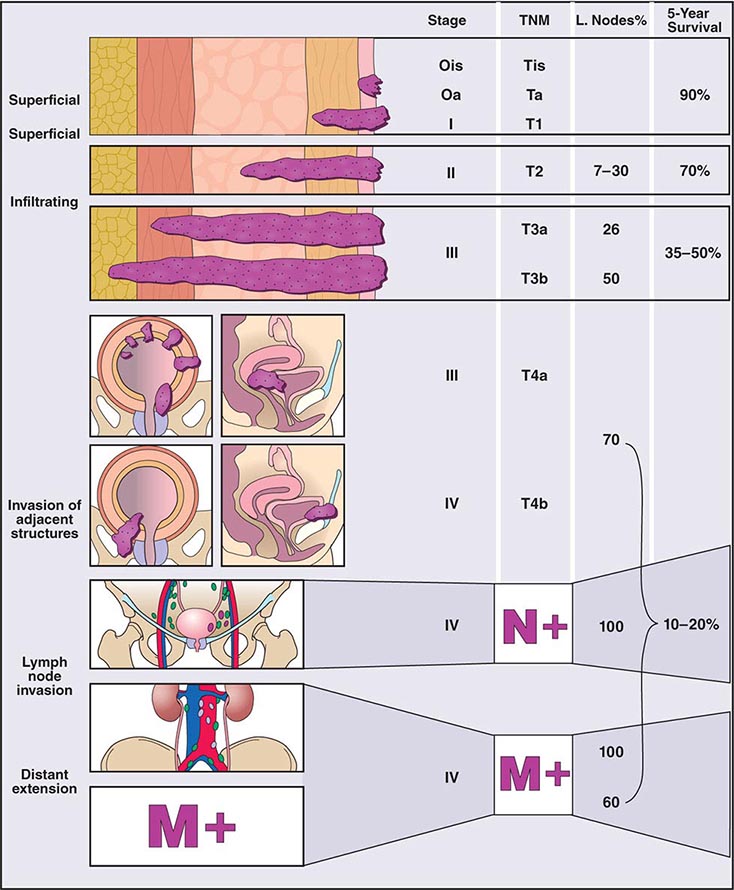
FIGURE 114-1 Bladder staging. TNM, tumor, node, metastasis.
More than 95% of urothelial tumors in the United States are transitional cell in origin. Pure squamous cancers with keratinization constitute 3%, adenocarcinomas 2%, and small cell tumors (often with paraneoplastic syndromes) <1%. Adenocarcinomas develop primarily in the urachal remnant in the dome of the bladder or in the periurethral tissues. Paragangliomas, lymphomas, and melanomas are rare. Of the transitional cell tumors, low-grade papillary lesions that grow on a central stalk are most common. These tumors are very friable, have a tendency to bleed, and have a high risk for recurrence, yet they rarely progress to the more lethal invasive variety. In contrast, carcinoma in situ (CIS) is a high-grade tumor that is considered a precursor of the more lethal muscle-invasive disease.
PATHOGENESIS
The multicentric nature of the disease and high recurrence suggests a field effect in the urothelium that results in a predisposition to develop cancer. Molecular genetic analyses suggest that the superficial and invasive lesions develop along distinct molecular pathways. Low-grade noninvasive papillary tumors harbor constitutive activation of the receptor tyrosine kinase-Ras signal transduction pathway and high frequencies of fibroblast growth factor receptor 3 and phosphoinositide-3 kinase α subunit mutations. In contrast, CIS and invasive tumors have a higher frequency of TP53 and RB gene alterations. Within all clinical stages, including Tis, T1, and T2 or greater lesions, tumors with alterations in p53, p21, and/or RB have a higher probability of recurrence, metastasis, and death from disease.
CLINICAL PRESENTATION, DIAGNOSIS, AND STAGING
Hematuria occurs in 80–90% of patients and often reflects exophytic tumors. The bladder is the most common source of gross hematuria (40%), but benign cystitis (22%) is a more common cause than bladder cancer (15%) (Chap. 61). Microscopic hematuria is more commonly of prostate origin (25%); only 2% of bladder cancers produce microscopic hematuria. Once hematuria is documented, a urinary cytology, visualization of the urothelial tract by computed tomography (CT) or magnetic resonance urogram or intravenous pyelogram, and cystoscopy are recommended if no other etiology is found. Screening asymptomatic individuals for hematuria increases the diagnosis of tumors at an early stage but has not been shown to prolong life. After hematuria, irritative symptoms are the next most common presentation. Ureteral obstruction may cause flank pain. Symptoms of metastatic disease are rarely the first presenting sign.
The endoscopic evaluation includes an examination under anesthesia to determine whether a palpable mass is present. A flexible endoscope is inserted into the bladder, and bladder barbotage for cytology is performed. Visual inspection includes mapping the location, size, and number of lesions, as well as a description of the growth pattern (solid vs papillary). All visible tumors should be resected, and a sample of the muscle underlying the tumor should be obtained to assess the depth of invasion. Normal-appearing areas are biopsied at random to ensure no CIS is present. A notation is made as to whether a tumor was completely or incompletely resected. Selective catheterization and visualization of the upper tracts should be performed if the cytology is positive and no disease is visible in the bladder. Ultrasonography, CT, and/or magnetic resonance imaging (MRI) are used to determine whether a tumor extends to perivesical fat (T3) and to document nodal spread. Distant metastases are assessed by CT of the chest and abdomen, MRI, or radionuclide imaging of the skeleton.
TREATMENT BLADDER CANCER
Management depends on whether the tumor invades muscle and whether it has spread to the regional lymph nodes and beyond. The probability of spread increases with increasing T stage.
NON–MUSCLE-INVASIVE DISEASE
At a minimum, the management is complete endoscopic resection with or without intravesical therapy. The decision to recommend intravesical therapy depends on the histologic subtype, number of lesions, depth of invasion, presence or absence of CIS, and antecedent history. Recurrences develop in upward of 50% of cases, of which 5–20% progress to a more advanced stage. In general, solitary papillary lesions are managed by transurethral surgery alone. CIS and recurrent disease are treated by transurethral surgery followed by intravesical therapy.
Intravesical therapies are used in two general contexts: as an adjuvant to a complete endoscopic resection to prevent recurrence or to eliminate disease that cannot be controlled by endoscopic resection alone. Intravesical treatments are advised for patients with diffuse CIS, recurrent disease, >40% involvement of the bladder surface by tumor, or T1 disease. The standard therapy, based on randomized comparisons, is Bacillus Calmette-Guérin (BCG) in six weekly instillations, often followed by maintenance administrations for ≥1 year. Other agents with activity include mitomycin C, interferon, and gemcitabine. The side effects of intravesical therapies include dysuria, urinary frequency, and, depending on the drug, myelosuppression or contact dermatitis. Rarely, intravesical BCG may produce a systemic illness associated with granulomatous infections in multiple sites requiring antituberculin therapy.
Following the endoscopic resection, patients are monitored for recurrence at 3-month intervals during the first year. Recurrence may develop anywhere along the urothelial tract, including the renal pelvis, ureter, or urethra. Persistent disease in the bladder and new tumors are treated with a second course of BCG or intravesical chemotherapy with valrubicin or gemcitabine. In some cases, cystectomy is recommended. Tumors in the ureter or renal pelvis are typically managed by resection during retrograde examination or, in some cases, by instillation through the renal pelvis. Prostatic urethral tumors may require cystoprostatectomy if the tumor cannot be resected completely.
MUSCLE-INVASIVE DISEASE
The treatment of a tumor that has invaded muscle can be separated into control of the primary tumor and systemic chemotherapy to treat micrometastatic disease. Radical cystectomy is the standard treatment in the United States, although in selected cases, a bladder-sparing approach is used. This approach includes complete endoscopic resection; partial cystectomy; or a combination of resection, systemic chemotherapy, and external beam radiation therapy. In some countries, external beam radiation therapy is considered standard. In the United States, it is generally limited to those patients deemed unfit for cystectomy, those with unresectable local disease, or as part of an experimental bladder-sparing approach.
Indications for cystectomy include muscle-invading tumors not suitable for segmental resection; non–muscle-invasive tumors unsuitable for conservative management (e.g., due to multicentric and frequent recurrences resistant to intravesical instillations); high-grade T1 tumors especially if associated with CIS; and bladder symptoms (e.g., frequency or hemorrhage) that impair quality of life.
Radical cystectomy is major surgery that requires appropriate preoperative evaluation and management. It involves removal of the bladder and pelvic lymph nodes and creation of a conduit or reservoir for urinary flow. Grossly abnormal lymph nodes are evaluated by frozen section. If metastases are confirmed, the procedure is often aborted. In males, radical cystectomy includes the removal of the prostate, seminal vesicles, and proximal urethra. Impotence is universal unless the nerves responsible for erectile function are preserved. In females, the procedure includes removal of the bladder, urethra, uterus, fallopian tubes, ovaries, anterior vaginal wall, and surrounding fascia.
Several options are frequently used for urinary diversion. Ileal conduits bring urine directly from the ureter to the abdominal wall. Some patients receive either a continent cutaneous reservoir constructed from detubularized bowel or an orthotopic neobladder. Approximately 25% of men receive a neobladder, leading to 85–90% continence during the day. Cutaneous reservoirs are drained by intermittent catheterization. Contraindications to a neobladder include renal insufficiency, an inability to self-catheterize, or CIS or an exophytic tumor in the urethra. Diffuse CIS in the bladder is a relative contraindication based on the risk of a urethral recurrence. Concurrent ulcerative colitis or Crohn’s disease may hinder the use of bowel.
A partial cystectomy may be considered when the disease is limited to the dome of the bladder, a ≥2 cm margin can be achieved, there is no associated CIS, and the bladder capacity is adequate after resection. This occurs in 5–10% of cases. Carcinomas in the ureter or in the renal pelvis are treated with nephroureterectomy with a bladder cuff to remove the tumor.
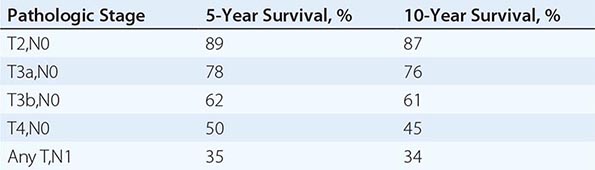
The probability of recurrence following surgery is based on pathologic stage, presence or absence of lymphatic or vascular invasion, and nodal spread. Among those whose cancers recur, the recurrence develops in a median of 1 year. Long-term outcomes vary by pathologic stage and histology (Table 114-1). The number of lymph nodes removed is also prognostic, whether or not the nodes contained tumor.
|
SURVIVAL FOLLOWING SURGERY FOR BLADDER CANCER |
Chemotherapy (described below) has been shown to prolong the survival of patients with muscle-invasive disease when combined with definitive treatment of the bladder by radical cystectomy or radiation therapy. Presurgical (or neoadjuvant) chemotherapy has been the most thoroughly explored, and increases the cure rate by 5–15%, whereas postsurgical (adjuvant) chemotherapy has not been proven definitively beneficial. For the majority of patients, chemotherapy alone is inadequate to eradicate the disease. Use of neoadjuvant chemotherapy is increasing, although it still remains underused. Experimental studies are evaluating bladder preservation strategies by combining chemotherapy and radiation therapy in patients whose tumors were endoscopically removed.
METASTATIC DISEASE
The primary goal of metastatic disease treatment is to achieve complete remission with chemotherapy alone or with a combined-modality approach of chemotherapy followed by surgical resection of residual disease. One can define a goal in terms of cure or palliation on the basis of the probability of achieving a complete response to chemotherapy using prognostic factors, such as Karnofsky performance status (KPS) (<80%) and whether the pattern of spread is nodal or visceral (liver, lung, or bone). For those with zero, one, or two risk factors, the probability of complete remission is 38, 25, and 5%, respectively, and median survival is 33, 13.4, and 9.3 months, respectively. Patients who have low KPS or who have visceral disease or bone metastases rarely achieve long-term survival. The toxicities also vary as a function of risk, and treatment-related mortality rates are as high as 3–4% using some combinations in these poor-risk patient groups. For most patients, treatment is palliative, aimed at delaying or relieving cancer-related symptoms, because few patients experience durable complete remissions.
CHEMOTHERAPY
A number of chemotherapeutic drugs have activity as single agents; cisplatin, paclitaxel, and gemcitabine are considered most active. Standard therapy consists of two-, three-, or four-drug combinations. Overall response rates of >50% have been reported using combinations such as methotrexate, vinblastine, doxorubicin, and cisplatin (MVAC); gemcitabine and cisplatin (GC); or gemcitabine, paclitaxel, and cisplatin (GPC). MVAC was considered standard, but the toxicities of neutropenia and fever, mucositis, diminished renal and auditory function, and peripheral neuropathy led to the development of alternative regimens. At present, GC is used more commonly than MVAC based on the results of a comparative trial of MVAC versus GC that showed less neutropenia and fever and less mucositis for the GC regimen with similar response rates and median overall survival. Anemia and thrombocytopenia were more common with GC. GPC is not more effective than GC.
Chemotherapy has also been tested in the neoadjuvant and adjuvant settings. In a randomized trial, patients receiving three cycles of neoadjuvant MVAC followed by cystectomy had a significantly better median (6.2 years) and 5-year survival (57%) compared to cystectomy alone (median survival 3.8 years; 5-year survival 42%). Similar results were obtained in an international study of three cycles of cisplatin, methotrexate, and vinblastine (CMV) followed by either radical cystectomy or radiation therapy. The decision to administer adjuvant therapy is based on recurrence risk after cystectomy. Studies of adjuvant chemotherapy have been underpowered, and most closed for lack of accrual. One underpowered study using the GPC regimen suggested that adjuvant treatment improved survival, although many patients never received chemotherapy for metastases. Another underpowered study did not show a benefit for GC chemotherapy. Therefore, preoperative chemotherapy is preferred when medically appropriate. Indications for adjuvant chemotherapy in patients who did not receive neoadjuvant treatment include nodal disease, extravesical tumor extension, or vascular invasion in the resected specimen.
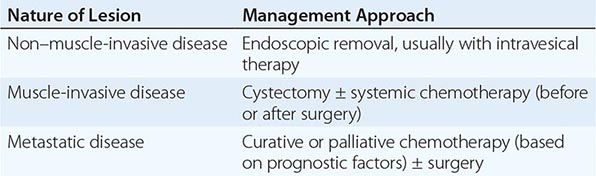
The management of bladder cancer is summarized in Table 114-2.
|
MANAGEMENT OF BLADDER CANCER |
CARCINOMA OF THE RENAL PELVIS AND URETER
About 5000 cases of renal pelvis and ureter cancer occur each year; nearly all are transitional cell carcinomas similar to bladder cancer in biology and appearance. This tumor is associated with chronic phenacetin abuse and aristolochic acid consumption in Chinese herbal preparations; aristolochic acid also seems to be associated with Balkan nephropathy, a chronic interstitial nephritis endemic in Bulgaria, Greece, Bosnia-Herzegovina, and Romania. In addition, upper tract urothelial carcinoma is linked to hereditary nonpolyposis colorectal cancer.
The most common symptom is painless gross hematuria, and the disease is usually detected on imaging during the workup for hematuria. Patterns of spread are like bladder cancer. For low-grade disease localized to the renal pelvis and ureter, nephroureterectomy (including excision of the distal ureter with a portion of the bladder) is associated with 5-year survival of 80–90%. More invasive or poorly differentiated tumors are more likely to recur locally and to metastasize. Metastatic disease is treated with the chemotherapy used in bladder cancer, and the outcome is similar to that of metastatic bladder cancer.
RENAL CELL CARCINOMA
Renal cell carcinomas account for 90–95% of malignant neoplasms arising from the kidney. Notable features include resistance to cytotoxic agents, infrequent responses to biologic response modifiers such as interleukin (IL) 2, robust activity to antiangiogenesis targeted agents, and a variable clinical course for patients with metastatic disease, including anecdotal reports of spontaneous regression.
EPIDEMIOLOGY
The incidence of renal cell carcinoma continues to rise and is now nearly 65,000 cases annually in the United States, resulting in 13,700 deaths. The male-to-female ratio is 2:1. Incidence peaks between the ages of 50 and 70 years, although this malignancy may be diagnosed at any age. Many environmental factors have been investigated as possible contributing causes; the strongest association is with cigarette smoking. Risk is also increased for patients who have acquired cystic disease of the kidney associated with end-stage renal disease and for those with tuberous sclerosis. Most cases are sporadic, although familial forms have been reported. One is associated with von Hippel-Lindau (VHL) syndrome. VHL syndrome is an autosomal dominant disorder. Genetic studies identified the VHL gene on the short arm of chromosome 3. Approximately 35% of individuals with VHL disease develop clear cell renal cell carcinoma. Other associated neoplasms include retinal hemangioma, hemangioblastoma of the spinal cord and cerebellum, pheochromocytoma, neuroendocrine tumors and cysts, and cysts in the epididymis of the testis in men and the broad ligament in women.
PATHOLOGY AND GENETICS
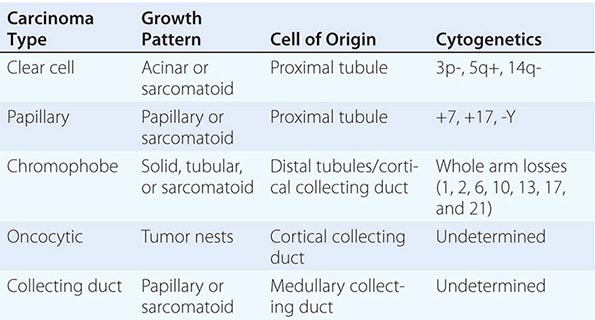
Renal cell neoplasia represents a heterogeneous group of tumors with distinct histopathologic, genetic, and clinical features ranging from benign to high-grade malignant (Table 114-3). They are classified on the basis of morphology and histology. Categories include clear cell carcinoma (60% of cases), papillary tumors (5–15%), chromophobe tumors (5–10%), oncocytomas (5–10%), and collecting or Bellini duct tumors (<1%). Papillary tumors tend to be bilateral and multifocal. Chromophobe tumors have a more indolent clinical course, and oncocytomas are considered benign neoplasms. In contrast, Bellini duct carcinomas, which are thought to arise from the collecting ducts within the renal medulla, are rare but often very aggressive. Clear cell tumors, the predominant histology, are found in >80% of patients who develop metastases. Clear cell tumors arise from the epithelial cells of the proximal tubules and usually show chromosome 3p deletions. Deletions of 3p21–26 (where the VHL gene maps) are identified in patients with familial as well as sporadic tumors. VHL encodes a tumor suppressor protein that is involved in regulating the transcription of vascular endothelial growth factor (VEGF), platelet-derived growth factor (PDGF), and a number of other hypoxia-inducible proteins. Inactivation of VHL leads to overexpression of these agonists of the VEGF and PDGF receptors, which promote tumor angiogenesis and tumor growth. Agents that inhibit proangiogenic growth factor activity show antitumor effects. Enormous genetic variability has been documented in tumors from individual patients. Although the tumors have a clear clonal origin and often contain VHL mutations in common, different portions of the primary tumor and different metastatic sites may have wide variation in genetic lesions they contain. This tumor heterogeneity may underlie the emergence of treatment resistance.
|
CLASSIFICATION OF EPITHELIAL NEOPLASMS ARISING FROM THE KIDNEY |
CLINICAL PRESENTATION
The presenting signs and symptoms include hematuria, abdominal pain, and a flank or abdominal mass. Other symptoms are fever, weight loss, anemia, and a varicocele. The tumor is most commonly detected as an incidental finding on a radiograph. Widespread use of radiologic cross-sectional imaging procedures (CT, ultrasound, MRI) contributes to earlier detection, including incidental renal masses detected during evaluation for other medical conditions. The increasing number of incidentally discovered low-stage tumors has contributed to an improved 5-year survival for patients with renal cell carcinoma and increased use of nephron-sparing surgery (partial nephrectomy). A spectrum of paraneoplastic syndromes has been associated with these malignancies, including erythrocytosis, hypercalcemia, nonmetastatic hepatic dysfunction (Stauffer’s syndrome), and acquired dysfibrinogenemia. Erythrocytosis is noted at presentation in only about 3% of patients. Anemia, a sign of advanced disease, is more common.
The standard evaluation of patients with suspected renal cell tumors includes a CT scan of the abdomen and pelvis, chest radiograph, urine analysis, and urine cytology. If metastatic disease is suspected from the chest radiograph, a CT of the chest is warranted. MRI is useful in evaluating the inferior vena cava in cases of suspected tumor involvement or invasion by thrombus. In clinical practice, any solid renal masses should be considered malignant until proven otherwise; a definitive diagnosis is required. If no metastases are demonstrated, surgery is indicated, even if the renal vein is invaded. The differential diagnosis of a renal mass includes cysts, benign neoplasms (adenoma, angiomyolipoma, oncocytoma), inflammatory lesions (pyelonephritis or abscesses), and other primary or metastatic cancers. Other malignancies that may involve the kidney include transitional cell carcinoma of the renal pelvis, sarcoma, lymphoma, and Wilms’ tumor. All of these are less common causes of renal masses than is renal cell cancer.
STAGING AND PROGNOSIS
Staging is based on the American Joint Committee on Cancer (AJCC) staging system (Fig. 114-2). Stage I tumors are <7 cm in greatest diameter and confined to the kidney, stage II tumors are ≥7 cm and confined to the kidney, stage III tumors extend through the renal capsule but are confined to Gerota’s fascia (IIIa) or involve a single hilar lymph node (N1), and stage IV disease includes tumors that have invaded adjacent organs (excluding the adrenal gland) or involve multiple lymph nodes or distant metastases. The 5-year survival rate varies by stage: >90% for stage I, 85% for stage II, 60% for stage III, and 10% for stage IV.
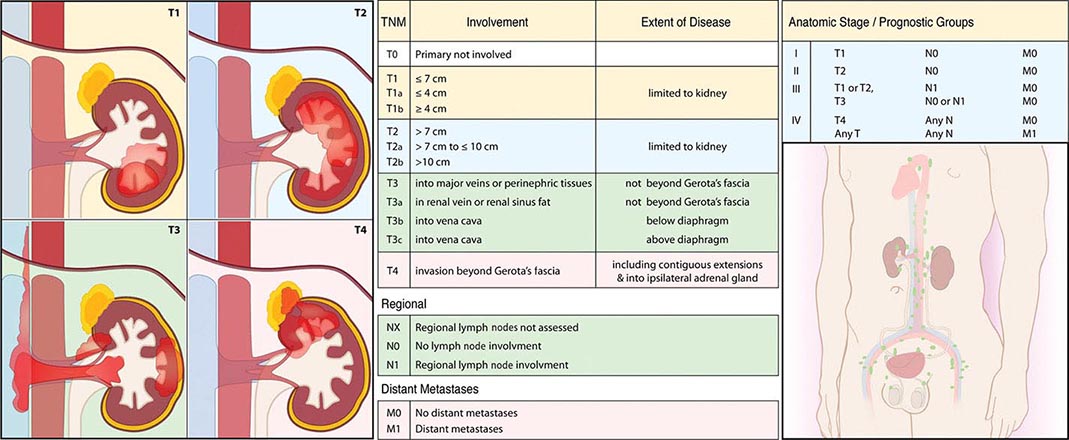
FIGURE 114-2 Renal cell carcinoma staging. TNM, tumor, node, metastasis.
115 |
Benign and Malignant Diseases of the Prostate |
Benign and malignant changes in the prostate increase with age. Autopsies of men in the eighth decade of life show hyperplastic changes in >90% and malignant changes in >70% of individuals. The high prevalence of these diseases among the elderly, who often have competing causes of morbidity and mortality, mandates a risk-adapted approach to diagnosis and treatment. This can be achieved by considering these diseases as a series of states. Each state represents a distinct clinical milestone for which therapy(ies) may be recommended based on current symptoms, the risk of developing symptoms, or death from disease in relation to death from other causes within a given time frame. For benign proliferative disorders, symptoms of urinary frequency, infection, and potential for obstruction are weighed against the side effects and complications of medical or surgical intervention. For prostate malignancies, the risks of developing the disease, symptoms, or death from cancer are balanced against the morbidities of the recommended treatments and preexisting comorbidities.
ANATOMY
The prostate is located in the pelvis and is surrounded by the rectum, the bladder, the periprostatic and dorsal vein complexes and neurovascular bundles that are responsible for erectile function, and the urinary sphincter that is responsible for passive urinary control. The prostate is composed of branching tubuloalveolar glands arranged in lobules surrounded by fibromuscular stroma. The acinar unit includes an epithelial compartment made up of epithelial, basal, and neuroendocrine cells and separated by a basement membrane, and a stromal compartment that includes fibroblasts and smooth-muscle cells. Prostate-specific antigen (PSA) and prostatic acid phosphatase (PAP) are produced in the epithelial cells. Both prostate epithelial cells and stromal cells express androgen receptors (ARs) and depend on androgens for growth. Testosterone, the major circulating androgen, is converted by the enzyme 5α-reductase to dihydrotestosterone in the gland.
The periurethral portion of the gland increases in size during puberty and after the age of 55 years due to the growth of nonmalignant cells in the transition zone of the prostate that surrounds the urethra. Most cancers develop in the peripheral zone, and cancers in this location may be palpated during a digital rectal examination (DRE).
PROSTATE CANCER
In 2013, approximately 238,590 prostate cancer cases were diagnosed, and 29,720 men died from prostate cancer in the United States. The absolute number of prostate cancer deaths has decreased in the past 5 years, which has been attributed by some to the widespread use of PSA-based detection strategies. However, the benefit of screening on survival is unclear. The paradox of management is that although 1 in 6 men will eventually be diagnosed with the disease, and the disease remains the second leading cause of cancer deaths in men, only 1 man in 30 with prostate cancer will die of his disease.
EPIDEMIOLOGY
Epidemiologic studies show that the risk of being diagnosed with prostate cancer increases by a factor of two if one first-degree relative is affected and by four if two or more are affected. Current estimates are that 40% of early-onset and 5–10% of all prostate cancers are hereditary. Prostate cancer affects ethnic groups differently. Matched for age, African-American males have both a higher incidence of prostate cancer and larger tumors and more worrisome histologic features than white males. Polymorphic variants of the AR, the cytochrome P450 C17, and the steroid 5α-reductase type II (SRD5A2) genes have been implicated in the variations in incidence.
The prevalence of autopsy-detected cancers is similar around the world, while the incidence of clinical disease varies. Thus, environmental and dietary factors may play a role in prostate cancer growth and progression. High consumption of dietary fats, such as α-linoleic acid or the polycyclic aromatic hydrocarbons that form when red meats are cooked, is believed to increase risk. Similar to breast cancer in Asian women, the risk of prostate cancer in Asian men increases when they move to Western environments. Protective factors include consumption of the isoflavonoid genistein (which inhibits 5α-reductase) found in many legumes, cruciferous vegetables that contain the isothiocyanate sulforaphane, retinoids such as lycopene found in tomatoes, and inhibitors of cholesterol biosynthesis (e.g., statin drugs). The development of prostate cancer is a multistep process. One early change is hypermethylation of the GSTP1 gene promoter, which leads to loss of function of a gene that detoxifies carcinogens. The finding that many prostate cancers develop adjacent to a lesion termed proliferative inflammatory atrophy (PIA) suggests a role for inflammation.
PREVENTION
Currently no drugs or dietary supplements are approved by the U.S. Food and Drug Administration (FDA) for prevention of prostate cancer, nor are any recommended by the major clinical guidelines. Although statins may have some protective effect, the potential risks outweigh the benefits given the small number of men who die of prostate cancer. The results from several large, double-blind, randomized chemoprevention trials established 5α-reductase inhibitors (5ARI) as the most likely therapy to reduce the future risk of a prostate cancer diagnosis. The Prostate Cancer Prevention Trial (PCPT), in which men older than age 55 years received placebo or the 5ARI finasteride, which inhibits the type 1 isoform, showed a 25% (95% confidence interval 19–31%) reduction in the period prevalence of prostate cancer across all age groups in favor of finasteride (18.4%) over placebo (24.4%). In the Reduction by Dutasteride of Prostate Cancer Events (REDUCE) trial, a similar 23% reduction in the 4-year period prevalence was observed in favor of dutasteride (p = .001). Dutasteride inhibits both the type 1 and type 2 5ARI isoforms. While both studies met their endpoint, there was concern that most of the cancers that were prevented were low risk and that there was a slightly higher rate of clinically significant cancers (those with higher Gleason score) in the treatment arm. Neither drug was FDA-approved for prostate cancer prevention. In comparison, the Selenium and Vitamin E Cancer Prevention Trial (SELECT), which enrolled African-American men age ≥50 years and others age ≥55 years, showed no difference in cancer incidence in patients receiving vitamin E (4.6%) or selenium (4.9%) alone or in combination (4.6%) relative to placebo (4.4%). A similar lack of benefit for vitamin E, vitamin C, and selenium was seen in the Physicians Health Study II.
THE CLINICAL STATES MODEL
The prostate cancer continuum—from the appearance of a preneoplastic and invasive lesion localized to the prostate, to a metastatic lesion that results in symptoms and, ultimately, mortality—can span decades. To facilitate disease management, competing risks are considered in the context of a series of clinical states (Fig. 115-1). The states are defined operationally on the basis of whether or not a cancer diagnosis has been established and, for those with a diagnosis, whether or not metastases are detectable on imaging studies and the measured level of testosterone in the blood. With this approach, an individual resides in only one state and remains in that state until he has progressed. At each assessment, the decision to offer treatment and the specific form of treatment are based on the risk posed by the cancer relative to competing causes of mortality that may be present in that individual. It follows that the more advanced the disease, the greater is the need for treatment.
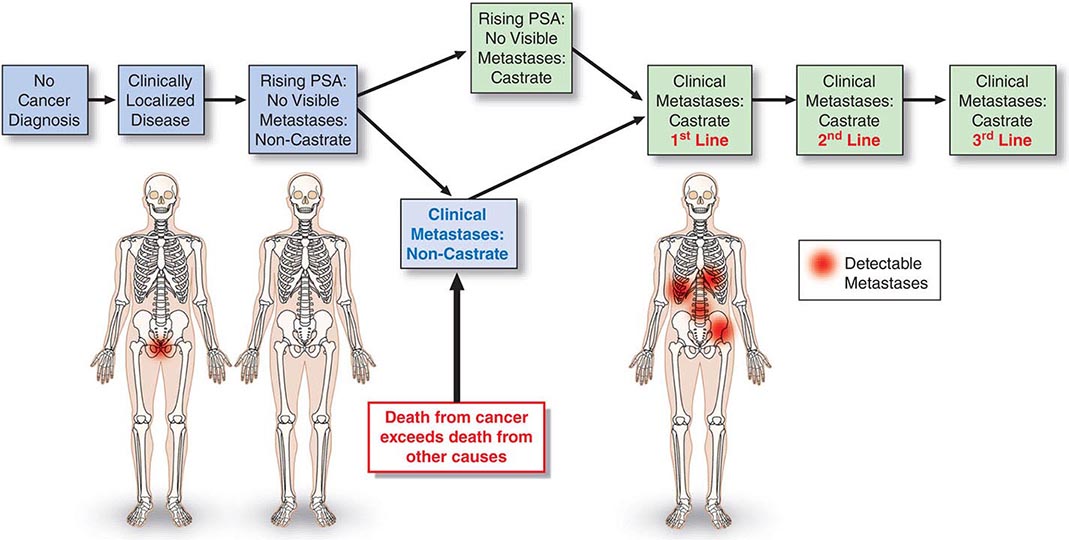
FIGURE 115-1 Clinical states of prostate cancer. PSA, prostate-specific antigen.
For those without a cancer diagnosis, the decision to undergo testing to detect a cancer is based on the individual’s estimated life expectancy and, separately, the probability that a clinically significant cancer may be present. For those with a prostate cancer diagnosis, the clinical states model considers the probability of developing symptoms or dying from prostate cancer. Thus, a patient with localized prostate cancer who has had all cancer removed surgically remains in the state of localized disease as long as the PSA remains undetectable. The time within a state becomes a measure of the efficacy of an intervention, although the effect may not be assessable for years. Because many men with active cancer are not at risk for metastases, symptoms, or death, the clinical states model allows a distinction between cure—the elimination of all cancer cells, the primary therapeutic objective when treating most cancers—and cancer control, in which the tempo of the illness is altered and symptoms are controlled until the patient dies of other causes. These can be equivalent therapeutically from a patient standpoint if the patient has not experienced symptoms of the disease or the treatment needed to control it. Even when a recurrence is documented, immediate therapy is not always necessary. Rather, as at the time of diagnosis, the need for intervention is based on the tempo of the illness as it unfolds in the individual, relative to the risk-to-benefit ratio of the therapy being considered.
SCREENING AND DIAGNOSIS
Physical Examination The need to pursue a diagnosis of prostate cancer is based on symptoms, an abnormal DRE, or, more typically, a change in or an elevated serum PSA. The urologic history should focus on symptoms of outlet obstruction, continence, potency, or change in ejaculatory pattern.
The DRE focuses on prostate size and consistency and abnormalities within or beyond the gland. Many cancers occur in the peripheral zone and may be palpated on DRE. Carcinomas are characteristically hard, nodular, and irregular, while induration may also be due to benign prostatic hypertrophy (BPH) or calculi. Overall, 20–25% of men with an abnormal DRE have cancer.
Prostate-Specific Antigen PSA (kallikrein-related peptidase 3; KLK3) is a kallikrein-related serine protease that causes liquefaction of seminal coagulum. It is produced by both nonmalignant and malignant epithelial cells and, as such, is prostate-specific, not prostate cancer–specific. Serum levels may also increase from prostatitis and BPH. Serum levels are not significantly affected by DRE, but the performance of a prostate biopsy can increase PSA levels up to tenfold for 8–10 weeks. PSA circulating in the blood is inactive and mainly occurs as a complex with the protease inhibitor α1-antichymotrypsin and as free (unbound) PSA forms. The formation of complexes between PSA, α2-macroglobulin, or other protease inhibitors is less significant. Free PSA is rapidly eliminated from the blood by glomerular filtration with an estimated half-life of 12–18 h. Elimination of PSA bound to α1-antichymotrypsin is slow (estimated half-life of 1–2 weeks) because it too is largely cleared by the kidneys. Levels should be undetectable after about 6 weeks if the prostate has been removed. Immunohistochemical staining for PSA can be used to establish a prostate cancer diagnosis.
PSA-BASED SCREENING AND EARLY DETECTION PSA testing was approved by the U.S. FDA in 1994 for early detection of prostate cancer, and the widespread use of the test has played a significant role in the proportion of men diagnosed with early-stage cancers: more than 70–80% of newly diagnosed cancers are clinically organ-confined. The level of PSA in blood is strongly associated with the risk and outcome of prostate cancer. A single PSA measured at age 60 is associated (area under the curve [AUC] of 0.90) with lifetime risk of death from prostate cancer. Most prostate cancer deaths (90%) occur among men with PSA levels in the top quartile (>2 ng/mL), although only a minority of men with PSA >2 ng/mL will develop lethal prostate cancer. Despite this and mortality rate reductions reported from large randomized prostate cancer screening trials, routine use of the test remains controversial.
The U.S. Preventive Services Task Force (USPSTF) reviewed the evidence for screening for prostate cancer and made a clear recommendation against screening. By giving a grade of “D” in the recommendation statement that was based on this review, the USPSTF concluded that “there is moderate or high certainty that this service has no net benefit or that the harms outweigh the benefits.” Whether the harms of screening, overdiagnosis, and overtreatment are justified by the benefits in terms of reduced prostate cancer mortality is open to reasonable doubt. In response to the USPSTF, the American Urological Association (AUA) updated their consensus statement regarding prostate cancer screening. They concluded that the quality of evidence for the benefits of screening was moderate, and evidence for harm was high for men age 55–69 years. For men outside this age range, evidence was lacking for benefit, but the harms of screening, including overdiagnosis and overtreatment, remained. The AUA recommends shared decision making considering PSA-based screening for men age 55–69, a target age group for whom benefits may outweigh harms. Outside this age range, PSA-based screening as a routine test was not recommended based on the available evidence. The entire guideline is available at www.AUAnet.org/education/guidelines/prostate-cancer-detection.cfm.
The PSA criteria used to recommend a diagnostic prostate biopsy have evolved over time. However, based on the commonly used cut point for prostate biopsy (a total PSA ≥4 ng/mL), most men with a PSA elevation do not have histologic evidence of prostate cancer at biopsy. In addition, many men with PSA levels below this cut point harbor cancer cells in their prostate. Information from the PCPT demonstrates that there is no PSA below which the risk of prostate cancer is zero. Thus, the PSA level establishes the likelihood that a man will harbor cancer if he undergoes a prostate biopsy. The goal is to increase the sensitivity of the test for younger men more likely to die of the disease and to reduce the frequency of detecting cancers of low malignant potential in elderly men more likely to die of other causes. Patients with symptomatic prostatitis should have a course of antibiotics before biopsy. However, the routine use of antibiotics in an asymptomatic man with an elevated PSA level is strongly discouraged.
Prostate Biopsy A diagnosis of cancer is established by an image-guided needle biopsy. Direct visualization by transrectal ultrasound (TRUS) or magnetic resonance imaging (MRI) assures that all areas of the gland are sampled. Contemporary schemas advise an extended-pattern 12-core biopsy that includes sampling from the peripheral zone as well as a lesion-directed palpable nodule or suspicious image-guided sampling. Men with an abnormal PSA and negative biopsy are advised to undergo a repeat biopsy.
BIOPSY PATHOLOGY Each core of the biopsy is examined for the presence of cancer, and the amount of cancer is quantified based on the length of the cancer within the core and the percentage of the core involved. Of the cancers identified, >95% are adenocarcinomas; the rest are squamous or transitional cell tumors or, rarely, carcinosarcomas. Metastases to the prostate are rare, but in some cases colon cancers or transitional cell tumors of the bladder invade the gland by direct extension.
When prostate cancer is diagnosed, a measure of histologic aggressiveness is assigned using the Gleason grading system, in which the dominant and secondary glandular histologic patterns are scored from 1 (well-differentiated) to 5 (undifferentiated) and summed to give a total score of 2–10 for each tumor. The most poorly differentiated area of tumor (i.e., the area with the highest histologic grade) often determines biologic behavior. The presence or absence of perineural invasion and extracapsular spread is also recorded.
Prostate Cancer Staging The tumor, node, metastasis (TNM) staging system includes categories for cancers identified solely on the basis of an abnormal PSA (T1c), those that are palpable but clinically confined to the gland (T2), and those that have extended outside the gland (T3 and T4) (Table 115-1, Fig. 115-2). DRE alone is inaccurate in determining the extent of disease within the gland, the presence or absence of capsular invasion, involvement of seminal vesicles, and extension of disease to lymph nodes. Because of the inadequacy of DRE for staging, the TNM staging system was modified to include the results of imaging. Unfortunately, no single test has proven to accurately indicate the stage or the presence of organ-confined disease, seminal vesicle involvement, or lymph node spread.
|
TNM CLASSIFICATION |
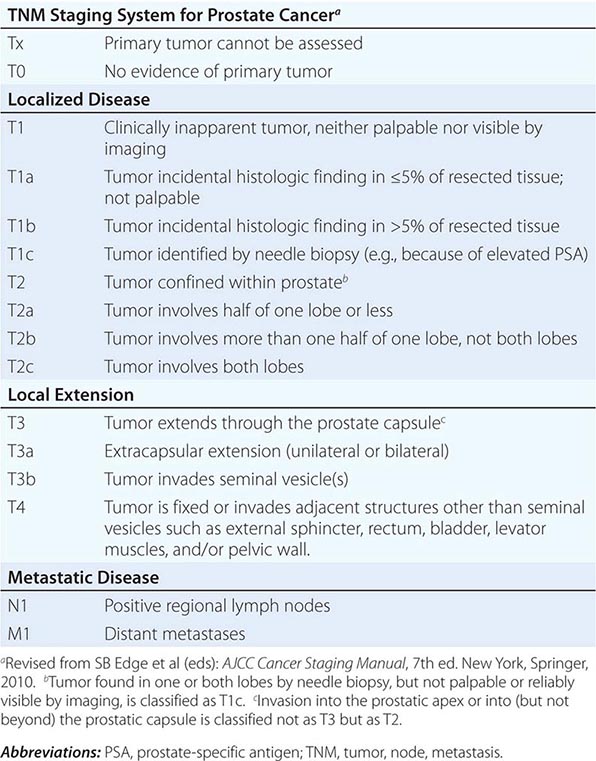
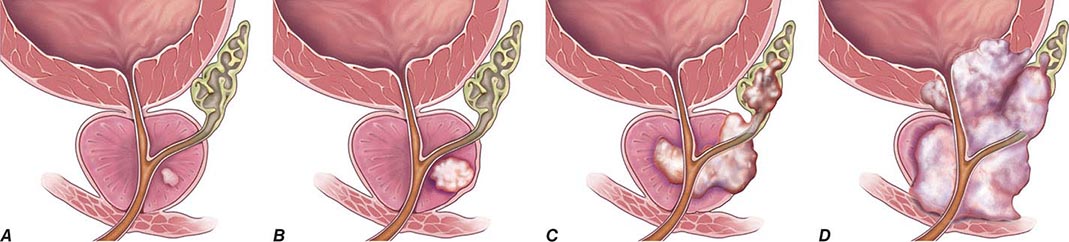
FIGURE 115-2 T stages of prostate cancer. (A) T1—Clinically inapparent tumor, neither palpable nor visible by imaging; (B) T2—Tumor confined within prostate; (C) T3—Tumor extends through prostate capsule and may invade the seminal vesicles; (D) T4—Tumor is fixed or invades adjacent structures. Eighty-one percent of patients present with local disease (T1 and T2), which is associated with a 5-year survival rate of 100%. An additional 12% of patients present with regional disease (T3 and T4 without metastases), which is also associated with a 100% survival rate after 5 years. Four percent of patients present with distant disease (T4 with metastases), which is associated with a 28% 5-year survival rate. (Three percent of patients are ungraded, and this group is associated with a 73% 5-year survival rate.) (Data from AJCC, http://seer.cancer.gov/statfacts/html/prost.html. Figure © 2014 Memorial Sloan-Kettering Cancer Center; used with permission.)
TRUS is the imaging technique most frequently used to assess the primary tumor, but its chief use is directing prostate biopsies, not staging. No TRUS finding consistently indicates cancer with certainty. Computed tomography (CT) lacks sensitivity and specificity to detect extraprostatic extension and is inferior to MRI in visualization of lymph nodes. In general, MRI performed with an endorectal coil is superior to CT to detect cancer in the prostate and to assess local disease extent. T1-weighted MRI produces a high signal in the periprostatic fat, periprostatic venous plexus, perivesicular tissues, lymph nodes, and bone marrow. T2-weighted MRI demonstrates the internal architecture of the prostate and seminal vesicles. Most cancers have a low signal, while the normal peripheral zone has a high signal, although the technique lacks sensitivity and specificity. MRI is also useful for the planning of surgery and radiation therapy.
Radionuclide bone scans (bone scintigraphy) are used to evaluate spread to osseous sites. This test is sensitive but relatively nonspecific because areas of increased uptake are not always related to metastatic disease. Healing fractures, arthritis, Paget’s disease, and other conditions will also cause abnormal uptake. True-positive bone scans are uncommon when the PSA is <10 ng/mL unless the tumor is high grade.
|
TREATMENT |
TREATMENT OF PROSTATE CANCER BY CLINICAL STATE |
CLINICALLY LOCALIZED PROSTATE CANCER
Clinically localized prostate cancers are those that appear to be nonmetastatic after staging studies are performed. Patients with clinically localized disease are managed by radical prostatectomy, radiation therapy, or active surveillance. Choice of therapy requires the consideration of several factors: the presence of symptoms, the probability that the untreated tumor will adversely affect the quality or duration of survival and thus require treatment, and the probability that the tumor can be cured by single-modality therapy directed at the prostate or that it will require both local and systemic therapy to achieve cure.
Data from the literature do not provide clear evidence for the superiority of any one treatment relative to another. Comparison of outcomes of various forms of therapy is limited by the lack of prospective trials, referral bias, the experience of the treating teams, and differences in endpoints and cancer control definitions. Often, PSA relapse–free survival is used because an effect on metastatic progression or survival may not be apparent for years. After radical surgery to remove all prostate tissue, PSA should become undetectable in the blood within 6 weeks. If PSA remains or becomes detectable after radical prostatectomy, the patient is considered to have persistent disease. After radiation therapy, in contrast, PSA does not become undetectable because the remaining nonmalignant elements of the gland continue to produce PSA even if all cancer cells have been eliminated. Similarly, cancer control is not well defined for a patient managed by active surveillance because PSA levels will continue to rise in the absence of therapy. Other outcomes are time to objective progression (local or systemic), cancer-specific survival, and overall survival; however, these outcomes may take years to assess.
The more advanced the disease, the lower the probability of local control and the higher the probability of systemic relapse. More important is that within the categories of T1, T2, and T3 disease are cancers with a range of prognoses. Some T3 tumors are curable with therapy directed solely at the prostate, and some T1 lesions have a high probability of systemic relapse that requires the integration of local and systemic therapy to achieve cure. For T1c cancers in particular, stage alone is inadequate to predict outcome and select treatment; other factors must be considered.
Nomograms To better assess risk and guide treatment selection, many groups have developed prognostic models or nomograms that use a combination of the initial clinical T stage, biopsy Gleason score, and baseline PSA. Some use discrete cut points (PSA <10 or ≥10 ng/mL; Gleason score of ≤6, 7, or ≥8); others employ nomograms that use PSA and Gleason score as continuous variables. More than 100 nomograms have been reported to predict the probability that a clinically significant prostate cancer is present, disease extent (organ-confined vs non–organ-confined, node-negative or -positive), or the probability of success of treatment for specific local therapies using pretreatment variables. Considerable controversy exists over what constitutes “high risk” based on a predicted probability of success or failure. In these situations, nomograms and predictive models can only go so far. Exactly what probability of success or failure would lead a physician to recommend and a patient to seek alternative approaches is controversial. As an example, it may be appropriate to recommend radical surgery for a younger patient with a low probability of cure. Nomograms are being refined continually to incorporate additional clinical parameters, biologic determinants, and year of treatment, which can also affect outcomes, making treatment decisions a dynamic process.
Treatment-Related Adverse Events The frequency of adverse events varies by treatment modality and the experience of the treating team. For example, following radical prostatectomy, incontinence rates range from 2–47% and impotence rates range from 25–89%. Part of the variability relates to how the complication is defined and whether the patient or physician is reporting the event. The time of the assessment is also important. After surgery, impotence is immediate but may reverse over time, while with radiation therapy impotence is not immediate but may develop over time. Of greatest concern to patients are the effects on continence, sexual potency, and bowel function.
Radical Prostatectomy The goal of radical prostatectomy is to excise the cancer completely with a clear margin, to maintain continence by preserving the external sphincter, and to preserve potency by sparing the autonomic nerves in the neurovascular bundle. The procedure is advised for patients with a life expectancy of 10 years or more and is performed via a retropubic or perineal approach or via a minimally invasive robotic-assisted or hand-held laparoscopic approach. Outcomes can be predicted using postoperative nomograms that consider pretreatment factors and the pathologic findings at surgery. PSA failure is usually defined as a value greater than 0.1 or 0.2 ng/mL. Specific criteria to guide the choice of one approach over another are lacking. Minimally invasive approaches offer the advantage of a shorter hospital stay and reduced blood loss. Rates of cancer control, recovery of continence, and recovery of erectile function are comparable between open and minimally invasive approaches. The individual surgeon rather than the surgical approach used is most important in determining outcomes after surgery.
Neoadjuvant hormonal therapy has also been explored in an attempt to improve the outcomes of surgery for high-risk patients, using a variety of definitions. The results of several large trials testing 3 or 8 months of androgen depletion before surgery showed that serum PSA levels decreased by 96%, prostate volumes decreased by 34%, and margin positivity rates decreased from 41% to 17%. Unfortunately, hormones did not produce an improvement in PSA relapse–free survival. Thus, neoadjuvant hormonal therapy is not recommended.
Factors associated with incontinence following radical prostatectomy include older age and urethral length, which impacts the ability to preserve the urethra beyond the apex and the distal sphincter. The skill and experience of the surgeon are also factors. Recovery of erectile function is associated with younger age, quality of erections before surgery, and the absence of damage to the neurovascular bundles. In general, erectile function begins to return about 6 months after surgery if both neurovascular bundles are preserved. Potency is reduced by half if at least one neurovascular bundle is sacrificed. Overall, with the availability of drugs such as phosphodiesterase-5 (PDE5) inhibitors, intraurethral inserts of alprostadil, and intracavernosal injections of vasodilators, many patients recover satisfactory sexual function.
Radiation Therapy Radiation therapy is given by external beam, by radioactive sources implanted into the gland, or by a combination of the two techniques.
EXTERNAL-BEAM RADIATION THERAPY Contemporary external-beam radiation therapy requires three-dimensional conformal treatment plans to maximize the dose to the prostate and to minimize the exposure of the surrounding normal tissue. Intensity-modulated radiation therapy (IMRT) permits shaping of the dose and allows the delivery of higher doses to the prostate and a further reduction in normal tissue exposure than three-dimensional conformal treatment alone. These advances have enabled the safe administration of doses >80 Gy and resulted in higher local control rates and fewer side effects.
Cancer control after radiation therapy has been defined by various criteria, including a decline in PSA to <0.5 or 1 ng/mL, “nonrising” PSA values, and a negative biopsy of the prostate 2 years after completion of treatment. The current standard definition of biochemical failure (the Phoenix definition) is a rise in PSA by ≥2 ng/mL higher than the lowest PSA achieved. The date of failure is “at call” and not backdated.
Radiation dose is critical to the eradication of prostate cancer. In a representative study, a PSA nadir of <1.0 ng/mL was achieved in 90% of patients receiving 75.6 or 81.0 Gy versus 76% and 56% of those receiving 70.2 and 64.8 Gy, respectively. Positive biopsy rates at 2.5 years were 4% for those treated with 81 Gy versus 27% and 36% for those receiving 75.6 and 70.2 Gy, respectively.
Overall, radiation therapy is associated with a higher frequency of bowel complications (mainly diarrhea and proctitis) than surgery. The frequency relates directly to the volume of the anterior rectal wall receiving full-dose treatment. In one series, grade 3 rectal or urinary toxicities were seen in 2.1% of patients who received a median dose of 75.6 Gy, whereas grade 3 urethral strictures requiring dilatation developed in 1% of cases, all of whom had undergone a transurethral resection of the prostate (TURP). Pooled data show that the frequency of grade 3 and 4 toxicities is 6.9% and 3.5%, respectively, for patients who received >70 Gy. The frequency of erectile dysfunction is related to the age of the patient, the quality of erections pretreatment, the dose administered, and the time of assessment. Postradiation erectile dysfunction is related to a disruption of the vascular supply and not the nerve fibers.
Neoadjuvant hormone therapy before radiation therapy has the aim of decreasing the size of the prostate and, consequently, reducing the exposure of normal tissues to full-dose radiation, increasing local control rates, and decreasing the rate of systemic failure. Short-term hormone therapy can reduce toxicities and improve local control rates, but long-term treatment (2–3 years) is needed to prolong the time to PSA failure and lower the risk of metastatic disease in men with high-risk cancers. The impact on survival has been less clear.
BRACHYTHERAPY Brachytherapy is the direct implantation of radioactive sources (seeds) into the prostate. It is based on the principle that the deposition of radiation energy in tissues decreases as a function of the square of the distance from the source (Chap. 103e). The goal is to deliver intensive irradiation to the prostate, minimizing the exposure of the surrounding tissues. The current standard technique achieves a more homogeneous dose distribution by placing seeds according to a customized template based on imaging assessment of the cancer and computer-optimized dosimetry. The implantation is performed transperineally as an outpatient procedure with real-time imaging.
Improvements in brachytherapy techniques have resulted in fewer complications and a marked reduction in local failure rates. In a series of 197 patients followed for a median of 3 years, 5-year actuarial PSA relapse–free survival for patients with pretherapy PSA levels of 0–4, 4–10, and >10 ng/mL were 98%, 90%, and 89%, respectively. In a separate report of 201 patients who underwent posttreatment biopsies, 80% were negative, 17% were indeterminate, and 3% were positive. The results did not change with longer follow-up. Nevertheless, many physicians feel that implantation is best reserved for patients with good or intermediate prognostic features.
Brachytherapy is well tolerated, although most patients experience urinary frequency and urgency that can persist for several months. Incontinence has been seen in 2–4% of cases. Higher complication rates are observed in patients who have undergone a prior TURP, whereas those with obstructive symptoms at baseline are at a higher risk for retention and persistent voiding symptoms. Proctitis has been reported in <2% of patients.
Active Surveillance Although prostate cancer is the most common form of cancer affecting men in the United States, patients are being diagnosed earlier and more frequently present with early-stage disease. Active surveillance, described previously as watchful waiting or deferred therapy, is the policy of monitoring the illness at fixed intervals with DREs, PSA measurements, and repeat prostate biopsies as indicated until histopathologic or serologic changes correlative of progression warrant treatment with curative intent. It evolved from studies that evaluated predominantly elderly men with well-differentiated tumors who demonstrated no clinically significant progression for protracted periods, recognition of the contrast between incidence and disease-specific mortality, the high prevalence of autopsy cancers, and an effort to reduce overtreatment. A recent screening study estimated that between 50–100 men with low-risk disease would need to be treated to prevent one prostate cancer–specific death.
Arguing against active surveillance are the results of a Swedish randomized trial of radical prostatectomy versus active surveillance. With a median follow-up of 6.2 years, men treated by radical surgery had a lower risk of prostate cancer death relative to active surveillance patients (4.6% vs 8.9%) and a lower risk of metastatic progression (hazard ratio 0.63). Case selection is critical, and determining clinical parameters predictive of cancer aggressiveness that can be used to reliably select men most likely to benefit from active surveillance is an area of intense study. In one prostatectomy series, it was estimated that 10–15% of those treated had “insignificant” disease. One set of criteria includes men with clinical T1c tumors that are biopsy Gleason grade 6 or less involving three or fewer cores, each of them having less than 50% involvement by tumor and a PSA density of less than 0.15.
Concerns about active surveillance include the limited ability to predict pathologic findings by needle biopsy even when multiple cores are obtained, the recognized multifocality of the disease, and the possibility of a missed opportunity to cure the disease. Nomograms to help predict which patients can safely be managed by active surveillance continue to be refined, and as their predictive accuracy improves, it can be anticipated that more patients will be candidates.
RISING PSA AFTER DEFINITIVE LOCAL THERAPY
This term is applied to a group of patients in whom the sole manifestation of disease is a rising PSA after surgery and/or radiation therapy. By definition, there is no evidence of disease on an imaging study. For these patients, the central issue is whether the rise in PSA results from persistent disease in the primary site, systemic disease, or both. In theory, disease in the primary site may still be curable by additional local treatment.
The decision to recommend radiation therapy after prostatectomy is guided by the pathologic findings at surgery, because imaging studies such as CT and bone scan are typically uninformative. Some recommend a choline-11 positron emission tomography (PET) scan, but availability in the United States is limited. Others recommend that a biopsy of the urethrovesical anastomosis be obtained before considering radiation, whereas others treat empirically based on risk. Factors that predict for response to salvage radiation therapy are a positive surgical margin, lower Gleason score in the radical prostatectomy specimen, long interval from surgery to PSA failure, slow PSA doubling time, absence of disease in the lymph nodes, and a low (<0.5–1 ng/mL) PSA value at the time of radiation treatment. Radiation therapy is generally not recommended if the PSA was persistently elevated after surgery, which usually indicates that the disease has spread outside of the area of the prostate bed and is unlikely to be controlled with radiation therapy. As is the case for other disease states, nomograms to predict the likelihood of success are available.
For patients with a rising PSA after radiation therapy, salvage local therapy can be considered if the disease was “curable” at the time of diagnosis, if persistent disease has been documented by a biopsy of the prostate, and if no metastatic disease is seen on imaging studies. Unfortunately, case selection is poorly defined in most series, and morbidities are significant. Options include salvage radical prostatectomy, salvage cryotherapy, salvage radiation therapy, and salvage irreversible electroporation.
The rise in PSA after surgery or radiation therapy may indicate subclinical or micrometastatic disease with or without local recurrence. In these cases, the need for treatment depends, in part, on the estimated probability that the patient will develop clinically detectable metastatic disease on a scan and in what time frame. That immediate therapy is not always required was shown in a series where patients who developed a biochemical recurrence after radical prostatectomy received no systemic therapy until metastatic disease was documented. Overall, the median time to metastatic progression by imaging was 8 years, and 63% of the patients with rising PSA values remained free of metastases at 5 years. Factors associated with progression included the Gleason score of the radical prostatectomy specimen, time to recurrence, and PSA doubling time. For those with Gleason grade ≥8, the probability of metastatic progression was 37%, 51%, and 71% at 3, 5, and 7 years, respectively. If the time to recurrence was <2 years and PSA doubling time was long (>10 months), the proportions with metastatic disease at the same time intervals were 23%, 32%, and 53%, versus 47%, 69%, and 79% if the doubling time was short (<10 months). PSA doubling times are also prognostic for survival. In one series, all patients who succumbed to disease had PSA doubling times of 3 months or less.
Most physicians advise treatment if the PSA doubling time is 12 months or less. A difficulty with predicting the risk of metastatic spread, symptoms, or death from disease in the rising PSA state is that most patients receive some form of therapy before the development of metastases. Nevertheless, predictive models continue to be refined.
METASTATIC DISEASE: NONCASTRATE
The state of noncastrate metastatic prostate cancer includes men with metastases visible on an imaging study and noncastrate levels of testosterone (>150 ng/dL). The patient may be newly diagnosed or have a recurrence after treatment for localized disease. Symptoms of metastatic disease include pain from osseous spread, although many patients are asymptomatic despite extensive spread. Less common are symptoms related to marrow compromise (myelophthisis), spinal cord compression, or a coagulopathy.
Standard treatment is to deplete/lower androgens by medical or surgical means and/or to block androgen binding to the AR with antiandrogens. More than 90% of male hormones originate in the testes; <10% are synthesized in the adrenal gland. Surgical orchiectomy is the “gold standard” but is rarely used due to the availability of effective medical therapies and the more widespread use of hormones on an intermittent basis by which patients are treated for defined periods of time, following which the treatments are intentionally discontinued (discussed further below) (Fig. 115-3).
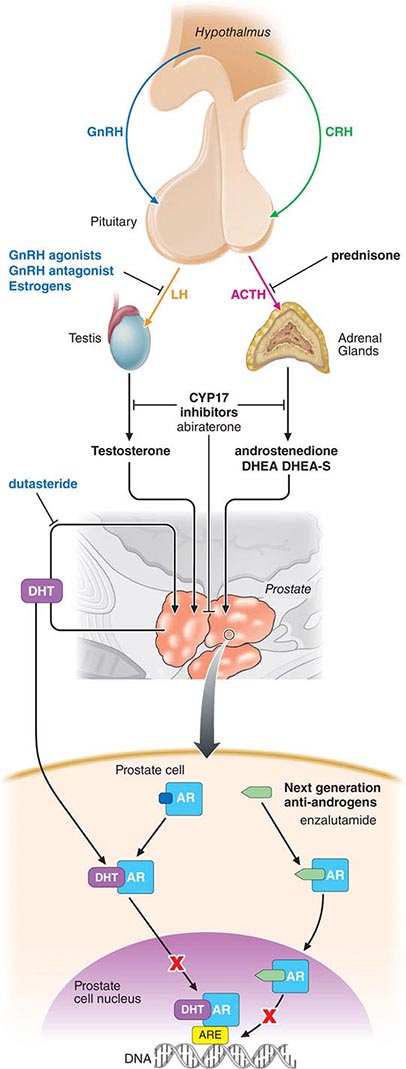
FIGURE 115-3 Sites of action of different hormone therapies. ACTH, adrenocorticotropic hormone; AR, androgen receptor; ARE, androgen-response element; CRH, corticotropin-releasing hormone; DHEA, dehydroepiandrosterone; DHEA-S, dehydroepiandrosterone sulphate; DHT, dihydrotestosterone; GnRH, gonadotropin-releasing hormone; LH, luteinizing hormone.
Testosterone-Lowering Agents Medical therapies that lower testosterone levels include the gonadotropin-releasing hormone (GnRH) agonists/antagonists, 17,20-lyase inhibitors, CYP17 inhibitors, estrogens, and progestational agents. Of these, GnRH analogues such as leuprolide acetate and goserelin acetate initially produce a rise in luteinizing hormone and follicle-stimulating hormone, followed by a downregulation of receptors in the pituitary gland, which effects a chemical castration. They were approved on the basis of randomized comparisons showing an improved safety profile (specifically, reduced cardiovascular toxicities) relative to diethylstilbestrol (DES), with equivalent potency. The initial rise in testosterone may result in a clinical flare of the disease. As such, these agents are relatively contraindicated in men with significant obstructive symptoms, cancer-related pain, or spinal cord compromise. GnRH antagonists such as degarelix achieve castrate levels of testosterone within 48 h without the initial rise in serum testosterone and do not cause a flare in the disease. Estrogens such as DES are rarely used due to the risk of vascular complications such as fluid retention, phlebitis, embolic events, and stroke. Progestational agents alone are less efficacious.
Agents that lower testosterone are associated with an androgen-depletion syndrome that includes hot flushes, weakness, fatigue, loss of libido, impotence, sarcopenia, anemia, change in personality, and depression. Changes in lipids, obesity, and insulin resistance, along with an increased risk of diabetes and cardiovascular disease, can also occur, mimicking the metabolic syndrome. A decrease in bone density may also result that worsens over time and results in an increased risk of clinical fractures. This is a particular concern, often underappreciated, for men with preexisting osteopenia secondary to hypogonadism or glucocorticoid or alcohol use. Baseline fracture risk can be assessed using the Fracture Risk Assessment Scale (FRAX), and to minimize fracture risk, patients are advised to take calcium and vitamin D supplementation, along with a bisphosphonate or the RANK ligand inhibitor, denosumab.
Antiandrogens First-generation nonsteroidal antiandrogens such as flutamide, bicalutamide, and nilutamide block ligand binding to the AR and were initially approved to block the disease flare that may occur with the rise in serum testosterone associated with GnRH agonist therapy. When antiandrogens are given alone, testosterone levels typically increase above baseline, but relative to testosterone-lowering therapies, they cause fewer hot flushes, less of an effect on libido, less muscle wasting, fewer personality changes, and less bone loss. Gynecomastia remains a significant problem but can be alleviated in part by tamoxifen.
Most reported randomized trials suggest that the cancer-specific outcomes are inferior when antiandrogens are used alone. Bicalutamide, even at 150 mg (three times the recommended dose), was associated with a shorter time to progression and inferior survival compared to surgical castration for patients with established metastatic disease. Nevertheless, some men may accept the trade-off of a potentially inferior cancer outcome for an improved quality of life.
Combined androgen blockade, the administration of an antiandrogen plus a GnRH analogue or surgical orchiectomy, and triple androgen blockade, which includes the addition of a 5ARI, have not been shown to be superior to androgen depletion monotherapies and are no longer recommended. In practice, most patients who are treated with a GnRH agonist receive an antiandrogen for the first 2–4 weeks of treatment to protect against the flare.
Intermittent Androgen Deprivation Therapy (IADT) The use of hormones in an “on-and-off” approach was initially proposed as a way to prevent the selection of cells that are resistant to androgen depletion and to reduce side effects. The hypothesis is that by allowing endogenous testosterone levels to rise, the cells that survive androgen depletion will induce a normal differentiation pathway. It is postulated that by allowing the surviving cells to proliferate in the presence of androgen, sensitivity to subsequent androgen depletion will be retained and the chance of developing a castration-resistant state will be reduced. Applied in the clinic, androgen depletion is continued for 2–6 months beyond the point of maximal response. Once treatment is stopped, endogenous testosterone levels increase, and the symptoms associated with hormone treatment abate. PSA levels also begin to rise, and at some level, treatment is restarted. With this approach, multiple cycles of regression and proliferation have been documented in individual patients. It is unknown whether the intermittent approach increases, decreases, or does not change the overall duration of sensitivity to androgen depletion. The approach is safe, but long-term data are needed to assess the course in men with low PSA levels. A randomized trial showed similar survival time between patients treated with intermittent versus continuous treatment, with a slightly higher risk of prostate cancer–specific mortality in the intermittent group, and higher cardiovascular mortality in patients on continuous therapy. The intermittent therapy was better tolerated.
Outcomes of Androgen Depletion The anti–prostate cancer effects of the various androgen depletion/blockade strategies are similar, and the outcomes predictable: an initial response, then a period of stability in which tumor cells are dormant and nonproliferative, followed after a variable period of time by a rise in PSA and tumor regrowth as a castration-resistant lesion that for most men is invariably lethal. Androgen depletion is not curative because cells that survive castration are present when the disease is first diagnosed. Considered by disease manifestation, PSA levels return to normal in 60–70% of cases, and measurable lesions regress in about 50%; improvements in bone scan occur in 25% of cases, but the majority of cases remain stable. The duration of response and survival is inversely proportional to disease extent at the time androgen depletion is first started, whereas the degree of PSA decline at 6 months has been shown to be prognostic. In a large-scale trial, PSA nadir proved prognostic.
An active question is whether hormones should be given in the adjuvant setting after surgery or radiation treatment of the primary tumor or whether to wait until PSA recurrence, metastatic disease, or symptoms are documented. Trials in support of early therapy have often been underpowered relative to the reported benefit or have been criticized on methodologic grounds. One trial showing a survival benefit for patients treated with radiation therapy and 3 years of androgen depletion, relative to radiation alone, was criticized for the poor outcomes of the control group. Another showing a survival benefit for patients with positive lymph nodes who were randomized to immediate medical or surgical castration compared to observation (p = .02) was criticized because the confidence intervals around the 5- and 8-year survival distributions for the two groups overlapped. A large randomized study comparing early to late hormone treatment (orchiectomy or GnRH analogue) in patients with locally advanced or asymptomatic metastatic disease showed that patients treated early were less likely to progress from M0 to M1 disease, to develop pain, and to die of prostate cancer. This trial was criticized because therapy was delayed “too long” in the late-treatment group. Noteworthy is that the American Society of Clinical Oncology Guidelines recommend deferring treatment until the disease has recurred and the prognosis has been reassessed. These guidelines do not support immediate therapy.
METASTATIC DISEASE: CASTRATE
Castration-resistant prostate cancer (CRPC) is defined as disease that progresses despite androgen suppression by medical or surgical therapies where the measured levels of testosterone are 50 ng/mL or lower. The rise in PSA indicates continued signaling through the AR signaling axis, the result of a series of oncogenic changes that include overexpression of androgen biosynthetic enzymes that can lead to increased intratumoral androgens, and overexpression of the receptor itself that enables signaling to occur even in the setting of low levels of androgen. The majority of CRPC cases are not “hormone-refractory,” and considering them as such can deny patients safe and effective treatment. CRPC can manifest in many ways. For some, it is a rise in PSA with no change in radiographs and no new symptoms. In others, it is a rising PSA and progression in bone with or without symptoms of disease. Still others will show soft tissue disease with or without osseous metastases, and others have visceral spread.
For the individual patient, it is first essential to ensure that a castrate status be documented. Patients receiving an antiandrogen alone, whose serum testosterone levels are elevated, should be treated first with a GnRH analogue or orchiectomy and observed for response. Patients on an antiandrogen in combination with a GnRH analogue should have the antiandrogen discontinued, because approximately 20% will respond to the selective discontinuation of the antiandrogen.
Chemotherapy and New Agents Through 2009, docetaxel was the only systemic therapy proven to prolong life. As a single agent, the drug produced PSA declines in 50% of patients, measurable disease regression in 25%, and improvement in both preexisting pain and prevention of future cancer-related pain. Since then, six agents with diverse mechanisms of action that target the tumor itself or other aspects of the metastatic process have been proven to prolong life and were FDA approved. The first was sipuleucel-T, the first biologic approach shown to prolong life in which antigen-presenting cells are activated ex vivo, pulsed with antigen, and reinfused. The second, cabazitaxel, a non–cross-resistant taxane, was shown to be superior to mitoxantrone in the post-docetaxel setting. This was followed by the CYP17 inhibitor abiraterone acetate, which lowers androgen levels in the tumor, adrenal glands, and testis, and the next-generation antiandrogen enzalutamide, which not only has a higher binding affinity to the AR relative to first-generation compounds, but uniquely inhibits nuclear location and DNA binding of the receptor complex. Both abiraterone acetate and enzalutamide were first approved for postchemotherapy treated patients on the basis of placebo-controlled phase III trials—a further indication that these tumors are not uniformly hormone-refractory. The indication for abiraterone acetate was later expanded to the prechemotherapy setting, based on a second trial using a co-primary endpoint of radiographic progression–free survival and overall survival. Similar results were seen with enzalutamide, for which an expanded indication is also anticipated. Alpharadin (radium-223 chloride), an alpha-emitting bone-seeking radioisotope, has been shown to prolong life in patients with symptoms related to osseous disease. The alpharadin result validated the bone microenvironment as a therapeutic target independent of direct effects on the tumor itself, as no declines in PSA were observed in the trial. Notable is that in addition to a survival benefit, the drug also reduced the development of significant skeletal events.
Other bone-targeted agents, such as the bisphosphonates and the RANK ligand inhibitor denosumab, protect against bone loss associated with androgen depletion and also reduce skeletal-related events by targeting bone osteoclasts. In one trial, denosumab was shown to be superior to zoledronic acid with respect to skeletal-related events, but had a slightly higher frequency of osteonecrosis of the jaw.
In clinical practice, most men seek to avoid chemotherapy and are first treated with a biologic agent and/or newer hormonal agent approved for this indication. It is crucial to the management of the individual patient to define therapeutic objectives before initiating treatment, as there are defined standards of care for different disease manifestations. For example, sipuleucel-T is not indicated for patients with symptoms or visceral disease because the effects on the disease occur late. Similarly, alpharadin is not indicated for patients with disease that is predominantly in soft tissue or who have osseous disease that is not causing symptoms.
Pain Management Management of pain secondary to osseous metastatic disease is a critical part of therapy. Optimal palliation requires assessing whether the symptoms are from metastases that threaten or that are already affecting the spinal cord, the cauda equina, or the base of the skull, which are best treated with external-beam radiation, as are single sites of pain. Neurologic symptoms require emergency evaluation because loss of function may be permanent if not addressed quickly. Because the disease is often diffuse, palliation at one site is often followed by the emergence of symptoms in a separate site that had not received radiation. In these cases, bone-seeking radioisotopes such as alpharadin or the beta emitter 153Sm-EDTMP (Quadramet) can be considered in addition to abiraterone acetate, docetaxel, and mitoxantrone, each of which is formally approved for the palliation of pain due to prostate cancer metastases.
BENIGN DISEASE
BENIGN PROSTATIC HYPERTROPHY
BPH is a pathologic process that contributes to the development of lower urinary tract symptoms in men. Such symptoms, arising from lower urinary tract dysfunction, are further subdivided into obstructive symptoms (urinary hesitancy, straining, weak stream, terminal dribbling, prolonged voiding, incomplete emptying) and irritative symptoms (urinary frequency, urgency, nocturia, urge incontinence, small voided volumes). Lower urinary tract symptoms and other sequelae of BPH are not just due to a mass effect, but are also likely due to a combination of the prostatic enlargement and age-related detrusor dysfunction.
TREATMENT BENIGN PROSTATIC HYPERTROPHY
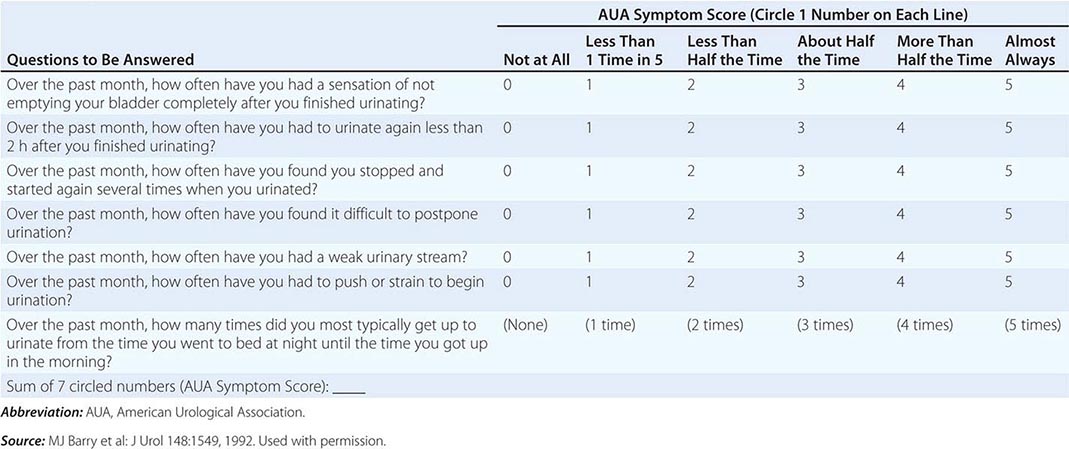
The symptoms are generally measured using a validated, reproducible index that is designed to determine disease severity and response to therapy—the AUA’s Symptom Index (AUASI), also adopted as the International Prostate Symptom Score (IPSS) (Table 115–2). Serial AUASI is particularly useful in following patients as they are treated with various forms of therapy. Asymptomatic patients do not require treatment regardless of the size of the gland, whereas patients with an inability to urinate, gross hematuria, recurrent infection, or bladder stones may require surgery. In patients with symptoms, uroflowmetry can identify those with normal flow rates who are unlikely to benefit from treatment, and bladder ultrasound can identify those with high postvoid residuals who may need intervention. Pressure-flow (urodynamic) studies detect primary bladder dysfunction. Cystoscopy is recommended if hematuria is documented and to assess the urinary outflow tract before surgery. Imaging of the upper tracts is advised for patients with hematuria, a history of calculi, or prior urinary tract problems.
|
AUA SYMPTOM INDEX |
Symptomatic relief is the most common reason men seek treatment for BPH, and therefore the goal of therapy for BPH is usually relief of these symptoms. Alpha-adrenergic receptor antagonists are thought to treat the dynamic aspect of BPH by reducing sympathetic tone of the bladder outlet, thereby decreasing resistance and improving urinary flow. 5ARIs are thought to treat the static aspect of BPH by reducing prostate volume and having a similar, albeit delayed effect. They have also proven to be beneficial in the prevention of BPH progression, as measured by prostate volume, the risk of developing acute urinary retention, and the risk of having BPH-related surgery. The use of an alpha-adrenergic receptor antagonist and a 5ARI as combination therapy seeks to provide symptomatic relief while preventing progression of BPH.
Another class of medications that has shown improvement in lower urinary tract symptoms secondary to BPH is PDE5 inhibitors, used currently in the treatment of erectile dysfunction. All three of the PDE5 inhibitors available in the United States, sildenafil, vardenafil, and tadalafil, appear to be effective in the treatment of symptoms secondary to BPH. The use of PDE5 inhibitors is not without controversy, however, given the fact that short-acting phosphodiesterase inhibitors such as sildenafil need to be dosed separately from alpha blockers such as tamsulosin because of potential hypotensive effects. Newer classes of pharmacologic agents have been used to treat symptoms secondary to BPH. Symptoms due to BPH often coexist with symptoms due to overactive bladder, and the most common pharmacologic agents for the treatment of overactive bladder symptoms are anticholinergics. This has led to multiple studies evaluating the efficacy of anticholinergics for the treatment of lower urinary tract symptoms secondary to BPH. Surgical therapy is now considered second-line therapy and is usually reserved for patients after a trial of medical therapy. The goal of surgical therapy is to reduce the size of the prostate, effectively reducing resistance to urine flow.
Surgical approaches include TURP, transurethral incision, or removal of the gland via a retropubic, suprapubic, or perineal approach. Also used are transurethral ultrasound-guided laser-induced prostatectomy (TULIP), stents, and hyperthermia.
116 |
Testicular Cancer |
Primary germ cell tumors (GCTs) of the testis arising by the malignant transformation of primordial germ cells constitute 95% of all testicular neoplasms. Infrequently, GCTs arise from an extragonadal site, including the mediastinum, retroperitoneum, and, very rarely, the pineal gland. This disease is notable for the young age of the afflicted patients, the totipotent capacity for differentiation of the tumor cells, and its curability; approximately 95% of newly diagnosed patients are cured. Experience in the management of GCTs leads to improved outcome.
INCIDENCE AND EPIDEMIOLOGY
![]() The incidence of testicular GCT is now approximately 8000 cases annually in the United States, resulting in nearly 400 deaths. The tumor occurs most frequently in men between the ages of 20 and 40 years. A testicular mass in a male ≥50 years should be regarded as a lymphoma until proved otherwise. GCT is at least four to five times more common in white than in African-American males, and a higher incidence has been observed in Scandinavia and New Zealand than in the United States.
The incidence of testicular GCT is now approximately 8000 cases annually in the United States, resulting in nearly 400 deaths. The tumor occurs most frequently in men between the ages of 20 and 40 years. A testicular mass in a male ≥50 years should be regarded as a lymphoma until proved otherwise. GCT is at least four to five times more common in white than in African-American males, and a higher incidence has been observed in Scandinavia and New Zealand than in the United States.
ETIOLOGY AND GENETICS
Cryptorchidism is associated with a several-fold higher risk of GCT. Abdominal cryptorchid testes are at a higher risk than inguinal cryptorchid testes. Orchiopexy should be performed before puberty, if possible. Early orchiopexy reduces the risk of GCT and improves the ability to save the testis. An abdominal cryptorchid testis that cannot be brought into the scrotum should be removed. Approximately 2% of men with GCTs of one testis will develop a primary tumor in the other testis. Testicular feminization syndromes and family history increase the risk of testicular GCT, and Klinefelter’s syndrome is associated with mediastinal GCT.
An isochromosome of the short arm of chromosome 12 [i(12p)] is pathognomonic for GCT. Excess 12p copy number, either in the form of i(12p) or as increased 12p on aberrantly banded marker chromosomes, occurs in nearly all GCTs, but the gene(s) on 12p involved in the pathogenesis are not yet defined.
CLINICAL PRESENTATION
A painless testicular mass is pathognomonic for a testicular malignancy. More commonly, patients present with testicular discomfort or swelling suggestive of epididymitis and/or orchitis. In this circumstance, a trial of antibiotics is reasonable. However, if symptoms persist or a residual abnormality remains, then testicular ultrasound examination is indicated.
Ultrasound of the testis is indicated whenever a testicular malignancy is considered and for persistent or painful testicular swelling. If a testicular mass is detected, a radical inguinal orchiectomy should be performed. Because the testis develops from the gonadal ridge, its blood supply and lymphatic drainage originate in the abdomen and descend with the testis into the scrotum. An inguinal approach is taken to avoid breaching anatomic barriers and permitting additional pathways of spread.
Back pain from retroperitoneal metastases is common and must be distinguished from musculoskeletal pain. Dyspnea from pulmonary metastases occurs infrequently. Patients with increased serum levels of human chorionic gonadotropin (hCG) may present with gynecomastia. A delay in diagnosis is associated with a more advanced stage and possibly worse survival.
The staging evaluation for GCT includes a determination of serum levels of α fetoprotein (AFP), hCG, and lactate dehydrogenase (LDH). After orchiectomy, a computed tomography (CT) scan of the chest, abdomen, and pelvis is generally performed. Stage I disease is limited to the testis, epididymis, or spermatic cord. Stage II disease is limited to retroperitoneal (regional) lymph nodes. Stage III disease is disease outside the retroperitoneum, involving supradiaphragmatic nodal sites or viscera. The staging may be “clinical”—defined solely by physical examination, blood marker evaluation, and radiographs—or “pathologic”—defined by an operative procedure.
The regional draining lymph nodes for the testis are in the retroperitoneum, and the vascular supply originates from the great vessels (for the right testis) or the renal vessels (for the left testis). As a result, the lymph nodes that are involved first by a right testicular tumor are the interaortocaval lymph nodes just below the renal vessels. For a left testicular tumor, the first involved lymph nodes are lateral to the aorta (para-aortic) and below the left renal vessels. In both cases, further retroperitoneal nodal spread is inferior, contralateral, and, less commonly, above the renal hilum. Lymphatic involvement can extend cephalad to the retrocrural, posterior mediastinal, and supraclavicular lymph nodes. Treatment is determined by tumor histology (seminoma versus nonseminoma) and clinical stage (Fig. 116-1).
FIGURE 116-1 Germ cell tumor staging and treatment. RPLND, retroperitoneal lymph node dissection; RT, radiotherapy.
PATHOLOGY
GCTs are divided into nonseminoma and seminoma subtypes. Nonseminomatous GCTs are most frequent in the third decade of life and can display the full spectrum of embryonic and adult cellular differentiation. This entity comprises four histologies: embryonal carcinoma, teratoma, choriocarcinoma, and endodermal sinus (yolk sac) tumor. Choriocarcinoma, consisting of both cytotrophoblasts and syncytiotrophoblasts, represents malignant trophoblastic differentiation and is invariably associated with secretion of hCG. Endodermal sinus tumor is the malignant counterpart of the fetal yolk sac and is associated with secretion of AFP. Pure embryonal carcinoma may secrete AFP or hCG, or both; this pattern is biochemical evidence of differentiation. Teratoma is composed of somatic cell types derived from two or more germ layers (ectoderm, mesoderm, or endoderm). Each of these histologies may be present alone or in combination with others. Nonseminomatous GCTs tend to metastasize early to sites such as the retroperitoneal lymph nodes and lung parenchyma. Sixty percent of patients present with disease limited to the testis (stage I), 20% with retroperitoneal metastases (stage II), and 20% with more extensive supradiaphragmatic nodal or visceral metastases (stage III).
Seminoma represents approximately 50% of all GCTs, has a median age in the fourth decade, and generally follows a more indolent clinical course. Eighty percent of patients present with stage I disease, approximately 10% with stage II disease, and 10% with stage III disease; lung or other visceral metastases are rare. When a tumor contains both seminoma and nonseminoma components, patient management is directed by the more aggressive nonseminoma component.
TUMOR MARKERS
Careful monitoring of the serum tumor markers AFP and hCG is essential in the management of patients with GCT, because these markers are important for diagnosis, as prognostic indicators, in monitoring treatment response, and in the early detection of relapse. Approximately 70% of patients presenting with disseminated nonseminomatous GCT have increased serum concentrations of AFP and/or hCG. Although hCG concentrations may be increased in patients with either nonseminoma or seminoma histology, the AFP concentration is increased only in patients with nonseminoma. The presence of an increased AFP level in a patient whose tumor shows only seminoma indicates that an occult nonseminomatous component exists, and the patient should be treated for nonseminomatous GCT. LDH levels are less specific than AFP or hCG but are increased in 50–60% patients with metastatic nonseminoma and in up to 80% of patients with advanced seminoma.
AFP, hCG, and LDH levels should be determined before and after orchiectomy. Increased serum AFP and hCG concentrations decay according to first-order kinetics; the half-life is 24–36 h for hCG and 5–7 days for AFP. AFP and hCG should be assayed serially during and after treatment. The reappearance of hCG and/or AFP or the failure of these markers to decline according to the predicted half-life is an indicator of persistent or recurrent tumor.
STAGE I NONSEMINOMA
Patients with radiographs and physical examination showing no evidence of disease and serum AFP and hCG concentrations that are either normal or declining to normal according to the known half-life have clinical stage I disease. Approximately 20–50% of such patients will have retroperitoneal lymph node metastases (pathologic stage II) but will still be cured in over 95% of cases. Depending on risk of relapse, which is determined by the pathology (see below), surveillance, a nerve-sparing retroperitoneal lymph node dissection (RPLND), or adjuvant chemotherapy (one to two cycles of bleomycin, etoposide, and cisplatin [BEP]) may be appropriate choices depending on the availability of surgical expertise and patient and physician preference. If the primary tumor shows no evidence for lymphatic or vascular invasion and is limited to the testis (T1, clinical stage IA), then the risk of relapse is only 10–20%. Because over 80% of patients with clinical stage IA nonseminoma are cured with orchiectomy alone and there is no survival advantage to RPLND (or adjuvant chemotherapy), surveillance is the preferred treatment option. This avoids overtreatment with the potential for both acute and long-term toxicities (see below). Surveillance requires patients to be carefully followed with periodic chest radiography, physical examination, CT scan of the abdomen, and serum tumor marker determinations. The median time to relapse is approximately 7 months, and late relapses (>2 years) are rare. Noncompliant patients can be considered for RPLND or adjuvant BEP.
If lymphatic or vascular invasion is present or the tumor extends through the tunica, spermatic cord, or scrotum (T2 through T4, clinical stage IB), then the risk of relapse is approximately 50%, and RPLND and adjuvant chemotherapy can be considered. Relapse rates are reduced to 3–5% after one to two cycles of adjuvant BEP. All three approaches (surveillance, RPLND, and adjuvant BEP) should cure >95% of patients with clinical stage IB disease.
RPLND is the standard operation for removal of the regional lymph nodes of the testis (retroperitoneal nodes). The operation removes the lymph nodes draining the primary site and the nodal groups adjacent to the primary landing zone. The standard (modified bilateral) RPLND removes all node-bearing tissue down to the bifurcation of the great vessels, including the ipsilateral iliac nodes. The major long-term effect of this operation is retrograde ejaculation with resultant infertility. Nerve-sparing RPLND can preserve anterograde ejaculation in ~90% of patients. Patients with pathologic stage I disease are observed, and only the <10% who relapse require additional therapy. If nodes are found to be involved at RPLND, then a decision regarding adjuvant chemotherapy is made on the basis of the extent of retroperitoneal disease (see “Stage II Nonseminoma” below). Hence, because less than 20% of patients require chemotherapy, of the three approaches, RPLND results in the lowest number of patients at risk for the late toxicities of chemotherapy.
STAGE II NONSEMINOMA
Patients with limited, ipsilateral retroperitoneal adenopathy ≤2 cm in largest diameter and normal levels of AFP and hCG can be treated with either a modified bilateral nerve-sparing RPLND or chemotherapy. The local recurrence rate after a properly performed RPLND is very low. Depending on the extent of disease, the postoperative management options include either surveillance or two cycles of adjuvant chemotherapy. Surveillance is the preferred approach for patients with resected “low-volume” metastases (tumor nodes ≤2 cm in diameter and <6 nodes involved) because the probability of relapse is one-third or less. For those who relapse, risk-directed chemotherapy is indicated (see section on advanced GCT below). Because relapse occurs in ≥50% of patients with “high-volume” metastases (>6 nodes involved, or any involved node >2 cm in largest diameter, or extranodal tumor extension), two cycles of adjuvant chemotherapy should be considered, as it results in a cure in ≥98% of patients. Regimens consisting of etoposide plus cisplatin (EP) with or without bleomycin every 3 weeks are effective and well tolerated.
Increased levels of either AFP or hCG imply metastatic disease outside the retroperitoneum; full-dose (not adjuvant) chemotherapy is used in this setting. Primary management with chemotherapy is also favored for patients with larger (>2 cm) or bilateral retroperitoneal nodes (see section on advanced GCT below).
STAGES I AND II SEMINOMA
Inguinal orchiectomy followed by immediate retroperitoneal radiation therapy or surveillance with treatment at relapse both result in cure in nearly 100% of patients with stage I seminoma. Historically, radiation was the mainstay of treatment, but the reported association between radiation and secondary malignancies and the absence of a survival advantage of radiation over surveillance has led many to favor surveillance for compliant patients. Approximately 15% of patients relapse, which is usually treated with chemotherapy. Long-term follow-up is essential, because approximately 30% of relapses occur after 2 years and 5% occur after 5 years. A single dose of carboplatin has also been investigated as an alternative to radiation therapy; the outcome was similar, but long-term safety data are lacking, and the retroperitoneum remained the most frequent site of relapse.
Generally, nonbulky retroperitoneal disease (stage IIA and small IIB) is treated with retroperitoneal radiation therapy. Approximately 90% of patients achieve relapse-free survival with retroperitoneal masses <3 cm in diameter. Due to higher relapse rates after radiation for bulkier disease, initial chemotherapy is preferred for all stage IIC and some stage IIB patients. Chemotherapy has been studied as an alternative to radiation for stage IIA and small stage IIB seminoma with lower recurrence rates compared with historical controls. These results, combined with studies demonstrating a threefold increase in the incidence of secondary malignancies and cardiovascular disease among patients who receive both radiation and chemotherapy (patients relapsing after radiation fall into this category), have led some experts to prefer chemotherapy for all stage II seminomas.
CHEMOTHERAPY FOR ADVANCED GCT
Regardless of histology, all patients with stage IIC and stage III and most with stage IIB GCT are treated with chemotherapy. Combination chemotherapy programs based on cisplatin at doses of 100 mg/m2 plus etoposide at doses of 500 mg/m2 per cycle cure 70–80% of such patients, with or without bleomycin, depending on risk stratification (see below). A complete response (the complete disappearance of all clinical evidence of tumor on physical examination and radiography plus normal serum levels of AFP and hCG for ≥1 month) occurs after chemotherapy alone in ~60% of patients, and another 10–20% become disease free with surgical resection of residual masses containing viable GCT. Lower doses of cisplatin result in inferior survival rates.
The toxicity of four cycles of the BEP is substantial. Nausea, vomiting, and hair loss occur in most patients, although nausea and vomiting have been markedly ameliorated by modern antiemetic regimens. Myelosuppression is frequent, and symptomatic bleomycin pulmonary toxicity occurs in ~5% of patients. Treatment-induced mortality due to neutropenia with septicemia or bleomycin-induced pulmonary failure occurs in 1–3% of patients. Dose reductions for myelosuppression are rarely indicated. Long-term permanent toxicities include nephrotoxicity (reduced glomerular filtration and persistent magnesium wasting), ototoxicity, peripheral neuropathy, and infertility. When bleomycin is administered by weekly bolus injection, Raynaud’s phenomenon appears in 5–10% of patients. Other evidence of small blood vessel damage, such as transient ischemic attacks and myocardial infarction, is seen less often.
RISK-DIRECTED CHEMOTHERAPY
Because not all patients are cured and treatment may cause significant toxicities, patients are stratified into “good-risk,” “intermediate-risk,” and “poor-risk” groups according to pretreatment clinical features established by the International Germ Cell Cancer Consensus Group (Table 116-1). For good-risk patients, the goal is to achieve maximum efficacy with minimal toxicity. For intermediate- and poor-risk patients, the goal is to identify more effective therapy with tolerable toxicity.
|
INTERNATIONAL GERM CELL CANCER CONSENSUS GROUP RISK CLASSIFICATION FOR ADVANCED GERM CELL TUMORS |
The marker cut offs are included in the TNM (primary tumor, regional nodes, metastasis) staging of GCT. Hence, TNM stage groupings are based on both anatomy (site and extent of disease) and biology (marker status and histology). Seminoma is either good- or intermediate-risk, based on the absence or presence of nonpulmonary visceral metastases. No poor-risk category exists for seminoma. Marker levels and primary site play no role in defining risk for seminoma. Nonseminomas have good-, intermediate-, and poor-risk categories based on the primary site of the tumor, the presence or absence of nonpulmonary visceral metastases, and marker levels.
For ~90% of patients with good-risk GCTs, four cycles of EP or three cycles of BEP produce durable complete responses, with minimal acute and chronic toxicity, and a low relapse rate. Pulmonary toxicity is absent when bleomycin is not used and is rare when therapy is limited to 9 weeks; myelosuppression with neutropenic fever is less frequent; and the treatment mortality rate is negligible. Approximately 75% of intermediate-risk patients and 50% of poor-risk patients achieve durable complete remission with four cycles of BEP, and no regimen has proved superior.
POSTCHEMOTHERAPY SURGERY Resection of residual metastases after the completion of chemotherapy is an integral part of therapy. If the initial histology is nonseminoma and the marker values have normalized, all sites of residual disease should be resected. In general, residual retroperitoneal disease requires a modified bilateral RPLND. Thoracotomy (unilateral or bilateral) and neck dissection are less frequently required to remove residual mediastinal, pulmonary parenchymal, or cervical nodal disease. Viable tumor (seminoma, embryonal carcinoma, yolk sac tumor, or choriocarcinoma) will be present in 15%, mature teratoma in 40%, and necrotic debris and fibrosis in 45% of resected specimens. The frequency of teratoma or viable disease is highest in residual mediastinal tumors. If necrotic debris or mature teratoma is present, no further chemotherapy is necessary. If viable tumor is present but is completely excised, two additional cycles of chemotherapy are given.
If the initial histology is pure seminoma, mature teratoma is rarely present, and the most frequent finding is necrotic debris. For residual retroperitoneal disease, a complete RPLND is technically difficult due to extensive postchemotherapy fibrosis. Observation is recommended when no radiographic abnormality exists on CT scan. Positive findings on a positron emission tomography (PET) scan correlate with viable seminoma in residua and mandate surgical excision or biopsy.
SALVAGE CHEMOTHERAPY
Of patients with advanced GCT, 20–30% fail to achieve a durable complete response to first-line chemotherapy. A combination of vinblastine, ifosfamide, and cisplatin (VeIP) will cure approximately 25% of patients as a second-line therapy. Patients are more likely to achieve a durable complete response if they had a testicular primary tumor and relapsed from a prior complete remission to first-line cisplatin-containing chemotherapy. Substitution of paclitaxel for vinblastine (TIP) in this setting was associated with durable remission in nearly two-thirds of patients. In contrast, for patients with a primary mediastinal nonseminoma or who did not achieve a complete response with first-line chemotherapy, then VeIP standard-dose salvage therapy is rarely beneficial. Such patients are usually managed with high-dose chemotherapy and/or surgical resection.
Chemotherapy consisting of dose-intensive, high-dose carboplatin plus high-dose etoposide, with peripheral blood stem cell support, induces a complete response in 25–40% of patients who have progressed after ifosfamide-containing salvage chemotherapy. Approximately one-half of the complete responses will be durable. High-dose therapy is standard of care for this patient population and has been suggested as the treatment of choice for all patients with relapsed or refractory disease. Paclitaxel is active when incorporated into high-dose combination programs. Cure is still possible in some relapsed patients.
EXTRAGONADAL GCT
The prognosis and management of patients with extragonadal GCT depends on the tumor histology and site of origin. All patients with a diagnosis of extragonadal GCT should have a testicular ultrasound examination. Nearly all patients with retroperitoneal or mediastinal seminoma achieve a durable complete response to BEP or EP. The clinical features of patients with primary retroperitoneal nonseminoma GCT are similar to those of patients with a primary tumor of testis origin, and careful evaluation will find evidence of a primary testicular GCT in about two-thirds of cases. In contrast, a primary mediastinal nonseminomatous GCT is associated with a poor prognosis; one-third of patients are cured with standard therapy (four cycles of BEP). Patients with newly diagnosed mediastinal nonseminoma are considered to have poor-risk disease and should be considered for clinical trials testing regimens of possibly greater efficacy. In addition, mediastinal nonseminoma is associated with hematologic disorders, including acute myelogenous leukemia, myelodysplastic syndrome, and essential thrombocytosis unrelated to previous chemotherapy. These hematologic disorders are very refractory to treatment. Nonseminoma of any primary site may change into other malignant histologies such as embryonal rhabdomyosarcoma or adenocarcinoma. This is called malignant transformation. i(12p) has been identified in the transformed cell type, indicating GCT clonal origin.
A group of patients with poorly differentiated tumors of unknown histogenesis, midline in distribution, and not associated with secretion of AFP or hCG has been described; a few (10–20%) are cured by standard cisplatin-containing chemotherapy. An i(12p) is present in ~25% of such tumors (the fraction that are cisplatin-responsive), confirming their origin from primitive germ cells. This finding is also predictive of the response to cisplatin-based chemotherapy and resulting long-term survival. These tumors are heterogeneous; neuroepithelial tumors and lymphoma may also present in this fashion.
FERTILITY
Infertility is an important consequence of the treatment of GCTs. Preexisting infertility or impaired fertility is often present. Azoospermia and/or oligospermia are present at diagnosis in at least 50% of patients with testicular GCTs. Ejaculatory dysfunction is associated with RPLND, and germ cell damage may result from cisplatin-containing chemotherapy. Nerve-sparing techniques to preserve the retroperitoneal sympathetic nerves have made retrograde ejaculation less likely in the subgroups of patients who are candidates for this operation. Spermatogenesis does recur in some patients after chemotherapy. However, because of the significant risk of impaired reproductive capacity, semen analysis and cryopreservation of sperm in a sperm bank should be recommended to all patients before treatment.
117 |
Gynecologic Malignancies |
OVARIAN CANCER
INCIDENCE AND PATHOLOGY
Cancer arising in or near the ovary is actually a collection of diverse malignancies. This collection of malignancies, often referred to as “ovary cancer,” is the most lethal gynecologic malignancy in the United States and other countries that routinely screen women for cervical neoplasia. In 2014, it was estimated that there were 21,980 cases of ovarian cancer with 14,270 deaths in the United States. The ovary is a complex and dynamic organ and, between the ages of approximately 11 and 50 years, is responsible for follicle maturation associated with egg maturation, ovulation, and cyclical sex steroid hormone production. These complex and linked biologic functions are coordinated through a variety of cells within the ovary, each of which possesses neoplastic potential. By far the most common and most lethal of the ovarian neoplasms arise from the ovarian epithelium or, alternatively, the neighboring specialized epithelium of the fallopian tube, uterine corpus, or cervix. Epithelial tumors may be benign (50%), malignant (33%), or of borderline malignancy (16%). Age influences risk of malignancy; tumors in younger women are more likely benign. The most common of the ovarian epithelial malignancies are serous tumors (50%); tumors of mucinous (25%), endometrioid (15%), clear cell (5%), and transitional cell histology or Brenner tumor (1%) represent smaller proportions of epithelial ovarian tumors. In contrast, stromal tumors arise from the steroid hormone–producing cells and likewise have different phenotypes and clinical presentations largely dependent on the type and quantity of hormone production. Tumors arising in the germ cell are most similar in biology and behavior to testicular tumors in males (Chap. 116).
Tumors may also metastasize to the ovary from breast, colon, appendiceal, gastric, and pancreatic primaries. Bilateral ovarian masses from metastatic mucin-secreting gastrointestinal cancers are termed Krukenberg tumors.
OVARIAN CANCER OF EPITHELIAL ORIGIN
Epidemiology and Pathogenesis A female has approximately a 1 in 72 lifetime risk (1.6%) of developing ovarian cancer, with the majority of affected women developing epithelial tumors. Each of the histologic variants of epithelial tumors is distinct with unique molecular features. As a group of malignancies, epithelial tumors of the ovary have a peak incidence in women in their sixties, although age at presentation can range across the extremes of adult life, with cases being reported in women in their twenties to nineties. Each histologic subtype of ovarian cancer likely has its own associated risk factors. Serous cancer, the most common type of epithelial ovarian cancer, is seen with increased frequency in women who are nulliparous or have a history of use of talc agents applied to the perineum; other risk factors include obesity and probably hormone replacement therapy. Protective factors include the use of oral contraceptives, multiparity, and breast-feeding. These protective factors are thought to work through suppression of ovulation and perhaps the associated reduction of ovulation associated inflammation of the ovarian epithelium or, alternatively, the serous epithelium located within the fimbriae of the fallopian tube. Other protective factors, such as fallopian tube ligation, are thought to protect the ovarian epithelium (or perhaps the distal fallopian tube fimbriae) from carcinogens that migrate from the vagina to the tubes and ovarian surface epithelium. Mucinous tumors are more frequent in women with a history of cigarette smoking, whereas endometrioid and clear cell tumors are more frequent in women with a history of endometriosis.
Considerable evidence now suggests that the precursor cell to serous carcinoma of the ovary might actually arise in the fimbria of the fallopian tube with extension or metastasis to the ovarian surface or capture of preneoplastic or neoplastic exfoliating tubal cells into an involuting ovarian follicle around the time of ovulation. Careful histologic and molecular analysis of tubal epithelium demonstrates molecular and histologic abnormalities, termed serous tubular intraepithelial carcinoma (STIC) lesions, in a high proportion of women undergoing risk-reducing salpingo-oophorectomies in the context of high-risk germline mutations in BRCA1 and BRCA2, as well as a modest proportion of women with ovarian cancer in the absence of such mutations.
![]() Genetic Risk Factors A variety of genetic syndromes substantially increase a woman’s risk of developing ovarian cancer. Approximately 10% of women with ovarian cancer have a germline mutation in one of two DNA repair genes: BRCA1 (chromosome 17q12-21) or BRCA2 (chromosome 13q12-13). Individuals inheriting a single copy of a mutant allele have a very high incidence of breast and ovarian cancer. Most of these women have a family history that is notable for multiple cases of breast and/or ovarian cancer, although inheritance through male members of the family can camouflage this genotype through several generations. The most common malignancy in these women is breast carcinoma, although women harboring germline BRCA1 mutations have a marked increased risk of developing ovarian malignancies in their forties and fifties with a 30–50% lifetime risk of developing ovarian cancer. Women harboring a mutation in BRCA2 have a lower penetrance of ovarian cancer with perhaps a 20–40% chance of developing this malignancy, with onset typically in their fifties or sixties. Women with a BRCA2 mutation also are at slightly increased risk of pancreatic cancer. Likewise women with mutations in the DNA mismatch repair genes associated with Lynch syndrome, type 2 (MSH2, MLH1, MLH6, PMS1, PMS2) may have a risk of ovarian cancer as high as 1% per year in their forties and fifties. Finally, a small group of women with familial ovarian cancer may have mutations in other BRCA-associated genes such as RAD51, CHK2, and others. Screening studies in this select population suggest that current screening techniques, including serial evaluation of the CA-125 tumor marker and ultrasound, are insufficient at detecting early-stage and curable disease, so women with these germline mutations are advised to undergo prophylactic removal of ovaries and fallopian tubes typically after completing childbearing and ideally before age 35–40 years. Early prophylactic oophorectomy also protects these women from subsequent breast cancer with a reduction of breast cancer risk of approximately 50%.
Genetic Risk Factors A variety of genetic syndromes substantially increase a woman’s risk of developing ovarian cancer. Approximately 10% of women with ovarian cancer have a germline mutation in one of two DNA repair genes: BRCA1 (chromosome 17q12-21) or BRCA2 (chromosome 13q12-13). Individuals inheriting a single copy of a mutant allele have a very high incidence of breast and ovarian cancer. Most of these women have a family history that is notable for multiple cases of breast and/or ovarian cancer, although inheritance through male members of the family can camouflage this genotype through several generations. The most common malignancy in these women is breast carcinoma, although women harboring germline BRCA1 mutations have a marked increased risk of developing ovarian malignancies in their forties and fifties with a 30–50% lifetime risk of developing ovarian cancer. Women harboring a mutation in BRCA2 have a lower penetrance of ovarian cancer with perhaps a 20–40% chance of developing this malignancy, with onset typically in their fifties or sixties. Women with a BRCA2 mutation also are at slightly increased risk of pancreatic cancer. Likewise women with mutations in the DNA mismatch repair genes associated with Lynch syndrome, type 2 (MSH2, MLH1, MLH6, PMS1, PMS2) may have a risk of ovarian cancer as high as 1% per year in their forties and fifties. Finally, a small group of women with familial ovarian cancer may have mutations in other BRCA-associated genes such as RAD51, CHK2, and others. Screening studies in this select population suggest that current screening techniques, including serial evaluation of the CA-125 tumor marker and ultrasound, are insufficient at detecting early-stage and curable disease, so women with these germline mutations are advised to undergo prophylactic removal of ovaries and fallopian tubes typically after completing childbearing and ideally before age 35–40 years. Early prophylactic oophorectomy also protects these women from subsequent breast cancer with a reduction of breast cancer risk of approximately 50%.
Presentation Neoplasms of the ovary tend to be painless unless they undergo torsion. Symptoms are therefore typically related to compression of local organs or due to symptoms from metastatic disease. Women with tumors localized to the ovary do have an increased incidence of symptoms including pelvic discomfort, bloating, and perhaps changes in a woman’s typical urinary or bowel pattern. Unfortunately, these symptoms are frequently dismissed by either the woman or her health care team. It is believed that high-grade tumors metastasize early in the neoplastic process. Unlike other epithelial malignancies, these tumors tend to exfoliate throughout the peritoneal cavity and thus present with symptoms associated with disseminated intraperitoneal tumors. The most common symptoms at presentation include a multimonth period of progressive complaints that typically include some combination of heartburn, nausea, early satiety, indigestion, constipation, and abdominal pain. Signs include the rapid increase in abdominal girth due to the accumulation of ascites that typically alerts the patient and her physician that the concurrent gastrointestinal symptoms are likely associated with serious pathology. Radiologic evaluation typically demonstrates a complex adnexal mass and ascites. Laboratory evaluation usually demonstrates a markedly elevated CA-125, a shed mucin (Muc 16) associated with, but not specific for, ovarian cancer. Hematogenous and lymphatic spread are seen but are not the typical presentation. Ovarian cancers are divided into four stages, with stage I tumors confined to the ovary, stage II malignancies confined to the pelvis, and stage III tumors confined to the peritoneal cavity (Table 117-1). These three stages are subdivided, with the most common presentation, stage IIIC, defined as tumors with bulky intraperitoneal disease. About 60% of women present with stage IIIC disease. Stage IV disease includes women with parenchymal metastases (liver, lung, spleen) or, alternatively, abdominal wall or pleural disease. The 40% not presenting with stage IIIC disease are roughly evenly distributed among the other stages, although mucinous and clear cell tumors are overrepresented in stage I tumors.
|
STAGING AND SURVIVAL IN GYNECOLOGIC MALIGNANCIES |
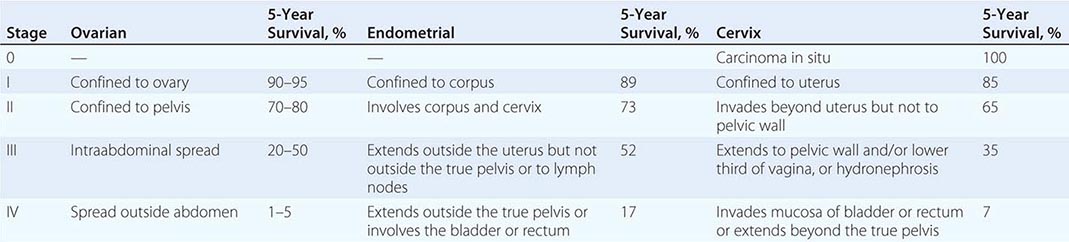
Screening Ovarian cancer is the fifth most lethal malignancy in women in the United States. It is curable in early stages, but seldom curable in advanced stages; hence, the development of effective screening strategies is of considerable interest. Furthermore, the ovary is well visualized with a variety of imaging techniques, most notably transvaginal ultrasound. Early-stage tumors often produce proteins that can be measured in the blood such as CA-125 and HE-4. Nevertheless, the incidence of ovarian cancer in the middle-aged female population is low, with only approximately 1 in 2000 women between the ages of 50 and 60 carrying an asymptomatic and undetected tumor. Thus effective screening techniques must be sensitive but, more importantly, highly specific to minimize the number of false-positive results. Even a screening test with 98% specificity and 50% sensitivity would have a positive predictive value of only about 1%. A large randomized study of active screening versus usual standard care demonstrated that a screening program consisting of six annual CA-125 measurements and four annual transvaginal ultrasounds in a population of women age 55–74 was not effective at reducing death from ovarian cancer and was associated with significant morbidity in the screened arm due to complications associated with diagnostic testing in the screened group. Although ongoing studies are evaluating the utility of alternative screening strategies, currently screening of normal-risk women is not recommended outside of a clinical trial.
OVARIAN SEX CORD AND STROMAL TUMORS
Epidemiology, Presentation, and Predisposing Syndromes Approximately 7% of ovarian neoplasms are stromal or sex cord tumors, with approximately 1800 cases expected each year in the United States. Ovarian stromal tumors or sex cord tumors are most common in women in their fifties or sixties, but tumors can present in the extremes of age, including the pediatric population. These tumors arise from the mesenchymal components of the ovary, including steroid-producing cells as well as fibroblasts. Essentially all of these tumors are of low malignant potential and present as unilateral solid masses. Three clinical presentations are common: the detection of an abdominal mass; abdominal pain due to ovarian torsion, intratumoral hemorrhage, or rupture; or signs and symptoms due to hormonal production by these tumors.
The most common hormone-producing tumors include thecomas, granulosa cell tumor, or juvenile granulosa tumors in children. These estrogen-producing tumors often present with breast tenderness as well as isosexual precocious pseudopuberty in children, menometrorrhagia, oligomenorrhea, or amenorrhea in premenopausal women, or alternatively as postmenopausal bleeding in older women. In some women, estrogen-associated secondary malignancies, such as endometrial or breast cancer, may present as synchronous malignancies. Alternatively, endometrial cancer may serve as the presenting malignancy with evaluation subsequently identifying a unilateral solid ovarian neoplasm that proves to be an occult granulosa cell tumor. Sertoli-Leydig tumors often present with hirsutism, virilization, and occasionally Cushing’s syndrome due to increased production of testosterone, androstenedione, or other 17-ketosteroids. Hormonally inert tumors include fibroma that presents as a solitary mass often in association with ascites and occasionally hydrothorax also known as Meigs’ syndrome. A subset of these tumors present in individuals with a variety of inherited disorders that predispose them to mesenchymal neoplasia. Associations include juvenile granulosa cell tumors and perhaps Sertoli-Leydig tumors with Ollier’s disease (multiple enchondromatosis) or Maffucci’s syndrome, ovarian sex cord tumors with annular tubules with Peutz-Jeghers syndrome, and fibromas with Gorlin’s disease. Essentially all granulosa tumors and a minority of juvenile granulosa cell tumors and thecomas have a defined somatic point mutation in the FOXL2 gene at C134W generated by replacement of cysteine with a guanine at position 402. About 30% of Sertoli-Leydig tumors harbor a mutation in the RNA-processing gene DICER in the RNAIIIb domain.
GERM CELL TUMORS OF THE OVARY
Germ cell tumors, like their counterparts in the testis, are cancers of germ cells. These totipotent cells contain the programming for differentiation to essentially all tissue types, and hence the germ cell tumors include a histologic menagerie of bizarre tumors, including benign teratomas and a variety of malignant tumors, such as immature teratomas, dysgerminomas, yolk sac malignancies, and choriocarcinomas. Benign teratoma (or dermoid cyst) is the most common germ cell neoplasm of the ovary and often presents in young woman. These tumors include a complex mixture of differentiated tissue including tissues from all three germ layers. In older women, these differentiated tumors can develop malignant transformation, most commonly squamous cell carcinomas. Malignant germ cell tumors include dysgerminomas, yolk sac tumors, immature teratomas, and embryonal carcinoma and choriocarcinomas. There are no known genetic abnormalities that unify these tumors. A subset of dysgerminomas harbor mutations in c-Kit oncogenes (as seen in gastrointestinal stromal tumors [GIST]), whereas a subset of germ cell tumors have isochromosome 12 abnormalities, as seen in testicular malignancies. In addition, a subset of dysgerminomas is associated with dysgenetic ovaries. Identification of a dysgerminoma arising in genotypic XY gonads is important in that it highlights the need to identify and remove the contralateral gonad due to risk of gonadoblastoma.
Presentation Germ cell tumors can present at all ages, but the peak age of presentation tends to be in females in their late teens or early twenties. Typically these tumors will become large ovarian masses, which eventually present as palpable low abdominal or pelvic masses. Like sex cord tumors, torsion or hemorrhage may present urgently or emergently as acute abdominal pain. Some of these tumors produce elevated levels of human chorionic gonadotropin (hCG), which can lead to isosexual precocious puberty when tumors present in younger girls. Unlike epithelial ovarian cancer, these tumors have a higher proclivity for nodal or hematogenous metastases. As with testicular tumors, some of these tumors tend to produce AFP (yolk sac tumors) or hCG (embryonal carcinoma, choriocarcinomas, and some dysgerminomas) that are reliable tumor markers.
FALLOPIAN TUBE CANCER
Transport of the egg to the uterus occurs via transit through the fallopian tube, with the distal ends of these tubes composed of fimbriae that drape about the ovarian surface and capture the egg as it erupts from the ovarian cortex. Fallopian tube malignancies are typically serous tumors. Previous teaching was that these malignancies were rare, but more careful histologic examination suggests that many “ovarian malignancies” might actually arise in the distal fimbria of the fallopian tube (see above). These women often present with adnexal masses, and like ovarian cancer, these tumors spread relatively early throughout the peritoneal cavity and respond to platinum and taxane therapy and have a natural history that is essentially identical to ovarian cancer (Table 117-1).
CERVICAL CANCER
GLOBAL CONSIDERATIONS
![]() Cervical cancer is the second most common and most lethal malignancy in women worldwide likely due to the widespread infection with high-risk strains of human papillomavirus (HPV) and limited utilization of or access to Pap smear screening in many nations throughout the world. Nearly 500,000 cases of cervical cancer are expected worldwide, with approximately 240,000 deaths annually. Cancer incidence is particularly high in women residing in Central and South America, the Caribbean, and southern and eastern Africa. Mortality rate is disproportionately high in Africa. In the United States, 12,360 women were diagnosed with cervical cancer and 4020 women died in 2014. Developed countries have looked at high-technology screening techniques for HPV involving automated polymerase chain reaction in thin preps that identify dysplastic cytology as well as high-risk HPV genetic material. Visual inspection of the cervix coated with acetic acid has demonstrated the ability to reduce mortality from cervical cancer with potential broad applicability in low-resource environments. The development of effective vaccines for high-risk HPV types makes it imperative to determine economical, socially acceptable, and logistically feasible strategies to deliver and distribute this vaccine to girls and boys before their engagement in sexual activity.
Cervical cancer is the second most common and most lethal malignancy in women worldwide likely due to the widespread infection with high-risk strains of human papillomavirus (HPV) and limited utilization of or access to Pap smear screening in many nations throughout the world. Nearly 500,000 cases of cervical cancer are expected worldwide, with approximately 240,000 deaths annually. Cancer incidence is particularly high in women residing in Central and South America, the Caribbean, and southern and eastern Africa. Mortality rate is disproportionately high in Africa. In the United States, 12,360 women were diagnosed with cervical cancer and 4020 women died in 2014. Developed countries have looked at high-technology screening techniques for HPV involving automated polymerase chain reaction in thin preps that identify dysplastic cytology as well as high-risk HPV genetic material. Visual inspection of the cervix coated with acetic acid has demonstrated the ability to reduce mortality from cervical cancer with potential broad applicability in low-resource environments. The development of effective vaccines for high-risk HPV types makes it imperative to determine economical, socially acceptable, and logistically feasible strategies to deliver and distribute this vaccine to girls and boys before their engagement in sexual activity.
HPV INFECTION AND PREVENTIVE VACCINATION
HPV is the primary neoplastic-initiating event in the vast majority of women with invasive cervical cancer. This double-strand DNA virus infects epithelium near the transformation zone of the cervix. More than 60 types of HPV are known, with approximately 20 types having the ability to generate high-grade dysplasia and malignancy. HPV-16 and -18 are the types most frequently associated with high-grade dysplasia and targeted by both U.S. Food and Drug Administration–approved vaccines. The large majority of sexually active adults are exposed to HPV, and most women clear the infection without specific intervention. The 8-kilobase HPV genome encodes seven early genes, most notably E6 and E7, which can bind to RB and p53, respectively. High-risk types of HPV encode E6 and E7 molecules that are particularly effective at inhibiting the normal cell cycle checkpoint functions of these regulatory proteins, leading to immortalization but not full transformation of cervical epithelium. A minority of woman will fail to clear the infection with subsequent HPV integration into the host genome. Over the course of as short as months but more typically years, some of these women develop high-grade dysplasia. The time from dysplasia to carcinoma is likely years to more than a decade and almost certainly requires the acquisition of other poorly defined genetic mutations within the infected and immortalized epithelium.
Risk factors for HPV infection and, in particular, dysplasia include a high number of sexual partners, early age of first intercourse, and history of venereal disease. Smoking is a cofactor; heavy smokers have a higher risk of dysplasia with HPV infection. HIV infection, especially when associated with low CD4+ T cell counts, is associated with a higher rate of high-grade dysplasia and likely a shorter latency period between infection and invasive disease. The administration of highly active antiretroviral therapy reduces the risk of high-grade dysplasia associated with HPV infection.
Currently approved vaccines include the recombinant proteins to the late proteins, L1 and L2, of HPV-16 and -18. Vaccination of women before the initiation of sexual activity dramatically reduces the rate of HPV-16 and -18 infection and subsequent dysplasia. There is also partial protection against other HPV types, although vaccinated women are still at risk for HPV infection and still require standard Pap smear screening. Although no randomized trial data demonstrate the utility of Pap smears, the dramatic drop in cervical cancer incidence and death in developed countries employing wide-scale screening provides strong evidence for its effectiveness. In addition, even visual inspection of the cervix with preapplication of acetic acid using a “see and treat” strategy has demonstrated a 30% reduction in cervical cancer death. The incorporation of HPV testing by polymerase chain reaction or other molecular techniques increases the sensitivity of detecting cervical pathology but at the cost of identifying many women with transient infections who require no specific medical intervention.
CLINICAL PRESENTATIONS
The majority of cervical malignancies are squamous cell carcinomas associated with HPV. Adenocarcinomas are also HPV-related and arise deep in the endocervical canal; they are typically not seen by visual inspection of the cervix and thus are often missed by Pap smear screening. A variety of rarer malignancies including atypical epithelial tumors, carcinoids, small cell carcinomas, sarcomas, and lymphomas have also been reported.
The principal role of Pap smear testing is the detection of asymptomatic preinvasive cervical dysplasia of squamous epithelial lining. Invasive carcinomas often have symptoms or signs including postcoital spotting or intermenstrual cycle bleeding or menometrorrhagia. Foul-smelling or persistent yellow discharge may also be seen. Presentations that include pelvic or sacral pain suggest lateral extension of the tumor into pelvic nerve plexus by either the primary tumor or a pelvic node and are signs of advanced-stage disease. Likewise, flank pain from hydronephrosis from ureteral compression or deep venous thrombosis from iliac vessel compression suggests either extensive nodal disease or direct extension of the primary tumor to the pelvic sidewall. The most common finding of physical exam is a visible tumor on the cervix.
TREATMENT CERVICAL CANCER
Scans are not part of the formal clinical staging of cervical cancer yet are very useful in planning appropriate therapy. CT can detect hydronephrosis indicative of pelvic sidewall disease but is not accurate at evaluating other pelvic structures. Magnetic resonance imaging (MRI) is more accurate at estimating uterine extension and paracervical extension of disease into soft tissues typically bordered by broad and cardinal ligaments that support the uterus in the central pelvis. Positron emission tomography (PET) scan is the most accurate technique for evaluating the pelvis and more importantly nodal (pelvic, para-aortic, and scalene) sites for disease. This technique seems more prognostic and accurate than CT, MRI, or lymphangiogram, especially in the para-aortic region.
Stage I cervical tumors are confined to the cervix, whereas stage II tumors extend into the upper vagina or paracervical soft tissue (Fig. 117-1). Stage III tumors extend to the lower vagina or the pelvic sidewalls, whereas stage IV tumors invade the bladder or rectum or have spread to distant sites. Very small stage I cervical tumors can be treated with a variety of surgical procedures. In young women desiring to maintain fertility, radical trachelectomy removes the cervix with subsequent anastomosis of the upper vagina to the uterine corpus. Larger cervical tumors confined to the cervix can be treated with either surgical resection or radiation therapy in combination with cisplatin-based chemotherapy with a high chance of cure. Larger tumors that extend regionally down the vagina or into the paracervical soft tissues or the pelvic sidewalls are treated with combination chemotherapy and radiation therapy. The treatment of recurrent or metastatic disease is unsatisfactory due to the relative resistance of these tumors to chemotherapy and currently available biological agents, although bevacizumab, a monoclonal antibody that is said to inhibit tumor-associated angiogenesis, has demonstrated clinically meaningful activity in the management of metastatic disease.
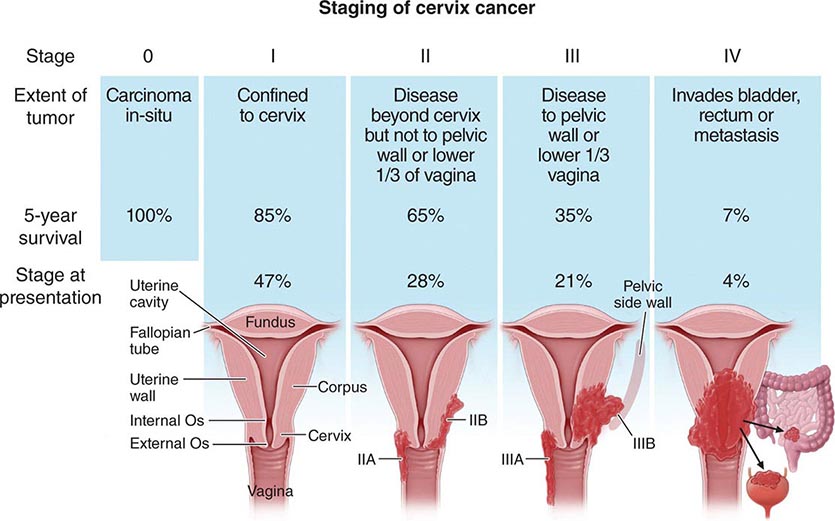
FIGURE 117-1 Anatomic display of the stages of cervix cancer defined by location, extent of tumor, frequency of presentation, and 5-year survival.
UTERINE CANCER
EPIDEMIOLOGY
Several different tumor types arise in uterine corpus. Most tumors arise in the glandular lining and are endometrial adenocarcinomas. Tumors can also arise in the smooth muscle; most are benign (uterine leiomyoma), with a small minority of tumors being sarcomas. The endometrioid histologic subtype of endometrial cancer is the most common gynecologic malignancy in the United States. In 2014, an estimated 52,630 women were diagnosed with cancer of the uterine corpus, with 8590 deaths from the disease. Development of these tumors is a multistep process, with estrogen playing an important early role in driving endometrial gland proliferation. Relative overexposure to this class of hormones is a risk factor for the subsequent development of endometrioid tumors. In contrast, progestins drive glandular maturation and are protective. Hence, women with high endogenous or pharmacologic exposure to estrogens, especially if unopposed by progesterone, are at high risk for endometrial cancer. Obese women, women treated with unopposed estrogens, or women with estrogen-producing tumors (such as granulosa cell tumors of the ovary) are at higher risk for endometrial cancer. In addition, treatment with tamoxifen, which has antiestrogenic effects in breast tissue but estrogenic effects in uterine epithelium, is associated with an increased risk of endometrial cancer. Events such as the loss of the PTEN tumor suppressor gene with activation and often additional mutations in the PIK-3CA/AKT pathways likely serve as secondary events in carcinogenesis. The Cancer Genome Atlas Research Network has demonstrated that endometrioid tumors can be divided into four subgroups: ultramutated, microsatellite instability hypermutated, copy number low, and copy number high subgroups. These groups have different natural histories; therapy for these subgroups may eventually be individualized. Serous tumors of the uterine corpus represent approximately 5–10% of epithelial tumors of the uterine corpus and possess distinct molecular characteristics that are most similar to those seen in serous tumors arising in the ovary or fallopian tube.
Women with a mutation in one of a series of DNA mismatch repair genes associated with the Lynch syndrome, also known as hereditary nonpolyposis colon cancer (HNPCC), are at increased risk for endometrioid endometrial carcinoma. These individuals have germline mutations in MSH2, MLH1, and in rare cases PMS1 and PMS2, with resulting microsatellite instability and hypermutation. Individuals who carry these mutations typically have a family history of cancer and are at markedly increased risk for colon cancer and modestly increased risk for ovarian cancer and a variety of other tumors. Middle-aged women with HNPCC carry a 4% annual risk of endometrial cancer and a relative overall risk of approximately 200-fold as compared to age-matched women without HNPCC.
PRESENTATIONS
The majority of women with tumors of the uterine corpus present with postmenopausal vaginal bleeding due to shedding of the malignant endometrial lining. Premenopausal women often will present with atypical bleeding between typical menstrual cycles. These signs typically bring a woman to the attention of a health care professional, and hence the majority of women present with early-stage disease with the tumor confined to the uterine corpus. Diagnosis is typically established by endometrial biopsy. Epithelial tumors may spread to pelvic or para-aortic lymph nodes. Pulmonary metastases can appear later in the natural history of this disease but are very uncommon at initial presentation. Serous tumors tend to have patterns of spread much more reminiscent of ovarian cancer with many patients presenting with disseminated peritoneal disease and sometimes ascites. Some women presenting with uterine sarcomas will present with pelvic pain. Nodal metastases are uncommon with sarcomas, which are more likely to present with either intraabdominal disease or pulmonary metastases.
GESTATIONAL TROPHOBLASTIC TUMORS
GLOBAL CONSIDERATIONS
![]() Gestational trophoblastic diseases represent a spectrum of neoplasia from benign hydatidiform mole to choriocarcinoma due to persistent trophoblastic disease associated most commonly with molar pregnancy but occasionally seen after normal gestation. The most common presentations of trophoblastic tumors are partial and complete molar pregnancies. These represent approximately 1 in 1500 conceptions in developed Western countries. The incidence widely varies globally, with areas in Southeast Asia having a much higher incidence of molar pregnancy. Regions with high molar pregnancy rates are often associated with diets low in carotene and animal fats.
Gestational trophoblastic diseases represent a spectrum of neoplasia from benign hydatidiform mole to choriocarcinoma due to persistent trophoblastic disease associated most commonly with molar pregnancy but occasionally seen after normal gestation. The most common presentations of trophoblastic tumors are partial and complete molar pregnancies. These represent approximately 1 in 1500 conceptions in developed Western countries. The incidence widely varies globally, with areas in Southeast Asia having a much higher incidence of molar pregnancy. Regions with high molar pregnancy rates are often associated with diets low in carotene and animal fats.
RISK FACTORS
Trophoblastic tumors result from the outgrowth or persistence of placental tissue. They arise most commonly in the uterus but can also arise in other sites such as the fallopian tubes due to ectopic pregnancy. Risk factors include poorly defined dietary and environmental factors as well as conceptions at the extremes of reproductive age, with the incidence particularly high in females conceiving younger than age 16 or older than age 50. In older women, the incidence of molar pregnancy might be as high as one in three, likely due to increased risk of abnormal fertilization of the aged ova. Most trophoblastic neoplasms are associated with complete moles, diploid tumors with all genetic material from the paternal donor (known as parental disomy). This is thought to occur when a single sperm fertilizes an enucleate egg that subsequently duplicates the paternal DNA. Trophoblastic proliferation occurs with exuberant villous stroma. If pseudopregnancy extends out past the 12th week, fluid progressively accumulates within the stroma, leading to “hydropic changes.” There is no fetal development in complete moles.
Partial moles arise from the fertilization of an egg with two sperm; hence two-thirds of genetic material is paternal in these triploid tumors. Hydropic changes are less dramatic, and fetal development can often occur through late first trimester or early second trimester at which point spontaneous abortion is common. Laboratory findings will include excessively high hCG and high AFP. The risk of persistent gestational trophoblastic disease after partial mole is approximately 5%. Complete and partial moles can be noninvasive or invasive. Myometrial invasion occurs in no more than one in six complete moles and a lower portion of partial moles.
PRESENTATION OF INVASIVE TROPHOBLASTIC DISEASE
The clinical presentation of molar pregnancy is changing in developed countries due to the early detection of pregnancy with home pregnancy kits and the very early use of Doppler and ultrasound to evaluate the early fetus and uterine cavity for evidence of a viable fetus. Thus, in these countries, the majority of women presenting with trophoblastic disease have their moles detected early and have typical symptoms of early pregnancy including nausea, amenorrhea, and breast tenderness. With uterine evacuation of early complete and partial moles, most women experience spontaneous remission of their disease as monitored by serial hCG levels. These women require no chemotherapy. Patients with persistent elevation of hCG or rising hCG after evacuation have persistent or actively growing gestational trophoblastic disease and require therapy. Most series suggest that between 15 and 25% of women will have evidence of persistent gestational trophoblastic disease after molar evacuation.
In women who lack access to prenatal care, presenting symptoms can be life threatening including the development of preeclampsia or even eclampsia. Hyperthyroidism can also be seen. Evacuation of large moles can be associated with life-threatening complications including uterine perforation, volume loss, high-output cardiac failure, and adult respiratory distress syndrome (ARDS).
For women with evidence of rising hCG or radiologic confirmation of metastatic or persistent regional disease, prognosis can be estimated through a variety of scoring algorithms that identify those women at low, intermediate, and high risk for requiring multiagent chemotherapy. In general, women with widely metastatic nonpulmonary disease, very elevated hCG, and prior normal antecedent term pregnancy are considered at high risk and typically require multiagent chemotherapy for cure.
118 |
Primary and Metastatic Tumors of the Nervous System |
Primary brain tumors are diagnosed in approximately 52,000 people each year in the United States. At least one-half of these tumors are malignant and associated with a high mortality. Glial tumors account for about 30% of all primary brain tumors, and 80% of those are malignant. Meningiomas account for 35%, vestibular schwannomas 10%, and central nervous system (CNS) lymphomas about 2%. Brain metastases are three times more common than all primary brain tumors combined and are diagnosed in approximately 150,000 people each year. Metastases to the leptomeninges and epidural space of the spinal cord each occur in approximately 3–5% of patients with systemic cancer and are also a major cause of neurologic disability.
APPROACH TO THE PATIENT:
Primary and Metastatic Tumors of the Nervous System
CLINICAL FEATURES
Brain tumors of any type can present with a variety of symptoms and signs that fall into two categories: general and focal; patients often have a combination of the two (Table 118-1). General or nonspecific symptoms include headache, with or without nausea or vomiting, cognitive difficulties, personality change, and gait disorder. Generalized symptoms arise when the enlarging tumor and its surrounding edema cause an increase in intracranial pressure or direct compression of cerebrospinal fluid (CSF) circulation leading to hydrocephalus. The classic headache associated with a brain tumor is most evident in the morning and improves during the day, but this particular pattern is actually seen in a minority of patients. Headaches are often holocephalic but can be ipsilateral to the side of a tumor. Occasionally, headaches have features of a typical migraine with unilateral throbbing pain associated with visual scotoma. Personality changes may include apathy and withdrawal from social circumstances, mimicking depression. Focal or lateralizing findings include hemiparesis, aphasia, or visual field defect. Lateralizing symptoms are typically subacute and progressive. A visual field defect is often unnoticed by the patient; its presence may only be revealed after it leads to an injury such as an automobile accident occurring in the blind visual field. Language difficulties may be mistaken for confusion. Seizures are a common presentation of brain tumors, occurring in about 25% of patients with brain metastases or malignant gliomas but can be the presenting symptom in up to 90% of patients with a low-grade glioma. All seizures that arise from a brain tumor will have a focal onset whether or not it is apparent clinically.
|
SYMPTOMS AND SIGNS AT PRESENTATION OF BRAIN TUMORS |
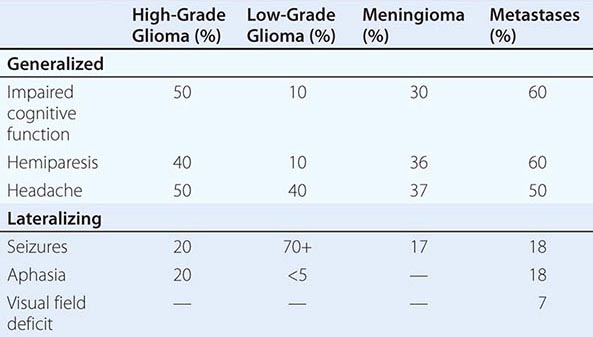
NEUROIMAGING
Cranial MRI is the preferred diagnostic test for any patient suspected of having a brain tumor and should be performed with gadolinium contrast administration. Computed tomography (CT) scan should be reserved for those patients unable to undergo magnetic resonance imaging (MRI; e.g., pacemaker). Malignant brain tumors—whether primary or metastatic—typically enhance with gadolinium and may have central areas of necrosis; they are characteristically surrounded by edema of the neighboring white matter. Low-grade gliomas usually do not enhance with gadolinium and are best appreciated on fluid-attenuated inversion recovery (FLAIR) MRIs. Meningiomas have a characteristic appearance on MRI because they are dural-based with a dural tail and compress but do not invade the brain. Dural metastases or a dural lymphoma can have a similar appearance. Imaging is characteristic for many primary and metastatic tumors and sometimes will suffice to establish a diagnosis when the location precludes surgical intervention (e.g., brainstem glioma). Functional MRI is useful in presurgical planning to define eloquent sensory, motor, or language cortex. Positron emission tomography (PET) is useful in determining the metabolic activity of the lesions seen on MRI; MR perfusion and spectroscopy can provide information on blood flow or tissue composition. These techniques may help distinguish tumor progression from necrotic tissue as a consequence of treatment with radiation and chemotherapy or identify foci of high-grade tumor in an otherwise low-grade-appearing glioma.
Neuroimaging is the only test necessary to diagnose a brain tumor. Laboratory tests are rarely useful, although patients with metastatic disease may have elevation of a tumor marker in their serum that reflects the presence of brain metastases (e.g., β human chorionic gonadotropin [β-hCG] from testicular cancer). Additional testing such as cerebral angiogram, electroencephalogram (EEG), or lumbar puncture is rarely indicated or helpful.
PRIMARY BRAIN TUMORS
PATHOGENESIS
No underlying cause has been identified for the majority of primary brain tumors. The only established risk factors are exposure to ionizing radiation (meningiomas, gliomas, and schwannomas) and immunosuppression (primary CNS lymphoma). Evidence for an association with exposure to electromagnetic fields including cellular telephones, head injury, foods containing N-nitroso compounds, or occupational risk factors are unproven. A small minority of patients have a family history of brain tumors. Some of these familial cases are associated with genetic syndromes (Table 118-2).
|
GENETIC SYNDROMES ASSOCIATED WITH PRIMARY BRAIN TUMORS |
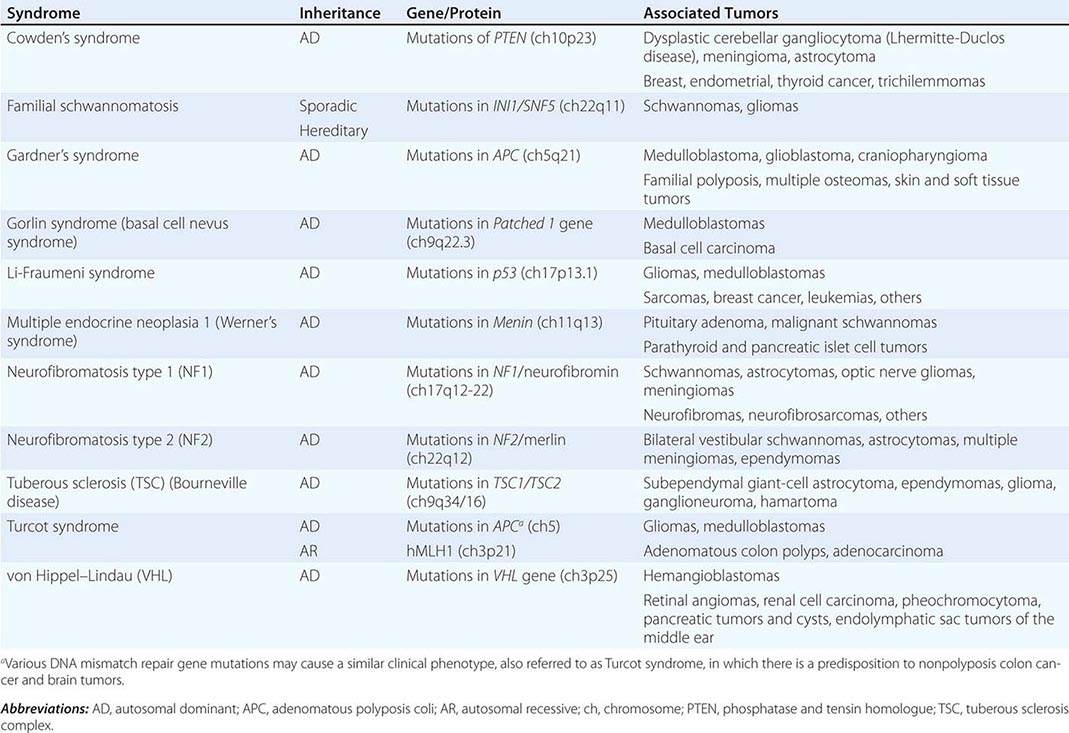
As with other neoplasms, brain tumors arise as a result of a multistep process driven by the sequential acquisition of genetic alterations. These include loss of tumor-suppressor genes (e.g., p53 and phosphatase and tensin homolog on chromosome 10 [PTEN]) and amplification and overexpression of protooncogenes such as the epidermal growth factor receptor (EGFR) and the platelet-derived growth factor receptors (PDGFR). The accumulation of these genetic abnormalities results in uncontrolled cell growth and tumor formation.
Important progress has been made in understanding the molecular pathogenesis of several types of brain tumors, including glioblastoma and medulloblastoma. Morphologically indistinguishable glioblastomas can be separated into four subtypes defined by molecular profiling: (1) classical, characterized by overactivation of the EGFR pathway; (2) proneural, characterized by overexpression of PDGFRA, mutations of the isocitrate dehydrogenase (IDH) 1 and 2 genes, and expression of neural markers; (3) mesenchymal, defined by expression of mesenchymal markers and loss of NF1; and (4) neural, characterized by overactivity of EGFR and expression of neural markers. The clinical implications of these subtypes are under study. Medulloblastoma is the other primary brain tumor that has been highly analyzed, and four molecular subtypes have also been identified: (1) the Wnt subtype is defined by a mutation in β-catenin and has an excellent prognosis; (2) the SHH subtype has mutations in PTCH1, SMO, GLI2, or SUFU and has an intermediate prognosis; (3) group 3 has elevated MYC expression and has the worst prognosis; and (4) group 4 is characterized by isochromosome 17q. Targeted therapeutics are under development for some of the medulloblastoma subtypes, especially the SHH group.
INTRINSIC “MALIGNANT” TUMORS
ASTROCYTOMAS
These are infiltrative tumors with a presumptive glial cell of origin. The World Health Organization (WHO) classifies astrocytomas into four prognostic grades based on histologic features: grade I (pilocytic astrocytoma, subependymal giant cell astrocytoma); grade II (diffuse astrocytoma); grade III (anaplastic astrocytoma); and grade IV (glioblastoma). Grades I and II are considered low-grade astrocytomas, and grades III and IV are considered high-grade astrocytomas.
Low-Grade Astrocytoma These tumors occur predominantly in children and young adults.
GRADE I ASTROCYTOMAS Pilocytic astrocytomas (WHO grade I) are the most common tumor of childhood. They occur typically in the cerebellum but may also be found elsewhere in the neuraxis, including the optic nerves and brainstem. Frequently they appear as cystic lesions with an enhancing mural nodule. These are well-demarcated lesions that are potentially curable if they can be resected completely. Giant-cell subependymal astrocytomas are usually found in the ventricular wall of patients with tuberous sclerosis. They often do not require intervention but can be treated surgically or with inhibitors of the mammalian target of rapamycin (mTOR).
GRADE II ASTROCYTOMAS These are infiltrative tumors that usually present with seizures in young adults. They appear as nonenhancing tumors with increased T2/FLAIR signal (Fig. 118-1). If feasible, patients should undergo maximal surgical resection, although complete resection is rarely possible because of the invasive nature of the tumor. Radiation therapy (RT) is helpful, but there is no difference in overall survival between RT administered postoperatively or delayed until the time of tumor progression. There is increasing evidence that chemotherapeutic agents such as temozolomide, an oral alkylating agent, can be helpful in some patients. The tumor transforms to a malignant astrocytoma in the majority of patients, leading to variable survival with a median of about 5 years.
FIGURE 118-1 Fluid-attenuated inversion recovery (FLAIR) MRI of a left frontal low-grade astrocytoma. This lesion did not enhance.
High-Grade Astrocytoma
GRADE III (ANAPLASTIC) ASTROCYTOMA These account for approximately 15–20% of high-grade astrocytomas. They generally present in the fourth and fifth decades of life as variably enhancing tumors. Treatment is the same as for glioblastoma, consisting of maximal safe surgical resection followed by RT with concurrent and adjuvant temozolomide or by RT and adjuvant temozolomide alone.
GRADE IV ASTROCYTOMA (GLIOBLASTOMA) Glioblastoma accounts for the majority of high-grade astrocytomas. They are the most common malignant primary brain tumor, with over 10,000 cases diagnosed each year in the United States. Patients usually present in the sixth and seventh decades of life with headache, seizures, or focal neurologic deficits. The tumors appear as ring-enhancing masses with central necrosis and surrounding edema (Fig. 118-2). These are highly infiltrative tumors, and the areas of increased T2/FLAIR signal surrounding the main tumor mass contain invading tumor cells. Treatment involves maximal surgical resection followed by partial-field external-beam RT (6000 cGy in thirty 200-cGy fractions) with concomitant temozolomide, followed by 6–12 months of adjuvant temozolomide. With this regimen, median survival is increased to 14.6 months compared to only 12 months with RT alone, and 2-year survival is increased to 27%, compared to 10% with RT alone. Patients whose tumor contains the DNA repair enzyme O6-methylguanine-DNA methyltransferase (MGMT) are relatively resistant to temozolomide and have a worse prognosis compared to those whose tumors contain low levels of MGMT as a result of silencing of the MGMT gene by promoter hypermethylation. Implantation of biodegradable polymers containing the chemotherapeutic agent carmustine into the tumor bed after resection of the tumor also produces a modest improvement in survival.
FIGURE 118-2 Postgadolinium T1 MRI of a large cystic left frontal glioblastoma.
Despite optimal therapy, glioblastomas invariably recur. Treatment options for recurrent disease may include reoperation, carmustine wafers, and alternate chemotherapeutic regimens. Reirradiation is rarely helpful. Bevacizumab, a humanized vascular endothelial growth factor (VEGF) monoclonal antibody, has activity in recurrent glioblastoma, increasing progression-free survival and reducing peritumoral edema and glucocorticoid use (Fig. 118-3). Treatment decisions for patients with recurrent glioblastoma must be made on an individual basis, taking into consideration such factors as previous therapy, time to relapse, performance status, and quality of life. Whenever feasible, patients with recurrent disease should be enrolled in clinical trials. Novel therapies undergoing evaluation in patients with glioblastoma include targeted molecular agents directed at receptor tyrosine kinases and signal transduction pathways; antiangiogenic agents, especially those directed at the VEGF receptors; chemotherapeutic agents that cross the blood-brain barrier more effectively than currently available drugs; gene therapy; immunotherapy; and infusion of radiolabeled drugs and targeted toxins into the tumor and surrounding brain by means of convection-enhanced delivery.
FIGURE 118-3 Postgadolinium T1 MRI of a recurrent glioblastoma before (A) and after (B) administration of bevacizumab. Note the decreased enhancement and mass effect.
The most important adverse prognostic factors in patients with high-grade astrocytomas are older age, histologic features of glioblastoma, poor Karnofsky performance status, and unresectable tumor. Patients whose tumor contains an unmethylated MGMT promoter resulting in the presence of the repair enzyme in tumor cells and resistance to temozolomide also have a worse prognosis.
Gliomatosis Cerebri Rarely, patients may present with a highly infiltrating, nonenhancing tumor of variable histologic grade involving more than two lobes of the brain. These tumors may be indolent initially, but will eventually behave aggressively and have a poor outcome. Treatment involves RT and temozolomide chemotherapy.
OLIGODENDROGLIOMA
Oligodendrogliomas account for approximately 15–20% of gliomas. They are classified by the WHO into well-differentiated oligodendrogliomas (grade II) or anaplastic oligodendrogliomas (AOs) (grade III). Tumors with oligodendroglial components have distinctive pathologic features such as perinuclear clearing—giving rise to a “fried-egg” appearance—and a reticular pattern of blood vessel growth. Some tumors have both an oligodendroglial as well as an astrocytic component. These mixed tumors, or oligoastrocytomas (OAs), are also classified into well-differentiated OA (grade II) or anaplastic oligoastrocytomas (AOAs) (grade III).
Grade II oligodendrogliomas and OAs are generally more responsive to therapy and have a better prognosis than pure astrocytic tumors. These tumors present similarly to grade II astrocytomas in young adults. The tumors are nonenhancing and often partially calcified. They should be treated with surgery and, if necessary, RT and chemotherapy. Patients with oligodendrogliomas have a median survival in excess of 10 years.
AOs and AOAs present in the fourth and fifth decades as variably enhancing tumors. They are more responsive to therapy than grade III astrocytomas. Co-deletion of chromosomes 1p and 19q, mediated by an unbalanced translocation of 19p to 1q, occurs in 61–89% of patients with AO and 14–20% of patients with AOA. Tumors with the 1p and 19q co-deletion are particularly sensitive to chemotherapy with procarbazine, lomustine (cyclohexylchloroethylnitrosourea [CCNU]), and vincristine (PCV) or temozolomide, as well as to RT. Median survival of patients with AO or AOA is approximately 3–6 years, but those with co-deleted tumors can have a median survival of 10–14 years if treated with RT and chemotherapy.
EPENDYMOMAS
Ependymomas are tumors derived from ependymal cells that line the ventricular surface. They account for approximately 5% of childhood tumors and frequently arise from the wall of the fourth ventricle in the posterior fossa. Although adults can have intracranial ependymomas, they occur more commonly in the spine, especially in the filum terminale of the spinal cord where they have a myxopapillary histology. Ependymomas that can be completely resected are potentially curable. Partially resected ependymomas will recur and require irradiation. The less common anaplastic ependymoma is more aggressive and is treated with resection and RT; chemotherapy has limited efficacy. Subependymomas are slow-growing benign lesions arising in the wall of ventricles that often do not require treatment.
OTHER LESS COMMON GLIOMAS
Gangliogliomas and pleomorphic xanthoastrocytomas occur in young adults. They behave as more indolent forms of grade II gliomas and are treated in the same way. Brainstem gliomas usually occur in children or young adults. Despite treatment with RT and chemotherapy, the prognosis is poor, with a median survival of only 1 year. Gliosarcomas contain both an astrocytic as well as a sarcomatous component and are treated in the same way as glioblastomas.
PRIMARY CENTRAL NERVOUS SYSTEM LYMPHOMA
Primary central nervous system lymphoma (PCNSL) is a rare non-Hodgkin lymphoma accounting for less than 3% of primary brain tumors. For unclear reasons, its incidence is increasing, particularly in immunocompetent individuals.
PCNSL in immunocompetent patients usually consists of a diffuse large B cell lymphoma. PCNSL may also occur in immunocompromised patients, usually those infected with the human immunodeficiency virus (HIV) or organ transplant recipients on immunosuppressive therapy. PCNSL in immunocompromised patients is typically large cell with immunoblastic and more aggressive features. These patients are usually severely immunocompromised, with CD4 counts of less than 50/mL. The Epstein-Barr virus (EBV) frequently plays an important role in the pathogenesis of HIV-related PCNSL.
Immunocompetent patients with PCNSL are older (median 60 years) compared to patients with HIV-related PCNSL (median 31 years). PCNSL usually presents as a mass lesion, with neuropsychiatric symptoms, symptoms of increased intracranial pressure, lateralizing signs, or seizures.
On contrast-enhanced MRI, PCNSL usually appears as a densely enhancing tumor (Fig. 118-4). Immunocompetent patients have solitary lesions more often than immunosuppressed patients. Frequently there is involvement of the basal ganglia, corpus callosum, or periventricular region. Although the imaging features are often characteristic, PCNSL can sometimes be difficult to differentiate from high-grade gliomas, infections, or demyelination. Stereotactic biopsy is necessary to obtain a histologic diagnosis. Whenever possible, glucocorticoids should be withheld until after the biopsy has been obtained because they have a cytolytic effect on lymphoma cells and may lead to nondiagnostic tissue. In addition, patients should be tested for HIV and the extent of disease should be assessed by performing PET or CT of the body, MRI of the spine, CSF analysis, and slit-lamp examination of the eye. Bone marrow biopsy and testicular ultrasound are occasionally performed.
FIGURE 118-4 Postgadolinium T1 MRI demonstrating a large bifrontal primary central nervous system lymphoma (PCNSL). The periventricular location and diffuse enhancement pattern are characteristic of lymphoma.
MEDULLOBLASTOMAS
Medulloblastomas are the most common malignant brain tumor of childhood, accounting for approximately 20% of all primary CNS tumors among children. They arise from granule cell progenitors or from multipotent progenitors from the ventricular zone. Approximately 5% of children have inherited disorders with germline mutations of genes that predispose to the development of medulloblastoma. Gorlin syndrome, the most common of these inherited disorders, is due to mutations in the patched-1 (PTCH-1) gene, a key component in the sonic hedgehog pathway. Turcot syndrome, caused by mutations in the adenomatous polyposis coli (APC) gene and familial adenomatous polyposis, has also been associated with an increased incidence of medulloblastoma. Histologically, medulloblastomas are highly cellular tumors with abundant dark staining, round nuclei, and rosette formation (Homer-Wright rosettes). They present with headache, ataxia, and signs of brainstem involvement. On MRI they appear as densely enhancing tumors in the posterior fossa, sometimes associated with hydrocephalus. Seeding of the CSF is common. Treatment involves maximal surgical resection, craniospinal irradiation, and chemotherapy with agents such as cisplatin, lomustine, cyclophosphamide, and vincristine. Approximately 70% of patients have long-term survival but usually at the cost of significant neurocognitive impairment. A major goal of current research is to improve survival while minimizing long-term complications.
PINEAL REGION TUMORS
A large number of tumors can arise in the region of the pineal gland. These typically present with headache, visual symptoms, and hydrocephalus. Patients may have Parinaud syndrome characterized by impaired upgaze and accommodation. Some pineal tumors such as pineocytomas and benign teratomas can be treated simply by surgical resection. Germinomas respond to irradiation, whereas pineoblastomas and malignant germ cell tumors require craniospinal radiation and chemotherapy.
EXTRINSIC “BENIGN” TUMORS
MENINGIOMAS
Meningiomas are diagnosed with increasing frequency as more people undergo neuroimaging for various indications. They are now the most common primary brain tumor, accounting for approximately 35% of the total. Their incidence increases with age. They tend to be more common in women and in patients with neurofibromatosis type 2. They also occur more commonly in patients with a past history of cranial irradiation.
Meningiomas arise from the dura mater and are composed of neoplastic meningothelial (arachnoidal cap) cells. They are most commonly located over the cerebral convexities, especially adjacent to the sagittal sinus, but can also occur in the skull base and along the dorsum of the spinal cord. Meningiomas are classified by the WHO into three histologic grades of increasing aggressiveness: grade I (benign), grade II (atypical), and grade III (malignant).
Many meningiomas are found incidentally following neuroimaging for unrelated reasons. They can also present with headaches, seizures, or focal neurologic deficits. On imaging studies they have a characteristic appearance usually consisting of a partially calcified, densely enhancing extraaxial tumor arising from the dura (Fig. 118-5). Occasionally they may have a dural tail, consisting of thickened, enhanced dura extending like a tail from the mass. The main differential diagnosis of meningioma is a dural metastasis.
FIGURE 118-5 Postgadolinium T1 MRI demonstrating multiple meningiomas along the falx and left parietal cortex.
If the meningioma is small and asymptomatic, no intervention is necessary and the lesion can be observed with serial MRI studies. Larger, symptomatic lesions should be resected. If complete resection is achieved, the patient is cured. Incompletely resected tumors tend to recur, although the rate of recurrence can be very slow with grade I tumors. Tumors that cannot be resected, or can only be partially removed, may benefit from treatment with external-beam RT or stereotactic radiosurgery (SRS). These treatments may also be helpful in patients whose tumor has recurred after surgery. Hormonal therapy and chemotherapy are currently unproven.
Rarer tumors that resemble meningiomas include hemangiopericytomas and solitary fibrous tumors. These are treated with surgery and RT but have a higher propensity to recur locally or metastasize systemically.
SCHWANNOMAS
These are generally benign tumors arising from the Schwann cells of cranial and spinal nerve roots. The most common schwannomas, termed vestibular schwannomas or acoustic neuromas, arise from the vestibular portion of the eighth cranial nerve and account for approximately 9% of primary brain tumors. Patients with neurofibromatosis type 2 have a high incidence of vestibular schwannomas that are frequently bilateral. Schwannomas arising from other cranial nerves, such as the trigeminal nerve (cranial nerve V), occur with much lower frequency. Neurofibromatosis type 1 is associated with an increased incidence of schwannomas of the spinal nerve roots.
Vestibular schwannomas may be found incidentally on neuroimaging or present with progressive unilateral hearing loss, dizziness, tinnitus, or less commonly, symptoms resulting from compression of the brainstem and cerebellum. On MRI they appear as densely enhancing lesions, enlarging the internal auditory canal and often extending into the cerebellopontine angle (Fig. 118-6). The differential diagnosis includes meningioma. Very small, asymptomatic lesions can be observed with serial MRIs. Larger lesions should be treated with surgery or SRS. The optimal treatment will depend on the size of the tumor, symptoms, and the patient’s preference. In patients with small vestibular schwannomas and relatively intact hearing, early surgical intervention increases the chance of preserving hearing.
FIGURE 118-6 Postgadolinium MRI of a right vestibular schwannoma. The tumor can be seen to involve the internal auditory canal.
PITUITARY TUMORS (CHAP. 401e)
These account for approximately 9% of primary brain tumors. They can be divided into functioning and nonfunctioning tumors. Functioning tumors are usually microadenomas (<1 cm in diameter) that secrete hormones and produce specific endocrine syndromes (e.g., acromegaly for growth hormone–secreting tumors, Cushing syndrome for adrenocorticotropic hormone [ACTH]-secreting tumors, and galactorrhea, amenorrhea, and infertility for prolactin-secreting tumors). Nonfunctioning pituitary tumors tend to be macroadenomas (>1 cm) that produce symptoms by mass effect, giving rise to headaches, visual impairment (such as bitemporal hemianopia), and hypopituitarism. Prolactin-secreting tumors respond well to dopamine agonists such as bromocriptine and cabergoline. Other pituitary tumors usually require treatment with surgery and sometimes RT or radiosurgery and hormonal therapy.
CRANIOPHARYNGIOMAS
Craniopharyngiomas are rare, usually suprasellar, partially calcified, solid, or mixed solid-cystic benign tumors that arise from remnants of Rathke’s pouch. They have a bimodal distribution, occurring predominantly in children but also between the ages of 55 and 65 years. They present with headaches, visual impairment, and impaired growth in children and hypopituitarism in adults. Treatment involves surgery, RT, or a combination of the two.
OTHER BENIGN TUMORS
Dysembryoplastic Neuroepithelial Tumors (DNTs) These are benign, supratentorial tumors, usually in the temporal lobe. They typically occur in children and young adults with a long-standing history of seizures. Surgical resection is curative.
Epidermoid Cysts These consist of squamous epithelium surrounding a keratin-filled cyst. They are usually found in the cerebellopontine angle and the intrasellar and suprasellar regions. They may present with headaches, cranial nerve abnormalities, seizures, or hydrocephalus. Imaging studies demonstrate extraaxial lesions with characteristics that are similar to CSF but have restricted diffusion. Treatment involves surgical resection.
Dermoid Cysts Like epidermoid cysts, dermoid cysts arise from epithelial cells that are retained during closure of the neural tube. They contain both epidermal and dermal structures such as hair follicles, sweat glands, and sebaceous glands. Unlike epidermoid cysts, these tumors usually have a midline location. They occur most frequently in the posterior fossa, especially the vermis, fourth ventricle, and suprasellar cistern. Radiographically, dermoid cysts resemble lipomas, demonstrating T1 hyperintensity and variable signal on T2. Symptomatic dermoid cysts can be treated with surgery.
Colloid Cysts These usually arise in the anterior third ventricle and may present with headaches, hydrocephalus, and, very rarely, sudden death. Surgical resection is curative, or a third ventriculostomy may relieve the obstructive hydrocephalus and be sufficient therapy.
NEUROCUTANEOUS SYNDROMES (PHAKOMATOSES)
A number of genetic disorders are characterized by cutaneous lesions and an increased risk of brain tumors. Most of these disorders have an autosomal dominant inheritance with variable penetrance.
NEUROFIBROMATOSIS TYPE 1 (NF1) (von RECKLINGHAUSEN’S DISEASE)
NF1 is an autosomal dominant disorder with an incidence of approximately 1 in 2600–3000. Approximately one-half the cases are familial; the remainder are caused by new mutations arising in patients with unaffected parents. The NF1 gene on chromosome 17q11.2 encodes a protein, neurofibromin, a guanosine triphosphatase (GTPase)-activating protein (GAP) that modulates signaling through the ras pathway. Mutations of NF1 result in a large number of nervous system tumors including neurofibromas, plexiform neurofibromas, optic nerve gliomas, astrocytomas, and meningiomas. In addition to neurofibromas, which appear as multiple, soft, rubbery cutaneous tumors, other cutaneous manifestations of NF1 include café-au-lait spots and axillary freckling. NF1 is also associated with hamartomas of the iris termed Lisch nodules, pheochromocytomas, pseudoarthrosis of the tibia, scoliosis, epilepsy, and mental retardation.
NEUROFIBROMATOSIS TYPE 2 (NF2)
NF2 is less common than NF1, with an incidence of 1 in 25,000–40,000. It is an autosomal dominant disorder with full penetrance. As with NF1, approximately one-half the cases arise from new mutations. The NF2 gene on 22q encodes a cytoskeletal protein, merlin (moesin, ezrin, radixin-like protein) that functions as a tumor suppressor. NF2 is characterized by bilateral vestibular schwannomas in over 90% of patients, multiple meningiomas, and spinal ependymomas and astrocytomas. Treatment of bilateral vestibular schwannomas can be challenging because the goal is to preserve hearing for as long as possible. These patients may also have diffuse schwannomatosis that may affect the cranial, spinal, or peripheral nerves; posterior subcapsular lens opacities; and retinal hamartomas.
TUBEROUS SCLEROSIS (BOURNEVILLE DISEASE)
This is an autosomal dominant disorder with an incidence of approximately 1 in 5000–10,000 live births. It is caused by mutations in either the TSC1 gene, which maps to chromosome 9q34 and encodes a protein termed hamartin, or the TSC2 gene, which maps to chromosome 16p13.3 and encodes the protein tuberin. Hamartin forms a complex with tuberin, which inhibits cellular signaling through the mTOR, and acts as a negative regulator of the cell cycle. Patients with tuberous sclerosis may have seizures, mental retardation, adenoma sebaceum (facial angiofibromas), shagreen patch, hypomelanotic macules, periungual fibromas, renal angiomyolipomas, and cardiac rhabdomyomas. These patients have an increased incidence of subependymal nodules, cortical tubers, and subependymal giant-cell astrocytomas (SEGA). Patients frequently require anticonvulsants for seizures. SEGAs do not always require therapeutic intervention, but the most effective therapy is with the mTOR inhibitors sirolimus or everolimus, which often decrease seizures as well as SEGA size.
TUMORS METASTATIC TO THE BRAIN
Brain metastases arise from hematogenous spread and frequently either arise from a lung primary or are associated with pulmonary metastases. Most metastases develop at the gray matter–white matter junction in the watershed distribution of the brain where intravascular tumor cells lodge in terminal arterioles. The distribution of metastases in the brain approximates the proportion of blood flow such that about 85% of all metastases are supratentorial and 15% occur in the posterior fossa. The most common sources of brain metastases are lung and breast carcinomas; melanoma has the greatest propensity to metastasize to the brain, being found in 80% of patients at autopsy Table 118-3). Other tumor types such as ovarian and esophageal carcinoma rarely metastasize to the brain. Prostate and breast cancer also have a propensity to metastasize to the dura and can mimic meningioma. Leptomeningeal metastases are common from hematologic malignancies and also breast and lung cancers. Spinal cord compression primarily arises in patients with prostate and breast cancer, tumors with a strong propensity to metastasize to the axial skeleton.
|
FREQUENCY OF NERVOUS SYSTEM METASTASES BY COMMON PRIMARY TUMORS |
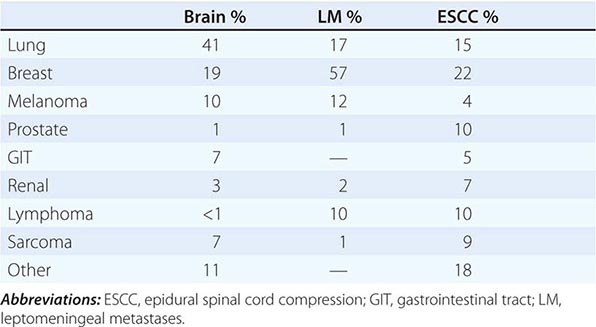
DIAGNOSIS OF METASTASES
Brain metastases are best visualized on MRI, where they usually appear as well-circumscribed lesions (Fig. 118-7). The amount of perilesional edema can be highly variable, with large lesions causing minimal edema and sometimes very small lesions causing extensive edema. Enhancement may be in a ring pattern or diffuse. Occasionally, intracranial metastases will hemorrhage; although melanoma, thyroid, and kidney cancer have the greatest propensity to hemorrhage, the most common cause of a hemorrhagic metastasis is lung cancer because it accounts for the majority of brain metastases. The radiographic appearance of brain metastasis is nonspecific, and similar-appearing lesions can occur with infection including brain abscesses and also with demyelinating lesions, sarcoidosis, radiation necrosis in a previously treated patient, or a primary brain tumor that may be a second malignancy in a patient with systemic cancer. However, biopsy is rarely necessary for diagnosis in most patients because imaging alone in the appropriate clinical situation usually suffices. This is straightforward for the majority of patients with brain metastases because they have a known systemic cancer. However, in approximately 10% of patients, a systemic cancer may present with a brain metastasis, and if there is not an easily accessible systemic site to biopsy, then a brain lesion must be removed for diagnostic purposes.
FIGURE 118-7 Postgadolinium T1 MRI of multiple brain metastases from non-small-cell lung cancer involving the right frontal (A) and right cerebellar (B) hemispheres. Note the diffuse enhancement pattern and absence of central necrosis.
LEPTOMENINGEAL METASTASES
Leptomeningeal metastases are also identified as carcinomatous meningitis, meningeal carcinomatosis, or in the case of specific tumors, leukemic or lymphomatous meningitis. Among the hematologic malignancies, acute leukemia is the most common to metastasize to the subarachnoid space, and in lymphomas the aggressive diffuse lymphomas can metastasize to the subarachnoid space frequently as well. Among solid tumors, breast and lung carcinomas and melanoma most frequently spread in this fashion. Tumor cells reach the subarachnoid space via the arterial circulation or occasionally through retrograde flow in venous systems that drain metastases along the bony spine or cranium. In addition, leptomeningeal metastases may develop as a direct consequence of prior brain metastases and can develop in almost 40% of patients who have a metastasis resected from the cerebellum.
CLINICAL FEATURES
Leptomeningeal metastases are characterized clinically by multilevel symptoms and signs along the neuraxis. Combinations of lumbar and cervical radiculopathies, cranial neuropathies, seizures, confusion, and encephalopathy from hydrocephalus or raised intracranial pressure can be present. Focal deficits such as hemiparesis or aphasia are rarely due to leptomeningeal metastases unless there is direct brain infiltration, and they are more often associated with coexisting brain lesions. New-onset limb pain in patients with breast cancer, lung cancer, or melanoma should prompt consideration of leptomeningeal spread.
LABORATORY AND IMAGING DIAGNOSIS
Leptomeningeal metastases are particularly challenging to diagnose because identification of tumor cells in the subarachnoid compartment may be elusive. MRI can be definitive in patients when there are clear tumor nodules adherent to the cauda equina or spinal cord, enhancing cranial nerves, or subarachnoid enhancement on brain imaging (Fig. 118-8). Imaging is diagnostic in approximately 75% of patients and is more often positive in patients with solid tumors. Demonstration of tumor cells in the CSF is definitive and often considered the gold standard. However, CSF cytologic examination is positive in only 50% of patients on the first lumbar puncture and still misses 10% after three CSF samples. CSF cytologic examination is most useful in hematologic malignancies. Accompanying CSF abnormalities include an elevated protein concentration and an elevated white count. Hypoglycorrhachia is noted in less than 25% of patients but is useful when present. Identification of tumor markers or molecular confirmation of clonal proliferation with techniques such as flow cytometry within the CSF can also be definitive when present. Tumor markers are usually specific to solid tumors, and chromosomal or molecular markers are most useful in patients with hematologic malignancies. New technologies, such as rare cell capture, may enhance identification of tumor cells in the CSF.
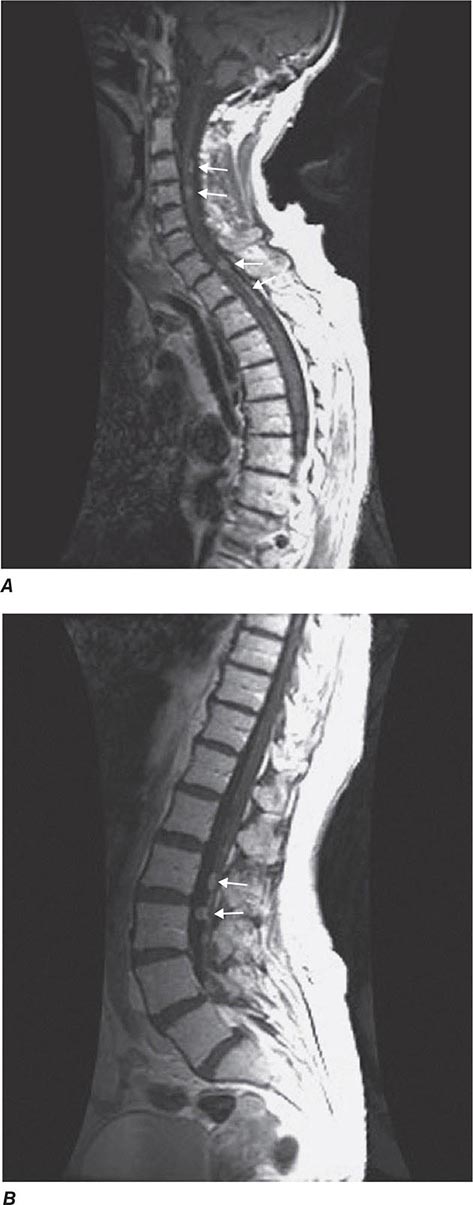
FIGURE 118-8 Postgadolinium MRI images of extensive leptomeningeal metastases from breast cancer. Nodules along the dorsal surface of the spinal cord (A) and cauda equina (B) are seen.
EPIDURAL METASTASIS
Epidural metastasis occurs in 3–5% of patients with a systemic malignancy and causes neurologic compromise by compressing the spinal cord or cauda equina. The most common cancers that metastasize to the epidural space are those malignancies that spread to bone, such as breast and prostate. Lymphoma can cause bone involvement and compression, but it can also invade the intervertebral foramens and cause spinal cord compression without bone destruction. The thoracic spine is affected most commonly, followed by the lumbar and then cervical spine.
CLINICAL FEATURES
Back pain is the presenting symptom of epidural metastasis in virtually all patients; the pain may precede neurologic findings by weeks or months. The pain is usually exacerbated by lying down; by contrast, arthritic pain is often relieved by recumbency. Leg weakness is seen in about 50% of patients, as is sensory dysfunction. Sphincter problems are present in about 25% of patients at diagnosis.
DIAGNOSIS
Diagnosis is established by imaging, with MRI of the complete spine being the best test (Fig. 118-9). Contrast is not needed to identify spinal or epidural lesions. Any patient with cancer who has severe back pain should undergo an MRI. Plain films, bone scans, or even CT scans may show bone metastases, but only MRI can reliably delineate epidural tumor. For patients unable to have an MRI, CT myelography should be performed to outline the epidural space. The differential diagnosis of epidural tumor includes epidural abscess, acute or chronic hematomas, and rarely, extramedullary hematopoiesis.
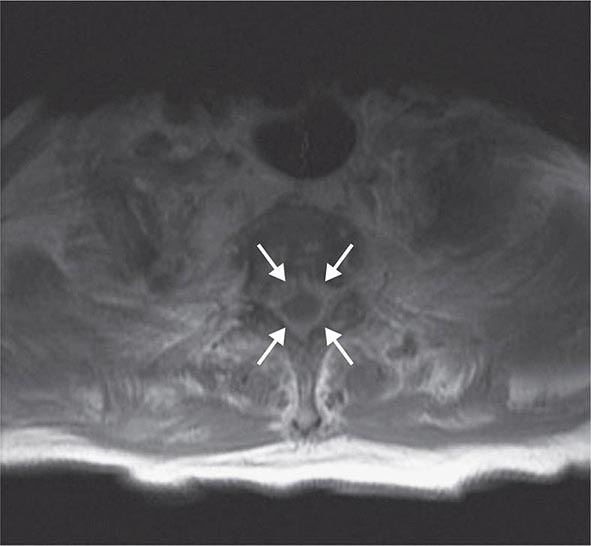
FIGURE 118-9 Postgadolinium T1 MRI showing circumferential epidural tumor around the thoracic spinal cord from esophageal cancer.
NEUROLOGIC TOXICITY OF THERAPY
TOXICITY FROM RADIOTHERAPY
RT can cause a variety of toxicities in the CNS. These are usually described based on their relationship in time to the administration of RT: acute (occurring within days of RT), early delayed (months), or late delayed (years). In general, the acute and early delayed syndromes resolve and do not result in persistent deficits, whereas the late delayed toxicities are usually permanent and sometimes progressive.
Acute Toxicity Acute cerebral toxicity usually occurs during RT to the brain. RT can cause a transient disruption of the blood-brain barrier, resulting in increased edema and elevated intracranial pressure. This is usually manifest as headache, lethargy, nausea, and vomiting and can be both prevented and treated with the administration of glucocorticoids. There is no acute RT toxicity that affects the spinal cord.
Early Delayed Toxicity Early delayed toxicity is usually apparent weeks to months after completion of cranial irradiation and is likely due to focal demyelination. Clinically it may be asymptomatic or take the form of worsening or reappearance of a preexisting neurologic deficit. At times a contrast-enhancing lesion can be seen on MRI/CT that can mimic the tumor for which the patient received the RT. For patients with a malignant glioma, this has been described as “pseudoprogression” because it mimics tumor recurrence on MRI but actually represents inflammation and necrotic debris engendered by effective therapy. This is seen with increased frequency when chemotherapy, particularly temozolomide, is given concurrently with RT. Pseudoprogression can resolve on its own or, if very symptomatic, may require resection. A rare form of early delayed toxicity is the somnolence syndrome that occurs primarily in children and is characterized by marked sleepiness.
In the spinal cord, early delayed RT toxicity is manifest as a Lhermitte symptom with paresthesias of the limbs or along the spine when the patient flexes the neck. Although frightening, it is benign, resolves on its own, and does not portend more serious problems.
Late Delayed Toxicity Late delayed toxicities are the most serious because they are often irreversible and cause severe neurologic deficits. In the brain, late toxicities can take several forms, the most common of which include radiation necrosis and leukoencephalopathy. Radiation necrosis is a focal mass of necrotic tissue that is contrast enhancing on CT/MRI and may be associated with significant edema. This may appear identical to pseudoprogression but is seen months to years after RT and is always symptomatic. Clinical symptoms and signs include seizure and lateralizing findings referable to the location of the necrotic mass. The necrosis is caused by the effect of RT on cerebral vasculature with resultant fibrinoid necrosis and occlusion of the blood vessels. It can mimic tumor radiographically, but unlike tumor, it is typically hypometabolic on a PET scan and has reduced perfusion on perfusion MR sequences. It may require resection for diagnosis and treatment unless it can be managed with glucocorticoids. There are rare reports of improvement with hyperbaric oxygen or anticoagulation, but the usefulness of these approaches is questionable.
Leukoencephalopathy is seen most commonly after WBRT as opposed to focal RT. On T2 or FLAIR MR sequences, there is diffuse increased signal seen throughout the hemispheric white matter, often bilaterally and symmetrically. There tends to be a periventricular predominance that may be associated with atrophy and ventricular enlargement. Clinically, patients develop cognitive impairment, gait disorder, and later urinary incontinence, all of which can progress over time. These symptoms mimic those of normal pressure hydrocephalus, and placement of a ventriculoperitoneal shunt can improve function in some patients but does not reverse the deficits completely. Increased age is a risk factor for leukoencephalopathy but not for radiation necrosis. Necrosis appears to depend on an as yet unidentified predisposition.
Other late neurologic toxicities include endocrine dysfunction if the pituitary or hypothalamus was included in the RT port. An RT-induced neoplasm can occur many years after therapeutic RT for either a prior CNS tumor or a head and neck cancer; accurate diagnosis requires surgical resection or biopsy. In addition, RT causes accelerated atherosclerosis, which can cause stroke either from intracranial vascular disease or carotid plaque from neck irradiation.
The peripheral nervous system is relatively resistant to RT toxicities. Peripheral nerves are rarely affected by RT, but the plexus is more vulnerable. Plexopathy develops more commonly in the brachial distribution than in the lumbosacral distribution. It must be differentiated from tumor progression in the plexus, which is usually accomplished with CT/MR imaging of the area or PET scan demonstrating tumor infiltrating the region. Clinically, tumor progression is usually painful, whereas RT-induced plexopathy is painless. Radiation plexopathy is also more commonly associated with lymphedema of the affected limb. Sensory loss and weakness are seen in both.
TOXICITY FROM CHEMOTHERAPY
Neurotoxicity is second to myelosuppression as the dose-limiting toxicity of chemotherapeutic agents (Table 118-4). Chemotherapy causes peripheral neuropathy from a number of commonly used agents, and the type of neuropathy can differ, depending on the drug. Vincristine causes paresthesias but little sensory loss and is associated with motor dysfunction, autonomic impairment (frequently ileus), and rarely cranial nerve compromise. Cisplatin causes large fiber sensory loss resulting in sensory ataxia but little cutaneous sensory loss and no weakness. The taxanes also cause a predominately sensory neuropathy. Agents such as bortezomib and thalidomide also cause neuropathy.
|
NEUROLOGIC SIGNS CAUSED BY AGENTS COMMONLY USED IN PATIENTS WITH CANCER |
Abbreviations: IT, intrathecal; IV, intravenous; PRES, posterior reversible encephalopathy syndrome.
Encephalopathy and seizures are common toxicities from chemotherapeutic drugs. Ifosfamide can cause a severe encephalopathy, which is reversible with discontinuation of the drug and the use of methylene blue for severely affected patients. Fludarabine also causes a severe global encephalopathy that may be permanent. Bevacizumab and other anti-VEGF agents can cause posterior reversible encephalopathy syndrome. Cisplatin can cause hearing loss and less frequently vestibular dysfunction. Immunotherapy with anti-CTLA-4 monoclonal antibodies, such as ipilimumab, can cause an autoimmune hypophysitis.
119e |
Soft Tissue and Bone Sarcomas and Bone Metastases |
Sarcomas are rare (<1% of all malignancies) mesenchymal neoplasms that arise in bone and soft tissues. These tumors are usually of mesodermal origin, although a few are derived from neuroectoderm, and they are biologically distinct from the more common epithelial malignancies. Sarcomas affect all age groups; 15% are found in children <15 years of age, and 40% occur after age 55 years. Sarcomas are one of the most common solid tumors of childhood and are the fifth most common cause of cancer deaths in children. Sarcomas may be divided into two groups, those derived from bone and those derived from soft tissues.
SOFT TISSUE SARCOMAS
Soft tissues include muscles, tendons, fat, fibrous tissue, synovial tissue, vessels, and nerves. Approximately 60% of soft tissue sarcomas arise in the extremities, with the lower extremities involved three times as often as the upper extremities. Thirty percent arise in the trunk, the retroperitoneum accounting for 40% of all trunk lesions. The remaining 10% arise in the head and neck.
INCIDENCE
Approximately 11,410 new cases of soft tissue sarcomas occurred in the United States in 2013. The annual age-adjusted incidence is 3 per 100,000 population, but the incidence varies with age. Soft tissue sarcomas constitute 0.7% of all cancers in the general population and 6.5% of all cancers in children.
EPIDEMIOLOGY
Malignant transformation of a benign soft tissue tumor is extremely rare, with the exception that malignant peripheral nerve sheath tumors (neurofibrosarcoma, malignant schwannoma) can arise from neurofibromas in patients with neurofibromatosis. Several etiologic factors have been implicated in soft tissue sarcomas.
Environmental Factors Trauma or previous injury is rarely involved, but sarcomas can arise in scar tissue resulting from a prior operation, burn, fracture, or foreign body implantation. Chemical carcinogens such as polycyclic hydrocarbons, asbestos, and dioxin may be involved in the pathogenesis.
Iatrogenic Factors Sarcomas in bone or soft tissues occur in patients who are treated with radiation therapy. The tumor nearly always arises in the irradiated field. The risk increases with time.
Viruses Kaposi’s sarcoma (KS) in patients with HIV type 1, classic KS, and KS in HIV-negative homosexual men is caused by human herpesvirus (HHV) 8 (Chap. 219). No other sarcomas are associated with viruses.
Immunologic Factors Congenital or acquired immunodeficiency, including therapeutic immunosuppression, increases the risk of sarcoma.
GENETIC CONSIDERATIONS
![]() Li-Fraumeni syndrome is a familial cancer syndrome in which affected individuals have germline abnormalities of the tumor-suppressor gene p53 and an increased incidence of soft tissue sarcomas and other malignancies, including breast cancer, osteosarcoma, brain tumor, leukemia, and adrenal carcinoma (Chap. 101e). Neurofibromatosis 1 (NF-1, peripheral form, von Recklinghausen’s disease) is characterized by multiple neurofibromas and café-au-lait spots. Neurofibromas occasionally undergo malignant degeneration to become malignant peripheral nerve sheath tumors. The gene for NF-1 is located in the pericentromeric region of chromosome 17 and encodes neurofibromin, a tumor-suppressor protein with guanosine 5′-triphosphate (GTP)ase-activating activity that inhibits Ras function (Chap. 118). Germline mutation of the Rb-1 locus (chromosome 13q14) in patients with inherited retinoblastoma is associated with the development of osteosarcoma in those who survive the retinoblastoma and of soft tissue sarcomas unrelated to radiation therapy. Other soft tissue tumors, including desmoid tumors, lipomas, leiomyomas, neuroblastomas, and paragangliomas, occasionally show a familial predisposition.
Li-Fraumeni syndrome is a familial cancer syndrome in which affected individuals have germline abnormalities of the tumor-suppressor gene p53 and an increased incidence of soft tissue sarcomas and other malignancies, including breast cancer, osteosarcoma, brain tumor, leukemia, and adrenal carcinoma (Chap. 101e). Neurofibromatosis 1 (NF-1, peripheral form, von Recklinghausen’s disease) is characterized by multiple neurofibromas and café-au-lait spots. Neurofibromas occasionally undergo malignant degeneration to become malignant peripheral nerve sheath tumors. The gene for NF-1 is located in the pericentromeric region of chromosome 17 and encodes neurofibromin, a tumor-suppressor protein with guanosine 5′-triphosphate (GTP)ase-activating activity that inhibits Ras function (Chap. 118). Germline mutation of the Rb-1 locus (chromosome 13q14) in patients with inherited retinoblastoma is associated with the development of osteosarcoma in those who survive the retinoblastoma and of soft tissue sarcomas unrelated to radiation therapy. Other soft tissue tumors, including desmoid tumors, lipomas, leiomyomas, neuroblastomas, and paragangliomas, occasionally show a familial predisposition.
Ninety percent of synovial sarcomas contain a characteristic chromosomal translocation t(X;18)(p11;q11) involving a nuclear transcription factor on chromosome 18 called SYT and two breakpoints on X. Patients with translocations to the second × breakpoint (SSX2) may have longer survival than those with translocations involving SSX1.
Insulin-like growth factor (IGF) type II is produced by some sarcomas and may act as an autocrine growth factor and as a motility factor that promotes metastatic spread. IGF-II stimulates growth through IGF-I receptors, but its effects on motility are through different receptors. If secreted in large amounts, IGF-II may produce hypoglycemia (Chaps. 121 and 420).
CLASSIFICATION
Approximately 20 different groups of sarcomas are recognized on the basis of the pattern of differentiation toward normal tissue. For example, rhabdomyosarcoma shows evidence of skeletal muscle fibers with cross-striations; leiomyosarcomas contain interlacing fascicles of spindle cells resembling smooth muscle; and liposarcomas contain adipocytes. When precise characterization of the group is not possible, the tumors are called unclassified sarcomas. All of the primary bone sarcomas can also arise from soft tissues (e.g., extraskeletal osteosarcoma). The entity malignant fibrous histiocytoma (MFH) includes many tumors previously classified as fibrosarcomas or as pleomorphic variants of other sarcomas and is characterized by a mixture of spindle (fibrous) cells and round (histiocytic) cells arranged in a storiform pattern with frequent giant cells and areas of pleomorphism. As immunohistochemical suggestion of differentiation, particularly myogenic differentiation, may be found in a significant fraction of these patients, many are now characterized as poorly differentiated leiomyosarcomas, and the terms undifferentiated pleomorphic sarcoma (UPS) and myxofibrosarcoma are replacing MFH and myxoid MFH.
For purposes of treatment, most soft tissue sarcomas can be considered together. However, some specific tumors have distinct features. For example, liposarcoma can have a spectrum of behaviors. Pleomorphic liposarcomas and dedifferentiated liposarcomas behave like other high-grade sarcomas; in contrast, well-differentiated liposarcomas (better termed atypical lipomatous tumors) lack metastatic potential, and myxoid liposarcomas metastasize infrequently, but, when they do, they have a predilection for unusual metastatic sites containing fat, such as the retroperitoneum, mediastinum, and subcutaneous tissue. Rhabdomyosarcomas, Ewing’s sarcoma, and other small-cell sarcomas tend to be more aggressive and are more responsive to chemotherapy than other soft tissue sarcomas.
Gastrointestinal stromal cell tumors (GISTs), previously classified as gastrointestinal leiomyosarcomas, are now recognized as a distinct entity within soft tissue sarcomas. Its cell of origin resembles the interstitial cell of Cajal, which controls peristalsis. The majority of malignant GISTs have activating mutations of the c-kit gene that result in ligand-independent phosphorylation and activation of the KIT receptor tyrosine kinase, leading to tumorigenesis. Approximately 5–10% of tumors will have a mutation in the platelet-derived growth factor receptor α (PDGFRA). GISTs that are wild type for both KIT and PDGFRA mutations may show mutations in SDH B, C, or D and may be driven by the IGF-I pathway.
DIAGNOSIS
The most common presentation is an asymptomatic mass. Mechanical symptoms referable to pressure, traction, or entrapment of nerves or muscles may be present. All new and persistent or growing masses should be biopsied, either by a cutting needle (core-needle biopsy) or by a small incision, placed so that it can be encompassed in the subsequent excision without compromising a definitive resection. Lymph node metastases occur in 5%, except in synovial and epithelioid sarcomas, clear-cell sarcoma (melanoma of the soft parts), angiosarcoma, and rhabdomyosarcoma, where nodal spread may be seen in 17%. The pulmonary parenchyma is the most common site of metastases. Exceptions are GISTs, which metastasize to the liver; myxoid liposarcomas, which seek fatty tissue; and clear-cell sarcomas, which may metastasize to bones. Central nervous system metastases are rare, except in alveolar soft part sarcoma.
Radiographic Evaluation Imaging of the primary tumor is best with plain radiographs and magnetic resonance imaging (MRI) for tumors of the extremities or head and neck and by computed tomography (CT) for tumors of the chest, abdomen, or retroperitoneal cavity. A radiograph and CT scan of the chest are important for the detection of lung metastases. Other imaging studies may be indicated, depending on the symptoms, signs, or histology.
STAGING AND PROGNOSIS
The histologic grade, relationship to fascial planes, and size of the primary tumor are the most important prognostic factors. The current American Joint Committee on Cancer (AJCC) staging system is shown in Table 119e-1. Prognosis is related to the stage. Cure is common in the absence of metastatic disease, but a small number of patients with metastases can also be cured. Most patients with stage IV disease die within 12 months, but some patients may live with slowly progressive disease for many years.
|
AMERICAN JOINT COMMITTEE ON CANCER STAGING SYSTEM FOR SARCOMAS |
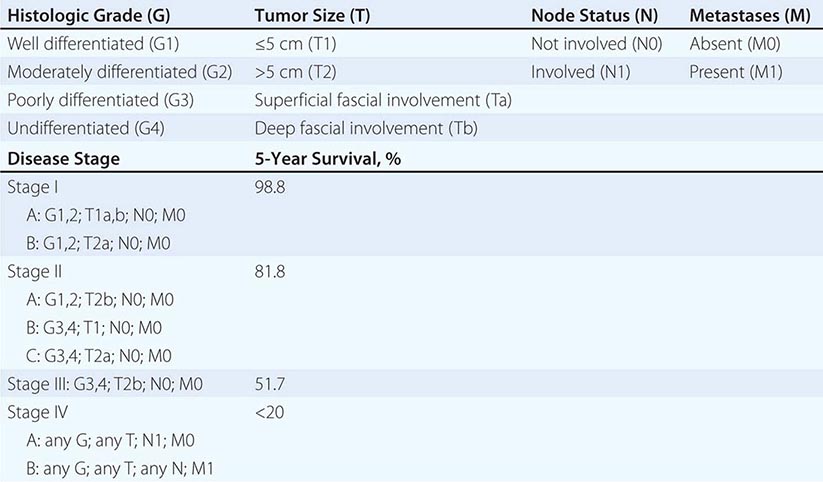
BONE SARCOMAS
INCIDENCE AND EPIDEMIOLOGY
Bone sarcomas are rarer than soft tissue sarcomas; they accounted for only 0.2% of all new malignancies and 2890 new cases in the United States in 2013. Several benign bone lesions have the potential for malignant transformation. Enchondromas and osteochondromas can transform into chondrosarcoma; fibrous dysplasia, bone infarcts, and Paget’s disease of bone can transform into either UPS or osteosarcoma.
CLASSIFICATION
Benign Tumors The common benign bone tumors include enchondroma, osteochondroma, chondroblastoma, and chondromyxoid fibroma, of cartilage origin; osteoid osteoma and osteoblastoma, of bone origin; fibroma and desmoplastic fibroma, of fibrous tissue origin; hemangioma, of vascular origin; and giant-cell tumor, of unknown origin.
Malignant Tumors The most common malignant tumors of bone are plasma cell tumors (Chap. 136). The four most common malignant nonhematopoietic bone tumors are osteosarcoma, chondrosarcoma, Ewing’s sarcoma, and UPS. Rare malignant tumors include chordoma (of notochordal origin), malignant giant-cell tumor and adamantinoma (of unknown origin), and hemangioendothelioma (of vascular origin).
Musculoskeletal Tumor Society Staging System Sarcomas of bone are staged according to the Musculoskeletal Tumor Society staging system based on grade and compartmental localization. A Roman numeral reflects the tumor grade: stage I is low grade, stage II is high grade, and stage III includes tumors of any grade that have lymph node or distant metastases. In addition, the tumor is given a letter reflecting its compartmental localization. Tumors designated A are intracompartmental (i.e., confined to the same soft tissue compartment as the initial tumor), and tumors designated B are extracompartmental (i.e., extending into the adjacent soft tissue compartment or into bone). The tumor-node-metastasis (TNM) staging system is shown in Table 119e-2.
|
STAGING SYSTEM FOR BONE SARCOMAS |
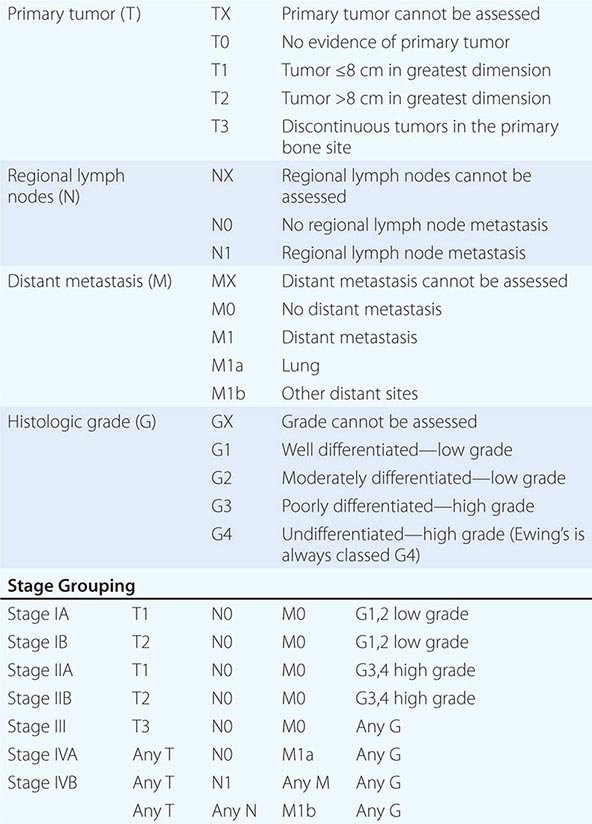
OSTEOSARCOMA
Osteosarcoma, accounting for almost 45% of all bone sarcomas, is a spindle cell neoplasm that produces osteoid (unmineralized bone) or bone. Approximately 60% of all osteosarcomas occur in children and adolescents in the second decade of life, and approximately 10% occur in the third decade of life. Osteosarcomas in the fifth and sixth decades of life are frequently secondary to either radiation therapy or transformation in a preexisting benign condition, such as Paget’s disease. Males are affected 1.5–2 times as often as females. Osteosarcoma has a predilection for metaphyses of long bones; the most common sites of involvement are the distal femur, proximal tibia, and proximal humerus. The classification of osteosarcoma is complex, but 75% of osteosarcomas fall into the “classic” category, which include osteoblastic, chondroblastic, and fibroblastic osteosarcomas. The remaining 25% are classified as “variants” on the basis of (1) clinical characteristics, as in the case of osteosarcoma of the jaw, postradiation osteosarcoma, or Paget’s osteosarcoma; (2) morphologic characteristics, as in the case of telangiectatic osteosarcoma, small-cell osteosarcoma, or epithelioid osteosarcoma; or (3) location, as in parosteal or periosteal osteosarcoma. Diagnosis usually requires a synthesis of clinical, radiologic, and pathologic features. Patients typically present with pain and swelling of the affected area. A plain radiograph reveals a destructive lesion with a moth-eaten appearance, a spiculated periosteal reaction (sunburst appearance), and a cuff of periosteal new bone formation at the margin of the soft tissue mass (Codman’s triangle). A CT scan of the primary tumor is best for defining bone destruction and the pattern of calcification, whereas MRI is better for defining intramedullary and soft tissue extension. A chest radiograph and CT scan are used to detect lung metastases. Metastases to the bony skeleton should be imaged by a bone scan or by fluorodeoxyglucose positron emission tomography (FDG-PET). Almost all osteosarcomas are hypervascular. Angiography is not helpful for diagnosis, but it is the most sensitive test for assessing the response to preoperative chemotherapy. Pathologic diagnosis is established either with a core-needle biopsy, where feasible, or with an open biopsy with an appropriately placed incision that does not compromise future limb-sparing resection. Most osteosarcomas are high-grade. The most important prognostic factor for long-term survival is response to chemotherapy. Preoperative chemotherapy followed by limb-sparing surgery (which can be accomplished in >80% of patients) followed by postoperative chemotherapy is standard management. The effective drugs are doxorubicin, ifosfamide, cisplatin, and high-dose methotrexate with leucovorin rescue. The various combinations of these agents that have been used have all been about equally successful. Long-term survival rates in extremity osteosarcoma range from 60 to 80%. Osteosarcoma is radioresistant; radiation therapy has no role in the routine management. UPS is considered a part of the spectrum of osteosarcoma and is managed similarly.
CHONDROSARCOMA
Chondrosarcoma, which constitutes ~20–25% of all bone sarcomas, is a tumor of adulthood and old age with a peak incidence in the fourth to sixth decades of life. It has a predilection for the flat bones, especially the shoulder and pelvic girdles, but can also affect the diaphyseal portions of long bones. Chondrosarcomas can arise de novo or as a malignant transformation of an enchondroma or, rarely, of the cartilaginous cap of an osteochondroma. Chondrosarcomas have an indolent natural history and typically present as pain and swelling. Radiographically, the lesion may have a lobular appearance with mottled or punctate or annular calcification of the cartilaginous matrix. It is difficult to distinguish low-grade chondrosarcoma from benign lesions by x-ray or histologic examination. The diagnosis is therefore influenced by clinical history and physical examination. A new onset of pain, signs of inflammation, and progressive increase in the size of the mass suggest malignancy. The histologic classification is complex, but most tumors fall within the classic category. Like other bone sarcomas, high-grade chondrosarcomas spread to the lungs. Most chondrosarcomas are resistant to chemotherapy, and surgical resection of primary or recurrent tumors, including pulmonary metastases, is the mainstay of therapy. This rule does not hold for two histologic variants. Dedifferentiated chondrosarcoma has a high-grade osteosarcoma or a malignant fibrous histiocytoma component that responds to chemotherapy. Mesenchymal chondrosarcoma, a rare variant composed of a small-cell element, also is responsive to systemic chemotherapy and is treated like Ewing’s sarcoma.
EWING’S SARCOMA
Ewing’s sarcoma, which constitutes ~10–15% of all bone sarcomas, is common in adolescence and has a peak incidence in the second decade of life. It typically involves the diaphyseal region of long bones and also has an affinity for flat bones. The plain radiograph may show a characteristic “onion peel” periosteal reaction with a generous soft tissue mass, which is better demonstrated by CT or MRI. This mass is composed of sheets of monotonous, small, round, blue cells and can be confused with lymphoma, embryonal rhabdomyosarcoma, and small-cell carcinoma. The presence of p30/32, the product of the mic-2 gene (which maps to the pseudoautosomal region of the × and Y chromosomes), is a cell-surface marker for Ewing’s sarcoma (and other members of the Ewing’s family of tumors, sometimes called PNETs). Most PNETs arise in soft tissues; they include peripheral neuroepithelioma, Askin’s tumor (chest wall), and esthesioneuroblastoma. Glycogen-filled cytoplasm detected by staining with periodic acid–Schiff is also characteristic of Ewing’s sarcoma cells. The classic cytogenetic abnormality associated with this disease (and other PNETs) is a reciprocal translocation of the long arms of chromosomes 11 and 22, t(11;22), which creates a chimeric gene product of unknown function with components from the fli-1 gene on chromosome 11 and ews on 22. This disease is very aggressive, and it is therefore considered a systemic disease. Common sites of metastases are lung, bones, and bone marrow. Systemic chemotherapy is the mainstay of therapy, often being used before surgery. Doxorubicin, cyclophosphamide or ifosfamide, etoposide, vincristine, and dactinomycin are active drugs. Topotecan or irinotecan in combination with an alkylating agent is often used in relapsed patients. Targeted therapy with an anti-IGF-I receptor antibody in combination with an inhibitor of mammalian target of rapamycin (mTOR) appears to have promising activity in refractory cases. Local treatment for the primary tumor includes surgical resection, usually with limb salvage or radiation therapy. Patients with lesions below the elbow and below the mid-calf have a 5-year survival rate of 80% with effective treatment. Ewing’s sarcoma at first presentation is a curable tumor, even in the presence of obvious metastatic disease, especially in children <11 years old.
TUMORS METASTATIC TO BONE
Bone is a common site of metastasis for carcinomas of the prostate, breast, lung, kidney, bladder, and thyroid and for lymphomas and sarcomas. Prostate, breast, and lung primaries account for 80% of all bone metastases. Metastatic tumors of bone are more common than primary bone tumors. Tumors usually spread to bone hematogenously, but local invasion from soft tissue masses also occurs. In descending order of frequency, the sites most often involved are the vertebrae, proximal femur, pelvis, ribs, sternum, proximal humerus, and skull. Bone metastases may be asymptomatic or may produce pain, swelling, nerve root or spinal cord compression, pathologic fracture, or myelophthisis (replacement of the marrow). Symptoms of hypercalcemia may be noted in cases of bony destruction.
Pain is the most frequent symptom. It usually develops gradually over weeks, is usually localized, and often is more severe at night. When patients with back pain develop neurologic signs or symptoms, emergency evaluation for spinal cord compression is indicated (Chap. 331). Bone metastases exert a major adverse effect on quality of life in cancer patients.
Cancer in the bone may produce osteolysis, osteogenesis, or both. Osteolytic lesions result when the tumor produces substances that can directly elicit bone resorption (vitamin D–like steroids, prostaglandins, or parathyroid hormone–related peptide) or cytokines that can induce the formation of osteoclasts (interleukin 1 and tumor necrosis factor). Osteoblastic lesions result when the tumor produces cytokines that activate osteoblasts. In general, purely osteolytic lesions are best detected by plain radiography, but they may not be apparent until they are >1 cm. These lesions are more commonly associated with hypercalcemia and with the excretion of hydroxyproline-containing peptides indicative of matrix destruction. When osteoblastic activity is prominent, the lesions may be readily detected using radionuclide bone scanning (which is sensitive to new bone formation), and the radiographic appearance may show increased bone density or sclerosis. Osteoblastic lesions are associated with higher serum levels of alkaline phosphatase and, if extensive, may produce hypocalcemia. Although some tumors may produce mainly osteolytic lesions (e.g., kidney cancer) and others mainly osteoblastic lesions (e.g., prostate cancer), most metastatic lesions produce both types of lesion and may go through stages where one or the other predominates.
In older patients, particularly women, it may be necessary to distinguish metastatic disease of the spine from osteoporosis. In osteoporosis, the cortical bone may be preserved, whereas cortical bone destruction is usually noted with metastatic cancer.
[/level-membership-for-internal-medicine-category][not-level-membership-for-internal-medicine-category]
114 |
Bladder and Renal Cell Carcinomas |
BLADDER CANCER
Transitional cell epithelium lines the urinary tract from the renal pelvis to the ureter, urinary bladder, and the proximal two-thirds of the urethra. Cancers can occur at any point: 90% of malignancies develop in the bladder, 8% in the renal pelvis, and 2% in the ureter or urethra. Bladder cancer is the fourth most common cancer in men and the thirteenth in women, with an estimated 72,570 new cases and 15,210 deaths in the United States predicted for the year 2013. The almost 5:1 ratio of incidence to mortality reflects the higher frequency of the less lethal superficial variants compared to the more lethal invasive and metastatic variants. The incidence is roughly four times higher in men than in women and twofold higher in white men than in black men, with a median age of 65 years.
Once diagnosed, urothelial tumors exhibit polychronotropism, which is the tendency to recur over time in new locations in the urothelial tract. As long as urothelium is present, continuous monitoring is required.
EPIDEMIOLOGY
Cigarette smoking is believed to contribute to up to 50% of urothelial cancers in men and nearly 40% in women. The risk of developing a urothelial cancer in male smokers is increased two- to fourfold relative to nonsmokers and continues for 10 years or longer after cessation. Other implicated agents include aniline dyes, the drugs phenacetin and chlornaphazine, and external beam radiation. Chronic cyclophosphamide exposure also increases risk, whereas vitamin A supplements appear to be protective. Exposure to Schistosoma haematobium, a parasite found in many developing countries, is associated with an increase in both squamous and transitional cell carcinomas of the bladder.
PATHOLOGY
Clinical subtypes are grouped into three categories: 75% are superficial, 20% invade muscle, and 5% are metastatic at presentation. Staging of the tumor within the bladder is based on the pattern of growth and depth of invasion. The revised tumor, node, metastasis (TNM) staging system is illustrated in Fig. 114-1. About half of invasive tumors presented originally as superficial lesions that later progressed. Tumors are also rated by grade. Low-grade (highly differentiated) tumors rarely progress to a higher stage, whereas high-grade tumors do.
FIGURE 114-1 Bladder staging. TNM, tumor, node, metastasis.
More than 95% of urothelial tumors in the United States are transitional cell in origin. Pure squamous cancers with keratinization constitute 3%, adenocarcinomas 2%, and small cell tumors (often with paraneoplastic syndromes) <1%. Adenocarcinomas develop primarily in the urachal remnant in the dome of the bladder or in the periurethral tissues. Paragangliomas, lymphomas, and melanomas are rare. Of the transitional cell tumors, low-grade papillary lesions that grow on a central stalk are most common. These tumors are very friable, have a tendency to bleed, and have a high risk for recurrence, yet they rarely progress to the more lethal invasive variety. In contrast, carcinoma in situ (CIS) is a high-grade tumor that is considered a precursor of the more lethal muscle-invasive disease.
PATHOGENESIS
The multicentric nature of the disease and high recurrence suggests a field effect in the urothelium that results in a predisposition to develop cancer. Molecular genetic analyses suggest that the superficial and invasive lesions develop along distinct molecular pathways. Low-grade noninvasive papillary tumors harbor constitutive activation of the receptor tyrosine kinase-Ras signal transduction pathway and high frequencies of fibroblast growth factor receptor 3 and phosphoinositide-3 kinase α subunit mutations. In contrast, CIS and invasive tumors have a higher frequency of TP53 and RB gene alterations. Within all clinical stages, including Tis, T1, and T2 or greater lesions, tumors with alterations in p53, p21, and/or RB have a higher probability of recurrence, metastasis, and death from disease.
CLINICAL PRESENTATION, DIAGNOSIS, AND STAGING
Hematuria occurs in 80–90% of patients and often reflects exophytic tumors. The bladder is the most common source of gross hematuria (40%), but benign cystitis (22%) is a more common cause than bladder cancer (15%) (Chap. 61). Microscopic hematuria is more commonly of prostate origin (25%); only 2% of bladder cancers produce microscopic hematuria. Once hematuria is documented, a urinary cytology, visualization of the urothelial tract by computed tomography (CT) or magnetic resonance urogram or intravenous pyelogram, and cystoscopy are recommended if no other etiology is found. Screening asymptomatic individuals for hematuria increases the diagnosis of tumors at an early stage but has not been shown to prolong life. After hematuria, irritative symptoms are the next most common presentation. Ureteral obstruction may cause flank pain. Symptoms of metastatic disease are rarely the first presenting sign.
The endoscopic evaluation includes an examination under anesthesia to determine whether a palpable mass is present. A flexible endoscope is inserted into the bladder, and bladder barbotage for cytology is performed. Visual inspection includes mapping the location, size, and number of lesions, as well as a description of the growth pattern (solid vs papillary). All visible tumors should be resected, and a sample of the muscle underlying the tumor should be obtained to assess the depth of invasion. Normal-appearing areas are biopsied at random to ensure no CIS is present. A notation is made as to whether a tumor was completely or incompletely resected. Selective catheterization and visualization of the upper tracts should be performed if the cytology is positive and no disease is visible in the bladder. Ultrasonography, CT, and/or magnetic resonance imaging (MRI) are used to determine whether a tumor extends to perivesical fat (T3) and to document nodal spread. Distant metastases are assessed by CT of the chest and abdomen, MRI, or radionuclide imaging of the skeleton.
TREATMENT BLADDER CANCER
Management depends on whether the tumor invades muscle and whether it has spread to the regional lymph nodes and beyond. The probability of spread increases with increasing T stage.
NON–MUSCLE-INVASIVE DISEASE
At a minimum, the management is complete endoscopic resection with or without intravesical therapy. The decision to recommend intravesical therapy depends on the histologic subtype, number of lesions, depth of invasion, presence or absence of CIS, and antecedent history. Recurrences develop in upward of 50% of cases, of which 5–20% progress to a more advanced stage. In general, solitary papillary lesions are managed by transurethral surgery alone. CIS and recurrent disease are treated by transurethral surgery followed by intravesical therapy.
Intravesical therapies are used in two general contexts: as an adjuvant to a complete endoscopic resection to prevent recurrence or to eliminate disease that cannot be controlled by endoscopic resection alone. Intravesical treatments are advised for patients with diffuse CIS, recurrent disease, >40% involvement of the bladder surface by tumor, or T1 disease. The standard therapy, based on randomized comparisons, is Bacillus Calmette-Guérin (BCG) in six weekly instillations, often followed by maintenance administrations for ≥1 year. Other agents with activity include mitomycin C, interferon, and gemcitabine. The side effects of intravesical therapies include dysuria, urinary frequency, and, depending on the drug, myelosuppression or contact dermatitis. Rarely, intravesical BCG may produce a systemic illness associated with granulomatous infections in multiple sites requiring antituberculin therapy.
Following the endoscopic resection, patients are monitored for recurrence at 3-month intervals during the first year. Recurrence may develop anywhere along the urothelial tract, including the renal pelvis, ureter, or urethra. Persistent disease in the bladder and new tumors are treated with a second course of BCG or intravesical chemotherapy with valrubicin or gemcitabine. In some cases, cystectomy is recommended. Tumors in the ureter or renal pelvis are typically managed by resection during retrograde examination or, in some cases, by instillation through the renal pelvis. Prostatic urethral tumors may require cystoprostatectomy if the tumor cannot be resected completely.
MUSCLE-INVASIVE DISEASE
The treatment of a tumor that has invaded muscle can be separated into control of the primary tumor and systemic chemotherapy to treat micrometastatic disease. Radical cystectomy is the standard treatment in the United States, although in selected cases, a bladder-sparing approach is used. This approach includes complete endoscopic resection; partial cystectomy; or a combination of resection, systemic chemotherapy, and external beam radiation therapy. In some countries, external beam radiation therapy is considered standard. In the United States, it is generally limited to those patients deemed unfit for cystectomy, those with unresectable local disease, or as part of an experimental bladder-sparing approach.
Indications for cystectomy include muscle-invading tumors not suitable for segmental resection; non–muscle-invasive tumors unsuitable for conservative management (e.g., due to multicentric and frequent recurrences resistant to intravesical instillations); high-grade T1 tumors especially if associated with CIS; and bladder symptoms (e.g., frequency or hemorrhage) that impair quality of life.
Radical cystectomy is major surgery that requires appropriate preoperative evaluation and management. It involves removal of the bladder and pelvic lymph nodes and creation of a conduit or reservoir for urinary flow. Grossly abnormal lymph nodes are evaluated by frozen section. If metastases are confirmed, the procedure is often aborted. In males, radical cystectomy includes the removal of the prostate, seminal vesicles, and proximal urethra. Impotence is universal unless the nerves responsible for erectile function are preserved. In females, the procedure includes removal of the bladder, urethra, uterus, fallopian tubes, ovaries, anterior vaginal wall, and surrounding fascia.
Several options are frequently used for urinary diversion. Ileal conduits bring urine directly from the ureter to the abdominal wall. Some patients receive either a continent cutaneous reservoir constructed from detubularized bowel or an orthotopic neobladder. Approximately 25% of men receive a neobladder, leading to 85–90% continence during the day. Cutaneous reservoirs are drained by intermittent catheterization. Contraindications to a neobladder include renal insufficiency, an inability to self-catheterize, or CIS or an exophytic tumor in the urethra. Diffuse CIS in the bladder is a relative contraindication based on the risk of a urethral recurrence. Concurrent ulcerative colitis or Crohn’s disease may hinder the use of bowel.
A partial cystectomy may be considered when the disease is limited to the dome of the bladder, a ≥2 cm margin can be achieved, there is no associated CIS, and the bladder capacity is adequate after resection. This occurs in 5–10% of cases. Carcinomas in the ureter or in the renal pelvis are treated with nephroureterectomy with a bladder cuff to remove the tumor.
The probability of recurrence following surgery is based on pathologic stage, presence or absence of lymphatic or vascular invasion, and nodal spread. Among those whose cancers recur, the recurrence develops in a median of 1 year. Long-term outcomes vary by pathologic stage and histology (Table 114-1). The number of lymph nodes removed is also prognostic, whether or not the nodes contained tumor.
|
SURVIVAL FOLLOWING SURGERY FOR BLADDER CANCER |
Chemotherapy (described below) has been shown to prolong the survival of patients with muscle-invasive disease when combined with definitive treatment of the bladder by radical cystectomy or radiation therapy. Presurgical (or neoadjuvant) chemotherapy has been the most thoroughly explored, and increases the cure rate by 5–15%, whereas postsurgical (adjuvant) chemotherapy has not been proven definitively beneficial. For the majority of patients, chemotherapy alone is inadequate to eradicate the disease. Use of neoadjuvant chemotherapy is increasing, although it still remains underused. Experimental studies are evaluating bladder preservation strategies by combining chemotherapy and radiation therapy in patients whose tumors were endoscopically removed.
METASTATIC DISEASE
The primary goal of metastatic disease treatment is to achieve complete remission with chemotherapy alone or with a combined-modality approach of chemotherapy followed by surgical resection of residual disease. One can define a goal in terms of cure or palliation on the basis of the probability of achieving a complete response to chemotherapy using prognostic factors, such as Karnofsky performance status (KPS) (<80%) and whether the pattern of spread is nodal or visceral (liver, lung, or bone). For those with zero, one, or two risk factors, the probability of complete remission is 38, 25, and 5%, respectively, and median survival is 33, 13.4, and 9.3 months, respectively. Patients who have low KPS or who have visceral disease or bone metastases rarely achieve long-term survival. The toxicities also vary as a function of risk, and treatment-related mortality rates are as high as 3–4% using some combinations in these poor-risk patient groups. For most patients, treatment is palliative, aimed at delaying or relieving cancer-related symptoms, because few patients experience durable complete remissions.
CHEMOTHERAPY
A number of chemotherapeutic drugs have activity as single agents; cisplatin, paclitaxel, and gemcitabine are considered most active. Standard therapy consists of two-, three-, or four-drug combinations. Overall response rates of >50% have been reported using combinations such as methotrexate, vinblastine, doxorubicin, and cisplatin (MVAC); gemcitabine and cisplatin (GC); or gemcitabine, paclitaxel, and cisplatin (GPC). MVAC was considered standard, but the toxicities of neutropenia and fever, mucositis, diminished renal and auditory function, and peripheral neuropathy led to the development of alternative regimens. At present, GC is used more commonly than MVAC based on the results of a comparative trial of MVAC versus GC that showed less neutropenia and fever and less mucositis for the GC regimen with similar response rates and median overall survival. Anemia and thrombocytopenia were more common with GC. GPC is not more effective than GC.
Chemotherapy has also been tested in the neoadjuvant and adjuvant settings. In a randomized trial, patients receiving three cycles of neoadjuvant MVAC followed by cystectomy had a significantly better median (6.2 years) and 5-year survival (57%) compared to cystectomy alone (median survival 3.8 years; 5-year survival 42%). Similar results were obtained in an international study of three cycles of cisplatin, methotrexate, and vinblastine (CMV) followed by either radical cystectomy or radiation therapy. The decision to administer adjuvant therapy is based on recurrence risk after cystectomy. Studies of adjuvant chemotherapy have been underpowered, and most closed for lack of accrual. One underpowered study using the GPC regimen suggested that adjuvant treatment improved survival, although many patients never received chemotherapy for metastases. Another underpowered study did not show a benefit for GC chemotherapy. Therefore, preoperative chemotherapy is preferred when medically appropriate. Indications for adjuvant chemotherapy in patients who did not receive neoadjuvant treatment include nodal disease, extravesical tumor extension, or vascular invasion in the resected specimen.
The management of bladder cancer is summarized in Table 114-2.
|
MANAGEMENT OF BLADDER CANCER |
CARCINOMA OF THE RENAL PELVIS AND URETER
About 5000 cases of renal pelvis and ureter cancer occur each year; nearly all are transitional cell carcinomas similar to bladder cancer in biology and appearance. This tumor is associated with chronic phenacetin abuse and aristolochic acid consumption in Chinese herbal preparations; aristolochic acid also seems to be associated with Balkan nephropathy, a chronic interstitial nephritis endemic in Bulgaria, Greece, Bosnia-Herzegovina, and Romania. In addition, upper tract urothelial carcinoma is linked to hereditary nonpolyposis colorectal cancer.
The most common symptom is painless gross hematuria, and the disease is usually detected on imaging during the workup for hematuria. Patterns of spread are like bladder cancer. For low-grade disease localized to the renal pelvis and ureter, nephroureterectomy (including excision of the distal ureter with a portion of the bladder) is associated with 5-year survival of 80–90%. More invasive or poorly differentiated tumors are more likely to recur locally and to metastasize. Metastatic disease is treated with the chemotherapy used in bladder cancer, and the outcome is similar to that of metastatic bladder cancer.
RENAL CELL CARCINOMA
Renal cell carcinomas account for 90–95% of malignant neoplasms arising from the kidney. Notable features include resistance to cytotoxic agents, infrequent responses to biologic response modifiers such as interleukin (IL) 2, robust activity to antiangiogenesis targeted agents, and a variable clinical course for patients with metastatic disease, including anecdotal reports of spontaneous regression.
EPIDEMIOLOGY
The incidence of renal cell carcinoma continues to rise and is now nearly 65,000 cases annually in the United States, resulting in 13,700 deaths. The male-to-female ratio is 2:1. Incidence peaks between the ages of 50 and 70 years, although this malignancy may be diagnosed at any age. Many environmental factors have been investigated as possible contributing causes; the strongest association is with cigarette smoking. Risk is also increased for patients who have acquired cystic disease of the kidney associated with end-stage renal disease and for those with tuberous sclerosis. Most cases are sporadic, although familial forms have been reported. One is associated with von Hippel-Lindau (VHL) syndrome. VHL syndrome is an autosomal dominant disorder. Genetic studies identified the VHL gene on the short arm of chromosome 3. Approximately 35% of individuals with VHL disease develop clear cell renal cell carcinoma. Other associated neoplasms include retinal hemangioma, hemangioblastoma of the spinal cord and cerebellum, pheochromocytoma, neuroendocrine tumors and cysts, and cysts in the epididymis of the testis in men and the broad ligament in women.
PATHOLOGY AND GENETICS
Renal cell neoplasia represents a heterogeneous group of tumors with distinct histopathologic, genetic, and clinical features ranging from benign to high-grade malignant (Table 114-3). They are classified on the basis of morphology and histology. Categories include clear cell carcinoma (60% of cases), papillary tumors (5–15%), chromophobe tumors (5–10%), oncocytomas (5–10%), and collecting or Bellini duct tumors (<1%). Papillary tumors tend to be bilateral and multifocal. Chromophobe tumors have a more indolent clinical course, and oncocytomas are considered benign neoplasms. In contrast, Bellini duct carcinomas, which are thought to arise from the collecting ducts within the renal medulla, are rare but often very aggressive. Clear cell tumors, the predominant histology, are found in >80% of patients who develop metastases. Clear cell tumors arise from the epithelial cells of the proximal tubules and usually show chromosome 3p deletions. Deletions of 3p21–26 (where the VHL gene maps) are identified in patients with familial as well as sporadic tumors. VHL encodes a tumor suppressor protein that is involved in regulating the transcription of vascular endothelial growth factor (VEGF), platelet-derived growth factor (PDGF), and a number of other hypoxia-inducible proteins. Inactivation of VHL leads to overexpression of these agonists of the VEGF and PDGF receptors, which promote tumor angiogenesis and tumor growth. Agents that inhibit proangiogenic growth factor activity show antitumor effects. Enormous genetic variability has been documented in tumors from individual patients. Although the tumors have a clear clonal origin and often contain VHL mutations in common, different portions of the primary tumor and different metastatic sites may have wide variation in genetic lesions they contain. This tumor heterogeneity may underlie the emergence of treatment resistance.
|
CLASSIFICATION OF EPITHELIAL NEOPLASMS ARISING FROM THE KIDNEY |
CLINICAL PRESENTATION
The presenting signs and symptoms include hematuria, abdominal pain, and a flank or abdominal mass. Other symptoms are fever, weight loss, anemia, and a varicocele. The tumor is most commonly detected as an incidental finding on a radiograph. Widespread use of radiologic cross-sectional imaging procedures (CT, ultrasound, MRI) contributes to earlier detection, including incidental renal masses detected during evaluation for other medical conditions. The increasing number of incidentally discovered low-stage tumors has contributed to an improved 5-year survival for patients with renal cell carcinoma and increased use of nephron-sparing surgery (partial nephrectomy). A spectrum of paraneoplastic syndromes has been associated with these malignancies, including erythrocytosis, hypercalcemia, nonmetastatic hepatic dysfunction (Stauffer’s syndrome), and acquired dysfibrinogenemia. Erythrocytosis is noted at presentation in only about 3% of patients. Anemia, a sign of advanced disease, is more common.
The standard evaluation of patients with suspected renal cell tumors includes a CT scan of the abdomen and pelvis, chest radiograph, urine analysis, and urine cytology. If metastatic disease is suspected from the chest radiograph, a CT of the chest is warranted. MRI is useful in evaluating the inferior vena cava in cases of suspected tumor involvement or invasion by thrombus. In clinical practice, any solid renal masses should be considered malignant until proven otherwise; a definitive diagnosis is required. If no metastases are demonstrated, surgery is indicated, even if the renal vein is invaded. The differential diagnosis of a renal mass includes cysts, benign neoplasms (adenoma, angiomyolipoma, oncocytoma), inflammatory lesions (pyelonephritis or abscesses), and other primary or metastatic cancers. Other malignancies that may involve the kidney include transitional cell carcinoma of the renal pelvis, sarcoma, lymphoma, and Wilms’ tumor. All of these are less common causes of renal masses than is renal cell cancer.
STAGING AND PROGNOSIS
Staging is based on the American Joint Committee on Cancer (AJCC) staging system (Fig. 114-2). Stage I tumors are <7 cm in greatest diameter and confined to the kidney, stage II tumors are ≥7 cm and confined to the kidney, stage III tumors extend through the renal capsule but are confined to Gerota’s fascia (IIIa) or involve a single hilar lymph node (N1), and stage IV disease includes tumors that have invaded adjacent organs (excluding the adrenal gland) or involve multiple lymph nodes or distant metastases. The 5-year survival rate varies by stage: >90% for stage I, 85% for stage II, 60% for stage III, and 10% for stage IV.
FIGURE 114-2 Renal cell carcinoma staging. TNM, tumor, node, metastasis.
115 |
Benign and Malignant Diseases of the Prostate |
Benign and malignant changes in the prostate increase with age. Autopsies of men in the eighth decade of life show hyperplastic changes in >90% and malignant changes in >70% of individuals. The high prevalence of these diseases among the elderly, who often have competing causes of morbidity and mortality, mandates a risk-adapted approach to diagnosis and treatment. This can be achieved by considering these diseases as a series of states. Each state represents a distinct clinical milestone for which therapy(ies) may be recommended based on current symptoms, the risk of developing symptoms, or death from disease in relation to death from other causes within a given time frame. For benign proliferative disorders, symptoms of urinary frequency, infection, and potential for obstruction are weighed against the side effects and complications of medical or surgical intervention. For prostate malignancies, the risks of developing the disease, symptoms, or death from cancer are balanced against the morbidities of the recommended treatments and preexisting comorbidities.
ANATOMY
The prostate is located in the pelvis and is surrounded by the rectum, the bladder, the periprostatic and dorsal vein complexes and neurovascular bundles that are responsible for erectile function, and the urinary sphincter that is responsible for passive urinary control. The prostate is composed of branching tubuloalveolar glands arranged in lobules surrounded by fibromuscular stroma. The acinar unit includes an epithelial compartment made up of epithelial, basal, and neuroendocrine cells and separated by a basement membrane, and a stromal compartment that includes fibroblasts and smooth-muscle cells. Prostate-specific antigen (PSA) and prostatic acid phosphatase (PAP) are produced in the epithelial cells. Both prostate epithelial cells and stromal cells express androgen receptors (ARs) and depend on androgens for growth. Testosterone, the major circulating androgen, is converted by the enzyme 5α-reductase to dihydrotestosterone in the gland.
The periurethral portion of the gland increases in size during puberty and after the age of 55 years due to the growth of nonmalignant cells in the transition zone of the prostate that surrounds the urethra. Most cancers develop in the peripheral zone, and cancers in this location may be palpated during a digital rectal examination (DRE).
PROSTATE CANCER
In 2013, approximately 238,590 prostate cancer cases were diagnosed, and 29,720 men died from prostate cancer in the United States. The absolute number of prostate cancer deaths has decreased in the past 5 years, which has been attributed by some to the widespread use of PSA-based detection strategies. However, the benefit of screening on survival is unclear. The paradox of management is that although 1 in 6 men will eventually be diagnosed with the disease, and the disease remains the second leading cause of cancer deaths in men, only 1 man in 30 with prostate cancer will die of his disease.
EPIDEMIOLOGY
Epidemiologic studies show that the risk of being diagnosed with prostate cancer increases by a factor of two if one first-degree relative is affected and by four if two or more are affected. Current estimates are that 40% of early-onset and 5–10% of all prostate cancers are hereditary. Prostate cancer affects ethnic groups differently. Matched for age, African-American males have both a higher incidence of prostate cancer and larger tumors and more worrisome histologic features than white males. Polymorphic variants of the AR, the cytochrome P450 C17, and the steroid 5α-reductase type II (SRD5A2) genes have been implicated in the variations in incidence.
The prevalence of autopsy-detected cancers is similar around the world, while the incidence of clinical disease varies. Thus, environmental and dietary factors may play a role in prostate cancer growth and progression. High consumption of dietary fats, such as α-linoleic acid or the polycyclic aromatic hydrocarbons that form when red meats are cooked, is believed to increase risk. Similar to breast cancer in Asian women, the risk of prostate cancer in Asian men increases when they move to Western environments. Protective factors include consumption of the isoflavonoid genistein (which inhibits 5α-reductase) found in many legumes, cruciferous vegetables that contain the isothiocyanate sulforaphane, retinoids such as lycopene found in tomatoes, and inhibitors of cholesterol biosynthesis (e.g., statin drugs). The development of prostate cancer is a multistep process. One early change is hypermethylation of the GSTP1 gene promoter, which leads to loss of function of a gene that detoxifies carcinogens. The finding that many prostate cancers develop adjacent to a lesion termed proliferative inflammatory atrophy (PIA) suggests a role for inflammation.
PREVENTION
Currently no drugs or dietary supplements are approved by the U.S. Food and Drug Administration (FDA) for prevention of prostate cancer, nor are any recommended by the major clinical guidelines. Although statins may have some protective effect, the potential risks outweigh the benefits given the small number of men who die of prostate cancer. The results from several large, double-blind, randomized chemoprevention trials established 5α-reductase inhibitors (5ARI) as the most likely therapy to reduce the future risk of a prostate cancer diagnosis. The Prostate Cancer Prevention Trial (PCPT), in which men older than age 55 years received placebo or the 5ARI finasteride, which inhibits the type 1 isoform, showed a 25% (95% confidence interval 19–31%) reduction in the period prevalence of prostate cancer across all age groups in favor of finasteride (18.4%) over placebo (24.4%). In the Reduction by Dutasteride of Prostate Cancer Events (REDUCE) trial, a similar 23% reduction in the 4-year period prevalence was observed in favor of dutasteride (p = .001). Dutasteride inhibits both the type 1 and type 2 5ARI isoforms. While both studies met their endpoint, there was concern that most of the cancers that were prevented were low risk and that there was a slightly higher rate of clinically significant cancers (those with higher Gleason score) in the treatment arm. Neither drug was FDA-approved for prostate cancer prevention. In comparison, the Selenium and Vitamin E Cancer Prevention Trial (SELECT), which enrolled African-American men age ≥50 years and others age ≥55 years, showed no difference in cancer incidence in patients receiving vitamin E (4.6%) or selenium (4.9%) alone or in combination (4.6%) relative to placebo (4.4%). A similar lack of benefit for vitamin E, vitamin C, and selenium was seen in the Physicians Health Study II.
THE CLINICAL STATES MODEL
The prostate cancer continuum—from the appearance of a preneoplastic and invasive lesion localized to the prostate, to a metastatic lesion that results in symptoms and, ultimately, mortality—can span decades. To facilitate disease management, competing risks are considered in the context of a series of clinical states (Fig. 115-1). The states are defined operationally on the basis of whether or not a cancer diagnosis has been established and, for those with a diagnosis, whether or not metastases are detectable on imaging studies and the measured level of testosterone in the blood. With this approach, an individual resides in only one state and remains in that state until he has progressed. At each assessment, the decision to offer treatment and the specific form of treatment are based on the risk posed by the cancer relative to competing causes of mortality that may be present in that individual. It follows that the more advanced the disease, the greater is the need for treatment.
FIGURE 115-1 Clinical states of prostate cancer. PSA, prostate-specific antigen.
For those without a cancer diagnosis, the decision to undergo testing to detect a cancer is based on the individual’s estimated life expectancy and, separately, the probability that a clinically significant cancer may be present. For those with a prostate cancer diagnosis, the clinical states model considers the probability of developing symptoms or dying from prostate cancer. Thus, a patient with localized prostate cancer who has had all cancer removed surgically remains in the state of localized disease as long as the PSA remains undetectable. The time within a state becomes a measure of the efficacy of an intervention, although the effect may not be assessable for years. Because many men with active cancer are not at risk for metastases, symptoms, or death, the clinical states model allows a distinction between cure—the elimination of all cancer cells, the primary therapeutic objective when treating most cancers—and cancer control, in which the tempo of the illness is altered and symptoms are controlled until the patient dies of other causes. These can be equivalent therapeutically from a patient standpoint if the patient has not experienced symptoms of the disease or the treatment needed to control it. Even when a recurrence is documented, immediate therapy is not always necessary. Rather, as at the time of diagnosis, the need for intervention is based on the tempo of the illness as it unfolds in the individual, relative to the risk-to-benefit ratio of the therapy being considered.
SCREENING AND DIAGNOSIS
Physical Examination The need to pursue a diagnosis of prostate cancer is based on symptoms, an abnormal DRE, or, more typically, a change in or an elevated serum PSA. The urologic history should focus on symptoms of outlet obstruction, continence, potency, or change in ejaculatory pattern.
The DRE focuses on prostate size and consistency and abnormalities within or beyond the gland. Many cancers occur in the peripheral zone and may be palpated on DRE. Carcinomas are characteristically hard, nodular, and irregular, while induration may also be due to benign prostatic hypertrophy (BPH) or calculi. Overall, 20–25% of men with an abnormal DRE have cancer.
Prostate-Specific Antigen PSA (kallikrein-related peptidase 3; KLK3) is a kallikrein-related serine protease that causes liquefaction of seminal coagulum. It is produced by both nonmalignant and malignant epithelial cells and, as such, is prostate-specific, not prostate cancer–specific. Serum levels may also increase from prostatitis and BPH. Serum levels are not significantly affected by DRE, but the performance of a prostate biopsy can increase PSA levels up to tenfold for 8–10 weeks. PSA circulating in the blood is inactive and mainly occurs as a complex with the protease inhibitor α1-antichymotrypsin and as free (unbound) PSA forms. The formation of complexes between PSA, α2-macroglobulin, or other protease inhibitors is less significant. Free PSA is rapidly eliminated from the blood by glomerular filtration with an estimated half-life of 12–18 h. Elimination of PSA bound to α1-antichymotrypsin is slow (estimated half-life of 1–2 weeks) because it too is largely cleared by the kidneys. Levels should be undetectable after about 6 weeks if the prostate has been removed. Immunohistochemical staining for PSA can be used to establish a prostate cancer diagnosis.
PSA-BASED SCREENING AND EARLY DETECTION PSA testing was approved by the U.S. FDA in 1994 for early detection of prostate cancer, and the widespread use of the test has played a significant role in the proportion of men diagnosed with early-stage cancers: more than 70–80% of newly diagnosed cancers are clinically organ-confined. The level of PSA in blood is strongly associated with the risk and outcome of prostate cancer. A single PSA measured at age 60 is associated (area under the curve [AUC] of 0.90) with lifetime risk of death from prostate cancer. Most prostate cancer deaths (90%) occur among men with PSA levels in the top quartile (>2 ng/mL), although only a minority of men with PSA >2 ng/mL will develop lethal prostate cancer. Despite this and mortality rate reductions reported from large randomized prostate cancer screening trials, routine use of the test remains controversial.
The U.S. Preventive Services Task Force (USPSTF) reviewed the evidence for screening for prostate cancer and made a clear recommendation against screening. By giving a grade of “D” in the recommendation statement that was based on this review, the USPSTF concluded that “there is moderate or high certainty that this service has no net benefit or that the harms outweigh the benefits.” Whether the harms of screening, overdiagnosis, and overtreatment are justified by the benefits in terms of reduced prostate cancer mortality is open to reasonable doubt. In response to the USPSTF, the American Urological Association (AUA) updated their consensus statement regarding prostate cancer screening. They concluded that the quality of evidence for the benefits of screening was moderate, and evidence for harm was high for men age 55–69 years. For men outside this age range, evidence was lacking for benefit, but the harms of screening, including overdiagnosis and overtreatment, remained. The AUA recommends shared decision making considering PSA-based screening for men age 55–69, a target age group for whom benefits may outweigh harms. Outside this age range, PSA-based screening as a routine test was not recommended based on the available evidence. The entire guideline is available at www.AUAnet.org/education/guidelines/prostate-cancer-detection.cfm.
The PSA criteria used to recommend a diagnostic prostate biopsy have evolved over time. However, based on the commonly used cut point for prostate biopsy (a total PSA ≥4 ng/mL), most men with a PSA elevation do not have histologic evidence of prostate cancer at biopsy. In addition, many men with PSA levels below this cut point harbor cancer cells in their prostate. Information from the PCPT demonstrates that there is no PSA below which the risk of prostate cancer is zero. Thus, the PSA level establishes the likelihood that a man will harbor cancer if he undergoes a prostate biopsy. The goal is to increase the sensitivity of the test for younger men more likely to die of the disease and to reduce the frequency of detecting cancers of low malignant potential in elderly men more likely to die of other causes. Patients with symptomatic prostatitis should have a course of antibiotics before biopsy. However, the routine use of antibiotics in an asymptomatic man with an elevated PSA level is strongly discouraged.
Prostate Biopsy A diagnosis of cancer is established by an image-guided needle biopsy. Direct visualization by transrectal ultrasound (TRUS) or magnetic resonance imaging (MRI) assures that all areas of the gland are sampled. Contemporary schemas advise an extended-pattern 12-core biopsy that includes sampling from the peripheral zone as well as a lesion-directed palpable nodule or suspicious image-guided sampling. Men with an abnormal PSA and negative biopsy are advised to undergo a repeat biopsy.
BIOPSY PATHOLOGY Each core of the biopsy is examined for the presence of cancer, and the amount of cancer is quantified based on the length of the cancer within the core and the percentage of the core involved. Of the cancers identified, >95% are adenocarcinomas; the rest are squamous or transitional cell tumors or, rarely, carcinosarcomas. Metastases to the prostate are rare, but in some cases colon cancers or transitional cell tumors of the bladder invade the gland by direct extension.
When prostate cancer is diagnosed, a measure of histologic aggressiveness is assigned using the Gleason grading system, in which the dominant and secondary glandular histologic patterns are scored from 1 (well-differentiated) to 5 (undifferentiated) and summed to give a total score of 2–10 for each tumor. The most poorly differentiated area of tumor (i.e., the area with the highest histologic grade) often determines biologic behavior. The presence or absence of perineural invasion and extracapsular spread is also recorded.
Prostate Cancer Staging The tumor, node, metastasis (TNM) staging system includes categories for cancers identified solely on the basis of an abnormal PSA (T1c), those that are palpable but clinically confined to the gland (T2), and those that have extended outside the gland (T3 and T4) (Table 115-1, Fig. 115-2). DRE alone is inaccurate in determining the extent of disease within the gland, the presence or absence of capsular invasion, involvement of seminal vesicles, and extension of disease to lymph nodes. Because of the inadequacy of DRE for staging, the TNM staging system was modified to include the results of imaging. Unfortunately, no single test has proven to accurately indicate the stage or the presence of organ-confined disease, seminal vesicle involvement, or lymph node spread.
|
TNM CLASSIFICATION |
FIGURE 115-2 T stages of prostate cancer. (A) T1—Clinically inapparent tumor, neither palpable nor visible by imaging; (B) T2—Tumor confined within prostate; (C) T3—Tumor extends through prostate capsule and may invade the seminal vesicles; (D) T4—Tumor is fixed or invades adjacent structures. Eighty-one percent of patients present with local disease (T1 and T2), which is associated with a 5-year survival rate of 100%. An additional 12% of patients present with regional disease (T3 and T4 without metastases), which is also associated with a 100% survival rate after 5 years. Four percent of patients present with distant disease (T4 with metastases), which is associated with a 28% 5-year survival rate. (Three percent of patients are ungraded, and this group is associated with a 73% 5-year survival rate.) (Data from AJCC, http://seer.cancer.gov/statfacts/html/prost.html. Figure © 2014 Memorial Sloan-Kettering Cancer Center; used with permission.)
TRUS is the imaging technique most frequently used to assess the primary tumor, but its chief use is directing prostate biopsies, not staging. No TRUS finding consistently indicates cancer with certainty. Computed tomography (CT) lacks sensitivity and specificity to detect extraprostatic extension and is inferior to MRI in visualization of lymph nodes. In general, MRI performed with an endorectal coil is superior to CT to detect cancer in the prostate and to assess local disease extent. T1-weighted MRI produces a high signal in the periprostatic fat, periprostatic venous plexus, perivesicular tissues, lymph nodes, and bone marrow. T2-weighted MRI demonstrates the internal architecture of the prostate and seminal vesicles. Most cancers have a low signal, while the normal peripheral zone has a high signal, although the technique lacks sensitivity and specificity. MRI is also useful for the planning of surgery and radiation therapy.
Radionuclide bone scans (bone scintigraphy) are used to evaluate spread to osseous sites. This test is sensitive but relatively nonspecific because areas of increased uptake are not always related to metastatic disease. Healing fractures, arthritis, Paget’s disease, and other conditions will also cause abnormal uptake. True-positive bone scans are uncommon when the PSA is <10 ng/mL unless the tumor is high grade.
|
TREATMENT |
TREATMENT OF PROSTATE CANCER BY CLINICAL STATE |
CLINICALLY LOCALIZED PROSTATE CANCER
Clinically localized prostate cancers are those that appear to be nonmetastatic after staging studies are performed. Patients with clinically localized disease are managed by radical prostatectomy, radiation therapy, or active surveillance. Choice of therapy requires the consideration of several factors: the presence of symptoms, the probability that the untreated tumor will adversely affect the quality or duration of survival and thus require treatment, and the probability that the tumor can be cured by single-modality therapy directed at the prostate or that it will require both local and systemic therapy to achieve cure.
Data from the literature do not provide clear evidence for the superiority of any one treatment relative to another. Comparison of outcomes of various forms of therapy is limited by the lack of prospective trials, referral bias, the experience of the treating teams, and differences in endpoints and cancer control definitions. Often, PSA relapse–free survival is used because an effect on metastatic progression or survival may not be apparent for years. After radical surgery to remove all prostate tissue, PSA should become undetectable in the blood within 6 weeks. If PSA remains or becomes detectable after radical prostatectomy, the patient is considered to have persistent disease. After radiation therapy, in contrast, PSA does not become undetectable because the remaining nonmalignant elements of the gland continue to produce PSA even if all cancer cells have been eliminated. Similarly, cancer control is not well defined for a patient managed by active surveillance because PSA levels will continue to rise in the absence of therapy. Other outcomes are time to objective progression (local or systemic), cancer-specific survival, and overall survival; however, these outcomes may take years to assess.
The more advanced the disease, the lower the probability of local control and the higher the probability of systemic relapse. More important is that within the categories of T1, T2, and T3 disease are cancers with a range of prognoses. Some T3 tumors are curable with therapy directed solely at the prostate, and some T1 lesions have a high probability of systemic relapse that requires the integration of local and systemic therapy to achieve cure. For T1c cancers in particular, stage alone is inadequate to predict outcome and select treatment; other factors must be considered.
Nomograms To better assess risk and guide treatment selection, many groups have developed prognostic models or nomograms that use a combination of the initial clinical T stage, biopsy Gleason score, and baseline PSA. Some use discrete cut points (PSA <10 or ≥10 ng/mL; Gleason score of ≤6, 7, or ≥8); others employ nomograms that use PSA and Gleason score as continuous variables. More than 100 nomograms have been reported to predict the probability that a clinically significant prostate cancer is present, disease extent (organ-confined vs non–organ-confined, node-negative or -positive), or the probability of success of treatment for specific local therapies using pretreatment variables. Considerable controversy exists over what constitutes “high risk” based on a predicted probability of success or failure. In these situations, nomograms and predictive models can only go so far. Exactly what probability of success or failure would lead a physician to recommend and a patient to seek alternative approaches is controversial. As an example, it may be appropriate to recommend radical surgery for a younger patient with a low probability of cure. Nomograms are being refined continually to incorporate additional clinical parameters, biologic determinants, and year of treatment, which can also affect outcomes, making treatment decisions a dynamic process.
Treatment-Related Adverse Events The frequency of adverse events varies by treatment modality and the experience of the treating team. For example, following radical prostatectomy, incontinence rates range from 2–47% and impotence rates range from 25–89%. Part of the variability relates to how the complication is defined and whether the patient or physician is reporting the event. The time of the assessment is also important. After surgery, impotence is immediate but may reverse over time, while with radiation therapy impotence is not immediate but may develop over time. Of greatest concern to patients are the effects on continence, sexual potency, and bowel function.
Radical Prostatectomy The goal of radical prostatectomy is to excise the cancer completely with a clear margin, to maintain continence by preserving the external sphincter, and to preserve potency by sparing the autonomic nerves in the neurovascular bundle. The procedure is advised for patients with a life expectancy of 10 years or more and is performed via a retropubic or perineal approach or via a minimally invasive robotic-assisted or hand-held laparoscopic approach. Outcomes can be predicted using postoperative nomograms that consider pretreatment factors and the pathologic findings at surgery. PSA failure is usually defined as a value greater than 0.1 or 0.2 ng/mL. Specific criteria to guide the choice of one approach over another are lacking. Minimally invasive approaches offer the advantage of a shorter hospital stay and reduced blood loss. Rates of cancer control, recovery of continence, and recovery of erectile function are comparable between open and minimally invasive approaches. The individual surgeon rather than the surgical approach used is most important in determining outcomes after surgery.
Neoadjuvant hormonal therapy has also been explored in an attempt to improve the outcomes of surgery for high-risk patients, using a variety of definitions. The results of several large trials testing 3 or 8 months of androgen depletion before surgery showed that serum PSA levels decreased by 96%, prostate volumes decreased by 34%, and margin positivity rates decreased from 41% to 17%. Unfortunately, hormones did not produce an improvement in PSA relapse–free survival. Thus, neoadjuvant hormonal therapy is not recommended.
Factors associated with incontinence following radical prostatectomy include older age and urethral length, which impacts the ability to preserve the urethra beyond the apex and the distal sphincter. The skill and experience of the surgeon are also factors. Recovery of erectile function is associated with younger age, quality of erections before surgery, and the absence of damage to the neurovascular bundles. In general, erectile function begins to return about 6 months after surgery if both neurovascular bundles are preserved. Potency is reduced by half if at least one neurovascular bundle is sacrificed. Overall, with the availability of drugs such as phosphodiesterase-5 (PDE5) inhibitors, intraurethral inserts of alprostadil, and intracavernosal injections of vasodilators, many patients recover satisfactory sexual function.
Radiation Therapy Radiation therapy is given by external beam, by radioactive sources implanted into the gland, or by a combination of the two techniques.
EXTERNAL-BEAM RADIATION THERAPY Contemporary external-beam radiation therapy requires three-dimensional conformal treatment plans to maximize the dose to the prostate and to minimize the exposure of the surrounding normal tissue. Intensity-modulated radiation therapy (IMRT) permits shaping of the dose and allows the delivery of higher doses to the prostate and a further reduction in normal tissue exposure than three-dimensional conformal treatment alone. These advances have enabled the safe administration of doses >80 Gy and resulted in higher local control rates and fewer side effects.
Cancer control after radiation therapy has been defined by various criteria, including a decline in PSA to <0.5 or 1 ng/mL, “nonrising” PSA values, and a negative biopsy of the prostate 2 years after completion of treatment. The current standard definition of biochemical failure (the Phoenix definition) is a rise in PSA by ≥2 ng/mL higher than the lowest PSA achieved. The date of failure is “at call” and not backdated.
Radiation dose is critical to the eradication of prostate cancer. In a representative study, a PSA nadir of <1.0 ng/mL was achieved in 90% of patients receiving 75.6 or 81.0 Gy versus 76% and 56% of those receiving 70.2 and 64.8 Gy, respectively. Positive biopsy rates at 2.5 years were 4% for those treated with 81 Gy versus 27% and 36% for those receiving 75.6 and 70.2 Gy, respectively.
Overall, radiation therapy is associated with a higher frequency of bowel complications (mainly diarrhea and proctitis) than surgery. The frequency relates directly to the volume of the anterior rectal wall receiving full-dose treatment. In one series, grade 3 rectal or urinary toxicities were seen in 2.1% of patients who received a median dose of 75.6 Gy, whereas grade 3 urethral strictures requiring dilatation developed in 1% of cases, all of whom had undergone a transurethral resection of the prostate (TURP). Pooled data show that the frequency of grade 3 and 4 toxicities is 6.9% and 3.5%, respectively, for patients who received >70 Gy. The frequency of erectile dysfunction is related to the age of the patient, the quality of erections pretreatment, the dose administered, and the time of assessment. Postradiation erectile dysfunction is related to a disruption of the vascular supply and not the nerve fibers.
Neoadjuvant hormone therapy before radiation therapy has the aim of decreasing the size of the prostate and, consequently, reducing the exposure of normal tissues to full-dose radiation, increasing local control rates, and decreasing the rate of systemic failure. Short-term hormone therapy can reduce toxicities and improve local control rates, but long-term treatment (2–3 years) is needed to prolong the time to PSA failure and lower the risk of metastatic disease in men with high-risk cancers. The impact on survival has been less clear.
BRACHYTHERAPY Brachytherapy is the direct implantation of radioactive sources (seeds) into the prostate. It is based on the principle that the deposition of radiation energy in tissues decreases as a function of the square of the distance from the source (Chap. 103e). The goal is to deliver intensive irradiation to the prostate, minimizing the exposure of the surrounding tissues. The current standard technique achieves a more homogeneous dose distribution by placing seeds according to a customized template based on imaging assessment of the cancer and computer-optimized dosimetry. The implantation is performed transperineally as an outpatient procedure with real-time imaging.
Improvements in brachytherapy techniques have resulted in fewer complications and a marked reduction in local failure rates. In a series of 197 patients followed for a median of 3 years, 5-year actuarial PSA relapse–free survival for patients with pretherapy PSA levels of 0–4, 4–10, and >10 ng/mL were 98%, 90%, and 89%, respectively. In a separate report of 201 patients who underwent posttreatment biopsies, 80% were negative, 17% were indeterminate, and 3% were positive. The results did not change with longer follow-up. Nevertheless, many physicians feel that implantation is best reserved for patients with good or intermediate prognostic features.
Brachytherapy is well tolerated, although most patients experience urinary frequency and urgency that can persist for several months. Incontinence has been seen in 2–4% of cases. Higher complication rates are observed in patients who have undergone a prior TURP, whereas those with obstructive symptoms at baseline are at a higher risk for retention and persistent voiding symptoms. Proctitis has been reported in <2% of patients.
Active Surveillance Although prostate cancer is the most common form of cancer affecting men in the United States, patients are being diagnosed earlier and more frequently present with early-stage disease. Active surveillance, described previously as watchful waiting or deferred therapy, is the policy of monitoring the illness at fixed intervals with DREs, PSA measurements, and repeat prostate biopsies as indicated until histopathologic or serologic changes correlative of progression warrant treatment with curative intent. It evolved from studies that evaluated predominantly elderly men with well-differentiated tumors who demonstrated no clinically significant progression for protracted periods, recognition of the contrast between incidence and disease-specific mortality, the high prevalence of autopsy cancers, and an effort to reduce overtreatment. A recent screening study estimated that between 50–100 men with low-risk disease would need to be treated to prevent one prostate cancer–specific death.
Arguing against active surveillance are the results of a Swedish randomized trial of radical prostatectomy versus active surveillance. With a median follow-up of 6.2 years, men treated by radical surgery had a lower risk of prostate cancer death relative to active surveillance patients (4.6% vs 8.9%) and a lower risk of metastatic progression (hazard ratio 0.63). Case selection is critical, and determining clinical parameters predictive of cancer aggressiveness that can be used to reliably select men most likely to benefit from active surveillance is an area of intense study. In one prostatectomy series, it was estimated that 10–15% of those treated had “insignificant” disease. One set of criteria includes men with clinical T1c tumors that are biopsy Gleason grade 6 or less involving three or fewer cores, each of them having less than 50% involvement by tumor and a PSA density of less than 0.15.
Concerns about active surveillance include the limited ability to predict pathologic findings by needle biopsy even when multiple cores are obtained, the recognized multifocality of the disease, and the possibility of a missed opportunity to cure the disease. Nomograms to help predict which patients can safely be managed by active surveillance continue to be refined, and as their predictive accuracy improves, it can be anticipated that more patients will be candidates.
RISING PSA AFTER DEFINITIVE LOCAL THERAPY
This term is applied to a group of patients in whom the sole manifestation of disease is a rising PSA after surgery and/or radiation therapy. By definition, there is no evidence of disease on an imaging study. For these patients, the central issue is whether the rise in PSA results from persistent disease in the primary site, systemic disease, or both. In theory, disease in the primary site may still be curable by additional local treatment.
The decision to recommend radiation therapy after prostatectomy is guided by the pathologic findings at surgery, because imaging studies such as CT and bone scan are typically uninformative. Some recommend a choline-11 positron emission tomography (PET) scan, but availability in the United States is limited. Others recommend that a biopsy of the urethrovesical anastomosis be obtained before considering radiation, whereas others treat empirically based on risk. Factors that predict for response to salvage radiation therapy are a positive surgical margin, lower Gleason score in the radical prostatectomy specimen, long interval from surgery to PSA failure, slow PSA doubling time, absence of disease in the lymph nodes, and a low (<0.5–1 ng/mL) PSA value at the time of radiation treatment. Radiation therapy is generally not recommended if the PSA was persistently elevated after surgery, which usually indicates that the disease has spread outside of the area of the prostate bed and is unlikely to be controlled with radiation therapy. As is the case for other disease states, nomograms to predict the likelihood of success are available.
For patients with a rising PSA after radiation therapy, salvage local therapy can be considered if the disease was “curable” at the time of diagnosis, if persistent disease has been documented by a biopsy of the prostate, and if no metastatic disease is seen on imaging studies. Unfortunately, case selection is poorly defined in most series, and morbidities are significant. Options include salvage radical prostatectomy, salvage cryotherapy, salvage radiation therapy, and salvage irreversible electroporation.
The rise in PSA after surgery or radiation therapy may indicate subclinical or micrometastatic disease with or without local recurrence. In these cases, the need for treatment depends, in part, on the estimated probability that the patient will develop clinically detectable metastatic disease on a scan and in what time frame. That immediate therapy is not always required was shown in a series where patients who developed a biochemical recurrence after radical prostatectomy received no systemic therapy until metastatic disease was documented. Overall, the median time to metastatic progression by imaging was 8 years, and 63% of the patients with rising PSA values remained free of metastases at 5 years. Factors associated with progression included the Gleason score of the radical prostatectomy specimen, time to recurrence, and PSA doubling time. For those with Gleason grade ≥8, the probability of metastatic progression was 37%, 51%, and 71% at 3, 5, and 7 years, respectively. If the time to recurrence was <2 years and PSA doubling time was long (>10 months), the proportions with metastatic disease at the same time intervals were 23%, 32%, and 53%, versus 47%, 69%, and 79% if the doubling time was short (<10 months). PSA doubling times are also prognostic for survival. In one series, all patients who succumbed to disease had PSA doubling times of 3 months or less.
Most physicians advise treatment if the PSA doubling time is 12 months or less. A difficulty with predicting the risk of metastatic spread, symptoms, or death from disease in the rising PSA state is that most patients receive some form of therapy before the development of metastases. Nevertheless, predictive models continue to be refined.
METASTATIC DISEASE: NONCASTRATE
The state of noncastrate metastatic prostate cancer includes men with metastases visible on an imaging study and noncastrate levels of testosterone (>150 ng/dL). The patient may be newly diagnosed or have a recurrence after treatment for localized disease. Symptoms of metastatic disease include pain from osseous spread, although many patients are asymptomatic despite extensive spread. Less common are symptoms related to marrow compromise (myelophthisis), spinal cord compression, or a coagulopathy.
Standard treatment is to deplete/lower androgens by medical or surgical means and/or to block androgen binding to the AR with antiandrogens. More than 90% of male hormones originate in the testes; <10% are synthesized in the adrenal gland. Surgical orchiectomy is the “gold standard” but is rarely used due to the availability of effective medical therapies and the more widespread use of hormones on an intermittent basis by which patients are treated for defined periods of time, following which the treatments are intentionally discontinued (discussed further below) (Fig. 115-3).
FIGURE 115-3 Sites of action of different hormone therapies. ACTH, adrenocorticotropic hormone; AR, androgen receptor; ARE, androgen-response element; CRH, corticotropin-releasing hormone; DHEA, dehydroepiandrosterone; DHEA-S, dehydroepiandrosterone sulphate; DHT, dihydrotestosterone; GnRH, gonadotropin-releasing hormone; LH, luteinizing hormone.
Testosterone-Lowering Agents Medical therapies that lower testosterone levels include the gonadotropin-releasing hormone (GnRH) agonists/antagonists, 17,20-lyase inhibitors, CYP17 inhibitors, estrogens, and progestational agents. Of these, GnRH analogues such as leuprolide acetate and goserelin acetate initially produce a rise in luteinizing hormone and follicle-stimulating hormone, followed by a downregulation of receptors in the pituitary gland, which effects a chemical castration. They were approved on the basis of randomized comparisons showing an improved safety profile (specifically, reduced cardiovascular toxicities) relative to diethylstilbestrol (DES), with equivalent potency. The initial rise in testosterone may result in a clinical flare of the disease. As such, these agents are relatively contraindicated in men with significant obstructive symptoms, cancer-related pain, or spinal cord compromise. GnRH antagonists such as degarelix achieve castrate levels of testosterone within 48 h without the initial rise in serum testosterone and do not cause a flare in the disease. Estrogens such as DES are rarely used due to the risk of vascular complications such as fluid retention, phlebitis, embolic events, and stroke. Progestational agents alone are less efficacious.
Agents that lower testosterone are associated with an androgen-depletion syndrome that includes hot flushes, weakness, fatigue, loss of libido, impotence, sarcopenia, anemia, change in personality, and depression. Changes in lipids, obesity, and insulin resistance, along with an increased risk of diabetes and cardiovascular disease, can also occur, mimicking the metabolic syndrome. A decrease in bone density may also result that worsens over time and results in an increased risk of clinical fractures. This is a particular concern, often underappreciated, for men with preexisting osteopenia secondary to hypogonadism or glucocorticoid or alcohol use. Baseline fracture risk can be assessed using the Fracture Risk Assessment Scale (FRAX), and to minimize fracture risk, patients are advised to take calcium and vitamin D supplementation, along with a bisphosphonate or the RANK ligand inhibitor, denosumab.
Antiandrogens First-generation nonsteroidal antiandrogens such as flutamide, bicalutamide, and nilutamide block ligand binding to the AR and were initially approved to block the disease flare that may occur with the rise in serum testosterone associated with GnRH agonist therapy. When antiandrogens are given alone, testosterone levels typically increase above baseline, but relative to testosterone-lowering therapies, they cause fewer hot flushes, less of an effect on libido, less muscle wasting, fewer personality changes, and less bone loss. Gynecomastia remains a significant problem but can be alleviated in part by tamoxifen.
Most reported randomized trials suggest that the cancer-specific outcomes are inferior when antiandrogens are used alone. Bicalutamide, even at 150 mg (three times the recommended dose), was associated with a shorter time to progression and inferior survival compared to surgical castration for patients with established metastatic disease. Nevertheless, some men may accept the trade-off of a potentially inferior cancer outcome for an improved quality of life.
Combined androgen blockade, the administration of an antiandrogen plus a GnRH analogue or surgical orchiectomy, and triple androgen blockade, which includes the addition of a 5ARI, have not been shown to be superior to androgen depletion monotherapies and are no longer recommended. In practice, most patients who are treated with a GnRH agonist receive an antiandrogen for the first 2–4 weeks of treatment to protect against the flare.
Intermittent Androgen Deprivation Therapy (IADT) The use of hormones in an “on-and-off” approach was initially proposed as a way to prevent the selection of cells that are resistant to androgen depletion and to reduce side effects. The hypothesis is that by allowing endogenous testosterone levels to rise, the cells that survive androgen depletion will induce a normal differentiation pathway. It is postulated that by allowing the surviving cells to proliferate in the presence of androgen, sensitivity to subsequent androgen depletion will be retained and the chance of developing a castration-resistant state will be reduced. Applied in the clinic, androgen depletion is continued for 2–6 months beyond the point of maximal response. Once treatment is stopped, endogenous testosterone levels increase, and the symptoms associated with hormone treatment abate. PSA levels also begin to rise, and at some level, treatment is restarted. With this approach, multiple cycles of regression and proliferation have been documented in individual patients. It is unknown whether the intermittent approach increases, decreases, or does not change the overall duration of sensitivity to androgen depletion. The approach is safe, but long-term data are needed to assess the course in men with low PSA levels. A randomized trial showed similar survival time between patients treated with intermittent versus continuous treatment, with a slightly higher risk of prostate cancer–specific mortality in the intermittent group, and higher cardiovascular mortality in patients on continuous therapy. The intermittent therapy was better tolerated.
Outcomes of Androgen Depletion The anti–prostate cancer effects of the various androgen depletion/blockade strategies are similar, and the outcomes predictable: an initial response, then a period of stability in which tumor cells are dormant and nonproliferative, followed after a variable period of time by a rise in PSA and tumor regrowth as a castration-resistant lesion that for most men is invariably lethal. Androgen depletion is not curative because cells that survive castration are present when the disease is first diagnosed. Considered by disease manifestation, PSA levels return to normal in 60–70% of cases, and measurable lesions regress in about 50%; improvements in bone scan occur in 25% of cases, but the majority of cases remain stable. The duration of response and survival is inversely proportional to disease extent at the time androgen depletion is first started, whereas the degree of PSA decline at 6 months has been shown to be prognostic. In a large-scale trial, PSA nadir proved prognostic.
An active question is whether hormones should be given in the adjuvant setting after surgery or radiation treatment of the primary tumor or whether to wait until PSA recurrence, metastatic disease, or symptoms are documented. Trials in support of early therapy have often been underpowered relative to the reported benefit or have been criticized on methodologic grounds. One trial showing a survival benefit for patients treated with radiation therapy and 3 years of androgen depletion, relative to radiation alone, was criticized for the poor outcomes of the control group. Another showing a survival benefit for patients with positive lymph nodes who were randomized to immediate medical or surgical castration compared to observation (p = .02) was criticized because the confidence intervals around the 5- and 8-year survival distributions for the two groups overlapped. A large randomized study comparing early to late hormone treatment (orchiectomy or GnRH analogue) in patients with locally advanced or asymptomatic metastatic disease showed that patients treated early were less likely to progress from M0 to M1 disease, to develop pain, and to die of prostate cancer. This trial was criticized because therapy was delayed “too long” in the late-treatment group. Noteworthy is that the American Society of Clinical Oncology Guidelines recommend deferring treatment until the disease has recurred and the prognosis has been reassessed. These guidelines do not support immediate therapy.
METASTATIC DISEASE: CASTRATE
Castration-resistant prostate cancer (CRPC) is defined as disease that progresses despite androgen suppression by medical or surgical therapies where the measured levels of testosterone are 50 ng/mL or lower. The rise in PSA indicates continued signaling through the AR signaling axis, the result of a series of oncogenic changes that include overexpression of androgen biosynthetic enzymes that can lead to increased intratumoral androgens, and overexpression of the receptor itself that enables signaling to occur even in the setting of low levels of androgen. The majority of CRPC cases are not “hormone-refractory,” and considering them as such can deny patients safe and effective treatment. CRPC can manifest in many ways. For some, it is a rise in PSA with no change in radiographs and no new symptoms. In others, it is a rising PSA and progression in bone with or without symptoms of disease. Still others will show soft tissue disease with or without osseous metastases, and others have visceral spread.
For the individual patient, it is first essential to ensure that a castrate status be documented. Patients receiving an antiandrogen alone, whose serum testosterone levels are elevated, should be treated first with a GnRH analogue or orchiectomy and observed for response. Patients on an antiandrogen in combination with a GnRH analogue should have the antiandrogen discontinued, because approximately 20% will respond to the selective discontinuation of the antiandrogen.
Chemotherapy and New Agents Through 2009, docetaxel was the only systemic therapy proven to prolong life. As a single agent, the drug produced PSA declines in 50% of patients, measurable disease regression in 25%, and improvement in both preexisting pain and prevention of future cancer-related pain. Since then, six agents with diverse mechanisms of action that target the tumor itself or other aspects of the metastatic process have been proven to prolong life and were FDA approved. The first was sipuleucel-T, the first biologic approach shown to prolong life in which antigen-presenting cells are activated ex vivo, pulsed with antigen, and reinfused. The second, cabazitaxel, a non–cross-resistant taxane, was shown to be superior to mitoxantrone in the post-docetaxel setting. This was followed by the CYP17 inhibitor abiraterone acetate, which lowers androgen levels in the tumor, adrenal glands, and testis, and the next-generation antiandrogen enzalutamide, which not only has a higher binding affinity to the AR relative to first-generation compounds, but uniquely inhibits nuclear location and DNA binding of the receptor complex. Both abiraterone acetate and enzalutamide were first approved for postchemotherapy treated patients on the basis of placebo-controlled phase III trials—a further indication that these tumors are not uniformly hormone-refractory. The indication for abiraterone acetate was later expanded to the prechemotherapy setting, based on a second trial using a co-primary endpoint of radiographic progression–free survival and overall survival. Similar results were seen with enzalutamide, for which an expanded indication is also anticipated. Alpharadin (radium-223 chloride), an alpha-emitting bone-seeking radioisotope, has been shown to prolong life in patients with symptoms related to osseous disease. The alpharadin result validated the bone microenvironment as a therapeutic target independent of direct effects on the tumor itself, as no declines in PSA were observed in the trial. Notable is that in addition to a survival benefit, the drug also reduced the development of significant skeletal events.
Other bone-targeted agents, such as the bisphosphonates and the RANK ligand inhibitor denosumab, protect against bone loss associated with androgen depletion and also reduce skeletal-related events by targeting bone osteoclasts. In one trial, denosumab was shown to be superior to zoledronic acid with respect to skeletal-related events, but had a slightly higher frequency of osteonecrosis of the jaw.
In clinical practice, most men seek to avoid chemotherapy and are first treated with a biologic agent and/or newer hormonal agent approved for this indication. It is crucial to the management of the individual patient to define therapeutic objectives before initiating treatment, as there are defined standards of care for different disease manifestations. For example, sipuleucel-T is not indicated for patients with symptoms or visceral disease because the effects on the disease occur late. Similarly, alpharadin is not indicated for patients with disease that is predominantly in soft tissue or who have osseous disease that is not causing symptoms.
Pain Management Management of pain secondary to osseous metastatic disease is a critical part of therapy. Optimal palliation requires assessing whether the symptoms are from metastases that threaten or that are already affecting the spinal cord, the cauda equina, or the base of the skull, which are best treated with external-beam radiation, as are single sites of pain. Neurologic symptoms require emergency evaluation because loss of function may be permanent if not addressed quickly. Because the disease is often diffuse, palliation at one site is often followed by the emergence of symptoms in a separate site that had not received radiation. In these cases, bone-seeking radioisotopes such as alpharadin or the beta emitter 153Sm-EDTMP (Quadramet) can be considered in addition to abiraterone acetate, docetaxel, and mitoxantrone, each of which is formally approved for the palliation of pain due to prostate cancer metastases.
BENIGN DISEASE
BENIGN PROSTATIC HYPERTROPHY
BPH is a pathologic process that contributes to the development of lower urinary tract symptoms in men. Such symptoms, arising from lower urinary tract dysfunction, are further subdivided into obstructive symptoms (urinary hesitancy, straining, weak stream, terminal dribbling, prolonged voiding, incomplete emptying) and irritative symptoms (urinary frequency, urgency, nocturia, urge incontinence, small voided volumes). Lower urinary tract symptoms and other sequelae of BPH are not just due to a mass effect, but are also likely due to a combination of the prostatic enlargement and age-related detrusor dysfunction.
