19 Benign and Borderline Tumors of the Lungs and Pleura
Malignant neoplasms are more common, by far, than benign tumors in the lower respiratory tract. In the United States, lung carcinoma is the leading cause of cancer-related death in both sexes, and it accounts for more than 0.7% of new malignancies each year in men.1 In Europe, the situation is even worse; for example, more than 3.0% of newly diagnosed malignant neoplasms in Germany are lung cancers.2 These data reflect the continuing use of cigarettes worldwide and the relative potency of tobacco smoke as a carcinogenic agent.
Benign Pleuropulmonary Neoplasms
Clinical Features
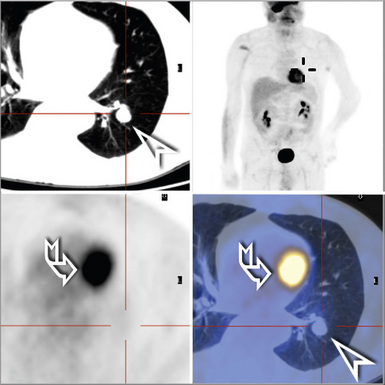
The clinical characteristics of benign tumors in the lower respiratory tract can be considered in an overview, because they are generally not specific to any particular diagnosis. Most benign intrapulmonary lesions are associated with no symptoms or signs whatsoever, and are found incidentally with screening radiographic studies. As discussed elsewhere in this book, hamartomas are often separable from other benign but truly neoplastic masses of the lung on imaging studies because of their common content of distinctive calcifications, fat densities, or both3 (Fig. 19-1). Otherwise, the radiologist is not typically able to distinguish between specific histologic entities in this context. Endotracheal and endobronchial tumors are more often related to clinical symptoms such as wheezing, hemoptysis, obstructive pneumonia, and postobstructive pulmonary hyperinflation, depending on their anatomic location.
In the past, radiologists were comfortable simply observing a pulmonary mass over time, if it had reassuring morphologic characteristics. In fact, statistical paradigms have been constructed to aid in this process.4 However, because of the unfortunate pressure of litigation involving putative “delays in diagnosis” of malignant neoplasms at various sites5—together with the modern availability of techniques such as video-assisted thoracoscopic surgery6—many more benign lesions of the lungs are excised today than in previous years.
Benign Tumors That Are Principally Tracheal and Endobronchial
Solitary Tracheobronchial Papilloma
A solitary papilloma of the tracheobronchial tree (SPTT) most often arises in adults—typically middle-aged—with a slight male predominance.7–22 Occasionally, even though only one papillomatous lesion of the lower respiratory tract may be present, it may coexist with papillary squamous proliferations of the larynx or oropharynx.
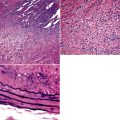
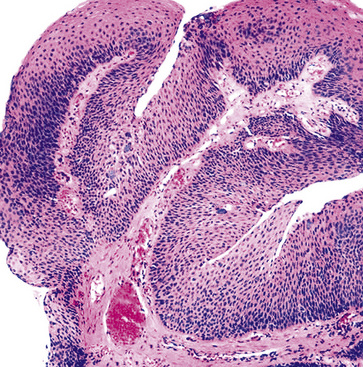
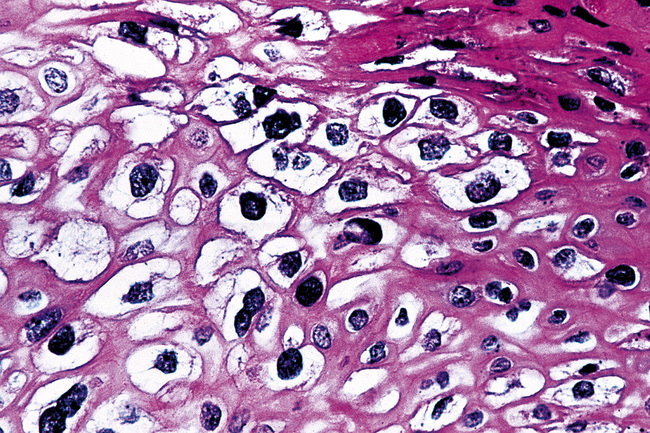
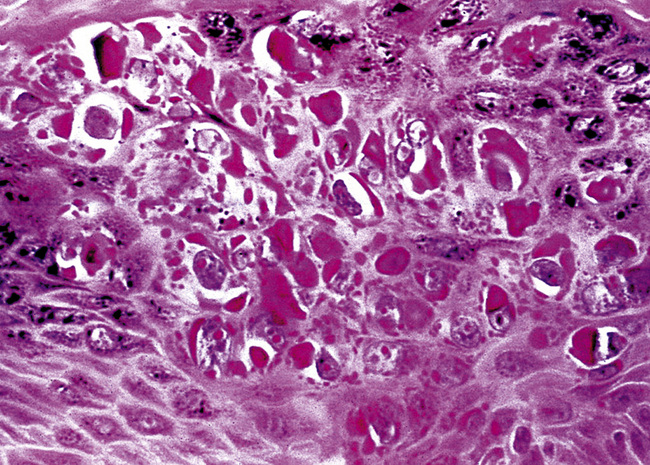
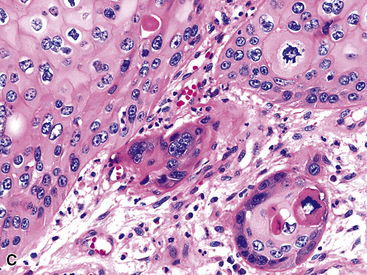
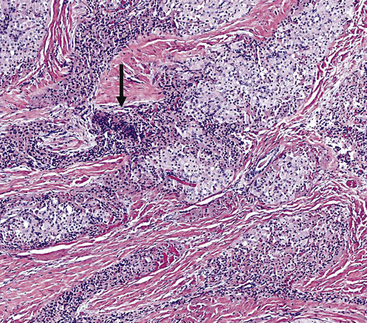
Microscopically, SPTTs are similar to viral papillomas elsewhere in the body, particularly in the genitoperineal region. They are constituted by arborizing fronds with fibrovascular cores, mantled by relatively bland squamous epithelial cells (Fig. 19-2). These often exhibit nuclear hyperchromasia and crenation, but the nuclear chromatin may be homogenized and glassy. The cytoplasm is commonly unremarkable; alternatively, it may show perinuclear clearing, eosinophilic globular inclusions, or hypergranulation with clumping of keratohyaline granules (Figs. 19-3 and 19-4). Nuclear atypia has been observed in squamous SPTT, and squamous carcinoma may develop in this setting.23,24
Another even rarer variant of SPTT is the columnar papilloma, composed of columnar epithelial cells rather than squamous elements.14 Putatively, it has no potential for malignant change.
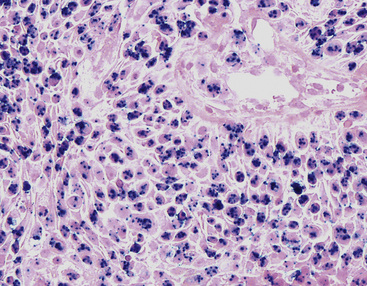
Interestingly, the biologic associations between squamous SPTT and human papillomavirus (HPV) types are comparable to those in the genital tract. Specifically, HPV types 7 and 11 are most commonly seen in uncomplicated SPTT; in contrast, HPV types 16 and 18 are associated with dysplastic nuclear features and a higher risk of carcinomatous transformation.23,24 The presence of these viral agents can be evaluated using in situ hybridization (Fig. 19-5), the hybrid capture method, or polymerase chain reaction.23–27
In light of the usual behavior of SPTT, conservative therapeutic approaches, such as endoscopic removal, cryotherapy, or fulguration, are typically applied. Lesions in which malignancy develops must be treated in a manner appropriate for “ordinary” lung cancers (Fig. 19-6).28–31
Multifocal Respiratory Tract Papillomatosis
Respiratory tract papillomatosis (RTP) is the multifocal form of viral papillomagenesis, as described earlier. Its onset is early in life because the mode of transmission is natal inhalation of virally infected genital tract secretions during vaginal delivery.32–41 Children with RTP typically have oropharyngeal or laryngeal disease initially; it may remain localized or spread to involve the lower airways as well, as seen in 5% of cases.40 In the latter instance, growth into the lung parenchyma may supervene, with the eventual appearance of cavitary lesions that can simulate carcinomas radiographically.32,41 Obstruction of the tracheal or bronchial lumina is associated with recurrent pneumonia, hemoptysis, and asthma-like symptoms. Even though RTP is often said to be “recurring,” the entire respiratory tract is at risk for infection by HPV in this disorder. Thus, it is more apropos to consider separate lesions as metachronously or synchronously independent of one another pathogenetically.
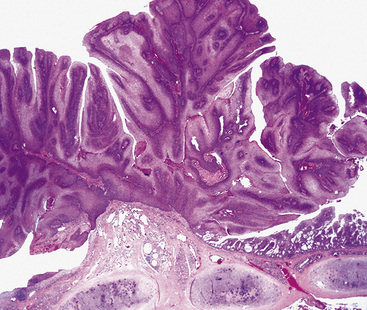
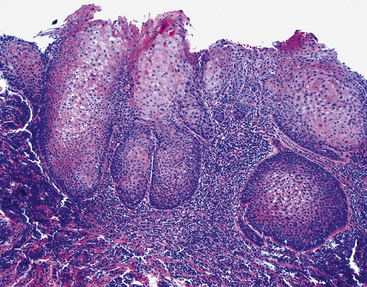
The gross and histologic features of RTP are largely the same as those of SSTP (Figs. 19-7 and 19-8). Exceptions include the multiplicity of lesions and the greater tendency for “inverting” or tissue-destructive growth of the papillomatous lesions in RTP (Fig. 19-9). The HPV profiles of RTP and SSTP are comparable, with the notable proviso that HPV-11 is overwhelmingly the most common viral type seen in RTP. Mutations of the p53 gene have been linked to malignant transformation of individual lesions in both conditions.23,24,27,41–46
It does not appear as though treatment with HPV-targeted antiviral medications (specifically, cidofovir) produced morphologic changes in persistent papillomatous lesions of the airway.47
The incidence of previous HPV integration was assessed by Clavel and colleagues25 using the hybrid capture method in a series of bronchopulmonary carcinomas. They found evidence of oncogenic HPV integration in only 2.7% of those tumors. On the other hand, Yousem and colleagues48 demonstrated similar positivity by in situ hybridization in 30% of squamous carcinomas and 17% of large cell undifferentiated carcinomas of the lung; Syrjanen28 likewise observed histologic viral changes in adjacent metaplastic bronchial mucosa in 26 of 104 squamous carcinomas (25%). Based on these data, it must be acknowledged that HPV may play a greater role in the etiology of lung carcinomas (particularly of the squamous type) than previously thought.
Bronchial Mucous Gland Adenoma
The term “bronchial adenoma” has been plagued by many misconceptions and misapplications since its introduction by Liebow in 1952.49 However, two benign neoplastic entities could still properly be called “bronchial adenomas”—mucous gland adenoma (MGA) and mixed tumor (MT; pleomorphic adenoma; discussed later).
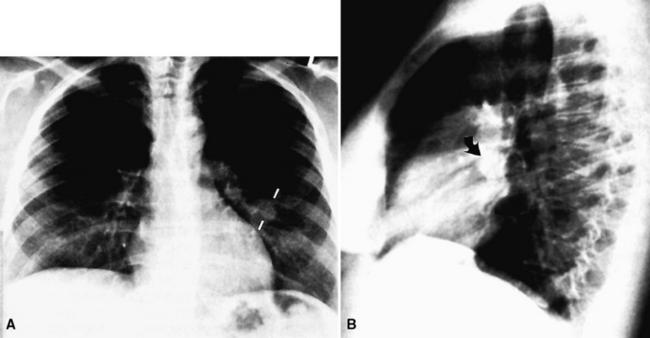
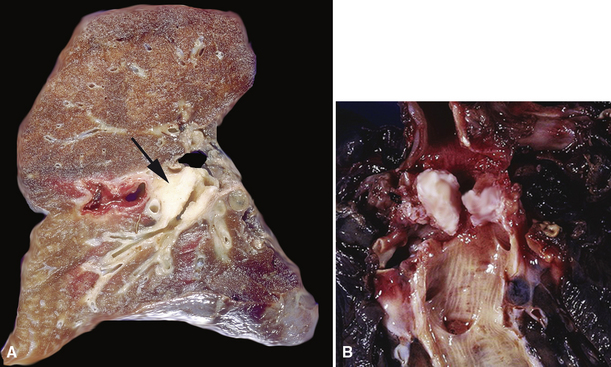
Mucous gland adenoma is an extraordinarily rare lesion. England and Hochholzer50 noted that one series of more than 3000 pulmonary tumors included no examples of MGA,51 and only 1 MGA was represented in another report of 130 benign neoplasms of the lung seen at a large referral center.52 In the vast experience of the U.S. Armed Forces Institute of Pathology, only 10 examples of MGA were found. Men and women were equally affected, and the patients ranged in age from 25 to 67 years.50,53 There is no particular predilection for anatomic location; MGAs may arise in any of the major lobar or segmental bronchi. Radiographs either show changes of postobstructive pneumonia or hyperinflation or demonstrate a discrete nodule or coin lesion that is centered on a bronchus54–66 (Fig. 19-10). Kwon and coworkers67 have noted that MGA may produce the “air meniscus sign” on computed tomogram (CT) of the airways.
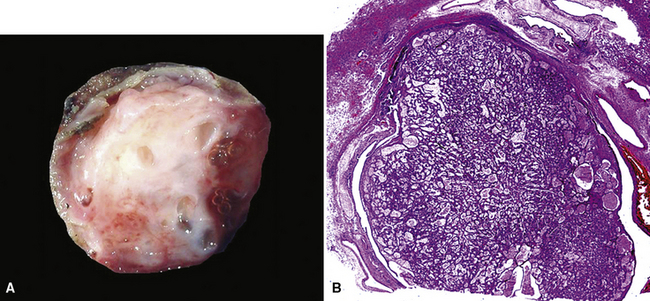
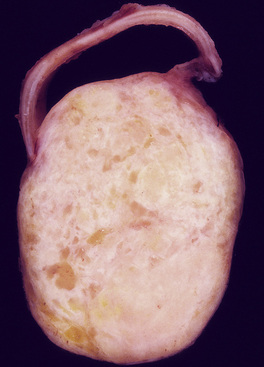
Mucous gland adenomas measure between 0.5 and 1 cm in maximal dimension. They often demonstrate encapsulation, with mucoid cut surfaces; internal fibrous septations may yield a loculated appearance as well. Occasional lesions may completely occlude the bronchial lumen (Fig. 19-11).
As succinctly stated by England and Hochholzer,50 “cystic change [is] the cardinal feature of MGA of the bronchus.” Microcystic arrays of cuboidal or columnar tumor cells may permeate the bronchial wall to the level of the cartilaginous plates; however, growth beyond that point is absent. Cystic contents are either overtly mucinous or more serous. Secondary formation of cholesterol clefts and dystrophic calcifications may also be seen, and squamous metaplasia in the most luminal aspect of the lesion may occur. Nuclei are generally bland, with small nucleoli, and cytoplasm may be amphophilic, oxyphilic, clear, or foamy. Mitotic figures are scarce.
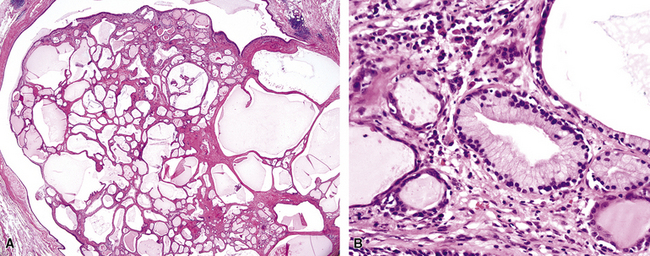
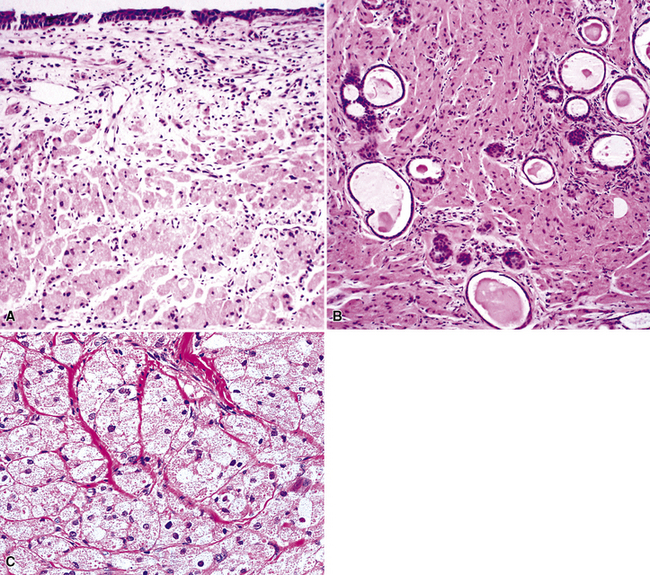
The internal substructure of MGA has been divided into two morphologic patterns: glandular-tubulocystic (Fig. 19-12) and papillocystic (Fig. 19-13). Within each of these categories, there may be a spectrum of appearances, ranging from a monotonous tubular pattern (Fig. 19-14) to a complex arborizing aggregation of papillary structures.50 Stromal sclerosis may be present.
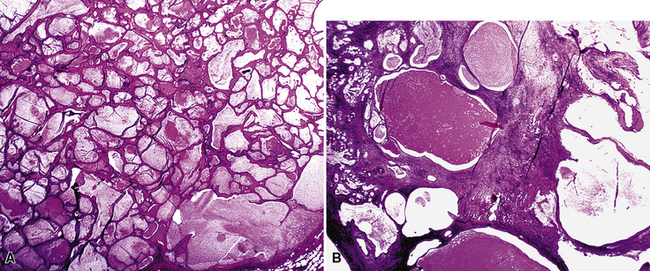
Figure 19-12 A and B, A glandular-tubulocystic growth pattern is present in this mucous gland adenoma of the bronchus.
(Courtesy of Dr. Douglas England, Madison, WI.)
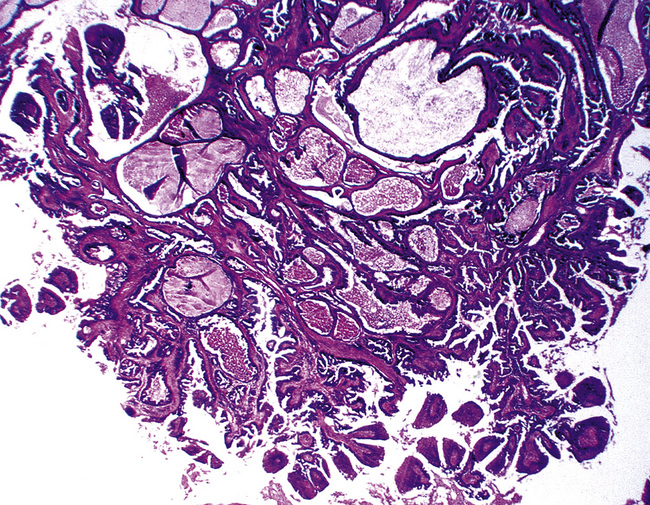
Figure 19-13 This mucous gland bronchial adenoma shows a papillocystic configuration.
(Courtesy of Dr. Douglas England, Madison, WI.)
Immunohistologic features of MGA are comparable to those of non-neoplastic bronchial glands. The constituent cells are consistently labeled for cytokeratin, epithelial membrane antigen, and blood group isoantigens, with less uniform reactivity for carcinoembryonic antigen. Stromal cells show myoepithelial features, with concurrent staining for keratin, actin, and S-100 protein.50 Squamous-type (“high molecular-weight”) keratins may be expressed in MGAs.68 This could be a diagnostic trap vis-à-vis the alternative interpretation of mucoepidermoid carcinoma (discussed later).
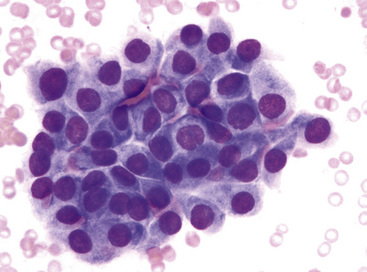
The differential diagnosis includes predominantly cystic mucoepidermoid carcinoma, MT, “sclerosing hemangioma” (pneumocytoma), and primary or metastatic mucinous (“colloid”) adenocarcinoma. Of these possibilities, the first two are the most problematic, necessitating adequate biopsies for visualization of the tumor architecture. Cytologically, MGA shows bland nests and sheets of monotonous epithelioid cells69 (Fig. 19-15). Diagnostic separation from mucoepidermoid carcinoma or MT usually is not possible using fine-needle aspiration (FNA) biopsy or bronchial brushing specimens.
An admixture of squamous elements throughout the mass, infiltrative growth through the bronchial wall, or both would tend to argue for a diagnosis of mucoepidermoid carcinoma. Although MGA does not manifest chondromyxoid stroma or immunoreactivity for glial fibrillary acidic protein, as seen in some MTs,70 the possibility that MGA is related nosologically to monomorphic adenoma—a variant of MT—cannot be dismissed. In any event, it represents a distinctive clinicopathologic entity that is believed to deserve its own diagnostic designation.
Treatment of MGA can be conservative, with sleeve resection of the bronchus and reconstruction when clinically feasible.50,57
Salivary Gland Analog Tumors
Mixed Tumor (Pleomorphic Adenoma)
Pleomorphic adenoma—or MT—occurs predominantly in adults, with a slight preference for women.71–75 The age range is 8 to 75 years.76 The lesion may present as either a central or a peripheral mass77–79 (Fig. 19-16).
Grossly, central main stem bronchial lesions are commonly polypoid, whereas those in the periphery present as well-circumscribed tumors usually attached to bronchi (Fig. 19-17). The size of the neoplasms varies from 1 to 16 cm in greatest diameter. The cut surfaces of MTs may be chondroid and firm, or may have a soft consistency, with only focal induration.
By definition, pleomorphic adenomas show at least a biphasic microscopic image; they are characteristically composed of epithelial tubules and nests that are embedded in a chondromyxoid stroma (Figs. 19-18 and 19-19). Interestingly, intrapulmonary MTs rarely show the amount of mature cartilaginous stroma present in MTs of the salivary glands. In some lesions, the predominant growth pattern may be solid myoepithelial proliferation that approximates that of “cellular” MTs in the head and neck. Those neoplasms comprise compact epithelioid cells with round or oval nuclei, which generally lack nuclear atypia, necrosis, hemorrhage, and mitotic activity (Fig. 19-20). Notably, there have been no well-documented cases of carcinoma arising from preexisting MTs of the lung, as may rarely occur in salivary glandular sites. Other variants of MT include a “myoepitheliomatous” subtype that may manifest either a spindle cell composition80 (Fig. 19-21) or a plasmacytoid constituency; a form with extensive squamous metaplasia (Fig. 19-22); a subtype in which sizable zones of the lesion demonstrate an adenoid cystic carcinoma-like cribriform architecture; and a chondroid-rich form that simulates pulmonary chondroma or chondromatous hamartoma.
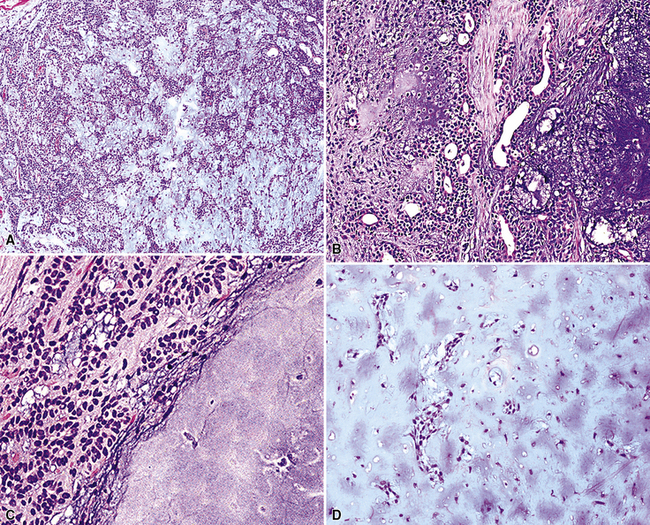
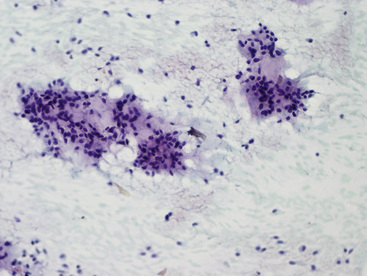
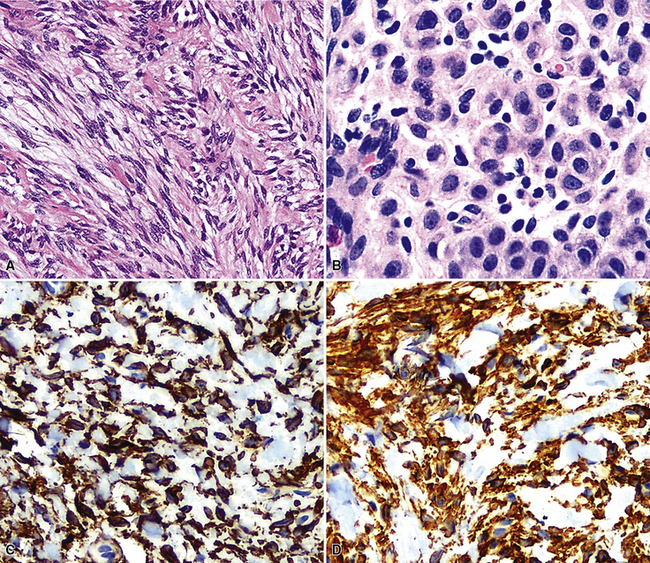
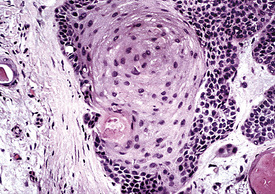
Figure 19-22 Prominent squamous metaplasia is apparent in the epithelial element of this bronchial mixed tumor.
The differential diagnosis depends on whether one is dealing with a biopsy specimen or a complete resection of the tumor. In the former instance, MTs can be confused with other salivary gland-like tumors, such as adenoid cystic carcinoma, as well as with hamartoma/chondroma, squamous cell carcinoma (when squamous metaplasia is dominant), and biphasic malignancies, such as sarcomatoid carcinoma. In resection specimens, the diagnosis is typically straightforward. However, if MTs demonstrate an overwhelmingly prominent solid pattern with spindle cell (“myoepitheliomatous”) differentiation, sarcomas may also be considered. In this setting, concurrent immunoreactivity for keratin, vimentin, S-100 protein, actin or caldesmon, p63 protein, and glial fibrillary acidic protein81,82 provides the necessary evidence for a conclusive diagnosis of MT.
Mixed tumors of the lung behave in an indolent fashion. There are only anecdotal reports of metastasizing lesions of this type,83 in analogy to rare examples in the salivary glands. No particular pathologic features of such neoplasms can be used to predict this unusual adverse behavior. Complete but conservative excision is the treatment of choice.77,78
Oncocytoma
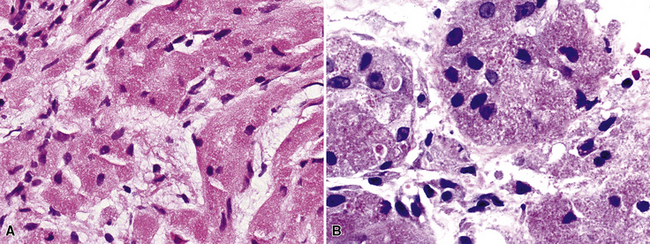
There are only a few reported cases of pulmonary oncocytoma.84–93 These tumors exhibit morphologic similarities to comparable tumors in the salivary glands, showing a brownish gross appearance (Fig. 19-23). They are composed of nests of uniformly large polygonal cells with prominently eosinophilic granular cytoplasm and bland nuclei (Figs. 19-24 and 19-25). In view of the existence of other, more common pulmonary tumors that can show oncocytic changes, it is important to properly exclude those other possibilities by adjunctive studies. Neuroendocrine tumors showing oncocytic features (oncocytic “carcinoids;” grade I neuroendocrine carcinomas) are far more common than oncocytomas and are recognizable by their immunoreactivity for chromogranin-A, synaptophysin, and CD56.94,95 Metastatic tumors from the salivary glands and kidneys also must be considered, particularly in rare cases where pulmonary oncocytomas appear to be synchronously multifocal.88 Clinical information is important in this context because the electron microscopic and immunophenotypic properties of primary and metastatic oncocytic neoplasms may be very similar (see Chapter 17).
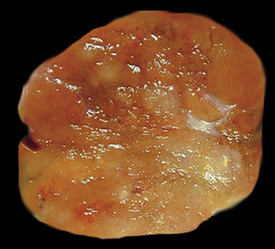
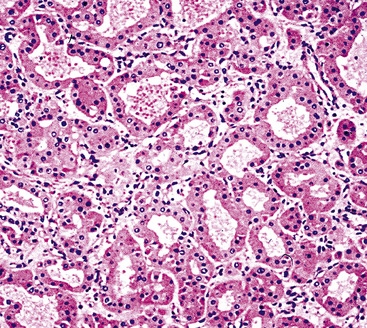
Figure 19-24 Either solid or tubular profiles of oxyphilic polygonal cells can be seen in oncocytoma.
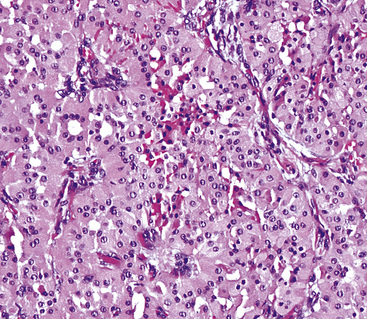
In general, oncocytomas in the lung and other anatomic sites are epithelial, demonstrating immunoreactivity for keratins and epithelial membrane antigen; vimentin is inconsistently present, but markers of muscular, neural, or neuroendocrine lineages are absent.88,96 Immunolabeling with antibodies to mitochondrial proteins is common,97 corresponding to the ultrastructural hallmark of these neoplasms.
Fine-needle aspiration biopsy of oncocytic neoplasms yields a monomorphic population of large epithelioid cells with round to oval nuclei, dispersed chromatin, small nucleoli, and amphophilic to acidophilic cytoplasm. They are variably cohesive and typically show little nuclear pleomorphism.88
Because of problems with the previous definition of “pulmonary oncocytoma,” as noted earlier, meaningful comments on its behavior are difficult. However, we have seen cases that obviously invaded the lung parenchyma and had atypical morphologic features, such as nuclear pleomorphism and atypical mitoses (Fig. 19-26). Accordingly, such lesions are defensibly labeled as “malignant” oncocytomas.85
Peripheral Nerve Sheath Tumors
Primary neoplasms of the lung that demonstrate schwannian or perineurial differentiation are more often located in the walls of the major bronchi than in the peripheral lung.98–113 Chest radiographs show nodular or irregular masses associated with bronchi; secondary atelectasis is sometimes noted as well98 (Fig. 19-27). Some patients with primary neurogenic pulmonary tumors have neurofibromatosis type 1 (NF1; von Recklinghausen’s disease), and that is true for both neurofibroma and neurilemmoma.114 Neurogenic sarcomas also arise in the lungs,115–117 but it is unclear how many have occurred in the context of NF1 in association with preexisting pulmonary neurofibromas.
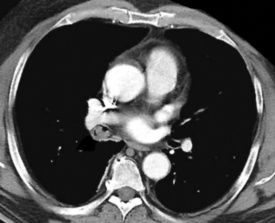
Figure 19-27 A computed tomographic image of a bronchial neurilemmoma showing a mass in the lumen of the right mainstem bronchus.
Grossly, peripheral nerve sheath tumors (PNSTs) of the bronchus are well-demarcated yellow-white masses centered on the bronchial wall and often protruding into the bronchial lumen (Figs. 19-28 and 19-29). Gross foci of hemorrhage, necrosis, or cystification are absent in benign lesions of this type.
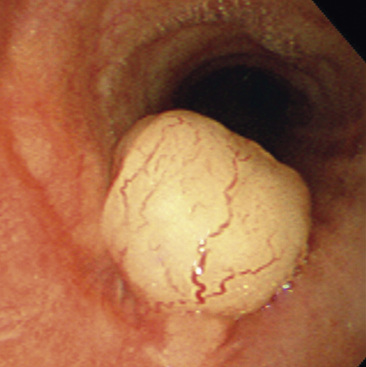
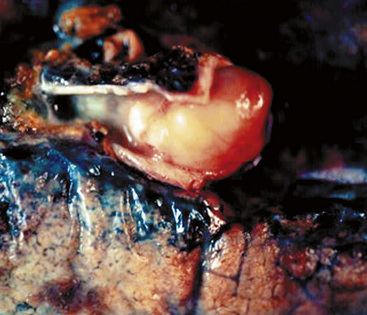
The histologic images associated with benign PNSTs are varied. Prototypical neurofibromas are “plexiform” in NF1, putatively reflecting a neoplastic attempt to recapitulate a neural plexus. Constituent cells are serpiginous, with attenuated fusiform nuclei and delicately fibrillar cytoplasm (Fig. 19-30). The supporting matrix is myxedematous or collagenized, and it may contain scattered foam cells, mast cells, and lymphocytes. Mitotic activity is typically absent. Neurilemmomas—also termed “schwannomas”—are biphasic in their classic form, with compactly cellular (“Antoni A”) areas alternating with zones of loose cellularity and myxoid change (“Antoni B” foci) (Figs. 19-31 and 19-32). Nuclei may be aligned in register in Antoni A areas, representing “Verocay bodies” (Fig. 19-33). Intralesional blood vessels have thick walls in many neurilemmomas, and a well-defined peripheral tumor capsule may be identified in some instances. Variants of neurilemmoma include “cellular” schwannoma, which manifests intersecting bundles of densely apposed, focally mitotic spindle cells in the context of an encapsulated mass111 (Fig. 19-34); “glandular” schwannoma; “ancient” schwannoma, showing scattered pleomorphic hyperchromatic nuclei in degenerative cells (Fig. 19-35); plexiform schwannoma, in which broad plexiform fascicles of tumor cells show the substructure of neurilemmoma; and melanotic psammomatous schwannoma, exhibiting microcalcifications and melanin pigmentation118 (Fig. 19-36). The last of these subtypes may be associated with myxomas of the skin, heart, and breast; the presence of cutaneous ephelides; and overactivity of the endocrine glands.
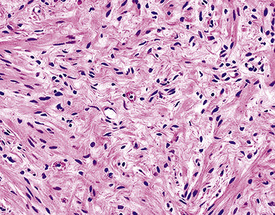
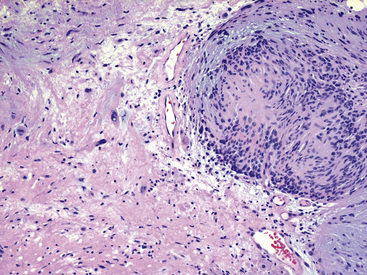
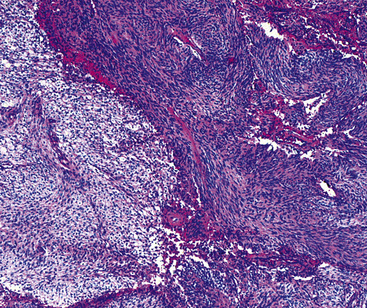
Figure 19-32 An Antoni B focus in a bronchial neurilemmoma (lower left) with a myxoid intercellular matrix.
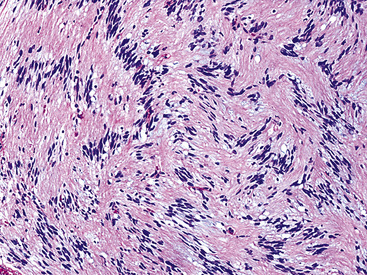
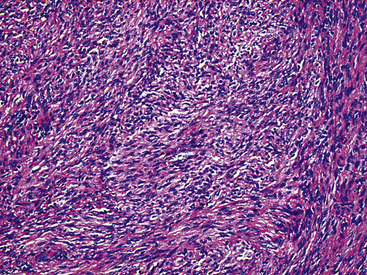
Figure 19-34 Cellular schwannoma (neurilemmoma) demonstrating densely agglomerated spindle cells with focal mitotic activity.
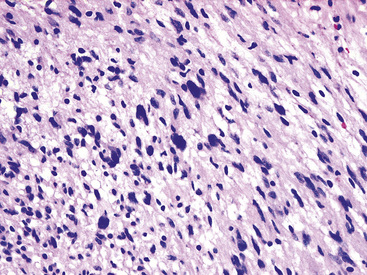
Figure 19-35 “Ancient” neurilemmoma showing degenerative atypia of tumor cell nuclei with mild pleomorphism and hyperchromasia.
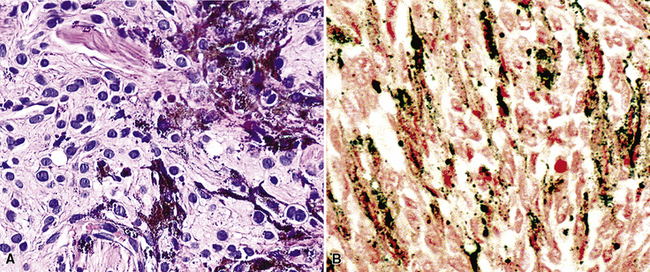
The immunophenotype of PNSTs features reactivity for vimentin and variable labeling for S-100 protein (Fig. 19-37), CD56, CD57, glial fibrillary acidic protein, and epithelial membrane antigen. The last of these markers is believed to represent perineurial differentiation in this context. Keratin positivity is absent, except in the glands of glandular schwannoma, and myogenous markers should be negative.
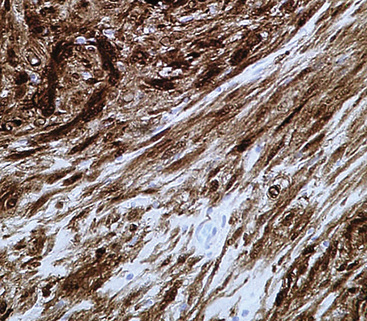
If the identification of PNST can be established in evaluations of bronchial biopsy specimens and radiographic findings support a benign diagnosis, the surgeon can undertake a conservative approach to the removal of such lesions.102,105 However, small samples of neurogenic neoplasms are notoriously unreliable in predicting the biologic potential of PNSTs, and they should not be used in isolation to govern decisions on therapy.
Granular Cell Tumors
Granular cell tumor (GCT; also known as “Abrikossoff’s tumor”119) may be seen in many anatomic locations.120 Fewer than 200 have been reported in the trachea and lungs.121–139 Patients of any age may have such lesions, and multiple tumors in the same individual have been reported.132,135,138,140,141
Endoscopic examination often reveals a sessile polypoid endoluminal component, with intact overlying mucosa131 (Fig. 19-38). Radiographic studies confirm that characteristic, but also demonstrate an infiltrative aspect to many GCTs that may lead to a mistaken preoperative diagnosis of malignancy.130–136 GCTs in the peripheral lung parenchyma are unusual,138 but these likewise may assume a spiculated appearance that closely simulates that of adenocarcinoma. This situation is further complicated by occasional case reports of GCTs that coexisted with malignant neoplasms.142,143
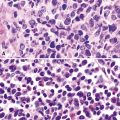
Microscopically, these tumors comprise a uniform population of polygonal or fusiform cells that contain small ovoid hyperchromatic nuclei with indistinct nucleoli (Fig. 19-39). The cytoplasm is abundant, eosinophilic or amphophilic, and coarsely granulated, often with the additional presence of rounded inclusions that superficially resemble Michaelis-Gutman bodies (Fig. 19-40). Like Michaelis-Gutman bodies, these may also have a targetoid configuration. Mitotic figures are typically scarce and physiologic; necrosis and vascular invasion are absent. The advancing border of granular cell tumors may be “pushing” or irregular and permeative. A proportion of these lesions infiltrate deeply into the bronchial wall or even through it into the adjacent parenchyma. In nonpulmonary sites, such a growth pattern has been linked to a greater risk of recurrence,144 but that correlation does not seem to apply to GCTs in the airways or lungs. The existence of primary malignant GCT of the respiratory tract has never been convincingly documented, and recurrent neoplasms are typically those that have never been completely excised.
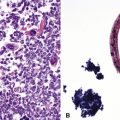
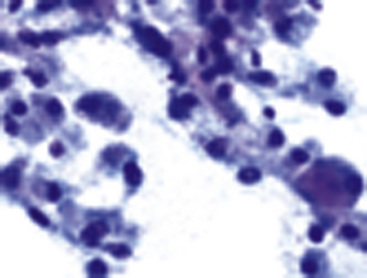
Fine-needle aspiration biopsy of GCT yields a monotonous population of large epithelioid cells that are variably cohesive. They contain ovoid nuclei, distinct chromocenters, and abundant granular cytoplasm that is best seen in Romanowsky-stained slides (Fig. 19-41). A plasmacytoid appearance is common.145
Electron microscopy of GCTs shows a distinctive multiplicity of secondary and tertiary lysosomes in the cytoplasm of the tumor cells, virtually to the exclusion of other organelles146,147 (Fig. 19-42). Immunohistologic evaluation most often reveals evidence of schwannian differentiation, with reactivity for S-100 protein, CD56, CD57, myelin basic protein, and combinations thereof, in 80% to 85% of cases147–150 (Fig. 19-43). The abundance of lysosomes is reflected by their CD68 reactivity. Recently, the presence of calretinin and alpha-inhibin has also been reported in GCT.151
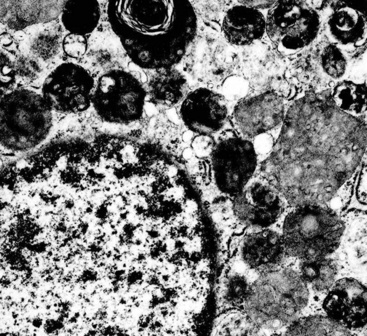
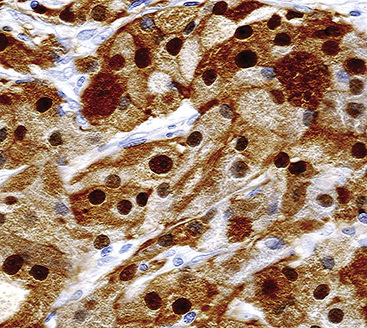
An important caveat regarding the immunophenotype of this tumor type is that non-schwannian lesions containing granular cells are a heterogenous group.152 Carcinomas (both neuroendocrine and nonendocrine),153,154 smooth muscle tumors, endothelial proliferations, and neoplasms of uncommitted lineage have all been described with a granular cell phenotype. Therefore, it is important to include immunohistologic evaluations to address these possibilities in differential diagnosis, with or without ultrastructural studies. Oncocytoid granular cell grade 1 neuroendocrine carcinoma (“carcinoid”) of the lung is the most common simulator of GCT,155,156 making chromogranin-A and synaptophysin important markers in this context.
Benign Lesions Affecting Either the Airways or the Lung Parenchyma
Alveolar Adenoma
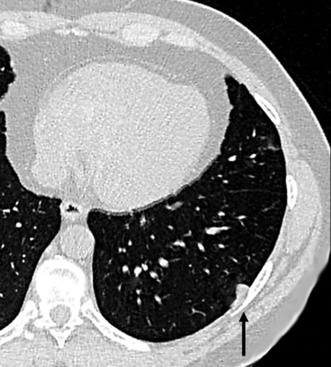
Alveolar adenoma (AA)157 is typically a solitary peripheral parenchymal nodule found incidentally in asymptomatic adult patients, with no sex predilection.158 On imaging studies, AA presents a rounded configuration and measures 1 to 6 cm in greatest dimension (Fig. 19-44).159 The surgeon is likely to report that it “shells out” from the surrounding lung tissue.160
Grossly, AA is circumscribed, with a spongy gray-white multilocular cut surface. Microscopically, it comprises many cystic spaces of variable sizes, which are mantled by low-cuboidal epithelial cells (Fig. 19-45).157,158 Eosinophilic granular material is commonly present in the cyst lumina. The tissue between the cysts is represented by cytologically bland and closely apposed bluntly fusiform cells in a loose fibromyxoid matrix. Mitotic activity, nuclear pleomorphism, and necrosis are absent.161 The immunohistochemical features of AA closely parallel those of sclerosing hemangioma (“pneumocytoma”; discussed later), and it is our belief—shared by other authors as well162—that the two tumors are likely part of a neoplastic family rather than entirely distinct entities. One sees epithelial markers—including thyroid transcription factor 1—in the cuboidal cells lining tumoral microcysts, but not in the stromal elements.158,161–164 On the other hand, the latter components are reactive for vimentin, and often for CD34.158
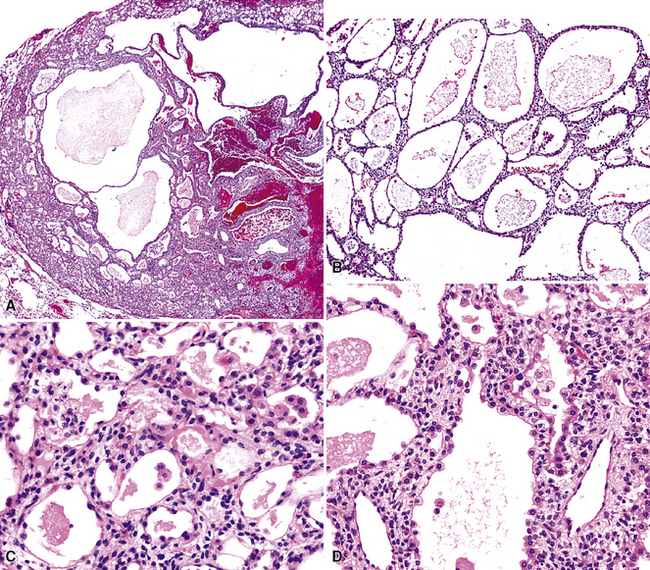
The differential diagnosis of AA is limited. Aside from sclerosing hemangioma, which is usually larger than AA and does not have its prominently cystic substructure,162 the principal considerations are pulmonary hemangioma-lymphangioma and atypical adenomatous hyperplasia (see Chapter 16). The vascular lesions can be identified by appropriate immunohistochemical stains.165,166 Atypical adenomatous hyperplasia lacks microcysts and does not contain a mesenchymal stromal cell component.167
Even though fewer than 50 cases of AA have been reported, the behavior of this tumor has been uniformly favorable. Hence, conservative wedge excision appears to represent adequate treatment.158,160,168,169
Papillary Adenoma
Another uncommon neoplasm that is likely related to sclerosing hemangioma has been reported separately as “papillary adenoma” of the lung (PAL). It may be seen in children and adults alike, as a nondescript and asymptomatic parenchymal nodule in imaging studies.170–180 Multifocality occurs, and in one case, such multiplicity was seen in a patient with neurofibromatosis.176 Grossly, this tumor usually shows sharp circumscription from the surrounding lung tissue in most cases, with a solid tan-white cut surface (Fig. 19-46). Nonetheless, a few cases have had infiltrative characteristics.172,181,182
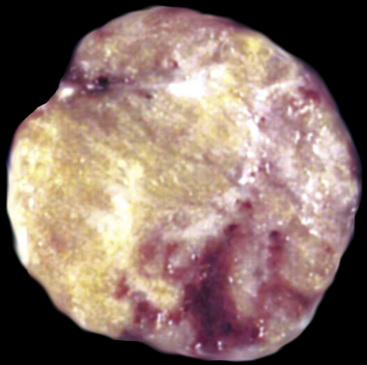
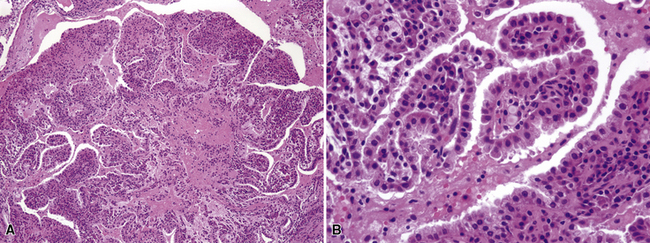
Histologically, PAL shows papillary profiles of cuboidal to low columnar, nonciliated, focally vacuolated epithelial cells that mantle well-formed fibrovascular cores (Fig. 19-47). The stroma in the latter structures may contain mixed inflammatory infiltrates, potentially including lymphocytes, mast cells, plasma cells, and eosinophils. The surrounding lung typically demonstrates a fibroblastic response to the lesion, sometimes with formation of a circumferential pseudocapsule. Mitotic activity is limited, and necrosis is absent.
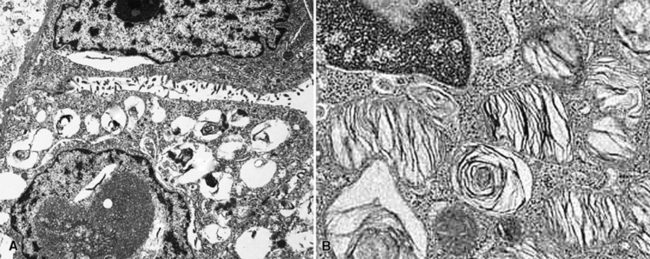
Immunohistochemical studies of PAL show consistent reactivity for keratin, surfactant-related apoproteins, thyroid transcription factor 1, and napsin-A in the neoplastic cells.172,182 In addition, labeling for Clara cell antigen and carcinoembryonic antigen may be present. Ultrastructural analysis shows microvillous differentiation of the plasmalemmae and the presence of lamellar bodies in the tumor cell cytoplasm (Fig. 19-48).170–173,182
Because of the sometimes-infiltrative nature of PAL, some authors have suggested that it be considered borderline malignant.172,181,182 Nevertheless, there have been no reports of recurrence or metastasis of this lesion, and conservative surgical removal appears sufficient.
Leiomyoma
Solitary leiomyoma of the respiratory tract has been reported as an independent entity, distinct from hamartomas. These tumors are most often located in the wall of the trachea or bronchi (Fig. 19-49), with pulmonary parenchymal or pleural origins rare.183–195 Bronchoscopic and gross evaluation of central lesions shows a dome-shaped endoluminal mass, usually with a smooth mucosal surface (Figs. 19-50 and 19-51).
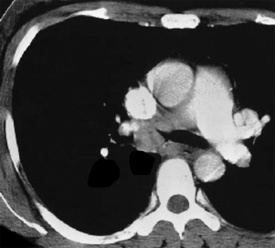
Figure 19-49 An endoluminal mass evident in the right bronchus intermedius proved to be a leiomyoma.
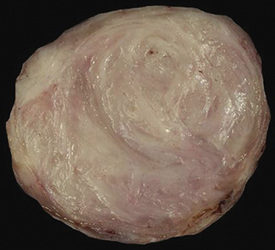
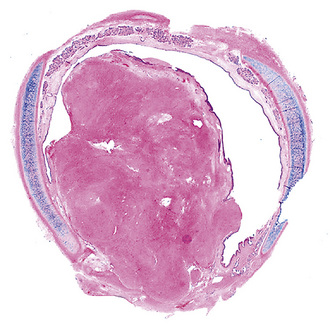
Figure 19-51 Low-power micrograph demonstrates the relationship of an endoluminal bronchial leiomyoma to the wall of the airway.
Histologically, bronchial leiomyoma is identical to benign smooth muscle tumors elsewhere in the body, comprising intertwining fascicles of spindle cells with fusiform nuclear contours, perinuclear cytoplasmic vacuolization, and finely fibrillary eosinophilic cytoplasm (Fig. 19-52). Mitotic activity is limited, there is no infiltration of adjacent tissues, and necrosis is absent.
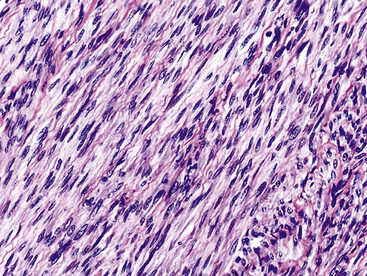
The fine structural features of smooth muscle proliferations in the lung include pericellular basal lamina, plasmalemmal hemidesmosomes and micropinocytotic vesicles, skeins of cytoplasmic thin filaments, and intrafilamentous dense bodies.196–198 Immunohistologically, one typically sees reactivity for muscle-specific actin, alpha-isoform (“smooth muscle”) actin, desmin, calponin, and caldesmon, with an absence of S-100 protein, CD56, CD57, and keratin.188
Solitary smooth muscle tumors are treated with simple but complete excision, if thorough clinical evaluation has excluded an extrapulmonary primary lesion of the same type. The latter proviso relates to the fact that some leiomyosarcomas (particularly in the retroperitoneum) are extremely low-grade proliferations that may produce “pseudoleiomyomatous” metastases.199
Glomus Tumor and Glomangioma
Glomus tumor and glomangioma (GTG)200–205 are most often seen in the skin and superficial soft tissue,206 but also occur with relative frequency in the alimentary tract207 and rarely the respiratory system.204 Pulmonary GTG affects adults, with an age range of 20 to 68 years204 (Fig. 19-53).
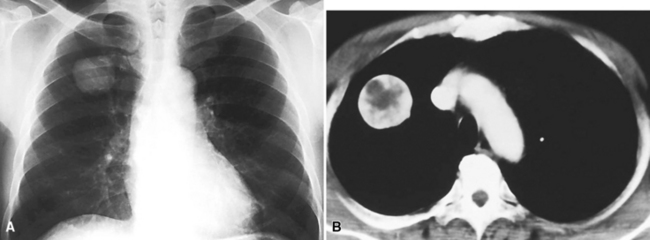
The gross features of GTG include a nodular configuration, with uniform gray-white or yellow cut surfaces and a maximum dimension of 6.5 cm, sometimes mimicking a carcinoid tumor.205
Microscopically, the lesions include compact polygonal or round cells that are closely apposed (Fig. 19-54). Nuclei are round or oval, with dispersed chromatin, scarce mitotic activity, and little if any pleomorphism. Cytoplasm is modest in amount and amphophilic or eosinophilic (Fig. 19-55). Supporting stroma consists of delicate fibrovascular septations. In lesions that lie toward the “glomangioma” pole of the spectrum, dilated vascular spaces also punctuate the lesions (Fig. 19-56), and some of these may assume a “moose antler” shape, as in the vessels seen in hemangiopericytomas. Although most are well circumscribed, a proportion of pulmonary GTGs are infiltrative.
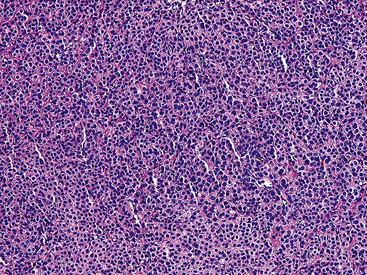
Figure 19-54 A low-power micrograph of a pulmonary glomus tumor showing a monotonous sheet-like proliferation of cells.
One exception to this description was represented by a tumor in the series of Gaertner and associates.204 This tumor demonstrated infiltrative growth, obvious nuclear atypia with prominent nucleoli, necrosis, and brisk mitotic activity. It metastasized to several visceral sites and soft tissue, causing death in 1.3 years. Thus, it was classified as a glomangiosarcoma.
Electron microscopic examination of GTGs shows evidence of specialized smooth muscle differentiation (discussed earlier), as expected in pericytic perivascular cells.203,204 The immunophenotype of GTG includes reactivity for vimentin, actin, laminin, and collagen type IV, with an absence of keratin, neuroendocrine markers, CD31, and CD34.204
Chondroma, Myxoma, and Fibromyxoma
Several authors have posited that true chondromas and fibromyxomas of the lung208,209 exist apart from pulmonary hamartomas210–213 (see Chapter 18). In particular, Carney214 and Wick and colleagues215 have documented a constellation of masses in young women wherein multifocal cartilaginous neoplasms—apparently true chondromas—are part of a syndromic triad that includes extra-adrenal paragangliomas and gastric epithelioid stromal tumors. These are histologically different from “usual” chondroid hamartomas of the lung.216 They are frequently multiple, although they may occur singly (Fig. 19-57) in young women, more often than in elderly men (as is true for hamartomas). Chondromas lack the epithelial entrapment and “uncommitted” fibroblastic mesenchymal component that is observed in chondroid hamartomas. Instead, the interface between chondromas and the surrounding lung parenchyma is sharply demarcated (Fig. 19-58). Constituent chondrocytes are hyaline, and metaplastic osteoid is common in such lesions, more than in pulmonary chondroid hamartomas.
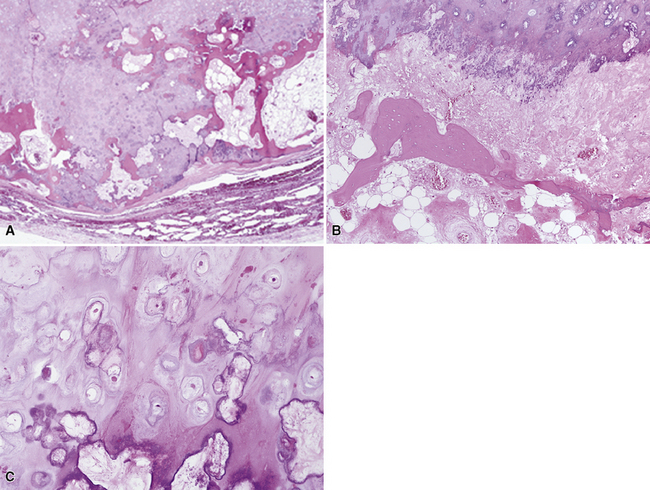
Solitary Pulmonary Hemangioma and “Hemangiomatosis”
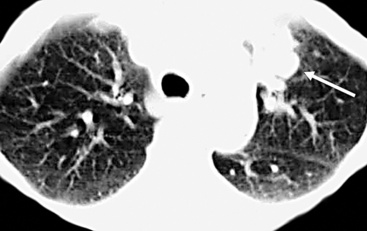
True hemangiomas of the tracheobronchial tree and lung are vanishingly rare. They are usually detected in childhood.217 Discrete, variably dense and sometimes-cystic masses are present on chest radiographs (especially high-resolution CT)218 (Fig. 19-59); they may be multiple. The maximum size of individual lesions can be several centimeters. In at least one case, an intrapulmonary hemangioma was interpreted radiographically as an intrapulmonary bronchogenic cyst.217 Simple excision has been curative.
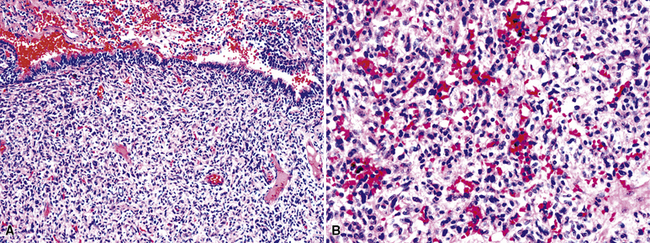
Microscopically, true hemangiomas of the airways and lungs completely replace a portion of the native tissue, with a proliferation of tubular vascular spaces lined by bland endothelial cells. A “capillary hemangioma” pattern appears to be most common, in which the caliber of the neoplastic vessels approximates that of normal small venules or capillaries (Fig. 19-60). It is implicit in this diagnosis that no other tumoral elements be identified; this is an important caveat because several other neoplasms in the lung—including some carcinomas—may contain blood lakes or pseudovascular foci that resemble the image of vascular proliferations.219
Another salient differential diagnostic consideration is “pulmonary hemangiomatosis” (see Chapter 11). That is a condition in which capillary-sized blood vessels proliferate throughout the pre-existing pulmonary parenchyma and stroma, including the alveolar septa, interlobular and interlobar septa, bronchial and bronchiolar walls, intrapulmonary vasa vasorum, and pleural surfaces.220,221 In reality, it is probably not a neoplastic process at all, but rather a malformative or reactive proliferation. For example, some cases of pulmonary hemangiomatosis have been associated with longstanding passive congestion of the lungs, as seen in left-sided heart failure.221
Lipoma and Lipoblastoma
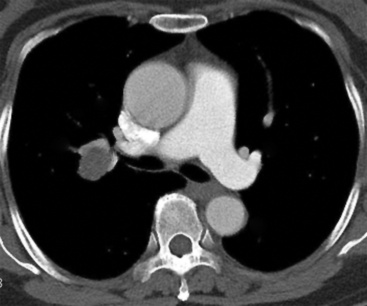
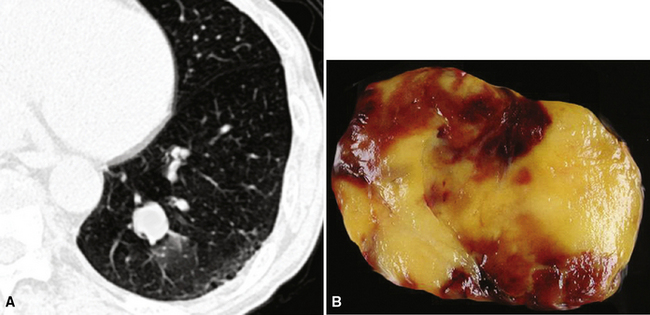
Separating examples of lipoma of the trachea and major bronchi and lung parenchyma (LTMB)222–235 from adipocytic predominant hamartomas may be difficult, and pathologic criteria for doing so are arbitrary.228 Patients with LTMB are adults, usually in the fifth or sixth decade of life.225,229 Endoscopic examination shows a variably polypoid submucosal nodule in one of the first three subdivisions of the tracheobronchial tree; radiographs demonstrate secondary atelectasis, obstructive pneumonia, or a mass in 80% of cases, but the remainder have no radiologic abnormalities on plain films.223 CTs are more sensitive in revealing the presence of lesions in airway lumina227 (Figs. 19-61 and 19-62). Current treatment recommendations are for conservative endoscopic resection after a transbronchial biopsy has established the adipocytic nature of the lesion.
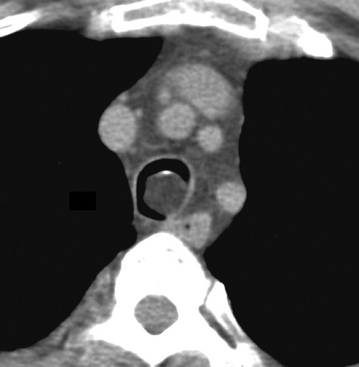
Figure 19-61 This computed tomogram demonstrates an endoluminal tracheal mass that proved to be a lipoma.
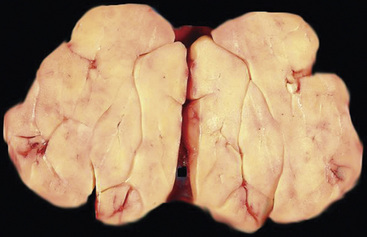
Figure 19-62 This resection specimen of the lesion shown in Figure 19-61 shows uniform yellow tissue with internal lobulation.
Intrapulmonary lipomas are often subpleural (Fig. 19-63) and may even protrude into the pleural space. They are typically detected incidentally on chest radiographs. CT shows that lipomas of the lung have a density approximating that of pleural adipose tissue.227 Occasional lesions of this type have been found coincidentally in patients with pulmonary carcinomas.224 If the lipomas are multiple, small, and subpleural, they may also imitate metastatic lesions.234,235
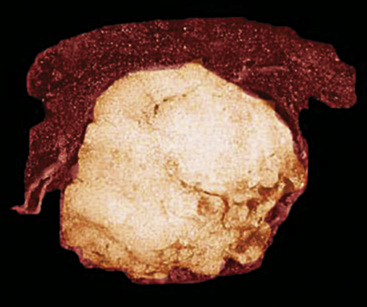
Figure 19-63 A peripheral intrapulmonary lipoma, represented by a large globular tan-yellow lesion beneath the pleural surface.
Lipoblastoma, a neoplasm of young children236 may affect the pleura and chest wall, but only anecdotal reports exist of intrapulmonary lipoblastomas.237,238 They may be extremely large, filling almost an entire hemithorax.237 Excision is curative in the few cases with meaningful follow-up.
Liposarcoma is the rarest of primary pulmonary adipocytic neoplasms, and it has usually presented as a large single peripheral mass.239,240 Liposarcoma-like components can be seen as part of sarcomatoid carcinomas,241 and the latter lesions are far more common than primary liposarcomas of the lung.
The principal gross feature of intrapulmonary adipocytic tumors is a yellow, globular, relatively soft mass in the parenchyma (see Fig. 19-63). Internal fibrous septation may be apparent, as may a circumferential capsule or small foci of intralesional sclerosis. There is no necrosis or hemorrhage.
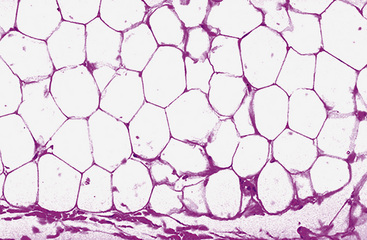
Microscopically, one sees sheets of fully mature adipocytes in ordinary lipomas (Fig. 19-64), supported by delicate fibrovascular stroma. Atypical intrapulmonary lipomas, showing scattered multinucleated “floret” cells, have been rarely reported231; however, this does not seem to have any bearing on prognosis. Lipoblastomas have a more lobulated substructure, with an admixture of uncommitted fibromyxoid tissue among mature fat cells236,242 (Fig. 19-65). True lipoblasts may be apparent as well, rare mitotic figures may be evident, and the supporting stromal tissue contains a delicately arborizing capillary network (Fig. 19-66). Therefore, the overall image of lipoblastoma may be quite similar to that of myxoid liposarcoma, and the mutually exclusive occurrence of those tumors in different age groups—with liposarcomas being seen in adults—is important to their proper identification.
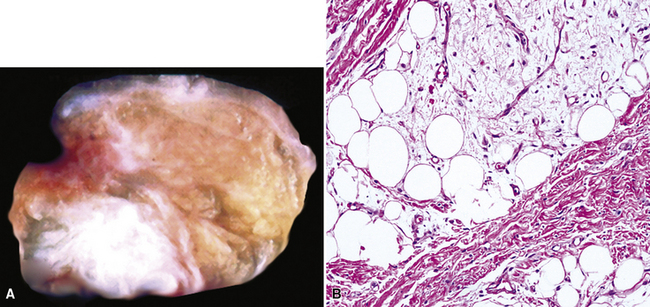
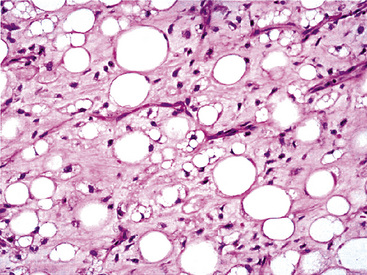
Liposarcoma in the lung may assume any of the recognized morphotypes of that tumor (e.g., lipoma-like, sclerosing, myxoid, round cell, pleomorphic, and “dedifferentiated”).239,240 Those variants are recapitulated in metastatic intrapulmonary liposarcomas from primary soft tissue sites.243
Karyotypic analysis using fluorescent in situ hybridization may be useful in the differential diagnosis. Abnormalities in chromosome 8q are most common in lipoblastomas,242 whereas myxoid liposarcoma demonstrates a reproducible t(12;16) chromosomal translocation.244 Parenthetically, cytogenetic profiles are somewhat less helpful in the distinction between lipomas of the lung and lipomatous hamartomas, both of which may show abnormalities in chromosome 12.232,245 However, exchanges of material between chromosomes 6 and 14 are also potentially observed in hamartomas but not lipomas.246
Immunohistology of fatty tumors of the lungs shows consistent reactivity for S-100 protein in the adipocytic elements as well as “high-mobility-group” proteins.245 Lipoblastoma may also show positivity for factor XIIIa in its stellate and fusiform cells.242
Angiomyolipoma
In recent years, it has become apparent that a family of neoplasms—which may arise in the kidney, pancreas, alimentary system, liver, soft tissue, or respiratory tract—demonstrates differentiation toward a unique cell type that has both myogenous and melanocytic features.247–250 These tumors have been variously called “angiomyolipomas,” “perivascular epithelioid cell neoplasms” (PEComas), and “myomelanocytomas.”247–251 If a typical morphologic appearance, including smooth muscle, fat, and vascular proliferation, is observed, the first of these terms is still preferred; a few lesions with such characteristics have been reported in the lung.252–257 Another member of this nosologic group—the pulmonary clear cell or “sugar tumor”—is discussed later. All of these proliferations potentially share immunoreactivity with the melanocyte-related antibody human melanin black (HMB)-45, as well as anti-actins.248 Other immunohistochemical markers of melanocytic and smooth muscle differentiation—such as antibodies to S-100 protein, tyrosinase, HMB-50, melanoma antigen recognized by T cells 1 (MART-1), caldesmon, calponin, and desmin—may also label them.249
A proportion of patients with angiomyolipomas, regardless of anatomic location, will have phakomatosis tuberous sclerosis (Bourneville syndrome). It classically includes nodular periventricular glial proliferations and subependymal giant cell astrocytomas in the brain; cutaneous connective tissue nevi; often-multifocal renal angiomyolipomas; and pulmonary lymphangioleiomyomatosis, with or without multifocal micronodular pneumocytic hyperplasia (see Chapter 7).253,258–260
Microscopically, pulmonary angiomyolipoma is identical to its better-known renal counterpart. Prototypically, it manifests a concatenation of mature fat, variably sized and haphazardly arranged blood vessels with muscular walls, and nests or skeins of fusiform or epithelioid smooth muscle elements (Fig. 19-67). Either the fat or the smooth muscle may dominate the histologic picture and cause diagnostic confusion with either pure adipocytic neoplasms or sarcomas, respectively. Nuclear atypia has been seen in the smooth muscle elements of angiomyolipomas in extrapulmonary sites, and rare cases outside the lung have even shown overtly malignant transformation with necrosis and atypical mitotic activity.261 Those attributes have not been seen in pulmonary lesions of this type.
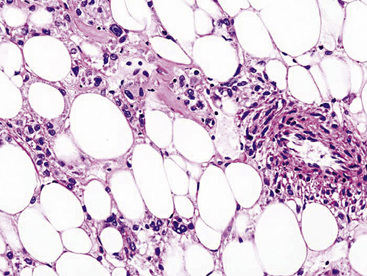
Electron microscopic analysis of angiomyolipoma demonstrates findings that recall the characteristics of smooth muscle cells, including pericellular basal lamina and plasmalemma-associated pinocytotic vesicles.252 Cytoplasmic bundles of thin filaments have not been found. Approximately 50% of such neoplasms show cytoplasmic premelanosomes.249
The principal immunohistologic findings in angiomyolipomas have been described earlier. Adachi and coworkers262 have reported immunoreactivity for CD1a in PEComas, including angiomyolipoma, but we have been unable to reproduce this finding.
In its “classic” form, angiomyolipoma has no realistic differential diagnostic alternatives. Nevertheless, its morphologic variants may be difficult to distinguish from metastatic melanomas, carcinomas, or sarcomas in the lung. Before an unqualified diagnosis of a pure pulmonary smooth muscle tumor or melanoma is made, additional immunocytochemical evaluation with HMB-45 (Fig. 19-68) and anti-actins is probably a prudent step.

Myelolipoma
Myelolipoma is a peculiar lesion that is typically located in the adrenal glands or the retroperitoneum.263,264 As its name suggests, it comprises a tumefactive admixture of mature adipose tissue and hematopoietic precursor cells, including megakaryocytes (Figs. 19-69 and 19-70). It is still unclear whether myelolipoma represents a true neoplasm or a peculiar form of extramedullary hematopoiesis; the difference between those entities may sometimes be only semantic. Alternatively, myelolipoma could be regarded as a lipoma that has been secondarily populated by bone marrow elements.
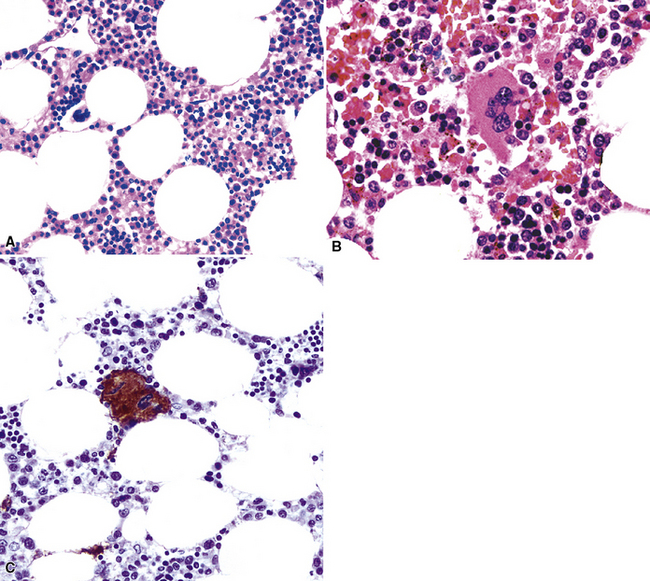
In any event, anecdotal reports have been made of primary pulmonary myelolipoma, all of which occurred in adult patients.265–269 They may occur multiply and mimic metastases.265
Benign Tumors of the Pleura
Adenomatoid Tumor
It is an unfortunate truism that completely benign neoplasms of the pleura are a distinct rarity. Indeed, the only representative of that diagnostic category is the adenomatoid tumor. That neoplasm is a lesion with mesothelial differentiation that is most often encountered in the adnexal soft tissue surrounding the uterus and testes.270,271 Only five cases have been described in the pleura.272–275 These lesions occurred in a middle-aged man, two middle-aged women, an elderly woman, and an elderly man. They were all incidental findings during surgical procedures that were done for unrelated lesions (pulmonary squamous cell carcinoma, mesothelioma, pulmonary adenosquamous carcinoma, esophageal adenocarcinoma, and pulmonary histoplasmoma), and they showed no subsequent evidence of aggressive behavior.
Pleural adenomatoid tumors are unencapsulated and measure 0.5 to 2.5 cm in greatest dimension. Microscopically, the lesions are composed of epithelioid cells organized into compact gland-like profiles (Fig. 19-71). They have vesicular nuclear chromatin, inconspicuous nucleoli, and relatively abundant eosinophilic or vacuolated cytoplasm. No areas of nuclear pleomorphism should be present, mitotic figures are rare, and they do not invade the subjacent lung parenchyma.
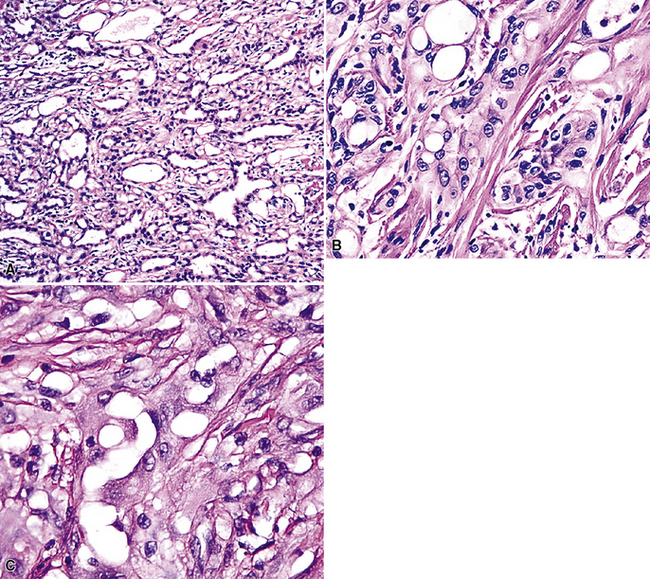
Figure 19-71 A to C, Pleural adenomatoid tumor comprised of microcystic arrays of bland cuboidal epithelioid cells.
Electron microscopy272 shows branching plasmalemmal microvilli and prominent intercellular attachment complexes, typical of mesothelial lesions. Immunohistologically, the tumor cells label for keratin and calretenin, but are negative for carcinoembryonic antigen, CD15, CD34, Ber-EP4 antigen, and tumor-associated glycoprotein 72.
The principal components of the differential diagnosis for pleural adenomatoid tumor are metastatic adenocarcinoma and malignant mesothelioma. Both of those possibilities are rendered unlikely by the bland histologic images of pleural adenomatoid tumor, with no evidence of infiltrative growth (see Chapter 20). Ki-67 (MIB-1) staining may also be helpful in this setting275 because the labeling index in adenomatoid tumor is only 1% to 2%. In contrast, “microcystic” (adenomatoid tumor–like) mesothelioma of the pleura shows a high Ki-67 index (>50%).274
Calcifying Fibrous Pseudotumor
An unusual entity known as calcifying fibrous “pseudotumor” (CFPT) has been described in the pleura.276–279 This lesion is histologically similar, if not identical, to calcifying fibrous “pseudotumors” of the soft tissue.280
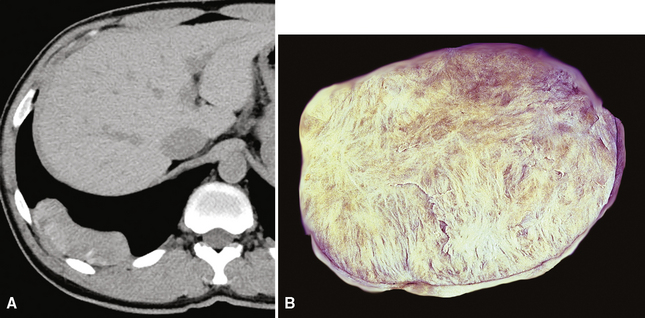
The documented cases of pleural CFPT have occurred in adults (age range, 23–46 years), most of whom were women. Pleural-based masses281,282 are present on chest radiographs, and CT scans demonstrate well-demarcated, partially calcified nodular lesions, measuring up to 12 cm in diameter (Fig. 19-72). An intrapulmonary CFPT has been reported.283
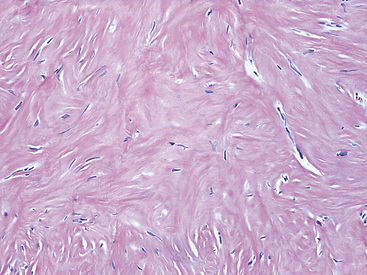
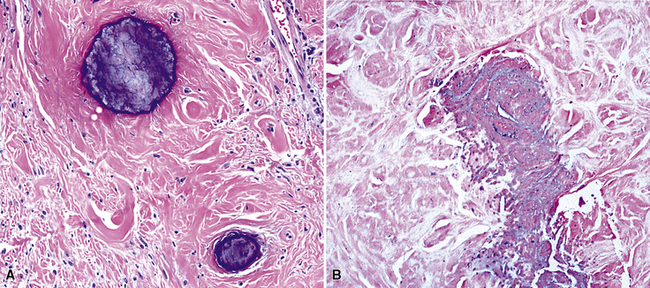
Histologically, the masses are circumscribed nonencapsulated fibrous lesions composed of dense, hyalinized, collagenous tissue, with interspersed bland spindle cells (Fig. 19-73). They are generally hypocellular, particularly at the periphery, without nuclear atypia. A scant chronic inflammatory infiltrate may be seen, without lymphoid aggregates, giant cells, or necrosis. All lesions also feature calcifications of the psammomatous (Fig. 19-74) as well as dystrophic types. These histologic features are identical to those of calcifying fibrous pseudotumors of soft tissue.284–286
Biologically Borderline Tumors of the Lung and Pleura
Inflammatory Myofibroblastic Tumor (“Inflammatory Pseudotumor”)
In 1939, Brunn289 described two pulmonary lesions that were composed of spindle cells admixed with inflammatory elements in patients with fever and weight loss. The systemic symptoms disappeared after surgical removal of the lung tumors. Although they were believed to be true neoplasms of probable smooth muscle lineage, subsequent authors espoused the theory that masses with the same attributes were inflammatory and reparative lesions.290,291 Hence, the term “inflammatory pseudotumor” gained favor. However, Spencer292 noted that a proportion of such proliferations behaved in a biologically malignant fashion, and other reports also documented the presence of vascular invasion and recurrences.293–295 Molecular analyses done in the 1990s revealed a clonal character of some “inflammatory pseudotumors” in the lungs and other anatomic sites,296–298 and, together with aggregated clinicopathologic information, these data resulted in amendment of their name to “inflammatory myofibroblastic tumors.”299 It is now accepted that they are separate from inflammatory pseudotumors and that IMTs form a conceptual continuum with “inflammatory fibrosarcoma” or “myofibrosarcoma,”300–303 Because they may recur and occasionally metastasize, IMTs are best considered biologically “borderline” neoplasms.
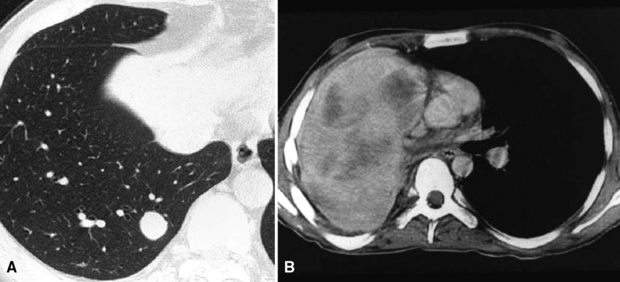
The lung is most frequently affected by IMT, among all potential topographic locations.295 Although the majority of IMTs are seen in children and young adults, they may be encountered in patients of all ages, with no sex predilection.295 Some IMTs are situated in the large airways, with no parenchymal involvement, but they are exceptional; most arise in the peripheral lung fields. Systemic symptoms of a paraneoplastic nature have been reported in up to 50% of cases, including such findings as anemia, fever, weight loss, hyperglobulinemia, leukothrombocytosis, and elevations in the erythrocyte sedimentation rate.301,303 As discussed earlier, those abnormalities typically remit when the IMT is removed. The remaining cases present with cough, vague chest discomfort, or hemoptysis, or are asymptomatic, with the lesions found incidentally on chest radiographs.299 On imaging studies, IMT is typically a lobulated or globoid mass (Fig. 19-75), usually measuring less than 5 cm in maximum dimension. Occasionally, it may attain a size of greater than 10 cm; internal calcifications are commonly seen. Infiltration of great vessels, mediastinal soft tissue, or chest wall is apparent radiographically in a minority of cases.
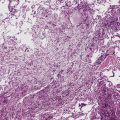
The histologic profile of IMT features a proliferation of relatively bland spindle cells arranged haphazardly or in vague fascicles299,301,304 (Figs. 19-76 and 19-77). In contrast to the gross appearance, this lesion has an irregular peripheral zone of growth microscopically, with tongues of tumor that comingle with adjacent normal tissues. Not surprisingly, infiltration of blood vessels, bronchi, and pleura may be evident. Nuclei in the neoplastic fusiform elements usually contain dispersed or vesicular chromatin with compact nucleoli (Fig. 19-78). Mitotic activity is typically easily found, but without pathologic division figures. Cytoplasm is amphophilic or lightly eosinophilic and may show a faintly fibrillar character. Admixed inflammatory cells are greatly variable in density and type, and some cases of IMT show virtually none. Lymphocytes, plasma cells, macrophages, eosinophils, and neutrophils are also potentially represented (Fig. 19-79), and intralesional aggregates of xanthomatized foam cells are sometimes apparent. Some cases demonstrate rather striking zones of sclerosis, and these may even be hyalinized. On the other hand, 20% to 30% of these lesions show dense cellularity with focal or global nuclear atypia, relatively high nuclear-to-cytoplasmic ratios, nuclear hyperchromasia, mild nuclear pleomorphism, and zones of necrosis.295
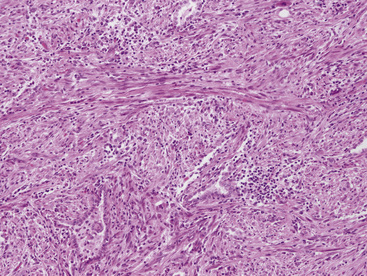
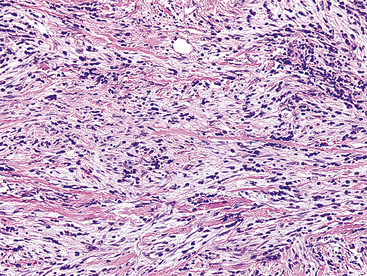
Figure 19-77 The spindle cells in this pulmonary inflammatory myofibroblastic tumor are arranged in interweaving bundles.
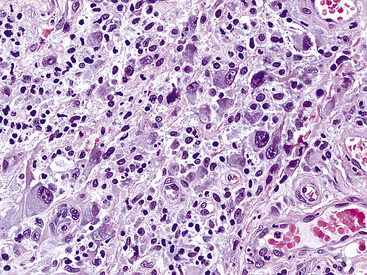
Figure 19-78 This area in an inflammatory myofibroblastic tumor demonstrates moderate pleomorphism of the constituent tumor cells.
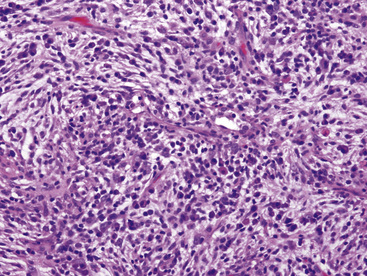
Figure 19-79 Intralesional lymphocytes and plasma cells are numerous in this pulmonary inflammatory myofibroblastic tumor.
Electron microscopic evaluation of IMTs shows that the tumor cells possess some features associated with smooth muscle, such as plasmalemmal dense patches, pinocytotic vesicles, and cytoplasmic bundles of thin (actin-type) filaments. Pericellular basal lamina is variably present, but there are no intercellular attachment complexes.299
By immunohistochemical assessment, the spindle cells react with antibodies to vimentin, alpha-isoform and muscle-specific actins, and calponin, but usually not with desmin, caldesmon, CD34, or keratin.301 Mutant p53 protein is detectable immunohistologically in fewer than 10% of cases.298 On the other hand, two proteins that relate to reproducible cytogenetic abnormalities in chromosome 2p23 in IMTs are detectable in approximately 40% of these lesions. These are known as “anaplastic lymphoma kinase 1” (ALK-1; Fig. 19-80) and “p80,” and were originally studied in anaplastic large-cell (“Ki-1”) lymphomas.305 Coffin and colleagues306 found that ALK-1 reactivity predominated in IMTs of patients 20 years of age and older; those lesions also were more likely to demonstrate cytomorphologic atypia.
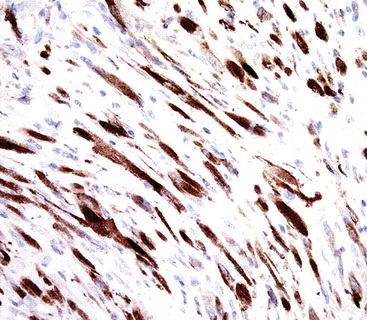
Figure 19-80 Diffuse immunoreactivity is present for ALK-1 protein in this inflammatory myofibroblastic tumor of the lung.
The differential diagnosis of pulmonary IMT includes tumefactive organizing pneumonia304; “true inflammatory pseudotumor” or “plasma cell granuloma” (see Chapter 18); immunoglobulin G4 (IgG4)-predominant lymphoplasmacytic lesions307; smooth muscle proliferations in the lung; inflammatory sarcomatoid carcinoma295 (Fig. 19-81); and inflammatory malignant fibrous histiocytoma. ALK-1/p80 reactivity is diagnostic of IMT in this setting, but as mentioned earlier, is seen in only a minority of cases. The spindle cells of inflammatory sarcomatoid carcinoma can be labeled for keratin and epithelial membrane antigen, in contrast to those of IMT.295,308 Inflammatory malignant fibrous histiocytoma is typically nonreactive for actins or calponin, as seen in IMT. Spindle cells in organizing pneumonia typically form smaller whorls of spindle cells than IMT.302 IgG4-predominant lymphoplasmacytic lesions are, by definition, recognized by a preponderance of IgG4-immunoreactive cells in the inflammatory components.307
There are few if any pathologic variables that can be used successfully in any given case of IMT to predict its biologic behavior with certainty.309 Some examples of pulmonary IMT have been observed for extended periods with minimal growth. Spontaneous complete resolution has also been recorded, and a few lesions have diminished in size after nonsurgical therapy.301 Operative removal is usually necessary to establish a firm diagnosis of IMT, and if the lesion has been completely excised, no further intervention is required. On the other hand, it is logical to expect that incomplete removal—particularly if invasion of adjacent extrapulmonary tissues is present—might, in some instances, be followed by continued growth.310 In particular, involvement of the pleura, pulmonary hilum, diaphragm, or mediastinum is associated with morbidity from IMTs of the lung. They may even, exceptionally, prove fatal if they are massive and infiltrate the mediastinum extensively, or if distant spread occurs. Overall, recurrence of IMT is seen in approximately 55% of cases, and roughly 10% metastasize.306
Sclerosing Hemangioma (“Pneumocytoma”)
More than 50 years ago, Liebow and Hubbell311 described a peculiar tumor of the peripheral lung parenchyma with a sclerosing, angiomatoid, and papillary architecture. They gave it the name “sclerosing hemangioma,” but this designation was chosen in unfortunate mimicry of a convention then extant in dermatopathology. In the 1950s, the lesion now known as “dermatofibroma” or “cutaneous fibrous histiocytoma” was likewise commonly called “sclerosing hemangioma,”312 because it sometimes showed internal blood lakes and hemosiderosis. In their seminal description of “sclerosing hemangioma” of the lung, Liebow and Hubbell disavowed an endothelial derivation for the tumor and appended the alternative appellations of “histiocytoma” and “xanthoma,” perhaps in deference to the true identity of its alleged dermal analog. This enigmatic series of nosologic and terminologic choices set the stage for confusion in the succeeding decades. Various studies of the cellular nature of pulmonary “sclerosing hemangioma” have suggested vascular, fibrohistiocytic, mesothelial, and epithelial lineages,313–317 and controversies over such proposals persist to some extent.
Based on the aggregated data,318,319 this tumor is properly considered as a neoplasm showing differentiation that is most like that of embryonic (uncommitted) respiratory epithelium. Therefore, the entity in question is best designated as “pneumocytoma,” as suggested by Shimosato320; however, the World Health Organization has persisted in using the historic name “sclerosing hemangioma.”
Sclerosing hemangioma can affect patients of almost any age, from early childhood321 through the end of life. Females predominate by a factor of 5:1.322–332 Only approximately 20% of patients have any respiratory symptoms at the time their tumors are found. It may arise in any of the pulmonary lobes; localization in the large airways, pleura, and mediastinum has been rarely reported.322,333
Radiographically, patients with sclerosing hemangioma usually have solitary peripheral masses with well-defined homogeneous round or oval profiles on chest roentgenograms (Fig. 19-82). CT shows homogeneity and high density of the lesions in the great majority of cases (Fig. 19-83); low-density areas may be apparent in some tumors because of cystic change. In addition, the “air meniscus sign” (also known as the “air trapping” or “air crescent” sign)327,328,334—a rim of air that partially or circumferentially surrounds a mass—is evident in the lung parenchyma adjacent to some lesions of this type.
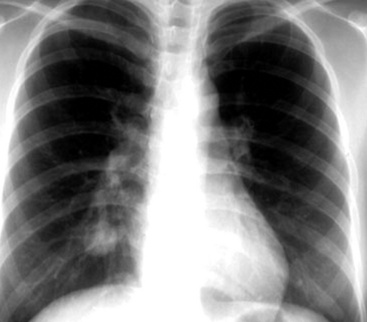
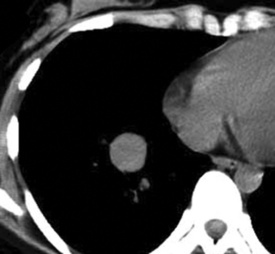
Figure 19-83 A sclerosing hemangioma. This computed tomogram of the lesion shown in Figure 19-82 shows a circumscribed spherical tumor with a uniform internal density.
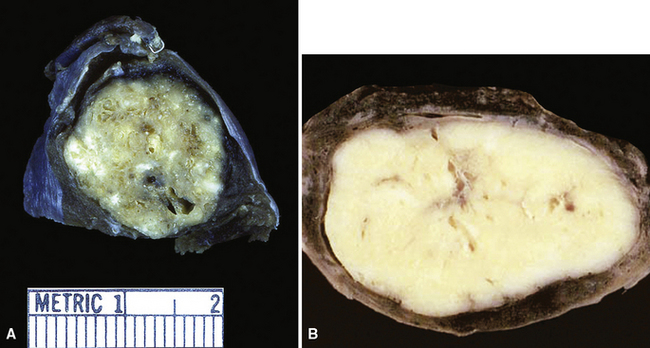
Grossly, most sclerosing hemangiomas are less than 3 cm in maximum dimension (Fig. 19-84), but sometimes they attain a size of up to 10 cm. Multifocality, sometimes featuring a satellitotic configuration of small nodules around a dominant central nodule,322 is apparent in approximately 4% of cases.335 Roughly the same proportion are pleural-based and may be polypoid, potentially simulating the appearance of solitary fibrous tumors.322 Sharp circumscription from the surrounding lung parenchyma is the rule, so much so that these tumors are sometimes “shelled out” by the surgeon with little if any attached alveolated tissue. Their cut surfaces are tan-gray, yellow, or mottled. Cystic areas are evident in roughly 3% of cases; 20% show areas of intralesional hemorrhage, which may be extensive.
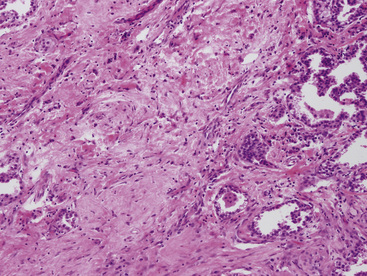
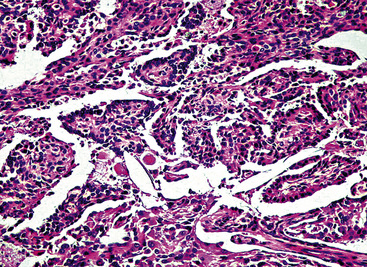
The histologic images of sclerosing hemangioma include varying admixtures of four basic growth patterns—sclerotic (Fig. 19-85), papillary (Fig. 19-86), solid (Fig. 19-87), and hemorrhagic/angiomatoid (Fig. 19-88). Approximately 15% of cases are monomorphic, and roughly 20% contain areas representing all of the patterns in the same lesion. Two basic cell types comprise the neoplastic elements in sclerosing hemangioma—“surface” cells, which mantle papillary projections, and “round” cells, seen in the cores of papillary structures and in solid sheets within the lesions. Surface cells often show intranuclear invaginations of cytoplasm, yielding “pseudoinclusions” such as those seen in type II pneumocytes and Clara cells. They also may be multinucleated. Round cells are actually polygonal, with oval nuclei, dispersed chromatin, indiscernible nucleoli, and extremely rare mitotic figures. They may focally exhibit cytoplasmic vacuolation and even assume the morphologic features of a signet ring cell. Rarely, nuclear hyperchromasia and moderate pleomorphism are seen in the round cell element.322,336
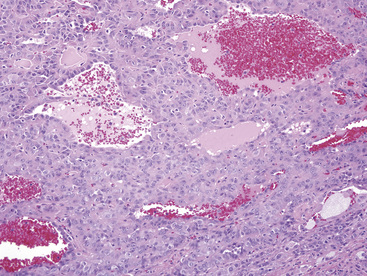
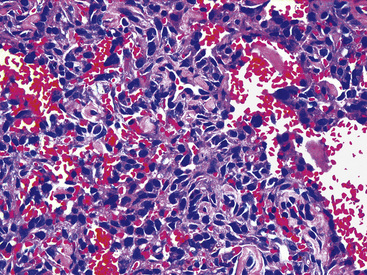
Secondary features of sclerosing hemangioma include blood lakes in angiomatoid foci; limited areas of necrosis; stromal hemosiderosis, calcification, cholesterolosis, or combinations thereof; internal cystification, with mucinous contents; granulomatous inflammation; and a potential association with foci of neuroendocrine hyperplasia (“tumorlets”) in the surrounding lung parenchyma.322,323,336,337
We have previously stated our opinion that pneumocytoma/sclerosing hemangioma, alveolar adenoma, and papillary adenoma comprise a biologically interrelated family of pulmonary lesions. Nevertheless, these entities differ morphologically, in that sclerosing hemangioma lacks the “Swiss cheese” microcystic structure of alveolar adenoma and papillary adenoma does not contain the “round cell” elements of sclerosing hemangioma.158
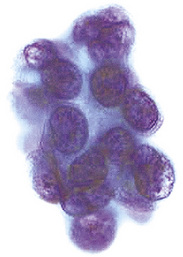
Several publications have addressed FNA biopsy findings in sclerosing hemangioma, and most have noted substantial morphologic overlap between those lesions and well-differentiated adenocarcinomas (particularly of the bronchioloalveolar type).326,329,330,338–340 A shared potential for nuclear atypia, intranuclear pseudoinclusions, and micropapillary growth in both tumor entities makes the definitive cytologic recognition of sclerosing hemangioma very difficult (Fig. 19-89). Despite assertions that it can be accomplished,341 we believe that an unqualified interpretation of sclerosing hemangioma by FNA biopsy is unwise. In appropriate cases, that possibility can be included in the diagnostic report, prompting an intraoperative frozen-section examination to further guide the surgeon’s choice of procedure. This proviso relates to the very close cytologic similarity we have noted between pneumocytoma and selected adenocarcinomas.
Electron microscopy of sclerosing hemangiomas has shown that both the surface cell and round cell components demonstrate type II pneumocytic features, containing cytoplasmic lamellar bodies with variable levels of maturation.315,322 Other organelles are nonspecific. These findings support the interrelatedness of sclerosing hemangioma and papillary adenoma.
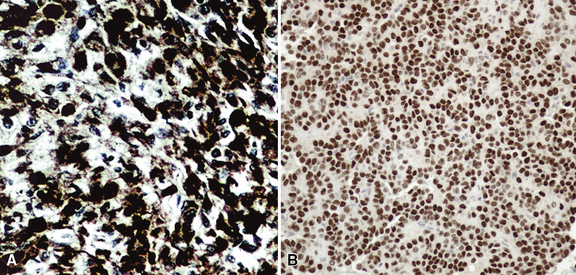
Immunohistologically, variable reactivity has been seen for keratin, epithelial membrane antigen (EMA), surfactant-related proteins, Clara cell antigen, and thyroid transcription factor 1 in both cellular components of sclerosing hemangioma322,331,332,338,342–349 (Fig. 19-90). Receptors for estrogen and progesterone are also demonstrable in a minority of cases.322 On the other hand, mesothelial markers, such as calretinin, HBME-1, cytokeratin 5/6, and WT-1 protein are absent in these lesions, as are neuroendocrine, neural, and myogenous determinants.
Justification for inclusion of sclerosing hemangioma as a “borderline” lesion stems from reports of its recurrence341,350 and the finding of lymph nodal metastases in 10 patients to date.322,324,351–353 In the U.S. Armed Forces Institute of Pathology series, this behavior was observed in 1% of cases.322 Despite that observation, no patient with metastatic tumor has died of the tumor, and lymph node involvement324 does not appear to affect survival.
As mentioned earlier, the principal differential diagnostic alternative to sclerosing hemangioma is low-grade adenocarcinoma of bronchioloalveolar or conventional types. 330,339,340 An adequate tissue sample—allowing for evaluation of the architecture of the entire mass—is essential to making that distinction. Another diagnostic possibility is the papillary variant of low-grade mucoepidermoid carcinoma,354 but its particular histologic pattern, with an intimate admixture of mucinous and squamous epithelium, should allow for a distinction from sclerosing hemangioma.
Pulmonary Mucinous Cystadenoma and Borderline Mucinous Tumor
Mucinous cystadenomas and cystic mucinous tumors of low malignant potential—“borderline” mucinous neoplasms (BMTs)—are well known to gynecologic and gastrointestinal pathologists because they rather commonly arise in the ovaries, gut, and pancreas.355,356 A primary origin in the lung for such lesions is unusual, but possible, and approximately 50 cases have been reported.357–369 These neoplasms occur in adults as solitary lesions in the peripheral parenchyma. An association with von Hippel-Lindau disease has been reported.369 Both plain films and CTs of the thorax clearly show the cystic nature of these tumors (Fig. 19-91), which generally measure several centimeters in diameter.
Figure 19-91 This chest radiograph shows a large ovoid mass in the right lower lung field, representing a cystic mucinous tumor.
Grossly, mucinous cystadenomas and BMTs are sharply demarcated from the surrounding lung parenchyma (Fig. 19-92) and may even “shell out” for the surgeon. Their walls are relatively thick and fibrous, enclosing locules that contain thick viscous mucoid material. Foci of granular tissue are present on the internal aspects of the cysts, representing epithelial projections.
Histologically, the latter structures comprise cytologically bland mucin-containing tumor cells that are cuboidal or low columnar (Fig. 19-93). Nuclei are generally basally located and relatively banal, with no pleomorphism or mitotic activity (Fig. 19-94). Cystadenomas do not demonstrate invasive growth into the fibrous walls of the lesions, but BMTs may do so, and may manifest a “piling up” of lesional epithelium that yields complex micropapillary structures (Fig. 19-95). Cases in which frank adenocarcinoma evolved from pre-existing pulmonary mucinous cystadenoma have been reported (Fig. 19-96).358,364,368
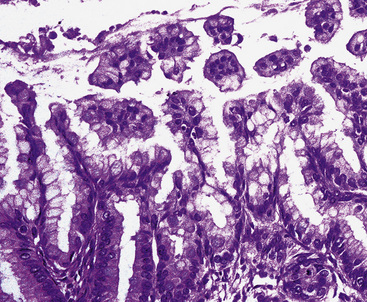
Figure 19-95 Micropapillary growth is evident in some cystic mucinous tumors of the lung, justifying use of the term “borderline.”
The classification of pulmonary BMT as a neoplasm with low malignant potential is supported by the observations of Gao and Urbanski.368 Those authors documented a mortality rate (due to metastatic tumor) of 30% in their series, with up to 10 years of postoperative surveillance, raising the possibility that these should be considered frank carcinomas rather than borderline lesions. They further described a spectrum of histologic appearances in the epithelium of BMT, ranging from completely bland to overtly malignant. Pathologic findings that were proposed as adverse prognosticators included solid epithelial growth at the periphery of the lesion, adhesion of the tumor to the pleura, crossing of the intrapulmonary septa by the tumor, obvious nuclear atypicality, and invasive growth into the surrounding lung tissue.368
Solitary Fibrous Tumor
Solitary fibrous tumor (SFT; formerly called “fibroma”)370,371 of the lung and pleura is still confused by some clinicians with mesothelial neoplasms. That is because of a nosologic scheme advanced by Klemperer and Rabin in 1931372 that was used for many years thereafter and gave SFT the designation of “localized fibrous mesothelioma.” Particularly in the last two decades, much work has been done that unequivocally shows that SFT lacks mesothelial differentiation373; instead, this tumor is composed of facultative fibroblastic elements such as those seen in the submesothelial zone of the normal lung. Even though it may occasionally recur and sometimes demonstrates locally aggressive growth—justifying its classification as a “borderline” mesenchymal tumor—SFT generally has a favorable prognosis that differs markedly from that of mesothelioma.373–383 Likewise, pleural fibrous tumors have no etiologic relationship whatsoever to occupational-level asbestos exposure, in contrast to a proportion of mesotheliomas.381
Approximately two mesotheliomas are encountered for every SFT in general thoracic surgical practice.373 Outside of infancy, patients of any age may have an SFT, but they are most often seen in patients older than 40 years old, with no sex predilection. The great majority of cases present with an asymptomatic pleuropulmonary mass that is seen on screening radiographs. Rarely, one of two paraneoplastic complexes accompanies SFT; those are represented by hypertrophic osteoarthropathy and tumor-associated hypoglycemia (Doege-Potter syndrome).381 The cause of the first condition is unknown; the second is related to an insulin-like growth factor that is produced by the neoplastic cells.
Radiographically, SFT may be as small as 1 cm in maximal dimension or as large as 36 cm. Occasional lesions of this type have occupied virtually an entire hemithorax and weighed in excess of 5 kg.380 They are usually globoid neoplasms with a relatively homogeneous internal density, but cystic change, calcification, or foci of necrosis may be sometimes apparent (Fig. 19-97). A minority of cases demonstrate a pedicle that attaches an SFT in the pleural space to the pleura itself. Conversely, other lesions “invert” into the lung parenchyma or may arise from interlobar pleural reflections, producing the appearance of a peripheral intrapulmonary mass370,371,384–388; large tumors also may displace the trachea or intramediastinal structures.373 Rarely, overt invasion of the chest wall, vertebral bodies, or adjacent lung tissue is apparent on imaging studies.384,389 A small number of SFTs show regional satellitotic growth in the pleura.388
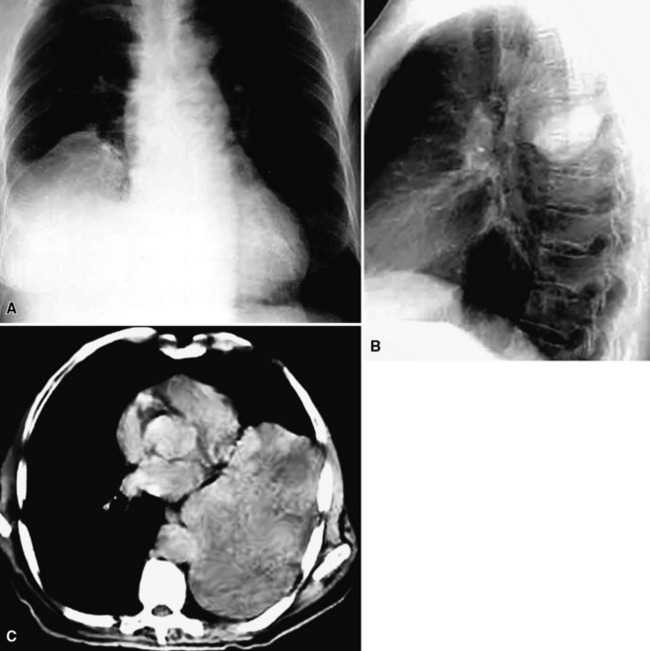
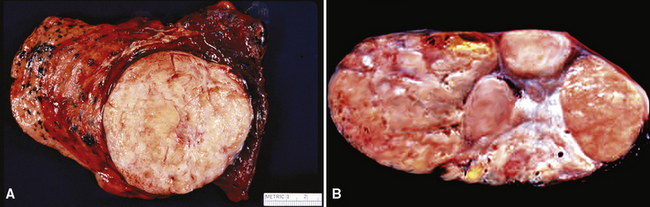
Gross examination shows a lobulated mass that is invested by pleural tissue (Fig. 19-98). Cut surfaces can be homogeneous, tan-gray, and vaguely “whorled,” or may show distinct internal septa and foci of degeneration, mucoid change, hemorrhage, calcification, or necrosis.383,384 Intralesional cystic change can also be present.
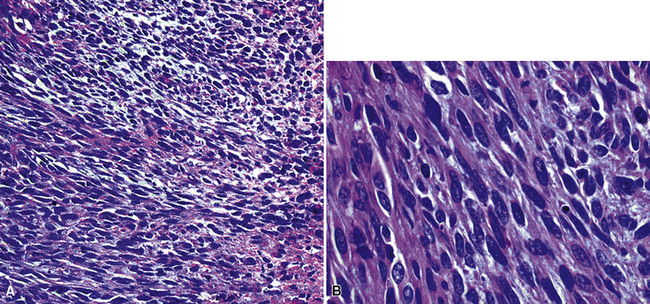
The microscopic spectrum of pleuropulmonary SFT is broad.383 A solid spindle cell growth and a diffuse sclerosing pattern are most frequent. In the first of these configurations, fusiform cells are arranged in random arrays (a “patternless pattern”) (Fig. 19-99), with fascicular groupings, storiform or herringbone patterns, or regimented arrays with nuclear palisading. SFT may also have epithelioid cells (Fig. 19-100) and branched intralesional blood vessels resembling “moose antlers,” as seen in hemangiopericytoma (which has been merged nosologically with SFT) or synovial sarcoma. Zones of fibrosis may be interspersed with cellular zones and dominate in lesions classified as “diffuse sclerosing” SFT (Fig. 19-101). Focal collagenous degeneration is occasionally apparent, simulating true necrosis. Less frequent features include multinucleated tumor giant cells, “amianthoid” arrays of collagen fibers, myxoid stromal change, limited areas of spontaneous necrosis, hemorrhage, and metaplastic ossification. Mitotic activity in most SFTs is present but not prominent, and division figures are normal.
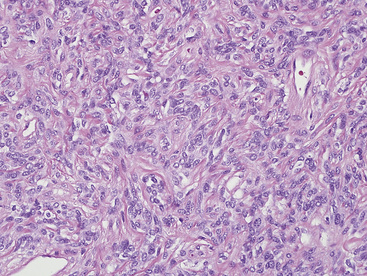
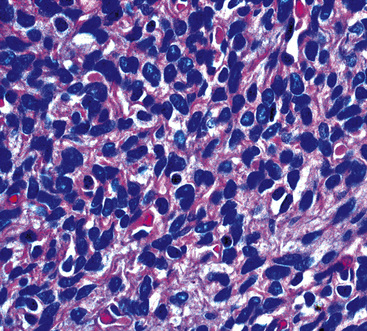
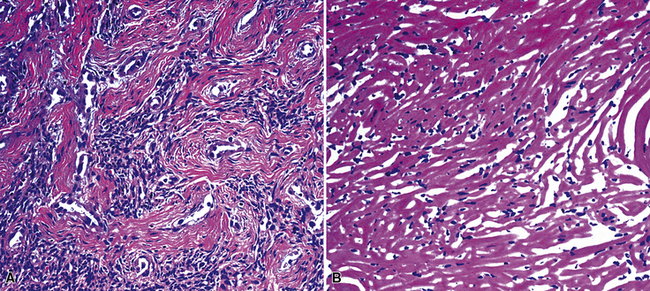
Figure 19-101 A and B, Hyalinizing collagenous stroma is prominent in this solitary fibrous pleural tumor.
England and associates384 attempted a codification of criteria for malignancy in SFT. These included dense cellularity with overlapping nuclei; nuclear hyperchromasia and pleomorphism (Fig. 19-102); mitotic activity greater than four division figures per 10 high-power (×400) microscopic fields; and necrosis and hemorrhage. However, only 55% of the lesions with such features actually had an aggressive course, with recurrence, metastasis, or both. Harrison-Phipps and colleagues390 found that approximately 13% of SFTs showed the “malignant” attributes described earlier, and they also noted that the likelihood of morphologic atypia correlated directly with tumor size. In that series, “malignant” SFTs averaged 12 cm in maximal dimension, compared with 4.5 cm for tumors that lacked histologic atypia. Vallat-Decouvelaere and coworkers375 also emphasized that histologic findings in SFTs may not be accurate predictors of their behavior. In line with this admonition, a small proportion of histologically banal SFTs manifest untoward behavior, typically with invasion of the bony structures and soft tissues of the thorax, or recurrence.373,381 Because of these attributes, it is best to consider SFT a borderline neoplasm. As Vallat-Decouvelaere and colleagues375 sagely stated, “it is probably unwise to regard any such lesion as definitely benign.”
In FNA biopsy specimens,389,391,392 smears show variably cohesive and pleomorphic spindle cells in a bloody background, representing an image that corresponds to a sizable differential diagnosis. Preparation of cell block sections and procurement of adjunctive pathologic studies is essential to a specific diagnosis.
Electron microscopic analysis of SFT has shown that the tumor cells are fibroblast-like. They lack basal lamina, intercellular junctions, and plasmalemmal microvilli, as expected in epithelial or mesothelial cells, and instead contain only basic intracellular organelles.381 Occasionally, intrareticular collagen fibrils are apparent within profiles of endoplasmic reticulum.
Immunophenotypically, SFT demonstrates reactivity for vimentin, CD34, CD99, and bcl-2 protein in more than 85% of cases375,382,384,389,392–396 (Fig. 19-103). There is typically no labeling for keratin, EMA, desmin, actins, S-100 protein, collagen type IV, CD31, or CD57. Anecdotally, lesions with malignant histologic features may exhibit mutant p53 protein immunoreactivity,397 but this relationship has not been subjected to rigorous evaluation.
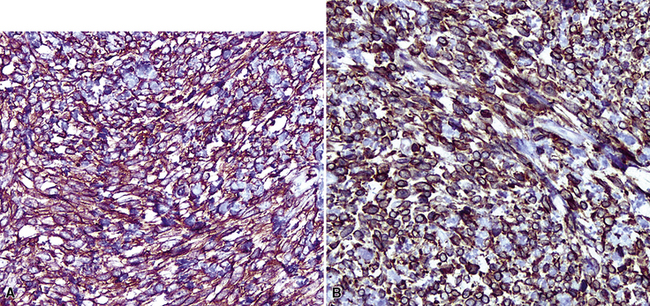
Figure 19-103 Diffuse immunoreactivity for CD34 (A) and CD99 (B) is characteristic of solitary fibrous tumors.
Cytogenetic assessment of SFT is still developmental. However, the most frequent defects reported in this tumor type have involved chromosomes 4q, 8, 13q, 15q, and 21q.398,399 One case has shown a balanced t(4;15)(q13;q26) translocation.399
The differential diagnosis of pleuropulmonary SFT includes primary and secondary sarcomatoid carcinoma, sarcomatoid or desmoplastic mesothelioma, fibrosarcoma, storiform malignant fibrous histiocytoma, synovial sarcoma, hemangiopericytoma, and metastatic endometrial stromal sarcoma.383 Among these possibilities, carcinomas, mesotheliomas, and synovial sarcomas may be excluded because of their immunoreactivity for keratin, EMA, or both. In addition, synovial sarcoma reproducibly shows a t(X;18) chromosomal translocation400 and nuclear immunoreactivity for transducin-like enhancer of split 1 (TLE1),401 both of which are absent in SFT. Consistent CD10 reactivity in metastatic endometrial stromal sarcoma differs from the properties of SFT.402 Fibrosarcoma and malignant fibrous histiocytoma lack CD34, CD99, and bcl-2 protein, all of which are typically manifest in SFT.
The biologic attributes of SFT have been largely discussed previously. However, some of its characteristics merit reiteration. Approximately 10% of “ordinary” intrathoracic SFTs (lacking histologic indicators of possible malignancy) recur, sometimes massively.373,403 This behavior has most often been linked to incomplete lesional excision at initial surgery, and salvage of the patient is still a distinct possibility if total removal of the tumor (e.g., by extrapleural pneumonectomy)404 can be accomplished.295,297 Metastastic disease is very unusual,405 and it tends to be refractory to oncologic intervention.
Desmoid Tumor
“Desmoid” tumor (DT) is best known as a borderline neoplasm of the deep soft tissues and as a member of the family of fibromatoses.406,407 It can be seen sporadically in patients of virtually any age, but also has a biologic linkage to familial adenomatous polyposis syndrome, in which it is over-represented.408 Wherever it arises in the body, DT pursues a slowly progressive, infiltrative course of growth, eventually impinging on nerves, blood vessels, and other anatomic structures. That property accounts for associated symptoms and signs, which reflect secondary compressive effects of the mass.406
Several examples of DT have been reported as apparently primary pleural neoplasms.409–413 Demographic, radiologic, and clinical findings in such cases are superimposable on those associated with SFT.409,413 Pleural desmoids are deceptively circumscribed in imaging studies, and they typically have a homogeneous radiodensity (Fig. 19-104).
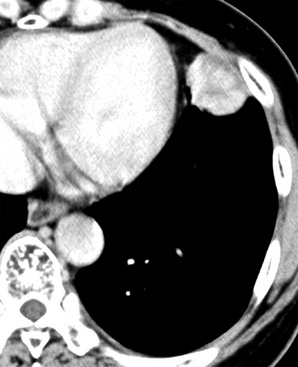
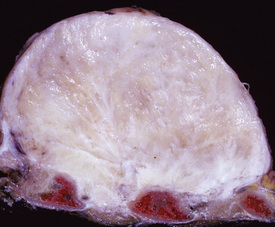
Grossly, DTs of the pleura show a fleshy, uniform, tan-gray cut surface with subtly incorporated bands of internal fibrous tissue (Fig. 19-105). They often appear to have a distinct interface with surrounding tissues, but this attribute is misleading because microscopic study often shows tumor growth beyond the gross boundaries of the lesion.
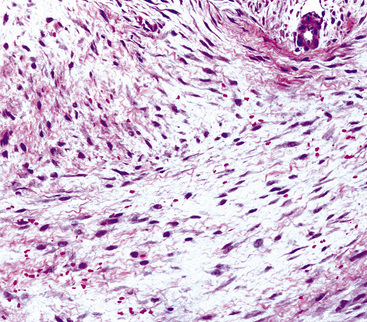
Histologically, the usual appearance of pleural DT is reproducible. It is a hypocellular neoplasm that composes bland fusiform and stellate cells, set in a fibromyxoid stroma (Fig. 19-106). Mitoses are absent or rare; nuclear atypia and necrosis are lacking.406,407 A helpful diagnostic feature is the presence of regularly spaced small blood vessels throughout the lesion; these have open round or oval lumina and relatively thick pericytic cuffs. As mentioned earlier, permeative infiltration of surrounding tissues by the tumor is common.
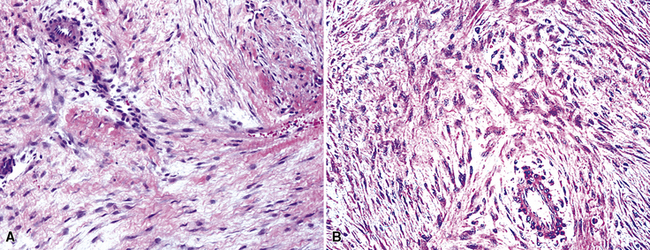
A potential diagnostic trap is represented by selected examples of DT that have a markedly myxedematous stroma, with or without extravasated erythrocytes (Fig. 19-107). The resulting microscopic image can strongly resemble that of nodular fasciitis, a completely innocuous pseudoneoplastic condition.414 Nodular fasciitis has not been reported in the pleura, however.
Fine-needle aspiration biopsy of DT may show either hypocellular or hypercellular foci, often in juxtaposition.415 The stromal staining characteristics are those of mature collagen. Individual tumor cells are dyshesive and morphologically bland, with tapered ends, fusiform nuclei, dispersed chromatin, and a lack of pleomorphism.
As a derivative of the association between desmoids and familial adenomatous polyposis syndrome, beta-catenin protein is usually dysregulated in DT. Instead of exclusively cytoplasmic immunolocalization of that moiety, it is instead seen as an intranuclear reactant (Fig. 19-108).409,415–417 Unfortunately, an important (if not the principal) differential diagnostic consideration—solitary fibrous tumor—also commonly demonstrates nuclear labeling for beta-catenin.417,418 Hence, one must rely on other immunohistochemical discriminants to separate those two neoplasms. DT is reactive for alpha-isoform and “muscle-specific” actins, and sometimes for desmin, whereas SFT is not.409 Conversely, reactivity for CD34 and CD99 is common in SFT but absent in DT. Diagnostic distinctions among DT, other myofibroblastic proliferations, leiomyomas, and low-grade leiomyosarcomas are not possible at an immunohistochemical level of analysis409,415 and must be made by other means.
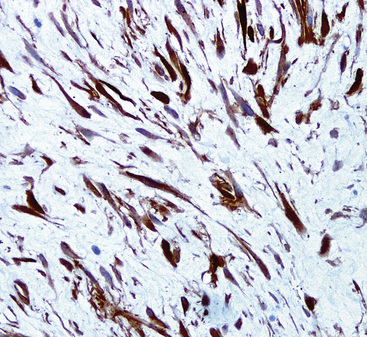
The biologic behavior of DT approximates that of low-grade sarcomas; local recurrences can be tenacious and even life-threatening, but distant metastasis does not occur.406,407,413 Curative surgical excision requires complete removal of the mass.419,420
Clear Cell Tumor
The general premises underlying the nosologic family of “myomelanocytomas” (MMCs) have been described previously in reference to angiomyolipomas. Another member of that conceptual group—clear cell “sugar” tumor of the lung (CCT)—is a purely epithelioid MMC,250 which, for reasons to be discussed, is probably best regarded as a biologically “borderline” neoplasm.
Pulmonary “sugar tumor” was first described noncommittally as “clear cell tumor of the lung” due to the presence of intracytoplasmic glycogen.421 It typically occurs in patients older than 40 years of age422–439 as a peripheral lung nodule. However, occasional examples have been reported in the trachea or large bronchi.425,436 Like angiomyolipoma, CCT has been found concurrently with lymphangioleiomyomatosis and multifocal micronodular pneumocytic hyperplasia in some patients with tuberous sclerosis.433 Tumors in such patients demonstrate a loss of heterozygosity in the TSC2 region of chromosome 16p13.440,441
The radiographic attributes of CCT are nondescript.424,429 It is a homogeneously dense, round to ovoid, peripheral lung mass that may be seen in any region of the lung (Fig. 19-109). CT has typically shown sharp circumscription.
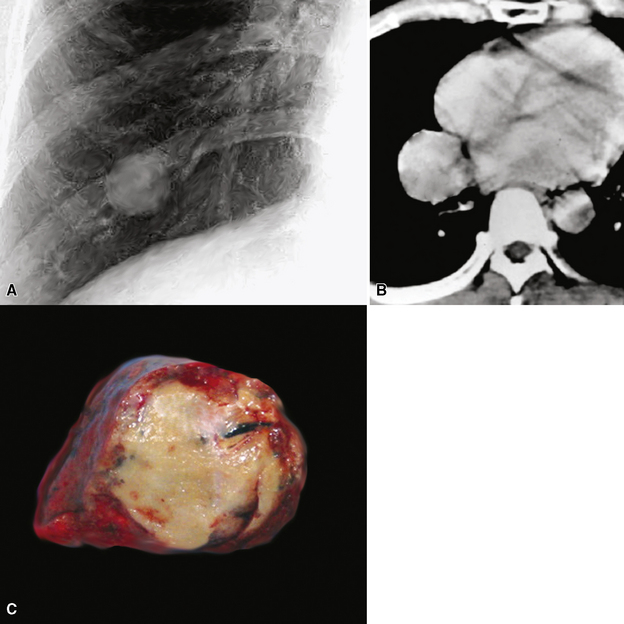
Histologic examination shows one of two general growth patterns in CCTs. The first is organoid, with broad cords and rounded nests of cells separated by a variably prominent fibrovascular stroma, mimicking renal cell carcinoma. The second configuration is a medullary image, showing sheets of epithelioid cells with little internal clustering (Fig. 19-110). Pre-existing small bronchi and bronchioles are often entrapped by the neoplastic cells. Nuclei are round to ovoid with small nucleoli; intranuclear invaginations of cytoplasm also may be apparent, but mitotic figures are usually difficult to find. Cytoplasm is clear or lightly eosinophilic, and may be finely granular. Histochemical evaluation with the periodic acid/Schiff method usually shows intense cytoplasmic labeling for glycogen in the tumor cells (Fig. 19-111). Intralesional blood vessels can be markedly sclerotic in some cases, spontaneous necrosis is occasionally seen, and sparse intratumoral chronic inflammatory cells may be identified.
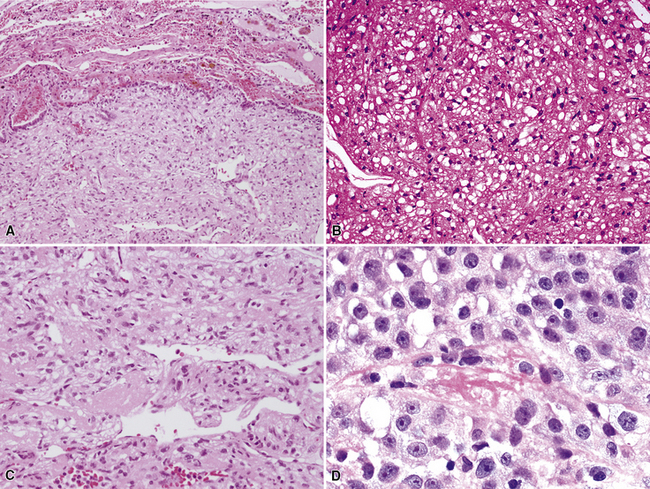
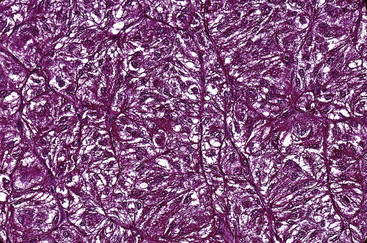
Ultrastructural studies of CCT have shown attributes suggesting a “hybrid” cell type that incorporates elements of modified perivascular smooth muscle and melanocytes. These include interdigitating cellular processes, pericellular basal lamina, primitive intercellular attachment complexes, plasmalemmal pinocytosis, membrane-bound and free cytoplasmic glycogen granules, and variant form premelanosomes.423,426,428,430,432,435
Immunohistologically, one sees a uniform lack of reactivity for epithelial markers in CCT, but consistent reactivity for vimentin, CD117, collagen type IV, HMB-45, MART-1, and micro-ophthalmia transcription factor 1 (with the last three determinants all related to melanocytes)246,427,428,432,434 (Fig. 19-112). Inconsistent but generally positive results are obtained for muscle-specific actin, S-100 protein, and neuron-specific enolase. Panizo-Santos and colleagues442 have described aberrant but virtually exclusive cytoplasmic immunoreactivity for Myo-D1 in members of the MMC family of tumors. This marker is a nuclear transcription factor related to striated muscle development, and bona fide labeling for it would not be expected in the cytoplasm. Hence, it appears that the cited pattern of staining probably represents a reproducible artifact, but if corroborated, it could prove to be useful diagnostically.
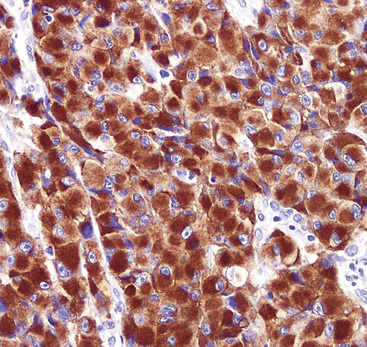
The differential diagnosis for CCT is a lengthy one, including primary carcinomas of the lung with clear cell features; metastatic clear cell carcinomas from the kidney, urogenital tract, breast, and other locations; metastatic clear cell sarcoma; and metastatic “balloon cell” melanoma.443 The generic possibility of carcinoma can be excluded by the lack of keratin and EMA in CCT, but clear cell sarcoma and melanoma are not as easily dismissed because they potentially share the entire complement of immunohistologic melanocyte markers with MMCs. Reactivity for actin or cytoplasmic Myo-D1 would strongly argue in favor of CCT in this setting, because those determinants have not been reported in metastatic melanomas with epithelioid features.
Biologically, CCT has traditionally been considered a benign pulmonary tumor. Nonetheless, at least two examples435,437 have metastasized to other visceral sites, and one proved fatal. This behavior parallels the biologic potential of MMCs in other organs, notably the kidney. Thus, it would seem appropriate to regard CCT as another borderline neoplasm of the lung. With that having been said, however, simple surgical removal of the lesion—with wedge excision of the peripheral lung parenchyma, if possible—is believed to represent adequate therapy.
Primary Pleuropulmonary Thymoma
Even though thymomas typically arise in the anterosuperior mediastinum, the literature contains ample evidence of their ability to develop in ectopic sites. These tumors have been described in the soft tissue of the lower neck, as well as the thyroid, pericardium, lungs, and pleura.444–449 Because of their ability to show a relatively wide morphologic spectrum, heterotopic thymic tumors may be quite difficult to recognize diagnostically.
Intrapulmonary thymoma is seen in adults between the ages of 20 and 80 years. In the minority of cases where the lesion is linked to one of several distinctive thymoma-related paraneoplastic syndromes— principally myasthenia gravis, pure red cell aplasia, and acquired hypogammaglobulinemia—its identity may be suspected clinically.450 However, the majority of these tumors present as asymptomatic masses that are found radiographically. They can be situated centrally, close to the hilum, and even endobronchially in rare instances, as well as in the mid-lung fields or beneath the pleural surfaces451–461 (Fig. 19-113). Occasionally, two or more intrapulmonary masses are seen concurrently. In analogy to mediastinal thymomas, those arising in the lungs may be either circumscribed or infiltrative, and some demonstrate prominent intralesional cystification.
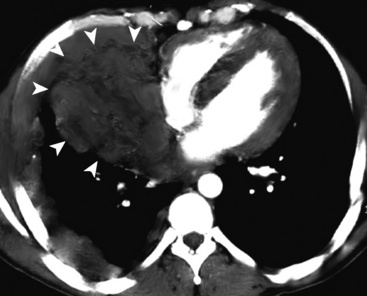
Pleural thymomas may simulate the appearance of any other solitary serosal neoplasm. In rare examples, they have diffusely effaced the pleural space in one or both hemithoraces, reproducing the radiographic and gross pathologic image of diffuse pleural mesothelioma or metastatic serosal carcinoma.459–461
These tumors have fleshy pink-tan cut surfaces that closely resemble those of lymphoreticular neoplasms, but areas of necrosis, hemorrhage, or cyst formation are much more common than they are in lymphomas. They may also contain internal fibrous septations, subdividing the masses into angulated tissue compartments (Fig. 19-114). Dystrophic calcification may be present.
Microscopically, circumferential fibrous encapsulation of pulmonary or pleural thymomas is unusual, in contrast to their intrathymic counterparts. As a result, ectopic lesions in the hemithoraces must commonly be classified as “invasive,” almost by definition (Fig. 19-115). As stated previously, intralesional fibrous septa intersect one another at acute angles (Fig. 19-116), differing from the obtuse connections that are seen in nodular sclerosing Hodgkin’s lymphoma or sclerosing non-Hodgkin’s lymphomas in the chest. At the University of Virginia, the classic Bernatz system of histologic classification for thymic epithelial tumors is used, which has five subdivisions:
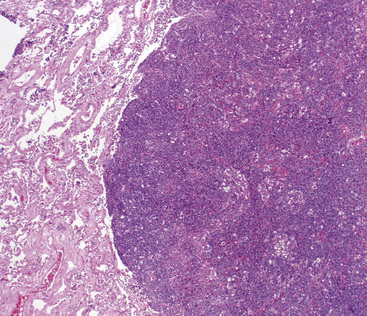
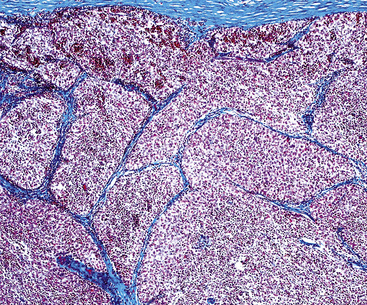
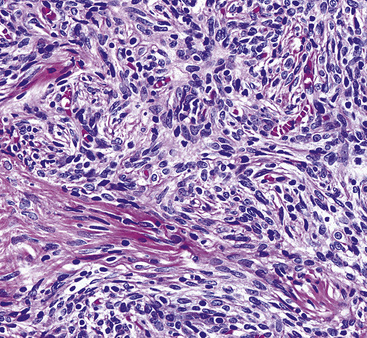
Figure 19-117 Predominantly spindle cell (medullary, World Health Organization type A) intrapulmonary thymoma.
Other alternative nosologic constructions for thymic tumors have entered common use as well, including the Marino-Muller-Hermelink scheme,463 the World Health Organization system,464 and the Suster-Moran codification.465 The last one can easily be linked with the Bernatz system to specifically address tumors that demonstrate a notable degree of nuclear atypia, yet whose features are insufficient for an outright diagnosis of carcinoma. Such neoplasms are termed “atypical” thymomas.
The epithelial cells of thymomas—not the lymphoid cells—are the neoplastic elements. They are characterized by a range of cytologic images. Some have vague cellular borders, oval nuclear contours, dispersed chromatin, and indistinct nucleoli. At the other end of the spectrum, one sees rather clear-cut plasmalemmal interfaces, moderate nuclear irregularity and hyperchromasia, and distinct nucleoli (Fig. 19-118). As mentioned earlier, spindle cell change is another potential image in thymomas, and the nuclei in such lesions tend to have bland and uniform morphologic characteristics. Mitotic activity in thymic tumors is greatly variable as well, and it achieves importance as a possible marker of thymic carcinoma only in neoplasms that also show nuclear abnormalities.
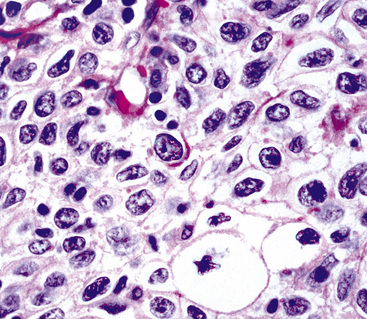
Secondary architectural changes in thymomas include the formation of perivascular “lakes” of serum, in which lymphoid cells are suspended; pseudoglandular arrays or pseudorosettes of epithelial cells; microcysts or areas of gross cystic change, with or without necrosis; stromal blood lakes and vascular dilatation; and “medullary differentiation,” in which loose and vaguely nodular aggregates of stromal lymphocytes are present.462 Hassall’s corpuscles are seen in only a small minority of cases. Some spindle cell thymomas contain a vascular network comprising numerous vessels with a “moose antler” configuration, yielding a pattern that virtually perfectly imitates that of hemangiopericytoma–solitary fibrous tumor.
Epithelial-predominant thymomas with nuclear atypia, an organoid growth pattern, and distinct cellular borders have been called “well-differentiated thymic carcinomas.”466 However, their generic clinicopathologic characteristics are clearly dissimilar to those of outright thymic carcinomas, and some observers believe that they are more properly termed “atypical thymomas.”465
The cytopathologic attributes of thymic epithelial tumors seen in FNA biopsy specimens are well described467–469 and reflect their histologic heterogeneity. Mature lymphocytes are admixed with thymic epithelial cells, which show a tendency toward cohesion. However, scattered single epithelial cells may also be evident. Epithelial cell nuclei are generally monomorphic, with dispersed chromatin and small chromocenters. Cytoplasm is amphophilic, with indistinct cell borders. A fusiform cellular shape may be encountered in spindle cell tumors, but the grouping of such cells into small clusters helps to distinguish them from mesenchymal proliferations. The lymphoid elements in thymoma are a potential diagnostic pitfall, and nuclear “activation” may be seen with increases in the nuclear-to-cytoplasmic ratios. Convolution of the nuclear borders can also be present, as may mitotic figures. The overall image of “activated” intratumoral lymphocytes is quite similar to that of lymphoblastic lymphoma, and a misdiagnosis may ensue unless adjunctive studies are performed to detect the epithelial elements of thymoma.454,467 Predictably, invasive thymomas cannot be distinguished from encapsulated tumors using the FNA technique.
Electron microscopy of thymomas shows elongated and interdigitating cytoplasmic processes emanating from constituent epithelial cells, and these are joined to one another by well-formed desmosomes into which broad tonofibrils insert. Plasmalemmal microvilli are absent, and intralesional lymphoid cells are usually intercalated between the epithelial elements.470
Cytogenetic analysis has shown no consistent karyotypic aberrations in thymomas. Several different abnormalities have been documented, including deletions of chromosome 6p; t(1;8) and t(15;22) chromosomal translocations; pseudodicentric (16;12)(q11;p11.2); and ring chromosome 6.471–473 However, none of these findings is typically associated with other primary pleuropulmonary tumors.
Immunohistologic studies are paramount in confirming the cellular nature of ectopic thymomas. Keratin immunostains show a characteristically arborizing network of reactivity, reflecting the presence of interconnecting cytoplasmic processes474 (Fig. 19-119). The keratin subtype 5/6 is also characteristic of thymic epithelium.475 A member of the p53 protein family, p63 protein, is likewise consistently present in thymoma (Fig. 19-120), as it is in squamous neoplasms.476 Finally, the lymphoid cells in thymomas are true thymocytes. They express CD1a, nuclear terminal deoxynucleotidyl transferase (TdT), and CD99.454 The latter phenotype is shared only by the tumor cells of lymphoblastic lymphomas, but these hematopoietic neoplasms are uniformly nonreactive for p63 and keratin. Thus, there is a truly diagnostic immunoprofile for thymoma. Parenthetically, it should be noted that “pseudomesotheliomatous” pleural thymomas may be reactive for calretinin, keratin 5/6, and thrombomodulin, all of which are commonly associated with mesothelial cells.461 That combination of results may easily lead to misdiagnosis if studies for p63, CD1a, TdT, and CD99 are omitted.
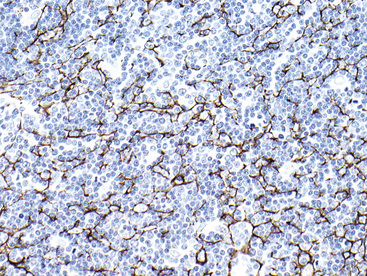
Figure 19-119 Interconnecting cell processes in a thymoma are labeled for keratin, yielding a “lacy” immunostaining pattern.
(Image courtesy of Dr. Anja Roden, Mayo Clinic, Rochester, MN.)
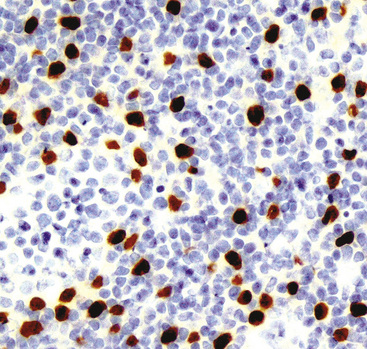
Behaviorally, ectopic thymomas in the lung and pleura are typically indolent lesions that can be cured by complete surgical resection if they are solitary.477 However, several examples have been reported in which recurrence, distant metastasis, or both, was observed.451–458 Moreover, lesions that simulate mesothelioma because of their encasement of the lungs or heart are associated with a greater risk of adverse behavior.459–461 Because a minority of all pleuropulmonary thymomas are fatal, they are justifiably considered borderline.
Heterotopic Meningeal Proliferations
Meningiomas are typically considered neoplasms of the main neural axis, but they are perhaps the most widely distributed of any tumor in that group. Tumefactive proliferations with meningothelial differentiation have been reported in a wide variety of locations, including the mediastinum and lungs, in the absence of involvement of the coverings of the brain and spinal cord.478 Patients with such lesions seem to be somewhat younger on average than those with intracranial meningiomas, most of whom are middle-aged or older. Because these neoplasms are clearly not expected outside of their usual confines, their clinical presentation in heterotopic locations is typically undiagnosed at a clinical level.479 For example, those in the lung are usually believed to represent granulomas or adenocarcinomas radiographically (Fig. 19-121). Ectopic meningiomas generally pursue a relatively favorable clinical course; although local recurrence is a possibility, these lesions are uncommonly associated with distant metastasis.480
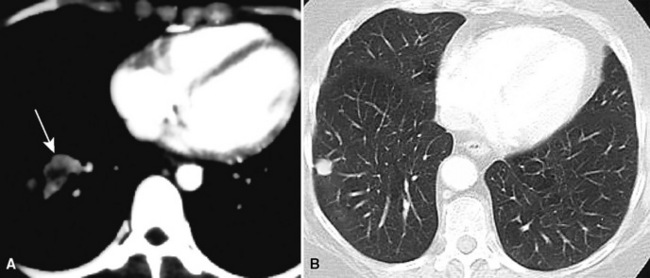
Grossly, meningioma of the lung is usually sharply marginated and easily dissected from the surrounding parenchyma (Fig. 19-122). It measures between 1 and 5 cm in diameter and has a globoid configuration and a uniformly white-gray cut surface. Those rare lesions that are malignant may show areas of necrosis and hemorrhage, as well as infiltration of the adjacent lung.480,481
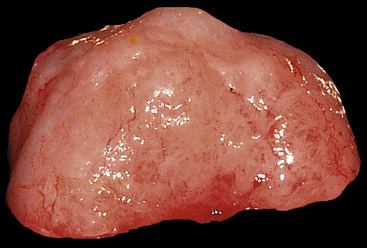
Histologically, heterotopic meningiomas show preferential expression of meningothelial, fibroblastic, or “transitional” growth patterns.482–493 As such, meningiomas of the lung are composed of polygonal or fusiform cells with monomorphic oval nuclei and dispersed chromatin (Fig. 19-123). Whorled aggregates of tumor cells may be observed multifocally, with or without secondary psammomatous microcalcifications. In some cases, the latter structures are numerous.
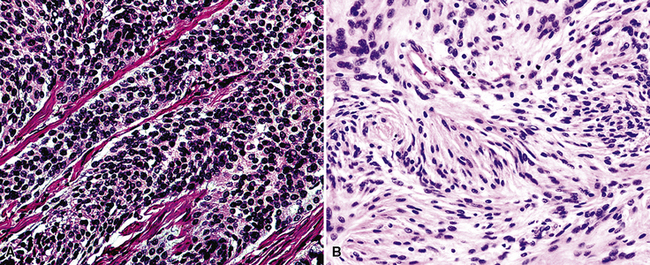
Figure 19-123 Intrapulmonary meningiomas with meningotheliomatous (A) and transitional (mixed) (B) features.
Purely meningotheliomatous lesions—comprising a pure population of polygonal cells—may simulate the appearance of a carcinoma; at the other pole of the spectrum, “fibroblastic” or solely spindle cell meningiomas can imitate the configuration of solitary fibrous tumor–hemangiopericytoma or other primary soft tissue neoplasms478,481 (Fig. 19-124). Mitotic activity is limited, nuclear atypia is only modest, and necrosis is usually absent in benign pulmonary meningiomas, but the presence of those findings should raise concern about the rare possibility of malignancy.494
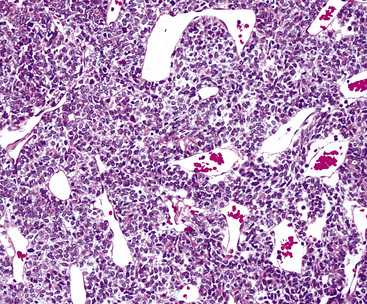
The premise has been advanced by some authors that primary pulmonary meningioma may arise from “meningothelial-like micronodules” in the lung482,484 (formerly and erroneously called “minute pulmonary chemodectomas”), which are related to parenchymal damage as a result of cardiac failure, chronic obstructive pulmonary disease, and thromboemboli495 (Fig. 19-125). The conclusion that they are the precursors of meningioma was derived from the fact that both lesions have been seen together in the same case; however, no molecular analyses have been done to further address that possibility. In our opinion, however, this is unlikely; pertinent data show that meningothelial-like micronodules are polyclonal and probably reactive.496 When multiple, the phenomenon has been called “menigotheliomatosis,”497 which may lead to radiologic confusion with interstitial lung disease or metastases (Fig. 19-126).
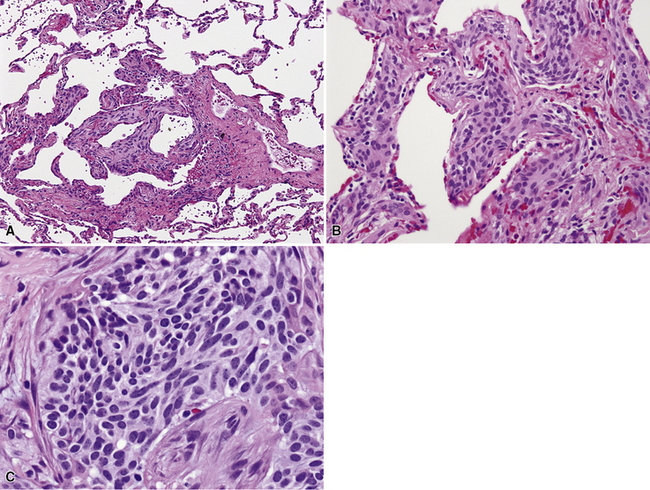
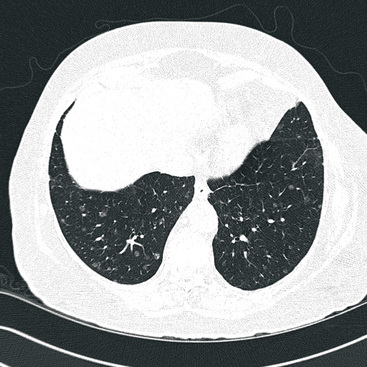
Fine-needle aspiration biopsy specimens of pulmonary meningioma482,486 show scanty material, represented by whorls of cells with a concentric internal configuration (Fig. 19-127), as well as isolated individual cells. Intranuclear inclusions may be apparent, and psammomatous calcifications can be observed in rare instances.
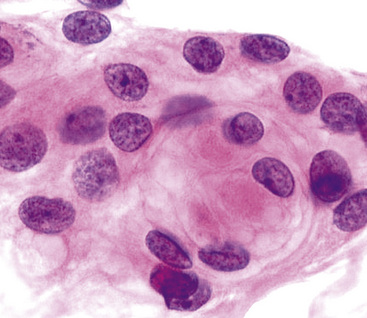
Electron microscopy is still an extremely useful tool for the resolution of differential diagnostic questions surrounding these tumors.498 Meningioma has a singular ultrastructural appearance that features numerous interdigitating cell processes attached by prominent desmosomes. Skeins of well-formed tonofibrils insert into those attachment complexes. To our knowledge, this particular constellation of findings is not recapitulated by any other tumor type.
Prototypically, meningiomas are reactive for vimentin to the exclusion of other intermediate filament proteins; EMA is also present universally, as is immunoreactivity for desmoplakin, a desmosomal constituent.478,481–483,498 Intracranial meningiomas are immunoreactive for progesterone receptor protein in the majority of cases.499,500 However, that marker has not yet been assessed in primary pulmonary meningothelial tumors. Immunophenotypic characteristics of ectopic meningiomas also differ from those of their neuraxis analogs, in that the former lesions appear to express keratin more often. Admittedly, this attribute is probably influenced by the site of origin. Additional markers that are germane to the differential diagnosis, such as CD34 and S-100 protein, have also been reported sporadically in heterotopic meningiomas in some sites.
Other pertinent diagnostic considerations for primary pulmonary meningiomas include carcinomas with or without spindle cell features, solitary fibrous tumors, hemangiopericytomas, schwannomas, and amelanotic malignant melanomas.481 Electron microscopy is still very effective in resolving such uncertainties because of the distinctive profile of meningioma. Immunohistochemical analyses require the application of antibodies to Ber-EP4, MOC-31, carcinoembryonic antigen, CD56, CD57, CD99, melan-A/Mart-1, and tyrosinase. Podoplanin (recognized by antibody D2-40), a relative newcomer to clinical immunohistochemistry, is not useful in this specific context. It may be seen in SFT, schwannoma, meningioma, and sarcomatoid melanoma.501–503 However, another useful modality of investigation is fluorescent in situ hybridization, which demonstrates monosomy of chromosome 22 in a majority of meningiomas,504 a cytogenetic abnormality that is not shared by other differential diagnostic possibilities.
None of these studies is effective in separating primary ectopic from metastatic neuraxis-based meningiomas.491,505 However, it is an exceedingly rare situation for the latter neoplasms to present in distant sites with no previous knowledge of a primary tumor within the cranial vault or spine.505
Intrapulmonary Teratomas
Examples of primary intrapulmonary teratoma have been reported with extraordinary rarity506–509 (Fig. 19-128). The overwhelming majority of teratoid tumors in the lungs and pleura represent altered metastases of gonadal germ cell tumors, in which malignant histologic components were formerly present but have been complete removed by chemotherapy.510 Another small group of cases comprises primary malignant teratoid tumors of the lung that contain seminoma, embryonal carcinoma, yolk sac carcinoma, or choriocarcinoma.511–513 Primary pulmonary teratomas without cytologically malignant components are extremely uncommon; they have principally been seen as variably cystic masses in young adults. One such lesion was associated with pyothorax, and others have presented with extrusion of hair (from the contents of the lesion) into a large airway.506,514,515
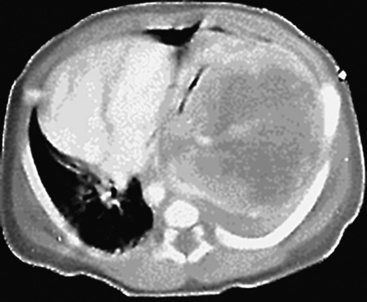
Grossly, the most obvious constituents of such neoplasms are squamous epithelium with associated keratinous debris and cartilage (Fig. 19-129). Mucoid contents may sometimes be seen in cystic areas, and nondescript solid foci may also be present.
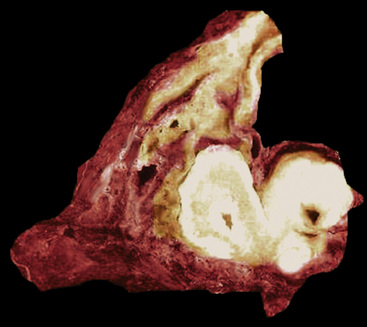
Microscopically, teratomas of the lung can be divided into mature and immature varieties, as is true of these tumors occurring elsewhere in the body. The first of those subtypes is self-explanatory, but the term “immature” in this context refers specifically to the presence of primitive neurectodermal tissue that resembles the developing neural tube. Otherwise, virtually any tissue in the body can be reflected in the constituents of teratoid lesions, including such unexpected elements as retina (Fig. 19-130). To qualify as teratomatous, the tumor must demonstrate derivatives from at least two of the three germinal lines (ectoderm, endoderm, and mesoderm).
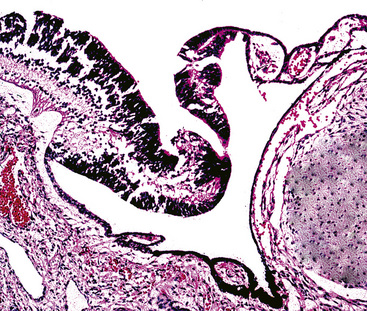
Cystic Fibrohistiocytic Tumor of the Lung
Joseph and colleagues516 described two adult patients who were found to have variably cystic lung lesions, represented by multiple parenchymal abnormalities on chest radiographs. One patient had pneumothorax and shortness of breath, and lesions in the other case were asymptomatic. Open-lung biopsies showed a proliferation of relatively bland spindle cells and histiocyte-like elements, mantling cystic spaces that were lined by metaplastic bronchiolar epithelium, squamous cells, or type II pneumocytes (Fig. 19-131). Some of the cysts contained erythrocytes and hemosiderin deposits. Another case has been illustrated anecdotally by Han.517
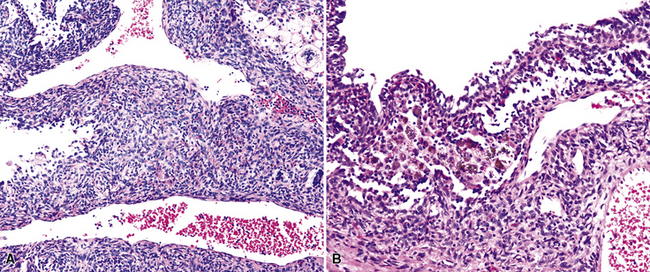
Although the lesions described by Joseph and coworkers516 enlarged very slowly over time, the authors suggested that the ultimate biologic potential and recommended treatment of pulmonary cystic fibrohistiocytic tumors was unclear. Currently, in accord with the opinions of Colome-Grimmer and Evans518 and the authors of the original report on this entity (T. V. Colby, personal communication), it is believed that cystic fibrohistiocytic tumors are actually not primary lesions of the lung. Instead, they likely represent metastases of extrapulmonary low-grade fibrohistiocytic tumors, including such tumors as cellular dermatofibroma of skin.
Other Lesions
Well-differentiated papillary mesothelioma of the pleura and intrapulmonary paraganglioma are other borderline lesions that are properly mentioned here. Because of its specialized nature and possible relationship to malignant mesotheliomas, well-differentiated papillary mesothelioma is discussed in Chapter 20. Intrapulmonary paraganglionic tumors are discussed in Chapter 13, dealing with neuroendocrine neoplasms. Because its biologic characteristics are more aggressive than those of most other “borderline lesions,” hemangioendothelioma is discussed in Chapter 14, on malignant mesenchymal tumors of the lung and pleura.
1 Wingo P.A., Cardinez C.J., Landis S.H., et al. Long-term trends in cancer mortality in the United States, 1930–1998. Cancer. 2003;97(12 suppl):3133-3275.
2 Lutz J.M., Francisci S., Mugno E., et al. Cancer prevalence in Central Europe: the EUROPREVAL study. Ann Oncol. 2003;14:313-322.
3 Oei T.K., Wouters E.F., Visser R., VanEngelshoven J.M., Greve L.H. The value of conventional radiography and computed tomography (CT) in diagnosis of pulmonary hamartoma. Rontgenblatter. 1983;36:324-327.
4 Erasmus J.J., Connolly J.E., McAdams H.P., Roggli V.L. Solitary pulmonary nodules: Part I. Morphologic evaluation for differentiation of benign and malignant lesions. Radiographics. 2000;20:43-58.
5 Petticrew M.P., Sowden A.J., Lister-Sharp D., Wright K. False-negative results in screening programs: systematic review of impact and implications. Health Technol Assess. 2000;4:1-120.
6 Cardillo G., Regal M., Sera F., et al. Videothoracoscopic management of the solitary pulmonary nodule: a single-institution study on 429 cases. Ann Thorac Surg. 2003;75:1607-1611.
7 Barzo P., Molnar L., Minik K. Bronchial papilloma of various origins. Chest. 1987;92:132-136.
8 Spencer H., Dail D.H., Arneaud J. Noninvasive bronchial epithelial papillary tumors. Cancer. 1980;45:1486-1497.
9 Zimmerman A., Lang H.R., Muhlberger F., Bachman M. Papilloma of the bronchus. Respiration. 1980;39:286-290.
10 Maxwell R.J., Gibbons J.R., O’Hara M.D. Solitary squamous papilloma of the bronchus. Thorax. 1985;40:68-71.
11 Laubscher F.A. Solitary squamous cell papilloma of bronchial origin. Am J Clin Pathol. 1969;52:599-603.
12 Greene J.G., Tassin L., Saberi A. Endobronchial epithelial papilloma associated with a foreign body. Chest. 1990;97:229-230.
13 Miura H., Tsuchida T., Kawate N., et al. Asymptomatic solitary papilloma of the bronchus: review of occurrence in Japan. Eur Respir J. 1993;6:1070-1073.
14 Basheda S., Gephardt G.N., Stoller J.K. Columnar papilloma of the bronchus: case report and literature review. Am Rev Respir Dis. 1991;144:1400-1402.
15 Trillo A., Guha A. Solitary condylomatous papilloma of the bronchus. Arch Pathol Lab Med. 1988;112:731-733.
16 Desai A.V., Scolyer R.A., McCaughan B., Torzillo P.J. Middle lobe syndrome due to obstructing solitary bronchial papilloma. Aust N Z J Med. 1999;29:745-747.
17 Hurt R. Benign tumors of the bronchus and trachea, 1951–1981. Ann R Coll Surg Engl. 1984;66:22-26.
18 Roviaro G.C., Varoli F., Pagnini C.A. Is the solitary papilloma of the bronchus always a benign tumor? J Otorhinolaryngol Relat Spec. 1981;43:301-308.
19 Katial R.K., Ranlett R., Whitlock W.L. Human papillomavirus associated with solitary squamous papilloma complicated by bronchiectasis and bronchial stenosis. Chest. 1994;106:1887-1889.
20 Roglic M., Jukic S., Damjanov I. Cytology of the solitary papilloma of the bronchus. Acta Cytol. 1975;19:11-13.
21 Flieder D.B., Koss M.N., Nicholson A., et al. Solitary pulmonary papillomas in adults: a clinicopathologic and in-situ hybridization study of 14 cases combined with 27 cases in the literature. Am J Surg Pathol. 1998;22:1328-1342.
22 Lee Y.O., Kim D.H., Kim C.H., Park T.I., Cho S. Rare tumor of the tracheobronchial tree: solitary squamous papilloma. Thorac Cardiovasc Surg. 2009;57:178-179.
23 Popper H.H., El-Shabrawi Y., Wockel W., et al. Prognostic importance of human papillomavirus typing in squamous cell papilloma of the bronchus: comparison of in-situ hybridization and the polymerase chain reaction. Hum Pathol. 1994;25:1191-1197.
24 Popper H.H., Wirnsberger G., Juttner-Smolle F.M., Pongratz M.G., Sommersgutter M. The predictive value of human papillomavirus (HPV) typing in the prognosis of bronchial squamous cell papillomas. Histopathology. 1992;21:323-330.
25 Clavel C.E., Nawrocki B., Bosseaux B., et al. Detection of human papillomavirus DNA in bronchopulmonary carcinomas by hybrid capture. II. A study of 185 tumors. Cancer. 2000;88:1347-1352.
26 Smith E., Pignatari S., Gray S., Haugen T., Turak L. Human papillomavirus infection in papillomas and nondiseased respiratory sites of patients with recurrent respiratory papillomatosis, using the polymerase chain reaction. Arch Otolaryngol Head Neck Surg. 1993;119:554-557.
27 Rady P.L., Schnadig V.J., Weiss R.L., Hughes T.K., Tyring S.K. Malignant transformation of recurrent respiratory papillomatosis associated with integrated human papillomavirus type 11 DNA and mutation of p53. Laryngoscope. 1998;108:735-740.
28 Syrjanen R.J. Epithelial lesions suggestive of a condylomatous origin found closely associated with invasive bronchial squamous cell carcinomas. Respiration. 1980;40:150-160.
29 DiMarco A.F., Montenegro H., Payne C.B.Jr, Kwon K.H. Papillomas of the tracheobronchial tree with malignant degeneration. Chest. 1978;74:464-465.
30 Bejui-Thivolet F., Chardonnet Y., Patricot L.M. Human papillomavirus type 11 DNA in papillary squamous cell lung carcinoma. Virchows Arch A. 1990;417:457-461.
31 Inoue Y., Oka M., Ishii H., et al. A solitary bronchial papilloma with malignant changes. Intern Med. 2001;40:56-60.
32 Kramer S., Wehunt W., Stocker J., Kashima H. Pulmonary manifestations of juvenile laryngotracheal papillomatosis. Am J Roentgenol. 1985;144:687-694.
33 Fechner R.E., Fitz-Hugh G. Invasive tracheal papillomatosis. Am J Surg Pathol. 1980;4:79-86.
34 Peterson B.L., Buchwald C., Gerstoft J., Bretlau P., Lindeberg H. An aggressive and invasive growth of juvenile papillomas involving the total respiratory tract. J Laryngol Otol. 1998;112:1101-1104.
35 Franzmann M.B., Buchwald C., Larsen P., Balle V. Tracheobronchial involvement of laryngeal papillomatosis at onset. J Laryngol Otol. 1994;108:164-165.
36 Shykhon M., Kuo M., Pearman R. Recurrent respiratory papillomatosis. Clin Otolaryngol. 2002;27:237-243.
37 Blackledge R.A., Anandi V.K. Tracheobronchial extension of recurrent respiratory papillomatosis. Ann Otol Rhinol Laryngol. 2000;109:812-818.
38 Harada H., Miura K., Tsutsui Y., et al. Solitary squamous cell papilloma of the lung in a 40 year old woman with recurrent laryngeal papillomatosis. Pathol Int. 2000;50:431-439.
39 Weingarten J. Cytologic and histologic findings in a case of tracheobronchial papillomatosis. Acta Cytol. 1981;25:167-170.
40 Kashima H., Mounts P., Leventhal B., Hruban R.H. Sites of predilection in recurrent respiratory papillomatosis. Ann Otol Rhinol Laryngol. 1993;102:580-583.
41 Al-Saleem T., Peale A.R., Norris C.M. Multiple papillomatosis of the lower respiratory tract: clinical and pathologic study of eleven cases. Cancer. 1968;22:1173-1184.
42 Rahman A., Ziment I. Tracheobronchial papillomatosis with malignant transformation. Arch Intern Med. 1983;143:577-578.
43 Batsakis J.G., Raymond A.K., Rice D.H. The pathology of head and neck tumors: papillomas of the upper aerodigestive tracts, Part 18. Head Neck Surg. 1983;5:332-344.
44 Zarod A., Rutherford J., Corbitt G. Malignant progression of laryngeal papilloma associated with human papillomavirus type 6 (HPV-6). J Clin Pathol. 1988;41:280-283.
45 Byrne J., Tsao M.S., Fraser R., Howley P. Human papillomavirus-11 DNA in a patient with chronic laryngotracheobronchial papillomatosis and metastatic squamous cell carcinoma of the lung. N Engl J Med. 1987;317:873-878.
46 Wilde E., Duggan M., Field S. Bronchogenic squamous cell carcinoma complicating localized recurrent respiratory papillomatosis. Chest. 1994;105:1887-1888.
47 Lindsay F., Bloom D., Pransky S., Stabley R., Shick P. Histologic review of cidofovir-treated recurrent respiratory papillomatosis. Ann Otol Rhinol Laryngol. 2008;117:113-117.
48 Yousem S.A., Ohori N.P., Sonmez-Alpan E. Occurrence of human papillomavirus DNA in primary lung neoplasms. Cancer. 1992;69:693-697.
49 Liebow A.A. Tumors of the lower respiratory tract. In: Atlas of Tumor Pathology, Series 1, Fascicle 17. Washington, DC: Armed Forces Institute of Pathology; 1952:22.
50 England D.M., Hochholzer L. Truly benign “bronchial adenoma:” report of 10 cases of mucous gland adenoma with immunohistochemical and ultrastructural findings. Am J Surg Pathol. 1995;19:887-899.
51 Markel S.F., Abell M.R., Haight C., French A.J. Neoplasms of the bronchus commonly designated as adenomas. Cancer. 1964;17:590-608.
52 Payne W.S., Fontana R.S., Woolner L.B. Bronchial tumors originating from mucous glands: current classification and unusual manifestations. Med Clin North Am Dis Respir. 1964;48:945-960.
53 Milenkovic B., Stojsic J., Mandaric D., Stevic R. Mucous gland adenoma simulating bronchial asthma: case report and literature review. J Asthma. 2007;44:789-793.
54 Spencer H. Bronchial mucous gland tumors. Virchows Arch Pathol Anat. 1979;383:101-115.
55 Allen M.S.Jr, Marsh W.L.Jr, Greissinger W.T. Mucous gland adenoma of the bronchus. J Thorac Cardiovasc Surg. 1974;67:966-968.
56 Emory W.B., Mitchel W.T.Jr, Hatch H.G.Jr. Mucous gland adenoma of the bronchus. Am Rev Respir Dis. 1973;108:1407-1410.
57 Edwards C.W., Matthews H.R. Mucous gland adenoma of the bronchus. Thorax. 1981;36:147-148.
58 Kroe D.J., Pitcock J.A. Benign mucous gland adenoma of the bronchus. Arch Pathol. 1967;84:539-540.
59 Heard B.E., Corrin B., Dewar A. Pathology of seven mucous cell adenomas of the bronchial glands with particular reference to ultrastructure. Histopathology. 1985;9:687-701.
60 Akhtar M., Young I., Reyes F. Bronchial adenoma with polymorphous features. Cancer. 1974;33:1572-1576.
61 Rosenblum P., Klein R.I. Adenomatous polyp of the right main bronchus producing atelectasis. Pediatrics. 1936;78:791-796.
62 Smith A.G. Benign epithelial tumors of the bronchus. South Med J. 1965;58:1535-1541.
63 Courtin P., Janin A., Sault M.C., et al. Pure bronchial adenoma. Anatomic, clinical, and ultrastructural study of a case. Ann Pathol. 1987;7:315-319.
64 Weinberger M.A., Katz S., Davis E.W. Peripheral bronchial adenoma of mucous gland type: clinical and pathological aspects. J Thorac Surg. 1955;29:626-635.
65 Key B.M., Pritchett P.S. Mucous gland adenoma of the bronchus. South Med J. 1979;72:83-85.
66 Arrigoni M.G., Woolner L.B., Bernatz P.E., Miller W.E., Fontana R.S. Benign tumors of the lung: a ten-year experience. J Thorac Cardiovasc Surg. 1970;60:589-599.
67 Kwon J.W., Goo J.M., Seo J.B., Seo J.W., Im J.G. Mucous gland adenoma of the bronchus: CT findings in two patients. J Comput Assist Tomogr. 1999;23:758-760.
68 Mejean-Lebreton F., Barnoud R., De la Roche E., Devouassoux-Shisheboran M. Benign salivary gland-type tumors of the bronchus: expression of high molecular-weight cytokeratins. Ann Pathol. 2006;26:30-34.
69 Delpiano C., Claren R., Sironi M., Cenacchi G., Spinelli M. Cytological appearance of papillary mucous gland adenoma of the left lobar bronchus with histological confirmation. Cytopathology. 2000;11:193-196.
70 Moran C.A., Suster S., Askin F.B., Koss M.N. Benign and malignant salivary gland-type mixed tumors of the lung: clinicopathologic and immunohistochemical study of eight cases. Cancer. 1994;73:2481-2490.
71 Davis P.W., Briggs J.C., Leal R.M.E., et al. Benign and malignant mixed tumors of the lung. Thorax. 1972;27:657-673.
72 Hayes M.M., Van der Westhuizen N.G., Forgie R. Malignant mixed tumors of the bronchus: a biphasic neoplasm of epithelial and myoepithelial cells. Mod Pathol. 1993;6:85-88.
73 Payne W.S., Scier J., Woolner L.B. Mixed tumors of the bronchus (salivary gland type). J Thorac Cardiovasc Surg. 1965;49:663-668.
74 Sakamoto H., Uda H., Tanaka T., et al. Pleomorphic adenoma in the periphery of the lung: report of a case and review of the literature. Arch Pathol Lab Med. 1991;115:393-396.
75 Moran C.A., Suster S., Askin F.B., Koss M.N. Benign and malignant salivary gland-type tumors of the lung: clinicopathologic and immunohistochemical study of eight cases. Cancer. 1994;73:2481-2490.
76 Baghai-Wadji M., Sianati M., Nikpour H., Koochekpour S. Pleomorphic adenoma of the trachea in an 8 year-old boy: a case report. J Pediatr Surg. 2006;41:e23-e26. (August 2006)
77 Kamiyoshihara M., Ibe T., Takeyoshi I. Pleomorphic adenoma of the main bronchus in an adult treated using a wedge bronchiectomy. Gen Thorac Cardiovasc Surg. 2009;57:43-45.
78 Fitchett J., Luckraz H., Gibbs A., O’Keefe P. A rare case of primary pleomorphic adenoma in main bronchus. Ann Thorac Surg. 2008;86:1025-1026.
79 Jin H.Y., Park T.S. Pulmonary pleomorphic adenoma: report of a rare case. Korean J Intern Med. 2007;22:122-124.
80 Kourda J., Ismail O., Smati B.H., Ayadi A., Kilani T., El-Mezni F. Benign myoepithelioma of the lung—a case report and review of the literature. Cases J. 2010;13:25.
81 Angelov A., Dikranian K., Trosheva M. Immunomorphological characteristics of pleomorphic adenoma of salivary glands. Bull Group Int Rech Sci Stomatol Odontol. 1996;39:67-75.
82 Ang K.L., Dhannapuneni V.R., Morgan W.E., Soomro I.N. Primary pulmonary pleomorphic adenoma: an immunohistochemical study and review of the literature. Arch Pathol Lab Med. 2003;127:621-622.
83 Takeuchi E., Shimizu E., Sano N., et al. A case of pleomorphic adenoma of the lung with multiple distant metastases—observations on its oncogene and tumor suppressor gene expression. Anticancer Res. 1998;18:2015-2020.
84 Fechner R.E., Bentnick B.R. Ultrastructure of bronchial oncocytoma. Cancer. 1973;31:1451-1457.
85 Nielsen A.L. Malignant bronchial oncocytoma: a case report and review of the literature. Hum Pathol. 1985;16:852-854.
86 Santos-Briz A., Jenron J., Sastre R., Romero L., Valle A. Oncocytoma of the lung. Cancer. 1977;40:1330-1336.
87 Burrah R., Kini U., Correa M., Srirangapatna S. pulmonary oncocytoma: a rare case. Asian Cardiovasc Thorac Ann. 2006;14:e113-e114.
88 Laforga J.B., Aranda F.I. Multicentric oncocytoma of the lung diagnosed by fine-needle aspiration. Diagn Cytopathol. 1999;21:51-54.
89 Tashiro Y., Iwata Y., Nabae T., Manage H. Pulmonary oncocytoma: report of a case in conjunction with an immunohistochemical and ultrastructural study. Pathol Int. 1995;45:448-451.
90 De Jesus M.G., Poon T.P., Chung K.Y. Pulmonary oncocytoma. N Y State J Med. 1989;89:477-480.
91 Tesluk H., Dajee A. Pulmonary oncocytoma. J Surg Oncol. 1985;29:173-175.
92 Fernandez M.A., Nyssen J. Oncocytoma of the lung. Can J Surg. 1982;25:332-333.
93 Black W.C.III. Pulmonary oncocytoma. Cancer. 1969;23:1347-1357.
94 Ritter J.H., Nappi O. Oxyphilic proliferations of the respiratory tract and paranasal sinuses. Semin Diagn Pathol. 1999;16:105-116.
95 Arora R., Mathur S.R., Aron M., et al. Oncocytic carcinoid tumor of the lung: a case report of diagnostic pitfall in filter membrane preparation of bronchial washings. Acta Cytol. 2007;51:907-910.
96 Zhou C.X., Gao Y. Oncocytoma of the salivary glands: a clinicopathologic and immunohistochemical study. Oral Oncol. 2009;45:e232-e238. (September 30, 2009)
97 Mete O., Kilicasian I., Gulluoglu M.G., Uysal V. Can renal oncocytoma be differentiated from its renal mimics? The utility of anti-mitochondrial, caveolin-1, CD63, and cytokeratin 14 antibodies in the differential diagnosis. Virchows Arch. 2005;447:938-946.
98 Bartley T.D., Arean V.M. Intrapulmonary neurogenic tumors. J Thorac Cardiovasc Surg. 1965;50:114-123.
99 Roviaro G., Montorsi M., Varoli F., Binda R., Cecchetto A. Primary pulmonary tumors of neurogenic origin. Thorax. 1983;38:942-945.
100 Silverman J.F., Leffers B.R., Kay S. Primary pulmonary neurilemmoma: report of a case with ultrastructural examination. Arch Pathol Lab Med. 1976;100:644-648.
101 Yamakawa H. Intrapulmonary schwannoma: a case report. J Jpn Assoc Chest Surg. 1993;7:165-169.
102 Sugita M., Fujimura S., Hasumi T., Kondo T., Sagawa M. Sleeve superior segmentectomy of the right lower lobe for endobronchial neurinoma: report of a case. Respiration. 1996;63:191-194.
103 McCluggage W.G., Bharucha H. Primary pulmonary tumors of nerve sheath origin. Histopathology. 1995;26:247-254.
104 Lin Y.C., Lin M.C., Chen T.C., Huang C.C., Lee C.H. Tracheal neurilemmoma mimicking bronchial asthma—a dilemma of difficult diagnosis: case report. Changgeng Yi Xue Za Zhi. 1999;22:525-529.
105 Tsukada H., Hsada H., Kojima K., Yamata N. Bronchial wall schwannoma removed by sleeve resection of the right mainstem bronchus without lung resection. J Cardiovasc Surg. 1998;39:511-513.
106 Ashkan K., Casey A.T. Pulmonary apex schwannoma. J Neurol Neurosurg Psych. 1997;63:719.
107 Bosch X., Ramirez J., Font J., et al. Primary intrapulmonary benign schwannoma: a case with ultrastructural and immunohistochemical confirmation. Eur Respir J. 1990;3:234-237.
108 Muhrer K.H., Fischer H.P. Primary pulmonary neurilemmoma. Thorac Cardiovasc Surg. 1983;31:313-316.
109 Imaizumi M., Takahashi T., Niimi T., et al. A case of primary intrapulmonary neurilemmoma and review of the literature. Jpn J Surg. 1989;19:740-746.
110 Feldhaus R.J., Anene C., Bogard P. A rare endobronchial neurilemmoma (schwannoma). Chest. 1989;95:461-462.
111 Nesbitt J.C., Vega D.M., Burke T., Mackay B. Cellular schwannoma of the bronchus. Ultrastruct Pathol. 1996;20:349-354.
112 Noah M.A., Gorecha M., Firmin R.K. Primary benign bronchial schwannoma presenting as asthma. J Asthma. 2009;46:856-857.
113 Onal M., Ernam D., Atikcan S., Memis L. Endobronchial schwannoma with massive hemoptysis. Tuberk Toraks. 2009;57:89-92.
114 Unger P.D., Geller S.A., Anderson P.J. Pulmonary lesions in a patient with neurofibromatosis. Arch Pathol Lab Med. 1984;108:654-657.
115 Bacha E.A., Wright C.D., Grillo H.C., et al. Surgical treatment of primary pulmonary sarcomas. Eur J Cardiothorac Surg. 1999;15:456-460.
116 Moran C.A., Suster S., Koss M.N. Primary malignant “triton” tumor of the lung. Histopathology. 1997;30:140-144.
117 Attanoos R.L., Appleton M.A., Gibbs A.R. Primary sarcomas of the lung: a clinicopathological and immunohistochemical study of 14 cases. Histopathology. 1996;29:29-36.
118 Simansky D.A., Aviel-Ronen S., Reder I., et al. Psammomatous melanotic schwannoma: presentation of a rare primary lung tumor. Ann Thorac Surg. 2000;70:671-672.
119 Abrikossoff A. Uber Myome ausgehend von der quergestreiften willkurlichen Muskulatur. Virchows Arch Pathol Anat Physiol Klin Med. 1926;260:215-233.
120 Lack E.E., Worsham G.F., Callihan M.D., et al. Granular cell tumor: a clinicopathologic study of 110 patients. J Surg Oncol. 1980;13:301-316.
121 Alvarez-Fernandez E., Carretero-Albinana L. Bronchial granular cell tumor: presentation of three cases with tissue culture and ultrastructural study. Arch Pathol Lab Med. 1987;111:1065-1069.
122 DeClerq D., Van der Straten M., Roels H. Granular cell myoblastoma of the bronchus. Eur J Respir Dis. 1983;64:72-76.
123 Hurwitz S.S., Conlan A.A., Gritzman M.C., Krut L.H. Granular cell myoblastoma. Thorax. 1982;37:392-393.
124 Hosaka T., Suzuki S., Niikawa H., et al. A rare case of a pulmonary granular cell tumor presenting as a coin lesion. Jpn J Thorac Cardiovasc Surg. 2003;51:107-109.
125 Abdulhamid I., Rabah R. Granular cell tumor of the bronchus. Pediatr Pulmonol. 2000;30:425-428.
126 Husain M., Nguyen G.K. Cytopathology of granular cell tumor of the lung. Diagn Cytopathol. 2000;23:294-295.
127 Al-Ghamdi A.M., Flint J.D., Muller N.L., Stewart K.C. Hilar pulmonary granular cell tumor: a case report and review of the literature. Ann Diagn Pathol. 2002;4:245-251.
128 Scala R., Naldi M., Fabianelli F., et al. Endobronchial granular cell tumor. Monaldi Arch Chest Dis. 1999;54:404-406.
129 Thomas de Montpreville V., Dulmet E.M. Granular cell tumors of the lower respiratory tract. Histopathology. 1995;27:257-262.
130 Deavers M., Guinee D., Koss M.N., Travis W.D. Granular cell tumors of the lung: clinicopathologic study of 20 cases. Am J Surg Pathol. 1995;19:627-635.
131 Hernandez O.G., Haponik E.F., Summer W.R. Granular cell tumor of the bronchus: bronchoscopic and clinical features. Thorax. 1986;41:927-931.
132 Mullen C.V.Jr, Hewan-Lowe K., Gilman M.J. Massive hemoptysis associated with granular cell tumor of the bronchus. South Med J. 1983;76:1452.
133 Lack E.E., Harris G.B., Eraklis A.J., Vawter G.F. Primary bronchial tumors in childhood: a clinicopathologic study of six cases. Cancer. 1983;51:492-497.
134 Guillou L., Gloor E., Anani P.A., Kaelin R. Bronchial granular cell tumor: report of a case with preoperative cytologic diagnosis on bronchial brushings and immunohistochemical studies. Acta Cytol. 1991;35:375-380.
135 Majmudar B., Thomas J., Gorelkin L., Symbas P.N. Respiratory obstruction caused by a multicentric granular cell tumor of the laryngotracheobronchial tree. Hum Pathol. 1981;12:283-286.
136 Oparah S.S., Subramanian V.A. Granular cell myoblastoma of the bronchus: report of 2 cases and review of the literature. Ann Thorac Surg. 1976;22:199-202.
137 Robinson J.M., Knoll R., Henry D.A. Intrathoracic giant cell myoblastoma. South Med J. 1988;81:1453-1457.
138 Schulster P.L., Khan F.A., Azueta V. Asymptomatic pulmonary granular cell tumor presenting as a coin lesion. Chest. 1975;68:256-258.
139 Chen K.T.K. Cytology of bronchial benign granular cell tumor. Acta Cytol. 1991;35:381-384.
140 Miyake M., Tateishi U., Maeda T., Arai Y., Hasegawa T., Sugimura K. Bronchial granular cell tumor: a case presenting secondary obstructive changes on CT. Radiat Med. 2006;24:154-157.
141 Fang H.Y., Wu C.Y., Huang H.J., Lin Y.M. Granular cell tumor of the lung. Lungs. 2010. Jan. 12 (E-pub ahead of print)
142 Muhammed A.A., Sikka P., Dhillon R.S., Gibbons W.J., Ahmed A. Coexisting granular cell tumor and adenocarcinoma of the lung: a case report and review of the literature. Respir Care. 2001;46:702-704.
143 Cutlan R.T., Eltorky M. Pulmonary granular cell tumor coexisting with bronchogenic carcinoma. Ann Diagn Pathol. 2001;5:74-79.
144 Althausen A.M., Kowalski D.P., Ludwig M.E., Curry S.L., Greene J.F. Granular cell tumors: a new clinically important histologic finding. Gynecol Oncol. 2000;77:310-313.
145 Elmberger P.G., Skold C.M., Collins B.T. Fine needle aspiration biopsy of intrabronchial granular cell tumor. Acta Cytol. 2005;49:223-224.
146 Sobel H.J., Marquet E., Avrin E., Schwarz R. Granular cell myoblastoma: an electron microscopic and cytochemical study illustrating the genesis of granules and aging of myoblastoma cells. Am J Pathol. 1971;65:59-78.
147 Miettinen M., Lehtonen E., Lehtola H., et al. Histogenesis of granular cell tumor: an immunohistochemical and ultrastructural study. J Pathol. 1984;142:221-229.
148 Mazur M., Shultz J.J., Myers J.L. Granular cell tumor: immunohistochemical analysis of 21 benign tumors and one malignant tumor. Arch Pathol Lab Med. 1990;114:692-696.
149 Liu Z., Mira J.L., Vu H. Diagnosis of malignant granular cell tumor by fine needle aspiration cytology. Acta Cytol. 2001;45:1011-1021.
150 Ordonez N.G., Mackay B. Granular cell tumor: a review of the pathology and histogenesis. Ultrastruct Pathol. 1999;23:207-222.
151 Fine S.W., Li M. Expression of calretinin and the alpha-subunit of inhibin in granular cell tumors. Am J Clin Pathol. 2003;119:259-264.
152 Nappi O., Ferrara G., Wick M.R. Neoplasms composed of eosinophilic polygonal cells: an overview with consideration of different cytomorphologic patterns. Semin Diagn Pathol. 1999;16:82-90.
153 Franzblau M.J., Manwaring M., Plumhof C., Listrom J.B., Burgdorf W.H.C. Metastatic breast carcinoma mimicking granular cell tumor. J Cutan Pathol. 1989;16:218-221.
154 Reuter V.E. Renal tumors exhibiting granular cytoplasm. Semin Diagn Pathol. 1999;16:135-145.
155 Ogino S., Al-Kaisi N., Abdul-Karim F.W. Cytopathology of oncocytic carcinoid tumor of the lung mimicking granular cell tumor: a case report. Acta Cytol. 2000;44:247-250.
156 Ritter J.H., Nappi O. Oxyphilic proliferations of the respiratory tract and paranasal sinuses. Semin Diagn Pathol. 1999;16:105-116.
157 Yousem S.A., Hochholzer L. Alveolar adenoma. Hum Pathol. 1986;17:1066-1071.
158 Sak S.D., Koseoglu R.D., Demirag F., Akbulut H., Gungor A. Alveolar adenoma of the lung: immunohistochemical and flow-cytometric characteristics of two new cases and a review of the literature. APMIS. 2007;115:1443-1449.
159 Fujimoto K., Muller N.L., Sadohara J., et al. Alveolar adenoma of the lung: computed tomography and magnetic resonance imaging findings. J Thorac Imaging. 2002;17:163-166.
160 Hartman M.S., Epstein D.M., Geyer S.J., Keenan R.J. Alveolar adenoma. Ann Thorac Surg. 2004;78:1842-1843.
161 Cavazza A., Paci M., De Marco L., et al. Alveolar adenoma of the lung: a clinicopathologic, immunohistochemical, and molecular study of an unusuasl case. Int J Surg Pathol. 2004;12:155-159.
162 Semeraro D., Gibbs A.R. Pulmonary adenoma: a variant of sclerosing hemangioma of lung? J Clin Pathol. 1989;42:1222-1223.
163 Oliveira P., Moura-Nunes J.F., Clode A.L., da Costa J.D., Almeida M.O. Alveolar adenoma of the lung: further characterization of this uncommon tumor. Virchows Arch. 1996;429:101-108.
164 Burke L.M., Rush W.I., Khoor A., et al. Alveolar adenoma: a histochemical, immunohistochemical, and ultrastructural analysis of 17 cases. Hum Pathol. 1999;30:158-167.
165 Al-Hilli F. Lymphangioma (of alveolar adenoma?) of the lung. Histopathology. 1987;11:979-986.
166 Wada A., Tateishi R., Terazawa T., Matsuda M., Hattori S. Lymphangioma of the lung. Arch Pathol. 1974;98:211-213.
167 Miller R.R. Bronchioloalveolar cell adenomas. Am J Surg Pathol. 1990;14:904-912.
168 Glaab R., Turina M., Achermann E., Maurer R., Went P., Schob G. Alveolar adenoma—a rare pulmonary mass: case report and review of the literature. Zenbralbl Chir. 2009;134:478-480.
169 Nakamura H., Adachi Y., Arai T., et al. A small alveolar adenoma resected by thoracoscopic surgery. Ann Thorac Surg. 2009;87:956-957.
170 Hegg C.A., Flint A., Singh G. Papillary adenoma of the lung. Am J Clin Pathol. 1992;97:393-397.
171 Fukuda T., Ohnishi Y., Kanai I., et al. Papillary adenoma of the lung: histological and ultrastructural findings in two cases. Acta Pathol Jpn. 1992;42:56-61.
172 Mori M., Chiba R., Tezuka F., et al. Papillary adenoma of type II pneumocytes might have malignant potential. Virchows Arch. 1996;428:195-200.
173 Yamamoto T., Horiguchi H., Shibagaki T., Kamma H., Ogata T., Mitsui K. Encapsulated type II pneumocyte adenoma: a case report & review of the literature. Respiration. 1993;60:373-377.
174 Noguchi M., Kodama T., Shimosato Y., et al. Papillary adenoma of type 2 pneumocytes. Am J Surg Pathol. 1986;10:134-139.
175 Fine G., Chang C.H. Adenoma of type 2 pneumocytes with oncocytic features. Arch Pathol Lab Med. 1991;115:797-801.
176 Kurotaki H., Kamata Y., Kimura M., Nagai K. Multiple papillary adenomas of type II pneumocytes found in a 13 year old boy with von Recklinghausen’s disease. Virchows Arch. 1993;423:319-322.
177 Spencer H., Dail D.H., Arneaud J. Noninvasive bronchial epithelial papillary tumors. Cancer. 1980;45:1486-1497.
178 Sanchez-Jimenez J., Ballester-Martinez A., Lodo-Besse J., et al. Papillary adenoma of type II pneumocytes. Pediatr Pulmonol. 1994;17:396-400.
179 Gesierich W., Diwersy C., Leinsinger G., et al. Papillary adenoma of type II pneumocytes as a rare differential diagnosis of a solitary pulmonary nodule. Pneumologie. 2007;61:697-699.
180 Papla B. Papillary adenoma of the lung. Pol J Pathol. 2009;60:49-51.
181 Fantone J.C., Geisinger K.R., Appelman H.D. Papillary adenoma of the lung with lamellar and electron-dense granules: an ultrastructural study. Cancer. 1982;50:2839-2844.
182 Dessy E., Braidotti P., Del Curto B., et al. Peripheral papillary tumor of type II pneumocytes: a rare neoplasm of undetermined malignant potential. Virchows Arch. 2000;436:289-295.
183 Tomashefski J.F. Benign endobronchial mesenchymal tumors: their relationship to parenchymal pulmonary hamartomas. Am J Surg Pathol. 1982;6:531-540.
184 Yellin A., Roserman Y., Lieberman Y. Review of smooth muscle tumors of the lower respiratory tract. Br J Dis Chest. 1984;78:337-351.
185 White S.H., Ibrahim N.B.N., Forrester-Wood C.P., Jeyasingham R. Leiomyomas of the lower respiratory tract. Thorax. 1985;40:306-311.
186 Orlowski T.M., Stasiak K., Kolodziej J. Leiomyoma of the lung. J Thorac Cardiovasc Surg. 1978;76:257-261.
187 Van den Bosch J.M., Wagenaar S.S., Corrin B., et al. Mesenchymoma of the lung (so-called hamartoma): a review of 154 parenchymal and endobronchial cases. Thorax. 1987;42:790-793.
188 Gal A.A., Brooks J.J., Pietra G.G. Leiomyomatous lung neoplasms: a clinical, histologic, and immunohistochemical study. Mod Pathol. 1989;2:209-216.
189 Van Way C.W., McCracken R.L., Carlisle B.B. Leiomyoma of the lower respiratory tract. Ann Thorac Surg. 1968;6:273-276.
190 Taylor T.L., Miller D.R. Leiomyoma of the bronchus. J Thorac Cardiovasc Surg. 1969;57:245-248.
191 Yamada H., Katoh O., Yamaguchi T., Natsuaki M., Itoh T. Intrabronchial leiomyoma treated by localized resection vis bronchotomy and bronchoplasty. Chest. 1987;91:283-284.
192 Shahian D.M., McEnahy M.R. Complete endobronchial resection of leiomyoma of the bronchus. J Thorac Cardiovasc Surg. 1979;77:87-91.
193 Kim K.H., Suh J.S., Han W.S. Leiomyoma of the bronchus treated by endoscopic resection. Ann Thorac Surg. 1993;56:1164-1166.
194 Mouveroux F.A., Bourcereau J., Fressinaud C., Bourras P. Bronchial leiomyoma: report of a case successfully treated by Nd-YAG laser. Thorac Cardiovasc Surg. 1988;95:536-537.
195 Sivalingam B., Somani K., Sreenivasan L., Collins F.J. Endobronchial leiomyoma: successful resection by sleeve lobectomy. Int J Thorac Cardiovasc Surg, www.ispub.com/ispub/ijtcvs/volume_7_number_2/endobronchial_leiomyoma_successful_resection_by_sleeve)lobectomy.
196 Silverman J.F., Kay S. Multiple pulmonary leiomyomatous hamartomas: report of case with ultrastructural examination. Cancer. 1976;38:1199-1204.
197 Hull M.T., Gonzalez-Crussi F., Grosfeld J.L. Multiple pulmonary fibroleiomyomatous hamartomata in childhood. J Pediatr Surg. 1979;14:428-431.
198 Itoh H., Yanagi M., Setoyama T., et al. Solitary fibroleiomyomatous hamartoma of the lung in a patient without a preexisting smooth muscle tumor. Pathol Int. 2001;51:661-665.
199 Shmookler B.M., Lauer D.H. Retroperitoneal leiomyosarcoma: a clinicopathologic analysis of 36 cases. Am J Surg Pathol. 1983;7:269-280.
200 Mackay B., Legha S.S. Coin lesion of the lung in a 19 year old male. Ultrastruct Pathol. 1981;2:289-294.
201 Alt B., Huffer W.E., Belchis D.A. A vascular lesion with smooth muscle differentiation presenting as a coin lesion in the lung: glomus tumor versus hemangiopericytoma. Am J Clin Pathol. 1983;80:765-771.
202 Fabich D.R., Hafez G.R. Glomangioma of the trachea. Cancer. 1980;45:2337-2341.
203 Tang C., Toker C.K., Foris N.P., Trump B.F. Glomangioma of the lung. Am J Surg Pathol. 1978;2:103-109.
204 Gaertner E.M., Steinberg D.M., Huber M., et al. Pulmonary and mediastinal glomus tumors: report of five cases including a pulmonary glomangiosarcoma. A clinicopathologic study with literature review. Am J Surg Pathol. 2000;24:1105-1114.
205 Yilmaz A., Bayramgurler B., Aksoy F., et al. Pulmonary glomus tumor: a case initially diagnosed as carcinoid tumor. Respirology. 2002;7:369-371.
206 Shugart R.R., Soule E.H., Johnson E.W. Glomus tumors. Surg Gynecol Obstet. 1963;117:334-340.
207 Miettinen M., Paal E., Lasota J., Sobin L.H. Gastrointestinal glomus tumors: a clinicopathologic, immunohistochemical, and molecular genetic study of 32 cases. Am J Surg Pathol. 2002;26:301-311.
208 Littlefield J.B., Drash E.C. Myxoma of the lung. J Thorac Surg. 1959;37:745-749.
209 Pollak E.R., Naunheim K.S., Little A.G. Fibromyxoma of the trachea. Arch Pathol Lab Med. 1985;109:926-929.
210 Butler C., Kleinerman J. Pulmonary hamartoma. Arch Pathol. 1969;88:584-592.
211 Gjevre J.A., Myers J.L., Prakash U.B.S. Pulmonary hamartomas. Mayo Clin Proc. 1996;71:14-20.
212 Hamper U.M., Khouri N.F., Stitik F.P., et al. Pulmonary hamartoma: diagnosis by transthoracic needle-aspiration biopsy. Radiology. 1985;155:15-18.
213 Wiatrowska B.A., Yazdi H.M., Matzinger F.R., MacDonald L.L. Fine needle aspiration biopsy of pulmonary hamartomas: radiologic, cytologic, and immunocytochemical study of 15 cases. Acta Cytol. 1995;39:1167-1174.
214 Carney J.A. The triad of gastric epithelioid leiomyosarcoma, functioning extra-adrenal paraganglioma, and pulmonary chondroma. Cancer. 1979;43:374-382.
215 Wick M.R., Ruebner B.H., Carney J.A. Gastric tumors in patients with pulmonary chondroma or extra-adrenal paraganglioma: an ultrastructural study. Arch Pathol Lab Med. 1981;105:527-531.
216 Rodriguez F.J., Aubry M.C., Tazelaar H.D., Slezak J., Carney J.A. Pulmonary chondroma: a tumor associated with Carney triad and different from pulmonary hamartoma. Am J Surg Pathol. 2007;31:1844-1853.
217 Abrahams N.A., Colby T.V., Pearl R.H., et al. Pulmonary hemangiomas of infancy and childhood: report of two cases and review of the literature. Pediatr Dev Pathol. 2002;5:283-292.
218 Fugo K., Matsuno Y., Okamoto K., et al. Solitary capillary hemangioma of the lung: report of 2 resected cases detected by high-resolution CT. Am J Surg Pathol. 2006;30:750-753.
219 Ritter J.H., Mills S.E., Nappi O., Wick M.R. Angiosarcoma-like neoplasms of epithelial organs: true endothelial tumors or variants of carcinoma? Semin Diagn Pathol. 1995;12:270-282.
220 Almagro P., Julia J., Sanjaume M., et al. Pulmonary capillary hemangiomatosis associated with primary pulmonary hypertension: report of 2 new cases and review of 35 cases from the literature. Medicine. 2002;81:417-424.
221 Havlik D.M., Massie L.W., Williams W.L., Crooks L.A. Pulmonary capillary hemangiomatosis-like foci: an autopsy study of 8 cases. Am J Clin Pathol. 2000;113:655-662.
222 Eastridge C.F., Young J.M., Steplock A.L. Endobronchial lipoma. South Med J. 1984;77:759-761.
223 Politis J., Funahashi A., Gehlsen J.A., et al. Intrathoracic lipomas: report of three cases and review of the literature with emphasis on endobronchial lipoma. J Thorac Cardiovasc Surg. 1979;77:550-556.
224 Kamiyoshihara M., Sakata K., Ohtani Y., et al. Endobronchial lipoma accompanied with primary lung cancer: report of a case. Surg Today. 2002;32:402-405.
225 Moran C.A., Suster S., Koss M.N. Endobronchial lipomas: a clinicopathologic study of four cases. Mod Pathol. 1994;7:212-214.
226 Muraoka M., Oka T., Akamine S., et al. Endobronchial lipoma: review of 64 cases reported in Japan. Chest. 2003;123:293-296.
227 Gaerte S.C., Meyer C.A., Winer-Muram H.T., Tarver R.D., Conces D.J.Jr. Fat-containing lesions of the chest. Radiographics. 2002;22(suppl):S61-S78.
228 Solli P., Rossi G., Carbagnani P., et al. Pulmonary abnormalities in Cowden’s disease. J Cardiovasc Surg. 1998;40:753-755.
229 Irani F., Kumar B., Reddy P., Narwal-Chadha R., Kasmani R., Tita J. An endobronchial lipoma mimicking asthma and malignancy. Prim Care Respir J. 2009. Dec. 17 pii: pcrj-2009-04-0033.R1 10.4104/pcrj.2009.00070 (E-publication)
230 Sekine I., Kodama T., Yokose T., et al. Rare pulmonary tumors—a review of 32 cases. Oncology. 1998;55:431-434.
231 Matsuba K., Saito T., Ando K., Shirakusa T. Atypical lipoma of the lung. Thorax. 1991;46:685.
232 Bridge J.A., Roberts C.A., Degenhardt J., et al. Low-level chromosome 12 amplification in a primary lipoma of the lung: evidence for a pathogenetic relationship with common adipose tissue tumors. Arch Pathol Lab Med. 1998;122:187-190.
233 Hirata J., Reshad K., Itoi K., Muro K., Akiyama J. Lipomas of the peripheral lung—a case report and review of the literature. Thorac Cardiovasc Surg. 1989;37:385-387.
234 Erkilic S., Kocer N.E., Tuncozgur B. Peripheral intrapulmonary lipoma: a case report. Acta Chir Belg. 2007;107:700-702.
235 Shimoyama T., Kimura B. Peripheral intrapulmonary lipoma: report of a case. Jpn J Thorac Surg. 2009;62:1186-1189.
236 Coffin C.M. Lipoblastoma: an embryonal tumor of soft tissue related to organogenesis. Semin Diagn Pathol. 1994;11:98-103.
237 Kanu A., Oermann C.M., Malicki D., Wagner M., Langston C. Pulmonary lipoblastoma in an 18-month-old child: a unique tumor in children. Pediatr Pulmonol. 2002;34:150-154.
238 Mathew J., Sen S., Chandi S.M., et al. Pulmonary lipoblastoma: a case report. Pediatr Surg Int. 2001;17:543-544.
239 Achir A., Ouadnouni Y., Smahi M., Bouchikh M., Msougar Y., Benosman A. Primary pulmonary liposarcoma—a case report. Thorac Cardiovasc Surg. 2009;57:119-120.
240 Loddenkempter C., Perez-Canto A., Leschber G., Stein H. Primary dedifferentiated liposarcoma of the lung. Histopathology. 2005;46:710-712.
241 Kitizawa R., Kitazawa S., Nishimura Y., Kondo T., Obayashi C. Lung carcinosarcoma with liposarcoma element: autopsy case. Pathol Int. 2006;56:449-452.
242 Hicks J., Dilley A., Patel D., et al. Lipoblastoma and lipoblastomatosis in infancy and childhood: histopathologic, ultrastructural, and cytogenetic features. Ultrastruct Pathol. 2001;25:321-333.
243 Nicolas M., Moran C.A., Suster S. Pulmonary metastasis from liposarcoma: a clinicopathologic and immunohistochemical study of 24 cases. Am J Clin Pathol. 2005;123:265-275.
244 Meis-Kindblom J.M., Sjogren H., Kindblom L.G., et al. Cytogenetic and molecular genetic analyses of liposarcoma and its soft tissue simulators: recognition of new variants and differential diagnosis. Virchows Arch. 2001;439:141-151.
245 Tallini G., Vanni R., Manfioletti G., et al. HMGI-C and HMGI(Y) immunoreactivity correlates with cytogenetic abnormalities in lipomas, pulmonary chondroid hamartomas, endometrial polyps, and uterine leiomyomas and is compatible with rearrangement of the HMGI-C and HMGI(Y) genes. Lab Invest. 2000;80:359-369.
246 Johansson M., Dietrich C., Mandahl N., et al. Recombinations of chromosomal bands 6p21 and 14q24 characterize pulmonary hamartomas. Br J Cancer. 1993;67:1236-1241.
247 Pea M., Bonetti F., Zamboni G., et al. Clear cell tumor and angiomyolipoma. Am J Surg Pathol. 1991;15:199-202.
248 Bacchi C.E., Bonetti F., Pea M., Martignoni G., Gown A.M. HMB-45: a review. Appl Immunohistochem Mol Morphol. 1996;4:73-85.
249 Bonetti F., Pea M., Martignoni G., et al. Clear cell (“sugar”) tumor of the lung is a lesion strictly related to angiomyolipoma—the concept of a family of lesions characterized by the presence of the perivascular epithelioid cell (PEC). Pathology. 1994;26:230-236.
250 Hornick J.L., Fletcher C.D.M. PEComa: what do we know so far? Histopathology. 2006;48:75-82.
251 Folpe A.L., McKenney J.K., Li Z., Smith S.J., Weiss S.W. Clear cell myomelanocytic tumor of the thigh: report of a unique case. Am J Surg Pathol. 2002;26:809-812.
252 Garcia T.R., Mestre-de-Juan M.J. Angiomyolipoma of the liver and lung: a case explained by the presence of perivascular epithelioid cells. Pathol Res Pract. 2002;198:363-367.
253 Wu K., Tazelaar H.D. Pulmonary angiomyolipoma and multifocal micronodular pneumocyte hyperplasia associated with tuberous sclerosis. Hum Pathol. 1999;30:1266-1268.
254 Papla B., Malinowski E. Angiomyolipoma of the lung. Pol J Pathol. 1999;50:47-50.
255 Ito M., Sugamura Y., Ikari H., Sekine I. Angiomyolipoma of the lung. Arch Pathol Lab Med. 1998;122:1023-1025.
256 Guinee D.G.Jr, Thornberry D.S., Azumi N., et al. Unique pulmonary presentation of an angiomyolipoma: analysis of clinical, radiographic, and histopathologic features. Am J Surg Pathol. 1995;19:476-480.
257 Marcheix B., Brouchet L., Lamarche Y., et al. Pulmonary angiomyolipoma. Ann Thorac Surg. 2006;82:1504-1506.
258 Chu S.C., Horiba K., Usuki J., et al. Comprehensive evaluation of 35 patients with lymphangioleiomyomatosis. Chest. 1999;115:1041-1052.
259 Kitaichi M., Nishimura K., Itoh H., Izumi T. Pulmonary lymphangioleiomyomatosis: a report of 46 patients including a clinicopathologic study of prognostic factors. Am J Respir Crit Care Med. 1995;151:527-533.
260 Matsumoto S., Nishioka T., Akiyama T. Renal angiomyolipoma associated with micronodular pneumocyte hyperplasia of the lung with tuberous sclerosis. Int J Urol. 2001;8:242-244.
261 Takahashi N., Kitihara R., Hishimoto Y., et al. Malignant transformation of renal angiomyolipoma. Int J Urol. 2003;10:271-273.
262 Adachi Y., Horie Y., Kitamura Y., et al. CD1a expression in PEComas. Pathol Int. 2008;58:169-173.
263 Gee W.F., Chikos P.M., Greaves J.P., Ikemoto N., Tremann J.A. Adrenal myelolipoma. Urology. 1975;5:562-566.
264 Singla A.K., Kechejian G., Lopez M.J. Giant presacral myelolipoma. Am Surg. 2003;69:334-338.
265 Hunter S.B., Schemankewitz E.H., Patterson C., Varma V.A. Extraadrenal myelolipoma: a report of two cases. Am J Clin Pathol. 1992;97:402-404.
266 Sabate C.J., Shahian D.M. Pulmonary myelolipoma. Ann Thorac Surg. 2002;74:573-575.
267 Piccinini L., Barbolini G. A case of lung myelolipomatosis in a patient with bronchial carcinoid. Panminerva Med. 1999;41:175-178.
268 Ziolkowski P., Muszczynska-Bernhard B., Dziegiel P. Myelolipoma: the report of two cases in a pulmonary location. Pol J Pathol. 1996;47:141-142.
269 Sato K., Ueda Y., Katsuda S., Tsuchihara K. Myelolipoma of the lung: a case report and brief review. J Clin Pathol. 2007;60:728-730.
270 Quigley J.C., Hart W.R. Adenomatoid tumors of the uterus. Am J Clin Pathol. 1981;76:627-635.
271 Srigley J.R., Hartwick R.W. Tumors and cysts of the paratesticular region. Pathol Annu. 1990;25:51-108.
272 Kaplan M.A., Tazelaar H.D., Hayashi T., Schroer K.R., Travis W.D. Adenomatoid tumors of the pleura. Am J Surg Pathol. 1996;20:1219-1223.
273 Handra-Luca A., Couvelard A., Abd-Alsamad I., et al. Adenomatoid tumor of the pleura: case report. Ann Pathol. 2000;20:369-372.
274 Umezu H., Kuwata K., Ebe Y., et al. Microcystic variant of localized malignant mesothelioma accompanying an adenomatoid tumor-like lesion. Pathol Int. 2002;52:416-422.
275 Minato H., Nojima T., Kurose N., Kinoshita E. Adenomatoid tumor of the pleura. Pathol Int. 2009;59:567-571.
276 Pinkard N.B., Wilson R.W., Lawless N., et al. Calcifying fibrous pseudotumor of the pleura: a report of three cases of a newly described entity involving the pleura. Am J Clin Pathol. 1996;105:189-194.
277 Murray J.G., Pinkard N.B. Calcifying fibrous pseudotumor of the pleura: radiologic features in three cases. J Comput Assist Tomogr. 1996;20:763-765.
278 Hainaut P., Lesage V., Weynand B., Coche E., Noirhomme P. Calcifying fibrous pseudotumor (CFPT): a patient presenting with multiple pleural lesions. Acta Clin Belg. 1999;54:162-164.
279 Cavazza A., Gelli M.C., Agostini L., et al. Calcified pseudotumor of the pleura: description of a case. Pathologica. 2002;94:201-205.
280 Rosenthal N.S., Abdul-Karim F.W. Childhood fibrous tumor with psammoma bodies: clinicopathologic features in two cases. Arch Pathol Lab Med. 1988;112:798-800.
281 Suh J.H., Shin O.R., Kim Y.H. Multiple calcifying fibrous pseudotumor of the pleura. J Thorac Oncol. 2008;3:1356-1358.
282 Shibata K., Yuki D., Sakata K. Multiple calcifying fibrous pseudotumors disseminated in the pleura. Ann Thorac Surg. 2008;85:e3-e5. (E-publication)
283 Soyer T., Ciftci A.O., Gucer S., Orhan D., Senocak M.E. Calcifying fibrous pseudotumor of lung: a previously-unreported entity. J Pediatr Surg. 2004;39:1729-1730.
284 Maeda T., Hirose T., Furuya K., Kameoka K. Calcifying fibrous pseudotumor: an ultrastructural study. Ultrastruct Pathol. 1999;23:189-192.
285 Hill K.A., Gonzalez-Crussi F., Chou P.M. Calcifying fibrous pseudotumor versus inflammatory myofibroblastic tumor: a histological and immunohistochemical comparison. Mod Pathol. 2001;14:784-790.
286 Nascimento A.F., Ruiz R., Hornick J.L., Fletcher C.D.M. Calcifying fibrous “pseudotumor”: clinicopathologic study of 15 cases and analysis of its relationship to inflammatory myofibroblastic tumor. Int J Surg Pathol. 2002;10:189-196.
287 Gibbs A.R. Smooth mucle tumors of the pleura. Histopathology. 1995;27:295-296.
288 Mochizuki H., Okada T., Yoshizawa H., Suzuki E., Gejyo F. A case of primary pleural leiomyoma. J Jpn Resp Soc. 2004;42:625-628.
289 Brunn H. Two interesting benign lung tumors of contradictory histopathology. J Thorac Cardiovasc Surg. 1939;9:119-131.
290 Umiker W.O., Iverson L. Postinflammatory “tumors” of the lung: report of four cases simulating xanthoma, fibroma, or plasma cell tumor. J Thorac Cardiovasc Surg. 1954;28:55-63.
291 Bahadori M., Liebow A.A. Plasma cell granulomas of the lung. Cancer. 1973;31:191-208.
292 Spencer H. The pulmonary plasma cell/histiocytoma complex. Histopathology. 1984;8:903-916.
293 Berardi R.S., Lee S.S., Chen H.P., et al. Inflammatory pseudotumors of the lung. Surg Gynecol Obstet. 1983;156:89-96.
294 Souid A.K., Ziemba M.C., Dubansky A.S., et al. Inflammatory myofibroblastic tumor in children. Cancer. 1993;72:2042-2048.
295 Wick M.R., Ritter J.H., Nappi O. Inflammatory sarcomatoid carcinoma of the lung: report of three cases with comparison with inflammatory pseudotumors in adult patients. Hum Pathol. 1995;26:1014-1021.
296 Su L.D., Atayde-Perez A., Sheldon S., Fletcher J.A., Weiss S.W. Inflammatory myofibroblastic tumor: cytogenetic evidence supporting a clonal origin. Mod Pathol. 1998;11:364-368.
297 Snyder C.S., Dell’Aquila M., Haghighi P., et al. Clonal changes in inflammatory pseudotumor of the lung: a case report. Cancer. 1995;76:1545-1549.
298 Yamamoto H., Oda Y., Saito T., et al. p53 mutation and MDM2 amplification in inflammatory myofibroblastic tumors. Histopathology. 2003;42:431-439.
299 Pettinato G., Manivel J.C., DeRosa N., et al. Inflammatory myofibroblastic tumor (plasma cell granuloma): a clinicopathologic study of 20 cases with immunohistochemical and ultrastructural observations. Am J Clin Pathol. 1990;94:538-546.
300 Meis J.M., Enzinger F.M. Inflammatory fibrosarcoma of the mesentery and retroperitoneum: a tumor closely simulating inflammatory pseudotumor. Am J Surg Pathol. 1991;15:1146-1156.
301 Coffin C.M., Dehner L.P., Meis-Kindblom J.M. Inflammatory myofibroblastic tumor, inflammatory fibrosarcoma, and related lesions: an historical review with differential diagnostic considerations. Semin Diagn Pathol. 1998;15:102-110.
302 Lioulias A., Misthos P., Neofotistos E., Legaki S. Primary myofibrosarcoma of the lung: a rare case with challenging diagnosis. J BUON. 2009;14:143-145.
303 Takeda S., Onishi Y., Kawamura T., Maeda H. Clinical spectrum of pulmonary inflammatory myofibroblastic tumor. Interact Cardiovasc Thorac Surg. 2008;7:629-633.
304 Matsubara O., Tan-Liu N.S., Kenney R.M., et al. Inflammatory pseudotumors of the lung: progression from organizing pneumonia to fibrous histiocytoma or to plasma cell granuloma in 32 cases. Hum Pathol. 1988;19:807-814.
305 Cessna M.H., Zhou H., Sanger W.G., et al. Expression of ALK1 and p80 in inflammatory myofibroblastic tumor and its mesenchymal mimics: a study of 135 cases. Mod Pathol. 2002;15:931-938.
306 Coffin C.M., Hornick J.L., Fletcher C.D.M. Inflammatory myofibroblastic tumor: comparison of clinicopathologic, histologic, and immunohistochemical features including ALK expression in atypical and aggressive cases. Am J Surg Pathol. 2007;31:509-520.
307 Yamamoto H., Yamaguchi H., Aishima S., et al. Inflammatory myofibroblastic tumor versus IgG4-related sclerosing disease and inflammatory pseudotumor: a comparative clinicopathologic study. Am J Surg Pathol. 2009;33:1330-1340.
308 Antic T., Kapur U., Vigneswaran W.T., Oshima K. Inflammatory sarcomatoid carcinoma: a case report and discussion of a malignant tumor with benign appearance. Arch Pathol Lab Med. 2005;129:1334-1337.
309 Hussong J.W., Brown M., Perkins S.L., Dehner L.P., Coffin C.M. Comparison of DNA ploidy, histologic, and immunohistochemical findings with clinical outcome in inflammatory myofibroblastic tumors. Mod Pathol. 1999;12:279-286.
310 Messineo A., Mognato G., D’Amore E.S., et al. Inflammatory pseudotumors of the lung in children: conservative or aggressive approach? Med Pediatr Oncol. 1998;31:100-104.
311 Liebow A.A., Hubbell D.S. Sclerosing hemangioma (histiocytoma, xanthoma) of the lung. Cancer. 1956;9:53-75.
312 Gross R.E., Wolbach S.B. Sclerosing hemangiomas: their relationship to dermatofibroma, histiocytoma, xanthoma, and to certain pigmented lesions of the skin. Am J Pathol. 1943;19:533-546.
313 Katzenstein A.L., Weise D., Fuilling K., Battifora H. So-called sclerosing hemangioma of the lung: evidence for mesothelial origin. Am J Surg Pathol. 1983;7:3-14.
314 Kennedy A. Sclerosing hemangioma” of the lung: an alternative view of its development. J Clin Pathol. 1973;26:792-799.
315 Hill G.S., Eggleston J.C. Electron microscopic study of so-called pulmonary “sclerosing hemangioma”: report of a case suggesting an epithelial origin. Cancer. 1972;30:1092-1106.
316 Kay S., Still W.J., Borochovitz D. Sclerosing hemangioma of the lung: an endothelial or epithelial neoplasm? Hum Pathol. 1977;8:468-474.
317 Spencer H., Nambu S. Sclerosing hemangiomas of the lung. Histopathology. 1986;10:477-487.
318 Keylock J.B., Galvin J.R., Franks T.J. Sclerosing hemangioma of the lung. Arch Pathol Lab Med. 2009;133:820-825.
319 Kalhor N., Staerkel G.A., Moran C.A. So-called sclerosing hemangioma of lung: current concepts. Ann Diagn Pathol. 2010;14:60-67.
320 Shimosato Y. Lung tumors of uncertain histogenesis. Semin Diagn Pathol. 1995;12:185-192.
321 Batinica S., Gunek G., Raos M., Jelasic D., Bogovic M. Sclerosing hemangioma of the lung in a 4-year-old child. Eur J Pediatr Surg. 2002;12:192-194.
322 Devouassoux-Shisheboran M., Hayashi T., Linnoila R., Koss M.N., Travis W.D. A clinicopathologic study of 100 cases of pulmonary sclerosing hemangioma with immunohistochemical studies: TTF-1 is expressed in both round and surface cells, suggesting an origin from primitive respiratory epithelium. Am J Surg Pathol. 2000;24:906-916.
323 Kuo K.T., Hsu W.H., Wu Y.C., Huang M.H., Li W.Y. Sclerosing hemangioma of the lung: an analysis of 44 cases. J Chin Med Assoc. 2003;66:33-38.
324 Miyagawa-Hayashino A., Tazelaar H.D., Langel D.J., Colby T.V. Pulmonary sclerosing hemangioma with lymph node metastases: report of 4 cases. Arch Pathol Lab Med. 2003;127:321-325.
325 Hayashi A., Takamori S., Mitsuoka M., et al. Unilateral progressive multiple sclerosing hemangioma in a young female successfully treated by pneumonectomy: report of a case. Int Surg. 2002;87:69-72.
326 Iyoda A., Baba M., Saitoh H., et al. Imprint cytologic features of pulmonary sclerosing hemangioma: comparison with well-differentiated papillary adenocarcinoma. Cancer. 2002;96:146-149.
327 Nam J.E., Ryu Y.H., Cho S.H., et al. Air-trapping zone surrounding sclerosing hemangioma of the lung. J Comput Assist Tomogr. 2002;26:358-361.
328 Yano M., Yamakawa Y., Kiriyama M., Hara M., Murase T. Sclerosing hemangioma with metastases to multiple nodal stations. Ann Thorac Surg. 2002;73:981-983.
329 Gal A.A., Nassar V.H., Miller J.I. Cytopathologic diagnosis of pulmonary sclerosing hemangioma. Diagn Cytopathol. 2002;26:163-166.
330 Ng W.K., Fu K.H., Wang E., Tang V. Sclerosing hemangioma of lung: a close cytologic mimicker of pulmonary adenocarcinoma. Diagn Cytopathol. 2001;25:316-320.
331 Illei P.B., Rosai J., Klimstra D.S. Expression of thyroid transcription factor-1 and other markers in sclerosing hemangioma of the lung. Arch Pathol Lab Med. 2001;125:1335-1339.
332 Chan A.C., Chan J.K.C. Pulmonary sclerosing hemangioma consistently expresses thyroid transcription factor-1 (TTF-1): a new clue to its histogenesis. Am J Surg Pathol. 2000;24:1531-1536.
333 Wani Y., Notohara K., Tsukayama C., Okumura N. Sclerosing hemangioma with florid endobronchial and endobronchiolar growth. Virchows Arch. 2007;450:221-223.
334 Guibaud L., Pracros J.P., Rode V., et al. Sclerosing hemangioma of the lung: radiological findings and pathological diagnosis. Pediatr Radiol. 1995;25(suppl 1):S207-S208.
335 Komatsu T., Fukuse T., Wada H., Sakurai T. Pulmonary sclerosing hemangioma with pulmonary metastasis. Thorac Cardiovasc Surg. 2006;54:348-349.
336 Katzenstein A.L., Gmelich J., Carrington C. Sclerosing hemangioma of the lung: a clinicopathologic study of 51 cases. Am J Surg Pathol. 1980;4:343-356.
337 Moran C.A., Zeren H., Koss M.N. Sclerosing hemangioma of the lung: granulomatous variant. Arch Pathol Lab Med. 1994;118:1028-1030.
338 Wojcik E., Sneige N., Lawrence D., Ordonez N.G. Fine needle aspiration cytology of sclerosing hemangioma of the lung: case report with immunohistochemical study. Diagn Cytopathol. 1993;9:304-309.
339 Gottschalk-Sabag S., Hadas-Halpern I., Glick T. Sclerosing hemangioma of lung mimicking carcinoma, diagnosed by fine needle aspiration (FNA) cytology. Cytopathology. 1995;6:115-120.
340 Krishnamurthy S.C., Naresh K.N., Soni M., Bhasin S.D. Sclerosing hemangioma of the lung: a potential source of error in fine needle aspiration cytology. Acta Cytol. 1995;38:111-112.
341 Islam S., Roustan-Delatour N.L., Salahdeen S.R., Mai K.T., Senterman M., Mokhtar G.A. Cytologic features of benign solitary pulmonary nodules with radiologic correlation and diagnostic pitfalls: a report of six cases. Acta Cytol. 2009;53:201-210.
342 Yousem S.A., Wick M.R., Singh G., et al. So-called sclerosing hemangiomas of lung: an immunohistochemical study supporting a respiratory epithelial origin. Am J Surg Pathol. 1988;12:582-590.
343 Satoh Y., Tsuchiya E., Weng S.Y., et al. Pulmonary sclerosing hemangioma of the lung: type II pneumocytoma by immunohistochemistry and immunoelectron microscopic studies. Cancer. 1989;64:1310-1317.
344 Alvarez-Fernandez B., Carretero-Albinana L., Menarquez-Palanca J. Sclerosing hemangioma of the lung: an immunohistochemical study of intermediate filaments and endothelial markers. Arch Pathol Lab Med. 1989;113:121-124.
345 Fukayama M., Mikoike M. So-called sclerosing hemangioma of the lung: an immunohistochemical, histochemical, and ultrastructural study. Acta Pathol Jpn. 1988;38:627-642.
346 Leong A.S.Y., Chan K.W., Seneviratne H.S. A morphological and immunohistochemical study of 25 cases of so-called sclerosing hemangioma of the lung. Histopathology. 1995;27:121-128.
347 Nagata N., Dairaku M., Sueishi K., Tanaka K. Sclerosing hemangioma of the lung: an epithelial tumor composed of immunohistochemically heterogeneous cells. Am J Clin Pathol. 1987;88:552-559.
348 Xu H.M., Li W.H., Hou N., et al. Neuroendocrine differentiation in 32 cases of so-called sclerosing hemangioma of the lung, identified by immunohistochemistry and ultrastructural study. Am J Surg Pathol. 1997;21:1013-1022.
349 Wu C.T., Chang Y.L., Lee Y.C. Expression of the estrogen receptor beta in 37 surgically treated pulmonary sclerosing hemangiomas in comparison with non-small-cell lung carcinomas. Hum Pathol. 2005;36:1108-1112.
350 Wei S., Tian J., Song X., Chen Y. Recurrence of pulmonary sclerosing hemangioma. Thorac Cardiovasc Surg. 2008;56:120-122.
351 Tanaka I., Inoue M., Matsui Y., et al. A case of pneumocytoma (so-called sclerosing hemangioma) with lymph node metastasis. Jpn J Clin Oncol. 1986;16:77-86.
352 Vaideeswar P. Sclerosing hemangioma with lymph node metastases. Indian J Pathol Microbiol. 2009;52:392-394.
353 Chien N.C., Lin C.W., Tzeng J.E. Sclerosing hemangioma with lymph node metasatasis. Respirology. 2009;14:614-616.
354 Guillou L., DeLuze P., Zysset F., Costa J. Papillary variant of low-grade mucoepidermoid carcinoma—an unusual bronchial neoplasm: a light microscopic, ultrastructural, and immunohistochemical study. Am J Clin Pathol. 1994;101:269-274.
355 Rodriguez I.M., Prat J. Mucinous tumors of the ovary: a clinicopathologic analysis of 75 borderline tumors (of intestinal type) and carcinomas. Am J Surg Pathol. 2002;26:139-152.
356 Izumo A., Yamaguchi K., Eguchi T., et al. Mucinous cystic tumor of the pancreas. Oncol Rep. 2003;10:515-525.
357 Sambrook-Gowar F.J. An unusual mucous cyst of the lung. Thorax. 1978;33:796-799.
358 Butnor K.J., Sporn T.A., Dodd L.G. Fine needle aspiration cytology of mucinous cystadenocarcinoma of the lung: report of a case with radiographic and histologic correlation. Acta Cytol. 2001;45:779-783.
359 Monaghan H., Salter D.M., Ferguson T. Pulmonary mucinous cystic tumor of borderline malignancy: a rare variant of adenocarcinoma. J Clin Pathol. 2002;55:156.
360 Divisi D., Battaglia C., Giusti L., et al. Mucinous cystadenoma of the lung. Acta Biomed Ateneo Parmense. 1997;68:115-118.
361 Papla B., Malinowski E., Harazda M. Pulmonary mucinous cystadenoma of borderline malignancy: a report of two cases. Pol J Pathol. 1996;47:87-90.
362 Roux F.J., Lantuejoul S., Brambilla E., Brambilla C. Mucinous cystadenoma of the lung. Cancer. 1995;76:1540-1544.
363 Pelletier B., Dubigeon P., Despins P., Dehajartre A.Y. Cystic mucinous pulmonary tumor of borderline malignancy: report of a case. Ann Pathol. 1993;13:405-408.
364 Davison A.M., Lowe J.W., DaCosta P. Adenocarcinoma arising in a mucinous cystadenoma of the lung. Thorax. 1992;47:129-130.
365 Traub B. Mucinous cystadenoma of the lung. Arch Pathol Lab Med. 1991;115:740-741.
366 Kragel P.J., Devaney K.O., Meth B.M., et al. Mucinous cystadenoma of the lung: a report of two cases with immunohistochemical and ultrastructural analysis. Arch Pathol Lab Med. 1990;114:1053-1056.
367 Matsuo T., Yusuke-Kimura N., Takamori S., Shirouzu K. Recurrent pulmonary mucinous cystadenoma. Eur J Cardiothorac Surg. 2005;28:176-177.
368 Gao Z.H., Urbanski S.J. The spectrum of pulmonary mucinous cystic neoplasia: a clinicopathologic & immunohistochemical study of ten cases and review of literature. Am J Clin Pathol. 2005;124:62-70.
369 Klein J., Zhuang Z., Lubensky I., Colby T.V., Martinez F.Jr, Leslie K.O. Multifocal microcysts and papillary cystadenoma of the lung in von Hippel-Lindau disease. Am J Surg Pathol. 2007;31:1292-1296.
370 Kleinert R., Popper H. Giant fibroma of the lung: a morphologic study. Virchows Arch A. 1987;410:363-367.
371 Sauk J.J., Pliego M., Anderson W.R. Primary pulmonary fibroma. Minn Med. 1972;55:220-223.
372 Klemperer P., Rabin C.B. Primary neoplasms of the pleura. Arch Pathol. 1931;11:385-412.
373 Briselli M.F., Mark E.J. Solitary fibrous tumors of the pleura and benign extrapleural tumors of mesothelial origin. In: Antman K., Aisner J., editors. Asbestos-Related Malignancy. Philadelphia: Grune & Stratton; 1986:165-178.
374 Dalton W., Zollike A., McCaughey W.T., Jacques J., Kannerstein M. Localized primary tumors of the pleura. Cancer. 1979;44:1465-1475.
375 Vallat-Decouvelaere A.V., Dry S.M., Fletcher C.D.M. Atypical and malignant solitary fibrous tumors in extrathoracic locations: evidence of their comparability to intrathoracic tumors. Am J Surg Pathol. 1998;22:1501-1511.
376 Magdeleinat P., Alifano M., Petino A., et al. Solitary fibrous tumors of the pleura: clinical characteristics, surgical treatment, and outcome. Eur J Cardiothorac Surg. 2002;21:1087-1093.
377 Cardillo G., Facciolo F., Cavazzana A.O., et al. Localized (solitary) fibrous tumors of the pleura: an analysis of 55 patients. Ann Thorac Surg. 2000;70:1808-1812.
378 Duster P., Mayer E., Kramm T., et al. Solitary fibrous pleural tumors—rare tumors with unpredictable clinical behavior. Pneumologie. 2000;54:16-19.
379 Meyer M., Krause U. Solitary fibrous tumors of the pleura. Chirurgie. 1999;70:949-952.
380 Khan J.H., Rahman S.B., Clary-Macy C., et al. Giant solitary fibrous tumor of the pleura. Ann Thorac Surg. 1998;65:1461-1464.
381 Briselli M.F., Mark E.J., Dickersin G.R. Solitary fibrous tumors of the pleura: eight new cases and review of 360 cases in the literature. Cancer. 1981;47:2678-2689.
382 Hanau C.A., Miettinen M. Solitary fibrous tumor: histological and immunohistochemical spectrum of benign and malignant variants presenting at different sites. Hum Pathol. 1995;26:440-449.
383 Moran C.A., Suster S., Koss M.N. The spectrum of histologic growth patterns in benign and malignant fibrous tumors of the pleura. Semin Diagn Pathol. 1992;9:169-180.
384 England D.M., Hochholzer L., McCarthy M.J. Localized benign and malignant fibrous tumors of the pleura. Am J Surg Pathol. 1989;13:640-658.
385 Patsios D., Hwang D.M., Chung T.B. Intraparenchymal solitary fibrous tumor of the lung: an uncommon cause of a pulmonary nodule. J Thorac Imaging. 2006;21:50-53.
386 Sakurai H., Tanaka W., Kaji M., Yamazaki K., Suemasu K. Intrapulmonary localized fibrous tumor of the lung: a very unusual presentation. Ann Thorac Surg. 2008;86:1360-1362.
387 Cardinale L., Ardissone F., Cataldi A., Familiari U., Solitro F., Fava C. Solitary fibrous tumor of the lung: three rare cases of intraparenchymal nodules. Acta Radiol. 2009;50:379-382.
388 Fujiu K., Miyamoto H., Sakuma H., Mori M. Solitary fibrous tumors of the pleura presenting satellite tumors. Gen Thorac Cardiovasc Surg. 2009;57:382-384.
389 Ali S.Z., Hoon V., Hoda S., Heelan R., Zakowski M.F. Solitary fibrous tumor: a cytologic–histologic study with clinical, radiologic, and immunohistochemical correlations. Cancer. 1997;81:116-121.
390 Harrison-Phipps K.M., Nichols F.C., Schleck C.D., et al. Solitary fibrous tumors of the pleura: results of surgical treatment and long-term prognosis. J Thorac Cardiovasc Surg. 2009;138:19-25.
391 Weynand B., Collard P., Galant C. Cytopathological features of solitary fibrous tumor of the pleura: a study of 5 cases. Diagn Cytopathol. 1998;18:118-124.
392 Caruso R.A., LaSpada F., Gaeta M., Minutoli I., Inferrera C. Report of an intrapulmonary solitary fibrous tumor: fine needle aspiration cytologic findings, clinicopathological, and immunohistochemical features. Diagn Cytopathol. 1996;14:64-67.
393 Khalifa M.A., Montgomery E.A., Azumi N., et al. Solitary fibrous tumors: a series of lesions, some in unusual sites. South Med J. 1997;90:793-799.
394 Van de Rijn M., Lombard C.M., Rouse R.V. Expression of CD34 by solitary fibrous tumors of the pleura, mediastinum, and lung. Am J Surg Pathol. 1994;18:814-820.
395 Suster S., Fisher C., Moran C.A. Expression of bcl-2 oncoprotein in benign and malignant spindle cell tumors of soft tissue, skin, serosal surfaces, and gastrointestinal tract. Am J Surg Pathol. 1998;22:863-872.
396 Flint A., Weiss S.W. CD34 and keratin expression distinguishes solitary fibrous tumor (fibrous mesothelioma) of pleura from desmoplastic mesothelioma. Hum Pathol. 1995;26:428-431.
397 Ledet S.C., Brown R.W., Cagle P.T. p53 immunostaining in the differentiation of inflammatory pseudotumor from sarcoma involving the lung. Mod Pathol. 1995;8:282-286.
398 Krismann M., Adams H., Jaworska M., Muller K.M., Johnen G. Patterns of chromosomal imbalances in benign solitary fibrous tumors of the pleura. Virchows Arch A. 2000;70:1808-1812.
399 Dal Cin P., Pauwels P., Van den Berghe H. Solitary fibrous tumor of the pleura with t(4;15)(q13;q26). Histopathology. 1999;35:94-95.
400 Carbone M., Rizzo P., Powers A., et al. Molecular analyses, morphology, and immunohistochemistry together differentiate pleural synovial sarcomas from mesotheliomas: clinical implications. Anticancer Res. 2002;22:3443-3448.
401 Knosel T., Hertsch S., Alendorf-Hofmann A., et al. TLE1 is a robust diagnostic biomarker for synovial sarcomas and correlates with t(X;18): analysis of 319 cases. Eur J Cancer. 2010;46:1170-1176.
402 Sumathi V.P., McCluggage W.G. CD10 is useful in demonstrating endometrial stroma at ectopic sites and in confirming a diagnosis of endometriosis. J Clin Pathol. 2002;55:391-392.
403 Takagi M., Kuwano K., Watanabe K., Akiba T. A case of recurrence and rapid growth of pleural solitary fibrous tumor 8 years after initial surgery. Ann Thorac Cardiovasc Surg. 2009;15:178-181.
404 Miyoshi N., Takami K., Okami J., et al. Extrapleural pneumonectomy for relapsed solitary fibrous tumors of the pleura with pleural dissemination. Jpn J Thorac Surg. 2007;60:800-805.
405 Liu C.C., Wang H.W., Li F.Y., et al. Solitary fibrous tumors of the pleura: clinicopathological characteristics, immunohistochemical profiles, and surgical outcomes with long-term followup. Thorac Cadiovasc Surg. 2008;56:291-297.
406 Anthony T., Rodriguez-Bigas M.A., Weber T.K., Petrelli N.J. Desmoid tumors. J Am Coll Surg. 1996;182:369-377.
407 Dashiell T.G., Payne W.S., Hepper N.G., Soule E.H. Desmoid tumors of the chest wall. Chest. 1978;74:157-162.
408 Rodriguez-Bigas M.A., Mahoney M.C., Karakousis C.P., Petrelli N.J. Desmoid tumors in patients with familial adenomatous polyposis. Cancer. 1994;74:1270-1274.
409 Andino L., Cagle P.T., Maurer B., et al. Pleuropulmonary desmoid tumors: immunohistochemical comparison with solitary fibrous tumors and assessment of beta-catenin and cyclin-D1 expression. Arch Pathol Lab Med. 2006;130:1503-1509.
410 Varghese T.K.Jr, Gupta R., Yeldandi A.V., Sundaresan S.R. Desmoid tumor of the chest wall with pleural involvement. Ann Thorac Surg. 2003;76:937-939.
411 Peled N., Babyn P.S., Manson D., de Nanassy J. Aggressive fibromatosis simulating congenital lung malformation. Can Assoc Radiol J. 1993;44:221-223.
412 Takeshima Y., Nakayori F., Nakano T., et al. Extra-abdominal desmoid tumor presenting as an intrathoracic tumor: case report and literature review. Pathol Int. 2001;51:824-828.
413 Wilson R.W., Gallateau-Salle F., Moran C.A. Desmoid tumors of the pleura: a clinicopathologic mimic of localized fibrous tumor. Mod Pathol. 1999;12:9-14.
414 Montgomery E.A., Meis J.M. Nodular fasciitis: its morphologic spectrum and immunohistochemical profile. Am J Surg Pathol. 1991;15:942-948.
415 Owens C.L., Sharma R., Ali S.Z. Deep fibromatosis (desmoid tumor): cytopathologic characteristics, clinicoradiologic features, and immunohistochemical findings on fine-need aspiration. Cancer. 2007;111:166-172.
416 Bhattacharya B., Dilworth H.P., Iacobuzio-Donahue C., et al. Nuclear beta-catenin expression distinguishes deep fibromatosis from other benign and malignant fibroblastic and myofibroblastic lesions. Am J Surg Pathol. 2005;29:653-659.
417 Ng T.L., Gown A.M., Barry T.S., et al. Nuclear beta-catenin in mesenchymal tumors. Mod Pathol. 2005;18:68-74.
418 Carlson J.W., Fletcher C.D.M. Immunohistochemistry for beta-catenin in the differential diagnosis of spindle-cell lesions: analysis of a series and review of the literature. Histopathology. 2007;51:509-514.
419 De Bree E., Keus R., Melissas J., Tsiftsis D., van Coevorden F. Desmoid tumors: need for an individualized approach. Expert Rev Anticancer Ther. 2009;9:525-535.
420 Abbas A.E., Deschamps C., Cassivi S.D., et al. Chest-wall desmoid tumors: results of surgical intervention. Ann Thorac Surg. 2004;78:1219-1223.
421 Liebow A.A., Castleman B. Benign “clear cell tumors” of the lung. Am J Pathol. 1963;43:13-14.
422 Liebow A.A., Castleman B. Benign clear cell (“sugar”) tumors of the lung. Yale J Biol Med. 1971;43:213-222.
423 Hoch W.S., Patchefsky A.S., Takeda M., Gordon G. Benign clear-cell tumor of the lung: an ultrastructural study. Cancer. 1974;33:1328-1336.
424 Ozdemir I.A., Zaman N.U., Rullis I., Webb W.R. Benign clear cell tumor of lung. J Thorac Cardiovasc Surg. 1974;68:131-133.
425 Kung M., Landa J.F., Lubin J. Benign clear cell tumor (“sugar tumor”) of the trachea. Cancer. 1984;54:517-519.
426 Hashimoto T., Oka K., Hakozaki H., et al. Benign clear cell tumor of the lung. Ultrastruct Pathol. 2001;25:479-483.
427 Slodkowska J., Suilkowska-Rowinska A., Dorosz P., Radomski P., Wasiutynski A. Benign clear cell tumor of the lung (“sugar tumor”): morphologic, immunohistochemical, and ultrastructural evaluation. Pneumonol Alergol Pol. 2000;68:60-64.
428 Gaffey M.J., Mills S.E., Ritter J.H. Clear cell tumors of the lower respiratory tract. Semin Diagn Pathol. 1997;14:222-232.
429 Jeanfaivre T., Savary L., Richard C. Benign clear cell tumor (“sugar tumor”): an unusual cause of intrapulmonary coin lesion. Rev Mal Respir. 1997;14:223-224.
430 Andrion A., Mazzucco G., Gugliotta P., Monga G. Benign clear-cell (“sugar”) tumor of the lung: a light microscopic, histochemical, and ultrastructural study with a review of the literature. Cancer. 1985;56:2657-2663.
431 Sale G.E., Kulander B.G. Benign clear-cell tumor of lung with necrosis. Cancer. 1976;37:2355-2358.
432 Lantuejoul S., Isaac S., Pinel N., et al. Clear cell tumor of the lung: an immunohistochemical and ultrastructural study supporting a pericytic differentiation. Mod Pathol. 1997;10:1001-1008.
433 Flieder D.B., Travis W.D. Clear cell “sugar” tumor of the lung associated with lymphangioleiomyomatosis and multifocal micronodular pneumocyte hyperplasia in a patient with tuberous sclerosis. Am J Surg Pathol. 1997;21:1242-1247.
434 Gal A.A., Koss M.N., Hochholzer L., Chejfec G. An immunohistochemical study of benign clear-cell (“sugar”) tumor of the lung. Arch Pathol Lab Med. 1991;115:1034-1038.
435 Gaffey M.J., Mills S.E., Askin F.B., et al. Clear cell tumor of the lung: a clinicopathologic, immunohistochemical, and ultrastructural study of eight cases. Am J Surg Pathol. 1991;15:199-202.
436 Stolz A.J., Schutzner J., Lischke R., et al. Benign clear cell tumors of the lung—sugar tumors. Rozhl Chir. 2003;82:149-151.
437 Sale G.E., Kulander B.G. Benign clear-cell tumor (sugar tumor) of the lung with hepatic metastases ten years after resection of pulmonary primary tumor. Arch Pathol Lab Med. 1988;112:1177-1178.
438 Cavazza A., Sgarbi G., Ferrari G., Putrino I., Gardini G. Clear cell tumor of the lung: description of a case 1 mm. in diameter (“micro-sugar tumor”). Pathologica. 2001;93:556-560.
439 Justrabo E., Mergey E., Piard F., Michiels R., Viard H. Benign clear cell tumour of the lung. Sem Hop. 1982;58:673-677.
440 Urban T. Clinical and molecular epidemiology of lymphangioleiomyomatosis and pulmonary pathology in tuberous sclerosis. Rev Mal Respir. 2000;17:597-603.
441 Knowles M.A., Hornigold N., Pitt E. Tuberous sclerosis complex (TSC) gene involvement in sporadic tumors. Biochem Soc Trans. 2003;31:597-602.
442 Panizo-Santos A., Sola I., DeAlava E., et al. Angiomyolipoma and PEComa are immunoreactive for Myo-D1 in a cell cytoplasmic staining pattern. Appl Immunohistochem Mol Morphol. 2003;11:156-160.
443 Nappi O., Mills S.E., Swanson P.E., Wick M.R. Clear-cell tumors of unknown nature and origin: a systematic approach to diagnosis. Semin Diagn Pathol. 1997;14:164-174.
444 Rosai J., Limas C., Husband E.M. Ectopic hamartomatous thymoma. A distinctive benign lesion of lower neck. Am J Surg Pathol. 1984;8:501-513.
445 Cohen J.B., Troxell M., Kong C.S., McDougall J.R. Ectopic intrathyroidal thymoma: a case report and review. Thyroid. 2003;13:305-308.
446 Ben-Hami B., Caulet-Maugendre S., Valla J., et al. Primary pericardial thymoma: an unusual etiology of neoplastic pericarditis. Ann Pathol. 1996;16:445-448.
447 Marchevsky A.M. Lung tumors derived from ectopic tissues. Semin Diagn Pathol. 1995;12:172-184.
448 Gong L., Li Y.H., He X.L., et al. Primary intrapulmonary thymomas: case report and review of the literature. J Int Med Res. 2009;37:1252-1257.
449 Sajwani R.A., Gowani S.A., Khowaja A.A., Khan A., Fatimi S.H. Extensive primary malignant thymoma involving pericardium, pleura, diaphragm, and lungs—a case report. J Pak Med Assoc. 2008;58:287-288.
450 Velojic D., Marsenic B., Nikolic D., Bosnic M., Djordjevic A. Ectopic thymoma with myasthenic and cardiac symptomatology. Plucne Bolesti Tuberk. 1972;24:199-204.
451 Veynovich B., Masetti P., Kaplan P.D., et al. Primary pulmonary thymoma. Ann Thorac Surg. 1997;64:1471-1473.
452 Moran C.A., Suster S., Fishback N.F., Koss M.N. Primary intrapulmonary thymoma: a clinicopathologic and immunohistochemical study of eight cases. Am J Surg Pathol. 1995;19:304-312.
453 James C.L., Iyer P.V., Leong A.S.Y. Intrapulmonary thymoma. Histopathology. 1992;21:175-177.
454 Fukayama M., Maeda Y., Funata N., et al. Pulmonary and pleural thymoma: diagnostic application of lymphocyte markers to the thymoma of unusual site. Am J Clin Pathol. 1988;89:617-621.
455 Green W.R., Pressoir R., Gumbs R.V., et al. Intrapulmonary thymoma. Arch Pathol Lab Med. 1987;111:1074-1076.
456 Kung I.T., Loke S.L., So S.Y., et al. Intrapulmonary thymoma: report of two cases. Thorax. 1985;40:471-474.
457 Yeoh C.B., Ford J.M., Lattes R., Wylie R.H. Intrapulmonary thymoma. J Thorac Cardiovasc Surg. 1966;51:131-136.
458 Shih D.F., Wang J.S., Tseng H.H., Tiao W.M. Primary pleural thymoma. Arch Pathol Lab Med. 1997;121:79-82.
459 Honma K., Shimada K. Metastasizing ectopic thymoma arising in the right thoracic cavity and mimicking diffuse pleural mesothelioma—an autopsy study of a case with review of literature. Wien Klin Wochenschr. 1986;98:14-20.
460 Fushimi H., Tanio Y., Kotoh K. Ectopic thymoma mimicking diffuse pleural mesothelioma: a case report. Hum Pathol. 1998;29:409-410.
461 Attanoos R.L., Galateau-Salle F., Gibbs A.R., et al. Primary thymic epithelial tumors of the pleura mimicking malignant mesothelioma. Histopathology. 2002;41:42-49.
462 Bernatz P.E., Harrison E.G., Clagett O.T. Thymoma: a clinicopathologic study. J Thorac Cardiovasc Surg. 1961;42:424-444.
463 Muller-Hermelink H.K., Marino M., Palestro G. Pathology of thymic epithelial tumors. Curr Top Pathol. 1986;75:207-268.
464 Dadmanesh F., Sekihara T., Rosai J. Histologic typing of thymoma according to the new World Health Organization classification. Chest Surg Clin N Am. 2001;11:407-420.
465 Suster S., Moran C.A. Thymoma, atypical thymoma, and thymic carcinoma: a novel conceptual approach to the classification of thymic epithelial neoplasms. Am J Clin Pathol. 1999;111:826-833.
466 Kirchner T., Schalke B., Buchwald J., et al. Well-differentiated thymic carcinoma: an organotypical low-grade carcinoma with relationship to cortical thymoma. Am J Surg Pathol. 1992;16:1153-1169.
467 Wakely P.E.Jr. Cytopathology–histopathology of the mediastinum: epithelial, lymphoproliferative, and germ cell neoplasms. Ann Diagn Pathol. 2002;6:30-43.
468 Chhieng D.C., Rose D., Ludwig M.E., Zukowski M.F. Cytology of thymomas: emphasis on morphology and correlation with histologic subtypes. Cancer. 2000;90:24-32.
469 Shabb N.S., Fahl M., Shabb B., Haswani P., Zaatari G. Fine needle aspiration of the mediastinum: a clinical, radiologic, cytologic, and histologic study of 42 cases. Diagn Cytopathol. 1998;19:428-436.
470 Levine G.D., Rosai J. Thymic hyperplasia and neoplasia: a review of current concepts. Hum Pathol. 1978;9:495-515.
471 Mirza I., Kazimi S.N., Ligi R., Burns J., Braza F. Cytogenetic profile of a thymoma: a case report and review of the literature. Arch Pathol Lab Med. 2000;124:1714-1716.
472 Goh S.G., Lau L.C., Sivaswaren C., et al. Pseudodicentric (16;12)(q11;p11.2) in a type AB (mixed) thymoma. Cancer Genet Cytogenet. 2001;131:42-47.
473 Van den Berghe I., Debiec-Rychter M., Proot L., Hagemeijer A., Michielssen P. Ring chromosome 6 may represent a cytogenetic subgroup in benign thymoma. Cancer Genet Cytogenet. 2002;137:75-77.
474 Kuo T.T. Cytokeratin profiles of the thymus and thymomas: histogenetic correlations and proposal for a histological classification of thymomas. Histopathology. 2000;36:403-414.
475 Chu P.G., Weiss L.M. Expression of cytokeratin 5/6 in epithelial neoplasms: an immunohistochemical study of 509 cases. Mod Pathol. 2002;15:6-10.
476 DiComo C.J., Urist M.J., Babayan I., et al. p63 expression profiles in human normal and tumor tissues. Clin Cancer Res. 2002;8:494-501.
477 Myers P.O., Kritikos N., Bongiovanni M., et al. Primary intrapulmonary thymoma: a systematic review. Eur J Surg Oncol. 2007;33:1137-1141.
478 Wick M.R., Nappi O. Ectopic neural and neuroendocrine neoplasms. Semin Diagn Pathol. 2003;20:305-323.
479 Cesario A., Galetta D., Margaritora S., Granone P. Unsuspected primary pulmonary meningioma. Eur J Cardiothorac Surg. 2002;21:553-555.
480 Prayson R.A., Farver C.F. Primary pulmonary malignant meningioma. Am J Surg Pathol. 1999;23:722-726.
481 Moran C.A., Hochholzer L., Rush W., Koss M.N. Primary intrapulmonary meningiomas: a clinicopathologic and immunohistochemical study of ten cases. Cancer. 1996;78:2328-2333.
482 Gomez-Aracil V., Mayayo E., Alvira R., Arraiza A., Ramon y Cajal S. Fine needle aspiration cytology of primary pulmonary meningioma associated with minute meningothelial-like nodules. Report of a case with histologic, immunohistochemical, and ultrastructural studies. Acta Cytol. 2002;46:899-903.
483 Falleni M., Roz E., Dessy E., et al. Primary intrathoracic meningioma: histopathological, immunohistochemical, and ultrastructural study of two cases. Virchows Arch A. 2001;439:196-200.
484 Spinelli M., Claren R., Colombi R., Sironi M. Primary pulmonary meningioma may arise from meningothelial-like nodules. Adv Clin Path. 2000;4:35-39.
485 DePerrot M., Kurt A.M., Robert J., Spiliopoulos A. Primary pulmonary meningioma presenting as lung metastasis. Scand Cardiovasc J. 1999;33:121-123.
486 Ueno M., Fujiyama J., Yamazaki I., et al. Cytology of primary pulmonary meningioma: report of the first multiple case. Acta Cytol. 1998;42:1424-1430.
487 Kaleem Z., Fitzpatrick M.M., Ritter J.H. Primary pulmonary meningioma: report of a case and review of the literature. Arch Pathol Lab Med. 1997;121:631-636.
488 Lockett L., Chiang V., Scully N. Primary pulmonary meningioma: report of a case and review of the literature. Am J Surg Pathol. 1997;21:453-460.
489 Maiorana A., Ficarra G., Fano R.A., Spagna G. Primary solitary meningioma of the lung. Pathologica. 1996;88:457-462.
490 Flynn S.D., Yousem S.A. Pulmonary meningiomas: report of two cases. Hum Pathol. 1991;22:469-474.
491 Kodama K., Doi O., Higashiyama M., et al. Primary and metastatic pulmonary meningioma. Cancer. 1991;67:1412-1417.
492 Strimlan C.V., Golembiewski R.S., Celko D.A., Fino G.J. Primary pulmonary meningioma. Surg Neurol. 1988;29:410-413.
493 Rowsell C., Sirbovan J., Rosenblum M.K., Perez-Ordonez B. Primary chordoid meningioma of lung. Virchows Arch. 2005;446:333-337.
494 Incarbone M., Ceresoli G.L., Di Tommaso L., et al. Primary pulmonary meningioma: report of a case and review of the literature. Lung Cancer. 2008;62:401-407.
495 Churg A.M., Warnock M.L. So-called “minute pulmonary chemodectoma:” a tumor not related to paragangliomas. Cancer. 1976;37:1759-1769.
496 Ionescu D.N., Sasatomi E., Aldeeb D., et al. Pulmonary meningothelial-like nodules: a genotypic comparison with meningiomas. Am J Surg Pathol. 2004;28:207-214.
497 Suster S., Moran C.A. Diffuse pulmonary meningotheliomatosis. Am J Surg Pathol. 2007;31:624-631.
498 Drlicek M., Grisold W., Lorber J., et al. Pulmonary meningioma: immunohistochemical and ultrastructural features. Am J Surg Pathol. 1991;15:455-459.
499 Omulecka A., Papierz W., Nawrocka-Kunecka A., Lewy-Trenda I. Immunohistochemical expression of progesterone and estrogen receptors in meningiomas. Folia Neuropathol. 2006;44:111-115.
500 Leaes C.G., Meurer R.T., Coutinho L.B., Ferreira N.P., Pereira-Lima J.F., da Costa-Oliveira M. Immunohistochemical expression of aromatase and estrogen, androgen, & progesterone receptors in normal and neoplastic human meningeal cells. Neuropathology. 2010;30:44-49.
501 Shintaku M., Honda T., Sakai T. Expression of podoplanin and calretinin in meningiomas: an immunohistochemical study. Brain Tumor Pathol. 2010;27:23-27.
502 Hu Y., Yang Q., McMahon L.A., Wang H.L., Xu H. Value of D2-40 in the differential diagnosis of pleural neoplasms, with emphasis on its positivity in solitary fibrous tumor. Appl Immunohistochem Mol Morphol. 2010. April 27 (E-pub ahead of print)
503 Jokinen C.H., Dadras S.S., Goldblum J.R., van de Rijn M., West R.B., Rubin B.P. Diagnostic implications of podoplanin expression in peripheral nerve sheath neoplasms. Am J Clin Pathol. 2008;129:886-893.
504 Ishino S., Hashimoto N., Fushiki S., et al. Loss of material from chromosome arm 1p during malignant progression of meningioma revealed by fluorescent in-situ hybridization. Cancer. 1998;83:360-366.
505 Adlakha A., Rao K., Adlakha H., et al. Meningioma metastatic to the lung. Mayo Clin Proc. 1999;74:1129-1133.
506 Tandon S., Kant S., Singh A.K., et al. Primary intrapulmonary teratoma presenting as pyothorax. Indian J Chest Dis Allied Sci. 1999;41:51-55.
507 Kayser K., Gabius H.J., Hagemeyer O. Malignant teratoma of the lung with lymph node metastasis of the ectodermal compartment: a case report. Ann Cell Pathol. 1993;5:31-37.
508 Eggerath A., Ammon J., Karstens J.H., Rubben H., Morales M.R. Extragonadal germ cell tumors—diagnostic and therapeutic aspects. Strahlentherapie. 1984;160:1-7.
509 Hartman G.E., Shochat S.J. Primary pulmonary neoplasms of childhood: a review. Ann Thorac Surg. 1983;36:108-119.
510 Moran C.A., Travis W.D., Carter D., Koss M.N. Metastatic mature teratoma in lung following testicular embryonal carcinoma and teratocarcinoma. Arch Pathol Lab Med. 1993;117:641-644.
511 Kakkar N., Vashishta R.K., Banerjee A.K., et al. Primary pulmonary malignant teratoma with yolk sac elements associated with hematologic neoplasia. Respiration. 1996;63:52-54.
512 Stair J.M., Stevenson D.R., Schaefer R.F., Fullenwider J.P., Campbell G.S. Primary teratocarcinoma of the lung. J Surg Oncol. 1986;33:262-267.
513 Berghout A., Mallens W.M., TeVelde J., Haak H.L. Teratoma of the lung in a hemophilic patient. Acta Hematol. 1983;70:330-334.
514 Turna A., Ozgul A., Kahraman S., urses A., Fener N., Yilmaz V. Primary pulmonary teratoma: report of a case and the proposition of “bronchotrichosis” as a new term. Ann Thorac Cardiovasc Surg. 2009;15:247-249.
515 Saini M.L., Krishnamurthy S., Kumar R.V. Intrapulmonary mature teratoma. Diagn Pathol. 2006;1:38.
516 Joseph M.B., Colby T.V., Swensen S.J., Mikus J.P., Gaensler E.A. Multiple cystic fibrohistiocytic tumors of the lung: report of two cases. Mayo Clin Proc. 1990;65:192-197.
517 Han J.H. Uncommon tumors of the lung (Internet presentation). www.pathology.or.kr/studygroup/cardiopulmonary/lecture/lenote/hjh.htm.
518 Colome-Grimmer M.I., Evans H.L. Metastasizing cellular dermatofibroma: a report of two cases. Am J Surg Pathol. 1996;20:1361-1367.