CHAPTER 75 Basic Science of Spinal Cord Injury
Pathophysiologic Response to Spinal Cord Injury
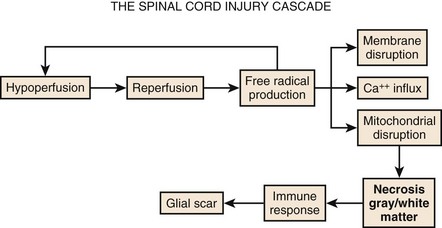
Although physical trauma to the spinal cord undoubtedly causes direct physical injury to the neural tissues, it has become clear that a cascade of events follows the inciting episode and can both inhibit recovery and cause additional neural damage.1,2 This concept of secondary injury has been validated in both animal and clinical studies, and secondary injury mechanisms have been the target of the bulk of pharmacologic interventions to date. These processes begin immediately following the injury and continue for weeks (Fig. 75–1).
Circulatory Collapse
The cascade of secondary injury often begins with microcirculatory insufficiency shortly after mechanical trauma.2 Vascular hypoperfusion due to capillary loss, capillary spasm, thrombosis, systemic hypotension, and autonomic regulatory interruption lead to cellular ischemia at the epicenter of cord injury. The gray matter, rich with neuronal cell bodies, is highly vulnerable to ischemia. The resultant shift in pH renders neuronal cell body and axonal membranes highly vulnerable to subsequent injury. The neurons are now primed to enter into what will become an increasingly self-destructive path.
Oxidative Damage
After a transient period of hypoperfusion and ischemia, a sudden and unregulated reperfusion of the injury epicenter occurs. The introduction of oxygen to the compromised cell membranes produces a highly toxic environment whereby the membrane lipid fatty acids undergo oxidation.3 This membrane lipid peroxidation produces several varieties of free radicals that in turn drive even further lipid peroxidation and free radical production. Some of the free radicals accumulate within the cell and denature deoxyribonucleic acid (DNA) and mitochondrial proteins, and eventually bring energy production to a halt resulting in irreversible damage and cell death.
Excitotoxicity
The remainder of the free radicals further disrupt and destabilize the neuronal membrane. The cellular release of the ubiquitous neurotransmitter glutamate changes the extracellular space into a hostile extracellular milieu.4 Glutamate activates various cell surface receptors that in turn mediate a large variety of intracellular processes. Excessive glutamate will drive these processes to the point of fatal overload to the cell. The most studied of the glutamic receptors is the N-methyl-D-aspartate (NMDA) receptor, which mediates entry of Ca++ into cellular cytoplasm from both extracellular and intracellular stores. Although calcium in physiologic amounts is the necessary component for many important enzyme-mediated cellular processes, pathophysiologic quantities of calcium lead to the persistent activity of destructive enzymes including lipoxygenases and phospholipases. These enzymes will again target the beleaguered cell membrane to generate free radicals from lipid oxidation. The radicals will disrupt cellular proteins and, in particular, those that mediate the ability of mitochondria to drive oxidative phosphorylation. The neuron, starved of ATP, will terminate itself via necrotic or apoptotic cell death mechanisms.
Neuroimmunologic Response
The events taking place moments after spinal trauma, beginning with microcirculatory failure within the cord and leading to free-radical-mediated cytotoxicity of the gray and white matter, do not go unnoticed by the immune system. The ever-vigilant inflammatory cells of the body are instantly attracted to the neuronal self-destruction. Over the next hours to weeks, they will lay the foundations for an extracellular environment that will inhibit axonal regeneration. The first of these cells to appear at the site of injury are circulating neutrophils. Once active, neutrophils will secrete cytokines that stimulate production of phospholipases and cyclooxygenase.5 The former will consume neural membranes to produce arachidonic acid, which the latter (cyclooxygenase) uses to produce prostaglandins and thromboxanes. Prostaglandins (PGE2, PGD2, PGF2a, PGI2) serve (1) to amplify the inflammatory response by increasing capillary permeability to allow additional inflammatory cells influx; (2) to increase neuronal calcium concentration, thus promoting excitotoxicity; and (3) to activate other inflammatory cells.1 Thromboxanes promote platelet aggregation within capillaries and thus worsen local tissue ischemia.
Astroglial Scar
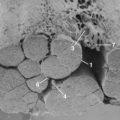
Trauma to the adult spinal cord is particularly devastating because of the inability of the central nervous system (CNS) to regenerate after injury. Unlike in the peripheral nervous system, axonal recovery in the spinal cord is thwarted by two fundamental obstacles: the inherently weak regenerative ability of CNS axons and a powerfully inhibitive postinjury milieu of physical and chemical factors.6 The most potent of these factors is the glial scar that develops after any CNS injury.7–9 The glial scar is a collection of reactive cells (astrocytes, microglia, oligodendrocyte precursors, meningeal fibroblasts) that express cell-surface and matrix molecules, which surround the area of injury and ultimately repel the advancement of regenerating axons (Fig. 75–2).
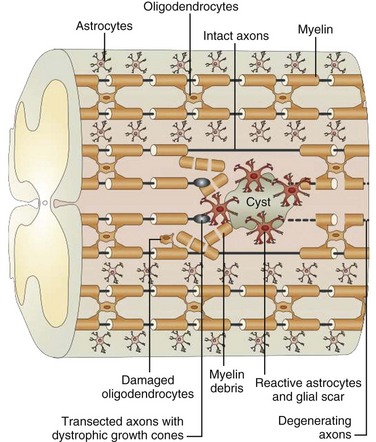
The scar features a core zone of meningeal cells and oligodendrocyte precursors, as well as a peripheral zone of astrocytes, oligodendrocyte precursors, and microglia. The core zone is separated from the surround zone by a basement membrane composed mostly of type IV collagen.10 Although some axons may regenerate through the surround zone, no axon can penetrate the core zone without some form of experimental manipulation.11
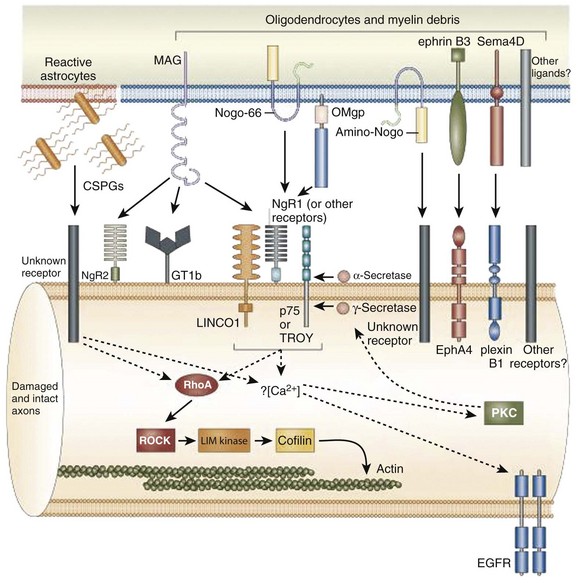
The inhibitory effects of the scar are conferred by three classes of molecules, all of which are expressed by one or more of the reactive cells in the glial scar. These include the chondroitin sulphate proteoglycans (CSPGs) (NG2, brevican, phosphacan, neurocan, versican), semaphorin 3 proteins, and eph/ephrin tyrosine kinases. Although the precise mechanisms of their actions are unclear, the molecules exert their inhibitory effects either by directly or indirectly binding to the axon cell surface or by binding and deactivating trophic factors, cell adhesion molecules, and extracellular matrix molecules that are a requisite for axonal growth and regeneration.12 The ultimate effect of the gliotic response to injury is the inhibition of successful axonal regeneration and remyelination by both physical and chemical means (Fig. 75–3).13
There is tremendous therapeutic potential in the ability to modulate the gliotic scarring response to CNS injury. In-vitro and in-vivo studies to date, though relatively limited, have demonstrated enhancement of axonal regeneration and functional recovery after inhibition of specific glial scar constituents. Enzymatic digestion of the glycosaminoglycan chains of CSPGs, for instance, stimulates axonal regeneration through the site of injury.14 Function-blocking antibodies to semaphorin receptors have allowed sensory axons to regenerate into the formidable core zone of the scar.15 Chelating agents that prevent collagen IV synthesis around the core zone have also allowed successful axonal regeneration in some animal models.16 Animals with clonal deletions of a certain eph molecule have almost no astroglial scar response and demonstrate unimpeded regeneration of motor axons through the zone of injury.17
Despite its multifaceted inhibitory influence, several recent studies suggest that the glial scar must offer some protective benefit to the injured spinal cord. The role of the gliotic response in mitigating the extension of cord injury beyond the initial site of trauma, for instance, has been proposed.18 In a study of transgenic mice, selective ablation of reactive astrocytes in the glial scar after both contusion and penetrating SCI led to markedly increased tissue disruption, cellular degeneration, cystic changes, and profound and persistent motor deficits relative to nonablated controls.19 It is likely that both the cellular and extracellular matrix elements of the gliotic scar play a critical role in biochemical protection and structural stabilization of cord integrity, and thus function, after spinal cord injury.
Basic Science of a Cure
Despite the confounding nature of the injury response, several glimpses of hope for a cure exist and they capitalize on our understanding of the basic science of spinal cord injury. Consistent with their basic science foundations, the human acute SCI therapies have one of two ambitions: (1) to limit secondary injury (neuroprotection, acute surgical intervention, rehabilitation trials) and (2) to reverse injury (regeneration trials) (Box 75–1).
Methylprednisolone
The first randomized, controlled multicenter trial of a neuroprotective agent was the National Acute Spinal Cord Injury (NASCIS) I Study, which attempted to establish the clinical efficacy of methylprednisolone.20,21 Though its precise mechanism of action is still unclear, methylprednisolone was thought to exert either a cell-stabilizing effect via the glucocorticoid receptor or a cord-stabilizing effect via free-radical inhibition.22–24 The trial was based on several animals studies that suggested improved neurologic recovery when the corticosteroid was administered promptly after experimental injury.25 Published in 1984, NASCIS I included 330 patients with acute SCI (defined as any loss of sensation or motor function below the level of injury) who were randomized into two groups within 48 hours of injury: a “low-dose group” receiving 100 mg IV methylprednisolone bolus and then 25 mg every 6 hours for 10 days and a “high-dose” group receiving bolus and maintenance doses 10 times those of the low-dose group every 6 hours for 10 days. Outcome measures consisted of motor and sensory indices of 14 muscle groups and 29 dermatomes. The follow-up periods were 6 weeks and 6 months. Though there was no difference between the two groups in terms of neurologic outcome, there was an increased incidence of wound infection and even fatality in the higher-dose group, with the former achieving statistical significance.
One year later, NASCIS II was launched as the first randomized, prospective, placebo-controlled trial of a candidate therapy for SCI.26,27 It was devised to address the lack of a placebo control in NASCIS I and to incorporate new basic-science findings regarding effective methylprednisolone dosing and mechanisms of action. The study involved 487 patients who were randomized into three groups within 12 hours of sustaining either complete or incomplete SCI: a methylprednisolone group receiving an unprecedented 30-mg/kg IV bolus, followed by a maintenance infusion of 5.4 mg/kg/hr over 23 hours; a placebo group; and a third group consisting of a 5.4 mg/kg bolus and 4 mg/kg/hr maintenance infusion of the opioid antagonist naloxone, whose neuroprotective effects had also been suggested by animal studies.28 The outcome methodology was similar to that in NASCIS I.
When all members of the methylprednisolone group were compared with placebo, there were no statistically significant improvements in sensory or motor function at 6 weeks. However, when the steroid group was stratified according to timing of administration, patients receiving treatment within 8 hours of injury demonstrated a significant improvement in sensory and motor function by 6 weeks versus placebo. The relative improvement was sustained through the 6-month follow-up point. Within the steroid group treated within 8 hours, further subgroup analysis with respect to American Spinal Injury Association (ASIA) scale injury severity revealed that class A patients had the greatest statistically significant improvement in motor and sensory measures versus placebo. Additionally, ASIA C&D patients had only motor improvement, whereas class B patients had neither sensory or motor improvement that reached statistical significance.26,29 The complication of wound infection was more frequent but statistically insignificant in the steroid group despite the heavy dosing. There were no differences in motor or sensory outcomes between the naloxone and placebo group at either of the follow-up points. NASCIS II established the now ubiquitous “steroid protocol,” despite controversies regarding possible nontransparency, data misinterpretation, and near-normal function of some participants.
The objectives of the third and final NASCIS were to investigate the interplay between timing of steroid administration and duration of therapy and to evaluate the efficacy of the 21-aminosteroid tirilazad mesylate, which purportedly had a better safety profile than methylprednisolone. Four-hundred ninety-nine patients were randomized into three treatment groups within 6 hours of injury: the first group received methylprednisolone according to the NASCIS II dosing for 24 hours, the second group received this dosing for 48 hours, and the third group received a methylprednisolone bolus of 5.4 mg/kg/hr followed by a maintenance infusion of tirilazad at 2.5 mg/kg IV every 6 hours for 48 hours.30 With outcome measures including motor function, sensory function, and functional independence; the NASCIS III revealed that increased duration of steroid administration (48 hours) resulted in statistically significant benefit only if treatment was initiated between 3 and 8 hours of injury. Infectious complications were more common in the 48-hour corticosteroid group but were statistically insignificant. There were no differences between the tirilazad group and the 24-hour methylprednisolone group.
Ganglioside GM-1
Trials of the glycosphingolipid ganglioside GM-1 followed closely on the heels of the methylprednisolone studies. Ganglioside are mammalian neuronal cell membrane constituents and play a substantial role in neuronal plasticity, axonal recovery after experimental SCI, and cell preservation after ischemia.31–34 The Maryland GM-1 ganglioside trial was a prospective, randomized, placebo-controlled pilot study of 37 patients with SCI who were assigned to one of two groups: GM-1 100 mg IV per day for 30 days, and placebo.35 Outcome measures were in terms of motor scores and Frankel grades, and the follow-up point was at 6 months. By 6 months, the authors demonstrated a two-grade improvement on the Frankel scale in 50% of patients receiving GM-1 but only in 7% of patients receiving placebo—a difference that was statistically significant. The pilot study was then expanded into the largest clinical trial of SCI therapy, known as the Sygen Multicenter Acute Spinal Cord Injury Study (SMASCIS).36 The study randomized 797 patients with SCI into two groups receiving GM-1 (300 mg IV bolus + 100 mg IV daily for 56 days vs. 600 mg IV bolus + 200 mg IV daily for 56 days) and NASCIS II protocol. Outcome measures included ASIA sensory and motor scores and the ASIA impairment scale rating. At 8 weeks, patients exhibited dose-related, statistically significant improvements in impairment scale ratings relative to placebo; but by the 6-month end point there were no differences in the number of patients achieving improvement (defined as at least a two-grade improvement in the impairment index) between any of the groups. SMASCIS had revealed that GM-1 ganglioside was ultimately no better than placebo in the setting of acute SCI.
Thyrotropin-Releasing Hormone
Thyrotropin-releasing hormone (TRH) and its analogs have been shown in multiple animal models of acute SCI to improve functional recovery by acting as neuroprotective partial opioid antagonists.27,37–39 The prospective trial evaluating its efficacy in human subjects involved 20 patients with acute SCI who were randomly assigned within 12 hours of injury into one of two groups: a TRH group receiving 0.2 mg/kg bolus IV dose followed by 0.2 mg/kg/hr infusion for 6 hours and a placebo group.40 NASCIS motor and sensory indices and the Sunnybrook system were used as outcome measures. Follow-up time points were at 24 hours, 72 hours, 7 days, 1 month, 4 months, and 1 year after injury. Patients with complete injuries did not demonstrate a benefit from TRH administration relative to placebo group patients; patients with incomplete injuries did show statistically significant improvement in outcome measure after TRH treatment versus placebo. However, the authors were cautious with their conclusions due to sample size issues and the number of patients lost to follow-up. A larger clinical trial of the hormone has not been completed.
Nimodipine and Gacyclidine
In a number of animal models of SCI, the calcium channel blocker nimodipine has been reported to improve neurologic recovery by increasing spinal cord blood flow and limiting vasospasm, ischemia, and secondary infarction.41,42 The prospective clinical trial of the antihypertensive involved 106 patients with complete or incomplete SCI who were randomized to one of four groups: nimodipine 0.015 mg/kg/hr IV loading dose for 2 hours + 0.03 mg/kg/hr infusion for 7 days, methylprednisolone according to NASCIS II protocol, both agents, and placebo.43 Outcome was measured with the ASIA grade and ASIA motor and sensory scores. At 1 year follow-up none of the treatment groups demonstrated efficacy over placebo. Interestingly, 80 of the 106 patients were surgically treated within 24 hours of injury; a subgroup analysis revealed that surgery within 8 hours of injury and between 8 and 24 hours of injury yielded identical outcomes.
Post-traumatic glutamate toxicity is the target of the novel N-methyl-D-aspartate antagonist (NMDA) receptor antagonist gacyclidine, which in rat models has been shown to enhance recovery after contusive SCI.44 In a prospective, controlled clinical trial 272 patients were randomized to one of four groups within 2 hours of SCI: gacyclidine at 0.005 mg/kg IV within 2 hours of trauma and once again at 6 hours after trauma, gacyclidine at 0.001 mg/kg IV × 2 doses, gacyclidine at 0.02 mg/kg × 2 doses, and placebo. Outcome measures included the ASIA motor and sensory scores. At 1-year follow-up, there were no statistical differences in outcomes between any of the groups.
Regeneration Strategies
Although neuroprotective strategies are directed primarily at limiting secondary neural injury, regeneration strategies involve repair of the damaged tissues. This, of course, is more theoretically appealing due to the potential for greater degrees of neurologic improvement. However, any such therapeutic strategy must also somehow modify the nonideal local milieu of the injured cord because biochemical, cellular, and extracellular matrix perturbations may limit the potential for axonal repair.6,45,46 Although a number of laboratory investigations have reported remarkable success using reparative strategies, translation to the clinical setting has been difficult and clinical trials of regenerative strategies are just beginning.
Immune-Mediated Neural Repair and Autologous Macrophage Transplantation
A controlled inflammatory response is essential to proper tissue repair in any part of the body including the CNS. The brain and spinal cord are naturally sequestered from inflammatory cells of the systemic circulation by highly selective basement membranes (the blood-brain barrier). Therefore the injured CNS often will not elicit a bona fide immune response, and its innate healing capacity is limited.47 The concept of immune-mediated neural repair is based on several laboratory studies that demonstrated protective autoimmunity with macrophages: Animals with transection SCI treated with autologous macrophages at the lesion site had enhanced functional and histologic recovery.48,49
This strategy was translated into the clinical setting in an Israeli pilot trial using autologous activated macrophages that were injected into SCI lesion sites. The macrophages were activated in vitro with autologous dermis. The results from the pilot trial demonstrated that three of eight ASIA A patients converted to ASIA C at 1 year of follow-up.50 On the basis of these encouraging results, a phase II randomized, controlled clinical trial of autologous incubated macrophage treatment of SCI patients complete injuries was initiated in 2003 at six SCI treatment centers in Israel and North America. The trial had enrolled 50 patients by the spring of 2006 but was stopped for financial reasons. This trial was remarkable for a number of reasons. The enrollment window of 14 days allowed patients who had been stabilized and received acute treatment at a referring trauma center to be transported to one of the study sites. At these facilities the patients were then consented, assessed, and randomized. They underwent blood and skin harvest for cell processing and then surgical implantation of the incubated autologous macrophages into the caudal boundary of the spinal cord contusion. In addition, the patient selection process included radiographic criteria: those with intramedullary lesions larger than 3 cm on MRI were excluded. This strategy of excluding patients with severe radiographic spinal cord abnormalities may be helpful for determining the efficacy of an intervention with smaller numbers of patients.
Rho Antagonist
Several inhibitors of nerve regeneration have been identified including Nogo, myelin-associated glycoprotein (MAG), and myelin-oligodendrocyte glycoprotein (OMgp). These factors act commonly through the GTPase Rho, an enzyme that orchestrates a potent inhibitory cascade after injury that culminates in growth cone disintegration, neurite sprouting inhibition, and neuronal and glial cell apoptosis.51,52 The naturally occurring inhibitor of the Rho enzyme, C3 transferase from Clostridium botulinum, has been shown to effect remarkably rapid functional recovery in hemisection SCI rats.53 Histologic analysis of C3-treated rats treated within 24 hours of injury showed increased neuronal sprouting, and functional analysis revealed enhanced locomotor recovery and limb coordination.
A recombinant pharmaceutic version of C3 transferase known as Cethrin (Alseres Pharmaceuticals, Hopkinton, MA), in which the enzyme is linked to a protein that assists blood-brain barrier penetration, has undergone phase I/IIa trials at nine North American centers to establish safety and efficacy. Preliminary results of this trial have been discussed at scientific meetings and published in abstract form.54 The study included 37 patients with complete (ASIA A) SCI at either the cervical or thoracic level who were given increasing doses of extradurally applied Cethrin (0.3, 1.0, 3.0, and 6.0 mg). Outcome measures (ASIA International Standards) were collected at 1.5, 3, 6, and 12 months. The investigators documented no adverse effects of the treatment and by the 1.5-month follow-up period observed improvement of at least one ASIA Impairment Scale grade in 30% of patients. Initial data from the 6-month follow-up period suggest similar rates of recovery.
A phase II/III multicenter clinical trial of the drug Cethrin is due to begin in 2008. The study is anticipating enrollment of complete SCI (ASIA A) patients to undergo surgery for epidural application of the soluble enzyme antagonist at the time of early surgical decompression.24 Study enrollment will incorporate a Bayesian trial design in an effort to determine optimal dosing while keeping sample numbers to a minimum.
Anti-Nogo Antibody
Nogo is an oligodendrocyte-expressed soluble factor that is potent in its ability to inhibit axonal regeneration. Investigators have been able to restore corticospinal axonal regeneration and locomotion in rat and primate SCI models in which Nogo was neutralized with the monoclonal antibody IN-1.55,56 A phase I clinical trial of intrathecally administered Nogo-A antibody (Novartis International AG, Basel, Switzerland) in acute, complete (ASIA A) SCI is currently under way in European SCI centers with expansion of the trial to Canadian centers anticipated.
Peripheral Nerve and Schwann Cell Transplantation
Inducing the CNS to mimic the natural regenerative ability of the peripheral nervous system (PNS) is a pillar of current SCI research. The injured peripheral nerve expresses a permissive environment for axonal regrowth that is engendered primarily by its myelin-forming cell, the Schwann cell, and its basal lamina. Schwann cells secrete trophic factors, express cell adhesion molecules, and produce extracellular matrix molecules that are requisite for axonal growth and regeneration.57,58 The permissive features of Schwann cells may prove valuable in the treatment of spinal cord injury. Several groups have demonstrated improved axonal regeneration and functional recovery after Schwann cell transplantation to the site of SCI in rats.59 Furthermore, the grafting of peripheral nerves with varying combinations of growth factors in SCI rats has resulted in significant axonal and functional recovery.60 Most of the ongoing clinical studies involving peripheral nerve grafts that are used to bridge lesional sites in the spinal cord are phase I trials and currently involve few patients. Some have shown neurologic improvement, whereas others have not; none of the studies are prospective, randomized trials yet.61 Other studies have involved the injection of Schwann cells to the area of cord lesion. A recent review of current clinical trials documents a Chinese clinical trial using a Schwann cell delivery strategy to treat 47 patients with SCI and documented improvement in ASIA motor and sensory scores.
Olfactory Ensheathing Glial Cell Transplantation
The olfactory ensheathing glial (OEG) cell spans both the PNS and CNS and thus has a unique role in regenerative SCI therapy; several preclinical studies have shown the OEG to have permissive capabilities similar to that of the Schwann cell.62 A nonrandomized, noncontrolled clinical trial of OEG transplantation in 171 patients with SCI showed modest improvement in ASIA motor and sensory scores after 8 weeks.63 The study was criticized for its short duration, potential for bias, and lack of control. Neurologic outcomes in the first seven chronic (>6 months postinjury) SCI patients treated with surgical implantation of olfactory mucosal autograft in an ongoing surgical case series in Lisbon, Portugal, were recently published.64 The investigators claimed to show a modest improvement in ASIA motor and sensory scores in the treated patients. Preliminary safety results of a small Australian-controlled clinical trial of surgical implantation of cultured autologous olfactory cells in patients with chronic SCI have recently been published. The investigators report no significant safety concerns at 1 year postimplantation and plan to report efficacy outcomes after 3 years of follow-up.
Summary
Key Points
1 Kwon BK, Tetzlaff W, Grauer JN, et al. Pathophysiology and pharmacologic treatment of acute spinal cord injury. Spine J. 2004;4:451-464.
2 Tator CH, Fehlings MG. Review of the secondary injury theory of acute spinal cord trauma with emphasis on vascular mechanisms. J Neurosurg. 1991;75:15-26.
3 Basu S, Hellberg A, Ulus AT, et al. Biomarkers of free radical injury during spinal cord ischemia. FEBS Lett. 2001;508:36-38.
4 Choi DW. Excitotoxic cell death. J Neurobiol. 1992;23:1261-1276.
5 Popovich PG, Wei P, Stokes BT. Cellular inflammatory response after spinal cord injury in Sprague-Dawley and Lewis rats. J Comp Neurol. 1997;377:443-464.
6 Fawcett JW. Overcoming inhibition in the damaged spinal cord. J Neurotrauma. 2006;23:371-383.
7 Bahr M, Przyrembel C, Bastmeyer M. Astrocytes from adult rat optic nerves are nonpermissive for regenerating retinal ganglion cell axons. Exp Neurol. 1995;131:211-220.
8 Davies SJ, Goucher DR, Doller C, Silver J. Robust regeneration of adult sensory axons in degenerating white matter of the adult rat spinal cord. J Neurosci. 1999;19:5810-5822.
9 Hermanns S, Klapka N, Muller HW. The collagenous lesion scar—an obstacle for axonal regeneration in brain and spinal cord injury. Restor Neurol Neurosci. 2001;19:139-148.
10 Stichel CC, Muller HW. The CNS lesion scar: new vistas on an old regeneration barrier. Cell Tissue Res. 1998;294:1-9.
11 Rasouli A, Bhatia N, Suryadevara S, et al. Transplantation of preconditioned schwann cells in peripheral nerve grafts after contusion in the adult spinal cord. Improvement of recovery in a rat model. J Bone Joint Surg Am. 2006;88:2400-2410.
12 Li Y, Raisman G: Schwann cells induce sprouting in motor and sensory axons in the adult rat spinal cord. J Neurosci 14:4050–4063.
13 Fawcett JW, Asher RA. The glial scar and central nervous system repair. Brain Res Bull. 1999;49:377-391.
14 Smith-Thomas LC, Stevens J, Fok-Seang J, et al: Increased axon regeneration in astrocytes grown in the presence of proteoglycan synthesis inhibitors. J Cell Sci 108(Pt 3):1307–1315.
15 Shearer MC, Niclou SP, Brown D, et al. The astrocyte/meningeal cell interface is a barrier to neurite outgrowth which can be overcome by manipulation of inhibitory molecules or axonal signalling pathways. Mol Cell Neurosci. 2003;24:913-925.
16 Klapka N, Hermanns S, Straten G, Masanneck C, et al. Suppression of fibrous scarring in spinal cord injury of rat promotes long-distance regeneration of corticospinal tract axons, rescue of primary motoneurons in somatosensory cortex and significant functional recovery. Eur J Neurosci. 2005;22:3047-3058.
17 Goldshmit Y, Galea MP, Bartlett PF, Turnley AM. EphA4 regulates central nervous system vascular formation. J Comp Neurol. 2006;497:864-875.
18 Yiu G, He Z. Glial inhibition of CNS axon regeneration. Nat Rev Neurosci. 2006;7:617-627.
19 Faulkner JR, Herrmann JE, Woo MJ, et al. Reactive astrocytes protect tissue and preserve function after spinal cord injury. J Neurosci. 2004;24:2143-2155.
20 Bracken MB, Shepard MJ, Hellenbrand KG, et al. Methylprednisolone and neurological function 1 year after spinal cord injury. Results of the National Acute Spinal Cord Injury Study. J Neurosurg. 1985;63:704-713.
21 Ducker TB, Hamit HF. Experimental treatments of acute spinal cord injury. J Neurosurg. 1969;30:693-697.
22 Campbell JB, DeCrescito V, Tomasula JJ, et al. Experimental treatment of spinal cord contusion in the cat. Surg Neurol. 1973;1:102-106.
23 Hall ED, Braughler JM. Glucocorticoid mechanisms in acute spinal cord injury: a review and therapeutic rationale. Surg Neurol. 1982;18:320-327.
24 Lammertse DP. Update on pharmaceutical trials in acute spinal cord injury. J Spinal Cord Med. 2004;27:319-325.
25 Bracken MB. Treatment of acute spinal cord injury with methylprednisolone: results of a multicenter, randomized clinical trial. J Neurotrauma. 1991;8(Suppl 1)):S47-S50. discussion S1-S2
26 Bracken MB, Shepard MJ, Collins WF, et al. A randomized, controlled trial of methylprednisolone or naloxone in the treatment of acute spinal-cord injury. Results of the Second National Acute Spinal Cord Injury Study. N Engl J Med. 1990;322:1405-1411.
27 Faden AI, Jacobs TP, Holaday JW. Thyrotropin-releasing hormone improves neurologic recovery after spinal trauma in cats. N Engl J Med. 1981;305:1063-1067.
28 Bracken MB, Shepard MJ, Collins WFJr, et al. Methylprednisolone or naloxone treatment after acute spinal cord injury: 1-year follow-up data. Results of the second National Acute Spinal Cord Injury Study. J Neurosurg. 1992;76:23-31.
29 Otani K, Abe H, Kadoya S, et al. Beneficial Effect of methylprednisolone sodium succinate in the treatment of acute spinal cord injury. Sekitsui Sekizui J. 1994;7:633-647.
30 Bracken MB, Shepard MJ, Holford TR, et al. Administration of methylprednisolone for 24 or 48 hours or tirilazad mesylate for 48 hours in the treatment of acute spinal cord injury. Results of the Third National Acute Spinal Cord Injury Randomized Controlled Trial. National Acute Spinal Cord Injury Study. JAMA. 1997;277:1597-1604.
31 Agnati LF, Fuxe K, Calza L, et al. Further studies on the effects of the GM1 ganglioside on the degenerative and regenerative features of mesostriatal dopamine neurons. Acta Physiol Scand Suppl. 1984;532:37-44.
32 Bose B, Osterholm JL, Kalia M. Ganglioside-induced regeneration and reestablishment of axonal continuity in spinal cord-transected rats. Neurosci Lett. 1986;63:165-169.
33 Toffano G, Agnati LF, Fuxe KG. The effect of the ganglioside GM1 on neuronal plasticity. Int J Dev Neurosci. 1986;4:97-100.
34 Toffano G, Savoini G, Moroni F, et al: GM1 ganglioside stimulates the regeneration of dopaminergic neurons in the central nervous system. Brain Res 261:63–166.
35 Geisler FH, Dorsey FC, Coleman WP. Recovery of motor function after spinal-cord injury–a randomized, placebo-controlled trial with GM-1 ganglioside. N Engl J Med. 1991;324:1829-1838.
36 Geisler FH, Coleman WP, Grieco G, Poonian D. The Sygen multicenter acute spinal cord injury study. Spine. 2001;26(24 Suppl):S87-S98.
37 Faden AI. New pharmacologic approaches to spinal cord injury: opiate antagonists and thyrotropin-releasing hormone. Cent Nerv Syst Trauma. 1985;2:5-8.
38 Faden AI. Opiate antagonists and thyrotropin-releasing hormone. II. Potential role in the treatment of central nervous system injury. JAMA. 1984;252:1452-1454.
39 Vink R, McIntosh TK, Faden AI. Treatment with the thyrotropin-releasing hormone analog CG3703 restores magnesium homeostasis following traumatic brain injury in rats. Brain Res. 1988;460:184-188.
40 Pitts LH, Ross A, Chase GA, Faden AI: Treatment with thyrotropin-releasing hormone (TRH) in patients with traumatic spinal cord injuries. J Neurotrauma 12:235–243.
41 Guha A, Tator CH, Piper I. Effect of a calcium channel blocker on posttraumatic spinal cord blood flow. J Neurosurg. 1987;66:423-430.
42 Pointillart V, Gense D, Gross C, et al. Effects of nimodipine on posttraumatic spinal cord ischemia in baboons. J Neurotrauma. 1993;10:201-213.
43 Gambardella G, Collufio D, Caruso GN, et al. Experimental incomplete spinal cord injury: treatment with a combination of nimodipine and adrenaline. J Neurosurg Sci. 1995;39:67-74.
44 Lepeintre JF, D’Arbigny P, Mathe JF, et al. Neuroprotective effect of gacyclidine. A multicenter double-blind pilot trial in patients with acute traumatic brain injury. Neurochirurgie. 2004;50(2-3 Pt 1):83-95.
45 Dinh P, Bhatia N, Rasouli A, et al. Transplantation of preconditioned Schwann cells following hemisection spinal cord injury. Spine. 2007;32:943-949.
46 Rasouli A, Bhatia N, Dinh P, et al: Resection of glial scar following spinal cord injury. J Orthop Res 27:931–936.
47 Perry VH, Brown MC, Gordon S. The macrophage response to central and peripheral nerve injury. A possible role for macrophages in regeneration. J Exp Med. 1987;165:1218-1223.
48 Rapalino O, Lazarov-Spiegler O, Agranov E, et al: Implantation of stimulated homologous macrophages results in partial recovery of paraplegic rats. Nat Med 4:814–821.
49 Schwartz M, Lazarov-Spiegler O, Rapalino O, et al. Potential repair of rat spinal cord injuries using stimulated homologous macrophages. Neurosurgery. 1999;44:1041-1045. discussion 5-6
50 Lazarov-Spiegler O, Solomon AS, Schwartz M. Peripheral nerve-stimulated macrophages simulate a peripheral nerve-like regenerative response in rat transected optic nerve. Glia. 1998;24:329-337.
51 Dubreuil CI, Winton MJ, McKerracher L. Rho activation patterns after spinal cord injury and the role of activated Rho in apoptosis in the central nervous system. J Cell Biol. 2003;162:233-243.
52 Sung JK, Miao L, Calvert JW, et al. A possible role of RhoA/Rho-kinase in experimental spinal cord injury in rat. Brain Res. 2003;959:29-38.
53 Dergham P, Ellezam B, Essagian C, et al. Rho signaling pathway targeted to promote spinal cord repair. J Neurosci. 2002;22:6570-6577.
54 Lehmann M, Fournier A, Selles-Navarro I, et al. Inactivation of Rho signaling pathway promotes CNS axon regeneration. J Neurosci. 1999;19:7537-7547.
55 Chen MS, Huber AB, van der Haar ME, et al. Nogo-A is a myelin-associated neurite outgrowth inhibitor and an antigen for monoclonal antibody IN-1. Nature. 2000;403:434-439.
56 Weinmann O, Schnell L, Ghosh A, Montani L, et al. Intrathecally infused antibodies against Nogo-A penetrate the CNS and downregulate the endogenous neurite growth inhibitor Nogo-A. Mol Cell Neurosci. 2006;32:161-173.
57 Bixby JL, Lilien J, Reichardt LF. Identification of the major proteins that promote neuronal process outgrowth on Schwann cells in vitro. J Cell Biol. 1988;107:353-361.
58 Bryan DJ, Wang KK, Chakalis-Haley DP. Effect of Schwann cells in the enhancement of peripheral-nerve regeneration. J Reconstr Microsurg. 12, 1996. 439–436
59 Bunge MB. Bridging the transected or contused adult rat spinal cord with Schwann cell and olfactory ensheathing glia transplants. Prog Brain Res. 2002;137:275-282.
60 Keirstead HS, Morgan SV, Wilby MJ, Fawcett JW. Enhanced axonal regeneration following combined demyelination plus Schwann cell transplantation therapy in the injured adult spinal cord. Exp Neurol. 1999;159:225-236.
61 Tadie M, Liu S, Robert R, et al. Partial return of motor function in paralyzed legs after surgical bypass of the lesion site by nerve autografts three years after spinal cord injury. J Neurotrauma. 2002;19:909-916.
62 Pearse DD, Sanchez AR, Pereira FC, et al. Transplantation of Schwann cells and/or olfactory ensheathing glia into the contused spinal cord: Survival, migration, axon association, and functional recovery. Glia. 2007;55:976-1000.
63 Huang H, Chen L, Wang H, et al. Influence of patients’ age on functional recovery after transplantation of olfactory ensheathing cells into injured spinal cord injury. Chin Med J (Engl). 2003;116:1488-1491.
64 Lima C, Pratas-Vital J, Escada P, et al. Olfactory mucosa autografts in human spinal cord injury: a pilot clinical study. J Spinal Cord Med. 2006;29:191-203. discussion 4-6