[level-membership-for-pulmolory-and-respiratory-category]
Chapter 2 Basic Aspects of Cellular and Molecular Biology
Molecular Biology
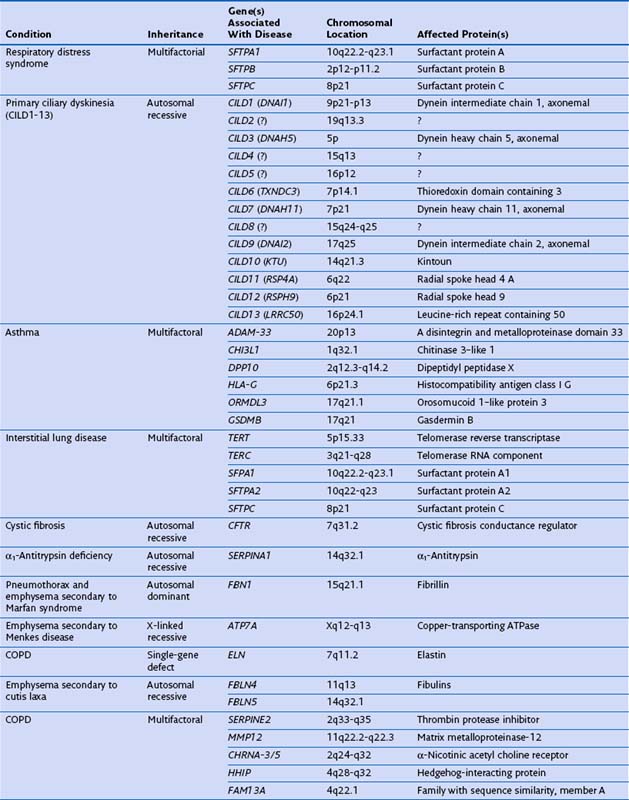
The Genetic Basis of Lung Disease
Genetic factors play an important role in diseases that affect the airways (asthma, chronic obstructive pulmonary disease [COPD], cystic fibrosis, primary ciliary dyskinesia), parenchyma (pulmonary fibrosis, Birt-Hogg-Dubé syndrome, tuberous sclerosis), and vasculature (hereditary hemorrhagic telangiectasia) of the lung (Table 2-1). Such conditions include simple monogenic disorders such as Kartagener syndrome and α1-antitrypsin deficiency, in which mutations of critical genes are sufficient to induce well-defined disease phenotypes. By contrast, many other disease processes affecting the lung are complex genetic traits in which inheritance subtly affects pathogenesis. This group of entities includes COPD, asthma, and idiopathic pulmonary fibrosis. Extending current understanding of the genetic basis of pulmonary conditions will be essential to provide new insights into their underlying pathophysiology, to make predictions about outcome, and to develop novel therapeutic strategies.
Single-Gene Disorders and Respiratory Disease
Many single-gene disorders have been linked with respiratory disease (see Table 2-1). They are perhaps best typified by the autosomal recessive condition α1-antitrypsin deficiency. This condition shows a clear genotype-phenotype correlation with current understanding of the molecular basis providing new insights into the pathogenesis of disease. α1-Antitrypsin is the archetypal member of the serine proteinase inhibitor (“serpin”) superfamily. It is synthesized in the liver and secreted into the plasma, where it is the most abundant circulating proteinase inhibitor. Most people of North European descent carry the normal M allele, but 1 in 25 carries the Z variant (Glu342Lys), which results in plasma α1-antitrypsin levels in the homozygote that are 10% to 15% of the normal M allele. The Z mutation causes the accumulation of α1-antitrypsin in the rough endoplasmic reticulum of the liver, predisposing the homozygote to the development of juvenile hepatitis, cirrhosis, and hepatocellular carcinoma. The greatly reduced circulating levels of α1-antitrypsin are unable to protect the lungs against proteolytic damage by neutrophil elastase, predisposing the Z homozygote to the development of early-onset emphysema.
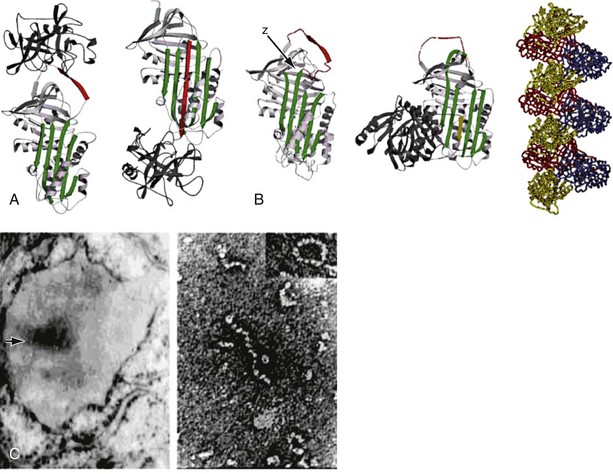
The structure of α1-antitrypsin is based on a dominant β-pleated sheet A and nine α-helices (Figure 2-1). This scaffold supports an exposed mobile reactive loop that presents a peptide sequence as a pseudosubstrate for the target proteinase. After docking, the proteinase is inactivated by a mousetrap-type action that swings it from the top to the bottom of the serpin in association with the insertion of an extra strand into β-sheet A (see Figure 2-1). This six-stranded protein bound to its target enzyme is then recognized by hepatic receptors and cleared from the circulation. The structure of α1-antitrypsin is central to its role as an effective antiproteinase but also renders it liable to undergo conformational change in association with disease. The Z mutation is at residue P17 (17 residues proximal to the key P1 amino acid that defines the inhibitory specificity of α1-antitrypsin) at the head of a strand of β-sheet A and the base of the mobile reactive loop (see Figure 2-1). The mutation opens β-sheet A, thereby favoring the insertion of the reactive loop of a second α1-antitrypsin molecule to form a dimer (see Figure 2-1). This dimer can then extend to form polymers that tangle in the endoplasmic reticulum of the liver to form the inclusion bodies resulting in liver disease. Support for this pathomechanism comes from the demonstration that Z α1-antitrypsin formed chains of polymers when incubated under physiologic conditions. The rate was accelerated by raising the temperature to 41° C and could be blocked by peptides that compete with the loop for annealing to β-sheet A. The role of polymerization in vivo was clarified by the finding of α1-antitrypsin polymers in inclusion bodies from the livers of Z α1-antitrypsin homozygotes (see Figure 2-1).
Gene Hunting for Complex Genetic Diseases and Its Pitfalls: COPD and Asthma
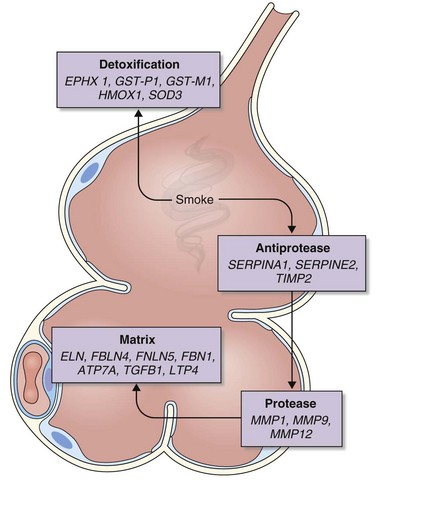
The analysis was made more complex in COPD by the inherent complexity of the disease phenotype—a heterogeneous mix of airway disease and emphysema. Indeed, larger family-based studies have shown the independent clustering of the airway disease and emphysema components of COPD within families. This finding suggests that different genetic factors predispose to each of these components of the phenotype. The only way to overcome the inherent variation in COPD is to focus on groups of patients with well-characterized disease components or to undertake studies with large sample sizes and then to replicate any positive findings in other cohorts. This is now the case with candidate gene studies, and good evidence has emerged to show that heterozygosity for α1-antitrypsin deficiency (phenotype PiMZ) and polymorphisms in genes involved in oxidative stress—those encoding microsomal epoxide hydrolase (EPHX1), glutathione S-transferase (GST-P1 and GST-M1), heme oxygenase (HMOX1), and superoxide dismutase 3 (SOD3)—are associated with an increased risk of COPD (Figure 2-2). More recently, a minor allele of an SNP in the matrix metalloprotease-12 gene (MMP12) has been shown to protect against COPD in adult smokers.
Familial clustering of asthma has also been recognized for many years, and comparisons between monozygotic and dizygotic twins suggest that 70% of asthma-related population variance is accounted for by genetic factors. Classical positional cloning using linkage analysis of large families has identified several candidates, including ADAM33, CHI3L1, DPP10, and HLA-G, and more recently, asthma-specific genome-wide association studies have identified further disease-associated loci (see Table 2-1). The first of these was in the long arm of chromosome 17 and found to contain two genes, ORMDL3 and GSDMB, whose expression levels are altered in asthmatic persons. The pathways implicated by such studies can now be tested to determine what role they play in disease pathogenesis.
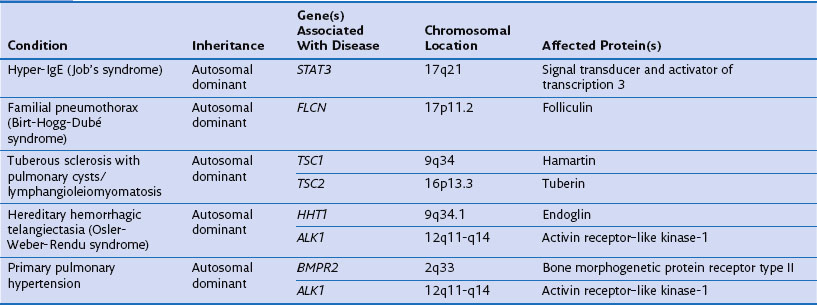
Cell Biology
Intracellular Signals
Oxidative Stress
A number of genes have been studied that might plausibly modify the cells’ responses to cigarette smoke and ROS. These include genes involved in detoxification of toxins and genes involved in neutralization of ROS. Many toxins in cigarette smoke are subject to first-pass metabolism in the liver, and one of the enzymes involved in this is microsomal epoxide hydrolase (encoded by EPHX1) localized to 1q42.1, which has been studied intensely in the context of COPD (see Figure 2-2). Several EPHX1 SNPs have been described that affect its activity. One of these leads to a 40% loss of in vitro activity (Tyr113His, the “slow” allele), whereas another increases activity by 25% (His139Arg, the “fast” allele). A recent systematic metaanalysis found homozygosity for the “slow” (Tyr113His) allele to be protective against COPD (odds ratio, 0.5). Analysis of the National Emphysema Treatment Trial (NETT) dataset has suggested a role for EPHX1 polymorphism in both severity of COPD and the distribution of emphysematous changes. In addition to EPHX1, glutathione S-transferase (GST) comprises a large family of enzymes capable of catalyzing the conjugation of GSH to noxious compounds. The GSTs are highly polymorphic, and SNPs in GSTP1 have been associated with COPD, the distribution of emphysema, and more rapid decline in lung function. The null mutation of GSTM1 (localized to 1p13.1) also has been associated with COPD.
Endoplasmic Reticulum Stress
The early steps in the biogenesis of secreted and membrane proteins occur in the lumen of the endoplasmic reticulum, where resident proteins that make up the endoplasmic reticulum machinery assist in their folding, maturation, and complex assembly (Figure 2-3). Variation in the load of endoplasmic reticulum client proteins and in the function of its protein-folding machinery can lead to an imbalance between the two that is referred to as endoplasmic reticulum stress. This imbalance triggers a cellular response, mediated by signaling pathways that restore balance between the protein-folding environment in the organelle by increasing the expression of genes that enhance most aspects of endoplasmic reticulum function and by transiently repressing the biosynthesis of new client proteins. This response has been termed the unfolded protein response (UPR) and is mediated by three signaling molecules, PERK, IRE1, and ATF6, located in the endoplasmic reticulum membrane (see Figure 2-3). It is now clear that the UPR plays a role in many human diseases, including many that affect the lung.
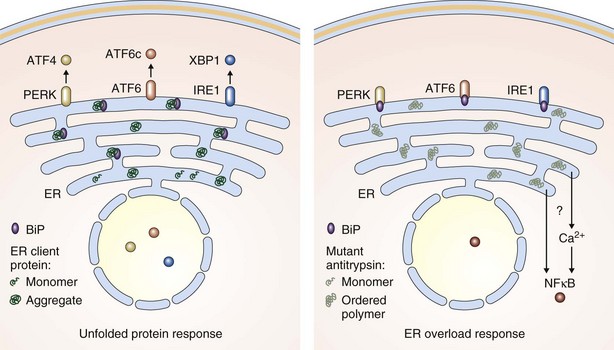
Endoplasmic Reticulum Overload
Remarkably, when Z α1-antitrypsin polymerizes within the endoplasmic reticulum of its cell of synthesis, it fails to induce a strong UPR. Instead, the predominant signaling response appears to be activation of NFκB through a poorly understood mechanism that has variously been termed the endoplasmic reticulum overload response or the ordered polymer response (Figure 2-3). In the liver, this ultimately can lead to cirrhosis. Z α1-antitrypsin appears also to be synthesized locally in the lung by some cell types, including bronchial and alveolar epithelial cells and macrophages. These too are likely to activate NFκB signaling cascades that would increase the production of inflammatory mediators and further amplify neutrophil recruitment and tissue damage. Chronic activation of NFκB would accelerate apoptosis within alveolar cells and thus contribute to the pathogenesis of emphysema. Because this effect would occur in all alveolar cells, it provides another explanation for the panlobular distribution of emphysema that characterizes α1-antitrypsin deficiency. Although Z α1-antitrypsin serves as an excellent model disease to study endoplasmic reticulum overload, it is likely that more common diseases such as some viral infections involve this form of signaling.
Maintenance of the Extracellular Matrix
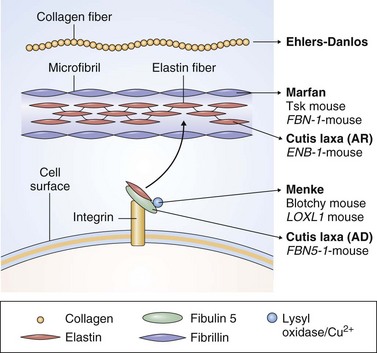
As in all tissues, cells of the lung communicate with one another through direct contact and by way of released diffusible and matrix molecules. This communication network is important during the inflammatory response but also is required for the maintenance of normal lung architecture. The extracellular matrix comprises a complex network of scaffolding proteins, principally elastin and collagen. The elastin filaments form from tropoelastin monomers that self-assemble into aggregates and then fuse with microfilaments. Multiple covalent cross-links between the lysines in neighboring filaments provide stability. Cutis laxa is a family of autosomal dominant, X-linked, and recessive human diseases characterized by excessively slack connective tissues. Several families with the milder autosomal dominant form show early-onset pulmonary pathology including emphysema, particularly if inherited with the Z allele of α1-antitrypsin. Mutations have been identified within the ELN (elastin) gene that cause mild cutis laxa and early-onset COPD (see Table 2-1). The ELN gene maps to 7q11.23 in humans, but because chromosome 7 has not been identified in linkage analysis as a site associated with COPD, it is likely that ELN mutations are a rare cause of this disease.
Elastin fibers bind other proteins, including fibulins, which in turn bind multiple extracellular matrix components and the basement membrane (Figure 2-4). The fibulins are a family of six proteins, at least two of which (those encoded by FBLN4, mapped to 11q13, and FBLN5, on 14q32.1) are mutated in severe autosomal recessive forms of cutis laxa and whose phenotype often includes early-onset emphysema. Both pathogenic mutations are located within an epidermal growth factor–like domain of each protein, suggesting these are critical for fibulins to maintain the integrity of the extracellular matrix within the lung. Of interest, analogous mutations in fibrillin, which bares homology to the fibulins, cause Marfan syndrome. Moreover, mutations of fibrillin (encoded by FBN1, localized to 15q21.1) have been described in neonatal Marfan syndrome with very-early-onset emphysema.
Repair
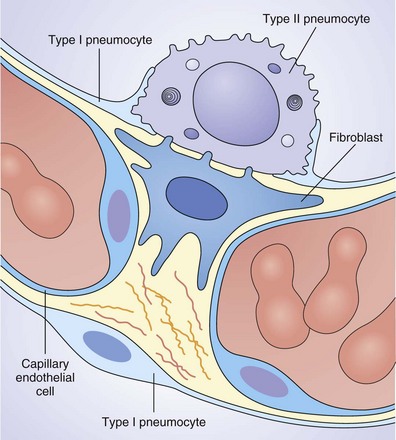
Alveoli are composed of capillaries and lymphatics encased in a thin epithelial layer. More than 90% of the alveolar surface is composed of type I pneumocytes (Figure 2-5). These are terminally differentiated, large flat squamous epithelial cells that possess a relatively simple ultrastructure. Their function is to allow gaseous exchange between the alveolar gas and the bloodstream; consequently, they require little more than a nucleus and cell membrane with a few mitochondria and a limited secretory pathway. They are unable to replicate and are susceptible to noxious insults from inhaled toxins such as cigarette smoke and therefore must be replenished by the major stem cell found within the alveolus, which is the type II pneumocyte. These are small cuboidal cells located predominantly at the alveolar septal junctions. Although contributing little surface area to the lung, they are abundant, making up half of the alveolar cells by number. Tight gap junctions separate their polarized apical and basolateral domains, enabling selective secretion toward their apical surface, readily identified by its many microvilli.
Pitfalls and Controversies
Barnes KC. Genetic studies of the etiology of asthma. Proc Am Thorac Soc. 2011;8:143–148.
Gooptu B, Lomas DA. Conformational pathology of the serpins—themes, variations and therapeutic strategies. Annu Rev Biochem. 2009;78:147–176.
Marciniak SJ, Lomas DA. α1-Antitrypsin deficiency and autophagy. N Engl J Med. 2010;363:1863–1864.
Marciniak SJ, Lomas DA. What can naturally occurring mutations tell us about the pathogenesis of COPD? Thorax. 2009;64:359–364.
Marciniak SJ, Ron D. The unfolded protein response in lung disease. Proc Am Thorac Soc. 2010;7:356–362.
Miranda E, Pérez J, Ekeowa UI, et al. A novel monoclonal antibody to characterize pathogenic polymers in liver disease associated with α1-antitrypsin deficiency. Hepatology. 2010;52:1078–1088.
Online Mendelian Inheritance in Man (OMIM) (website) http://www.ncbi.nlm.nih.gov/omim accessed March 5, 2011
Pearson H. Human genetics: one gene, twenty years. Nature. 2009;460:164–169.
Pillai SG, Ge D, Zhu G, et al A genome-wide association study in chronic obstructive pulmonary disease (COPD): identification of two major susceptibility loci. PLoS Genet. 2009(5):e1000421.
Stinchcombe SV, Maden M. Retinoic acid–induced alveolar regeneration: critical differences in strain sensitivity. Am J Respir Cell Mol Biol. 2008;38:185–191.
[/level-membership-for-pulmolory-and-respiratory-category][not-level-membership-for-pulmolory-and-respiratory-category]
Chapter 2 Basic Aspects of Cellular and Molecular Biology
Molecular Biology
The Genetic Basis of Lung Disease
Genetic factors play an important role in diseases that affect the airways (asthma, chronic obstructive pulmonary disease [COPD], cystic fibrosis, primary ciliary dyskinesia), parenchyma (pulmonary fibrosis, Birt-Hogg-Dubé syndrome, tuberous sclerosis), and vasculature (hereditary hemorrhagic telangiectasia) of the lung (Table 2-1). Such conditions include simple monogenic disorders such as Kartagener syndrome and α1-antitrypsin deficiency, in which mutations of critical genes are sufficient to induce well-defined disease phenotypes. By contrast, many other disease processes affecting the lung are complex genetic traits in which inheritance subtly affects pathogenesis. This group of entities includes COPD, asthma, and idiopathic pulmonary fibrosis. Extending current understanding of the genetic basis of pulmonary conditions will be essential to provide new insights into their underlying pathophysiology, to make predictions about outcome, and to develop novel therapeutic strategies.
Single-Gene Disorders and Respiratory Disease
Many single-gene disorders have been linked with respiratory disease (see Table 2-1). They are perhaps best typified by the autosomal recessive condition α1-antitrypsin deficiency. This condition shows a clear genotype-phenotype correlation with current understanding of the molecular basis providing new insights into the pathogenesis of disease. α1-Antitrypsin is the archetypal member of the serine proteinase inhibitor (“serpin”) superfamily. It is synthesized in the liver and secreted into the plasma, where it is the most abundant circulating proteinase inhibitor. Most people of North European descent carry the normal M allele, but 1 in 25 carries the Z variant (Glu342Lys), which results in plasma α1-antitrypsin levels in the homozygote that are 10% to 15% of the normal M allele. The Z mutation causes the accumulation of α1-antitrypsin in the rough endoplasmic reticulum of the liver, predisposing the homozygote to the development of juvenile hepatitis, cirrhosis, and hepatocellular carcinoma. The greatly reduced circulating levels of α1-antitrypsin are unable to protect the lungs against proteolytic damage by neutrophil elastase, predisposing the Z homozygote to the development of early-onset emphysema.
The structure of α1-antitrypsin is based on a dominant β-pleated sheet A and nine α-helices (Figure 2-1). This scaffold supports an exposed mobile reactive loop that presents a peptide sequence as a pseudosubstrate for the target proteinase. After docking, the proteinase is inactivated by a mousetrap-type action that swings it from the top to the bottom of the serpin in association with the insertion of an extra strand into β-sheet A (see Figure 2-1). This six-stranded protein bound to its target enzyme is then recognized by hepatic receptors and cleared from the circulation. The structure of α1-antitrypsin is central to its role as an effective antiproteinase but also renders it liable to undergo conformational change in association with disease. The Z mutation is at residue P17 (17 residues proximal to the key P1 amino acid that defines the inhibitory specificity of α1-antitrypsin) at the head of a strand of β-sheet A and the base of the mobile reactive loop (see Figure 2-1). The mutation opens β-sheet A, thereby favoring the insertion of the reactive loop of a second α1-antitrypsin molecule to form a dimer (see Figure 2-1). This dimer can then extend to form polymers that tangle in the endoplasmic reticulum of the liver to form the inclusion bodies resulting in liver disease. Support for this pathomechanism comes from the demonstration that Z α1-antitrypsin formed chains of polymers when incubated under physiologic conditions. The rate was accelerated by raising the temperature to 41° C and could be blocked by peptides that compete with the loop for annealing to β-sheet A. The role of polymerization in vivo was clarified by the finding of α1-antitrypsin polymers in inclusion bodies from the livers of Z α1-antitrypsin homozygotes (see Figure 2-1).
Gene Hunting for Complex Genetic Diseases and Its Pitfalls: COPD and Asthma
The analysis was made more complex in COPD by the inherent complexity of the disease phenotype—a heterogeneous mix of airway disease and emphysema. Indeed, larger family-based studies have shown the independent clustering of the airway disease and emphysema components of COPD within families. This finding suggests that different genetic factors predispose to each of these components of the phenotype. The only way to overcome the inherent variation in COPD is to focus on groups of patients with well-characterized disease components or to undertake studies with large sample sizes and then to replicate any positive findings in other cohorts. This is now the case with candidate gene studies, and good evidence has emerged to show that heterozygosity for α1-antitrypsin deficiency (phenotype PiMZ) and polymorphisms in genes involved in oxidative stress—those encoding microsomal epoxide hydrolase (EPHX1), glutathione S-transferase (GST-P1 and GST-M1), heme oxygenase (HMOX1), and superoxide dismutase 3 (SOD3)—are associated with an increased risk of COPD (Figure 2-2). More recently, a minor allele of an SNP in the matrix metalloprotease-12 gene (MMP12) has been shown to protect against COPD in adult smokers.
[/not-level-membership-for-pulmolory-and-respiratory-category]
