[level-membership-for-surgery-category]15
Barrett’s oesophagus
Definition
Barrett’s oesophagus is a change in any portion of the normal squamous oesophageal epithelium to a metaplastic columnar epithelium that is visible endoscopically and can be confirmed or corroborated histologically.1,2 There are three histologically distinct types of columnar metaplasia: intestinal (IM), cardiac (CM) and fundic. In the USA, unlike the UK and Japan, the diagnosis of Barrett’s oesophagus requires the identification of intestinalisation characterised by the presence of goblet cells. However, the UK definition considers that Barrett’s oesophagus is analogous to a ‘columnar lined oesophagus’ and does not require identification of goblet cells due to fears that sampling bias could lead to under-diagnosis and potentially exclude patients from surveillance programmes. It has been reported that a minimum of eight biopsies are required to confidently exclude intestinal metaplasia – if only four biopsies are taken the diagnostic yield is only 35%.3
Epidemiology
The exact population prevalence of Barrett’s oesophagus is unclear. Data described in post-mortem and endoscopic series range from 0.9% to 5.6% depending on the precise definition used and the type of study.4–7 It is likely that the true prevalence in the West is around 2%. When extrapolated to the UK and US populations, conservative estimates of prevalence are 1 million and 4 million affected individuals, respectively.8 There is also some evidence that the incidence of Barrett’s oesophagus in the West is increasing by up to 2% per year.5,9–11 Data from the Netherlands demonstrated an increase in the number of cases of Barrett’s oesophagus despite a decrease in the number of endoscopies being performed over the same period, suggesting a true increase in incidence.11

A population-based study recruited a representative sample of 1000 people from two communities in northern Sweden to undergo upper endoscopy and confirmed the presence of intestinal metaplasia in 1.6% of the population studied.9
The incidence of Barrett’s oesophagus increases with age, the mean age at diagnosis being approximately 62 years for men and 68 years for women. It predominantly affects Caucasians12 and is more common in men than women, with a ratio of approximately 1.7:1.13
The risk of developing Barrett’s is related to increased frequency and duration of reflux symptoms.14 This appears to correlate with the well-known association between increased frequency, duration and severity of reflux symptoms, and increased risk of adenocarcinoma of the oesophagus. The incidence of Barrett’s oesophagus in patients with symptomatic gastro-oesophageal reflux disease (GORD) is between 5% and 12%.9,15 Evidence from one case series suggests that more than 60% of patients with Barrett’s oesophagus develop the condition secondary to chronic GORD, although other causes of oesophagitis, including non-steroidal anti-inflammatory drugs (NSAIDs), chemotherapy and viral infections are also associated with the disease. It does raise an intriguing possibility that a smaller proportion of patients can develop Barrett’s de novo in the absence of obvious symptomatic or perhaps even pathological reflux. Therefore, other factors that may catalyse changes at the oesophagogastric junction (OGJ) are obesity and cigarette smoking,which have been identified as risk factors for both Barrett’s oesophagus and progression to malignancy.16
A Swedish case–control study demonstrated that patients with recurrent reflux symptoms, when compared with asymptomatic patients, had an odds ratio of 7.7 for oesophageal adenocarcinoma and 2.0 for adenocarcinoma of the gastric cardia. Patients with severe long-standing symptoms had an odds ratio of 43.5 and 4.4 for oesophageal and cardia adenocarcinoma, respectively.17
Endoscopic assessment

The ‘Prague C and M criteria’, defined by an International Working Group on Barrett’s oesophagus, offers a validated method of disease classification based on endoscopic appearance.18 The extent of circumferential involvement (C value) in centimetres from the OGJ should be recorded, as should the maximum length (M value) of the Barrett’s segment, including tongue extensions but excluding isolated ‘islands’. These criteria have been shown to have a high degree of reliability between different endoscopists. The use of the terms long-segment Barrett’s (> 3 cm) and short-segment Barrett’s (< 3 cm) should now be discouraged.
It is crucial to make a thorough and systematic inspection of the mucosa in order to identify any macroscopic neoplastic disease. Water or 1% acetylcysteine should be used to remove blood, saliva and refluxate from the oesophagus, and sufficient insufflation should be ensured to clearly visualise any mucosal abnormalities. Particular care must be taken to identify the OGJ in patients with a hiatus hernia as it is easy to miss the distal extent of a Barrett’s segment in these patients. Clinicians should be aware that at endoscopic inspection most areas of early neoplasia and cancer are detected in an area around the 2 to 4 o’clock position in the endoscopist’s view.19

Despite meticulous inspection during white light video endoscopy, recognition of dysplasia and intramucosal cancer is difficult and subjective, even for experienced endoscopists. Guidelines therefore recommend that quadrantic biopsies are taken every 2 cm of Barrett’s oesophagus in addition to further biopsies from any areas of visible mucosal abnormality.1 Currently, the use of jumbo biopsy forceps is not recommended routinely.
This rigorous biopsy protocol, which is often poorly adhered to outside of specialist centres, samples less than 5% of the mucosa and may miss up to 57% of dysplasia.20,21 Advanced endoscopic imaging techniques may allow targeted biopsies from high-risk areas, improving diagnostic yield (Table 15.1).22–26 Potentially, these imaging tools may also facilitate targeted endoscopic resection of high-grade dysplasia (HGD) and intramucosal cancer.
Table 15.1
Advanced endoscopic imaging modalities being investigated for use in Barrett’s oesophagus surveillance programmes and for facilitation of targeted endoscopic resection
| Imaging modality | Concept | Reference |
| White light endoscopy | ||
| High-resolution magnification endoscopy (HRME) | Greater magnification and resolution than normal endoscopy allowing more detailed visualisation of the mucosa | May et al. (2004)137 |
| Chromoendoscopy | Topical application of dyes improves visualisation of mucosal surfaces. Examples: methylene blue – absorbed with different patterns into different types of mucosa; indigo carmine – accumulates in mucosal fissures accentuating surface topography | Canto et al. (2006)138 |
| Optical endoscopy | ||
| Autofluorescence imaging (AFI) | Short-wavelength light causes excitation of endogenous biological tissues with subsequent release of longer wavelength fluorescent light | Kara et al. (2005)139 |
| Narrow-band imaging (NBI) | Narrow-bandwidth green and blue light (with exclusion of red light) only superficially penetrates mucosa, improving visualisation of mucosal microvasculature and surface morphology | Curvers et al. (2008)25 |
| Confocal microscopy (CM) | Real-time magnification of the mucosa up to 1000-fold enables visualisation of cellular structures | Dunbar and Canto (2010)22 |
| Elastic scattering spectroscopy (ESS) | Elastic scattering of white light generates real-time morphological information about the size and shape of the cell nuclei and the degree of cellular crowding in the mucosa and submucosa | Qiu et al. (2010)23 |
| Trimodal imaging | Incorporates HRME, AFI and NBI in a single endoscope with ability to switch between modalities during procedure | Curvers et al. (2010, 2011)140,141 |
| Molecular imaging | Fluorescently tagged molecular probes bind selectively to metaplastic or dysplastic cells | Bird-Lieberman et al. (2012)142 |
Pathophysiology of Barrett’s oesophagus and progression to adenocarcinoma
It is currently believed that Barrett’s metaplasia develops as a mucosal ‘adaptive’ response to increased cell loss as a result of chronic inflammation, secondary to GORD. Oesophageal squamous epithelium is highly sensitive to acid, alkaline and biliary reflux, which all cause inflammation, with cell loss, necrosis and ulceration. There is strong evidence that the site of origin of Barrett’s metaplasia is a progenitor stem cell located in the submucosal oesophageal gland ducts, following demonstration that a p16 point mutation originating in microdissected squamous duct tissue was also present in adjoining metaplastic crypts.27 Duodenal and gastric reflux-induced ulceration and inflammation is believed to induce tumour suppressor gene mutations, typically p53 and p16, in some of the stem cell populations located in oesophageal gland squamous ducts, which are present throughout the entire length of the oesophagus. Following this initiation phase, multiple distinct clones of metaplastic tissue compete to colonise the oesophagus, creating a mosaic pattern of clones across the segment. Clonal expansion of populations with greater selective advantage, such as ability to survive in a markedly acid- or bile-rich environment, leads to dominant and widespread clones. Once initiated, the promotion and propagation of metaplastic clones is dependent on the surrounding microenvironment, particularly the presence of a chronic inflammatory cell infiltrate, characterised by T lymphocytes, and cytokines such as interleukin-1, tumour necrosis factor-α and transforming growth factor-β. These lead to an increase in cyclo-oxygenase-2, c-myc and cyclin D1, which increase proliferation and decrease apoptosis, and a reduction in E-cadherin, with resultant loss of cell adhesion and localisation of β-catenin to the nucleus.28 These molecular changes underlie the progression of Barrett’s oesophagus to cancer via the metaplasia–dysplasia–adenocarcinoma sequence (see Fig. 15.1).
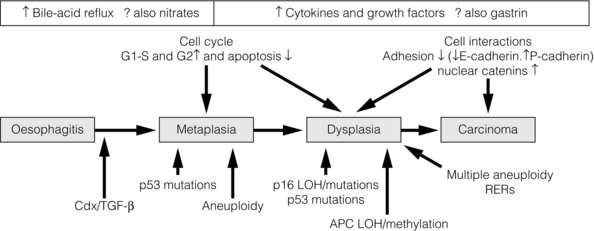
Figure 15.1 The metaplasia–dysplasia–adenocarcinoma sequence. There are histological stages of progression (shaded rectangles representing the clonal expansion of competing stem cells). In addition there are structural genetic changes in the form of mutations (vertical arrows) and environmental changes (white rectangles) driving cell cycle and cell adhesion biological sequelae. APC, adenomatous polyposis coli gene; Cdx, CauDal protein gene; LOH, loss of heterozygosity; RERs, random errors of replication; TGF-β, transforming growth factor-β. Adapted from Jankowski J, Harrison RF, Perry I et al. Barrett’s metaplasia. Lancet 2000; 356:2079–85. With permission from Elsevier.
Although traditionally thought of as an acquired condition, genetic factors may play a part in a small proportion of patients with Barrett’s metaplasia, as family and twin studies suggest a subgroup of individuals with a strong familial tendency to Barrett’s oesophagus.29,30 A family in the UK has been identified with a male index case with oesophageal adenocarcinoma, three brothers with Barrett’s-associated cancer or HGD, and six children with Barrett’s oesophagus.31 Linkage studies are being undertaken in order to further our understanding of this genetic inheritance. However, there are data to suggest Barrett’s is a polygenic disease with multiple contributing genes acting together.
Risk of cancer and mortality in Barrett’s oesophagus
Barrett’s oesophagus is accepted as a significant risk factor for adenocarcinoma of the oesophagus, although the risk of progression to adenocarcinoma and the risk of disease-specific mortality is low. A large number of studies have estimated the risk of adenocarcinoma arising from Barrett’s oesophagus, with very variable results.32–42 Former studies included small numbers of patients and were likely subject to publication bias, with results only being published if they showed a high incidence of cancer, leading to an overestimate of risk.43 Recently, two large population-based cohort studies have reported much lower annual rates of progression to adenocarcinoma (0.12–0.13% per year) than in former series (Table 15.2). It should be noted that these figures exclude carcinomas of the gastric cardia and also do not reflect progression to HGD. In addition, the two studies used different approaches to select patients with Barrett’s oesophagus. The study by Hvid-Jensen et al.33 identified patients with intestinal metaplasia (IM) from the Danish National Pathology Registry without corroboration with endoscopic findings. Therefore, potentially, patients may have been included who had a diagnosis of cardiac IM rather than true Barrett’s oesophagus, producing an incorrect denominator and leading to a slight underestimate of the risk of disease progression. Bhat et al.32 included patients with columnar-lined oesophagus (CLO) at endoscopy (although the validated Prague system was not used), which was corroborated histologically, and demonstrated an increased risk of progression in patients who had IM confirmed histologically at index endoscopy, compared to those with CLO without IM. This finding is in keeping with previous studies demonstrating a higher risk of disease progression in patients with confirmed IM.1,44,45
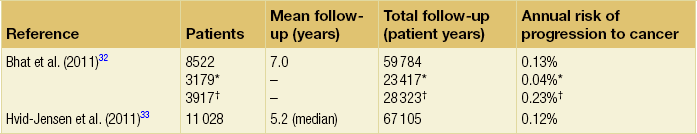
Previous studies have, however, suggested a significant geographical variation in the incidence of carcinoma arising in Barrett’s oesophagus in Western countries, with incidence rates in the UK almost double those in the USA.46 It is also worth noting that the population demographic in Denmark differs somewhat from the USA and UK, where rates of obesity are significantly higher and where a greater proportion of men, who are at higher risk of malignant progression, develop Barrett’s oesophagus.

A Danish population-based case–control study identified 11 028 patients from the national pathology registry with a diagnosis of intestinal metaplasia following oesophageal biopsy.33 Patients were followed for a median of 5.2 years. Compared to the general population, patients with Barrett’s oesophagus were found to have a relative risk of 11.3 for developing adenocarcinoma with an annual risk of 0.12%. Only 7.6% of the total oesophageal adenocarcinomas diagnosed nationwide over the study period had a previous diagnosis of Barrett’s oesophagus.
A recent meta-analysis reported a pooled estimate of the annual risk of cancer progression in non-dysplastic Barrett’s of 0.39% per year. Importantly, only eight of 47 studies that met all three quality criteria were included in this analysis; inclusion of the remaining studies significantly increased this figure.35 The risk in the UK following a meta-analysis is indicated as closer to 1%, higher than is reported in the USA.46

It is important to appreciate that while patients with Barrett’s oesophagus have an increased relative risk of adenocarcinoma, the majority of patients will die from other causes. A UK study has demonstrated an increase in both overall mortality rate and oesophageal cancer mortality rate in Barrett’s patients compared with the age- and sex-matched general population. However, only 10% of deaths were due to oesophageal cancer, while 49% were due to cardiorespiratory disease, especially ischaemic heart disease and bronchopneumonia, and 18% of deaths were due to other cancers.47
Natural history of dysplasia in Barrett’s oesophagus
When considering the natural history of dysplasia in Barrett’s oesophagus we must remember that in addition to potential problems with length of follow-up and sampling error at endoscopy, there is considerable inter- and intra-observer variation among experienced pathologists in the histological diagnosis of dysplasia. While pathologists can demonstrate acceptable levels of agreement in distinguishing HGD combined with carcinoma from no dysplasia combined with indefinite and low-grade dysplasia (kappa values of 0.8), there are much poorer levels of agreement in distinguishing between the four groups: indefinite for dysplasia, LGD, HGD and carcinoma (intra-observer kappa values of 0.43–0.64).48 Pathologists find it particularly difficult to separate inflammation in Barrett’s oesophagus from LGD. In this situation pathologists should be encouraged to make use of the indefinite for dysplasia category: such a diagnosis does not mean that the pathologist is uncertain, but rather that it is not possible, with confidence, to exclude LGD in inflamed material. The diagnosis of HGD has serious implications for patient management and the diagnosis should be confirmed by two expert pathologists.

A systematic review involving a total of 1488 patients with Barrett’s oesophagus reported that LGD was present at initial endoscopy in 169 patients (11%) and HGD in 18 patients (1.2%); 1301 (87%) had metaplasia with no dysplasia.49
Low-grade dysplasia
The natural history of LGD is not fully understood and reported rates of regression/progression vary considerably, reflecting the diagnostic difficulties discussed above. Sharma et al.50 followed 156 patients for a mean of 4.1 years and reported progression to HGD or cancer in 13%, regression in 66% and stable LGD in 21%. A more recent prospective cohort study of 713 Barrett’s patients, including 111 with LGD, reported that compared to non- dysplastic disease, LGD was a significant risk factor for progression to HGD or adenocarcinoma (relative risk (RR) 9.7; 95% confidence interval (CI) 4.4–21.5).51 Similarly, in their large population- based study, Hvid-Jensen et al.33 reported that the relative risk of oesophageal cancer among those who had LGD at baseline, as compared to those without LGD at baseline, was 4.8 (95% CI 2.6–8.8). The annual risk of progression to HGD or cancer was found to be 1.27% for those with LGD at baseline. Bhat et al.32 reported a hazard ratio of 5.67% (95% CI 3.77–8.33) for patients with LGD compared to no dysplasia. However, a recent study by Wani et al.,52 which followed up 210 patients with Barrett’s oesophagus with or without LGD for a mean of 6.2 years, found no associations of presence of prevalent, incident or persistent LGD, or the extent of LGD, with progression rates.
Bergman and colleagues recently demonstrated that LGD is over-diagnosed by non-specialist pathologists and argued that its true significance might have been underestimated by many reported series.53 In their study, 1198 patients underwent Barrett’s surveillance at six non-specialist hospitals, identifying 147 (12.5%) patients with LGD. However, only eight (0.7%) patients were deemed to have LGD following histological review by two external expert gastrointestinal pathologists. The majority of diagnoses were reclassified as non- dysplastic Barrett’s oesophagus. During a mean follow-up period of 51 months, 42% of patients with LGD diagnosed by consensus expert pathologists demonstrated progression to either carcinoma or HGD, and 2.2% regressed to non-dysplastic Barrett’s oesophagus.53

High-grade dysplasia
Studies reporting the natural history of HGD have also reported widely differing results. Reid et al.54 followed 76 patients for 5 years and reported that 59% developed adenocarcinoma. In a study of 100 patients with HGD, 66 of whom underwent surveillance, 3 of 24 patients (13%) with focal HGD and 17 of 42 patients (40%) with diffuse HGD developed carcinoma after a mean follow-up of 41 and 23 months, respectively.55
An important question to consider is what proportion of patients with a diagnosis of HGD who undergo oesophagectomy have an occult cancer detected in the resected specimen? Table 15.3 shows reported rates in the literature of 0–73%: overall the rate appears to be approximately 40%.56–72 Patients with visible, nodular HGD appear at greatest risk of harbouring coexisting cancer.73,74 This emphasises the fact that patients with HGD may be harbouring an undetected cancer and confirms the need for complete staging in these patients.
Table 15.3
Studies reporting the incidence of adenocarcinoma in resected specimens following oesophagectomy for high-grade dysplasia
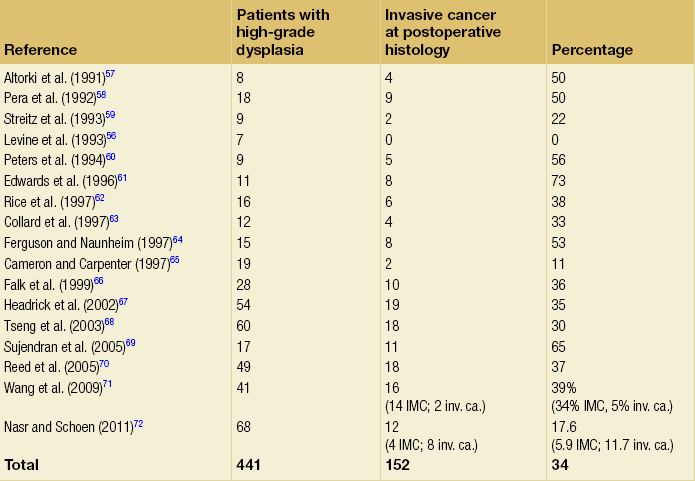
IMC, intramucosal cancer; inv. ca., invasive cancer (denotes invasion into submucosa or beyond).
Given that endotherapy is becoming a recognised treatment option for focal intramucosal cancers (T1a), a more pertinent question to ask might be: what is the prevalence of submucosal invasive cancer at oesophagectomy for HGD? The majority of studies in Table 15.3 make no attempt to separate intramucosal cancer (IMC) from more advanced lesions; however, some more recent reports suggest that rates of invasive cancer (submucosa or beyond) are considerably lower than 40%. Wang et al.71 retrospectively assessed 60 patients (41 with preoperative HGD and 19 with preoperative IMC) who underwent oesophagectomy. The overall rate of submucosal cancer was 6.7%, with a rate of 5% in patients with preoperative HGD and 11% in patients with preoperative IMC. Only one patient (1.7%) had nodal metastasis. Another recent study found the rate of invasive adenocarcinoma (excluding IMC) in association with Barrett’s HGD to be 11.7% (8/68), with 5.9% having occult cancer.75
Although some HGD may be stable or even regress, between 15% and 59% will progress to adenocarcinoma over 5 years. However, if detailed biopsy mapping endoscopies showed no previous HGD (prevalent HGD), then the detection of new HGD (incident HGD) is associated with a risk of subsequent progression to cancer of only between 3% and 5% per year.73,76 This area is being actively discussed in the Barrett’s Dysplasia and Cancer Taskforce (BAD CAT) group.
Risk factors for progression to cancer
The length of Barrett’s segment has been shown to be a significant risk factor for progression to cancer, a doubling of length increasing the risk 1.7-fold.77 The extent of HGD and/or LGD also appears to be a risk factor for progression to adenocarcinoma.55,78
Importantly, in a prospective longitudinal cohort study, individuals with Barrett’s oesophagus who were regularly taking aspirin or other NSAIDs were found to have a significantly lower 5-year cumulative incidence of adenocarcinoma compared with individuals not taking NSAIDs (6.6% and 14.3%, respectively), suggesting that this may be an effective chemotherapeutic intervention.79 An ongoing phase III multicentre randomised controlled trial (RCT), the AspECT trial (Aspirin and Esomeprazole Chemoprevention in Barrett’s Metaplasia), designed to test this hypothesis is due to report in 2016. The primary aim of this study is to determine whether acid suppression with proton-pump inhibition (high dose vs. low dose) with or without aspirin can reduce mortality or the conversion from Barrett’s metaplasia to HGD or adenocarcinoma. Both high- and low-dose acid suppression are being investigated as there remains doubt about the optimal dose of proton-pump inhibitor (PPI) to use, especially given the fact that Barrett’s mucosa is relatively insensitive, thus rendering symptoms unreliable. There is an argument that incomplete acid suppression might increase the risk of cancer by exposing the mucosa to short pulses of acid, thus stimulating the proliferation of abnormal cells. In contrast, there is some epidemiological evidence that high-dose proton-pump inhibition might increase the risk of cancer as bile acid might become cytotoxic at neutral pH. In addition, there have been fears that PPI-induced hypergastrinaemia could stimulate hyperproliferation of Barrett’s epithelium.80,81 Although this risk is yet to be evaluated in vivo, it appears more likely that gastrin induces epithelial restitution in Barrett’s oesophagus, without stimulation of clonal expansion or disease progression.82

Screening for Barrett’s oesophagus and adenocarcinoma using molecular markers
It is accepted that GORD is a significant risk factor for the development of adenocarcinoma, with a well-known Swedish case–control study demonstrating a 44-fold increased relative risk in individuals with frequent heartburn of greater than 20 years’ duration.17 This has led to the suggestion that screening individuals with chronic reflux symptoms to detect Barrett’s oesophagus and cancer may be of benefit. However, it is important to appreciate two flaws in this concept: firstly, approximately 40% of individuals with cancer in the series mentioned above denied frequent heartburn; secondly, a significant proportion of individuals with Barrett’s oesophagus are asymptomatic. In addition, Barrett’s patients experience less heartburn and use PPIs less frequently compared with controls.2,83,84
The endoscopic screening of individuals with chronic reflux symptoms to detect either Barrett’s or cancer is not currently recommended in the UK or USA.1,2 This is because of the low absolute risk of developing adenocarcinoma in individuals with chronic reflux, combined with the knowledge that most individuals with Barrett’s oesophagus die from causes other than oesophageal cancer. There are also concerns about the cost-effectiveness and invasiveness of endoscopy as a screening tool.
Several attempts have been made to develop a scoring system using patient demographics and symptoms to predict the presence of Barrett’s oesophagus for screening purposes.85,86 However, interest in these risk prediction strategies has declined due to inability to generate sufficient sensitivity and specificity.
Mutations in the p53 tumour suppressor gene are widely found in dysplastic Barrett’s oesophagus and oesophageal cancer. Younes et al.87 found p53 mutation in 9% of Barrett’s patients with LGD, 55% of patients with HGD and 87% of patients with carcinoma: no patients without dysplasia had a p53 mutation. Importantly, in a further study, 56% of patients with LGD and p53 mutation progressed to HGD or carcinoma, whereas no patient with LGD without p53 mutation progressed.88 Similarly, Reid et al.89 demonstrated that loss of heterozygosity of gene 17 (p53) was found in 6% of patients without dysplasia, 20% of patients with LGD and 57% of patients with HGD. Patients with loss of heterozygosity had a 16-fold increased risk of cancer after 3 years. These results have led to the suggestion that the subgroup of patients with low-grade or indefinite dysplasia and p53 mutation should be subjected to more rigorous surveillance protocols. However, it is important to remember that not all oesophageal adenocarcinomas express p53, and patients without expression can progress to cancer.
Other markers that have been identified as conferring a high risk of progression are p16 mutations,90 cyclin D1 overexpression,91 flow cytometry abnormalities such as aneuploidy and increase in the G2/tetraploidy fraction of DNA content,92 and reduced expression of E-cadherin, with resultant loss of cell adhesion and localisation of β-catenin to the nucleus.93
Several clinical trials are under way, including the Chemoprevention of Premalignant Intestinal Neoplasia (ChOPIN) trial and the Barrett’s Oesophagus Screening Trial (BEST2) trial, which aim to explore non-invasive methods of screening for malignant progression. ChOPIN aims to detect a panel of predictive serum biomarkers, whereas BEST2 is a case–control trial investigating the potential of a non-endoscopic immunocytological device (Cytosponge).94,95 This trial requires patients to swallow a small capsule that dissolves into a 3-cm sponge in the stomach and is then withdrawn through the oesophagus. Oesophageal cells are assessed for a range of predictive biomarkers, including TFF3 positivity (the principal end-point) as well as ploidy, Mcm2, cyclin A, TP53 and methylation. Cost data and the impact of screening on psychosocial well-being are also being evaluated. It is hoped that non-invasive screening tests such as these could enable safe, accurate and cost-effective population-based screening in the future.

Surveillance of non-dysplastic disease
The central concept of surveillance is that regular endoscopic examination and biopsy will allow the detection of cancer at an early asymptomatic stage, thereby resulting in better treatment outcomes. Several small retrospective studies have demonstrated a survival benefit associated with surveillance-detected cancers.59,60,96–98 However, other series have failed to support these findings.99 These studies may be subject to both selection bias and length bias, concerns that prompted the ongoing Barrett’s Oesophagus Surveillance Study (BOSS), which aims to define the objective value of endoscopic surveillance and the most appropriate surveillance protocol. BOSS randomises patients with at least 1 cm of circumferential or 2 cm non-circumferential Barrett’s oesophagus to either endoscopic surveillance with protocol biopsy1 (n = 1250) or endoscopy at the time of need (n = 1250), the latter group being discharged unless they develop new symptoms or alarming symptoms.


Effect of medical therapy and antireflux surgery
It has been shown that long-term acid suppression with PPIs can lead to an improvement in Barrett’s metaplasia. A study of 23 patients following a regimen of omeprazole 40 mg daily for 2 years demonstrated a significant reduction in the length of columnar mucosa, an increase in squamous islands within the columnar epithelium and a reduction in the proportion of sulphomucin-rich intestinal metaplasia.100 More recently, a study of 188 patients followed for up to 13 years (mean 5 years) reported development of squamous islands in 48% of patients, although the mean length of Barrett’s segment was not reduced and no patients regressed to squamous mucosa.101

A randomised double-blind trial of omeprazole 80 mg daily versus ranitidine 300 mg daily in patients with proven Barrett’s oesophagus and gastro-oesophageal reflux disease demonstrated a reduction in the length and surface area of columnar metaplasia in the omeprazole group but not in the ranitidine group. Both treatments successfully controlled reflux symptoms.102
The effect of antireflux surgery on Barrett’s metaplasia has proved a controversial subject. Selected series have demonstrated regression in Barrett’s length in 14–35% of patients, with complete regression of LGD in 44–93% of patients.103–105 However, the RCT evidence has not supported these findings, at best demonstrating a reduction in Barrett’s length without achieving complete regression of dysplasia.105 In addition, studies have failed to show an absolute reduction in rates of oesophageal adenocarcinoma following antireflux surgery.

A meta-analysis comparing the reported incidence of adenocarcinoma in Barrett’s patients after antireflux surgery with patients treated medically found no statistically significant difference in the incidence rates of 3.8 and 5.3 per 1000 patient years, respectively.106 A recent systematic review reported a statistically significant lower incidence of adenocarcinoma after antireflux surgery compared with medical therapy (2.8 vs. 6.3 per 1000 patient years, P = 0.03); however, when uncontrolled case series were excluded and the analysis was confined to randomised trials and cohort studies there was no significant difference between the two treatments (4.4 vs. 6.5 per 1000 patient years, P = 0.32).49 Accordingly, at present there is insufficient evidence to recommend antireflux surgery over proton-pump inhibition as a cancer-preventing procedure.
Endotherapy
Endotherapy, including endoscopic resection and ablative therapies, is indicated in selected patients with HGD, intramucosal cancer (T1a) and early submucosal cancers (T1b). The potential role of endotherapy in early oesophageal cancer, including the important diagnostic role of endoscopic resection, is addressed in Chapter 6 and so will not be discussed further here.
Endoscopic resection
Endoscopic mucosal resection aims to remove the mucosa and submucosa down to the muscularis and for this reason the term endoscopic resection (ER) is now preferred. ER is indicated for removal of focal HGD. Piecemeal resection is required for lesions greater than 2 cm, with meticulous care being taken to ensure completeness of excision. Complications are uncommon – bleeding (3%) and perforation (0.1–5%) – and most can be managed endoscopically.107
ER has been shown to achieve remission in 82.5–95% of patients with HGD, but may be associated with metachronous lesions or disease recurrence in up to 14% of patients within 12 months, and 21.5% of patients over 5 years.108–111 Factors associated with recurrence include piecemeal resection, long-segment Barrett’s oesophagus (> 5 cm), delayed treatment of HGD (> 10 months), multifocal disease and omission of adjuvant ablative therapy.108 Recurrent disease necessitates re-treatment, which can be successful and provide long-term disease control but which may have higher complication rates.
Several trials have reported circumferential ER for removal of widespread multifocal disease; however, this practice has led to high rates of post-treatment stricture formation (17–26%) and higher rates of perforation (3%) and is therefore not widely recommended.112,113
Endoscopic ablation
RFA and PDT deliver an even distribution of treatment over a consistent therapeutic depth and can be readily applied to large areas of circumferential disease. Both have been shown to be highly efficacious at eradicating dysplastic Barrett’s oesophagus.114–124 However, RFA is now widely regarded as the first-line therapy due to its relative ease of administration, more favourable side-effect profile and low rates of recurrent dysplasia.123

Shaheen et al.115 randomised 127 patients with dysplastic Barrett’s oesophagus in a 2:1 ratio to receive either RFA or sham procedure. Complete eradication of LGD occurred in 90.5% (ablation group) compared to 22.7% (control group) (P < 0.001) at 1 year. Complete eradication of HGD occurred in 81.0% (ablation group) versus 19.0% (control group) (P < 0.001). RFA decreased both the likelihood of disease progression (3.6% vs. 16.3%, P = 0.03) and cancer (1.2% vs. 9.3%, P = 0.045). Recent follow-up data after 3 years have shown that this effect is durable.114
There is RCT evidence supporting the efficacy of both MPEC and APC for treatment of dysplastic Barrett’s although, compared to RFA, these techniques are less user-friendly for treating large Barrett’s segments and treatment depths are less consistent. In addition, they may be more likely to be complicated by strictures and the development of buried glandular mucosa beneath neosquamous epithelium.125 Currently these techniques are favoured for ‘touch-up’ therapy in patients with small patches of persistent metaplasia following previous RFA treatments. An RCT comparing the two thermal techniques, MPEC and APC, found no significant advantage with either technique.125
Management of LGD
The detection of LGD should prompt a course of high-dose acid suppression with a PPI for 8–12 weeks followed by repeat endoscopy with extensive biopsies. If LGD persists then surveillance endoscopy should be repeated at 6-monthly intervals and the patient should remain on a PPI. If regression to metaplasia without dysplasia occurs on two consecutive examinations then the surveillance interval may return to 2-yearly.1
Management of HGD
Following referral to specialist centres patients with HGD should be re-biopsied using a Seattle biopsy protocol: quadrantic biopsies every 1 cm with further targeted biopsies from suspicious areas. A large number of samples should be taken – up to 84 biopsies from a single patient have been reported.56 Patients should be carefully staged, including use of diagnostic ER, and patients with nodular disease should be considered at particular risk of harbouring occult invasive disease.73,74
Falk et al.66 demonstrated that 38% of cancers were missed when taking quadrantic biopsies every 2 cm from patients with HGD. Jumbo biopsy forceps made little difference to detection rates (67% vs. 62%). Similarly, Cameron and Carpenter65 found 2 of 19 (10.5%) unsuspected adenocarcinomas following quadrantic 2-cm biopsies in patients who subsequently underwent oesophagectomy. Reid et al.126 compared a quadrantic 2-cm biopsy protocol to biopsies taken at 1-cm intervals in 45 patients diagnosed with HGD who subsequently developed cancer. The 2-cm protocol missed 50% of the cancers that were detected by the 1-cm protocol in Barrett’s segments 2 cm or more without visible lesions. This more intensive biopsy regimen is recommended in patients with HGD, at 3-monthly intervals. However, it is important to consider that Barrett’s adenocarcinomas may still be missed in up to 29% of cases.127
Most units favour a policy of focal lesion resection using ER followed by ablation of the entire Barrett’s segment using RFA to destroy the neoplastic field change in adjacent metaplasia. ER improves histological assessment and aids detection of occult adenocarcinoma,128,129 but the need for subsequent ablation of the surrounding non-dysplastic Barrett’s oesophagus is controversial. There are no RCTs addressing this directly; however, there is some evidence to suggest lower recurrence rates if ER is used in conjunction with whole segment ablation.44,66
The decision between endotherapy and oesophagectomy is controversial and dependent on patient comorbidity and the nature of the disease: HGD versus intramucosal cancer; unifocal versus multifocal; long- versus short-segment Barrett’s; presence or absence of lymphovascular invasion; and grade of differentiation. Endotherapy can effectively eradicate HGD, with low rates of disease recurrence in the medium term (long-term data await ed).114,115,130 In addition, any recurrent disease can usually be managed endoscopically, or if necessary surgically, without excess mortality.131,132 However, advocates of surgery point to the risk of occult adenocarcinoma, particularly in nodular disease, and the possible risk of under-staging of disease using a non-operative approach. In addition, there is evidence that prophylactic oesophagectomy in HGD is associated with a lower risk of operative mortality than routine oesophagectomy as patients are typically younger with fewer comorbidities, and have not undergone neoadjuvant therapy.68
The Surveillance Epidemiology and End Results (SEER) database of the US National Cancer Institute found no difference in survival between patients with HGD or stage 1 (T1N0M0) tumours treated by endoscopic therapy compared to radical surgery.133,134 However, most studies directly comparing radical surgery and endoscopic therapy have been severely limited by selection bias.133–135 Well-designed multicentre randomised trials are awaited.

There are no randomised trials comparing surgery and endotherapy in the management of HGD and these studies should be undertaken as a matter of urgency. Endotherapy is a viable treatment option for patients with HGD, with close surveillance, early endoscopic treatment of recurrence, and progression to oesophagectomy if indicated. A recent Cochrane review was unable to recommend either surgery or endotherapy as first-line treatment in HGD due to the paucity of high-level evidence and the multitude of contributing factors.136
References
1. Watson A, Heading RC, Shepherd NA. A Report of the Working Party of the British Society of Gastroenterology. Guidelines for the diagnosis and management of Barrett’s columnar-lined oesophagus. 2005.
2. Wang, K.K., Sampliner, R.E., Practice Parameters Committee of the American College of Gastroenterology, Updated guidelines 2008 for the diagnosis, surveillance and therapy of Barrett’s esophagus. Am J Gastroenterol. 2008;103(3):788–797. 18341497
3. Harrison, R., Perry, I., Haddadin, W., et al, Detection of intestinal metaplasia in Barrett’s esophagus: an observational comparator study suggests the need for a minimum of eight biopsies. Am J Gastroenterol. 2007;102(6):1154–1161. 17433019
4. Rex, D.K., Cummings, O.W., Shaw, M., et al, Screening for Barrett’s esophagus in colonoscopy patients with and without heartburn. Gastroenterology. 2003;125(6):1670–1677. 14724819
5. Vakil, N., van Zanten, S.V., Kahrilas, P., Global Consensus Group, The Montreal definition and classification of gastroesophageal reflux disease: a global evidence-based consensus. Am J Gastroenterol. 2006;101(8):1900–1920 quiz 1943. 16928254
6. Cameron, A.J., Zinsmeister, A.R., Ballard, D.J., et al, Prevalence of columnar-lined (Barrett’s) esophagus. Comparison of population-based clinical and autopsy findings. Gastroenterology. 1990;99(4):918–922. 2394347
7. Cameron, A.J., Lomboy, C.T., Barrett’s esophagus: age, prevalence, and extent of columnar epithelium. Gastroenterology. 1992;103(4):1241–1245. 1397881
8. Jankowski, J., Barr, H., Wang, K., et al, Diagnosis and management of Barrett’s oesophagus. Br Med J 2010; 341:4551. 20833742
9. Ronkainen, J., Aro, P., Storskrubb, T., et al, Prevalence of Barrett’s esophagus in the general population: an endoscopic study. Gastroenterology. 2005;129(6):1825–1831. 16344051 This population-based study recruited a representative sample of 1000 people from two communities in northern Sweden to undergo upper endoscopy and confirmed the presence of intestinal metaplasia in 1.6% of the population studied.
10. Gerson, L.B., Banerjee, S., Screening for Barrett’s esophagus in asymptomatic women. Gastrointest Endosc. 2009;70(5):867–873. 19640517
11. van Soest, E.M., Dieleman, J.P., Siersema, P.D., et al, Increasing incidence of Barrett’s oesophagus in the general population. Gut. 2005;54(8):1062–1066. 15857935
12. Spechler, S.J., Zeroogian, J.M., Antonioli, D.A., et al, Prevalence of metaplasia at the gastro-oesophageal junction. Lancet. 1994;344(8936):1533–1536. 7983953
13. Caygill, C.P., Watson, A., Reed, P.I., et al, UK National Barrett’s Oesophagus Registry (UKBOR) and the 27 Participating Centres. Characteristics and regional variations of patients with Barrett’s oesophagus in the UK. Eur J Gastroenterol Hepatol. 2003;15(11):1217–1222. 14560156
14. Eisen, G.M., Sandler, R.S., Murray, S., et al, The relationship between gastroesophageal reflux disease and its complications with Barrett’s esophagus. Am J Gastroenterol. 1997;92(1):27–31. 8995932
15. Winters, C., Jr., Spurling, T.J., Chobanian, S.J., et al, Barrett’s esophagus. A prevalent, occult complication of gastroesophageal reflux disease. Gastroenterology. 1987;92(1):118–124. 3781178
16. Smith, K.J., O’Brien, S.M., Smithers, B.M., et al, Interactions among smoking, obesity, and symptoms of acid reflux in Barrett’s esophagus. Cancer Epidemiol Biomarkers Prev. 2005;14(11, Pt 1):2481–2486. 16284367
17. Lagergren, J., Bergstrom, R., Lindgren, A., et al, Symptomatic gastroesophageal reflux as a risk factor for esophageal adenocarcinoma. N Engl J Med. 1999;340(11):825–831. 10080844
18. Sharma, P., Dent, J., Armstrong, D., et al, The development and validation of an endoscopic grading system for Barrett’s esophagus: the Prague C & M criteria. Gastroenterology. 2006;131(5):1392–1399. 17101315
19. Curvers, W.L., Bansal, A., Sharma, P., et al, Endoscopic work-up of early Barrett’s neoplasia. Endoscopy. 2008;40(12):1000–1007. 19065483
20. Singh, R., Ragunath, K., Jankowski, J., Barrett’s esophagus: diagnosis, screening, surveillance, and controversies. Gut Liver. 2007;1(2):93–100. 20485625
21. Vieth, M., Ell, C., Gossner, L., et al, Histological analysis of endoscopic resection specimens from 326 patients with Barrett’s esophagus and early neoplasia. Endoscopy. 2004;36(9):776–781. 15326572
22. Dunbar, K.B., Canto, M.I., Confocal laser endomicroscopy in Barrett’s esophagus and endoscopically inapparent Barrett’s neoplasia: a prospective, randomized, double-blind, controlled, crossover trial. Gastrointest Endosc. 2010;72(3):668. 20801292
23. Qiu, L., Pleskow, D.K., Chuttani, R., et al, Multispectral scanning during endoscopy guides biopsy of dysplasia in Barrett’s esophagus. Nat Med. 2010;16(5):603–606. 20383155
24. Kara, M.A., Smits, M.E., Rosmolen, W.D., et al, A randomized crossover study comparing light-induced fluorescence endoscopy with standard videoendoscopy for the detection of early neoplasia in Barrett’s esophagus. Gastrointest Endosc. 2005;61(6):671–678. 15855970
25. Curvers, W., Baak, L., Kiesslich, R., et al, Chromoendoscopy and narrow-band imaging compared with high-resolution magnification endoscopy in Barrett’s esophagus. Gastroenterology. 2008;134(3):670–679. 18242603
26. Thomas, T., Singh, R., Ragunath, K., Trimodal imaging-assisted endoscopic mucosal resection of early Barrett’s neoplasia. Surg Endosc. 2009;23(7):1609–1613. 19296171
27. Leedham, S.J., Preston, S.L., McDonald, S.A., et al, Individual crypt genetic heterogeneity and the origin of metaplastic glandular epithelium in human Barrett’s oesophagus. Gut. 2008;57(8):1041–1048. 18305067
28. Jankowski, J.A., Harrison, R.F., Perry, I., et al, Barrett’s metaplasia. Lancet. 2000;356(9247):2079–2085. 11145505
29. Cameron, A.J., Lagergren, J., Henriksson, C., et al, Gastroesophageal reflux disease in monozygotic and dizygotic twins. Gastroenterology. 2002;122(1):55–59. 11781280
30. Mohammed, I., Cherkas, L.F., Riley, S.A., et al, Genetic influences in gastro-oesophageal reflux disease: a twin study. Gut. 2003;52(8):1085–1089. 12865263
31. Groves, C., Jankowski, J., Barker, F., et al, A family history of Barrett’s oesophagus: another risk factor? Scand J Gastroenterol. 2005;40(9):1127–1128. 16211720
32. Bhat, S., Coleman, H.G., Yousef, F., et al, Risk of malignant progression in Barrett’s esophagus patients: results from a large population-based study. J Natl Cancer Inst. 2011;103(13):1049–1057. 21680910
33. Hvid-Jensen, F., Pedersen, L., Drewes, A.M., et al, Incidence of adenocarcinoma among patients with Barrett’s esophagus. N Engl J Med. 2011;365(15):1375–1383. 21995385 This Danish population-based case–control study is the largest study following patients with intestinal metaplasia. Over a median of 5.2 years, patients with IM were found to have a relative risk of 11.3 for developing adenocarcinoma with an annual risk of just 0.12%. Only 7.6% of the total oesophageal adenocarcinomas diagnosed nationwide over the study period had a previous diagnosis of Barrett’s oesophagus.
34. Sikkema, M., de Jonge, P.J., Steyerberg, E.W., et al, Risk of esophageal adenocarcinoma and mortality in patients with Barrett’s esophagus: a systematic review and meta-analysis. Clin Gastroenterol Hepatol. 2010;8(3):235–244. 19850156
35. Yousef, F., Cardwell, C., Cantwell, M.M., et al, The incidence of esophageal cancer and high-grade dysplasia in Barrett’s esophagus: a systematic review and meta-analysis. Am J Epidemiol. 2008;168(3):237–249. 18550563
36. Robertson, C.S., Mayberry, J.F., Nicholson, D.A., et al, Value of endoscopic surveillance in the detection of neoplastic change in Barrett’s oesophagus. Br J Surg. 1988;75(8):760–763. 3167523
37. Miros, M., Kerlin, P., Walker, N., Only patients with dysplasia progress to adenocarcinoma in Barrett’s oesophagus. Gut. 1991;32(12):1441–1446. 1773946
38. Iftikhar, S.Y., James, P.D., Steele, R.J., et al, Length of Barrett’s oesophagus: an important factor in the development of dysplasia and adenocarcinoma. Gut. 1992;33(9):1155–1158. 1427364
39. Wright, T.A., Gray, M.R., Morris, A.I., et al, Cost effectiveness of detecting Barrett’s cancer. Gut. 1996;39(4):574–579. 8944568
40. Drewitz, D.J., Sampliner, R.E., Garewal, H.S., The incidence of adenocarcinoma in Barrett’s esophagus: a prospective study of 170 patients followed 4.8 years. Am J Gastroenterol. 1997;92(2):212–215. 9040193
41. Katz, D., Rothstein, R., Schned, A., et al, The development of dysplasia and adenocarcinoma during endoscopic surveillance of Barrett’s esophagus. Am J Gastroenterol. 1998;93(4):536–541. 9576444
42. Hage, M., Siersema, P.D., van Dekken, H., et al, Oesophageal cancer incidence and mortality in patients with long-segment Barrett’s oesophagus after a mean follow-up of 12.7 years. Scand J Gastroenterol. 2004;39(12):1175–1179. 15742992
43. Shaheen, N.J., Crosby, M.A., Bozymski, E.M., et al, Is there publication bias in the reporting of cancer risk in Barrett’s esophagus? Gastroenterology. 2000;119(2):333–338. 10930368
44. Das, D., Ishaq, S., Harrison, R., et al, Management of Barrett’s esophagus in the UK: overtreated and underbiopsied but improved by the introduction of a national randomized trial. Am J Gastroenterol. 2008;103(5):1079–1089. 18445097
45. Cook, M.B., Wild, C.P., Everett, S.M., et al, Risk of mortality and cancer incidence in Barrett’s esophagus. Cancer Epidemiol Biomarkers Prev. 2007;16(10):2090–2096. 17890521
46. Jankowski, J.A., Provenzale, D., Moayyedi, P., Esophageal adenocarcinoma arising from Barrett’s metaplasia has regional variations in the west. Gastroenterology. 2002;122(2):588–590. 11845804
47. Moayyedi, P., Burch, N., Akhtar-Danesh, N., et al, Mortality rates in patients with Barrett’s oesophagus. Aliment Pharmacol Ther. 2008;27(4):316–320. 18062791
48. Montgomery, E., Bronner, M.P., Greenson, J.K., et al, Are ulcers a marker for invasive carcinoma in Barrett’s esophagus? Data from a diagnostic variability study with clinical follow-up. Am J Gastroenterol. 2002;97(1):27–31. 11808966
49. Chang, E.Y., Morris, C.D., Seltman, A.K., et al, The effect of antireflux surgery on esophageal carcinogenesis in patients with Barrett esophagus: a systematic review. Ann Surg. 2007;246(1):11–21. 17592284 A systematic review that failed to demonstrate a lower incidence of adenocarcinoma after antireflux surgery compared with medical therapy after excluding uncontrolled case series from analysis.
50. Sharma, P., Falk, G.W., Weston, A.P., et al, Dysplasia and cancer in a large multicenter cohort of patients with Barrett’s esophagus. Clin Gastroenterol Hepatol. 2006;4(5):566–572. 16630761
51. Sikkema, M., Looman, C.W., Steyerberg, E.W., et al, Predictors for neoplastic progression in patients with Barrett’s esophagus: a prospective cohort study. Am J Gastroenterol. 2011;106(7):1231–1238. 21577245
52. Wani, S., Falk, G.W., Post, J., et al, Risk factors for progression of low-grade dysplasia in patients with Barrett’s esophagus. Am J Gastroenterol. 2011;141(4):1179–1186 Oct. 20461069
53. Curvers, W.L., ten Kate, F.J., Krishnadath, K.K., et al, Low-grade dysplasia in Barrett’s esophagus: overdiagnosed and underestimated. Am J Gastroenterol. 2010;105(7):1523–1530. 20461069
54. Reid, B.J., Levine, D.S., Longton, G., et al, Predictors of progression to cancer in Barrett’s esophagus: baseline histology and flow cytometry identify low- and high-risk patient subsets. Am J Gastroenterol. 2000;95(7):1669–1676. 10925966
55. Buttar, N.S., Wang, K.K., Sebo, T.J., et al, Extent of high-grade dysplasia in Barrett’s esophagus correlates with risk of adenocarcinoma. Gastroenterology. 2001;120(7):1630–1639. 11375945
56. Levine, D.S., Haggitt, R.C., Blount, P.L., et al, An endoscopic biopsy protocol can differentiate high-grade dysplasia from early adenocarcinoma in Barrett’s esophagus. Gastroenterology. 1993;105(1):40–50. 8514061
57. Altorki, N.K., Sunagawa, M., Little, A.G., et al, High-grade dysplasia in the columnar-lined esophagus. Am J Surg. 1991;161(1):97–100. 1987863
58. Pera, M., Trastek, V.F., Carpenter, H.A., et al, Barrett’s esophagus with high-grade dysplasia: an indication for esophagectomy? Ann Thorac Surg. 1992;54(2):199–204. 1637206
59. Streitz, J.M., Jr., Andrews, C.W., Jr., Ellis, F.H., Jr., Endoscopic surveillance of Barrett’s esophagus. Does it help? J Thorac Cardiovasc Surg. 1993;105(3):383–388. 8445916
60. Peters, J.H., Clark, G.W., Ireland, A.P., et al, Outcome of adenocarcinoma arising in Barrett’s esophagus in endoscopically surveyed and nonsurveyed patients. J Thorac Cardiovasc Surg. 1994;108(5):813–822. 7967662
61. Edwards, M.J., Gable, D.R., Lentsch, A.B., et al, The rationale for esophagectomy as the optimal therapy for Barrett’s esophagus with high-grade dysplasia. Ann Surg. 1996;223(5):585–591. 8651749
62. Rice, T.W., Adelstein, D.J., Zuccaro, G., et al, Advances in the treatment of esophageal carcinoma. Gastroenterologist. 1997;5(4):278–294. 9436004
63. Collard, J.M., Romagnoli, R., Hermans, B.P., et al, Radical esophageal resection for adenocarcinoma arising in Barrett’s esophagus. Am J Surg. 1997;174(3):307–311. 9324143
64. Ferguson, M.K., Naunheim, K.S., Resection for Barrett’s mucosa with high-grade dysplasia: implications for prophylactic photodynamic therapy. J Thorac Cardiovasc Surg. 1997;114(5):824–829. 9375613
65. Cameron, A.J., Carpenter, H.A., Barrett’s esophagus, high-grade dysplasia, and early adenocarcinoma: a pathological study. Am J Gastroenterol. 1997;92(4):586–591. 9128304
66. Falk, G.W., Rice, T.W., Goldblum, J.R., et al, Jumbo biopsy forceps protocol still misses unsuspected cancer in Barrett’s esophagus with high-grade dysplasia. Gastrointest Endosc. 1999;49(2):170–176. 9925694
67. Headrick, J.R., Nichols, F.C., 3rd., Miller, D.L., et al, High-grade esophageal dysplasia: long-term survival and quality of life after esophagectomy. Ann Thorac Surg. 2002;73(6):1697–1703. 12078755
68. Tseng, E.E., Wu, T.T., Yeo, C.J., et al, Barrett’s esophagus with high grade dysplasia: surgical results and long-term outcome – an update. J Gastrointest Surg. 2003;7(2):164–171. 12600440
69. Sujendran, V., Sica, G., Warren, B., et al, Oesophagectomy remains the gold standard for treatment of high-grade dysplasia in Barrett’s oesophagus. Eur J Cardiothorac Surg. 2005;28(5):763–766. 16188449
70. Reed, M.F., Tolis, G., Jr., Edil, B.H., et al, Surgical treatment of esophageal high-grade dysplasia. Ann Thorac Surg. 2005;79(4):1110–1115. 15797034
71. Wang, V.S., Hornick, J.L., Sepulveda, J.A., et al, Low prevalence of submucosal invasive carcinoma at esophagectomy for high-grade dysplasia or intramucosal adenocarcinoma in Barrett’s esophagus: a 20-year experience. Gastrointest Endosc. 2009;69(4):777–783. 19136106
72. Nasr, J.Y., Schoen, R.E., Prevalence of adenocarcinoma at esophagectomy for Barrett’s esophagus with high grade dysplasia. J Gastrointest Oncol. 2011;2(1):34–38. 22811825
73. Konda, V.J., Ross, A.S., Ferguson, M.K., et al, Is the risk of concomitant invasive esophageal cancer in high-grade dysplasia in Barrett’s esophagus overestimated? Clin Gastroenterol Hepatol. 2008;6(2):159–164. 18096439
74. Tharavej, C., Hagen, J.A., Peters, J.H., et al, Predictive factors of coexisting cancer in Barrett’s high-grade dysplasia. Surg Endosc. 2006;20(3):439–443. 16437272
75. Boustany, N.N., Crawford, J.M., Manoharan, R., et al, Analysis of nucleotides and aromatic amino acids in normal and neoplastic colon mucosa by ultraviolet resonance Raman spectroscopy. Lab Invest. 1999;79(10):1201–1214. 10532584
76. Schnell, T.G., Sontag, S.J., Chejfec, G., et al, Long-term nonsurgical management of Barrett’s esophagus with high-grade dysplasia. Gastroenterology. 2001;120(7):1607–1619. 11375943
77. Menke-Pluymers, M.B., Hop, W.C., Dees, J., et al, Risk factors for the development of an adenocarcinoma in columnar-lined (Barrett) esophagus. The Rotterdam Esophageal Tumor Study Group. Cancer. 1993;72(4):1155–1158. 8339208
78. Srivastava, A., Hornick, J.L., Li, X., et al, Extent of low-grade dysplasia is a risk factor for the development of esophageal adenocarcinoma in Barrett’s esophagus. Am J Gastroenterol. 2007;102(3):483–494. 17338734
79. Vaughan, T.L., Dong, L.M., Blount, P.L., et al, Non-steroidal anti-inflammatory drugs and risk of neoplastic progression in Barrett’s oesophagus: a prospective study. Lancet Oncol. 2005;6(12):945–952. 16321762
80. Abdalla, S.I., Lao-Sirieix, P., Novelli, M.R., et al, Gastrin-induced cyclooxygenase-2 expression in Barrett’s carcinogenesis. Clin Cancer Res. 2004;10(14):4784–4792. 15269153
81. Haigh, C.R., Attwood, S.E., Thompson, D.G., et al, Gastrin induces proliferation in Barrett’s metaplasia through activation of the CCK2 receptor. Gastroenterology. 2003;124(3):615–625. 12612900
82. Obszynska, J.A., Atherfold, P.A., Nanji, M., et al, Long-term proton pump induced hypergastrinaemia does induce lineage-specific restitution but not clonal expansion in benign Barrett’s oesophagus in vivo. Gut. 2010;59(2):156–163. 19651631
83. Trimble, K.C., Pryde, A., Heading, R.C., Lowered oesophageal sensory thresholds in patients with symptomatic but not excess gastro-oesophageal reflux: evidence for a spectrum of visceral sensitivity in GORD. Gut. 1995;37(1):7–12. 7672684
84. de Jonge, P.J., Steyerberg, E.W., Kuipers, E.J., et al, Risk factors for the development of esophageal adenocarcinoma in Barrett’s esophagus. Am J Gastroenterol. 2006;101(7):1421–1429. 16863542
85. Gerson, L.B., Edson, R., Lavori, P.W., et al, Use of a simple symptom questionnaire to predict Barrett’s esophagus in patients with symptoms of gastroesophageal reflux. Am J Gastroenterol. 2001;96(7):2005–2012. 11467625
86. Locke, G.R., Zinsmeister, A.R., Talley, N.J., Can symptoms predict endoscopic findings in GERD? Gastrointest Endosc. 2003;58(5):661–670. 14595298
87. Younes, M., Lebovitz, R.M., Lechago, L.V., et al, p53 protein accumulation in Barrett’s metaplasia, dysplasia, and carcinoma: a follow-up study. Gastroenterology. 1993;105(6):1637–1642. 8253340
88. Younes, M., Ertan, A., Lechago, L.V., et al, p53 protein accumulation is a specific marker of malignant potential in Barrett’s metaplasia. Dig Dis Sci. 1997;42(4):697–701. 9125634
89. Reid, B.J., Prevo, L.J., Galipeau, P.C., et al, Predictors of progression in Barrett’s esophagus II: baseline 17p (p53) loss of heterozygosity identifies a patient subset at increased risk for neoplastic progression. Am J Gastroenterol. 2001;96(10):2839–2848. 11693316
90. Wong, D.J., Paulson, T.G., Prevo, L.J., et al, p16 (INK4a) lesions are common, early abnormalities that undergo clonal expansion in Barrett’s metaplastic epithelium. Cancer Res. 2001;61(22):8284–8289. 11719461
91. Bani-Hani, K., Martin, I.G., Hardie, L.J., et al, Prospective study of cyclin D1 overexpression in Barrett’s esophagus: association with increased risk of adenocarcinoma. J Natl Cancer Inst. 2000;92(16):1316–1321. 10944553
92. Menke-Pluymers, M.B., Mulder, A.H., Hop, W.C., et al, Dysplasia and aneuploidy as markers of malignant degeneration in Barrett’s oesophagus. The Rotterdam Oesophageal Tumour Study Group. Gut. 1994;35(10):1348–1351. 7959183
93. Bailey, T., Biddlestone, L., Shepherd, N., et al, Altered cadherin and catenin complexes in the Barrett’s esophagus–dysplasia–adenocarcinoma sequence: correlation with disease progression and dedifferentiation. Am J Pathol. 1998;152(1):135–144. 9422531
94. Kadri, S., Lao-Sirieix, P., Fitzgerald, R.C., Developing a nonendoscopic screening test for Barrett’s esophagus. Biomark Med. 2011;5(3):397–404. 21657849
95. Kadri, S.R., Lao-Sirieix, P., O’Donovan, M., et al, Acceptability and accuracy of a non-endoscopic screening test for Barrett’s oesophagus in primary care: cohort study. Br Med J 2010; 341:c4372. 20833740
96. van Sandick, J.W., van Lanschot, J.J., Kuiken, B.W., et al, Impact of endoscopic biopsy surveillance of Barrett’s oesophagus on pathological stage and clinical outcome of Barrett’s carcinoma. Gut. 1998;43(2):216–222. 10189847
97. Fountoulakis, A., Zafirellis, K.D., Dolan, K., et al, Effect of surveillance of Barrett’s oesophagus on the clinical outcome of oesophageal cancer. Br J Surg. 2004;91(8):997–1003. 15286961
98. Corley, D.A., Levin, T.R., Habel, L.A., et al, Surveillance and survival in Barrett’s adenocarcinomas: a population-based study. Gastroenterology. 2002;122(3):633–640. 11874995
99. Wong, T., Tian, J., Nagar, A.B., Barrett’s surveillance identifies patients with early esophageal adenocarcinoma. Am J Med. 2010;123(5):462–467. 20399324
100. Gore, S., Healey, C.J., Sutton, R., et al, Regression of columnar lined (Barrett’s) oesophagus with continuous omeprazole therapy. Aliment Pharmacol Ther. 1993;7(6):623–628. 8161668
101. Cooper, B.T., Chapman, W., Neumann, C.S., et al, Continuous treatment of Barrett’s oesophagus patients with proton pump inhibitors up to 13 years: observations on regression and cancer incidence. Aliment Pharmacol Ther. 2006;23(6):727–733. 16556174
102. Peters, F.T., Ganesh, S., Kuipers, E.J., et al, Endoscopic regression of Barrett’s oesophagus during omeprazole treatment; a randomised double blind study. Gut. 1999;45(4):489–494. 10486353 A blinded randomised trial showing that acid suppression can result in alterations in Barrett’s metaplasia.
103. Hofstetter, W.L., Peters, J.H., DeMeester, T.R., et al, Long-term outcome of antireflux surgery in patients with Barrett’s esophagus. Ann Surg. 2001;234(4):532–539. 11573046
104. O’Riordan, J.M., Byrne, P.J., Ravi, N., et al, Long-term clinical and pathologic response of Barrett’s esophagus after antireflux surgery. Am J Surg. 2004;188(1):27–33. 15219481
105. Rees, J.R., Lao-Sirieix, P., Wong, A., et al. Treatment for Barrett’s oesophagus. Cochrane Database Syst Rev. 2011; 1:004060.
106. Corey, K.E., Schmitz, S.M., Shaheen, N.J., Does a surgical antireflux procedure decrease the incidence of esophageal adenocarcinoma in Barrett’s esophagus? A meta-analysis. Am J Gastroenterol. 2003;98(11):2390–2394. 14638338 A meta-analysis demonstrating no difference between the incidence rates of adenocarcinoma in patients with Barrett’s oesophagus treated medically or following antireflux surgery.
107. Peters, F.P., Kara, M.A., Curvers, W.L., et al, Multiband mucosectomy for endoscopic resection of Barrett’s esophagus: feasibility study with matched historical controls. Eur J Gastroenterol Hepatol. 2007;19(4):311–315. 17353695
108. Pech, O., Behrens, A., May, A., et al, Long-term results and risk factor analysis for recurrence after curative endoscopic therapy in 349 patients with high-grade intraepithelial neoplasia and mucosal adenocarcinoma in Barrett’s oesophagus. Gut. 2008;57(9):1200–1206. 18460553
109. ASGE Technology CommitteeKantsevoy, S.V., Adler, D.G., Conway, J.D., et al, Endoscopic mucosal resection and endoscopic submucosal dissection. Gastrointest Endosc. 2008;68(1):11–18. 18577472
110. Ciocirlan, M., Lapalus, M.G., Hervieu, V., et al, Endoscopic mucosal resection for squamous premalignant and early malignant lesions of the esophagus. Endoscopy. 2007;39(1):24–29. 17252456
111. Inoue, H., Fukami, N., Yoshida, T., et al, Endoscopic mucosal resection for esophageal and gastric cancers. J Gastroenterol Hepatol. 2002;17(4):382–388. 11982716
112. Ell, C., May, A., Pech, O., et al, Curative endoscopic resection of early esophageal adenocarcinomas (Barrett’s cancer). Gastrointest Endosc. 2007;65(1):3–10. 17185072
113. Seewald, S., Akaraviputh, T., Seitz, U., et al, Circumferential EMR and complete removal of Barrett’s epithelium: a new approach to management of Barrett’s esophagus containing high-grade intraepithelial neoplasia and intramucosal carcinoma. Gastrointest Endosc. 2003;57(7):854–859. 12776032
114. Shaheen, N.J., Overholt, B.F., Sampliner, R.E., et al, Durability of radiofrequency ablation in Barrett’s esophagus with dysplasia. Gastroenterology. 2011;141(2):460–468. 21679712 Follow-up report of patients with dysplastic Barrett’s oesophagus treated using RFA. The study demonstrated that the effect of RFA was durable after 3 years.
115. Shaheen, N.J., Sharma, P., Overholt, B.F., et al, Radiofrequency ablation in Barrett’s esophagus with dysplasia. N Engl J Med. 2009;360(22):2277–2288. 19474425 This RCT compared RFA to sham procedure in the management of dysplastic Barrett’s oesophagus. Complete eradication of LGD occurred in 90.5% (ablation group) compared to 22.7% (control group) (P < 0.001) at 1 year. Complete eradication of HGD occurred in 81.0% (ablation group) versus 19.0% (control group) (P < 0.001). RFA decreased both the likelihood of disease progression (3.6% vs. 16.3%, P = 0.03) and cancer (1.2% vs. 9.3%, P = 0.045).
116. Overholt, B.F., Wang, K.K., Burdick, J.S., et al, Five-year efficacy and safety of photodynamic therapy with Photofrin in Barrett’s high-grade dysplasia. Gastrointest Endosc. 2007;66(3):460–468. 17643436
117. Ragunath, K., Krasner, N., Raman, V.S., et al, Endoscopic ablation of dysplastic Barrett’s oesophagus comparing argon plasma coagulation and photodynamic therapy: a randomized prospective trial assessing efficacy and cost-effectiveness. Scand J Gastroenterol. 2005;40(7):750–758. 16118910
118. Biddlestone, L.R., Barham, C.P., Wilkinson, S.P., et al, The histopathology of treated Barrett’s esophagus: squamous reepithelialization after acid suppression and laser and photodynamic therapy. Am J Surg Pathol. 1998;22(2):239–245. 9500226
119. Overholt, B.F., Lightdale, C.J., Wang, K.K., et al, Photodynamic therapy with porfimer sodium for ablation of high-grade dysplasia in Barrett’s esophagus: international, partially blinded, randomized phase III trial. Gastrointest Endosc. 2005;62(4):488–498. 16185958
120. Bulsiewicz, W.J., Shaheen, N.J., The role of radiofrequency ablation in the management of Barrett’s esophagus. Gastrointest Endosc Clin North Am. 2011;21(1):95–109. 21112500
121. Herrero, L.A., van Vilsteren, F.G., Pouw, R.E., et al, Endoscopic radiofrequency ablation combined with endoscopic resection for early neoplasia in Barrett’s esophagus longer than 10 cm. Gastrointest Endosc. 2011;73(4):682–690. 21292262
122. Lyday, W.D., Corbett, F.S., Kuperman, D.A., et al, Radiofrequency ablation of Barrett’s esophagus: outcomes of 429 patients from a multicenter community practice registry. Endoscopy. 2010;42(4):272–278. 20146164
123. Semlitsch, T., Jeitler, K., Schoefl, R., et al, A systematic review of the evidence for radiofrequency ablation for Barrett’s esophagus. Surg Endosc. 2010;24(12):2935–2943. 20464420
124. van Vilsteren, F.G., Pouw, R.E., Seewald, S., et al, Stepwise radical endoscopic resection versus radiofrequency ablation for Barrett’s oesophagus with high-grade dysplasia or early cancer: a multicentre randomised trial. Gut. 2011;60(6):765–773. 21209124
125. Sharma, P., Wani, S., Weston, A.P., et al, A randomised controlled trial of ablation of Barrett’s oesophagus with multipolar electrocoagulation versus argon plasma coagulation in combination with acid suppression: long term results. Gut. 2006;55(9):1233–1239. 16905695
126. Reid, B.J., Blount, P.L., Feng, Z., et al, Optimizing endoscopic biopsy detection of early cancers in Barrett’s high-grade dysplasia. Am J Gastroenterol. 2000;95(11):3089–3096. 11095322
127. Williams, V.A., Watson, T.J., Herbella, F.A., et al, Esophagectomy for high grade dysplasia is safe, curative, and results in good alimentary outcome. J Gastrointest Surg. 2007;11(12):1589–1597. 17909921
128. Hull, M.J., Mino-Kenudson, M., Nishioka, N.S., et al, Endoscopic mucosal resection: an improved diagnostic procedure for early gastroesophageal epithelial neoplasms. Am J Surg Pathol. 2006;30(1):114–118. 16330950
129. Moss, A., Bourke, M.J., Hourigan, L.F., et al, Endoscopic resection for Barrett’s high-grade dysplasia and early esophageal adenocarcinoma: an essential staging procedure with long-term therapeutic benefit. Am J Gastroenterol. 2010;105(6):1276–1283. 20179694
130. Pouw, R.E., Wirths, K., Eisendrath, P., et al, Efficacy of radiofrequency ablation combined with endoscopic resection for Barrett’s esophagus with early neoplasia. Clin Gastroenterol Hepatol. 2010;8(1):23–29. 19602454
131. Badreddine, R.J., Prasad, G.A., Wang, K.K., et al, Prevalence and predictors of recurrent neoplasia after ablation of Barrett’s esophagus. Gastrointest Endosc. 2010;71(4):697–703. 19959164
132. Pech, O., May, A., Rabenstein, T., et al, Endoscopic resection of early oesophageal cancer. Gut. 2007;56(11):1625–1634. 17938435
133. Das, A., Singh, V., Fleischer, D.E., et al, A comparison of endoscopic treatment and surgery in early esophageal cancer: an analysis of surveillance epidemiology and end results data. Am J Gastroenterol. 2008;103(6):1340–1345. 18510606
134. Allum, W.H., Blazeby, J.M., Griffin, S.M., et al, Guidelines for the management of oesophageal and gastric cancer. Gut. 2011;60(11):1449–1472. 21705456
135. Prasad, G.A., Wang, K.K., Buttar, N.S., et al, Long-term survival following endoscopic and surgical treatment of high-grade dysplasia in Barrett’s esophagus. Gastroenterology. 2007;132(4):1226–1233. 17408660
136. Bennett, C., Green, S., Barr, H., et al, Surgery versus radical endotherapies for early cancer and high grade dysplasia in Barrett’s oesophagus. Cochrane Database Syst Rev. 2010;(5) CD007334. 20464752
137. May, A., Gunter, E., Roth, F., et al, Accuracy of staging in early oesophageal cancer using high resolution endoscopy and high resolution endosonography: a comparative, prospective, and blinded trial. Gut. 2004;53(5):634–640. 15082579
138. Canto, M.I., Kalloo, A., Chromoendoscopy for Barrett’s esophagus in the twenty-first century: to stain or not to stain? Gastrointest Endosc. 2006;64(2):200–205. 16860069
139. Kara, M.A., Peters, F.P., Ten Kate, F.J., et al, Endoscopic video autofluorescence imaging may improve the detection of early neoplasia in patients with Barrett’s esophagus. Gastrointest Endosc. 2005;61(6):679–685. 15855971
140. Curvers, W.L., Herrero, L.A., Wallace, M.B., et al, Endoscopic tri-modal imaging is more effective than standard endoscopy in identifying early-stage neoplasia in Barrett’s esophagus. Gastroenterology. 2010;139(4):1106–1114. 20600033
141. Curvers, W.L., van Vilsteren, F.G., Baak, L.C., et al, Endoscopic trimodal imaging versus standard video endoscopy for detection of early Barrett’s neoplasia: a multicenter, randomized, crossover study in general practice. Gastrointest Endosc. 2011;73(2):195–203. 21168835
142. Bird-Lieberman, E.L., Neves, A.A., Lao-Sirieix, P., et al, Molecular imaging using fluorescent lectins permits rapid endoscopic identification of dysplasia in Barrett’s esophagus. Nat Med. 2012;18(2):315–321. 22245781
143. Rice, T.W., Falk, G.W., Achkar, E., et al, Surgical management of high-grade dysplasia in Barrett’s esophagus. Am J Gastroenterol. 1993;88(11):1832–1836. 8237928
[/level-membership-for-surgery-category][not-level-membership-for-surgery-category]15
Barrett’s oesophagus
Definition
Barrett’s oesophagus is a change in any portion of the normal squamous oesophageal epithelium to a metaplastic columnar epithelium that is visible endoscopically and can be confirmed or corroborated histologically.1,2 There are three histologically distinct types of columnar metaplasia: intestinal (IM), cardiac (CM) and fundic. In the USA, unlike the UK and Japan, the diagnosis of Barrett’s oesophagus requires the identification of intestinalisation characterised by the presence of goblet cells. However, the UK definition considers that Barrett’s oesophagus is analogous to a ‘columnar lined oesophagus’ and does not require identification of goblet cells due to fears that sampling bias could lead to under-diagnosis and potentially exclude patients from surveillance programmes. It has been reported that a minimum of eight biopsies are required to confidently exclude intestinal metaplasia – if only four biopsies are taken the diagnostic yield is only 35%.3
Epidemiology
The exact population prevalence of Barrett’s oesophagus is unclear. Data described in post-mortem and endoscopic series range from 0.9% to 5.6% depending on the precise definition used and the type of study.4–7 It is likely that the true prevalence in the West is around 2%. When extrapolated to the UK and US populations, conservative estimates of prevalence are 1 million and 4 million affected individuals, respectively.8 There is also some evidence that the incidence of Barrett’s oesophagus in the West is increasing by up to 2% per year.5,9–11 Data from the Netherlands demonstrated an increase in the number of cases of Barrett’s oesophagus despite a decrease in the number of endoscopies being performed over the same period, suggesting a true increase in incidence.11
A population-based study recruited a representative sample of 1000 people from two communities in northern Sweden to undergo upper endoscopy and confirmed the presence of intestinal metaplasia in 1.6% of the population studied.9
The incidence of Barrett’s oesophagus increases with age, the mean age at diagnosis being approximately 62 years for men and 68 years for women. It predominantly affects Caucasians12 and is more common in men than women, with a ratio of approximately 1.7:1.13
The risk of developing Barrett’s is related to increased frequency and duration of reflux symptoms.14 This appears to correlate with the well-known association between increased frequency, duration and severity of reflux symptoms, and increased risk of adenocarcinoma of the oesophagus. The incidence of Barrett’s oesophagus in patients with symptomatic gastro-oesophageal reflux disease (GORD) is between 5% and 12%.9,15 Evidence from one case series suggests that more than 60% of patients with Barrett’s oesophagus develop the condition secondary to chronic GORD, although other causes of oesophagitis, including non-steroidal anti-inflammatory drugs (NSAIDs), chemotherapy and viral infections are also associated with the disease. It does raise an intriguing possibility that a smaller proportion of patients can develop Barrett’s de novo in the absence of obvious symptomatic or perhaps even pathological reflux. Therefore, other factors that may catalyse changes at the oesophagogastric junction (OGJ) are obesity and cigarette smoking,which have been identified as risk factors for both Barrett’s oesophagus and progression to malignancy.16
A Swedish case–control study demonstrated that patients with recurrent reflux symptoms, when compared with asymptomatic patients, had an odds ratio of 7.7 for oesophageal adenocarcinoma and 2.0 for adenocarcinoma of the gastric cardia. Patients with severe long-standing symptoms had an odds ratio of 43.5 and 4.4 for oesophageal and cardia adenocarcinoma, respectively.17
Endoscopic assessment
The ‘Prague C and M criteria’, defined by an International Working Group on Barrett’s oesophagus, offers a validated method of disease classification based on endoscopic appearance.18 The extent of circumferential involvement (C value) in centimetres from the OGJ should be recorded, as should the maximum length (M value) of the Barrett’s segment, including tongue extensions but excluding isolated ‘islands’. These criteria have been shown to have a high degree of reliability between different endoscopists. The use of the terms long-segment Barrett’s (> 3 cm) and short-segment Barrett’s (< 3 cm) should now be discouraged.
It is crucial to make a thorough and systematic inspection of the mucosa in order to identify any macroscopic neoplastic disease. Water or 1% acetylcysteine should be used to remove blood, saliva and refluxate from the oesophagus, and sufficient insufflation should be ensured to clearly visualise any mucosal abnormalities. Particular care must be taken to identify the OGJ in patients with a hiatus hernia as it is easy to miss the distal extent of a Barrett’s segment in these patients. Clinicians should be aware that at endoscopic inspection most areas of early neoplasia and cancer are detected in an area around the 2 to 4 o’clock position in the endoscopist’s view.19
Despite meticulous inspection during white light video endoscopy, recognition of dysplasia and intramucosal cancer is difficult and subjective, even for experienced endoscopists. Guidelines therefore recommend that quadrantic biopsies are taken every 2 cm of Barrett’s oesophagus in addition to further biopsies from any areas of visible mucosal abnormality.1 Currently, the use of jumbo biopsy forceps is not recommended routinely.
This rigorous biopsy protocol, which is often poorly adhered to outside of specialist centres, samples less than 5% of the mucosa and may miss up to 57% of dysplasia.20,21 Advanced endoscopic imaging techniques may allow targeted biopsies from high-risk areas, improving diagnostic yield (Table 15.1).22–26 Potentially, these imaging tools may also facilitate targeted endoscopic resection of high-grade dysplasia (HGD) and intramucosal cancer.
Table 15.1
Advanced endoscopic imaging modalities being investigated for use in Barrett’s oesophagus surveillance programmes and for facilitation of targeted endoscopic resection
| Imaging modality | Concept | Reference |
| White light endoscopy | ||
| High-resolution magnification endoscopy (HRME) | Greater magnification and resolution than normal endoscopy allowing more detailed visualisation of the mucosa | May et al. (2004)137 |
| Chromoendoscopy | Topical application of dyes improves visualisation of mucosal surfaces. Examples: methylene blue – absorbed with different patterns into different types of mucosa; indigo carmine – accumulates in mucosal fissures accentuating surface topography | Canto et al. (2006)138 |
| Optical endoscopy | ||
| Autofluorescence imaging (AFI) | Short-wavelength light causes excitation of endogenous biological tissues with subsequent release of longer wavelength fluorescent light | Kara et al. (2005)139 |
| Narrow-band imaging (NBI) | Narrow-bandwidth green and blue light (with exclusion of red light) only superficially penetrates mucosa, improving visualisation of mucosal microvasculature and surface morphology | Curvers et al. (2008)25 |
| Confocal microscopy (CM) | Real-time magnification of the mucosa up to 1000-fold enables visualisation of cellular structures | Dunbar and Canto (2010)22 |
| Elastic scattering spectroscopy (ESS) | Elastic scattering of white light generates real-time morphological information about the size and shape of the cell nuclei and the degree of cellular crowding in the mucosa and submucosa | Qiu et al. (2010)23 |
| Trimodal imaging | Incorporates HRME, AFI and NBI in a single endoscope with ability to switch between modalities during procedure | Curvers et al. (2010, 2011)140,141 |
| Molecular imaging | Fluorescently tagged molecular probes bind selectively to metaplastic or dysplastic cells | Bird-Lieberman et al. (2012)142 |
Pathophysiology of Barrett’s oesophagus and progression to adenocarcinoma
It is currently believed that Barrett’s metaplasia develops as a mucosal ‘adaptive’ response to increased cell loss as a result of chronic inflammation, secondary to GORD. Oesophageal squamous epithelium is highly sensitive to acid, alkaline and biliary reflux, which all cause inflammation, with cell loss, necrosis and ulceration. There is strong evidence that the site of origin of Barrett’s metaplasia is a progenitor stem cell located in the submucosal oesophageal gland ducts, following demonstration that a p16 point mutation originating in microdissected squamous duct tissue was also present in adjoining metaplastic crypts.27 Duodenal and gastric reflux-induced ulceration and inflammation is believed to induce tumour suppressor gene mutations, typically p53 and p16, in some of the stem cell populations located in oesophageal gland squamous ducts, which are present throughout the entire length of the oesophagus. Following this initiation phase, multiple distinct clones of metaplastic tissue compete to colonise the oesophagus, creating a mosaic pattern of clones across the segment. Clonal expansion of populations with greater selective advantage, such as ability to survive in a markedly acid- or bile-rich environment, leads to dominant and widespread clones. Once initiated, the promotion and propagation of metaplastic clones is dependent on the surrounding microenvironment, particularly the presence of a chronic inflammatory cell infiltrate, characterised by T lymphocytes, and cytokines such as interleukin-1, tumour necrosis factor-α and transforming growth factor-β. These lead to an increase in cyclo-oxygenase-2, c-myc and cyclin D1, which increase proliferation and decrease apoptosis, and a reduction in E-cadherin, with resultant loss of cell adhesion and localisation of β-catenin to the nucleus.28 These molecular changes underlie the progression of Barrett’s oesophagus to cancer via the metaplasia–dysplasia–adenocarcinoma sequence (see Fig. 15.1).
Figure 15.1 The metaplasia–dysplasia–adenocarcinoma sequence. There are histological stages of progression (shaded rectangles representing the clonal expansion of competing stem cells). In addition there are structural genetic changes in the form of mutations (vertical arrows) and environmental changes (white rectangles) driving cell cycle and cell adhesion biological sequelae. APC, adenomatous polyposis coli gene; Cdx, CauDal protein gene; LOH, loss of heterozygosity; RERs, random errors of replication; TGF-β, transforming growth factor-β. Adapted from Jankowski J, Harrison RF, Perry I et al. Barrett’s metaplasia. Lancet 2000; 356:2079–85. With permission from Elsevier.
Although traditionally thought of as an acquired condition, genetic factors may play a part in a small proportion of patients with Barrett’s metaplasia, as family and twin studies suggest a subgroup of individuals with a strong familial tendency to Barrett’s oesophagus.29,30 A family in the UK has been identified with a male index case with oesophageal adenocarcinoma, three brothers with Barrett’s-associated cancer or HGD, and six children with Barrett’s oesophagus.31 Linkage studies are being undertaken in order to further our understanding of this genetic inheritance. However, there are data to suggest Barrett’s is a polygenic disease with multiple contributing genes acting together.
Risk of cancer and mortality in Barrett’s oesophagus
Barrett’s oesophagus is accepted as a significant risk factor for adenocarcinoma of the oesophagus, although the risk of progression to adenocarcinoma and the risk of disease-specific mortality is low. A large number of studies have estimated the risk of adenocarcinoma arising from Barrett’s oesophagus, with very variable results.32–42 Former studies included small numbers of patients and were likely subject to publication bias, with results only being published if they showed a high incidence of cancer, leading to an overestimate of risk.43 Recently, two large population-based cohort studies have reported much lower annual rates of progression to adenocarcinoma (0.12–0.13% per year) than in former series (Table 15.2). It should be noted that these figures exclude carcinomas of the gastric cardia and also do not reflect progression to HGD. In addition, the two studies used different approaches to select patients with Barrett’s oesophagus. The study by Hvid-Jensen et al.33 identified patients with intestinal metaplasia (IM) from the Danish National Pathology Registry without corroboration with endoscopic findings. Therefore, potentially, patients may have been included who had a diagnosis of cardiac IM rather than true Barrett’s oesophagus, producing an incorrect denominator and leading to a slight underestimate of the risk of disease progression. Bhat et al.32 included patients with columnar-lined oesophagus (CLO) at endoscopy (although the validated Prague system was not used), which was corroborated histologically, and demonstrated an increased risk of progression in patients who had IM confirmed histologically at index endoscopy, compared to those with CLO without IM. This finding is in keeping with previous studies demonstrating a higher risk of disease progression in patients with confirmed IM.1,44,45
Previous studies have, however, suggested a significant geographical variation in the incidence of carcinoma arising in Barrett’s oesophagus in Western countries, with incidence rates in the UK almost double those in the USA.46 It is also worth noting that the population demographic in Denmark differs somewhat from the USA and UK, where rates of obesity are significantly higher and where a greater proportion of men, who are at higher risk of malignant progression, develop Barrett’s oesophagus.
A Danish population-based case–control study identified 11 028 patients from the national pathology registry with a diagnosis of intestinal metaplasia following oesophageal biopsy.33 Patients were followed for a median of 5.2 years. Compared to the general population, patients with Barrett’s oesophagus were found to have a relative risk of 11.3 for developing adenocarcinoma with an annual risk of 0.12%. Only 7.6% of the total oesophageal adenocarcinomas diagnosed nationwide over the study period had a previous diagnosis of Barrett’s oesophagus.
A recent meta-analysis reported a pooled estimate of the annual risk of cancer progression in non-dysplastic Barrett’s of 0.39% per year. Importantly, only eight of 47 studies that met all three quality criteria were included in this analysis; inclusion of the remaining studies significantly increased this figure.35 The risk in the UK following a meta-analysis is indicated as closer to 1%, higher than is reported in the USA.46
It is important to appreciate that while patients with Barrett’s oesophagus have an increased relative risk of adenocarcinoma, the majority of patients will die from other causes. A UK study has demonstrated an increase in both overall mortality rate and oesophageal cancer mortality rate in Barrett’s patients compared with the age- and sex-matched general population. However, only 10% of deaths were due to oesophageal cancer, while 49% were due to cardiorespiratory disease, especially ischaemic heart disease and bronchopneumonia, and 18% of deaths were due to other cancers.47
Natural history of dysplasia in Barrett’s oesophagus
When considering the natural history of dysplasia in Barrett’s oesophagus we must remember that in addition to potential problems with length of follow-up and sampling error at endoscopy, there is considerable inter- and intra-observer variation among experienced pathologists in the histological diagnosis of dysplasia. While pathologists can demonstrate acceptable levels of agreement in distinguishing HGD combined with carcinoma from no dysplasia combined with indefinite and low-grade dysplasia (kappa values of 0.8), there are much poorer levels of agreement in distinguishing between the four groups: indefinite for dysplasia, LGD, HGD and carcinoma (intra-observer kappa values of 0.43–0.64).48 Pathologists find it particularly difficult to separate inflammation in Barrett’s oesophagus from LGD. In this situation pathologists should be encouraged to make use of the indefinite for dysplasia category: such a diagnosis does not mean that the pathologist is uncertain, but rather that it is not possible, with confidence, to exclude LGD in inflamed material. The diagnosis of HGD has serious implications for patient management and the diagnosis should be confirmed by two expert pathologists.
A systematic review involving a total of 1488 patients with Barrett’s oesophagus reported that LGD was present at initial endoscopy in 169 patients (11%) and HGD in 18 patients (1.2%); 1301 (87%) had metaplasia with no dysplasia.49
Low-grade dysplasia
The natural history of LGD is not fully understood and reported rates of regression/progression vary considerably, reflecting the diagnostic difficulties discussed above. Sharma et al.50 followed 156 patients for a mean of 4.1 years and reported progression to HGD or cancer in 13%, regression in 66% and stable LGD in 21%. A more recent prospective cohort study of 713 Barrett’s patients, including 111 with LGD, reported that compared to non- dysplastic disease, LGD was a significant risk factor for progression to HGD or adenocarcinoma (relative risk (RR) 9.7; 95% confidence interval (CI) 4.4–21.5).51 Similarly, in their large population- based study, Hvid-Jensen et al.33 reported that the relative risk of oesophageal cancer among those who had LGD at baseline, as compared to those without LGD at baseline, was 4.8 (95% CI 2.6–8.8). The annual risk of progression to HGD or cancer was found to be 1.27% for those with LGD at baseline. Bhat et al.32 reported a hazard ratio of 5.67% (95% CI 3.77–8.33) for patients with LGD compared to no dysplasia. However, a recent study by Wani et al.,52 which followed up 210 patients with Barrett’s oesophagus with or without LGD for a mean of 6.2 years, found no associations of presence of prevalent, incident or persistent LGD, or the extent of LGD, with progression rates.
Bergman and colleagues recently demonstrated that LGD is over-diagnosed by non-specialist pathologists and argued that its true significance might have been underestimated by many reported series.53 In their study, 1198 patients underwent Barrett’s surveillance at six non-specialist hospitals, identifying 147 (12.5%) patients with LGD. However, only eight (0.7%) patients were deemed to have LGD following histological review by two external expert gastrointestinal pathologists. The majority of diagnoses were reclassified as non- dysplastic Barrett’s oesophagus. During a mean follow-up period of 51 months, 42% of patients with LGD diagnosed by consensus expert pathologists demonstrated progression to either carcinoma or HGD, and 2.2% regressed to non-dysplastic Barrett’s oesophagus.53
High-grade dysplasia
Studies reporting the natural history of HGD have also reported widely differing results. Reid et al.54 followed 76 patients for 5 years and reported that 59% developed adenocarcinoma. In a study of 100 patients with HGD, 66 of whom underwent surveillance, 3 of 24 patients (13%) with focal HGD and 17 of 42 patients (40%) with diffuse HGD developed carcinoma after a mean follow-up of 41 and 23 months, respectively.55
An important question to consider is what proportion of patients with a diagnosis of HGD who undergo oesophagectomy have an occult cancer detected in the resected specimen? Table 15.3 shows reported rates in the literature of 0–73%: overall the rate appears to be approximately 40%.56–72 Patients with visible, nodular HGD appear at greatest risk of harbouring coexisting cancer.73,74 This emphasises the fact that patients with HGD may be harbouring an undetected cancer and confirms the need for complete staging in these patients.
Table 15.3
Studies reporting the incidence of adenocarcinoma in resected specimens following oesophagectomy for high-grade dysplasia
IMC, intramucosal cancer; inv. ca., invasive cancer (denotes invasion into submucosa or beyond).
Given that endotherapy is becoming a recognised treatment option for focal intramucosal cancers (T1a), a more pertinent question to ask might be: what is the prevalence of submucosal invasive cancer at oesophagectomy for HGD? The majority of studies in Table 15.3 make no attempt to separate intramucosal cancer (IMC) from more advanced lesions; however, some more recent reports suggest that rates of invasive cancer (submucosa or beyond) are considerably lower than 40%. Wang et al.71 retrospectively assessed 60 patients (41 with preoperative HGD and 19 with preoperative IMC) who underwent oesophagectomy. The overall rate of submucosal cancer was 6.7%, with a rate of 5% in patients with preoperative HGD and 11% in patients with preoperative IMC. Only one patient (1.7%) had nodal metastasis. Another recent study found the rate of invasive adenocarcinoma (excluding IMC) in association with Barrett’s HGD to be 11.7% (8/68), with 5.9% having occult cancer.75
Although some HGD may be stable or even regress, between 15% and 59% will progress to adenocarcinoma over 5 years. However, if detailed biopsy mapping endoscopies showed no previous HGD (prevalent HGD), then the detection of new HGD (incident HGD) is associated with a risk of subsequent progression to cancer of only between 3% and 5% per year.73,76 This area is being actively discussed in the Barrett’s Dysplasia and Cancer Taskforce (BAD CAT) group.
Risk factors for progression to cancer
The length of Barrett’s segment has been shown to be a significant risk factor for progression to cancer, a doubling of length increasing the risk 1.7-fold.77 The extent of HGD and/or LGD also appears to be a risk factor for progression to adenocarcinoma.55,78
Importantly, in a prospective longitudinal cohort study, individuals with Barrett’s oesophagus who were regularly taking aspirin or other NSAIDs were found to have a significantly lower 5-year cumulative incidence of adenocarcinoma compared with individuals not taking NSAIDs (6.6% and 14.3%, respectively), suggesting that this may be an effective chemotherapeutic intervention.79 An ongoing phase III multicentre randomised controlled trial (RCT), the AspECT trial (Aspirin and Esomeprazole Chemoprevention in Barrett’s Metaplasia), designed to test this hypothesis is due to report in 2016. The primary aim of this study is to determine whether acid suppression with proton-pump inhibition (high dose vs. low dose) with or without aspirin can reduce mortality or the conversion from Barrett’s metaplasia to HGD or adenocarcinoma. Both high- and low-dose acid suppression are being investigated as there remains doubt about the optimal dose of proton-pump inhibitor (PPI) to use, especially given the fact that Barrett’s mucosa is relatively insensitive, thus rendering symptoms unreliable. There is an argument that incomplete acid suppression might increase the risk of cancer by exposing the mucosa to short pulses of acid, thus stimulating the proliferation of abnormal cells. In contrast, there is some epidemiological evidence that high-dose proton-pump inhibition might increase the risk of cancer as bile acid might become cytotoxic at neutral pH. In addition, there have been fears that PPI-induced hypergastrinaemia could stimulate hyperproliferation of Barrett’s epithelium.80,81 Although this risk is yet to be evaluated in vivo, it appears more likely that gastrin induces epithelial restitution in Barrett’s oesophagus, without stimulation of clonal expansion or disease progression.82
Screening for Barrett’s oesophagus and adenocarcinoma using molecular markers
It is accepted that GORD is a significant risk factor for the development of adenocarcinoma, with a well-known Swedish case–control study demonstrating a 44-fold increased relative risk in individuals with frequent heartburn of greater than 20 years’ duration.17 This has led to the suggestion that screening individuals with chronic reflux symptoms to detect Barrett’s oesophagus and cancer may be of benefit. However, it is important to appreciate two flaws in this concept: firstly, approximately 40% of individuals with cancer in the series mentioned above denied frequent heartburn; secondly, a significant proportion of individuals with Barrett’s oesophagus are asymptomatic. In addition, Barrett’s patients experience less heartburn and use PPIs less frequently compared with controls.2,83,84
The endoscopic screening of individuals with chronic reflux symptoms to detect either Barrett’s or cancer is not currently recommended in the UK or USA.1,2 This is because of the low absolute risk of developing adenocarcinoma in individuals with chronic reflux, combined with the knowledge that most individuals with Barrett’s oesophagus die from causes other than oesophageal cancer. There are also concerns about the cost-effectiveness and invasiveness of endoscopy as a screening tool.
Several attempts have been made to develop a scoring system using patient demographics and symptoms to predict the presence of Barrett’s oesophagus for screening purposes.85,86 However, interest in these risk prediction strategies has declined due to inability to generate sufficient sensitivity and specificity.
Mutations in the p53 tumour suppressor gene are widely found in dysplastic Barrett’s oesophagus and oesophageal cancer. Younes et al.87 found p53 mutation in 9% of Barrett’s patients with LGD, 55% of patients with HGD and 87% of patients with carcinoma: no patients without dysplasia had a p53
[/not-level-membership-for-surgery-category]
