[level-membership-for-gastroenterology-and-hepatology-category]
CHAPTER 44 Barrett’s Esophagus
Barrett’s esophagus is the condition in which an abnormal columnar epithelium that is predisposed to malignancy replaces the stratified squamous epithelium that normally lines the distal esophagus.1 The condition is named for Norman Barrett, an Australian surgeon who drew attention to the columnar-lined esophagus in 1950.2 Barrett’s esophagus is a consequence of chronic gastroesophageal reflux disease (GERD), which damages the esophageal squamous epithelium and causes it to heal through a metaplastic process in which columnar cells replace reflux-damaged squamous cells. The columnar-lined esophagus causes no symptoms, and the condition has clinical importance only because it is a risk factor for esophageal adenocarcinoma, a tumor whose frequency has increased more than six-fold over the past several decades.3
DIAGNOSIS
Barrett’s esophagus is diagnosed by endoscopic examination, and two criteria must be fulfilled. First, the endoscopist must ascertain that columnar-appearing epithelium lines the distal esophagus. Second, biopsy specimens of that columnar-appearing epithelium must show evidence of metaplasia, which is a change from one adult cell type to another. To ascertain that columnar-appearing epithelium lines the distal esophagus, the endoscopist first must locate the gastroesophageal junction (GEJ, which is recognized as the most proximal extent of the gastric folds), and then determine that columnar-appearing epithelium extends above the GEJ into the esophagus (Fig. 44-1). Endoscopically, columnar epithelium has a reddish color and velvet-like texture that can be distinguished readily from normal esophageal squamous epithelium, which is pale and glossy. There is disagreement among experts regarding the histologic type of epithelium required to confirm that there is evidence of metaplasia in the esophagus.4 Virtually all would agree that the finding of an intestinal-type epithelium with goblet cells (which has been called intestinal metaplasia, specialized intestinal metaplasia, or specialized columnar epithelium) is clear evidence of metaplasia. Most published studies on Barrett’s esophagus have used intestinal metaplasia as a requisite diagnostic criterion. However, some authorities argue that gastric cardiac-type epithelium, which is composed almost exclusively of mucus-secreting cells, also is metaplastic, has malignant predisposition, and can be considered diagnostic of Barrett’s esophagus.5,6 This debate remains unresolved.
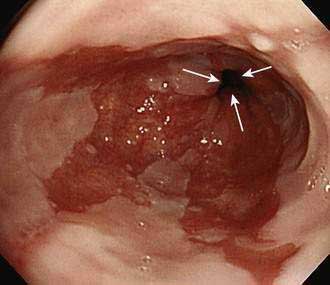
Barrett’s esophagus can be further categorized as long-segment (when the metaplastic epithelium extends at least 3 cm above the GEJ) or short-segment (when <3 cm of metaplastic epithelium lines the esophagus).7 Another more recently proposed system for categorizing Barrett’s esophagus, the Prague C and M criteria, identifies the circumferential (C) and the maximum extent (M) of Barrett’s metaplasia.8 Data suggest that the cancer risk in Barrett’s esophagus may vary with the extent of the metaplastic lining. However, the clinical value of the proposed classification systems has not been established and presently patients with any extent of Barrett’s metaplasia are managed similarly.
EPIDEMIOLOGY
Barrett’s esophagus typically is discovered during endoscopic examinations performed for the evaluation of GERD symptoms in middle-aged and older adults.9 The average age at the time of diagnosis is approximately 55 years. The condition is rare in children younger than age 10 and virtually nonexistent in children younger than age 5.10 White men predominate in most series and, for unknown reasons, Barrett’s esophagus is uncommon in black and Asian populations. Among adult patients who have endoscopic examinations because of GERD symptoms, long-segment Barrett’s esophagus is found in 3% to 5%, whereas 10% to 20% have short-segment Barrett’s esophagus.1 In the general adult population of Western countries, the prevalence of Barrett’s esophagus (predominantly short-segment) is between 1.6% and 6.8%.11,12
Published estimates on the annual incidence of cancer in patients with long-segment Barrett’s esophagus have ranged from 0.2% to 2.9%, but it has been shown that many of those estimates were based on older, small studies that suffered from publication bias. Modern, larger studies, which are less susceptible to such bias, suggest that the risk of cancer in the general population of patients with Barrett’s esophagus is approximately 0.5% per year.13
The epidemiology of esophageal adenocarcinoma is similar to that of Barrett’s esophagus. GERD is strongly associated with both conditions and, like Barrett’s esophagus, esophageal adenocarcinoma affects white men predominantly.9,14 Obesity, especially with central adiposity, predisposes to both Barrett’s esophagus and esophageal adenocarcinoma,14,15 and the dramatic rise in the frequency of obesity in the United States has paralleled a similar rise in the prevalence of Barrett’s cancer. The mechanisms underlying these associations with obesity are not clear, but may relate to the fact that central adiposity predisposes to GERD, perhaps by increasing intra-abdominal pressure16 (see also Chapters 43 and 46). Obesity also is associated with elevated serum levels of pro-proliferative hormones such as insulin-like growth factor I (IGF I) and leptin, and with decreased levels of the antiproliferative hormone adiponectin, factors that may contribute to carcinogenesis in Barrett’s esophagus.
It has been proposed that the declining frequency of infection with Helicobacter pylori in Western populations also may be contributing to the rising frequency of esophageal adenocarcinoma (see Chapter 46). A number of studies have suggested that H. pylori infection may protect against the development and neoplastic progression of Barrett’s esophagus, perhaps because, in a subset of patients, this infection may prevent GERD by decreasing gastric acid secretion.17 Other factors that appear to protect against the development of esophageal adenocarcinoma include the use of aspirin and other nonsteroidal anti-inflammatory drugs (NSAIDs),18,19 and the consumption of a diet high in fruits and vegetables.18 Although cigarette smoking and alcohol consumption are very strong risk factors for squamous cell carcinoma of the esophagus, cigarette smoking only modestly increases the risk for esophageal adenocarcinoma and alcohol does not appear to affect that risk at all.18
PATHOGENESIS
Patients with long-segment Barrett’s esophagus often have severe GERD (see Chapter 43). Table 44-1 lists some physiologic abnormalities that have been reported in Barrett’s patients, and suggests how those abnormalities might contribute to GERD severity. Individual patients may exhibit any, all, or none of those abnormalities, and their prevalence in Barrett’s esophagus is disputed. For example, some investigators have described normal gastric acid secretion in patients with long-segment Barrett’s esophagus.20 In addition, many patients with short-segment Barrett’s esophagus have no GERD symptoms and no endoscopic signs of esophagitis. Indeed, one large study has suggested that short-segment Barrett’s esophagus may affect approximately 5% of adults, irrespective of the presence of GERD symptoms.11 Studies have shown that even in healthy volunteers, the very distal esophagus can be exposed to acid for more than 10% of the day.21 Such acid exposure can damage the esophagus directly and indirectly when nitrite (generated from dietary nitrate) reacts with acid to produce nitric oxide. High concentrations of nitric oxide in the distal esophagus have been observed in patients with GERD who have ingested nitrate.22
Table 44-1 Proposed Physiologic Abnormalities Contributing to Gastroesophageal Reflux Disease in Patients with Barrett’s Esophagus*
| ABNORMALITY | POTENTIAL CONSEQUENCES |
|---|---|
| Extreme LES hypotension | Gastroesophageal reflux |
| Ineffective esophageal motility | Defective clearance of refluxed material |
| Gastric acid hypersecretion | Reflux of highly acidic gastric juice |
| Duodenogastric reflux | Esophageal injury caused by reflux of bile acids and pancreatic enzymes |
| Decreased salivary secretion of epidermal growth factor | Delayed healing of reflux-damaged esophageal mucosa |
| Decreased esophageal pain sensitivity | Loss of sensation to refluxed caustic material and resulting failure to initiate therapy |
LES, lower esophageal sphincter.
* See Chapter 43 for detailed discussion of these abnormalities.
The progenitor cells that give rise to Barrett’s metaplasia are not known. The prevailing hypothesis is that metaplasia results when GERD damages the esophageal squamous epithelium, thereby exposing multipotential stem cells in the basal layers to gastric juice, which stimulates their abnormal differentiation into columnar cells. Two other candidates for Barrett’s progenitor cells include stem cells in the ducts of the esophageal submucosal glands and circulating bone marrow stem cells.23 Genes that appear to play a key role in the squamous-to-columnar metaplasia of Barrett’s esophagus include certain Cdx genes, which are known to mediate the differentiation of intestinal epithelial cells, and the gene encoding bone morphogenetic protein (BMP)-4, which also is involved in columnar cell differentiation.24 Reflux esophagitis appears to up-regulate the expression of these genes by the squamous epithelium.
Barrett’s epithelial cells appear to be more capable of resisting reflux-induced esophageal injury than the native squamous epithelial cells. Unlike squamous cells, for example, Barrett’s cells secrete mucins and express the tight-junction protein claudin 18, features that render the epithelium more resistant to acid-peptic attack.25,26 Unfortunately, Barrett’s epithelium also is predisposed to neoplasia.
MOLECULAR BIOLOGY OF NEOPLASIA
During carcinogenesis, Barrett’s epithelial cells accumulate a series of genetic and epigenetic alterations that endow the cells with the physiological attributes of malignancy (see Chapter 3). Those include self-sufficiency in growth signals, insensitivity to anti-growth signals, evasion of apoptosis, limitless replicative potential, sustained angiogenesis, and the abilities to invade adjacent structures and to metastasize (Fig. 44-2).27 Numerous genetic alterations have been described during the neoplastic progression of Barrett’s esophagus. Although a single such alteration may have multiple disparate effects, conceptually it can be useful to classify the alteration according to the major physiologic cancer attributes that it endows (see Fig. 44-2).27 For example, the expression of oncogenes (e.g., cyclin D1, K-ras), growth factors (e.g., transforming growth factor-α [TGF-α]), and growth factor receptors (e.g., epidermal growth factor receptor [EGFR]) enable Barrett’s cells to acquire self-sufficiency in growth signals. Insensitivity to antigrowth signals occurs primarily through the inactivation of tumor suppressor genes (e.g., TP53 and p16). Inactivation of TP53 also enables cells to evade apoptosis. Reactivation of the enzyme telomerase, which enables the cells to replace telomeres needed for cell division, can endow the cells with limitless replicative potential.18 Neoplasms can increase their vascular supply by secreting angiogenic factors such as vascular endothelial growth factor (VEGF).18 Finally, for neoplastic cells to invade and metastasize, they must dissociate themselves from surrounding cells by disrupting cell adhesion proteins such as the cadherins and catenins, and by degrading the extracellular matrix through the secretion of enzymes such as matrix metalloproteases (MMPs).18
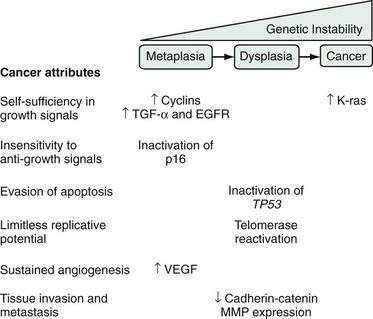
During carcinogenesis, Barrett’s epithelial cells display genetic instability manifested as gains or losses in segments of chromosomes, which alter the cells’ deoxyribonucleic acid (DNA) content. Aneuploidy is the condition in which there is abnormal cellular DNA content, and aneuploid cells are at increased risk for neoplastic progression.28 Aneuploidy can be detected by flow cytometry and by fluorescence in situ hybridization (FISH), and has been proposed as a biomarker for neoplastic progression in Barrett’s esophagus, as have a number of the genetic alterations discussed in the preceding paragraph.29 Although there have been some promising preliminary studies, molecular biomarkers are not yet ready for routine clinical use in patients with Barrett’s esophagus.
DYSPLASIA
Before neoplastic Barrett’s cells become malignant, some of the same genetic alterations that endow the physiologic attributes of malignancy also cause morphologic changes in the tissue that the pathologist recognizes as dysplasia (Fig. 44-3). Dysplasia (also called intraepithelial neoplasia) can be viewed as the histologic expression of genetic alterations that favor unregulated cell growth.30 Dysplasia is recognized by cytologic and architectural abnormalities in esophageal biopsy specimens that include (1) nuclear changes such as enlargement, pleomorphism, hyperchromatism, stratification, and atypical mitoses; (2) loss of cytoplasmic maturation; and (3) crowding of tubules and villiform surfaces. Dysplasia is categorized as low-grade or high-grade depending on the degree of histologic abnormalities, with more pronounced abnormalities assumed to reflect more severe genetic damage and greater potential for carcinogenesis. Pathologists have difficulty distinguishing low-grade dysplasia in Barrett’s esophagus from reactive changes caused by reflux esophagitis, and inter-observer agreement for the diagnosis of low-grade dysplasia may be less than 50%. Interobserver agreement is better (approximately 85%) for high-grade dysplasia, but there is substantial disagreement among pathologists in distinguishing high-grade dysplasia from intramucosal carcinoma (see Chapter 46).
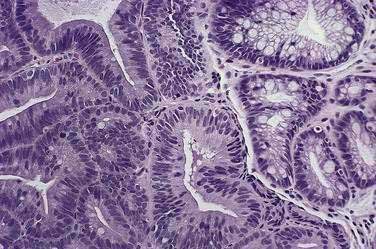
Dysplasia in Barrett’s esophagus often causes no endoscopically apparent abnormalities, and dysplasia can be patchy in its extent and severity. These factors contribute to the substantial problem of biopsy sampling error in identifying dysplasia. Although endoscopists traditionally have used a four-quadrant biopsy sampling system (essentially a random sampling technique) to find dysplasia in Barrett’s esophagus, this system can miss areas of dysplasia and even cancer. In series of patients who had esophagectomies because endoscopic examination with biopsies revealed high-grade dysplasia in Barrett’s esophagus, for example, a number of studies have found that invasive cancer is present in 30% to 40% of the resected esophagi.31 However, a critical review of those studies suggests that 13% is a more accurate estimate of the frequency of invasive cancer in this situation.32
Researchers have tried to develop endoscopic techniques for recognizing dysplasia and early cancer in Barrett’s esophagus including chromoendoscopy, autofluorescence endoscopy, magnification endoscopy, narrow band imaging, optical coherence tomography, Raman detection methods, and confocal laser endomicroscopy.33 There have been some promising preliminary results (see Chapter 46) but presently none of the techniques has provided sufficient clinical information to justify its routine application in clinical practice.
The overall incidence of cancer development in patients with Barrett’s esophagus is approximately 0.5% per year. A recent study suggests that patients who have non-neoplastic Barrett’s esophagus develop low-grade dysplasia at the rate of 4.3% per year, and high-grade dysplasia at the rate of 0.9% per year.34 Few meaningful data are available on the natural history of low-grade dysplasia in Barrett’s esophagus, but a recent study that included 156 patients with low-grade dysplasia found that they developed cancer at an incidence of 0.6% per year.35 For patients with high-grade dysplasia, however, the cancer risk is substantially higher. A recent critical review estimates that the rate of cancer development for patients with high-grade dysplasia in Barrett’s esophagus is 4% to 6% per year.30
MANAGEMENT
TREATMENT OF GASTROESOPHAGEAL REFLUX DISEASE
The general approach to the treatment of GERD for patients with Barrett’s esophagus is very similar to that recommended for patients who have GERD without Barrett’s esophagus (see Chapter 43). One important difference, however, is that modern authorities generally advocate initial and maintenance therapy with a proton pump inhibitor (PPI) for patients with Barrett’s esophagus, irrespective of symptoms and signs of esophagitis. This practice is based on indirect evidence suggesting that acid reflux promotes carcinogenesis in Barrett’s metaplasia, and that aggressive control of acid reflux may interfere with carcinogenesis.35
For patients with Barrett’s esophagus, the elimination of heartburn by antisecretory therapy should not be construed as evidence that acid reflux has normalized. Studies using esophageal pH monitoring have shown that Barrett’s patients may be rendered asymptomatic by PPIs given in dosages that fail to normalize esophageal acid exposure. In one such study of 48 patients with Barrett’s esophagus, for example, 24 of them had persistently abnormal acid reflux during PPI therapy that had abolished their GERD symptoms.36 It has been proposed that patients with long-segment Barrett’s esophagus might be unusually resistant to PPIs, but another study has suggested that this problem is not due to gastric resistance to the antisecretory effects of PPIs.37 In that study, patients with long-segment Barrett’s esophagus treated with high doses of esomeprazole exhibited normal degrees of gastric acid suppression, but up to 23% still had abnormal esophageal acid exposure. This suggests that the so-called PPI resistance of patients with Barrett’s esophagus is a consequence of their profound reflux diathesis.
Some surgeons have proposed that fundoplication might be more effective than antisecretory therapy for preventing cancer in Barrett’s esophagus, but a number of high quality studies on this issue refute that contention. These studies include one randomized controlled trial of medical and surgical GERD therapies, studies using large patient databases, and some meta-analyses.38–40 Available data suggest that antireflux surgery should not be performed solely for cancer prevention in patients with Barrett’s esophagus.
ENDOSCOPIC SURVEILLANCE FOR DYSPLASIA
Proponents of endoscopic surveillance counter the aforementioned arguments as follows41: (1) the concept that surveillance for Barrett’s esophagus can prevent deaths from esophageal adenocarcinoma seems reasonable, (2) no proof of efficacy in the form of a randomized controlled trial is likely to become available in the foreseeable future, (3) a number of observational studies suggest that surveillance is beneficial, (4) virtually all of the published computer models on this issue suggest that surveillance can be beneficial, and (5) the risks of endoscopy for otherwise healthy individuals with Barrett’s esophagus are very small, and no study has shown an overall survival disadvantage for patients in surveillance programs. The potentially adverse emotional and financial consequences of establishing a diagnosis of Barrett’s esophagus are regrettable, but less so than the failure to prevent an esophageal cancer. Therefore, the proponents argue, it is ethically wrong for physicians to forgo the potentially life-saving practice of performing endoscopic surveillance for Barrett’s esophagus while awaiting the results of a definitive study that may never appear.
There have been numerous debates at medical meetings and in medical journals regarding the utility of surveillance for Barrett’s esophagus, with no clear winners. This issue is likely to remain contentious for the foreseeable future. Likewise, whether certain individuals should be screened for Barrett’s esophagus is also controversial (see Chapter 46).
Some authorities also recommend a program of expectant management with intensive endoscopic surveillance (i.e., endoscopic examinations every three to six months) for patients with high-grade dysplasia in Barrett’s esophagus, withholding more invasive treatments (discussed following) until biopsy specimens reveal adenocarcinoma.30,42 Although this practice has been endorsed as a management option by the American College of Gastroenterology, few published data directly support the safety and efficacy of intensive surveillance for high-grade dysplasia. Available studies show that intensive endoscopic surveillance generally is safe, but even patients who are compliant with intensive surveillance programs can develop incurable cancers.30
TREATMENT OF DYSPLASIA
Esophagectomy
Esophagectomy is the most definitive and most hazardous of the treatments for dysplasia in Barrett’s esophagus. The average hospital stay for open esophagectomy is approximately two weeks, and 30% to 50% of patients develop at least one serious postoperative complication such as pneumonia, myocardial infarction, and wound infection.43 A number of series describe high operative mortality rates that in some series can exceed 20%. In addition to death and short-term complications, esophagectomy can be accompanied by substantial long-term morbidity including profound weight loss and dysphagia.
Interest in endoscopic therapies for dysplasia has been driven largely by the perception that esophagectomy has unacceptably high rates of mortality and morbidity. However, recent data suggest that for patients with Barrett’s esophagus those rates have been exaggerated. For example, mortality rates for esophagectomy are inversely related to the frequency with which the operation is performed. In a study of data from the Dutch National Medical Registry, the mortality rates for esophagectomy were 12.1%, 7.5%, and 4.9% at centers performing 1 to 10, 11 to 20, and more than 50 esophagectomies per year, respectively.44 Furthermore, estimates of mortality rates for esophagectomy generally have been based on series of patients with esophageal cancer who are often older adults and debilitated. Mortality rates for younger and otherwise healthy patients with dysplasia in Barrett’s esophagus may be substantially lower, especially when the operation is performed by experienced surgeons in a high-volume center. Finally, published data do not confirm the perception that most patients have an unacceptable quality of life after esophagectomy. In a study in which quality of life questionnaires were administered to 199 patients before and after esophagectomy, for example, quality of life was found to decline substantially immediately after the operation, but to return to baseline values within 2 years.45 Thus, the esophagectomy option still warrants serious consideration, especially for young and fit patients.
Endoscopic Ablative Therapies (see also Chapter 46 and Fig. 46-14)
An ideal ablative technique would inflict an injury deep enough to destroy all of the abnormal epithelium, but not so deep as to cause serious complications like esophageal hemorrhage, perforation, and stricture formation. So far, none of the ablative therapies has achieved this ideal, and all have been associated with serious complications. In addition, the procedures often leave behind residual foci of metaplastic epithelium.46 Partially ablated Barrett’s epithelium can heal with an overlying layer of squamous epithelium that “buries” metaplastic tissue (with its neoplastic potential) and hides it from the endoscopist. Even after apparent complete ablation, furthermore, Barrett’s metaplasia may recur over time.
To date, photodynamic therapy (PDT) has been the most extensively studied of the endoscopic ablative treatments for dysplasia in Barrett’s esophagus (see also Chapter 46 and Fig. 46-14). For PDT, patients are given a systemic dose of a light-activated chemical that is taken up by the esophageal cells. The esophagus is then irradiated using a low-power laser that activates the chemical, which transfers that acquired energy to molecular oxygen. This results in the formation of singlet oxygen, a toxic molecule that destroys the abnormal cells and their vasculature.
In a multicenter randomized trial of PDT using porfimer sodium for high-grade dysplasia in Barrett’s esophagus, 138 patients were treated with PDT plus omeprazole 20 mg twice daily, and 70 received omeprazole 20 mg twice daily alone.47,48 No dysplasia was found on repeat endoscopy with biopsy in 77% of the patients treated with PDT as compared with 39% of the patients who received omeprazole alone (P < 0.0001). During up to 5 years of follow-up, 15% of the PDT patients developed cancer, compared with 29% of those treated with omeprazole alone (P = 0.027). There was no procedure-related mortality, but 69% of the patients who received PDT developed photosensitivity reactions and 36% developed esophageal strictures. Although this study documents the superiority of PDT over PPI alone for eradicating dysplasia and preventing cancer in Barrett’s esophagus, the frequency of serious complications is disconcerting, as is the fact that 15% of the patients who received PDT developed cancer nevertheless.
In an uncontrolled study, 142 patients with high-grade dysplasia in Barrett’s esophagus were treated with the HALO360 system.49 There were no serious adverse events reported during 229 total ablation sessions, although 1 patient was found to have developed an asymptomatic esophageal stricture on follow-up endoscopic examination. At least one postablation endoscopy was performed during a median follow-up period of 12 months in 92 patients, only 9 of whom (10%) had high-grade dysplasia found in follow-up esophageal biopsy specimens. However, persistent low-grade dysplasia was found in another 9 patients (10%), and 42 patients (46%) had residual foci of nondysplastic intestinal metaplasia in the esophagus.
The preliminary results of a randomized sham-controlled trial of radiofrequency ablation for patients with dysplasia in Barrett’s esophagus have been presented in abstract form.50 The trial included 64 patients with low-grade dysplasia and 63 with high-grade dysplasia who were randomized to receive either radiofrequency ablation with the HALO360 system or sham ablation. Twelve months after the procedure, no dysplasia was found in esophageal biopsy specimens for 80% of the HALO-treated patients who had high-grade dysplasia at baseline, and for 11% of the patients who received sham treatment (P < 0.001). Among the patients with low-grade dysplasia at baseline, 90% of the HALO-treated patients had no dysplasia at 12 months, compared with 37% of the sham-treated patients (P < 0.001). Complications were few and easily managed. Five patients developed esophageal strictures that resolved with dilation. One patient experienced upper gastrointestinal bleeding, and 2 developed chest pain following the procedure that resulted in overnight hospitalizations.
Endoscopic Mucosal Resection (see also Chapter 46)
EMR commonly is performed using a “suck and cut” method in which the endoscopist elevates the dysplastic area by injecting fluid into the submucosa, after which the elevated mucosa is suctioned into a cap that fits over the tip of the endoscope.51 A polypectomy snare is then deployed around the suctioned area to remove it. A recent variation on this technique is the “band and snare” method that uses a ligating device, similar to that used for endoscopic variceal ligation, which deploys elastic bands around the suctioned mucosal segment without the requirement for prior submucosal fluid injection.52 The banded segment is removed using a polypectomy snare (see Fig. 46-11).
Available reports on limited (noncircumferential) EMR describe few serious complications and virtually no procedure-related mortality. However, esophageal stricturing occurs frequently if EMR is used to remove the entire circumferential extent of Barrett’s epithelium in a single endoscopic session.53 If the EMR specimen shows that there is no submucosal invasion and the margins of the specimen are free of neoplastic cells, then the patient may be cured and an esophagectomy is unlikely to show residual tumor.54 However, limited data suggest that a single cap-assisted EMR leaves neoplastic cells behind in the large majority of cases.
The long-term data that are available on the efficacy of EMR are limited but impressive (see also Chapter 46). Ell and colleagues performed EMR on 100 patients with early adenocarcinomas in Barrett’s esophagus (tumor diameter <20 mm, well-differentiated histology, no invasion of lymphatics or blood vessels, and no evidence of metastases, submucosal invasion, or lymph node involvement).55 There were no serious complications, and the calculated five-year survival rate was an extraordinary 98%. However, recurrent or metachronous cancers were found in 11% of the patients during a mean follow-up period of 37 months. The recurrent tumors were treated successfully with more endoscopic therapy, but this high recurrence rate shows that EMR often leaves behind cells with neoplastic potential.
A study from the Mayo Clinic compared long-term survivals in patients with high-grade dysplasia who were treated either with esophagectomy or with a combination of EMR and PDT.56 There was no statistically significant difference in survival for patients treated with either of the therapies, even though 6.2% of the patients treated with PDT and EMR were found to have a metachronous esophageal cancer during the follow-up period.
Another recent report describes the long-term results of endoscopic therapies in 349 patients who had high-grade dysplasia or mucosal adenocarcinoma in Barrett’s esophagus.57 Endoscopic treatments included EMR alone for 279 patients, PDT alone for 55, EMR and PDT combined for 13, and argon plasma coagulation alone for 2 patients. Serious complications of endoscopic therapy occurred in 5% of cases (important bleeding in 2 patients, esophageal stricture in 15 patients). During a mean follow-up of 64 months, a complete remission (defined as complete elimination of the neoplastic lesion and at least one follow-up endoscopy showing no neoplasia) was achieved in 97%. However, metachronous neoplasms were found during the follow-up period in 21%. The calculated five-year survival rate was 84%, and none of the deaths were from esophageal cancer.
RECOMMENDATIONS
The management strategy that has been endorsed by the American College of Gastroenterology is arguably the most complete and widely followed of the published guidelines to date for the management of patients with Barrett’s esophagus.58 Their guidelines are as follows:
Ell C, May A, Pech O, et al. Curative endoscopic resection of early esophageal adenocarcinomas (Barrett’s cancer). Gastrointest Endosc. 2007;65:3-10. (Ref 55.)
Iijima K, Henry E, Moriya A, et al. Dietary nitrate generates potentially mutagenic concentrations of nitric oxide at the gastroesophageal junction. Gastroenterology. 2002;122:1248-57. (Ref 22.)
Lagergren J, Bergstrom R, Lindgren A, Nyren O. Symptomatic gastroesophageal reflux as a risk factor for esophageal adenocarcinoma. N Engl J Med. 1999;340:825-31. (Ref 14.)
Overholt BF, Wang KK, Burdick JS, et al. on behalf of the International Photodynamic Group for High-Grade Dysplasia in Barrett’s Esophagus. Five-year efficacy and safety of photodynamic therapy with Photofrin in Barrett’s high-grade dysplasia. Gastrointest Endosc. 2007;66:460-8. (Ref 48.)
Pech O, Behrens A, May A, et al. Long-term results and risk factor analysis for recurrence after curative endoscopic therapy in 349 patients with high-grade intraepithelial neoplasia and mucosal adenocarcinoma in Barrett’s oesophagus. Gut. 2008;57:1200-6. (Ref 57.)
Pohl H, Welch HG. The role of overdiagnosis and reclassification in the marked increase of esophageal adenocarcinoma incidence. J Natl Cancer Inst. 2005;97:142-6. (Ref 3.)
Prasad GA, Wang KK, Buttar NS, et al. Long-term survival following endoscopic and surgical treatment of high-grade dysplasia in Barrett’s esophagus. Gastroenterology. 2007;132:1226-33. (Ref 56.)
Rex DK, Cummings OW, Shaw M, et al. Screening for Barrett’s esophagus in colonoscopy patients with and without heartburn. Gastroenterology. 2003;125:1670-7. (Ref 11.)
Shaheen NJ, Crosby MA, Bozymski EM, Sandler RS. Is there publication bias in the reporting of cancer risk in Barrett’s esophagus? Gastroenterology. 2000;119:333-8. (Ref 13.)
Souza RF, Krishnan K, Spechler SJ. Acid, bile and CDX: The ABCs of making Barrett’s metaplasia. Am J Physiol Gastrointest Liver Physiol. 2008;295:G211-18. (Ref 24.)
Souza RF, Morales CP, Spechler SJ. Review article: A conceptual approach to understanding the molecular mechanisms of cancer development in Barrett’s oesophagus. Aliment Pharmacol Ther. 2001;15:1087-100. (Ref 28.)
Souza RF, Spechler SJ. Concepts in the prevention of adenocarcinoma of the distal esophagus and proximal stomach. CA Cancer J Clin. 2005;55:334-51. (Ref 18.)
Spechler SJ. Barrett’s esophagus. N Engl J Med. 2002;346:836-42. (Ref 1.)
Spechler SJ. Dysplasia in Barrett’s esophagus: Limitations of current management strategies. Am J Gastroenterol. 2005;100:927-35. (Ref 30.)
Wang KK, Sampliner RE, Practice Parameters Committee of the American College of Gastroenterology. Updated guidelines 2008 for the diagnosis, surveillance and therapy of Barrett’s esophagus. Am J Gastroenterol. 2008;103:788-97. (Ref 58.)
1. Spechler SJ. Barrett’s esophagus. N Engl J Med. 2002;346:836-42.
2. Barrett NR. Chronic peptic ulcer of the oesophagus and “oesophagitis.”. Br J Surg. 1950;38:175-82.
3. Pohl H, Welch HG. The role of overdiagnosis and reclassification in the marked increase of esophageal adenocarcinoma incidence. J Natl Cancer Inst. 2005;97:142-6.
4. Spechler SJ, Goyal RK. The columnar lined esophagus, intestinal metaplasia, and Norman Barrett. Gastroenterology. 1996;110:614-21.
5. Chandrasoma P. Controversies of the cardiac mucosa and Barrett’s oesophagus. Histopathology. 2005;46:361-73.
6. British Society of Gastroenterology. Guidelines for the diagnosis and management of Barrett’s columnar-lined oesophagus. A Report of the working party of the British Society of Gastroenterology www.bsg.org.uk, August 2005.
7. Sharma P, Morales TG, Sampliner RE. Short segment Barrett’s esophagus. The need for standardization of the definition and of endoscopic criteria. Am J Gastroenterol. 1998;93:1033-6.
8. Sharma P, Dent J, Armstrong D, et al. The development and validation of an endoscopic grading system for Barrett’s esophagus: The Prague C & M criteria. Gastroenterology. 2006;131:1392-9.
9. Cameron AJ. Epidemiology of columnar-lined esophagus and adenocarcinoma. Gastroenterol Clin North Am. 1997;26:487-94.
10. Hassall E. Esophageal metaplasia: Definition and prevalence in childhood. Gastrointest Endosc. 2006;64:676-7.
11. Rex DK, Cummings OW, Shaw M, et al. Screening for Barrett’s esophagus in colonoscopy patients with and without heartburn. Gastroenterology. 2003;125:1670-7.
12. Ronkainen J, Aro P, Storskrubb T, et al. Prevalence of Barrett’s esophagus in the general population: An endoscopic study. Gastroenterology. 2005;129:1825-31.
13. Shaheen NJ, Crosby MA, Bozymski EM, Sandler RS. Is there publication bias in the reporting of cancer risk in Barrett’s esophagus? Gastroenterology. 2000;119:333-8.
14. Lagergren J, Bergstrom R, Lindgren A, Nyren O. Symptomatic gastroesophageal reflux as a risk factor for esophageal adenocarcinoma. N Engl J Med. 1999;340:825-31.
15. Hampel H, Abraham NS, El-Serag HB. Meta-analysis: obesity and the risk for gastroesophageal reflux disease and its complications. Ann Intern Med. 2005;143:199-211.
16. El-Serag HB, Ergun GA, Pandolfino J, et al. Obesity increases oesophageal acid exposure. Gut. 2007;56:749-55.
17. Delaney B, McColl K. Review article: Helicobacter pylori and gastro-oesophageal reflux disease. Aliment Pharmacol Ther. 2005;22:32-40.
18. Souza RF, Spechler SJ. Concepts in the prevention of adenocarcinoma of the distal esophagus and proximal stomach. CA Cancer J Clin. 2005;55:334-51.
19. Corley DA, Kerlikowske K, Verma R, Buffler P. Protective association of aspirin/NSAIDs and esophageal cancer: A systematic review and meta-analysis. Gastroenterology. 2003;124:47-56.
20. Hirschowitz BI. Gastric acid and pepsin secretion in patients with Barrett’s esophagus and appropriate controls. Dig Dis Sci. 1996;41:1384-91.
21. Fletcher J, Wirz A, Henry E, McColl KEL. Studies of acid exposure immediately above the gastro-oesophageal junction: Evidence of short segment reflux. Gut. 2004;53:168-73.
22. Iijima K, Henry E, Moriya A, et al. Dietary nitrate generates potentially mutagenic concentrations of nitric oxide at the gastroesophageal junction. Gastroenterology. 2002;122:1248-57.
23. Sarosi G, Brown G, Jaiswal K, et al. Bone marrow progenitor cells contribute to esophageal regeneration and metaplasia in a rat model of Barrett’s esophagus. Dis Esophagus. 2008;21:43-50.
24. Souza RF, Krishnan K, Spechler SJ. Acid, bile and CDX: The ABCs of making Barrett’s metaplasia. Am J Physiol Gastrointest Liver Physiol. 2008;295:G211-18.
25. Dixon J, Strugala V, Griffin SM, et al. Esophageal mucin: An adherent mucus gel barrier is absent in the normal esophagus but present in columnar-lined Barrett’s esophagus. Am J Gastroenterol. 2001;96:2575-83.
26. Jovov B, Van Itallie CM, Shaheen NJ, et al. Claudin-18: A dominant tight junction protein in Barrett’s esophagus and likely contributor to its acid resistance. Am J Physiol Gastrointest Liver Physiol. 2007;293:G1106-13.
27. Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57-70.
28. Souza RF, Morales CP, Spechler SJ. Review article: A conceptual approach to understanding the molecular mechanisms of cancer development in Barrett’s oesophagus. Aliment Pharmacol Ther. 2001;15:1087-100.
29. Fritcher EG, Brankley SM, Kipp BR, et al. A comparison of conventional cytology, DNA ploidy analysis, and fluorescence in situ hybridization for the detection of dysplasia and adenocarcinoma in patients with Barrett’s esophagus. Hum Pathol. 2008;39:1128-35.
30. Spechler SJ. Dysplasia in Barrett’s esophagus: Limitations of current management strategies. Am J Gastroenterol. 2005;100:927-35.
31. Collard JM. High-grade dysplasia in Barrett’s esophagus. The case for esophagectomy. Chest Surg Clin North Am. 2002;12:77-92.
32. Konda VJ, Ross AS, Ferguson MK, et al. Is the risk of concomitant invasive esophageal cancer in high-grade dysplasia in Barrett’s esophagus overestimated? Clin Gastroenterol Hepatol. 2008;6:159-64.
33. Curvers WL, Bergman JJ. Multimodality imaging in Barrett’s esophagus: Looking longer, seeing better, and recognizing more. Gastroenterology. 2008;135:297-9.
34. Sharma P, Falk GW, Weston AP, et al. Dysplasia and cancer in a large multicenter cohort of patients with Barrett’s esophagus. Clin Gastroenterol Hepatol. 2006;4:566-72.
35. Feagins LA, Zhang HY, Hormi-Carver K, et al. Acid has antiproliferative effects in nonneoplastic Barrett’s epithelial cells. Am J Gastroenterol. 2007;102:10-20.
36. Gerson LB, Boparai V, Ullah N, et al. Oesophageal and gastric pH profiles in patients with gastro-oesophageal reflux disease and Barrett’s oesophagus treated with proton pump inhibitors. Aliment Pharmacol Ther. 2004;20:637-43.
37. Spechler SJ, Sharma P, Traxler B, et al. Gastric and esophageal pH in patients with Barrett’s esophagus treated with three esomeprazole dosages: A randomized, double-blind, crossover trial. Am J Gastroenterol. 2006;101:1964-71.
38. Spechler SJ, Lee E, Ahnen D, et al. Long-term outcome of medical and surgical treatments for gastroesophageal reflux disease. Follow-up of a randomized controlled trial. JAMA. 2001;285:2331-8.
39. Ye W, Chow WH, Lagergren J, et al. Risk of adenocarcinoma of the esophagus and gastric cardia in patients with gastroesophageal reflux diseases and after antireflux surgery. Gastroenterology. 2001;121:1286-93.
40. Tran T, Spechler SJ, El-Serag HB. Fundoplication and the risk of cancer in gastroesophageal reflux disease: A veterans affairs cohort study. Am J Gastroenterol. 2005;100:1002-8.
41. Spechler SJ. Screening and surveillance for Barrett’s esophagus—An unresolved dilemma. Natl Clin Pract Gastroenterol Hepatol. 2007;4:470-1.
42. Schnell TG, Sontag SJ, Chejfec G, et al. Long-term nonsurgical management of Barrett’s esophagus with high-grade dysplasia. Gastroenterology. 2001;120:1607-19.
43. Karl RC, Schreiber R, Boulware D, et al. Factors affecting morbidity, mortality, and survival in patients undergoing Ivor-Lewis esophagogastrectomy. Ann Surg. 2000;231:635-43.
44. Van Lanschot JJB, Hulscher JBF, Buskens CJ, et al. Hospital volume and hospital mortality for esophagectomy. Cancer. 2001;91:1574-8.
45. de Boer AG, van Lanschot JJ, van Sandick JW, et al. Quality of life after transhiatal compared with extended transthoracic resection for adenocarcinoma of the esophagus. J Clin Oncol. 2004;22:4202-8.
46. Bergman JJ. Latest developments in the endoscopic management of gastroesophageal reflux disease and Barrett’s esophagus: An overview of the year’s literature. Endoscopy. 2006;8:122-32.
47. Overholt BF, Lightdale CJ, Wang KK, et al. Photodynamic therapy with porfimer sodium for ablation of high-grade dysplasia in Barrett’s esophagus: International, partially blinded, randomized phase III trial. Gastrointest Endosc. 2005;62:488-98.
48. Overholt BF, Wang KK, Burdick JS, et al. on behalf of the International Photodynamic Group for High-Grade Dysplasia in Barrett’s Esophagus. Five-year efficacy and safety of photodynamic therapy with Photofrin in Barrett’s high-grade dysplasia. Gastrointest Endosc. 2007;66:460-8.
49. Ganz RA, Overholt BF, Sharma VK, et al. U.S. Multicenter Registry. Circumferential ablation of Barrett’s esophagus that contains high-grade dysplasia: A U.S. Multicenter Registry. Gastrointest Endosc. 2008;68:35-40.
50. Shaheen NJ, Sharma P, Overholt BF, et al. Radiofrequency ablation in Barrett’s esophagus with dysplasia. New Engl J Med. 2009;360:2277-88.
51. Ahmadi A, Draganov P. Endoscopic mucosal resection in the upper gastrointestinal tract. World J Gastroenterol. 2008;14:1984-9.
52. Peters FP, Kara MA, Curvers WL, et al. Multiband mucosectomy for endoscopic resection of Barrett’s esophagus: Feasibility study with matched historical controls. Eur J Gastroenterol Hepatol. 2007;19:311-15.
53. Soehendra N, Seewald S, Groth S, et al. Use of modified multiband ligator facilitates circumferential EMR in Barrett’s esophagus (with video). Gastrointest Endosc. 2006;63:847-52.
54. Prasad GA, Buttar NS, Wongkeesong LM, et al. Significance of neoplastic involvement of margins obtained by endoscopic mucosal resection in Barrett’s esophagus. Am J Gastroenterol. 2007;102:2380-6.
55. Ell C, May A, Pech O, et al. Curative endoscopic resection of early esophageal adenocarcinomas (Barrett’s cancer). Gastrointest Endosc. 2007;65:3-10.
56. Prasad GA, Wang KK, Buttar NS, et al. Long-term survival following endoscopic and surgical treatment of high-grade dysplasia in Barrett’s esophagus. Gastroenterology. 2007;132:1226-33.
57. Pech O, Behrens A, May A, et al. Long-term results and risk factor analysis for recurrence after curative endoscopic therapy in 349 patients with high-grade intraepithelial neoplasia and mucosal adenocarcinoma in Barrett’s oesophagus. Gut. 2008;57:1200-6.
58. Wang KK, Sampliner RE. Practice Parameters Committee of the American College of Gastroenterology. Updated guidelines 2008 for the diagnosis, surveillance and therapy of Barrett’s esophagus. Am J Gastroenterol. 2008;103:788-97.
[/level-membership-for-gastroenterology-and-hepatology-category][not-level-membership-for-gastroenterology-and-hepatology-category]
CHAPTER 44 Barrett’s Esophagus
Barrett’s esophagus is the condition in which an abnormal columnar epithelium that is predisposed to malignancy replaces the stratified squamous epithelium that normally lines the distal esophagus.1 The condition is named for Norman Barrett, an Australian surgeon who drew attention to the columnar-lined esophagus in 1950.2 Barrett’s esophagus is a consequence of chronic gastroesophageal reflux disease (GERD), which damages the esophageal squamous epithelium and causes it to heal through a metaplastic process in which columnar cells replace reflux-damaged squamous cells. The columnar-lined esophagus causes no symptoms, and the condition has clinical importance only because it is a risk factor for esophageal adenocarcinoma, a tumor whose frequency has increased more than six-fold over the past several decades.3
DIAGNOSIS
Barrett’s esophagus is diagnosed by endoscopic examination, and two criteria must be fulfilled. First, the endoscopist must ascertain that columnar-appearing epithelium lines the distal esophagus. Second, biopsy specimens of that columnar-appearing epithelium must show evidence of metaplasia, which is a change from one adult cell type to another. To ascertain that columnar-appearing epithelium lines the distal esophagus, the endoscopist first must locate the gastroesophageal junction (GEJ, which is recognized as the most proximal extent of the gastric folds), and then determine that columnar-appearing epithelium extends above the GEJ into the esophagus (Fig. 44-1). Endoscopically, columnar epithelium has a reddish color and velvet-like texture that can be distinguished readily from normal esophageal squamous epithelium, which is pale and glossy. There is disagreement among experts regarding the histologic type of epithelium required to confirm that there is evidence of metaplasia in the esophagus.4 Virtually all would agree that the finding of an intestinal-type epithelium with goblet cells (which has been called intestinal metaplasia, specialized intestinal metaplasia, or specialized columnar epithelium) is clear evidence of metaplasia. Most published studies on Barrett’s esophagus have used intestinal metaplasia as a requisite diagnostic criterion. However, some authorities argue that gastric cardiac-type epithelium, which is composed almost exclusively of mucus-secreting cells, also is metaplastic, has malignant predisposition, and can be considered diagnostic of Barrett’s esophagus.5,6 This debate remains unresolved.
Barrett’s esophagus can be further categorized as long-segment (when the metaplastic epithelium extends at least 3 cm above the GEJ) or short-segment (when <3 cm of metaplastic epithelium lines the esophagus).7 Another more recently proposed system for categorizing Barrett’s esophagus, the Prague C and M criteria, identifies the circumferential (C) and the maximum extent (M) of Barrett’s metaplasia.8 Data suggest that the cancer risk in Barrett’s esophagus may vary with the extent of the metaplastic lining. However, the clinical value of the proposed classification systems has not been established and presently patients with any extent of Barrett’s metaplasia are managed similarly.
EPIDEMIOLOGY
Barrett’s esophagus typically is discovered during endoscopic examinations performed for the evaluation of GERD symptoms in middle-aged and older adults.9 The average age at the time of diagnosis is approximately 55 years. The condition is rare in children younger than age 10 and virtually nonexistent in children younger than age 5.10 White men predominate in most series and, for unknown reasons, Barrett’s esophagus is uncommon in black and Asian populations. Among adult patients who have endoscopic examinations because of GERD symptoms, long-segment Barrett’s esophagus is found in 3% to 5%, whereas 10% to 20% have short-segment Barrett’s esophagus.1 In the general adult population of Western countries, the prevalence of Barrett’s esophagus (predominantly short-segment) is between 1.6% and 6.8%.11,12
Published estimates on the annual incidence of cancer in patients with long-segment Barrett’s esophagus have ranged from 0.2% to 2.9%, but it has been shown that many of those estimates were based on older, small studies that suffered from publication bias. Modern, larger studies, which are less susceptible to such bias, suggest that the risk of cancer in the general population of patients with Barrett’s esophagus is approximately 0.5% per year.13
The epidemiology of esophageal adenocarcinoma is similar to that of Barrett’s esophagus. GERD is strongly associated with both conditions and, like Barrett’s esophagus, esophageal adenocarcinoma affects white men predominantly.9,14 Obesity, especially with central adiposity, predisposes to both Barrett’s esophagus and esophageal adenocarcinoma,14,15 and the dramatic rise in the frequency of obesity in the United States has paralleled a similar rise in the prevalence of Barrett’s cancer. The mechanisms underlying these associations with obesity are not clear, but may relate to the fact that central adiposity predisposes to GERD, perhaps by increasing intra-abdominal pressure16 (see also Chapters 43 and 46). Obesity also is associated with elevated serum levels of pro-proliferative hormones such as insulin-like growth factor I (IGF I) and leptin, and with decreased levels of the antiproliferative hormone adiponectin, factors that may contribute to carcinogenesis in Barrett’s esophagus.
It has been proposed that the declining frequency of infection with Helicobacter pylori in Western populations also may be contributing to the rising frequency of esophageal adenocarcinoma (see Chapter 46). A number of studies have suggested that H. pylori infection may protect against the development and neoplastic progression of Barrett’s esophagus, perhaps because, in a subset of patients, this infection may prevent GERD by decreasing gastric acid secretion.17 Other factors that appear to protect against the development of esophageal adenocarcinoma include the use of aspirin and other nonsteroidal anti-inflammatory drugs (NSAIDs),18,19 and the consumption of a diet high in fruits and vegetables.18 Although cigarette smoking and alcohol consumption are very strong risk factors for squamous cell carcinoma of the esophagus, cigarette smoking only modestly increases the risk for esophageal adenocarcinoma and alcohol does not appear to affect that risk at all.18
PATHOGENESIS
Patients with long-segment Barrett’s esophagus often have severe GERD (see Chapter 43). Table 44-1 lists some physiologic abnormalities that have been reported in Barrett’s patients, and suggests how those abnormalities might contribute to GERD severity. Individual patients may exhibit any, all, or none of those abnormalities, and their prevalence in Barrett’s esophagus is disputed. For example, some investigators have described normal gastric acid secretion in patients with long-segment Barrett’s esophagus.20 In addition, many patients with short-segment Barrett’s esophagus have no GERD symptoms and no endoscopic signs of esophagitis. Indeed, one large study has suggested that short-segment Barrett’s esophagus may affect approximately 5% of adults, irrespective of the presence of GERD symptoms.11 Studies have shown that even in healthy volunteers, the very distal esophagus can be exposed to acid for more than 10% of the day.21 Such acid exposure can damage the esophagus directly and indirectly when nitrite (generated from dietary nitrate) reacts with acid to produce nitric oxide. High concentrations of nitric oxide in the distal esophagus have been observed in patients with GERD who have ingested nitrate.22
Table 44-1 Proposed Physiologic Abnormalities Contributing to Gastroesophageal Reflux Disease in Patients with Barrett’s Esophagus*
| ABNORMALITY | POTENTIAL CONSEQUENCES |
|---|---|
| Extreme LES hypotension | Gastroesophageal reflux |
| Ineffective esophageal motility | Defective clearance of refluxed material |
| Gastric acid hypersecretion | Reflux of highly acidic gastric juice |
| Duodenogastric reflux | Esophageal injury caused by reflux of bile acids and pancreatic enzymes |
| Decreased salivary secretion of epidermal growth factor | Delayed healing of reflux-damaged esophageal mucosa |
| Decreased esophageal pain sensitivity | Loss of sensation to refluxed caustic material and resulting failure to initiate therapy |
LES, lower esophageal sphincter.
* See Chapter 43 for detailed discussion of these abnormalities.
The progenitor cells that give rise to Barrett’s metaplasia are not known. The prevailing hypothesis is that metaplasia results when GERD damages the esophageal squamous epithelium, thereby exposing multipotential stem cells in the basal layers to gastric juice, which stimulates their abnormal differentiation into columnar cells. Two other candidates for Barrett’s progenitor cells include stem cells in the ducts of the esophageal submucosal glands and circulating bone marrow stem cells.23 Genes that appear to play a key role in the squamous-to-columnar metaplasia of Barrett’s esophagus include certain Cdx genes, which are known to mediate the differentiation of intestinal epithelial cells, and the gene encoding bone morphogenetic protein (BMP)-4, which also is involved in columnar cell differentiation.24 Reflux esophagitis appears to up-regulate the expression of these genes by the squamous epithelium.
Barrett’s epithelial cells appear to be more capable of resisting reflux-induced esophageal injury than the native squamous epithelial cells. Unlike squamous cells, for example, Barrett’s cells secrete mucins and express the tight-junction protein claudin 18, features that render the epithelium more resistant to acid-peptic attack.25,26 Unfortunately, Barrett’s epithelium also is predisposed to neoplasia.
MOLECULAR BIOLOGY OF NEOPLASIA
During carcinogenesis, Barrett’s epithelial cells accumulate a series of genetic and epigenetic alterations that endow the cells with the physiological attributes of malignancy (see Chapter 3). Those include self-sufficiency in growth signals, insensitivity to anti-growth signals, evasion of apoptosis, limitless replicative potential, sustained angiogenesis, and the abilities to invade adjacent structures and to metastasize (Fig. 44-2).27 Numerous genetic alterations have been described during the neoplastic progression of Barrett’s esophagus. Although a single such alteration may have multiple disparate effects, conceptually it can be useful to classify the alteration according to the major physiologic cancer attributes that it endows (see Fig. 44-2).27 For example, the expression of oncogenes (e.g., cyclin D1, K-ras), growth factors (e.g., transforming growth factor-α [TGF-α]), and growth factor receptors (e.g., epidermal growth factor receptor [EGFR]) enable Barrett’s cells to acquire self-sufficiency in growth signals. Insensitivity to antigrowth signals occurs primarily through the inactivation of tumor suppressor genes (e.g., TP53 and p16). Inactivation of TP53 also enables cells to evade apoptosis. Reactivation of the enzyme telomerase, which enables the cells to replace telomeres needed for cell division, can endow the cells with limitless replicative potential.18 Neoplasms can increase their vascular supply by secreting angiogenic factors such as vascular endothelial growth factor (VEGF).18 Finally, for neoplastic cells to invade and metastasize, they must dissociate themselves from surrounding cells by disrupting cell adhesion proteins such as the cadherins and catenins, and by degrading the extracellular matrix through the secretion of enzymes such as matrix metalloproteases (MMPs).18
During carcinogenesis, Barrett’s epithelial cells display genetic instability manifested as gains or losses in segments of chromosomes, which alter the cells’ deoxyribonucleic acid (DNA) content. Aneuploidy is the condition in which there is abnormal cellular DNA content, and aneuploid cells are at increased risk for neoplastic progression.28 Aneuploidy can be detected by flow cytometry and by fluorescence in situ hybridization (FISH), and has been proposed as a biomarker for neoplastic progression in Barrett’s esophagus, as have a number of the genetic alterations discussed in the preceding paragraph.29 Although there have been some promising preliminary studies, molecular biomarkers are not yet ready for routine clinical use in patients with Barrett’s esophagus.
DYSPLASIA
Before neoplastic Barrett’s cells become malignant, some of the same genetic alterations that endow the physiologic attributes of malignancy also cause morphologic changes in the tissue that the pathologist recognizes as dysplasia (Fig. 44-3). Dysplasia (also called intraepithelial neoplasia) can be viewed as the histologic expression of genetic alterations that favor unregulated cell growth.30 Dysplasia is recognized by cytologic and architectural abnormalities in esophageal biopsy specimens that include (1) nuclear changes such as enlargement, pleomorphism, hyperchromatism, stratification, and atypical mitoses; (2) loss of cytoplasmic maturation; and (3) crowding of tubules and villiform surfaces. Dysplasia is categorized as low-grade or high-grade depending on the degree of histologic abnormalities, with more pronounced abnormalities assumed to reflect more severe genetic damage and greater potential for carcinogenesis. Pathologists have difficulty distinguishing low-grade dysplasia in Barrett’s esophagus from reactive changes caused by reflux esophagitis, and inter-observer agreement for the diagnosis of low-grade dysplasia may be less than 50%. Interobserver agreement is better (approximately 85%) for high-grade dysplasia, but there is substantial disagreement among pathologists in distinguishing high-grade dysplasia from intramucosal carcinoma (see Chapter 46).
Dysplasia in Barrett’s esophagus often causes no endoscopically apparent abnormalities, and dysplasia can be patchy in its extent and severity. These factors contribute to the substantial problem of biopsy sampling error in identifying dysplasia. Although endoscopists traditionally have used a four-quadrant biopsy sampling system (essentially a random sampling technique) to find dysplasia in Barrett’s esophagus, this system can miss areas of dysplasia and even cancer. In series of patients who had esophagectomies because endoscopic examination with biopsies revealed high-grade dysplasia in Barrett’s esophagus, for example, a number of studies have found that invasive cancer is present in 30% to 40% of the resected esophagi.31 However, a critical review of those studies suggests that 13% is a more accurate estimate of the frequency of invasive cancer in this situation.32
Researchers have tried to develop endoscopic techniques for recognizing dysplasia and early cancer in Barrett’s esophagus including chromoendoscopy, autofluorescence endoscopy, magnification endoscopy, narrow band imaging, optical coherence tomography, Raman detection methods, and confocal laser endomicroscopy.33 There have been some promising preliminary results (see Chapter 46) but presently none of the techniques has provided sufficient clinical information to justify its routine application in clinical practice.
The overall incidence of cancer development in patients with Barrett’s esophagus is approximately 0.5% per year. A recent study suggests that patients who have non-neoplastic Barrett’s esophagus develop low-grade dysplasia at the rate of 4.3% per year, and high-grade dysplasia at the rate of 0.9% per year.34 Few meaningful data are available on the natural history of low-grade dysplasia in Barrett’s esophagus, but a recent study that included 156 patients with low-grade dysplasia found that they developed cancer at an incidence of 0.6% per year.35
[/not-level-membership-for-gastroenterology-and-hepatology-category]
