Chapter 28 Autoimmunity
OVERVIEW
Autoimmunity is a normal event, while autoimmune diseases result from an aberration of this normal phenomenon.1 Autoimmune diseases are characterised by chronic inflammation with a loss of tolerance to ‘self’ or ‘auto’ antigens. The causes for the loss of tolerance, the shift from normal immune function to autoimmune pathology, are poorly understood but are generally agreed to be multifactorial, involving a combination of genetic, environmental, hormonal and immune factors.1,2 Probable key factors include abnormal cytokine biology and the direct activation of larger than normal quantities of auto- or self-reactive CD4 positive T-cells. Conventionally, autoimmune disorders are diagnosed and treated by physicians specialising in the particular system involved, as shown in Table 28.1. A new paradigm is emerging that groups the pathogeneses of the wide spectrum of autoimmune diseases by their common underlying mechanism: immune system dysregulation, mediated by an imbalance of pro-inflammatory and regulatory cytokines.3
| AUTOIMMUNE DISEASE | SYSTEM AFFECTED | MAIN ORGANS AFFECTED |
|---|---|---|
| Systemic lupus erythematosus (SLE) | Systemic | Skin, joints, kidneys, lungs, heart, brain and blood cells |
| Rheumatoid arthritis | Systemic; muscular skeletal | Connective tissue in joints |
| Dermatitis herpetiformis | Integumentary | Skin, particularly elbows, knees, back and back of neck |
| Multiple sclerosis | Nervous | Myelin sheath in neurons of brain and spinal chord |
| Myasthenia gravis | Nervous; neuromuscularjunction | Muscles , particularly the muscles around the eyes |
| Pernicious anaemia | Blood (haematologic); gastrointestinal | Parietal cells in stomach |
| Goodpasture’s disease | Renal; respiratory | Kidney and lungs |
| Graves’ disease (hyperthyroidism) | Endocrine | Thyroid gland |
| Hashimoto’s thyroiditis | Endocrine | Thyroid gland |
| Type I diabetes mellitus | Endocrine | Pancreatic beta cells |
| Coeliac disease | Gastrointestinal | Villi in small intestine |
| Ulcerative colitis (inflammatory bowel disease) | Gastrointestinal | Mucosa of colon, particularly large colon |
| Crohn’s disease | Gastrointestinal | Can affect entire colon wall, most commonly the lower ileum |
AETIOLOGY
Genetic factors
In the immune system, cell surface protein molecules called human leukocyte antigens (HLA) recognise self from non-self. These antigens are unique for each individual and are encoded by a group of genes on the sixth chromosome called the major histocompatibility complex (MHC).5 On exposure to a foreign antigen, MHC antigens present a component of the invading antigen to circulating T-cells. MHC class I interact with cytotoxic T (CD8+) cells, which track down and kill specific cells, and MHC class II interact with helper T (CD4+) cells which, in the presence of pro-inflammatory cytokines, activate a full immune response (discussed further below under ‘T-cells’).6
Certain MHC class II genotypes are associated with increased susceptibility to autoimmune diseases. This susceptibility may be clustered within specific populations; for instance, increased risk of rheumatoid arthritis is associated with HLA-DR1 (in Asians, Spanish and Jewish Israeli nationals) and HLA-DR4 (in Caucasians).6
Some autoimmune diseases are associated with a single mutation, such as autoimmune lymphoproliferative syndrome. Importantly, not everyone that has the mutant gene manifests the disease. Of those that do, there is a wide variation in the progression and severity of the disease. This implies that there are other factors controlling and regulating the pathogenesis of autoimmune disease.7
Most autoimmune diseases involve a combination of several genetic mutations. The net effect of a combination of mutations may manifest as a pathological phenotype (that is, the way a genotype actually manifests in an individual), depending on the presence or absence of protective factors, both genetic and non-genetic.7
Certain HLA alleles are protective against autoimmune disease. Protective genes and susceptibility genes may both be present in an individual. The net effect of protective and susceptibility genes determine an individual’s genetic susceptibility to autoimmune diseases.8
Specific gene-environment interactions are increasingly being discovered as research into the human genome progresses.9 It is estimated that there may be as many as 20 mutations associated with autoimmune diseases; these mutations are expected to be fully elucidated over the next few years as a result of work on the human genome project.10 However, it is important to note that not all individuals bearing susceptibility genes actually manifest the disease conditions. Gene–gene and gene–environment interactions that may help our understanding of the pathogenesis of autoimmune disease are gradually being discovered.
Environmental factors
Viral and bacterial infections, certain chemicals and drugs and mechanical injury have been implicated as potential triggers of autoimmune diseases in susceptible humans and animals.7,10,11 Two mechanisms that are widely accepted explanations of how an infection may cause autoimmunity are known as molecular mimicry and bystander activation
‘Molecular mimicry’ is the term given to the process of activation of autoreactive helper T (Th or CD4+) cells through cross-reactivity between foreign antigens and self antigens. The foreign antigen may so closely resemble a self antigen that the immune system, having reacted successfully to this foreign antigen, starts reacting to the self antigen as well.5 ‘Bystander activation’ refers to the spontaneous activation of local autoreactive Th cells as part of a coordinated inflammatory response, which may occur as a result of any threat to homeostasis (such as infection).12
Recent research has identified another potential mechanism whereby bacteria may elicit autoimmune pathology. A group of bacteria called superantigens, which includes streptococcal and staphylococcal exotoxins, seem to short-cut the usual immune mechanisms of presentation and directly trigger CD4 cells to launch a full inflammatory response. The result is an enormous release of pro-inflammatory cytokines from T-cells, especially tumour necrosis factor-alpha (TNF-α) and IL-2. Exposure to superantigens has been shown to drive the intense inflammatory responses that result in acute toxic shock or chronic inflammatory disease, such as rheumatic fever.5
Antigen triggers need not come from the external environment—they may be endogenous proteins. Endogenous proteins that have been implicated in the pathogenesis of rheumatoid arthritis include human cartilage glycoprotein 39, citrullinated protein and heavy-chain binding protein.13
Dietary antigens have also been implicated in the pathogenesis of autoimmune pathology, especially the gastrointestinal autoimmune diseases.14 For instance, cereal grains are known inducers of two autoimmune diseases (coeliac disease and dermatitis herpetiformis); removal of gluten from the diet ameliorates symptoms in these conditions. A process of molecular mimicry is strongly suspected in the pathophysiology of these conditions.15 An inflammatory environment in the intestine (from dietary antigens) is associated with increased permeability and increased frequency of potentially
pathogenic antigens crossing the intestinal epithelium and entering the internal environment (‘leaky gut syndrome’). This is mediated by IFN-γ and TNF-α, both altering the tight junctions between cells, while IFN-γ increases the uptake of proteins for transportation into the mucosal cells.16
Recent in vitro work has postulated a role for certain internal environmental conditions that may favour the recognition and binding of large numbers of autoantigens by antibodies. After transient exposure to protein destabilising (but not denaturing) conditions, such as low or high pH, high-salt environments and redox-reactive agents, a small percentage of autoantibodies become extremely autoreactive.17 High salt and its sequelae, widespread low-level metabolic acidity, are hallmarks of the Western diet and are suspected as having an involvement in many so-called modern lifestyle diseases.18
Whatever the environmental trigger, the process of antibody autoreactivity is mediated by cytokines, which turn on and off the signals for T-cells specific for self antigens.19 Thus, in the pathogenesis of autoimmunity, cytokine biology may provide the weakest link in the chain of events that result in pathology.11
Inflammatory mechanisms
Cytokines
Cytokines are found in tissues and in the blood and act as in autocrine, paracrine and endocrine ways. At their target cells cytokines bind to membrane receptors that use second messenger systems. The ensuing enzymatic cascade then results in stimulation or inhibition of cell functions. They can regulate the expression of membrane proteins, including cytokine receptors, and they can stimulate the expression and secretion of their own and other kinds of cytokines.20 In the immune system, cytokines are mainly secreted from activated macrophages and helper T lymphocytes (CD4+ cells). T helper (Th) cells are central to the regulation of the adaptive immune system, both the humoral and cellular arms.21 T helper cells are primarily regulated by T regulatory (Treg) cells (once known as suppressor T lymphocytes). An important role of Treg cells is the inhibition of B lymphocytes from differentiating into plasma cells, the precursors of antibodies. Altered regulatory T-cell functioning has been associated with autoimmune disease.22
T-cells
Antigen-specific cytotoxic T-cells hunt down and destroy the infected cells. Ligands that bind to MCH class II proteins alert T-cells to a threat that requires a coordinated immune system response, to be mediated by the cloning of antigen-specific helper T (CD4) cells.23
Depending on the nature of the antigen, T helper cells differentiate into distinct subsets, which tailor the adaptive immune system to suit the threat at hand. For instance, bacteria and viruses typically activate the cellular arm whereas helminths activate the humoral arm of adaptive immunity.24
CD4+ and CD8+ T-cells
MHC classes I and II antigens are recognised by two distinct subsets of T-cells. These T-cell subsets are distinguished by their respective cell surface proteins.25 Cell surface molecules have been designated as CD or ‘cluster of differentiation’, as they were identified using statistical cluster analysis to identify the specific cells that differentiated after being stimulated by certain antigens.
A numeric system was designated to CD cells as they were identified.26 The T-cell subsets that express CD4 molecules (CD4+ T-cells) may belong to either the helper or regulatory T-cell subsets, where those T-cells that express CD8 molecules (CD8+ T-cells) are cytotoxic T-cells.23 The CD4 and CD8 proteins function as co-receptors in that they cooperate with the T-cell receptor (TCR) to recognise and respond to an antigen bound to MHC classes II and I, respectively.
A portion of the CD4 and CD8 receptor sits outside the cell and directly interacts with the MHC on the antigen-presenting cell. The combination of the TCR and CD4 or CD8 binding to MHC is the first signal required for T-cell activation.27
T helper 1/T helper 2 hypothesis
For 20 years the study of immunology has used the Th1:Th2 hypothesis paradigm, which depicts many disease states to be characterised by the relationship or balance of T helper type 1 (CD4+Th1 or simply Th1) cells and T helper type 2 (CD4+Th2 or Th2) cells.28
During Th1 differentiation, activated antigen-presenting cells such as macrophages and dendritic cells (DCs) produce IL-12. IL-12 signals CD4+ cells to differentiate into Th1 cells, which secrete IL-2, a pro-inflammatory mediator. Interleukin-12 in the presence of IL-18 signals natural killer (NK) cells to secrete IFN-γ,28 another potent pro-inflammatory mediator.
Th1 cells augment a predominately cellular immune response by stimulating T-cytotoxic activity and NK cells, activating macrophages and stimulating the production of other key inflammatory mediators such as nitric oxide (NO).28 Th1 differentiation mediates a potent inflammatory response and an ongoing predominance of Th1 cytokines drives chronic inflammatory activity.3
During Th2 differentiation IL-4 induces naive T helper cells to differentiate into T helper type 2 (Th2) cells. Th2 cells secrete the cytokines IL-4, IL-5, IL-6, IL-10 and IL-13. This subset of cells activates and coordinates a predominately humoral immune response by stimulating the activity of oesinophils, mast cells and B-cell differentiation into plasma cells, which secrete antibodies.28
Cytokines derived from Th1 and Th2 have been shown to regulate each other by antagonistic and mutually inhibitive activity.29 For instance, IL-10 inhibits IL-12 secretion from antigen-presenting cells, but even after IL-12 has been induced in vivo, IL-10 down-regulates its receptors, thereby limiting the effectiveness of IL-12.30 Th2 cytokines are known to suppress the activation of macrophages, the proliferation of T-cells and the production of pro-inflammatory cytokines.28 The Th2 cytokines, IL-4 and IL-10, are considered the major anti-inflammatory cytokines. Within this Th1:Th2 paradigm, most autoimmune diseases have been viewed as Th1-driven disorders.28,31 The typical cytokine profile in Th1-mediated autoimmune diseases, such as rheumatoid arthritis, multiple sclerosis, autoimmune thyroid disease, type 1 diabetes and Crohn’s disease, is an overproduction of IL-12, IFN-γ, TNF-α and under-expression of the immunoregulatory Th2 cytokine, IL-10.28
Most researchers recognise the oversimplicity and rigidity of the Th1:Th2 phenotypes paradigm, but as a simplistic model it is exceedingly useful and has rejuvenated enormous clinical interest and research in the field of helper T-cell immunology. However, the story continues to unfold as new and exciting research suggests much more plasticity and diversity in CD4+Tcell subsets in vivo, previously underestimated as it was not easily demonstrated in vitro.32
The emerging paradigm in understanding T-helper cell subsets
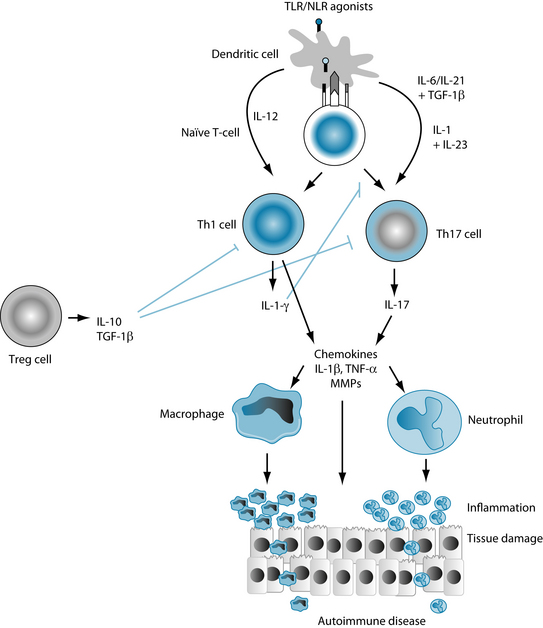
The recent discovery of Th-17 cells uncovers a whole new arm of adaptive immunity, specific for inflammation and autoimmunity as illustrated in Figure 28.1.24 A potent pro-inflammatory cytokine, IL-17, stimulates the production of TNF-α, IL-1β, IL-6, IL-8 and G-CSF (the colony-stimulating factor that increases neutrophils).27
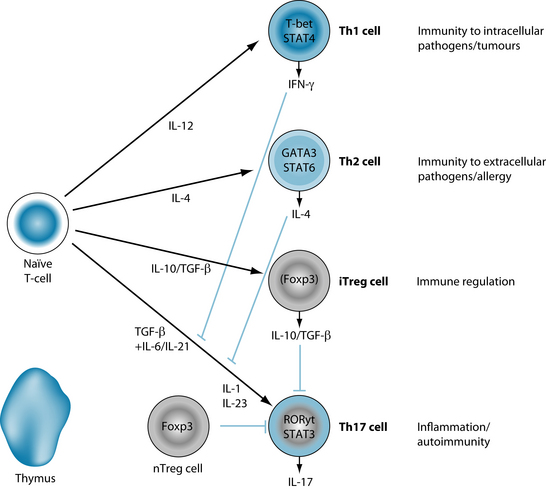
Increased levels of Th17 cells have been found to be associated with both animal and human autoimmune diseases.32–36 Many cytokines, such as IL-1, IL-6, IL-21, TNF-α, IL-23 and TGF-β, are known to stimulate IL-17 production from naive CD4+ T-cells in the presence of inflammatory stimuli such as toll-like receptors.32
Toll-like receptors are pattern-recognition receptors that have evolved to recognise specific bacteria and are found on many antigen-presenting cells, such as dendritic cells, in the innate immune system. When activated, toll-like receptors induce inflammation via activation of inflammatory transcription factors such as NF-kB, which up-regulate the production of pro-inflammatory cytokines, such as IL-17 from Th17 cells and IFG-γ from Th1 cells, as shown in Figure 28.2.
Cytokines as the third signal
Recently, a ‘third signal’, provided by pro-inflammatory cytokines, has been postulated as a necessary requirement for full T-cell activation.37 For instance, a soluble antigen injected into mice resulted in an increase in antigen-specific CD4+T-cells in lymph nodes and follicles. However, CD4+ cells activated in this way simply died off and did not react when re-exposed to the antigen. Although the CD4+ cells differentiated into antigen-specific T-cells in the first instance, when they received no further signal within a week after exposure to the antigen, they became hypo-responsive to the antigen on re-exposure.37,38
An important observation was made during these studies. It was noted that the effect of the bacteria was mediated by the secretion of the cytokine, IFN-γ, from T-cells.39 More importantly, the enhanced effect on T-cell activation by the bacteria was replicated when the bacteria were altogether replaced by the pro-inflammatory cytokines, tumour necrosis factors and IL-1. In addition, the bacterial-induced IFN-γ was replicated by IL-12. Thus, successful activation of T-cells in vivo after exposure to an antigen requires the presence of certain pro-inflammatory cytokines even in the absence of bacteria or other infecting agent.39
This ‘three-signal model’ has been further developed to specify IL-1 as the actual third signal for the antigen-induced activation of naive CD4+ T-cells, and IL-12 as that for CD8+ T-cells.37 It has been proposed that the first two signals are adequate for a transient, localised response but that a long-term, sustained response depends on the presence of certain pro-inflammatory cytokines. IL-1 and TNF-α appear to have significant roles in the cytokine-driven longevity of antigen-specific memory T-cell populations.38
Autoreactive CD4+ T-cells
Low levels of auto-reactive (or self-reactive) CD4+ T-cells naturally circulate in the blood of healthy people and are regulated by regulatory CD4+ T (Treg) cells.40 Naturally occurring regulatory T-cells such as CD4+CD25+ suppress the proliferation of autoreactive T-cells and are therefore critical in the maintenance of peripheral tolerance.41
The regulatory cytokines IL-4, IL-10 and TFB-β are believed to be involved in the suppression of activated autoreactive T helper cells by regulatory T-cells.42 Activation of autoreactive T helper cells and their subsequent differentiation into autoantigen-specific pro-inflammatory Th1 subsets is a pivotal step in the pathogenesis and progression of autoimmune diseases, such as multiple sclerosis.12
Cytokines are the signalling molecules that activate and deactivate autoreactive T-cells.19 However, pro-inflammatory cytokines such as TNF-α, which have been heavily implicated in the pathogenesis of autoimmune diseases, have also been shown to have immunoregulatory activity. It may be that in the short term TNF-α is pro-inflammatory, but chronic exposure leads to down-regulation of the immune response.43
One known mechanism is via pituitary-stimulated secretion of glucocorticoids (see Section 5 on the endocrine system for discussion of the HPA axis), which may constitute a homeostatic role for TNF-α in inflammation. It may also be that other cytokines more specific to autoimmune pathology, such as IL-17, are more critical to the long-term sustained activation of the immune system as observed in autoimmune diseases.44
Pro-inflammatory cytokines activate local autoreactive T-cells as a normal part of the coordinated response to a threat to homeostasis, such as infection or cell damage. In some cases, however, autoreactive T-cells may induce IL-12 secretion from antigen-presenting cells in the absence of infection or other threats to homeostasis, launching an unnecessary inflammatory response and enhancing susceptibility to autoimmune disease.30 This appears to be mediated by abnormal cytokine biology.
RISK FACTORS
As work on the human genome continues, so does our knowledge of how multiple susceptibility and protective genes combine to contribute towards an autoimmune-prone phenotype.45 However, the presence of a genetic susceptibility does not necessarily mean it will manifest as an autoimmune disease. Environmental factors may interact with genetic susceptibility to alter immunity to result in the production of autoantibodies.
Once genetically susceptible individuals have been identified, disease may be prevented, delayed or mitigated by avoiding or minimising exposure to known triggers.46 In a large Danish cohort study involving 37,338 twins, genetic factors were found to be less important than environmental factors.47 Environmental factors include exposure to infections and environmental chemicals but also include the internal environment, such as sex and stress hormones, and immune dysfunction.
Altered immune function
Ageing of the immune system has been associated with a decline of its ability to recognise self from non-self, thereby increasing the risk of the development of autoimmune diseases.48 People with autoimmune diseases have immune systems that resemble those in the elderly, such as involution of the thymus, and premature ageing has been postulated as a risk factor for autoimmune disease.49 On the other hand, young age is a risk factor for multiple sclerosis, with 70% of people with multiple sclerosis aged between 20 and 40 years.50
Viruses have been shown in animal models to be potent triggers of autoimmunity through molecular mimicry and promoters of pathogenic responses through bystander activation. Humans with rheumatoid arthritis and SLE have an altered virus-specific response to the Epstein-Barr virus, for example. Of particular note, the virus-specific T-cells that drive autoimmune pathology appear to be directed by cytokines rather than by the virus itself.51 In genetically susceptible individuals, a combination of a latent Epstein-Barr viral load and altered immune regulation, for instance a functional deficit of Epstein-Barr virus-specific T-cells, may trigger rheumatoid arthritis and SLE.52 The occurrence of chronic recurrent infections is a known risk factor for rheumatoid arthritis.53 A partial or complete deficiency in the complement proteins is a risk factor for SLE.54,55 Immunodeficiency, for any reason (congenital or iatrogenic), increases the risk of non-Hodgkin lymphoma. Inflammatory diseases also increase the risk of non-Hodgkin lymphoma, with more severe inflammation increasing risk further.56 Interestingly, a prospective cohort of 27,290 postmenopausal women found that NSAID use was associated with an increased risk of non-Hodgkin lymphoma.57 It is likely that the NSAID use indicates an unresolved underlying inflammation, which is the actual risk factor for non-Hodgkin lymphoma.
Exposure to chemicals
There is conclusive evidence that smoking is a risk factor for rheumatoid arthritis.58 There is also evidence that mercury from dental amalgam may increase the production of autoantibodies in mercury-sensitive patients with autoimmune thyroiditis.59 Occupational and environmental exposure to asbestos and silica, respectively, increases the risk of developing an autoimmune disease.60,61 Occupational exposure to mineral oil increased the risk of autoimmunity by 30%.58 Epidemiological studies that have looked at vaccinations and exposure to environmental toxins have failed to show them as strong risk factors for autoimmunity, but because there is evidence for an association in animal and case-controlled studies there has been a call for more research in this area.62,63
Diet and lifestyle
Many dietary and lifestyle factors may exacerbate autoimmune diseases. Early exposure to cow’s milk has been associated with increased risk of autoimmune diseases, especially type I diabetes.64 Antigens from milk, grains and legumes have also been found to contain peptides that mimic those found in the joints of people with rheumatoid arthritis.15 Vitamin D deficiency is a risk factor for SLE and multiple sclerosis.65 Dietary fatty acids is a possible risk factor in multiple sclerosis.66 Sleep deprivation was shown to be a risk factor for disease in mice with a genetic susceptibility to develop SLE.67 Ultraviolet radiation and geographic location are known risk factors for multiple sclerosis.66
Hormones
Sex is a risk factor for autoimmune diseases. The prevalence of many autoimmune diseases, such as multiple sclerosis, rheumatoid arthritis and SLE, are higher in women than men. For instance, 90% of SLE cases occur in women.68 On average, women experience their first symptoms of rheumatoid arthritis during menopause, around the time of the decline of ovarian oestrogens.69 In a randomised controlled trial of hormone replacement therapy (HRT) in rheumatoid arthritis of 200 women, those that responded well to the HRT (they had significantly increased serum oestrogen levels) demonstrated improvements in disease symptoms and progression compared with controls.70
Oestrogen appears to have a dual role in immune function, suppressing inflammation while stimulating antibody production.71 Oestrogen receptors have been found on cells from many different systems, including immune cells. In a mouse model of rheumatoid arthritis, when oestrogen receptors were blocked there was an earlier onset of the disease.72
Phytoestrogens are very interesting compounds from plants and can produce mild oestrogenic activity in humans. One such example is quercetin from soybeans. It was demonstrated that quercetin could block antigen-specific Th 1 differentiation in neurons and ameliorate clinical severity and duration of disease in a mouse model of multiple sclerosis by binding to oestrogen receptors.73 This suggests a potentially promising role for phytoestrogens in autoimmune disease.
Stress
Stress is a risk factor for many autoimmune diseases.53,74,75 Stress is also known to worsen symptoms in many autoimmune diseases, particularly rheumatoid arthritis and multiple sclerosis.76,77 Animal and human studies have linked defective neuroendocrine function with enhanced susceptibility of autoimmune diseases, such as rheumatoid arthritis, SLE, type 1 diabetes and Sjögren’s syndrome.78–81 In animal models of autoimmune disease, the onset of the disease is associated with a lack of HPA axis responsiveness (see Section 5 on the endocrine system for discussion) to IL-1, where resistance to autoimmune pathology was maintained by normal HPA axis–immune system responsiveness.82
CONVENTIONAL TREATMENTS
Conventional treatments focus on suppressing and managing the symptoms. Treatment will depend on the type and severity of the autoimmune disease. The conventional treatments for three major autoimmune diseases are given in Table 28.2.
Table 28.2 Conventional treatment for common autoimmune diseases81,82,84,87
| AUTOIMMUNE CONDITION | THERAPEUTIC AGENTS | METHOD OF ACTION |
|---|---|---|
| Rheumatoid arthritis |
Simple analgesics such as paracetamol and aspirin, and non-steroidal anti-inflammatory drugs (NSAIDs) are given in the first instance for symptomatic relief.
|
Research into new drugs for autoimmune diseases are primarily focused on biological agents in inflammatory pathways.83 In order to suppress inflammation and prevent damage, the inhibition of pro-inflammatory cytokines, especially TNF-α and IL-1, would appear to be the most useful approach.84 However, these are new and expensive drugs, and need to be monitored for potential adverse effects.85
In a review of the literature comparing clinical trial outcomes to clinical practice86 anti-TNF-α therapies were shown to produce remarkable improvements in clinical trials, reducing clinical symptoms and slowing disease progression; however, in clinical practice these improvements were found to be much more modest. It should be noted that while anti-TNF-α therapy has enjoyed spectacular success in clinical trials and modest success in clinical practice, it has also been associated with some serious side effects such as the induction of a reversible syndrome very similar to lupus (SLE is associated with the under-expression of TNF-α).7
KEY TREATMENT PROTOCOLS
It is essential that the imbalances of the background diet be fully understood and corrected as a priority in dealing with chronic inflammation. Correcting dietary imbalances has been shown to augment medical interventions, and may reduce reliance upon them.89,90 Dietary modifications that favour balanced immune function should, for the most part, be sustained throughout life.
Lifestyle factors must also be explored thoroughly for potential pro-inflammatory and protective influences. Pro-inflammatory influences need to be identified and minimised. Psychological stress induces an inflammatory state in the immune system by generating the production of pro-inflammatory cytokines.91 Chronic stress results in a chronically imbalanced and under-functioning immune system (see Section 5 on the endocrine system).
The perception of stress is the net result of the perceptions of the actual stressor minus available coping resources. Potential stressors need to be identified and reduced, and available coping resources need to be increased. This may require referral for psychological counselling, meditation or relaxation training, relationship counselling, or other integrative health-care stress and mood management interventions.
NATUROPATHIC TREATMENT AIMS
Protective lifestyle factors need to be instigated and sustained throughout life in all individuals interested in balancing immune function, and especially those with chronic inflammation. For instance, physical exercise has a protective influence on the immune system through several known mechanisms. Interestingly, moderate physical activity actually provides an essential nutrient, glutamine, for immune cells that is stored in skeletal muscles and released through muscle contractions. Glutamine is the precursor for an essential intracellular antioxidant, glutathione, which protects the cell from damage and subsequent inflammation. Exercise also has a favourable effect on cytokine biology, inducing anti-inflammatory factors.
Research into the newly established field of psychoneuroimmunology has greatly advanced the understanding of the psychological and emotional factors that influence immune function. It is known that negative and stressful emotions directly stimulate the production of pro-inflammatory cytokines that mediate and intensify disease.92 The idea that emotions affect health outcomes is not new and can be dated as far back as Hippocrates. What is new is that, through psychoneuroimmunology, science is providing evidence to support the ‘mind–body’ link.93 Laughter,94 meditation,95,96 spiritual beliefs,97 positive thinking98 and even choir singing and listening99 have all been shown to beneficially influence immune parameters. For more detail on this exceedingly interesting field of research, see the discussion in Chapter 6 on respiratory infections and immune insufficiency.
Dietary modulation to reduce inflammation
In human evolution, there have been two major changes that have resulted in an increased consumption of grain (the agricultural and industrial revolutions) and have dramatically increased the availability of omega-6 rich vegetable oils.100 Further, the domestication of livestock and poultry, with its associated decrease in omega-3-rich grass feeding and increase in omega-6 rich grain-supplemented feeding, resulted in a shift in the polyunsaturated compartment of animal products to higher levels of omega-6 fatty acids and lower levels of omega-3 fatty acids.101 During the 1960s the epidemic in coronary heart disease was attributed to the increased consumption of saturated fatty acids from animal products. High cholesterol was identified as a risk factor and every attempt was made to reduce cholesterol.102 There was a public health campaign that declared a ‘war on fat’. All Australians were urged to reduce fat consumption or at least to replace saturated fats with omega-6 rich polyunsaturated fatty acids, such as that from readily available vegetable oils.
The focus on reducing cholesterol by reducing dietary fat intake led to a new market range of ‘low-fat’ products. Natural products such as yoghurt were stripped of their fat content, which was replaced by a whole new range of synthetic additives, usually carbohydrate based. But this new Western diet, the combination of low fat and high carbohydrate intakes, over time has fed an epidemic of obesity and diabetes and the incidence of autoimmune diseases, especially type 1 diabetes, is rapidly rising.103
Carbohydrate intake in the Western diet is much higher than that of most traditional diets and there is a compelling argument that the modern high-carbohydrate diet is associated with negative health outcomes, including increased risk of various autoimmune diseases.15 Grains may contain antinutrients, such as lectins, which not only reduce the absorption of micronutrients from the small intestine but have been shown to induce intestinal secretion of IFN-γ and up-regulate MHC class II expression in enterocytes, thereby exacerbating inflammation in coeliac disease.104 The wheat protein gliadin was shown to inappropriately up-regulate MHC class II presentation in the presence of IFN-γ in inflammatory bowel disease.105 Several gliadins are suspected of
DIETARY CONSIDERATIONS TO REDUCE PRO-INFLAMMATORY CYTOKINES
molecular mimicry. Certain self-proteins, such as alpha-gliadin and the autoantigen BM 180, which has been isolated from the basement membrane of affected exocrine glands in Sjögren’s syndrome, share an almost identical amino acid sequence.15
Low levels of omega-3 intake, together with a high level of omega-6 intakes, creates an imbalance between n-6 and n-3 fatty acids that has been related to many modern-day diseases, from mental health problems to chronic inflammation and autoimmune disease. For instance, in habitually violent and impulsive male offenders with antisocial personality, plasma phospholipid DHA levels were significantly lower than controls, while the arachidonic acid metabolites PGE2 and TXB2 levels were elevated.106 The raised level of arachidonic acid metabolites are evidence that the inflammatory pathways related to NF-κB and COX-2 have been activated, the former stimulating the up-regulation of the expression and activity of genes that code for the pro-inflammatory cytokines. According to one study,107 the Palaeolithic diet consisted of an equal ratio of omega-6 and omega-3 fatty acids. This ratio is currently estimated at 10–15:1 in Australia, and 20–30:1 in the USA. The correction of the imbalance between the essential fatty acids has major clinical implications. For instance, the author of this study states that inflammation was reduced in patients with rheumatoid arthritis by reducing the ratio of n-6 to n-3 fatty acids to 2–3:1.
Early work in nutritional immunology showed that protein- and calorie-restricted diets were associated with dramatic and significant alterations in immune function, readily demonstrated in several animal models of autoimmune diseases.108 In animal studies low-protein diets have delayed the onset of autoimmunity and blunted the clinical progression of autoimmune disease, and caloric restriction has more than doubled life span.109 Protection from autoimmune disease by calorie restriction is thought to be associated with a reduction in T-cell proliferation and B-cell antigen formation when compared to a normal diet.110 Based on these findings, a 25–40% caloric reduction has been recommended as a safe and effective preventive strategy and treatment for autoimmune diseases in humans. This is, however, a huge undertaking for most people and requires an extraordinary commitment over a very long period of time.
Omega-3 fatty acids and the inflammatory cascade
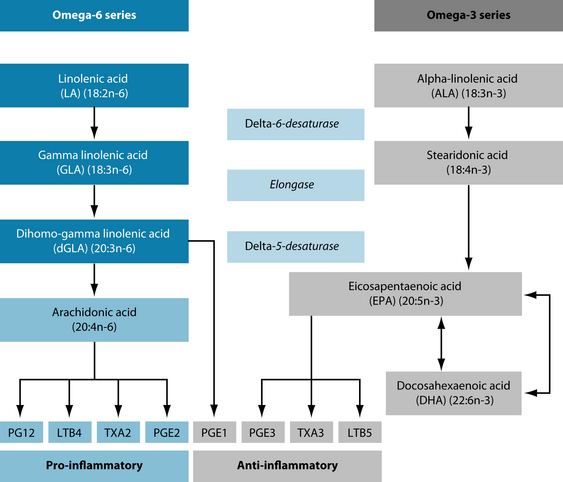
Omega-3 fatty acids influence the production of inflammatory metabolites from the omega-6 polyunsaturated fatty acid, arachidonic acid. For instance, leukotriene B4 (LTB4) is derived from arachidonic acid and is a potent stimulator of pro-inflammatory cytokine production.111 One mechanism that EPA uses to suppress pro-inflammatory cytokine production is via the suppression of the production of LTB4.112 Recently, the long-chain fatty acids have been shown to directly activate gene-transcription factors responsible for the regulation of gene expression and activity for cytokines, such as NF-κB and peroxisome proliferator activated receptors, respectively.
As a general rule omega-6 fatty acids promote while omega-3 fatty acids inhibit the expression, production and secretion, and target cell responsiveness, of pro-inflammatory cytokines, as illustrated in Figure 28.3.113 When high levels of omega-6 fatty acids are present, however, the enzyme shifts its preference to favour omega-6 fatty acid metabolism.114 This means that diets rich in omega-6 fatty acids reduce omega-3 metabolism while promoting the production of arachidonic acid derived eicosanoids.115 Conversely, fish oil has also been shown to decrease the enzyme activity, suggesting the possibility of antagonistic inhibition of omega-6 metabolism by high levels of omega-3 fatty acids.116 This has particular implications for vegetarians that rely on land-based omega-3 fatty acids.
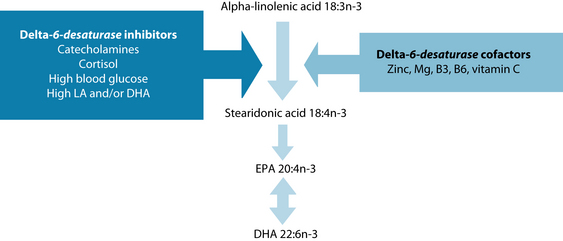
Vegetarians and vegans rely heavily on the activity of these liver enzymes for the production of the long chain omega-3 fatty acids, as they do not consume them in their diet. Figure 28.4 illustrates some of the known influences on delta-6-desaturase. Essential cofactors include zinc, magnesium, niacin (vitamin B3), pyridoxine (vitamin B6) and vitamin C. The stress hormones, adrenaline, noradrenaline and cortisol, inhibit the enzyme as do high blood sugar levels and high levels of linoleic acid, as in the modern Western diet. DHA, as in those taking fish-oil supplements, also inhibits these enzymes, inhibiting the production of arachidonic-derived inflammatory mediators, such as prostaglandins and leukotrienes. The enzyme delta-6-desaturase is the rate-limiting step in eicosanoid synthesis (see Figure 28.4). The stress hormones cortisol and the catecholamines (adrenaline and noradrenaline) inhibit the enzyme. Dietary cofactors, such as zinc, magnesium, vitamins B3 and B6 and vitamin C, are also required for optimal enzyme functioning.
Recently, the National Health and Medical Research Council (NHMRC) set targets for the daily intake of the long chain omega-3 fatty acids based on strong evidence of a protective effect from chronic disease at 610 mg for men and 430 mg for women (see the box below). In 2006, the Australian average intakes of omega-3 fatty acids were estimated from data collected during the 1995 National Nutrition Survey, taking into account the contribution of omega-3 fatty acids contained in red meat.117 The average estimate for Australians was 246 mg per day, but the median daily intake was only 121 mg per day.118 These data are alarming as they indicate that most Australians consume only about one-quarter of the daily requirements of these essential nutrients recommended for the prevention of chronic diseases.
In clinical trials, fish-oil supplements have shown beneficial outcomes in a range of autoimmune diseases. For instance, in a randomised, placebo-controlled, double-blind study of fish oil in systemic lupus erythematosus (SLE), 59 subjects were allocated into one of four groups. Subjects were given 3 × 1 g MaxEPA (180 mg EPA + 120 mg DHA) per day or 3 mg of copper (as copper di-glycinate amine acid complex) per day or both copper and fish oil or placebo (olive oil). The primary outcome measure was scores on the Systemic Lupus Activity Measure (SLAM-R), a routinely used, reliable and validated measure of disease activity. After 24 weeks of supplementation, participants in the fish-oil groups had significantly reduced scores on the SLAM-R (p < 0.05).119 This study demonstrated that symptoms of SLE improved significantly compared with placebo after 6 months of high doses of fish oil.
Inhibition of nuclear factor-kappa B (NF-κB)
Nuclear transcription factor kappa B (NF-κB) is a protein that resides in the cytoplasm of B and T lymphocytes. In its latent form it is bound to an inhibitory protein (iκB). After activation by an inducer, iκB is degraded and NF-κB is released. Immediately, it moves quickly into the cell nucleus. Within minutes it up-regulates the expression of the genes for many pro-inflammatory mediators, such as the cytokines IL-2, IL-6, and IL-8. Inducers of NF-κB include, but are not limited to, the reactive oxygen species, pro-inflammatory cytokines, TNF-α and IL-1, bacteria, viruses, UV radiation, hypoxia and growth factors.120
Some nutritional factors may predispose an organism towards a pro-inflammatory state by up-regulation of NF-κB. Zinc deficiency is associated with increased NF-κB activation and subsequent increased incidences of diseases in the elderly.121 These findings have been supported by animal studies that demonstrated that zinc was required for normal anti-inflammatory function of the cell.122 However, some conflicting data have also been published, where zinc deficiency was shown to delay the onset and progression of autoimmune disease in early stage SLE in mice.123 Until more research is conducted into the effects of zinc on the immune system in autoimmune disorders, it is probably best to ensure that zinc levels are within the normal range. In addition to zinc levels, normal range is also best advised for iron levels. Extremely low and high intakes of iron are associated with increased morbidity in mice models of SLE. While low iron levels are associated with immune dysfunction, high levels contribute to oxidative stress.124 Both zinc and iron levels should therefore be maintained within normal range.
Activation of NF-kB is involved in the pathogenesis of autoimmune diseases such as rheumatoid arthritis. Thus, agents which can block NF-kB activation, such as the glucocorticoids, are effective interventions in autoimmune diseases.120 Docosahexaenoic acid (DHA) is an omega-3 fatty acid that was shown to block NF-kB activation.125 Mice macrophage cells that were stimulated with the bacteria, lipopolysaccharides and IFN-γ increased NO production. Pre-treatment with various polyunsaturated fatty acids demonstrated that DHA had a marked dose-dependent inhibitory effect on NO production. The mechanism of effect for DHA was up-regulation of glutathione to a level high enough to effectively balance the oxidative stress and prevent the activation of NF-kB. It is interesting to note that the long-chain omega-3 fatty acids up-regulate antioxidant expression inside cells and assist the antioxidant protective mechanisms.
Antioxidants may mediate anti-inflammatory effects by preventing the activation of NF-kB in T-cells or via other mechanisms. The main polyphenol in green tea, epigallocatechin-3-gallate (EGCG), has been shown to down regulate TNF-α gene expression and activity by preventing proteolysis of ikB, thus effectively blocking the activation of NF-kB. This coincided with reduced activation of autoantigen-specific T-cells in humans with multiple sclerosis.126,127
High levels of the amino acids arginine and glutamine were shown to inhibit NF-kB. In 10 patients with Crohn’s disease, colonic biopsies were cultured with low or high (that is, physiological or pharmacological) amounts of arginine and glutamine, respectively. The formula with the pharmacological levels of both amino acids significantly reduced the production of the pro-inflammatory gene transcription factor NF-kB and the cytokines TNF-α, IL-1β, IL-6 and IL-8 in the culture medium.128 The citrus bioflavonoids luteolin,129 quercetin130 and 3’-hydroxy-flavone have been shown to block activation of NF-kB.131
Antioxidants and pro-inflammatory cytokines
The build up of free radicals (reactive oxygen species) activates NF-kB to up-regulate the expression of pro-inflammatory cytokines such as TNF-α, IL-1β and IL-6. Antioxidants inhibit the production of such pro-inflammatory cytokines by blocking reactive oxygen intermediate-induced activation of NF-kB.132 This prevents many major downstream inflammatory events, including the activation of COX-2, the subsequent production of the eicosanoids, especially LTB4, and the increased secretion of pro-inflammatory cytokines.133
ROLE OF OMEGA-3 FATTY ACIDS
Omega-3 fatty acids up-regulate antioxidants inside cells and thus prevent the activation of NF-kB.
response is via the generation of reactive oxygen species; and that disruption of the reactive oxygen species–cytokine pathway to the activation of the third signal has been shown to be an effective prevention of autoimmune disease.134
In a mouse model of type 1 diabetes, 10 mg/kg of antioxidants or placebo were injected several hours prior to immunisation with antigen and every day for 7 days post-immunisation. On day 8 the animals were sacrificed and spleen cells were incubated with antigen and TNF-α. The TNF-α was reduced by half in the presence of antioxidants compared with placebo. Antigen-specific T-cell proliferation and IFN-γ were also dramatically reduced compared with placebo. Spleen cells incubated in vitro with antigen demonstrated that both antigen-presenting cells and T-cells generate reactive oxygen species after exposure to an antigen, an effect that was substantially and significantly reduced by pre-treatment with antioxidants. The reduction of reactive oxygen species by antioxidants corresponded to reduced levels of TNF-α secretion from both T-cells and antigen-presenting cells. In vitro stimulation of spleen cells with antigen in the presence of TNF-α significantly reduced production of IFN-γ.134
Antioxidants therefore were able to substantially and significantly down-regulate the cytokine signals required for the activation of the autoimmune disease. Autoimmune-prone mice treated with antioxidants demonstrated reduced activation of naive T-cells in vivo, demonstrated by reduced levels of antigen-specific CD4+ T-cell proliferation and a 10-fold reduction of IFN-γ level in mice, compared with autoimmune prone mice that did not receive the antioxidants.134 In autoimmune-prone mice, dietary antioxidants induced hyporesponsiveness of autoantigen-specific CD4+ T-cells, even in the presence of pro-inflammatory cytokines! Antioxidants therefore were able to prevent the full activation of an autoimmune response even in the presence of cytokine signalling.
Alpha-lipoic acid is a potent antioxidant that appears to work in a multitude of ways. It is a free radical scavenger, it regenerates other antioxidants such as vitamin C and E, and it raises intracellular glutathione. Alpha-lipoic acid is a cofactor in several enzyme systems during aerobic metabolism. As a type of fatty acid, it readily crosses cell membranes and the blood–brain barrier, and has been shown to be neuroprotective in cerebral ischaemia and other brain injury caused by free radicals.135 Alpha-lipoic acid also has important anti-inflammatory properties.
In autoimmunity, alpha-lipoic acid has been shown to prevent the onset of disease in the mouse model of multiple sclerosis.136 Mice were treated with alpha-lipoic acid in the drinking water from the same day the mice were inoculated with a peptide that activates myelin-specific autoreactive T-cells. When compared with controls that had only drinking water, alpha-lipoic acid mice had delayed onset of the disease and reduced severity of its progression.137 This protective effect in clinical scores corresponded with a reduction in the infiltration of T-cells into the central nervous system and reduced demyelination of the central nervous system. In addition, a second group of mice were given alpha-lipoic acid or placebo from the onset of the first clinical signs; antigen-specific T-cells in the spleen and lymph nodes were significantly and substantially reduced by alpha-lipoic acid treatment. This demonstrates that alpha-lipoic acid is effective as both a prevention and an intervention in the animal model of multiple sclerosis.
While animal studies are indicative of possible therapeutic interventions, it is difficult to extrapolate these results to human studies, especially in terms of effective dosages and potential side effects. A pilot clinical trial of alpha-lipoic acid randomised subjects with multiple sclerosis into one of four treatment groups: a placebo group, and various doses of alpha-lipoic acid with 1200 mg/day as the highest dose. Subjects took the study medication before meals for 2 weeks.138 Alpha-lipoic acid was quite well tolerated, with some reports of mild self-limiting adverse effects, such as nausea, on the gastrointestinal system. Two markers of leukocyte trafficking were used as outcome measures: matrix metalloproteinase-9 (MMP-9), which is produced by activated T-cells to facilitate leukocyte migration into the central nervous system, and intercellular adhesion molecule-1 (ICAM-1), which binds leukocytes and facilitates their migration into the brain. Stimulation of the production of MMP-9 and ICAM-1 is mediated by pro-inflammatory cytokines, and increased levels are associated with multiple sclerosis.
Alpha-lipoic acid was shown to suppress both molecules in a dose-dependent manner, with subjects on the highest dose showing the lowest levels of ICAM-1. There was also a negative correlation between peak alpha-lipoic acid serum levels and MMP-9. Therefore, this pilot study indicated that high doses of alpha-lipoic acid were able to reduce leukocyte trafficking into the brain of people with multiple sclerosis and may thus constitute a valid dietary intervention in multiple sclerosis.136 While a pilot study does not have the statistical power to demonstrate conclusively that alpha-lipoic acid will be effective in people with multiple sclerosis, it provides good preliminary evidence of its safety and efficacy.
Genistein is an isoflavone with anti-inflammatory, antioxidant and apoptotic properties. Genistein binds to human oestrogen receptors on immune cells.139 In a study using the mouse model for multiple sclerosis, mice were fed genistein (200 mg/kg body weight per day) for 7 days. In autopsied brain tissue, IL-12, IFN-γ and TNF-α were significantly lower and IL-10 was higher in genistein-fed mice compared with non-treated mice.139 Thus genistein decreased inflammatory mediators and increased anti-inflammatory mediators during autoimmune pathology. This beneficial shift in cytokine biology corresponded to significant improvements in clinical signs of disease progression for the genistein-treated group, evident in less weight loss and lower total clinical score, reduced leukocyte trafficking and less endothelial adherence into the brain. Such animal studies suggest that genistein may offer effective protection at the blood-brain barrier by regulating cytokine profiles.
Quercetin is a phytoestrogenic flavanol with anti-inflammatory properties.140 Quercetin reduced the severity and duration of paralysis as well as reductions of central nervous system demyelination and inflammation in animal models of multiple sclerosis.73
Resveratrol is a polyphenol compound with oestrogen-modulating, antioxidant and anti-inflammatory properties.141 Its mechanisms of action includes inhibition of the production of pro-inflammatory cytokines and COX-2 via inhibition of NF-kB and the activator protein-1 (AP-1).142 Resveratrol significantly delayed the onset of autoimmune pathology and halved the clinical symptoms in a dose-dependent manner in animal models. The reduction in the clinical scores corresponded with less inflammatory-related damage in the central nervous system. Pro-inflammatory cytokines in serum, including TNF-α, IFN-γ and IL-2, were reduced, but not significantly.141 Serum IL-17 was significantly reduced in the resveratrol group, compared with placebos. Resveratrol also increased IL-6, compared with controls. Resveratrol was also found to induce cell death in activated autoreactive T-cells.141
Physical exercise and the immune system
There appears to be many mechanisms by which exercise mediates beneficial effects on the immune system.143 Perhaps the most interesting is that skeletal muscle acts as a storehouse of the branched-chain amino acids, which supply nitrogen for the synthesis of glutamine by skeletal muscle. Skeletal glutamine supplies the rest of the body with glutamine as a precursor for the antioxidant, glutathione.
Cells that rapidly proliferate, such as immune cells especially during inflammation, are highly dependent on skeletal glutamine release via muscle contractions during physical exercise. This has particular implications for autoimmune diabetes, as pancreatic beta cells have reduced expression and activity of glutathione, and rely heavily on a systemic supply. Skeletal muscles release glutamine into the bloodstream after physical exercise at a very high rate.144 Muscle contractions during physical exercise thus may offset pancreatic β-cell glutamine depletion, and subsequent increase in glutathione levels and antioxidant protective mechanisms.
Depletion of glutamine may cause the immunosuppression often observed after significant stressors such as major surgery or excessive exercise. Patients undergoing major surgery (coronary, vascular and colon) that received glutamine supplementation had significantly lower postoperative infection rates.145 Studies using the stress of elite athletics found that glutamine supplementation immediately after a race and again 2 hours after the race protected against the immunosuppression that often follows a race.146 In those athletes that drank the glutamine beverage, 81% reported no upper respiratory tract infection episodes in the 7 days after the race, compared with those that had the placebo drink, which contained maltodextrin, where only 49% reported no upper respiratory tract infections.
Metabolic acidosis may precipitate glutamate depletion as it increases the utilisation of glutamine in the kidney. Stress, strenuous physical exercise, starvation and major surgery are associated with depletion of plasma glutamine.147 The stress hormones cortisol and adrenaline are both associated with increased glutamine utilisation and depletion.
Physical exercise elicits a cytokine response in the body. The type of physical exercise is a determinant of the particular cytokine response. The take-home message is that moderate physical exercise has been shown to induce anti-inflammatory cytokines (IL-1 receptor antagonist and IL-10), and offers protection against inflammation and autoimmune diseases.148
Psychological stress
Psychological and emotional stress increases the secretion of pro-inflammatory cytokines.91,149–151 The clinical implications are that it is essential to address stress in order to effectively regulate cytokine biology. This may mean referral for counselling, meditation or relaxation training. Adaptogenic herbs and nutritional supplements are those that have been shown to be effective interventions in helping the organism cope with or adapt to stress. Botanical adaptogens and tonics that have received the most research attention include Panax ginseng, Eleutherococcus senticosus,152–159 Withania somnifera,160,161 Bacopa monnieri162 and Glycyrrhyza glabra163 (see the endocrine and nervous system sections for more detail). A multivitamin and mineral supplement also improved several parameters of stress in a randomised, placebo-controlled, double-blinded clinical trial.164 Lecithin has demonstrated adaptogenic qualities in rats.165 Omega-3 fatty acids have also been shown to inhibit cytokine production.111,166,167
Herbal medicine and inflammatory cytokines
Boswellia serrata has been used traditionally for treating a range of conditions, including inflammation. The anti-inflammatory agents in Boswellia serrata resin are the boswellic acids which have been shown to act as non-steroidal anti-inflammatory agents, inhibiting 5-lipoxygenase activity and thereby suppressing leukotriene production, such as the potent LTB4.168–170 A systematic review of all randomised controlled trials (RCTs) of Boswellia serrata in inflammatory disease states has been published recently.171 This review found seven RCTs using Boswellia serrata in inflammatory conditions. Of these, three were for osteoarthritis of the knee and one was for rheumatoid arthritis. The review concluded that there was ‘encouraging but not compelling’ evidence for the use of Boswellia serrata for inflammatory conditions. Adverse effects reported in the clinical trials were mild and self-limiting, and included nausea, heartburn and gastrointestinal disturbances. Although the study of rheumatoid arthritis patients did not find any significant differences between groups, the Boswellia serrata group reduced their NSAID use more than did the placebo group.172 For more discussion on herbs in osteoarthritis of the knee, see Chapter 22 on osteoarthritis.
Indian trials in rheumatoid arthritis and CD demonstrated significant abatement of symptoms with administration of Boswellia serrata and, although the trials are difficult to obtain, they have been included in an Indian review of the literature.173 The Indian review reported that in a series of clinical trials of moderate to severely affected patients with rheumatoid arthritis or aortic stenosis most (either bed-ridden or could not perform normal daily tasks) showed improvement in symptoms between 2 and 4 weeks of administration of Boswellia serrata extract.174 When 17 patients were changed to the placebo, they all suffered re-onset of symptoms within 10 days.173 Although it is not possible to fully evaluate the literature from a review, taken together these studies provide encouraging preliminary evidence for the use of Boswellia serrata as a potential intervention in autoimmune rheumatic conditions.
A note of caution: Boswellia serrata has been shown to inhibit in vitro all the major human drug detoxification enzymes, cytochrome P450 1A2/2C8/2C9/2C19/2D6 and 3A4 enzymes.177 This has implications for herb–drug interactions, as these enzymes are involved in the metabolism of 95% of pharmaceutical drugs.178 By inhibiting the metabolism of pharmaceuticals, Boswellia serrata may potentiate an overdose of other drugs. Until further research can clarify the clinical relevance of these interactions for therapeutic doses of Boswellia serrata, it may be best to avoid this herb in patients taking other medications.
Harpagophytum procumbens has been used traditionally for centuries to treat arthritic conditions, pain and a range of other conditions.179 The extract from the roots is now a licensed medicine in Germany.180 A thorough systematic review found there is good scientific evidence for the use of Harpagophytum procumbens in degenerative
HERBAL CYTOKINE MODULATION
A review article175 showed that a number of herbs modulate one or more cytokines. While some herbs stimulated pro-inflammatory cytokine secretion, other herbs had suppressive actions on these cytokines, and some herbs were shown to do both, depending on dosage, cell type and type of stimulus.
Traditional herbal immune suppressants include Hemidesmus indicus, Tylophora indica and Stephania tetrandra.176
Herbal cytokine modulators are summarised in Chapter 6 on respiratory infections and immune insufficiency and in Appendix 10.
joint diseases and other chronic inflammatory diseases.179 The key actives are the iridoid glycosides, especially harpagoside; however, the level of pharmacological activity reportedly differs significantly depending on geographical location, extraction methods and the presence of other constituents that may be synergistic or antagonistic. For instance, harpagoside was shown to inhibit COX-2 expression comparable with ibuprofen but harpagide, another pharmacologically active compound found in the extract, was shown to significantly stimulate COX-2 activity by twofold.180 Therefore, the anti-inflammatory activity of an extract may depend on the ratios of the pharmacologically active constituents. These variables have been postulated as an explanation for inconsistency across studies aiming to demonstrate anti-inflammatory activity of extracts.180
The mechanism of action is not clear, but it has been shown to inhibit the secretion of the pro-inflammatory cytokines IL-1β and TNF-α, the pro-inflammatory enzymes COX-2 and iNOS, and other inflammatory mediators such as leukotriene C4, prostaglandin E2 and thromboxane B2.181 An open study in people with rheumatic conditions in the UK aimed to assess the safety and efficacy of 960 mg (480 mg twice daily) of Harpagophytum procumbens in 259 patients over 8 weeks.182 Significant improvements in global pain, stiffness and function were reported, and 66% of patients had reduced or stopped their pain medication by week 8.182 The treatment was considered safe and well tolerated, with no serious adverse events. While this study provides evidence for its safety, the methodology used does not allow conclusions to be drawn about the efficacy of the herb, as the placebo effect was not estimated by inclusion of a control group.
A word about the safety of use: it has been reported that Harpagophytum procumbens is contraindicated in people with gastric and duodenal ulcer and gallstones due to its bitter taste, which increases gastric acid secretion. This would make it contraindicated in Crohn’s disease and ulcerative colitis. As no data are available for pregnancy and lactation, it is best avoided during these times.183 There is also some evidence that Harpagophytum procumbens may interact with anti-arrhythmic medications. In osteoarthritis studies, adverse effects were very mild gastrointestinal disturbances even at high doses (8100 mg) of extract. Most clinical trial dosages in osteoarthritis studies range from 960 mg to 2610 mg extract/day.183
Uncaria tomentosa has been used for centuries in Peru to support immune function.184 It is widely used as an anti-inflammatory herb and has been shown to significantly inhibit the activation of NF-kB,185 and inhibit the stimulated production of TNF-α by 65–85%.186 There are two species, Uncaria tomentosa and U. guianensis, which are used interchangeably in South America, but most Western preparations contain U. tomentosa due to its ease of standardisation.187 In a study comparing the antioxidant and anti-inflammatory activity of the two species it was found that there were 35 times more alkaloids in U. tomentosa than in U. guianensis.188 Interestingly, U. guianensis was the better antioxidant and stronger anti-inflammatory, challenging the widespread belief that oxindole alkaloids are responsible for its therapeutic actions. Others have identified the presence of quinic acids as having anti-inflammatory properties.189 In a randomised, placebo-controlled, double-blinded study of freeze-dried U. tomentosa (1 capsule of 100 mg daily) in osteoarthritis of the knee, those taking the U. tomentosa had non-significant reductions in medically assessed osteoarthritis after 1 week of treatment that continued to become highly significant at the 2-week mark.190 A review of the toxicology associated with oral administration of U. tomentosa noted that it had a low potential for acute toxicity.191
Curcuma longa and curcumin, the active constituent of C. longa, have demonstrated therapeutic effects in a wide range of autoimmune diseases.192 In a randomised, placebo-controlled double-blinded clinical trial of curcumin compared with placebo in 89 patients with quiescent ulcerative colitis, 2 g/day curcumin was significantly better than placebo at preventing remissions, and improving the clinical activity index and endoscopic index.89 Curcumin has been described as a non-steroidal anti-inflammatory agent, and found to be twice as potent as the NSAID phenylbutazone during acute inflammation, but only half as powerful during chronic inflammation.193 Curcumin was shown to inhibit the ex vivo stimulated production of pro-inflammatory cytokines TNF-α by 58% and IL-8 by 30%.194
A study of 54 traditional Mexican Indian herbs found that only three were able to inhibit NF-kB, All belong to the Asteraceae family and are rich in sesquiterpene lactones.195 Tanacetum parthenium contains high levels of the sesquiterpene lactone parthenolide, which has been shown to be anti-inflammatory by inhibiting the activation of NF-kB.196,197 Urtica dioica has also been shown to be a potent inhibitor of NF-kB. A standardised extract used in Germany as a commercial drug preparation (IDS23, Rheuma-Hek) was shown to be effective as the inhibition of ex vivo TNF-α-induced NF-kB activation in human cells inhibits ikB degradation.198 After oral administration of IDS23 for 7 and 21 days in 20 healthy volunteers, there was an inhibition in the ex vivo LPS stimulated TNF-α release of 14.6% and 24.0%, respectively, while IL-1β was inhibited by 19.2% and 39.3%, respectively.199
The leaves of Tylophora indica have been used in Ayurvedic medicine for the treatment of respiratory disorders such as asthma. In a series of studies comparing the effects of Tylphora spp. and placebo in mice after contact sensitivity with a noxious agent, Tylphora spp. significantly and reproducibly prevented the onset of the delayed hypersensitivity response. Although cytokines were not measured, these studies demonstrated that the mechanism of action involved suppression of cellular (T-cell) immunity.200 For a review of studies into its effects in respiratory conditions see Chapter 6 on respiratory infections.
Hemidesmus indicus was shown to inhibit the ex vivo production of the pro-inflammatory cytokines TNF-α and IL-8 by 50%.194 Anethole, a constituent of Foeniculum vulgare, has been shown to block TNF-α stimulated NF-kB activation.201 In 14 renal patients with increased blood levels of TNF-α and reduced levels of IL-10, glomerular dysfunction and proteinuria, 900–1125 mg/day Ganoderma lucidum was shown to reduce TNF-α and increase IL-10 and correct renal dysfunction, including suppressing proteinuria.202
For a summary of a systematic review of herbal medicine on cytokines, see Appendix 2 at the end of the book.
Aetiology of SLE
Systemic lupus erythematosus (SLE) is a chronic inflammatory connective tissue disease that predominately affects women of childbearing age, although men and children may also be affected. Immunologically, there is impairment in T-cell function with abnormal cytokine production—specifically the over-expression of the pro-inflammatory cytokines IL-17, IFN-γ203 and TNF-α. Overproduction of IFN-γ has been linked with B-cell activation.204 Autoantibody production by activated B cells results in positive serum antinuclear antibodies, often anti-DNA antibodies.
Example treatment
The patient was advised to find out more about living with lupus from appropriate support groups. She was advised to become familiar with her own early symptoms and trigger factors in order to minimise future flares. For instance, sunlight exposure may induce flares, and she reports her skin feeling strange after exposure to sun. She was encouraged to find an indoor pool and continue her swimming. Physical activity is not only good for helping deal with stress and tension, but also helps to boost the immune system. She was also advised that her condition would not necessarily affect her ability to have a baby, although she was advised to stabilise her condition before trying to conceive.
Stress is a trigger factor for many people with lupus, so she was advised to consider stress management techniques and/or counselling. Stress management techniques will depend on the type of stress that she is experiencing, which needs to be thoroughly teased out, and a referral for a course of counselling may be advised (see the referral section). Yoga and meditation practices would be a great way to re-learn how to relax in the body, as yoga may help to strengthen muscles and meditation would help to calm the mind.
Herbal prescription
| Uncaria tomentosa 1:2 | 30 mL |
| Harpagophytum procumbens 1:2 | 30 mL |
| Hemidesmus indicus 1:2 | 25 mL |
| Glycyrrhiza glabra 1:1 | 15 mL |
| 5 mL t.d.s. before meals | 100 mL |
Boswellia complex tablets containing Boswellia serrata, turmeric, ginger, celery seed. Take 4 per day for 1 month, then review.
Dietary modifications were the primary intervention for the first consultation. The fatty acid profile in the background diet was manipulated in order to reduce production of arachidonic acid inflammatory metabolites and the pro-inflammatory cytokines. Herbal medicines were also prescribed to support her through the period of increased stress (Glycyrrhiza glabra) and also to help reduce the chronic inflammation (Boswellia serrata, Harpagophytum procumbens, Hemidesmus indicus and Uncaria tomentosa). The combination of herbs, diet and exercise would be assessed at another consultation, scheduled for 1 month’s time.
INTEGRATIVE MEDICAL CONSIDERATIONS
Counselling
The patient was referred for counselling for support in dealing with stress relating to her diagnosis and family planning issues. Strong emotions usually accompany such a diagnosis but must be managed as chronic stress and depression can lead to deterioration of the condition in terms of increasing inflammation and the risk of flares.205 Education about how to manage the disease, the medications and the role of emotions can help to cope with the diagnosis.206
Daily stress events have been shown to worsen symptoms of lupus more so than significant life events.207 The patient was counselled on new ways of dealing with daily stress and distress through these sessions.
A brief pain and stress management program incorporating biofeedback and cognitive behaviour therapy was significantly more effective than symptom monitoring (placebo) or usual care (no intervention controls) at reducing pain and physical symptoms in 92 SLE patients in an American randomised, placebo-controlled trial.208 Psychological functioning was improved in the active intervention group compared with placebo, while the usual care group (controls) actually deteriorated over time.
Physical exercise
The patient was recommended to think about getting fit on her own or getting a personal trainer to help her to develop an aerobic training program. In chronic inflammatory diseases, the conventional acute management strategy is to rest and medicate. However, in chronic diseases that wax and wane, it is important during times of remission to build muscle strength and increase range of motion.209 Exercise should start as low-impact and gradually increase. The specific type of exercise is not important; instead a range of types incorporating aerobic and anabolic should be encouraged. It should start with stretching and gradually be built up.
Most women with SLE suffer from poor quality of sleep, contributed to by corticosteroid use, lack of exercise and depressed mood.210 A clinical trial of a supervised exercise program showed significant improvement in quality of life, depression and aerobic fitness in 60 women with SLE that participated in either the training group (60 minutes’ supervised training, three mornings per week for 12 weeks) or controls (did not participate in the training program).211 The training session consisted of 10 minutes warm-up and stretching, 40 minutes walking and 10 minutes of cooling down. There is good preliminary evidence that physical exercise can help in a range of symptoms associated with SLE.212 Exercise also improves mood outcomes.213
Homoeopathy
In a Lancet published meta-analysis of homoeopathy in clinical trials there was an overall trend towards an effect for homoeopathy.214 Many people with autoimmune diseases believe that homoeopathy may be helpful. In a survey of 413 people with inflammatory bowel disease, 52% had tried CAM, and homoeopathy was the most frequently used (55% of CAM users).215 A survey of people with multiple sclerosis showed a similar trend.216 Part of the reasons for the common use of homoeopathy seems to be dissatisfaction with conventional treatment and its side effects, and the fact that homoeopathy can be used alongside conventional medicine.217
In a randomised, placebo-controlled trial of 112 patients using homoeopathy in rheumatoid arthritis, there were significant improvements in pain (18%) and articular index (24%) for both the placebo and active group. There was a huge drop-out rate in the 6-month study, causing some methodological issues. Most of the homoeopathic prescriptions were for Sulphur 30C and Rhus Tox 6C and 30C.218 In an earlier, randomised, controlled trial using homoeopathy or placebo in rheumatoid arthritis in patients that were also using their own medication, those in the homoeopathy group had significant improvement in pain, articular index, stiffness and grip compared with the placebo group.219
Expected outcomes and follow-up protocols
The diagnosis of a chronic autoimmune disease, such as SLE, can be a stressful and disempowering experience. Empowerment through education and self-help strategies has been shown to improve outcomes for people with SLE on a range of parameters including feelings of uncertainty, self-worth and depression.220 The major strategies that need to be monitored and continued are the stress management, the dietary modifications, the anti-inflammatory herbal intervention, the gradual build-up of aerobic fitness and the regular relaxation or meditation practice. Other activities that reduce stress and have beneficial outcomes on immune parameters should also be encouraged, such as laughter, meditation and activities such as choir singing and listening.
Most of the medications used to treat SLE involve a multitude of serious side effects that have to be monitored and minimised if possible. For instance NSAIDs are COX inhibitors, but cyclooygenase (COX) has two isoforms. The inhibition of COX-1 is associated with the serious side effects, particularly to the gut mucosa, causing nausea, and may lead to ulceration, which may be asymptomatic or may cause bleeding.87 Some of these side effects are noted in the box above.221 These effects may be minimised using slippery elm powder and probiotic supplementation. The traditional way is to take a teaspoon of slippery elm in yoghurt before breakfast.
During infection and inflammation, however, it is COX-2 that is substantially raised. COX-2 plays a key role in the mediation of inflammation in autoimmune diseases. Pro-inflammatory mediators are known to induce COX-2 and raise prostaglandin synthesis. It is COX-2 that the anti-inflammatory steroid hormones (the glucocorticoids) suppress. NSAIDs that selectively inhibit COX-2 inhibitors are just as effective as the traditional non-selective COX inhibitors, but without the serious side effects.87 Further, small bowel permeability (‘leaky gut’) has been shown to be increased by the COX-1 inhibitors, but remains unaffected by COX-2 inhibitors.87 Where possible, therefore, COX-2 inhibitors are the NSAID of choice.
A note of caution about COX-2 inhibitors is that the long-term use of certain COX-2 inhibitors is associated with increased risk of primary heart attacks and stroke (in people with no previous history of heart disease) and hence certain COX-2 drugs were withdrawn from the market in 2004–5.222 Fortunately, it has been demonstrated that these complications can be reduced by combining fish-oil supplementation with COX-2 inhibitors.223
Lupus flares
Many people with lupus experience periods when the symptoms return, or the disease ‘flares’ up. Indeed, 11% of all hospital admissions in the USA are due to SLE or lupus flares.224 What causes them is not clear. One study set out to see whether T-cell activation could predict them. In a 4-year follow-up study, 60 patients (57 females) with SLE were tested at baseline for a urinary marker of a T-cell proliferation (T-bet).225 By the 4-year mark 46.6% had experienced a flare; 28% were severe (organ involvement). Those with high urinary T-bet were eight times more likely to have a flare.
It is an important aspect of each individual’s journey with lupus to identify the triggers that may combine to cause a flare. It may be useful to keep a journal for a while until some trigger factors become known. Most often they are lifestyle factors such as stress, infection, sunlight and some medications.226
The patient was advised to develop a strategy to deal with fatigue in order to minimise the risk of a flare. It is important to not get over-tired, to pay attention to the body signals and to rest often.226 It was advised that the patient start a yoga or meditation practice and instigate a discipline of practice for 1 hour every morning and 1 hour every evening. This may mean a major restructuring of the patient’s day and life to fit this in, but its importance must not be underestimated. The patient was also encouraged to build aerobic fitness by starting an exercise training program and building up to at least 30 minutes three times per week.
There is a known seasonal variation to lupus flares, with exposure to extremes of temperature best avoided to minimise the risk of flare.227 Sunlight exposure is another known trigger of flare and can be minimised by wearing protective clothing and sunscreen. The patient was advised to cover up when walking or jogging outside in summer and to go early in the morning or later in the afternoon to avoid the midday sun. Similarly, sunscreen should be used while swimming outside.
Reducing infections
Infections may be viral, bacterial, fungal or parasitic, and acute or chronic.228 Patients on immune-suppressive drugs and some patients with SLE are genetically more susceptible to opportunistic infections.229 Chronic infections such as hepatitis C should be closely monitored.
It has to be stressed that all infections should be treated immediately. See Chapter 6 on respiratory infections for advice on treating respiratory tract infections, and Chapter 27 for advice on urinary tract infections and the fungal infection, candida. Probiotics may need to be supplemented from time to time to ensure healthy bowel flora and help to prevent infections. If the patient does not respond quickly to herbal medicine treatment (within a few days), then medical treatment for antibiotics may be required to reduce the risk of endocarditis (inflammation of the heart valves).230 Sulfur antibiotics should be avoided as they are known to cause allergic reactions in 30% of SLE patients, and increase photosensitivity and skin rash.230
Infection in the SLE patient requires a much more rigorous approach than people without the disease. It may be prudent to refer early to a GP for antibiotics if a bacterial infection is suspected, as the consequences of an untreated infection can be devastating. The side effects of antibiotic therapy on the gut flora should be minimised by simultaneous supplementation with probiotics (see Section 1 on the gastrointestinal system). Every attempt should be made to avoid infection (see the box below for some ideas of prevention strategies).230
Pregnancy is known to trigger lupus flare in around 60% of women.231,232 The patient was advised that, in women with stable SLE, pregnancy is safe for mother and child, although it does increase the risk of a flare.233 Pregnancy requires a management strategy commencing with an assessment of pre-gestational risk factors before conception. In most cases of a lupus flare during pregnancy, the disease can be successfully managed under medical supervision with a multidisciplinary approach.234 Hospitalisation may be required during the pregnancy. The miscarriage rate is about one in five in SLE, as opposed to one in 10 for normal pregnancies.235 The contraceptive pill, on the other hand, has been shown in a double-blind, randomised, placebo-controlled clinical trial of 183 women using the lupus flare as the end point to not trigger a flare.236
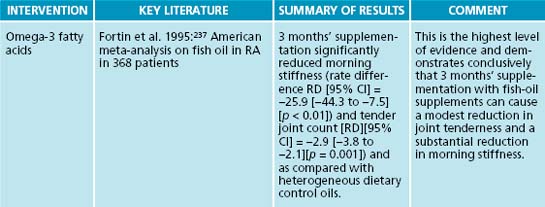
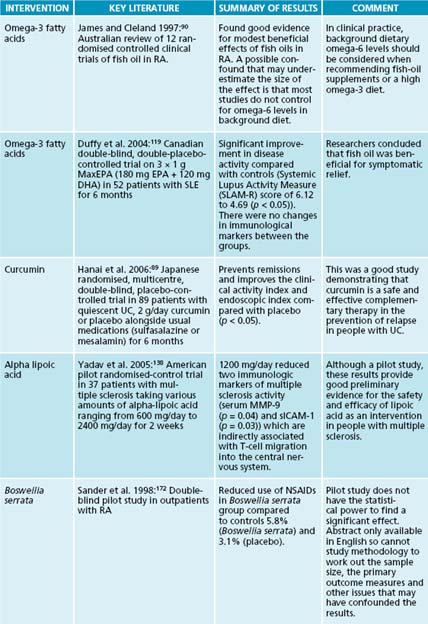
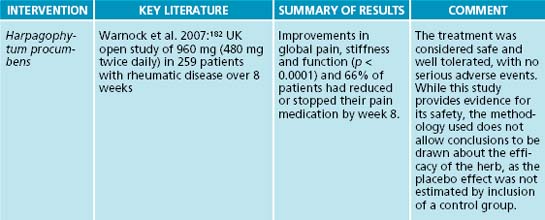
Brown A.C. Lupus erythematosus and nutrition: a review of the literature. J Ren Nutr [Review]. 2000;10(4):170-183.
Lupus Association, NSW. http://www.lupusnsw.org.au/
Lupus Foundation of America, Inc. http://www.lupus.org
Petri M. Treatment of systemic lupus erythematosus: an update. Am Fam Physician. 1998;57(11):2753-2760.
Vliet Vlieland T.P.M. Non-drug care for RA—is the era of evidence-based practice approaching? Rheumatology (Oxford) [Review]. 2007;46(9):1397-1404.
Wallace D.J. The lupus book, 3rd edn. New York: Oxford University Press; 2005.
1. Brickman C.M., Shoenfeld Y. The mosaic of autoimmunity. Scand J Clin Lab Invest. 2001;61(7):3-15.
2. Shoenfeld Y., et al. The mosaic of autoimmunity: hormonal and environmental factors involved in autoimmune diseases—2008. Isr Med Assoc J. 2008;10(1):8-12.
3. Kuek A., et al. Immune-mediated inflammatory diseases (IMIDs) and biologic therapy: a medical revolution. Postgrad Med J. 2007;83(978):251-260.
4. Kumar P., Clark M., editors. Clinical medicine, 5th edn., London: W.B. Saunders, 2002.
5. Porth C.M. Pathophysiology: concepts of altered health states, 4 edn. Philadelphia: J.B. Lippincott Company; 1994.
6. GPnotebook. Major histocompatibility antigen class II. Online. Available: http://www.gpnotebook.co.uk/simplepage.cfm?ID = -328531924&linkID = 21624&cook = yes. Accessed 4 February 2009.
7. Davidson A., Diamond B. Autoimmune diseases. N Engl J Med. 2001;345(5):340-350.
8. GPnotebook. Hashimoto’s thyroiditis. Online. Available: http://www.gpnotebook.co.uk/simplepage.cfm?ID = 342556680. Accessed 4 February 2009.
9. Ramagopalan S.V., et al. Expression of the multiple sclerosis-associated MHC class II Allele HLA-DRB1∗1501 is regulated by vitamin D. PLoS Genet. 5(2), 2009. e1000369
10. Mackay I.R. Science, medicine, and the future: tolerance and autoimmunity. BMJ. 2000;321(7253):93-96.
11. Hill N., Sarvetnick N. Cytokines: promoters and dampeners of autoimmunity. Curr Opin Immunol. 2002;14(6):791-797.
12. Sospedra M., Martin R. Immunology of multiple sclerosis. Annu Rev Immunol. 2005;23(1):683-747.
13. Blass S., et al. The immunologic homunculus in rheumatoid arthritis. Arthritis Rheum. 1999;42(12):2499-2506.
14. Head K.A., Jurenka J.S. Inflammatory bowel disease part 1: ulcerative colitis—pathophysiology and conventional and alternative treatment options. Altern Med Rev. 2003;8(3):247-283.
15. Cordain L. Cereal grains: humanity’s double-edged sword. World Rev Nutr Diet. 1999;84:19-73.
16. Heyman M., Desjeux J.F. Cytokine-induced alteration of the epithelial barrier to food antigens in disease. Ann N Y Acad Sci. 2000;915:304-311.
17. Dimitrov J.D., et al. Insight into the mechanism of the acquired antibody auto-reactivity. Autoimmun Rev. 2008;7:410-414.
18. Frassetto L., et al. Diet, evolution and aging—the pathophysiologic effects of the post-agricultural inversion of the potassium-to-sodium and base-to-chloride ratios in the human diet. Eur J Nutr. 2001;40(5):200-213.
19. Falcone M., Sarvetnick N. Cytokines that regulate autoimmune responses. Curr Opin Immunol. 1999;11(6):670-676.
20. Grimble R.F. Nutritional modulation of cytokine biology. Nutrition. 1998;14(7–8):634-640.
21. Pert C. Molecules of emotion. USA: Simon & Schuster; 1997.
22. Leonard B. Stress and the immune system: immunological aspects of depressive illness. In: Schmoll H.J., Tewes U., editors. Psychoneuroimmunology—interactions between brain, nervous system, behaviour, endocrine and immune system. New York: Hogrefe & Huber Publishers; 1999:114-136.
23. Germain R.N. T-cell development and the CD4-CD8 lineage decision. Nat Rev Immunol. 2002;2(5):309-322.
24. Weaver C.T., et al. IL-17 family cytokines and the expanding diversity of effector T cell lineages. Annu Rev Immunol. 2007;25(1):821-852.
25. Swain S.L. T cell subsets and the recognition of MHC class. Immunol Rev. 1983;74(1):129-142.
26. Zola H., et al. CD molecules 2005: human cell differentiation molecules. Blood. 2005;106(9):3123-3126.
27. Merck. Components of the immune system. Online. Available: http://www.merck.com/mmpe/sec13/ch163/ch163b.html#. Accessed 13 February 2009.
28. Elenkov I.J., et al. Cytokine dysregulation, inflammation and well-being. Neuroimmunomodulation. 2005;12:255-269.
29. McEwen B.S., et al. The role of adrenocorticoids as modulators of immune function in health and disease: neural, endocrine and immune interactions. Brain Res Brain Res Rev. 1997;23(1–2):79-133.
30. Chang J.T., et al. Regulation of interleukin (IL)-12 receptor beta 2 subunit expression by endogenous IL-12: a critical step in the differentiation of pathogenic autoreactive T cells. J Exp Med. 1999;189(6):969-978.
31. O’Shea J.J., et al. Cytokines and autoimmunity. Nat Rev Immunol. 2002;2(1):37-45.
32. Mills K.H.G. Induction, function and regulation of IL-17-producing T cells. Eur J Immunol. 2008;38(10):2636-2649.
33. Annunziato F., et al. Phenotypic and functional features of human Th17 cells. J Exp Med. 2007;204(8):1849-1861.
34. Daniels M.D., et al. Recombinant cardiac myosin fragment induces experimental autoimmune myocarditis via activation of Th1 and Th17 immunity. Autoimmunity. 2008;41(6):490-499.
35. Gaffen S.L. An overview of IL-17 function and signaling. Cytokine. 2008;43(3):402-407.
36. Ogura H., et al. Interleukin-17 promotes autoimmunity by triggering a positive-feedback loop via interleukin-6 induction. Immunity. 2008;29(4):628-636.
37. Curtsinger J.M., et al. Inflammatory cytokines provide a third signal for activation of naive CD4+ and CD8+ T cells. J Immunol. 1999;162(6):3256-3262.
38. Vella A.T., et al. CD28 engagement and proinflammatory cytokines contribute to T cell expansion and long-term survival in vivo. J Immunol. 1997;158(10):4714-4720.
39. Pape K.A., et al. Inflammatory cytokines enhance the in vivo clonal expansion and differentiation of antigen-activated CD4+ T cells. J Immunol. 1997;159(2):591-598.
40. Danke N.A., et al. Autoreactive T cells in healthy individuals. J Immunol. 2004;172(10):5967-5972.
41. Piccirillo C.A., et al. CD4+Foxp3+ regulatory T cells in the control of autoimmunity: in vivo veritas. Curr Opin Immunol. 2008;20(6):655-662.
42. Maloy K.J., Powrie F. Regulatory T cells in the control of immune pathology. Nat Immunol. 2001;2(9):816-822.
43. Cope A.P. Regulation of autoimmunity by proinflammatory cytokines. Curr Opin Immunol. 1998;10(6):669-676.
44. Strom T.B., Koulmanda M. Cytokine related therapies for autoimmune disease. Curr Opin Immunol. 2008;20(6):676-681.
45. Cooper G.S., et al. The role of genetic factors in autoimmune disease: implications for environmental research. Environ Health Perspect. 1999;107(Suppl. 5):693-700.
46. Nancy A.L., et al. Prediction and prevention of autoimmune skin disorders. Arch Dermatol Res. 2009;301(1):57-64.
47. Svendsen A.J., et al. Relative importance of genetic effects in rheumatoid arthritis: historical cohort study of Danish nationwide twin population. BMJ. 2002;324(7332):264.
48. Prelog M. Aging of the immune system: a risk factor for autoimmunity? Autoimmun Rev. 2006;5(2):136-139.
49. Thewissen M., et al. Premature immunosenescence in rheumatoid arthritis and multiple sclerosis patients. Ann N Y Acad Sci. 2005;1051:255-262.
50. University of maryland medical center. Multiple sclerosis—risk factors. Online. Available: http:www.umm.edu/patiented/articles/who_gets_multiple_sclerosis_000017_5.htm
51. Münz C., et al. Antiviral immune responses: triggers of or triggered by autoimmunity? Nat Rev Immunol. 2009;9(4):246-258.
52. Niller H.H., et al. Regulation and dysregulation of Epstein-Barr virus latency: implications for the development of autoimmune diseases. Autoimmunity. 2008;41(4):298-328.
53. Cutolo M., Straub R.H. Stress as a risk factor in the pathogenesis of rheumatoid arthritis. Neuroimmunomodulation. 2006;13(5–6):277-282.
54. Atkinson J.P. Complement deficiency. Predisposing factor to autoimmune syndromes. Am J Med. 1988;85(6A):45-47.
55. Tsutsumi A., et al. Mannose binding lectin: genetics and autoimmune disease. Autoimmun Rev. 2005;4(6):364-372.
56. Grulich A.E., et al. Altered immunity as a risk factor for non-Hodgkin lymphoma. Cancer Epidemiol Biomarkers Prev. 2007;16(3):405-408.
57. Cerhan J.R., et al. Association of aspirin and other non-steroidal anti-inflammatory drug use with incidence of non-Hodgkin lymphoma. Int J Cancer. 2003;106(5):784-788.
58. Yavari N., et al. What role do occupational exposures play in RA? Studies have found significant associations and increased risk. J Musculoskeletal Med. 2008;25(3):130.
59. Bártová J., et al. Dental amalgam as one of the risk factors in autoimmune diseases. Neuro Endocrinol Lett. 2003;24(1–2):65-67.
60. Noonan C.W., et al. Nested case-control study of autoimmune disease in an asbestos-exposed population. Environ Health Perspect. 2006;114(8):1243-1247.
61. Parks C.G., et al. Occupational exposure to crystalline silica and autoimmune disease. Environ Health Perspect. 1999;107(Suppl. 5):793-802.
62. Chen R.T., et al. Epidemiology of autoimmune reactions induced by vaccination. J Autoimmun. 2001;16(3):309-318.
63. Mayes M.D. Epidemiologic studies of environmental agents and systemic autoimmune diseases. Environ Health Perspect. 1999;107(Suppl. 5):743-748.
64. Dow C.T. Paratuberculosis and type I diabetes: is this the trigger? Med Hypotheses. 2006;67(4):782-785.
65. Kamen D.L., Aranow C. The link between vitamin D deficiency and systemic lupus erythematosus. Curr Rheumatol Rep. 2008;10(4):273-280.
66. Coo H., Aronson K.J. A systematic review of several potential non-genetic risk factors for multiple sclerosis. Neuroepidemiology. 2004;23(1–2):1-12.
67. Palma B.D., et al. Effects of sleep deprivation on the development of autoimmune disease in an experimental model of systemic lupus erythematosus. Am J Physiol Regul Integr Comp Physiol. 2006;291(5):R1527-R1532.
68. Beers M.H., Berkow R., editors. The Merck manual of diagnosis and therapy, 17th edn, New Jersey: Merck Research Laboratories, Division of Merck & Co., Inc., 1999.
69. Goemaere S., et al. Onset of symptoms of rheumatoid arthritis in relation to age, sex and menopausal transition. J Rheumatol. 1990;17(12):1620-1622.
70. Hall G.M., et al. A randomised controlled trial of the effect of hormone replacement therapy on disease activity in postmenopausal rheumatoid arthritis. Ann Rheum Dis. 1994;53(2):112-116.
71. Carlsten H., Carlsten H. Immune responses and bone loss: the estrogen connection. Immunol Rev. 2005;208:194-206.
72. Jansson L., Holmdahl R. Enhancement of collagen-induced arthritis in female mice by estrogen receptor blockage. Arthritis Rheum. 2001;44(9):2168-2175.
73. Muthian G., Bright J.J. Quercetin, a flavonoid phytoestrogen, ameliorates experimental allergic encephalomyelitis by blocking IL-12 signaling through JAK-STAT pathway in T lymphocyte. J Clin Immunol. 2004;24(5):542-552.
74. Klecha A.J., et al. Immune-endocrine interactions in autoimmune thyroid diseases. Neuroimmunomodulation. 2008;15(1):68-75.
75. Matos-Santos A., et al. Relationship between the number and impact of stressful life events and the onset of Graves disease and toxic nodular goitre [see comment]. Clin Endocrinol (Oxf). 2001;55(1):15-19.
76. Gold S.M., et al. Stress and disease progression in multiple sclerosis and its animal models. Neuroimmunomodulation. 2006;13(5–6):318-326.
77. Chida Y., et al. Social isolation stress exacerbates autoimmune disease in MRL/LPR mice. J Neuroimmunol. 2005;158(1–2):138-144.
78. Sternberg E.M. Neuroendocrine regulation of autoimmune/inflammatory disease. J Endocrinol. 2001;169(3):429-435.
79. Jara L.J., et al. Immune-neuroendocrine interactions and autoimmune diseases. Clin Dev Immunol. 2006;13(2–4):109-123.
80. Homo-Delarche F. Neuroendocrine immuno-ontogeny of the pathogenesis of autoimmune disease in the nonobese diabetic (NOD) mouse. ILAR J. 2004;45(3):237-258.
81. Kumar R., et al. Immunoregulatory role of stress mediators in rheumatoid arthritis. Z Rheumatoly. 1981;40:122-125.
82. Sternberg E.M., et al. Inflammatory mediator-induced hypothalamic-pituitary-adrenal axis activation is defective in streptococcal cell wall arthritis-susceptible Lewis rats. Proc Natl Acad Sci U S A. 1989;86:2374-2378.
83. Vally M., Pillarisetti S. Disease modification in rheumatoid arthritis and osteoarthritis current and emerging targets and therapeutics. Cur Med Chem Immunol Endocr Metab Agents. 2005;5:259-268.
84. Choy E.H., Panayi G.S. Cytokine pathways and joint inflammation in rheumatoid arthritis. N Engl J Med. 2001;344(12):907-916.
85. Brooks P. Rheumatoid arthritis. Herston: CMPMedica Australia Pty Ltd; 2006.
86. Kievit W., et al. The efficacy of anti-TNF in rheumatoid arthritis, a comparison between randomised controlled trials and clinical practice. Ann Rheum Dis. 2007;66(11):1473-1478.
87. Brooks P.M., Day R.O. COX-2 inhibitors. Med J Aust. 2000;173(8):433-436.
88. Fox R.I. Mechanism of action of hydroxychloroquine as an antirheumatic drug. Semin Arthritis Rheum. 1993;23(2 Suppl. 1):82-91.
89. Hanai H., et al. Curcumin maintenance therapy for ulcerative colitis: randomized, multicenter, double-blind, placebo-controlled trial. Clin Gastroenterol Hepatol. 2006;4(12):1502-1506.
90. James M.J., Cleland L.G. Dietary n-3 fatty acids and therapy for rheumatoid arthritis. Semin Arthritis Rheum. 1997;27(2):85-97.
91. Maes M., et al. The effects of psychological stress on humans: increased production of pro-inflammatory cytokines and a Th1-like response in stress-induced anxiety. Cytokine. 1998;10(4):313-318.
92. Kiecolt-Glaser J.K., et al. Emotions, morbidity, and mortality: new perspectives from psychoneuroimmunology. Annu Rev Psychol. 2002;53:83-107.
93. Salovey P., et al. Emotional states and physical health. Am Psychol. 2000;55(1):110-121.
94. Yoshino S., et al. Effects of mirthful laughter on neuroendocrine and immune systems in patients with rheumatoid arthritis. J Rheumatol. 1996;23(4):793-794.
95. Davidson R.J., et al. Alterations in brain and immune function produced by mindfulness meditation [see comment]. Psychosom Med. 2003;65(4):564-570.
96. Solberg E.E., et al. Meditation: a modulator of the immune response to physical stress? A brief report. Br J Sports Med. 1995;29(4):255-257.
97. Seeman T.E., et al. Religiosity/spirituality and health. A critical review of the evidence for biological pathways. Am Psychol. 2003;58(1):53-63.
98. Prather A.A., et al. Positive affective style covaries with stimulated IL-6 and IL-10 production in a middle-aged community sample. Brain Behav Immun. 2007;21(8):1033-1037.
99. Kreutz G., et al. Effects of choir singing or listening on secretory immunoglobulin A, cortisol, and emotional state. J Behav Med. 2004;27(6):623-635.
100. Miller J.B., et al. The GI factor: the glucose revolution, 2nd edn. Sydney: Hodder Headline; 1996.
101. Simopoulos A. Essential fatty acids in health and chronic disease. Am J Clin Nutr. 1999;70(3):560S-569S.
102. Muldoon M.F., et al. Lowering cholesterol concentration and mortality: a quantitative review of primary prevention trials. BMJ. 1990;301:309-314.
103. ABS. Diabetes mellitus. 41020—Australian Social Trends, 2007. Online. Available: http://www.abs.gov.au/ausstats/abs@.nsf/latestproducts/62051E65E97FF67BCA25732C002076F3?opendocument. Accessed 4 May 2009.
104. Lowes J.R., et al. Characterisation and quantification of mucosal cytokine that induces epithelial histocompatibility locus antigen-DR expression in inflammatory bowel disease. Gut. 1992;33(3):315-319.
105. Mothes T., et al. Effect of gliadin and other food peptides on expression of MHC class II molecules by HT-29 cells. Gut. 1995;36(4):548-552.
106. Virkkunen M.F., et al. Plasma phospholipid essential fatty acids and prostaglandins in alcoholic, habitually violent and impulsive offenders. Biol Psychiatry. 1987;22:1087-1096.
107. Simopoulos A.P. Omega-3 fatty acids in inflammation and autoimmune diseases. J Am Coll Nutr. 2002;21(6):495-505.
108. Fernandes G. Progress in nutritional immunology. Immunol Res. 2008;40(3):244-261.
109. Good R.A., et al. Nutritional deficiency, immunologic function, and disease. Am J Pathol. 1976;84(3):599-614.
110. Friend P.S., et al. Dietary restrictions early and late: effects on the nephropathy of the NZB X NZW mouse. Lab Invest. 1978;38(6):629-632.
111. Calder P. n-3 polyunsaturated fatty acids and cytokine production in health and disease. Ann Nutr Metab. 1997;41:203-234.
112. Calder P.C. Polyunsaturated fatty acids and inflammation. Prostaglandins Leukot Essent Fatty Acids. 2006;75(3):197-202.
113. Endres S., von Schacky C. n-3 polyunsaturated fatty acids and human cytokine synthesis. Curr Opin Lipidol. 1996;7(1):48-52.
114. Huang Y.S., Nassar B.A. Modulation of tissue fatty acid composition, prostaglandin production and cholesterol levels by dietary manipulation of n-3 and n-6 essential fatty acid metabolites. In: Horrobin D., editor. Omega 6 essential fatty acids: pathophysiology and roles in clinical medicine. USA: Wiley-Liss; 1990:127-144.
115. Brenner R. Nutritional and hormonal factors influencing desaturation of essential fatty acids. Prog Lipid Res. 1981;20:41-47.
116. Brenner R.R., Peluffo R.O. Inhibitory effect of docosa-4,7,10,13,16,19-hexaenoic acid upon the oxidative desaturation of linoleic into gamma-linolenic and of alpha-linolenic into octadeca-6,9,12,15-tetraenoic acid. Biochim Biophys Acta. 1967;137:184-186.
117. Howe P., et al. Dietary intake of long-chain [omega]-3 polyunsaturated fatty acids: contribution of meat sources. Nutrition. 2006;22(1):47-53.
118. Meyer B.J., et al. Dietary intakes and food sources of omega-6 and omega-3 polyunsaturated fatty acids. Lipids. 2003;38(4):391-398.
119. Duffy E.M., et al. The clinical effect of dietary supplementation with omega-3 fish oils and/or copper in systemic lupus erythematosus. J Rheumatol. 2004;31(8):1551-1556.
120. Baldwin A.S. The NF-kappa B and l kappa B proteins: new discoveries and insights. Annu Rev Immunol. 1996;14(1):649-681.
121. Vasto S., et al. Inflammation, genes and zinc in ageing and age-related diseases. Biogerontology. 2006;7(5–6):315-327.
122. Shen H., et al. Zinc deficiency induces vascular pro-inflammatory parameters associated with NF-kappaB and PPAR signaling. J Am Coll Nutr. 2008;27(5):577-587.
123. Beach R.S., et al. Nutritional factors and autoimmunity. III. Zinc deprivation versus restricted food intake in MRL/1 mice—the distinction between interacting dietary influences. J Immunol. 1982;129(6):2686-2692.
124. Leiter L.M., et al. Iron status alters murine systemic lupus erythematosus. J Nutr. 1995;125(3):474-484.
125. Komatsu W., et al. Docosahexaenoic acid suppresses nitric oxide production and inducible nitric oxide synthase expression in interferon-[gamma] plus lipopolysaccharide-stimulated murine macrophages by inhibiting the oxidative stress. Free Radic Biol Med. 2003;34(8):1006-1016.
126. Aktas O., et al. Green tea epigallocatechin-3-gallate mediates T cellular NF-kappa B inhibition and exerts neuroprotection in autoimmune encephalomyelitis. J Immunol. 2004;173(9):5794-5800.
127. Wheeler D.S., et al. Epigallocatechin-3-gallate, a green tea-derived polyphenol, inhibits IL-1beta-dependent proinflammatory signal transduction in cultured respiratory epithelial cells. J Nutr. 2004;134(5):1039-1044.
128. Schreiber S., et al. Activation of nuclear factor kappa B in inflammatory bowel disease. Gut. 1998;42(4):477-484.
129. Kim S.H., et al. Luteolin inhibits the nuclear factor-kappa B transcriptional activity in Rat-1 fibroblasts. Biochem Pharmacol. 2003;66(6):955-963.
130. Ruiz P.A., et al. Quercetin inhibits TNF-induced NF-kappaB transcription factor recruitment to proinflammatory gene promoters in murine intestinal epithelial cells. J Nutr. 2007;137(5):1208-1215.
131. Ruiz P.A., Haller D. Functional diversity of flavonoids in the inhibition of the proinflammatory NF-kappaB, IRF, and AKT signaling pathways in murine intestinal epithelial cells. J Nutr. 2006;136(3):664-671.
132. Ziegler-Heitbrock H.W., et al. Pyrrolidine dithiocarbamate inhibits NF-kappa B mobilization and TNF production in human monocytes. J Immunol. 1993;151(12):6986-6993.
133. Merrill J.E., Murphy S.P. Inflammatory events at the blood brain barrier: regulation of adhesion molecules, cytokines, and chemokines by reactive nitrogen and oxygen species. Brain Behav Immun. 1997;11(4):245-263.
134. Tse H.M., et al. Disruption of innate-mediated proinflammatory cytokine and reactive oxygen species third signal leads to antigen-specific hyporesponsiveness. J Immunol. 2007;178(2):908-917.
135. Packer L., et al. Neuroprotection by the metabolic antioxidant alpha-lipoic acid. Free Radic Biol Med. 1997;22(1–2):359-378.
136. Marracci G.H., et al. Alpha lipoic acid inhibits T cell migration into the spinal cord and suppresses and treats experimental autoimmune encephalomyelitis. J Neuroimmunol. 2002;131(1–2):104-114.
137. Morini M., et al. Alpha-Lipoic acid is effective in prevention and treatment of experimental autoimmune encephalomyelitis. J Neuroimmunol. 2004;148(1–2):146-153.
138. Yadav V., et al. Lipoic acid in multiple sclerosis: a pilot study. Mult Scler. 2005;11(2):159-165.
139. De Paula M.L., et al. Genistein down-modulates pro-inflammatory cytokines and reverses clinical signs of experimental autoimmune encephalomyelitis. Int Immunopharmacol. 2008;8(9):1291-1297.
140. Miodini P., et al. The two phyto-oestrogens genistein and quercetin exert different effects on oestrogen receptor function. Br J Cancer. 1999;80(8):1150-1155.
141. Singh N.P., et al. Resveratrol (trans-3,5,4’-Trihydroxystilbene) ameliorates experimental allergic encephalomyelitis, primarily via induction of apoptosis in T cells involving activation of aryl hydrocarbon receptor and estrogen receptor. Mol Pharmacol. 2007;72(6):1508-1521.
142. de la Lastra C.A., Villegas I. Resveratrol as an anti-inflammatory and anti-aging agent: mechanisms and clinical implications. Mol Nutr Food Res. 2005;49(5):405-430.
143. Pedersen B.K., Toft A.D. Effects of exercise on lymphocytes and cytokines. Br J Sports Med. 2000;34(4):246-251.
144. Costa Rosa L.F.B.P. Exercise as a time-conditioning effector in chronic disease: a complementary treatment strategy. Evid Based Complement Alternat Med. 2004;1(1):63-70.
145. Estivariz C.F., et al. Efficacy of parenteral nutrition supplemented with glutamine dipeptide to decrease hospital infections in critically ill surgical patients. J Parenter Enteral Nutr. 2008;32(4):389-402.
146. Castell L.M., et al. Does glutamine have a role in reducing infections in athletes? Eur J Appl Physiol. 1996;73(5):488-490.
147. Castell L.M. Glutamine supplementation in vitro and in vivo, in exercise and in immunodepression. Sports Med. 2003;33(5):323-345.
148. da Silva Krause M., de Bittencourt P.I.H. Type 1 diabetes: can exercise impair the autoimmune event? The L-arginine/glutamine coupling hypothesis. Cell Biochem Funct. 2008;26(4):406-433.
149. Anisman H., et al. Cytokines as a stressor: implications for depressive illness. Int J Neuropsychopharmacol. 2002;5(4):357-373.
150. Arimura A., et al. Interactions between cytokines and the hypothalamic-pituitary-adrenal axis during stress. Ann N Y Acad Sci. 1994;739:270-281.
151. De Kloet E.R., et al. Cytokines and the brain corticosteroid receptor balance: relevance to pathophysiology of neuroendocrine-immune communication. Psychoneuroendocrinology. 1994;19(2):121-134.
152. Banerjee U., Izquierdo J.A. Antistress and antifatigue properties of Panax ginseng: comparison with piracetam. Acta Physiol Lat Am. 1982;32(4):277-285.
153. Caso M.A., et al. Double-blind study of a multivitamin complex supplemented with ginseng extract. Drugs Exp Clin Res. 1996;22(6):323-329.
154. Dua P.R., et al. Adaptogenic activity of Indian Panax pseudoginseng. Indian J Exp Biol. 1989;27(7):631-634.
155. Grandhi A., et al. A comparative pharmacological investigation of ashwagandha and ginseng. J Ethnopharmacol. 1994;44:131-135.
156. Kumar R., et al. Enhanced thermogenesis in rats by Panax ginseng, multivitamins and minerals. Int J Biometeorol. 1996;39(4):187-191.
157. Leung K.W., et al. Angiomodulatory and neurological effects of ginsenosides. Curr Med Chem. 2007;14(12):1371-1380.
158. Nocerino E., et al. The aphrodisiac and adaptogenic properties of ginseng. Fitoterapia. 2000;71(Suppl. 1):S1-S5.
159. Saito H., et al. Effect of Panax ginseng root on exhaustive exercise in mice. Jpn J Pharmacol. 1974;24:119-127.
160. Bhattacharya S.K., Muruganandam A.V. Adaptogenic activity of Withania somnifera: an experimental study using a rat model of chronic stress. Pharmacol Biochem Behav. 2003;75(3):547-555.
161. Dhuley J.N. Adaptogenic and cardioprotective action of ashwagandha in rats and frogs. J Ethnopharmacol. 2000;70(1):57-63.
162. Rai D., et al. Adaptogenic effect of Bacopa monniera (brahmi). Pharmacol Biochem Behav. 2003;75(4):823-830.
163. Armanini D., et al. Affinity of licorice derivatives for mineralocorticoid and glucocorticoid receptors. Clin Endocrinol. 1983;19:609-612.
164. Schlebusch L., et al. A double-blind, placebo-controlled, double-centre study of the effects of an oral multivitamin-mineral combination on stress. S Afr Med J. 2000;90(12):1216-1223.
165. Kumar R., et al. Antistress and adaptogenic activity of lecithin supplementation. J Altern Complement Med. 2002;8(4):487-492.
166. De Caterina R., et al. The omega-3 fatty acid docosahexaenoate reduces cytokine-induced expression of proatherogenic and proinflammatory proteins in human endothelial cells. Arterioscler Thromb. 1994;14(11):1829-1836.
167. Maes M., et al. In humans, serum polyunsaturated fatty acid levels predict the response of proinflammatory cytokines to psychologic stress. Biol Psychiatry. 2000;47(10):910-920.
168. Safayhi H., et al. Mechanism of 5-lipoxygenase inhibition by acetyl-11-keto-beta-boswellic acid. Mol Pharmacol. 1995;47(6):1212-1216.
169. Ammon H.P., et al. Inhibition of leukotriene B4 formation in rat peritoneal neutrophils by an ethanolic extract of the gum resin exudate of Boswellia serrata. Planta Med. 1991;57(3):203-207.
170. Singh G.B., Atal C.K. Pharmacology of an extract of salai guggal ex- Boswellia serrata, a new non-steroidal anti-inflammatory agent. Inflamm Res. 1986;18(3):407-412.
171. Ernst E. Frankincense: systematic review. BMJ. 337, 2008. a2813
172. Sander O., et al. [Is H15 (resin extract of Boswellia serrata, ‘incense’) a useful supplement to established drug therapy of chronic polyarthritis? Results of a double-blind pilot study]. Z Rheumatol. 1998;57(1):11-16.
173. Shah B.A., et al. Boswellic acids: a group of medicinally important compounds. Nat Prod Rep. 2009;26(1):72-89.
174. Pachnanda V.K., et al. Clinical evaluation of salai guggul in patients of arthritis. Indian J Pharmacol. 1981;13:63.
175. Spelman K., et al. Modulation of cytokine expression by traditional medicines: a review of herbal immunomodulators. Altern Med Rev. 2006;11(2):128-150.
176. Bone K. Clinical applications of Ayurvedic and Chinese herbs. Warwick: Phytotherapy Press; 1996.
177. Frank A., Unger M. Analysis of frankincense from various Boswellia species with inhibitory activity on human drug metabolising cytochrome P450 enzymes using liquid chromatography mass spectrometry after automated on-line extraction. J Chromatogr A. 2006;1112(1–2):255-262.
178. Guengerich F.P. Cytochromes P450, drugs, and diseases. Mol Interv. 2003;3(4):194-204.
179. Brendler T., et al. Devil’s claw (Harpagophytum procumbens DC): an evidence-based systematic review by the Natural Standard Research Collaboration. J Herb Pharmacother. 2006;6(1):89-126.
180. Abdelouahab N., Heard C. Effect of the major glycosides of Harpagophytum procumbens (devil’s claw) on epidermal cyclooxygenase-2 (COX-2) in vitro. J Nat Prod. 2008;71(5):746-749.
181. Grant L., et al. A review of the biological and potential therapeutic actions of Harpagophytum procumbens. Phytother Res. 2007;21(3):199-209.
182. Warnock M., et al. Effectiveness and safety of devil’s claw tablets in patients with general rheumatic disorders. Phytother Res. 2007;21(12):1228-1233.
183. Brien S., et al. Devil’s claw (Harpagophytum procumbens) as a treatment for osteoarthritis: a review of efficacy and safety. J Altern Complement Med. 2006;12(10):981-993.
184. Cat’s Claw. Online. Available: http://nccam.nih.gov/health/catclaw/. Accessed 15 April 2009.
185. Sandoval-Chacon M., et al. Antiinflammatory actions of cat’s claw: the role of NF-kappaB. Aliment Pharmacol Ther. 1998;12(12):1279-1289.
186. Sandoval M., et al. Cat’s claw inhibits TNF alpha production and scavenges free radicals: role in cytoprotection. Free Radic Biol Med. 2000;29(1):71-78.
187. Hardin S.R., Hardin S.R. Cat’s claw: an Amazonian vine decreases inflammation in osteoarthritis. Complement Ther Clin Pract. 2007;13(1):25-28.
188. Sandoval M., et al. Anti-inflammatory and antioxidant activities of cat’s claw (Uncaria tomentosa and Uncaria guianensis) are independent of their alkaloid content. Phytomedicine. 2002;9(4):325-337.
189. Sheng Y., et al. An active ingredient of Cat’s Claw water extracts identification and efficacy of quinic acid. J Ethnopharmacol. 2005;96(3):577-584.
190. Piscoya J., et al. Efficacy and safety of freeze-dried cat’s claw in osteoarthritis of the knee: mechanisms of action of the species Uncaria guianensis. Inflamm Res. 2001;50(9):442-448.
191. Valerio Jnr L.G., et al. Toxicological aspects of the South American herbs cat’s claw (Uncaria tomentosa) and maca (Lepidium meyenii): a critical synopsis. Toxicol Rev. 2005;24(1):11-35.
192. Jagetia G.C., Aggarwal B.B. Spicing up of the immune system by curcumin. J Clin Immunol. 2007;27(1):19-35.
193. Srimal R.C., Dhawan B.N. Pharmacology of diferuloyl methane (curcumin), a non-steroidal anti-inflammatory agent. J Pharm Pharmacol. 1973;25(6):447-452.
194. Jain A., Basal E. Inhibition of Propionibacterium acnes-induced mediators of inflammation by Indian herbs. Phytomedicine. 2003;10(1):34-38.
195. Bork P.M., et al. Sesquiterpene lactone containing Mexican Indian medicinal plants and pure sesquiterpene lactones as potent inhibitors of transcription factor NF-kappaB. FEBS Lett. 1997;402(1):85-90.
196. Hehner S.P., et al. The antiinflammatory sesquiterpene lactone parthenolide inhibits NF-kappaB by targeting the I kappaB kinase complex. J Immunol. 1999;163(10):5617-5623.
197. Kwok B.H., et al. The anti-inflammatory natural product parthenolide from the medicinal herb feverfew directly binds to and inhibits I kappaB kinase. Chem Biol. 2001;8(8):759-766.
198. Riehemann K., et al. Plant extracts from stinging nettle (Urtica dioica), an antirheumatic remedy, inhibit the proinflammatory transcription factor NF-kappaB. FEBS Lett. 1999;442(1):89-94.
199. Teucher T., et al. Cytokine secretion in whole blood of healthy volunteers after oral ingestion of an Urtica dioica L. leaf extract. Zytokin-Sekretion im Vollblut gesunder Probanden nach oraler Einnahme eines Urtica dioica L-Blattextraktes. 1996;46(9):906-910.
200. Ganguly T., Sainis K.B. Inhibition of cellular immune responses by Tylophora indica in experimental models. Phytomedicine. 2001;8(5):348-355.
201. Chainy G.B., et al. Anethole blocks both early and late cellular responses transduced by tumor necrosis factor: effect on NF-kappaB, AP-1, JNK, MAPKK and apoptosis. Oncogene. 2000;19(25):2943-2950.
202. Futrakul N., et al. Ganoderma lucidum suppresses endothelial cell cytotoxicity and proteinuria in persistent proteinuric focal segmental glomerulosclerosis (FSGS) nephrosis. Clin Hemorheol Microcirc. 2004;31(4):267-272.
203. Crispin J.C., et al. Expanded double negative T cells in patients with systemic lupus erythematosus produce IL-17 and infiltrate the kidneys. J Immunol. 2008;181(12):8761-8766.
204. Harigai M., et al. Excessive production of IFN-gamma in patients with systemic lupus erythematosus and its contribution to induction of B lymphocyte stimulator/B cell-activating factor/TNF ligand superfamily-13B. J Immunol. 2008;181(3):2211-2219.
205. Frieri M. Neuroimmunology and inflammation: implications for therapy of allergic and autoimmune diseases. Ann Allergy Asthma Immunol. 2003;90(6 Suppl. 3):34-40.
206. Halverson P.B., Holmes S.B. Systemic lupus erythematosus: medical and nursing treatments. Orthop Nurs. 1992;11(6):17-24.
207. Peralta-Ramirez M.I., et al. The effects of daily stress and stressful life events on the clinical symptomatology of patients with lupus erythematosus. Psychosom Med. 2004;66(5):788-794.
208. Greco C.M., et al. Effects of a stress-reduction program on psychological function, pain and physical function of systemic lupus erythematosus patients: a randomized controlled trial. Arthritis Rheum. 2004;51(4):625-634.
209. Minor M.A., Lane N.E. Recreational exercise in arthritis. Rheum Dis Clin North Am. 1996;22(3):563-577.
210. Costa D.D., et al. Determinants of sleep quality in women with systemic lupus erythematosus. Arthritis Rheum. 2005;53(2):272-278.
211. Carvalho M.R., et al. Effects of supervised cardiovascular training program on exercise tolerance, aerobic capacity and quality of life in patients with systemic lupus erythematosus. Arthritis Rheum. 2005;53(6):838-844.
212. Ayan C., Martin V. Systemic lupus erythematosus and exercise. Lupus. 2007;16(1):5-9.
213. Dua J., Hargreaves L. Effect of aerobic exercise on negative affect, positive affect, stress, and depression. PerceptMotSkills. 1992;75(2):355-361.
214. Linde K., et al. Are the clinical effects of homoeopathy placebo effects? A meta-analysis of placebo-controlled trials. Lancet. 1997;350(9081):834-843.
215. Joos S., et al. Use of complementary and alternative medicine in Germany—a survey of patients with inflammatory bowel disease. BMC Complement Altern Med. 2006;6:19.
216. Schwarz S., et al. Complementary and alternative medicine for multiple sclerosis. Mult Scler. 2008;14(8):1113-1119.
217. Quattropani C., et al. Complementary alternative medicine in patients with inflammatory bowel disease: use and attitudes. Scand J Gastroenterol. 2003;38(3):277-282.
218. Fisher P., Scott D.L. A randomized controlled trial of homeopathy in rheumatoid arthritis. Rheumatology (Oxford). 2001;40(9):1052-1055.
219. Gibson R.G., et al. Homeopathic therapy in rheumatoid arthritis: evaluation by double blind clinical therapeutic trial. Br J Clin Pharmacol. 1980;9(5):453-459.
220. Braden C.J. Patterns of change over time in learned response to chronic illness among participants in a systemic lupus erythematosus self-help course. Arthritis Care Res. 1991;4(4):158-167.
221. NonSteroidal Antiinflammatory Drugs (NSAIDs). MedicineNetcom. Online. Available: http://www.medicinenet.com/nonsteroidal_antiinflammatory_drugs/article.htm.
222. Foster M., et al. COX-2 inhibitor safety in the workers’ compensation market. Lippincott’s Case Manag. 2005;10(4):217-220.
223. Das U.N. Can COX-2 inhibitor-induced increase in cardiovascular disease risk be modified by essential fatty acids? J Assoc Physicians India. 2005;53:623-627.
224. Krishnan E. Hospitalization and mortality of patients with systemic lupus erythematosus. J Rheumatol. 2006;33(9):1770-1774.
225. Chan R.W., et al. Expression of T-bet, a type 1 T-helper cell transcription factor, in the urinary sediment of lupus patients predicts disease flare. Rheumatology (Oxford). 2007;46(1):44-48.
226. Living With lupus. Online. Available: http://www.lupusnsw.org.au/lwlupus.html. Accessed 8 July 2009.
227. Szeto C.C., et al. Climatic influence on the prevalence of noncutaneous disease flare in systemic lupus erythematosus in Hong Kong. J Rheumatol. 2008;35(6):1031-1037.
228. Ramos-Casals M., et al. Acute viral infections in patients with systemic lupus erythematosus: description of 23 cases and review of the literature. Medicine (Baltimore). 2008;87(6):311-318.
229. Zandman-Goddard G., et al. Infections and SLE. Autoimmunity. 2005;38(7):473-485.
230. Lupus and Infections. Better Health channel: healthier living. Online. Available: http://www.betterhealth.vic.gov.au/bhcv2/bhcarticles.nsf/pages/Lupus_and_infections. Accessed 11 July 2009.
231. Tincani A., et al. Pregnancy and autoimmunity: maternal treatment and maternal disease influence on pregnancy outcome. Autoimmun Rev. 2005;4(7):423-428.
232. Petri M., et al. Frequency of lupus flare in pregnancy. The Hopkins Lupus Pregnancy Center experience [see comment]. Arthritis Rheum. 1991;34(12):1538-1545.
233. Molad Y. Systemic lupus erythematosus and pregnancy. Curr Opin Obstet Gynecol. 2006;18(6):613-617.
234. Khamashta M.A. Systemic lupus erythematosus and pregnancy. Best Pract Res Clin Rheumatol. 2006;20(4):685-694.
235. Lupus and Pregnancy. Better health channel: healthier living. Online. Available: http://www.betterhealth.vic.gov.au/bhcv2/bhcarticles.nsf/pages/Lupus_and_pregnancy. Accessed 11 July 2009.
236. Petri M., et al. Combined oral contraceptives in women with systemic lupus erythematosus [see comment]. N Engl J Med. 2005;353(24):2550-2558.
237. Fortin P.R., et al. Validation of a meta-analysis: the effects of fish oil in rheumatoid arthritis. J Clin Epidemiol. 1995;48(11):1379-1390.