Autoimmune Disorders
At the conclusion of this chapter, the reader should be able to:
• Describe the nature of autoimmune disorders.
• Compare organ-specific and organ-nonspecific characteristics.
• Describe organ-specific and midspectrum disorders.
• Analyze representative case studies.
• Correctly answer case study related multiple choice questions.
• Be prepared to participate in a discussion of case study related critical thinking questions.
• Describe the principle, sources of error, limitations, and application of the antinucleoprotein slide test.
The major autoimmune diseases, e.g., systemic lupus erythematosus (see Chapter 29), rheumatoid arthritis (see Chapter 30), diabetes type 1, and multiple sclerosis share many common features. Chronic and other intermittent inflammation contributes over time to the destruction of target organs that contain inciting antigens or are the sites of immune-complex deposition. Although the adaptive immune system has long been the focus of attention, innate immune mechanisms are now viewed as central to the pathogenesis of these disorders. New genetic findings emphasize the identification of environmental components that interact with host genetic factors are being important to developing a deeper understanding of autoimmunity.
What is autoimmunity?
The term autoimmune disorder is used when demonstrable immunoglobulins (autoantibodies) or cytotoxic T cells display specificity for self antigens, or autoantigens, and contribute to the pathogenesis of the disorder (Table 28-1). Autoimmune disorders are characterized by the persistent activation of immunologic effector mechanisms that alter the function and integrity of individual cells and organs. The sites of organ or tissue damage depend on the location of the immune reaction. The variety of signs and symptoms seen in patients with autoimmune disorders reflects the various forms of the immune response.
Table 28-1
Autoimmune Disorders and Associated Abnormalities
| Clinical Diagnosis | Autoantigen |
| Addison’s disease | P-450 enzymes |
| Crohn’s disease | p-ANCA, pancreatic acinar cells |
| Ovarian failure/infertility | P-450 enzymes |
| Pernicious anemia | Parietal cells |
| Ulcerative colitis | p-ANCA |
Spectrum of Autoimmune Disorders
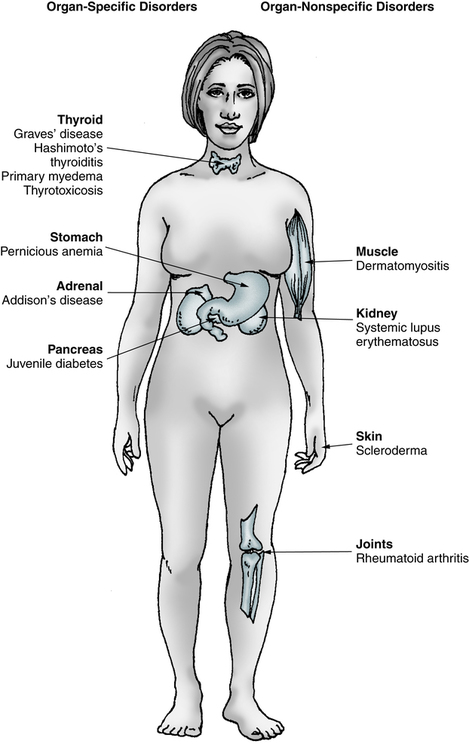
Many disorders are believed to be related to immunologic abnormalities and additional diseases are continually being identified (Box 28-1). Autoimmune disorders exhibit a full spectrum of tissue reactivity (Fig. 28-1). At one extreme are organ-specific disorders such as Hashimoto’s disease of the thyroid; at the other extreme are disorders that manifest as organ-nonspecific diseases, such as systemic lupus erythematosus (SLE; see Chapter 29) and rheumatoid arthritis (RA; see Chapter 30; Table 28-2).
Questionable abnormalities in immunoglobulin synthesis in relatives.
Immunopathogenic Mechanisms
An individual may develop an autoimmune response to a variety of immunogenic stimuli (Table 28-3). These responses may be caused by the following:
Table 28-3
Antigens Implicated in Autoimmune Endocrine Diseases
| Disorder | Antigen |
| Hashimoto’s disease | Thyroglobulin Thyroid peroxidase Thyrotropin receptor |
| Graves’ disease | Thyrotropin receptor Thyroid peroxidase Thyroglobulin 64-kDa antigen 70-kDa heat shock protein |
| Type 1 diabetes | Insulin/proinsulin Insulin receptor Glutamic acid decarboxylase B cell release granule Pancreatic cytokeratin 64-kDa antigen Glucagon 65-kDa heat shock protein |
| Addison’s disease | Adrenal cortical cells 55-kDa microsomal antigen |
| Idiopathic hypoparathyroidism | 130- and 200-kDa antigens Endothelial antigen Mitochondrial antigen |
• Antigens that do not normally circulate in the blood
The hidden antigen (sequestered antigen) theory is one of the earliest views of organ-specific antibodies. Antigens are sequestered within the organ and, because of the lack of contact with the mononuclear phagocyte system, they fail to establish immunologic tolerance. Any conditions producing a release of antigen would then provide an opportunity for autoantibody formation. This situation occurs when sperm cells or lens and heart tissues are released directly into the circulation, and autoantibodies are formed. Unmodified extracts of tissues involved in organ-specific autoimmune disorders, however, do not readily elicit antibody formation.
• Altered antigens that arise because of chemical, physical, or biological processes (e.g., hapten complexing, physical denaturation, mutation)
• A foreign antigen that is shared or cross-reactive with self antigens or tissue components
• Mutation of immunocompetent cells to acquire a response to self antigens
• Loss of the immunoregulatory function by T lymphocyte subsets
Self-Recognition (Tolerance)
Major Autoantibodies
Major autoantibodies can be detected in different disorders. Many diagnostic laboratory tests (Box 28-2) are based on detecting these autoimmune responses. Common autoantibodies include thyroid, gastric, adrenocortical, striated muscle, acetylcholine receptor, smooth muscle, salivary gland, mitochondrial, reticulin, myelin, islet cell, and skin. Antibodies to antinuclear antibodies (ANAs) include deoxyribonucleic acid (DNA), histone, and nonhistone protein antibodies.
Organ-Specific and Midspectrum Disorders
Cardiovascular Disorders
Vasculitis
Vasculitis is characterized by inflammation within blood vessels, which often results in a compromise of the vessel lumen with ischemia. Ischemia causes the major manifestations of the vasculitic syndromes and determines the prognosis. Any size and type of blood vessel may be involved. Therefore, the vasculitic syndromes are a heterogeneous group of diseases (Box 28-3).
Endocrine Gland Disorders: Thyroid Disease
Lymphoid (Hashimoto’s) Chronic Thyroiditis
Signs and Symptoms
Lymphoid thyroiditis is believed to be the most common cause of sporadic goiter. Characteristically, there is a firm, diffusely enlarged, nontender thyroid gland that may be lobulated. Hypothyroidism, however, is a common late sequela of lymphoid thyroiditis, and patients are usually euthyroid when first seen by a physician. Some individuals have clinical and pathologic evidence of the coexistence of Graves’ disease and lymphoid (Hashimoto’s) thyroiditis. Histologically, Hashimoto’s thyroiditis is characterized by diffuse lymphocytic infiltration (Fig. 28-2).
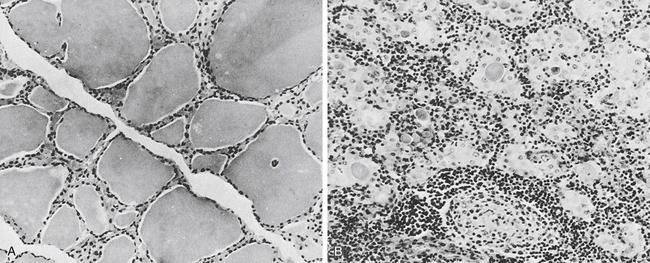
A, In the normal thyroid, colloid fills the vesicles, but in a diseased gland (B), only isolated deposits of colloid are seen. The cell infiltrate is lymphoid in nature. Note germinal center in lower middle (B). (From Anderson JR, Buchanan WW, Goudie RB: Autoimmunity, Springfield, Ill, 1967, Charles C Thomas.)
Diagnostic Evaluation
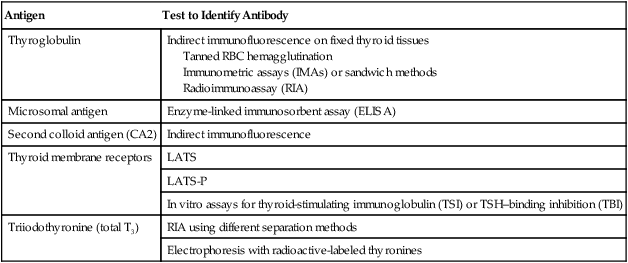
Antibodies directed against thyroid microsomal antigen (thyroid peroxidase antibody [anti-TPO]) can be detected by various techniques (Table 28-4). Chemiluminescent immunoassay is typically performed to detect anti-TPO autoantibodies. TPO plays a significant role in the biosynthesis of thyroid hormones by catalyzing the iodination of tyrosyl residues in thyroglobulin and the coupling of iodotyrosyl residues to form T4 and T3. Autoantibodies produced against TPO are capable of inhibiting enzyme activity. They are also complement-fixing antibodies that can induce cytotoxic changes in cells and consequently cause thyroid dysfunction. More than 90% of patients with autoimmune thyroiditis (Hashimoto’s thyroiditis) have anti-TPO. Antibodies to TPO have also been found in most patients with idiopathic hypothyroidism (85%) and Graves’ disease (50%).
Table 28-4
| Antigen | Test to Identify Antibody |
| Thyroglobulin | Indirect immunofluorescence on fixed thyroid tissues Tanned RBC hemagglutination Immunometric assays (IMAs) or sandwich methods Radioimmunoassay (RIA) |
| Microsomal antigen | Enzyme-linked immunosorbent assay (ELISA) |
| Second colloid antigen (CA2) | Indirect immunofluorescence |
| Thyroid membrane receptors | LATS |
| LATS-P | |
| In vitro assays for thyroid-stimulating immunoglobulin (TSI) or TSH–binding inhibition (TBI) | |
| Triiodothyronine (total T3) | RIA using different separation methods |
| Electrophoresis with radioactive-labeled thyronines |
Pancreatic Disorders
Insulin-Dependent Diabetes Mellitus
Immunologic Manifestations
T cells of the CD4+ type are responsible for initiating the immune response to the islets that results in islet cell autoantibodies and B cell destruction. Patients with T1D have the following types of autoantibodies (Box 28-4):
IA-2 is directed against a phosphatase-type transmembrane 37-kDa islet beta cell antigen (ICA512).
Autoimmune Pancreatitis
Immunologic Manifestations
• Hypergammaglobulinemia (elevated serum IgG or gamma globulin level) in patients with enhanced peripheral rim halo of the pancreas on CT
• Elevated serum IgG4 concentrations in patients with a diffusely enlarged pancreas
• Autoantibodies against carbonic anhydrase II (ACA II), lactoferrin (antilactoferrin antibody [ALA]), anti–smooth muscle antibody (ASMA), or ANA
• Increased number of CD4+ T lymphocytes in peripheral blood
Exocrine Gland Disorder
Sjögren’s Syndrome
Signs and Symptoms
The main clinical manifestations of Sjögren’s syndrome are dry eyes, dry mouth, and recurrent salivary gland pain and swelling (Table 28-5). Hoarseness, chronic cough, and increased incidence of infection have been observed. Dryness of the vagina leads to dyspareunia and itching. Dysphagia and atrophic gastritis can also be present. Extraglandular involvement results in interstitial pneumonitis and fibrosis. Renal tubular acidosis and vasculitis involving the peripheral nerves and central nervous system (CNS) can also result from Sjögren’s syndrome.
Table 28-5
Criteria for Diagnosis Sjögren’s Syndrome∗
| Ocular symptoms | Dry eyes daily for 3 mo, sand or gravel feeling in eyes |
| Oral symptoms | Dry mouth daily for 3 mo or recurrent or persistent swollen glands |
| Ocular signs | Post-Schirmer test or rose bengal score >4 |
| Histopathology | Aggregates of ≥50 mononuclear cells/4 mm2 of glandular tissue |
| Autoantibodies | Presence of anti-Ro (SS-A), anti-La (SS-B), ANAs, or rheumatoid factor |
∗Four or more of these criteria must be present.
From Vitali C, Bombardieri S, Moutsopoulos HM, et al: Preliminary criteria for the classification of Sjögren’s syndrome: results of a prospective concerted action supported by the European community, Arthritis Rheum 36:340–347, 1993.
Gastrointestinal Disorders
Atrophic Gastritis and Pernicious Anemia
Vitamin B12 (Cobalamin) Transport
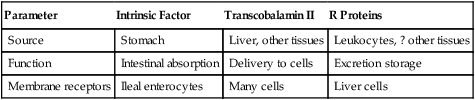
Cobalamin transport is mediated by three different binding proteins capable of binding the vitamin at its required physiologic concentrations—intrinsic factor, transcobalamin II, and the R proteins (Table 28-6).
Table 28-6
Vitamin B12 (Cobalamin)–Binding Proteins
| Parameter | Intrinsic Factor | Transcobalamin II | R Proteins |
| Source | Stomach | Liver, other tissues | Leukocytes, ? other tissues |
| Function | Intestinal absorption | Delivery to cells | Excretion storage |
| Membrane receptors | Ileal enterocytes | Many cells | Liver cells |
Autoimmune Liver Disease
Antinuclear autoantibody using HEp-2 cells will have differing levels of reactivity depending on factors such as the disease activity or multiple ANA specificities. A homogeneous staining pattern (see Color Plate 13) is the most frequent pattern particularly in active AIH. The frequency of positive ANA tests is about 70% in AIH. In remission, the frequency of ANA positivity decreases and the ANA pattern is replaced by a speckled pattern in almost 40% of cases. Other significant antibodies can include an atypical perinuclear ANCA (pANCA) in one type of AIH with a frequency of 65% positivity. In addition, AIH is characterized by autoantibodies to cytoskeletal proteins that support cellular structure, contractility, and locomotion: microfilaments. These autoantibodies to cytoskeleton can be studied by immunofluorescent light (IFL) methodology.
Inflammatory Bowel Disease
• Presence of increased levels of inflammation-promoting cytokines
• Protein molecules used by cells of the immune system to communicate with each other
Immune Markers
• Deoxyribonuclease (DNase I)–sensitive perinuclear antineutrophil cytoplasmic autoantibody (p-ANCA). IBD-associated p-ANCA defines an antibody to a nuclear antigen that is sensitive to DNase I.
• Anti–Saccharomyces cervisiae antibody (ASCA). This is present in the sera of up to 70% of CD patients.
• Pancreatic antibody. This is observed in approximately 30% of CD patients.
• Anti–outer membrane porin from Escherichia coli (anti-OmpC). An IgA response to OmpC is observed in 55% of CD patients.
Celiac Disease
Celiac disease is a lifelong autoimmune intestinal disorder found in individuals who are genetically susceptible. There are also associated clinical disorders of an immune basis (Box 28-5). Damage to the mucosal surface of the small intestine is caused by an immunologically toxic reaction to the ingestion of gluten and interferes with the absorption of nutrients. Celiac disease is unique in that a specific food component, gluten, has been identified as the trigger. Gluten is the common name for the offending proteins in specific cereal grains that are harmful to those with celiac disease. These proteins are found in all forms of wheat (e.g., durum, semolina, spelt, kamut, einkorn, faro) and related grains (rye, barley, triticale) and must be eliminated.
Other Gastrointestinal Tract Immunologic Disorders
Examples of other immunologic disorders related to the GI and hepatobiliary tracts include GI allergy, Whipple’s disease, immunoproliferative intestinal disease (alpha heavy-chain disease), and infectious hepatitis (see Chapter 23). Allergy of the GI tract is an IgE-mediated hypersensitivity to food substances that involves the GI tract and, in some cases, the skin and lungs. Examples of systemic autoimmune disease caused by mucosal immune abnormalities are IgA nephropathy (Berger’s disease), Henoch-Schönlein purpura, and diseases associated with circulating IgA complexes in the kidney and vasculature. Immunoproliferative intestinal disease is characterized by monoclonal B cells that produce an aberrant alpha heavy chain.
Autoimmune Hematologic Disorders
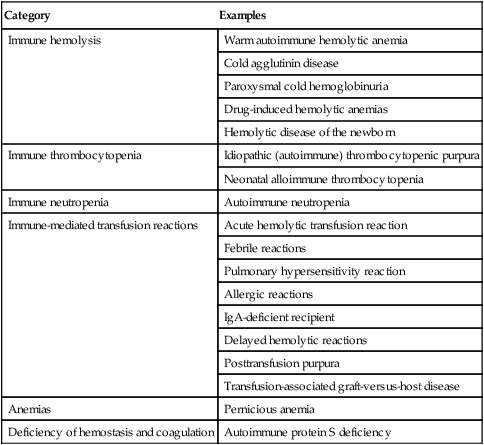
Various hematologic conditions can be caused by alloantibodies and autoantibodies (Table 28-7).
Table 28-7
| Category | Examples |
| Immune hemolysis | Warm autoimmune hemolytic anemia |
| Cold agglutinin disease | |
| Paroxysmal cold hemoglobinuria | |
| Drug-induced hemolytic anemias | |
| Hemolytic disease of the newborn | |
| Immune thrombocytopenia | Idiopathic (autoimmune) thrombocytopenic purpura |
| Neonatal alloimmune thrombocytopenia | |
| Immune neutropenia | Autoimmune neutropenia |
| Immune-mediated transfusion reactions | Acute hemolytic transfusion reaction |
| Febrile reactions | |
| Pulmonary hypersensitivity reaction | |
| Allergic reactions | |
| IgA-deficient recipient | |
| Delayed hemolytic reactions | |
| Posttransfusion purpura | |
| Transfusion-associated graft-versus-host disease | |
| Anemias | Pernicious anemia |
| Deficiency of hemostasis and coagulation | Autoimmune protein S deficiency |
Autoimmune Hemolytic Anemia
Autoimmune hemolytic anemia can be classified into the following four groups:
• Warm-reactive autoantibodies (most common)
• Cold-reactive autoantibodies (<20% of cases)
Drug-Induced Hemolysis
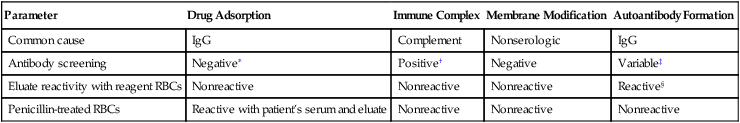
Coating of RBCs demonstrated by a positive direct anti–human globulin test (DAT) result may be drug induced and accompanied by hemolysis (Table 28-8). The reactivity has been described as being caused by four basic mechanisms: (1) drug adsorption; (2) immune complexing; (3) membrane modification; and (4) autoantibody formation.
Table 28-8
Drug-Induced Positive Direct Antiglobulin Test
| Parameter | Drug Adsorption | Immune Complex | Membrane Modification | Autoantibody Formation |
| Common cause | IgG | Complement | Nonserologic | IgG |
| Antibody screening | Negative∗ | Positive† | Negative | Variable‡ |
| Eluate reactivity with reagent RBCs | Nonreactive | Nonreactive | Nonreactive | Reactive§ |
| Penicillin-treated RBCs | Reactive with patient’s serum and eluate | Nonreactive | Nonreactive | Nonreactive |
∗Unless irregular antibodies are present in the sample.
†If the drug and complement are present in the test system.
‡If the autoantibody is high enough in titer, screening tests may be positive with all cells tested.
§Will react with all normal cells tested, occasionally showing Rh-like specificity.
Pernicious Anemia
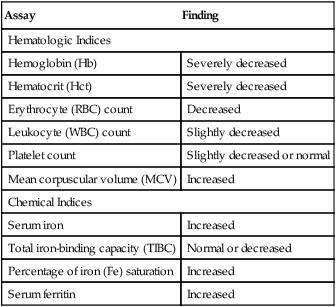
Pernicious anemia is a megaloblastic anemia characterized by a variety of hematologic and chemical manifestations (Table 28-9). PA is caused by a deficiency of vitamin B12 that results from the patient’s inability to secrete intrinsic factor. In autoimmune cases of PA, anti-IF or antiparietal antibodies have been reported. Demonstration of these antibodies supports the theory that PA is an autoimmune disorder. Nutritional disorders (e.g., vegan diet, gastric bypass surgery, AIDS, small bowel disorders, and competition for vitamin B12) can be nonimmunological causes of PA.
Table 28-9
Hematologic and Chemical Findings in Pernicious Anemia
| Assay | Finding |
| Hematologic Indices | |
| Hemoglobin (Hb) | Severely decreased |
| Hematocrit (Hct) | Severely decreased |
| Erythrocyte (RBC) count | Decreased |
| Leukocyte (WBC) count | Slightly decreased |
| Platelet count | Slightly decreased or normal |
| Mean corpuscular volume (MCV) | Increased |
| Chemical Indices | |
| Serum iron | Increased |
| Total iron-binding capacity (TIBC) | Normal or decreased |
| Percentage of iron (Fe) saturation | Increased |
| Serum ferritin | Increased |
Neuromuscular Disorders
Myasthenia Gravis
Myasthenia gravis is a disorder of the neuromuscular junction characterized by neurophysiologic and immunologic abnormalities (Box 28-6). A postsynaptic defect is caused by a decrease in receptors for acetylcholine and frequently an anatomic defect in the neuromuscular junction plate. Acetylcholine receptor (AChR)–binding antibody is directed against acetylcholine receptors at neuromuscular junctions of skeletal muscle and AChR-blocking antibodies. The ligand bungarotoxin or acetylcholine is important in producing a neuromuscular block. About one third of patients with myasthenia gravis demonstrate AChR-blocking antibodies.
Multiple Sclerosis
Immunologic Manifestations
Box 28-7 presents immunologic manifestations of MS suggestive of its autoimmune nature. Antimyelin antibodies directed against components of the myelin sheath of nerves or myelin basic protein can be demonstrated in patients with MS or other neurologic diseases. However, myelin antibodies are not detectable in the CSF of MS patients.
Neuropathies
In the autoimmune neuropathies, antibodies directed against peripheral nerve components are associated with specific clinical syndromes (Table 28-10). Knowledge of these syndromes and antibody tests can be used to identify a treatable neuropathy. In addition, many autoimmune neuropathic syndromes are associated with malignancies, which they often precede. Recognition of these syndromes can lead to early identification and treatment.
Table 28-10
Neuropathy Syndromes Associated With Antibodies Directed Against Peripheral Nerve Components
| Clinical Syndrome | Antibodies |
| Chronic sensorimotor demyelinating neuropathy | Antimyelin-associated glycoprotein |
| Chronic axonal sensory neuropathy | Antisulfatide or anti–chondroitin sulfate |
| Multifocal motor neuropathy | Anti-GM1 (IgM) |
| Acute axonal motor neuropathy | Anti-GM1 (IgG) |
| Fisher syndrome | Anti-GQ1b |
| Guillain-Barré syndrome | Anti-LM1, GD1b, GD1A, GT1b, sulfatide, B tubulin |
| Large-fiber sensory neuropathy with ataxia | Anti-GQ1b, GD3, GD1b, GT1b |
| Subacute sensory neuropathy/encephalomyelitis | Antineuronal nuclear antibody type 1 (anti-Hu) |
From Cohen B, Mitsumoto H: Neuropathy syndromes associated with antibodies against the peripheral nerve, Lab Med 26:459–463, 1995.
Systemic sclerosis (scleroderma) is an autoimmune disease characterized by a wide spectrum of clinical, pathologic, and serologic abnormalities. More than 90% of patients with systemic sclerosis spontaneously produce ANA. The structure and function of the intracellular antigens to which these ANAs are directed have been characterized. These serum autoantibodies are helpful markers because they correlate with certain clinical features of systemic sclerosis (Table 28-11). A more recently developed marker autoantibody, anti–RNA polymerase III antibody, has been identified in many patients who have systemic sclerosis with diffuse or extensive cutaneous involvement.
Table 28-11
Clinical Types of Systemic Sclerosis (SSc) and Associated Antibody Markers
| Clinical Type | Antibodies |
| SSc with diffuse cutaneous involvement (dcSsc) | Anti–RNA polymerase I, anti–topoisomerase I |
| SSc with limited cutaneous involvement (lcSSc) | Anti–Th ribonucleoprotein anticentromere antibody |
| SSc-polymyositis overlap syndrome | Anti–PM-Scl |
Renal Disorders
It is generally accepted that most immunologically mediated renal diseases fall into several categories (Box 28-9).
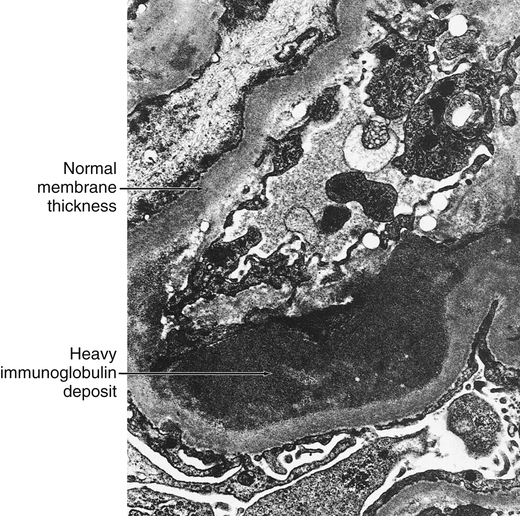
Renal Disease Associated With Anti–Glomerular Basement Membrane Antibody
Anti–glomerular basement membrane (GBM) antibodies are directed against GBM of the glomerulus of the kidney (Fig. 28-3). These antibodies are induced in vivo against the basement membrane of the glomerulus and possibly that of the renal tubule or lung. The factors that stimulate antibody production are not well defined, but it appears likely that binding of drugs (e.g., methicillin), certain infectious agents, or renal damage caused by other immune mechanisms may lead to an immune antibody response. The end result may be direct damage to the bone marrow, with or without complement activation. Production of anti–bone marrow antibodies, however, appears to be self-limited and lasts for several weeks to months after removal of the inciting agent (i.e., by the kidney).
Skeletal Muscle Disorders
Inflammatory Myopathy
• Primary idiopathic polymyositis
• Primary idiopathic dermatomyositis
• Polymyositis or dermatomyositis associated with neoplasia
• Childhood polymyositis or dermatomyositis
• Dermatomyositis or polymyositis associated with collagen vascular disease
• Polymyositis or dermatomyositis associated with infections
Patients with myositis have many immunologic abnormalities. One unique immunologic feature is the targeting by autoantibodies of certain cytoplasmic proteins and ribonucleic acids (RNAs) involved in the process of protein synthesis. These autoantibodies are found only in patients with myositis and are known as myositis-specific autoantibodies (MSAs; Box 28-10). The MSAs are antigen-driven, arise months before the onset of myositis, correlate in titer with disease activity, disappear after prolonged complete remission, and bind to and inhibit the function of targeted human autoantigenic enzymes on in vitro assays.
Skin Disorders: Bullous Disease and Other Conditions
A wide variety of autoimmune disorders are associated with skin manifestations (Box 28-11).
CASE STUDY 1
History and Physical Examination
Physical examination revealed slight hepatomegaly. Her physician ordered a complete blood count and urinalysis. See Table 28-12 for the results of these tests.
Table 28-12
| Patient’s Results | Reference Range | |
| Complete Blood Count | ||
| Hemoglobin (Hb) | 6.2 g/dL | 11.5-16.0 g/dL |
| Hematocrit (Hct) | 0.22 L/L | 0.37-0.47 L/L |
| RBC count | 1.7 × 1012/L | 4.2-5.4 × 1012/L |
| WBC count | 3.8 × 109/L | 4.5-11.0 × 109/L |
| Red Blood Cell Indices | ||
| Mean corpuscular volume (MCV) | 129.4 fL | 80-96 fL |
| Mean corpuscular hemoglobin (MCH) | 36.5 pg | 27-32 pg |
| Mean corpuscular hemoglobin concentration (MCHC) | 28% | 32%-36% |
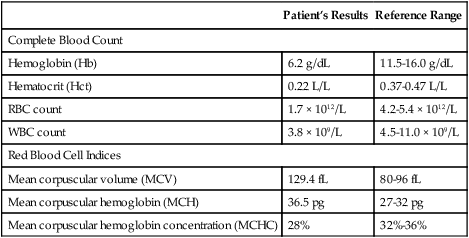
∗Blood smear comments: 3 macrocytic RBCs, polychromatophilia, a few nucleated RBCs.
See Appendix A for the answers to multiple choice questions.
Critical Thinking Group Discussion Questions
1. Which chemical and immunologic assays would be helpful in establishing a diagnosis for this patient?
2. What is the prevalence of anti–intrinsic factor in patients with pernicious anemia?
3. What is the prevalence of parietal cell antibodies in patients with pernicious anemia?
See instructor site  website for the discussion of the answers to these questions.
website for the discussion of the answers to these questions.
CASE STUDY 2
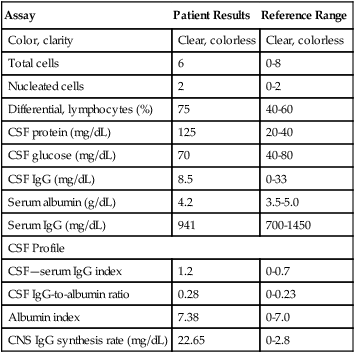
Laboratory and Medical Imaging Data: Laboratory Findings
See Table 28-13 for these findings.
Table 28-13
Cerebrospinal Fluid Examination
| Assay | Patient Results | Reference Range |
| Color, clarity | Clear, colorless | Clear, colorless |
| Total cells | 6 | 0-8 |
| Nucleated cells | 2 | 0-2 |
| Differential, lymphocytes (%) | 75 | 40-60 |
| CSF protein (mg/dL) | 125 | 20-40 |
| CSF glucose (mg/dL) | 70 | 40-80 |
| CSF IgG (mg/dL) | 8.5 | 0-33 |
| Serum albumin (g/dL) | 4.2 | 3.5-5.0 |
| Serum IgG (mg/dL) | 941 | 700-1450 |
| CSF Profile | ||
| CSF—serum IgG index | 1.2 | 0-0.7 |
| CSF IgG-to-albumin ratio | 0.28 | 0-0.23 |
| Albumin index | 7.38 | 0-7.0 |
| CNS IgG synthesis rate (mg/dL) | 22.65 | 0-2.8 |
• CSF agarose electrophoresis: Positive for oligoclonal bands (reference range negative)
• CSF isoelectric focusing: Positive for oligoclonal bands (reference range negative)
• Serum protein electrophoresis interpretation: No apparent monoclonal peak (reference range, no apparent monoclonal peak)
• Serum immunofixation: No paraprotein detected (reference range, no paraprotein detected)
See Appendix A for the answers to multiple choice questions.
Critical Thinking Group Discussion Questions
1. What is the cause of the patient’s symptoms?
2. Does the patient’s age provide a clue to the diagnosis?
3. What is the significance of the laboratory analysis of the CSF?
See instructor site  website for the discussion of the answers to these questions.
website for the discussion of the answers to these questions.
 Rapid Slide Test for Antinucleoprotein
Rapid Slide Test for Antinucleoprotein website for the procedural protocol.
website for the procedural protocol.Chapter Highlights
• Autoimmunity represents a breakdown of the immune system in its ability to discriminate between self and nonself.
• The term autoimmune disorder is used when demonstrable immunoglobulins, autoantibodies, or cytotoxic T cells display a specificity for self antigens and contribute to disease pathogenesis.
• At one extreme are organ-specific disorders; at the other end of the spectrum are disorders that manifest as organ-nonspecific diseases. Midspectrum disorders are characterized by localized lesions in a single organ and organ-nonspecific autoantibodies.
• The potential for autoimmunity is always present in every immunocompetent individual because lymphocytes that are potentially reactive with self antigens exist in the body.
• Antibody expression appears to be regulated by complex interactions that include genetic factors, patient age, and exogenous factors.
• Self-recognition (tolerance) is induced by at least two mechanisms, elimination of a small clone of immunocompetent cells programmed to react with antigen (Burnet’s clonal selection theory) or induction of unresponsiveness in immunocompetent cells through excessive antigen binding to them and through triggering of a suppressor mechanism.
• Major autoantibodies can be detected in different disorders. Many diagnostic laboratory tests are based on detecting these autoimmune responses.