[level-membership-for-internal-medicine-category]
FIGURE 76e-21 Non-Hodgkin’s lymphoma involving the skin, with typical violaceous, “plum-colored” nodules. (Courtesy of Jean Bolognia, MD; with permission.)
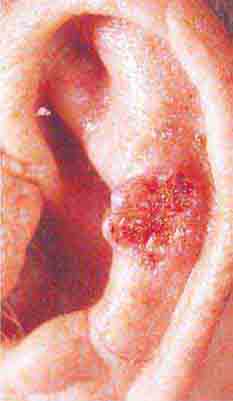
FIGURE 76e-22 Basal cell carcinoma, with central ulceration and a pearly, rolled, telangiectatic tumor border.
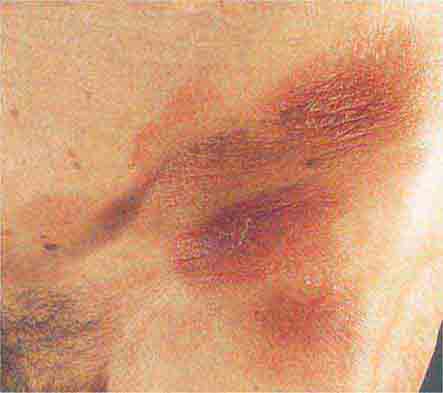
FIGURE 76e-23 Mycosis fungoides is a cutaneous T cell lymphoma. Plaque-stage lesions are seen in this patient.
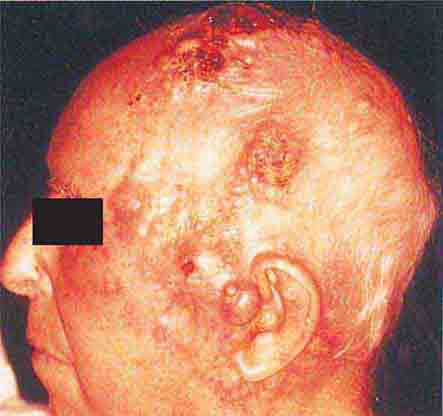
FIGURE 76e-24 Metastatic carcinoma to the skin is characterized by inflammatory, often ulcerated dermal nodules.
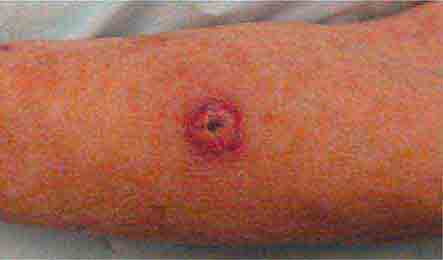
FIGURE 76e-25 Keratoacanthoma is a low-grade squamous cell carcinoma that presents as an exophytic nodule with central keratinous debris.
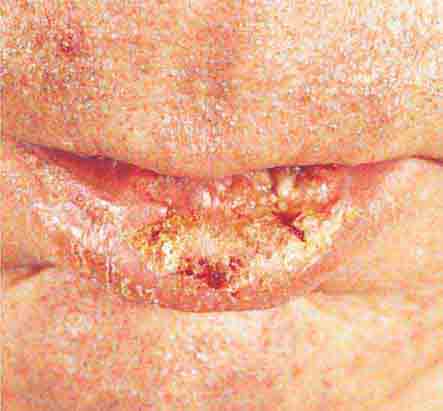
FIGURE 76e-26 Squamous cell carcinoma is seen here as a hyperkeratotic, crusted, and somewhat eroded plaque on the lower lip. Sun-exposed skin of the head, neck, hands, and arms are other typical sites of involvement.
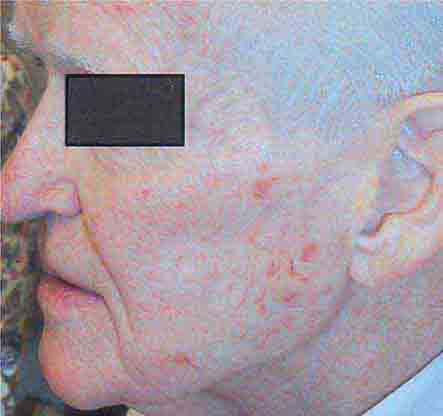
FIGURE 76e-27 Actinic keratoses consist of hyperkeratotic erythematous papules and patches on sun-exposed skin. They arise in middle-aged to older adults and have some potential for malignant transformation. (Courtesy of Robert Swerlick, MD; with permission.)
MELANOMA AND BENIGN PIGMENTED LESIONS
(Figs. 76e-28 to 76e-33) As the prognosis of melanoma is related primarily to the microscopic depth of invasion, and as early detection with surgical treatment can be curative in a high percentage of patients, it is essential that all clinicians acquire some facility in evaluating pigmented lesions. Three clinicopathologic subtypes of melanoma—superficial spreading, lentigo maligna, and acral lentiginous melanoma—typically display features noted in the “ABCD rule”: asymmetry (one half of the lesion varies from the other half); border irregularity (the circumferential border exhibits an irregular, sometimes jagged appearance); color (there is uneven coloration and tone to the pigmented lesion, with various shades of brown, black, red, and white in different areas); and diameter (the diameter is typically >6 mm). The more uncommon subtype, nodular melanoma, may not manifest all these features but rather may present as a more symmetric, evenly pigmented, or amelanotic lesion. Dysplastic (atypical) melanocytic nevi may occur as solitary or multiple lesions as well as in the setting of familial melanoma. These nevi display some degree of asymmetry, border irregularity, and color variation. Ordinary nevi may be acquired or congenital and are quite common.
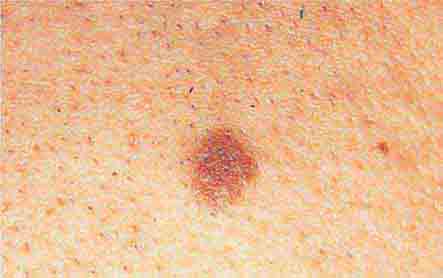
FIGURE 76e-28 Nevi are benign proliferations of nevomelanocytes characterized by regularly shaped hyperpigmented macules or papules of a uniform color.
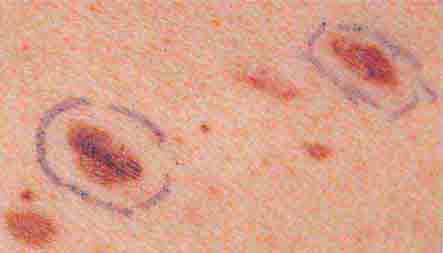
FIGURE 76e-29 Dysplastic nevi are irregularly pigmented and shaped nevomelanocytic lesions that may be associated with familial melanoma.
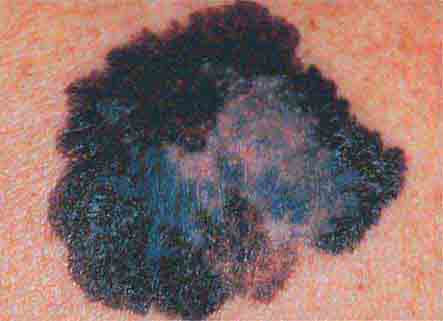
FIGURE 76e-30 Superficial spreading melanoma, the most common type of malignant melanoma, is characterized by color variegation (black, blue, brown, pink, and white) and irregular borders.
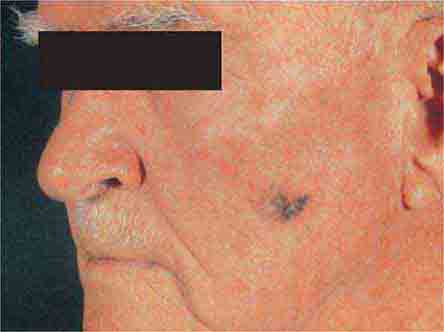
FIGURE 76e-31 Lentigo maligna melanoma occurs on sun-exposed skin as a large, hyperpigmented macule or plaque with irregular borders and variable pigmentation. (Courtesy of Alvin Solomon, MD; with permission.)

FIGURE 76e-32 Nodular melanoma most commonly manifests as a rapidly growing, often ulcerated or crusted black nodule. (Courtesy of S. Wright Caughman, MD; with permission.)
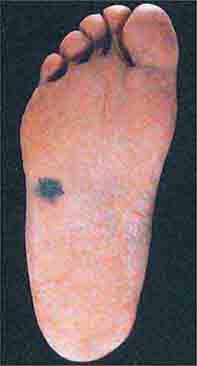
FIGURE 76e-33 Acral lentiginous melanoma is more common among blacks, Asians, and Hispanics and occurs as an enlarging hyperpigmented macule or plaque on the palms or soles. Lateral pigment diffusion is present.
INFECTIOUS DISEASE AND THE SKIN
(Figs. 76e-34 to 76e-58) One of the roles of the skin is to function as a barrier from the outside world. In this capacity, exposure to infectious agents occurs, and bacterial, viral, fungal, and parasitic infections may result. In addition, the skin may be secondarily involved and provides diagnostic clues to systemic infections such as meningococcemia, Rocky Mountain spotted fever, Lyme disease, and septic emboli. Most sexually transmitted bacterial and viral diseases exhibit cutaneous involvement; examples include primary and secondary syphilis, chancroid, genital herpes simplex, and condyloma acuminatum.
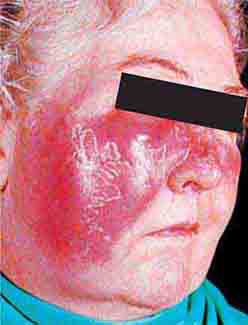
FIGURE 76e-34 Erysipelas is a streptococcal infection of the superficial dermis and consists of well-demarcated, erythematous, edematous, warm plaques.
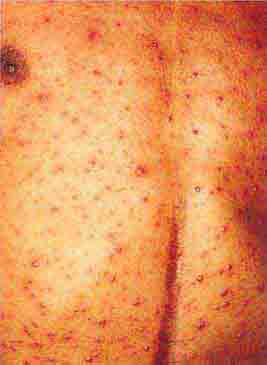
FIGURE 76e-35 Varicella, with numerous lesions in various stages of evolution: vesicles on an erythematous base, umbilicated vesicles, and crusts. (Courtesy of Robert Hartman, MD; with permission.)
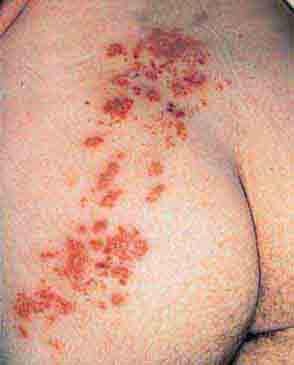
FIGURE 76e-36 Herpes zoster is seen in this HIV-infected patient as hemorrhagic vesicles and pustules on an erythematous base in a dermatomal distribution. (Courtesy of Robert Swerlick, MD; with permission.)
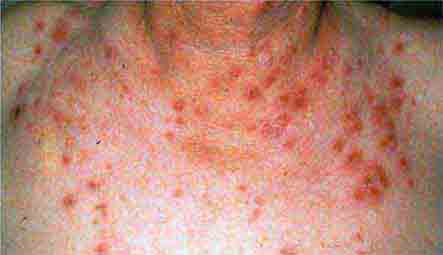
FIGURE 76e-37 Impetigo contagiosa is a superficial streptococcal or Staphylococcus aureus infection consisting of honey-colored crusts and erythematous weeping erosions. Bullous lesions are occasionally seen.
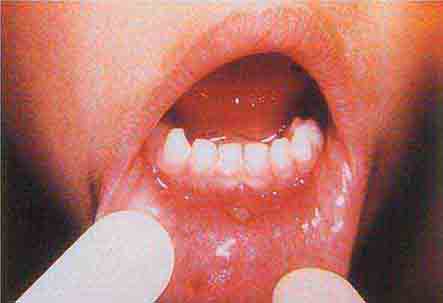
FIGURE 76e-38 Tender vesicles and erosions in the mouth of a patient with hand-foot-and-mouth disease. (Courtesy of Stephen D. Gellis, MD; with permission.)
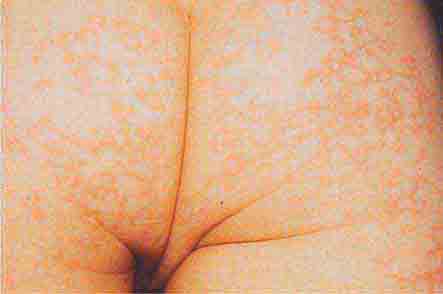
FIGURE 76e-39 Lacy reticular rash of erythema infectiosum (fifth disease).
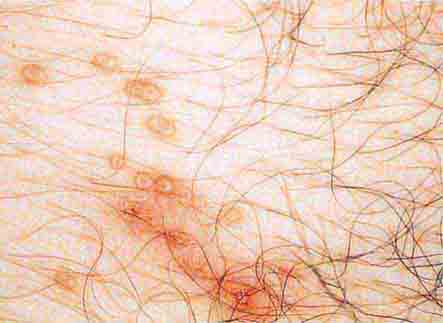
FIGURE 76e-40 Molluscum contagiosum is a cutaneous poxvirus infection characterized by multiple umbilicated flesh-colored or hypopigmented papules. (Courtesy of Yale Resident’s Slide Collection; with permission.)
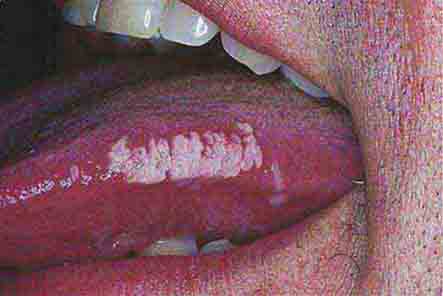
FIGURE 76e-41 Oral hairy leukoplakia often presents as white plaques on the lateral tongue and is associated with Epstein-Barr virus infection. (From K Wolff et al: Fitzpatrick’s Color Atlas & Synopsis of Clinical Dermatology, 5th ed. New York, McGraw-Hill, 2005. www.accessmedicine.com.)
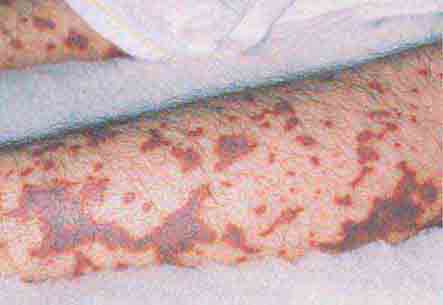
FIGURE 76e-42 Fulminant meningococcemia, with extensive angular purpuric patches. (Courtesy of Stephen D. Gellis, MD; with permission.)
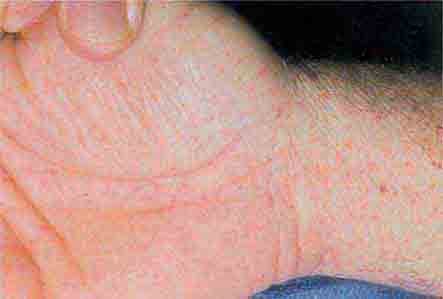
FIGURE 76e-43 Rocky Mountain spotted fever, with pinpoint petechial lesions on the palm and volar aspect of the wrist. (Courtesy of Robert Swerlick, MD; with permission.)
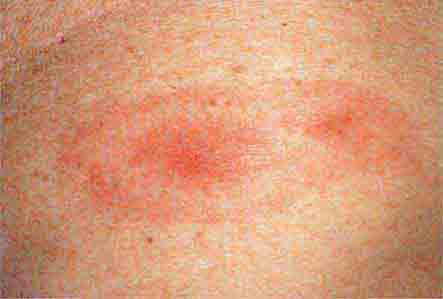
FIGURE 76e-44 Erythema migrans, the early cutaneous manifestation of Lyme disease, is characterized by erythematous annular patches, often with a central erythematous papule at the tick-bite site. (Courtesy of Yale Resident’s Slide Collection; with permission.)
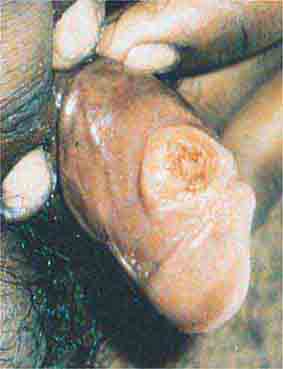
FIGURE 76e-45 Primary syphilis, with a firm, nontender chancre. (Courtesy of Gregory Cox, MD; with permission.)
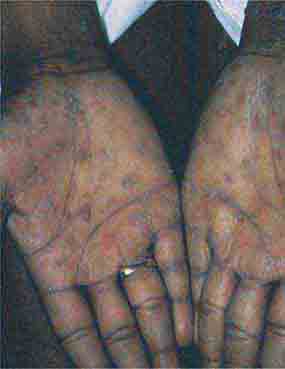
FIGURE 76e-46 Secondary syphilis commonly affects the palms and soles, with scaling, firm, red-brown papules. (Courtesy of Alvin Solomon, MD; with permission.)
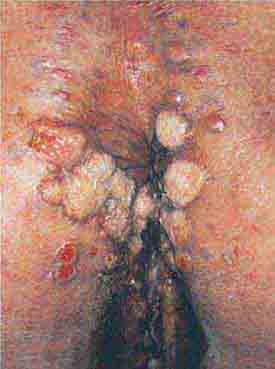
FIGURE 76e-47 Condylomata lata are moist, somewhat verrucous inter-triginous plaques seen in secondary syphilis. (Courtesy of Yale Resident’s Slide Collection; with permission.)
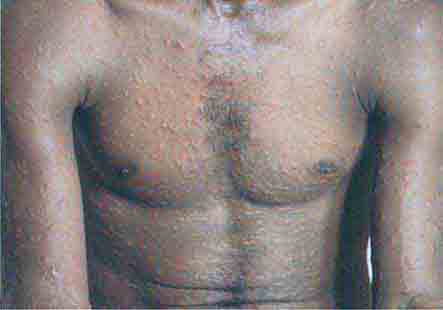
FIGURE 76e-48 Secondary syphilis, with the characteristic papulosquamous truncal eruption.
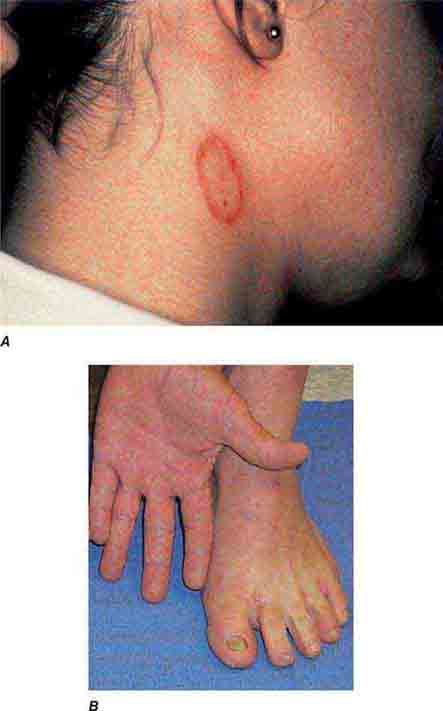
FIGURE 76e-49 A. Tinea corporis is a superficial fungal infection, seen here as an erythematous annular scaly plaque with central clearing. B. A common presentation of chronic dermatophyte infection involves the feet (tinea pedis), hands (tinea manum), and nails (tinea unguium).
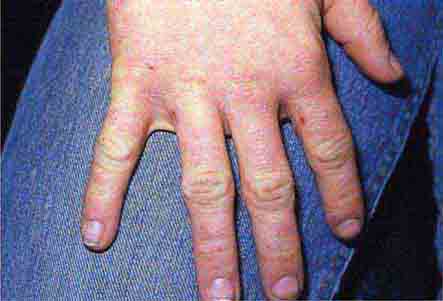
FIGURE 76e-50 Scabies, with typical scaling erythematous papules and few linear burrows.
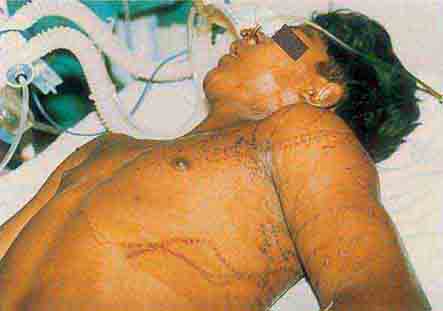
FIGURE 76e-51 Skin lesions caused by Chironex fleckeri sting. (Courtesy of V. Pranava Murthy, MD; with permission.)
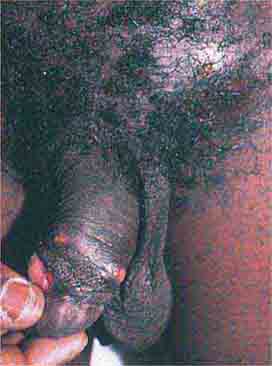
FIGURE 76e-52 Chancroid, with characteristic penile ulcers and associated left inguinal adenitis (bubo).
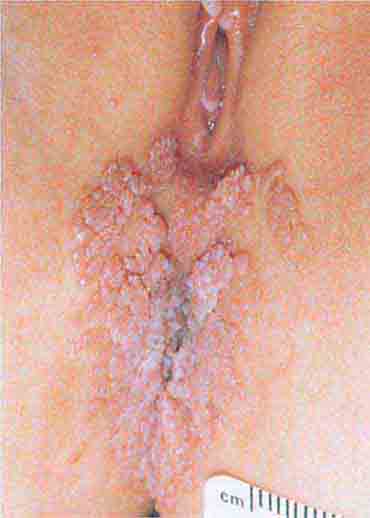
FIGURE 76e-53 Condylomata acuminata are lesions induced by human papillomavirus and in this patient are seen as multiple verrucous papules coalescing into plaques. (Courtesy of S. Wright Caughman, MD; with permission.)
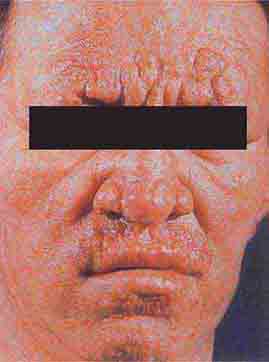
FIGURE 76e-54 A patient with features of polar lepromatous leprosy: multiple nodular skin lesions, particularly of the forehead, and loss of eyebrows. (Courtesy of Robert Gelber, MD; with permission.)
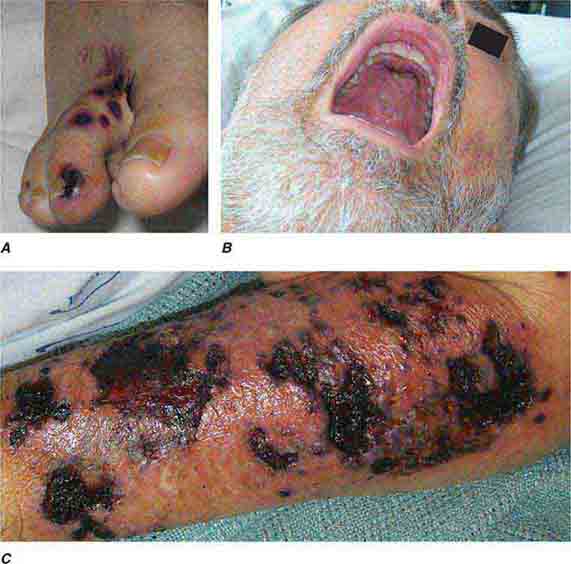
FIGURE 76e-55 Skin lesions of neutropenic patients. A. Hemorrhagic papules on the foot of a patient undergoing treatment for multiple myeloma. Biopsy and culture demonstrated Aspergillosis species. B. Eroded nodule on the hard palate of a patient undergoing chemotherapy. Biopsy and culture demonstrated Mucor species. C. Ecthyma gangrenosum in a neutropenic patient with Pseudomonas aeruginosa bacteremia.
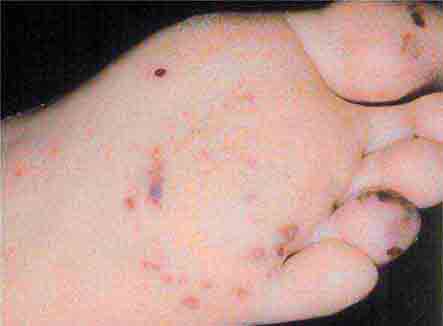
FIGURE 76e-56 Septic emboli, with hemorrhage and infarction due to acute Staphylococcus aureus endocarditis. (Courtesy of L. Baden, MD; with permission.)
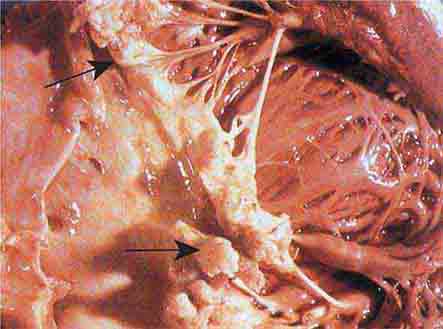
FIGURE 76e-57 Vegetations (arrows) due to viridans streptococcal endocarditis involving the mitral valve. (Courtesy of AW Karchmer, MD; with permission.)
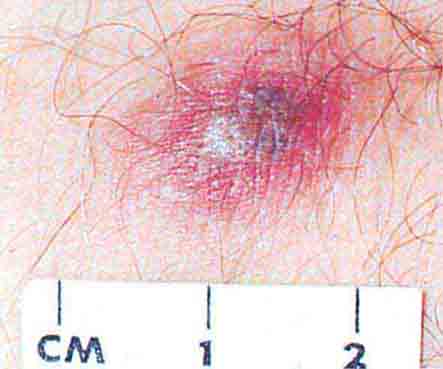
FIGURE 76e-58 Disseminated gonococcemia in the skin is seen as hemorrhagic papules and pustules with purpuric centers in an acral distribution. (Courtesy of Daniel M. Musher, MD; with permission.)
IMMUNOLOGICALLY MEDIATED SKIN DISEASE
(Figs. 76e-59 to 76e-70) Immunologically mediated skin disease may be largely localized to skin and mucous membranes and manifest with blisters and erosions such as pemphigus, pemphigoid, and dermatitis herpetiformis. In diseases such as systemic lupus erythematosus, dermatomyositis, and vasculitis, skin manifestations are often only one element of a widespread process.
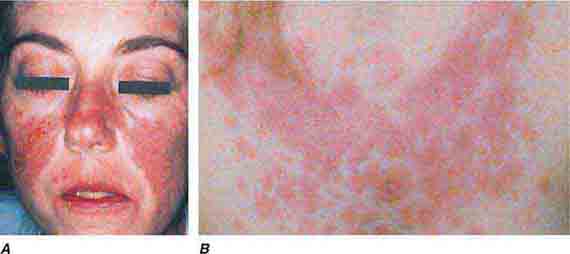
FIGURE 76e-59 Lupus erythematosus. A. Systemic lupus erythematosus, with prominent, scaly malar erythema. Involvement of other sun-exposed sites is also common. B. Acute lupus erythematosus on the upper chest, with brightly erythematous and slightly edematous coalescence of papules and plaques. (B: Courtesy of Robert Swerlick, MD; with permission.)
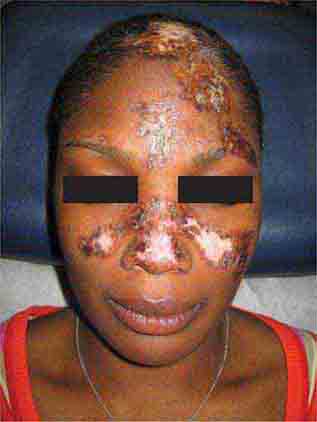
FIGURE 76e-60 Discoid lupus erythematosus. Atrophic, depigmented plaques and patches surrounded by hyperpigmentation and erythema in association with scarring and alopecia are characteristic of this cutaneous form of lupus.
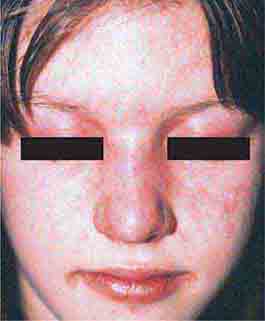
FIGURE 76e-61 Dermatomyositis. Periorbital violaceous erythema characterizes the classic heliotrope rash. (Courtesy of James Krell, MD; with permission.)
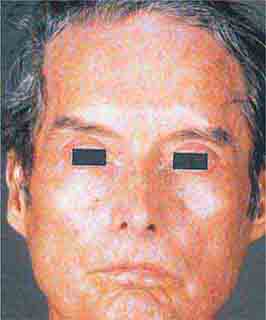
FIGURE 76e-62 Scleroderma characterized by typical expressionless, mask-like facies.
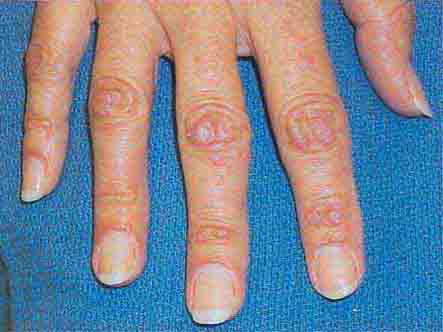
FIGURE 76e-63 Scleroderma, with acral sclerosis and focal digital ulcers.
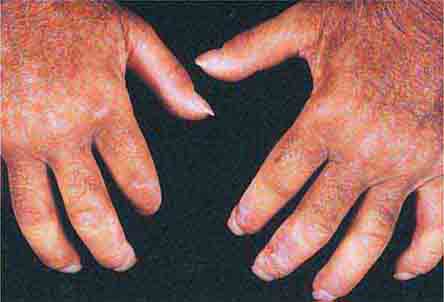
FIGURE 76e-64 Dermatomyositis often involves the hands as erythematous flat-topped papules over the knuckles (Gottron’s sign) and periungual telangiectasias.
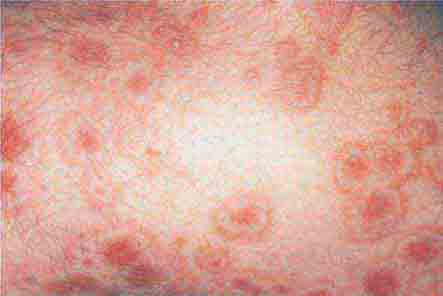
FIGURE 76e-65 Erythema multiforme is characterized by multiple erythematous plaques with a target or iris morphology and usually represents a hypersensitivity reaction to drugs or infections (especially herpes simplex virus). (Courtesy of Yale Resident’s Slide Collection; with permission.)
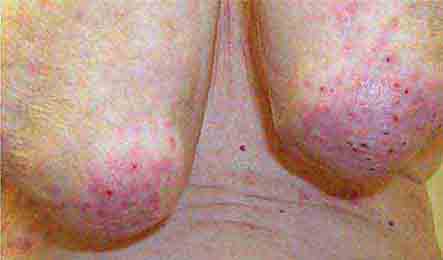
FIGURE 76e-66 Dermatitis herpetiformis, manifested by pruritic, grouped vesicles in a typical location. The vesicles are often excoriated and may also occur on the knees, buttocks, elbows, and posterior scalp.
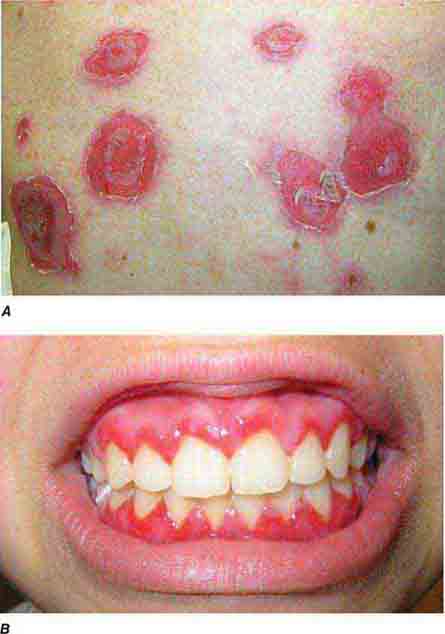
FIGURE 76e-67 Pemphigus vulgaris. A. Eroded bullae on the back. B. The oral mucosa is almost invariably involved, sometimes with erosions on the gingiva, buccal mucosa, palate, posterior pharynx, or tongue. (B: Courtesy of Robert Swerlick, MD; with permission.)
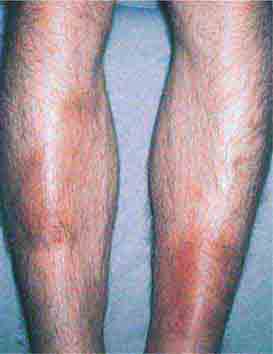
FIGURE 76e-68 Erythema nodosum is a panniculitis characterized by tender deep-seated nodules and plaques, usually located on the lower extremities. (Courtesy of Robert Swerlick, MD; with permission.)
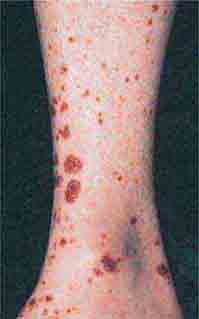
FIGURE 76e-69 Vasculitis. Palpable purpuric papules on the lower legs are seen in this patient with cutaneous small-vessel vasculitis. (Courtesy of Robert Swerlick, MD; with permission.)
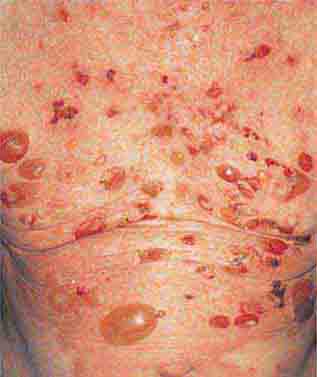
FIGURE 76e-70 Bullous pemphigoid, with tense vesicles and bullae on an erythematous, urticarial base. (Courtesy of Yale Resident’s Slide Collection; with permission.)
SKIN MANIFESTATIONS OF INTERNAL DISEASE
(Figs. 76e-71 to 76e-78) While many systemic diseases also have cutaneous manifestations, there are well-recognized dermatologic markers of internal disease, some of which are shown in this section. Many of these dermatologic markers may precede, accompany, or follow diagnosis of systemic disease. Acanthosis nigricans is a prototypical dermatologic process that often occurs in association with underlying systemic abnormalities, most commonly obesity and insulin resistance. It may also be associated with other endocrine disorders and several rare genetic syndromes. Malignant acanthosis nigricans may occur in association with several malignancies, especially adenocarcinoma of the gastrointestinal tract, lung, and breast. Other markers of internal disease in this section include pretibial myxedema, which is associated with thyroid disease, and Sweet syndrome, which may be associated with hematologic malignancies, solid tumors, infections, or inflammatory bowel disease. The skin is also involved in many systemic inflammatory diseases such as sarcoidosis, rheumatoid arthritis, and lupus erythematosus.
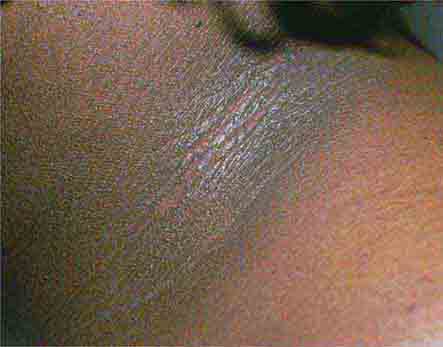
FIGURE 76e-71 Acanthosis nigricans, with typical hyperpigmented plaques on a velvet-like, verrucous surface on the neck.
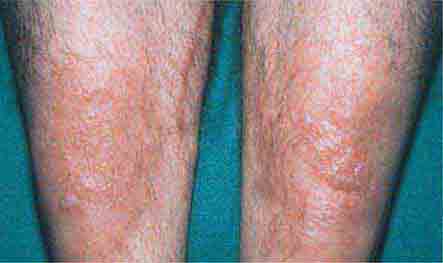
FIGURE 76e-72 Pretibial myxedema manifesting as waxy, infiltrated plaques in a patient with Graves’ disease.
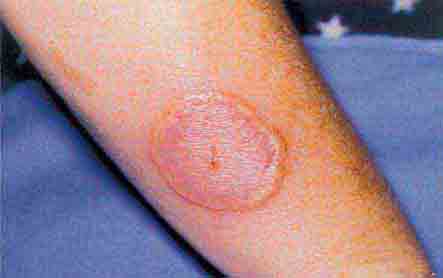
FIGURE 76e-73 Erythematous, indurated plaque of Sweet syndrome, with a pseudovesicular border. (Courtesy of Robert Swerlick, MD, with permission.)
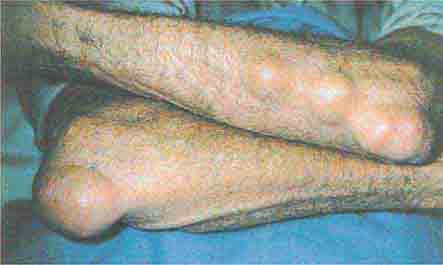
FIGURE 76e-74 Bilateral rheumatoid nodules of the upper extremities. (Courtesy of Robert Swerlick, MD; with permission.)
FIGURE 76e-75 Neurofibromatosis, with numerous flesh-colored cutaneous neurofibromas.
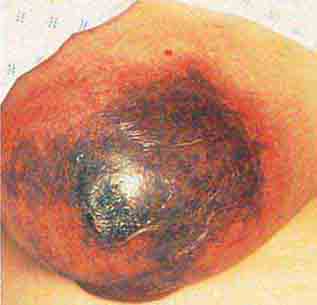
FIGURE 76e-76 Coumarin necrosis. Shown is cutaneous and subcutaneous necrosis of a breast. Other fatty areas, such as buttocks and thighs, are also common sites of involvement. (Courtesy of Kim Yancey, MD; with permission.)
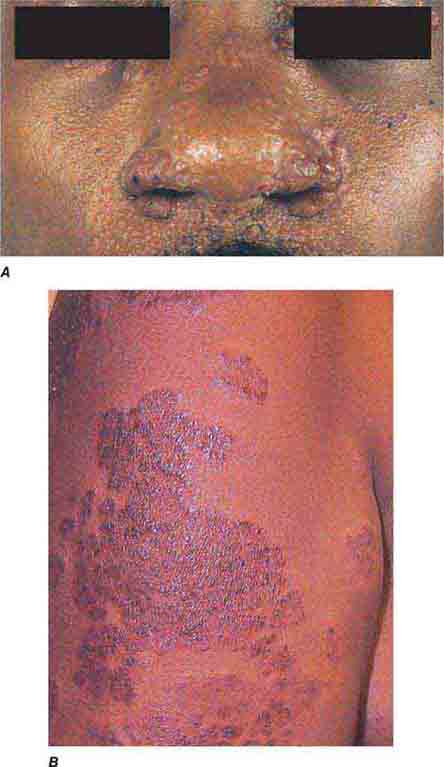
FIGURE 76e-77 Sarcoid. A. Infiltrated papules and plaques of variable color are seen in a typical paranasal and periorbital location. B. Infiltrated, hyperpigmented, and slightly erythematous coalescent papules and plaques on the upper arm. (B: Courtesy of Robert Swerlick, MD; with permission.)
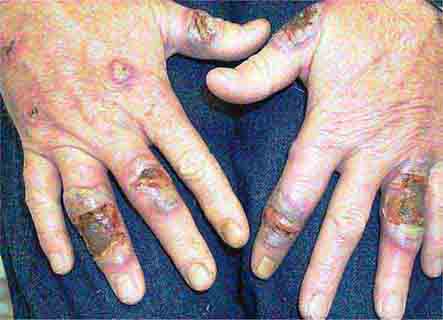
FIGURE 76e-78 Pyoderma gangrenosum on the dorsal aspect of both hands. Multiple necrotic ulcers are surrounded by a violaceous and undermined border. (Courtesy of Robert Swerlick, MD; with permission.)
SECTION 10 |
HEMATOLOGIC ALTERATIONS |
77 |
Anemia and Polycythemia |
HEMATOPOIESIS AND THE PHYSIOLOGIC BASIS OF RED CELL PRODUCTION
Hematopoiesis is the process by which the formed elements of blood are produced. The process is regulated through a series of steps beginning with the hematopoietic stem cell. Stem cells are capable of producing red cells, all classes of granulocytes, monocytes, platelets, and the cells of the immune system. The precise molecular mechanism—either intrinsic to the stem cell itself or through the action of extrinsic factors—by which the stem cell becomes committed to a given lineage is not fully defined. However, experiments in mice suggest that erythroid cells come from a common erythroid/megakaryocyte progenitor that does not develop in the absence of expression of the GATA-1 and FOG-1 (friend of GATA-1) transcription factors (Chap. 89e). Following lineage commitment, hematopoietic progenitor and precursor cells come increasingly under the regulatory influence of growth factors and hormones. For red cell production, erythropoietin (EPO) is the primary regulatory hormone. EPO is required for the maintenance of committed erythroid progenitor cells that, in the absence of the hormone, undergo programmed cell death (apoptosis). The regulated process of red cell production is erythropoiesis, and its key elements are illustrated in Fig. 77-1.
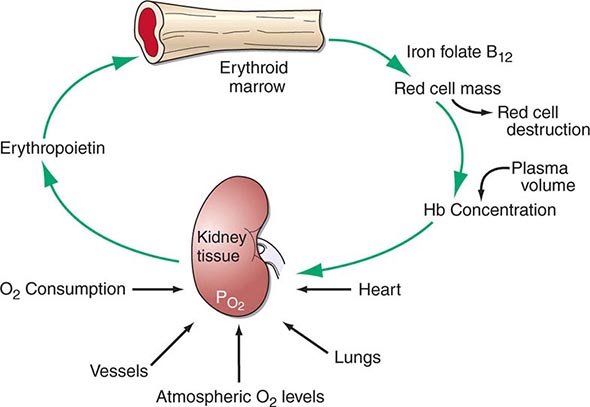
FIGURE 77-1 The physiologic regulation of red cell production by tissue oxygen tension. Hb, hemoglobin.
In the bone marrow, the first morphologically recognizable erythroid precursor is the pronormoblast. This cell can undergo four to five cell divisions, which result in the production of 16–32 mature red cells. With increased EPO production, or the administration of EPO as a drug, early progenitor cell numbers are amplified and, in turn, give rise to increased numbers of erythrocytes. The regulation of EPO production itself is linked to tissue oxygenation.
In mammals, O2 is transported to tissues bound to the hemoglobin contained within circulating red cells. The mature red cell is 8 μm in diameter, anucleate, discoid in shape, and extremely pliable in order to traverse the microcirculation successfully; its membrane integrity is maintained by the intracellular generation of ATP. Normal red cell production results in the daily replacement of 0.8–1% of all circulating red cells in the body, since the average red cell lives 100–120 days. The organ responsible for red cell production is called the erythron. The erythron is a dynamic organ made up of a rapidly proliferating pool of marrow erythroid precursor cells and a large mass of mature circulating red blood cells. The size of the red cell mass reflects the balance of red cell production and destruction. The physiologic basis of red cell production and destruction provides an understanding of the mechanisms that can lead to anemia.
The physiologic regulator of red cell production, the glycoprotein hormone EPO, is produced and released by peritubular capillary lining cells within the kidney. These cells are highly specialized epithelial-like cells. A small amount of EPO is produced by hepatocytes. The fundamental stimulus for EPO production is the availability of O2 for tissue metabolic needs. Key to EPO gene regulation is hypoxia-inducible factor (HIF)-1α. In the presence of O2, HIF-1α is hydroxylated at a key proline, allowing HIF-1α to be ubiquitinated and degraded via the proteasome pathway. If O2 becomes limiting, this critical hydroxylation step does not occur, allowing HIF-1α to partner with other proteins, translocate to the nucleus, and upregulate the expression of the EPO gene, among others.
Impaired O2 delivery to the kidney can result from a decreased red cell mass (anemia), impaired O2 loading of the hemoglobin molecule or a high O2 affinity mutant hemoglobin (hypoxemia), or, rarely, impaired blood flow to the kidney (renal artery stenosis). EPO governs the day-to-day production of red cells, and ambient levels of the hormone can be measured in the plasma by sensitive immunoassays—the normal level being 10–25 U/L. When the hemoglobin concentration falls below 100–120 g/L (10–12 g/dL), plasma EPO levels increase in proportion to the severity of the anemia (Fig. 77-2). In circulation, EPO has a half-clearance time of 6–9 h. EPO acts by binding to specific receptors on the surface of marrow erythroid precursors, inducing them to proliferate and to mature. With EPO stimulation, red cell production can increase four- to fivefold within a 1- to 2-week period, but only in the presence of adequate nutrients, especially iron. The functional capacity of the erythron, therefore, requires normal renal production of EPO, a functioning erythroid marrow, and an adequate supply of substrates for hemoglobin synthesis. A defect in any of these key components can lead to anemia. Generally, anemia is recognized in the laboratory when a patient’s hemoglobin level or hematocrit is reduced below an expected value (the normal range). The likelihood and severity of anemia are defined based on the deviation of the patient’s hemoglobin/hematocrit from values expected for age- and sex-matched normal subjects. The hemoglobin concentration in adults has a Gaussian distribution. The mean hematocrit value for adult males is 47% (standard deviation, ±7%) and that for adult females is 42% (±5%). Any single hematocrit or hemoglobin value carries with it a likelihood of associated anemia. Thus, a hematocrit of <39% in an adult male or <35% in an adult female has only about a 25% chance of being normal. Hematocrit levels are less useful than hemoglobin levels in assessing anemia because they are calculated rather than measured directly. Suspected low hemoglobin or hematocrit values are more easily interpreted if previous values for the same patient are known for comparison. The World Health Organization (WHO) defines anemia as a hemoglobin level <130 g/L (13 g/dL) in men and <120 g/L (12 g/dL) in women.
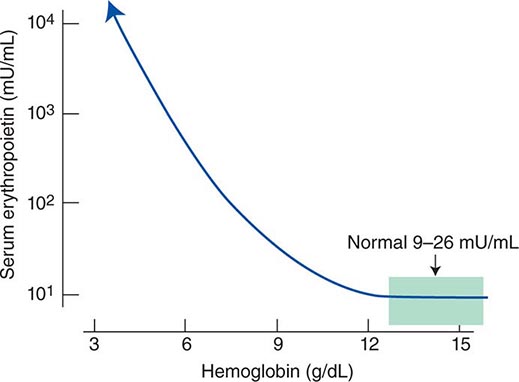
FIGURE 77-2 Erythropoietin (EPO) levels in response to anemia. When the hemoglobin level falls to 120 g/L (12 g/dL), plasma EPO levels increase logarithmically. In the presence of chronic kidney disease or chronic inflammation, EPO levels are typically lower than expected for the degree of anemia. As individuals age, the level of EPO needed to sustain normal hemoglobin levels appears to increase. (From RS Hillman et al: Hematology in Clinical Practice, 5th ed. New York, McGraw-Hill, 2010.)
The critical elements of erythropoiesis—EPO production, iron availability, the proliferative capacity of the bone marrow, and effective maturation of red cell precursors—are used for the initial classification of anemia (see below).
ANEMIA
CLINICAL PRESENTATION OF ANEMIA
Signs and Symptoms Anemia is most often recognized by abnormal screening laboratory tests. Patients less commonly present with advanced anemia and its attendant signs and symptoms. Acute anemia is due to blood loss or hemolysis. If blood loss is mild, enhanced O2 delivery is achieved through changes in the O2–hemoglobin dissociation curve mediated by a decreased pH or increased CO2 (Bohr effect). With acute blood loss, hypovolemia dominates the clinical picture, and the hematocrit and hemoglobin levels do not reflect the volume of blood lost. Signs of vascular instability appear with acute losses of 10–15% of the total blood volume. In such patients, the issue is not anemia but hypotension and decreased organ perfusion. When >30% of the blood volume is lost suddenly, patients are unable to compensate with the usual mechanisms of vascular contraction and changes in regional blood flow. The patient prefers to remain supine and will show postural hypotension and tachycardia. If the volume of blood lost is >40% (i.e., >2 L in the average-sized adult), signs of hypovolemic shock including confusion, dyspnea, diaphoresis, hypotension, and tachycardia appear (Chap. 129). Such patients have significant deficits in vital organ perfusion and require immediate volume replacement.
With acute hemolysis, the signs and symptoms depend on the mechanism that leads to red cell destruction. Intravascular hemolysis with release of free hemoglobin may be associated with acute back pain, free hemoglobin in the plasma and urine, and renal failure. Symptoms associated with more chronic or progressive anemia depend on the age of the patient and the adequacy of blood supply to critical organs. Symptoms associated with moderate anemia include fatigue, loss of stamina, breathlessness, and tachycardia (particularly with physical exertion). However, because of the intrinsic compensatory mechanisms that govern the O2–hemoglobin dissociation curve, the gradual onset of anemia—particularly in young patients—may not be associated with signs or symptoms until the anemia is severe (hemoglobin <70–80 g/L [7–8 g/dL]). When anemia develops over a period of days or weeks, the total blood volume is normal to slightly increased, and changes in cardiac output and regional blood flow help compensate for the overall loss in O2-carrying capacity. Changes in the position of the O2–hemoglobin dissociation curve account for some of the compensatory response to anemia. With chronic anemia, intracellular levels of 2,3-bisphosphoglycerate rise, shifting the dissociation curve to the right and facilitating O2 unloading. This compensatory mechanism can only maintain normal tissue O2 delivery in the face of a 20–30 g/L (2–3 g/dL) deficit in hemoglobin concentration. Finally, further protection of O2 delivery to vital organs is achieved by the shunting of blood away from organs that are relatively rich in blood supply, particularly the kidney, gut, and skin.
Certain disorders are commonly associated with anemia. Chronic inflammatory states (e.g., infection, rheumatoid arthritis, cancer) are associated with mild to moderate anemia, whereas lymphoproliferative disorders, such as chronic lymphocytic leukemia and certain other B cell neoplasms, may be associated with autoimmune hemolysis.
APPROACH TO THE PATIENT:
Anemia
The evaluation of the patient with anemia requires a careful history and physical examination. Nutritional history related to drugs or alcohol intake and family history of anemia should always be assessed. Certain geographic backgrounds and ethnic origins are associated with an increased likelihood of an inherited disorder of the hemoglobin molecule or intermediary metabolism. Glucose-6-phosphate dehydrogenase (G6PD) deficiency and certain hemoglobinopathies are seen more commonly in those of Middle Eastern or African origin, including African Americans who have a high frequency of G6PD deficiency. Other information that may be useful includes exposure to certain toxic agents or drugs and symptoms related to other disorders commonly associated with anemia. These include symptoms and signs such as bleeding, fatigue, malaise, fever, weight loss, night sweats, and other systemic symptoms. Clues to the mechanisms of anemia may be provided on physical examination by findings of infection, blood in the stool, lymphadenopathy, splenomegaly, or petechiae. Splenomegaly and lymphadenopathy suggest an underlying lymphoproliferative disease, whereas petechiae suggest platelet dysfunction. Past laboratory measurements are helpful to determine a time of onset.
In the anemic patient, physical examination may demonstrate a forceful heartbeat, strong peripheral pulses, and a systolic “flow” murmur. The skin and mucous membranes may be pale if the hemoglobin is <80–100 g/L (8–10 g/dL). This part of the physical examination should focus on areas where vessels are close to the surface such as the mucous membranes, nail beds, and palmar creases. If the palmar creases are lighter in color than the surrounding skin when the hand is hyperextended, the hemoglobin level is usually <80 g/L (8 g/dL).
LABORATORY EVALUATION
Table 77-1 lists the tests used in the initial workup of anemia. A routine complete blood count (CBC) is required as part of the evaluation and includes the hemoglobin, hematocrit, and red cell indices: the mean cell volume (MCV) in femtoliters, mean cell hemoglobin (MCH) in picograms per cell, and mean concentration of hemoglobin per volume of red cells (MCHC) in grams per liter (non-SI: grams per deciliter). The red cell indices are calculated as shown in Table 77-2, and the normal variations in the hemoglobin and hematocrit with age are shown in Table 77-3. A number of physiologic factors affect the CBC, including age, sex, pregnancy, smoking, and altitude. High-normal hemoglobin values may be seen in men and women who live at altitude or smoke heavily. Hemoglobin elevations due to smoking reflect normal compensation due to the displacement of O2 by CO in hemoglobin binding. Other important information is provided by the reticulocyte count and measurements of iron supply including serum iron, total iron-binding capacity (TIBC; an indirect measure of serum transferrin), and serum ferritin. Marked alterations in the red cell indices usually reflect disorders of maturation or iron deficiency. A careful evaluation of the peripheral blood smear is important, and clinical laboratories often provide a description of both the red and white cells, a white cell differential count, and the platelet count. In patients with severe anemia and abnormalities in red blood cell morphology and/or low reticulocyte counts, a bone marrow aspirate or biopsy can assist in the diagnosis. Other tests of value in the diagnosis of specific anemias are discussed in chapters on specific disease states.
|
LABORATORY TESTS IN ANEMIA DIAGNOSIS |
aM/E ratio, ratio of myeloid to erythroid precursors.
|
RED BLOOD CELL INDICES |

|
CHANGES IN NORMAL HEMOGLOBIN/HEMATOCRIT VALUES WITH AGE, SEX, AND PREGNANCY |
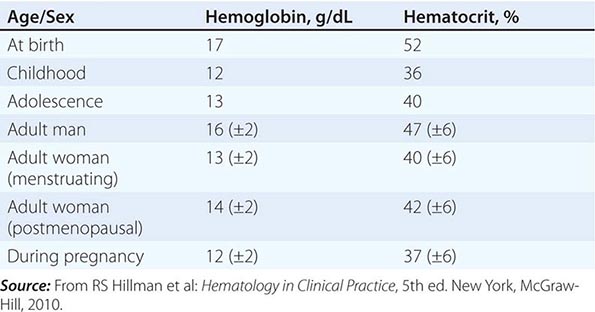
The components of the CBC also help in the classification of anemia. Microcytosis is reflected by a lower than normal MCV (<80), whereas high values (>100) reflect macrocytosis. The MCH and MCHC reflect defects in hemoglobin synthesis (hypochromia). Automated cell counters describe the red cell volume distribution width (RDW). The MCV (representing the peak of the distribution curve) is insensitive to the appearance of small populations of macrocytes or microcytes. An experienced laboratory technician will be able to identify minor populations of large or small cells or hypochromic cells before the red cell indices change.
Peripheral Blood Smear The peripheral blood smear provides important information about defects in red cell production (Chap. 81e). As a complement to the red cell indices, the blood smear also reveals variations in cell size (anisocytosis) and shape (poikilocytosis). The degree of anisocytosis usually correlates with increases in the RDW or the range of cell sizes. Poikilocytosis suggests a defect in the maturation of red cell precursors in the bone marrow or fragmentation of circulating red cells. The blood smear may also reveal polychromasia—red cells that are slightly larger than normal and grayish blue in color on the Wright-Giemsa stain. These cells are reticulocytes that have been prematurely released from the bone marrow, and their color represents residual amounts of ribosomal RNA. These cells appear in circulation in response to EPO stimulation or to architectural damage of the bone marrow (fibrosis, infiltration of the marrow by malignant cells, etc.) that results in their disordered release from the marrow. The appearance of nucleated red cells, Howell-Jolly bodies, target cells, sickle cells, and others may provide clues to specific disorders (Figs. 77-3 to 77-11).
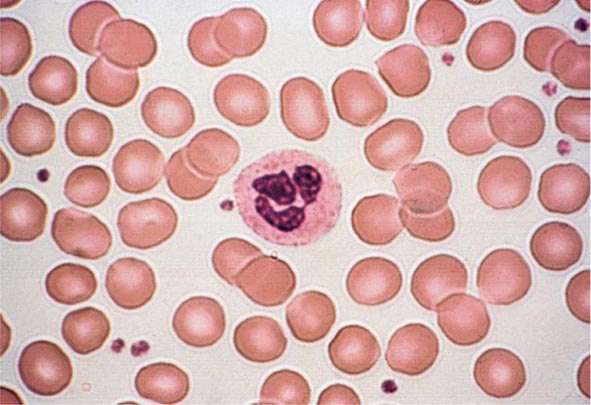
FIGURE 77-3 Normal blood smear (Wright stain). High-power field showing normal red cells, a neutrophil, and a few platelets. (From RS Hillman et al: Hematology in Clinical Practice, 5th ed. New York, McGraw-Hill, 2010.)
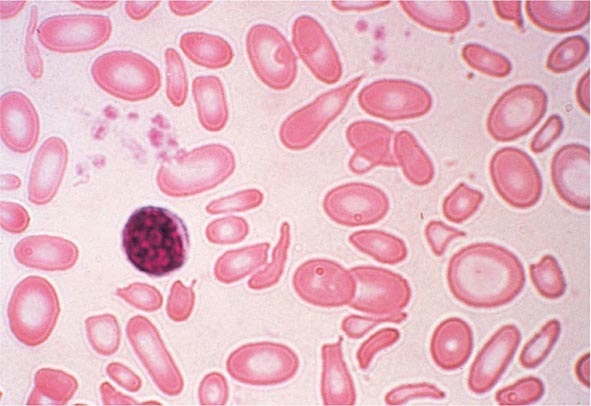
FIGURE 77-4 Severe iron-deficiency anemia. Microcytic and hypochromic red cells smaller than the nucleus of a lymphocyte associated with marked variation in size (anisocytosis) and shape (poikilocytosis). (From RS Hillman et al: Hematology in Clinical Practice, 5th ed. New York, McGraw-Hill, 2010.)
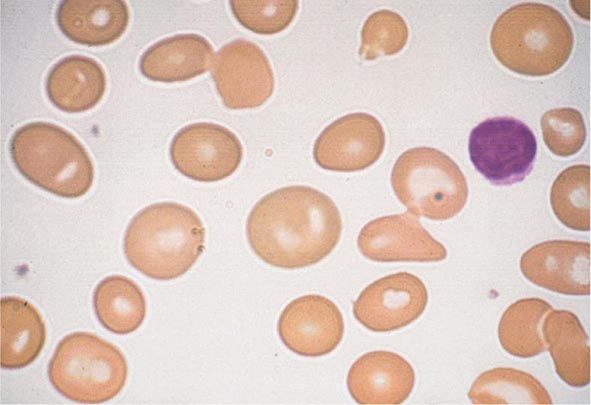
FIGURE 77-5 Macrocytosis. Red cells are larger than a small lymphocyte and well hemoglobinized. Often macrocytes are oval shaped (macro-ovalocytes).
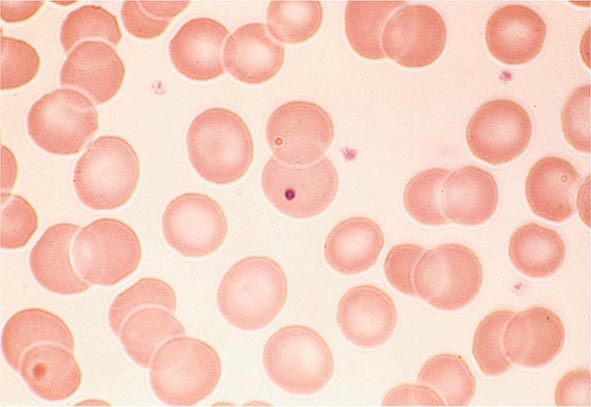
FIGURE 77-6 Howell-Jolly bodies. In the absence of a functional spleen, nuclear remnants are not culled from the red cells and remain as small homogeneously staining blue inclusions on Wright stain. (From RS Hillman et al: Hematology in Clinical Practice, 5th ed. New York, McGraw-Hill, 2010.)
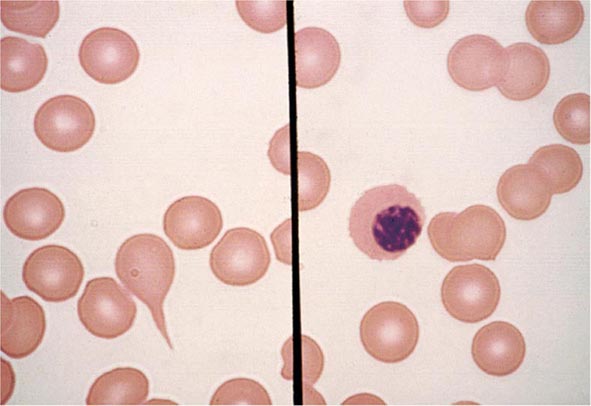
FIGURE 77-7 Red cell changes in myelofibrosis. The left panel shows a teardrop-shaped cell. The right panel shows a nucleated red cell. These forms can be seen in myelofibrosis.
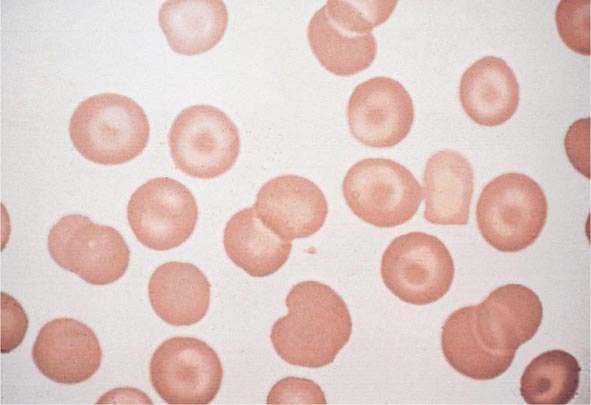
FIGURE 77-8 Target cells. Target cells have a bull’s-eye appearance and are seen in thalassemia and in liver disease. (From RS Hillman et al: Hematology in Clinical Practice, 5th ed. New York, McGraw-Hill, 2010.)
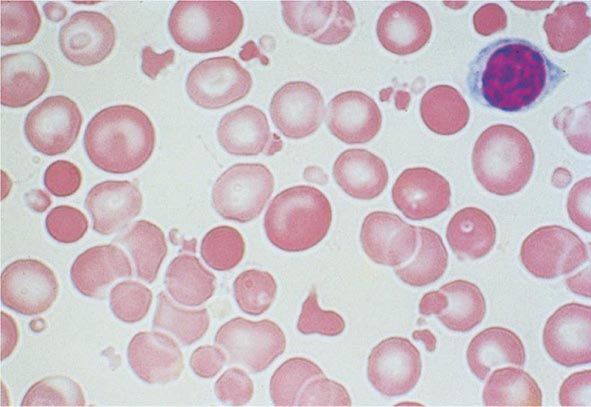
FIGURE 77-9 Red cell fragmentation. Red cells may become fragmented in the presence of foreign bodies in the circulation, such as mechanical heart valves, or in the setting of thermal injury. (From RS Hillman et al: Hematology in Clinical Practice, 5th ed. New York, McGraw-Hill, 2010.)
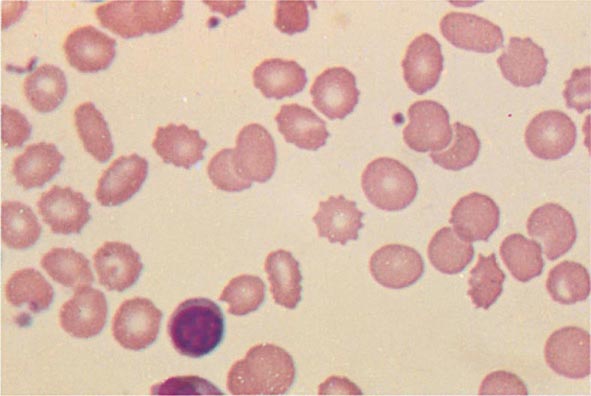
FIGURE 77-10 Uremia. The red cells in uremia may acquire numerous regularly spaced, small, spiny projections. Such cells, called burr cells or echinocytes, are readily distinguishable from irregularly spiculated acanthocytes shown in Fig. 77-11.
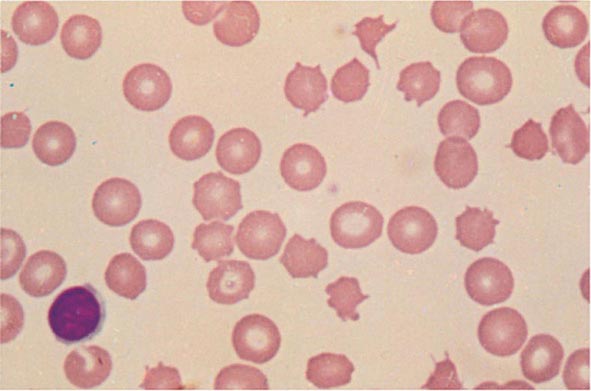
FIGURE 77-11 Spur cells. Spur cells are recognized as distorted red cells containing several irregularly distributed thornlike projections. Cells with this morphologic abnormality are also called acanthocytes. (From RS Hillman et al: Hematology in Clinical Practice, 5th ed. New York, McGraw-Hill, 2010.)
Reticulocyte Count An accurate reticulocyte count is key to the initial classification of anemia. Reticulocytes are red cells that have been recently released from the bone marrow. They are identified by staining with a supravital dye that precipitates the ribosomal RNA (Fig. 77-12). These precipitates appear as blue or black punctate spots and can be counted manually or, currently, by fluorescent emission of dyes that bind to RNA. This residual RNA is metabolized over the first 24–36 h of the reticulocyte’s life span in circulation. Normally, the reticulocyte count ranges from 1 to 2% and reflects the daily replacement of 0.8–1.0% of the circulating red cell population. A corrected reticulocyte count provides a reliable measure of effective red cell production.
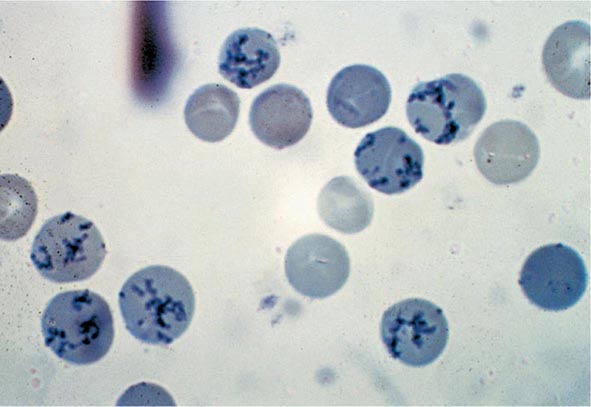
FIGURE 77-12 Reticulocytes. Methylene blue stain demonstrates residual RNA in newly made red cells. (From RS Hillman et al: Hematology in Clinical Practice, 5th ed. New York, McGraw-Hill, 2010.)
In the initial classification of anemia, the patient’s reticulocyte count is compared with the expected reticulocyte response. In general, if the EPO and erythroid marrow responses to moderate anemia [hemoglobin <100 g/L (10 g/dL)] are intact, the red cell production rate increases to two to three times normal within 10 days following the onset of anemia. In the face of established anemia, a reticulocyte response less than two to three times normal indicates an inadequate marrow response.
To use the reticulocyte count to estimate marrow response, two corrections are necessary. The first correction adjusts the reticulocyte count based on the reduced number of circulating red cells. With anemia, the percentage of reticulocytes may be increased while the absolute number is unchanged. To correct for this effect, the reticulocyte percentage is multiplied by the ratio of the patient’s hemoglobin or hematocrit to the expected hemoglobin/hematocrit for the age and sex of the patient (Table 77-4). This provides an estimate of the reticulocyte count corrected for anemia. To convert the corrected reticulocyte count to an index of marrow production, a further correction is required, depending on whether some of the reticulocytes in circulation have been released from the marrow prematurely. For this second correction, the peripheral blood smear is examined to see if there are polychromatophilic macrocytes present.
|
CALCULATION OF RETICULOCYTE PRODUCTION INDEX |
Correction #2 for Longer Life of Prematurely Released Reticulocytes in the Blood:
This correction produces the reticulocyte production index.
In a person whose reticulocyte count is 9%, hemoglobin 7.5 gm/dL, and hematocrit 23%, the reticulocyte production index

These cells, representing prematurely released reticulocytes, are referred to as “shift” cells, and the relationship between the degree of shift and the necessary shift correction factor is shown in Fig. 77-13. The correction is necessary because these prematurely released cells survive as reticulocytes in circulation for >1 day, thereby providing a falsely high estimate of daily red cell production. If polychromasia is increased, the reticulocyte count, already corrected for anemia, should be divided again by 2 to account for the prolonged reticulocyte maturation time. The second correction factor varies from 1 to 3 depending on the severity of anemia. In general, a correction of 2 is simply used. An appropriate correction is shown in Table 77-4. If polychromatophilic cells are not seen on the blood smear, the second correction is not required. The now doubly corrected reticulocyte count is the reticulocyte production index, and it provides an estimate of marrow production relative to normal. In many hospital laboratories, the reticulocyte count is reported not only as a percentage but also in absolute numbers. If so, no correction for dilution is required. A summary of the appropriate marrow response to varying degrees of anemia is shown in Table 77-5.
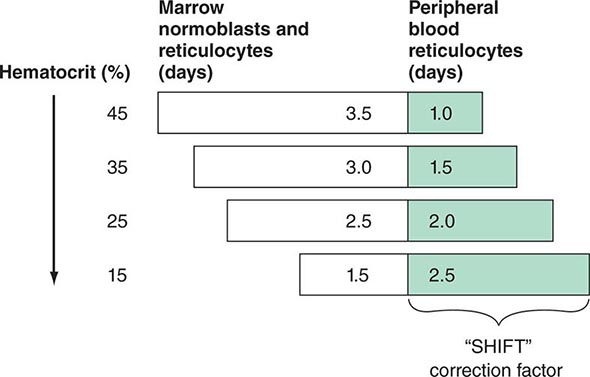
FIGURE 77-13 Correction of the reticulocyte count. To use the reticulocyte count as an indicator of effective red cell production, the reticulocyte percentage must be corrected based on the level of anemia and the circulating life span of the reticulocytes. Erythroid cells take ~4.5 days to mature. At a normal hemoglobin, reticulocytes are released to the circulation with ~1 day left as reticulocytes. However, with different levels of anemia, reticulocytes (and even earlier erythroid cells) may be released from the marrow prematurely. Most patients come to clinical attention with hematocrits in the mid-20s, and thus a correction factor of 2 is commonly used because the observed reticulocytes will live for 2 days in the circulation before losing their RNA.
|
NORMAL MARROW RESPONSE TO ANEMIA |
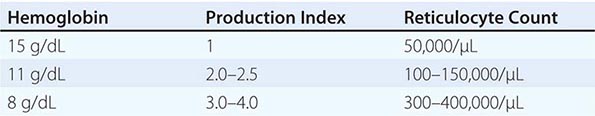
Premature release of reticulocytes is normally due to increased EPO stimulation. However, if the integrity of the bone marrow release process is lost through tumor infiltration, fibrosis, or other disorders, the appearance of nucleated red cells or polychromatophilic macrocytes should still invoke the second reticulocyte correction. The shift correction should always be applied to a patient with anemia and a very high reticulocyte count to provide a true index of effective red cell production. Patients with severe chronic hemolytic anemia may increase red cell production as much as six- to sevenfold. This measure alone confirms the fact that the patient has an appropriate EPO response, a normally functioning bone marrow, and sufficient iron available to meet the demands for new red cell formation. If the reticulocyte production index is <2 in the face of established anemia, a defect in erythroid marrow proliferation or maturation must be present.
Tests of Iron Supply and Storage The laboratory measurements that reflect the availability of iron for hemoglobin synthesis include the serum iron, the TIBC, and the percent transferrin saturation. The percent transferrin saturation is derived by dividing the serum iron level (× 100) by the TIBC. The normal serum iron ranges from 9 to 27 μmol/L (50–150 μg/dL), whereas the normal TIBC is 54–64 μmol/L (300–360 μg/dL); the normal transferrin saturation ranges from 25 to 50%. A diurnal variation in the serum iron leads to a variation in the percent transferrin saturation. The serum ferritin is used to evaluate total body iron stores. Adult males have serum ferritin levels that average ~100 μg/L, corresponding to iron stores of ~1 g. Adult females have lower serum ferritin levels averaging 30 μg/L, reflecting lower iron stores (~300 mg). A serum ferritin level of 10–15 μg/L indicates depletion of body iron stores. However, ferritin is also an acute-phase reactant and, in the presence of acute or chronic inflammation, may rise several-fold above baseline levels. As a rule, a serum ferritin >200 μg/L means there is at least some iron in tissue stores.
Bone Marrow Examination A bone marrow aspirate and smear or a needle biopsy can be useful in the evaluation of some patients with anemia. In patients with hypoproliferative anemia and normal iron status, a bone marrow is indicated. Marrow examination can diagnose primary marrow disorders such as myelofibrosis, a red cell maturation defect, or an infiltrative disease (Figs. 77-14 to 77-16). The increase or decrease of one cell lineage (myeloid vs erythroid) compared to another is obtained by a differential count of nucleated cells in a bone marrow smear (the myeloid/erythroid [M/E] ratio). A patient with a hypoproliferative anemia (see below) and a reticulocyte production index <2 will demonstrate an M/E ratio of 2 or 3:1. In contrast, patients with hemolytic disease and a production index >3 will have an M/E ratio of at least 1:1. Maturation disorders are identified from the discrepancy between the M/E ratio and the reticulocyte production index (see below). Either the marrow smear or biopsy can be stained for the presence of iron stores or iron in developing red cells. The storage iron is in the form of ferritin or hemosiderin. On carefully prepared bone marrow smears, small ferritin granules can normally be seen under oil immersion in 20–40% of developing erythroblasts. Such cells are called sideroblasts.
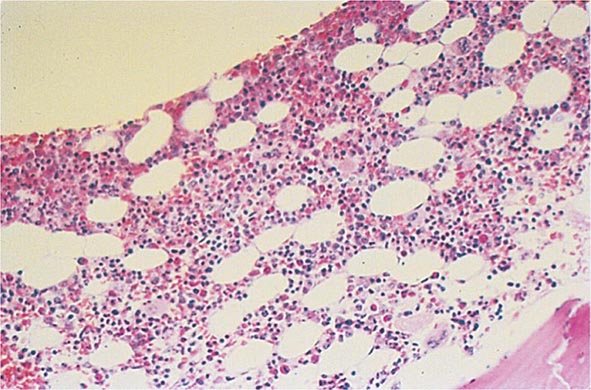
FIGURE 77-14 Normal bone marrow. This is a low-power view of a section of a normal bone marrow biopsy stained with hematoxylin and eosin (H&E). Note that the nucleated cellular elements account for ~40–50% and the fat (clear areas) accounts for ~50–60% of the area. (From RS Hillman et al: Hematology in Clinical Practice, 5th ed. New York, McGraw-Hill, 2010.)
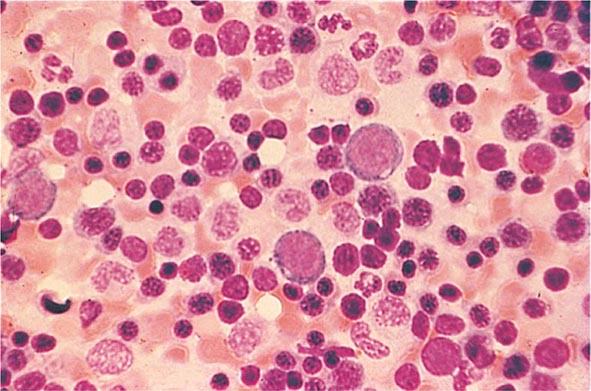
FIGURE 77-15 Erythroid hyperplasia. This marrow shows an increase in the fraction of cells in the erythroid lineage as might be seen when a normal marrow compensates for acute blood loss or hemolysis. The myeloid/erythroid (M/E) ratio is about 1:1. (From RS Hillman et al: Hematology in Clinical Practice, 5th ed. New York, McGraw-Hill, 2010.)
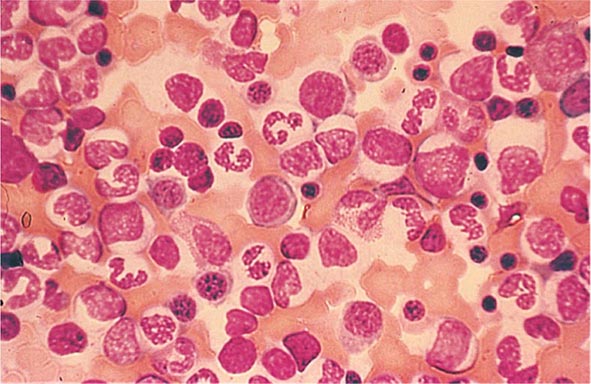
FIGURE 77-16 Myeloid hyperplasia. This marrow shows an increase in the fraction of cells in the myeloid or granulocytic lineage as might be seen in a normal marrow responding to infection. The myeloid/erythroid (M/E) ratio is >3:1. (From RS Hillman et al: Hematology in Clinical Practice, 5th ed. New York, McGraw-Hill, 2010.)
Additional laboratory tests may be of value in confirming specific diagnoses. For details of these tests and how they are applied in individual disorders, see Chaps. 126 to 130.
DEFINITION AND CLASSIFICATION OF ANEMIA
Initial Classification of Anemia The functional classification of anemia has three major categories. These are (1) marrow production defects (hypoproliferation), (2) red cell maturation defects (ineffective erythropoiesis), and (3) decreased red cell survival (blood loss/hemolysis). The classification is shown in Fig. 77-17. A hypoproliferative anemia is typically seen with a low reticulocyte production index together with little or no change in red cell morphology (a normocytic, normochromic anemia) (Chap. 126). Maturation disorders typically have a slight to moderately elevated reticulocyte production index that is accompanied by either macrocytic (Chap. 128) or microcytic (Chaps. 126, 127) red cell indices. Increased red blood cell destruction secondary to hemolysis results in an increase in the reticulocyte production index to at least three times normal (Chap. 129), provided sufficient iron is available. Hemorrhagic anemia does not typically result in production indices of more than 2.0–2.5 times normal because of the limitations placed on expansion of the erythroid marrow by iron availability.
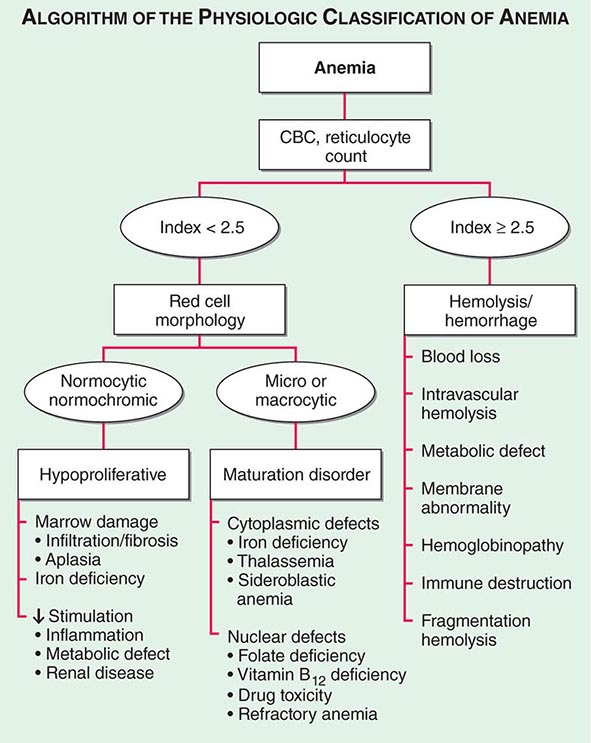
FIGURE 77-17 The physiologic classification of anemia. CBC, complete blood count.
In the first branch point of the classification of anemia, a reticulocyte production index >2.5 indicates that hemolysis is most likely. A reticulocyte production index <2 indicates either a hypoproliferative anemia or maturation disorder. The latter two possibilities can often be distinguished by the red cell indices, by examination of the peripheral blood smear, or by a marrow examination. If the red cell indices are normal, the anemia is almost certainly hypoproliferative in nature. Maturation disorders are characterized by ineffective red cell production and a low reticulocyte production index. Bizarre red cell shapes—macrocytes or hypochromic microcytes—are seen on the peripheral blood smear. With a hypoproliferative anemia, no erythroid hyperplasia is noted in the marrow, whereas patients with ineffective red cell production have erythroid hyperplasia and an M/E ratio <1:1.
Hypoproliferative Anemias At least 75% of all cases of anemia are hypoproliferative in nature. A hypoproliferative anemia reflects absolute or relative marrow failure in which the erythroid marrow has not proliferated appropriately for the degree of anemia. The majority of hypoproliferative anemias are due to mild to moderate iron deficiency or inflammation. A hypoproliferative anemia can result from marrow damage, iron deficiency, or inadequate EPO stimulation. The last may reflect impaired renal function, suppression of EPO production by inflammatory cytokines such as interleukin 1, or reduced tissue needs for O2 from metabolic disease such as hypothyroidism. Only occasionally is the marrow unable to produce red cells at a normal rate, and this is most prevalent in patients with renal failure. With diabetes mellitus or myeloma, the EPO deficiency may be more marked than would be predicted by the degree of renal insufficiency. In general, hypoproliferative anemias are characterized by normocytic, normochromic red cells, although microcytic, hypochromic cells may be observed with mild iron deficiency or long-standing chronic inflammatory disease. The key laboratory tests in distinguishing between the various forms of hypoproliferative anemia include the serum iron and iron-binding capacity, evaluation of renal and thyroid function, a marrow biopsy or aspirate to detect marrow damage or infiltrative disease, and serum ferritin to assess iron stores. An iron stain of the marrow will determine the pattern of iron distribution. Patients with the anemia of acute or chronic inflammation show a distinctive pattern of serum iron (low), TIBC (normal or low), percent transferrin saturation (low), and serum ferritin (normal or high). These changes in iron values are brought about by hepcidin, the iron regulatory hormone that is produced by the liver and is increased in inflammation (Chap. 126). A distinct pattern of results is noted in mild to moderate iron deficiency (low serum iron, high TIBC, low percent transferrin saturation, low serum ferritin) (Chap. 126). Marrow damage by drugs, infiltrative disease such as leukemia or lymphoma, or marrow aplasia is diagnosed from the peripheral blood and bone marrow morphology. With infiltrative disease or fibrosis, a marrow biopsy is required.
Maturation Disorders The presence of anemia with an inappropriately low reticulocyte production index, macro- or microcytosis on smear, and abnormal red cell indices suggests a maturation disorder. Maturation disorders are divided into two categories: nuclear maturation defects, associated with macrocytosis, and cytoplasmic maturation defects, associated with microcytosis and hypochromia usually from defects in hemoglobin synthesis. The inappropriately low reticulocyte production index is a reflection of the ineffective erythropoiesis that results from the destruction within the marrow of developing erythroblasts. Bone marrow examination shows erythroid hyperplasia.
Nuclear maturation defects result from vitamin B12 or folic acid deficiency, drug damage, or myelodysplasia. Drugs that interfere with cellular DNA synthesis, such as methotrexate or alkylating agents, can produce a nuclear maturation defect. Alcohol, alone, is also capable of producing macrocytosis and a variable degree of anemia, but this is usually associated with folic acid deficiency. Measurements of folic acid and vitamin B12 are critical not only in identifying the specific vitamin deficiency but also because they reflect different pathogenetic mechanisms (Chap. 128).
Cytoplasmic maturation defects result from severe iron deficiency or abnormalities in globin or heme synthesis. Iron deficiency occupies an unusual position in the classification of anemia. If the iron-deficiency anemia is mild to moderate, erythroid marrow proliferation is blunted and the anemia is classified as hypoproliferative. However, if the anemia is severe and prolonged, the erythroid marrow will become hyperplastic despite the inadequate iron supply, and the anemia will be classified as ineffective erythropoiesis with a cytoplasmic maturation defect. In either case, an inappropriately low reticulocyte production index, microcytosis, and a classic pattern of iron values make the diagnosis clear and easily distinguish iron deficiency from other cytoplasmic maturation defects such as the thalassemias. Defects in heme synthesis, in contrast to globin synthesis, are less common and may be acquired or inherited (Chap. 430). Acquired abnormalities are usually associated with myelodysplasia, may lead to either a macro- or microcytic anemia, and are frequently associated with mitochondrial iron loading. In these cases, iron is taken up by the mitochondria of the developing erythroid cell but not incorporated into heme. The iron-encrusted mitochondria surround the nucleus of the erythroid cell, forming a ring. Based on the distinctive finding of so-called ringed sideroblasts on the marrow iron stain, patients are diagnosed as having a sideroblastic anemia—almost always reflecting myelodysplasia. Again, studies of iron parameters are helpful in the differential diagnosis of these patients.
Blood Loss/Hemolytic Anemia In contrast to anemias associated with an inappropriately low reticulocyte production index, hemolysis is associated with red cell production indices ≥2.5 times normal. The stimulated erythropoiesis is reflected in the blood smear by the appearance of increased numbers of polychromatophilic macrocytes. A marrow examination is rarely indicated if the reticulocyte production index is increased appropriately. The red cell indices are typically normocytic or slightly macrocytic, reflecting the increased number of reticulocytes. Acute blood loss is not associated with an increased reticulocyte production index because of the time required to increase EPO production and, subsequently, marrow proliferation. Subacute blood loss may be associated with modest reticulocytosis. Anemia from chronic blood loss presents more often as iron deficiency than with the picture of increased red cell production.
The evaluation of blood loss anemia is usually not difficult. Most problems arise when a patient presents with an increased red cell production index from an episode of acute blood loss that went unrecognized. The cause of the anemia and increased red cell production may not be obvious. The confirmation of a recovering state may require observations over a period of 2–3 weeks, during which the hemoglobin concentration will rise and the reticulocyte production index fall (Chap. 129).
Hemolytic disease, while dramatic, is among the least common forms of anemia. The ability to sustain a high reticulocyte production index reflects the ability of the erythroid marrow to compensate for hemolysis and, in the case of extravascular hemolysis, the efficient recycling of iron from the destroyed red cells to support red cell production. With intravascular hemolysis, such as paroxysmal nocturnal hemoglobinuria, the loss of iron may limit the marrow response. The level of response depends on the severity of the anemia and the nature of the underlying disease process.
Hemoglobinopathies, such as sickle cell disease and the thalassemias, present a mixed picture. The reticulocyte index may be high but is inappropriately low for the degree of marrow erythroid hyperplasia (Chap. 127).
Hemolytic anemias present in different ways. Some appear suddenly as an acute, self-limited episode of intravascular or extravascular hemolysis, a presentation pattern often seen in patients with autoimmune hemolysis or with inherited defects of the Embden-Meyerhof pathway or the glutathione reductase pathway. Patients with inherited disorders of the hemoglobin molecule or red cell membrane generally have a lifelong clinical history typical of the disease process. Those with chronic hemolytic disease, such as hereditary spherocytosis, may actually present not with anemia but with a complication stemming from the prolonged increase in red cell destruction such as symptomatic bilirubin gallstones or splenomegaly. Patients with chronic hemolysis are also susceptible to aplastic crises if an infectious process interrupts red cell production.
The differential diagnosis of an acute or chronic hemolytic event requires the careful integration of family history, the pattern of clinical presentation, and—whether the disease is congenital or acquired—careful examination of the peripheral blood smear. Precise diagnosis may require more specialized laboratory tests, such as hemoglobin electrophoresis or a screen for red cell enzymes. Acquired defects in red cell survival are often immunologically mediated and require a direct or indirect antiglobulin test or a cold agglutinin titer to detect the presence of hemolytic antibodies or complement-mediated red cell destruction (Chap. 129).
POLYCYTHEMIA
Polycythemia is defined as an increase in the hemoglobin above normal. This increase may be real or only apparent because of a decrease in plasma volume (spurious or relative polycythemia). The term erythrocytosis may be used interchangeably with polycythemia, but some draw a distinction between them: erythrocytosis implies documentation of increased red cell mass, whereas polycythemia refers to any increase in red cells. Often patients with polycythemia are detected through an incidental finding of elevated hemoglobin or hematocrit levels. Concern that the hemoglobin level may be abnormally high is usually triggered at 170 g/L (17 g/dL) for men and 150 g/L (15 g/dL) for women. Hematocrit levels >50% in men or >45% in women may be abnormal. Hematocrits >60% in men and >55% in women are almost invariably associated with an increased red cell mass. Given that the machine that quantitates red cell parameters actually measures hemoglobin concentrations and calculates hematocrits, hemoglobin levels may be a better index.
Features of the clinical history that are useful in the differential diagnosis include smoking history; current living at high altitude; or a history of congenital heart disease, sleep apnea, or chronic lung disease.
Patients with polycythemia may be asymptomatic or experience symptoms related to the increased red cell mass or the underlying disease process that leads to the increased red cell mass. The dominant symptoms from an increased red cell mass are related to hyperviscosity and thrombosis (both venous and arterial), because the blood viscosity increases logarithmically at hematocrits >55%. Manifestations range from digital ischemia to Budd-Chiari syndrome with hepatic vein thrombosis. Abdominal vessel thromboses are particularly common. Neurologic symptoms such as vertigo, tinnitus, headache, and visual disturbances may occur. Hypertension is often present. Patients with polycythemia vera may have aquagenic pruritus and symptoms related to hepatosplenomegaly. Patients may have easy bruising, epistaxis, or bleeding from the gastrointestinal tract. Peptic ulcer disease is common. Patients with hypoxemia may develop cyanosis on minimal exertion or have headache, impaired mental acuity, and fatigue.
The physical examination usually reveals a ruddy complexion. Splenomegaly favors polycythemia vera as the diagnosis (Chap. 131). The presence of cyanosis or evidence of a right-to-left shunt suggests congenital heart disease presenting in the adult, particularly tetralogy of Fallot or Eisenmenger’s syndrome (Chap. 236). Increased blood viscosity raises pulmonary artery pressure; hypoxemia can lead to increased pulmonary vascular resistance. Together, these factors can produce cor pulmonale.
Polycythemia can be spurious (related to a decrease in plasma volume; Gaisbock’s syndrome), primary, or secondary in origin. The secondary causes are all associated with increases in EPO levels: either a physiologically adapted appropriate elevation based on tissue hypoxia (lung disease, high altitude, CO poisoning, high-affinity hemoglobinopathy) or an abnormal overproduction (renal cysts, renal artery stenosis, tumors with ectopic EPO production). A rare familial form of polycythemia is associated with normal EPO levels but hyperresponsive EPO receptors due to mutations.
APPROACH TO THE PATIENT:
Polycythemia
As shown in Fig. 77-18, the first step is to document the presence of an increased red cell mass using the principle of isotope dilution by administering 51Cr-labeled autologous red blood cells to the patient and sampling blood radioactivity over a 2-h period. If the red cell mass is normal (<36 mL/kg in men, <32 mL/kg in women), the patient has spurious or relative polycythemia. If the red cell mass is increased (>36 mL/kg in men, >32 mL/kg in women), serum EPO levels should be measured. If EPO levels are low or unmeasurable, the patient most likely has polycythemia vera. A mutation in JAK2 (Val617Phe), a key member of the cytokine intracellular signaling pathway, can be found in 90–95% of patients with polycythemia vera. Many of those without this particular JAK2 mutation have mutations in exon 12. As a practical matter, few centers assess red cell mass in the setting of an increased hematocrit. The short workup is to measure EPO levels, check for JAK2 mutation, and perform an abdominal ultrasound to assess spleen size. Tests that support the diagnosis of polycythemia vera include elevated white blood cell count, increased absolute basophil count, and thrombocytosis.
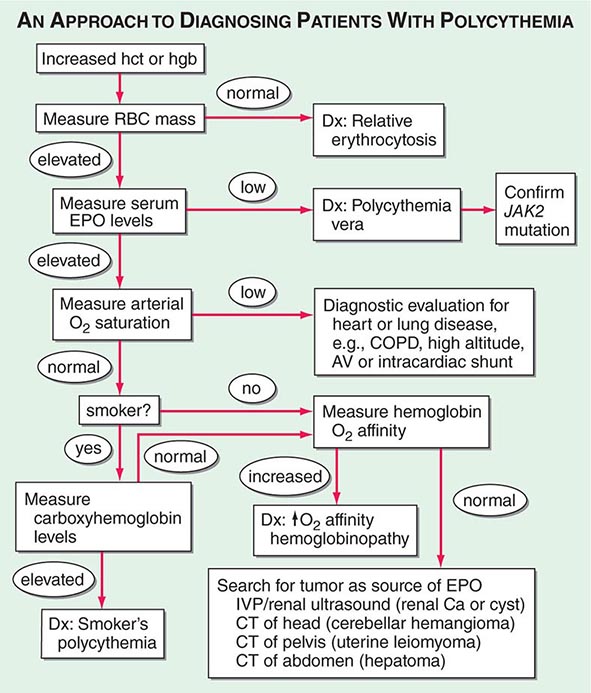
FIGURE 77-18 An approach to the differential diagnosis of patients with an elevated hemoglobin (possible polycythemia). AV, atrioventricular; COPD, chronic obstructive pulmonary disease; CT, computed tomography; EPO, erythropoietin; hct, hematocrit; hgb, hemoglobin; IVP, intravenous pyelogram; RBC, red blood cell.
If serum EPO levels are elevated, one needs to distinguish whether the elevation is a physiologic response to hypoxia or related to autonomous EPO production. Patients with low arterial O2 saturation (<92%) should be further evaluated for the presence of heart or lung disease, if they are not living at high altitude. Patients with normal O2 saturation who are smokers may have elevated EPO levels because of CO displacement of O2. If carboxyhemoglobin (COHb) levels are high, the diagnosis is “smoker’s polycythemia.” Such patients should be urged to stop smoking. Those who cannot stop smoking require phlebotomy to control their polycythemia. Patients with normal O2 saturation who do not smoke either have an abnormal hemoglobin that does not deliver O2 to the tissues (evaluated by finding elevated O2–hemoglobin affinity) or have a source of EPO production that is not responding to the normal feedback inhibition. Further workup is dictated by the differential diagnosis of EPO-producing neoplasms. Hepatoma, uterine leiomyoma, and renal cancer or cysts are all detectable with abdominopelvic computed tomography scans. Cerebellar hemangiomas may produce EPO, but they present with localizing neurologic signs and symptoms rather than polycythemia-related symptoms.
78 |
Bleeding and Thrombosis |
The human hemostatic system provides a natural balance between procoagulant and anticoagulant forces. The procoagulant forces include platelet adhesion and aggregation and fibrin clot formation; anticoagulant forces include the natural inhibitors of coagulation and fibrinolysis. Under normal circumstances, hemostasis is regulated to promote blood flow; however, it is also prepared to clot blood rapidly to arrest blood flow and prevent exsanguination. After bleeding is successfully halted, the system remodels the damaged vessel to restore normal blood flow. The major components of the hemostatic system, which function in concert, are (1) platelets and other formed elements of blood, such as monocytes and red cells; (2) plasma proteins (the coagulation and fibrinolytic factors and inhibitors); and (3) the vessel wall.
STEPS OF NORMAL HEMOSTASIS
PLATELET PLUG FORMATION
On vascular injury, platelets adhere to the site of injury, usually the denuded vascular intimal surface. Platelet adhesion is mediated primarily by Von Willebrand factor (VWF), a large multimeric protein present in both plasma and the extracellular matrix of the subendothelial vessel wall, which serves as the primary “molecular glue,” providing sufficient strength to withstand the high levels of shear stress that would tend to detach them with the flow of blood. Platelet adhesion is also facilitated by direct binding to subendothelial collagen through specific platelet membrane collagen receptors.
Platelet adhesion results in subsequent platelet activation and aggregation. This process is enhanced and amplified by humoral mediators in plasma (e.g., epinephrine, thrombin); mediators released from activated platelets (e.g., adenosine diphosphate, serotonin); and vessel wall extracellular matrix constituents that come in contact with adherent platelets (e.g., collagen, VWF). Activated platelets undergo the release reaction, during which they secrete contents that further promote aggregation and inhibit the naturally anticoagulant endothelial cell factors. During platelet aggregation (platelet-platelet interaction), additional platelets are recruited from the circulation to the site of vascular injury, leading to the formation of an occlusive platelet thrombus. The platelet plug is anchored and stabilized by the developing fibrin mesh.
The platelet glycoprotein (Gp) IIb/IIIa (αIIbβ3) complex is the most abundant receptor on the platelet surface. Platelet activation converts the normally inactive Gp IIb/IIIa receptor into an active receptor, enabling binding to fibrinogen and VWF. Because the surface of each platelet has about 50,000 Gp IIb/IIIa–binding sites, numerous activated platelets recruited to the site of vascular injury can rapidly form an occlusive aggregate by means of a dense network of intercellular fibrinogen bridges. Because this receptor is the key mediator of platelet aggregation, it has become an effective target for antiplatelet therapy.
FIBRIN CLOT FORMATION
Plasma coagulation proteins (clotting factors) normally circulate in plasma in their inactive forms. The sequence of coagulation protein reactions that culminate in the formation of fibrin was originally described as a waterfall or a cascade. Two pathways of blood coagulation have been described in the past: the so-called extrinsic, or tissue factor, pathway and the so-called intrinsic, or contact activation, pathway. We now know that coagulation is normally initiated through tissue factor (TF) exposure and activation through the classic extrinsic pathway but with critically important amplification through elements of the classic intrinsic pathway, as illustrated in Fig. 78-1. These reactions take place on phospholipid surfaces, usually the activated platelet surface. Coagulation testing in the laboratory can reflect other influences due to the artificial nature of the in vitro systems used (see below).
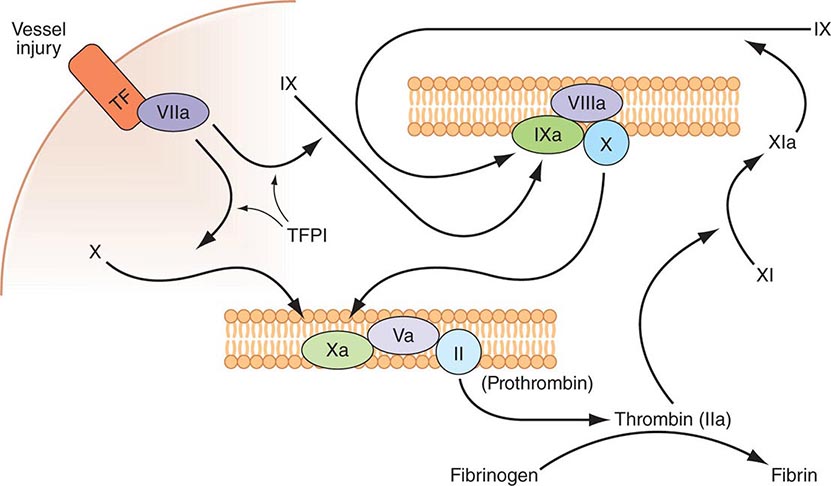
FIGURE 78-1 Coagulation is initiated by tissue factor (TF) exposure, which, with factor (F) VIIa, activates FIX and FX, which in turn, with FVIII and FV as cofactors, respectively, results in thrombin formation and subsequent conversion of fibrinogen to fibrin. Thrombin activates FXI, FVIII, and FV, amplifying the coagulation signal. Once the TF/FVIIa/FXa complex is formed, tissue factor pathway inhibitor (TFPI) inhibits the TF/FVIIa pathway, making coagulation dependent on the amplification loop through FIX/FVIII. Coagulation requires calcium (not shown) and takes place on phospholipid surfaces, usually the activated platelet membrane.
The immediate trigger for coagulation is vascular damage that exposes blood to TF that is constitutively expressed on the surfaces of subendothelial cellular components of the vessel wall, such as smooth muscle cells and fibroblasts. TF is also present in circulating microparticles, presumably shed from cells including monocytes and platelets. TF binds the serine protease factor VIIa; the complex activates factor × to factor Xa. Alternatively, the complex can indirectly activate factor × by initially converting factor IX to factor IXa, which then activates factor X. The participation of factor XI in hemostasis is not dependent on its activation by factor XIIa but rather on its positive feedback activation by thrombin. Thus, factor XIa functions in the propagation and amplification, rather than in the initiation, of the coagulation cascade.
Factor Xa can be formed through the actions of either the TF/factor VIIa complex or factor IXa (with factor VIIIa as a cofactor) and converts prothrombin to thrombin, the pivotal protease of the coagulation system. The essential cofactor for this reaction is factor Va. Like the homologous factor VIIIa, factor Va is produced by thrombin-induced limited proteolysis of factor V. Thrombin is a multifunctional enzyme that converts soluble plasma fibrinogen to an insoluble fibrin matrix. Fibrin polymerization involves an orderly process of intermolecular associations (Fig. 78-2). Thrombin also activates factor XIII (fibrin-stabilizing factor) to factor XIIIa, which covalently cross-links and thereby stabilizes the fibrin clot.
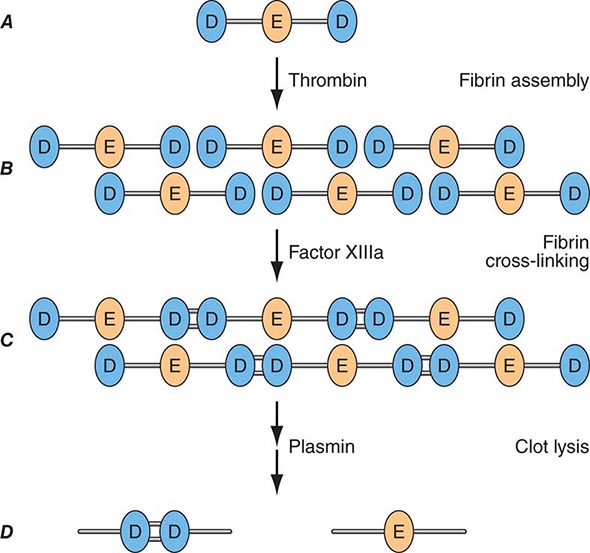
FIGURE 78-2 Fibrin formation and dissolution. (A) Fibrinogen is a trinodular structure consisting of two D domains and one E domain. Thrombin activation results in an ordered lateral assembly of protofibrils (B) with noncovalent associations. Factor XIIIa cross-links the D domains on adjacent molecules (C). Fibrin and fibrinogen (not shown) lysis by plasmin occurs at discrete sites and results in intermediary fibrin(ogen) degradation products (not shown). D-Dimers are the product of complete lysis of fibrin (D), maintaining the cross-linked D domains.
The assembly of the clotting factors on activated cell membrane surfaces greatly accelerates their reaction rates and also serves to localize blood clotting to sites of vascular injury. The critical cell membrane components, acidic phospholipids, are not normally exposed on resting cell membrane surfaces. However, when platelets, monocytes, and endothelial cells are activated by vascular injury or inflammatory stimuli, the procoagulant head groups of the membrane anionic phospholipids become translocated to the surfaces of these cells or released as part of microparticles, making them available to support and promote the plasma coagulation reactions.
ANTITHROMBOTIC MECHANISMS
Several physiologic antithrombotic mechanisms act in concert to prevent clotting under normal circumstances. These mechanisms operate to preserve blood fluidity and to limit blood clotting to specific focal sites of vascular injury. Endothelial cells have many antithrombotic effects. They produce prostacyclin, nitric oxide, and ectoADPase/CD39, which act to inhibit platelet binding, secretion, and aggregation. Endothelial cells produce anticoagulant factors including heparan proteoglycans, antithrombin, TF pathway inhibitor, and thrombomodulin. They also activate fibrinolytic mechanisms through the production of tissue plasminogen activator 1, urokinase, plasminogen activator inhibitor, and annexin-2. The sites of action of the major physiologic antithrombotic pathways are shown in Fig. 78-3.
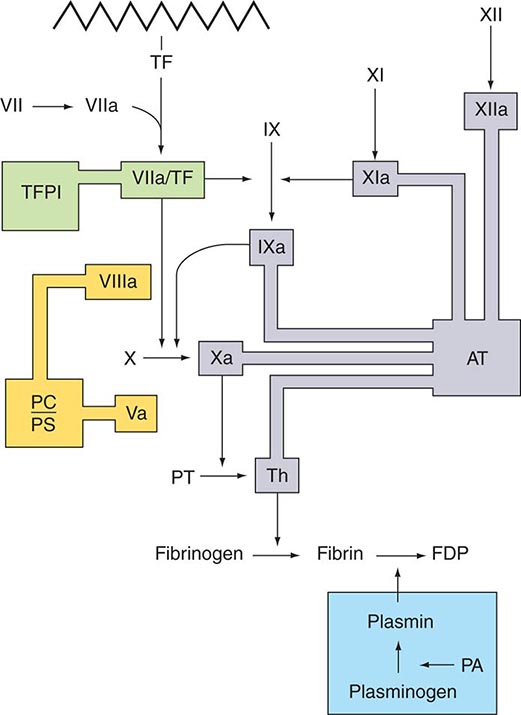
FIGURE 78-3 Sites of action of the four major physiologic antithrombotic pathways: antithrombin (AT); protein C/S (PC/PS); tissue factor pathway inhibitor (TFPI); and the fibrinolytic system, consisting of plasminogen, plasminogen activator (PA), and plasmin. PT, prothrombin; Th, thrombin; FDP, fibrin(ogen) degradation products. (Modified from BA Konkle, AI Schafer, in DP Zipes et al [eds]: Braunwald’s Heart Disease, 7th ed. Philadelphia, Saunders, 2005.)
Antithrombin (or antithrombin III) is the major plasma protease inhibitor of thrombin and the other clotting factors in coagulation. Antithrombin neutralizes thrombin and other activated coagulation factors by forming a complex between the active site of the enzyme and the reactive center of antithrombin. The rate of formation of these inactivating complexes increases by a factor of several thousand in the presence of heparin. Antithrombin inactivation of thrombin and other activated clotting factors occurs physiologically on vascular surfaces, where glycosoaminoglycans, including heparan sulfates, are present to catalyze these reactions. Inherited quantitative or qualitative deficiencies of antithrombin lead to a lifelong predisposition to venous thromboembolism.
Protein C is a plasma glycoprotein that becomes an anticoagulant when it is activated by thrombin. The thrombin-induced activation of protein C occurs physiologically on thrombomodulin, a transmembrane proteoglycan-binding site for thrombin on endothelial cell surfaces. The binding of protein C to its receptor on endothelial cells places it in proximity to the thrombin-thrombomodulin complex, thereby enhancing its activation efficiency. Activated protein C acts as an anticoagulant by cleaving and inactivating activated factors V and VIII. This reaction is accelerated by a cofactor, protein S, which, like protein C, is a glycoprotein that undergoes vitamin K–dependent posttranslational modification. Quantitative or qualitative deficiencies of protein C or protein S, or resistance to the action of activated protein C by a specific mutation at its target cleavage site in factor Va (factor V Leiden), lead to hypercoagulable states.
Tissue factor pathway inhibitor (TFPI) is a plasma protease inhibitor that regulates the TF-induced extrinsic pathway of coagulation. TFPI inhibits the TF/factor VIIa/factor Xa complex, essentially turning off the TF/factor VIIa initiation of coagulation, which then becomes dependent on the “amplification loop” via factor XI and factor VIII activation by thrombin. TFPI is bound to lipoprotein and can also be released by heparin from endothelial cells, where it is bound to glycosaminoglycans, and from platelets. The heparin-mediated release of TFPI may play a role in the anticoagulant effects of unfractionated and low-molecular-weight heparins.
THE FIBRINOLYTIC SYSTEM
Any thrombin that escapes the inhibitory effects of the physiologic anticoagulant systems is available to convert fibrinogen to fibrin. In response, the endogenous fibrinolytic system is then activated to dispose of intravascular fibrin and thereby maintain or reestablish the patency of the circulation. Just as thrombin is the key protease enzyme of the coagulation system, plasmin is the major protease enzyme of the fibrinolytic system, acting to digest fibrin to fibrin degradation products. The general scheme of fibrinolysis and its control is shown in Fig. 78-4.
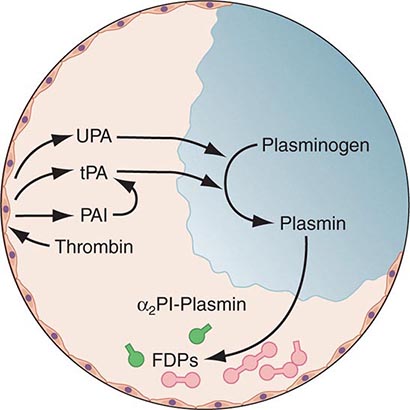
FIGURE 78-4 A schematic diagram of the fibrinolytic system. Tissue plasminogen activator (tPA) is released from endothelial cells, binds the fibrin clot, and activates plasminogen to plasmin. Excess fibrin is degraded by plasmin to distinct degradation products (FDPs). Any free plasmin is complexed with α2-antiplasmin (α2Pl). PAI, plasminogen activator inhibitor; UPA, urokinase-type plasminogen activator.
The plasminogen activators, tissue type plasminogen activator (tPA) and the urokinase-type plasminogen activator (uPA), cleave the Arg560-Val561 bond of plasminogen to generate the active enzyme plasmin. The lysine-binding sites of plasmin (and plasminogen) permit it to bind to fibrin, so that physiologic fibrinolysis is “fibrin specific.” Both plasminogen (through its lysine-binding sites) and tPA possess specific affinity for fibrin and thereby bind selectively to clots. The assembly of a ternary complex, consisting of fibrin, plasminogen, and tPA, promotes the localized interaction between plasminogen and tPA and greatly accelerates the rate of plasminogen activation to plasmin. Moreover, partial degradation of fibrin by plasmin exposes new plasminogen and tPA-binding sites in carboxy-terminus lysine residues of fibrin fragments to enhance these reactions further. This creates a highly efficient mechanism to generate plasmin focally on the fibrin clot, which then becomes plasmin’s substrate for digestion to fibrin degradation products.
Plasmin cleaves fibrin at distinct sites of the fibrin molecule, leading to the generation of characteristic fibrin fragments during the process of fibrinolysis (Fig. 78-2). The sites of plasmin cleavage of fibrin are the same as those in fibrinogen. However, when plasmin acts on covalently cross-linked fibrin, D-dimers are released; hence, D-dimers can be measured in plasma as a relatively specific test of fibrin (rather than fibrinogen) degradation. D-Dimer assays can be used as sensitive markers of blood clot formation and have been validated for clinical use to exclude the diagnosis of deep venous thrombosis (DVT) and pulmonary embolism in selected populations. In addition, D-dimer measurement can be used to stratify patients, particularly women, for risk of recurrent venous thromboembolism (VTE) when measured 1 month after discontinuation of anticoagulation given for treatment of an initial idiopathic event. D-Dimer levels may be elevated in the absence of VTE in elderly people.
Physiologic regulation of fibrinolysis occurs primarily at three levels: (1) plasminogen activator inhibitors (PAIs), specifically PAI-1 and PAI-2, inhibit the physiologic plasminogen activators; (2) the thrombin-activatable fibrinolysis inhibitor (TAFI) limits fibrinolysis; and (3) α2-antiplasmin inhibits plasmin. PAI-1 is the primary inhibitor of tPA and uPA in plasma. TAFI cleaves the N-terminal lysine residues of fibrin, which aid in localization of plasmin activity. α2-Antiplasmin is the main inhibitor of plasmin in human plasma, inactivating any nonfibrin clot-associated plasmin.
APPROACH TO THE PATIENT:
Bleeding and Thrombosis
CLINICAL PRESENTATION
Disorders of hemostasis may be either inherited or acquired. A detailed personal and family history is key in determining the chronicity of symptoms and the likelihood of the disorder being inherited, as well as providing clues to underlying conditions that have contributed to the bleeding or thrombotic state. In addition, the history can give clues as to the etiology by determining (1) the bleeding (mucosal and/or joint) or thrombosis (arterial and/or venous) site and (2) whether an underlying bleeding or clotting tendency was enhanced by another medical condition or the introduction of medications or dietary supplements.
History of Bleeding A history of bleeding is the most important predictor of bleeding risk. In evaluating a patient for a bleeding disorder, a history of at-risk situations, including the response to past surgeries, should be assessed. Does the patient have a history of spontaneous or trauma/surgery-induced bleeding? Spontaneous hemarthroses are a hallmark of moderate and severe factor VIII and IX deficiency and, in rare circumstances, of other clotting factor deficiencies. Mucosal bleeding symptoms are more suggestive of underlying platelet disorders or Von Willebrand disease (VWD), termed disorders of primary hemostasis or platelet plug formation. Disorders affecting primary hemostasis are shown in Table 78-1.
|
PRIMARY HEMOSTATIC (PLATELET PLUG) DISORDERS |
A bleeding score has been validated as a tool to predict patients more likely to have type 1 VWD (International Society on Thrombosis and Haemostasis Bleeding Assessment Tool [www.isth.org/resource/resmgr/ssc/isth-ssc_bleeding_assessment.pdf]). This is most useful tool in excluding the diagnosis of a bleeding disorder, and thus avoiding unnecessary testing. One study found that a low bleeding score (≤3) and a normal activated partial thromboplastin time (aPTT) had 99.6% negative predictive value for the diagnosis of VWD. Bleeding symptoms that appear to be more common in patients with bleeding disorders include prolonged bleeding with surgery, dental procedures and extractions, and/or trauma, menorrhagia or postpartum hemorrhage, and large bruises (often described with lumps).
Easy bruising and menorrhagia are common complaints in patients with and without bleeding disorders. Easy bruising can also be a sign of medical conditions in which there is no identifiable coagulopathy; instead, the conditions are caused by an abnormality of blood vessels or their supporting tissues. In Ehlers-Danlos syndrome, there may be posttraumatic bleeding and a history of joint hyperextensibility. Cushing’s syndrome, chronic steroid use, and aging result in changes in skin and subcutaneous tissue, and subcutaneous bleeding occurs in response to minor trauma. The latter has been termed senile purpura.
Epistaxis is a common symptom, particularly in children and in dry climates, and may not reflect an underlying bleeding disorder. However, it is the most common symptom in hereditary hemorrhagic telangiectasia and in boys with VWD. Clues that epistaxis is a symptom of an underlying bleeding disorder include lack of seasonal variation and bleeding that requires medical evaluation or treatment, including cauterization. Bleeding with eruption of primary teeth is seen in children with more severe bleeding disorders, such as moderate and severe hemophilia. It is uncommon in children with mild bleeding disorders. Patients with disorders of primary hemostasis (platelet adhesion) may have increased bleeding after dental cleanings and other procedures that involve gum manipulation.
Menorrhagia is defined quantitatively as a loss of >80 mL of blood per cycle, based on the quantity of blood loss required to produce iron-deficiency anemia. A complaint of heavy menses is subjective and has a poor correlation with excessive blood loss. Predictors of menorrhagia include bleeding resulting in iron-deficiency anemia or a need for blood transfusion, passage of clots >1 inch in diameter, and changing a pad or tampon more than hourly. Menorrhagia is a common symptom in women with underlying bleeding disorders and is reported in the majority of women with VWD, women with factor XI deficiency, and symptomatic carriers of hemophilia. Women with underlying bleeding disorders are more likely to have other bleeding symptoms, including bleeding after dental extractions, postoperative bleeding, and postpartum bleeding, and are much more likely to have menorrhagia beginning at menarche than women with menorrhagia due to other causes.
Postpartum hemorrhage (PPH) is a common symptom in women with underlying bleeding disorders. In women with type 1 VWD and symptomatic carriers of hemophilia A in whom levels of VWF and factor VIII usually normalize during pregnancy, PPH may be delayed. Women with a history of PPH have a high risk of recurrence with subsequent pregnancies. Rupture of ovarian cysts with intraabdominal hemorrhage has also been reported in women with underlying bleeding disorders.
Tonsillectomy is a major hemostatic challenge, because intact hemostatic mechanisms are essential to prevent excessive bleeding from the tonsillar bed. Bleeding may occur early after surgery or after approximately 7 days postoperatively, with loss of the eschar at the operative site. Similar delayed bleeding is seen after colonic polyp resection. Gastrointestinal (GI) bleeding and hematuria are usually due to underlying pathology, and procedures to identify and treat the bleeding site should be undertaken, even in patients with known bleeding disorders. VWD, particularly types 2 and 3, has been associated with angiodysplasia of the bowel and GI bleeding.
Hemarthroses and spontaneous muscle hematomas are characteristic of moderate or severe congenital factor VIII or IX deficiency. They can also be seen in moderate and severe deficiencies of fibrinogen, prothrombin, and factors V, VII, and X. Spontaneous hemarthroses occur rarely in other bleeding disorders except for severe VWD, with associated factor VIII levels <5%. Muscle and soft tissue bleeds are also common in acquired factor VIII deficiency. Bleeding into a joint results in severe pain and swelling, as well as loss of function, but is rarely associated with discoloration from bruising around the joint. Life-threatening sites of bleeding include bleeding into the oropharynx, where bleeding can obstruct the airway, into the central nervous system, and into the retroperitoneum. Central nervous system bleeding is the major cause of bleeding-related deaths in patients with severe congenital factor deficiencies.
Prohemorrhagic Effects of Medications and Dietary Supplements Aspirin and other nonsteroidal anti-inflammatory drugs (NSAIDs) that inhibit cyclooxygenase 1 impair primary hemostasis and may exacerbate bleeding from another cause or even unmask a previously occult mild bleeding disorder such as VWD. All NSAIDs, however, can precipitate GI bleeding, which may be more severe in patients with underlying bleeding disorders. The aspirin effect on platelet function as assessed by aggregometry can persist for up to 7 days, although it has frequently returned to normal by 3 days after the last dose. The effect of other NSAIDs is shorter, as the inhibitor effect is reversed when the drug is removed. Thienopyridines (clopidogrel and prasugrel) inhibit ADP-mediated platelet aggregation and, like NSAIDs, can precipitate or exacerbate bleeding symptoms.
Many herbal supplements can impair hemostatic function (Table 78-2). Some are more convincingly associated with a bleeding risk than others. Fish oil or concentrated omega-3 fatty acid supplements impair platelet function. They alter platelet biochemistry to produce more PGI3, a more potent platelet inhibitor than prostacyclin (PGI2), and more thromboxane A3, a less potent platelet activator than thromboxane A2. In fact, diets naturally rich in omega-3 fatty acids can result in a prolonged bleeding time and abnormal platelet aggregation studies, but the actual associated bleeding risk is unclear. Vitamin E appears to inhibit protein kinase C–mediated platelet aggregation and nitric oxide production. In patients with unexplained bruising or bleeding, it is prudent to review any new medications or supplements and discontinue those that may be associated with bleeding.
|
HERBAL SUPPLEMENTS ASSOCIATED WITH INCREASED BLEEDING |
Underlying Systemic Diseases That Cause or Exacerbate a Bleeding Tendency Acquired bleeding disorders are commonly secondary to, or associated with, systemic disease. The clinical evaluation of a patient with a bleeding tendency must therefore include a thorough assessment for evidence of underlying disease. Bruising or mucosal bleeding may be the presenting complaint in liver disease, severe renal impairment, hypothyroidism, paraproteinemias or amyloidosis, and conditions causing bone marrow failure. All coagulation factors are synthesized in the liver, and hepatic failure results in combined factor deficiencies. This is often compounded by thrombocytopenia from splenomegaly due to portal hypertension. Coagulation factors II, VII, IX, and × and proteins C, S, and Z are dependent on vitamin K for posttranslational modification. Although vitamin K is required in both procoagulant and anticoagulant processes, the phenotype of vitamin K deficiency or the warfarin effect on coagulation is bleeding.
The normal blood platelet count is 150,000–450,000/μL. Thrombocytopenia results from decreased production, increased destruction, and/or sequestration. Although the bleeding risk varies somewhat by the reason for the thrombocytopenia, bleeding rarely occurs in isolated thrombocytopenia at counts <50,000/μL and usually not until <10,000–20,000/μL. Coexisting coagulopathies, as is seen in liver failure or disseminated coagulation; infection; platelet-inhibitory drugs; and underlying medical conditions can all increase the risk of bleeding in the thrombocytopenic patient. Most procedures can be performed in patients with a platelet count of 50,000/μL. The level needed for major surgery will depend on the type of surgery and the patient’s underlying medical state, although a count of approximately 80,000/μL is likely sufficient.
HISTORY OF THROMBOSIS
The risk of thrombosis, like that of bleeding, is influenced by both genetic and environmental influences. The major risk factor for arterial thrombosis is atherosclerosis, whereas for venous thrombosis, the risk factors are immobility, surgery, underlying medical conditions such as malignancy, medications such as hormonal therapy, obesity, and genetic predispositions. Factors that increase risks for venous and for both venous and arterial thromboses are shown in Table 78-3.
|
RISK FACTORS FOR THROMBOSIS |
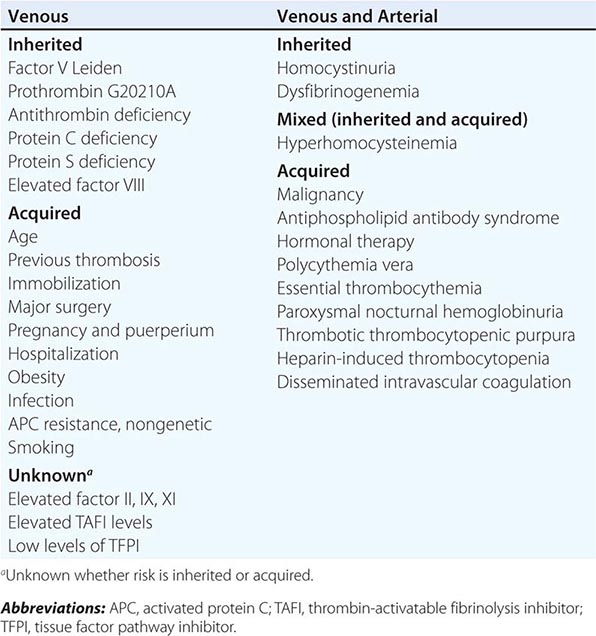
The most important point in a history related to venous thrombosis is determining whether the thrombotic event was idiopathic (meaning there was no clear precipitating factor) or was a precipitated event. In patients without underlying malignancy, having an idiopathic event is the strongest predictor of recurrence of VTE. In patients who have a vague history of thrombosis, a history of being treated with warfarin suggests a past DVT. Age is an important risk factor for venous thrombosis—the risk of DVT increases per decade, with an approximate incidence of 1/100,000 per year in early childhood to 1/200 per year among octogenarians. Family history is helpful in determining if there is a genetic predisposition and how strong that predisposition appears to be. A genetic thrombophilia that confers a relatively small increased risk, such as being a heterozygote for the prothrombin G20210A or factor V Leiden mutation, may be a minor determinant of risk in an elderly individual undergoing a high-risk surgical procedure. As illustrated in Fig. 78-5, a thrombotic event usually has more than one contributing factor. Predisposing factors must be carefully assessed to determine the risk of recurrent thrombosis and, with consideration of the patient’s bleeding risk, determine the length of anticoagulation. Similar consideration should be given in determining the need, if any, to test the patient and family members for thrombophilias.
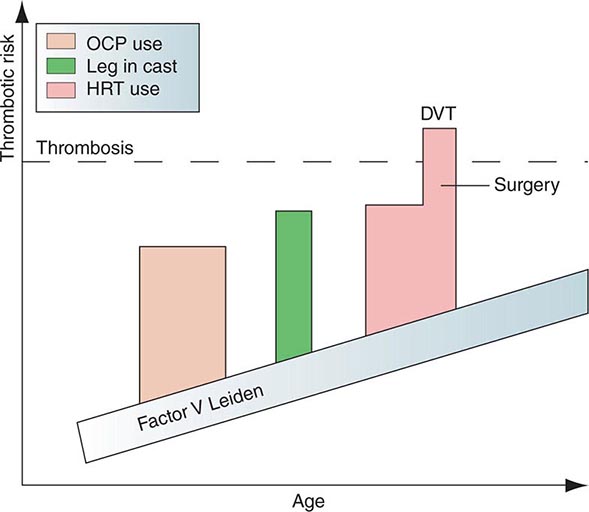
FIGURE 78-5 Thrombotic risk over time. Shown schematically is an individual’s thrombotic risk over time. An underlying factor V Leiden mutation provides a “theoretically” constant increased risk. The thrombotic risk increases with age and, intermittently, with oral contraceptive (OCP) or hormone replacement therapy (HRT) use; other events may increase the risk further. At some point, the cumulative risk may increase to the threshold for thrombosis and result in deep venous thrombosis (DVT). Note: The magnitude and duration of risk portrayed in the figure are meant for example only and may not precisely reflect the relative risk determined by clinical study. (From BA Konkle, A Schafer, in DP Zipes et al [eds]: Braunwald’s Heart Disease, 7th ed. Philadelphia, Saunders, 2005; modified with permission from FR Rosendaal: Venous thrombosis: A multicausal disease. Lancet 353:1167, 1999.)
LABORATORY EVALUATION
Careful history taking and clinical examination are essential components in the assessment of bleeding and thrombotic risk. The use of laboratory tests of coagulation complement, but cannot substitute for, clinical assessment. No test exists that provides a global assessment of hemostasis. The bleeding time has been used to assess bleeding risk; however, it does not predict bleeding risk with surgery and it is not recommended for this indication. The PFA-100, an instrument that measures platelet-dependent coagulation under flow conditions, is more sensitive and specific for VWD than the bleeding time; however, it is not sensitive enough to rule out mild bleeding disorders. PFA-100 closure times are prolonged in patients with some, but not all, inherited platelet disorders. Also, its utility in predicting bleeding risk has not been determined.
For routine preoperative and preprocedure testing, an abnormal prothrombin time (PT) may detect liver disease or vitamin K deficiency that had not been previously appreciated. Studies have not confirmed the usefulness of an aPTT in preoperative evaluations in patients with a negative bleeding history. The primary use of coagulation testing should be to confirm the presence and type of bleeding disorder in a patient with a suspicious clinical history.
Because of the nature of coagulation assays, proper sample acquisition and handling is critical to obtaining valid results. In patients with abnormal coagulation assays who have no bleeding history, repeat studies with attention to these factors frequently results in normal values. Most coagulation assays are performed in sodium citrate anticoagulated plasma that is recalcified for the assay. Because the anticoagulant is in liquid solution and needs to be added to blood in proportion to the plasma volume, incorrectly filled or inadequately mixed blood collection tubes will give erroneous results. Vacutainer tubes should be filled to >90% of the recommended fill, which is usually denoted by a line on the tube. An elevated hematocrit (>55%) can result in a false value due to a decreased plasma-to-anticoagulant ratio.
Screening Assays The most commonly used screening tests are the PT, aPTT, and platelet count. The PT assesses the factors I (fibrinogen), II (prothrombin), V, VII, and × (Fig. 78-6). The PT measures the time for clot formation of the citrated plasma after recalcification and addition of thromboplastin, a mixture of TF and phospholipids. The sensitivity of the assay varies by the source of thromboplastin. The relationship between defects in secondary hemostasis (fibrin formation) and coagulation test abnormalities is shown in Table 78-4. To adjust for this variability, the overall sensitivity of different thromboplastins to reduction of the vitamin K–dependent clotting factors II, VII, IX, and × in anticoagulation patients is now expressed as the International Sensitivity Index (ISI). An inverse relationship exists between ISI and thromboplastin sensitivity. The international normalized ratio (INR) is then determined based on the formula: INR = (PTpatient/PTnormal mean)ISI.
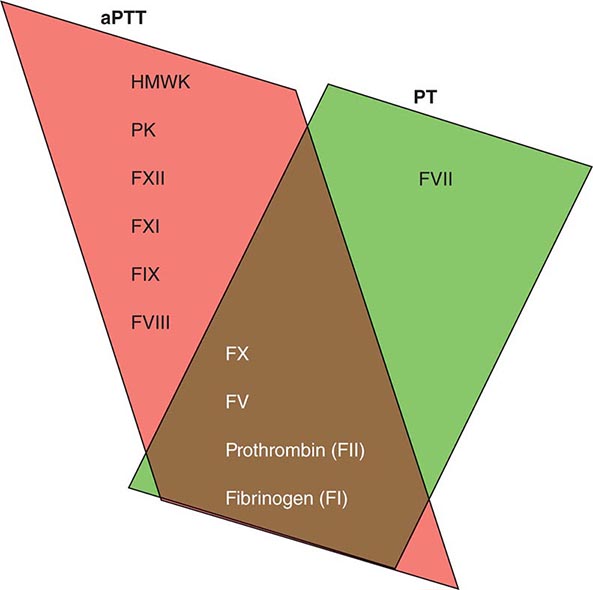
Figure 78-6 Coagulation factor activity tested in the activated partial thromboplastin time (aPTT) in red and prothrombin time (PT) in green, or both. F, factor; HMWK, high-molecular-weight kininogen; PK, prekallikrein.
|
HEMOSTATIC DISORDERS AND COAGULATION TEST ABNORMALITIES |
The INR was developed to assess stable anticoagulation due to reduction of vitamin K–dependent coagulation factors; it is commonly used in the evaluation of patients with liver disease. Although it does allow comparison between laboratories, reagent sensitivity as used to determine the ISI is not the same in liver disease as with warfarin anticoagulation. In addition, progressive liver failure is associated with variable changes in coagulation factors; the degree of prolongation of either the PT or the INR only roughly predicts the bleeding risk. Thrombin generation has been shown to be normal in many patients with mild to moderate liver dysfunction. Because the PT only measures one aspect of hemostasis affected by liver dysfunction, we likely overestimate the bleeding risk of a mildly elevated INR in this setting.
The aPTT assesses the intrinsic and common coagulation pathways; factors XI, IX, VIII, X, V, and II; fibrinogen; prekallikrein; high-molecular-weight kininogen; and factor XII (Fig. 78-6). The aPTT reagent contains phospholipids derived from either animal or vegetable sources that function as a platelet substitute in the coagulation pathways and includes an activator of the intrinsic coagulation system, such as nonparticulate ellagic acid or the particulate activators kaolin, celite, or micronized silica.
The phospholipid composition of aPTT reagents varies, which influences the sensitivity of individual reagents to clotting factor deficiencies and to inhibitors such as heparin and lupus anticoagulants. Thus, aPTT results will vary from one laboratory to another, and the normal range in the laboratory where the testing occurs should be used in the interpretation. Local laboratories can relate their aPTT values to the therapeutic heparin anticoagulation by correlating aPTT values with direct measurements of heparin activity (anti-Xa or protamine titration assays) in samples from heparinized patients, although correlation between these assays is often poor. The aPTT reagent will vary in sensitivity to individual factor deficiencies and usually becomes prolonged with individual factor deficiencies of 30–50%.
Mixing Studies Mixing studies are used to evaluate a prolonged aPTT or, less commonly PT, to distinguish between a factor deficiency and an inhibitor. In this assay, normal plasma and patient plasma are mixed in a 1:1 ratio, and the aPTT or PT is determined immediately and after incubation at 37°C for varying times, typically 30, 60, and/or 120 min. With isolated factor deficiencies, the aPTT will correct with mixing and stay corrected with incubation. With aPTT prolongation due to a lupus anticoagulant, the mixing and incubation will show no correction. In acquired neutralizing factor antibodies, notably an acquired factor VIII inhibitor, the initial assay may or may not correct immediately after mixing but will prolong or remain prolonged with incubation at 37°C. Failure to correct with mixing can also be due to the presence of other inhibitors or interfering substances such as heparin, fibrin split products, and paraproteins.
Specific Factor Assays Decisions to proceed with specific clotting factor assays will be influenced by the clinical situation and the results of coagulation screening tests. Precise diagnosis and effective management of inherited and acquired coagulation deficiencies necessitate quantitation of the relevant factors. When bleeding is severe, specific assays are urgently required to guide appropriate therapy. Individual factor assays are usually performed as modifications of the mixing study, where the patient’s plasma is mixed with plasma deficient in the factor being studied. This will correct all factor deficiencies to >50%, thus making prolongation of clot formation due to a factor deficiency dependent on the factor missing from the added plasma.
Testing for Antiphospholipid Antibodies Antibodies to phospholipids (cardiolipin) or phospholipid-binding proteins (β2-microglobulin and others) are detected by enzyme-linked immunosorbent assay (ELISA). When these antibodies interfere with phospholipid-dependent coagulation tests, they are termed lupus anticoagulants. The aPTT has variability sensitivity to lupus anticoagulants, depending in part on the aPTT reagents used. An assay using a sensitive reagent has been termed an LA-PTT. The dilute Russell viper venom test (dRVVT) and the tissue thromboplastin inhibition (TTI) test are modifications of standard tests with the phospholipid reagent decreased, thus increasing the sensitivity to antibodies that interfere with the phospholipid component. The tests, however, are not specific for lupus anticoagulants, because factor deficiencies or other inhibitors will also result in prolongation. Documentation of a lupus anticoagulant requires not only prolongation of a phospholipid-dependent coagulation test but also lack of correction when mixed with normal plasma and correction with the addition of activated platelet membranes or certain phospholipids (e.g., hexagonal phase).
Other Coagulation Tests The thrombin time and the reptilase time measure fibrinogen conversion to fibrin and are prolonged when the fibrinogen level is low (usually <80–100 mg/dL) or qualitatively abnormal, as seen in inherited or acquired dysfibrinogenemias, or when fibrin/fibrinogen degradation products interfere. The thrombin time, but not the reptilase time, is prolonged in the presence of heparin. The thrombin time is markedly prolonged in the presence of the direct thrombin inhibitor, dabigatran; a dilute thrombin time can be used to assess drug activity. Measurement of anti–factor Xa plasma inhibitory activity is a test frequently used to assess low-molecular-weight heparin (LMWH) levels, as a direct measurement of unfractionated heparin (UFH) activity, or to assess activity of the new direct Xa inhibitors rivaroxaban or apixaban. Drug in the patient sample inhibits the enzymatic conversion of an Xa-specific chromogenic substrate to colored product by factor Xa. Standard curves are created using multiple concentrations of drug and are used to calculate the concentration of anti-Xa activity in the patient plasma.
Laboratory Testing for Thrombophilia Laboratory assays to detect thrombophilic states include molecular diagnostics and immunologic and functional assays. These assays vary in their sensitivity and specificity for the condition being tested. Furthermore, acute thrombosis, acute illnesses, inflammatory conditions, pregnancy, and medications affect levels of many coagulation factors and their inhibitors. Antithrombin is decreased by heparin and in the setting of acute thrombosis. Protein C and S levels may be increased in the setting of acute thrombosis and are decreased by warfarin. Antiphospholipid antibodies are frequently transiently positive in acute illness. Testing for genetic thrombophilias should, in general, only be performed when there is a strong family history of thrombosis and results would affect clinical decision making.
Because thrombophilia evaluations are usually performed to assess the need to extend anticoagulation, testing should be performed in a steady state, remote from the acute event. In most instances, warfarin anticoagulation can be stopped after the initial 3–6 months of treatment, and testing can be performed at least 3 weeks later. As a sensitive marker of coagulation activation, the quantitative D-dimer assay, drawn 4 weeks after stopping anticoagulation, can be used to stratify risk of recurrent thrombosis in patients who have an idiopathic event.
Measures of Platelet Function The bleeding time has been used to assess bleeding risk; however, it has not been found to predict bleeding risk with surgery, and it is not recommended for use for this indication. The PFA-100 and similar instruments that measure platelet-dependent coagulation under flow conditions are generally more sensitive and specific for platelet disorders and VWD than the bleeding time; however, data are insufficient to support their use to predict bleeding risk or monitor response to therapy, and they will be normal in some patients with platelet disorders or mild VWD. When they are used in the evaluation of a patient with bleeding symptoms, abnormal results, as with the bleeding time, require specific testing, such as VWF assays and/or platelet aggregation studies. Because all of these “screening” assays may miss patients with mild bleeding disorders, further studies are needed to define their role in hemostasis testing.
For classic platelet aggregometry, various agonists are added to the patient’s platelet-rich plasma and platelet aggregation is measured. Tests of platelet secretion in response to agonists can also be measured. These tests are affected by many factors, including numerous medications, and the association between minor defects in aggregation or secretion in these assays and bleeding risk is not clearly established.
ACKNOWLEDGMENT
Robert I. Handin, MD, contributed this chapter in the 16th edition, and some material from that chapter has been retained here.
79 |
Enlargement of Lymph Nodes and Spleen |
This chapter is intended to serve as a guide to the evaluation of patients who present with enlargement of the lymph nodes (lymphadenopathy) or the spleen (splenomegaly). Lymphadenopathy is a rather common clinical finding in primary care settings, whereas palpable splenomegaly is less so.
LYMPHADENOPATHY
Lymphadenopathy may be an incidental finding in patients being examined for various reasons, or it may be a presenting sign or symptom of the patient’s illness. The physician must eventually decide whether the lymphadenopathy is a normal finding or one that requires further study, up to and including biopsy. Soft, flat, submandibular nodes (<1 cm) are often palpable in healthy children and young adults; healthy adults may have palpable inguinal nodes of up to 2 cm, which are considered normal. Further evaluation of these normal nodes is not warranted. In contrast, if the physician believes the node(s) to be abnormal, then pursuit of a more precise diagnosis is needed.
SPLENOMEGALY
STRUCTURE AND FUNCTION OF THE SPLEEN
The spleen is a reticuloendothelial organ that has its embryologic origin in the dorsal mesogastrium at about 5 weeks of gestation. It arises in a series of hillocks, migrates to its normal adult location in the left upper quadrant (LUQ), and is attached to the stomach via the gastrolienal ligament and to the kidney via the lienorenal ligament. When the hillocks fail to unify into a single tissue mass, accessory spleens may develop in around 20% of persons. The function of the spleen has been elusive. Galen believed it was the source of “black bile” or melancholia, and the word hypochondria (literally, beneath the ribs) and the idiom “to vent one’s spleen” attest to the beliefs that the spleen had an important influence on the psyche and emotions. In humans, its normal physiologic roles seem to be the following:
1. Maintenance of quality control over erythrocytes in the red pulp by removal of senescent and defective red blood cells. The spleen accomplishes this function through a unique organization of its parenchyma and vasculature (Fig. 79-1).
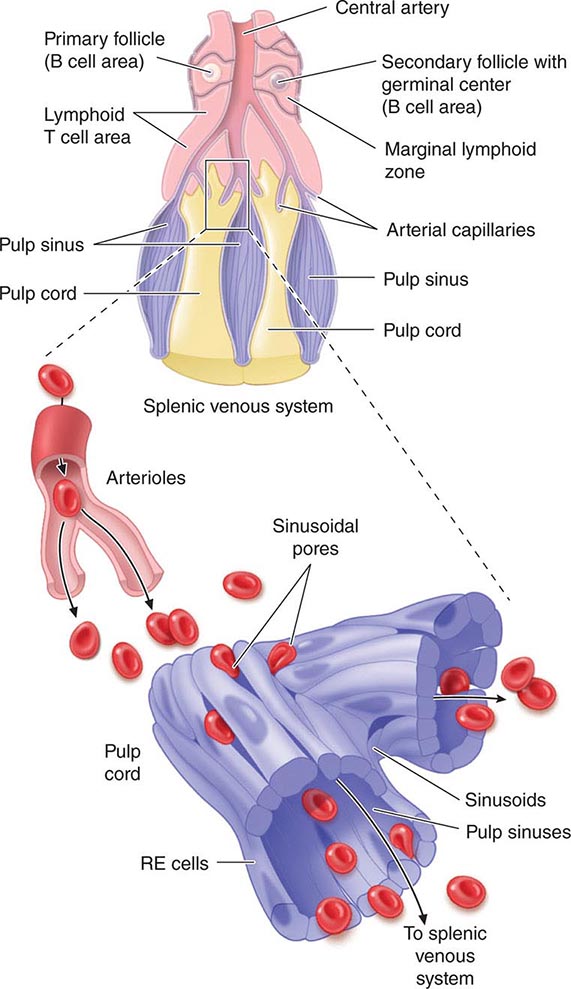
FIGURE 79-1 Schematic spleen structure. The spleen comprises many units of red and white pulp centered around small branches of the splenic artery, called central arteries. White pulp is lymphoid in nature and contains B cell follicles, a marginal zone around the follicles, and T cell–rich areas sheathing arterioles. The red pulp areas include pulp sinuses and pulp cords. The cords are dead ends. In order to regain access to the circulation, red blood cells must traverse tiny openings in the sinusoidal lining. Stiff, damaged, or old red cells cannot enter the sinuses. RE, reticuloendothelial. (Bottom portion of figure from RS Hillman, KA Ault: Hematology in Clinical Practice, 4th ed. New York, McGraw-Hill, 2005.)
2. Synthesis of antibodies in the white pulp.
3. The removal of antibody-coated bacteria and antibody-coated blood cells from the circulation.
An increase in these normal functions may result in splenomegaly.
The spleen is composed of red pulp and white pulp, which are Malpighi’s terms for the red blood–filled sinuses and reticuloendothelial cell–lined cords and the white lymphoid follicles arrayed within the red pulp matrix. The spleen is in the portal circulation. The reason for this is unknown but may relate to the fact that lower blood pressure allows less rapid flow and minimizes damage to normal erythrocytes. Blood flows into the spleen at a rate of about 150 mL/min through the splenic artery, which ultimately ramifies into central arterioles. Some blood goes from the arterioles to capillaries and then to splenic veins and out of the spleen, but the majority of blood from central arterioles flows into the macrophage-lined sinuses and cords. The blood entering the sinuses reenters the circulation through the splenic venules, but the blood entering the cords is subjected to an inspection of sorts. To return to the circulation, the blood cells in the cords must squeeze through slits in the cord lining to enter the sinuses that lead to the venules. Old and damaged erythrocytes are less deformable and are retained in the cords, where they are destroyed and their components recycled. Red cell–inclusion bodies such as parasites (Chaps. 248 and 250e), nuclear residua (Howell-Jolly bodies, see Fig. 77-6), or denatured hemoglobin (Heinz bodies) are pinched off in the process of passing through the slits, a process called pitting. The culling of dead and damaged cells and the pitting of cells with inclusions appear to occur without significant delay because the blood transit time through the spleen is only slightly slower than in other organs.
The spleen is also capable of assisting the host in adapting to its hostile environment. It has at least three adaptive functions: (1) clearance of bacteria and particulates from the blood, (2) the generation of immune responses to certain pathogens, and (3) the generation of cellular components of the blood under circumstances in which the marrow is unable to meet the needs (i.e., extramedullary hematopoiesis). The latter adaptation is a recapitulation of the blood-forming function the spleen plays during gestation. In some animals, the spleen also serves a role in the vascular adaptation to stress because it stores red blood cells (often hemoconcentrated to higher hematocrits than normal) under normal circumstances and contracts under the influence of β-adrenergic stimulation to provide the animal with an autotransfusion and improved oxygen-carrying capacity. However, the normal human spleen does not sequester or store red blood cells and does not contract in response to sympathetic stimuli. The normal human spleen contains approximately one-third of the total body platelets and a significant number of marginated neutrophils. These sequestered cells are available when needed to respond to bleeding or infection.
APPROACH TO THE PATIENT:
Splenomegaly
CLINICAL ASSESSMENT
The most common symptoms produced by diseases involving the spleen are pain and a heavy sensation in the LUQ. Massive splenomegaly may cause early satiety. Pain may result from acute swelling of the spleen with stretching of the capsule, infarction, or inflammation of the capsule. For many years, it was believed that splenic infarction was clinically silent, which, at times, is true. However, Soma Weiss, in his classic 1942 report of the self-observations by a Harvard medical student on the clinical course of subacute bacterial endocarditis, documented that severe LUQ and pleuritic chest pain may accompany thromboembolic occlusion of splenic blood flow. Vascular occlusion, with infarction and pain, is commonly seen in children with sickle cell crises. Rupture of the spleen, from either trauma or infiltrative disease that breaks the capsule, may result in intraperitoneal bleeding, shock, and death. The rupture itself may be painless.
A palpable spleen is the major physical sign produced by diseases affecting the spleen and suggests enlargement of the organ. The normal spleen weighs <250 g, decreases in size with age, normally lies entirely within the rib cage, has a maximum cephalocaudad diameter of 13 cm by ultrasonography or maximum length of 12 cm and/or width of 7 cm by radionuclide scan, and is usually not palpable. However, a palpable spleen was found in 3% of 2200 asymptomatic, male, freshman college students. Follow-up at 3 years revealed that 30% of those students still had a palpable spleen without any increase in disease prevalence. Ten-year follow-up found no evidence for lymphoid malignancies. Furthermore, in some tropical countries (e.g., New Guinea), the incidence of splenomegaly may reach 60%. Thus, the presence of a palpable spleen does not always equate with presence of disease. Even when disease is present, splenomegaly may not reflect the primary disease but rather a reaction to it. For example, in patients with Hodgkin’s disease, only two-thirds of the palpable spleens show involvement by the cancer.
Physical examination of the spleen uses primarily the techniques of palpation and percussion. Inspection may reveal fullness in the LUQ that descends on inspiration, a finding associated with a massively enlarged spleen. Auscultation may reveal a venous hum or friction rub.
Palpation can be accomplished by bimanual palpation, ballotment, and palpation from above (Middleton maneuver). For bimanual palpation, which is at least as reliable as the other techniques, the patient is supine with flexed knees. The examiner’s left hand is placed on the lower rib cage and pulls the skin toward the costal margin, allowing the fingertips of the right hand to feel the tip of the spleen as it descends while the patient inspires slowly, smoothly, and deeply. Palpation is begun with the right hand in the left lower quadrant with gradual movement toward the left costal margin, thereby identifying the lower edge of a massively enlarged spleen. When the spleen tip is felt, the finding is recorded as centimeters below the left costal margin at some arbitrary point, i.e., 10–15 cm, from the midpoint of the umbilicus or the xiphisternal junction. This allows other examiners to compare findings or the initial examiner to determine changes in size over time. Bimanual palpation in the right lateral decubitus position adds nothing to the supine examination.
Percussion for splenic dullness is accomplished with any of three techniques described by Nixon, Castell, or Barkun:
1. Nixon’s method: The patient is placed on the right side so that the spleen lies above the colon and stomach. Percussion begins at the lower level of pulmonary resonance in the posterior axillary line and proceeds diagonally along a perpendicular line toward the lower midanterior costal margin. The upper border of dullness is normally 6–8 cm above the costal margin. Dullness >8 cm in an adult is presumed to indicate splenic enlargement.
2. Castell’s method: With the patient supine, percussion in the lowest intercostal space in the anterior axillary line (eighth or ninth) produces a resonant note if the spleen is normal in size. This is true during expiration or full inspiration. A dull percussion note on full inspiration suggests splenomegaly.
3. Percussion of Traube’s semilunar space: The borders of Traube’s space are the sixth rib superiorly, the left midaxillary line laterally, and the left costal margin inferiorly. The patient is supine with the left arm slightly abducted. During normal breathing, this space is percussed from medial to lateral margins, yielding a normal resonant sound. A dull percussion note suggests splenomegaly.
Studies comparing methods of percussion and palpation with a standard of ultrasonography or scintigraphy have revealed sensitivity of 56–71% for palpation and 59–82% for percussion. Reproducibility among examiners is better for palpation than percussion. Both techniques are less reliable in obese patients or patients who have just eaten. Thus, the physical examination techniques of palpation and percussion are imprecise at best. It has been suggested that the examiner perform percussion first and, if positive, proceed to palpation; if the spleen is palpable, then one can be reasonably confident that splenomegaly exists. However, not all LUQ masses are enlarged spleens; gastric or colon tumors and pancreatic or renal cysts or tumors can mimic splenomegaly.
The presence of an enlarged spleen can be more precisely determined, if necessary, by liver-spleen radionuclide scan, CT, MRI, or ultrasonography. The latter technique is the current procedure of choice for routine assessment of spleen size (normal = a maximum cephalocaudad diameter of 13 cm) because it has high sensitivity and specificity and is safe, noninvasive, quick, mobile, and less costly. Nuclear medicine scans are accurate, sensitive, and reliable but are costly, require greater time to generate data, and use immobile equipment. They have the advantage of demonstrating accessory splenic tissue. CT and MRI provide accurate determination of spleen size, but the equipment is immobile and the procedures are expensive. MRI appears to offer no advantage over CT. Changes in spleen structure such as mass lesions, infarcts, inhomogeneous infiltrates, and cysts are more readily assessed by CT, MRI, or ultrasonography. None of these techniques is very reliable in the detection of patchy infiltration (e.g., Hodgkin’s disease).
DIFFERENTIAL DIAGNOSIS
Many of the diseases associated with splenomegaly are listed in Table 79-2. They are grouped according to the presumed basic mechanisms responsible for organ enlargement:
|
DISEASES ASSOCIATED WITH SPLENOMEGALY GROUPED BY PATHOGENIC MECHANISM |
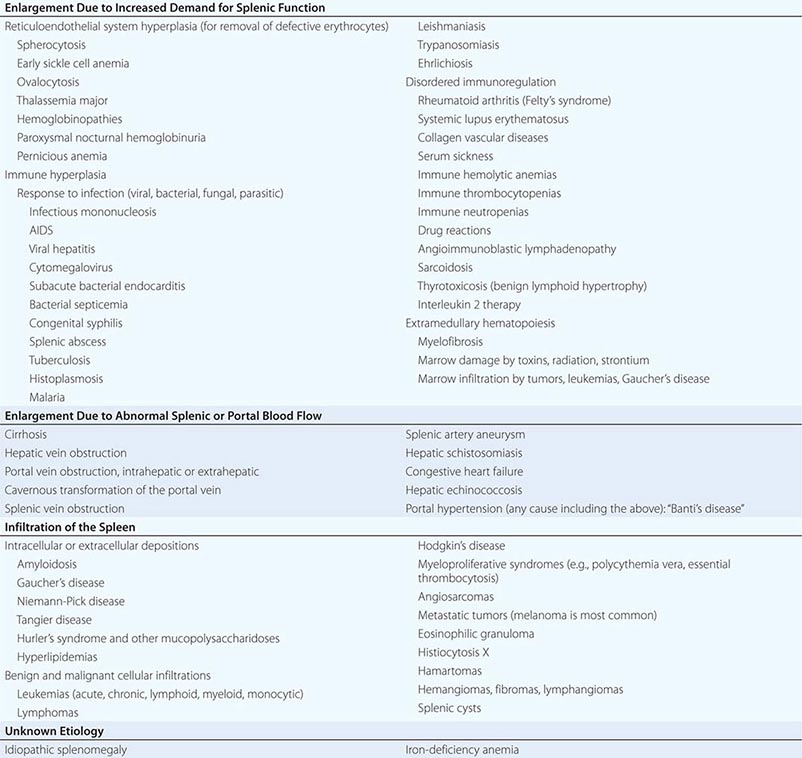
1. Hyperplasia or hypertrophy related to a particular splenic function such as reticuloendothelial hyperplasia (work hypertrophy) in diseases such as hereditary spherocytosis or thalassemia syndromes that require removal of large numbers of defective red blood cells; immune hyperplasia in response to systemic infection (infectious mononucleosis, subacute bacterial endocarditis) or to immunologic diseases (immune thrombocytopenia, SLE, Felty’s syndrome).
2. Passive congestion due to decreased blood flow from the spleen in conditions that produce portal hypertension (cirrhosis, Budd-Chiari syndrome, congestive heart failure).
3. Infiltrative diseases of the spleen (lymphomas, metastatic cancer, amyloidosis, Gaucher’s disease, myeloproliferative disorders with extramedullary hematopoiesis).
The differential diagnostic possibilities are much fewer when the spleen is “massively enlarged,” palpable more than 8 cm below the left costal margin or has a drained weight of ≥1000 g (Table 79-3). The vast majority of such patients will have non-Hodgkin’s lymphoma, chronic lymphocytic leukemia, hairy cell leukemia, chronic myeloid leukemia, myelofibrosis with myeloid metaplasia, or polycythemia vera.
|
DISEASES ASSOCIATED WITH MASSIVE SPLENOMEGALYa |

LABORATORY ASSESSMENT
The major laboratory abnormalities accompanying splenomegaly are determined by the underlying systemic illness. Erythrocyte counts may be normal, decreased (thalassemia major syndromes, SLE, cirrhosis with portal hypertension), or increased (polycythemia vera). Granulocyte counts may be normal, decreased (Felty’s syndrome, congestive splenomegaly, leukemias), or increased (infections or inflammatory disease, myeloproliferative disorders). Similarly, the platelet count may be normal, decreased when there is enhanced sequestration or destruction of platelets in an enlarged spleen (congestive splenomegaly, Gaucher’s disease, immune thrombocytopenia), or increased in the myeloproliferative disorders such as polycythemia vera.
The CBC may reveal cytopenia of one or more blood cell types, which should suggest hypersplenism. This condition is characterized by splenomegaly, cytopenia(s), normal or hyperplastic bone marrow, and a response to splenectomy. The latter characteristic is less precise because reversal of cytopenia, particularly granulocytopenia, is sometimes not sustained after splenectomy. The cytopenias result from increased destruction of the cellular elements secondary to reduced flow of blood through enlarged and congested cords (congestive splenomegaly) or to immune-mediated mechanisms. In hypersplenism, various cell types usually have normal morphology on the peripheral blood smear, although the red cells may be spherocytic due to loss of surface area during their longer transit through the enlarged spleen. The increased marrow production of red cells should be reflected as an increased reticulocyte production index, although the value may be less than expected due to increased sequestration of reticulocytes in the spleen.
The need for additional laboratory studies is dictated by the differential diagnosis of the underlying illness of which splenomegaly is a manifestation.
SPLENECTOMY
Splenectomy is infrequently performed for diagnostic purposes, especially in the absence of clinical illness or other diagnostic tests that suggest underlying disease. More often, splenectomy is performed for symptom control in patients with massive splenomegaly, for disease control in patients with traumatic splenic rupture, or for correction of cytopenias in patients with hypersplenism or immune-mediated destruction of one or more cellular blood elements. Splenectomy is necessary for staging of patients with Hodgkin’s disease only in those with clinical stage I or II disease in whom radiation therapy alone is contemplated as the treatment. Noninvasive staging of the spleen in Hodgkin’s disease is not a sufficiently reliable basis for treatment decisions because one-third of normal-sized spleens will be involved with Hodgkin’s disease and one-third of enlarged spleens will be tumor-free. The widespread use of systemic therapy to test all stages of Hodgkin’s disease has made staging laparotomy with splenectomy unnecessary. Although splenectomy in chronic myeloid leukemia (CML) does not affect the natural history of disease, removal of the massive spleen usually makes patients significantly more comfortable and simplifies their management by significantly reducing transfusion requirements. The improvements in therapy of CML have reduced the need for splenectomy for symptom control. Splenectomy is an effective secondary or tertiary treatment for two chronic B cell leukemias, hairy cell leukemia and prolymphocytic leukemia, and for the very rare splenic mantle cell or marginal zone lymphoma. Splenectomy in these diseases may be associated with significant tumor regression in bone marrow and other sites of disease. Similar regressions of systemic disease have been noted after splenic irradiation in some types of lymphoid tumors, especially chronic lymphocytic leukemia and prolymphocytic leukemia. This has been termed the abscopal effect. Such systemic tumor responses to local therapy directed at the spleen suggest that some hormone or growth factor produced by the spleen may affect tumor cell proliferation, but this conjecture is not yet substantiated. A common therapeutic indication for splenectomy is traumatic or iatrogenic splenic rupture. In a fraction of patients with splenic rupture, peritoneal seeding of splenic fragments can lead to splenosis—the presence of multiple rests of spleen tissue not connected to the portal circulation. This ectopic spleen tissue may cause pain or gastrointestinal obstruction, as in endometriosis. A large number of hematologic, immunologic, and congestive causes of splenomegaly can lead to destruction of one or more cellular blood elements. In the majority of such cases, splenectomy can correct the cytopenias, particularly anemia and thrombocytopenia. In a large series of patients seen in two tertiary care centers, the indication for splenectomy was diagnostic in 10% of patients, therapeutic in 44%, staging for Hodgkin’s disease in 20%, and incidental to another procedure in 26%. Perhaps the only contraindication to splenectomy is the presence of marrow failure, in which the enlarged spleen is the only source of hematopoietic tissue.
The absence of the spleen has minimal long-term effects on the hematologic profile. In the immediate postsplenectomy period, leukocytosis (up to 25,000/μL) and thrombocytosis (up to 1 × 106/μL) may develop, but within 2–3 weeks, blood cell counts and survival of each cell lineage are usually normal. The chronic manifestations of splenectomy are marked variation in size and shape of erythrocytes (anisocytosis, poikilocytosis) and the presence of Howell-Jolly bodies (nuclear remnants), Heinz bodies (denatured hemoglobin), basophilic stippling, and an occasional nucleated erythrocyte in the peripheral blood. When such erythrocyte abnormalities appear in a patient whose spleen has not been removed, one should suspect splenic infiltration by tumor that has interfered with its normal culling and pitting function.
The most serious consequence of splenectomy is increased susceptibility to bacterial infections, particularly those with capsules such as Streptococcus pneumoniae, Haemophilus influenzae, and some gram-negative enteric organisms. Patients under age 20 years are particularly susceptible to overwhelming sepsis with S. pneumoniae, and the overall actuarial risk of sepsis in patients who have had their spleens removed is about 7% in 10 years. The case–fatality rate for pneumococcal sepsis in splenectomized patients is 50–80%. About 25% of patients without spleens will develop a serious infection at some time in their life. The frequency is highest within the first 3 years after splenectomy. About 15% of the infections are polymicrobial, and lung, skin, and blood are the most common sites. No increased risk of viral infection has been noted in patients who have no spleen. The susceptibility to bacterial infections relates to the inability to remove opsonized bacteria from the bloodstream and a defect in making antibodies to T cell–independent antigens such as the polysaccharide components of bacterial capsules. Pneumococcal vaccine should be administered to all patients 2 weeks before elective splenectomy. The Advisory Committee on Immunization Practices recommends that these patients receive repeat vaccination 5 years after splenectomy. Efficacy has not been proven for this group, and the recommendation discounts the possibility that administration of the vaccine may actually lower the titer of specific pneumococcal antibodies. A more effective pneumococcal conjugate vaccine that involves T cells in the response is now available (Prevenar, 7-valent). The vaccine to Neisseria meningitidis should also be given to patients in whom elective splenectomy is planned. Although efficacy data for H. influenzae type b vaccine are not available for older children or adults, it may be given to patients who have had a splenectomy.
Splenectomized patients should be educated to consider any unexplained fever as a medical emergency. Prompt medical attention with evaluation and treatment of suspected bacteremia may be life-saving. Routine chemoprophylaxis with oral penicillin can result in the emergence of drug-resistant strains and is not recommended.
In addition to an increased susceptibility to bacterial infections, splenectomized patients are also more susceptible to the parasitic disease babesiosis. The splenectomized patient should avoid areas where the parasite Babesia is endemic (e.g., Cape Cod, MA).
Surgical removal of the spleen is an obvious cause of hyposplenism. Patients with sickle cell disease often suffer from autosplenectomy as a result of splenic destruction by the numerous infarcts associated with sickle cell crises during childhood. Indeed, the presence of a palpable spleen in a patient with sickle cell disease after age 5 suggests a coexisting hemoglobinopathy, e.g., thalassemia or hemoglobin C. In addition, patients who receive splenic irradiation for a neoplastic or autoimmune disease are also functionally hyposplenic. The term hyposplenism is preferred to asplenism in referring to the physiologic consequences of splenectomy because asplenia is a rare, specific, and fatal congenital abnormality in which there is a failure of the left side of the coelomic cavity (which includes the splenic anlagen) to develop normally. Infants with asplenia have no spleens, but that is the least of their problems. The right side of the developing embryo is duplicated on the left so there is liver where the spleen should be, there are two right lungs, and the heart comprises two right atria and two right ventricles.
80 |
Disorders of Granulocytes and Monocytes |
Leukocytes, the major cells comprising inflammatory and immune responses, include neutrophils, T and B lymphocytes, natural killer (NK) cells, monocytes, eosinophils, and basophils. These cells have specific functions, such as antibody production by B lymphocytes or destruction of bacteria by neutrophils, but in no single infectious disease is the exact role of the cell types completely established. Thus, whereas neutrophils are classically thought to be critical to host defense against bacteria, they may also play important roles in defense against viral infections.
The blood delivers leukocytes to the various tissues from the bone marrow, where they are produced. Normal blood leukocyte counts are 4.3–10.8 × 109/L, with neutrophils representing 45–74% of the cells, bands 0–4%, lymphocytes 16–45%, monocytes 4–10%, eosinophils 0–7%, and basophils 0–2%. Variation among individuals and among different ethnic groups can be substantial, with lower leukocyte numbers for certain African-American ethnic groups. The various leukocytes are derived from a common stem cell in the bone marrow. Three-fourths of the nucleated cells of bone marrow are committed to the production of leukocytes. Leukocyte maturation in the marrow is under the regulatory control of a number of different factors, known as colony-stimulating factors (CSFs) and interleukins (ILs). Because an alteration in the number and type of leukocytes is often associated with disease processes, total white blood cell (WBC) count (cells per μL) and differential counts are informative. This chapter focuses on neutrophils, monocytes, and eosinophils. Lymphocytes and basophils are discussed in Chaps. 372e and 376, respectively.
NEUTROPHILS
MATURATION
Important events in neutrophil life are summarized in Fig. 80-1. In normal humans, neutrophils are produced only in the bone marrow. The minimum number of stem cells necessary to support hematopoiesis is estimated to be 400–500 at any one time. Human blood monocytes, tissue macrophages, and stromal cells produce CSFs, hormones required for the growth of monocytes and neutrophils in the bone marrow. The hematopoietic system not only produces enough neutrophils (~1.3 × 1011 cells per 80-kg person per day) to carry out physiologic functions but also has a large reserve stored in the marrow, which can be mobilized in response to inflammation or infection. An increase in the number of blood neutrophils is called neutrophilia, and the presence of immature cells is termed a shift to the left. A decrease in the number of blood neutrophils is called neutropenia.
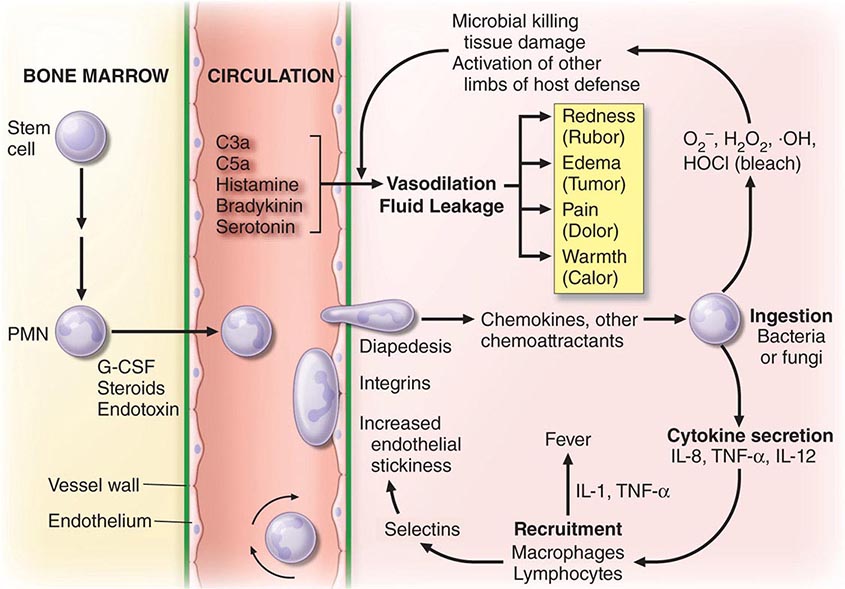
FIGURE 80-1 Schematic events in neutrophil production, recruitment, and inflammation. The four cardinal signs of inflammation (rubor, tumor, calor, dolor) are indicated, as are the interactions of neutrophils with other cells and cytokines. G-CSF, granulocyte colony-stimulating factor; IL, interleukin; PMN, polymorphonuclear leukocyte; TNF-α, tumor necrosis factor α.
Neutrophils and monocytes evolve from pluripotent stem cells under the influence of cytokines and CSFs (Fig. 80-2). The proliferation phase through the metamyelocyte takes about 1 week, while the maturation phase from metamyelocyte to mature neutrophil takes another week. The myeloblast is the first recognizable precursor cell and is followed by the promyelocyte. The promyelocyte evolves when the classic lysosomal granules, called the primary, or azurophil, granules are produced. The primary granules contain hydrolases, elastase, myeloperoxidase, cathepsin G, cationic proteins, and bactericidal/permeability-increasing protein, which is important for killing gram-negative bacteria. Azurophil granules also contain defensins, a family of cysteine-rich polypeptides with broad antimicrobial activity against bacteria, fungi, and certain enveloped viruses. The promyelocyte divides to produce the myelocyte, a cell responsible for the synthesis of the specific, or secondary, granules, which contain unique (specific) constituents such as lactoferrin, vitamin B12–binding protein, membrane components of the reduced nicotinamide-adenine dinucleotide phosphate (NADPH) oxidase required for hydrogen peroxide production, histaminase, and receptors for certain chemoattractants and adherence-promoting factors (CR3) as well as receptors for the basement membrane component, laminin. The secondary granules do not contain acid hydrolases and therefore are not classic lysosomes. Packaging of secondary granule contents during myelopoiesis is controlled by CCAAT/enhancer binding protein-ε. Secondary granule contents are readily released extracellularly, and their mobilization is important in modulating inflammation. During the final stages of maturation, no cell division occurs, and the cell passes through the metamyelocyte stage and then to the band neutrophil with a sausage-shaped nucleus (Fig. 80-3). As the band cell matures, the nucleus assumes a lobulated configuration. The nucleus of neutrophils normally contains up to four segments (Fig. 80-4). Excessive segmentation (more than five nuclear lobes) may be a manifestation of folate or vitamin B12 deficiency or the congenital neutropenia syndrome of warts, hypogammaglobulinemia, infections, and myelokathexis (WHIM) described below. The Pelger-Hüet anomaly (Fig. 80-5), an infrequent dominant benign inherited trait, results in neutrophils with distinctive bilobed nuclei that must be distinguished from band forms. Acquired bilobed nuclei, pseudo Pelger-Hüet anomaly, can occur with acute infections or in myelodysplastic syndromes. The physiologic role of the normal multilobed nucleus of neutrophils is unknown, but it may allow great deformation of neutrophils during migration into tissues at sites of inflammation.
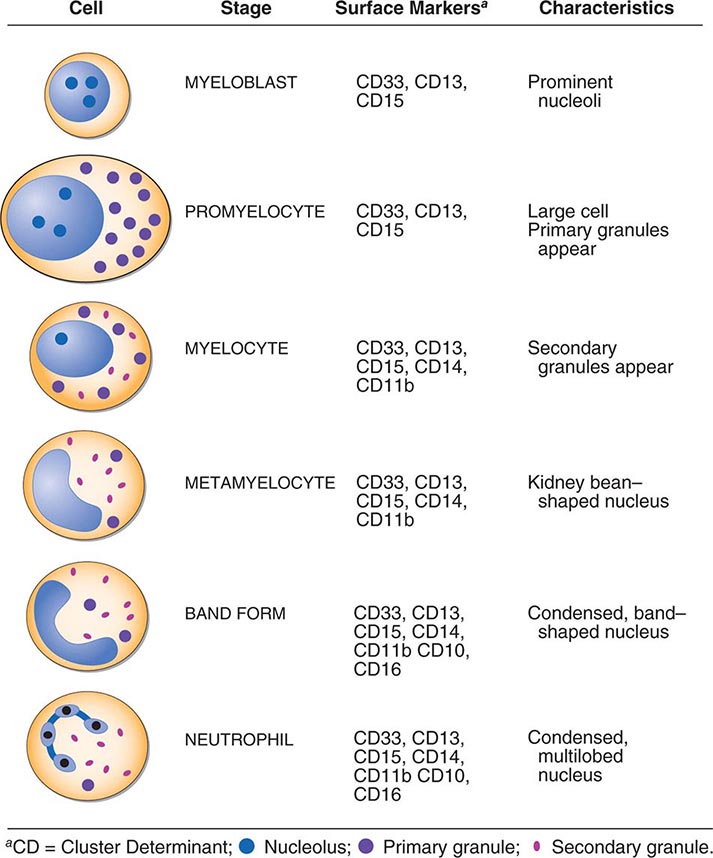
FIGURE 80-2 Stages of neutrophil development shown schematically. Granulocyte colony-stimulating factor (G-CSF) and granulocyte-macrophage colony-stimulating factor (GM-CSF) are critical to this process. Identifying cellular characteristics and specific cell-surface markers are listed for each maturational stage.
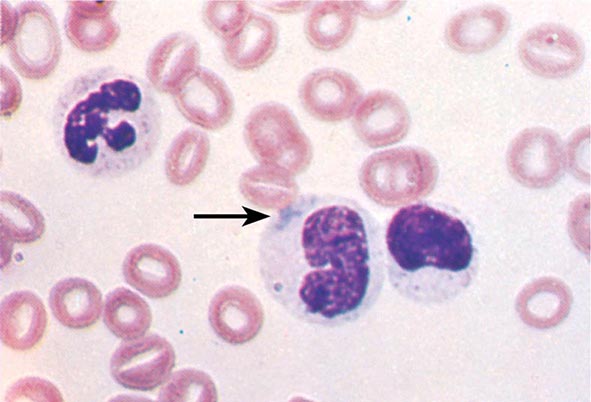
FIGURE 80-3 Neutrophil band with Döhle body. The neutrophil with a sausage-shaped nucleus in the center of the field is a band form. Döhle bodies are discrete, blue-staining, nongranular areas found in the periphery of the cytoplasm of the neutrophil in infections and other toxic states. They represent aggregates of rough endoplasmic reticulum.
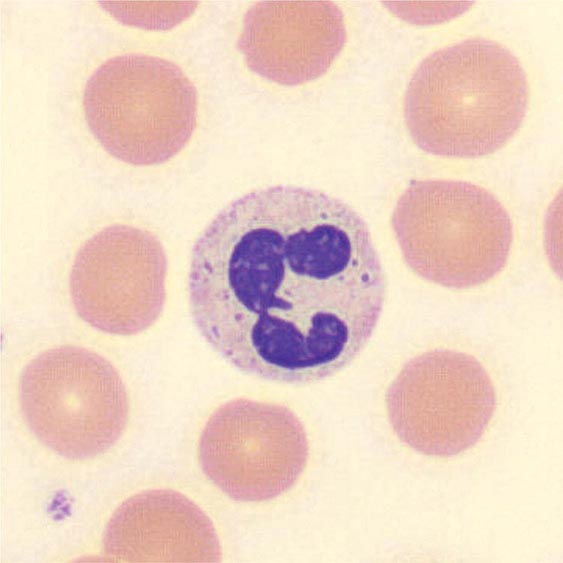
FIGURE 80-4 Normal granulocyte. The normal granulocyte has a segmented nucleus with heavy, clumped chromatin; fine neutrophilic granules are dispersed throughout the cytoplasm.
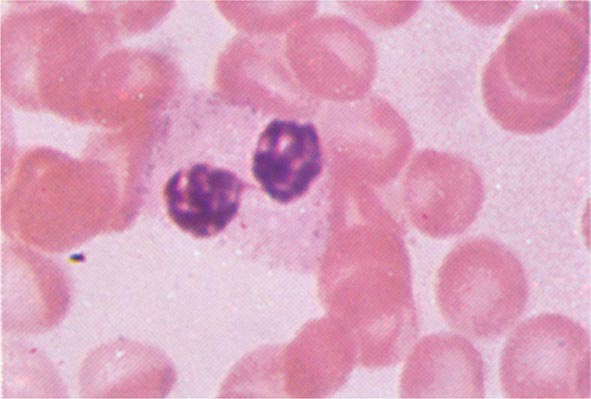
FIGURE 80-5 Pelger-Hüet anomaly. In this benign disorder, the majority of granulocytes are bilobed. The nucleus frequently has a spectacle-like, or “pince-nez,” configuration.
In severe acute bacterial infection, prominent neutrophil cytoplasmic granules, called toxic granulations, are occasionally seen. Toxic granulations are immature or abnormally staining azurophil granules. Cytoplasmic inclusions, also called Döhle bodies (Fig. 80-3), can be seen during infection and are fragments of ribosome-rich endoplasmic reticulum. Large neutrophil vacuoles are often present in acute bacterial infection and probably represent pinocytosed (internalized) membrane.
Neutrophils are heterogeneous in function. Monoclonal antibodies have been developed that recognize only a subset of mature neutrophils. The meaning of neutrophil heterogeneity is not known.
The morphology of eosinophils and basophils is shown in Fig. 80-6.
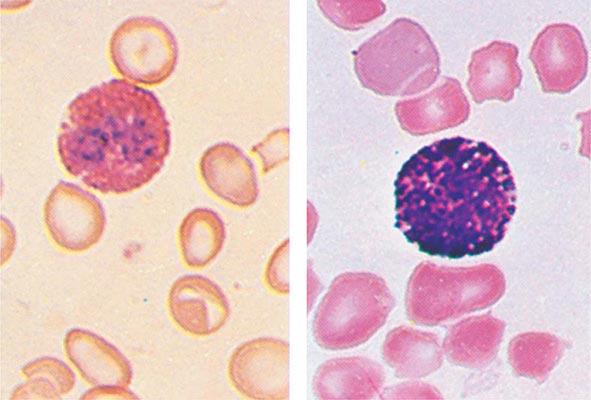
FIGURE 80-6 Normal eosinophil (left) and basophil (right). The eosinophil contains large, bright orange granules and usually a bilobed nucleus. The basophil contains large purple-black granules that fill the cell and obscure the nucleus.
MARROW RELEASE AND CIRCULATING COMPARTMENTS
Specific signals, including IL-1, tumor necrosis factor α (TNF-α), the CSFs, complement fragments, and chemokines, mobilize leukocytes from the bone marrow and deliver them to the blood in an unstimulated state. Under normal conditions, ~90% of the neutrophil pool is in the bone marrow, 2–3% in the circulation, and the remainder in the tissues (Fig. 80-7).
FIGURE 80-7 Schematic neutrophil distribution and kinetics between the different anatomic and functional pools.
The circulating pool exists in two dynamic compartments: one freely flowing and one marginated. The freely flowing pool is about one-half the neutrophils in the basal state and is composed of those cells that are in the blood and not in contact with the endothelium. Marginated leukocytes are those that are in close physical contact with the endothelium (Fig. 80-8). In the pulmonary circulation, where an extensive capillary bed (~1000 capillaries per alveolus) exists, margination occurs because the capillaries are about the same size as a mature neutrophil. Therefore, neutrophil fluidity and deformability are necessary to make the transit through the pulmonary bed. Increased neutrophil rigidity and decreased deformability lead to augmented neutrophil trapping and margination in the lung. In contrast, in the systemic postcapillary venules, margination is mediated by the interaction of specific cell-surface molecules called selectins. Selectins are glycoproteins expressed on neutrophils and endothelial cells, among others, that cause a low-affinity interaction, resulting in “rolling” of the neutrophil along the endothelial surface. On neutrophils, the molecule L-selectin (cluster determinant [CD] 62L) binds to glycosylated proteins on endothelial cells (e.g., glycosylation-dependent cell adhesion molecule [GlyCAM1] and CD34). Glycoproteins on neutrophils, most importantly sialyl-Lewisx (SLex, CD15s), are targets for binding of selectins expressed on endothelial cells (E-selectin [CD62E] and P-selectin [CD62P]) and other leukocytes. In response to chemotactic stimuli from injured tissues (e.g., complement product C5a, leukotriene B4, IL-8) or bacterial products (e.g., N-formylmethionylleucylphenylalanine [f-met-leu-phe]), neutrophil adhesiveness increases through mobilization of intracellular adhesion proteins stored in specific granules to the cell surface, and the cells “stick” to the endothelium through integrins. The integrins are leukocyte glycoproteins that exist as complexes of a common CD18 β chain with CD11a (LFA-1), CD11b (called Mac-1, CR3, or the C3bi receptor), and CD11c (called p150,95 or CR4). CD11a/CD18 and CD11b/CD18 bind to specific endothelial receptors (intercellular adhesion molecules [ICAM] 1 and 2).
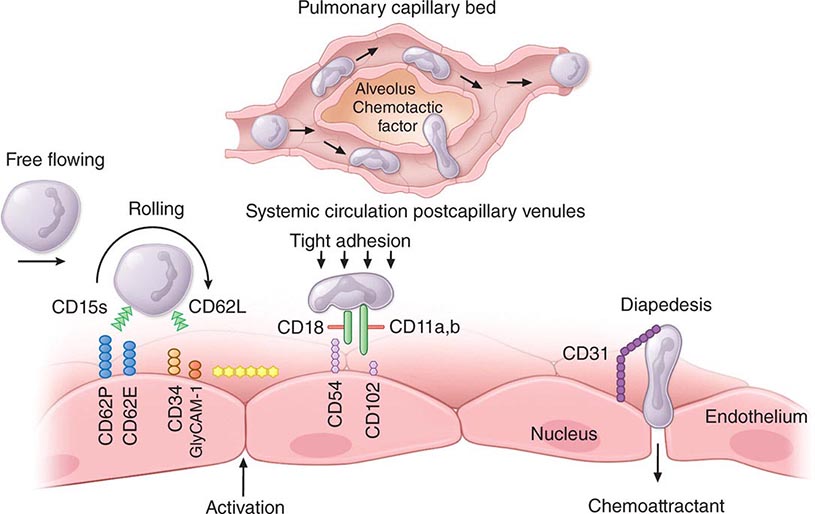
FIGURE 80-8 Neutrophil travel through the pulmonary capillaries is dependent on neutrophil deformability. Neutrophil rigidity (e.g., caused by C5a) enhances pulmonary trapping and response to pulmonary pathogens in a way that is not so dependent on cell-surface receptors. Intraalveolar chemotactic factors, such as those caused by certain bacteria (e.g., Streptococcus pneumoniae), lead to diapedesis of neutrophils from the pulmonary capillaries into the alveolar space. Neutrophil interaction with the endothelium of the systemic postcapillary venules is dependent on molecules of attachment. The neutrophil “rolls” along the endothelium using selectins: neutrophil CD15s (sialyl-Lewisx) binds to CD62E (E-selectin) and CD62P (P-selectin) on endothelial cells; CD62L (L-selectin) on neutrophils binds to CD34 and other molecules (e.g., GlyCAM-1) expressed on endothelium. Chemokines or other activation factors stimulate integrin-mediated “tight adhesion”: CD11a/CD18 (LFA-1) and CD11b/CD18 (Mac-1, CR3) bind to CD54 (ICAM-1) and CD102 (ICAM-2) on the endothelium. Diapedesis occurs between endothelial cells: CD31 (PECAM-1) expressed by the emigrating neutrophil interacts with CD31 expressed at the endothelial cell-cell junction. CD, cluster determinant; GlyCAM, glycosylation-dependent cell adhesion molecule; ICAM, intercellular adhesion molecule; PECAM, platelet/endothelial cell adhesion molecule.
On cell stimulation, L-selectin is shed from neutrophils, and E-selectin increases in the blood, presumably because it is shed from endothelial cells; receptors for chemoattractants and opsonins are mobilized; and the phagocytes orient toward the chemoattractant source in the extravascular space, increase their motile activity (chemokinesis), and migrate directionally (chemotaxis) into tissues. The process of migration into tissues is called diapedesis and involves the crawling of neutrophils between postcapillary endothelial cells that open junctions between adjacent cells to permit leukocyte passage. Diapedesis involves platelet/endothelial cell adhesion molecule (PECAM) 1 (CD31), which is expressed on both the emigrating leukocyte and the endothelial cells. The endothelial responses (increased blood flow from increased vasodilation and permeability) are mediated by anaphylatoxins (e.g., C3a and C5a) as well as vasodilators such as histamine, bradykinin, serotonin, nitric oxide, vascular endothelial growth factor (VEGF), and prostaglandins E and I. Cytokines regulate some of these processes (e.g., TNF-α induction of VEGF, interferon [IFN] γ inhibition of prostaglandin E).
In the healthy adult, most neutrophils leave the body by migration through the mucous membrane of the gastrointestinal tract. Normally, neutrophils spend a short time in the circulation (half-life, 6–7 h). Senescent neutrophils are cleared from the circulation by macrophages in the lung and spleen. Once in the tissues, neutrophils release enzymes, such as collagenase and elastase, which may help establish abscess cavities. Neutrophils ingest pathogenic materials that have been opsonized by IgG and C3b. Fibronectin and the tetrapeptide tuftsin also facilitate phagocytosis.
With phagocytosis comes a burst of oxygen consumption and activation of the hexose-monophosphate shunt. A membrane-associated NADPH oxidase, consisting of membrane and cytosolic components, is assembled and catalyzes the univalent reduction of oxygen to superoxide anion, which is then converted by superoxide dismutase to hydrogen peroxide and other toxic oxygen products (e.g., hydroxyl radical). Hydrogen peroxide + chloride + neutrophil myeloperoxidase generate hypochlorous acid (bleach), hypochlorite, and chlorine. These products oxidize and halogenate microorganisms and tumor cells and, when uncontrolled, can damage host tissue. Strongly cationic proteins, defensins, elastase, cathepsins, and probably nitric oxide also participate in microbial killing. Lactoferrin chelates iron, an important growth factor for microorganisms, especially fungi. Other enzymes, such as lysozyme and acid proteases, help digest microbial debris. After 1–4 days in tissues, neutrophils die. The apoptosis of neutrophils is also cytokine-regulated; granulocyte colony-stimulating factor (G-CSF) and IFN-γ prolong their life span. Under certain conditions, such as in delayed-type hypersensitivity, monocyte accumulation occurs within 6–12 h of initiation of inflammation. Neutrophils, monocytes, microorganisms in various states of digestion, and altered local tissue cells make up the inflammatory exudate, pus. Myeloperoxidase confers the characteristic green color to pus and may participate in turning off the inflammatory process by inactivating chemoattractants and immobilizing phagocytic cells.
Neutrophils respond to certain cytokines (IFN-γ, granulocyte-macrophage colony-stimulating factor [GM-CSF], IL-8) and produce cytokines and chemotactic signals (TNF-α, IL-8, macrophage inflammatory protein [MIP] 1) that modulate the inflammatory response. In the presence of fibrinogen, f-met-leu-phe or leukotriene B4 induces IL-8 production by neutrophils, providing autocrine amplification of inflammation. Chemokines (chemoattractant cytokines) are small proteins produced by many different cell types, including endothelial cells, fibroblasts, epithelial cells, neutrophils, and monocytes, that regulate neutrophil, monocyte, eosinophil, and lymphocyte recruitment and activation. Chemokines transduce their signals through heterotrimeric G protein–linked receptors that have seven cell membrane–spanning domains, the same type of cell-surface receptor that mediates the response to the classic chemoattractants f-met-leu-phe and C5a. Four major groups of chemokines are recognized based on the cysteine structure near the N terminus: C, CC, CXC, and CXXXC. The CXC cytokines such as IL-8 mainly attract neutrophils; CC chemokines such as MIP-1 attract lymphocytes, monocytes, eosinophils, and basophils; the C chemokine lymphotactin is T cell tropic; the CXXXC chemokine fractalkine attracts neutrophils, monocytes, and T cells. These molecules and their receptors not only regulate the trafficking and activation of inflammatory cells, but specific chemokine receptors also serve as co-receptors for HIV infection (Chap. 226) and have a role in other viral infections such as West Nile infection and atherogenesis.
NEUTROPHIL ABNORMALITIES
Defects in the neutrophil life cycle can lead to dysfunction and compromised host defenses. Inflammation is often depressed, and the clinical result is often recurrent, severe bacterial and fungal infections. Aphthous ulcers of mucous membranes (gray ulcers without pus) and gingivitis and periodontal disease suggest a phagocytic cell disorder. Patients with congenital phagocyte defects can have infections within the first few days of life. Skin, ear, upper and lower respiratory tract, and bone infections are common. Sepsis and meningitis are rare. In some disorders, the frequency of infection is variable, and patients can go for months or even years without major infection. Aggressive management of these congenital diseases has extended the life span of patients well beyond 30 years.
Neutropenia The consequences of absent neutrophils are dramatic. Susceptibility to infectious diseases increases sharply when neutrophil counts fall below 1000 cells/μL. When the absolute neutrophil count (ANC; band forms and mature neutrophils combined) falls to <500 cells/μL, control of endogenous microbial flora (e.g., mouth, gut) is impaired; when the ANC is <200/μL, the local inflammatory process is absent. Neutropenia can be due to depressed production, increased peripheral destruction, or excessive peripheral pooling. A falling neutrophil count or a significant decrease in the number of neutrophils below steady-state levels, together with a failure to increase neutrophil counts in the setting of infection or other challenge, requires investigation. Acute neutropenia, such as that caused by cancer chemotherapy, is more likely to be associated with increased risk of infection than neutropenia of long duration (months to years) that reverses in response to infection or carefully controlled administration of endotoxin (see “Laboratory Diagnosis and Management,” below).
Some causes of inherited and acquired neutropenia are listed in Table 80-1. The most common neutropenias are iatrogenic, resulting from the use of cytotoxic or immunosuppressive therapies for malignancy or control of autoimmune disorders. These drugs cause neutropenia because they result in decreased production of rapidly growing progenitor (stem) cells of the marrow. Certain antibiotics such as chloramphenicol, trimethoprim-sulfamethoxazole, flucytosine, vidarabine, and the antiretroviral drug zidovudine may cause neutropenia by inhibiting proliferation of myeloid precursors. Azathioprine and 6-mercaptopurine are metabolized by the enzyme thiopurine methyltransferase (TMPT), hypofunctional polymorphisms in which are found in 11% of whites and can lead to accumulation of 6-thioguanine and profound marrow toxicity. The marrow suppression is generally dose-related and dependent on continued administration of the drug. Cessation of the offending agent and recombinant human G-CSF usually reverse these forms of neutropenia.
|
CAUSES OF NEUTROPENIA |
Another important mechanism for iatrogenic neutropenia is the effect of drugs that serve as immune haptens and sensitize neutrophils or neutrophil precursors to immune-mediated peripheral destruction. This form of drug-induced neutropenia can be seen within 7 days of exposure to the drug; with previous drug exposure, resulting in preexisting antibodies, neutropenia may occur a few hours after administration of the drug. Although any drug can cause this form of neutropenia, the most frequent causes are commonly used antibiotics, such as sulfa-containing compounds, penicillins, and cephalosporins. Fever and eosinophilia may also be associated with drug reactions, but often these signs are not present. Drug-induced neutropenia can be severe, but discontinuation of the sensitizing drug is sufficient for recovery, which is usually seen within 5–7 days and is complete by 10 days. Readministration of the sensitizing drug should be avoided, because abrupt neutropenia will often result. For this reason, diagnostic challenge should be avoided.
Autoimmune neutropenias caused by circulating antineutrophil antibodies are another form of acquired neutropenia that results in increased destruction of neutrophils. Acquired neutropenia may also be seen with viral infections, including infection with HIV. Acquired neutropenia may be cyclic in nature, occurring at intervals of several weeks. Acquired cyclic or stable neutropenia may be associated with an expansion of large granular lymphocytes (LGLs), which may be T cells, NK cells, or NK-like cells. Patients with large granular lymphocytosis may have moderate blood and bone marrow lymphocytosis, neutropenia, polyclonal hypergammaglobulinemia, splenomegaly, rheumatoid arthritis, and absence of lymphadenopathy. Such patients may have a chronic and relatively stable course. Recurrent bacterial infections are frequent. Benign and malignant forms of this syndrome occur. In some patients, a spontaneous regression has occurred even after 11 years, suggesting an immunoregulatory defect as the basis for at least one form of the disorder. Glucocorticoids, cyclosporine, and methotrexate are commonly used to manage these cytopenias.
Hereditary Neutropenias Hereditary neutropenias are rare and may manifest in early childhood as a profound constant neutropenia or agranulocytosis. Congenital forms of neutropenia include Kostmann’s syndrome (neutrophil count <100/μL), which is often fatal and due to mutations in the antiapoptosis gene HAX-1; severe chronic neutropenia (neutrophil count of 300–1500/μL) due to mutations in neutrophil elastase (ELANE); hereditary cyclic neutropenia, or, more appropriately, cyclic hematopoiesis, also due to mutations in neutrophil elastase (ELANE); the cartilage-hair hypoplasia syndrome due to mutations in the mitochondrial RNA-processing endoribonuclease RMRP; Shwachman-Diamond syndrome associated with pancreatic insufficiency due to mutations in the Shwachman-Bodian-Diamond syndrome gene SBDS; the WHIM (warts, hypogammaglobulinemia, infections, myelokathexis [retention of WBCs in the marrow]) syndrome, characterized by neutrophil hypersegmentation and bone marrow myeloid arrest due to mutations in the chemokine receptor CXCR4; and neutropenias associated with other immune defects, such as X-linked agammaglobulinemia, Wiskott-Aldrich syndrome, and CD40 ligand deficiency. Mutations in the G-CSF receptor can develop in severe congenital neutropenia and are linked to leukemia. Absence of both myeloid and lymphoid cells is seen in reticular dysgenesis, due to mutations in the nuclear genome-encoded mitochondrial enzyme adenylate kinase-2 (AK2).
Maternal factors can be associated with neutropenia in the newborn. Transplacental transfer of IgG directed against antigens on fetal neutrophils can result in peripheral destruction. Drugs (e.g., thiazides) ingested during pregnancy can cause neutropenia in the newborn by either depressed production or peripheral destruction.
In Felty’s syndrome—the triad of rheumatoid arthritis, splenomegaly, and neutropenia (Chap. 380)—spleen-produced antibodies can shorten neutrophil life span, while large granular lymphocytes can attack marrow neutrophil precursors. Splenectomy may increase the neutrophil count in Felty’s syndrome and lower serum neutrophil-binding IgG. Some Felty’s syndrome patients also have neutropenia associated with an increased number of LGLs. Splenomegaly with peripheral trapping and destruction of neutrophils is also seen in lysosomal storage diseases and in portal hypertension.
Neutrophilia Neutrophilia results from increased neutrophil production, increased marrow release, or defective margination (Table 80-2). The most important acute cause of neutrophilia is infection. Neutrophilia from acute infection represents both increased production and increased marrow release. Increased production is also associated with chronic inflammation and certain myeloproliferative diseases. Increased marrow release and mobilization of the marginated leukocyte pool are induced by glucocorticoids. Release of epinephrine, as with vigorous exercise, excitement, or stress, will demarginate neutrophils in the spleen and lungs and double the neutrophil count in minutes. Cigarette smoking can elevate neutrophil counts above the normal range. Leukocytosis with cell counts of 10,000–25,000/μL occurs in response to infection and other forms of acute inflammation and results from both release of the marginated pool and mobilization of marrow reserves. Persistent neutrophilia with cell counts of ≥30,000–50,000/μL is called a leukemoid reaction, a term often used to distinguish this degree of neutrophilia from leukemia. In a leukemoid reaction, the circulating neutrophils are usually mature and not clonally derived.
|
CAUSES OF NEUTROPHILIA |
Abbreviation: G-CSF, granulocyte colony-stimulating factor.
Abnormal Neutrophil Function Inherited and acquired abnormalities of phagocyte function are listed in Table 80-3. The resulting diseases are best considered in terms of the functional defects of adherence, chemotaxis, and microbicidal activity. The distinguishing features of the important inherited disorders of phagocyte function are shown in Table 80-4.
|
TYPES OF GRANULOCYTE AND MONOCYTE DISORDERS |
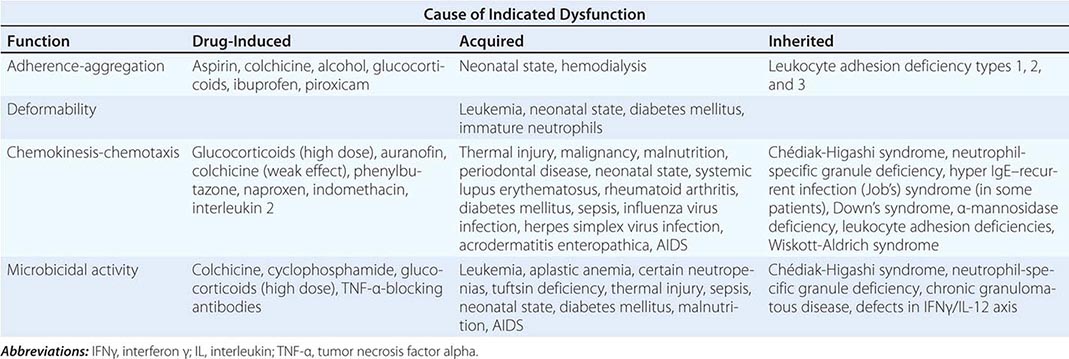
|
INHERITED DISORDERS OF PHAGOCYTE FUNCTION: DIFFERENTIAL FEATURES |
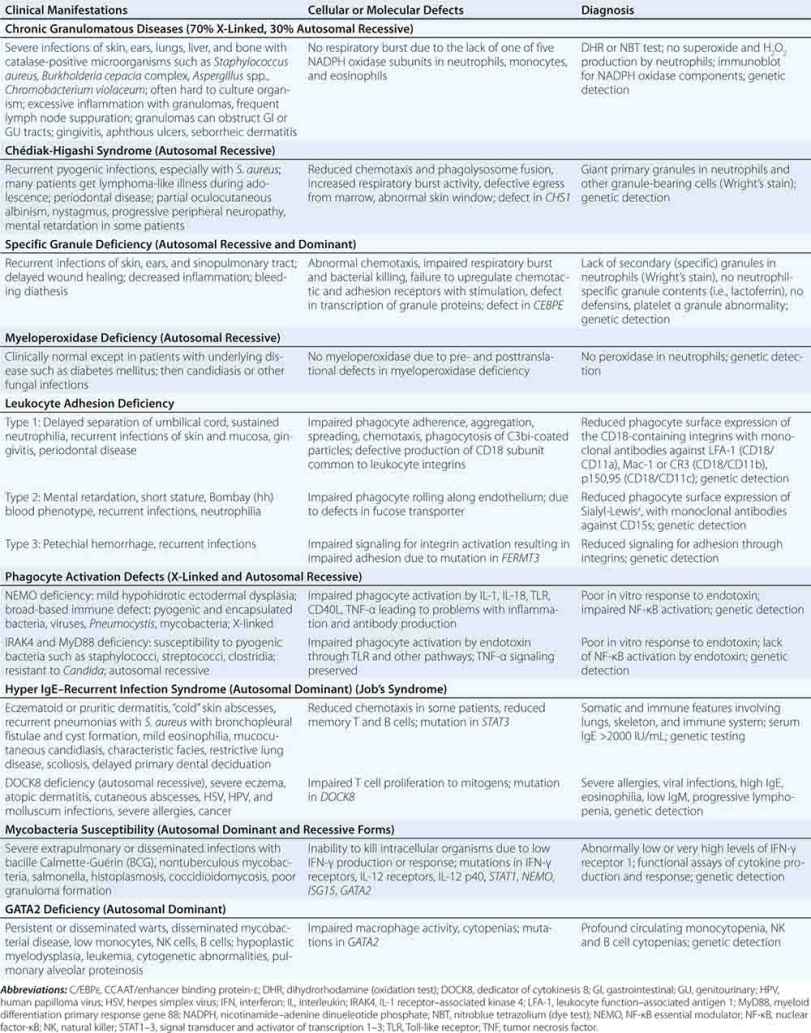
DISORDERS OF ADHESION Three main types of leukocyte adhesion deficiency (LAD) have been described. All are autosomal recessive and result in the inability of neutrophils to exit the circulation to sites of infection, leading to leukocytosis and increased susceptibility to infection (Fig. 80-8). Patients with LAD 1 have mutations in CD18, the common component of the integrins LFA-1, Mac-1, and p150,95, leading to a defect in tight adhesion between neutrophils and the endothelium. The heterodimer formed by CD18/CD11b (Mac-1) is also the receptor for the complement-derived opsonin C3bi (CR3). The CD18 gene is located on distal chromosome 21q. The severity of the defect determines the severity of clinical disease. Complete lack of expression of the leukocyte integrins results in a severe phenotype in which inflammatory stimuli do not increase the expression of leukocyte integrins on neutrophils or activated T and B cells. Neutrophils (and monocytes) from patients with LAD 1 adhere poorly to endothelial cells and protein-coated surfaces and exhibit defective spreading, aggregation, and chemotaxis. Patients with LAD 1 have recurrent bacterial infections involving the skin, oral and genital mucosa, and respiratory and intestinal tracts; persistent leukocytosis (resting neutrophil counts of 15,000–20,000/μL) because cells do not marginate; and, in severe cases, a history of delayed separation of the umbilical stump. Infections, especially of the skin, may become necrotic with progressively enlarging borders, slow healing, and development of dysplastic scars. The most common bacteria are Staphylococcus aureus and enteric gram-negative bacteria. LAD 2 is caused by an abnormality of fucosylation of SLex (CD15s), the ligand on neutrophils that interacts with selectins on endothelial cells and is responsible for neutrophil rolling along the endothelium. Infection susceptibility in LAD 2 appears to be less severe than in LAD 1. LAD 2 is also known as congenital disorder of glycosylation IIc (CDGIIc) due to mutation in a GDP-fucose transporter (SLC35C1). LAD 3 is characterized by infection susceptibility, leukocytosis, and petechial hemorrhage due to impaired integrin activation caused by mutations in the gene FERMT3.
DISORDERS OF NEUTROPHIL GRANULES The most common neutrophil defect is myeloperoxidase deficiency, a primary granule defect inherited as an autosomal recessive trait; the incidence is ~1 in 2000 persons. Isolated myeloperoxidase deficiency is not associated with clinically compromised defenses, presumably because other defense systems such as hydrogen peroxide generation are amplified. Microbicidal activity of neutrophils is delayed but not absent. Myeloperoxidase deficiency may make other acquired host defense defects more serious, and patients with myeloperoxidase deficiency and diabetes are more susceptible to Candida infections. An acquired form of myeloperoxidase deficiency occurs in myelomonocytic leukemia and acute myeloid leukemia.
Chédiak-Higashi syndrome (CHS) is a rare disease with autosomal recessive inheritance due to defects in the lysosomal transport protein LYST, encoded by the gene CHS1 at 1q42. This protein is required for normal packaging and disbursement of granules. Neutrophils (and all cells containing lysosomes) from patients with CHS characteristically have large granules (Fig. 80-9), making it a systemic disease. Patients with CHS have nystagmus, partial oculocutaneous albinism, and an increased number of infections resulting from many bacterial agents. Some CHS patients develop an “accelerated phase” in childhood with a hemophagocytic syndrome and an aggressive lymphoma requiring bone marrow transplantation. CHS neutrophils and monocytes have impaired chemotaxis and abnormal rates of microbial killing due to slow rates of fusion of the lysosomal granules with phagosomes. NK cell function is also impaired. CHS patients may develop a severe disabling peripheral neuropathy in adulthood that can lead to bed confinement.
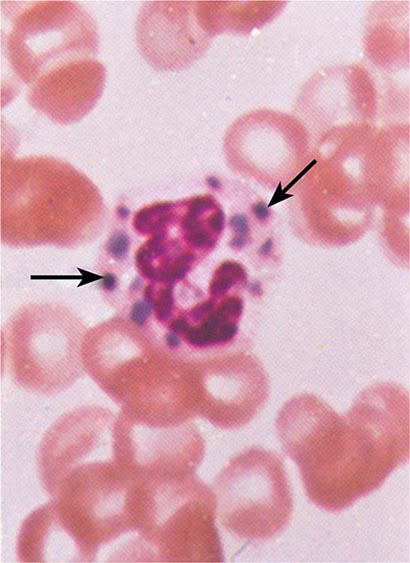
FIGURE 80-9 Chédiak-Higashi syndrome. The granulocytes contain huge cytoplasmic granules formed from aggregation and fusion of azurophilic and specific granules. Large abnormal granules are found in other granule-containing cells throughout the body.
Specific granule deficiency is a rare autosomal recessive disease in which the production of secondary granules and their contents, as well as the primary granule component defensins, is defective. The defect in killing leads to severe bacterial infections. One type of specific granule deficiency is due to a mutation in the CCAAT/enhancer binding protein-ε, a regulator of expression of granule components. A dominant mutation in C/EBP-ε has also been described.
CHRONIC GRANULOMATOUS DISEASE Chronic granulomatous disease (CGD) is a group of disorders of granulocyte and monocyte oxidative metabolism. Although CGD is rare, with an incidence of ~1 in 200,000 individuals, it is an important model of defective neutrophil oxidative metabolism. In about two-thirds of patients, CGD is inherited as an X-linked recessive trait; 30% of patients inherit the disease in an autosomal recessive pattern. Mutations in the genes for the five proteins that assemble at the plasma membrane account for all patients with CGD. Two proteins (a 91-kDa protein, abnormal in X-linked CGD, and a 22-kDa protein, absent in one form of autosomal recessive CGD) form the heterodimer cytochrome b-558 in the plasma membrane. Three other proteins (40, 47, and 67 kDa, abnormal in the other autosomal recessive forms of CGD) are cytoplasmic in origin and interact with the cytochrome after cell activation to form NADPH oxidase, required for hydrogen peroxide production. Leukocytes from patients with CGD have severely diminished hydrogen peroxide production. The genes involved in each of the defects have been cloned and sequenced and the chromosome locations identified. Patients with CGD characteristically have increased numbers of infections due to catalase-positive microorganisms (organisms that destroy their own hydrogen peroxide) such as S. aureus, Burkholderia cepacia, and Aspergillus species. When patients with CGD become infected, they often have extensive inflammatory reactions, and lymph node suppuration is common despite the administration of appropriate antibiotics. Aphthous ulcers and chronic inflammation of the nares are often present. Granulomas are frequent and can obstruct the gastrointestinal or genitourinary tracts. The excessive inflammation is due to failure to downregulate inflammation, reflecting failure to inhibit the synthesis of, degradation of, or response to chemoattractants or residual antigens, leading to persistent neutrophil accumulation. Impaired killing of intracellular microorganisms by macrophages may lead to persistent cell-mediated immune activation and granuloma formation. Autoimmune complications such as immune thrombocytopenic purpura and juvenile rheumatoid arthritis are also increased in CGD. In addition, for unexplained reasons, discoid lupus is more common in X-linked carriers. Late complications, including nodular regenerative hyperplasia and portal hypertension, are increasingly recognized in long-term survivors of severe CGD.
DISORDERS OF PHAGOCYTE ACTIVATION Phagocytes depend on cell-surface stimulation to induce signals that evoke multiple levels of the inflammatory response, including cytokine synthesis, chemotaxis, and antigen presentation. Mutations affecting the major pathway that signals through NF-κB have been noted in patients with a variety of infection susceptibility syndromes. If the defects are at a very late stage of signal transduction, in the protein critical for NF-κB activation known as the NF-κB essential modulator (NEMO), then affected males develop ectodermal dysplasia and severe immune deficiency with susceptibility to bacteria, fungi, mycobacteria, and viruses. If the defects in NF-κB activation are closer to the cell-surface receptors, in the proteins transducing Toll-like receptor signals, IL-1 receptor–associated kinase 4 (IRAK4), and myeloid differentiation primary response gene 88 (MyD88), then children have a marked susceptibility to pyogenic infections early in life but develop resistance to infection later.
MONONUCLEAR PHAGOCYTES
The mononuclear phagocyte system is composed of monoblasts, promonocytes, and monocytes, in addition to the structurally diverse tissue macrophages that make up what was previously referred to as the reticuloendothelial system. Macrophages are long-lived phagocytic cells capable of many of the functions of neutrophils. They are also secretory cells that participate in many immunologic and inflammatory processes distinct from neutrophils. Monocytes leave the circulation by diapedesis more slowly than neutrophils and have a half-life in the blood of 12–24 h.
After blood monocytes arrive in the tissues, they differentiate into macrophages (“big eaters”) with specialized functions suited for specific anatomic locations. Macrophages are particularly abundant in capillary walls of the lung, spleen, liver, and bone marrow, where they function to remove microorganisms and other noxious elements from the blood. Alveolar macrophages, liver Kupffer cells, splenic macrophages, peritoneal macrophages, bone marrow macrophages, lymphatic macrophages, brain microglial cells, and dendritic macrophages all have specialized functions. Macrophage-secreted products include lysozyme, neutral proteases, acid hydrolases, arginase, complement components, enzyme inhibitors (plasmin, α2-macroglobulin), binding proteins (transferrin, fibronectin, transcobalamin II), nucleosides, and cytokines (TNF-α; IL-1, -8, -12, -18). IL-1 (Chaps. 23 and 372e) has many functions, including initiating fever in the hypothalamus, mobilizing leukocytes from the bone marrow, and activating lymphocytes and neutrophils. TNF-α is a pyrogen that duplicates many of the actions of IL-1 and plays an important role in the pathogenesis of gram-negative shock (Chap. 325). TNF-α stimulates production of hydrogen peroxide and related toxic oxygen species by macrophages and neutrophils. In addition, TNF-α induces catabolic changes that contribute to the profound wasting (cachexia) associated with many chronic diseases.
Other macrophage-secreted products include reactive oxygen and nitrogen metabolites, bioactive lipids (arachidonic acid metabolites and platelet-activating factors), chemokines, CSFs, and factors stimulating fibroblast and vessel proliferation. Macrophages help regulate the replication of lymphocytes and participate in the killing of tumors, viruses, and certain bacteria (Mycobacterium tuberculosis and Listeria monocytogenes). Macrophages are key effector cells in the elimination of intracellular microorganisms. Their ability to fuse to form giant cells that coalesce into granulomas in response to some inflammatory stimuli is important in the elimination of intracellular microbes and is under the control of IFN-γ. Nitric oxide induced by IFN-γ is an important effector against intracellular parasites, including tuberculosis and Leishmania.
Macrophages play an important role in the immune response (Chap. 372e). They process and present antigen to lymphocytes and secrete cytokines that modulate and direct lymphocyte development and function. Macrophages participate in autoimmune phenomena by removing immune complexes and other substances from the circulation. Polymorphisms in macrophage receptors for immunoglobulin (FcγRII) determine susceptibility to some infections and autoimmune diseases. In wound healing, they dispose of senescent cells, and they contribute to atheroma development. Macrophage elastase mediates development of emphysema from cigarette smoking.
DISORDERS OF THE MONONUCLEAR PHAGOCYTE SYSTEM
Many disorders of neutrophils extend to mononuclear phagocytes. Monocytosis is associated with tuberculosis, brucellosis, subacute bacterial endocarditis, Rocky Mountain spotted fever, malaria, and visceral leishmaniasis (kala azar). Monocytosis also occurs with malignancies, leukemias, myeloproliferative syndromes, hemolytic anemias, chronic idiopathic neutropenias, and granulomatous diseases such as sarcoidosis, regional enteritis, and some collagen vascular diseases. Patients with LAD, hyperimmunoglobulin E–recurrent infection (Job’s) syndrome, CHS, and CGD all have defects in the mononuclear phagocyte system.
Monocyte cytokine production or response is impaired in some patients with disseminated nontuberculous mycobacterial infection who are not infected with HIV. Genetic defects in the pathways regulated by IFN-γ and IL-12 lead to impaired killing of intracellular bacteria, mycobacteria, salmonellae, and certain viruses (Fig. 80-10).
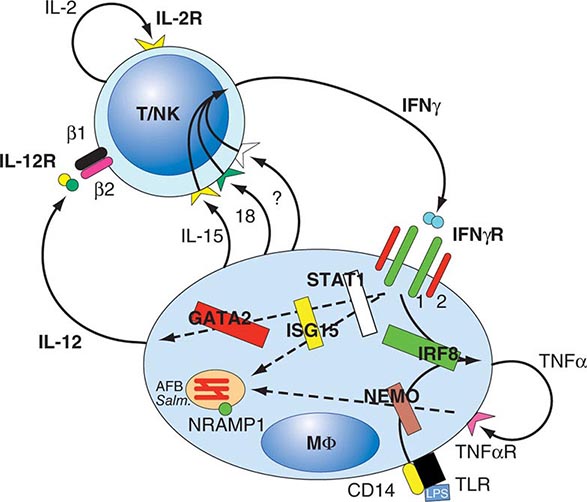
FIGURE 80-10 Lymphocyte-macrophage interactions underlying resistance to mycobacteria and other intracellular pathogens such as Salmonella, Histoplasma, and Coccidioides. Mycobacteria (and others) infect macrophages, leading to the production of IL-12, which activates T or NK cells through its receptor, leading to production of IL-2 and IFN-γ. IFN-γ acts through its receptor on macrophages to upregulate TNF-γ and IL-12 and kill intracellular pathogens. Other critical interacting molecules include signal transducer and activator of transcription 1 (STAT1), interferon regulatory factor 8 (IRF8), GATA2, and ISG15. Mutant forms of the cytokines and receptors shown in bold type have been found in severe cases of nontuberculous mycobacterial infection, salmonellosis and other intracellular pathogens. AFB, acid-fast bacilli; IFN, interferon; IL, interleukin; NEMO, nuclear factor-κB essential modulator; NK, natural killer; TLR, Toll-like receptor; TNF, tumor necrosis factor.
Certain viral infections impair mononuclear phagocyte function. For example, influenza virus infection causes abnormal monocyte chemotaxis. Mononuclear phagocytes can be infected by HIV using CCR5, the chemokine receptor that acts as a co-receptor with CD4 for HIV. T lymphocytes produce IFN-γ, which induces FcR expression and phagocytosis and stimulates hydrogen peroxide production by mononuclear phagocytes and neutrophils. In certain diseases, such as AIDS, IFN-γ production may be deficient, whereas in other diseases, such as T cell lymphomas, excessive release of IFN-γ may be associated with erythrophagocytosis by splenic macrophages.
Autoinflammatory diseases are characterized by abnormal cytokine regulation, leading to excess inflammation in the absence of infection. These diseases can mimic infectious or immunodeficient syndromes. Gain-of-function mutations in the TNF-α receptor cause TNF-α receptor–associated periodic syndrome (TRAPS), which is characterized by recurrent fever in the absence of infection, due to persistent stimulation of the TNF-α receptor (Chap. 392). Diseases with abnormal IL-1 regulation leading to fever include familial Mediterranean fever due to mutations in PYRIN. Mutations in cold-induced autoinflammatory syndrome 1 (CIAS1) lead to neonatal-onset multisystem autoinflammatory disease, familial cold urticaria, and Muckle-Wells syndrome. The syndrome of pyoderma gangrenosum, acne, and sterile pyogenic arthritis (PAPA syndrome) is caused by mutations in PSTPIP1. In contrast to these syndromes of overexpression of proinflammatory cytokines, blockade of TNF-α by the antagonists infliximab, adalimumab, certolizumab, golimumab, or etanercept has been associated with severe infections due to tuberculosis, nontuberculous mycobacteria, and fungi (Chap. 392).
Monocytopenia occurs with acute infections, with stress, and after treatment with glucocorticoids. Drugs that suppress neutrophil production in the bone marrow can cause monocytopenia. Persistent severe circulating monocytopenia is seen in GATA2 deficiency, even though macrophages are found at the sites of inflammation. Monocytopenia also occurs in aplastic anemia, hairy cell leukemia, acute myeloid leukemia, and as a direct result of myelotoxic drugs.
EOSINOPHILS
Eosinophils and neutrophils share similar morphology, many lysosomal constituents, phagocytic capacity, and oxidative metabolism. Eosinophils express a specific chemoattractant receptor and respond to a specific chemokine, eotaxin, but little is known about their required role. Eosinophils are much longer lived than neutrophils, and unlike neutrophils, tissue eosinophils can recirculate. During most infections, eosinophils appear unimportant. However, in invasive helminthic infections, such as hookworm, schistosomiasis, strongyloidiasis, toxocariasis, trichinosis, filariasis, echinococcosis, and cysticercosis, the eosinophil plays a central role in host defense. Eosinophils are associated with bronchial asthma, cutaneous allergic reactions, and other hypersensitivity states.
The distinctive feature of the red-staining (Wright’s stain) eosinophil granule is its crystalline core consisting of an arginine-rich protein (major basic protein) with histaminase activity, important in host defense against parasites. Eosinophil granules also contain a unique eosinophil peroxidase that catalyzes the oxidation of many substances by hydrogen peroxide and may facilitate killing of microorganisms.
Eosinophil peroxidase, in the presence of hydrogen peroxide and halide, initiates mast cell secretion in vitro and thereby promotes inflammation. Eosinophils contain cationic proteins, some of which bind to heparin and reduce its anticoagulant activity. Eosinophil-derived neurotoxin and eosinophil cationic protein are ribonucleases that can kill respiratory syncytial virus. Eosinophil cytoplasm contains Charcot-Leyden crystal protein, a hexagonal bipyramidal crystal first observed in a patient with leukemia and then in sputum of patients with asthma; this protein is lysophospholipase and may function to detoxify certain lysophospholipids.
Several factors enhance the eosinophil’s function in host defense. T cell–derived factors enhance the ability of eosinophils to kill parasites. Mast cell–derived eosinophil chemotactic factor of anaphylaxis (ECFa) increases the number of eosinophil complement receptors and enhances eosinophil killing of parasites. Eosinophil CSFs (e.g., IL-5) produced by macrophages increase eosinophil production in the bone marrow and activate eosinophils to kill parasites.
EOSINOPHILIA
Eosinophilia is the presence of >500 eosinophils per μL of blood and is common in many settings besides parasite infection. Significant tissue eosinophilia can occur without an elevated blood count. A common cause of eosinophilia is allergic reaction to drugs (iodides, aspirin, sulfonamides, nitrofurantoin, penicillins, and cephalosporins). Allergies such as hay fever, asthma, eczema, serum sickness, allergic vasculitis, and pemphigus are associated with eosinophilia. Eosinophilia also occurs in collagen vascular diseases (e.g., rheumatoid arthritis, eosinophilic fasciitis, allergic angiitis, and periarteritis nodosa) and malignancies (e.g., Hodgkin’s disease; mycosis fungoides; chronic myeloid leukemia; and cancer of the lung, stomach, pancreas, ovary, or uterus), as well as in Job’s syndrome, DOCK8 deficiency (see below), and CGD. Eosinophilia is commonly present in helminthic infections. IL-5 is the dominant eosinophil growth factor. Therapeutic administration of the cytokines IL-2 or GM-CSF frequently leads to transient eosinophilia. The most dramatic hypereosinophilic syndromes are Loeffler’s syndrome, tropical pulmonary eosinophilia, Loeffler’s endocarditis, eosinophilic leukemia, and idiopathic hypereosinophilic syndrome (50,000–100,000/μL). IL-5 is the dominant eosinophil growth factor and can be specifically inhibited with the monoclonal antibody mepolizumab.
The idiopathic hypereosinophilic syndrome represents a heterogeneous group of disorders with the common feature of prolonged eosinophilia of unknown cause and organ system dysfunction, including the heart, central nervous system, kidneys, lungs, gastrointestinal tract, and skin. The bone marrow is involved in all affected individuals, but the most severe complications involve the heart and central nervous system. Clinical manifestations and organ dysfunction are highly variable. Eosinophils are found in the involved tissues and likely cause tissue damage by local deposition of toxic eosinophil proteins such as eosinophil cationic protein and major basic protein. In the heart, the pathologic changes lead to thrombosis, endocardial fibrosis, and restrictive endomyocardiopathy. The damage to tissues in other organ systems is similar. Some cases are due to mutations involving the platelet-derived growth factor receptor, and these are extremely sensitive to the tyrosine kinase inhibitor imatinib. Glucocorticoids, hydroxyurea, and IFN-α each have been used successfully, as have therapeutic antibodies against IL-5. Cardiovascular complications are managed aggressively.
The eosinophilia-myalgia syndrome is a multisystem disease, with prominent cutaneous, hematologic, and visceral manifestations, that frequently evolves into a chronic course and can occasionally be fatal. The syndrome is characterized by eosinophilia (eosinophil count >1000/μL) and generalized disabling myalgias without other recognized causes. Eosinophilic fasciitis, pneumonitis, and myocarditis; neuropathy culminating in respiratory failure; and encephalopathy may occur. The disease is caused by ingesting contaminants in L-tryptophan–containing products. Eosinophils, lymphocytes, macrophages, and fibroblasts accumulate in the affected tissues, but their role in pathogenesis is unclear. Activation of eosinophils and fibroblasts and the deposition of eosinophil-derived toxic proteins in affected tissues may contribute. IL-5 and transforming growth factor β have been implicated as potential mediators. Treatment is withdrawal of products containing L-tryptophan and the administration of glucocorticoids. Most patients recover fully, remain stable, or show slow recovery, but the disease can be fatal in up to 5% of patients.
Eosinophilic neoplasms are discussed in Chapter 135e.
EOSINOPENIA
Eosinopenia occurs with stress, such as acute bacterial infection, and after treatment with glucocorticoids. The mechanism of eosinopenia of acute bacterial infection is unknown but is independent of endogenous glucocorticoids, because it occurs in animals after total adrenalectomy. There is no known adverse effect of eosinopenia.
HYPERIMMUNOGLOBULIN E–RECURRENT INFECTION SYNDROME
The hyperimmunoglobulin E–recurrent infection syndrome, or Job’s syndrome, is a rare multisystem disease in which the immune and somatic systems are affected, including neutrophils, monocytes, T cells, B cells, and osteoclasts. Autosomal dominant mutations in signal transducer and activator of transcription 3 (STAT3) lead to inhibition of normal STAT signaling with broad and profound effects. Patients have characteristic facies with broad nose, kyphoscoliosis, and eczema. The primary teeth erupt normally but do not deciduate, often requiring extraction. Patients develop recurrent sinopulmonary and cutaneous infections that tend to be much less inflamed than appropriate for the degree of infection and have been referred to as “cold abscesses.” Characteristically, pneumonias cavitate, leading to pneumatoceles. Coronary artery aneurysms are common, as are cerebral demyelinated plaques that accumulate with age. Importantly, IL-17–producing T cells, which are thought responsible for protection against extracellular and mucosal infections, are profoundly reduced in Job’s syndrome. Despite very high IgE levels, these patients do not have elevated levels of allergy. An important syndrome with clinical overlap with STAT3 deficiency is due to autosomal recessive defects in dedicator of cytokinesis 8 (DOCK8). In DOCK8 deficiency, IgE elevation is joined to severe allergy, viral susceptibility, and increased rates of cancer.
LABORATORY DIAGNOSIS AND MANAGEMENT
Initial studies of WBC and differential and often a bone marrow examination may be followed by assessment of bone marrow reserves (steroid challenge test), marginated circulating pool of cells (epinephrine challenge test), and marginating ability (endotoxin challenge test) (Fig. 80-7). In vivo assessment of inflammation is possible with a Rebuck skin window test or an in vivo skin blister assay, which measures the ability of leukocytes and inflammatory mediators to accumulate locally in the skin. In vitro tests of phagocyte aggregation, adherence, chemotaxis, phagocytosis, degranulation, and microbicidal activity (for S. aureus) may help pinpoint cellular or humoral lesions. Deficiencies of oxidative metabolism are detected with either the nitroblue tetrazolium (NBT) dye test or the dihydrorhodamine (DHR) oxidation test. These tests are based on the ability of products of oxidative metabolism to alter the oxidation states of reporter molecules so that they can be detected microscopically (NBT) or by flow cytometry (DHR). Qualitative studies of superoxide and hydrogen peroxide production may further define neutrophil oxidative function.
Patients with leukopenias or leukocyte dysfunction often have delayed inflammatory responses. Therefore, clinical manifestations may be minimal despite overwhelming infection, and unusual infections must always be suspected. Early signs of infection demand prompt, aggressive culturing for microorganisms, use of antibiotics, and surgical drainage of abscesses. Prolonged courses of antibiotics are often required. In patients with CGD, prophylactic antibiotics (trimethoprim-sulfamethoxazole) and antifungals (itraconazole) markedly diminish the frequency of life-threatening infections. Glucocorticoids may relieve gastrointestinal or genitourinary tract obstruction by granulomas in patients with CGD. Although TNF-α-blocking agents may markedly relieve inflammatory bowel symptoms, extreme caution must be exercised in their use in CGD inflammatory bowel disease, because it profoundly increases these patients’ already heightened susceptibility to infection. Recombinant human IFN-γ, which nonspecifically stimulates phagocytic cell function, reduces the frequency of infections in patients with CGD by 70% and reduces the severity of infection. This effect of IFN-γ in CGD is additive to the effect of prophylactic antibiotics. The recommended dose is 50 μg/m2 subcutaneously three times weekly. IFN-γ has also been used successfully in the treatment of leprosy, nontuberculous mycobacteria, and visceral leishmaniasis.
Rigorous oral hygiene reduces but does not eliminate the discomfort of gingivitis, periodontal disease, and aphthous ulcers; chlorhexidine mouthwash and tooth brushing with a hydrogen peroxide–sodium bicarbonate paste help many patients. Oral antifungal agents (fluconazole, itraconazole, voriconazole, posaconazole) have reduced mucocutaneous candidiasis in patients with Job’s syndrome. Androgens, glucocorticoids, lithium, and immunosuppressive therapy have been used to restore myelopoiesis in patients with neutropenia due to impaired production. Recombinant G-CSF is useful in the management of certain forms of neutropenia due to depressed neutrophil production, including those related to cancer chemotherapy. Patients with chronic neutropenia with evidence of a good bone marrow reserve need not receive prophylactic antibiotics. Patients with chronic or cyclic neutrophil counts <500/μL may benefit from prophylactic antibiotics and G-CSF during periods of neutropenia. Oral trimethoprim-sulfamethoxazole (160/800 mg) twice daily can prevent infection. Increased numbers of fungal infections are not seen in patients with CGD on this regimen. Oral quinolones such as levofloxacin and ciprofloxacin are alternatives.
In the setting of cytotoxic chemotherapy with severe, persistent lymphocyte dysfunction, trimethoprim-sulfamethoxazole prevents Pneumocystis jiroveci pneumonia. These patients, and patients with phagocytic cell dysfunction, should avoid heavy exposure to airborne soil, dust, or decaying matter (mulch, manure), which are often rich in Nocardia and the spores of Aspergillus and other fungi. Restriction of activities or social contact has no proven role in reducing risk of infection for phagocyte defects.
Although aggressive medical care for many patients with phagocytic disorders can allow them to go for years without a life-threatening infection, there may still be delayed effects of prolonged antimicrobials and other inflammatory complications. Cure of most congenital phagocyte defects is possible by bone marrow transplantation, and rates of success are improving (Chap. 139e). The identification of specific gene defects in patients with LAD 1, CGD, and other immunodeficiencies has led to gene therapy trials in a number of genetic white cell disorders.
81e |
Atlas of Hematology and Analysis of Peripheral Blood Smears |
Some of the relevant findings in peripheral blood, enlarged lymph nodes, and bone marrow are illustrated in this chapter. Systematic histologic examination of the bone marrow and lymph nodes is beyond the scope of a general medicine textbook. However, every internist should know how to examine a peripheral blood smear.
The examination of a peripheral blood smear is one of the most informative exercises a physician can perform. Although advances in automated technology have made the examination of a peripheral blood smear by a physician seem less important, the technology is not a completely satisfactory replacement for a blood smear interpretation by a trained medical professional who also knows the patient’s clinical history, family history, social history, and physical findings. It is useful to ask the laboratory to generate a Wright’s-stained peripheral blood smear and examine it.
The best place to examine blood cell morphology is the feathered edge of the blood smear where red cells lie in a single layer, side by side, just barely touching one another but not overlapping. The author’s approach is to look at the smallest cellular elements, the platelets, first and work his way up in size to red cells and then white cells.
Using an oil immersion lens that magnifies the cells 100-fold, one counts the platelets in five to six fields, averages the number per field, and multiplies by 20,000 to get a rough estimate of the platelet count. The platelets are usually 1–2 μm in diameter and have a blue granulated appearance. There is usually 1 platelet for every 20 or so red cells. Of course, the automated counter is much more accurate, but gross disparities between the automated and manual counts should be assessed. Large platelets may be a sign of rapid platelet turnover, as young platelets are often larger than old ones; alternatively, certain rare inherited syndromes can produce large platelets. Platelet clumping visible on the smear can be associated with falsely low automated platelet counts. Similarly, neutrophil fragmentation can be a source of falsely elevated automated platelet counts.
Next one examines the red blood cells. One can gauge their size by comparing the red cell to the nucleus of a small lymphocyte. Both are normally about 8 μm wide. Red cells that are smaller than the small lymphocyte nucleus may be microcytic; those larger than the small lymphocyte nucleus may be macrocytic. Macrocytic cells also tend to be more oval than spherical in shape and are sometimes called macroovalocytes. The automated mean corpuscular volume (MCV) can assist in making a classification. However, some patients may have both iron and vitamin B12 deficiency, which will produce an MCV in the normal range but wide variation in red cell size. When the red cells vary greatly in size, anisocytosis is said to be present. When the red cells vary greatly in shape, poikilocytosis is said to be present. The electronic cell counter provides an independent assessment of variability in red cell size. It measures the range of red cell volumes and reports the results as “red cell distribution width” (RDW). This value is calculated from the MCV; thus, cell width is not being measured but cell volume is. The term is derived from the curve displaying the frequency of cells at each volume, also called the distribution. The width of the red cell volume distribution curve is what determines the RDW. The RDW is calculated as follows: RDW = (standard deviation of MCV ÷ mean MCV) × 100. In the presence of morphologic anisocytosis, RDW (normally 11–14%) increases to 15–18%. The RDW is useful in at least two clinical settings. In patients with microcytic anemia, the differential diagnosis is generally between iron deficiency and thalassemia. In thalassemia, the small red cells are generally of uniform size with a normal small RDW. In iron deficiency, the size variability and the RDW are large. In addition, a large RDW can suggest a dimorphic anemia when a chronic atrophic gastritis can produce both vitamin B12 malabsorption to produce macrocytic anemia and blood loss to produce iron deficiency. In such settings, RDW is also large. An elevated RDW also has been reported as a risk factor for all-cause mortality in population-based studies (Patel KV et al: Arch Intern Med 169:515, 2009), a finding that is unexplained currently.
After red cell size is assessed, one examines the hemoglobin content of the cells. They are either normal in color (normochromic) or pale in color (hypochromic). They are never “hyperchromic.” If more than the normal amount of hemoglobin is made, the cells get larger—they do not become darker. In addition to hemoglobin content, the red cells are examined for inclusions. Red cell inclusions are the following:
1. Basophilic stippling—diffuse fine or coarse blue dots in the red cell usually representing RNA residue—especially common in lead poisoning
2. Howell-Jolly bodies—dense blue circular inclusions that represent nuclear remnants—their presence implies defective splenic function
3. Nuclei—red cells may be released or pushed out of the marrow prematurely before nuclear extrusion—often implies a myelophthisic process or a vigorous narrow response to anemia, usually hemolytic anemia
4. Parasites—red cell parasites include malaria and babesia (Chap. 250e)
5. Polychromatophilia—the red cell cytoplasm has a bluish hue, reflecting the persistence of ribosomes still actively making hemoglobin in a young red cell
Vital stains are necessary to see precipitated hemoglobin called Heinz bodies.
Red cells can take on a variety of different shapes. All abnormally shaped red cells are poikilocytes. Small red cells without the central pallor are spherocytes; they can be seen in hereditary spherocytosis, hemolytic anemias of other causes, and clostridial sepsis. Dacrocytes are teardrop-shaped cells that can be seen in hemolytic anemias, severe iron deficiency, thalassemias, myelofibrosis, and myelodysplastic syndromes. Schistocytes are helmet-shaped cells that reflect microangiopathic hemolytic anemia or fragmentation on an artificial heart valve. Echinocytes are spiculated red cells with the spikes evenly spaced; they can represent an artifact of abnormal drying of the blood smear or reflect changes in stored blood. They also can be seen in renal failure and malnutrition and are often reversible. Acanthocytes are spiculated red cells with the spikes irregularly distributed. This process tends to be irreversible and reflects underlying renal disease, abetalipoproteinemia, or splenectomy. Elliptocytes are elliptical-shaped red cells that can reflect an inherited defect in the red cell membrane, but they also are seen in iron deficiency, myelodysplastic syndromes, megaloblastic anemia, and thalassemias. Stomatocytes are red cells in which the area of central pallor takes on the morphology of a slit instead of the usual round shape. Stomatocytes can indicate an inherited red cell membrane defect and also can be seen in alcoholism. Target cells have an area of central pallor that contains a dense center, or bull’s-eye. These cells are seen classically in thalassemia, but they are also present in iron deficiency, cholestatic liver disease, and some hemoglobinopathies. They also can be generated artifactually by improper slide making.
One last feature of the red cells to assess before moving to the white blood cells is the distribution of the red cells on the smear. In most individuals, the cells lie side by side in a single layer. Some patients have red cell clumping (called agglutination) in which the red cells pile upon one another; it is seen in certain paraproteinemias and autoimmune hemolytic anemias. Another abnormal distribution involves red cells lying in single cell rows on top of one another like stacks of coins. This is called rouleaux formation and reflects abnormal serum protein levels.
Finally, one examines the white blood cells. Three types of granulocytes are usually present: neutrophils, eosinophils, and basophils, in decreasing frequency. Neutrophils are generally the most abundant white cell. They are round, are 10–14 μm wide, and contain a lobulated nucleus with two to five lobes connected by a thin chromatin thread. Bands are immature neutrophils that have not completed nuclear condensation and have a U-shaped nucleus. Bands reflect a left shift in neutrophil maturation in an effort to make more cells more rapidly. Neutrophils can provide clues to a variety of conditions. Vacuolated neutrophils may be a sign of bacterial sepsis. The presence of 1- to 2-μm blue cytoplasmic inclusions, called Döhle bodies, can reflect infections, burns, or other inflammatory states. If the neutrophil granules are larger than normal and stain a darker blue, “toxic granulations” are said to be present, and they also suggest a systemic inflammation. The presence of neutrophils with more than five nuclear lobes suggests megaloblastic anemia. Large misshapen granules may reflect the inherited Chédiak-Higashi syndrome.
Eosinophils are slightly larger than neutrophils, have bilobed nuclei, and contain large red granules. Diseases of eosinophils are associated with too many of them rather than any morphologic or qualitative change. They normally total less than one-thirtieth the number of neutrophils. Basophils are even rarer than eosinophils in the blood. They have large dark blue granules and may be increased as part of chronic myeloid leukemia.
Lymphocytes can be present in several morphologic forms. Most common in healthy individuals are small lymphocytes with a small dark nucleus and scarce cytoplasm. In the presence of viral infections, more of the lymphocytes are larger, about the size of neutrophils, with abundant cytoplasm and a less condensed nuclear chromatin. These cells are called reactive lymphocytes. About 1% of lymphocytes are larger and contain blue granules in a light blue cytoplasm; they are called large granular lymphocytes. In chronic lymphoid leukemia, the small lymphocytes are increased in number, and many of them are ruptured in making the blood smear, leaving a smudge of nuclear material without a surrounding cytoplasm or cell membrane; they are called smudge cells and are rare in the absence of chronic lymphoid leukemia.
Monocytes are the largest white blood cells, ranging from 15 to 22 μm in diameter. The nucleus can take on a variety of shapes but usually appears to be folded; the cytoplasm is gray.
Abnormal cells may appear in the blood. Most often the abnormal cells originate from neoplasms of bone marrow–derived cells, including lymphoid cells, myeloid cells, and occasionally red cells. More rarely, other types of tumors can get access to the bloodstream, and rare epithelial malignant cells may be identified. The chances of seeing such abnormal cells is increased by examining blood smears made from buffy coats, the layer of cells that is visible on top of sedimenting red cells when blood is left in the test tube for an hour. Smears made from finger sticks may include rare endothelial cells.
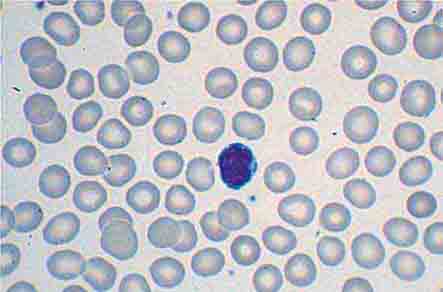
FIGURE 81e-1 Normal peripheral blood smear. Small lymphocyte in center of field. Note that the diameter of the red blood cell is similar to the diameter of the small lymphocyte nucleus.
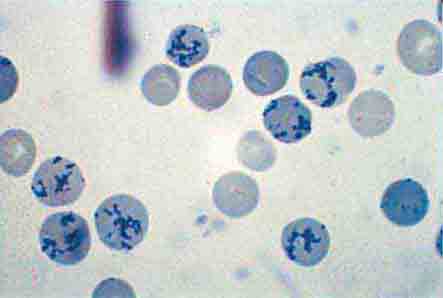
FIGURE 81e-2 Reticulocyte count preparation. This new methylene blue–stained blood smear shows large numbers of heavily stained reticulocytes (the cells containing the dark blue–staining RNA precipitates).
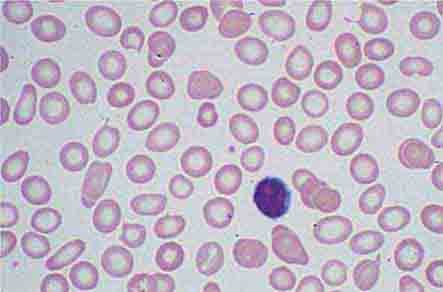
FIGURE 81e-3 Hypochromic microcytic anemia of iron deficiency. Small lymphocyte in field helps assess the red blood cell size.
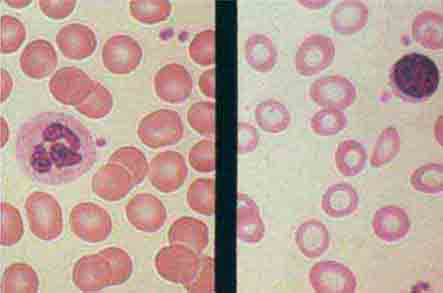
FIGURE 81e-4 Iron deficiency anemia next to normal red blood cells. Microcytes (right panel) are smaller than normal red blood cells (cell diameter <7 μm) and may or may not be poorly hemoglobinized (hypochromic).
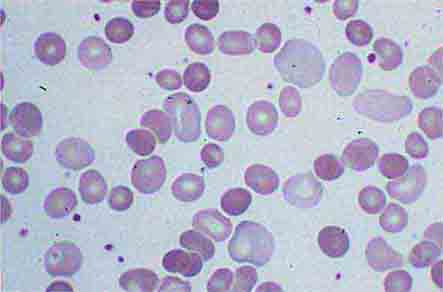
FIGURE 81e-5 Polychromatophilia. Note large red cells with light purple coloring.
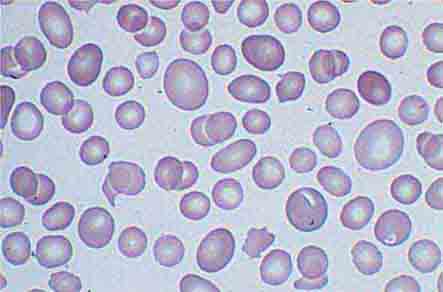
FIGURE 81e-6 Macrocytosis. These cells are both larger than normal (mean corpuscular volume >100) and somewhat oval in shape. Some morphologists call these cells macroovalocytes.
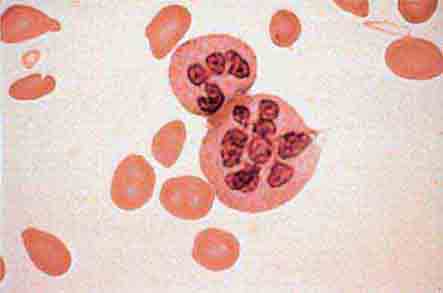
FIGURE 81e-7 Hypersegmented neutrophils. Hypersegmented neutrophils (multilobed polymorphonuclear leukocytes) are larger than normal neutrophils with five or more segmented nuclear lobes. They are commonly seen with folic acid or vitamin B12 deficiency.
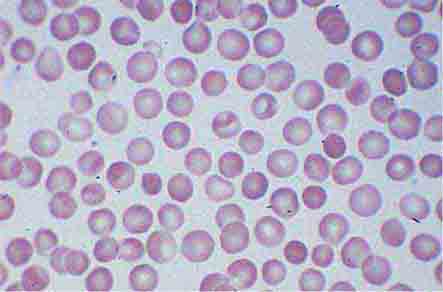
FIGURE 81e-8 Spherocytosis. Note small hyperchromatic cells without the usual clear area in the center.
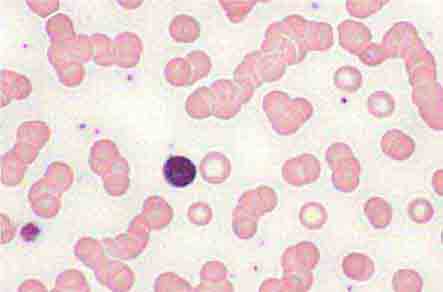
FIGURE 81e-9 Rouleaux formation. Small lymphocyte in center of field. These red cells align themselves in stacks and are related to increased serum protein levels.
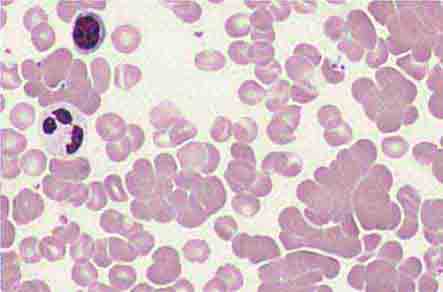
FIGURE 81e-10 Red cell agglutination. Small lymphocyte and segmented neutrophil in upper left center. Note irregular collections of aggregated red cells.
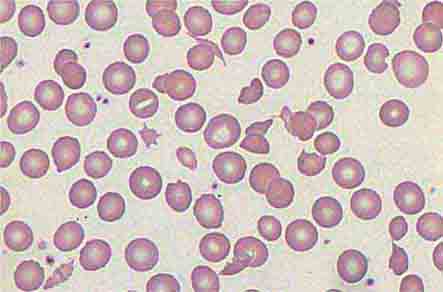
FIGURE 81e-11 Fragmented red cells. Heart valve hemolysis.
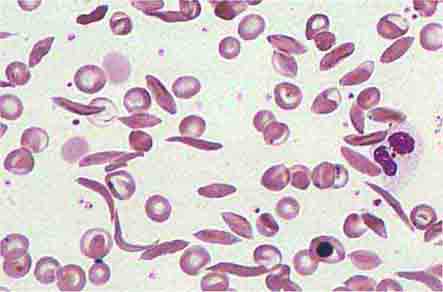
FIGURE 81e-12 Sickle cells. Homozygous sickle cell disease. A nucleated red cell and neutrophil are also in the field.
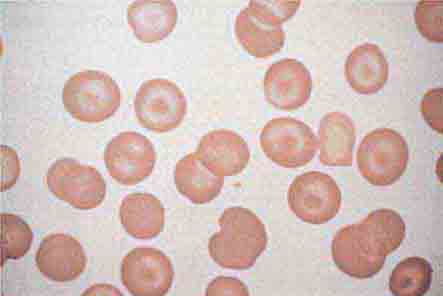
FIGURE 81e-13 Target cells. Target cells are recognized by the bull’s-eye appearance of the cell. Small numbers of target cells are seen with liver disease and thalassemia. Larger numbers are typical of hemoglobin C disease.
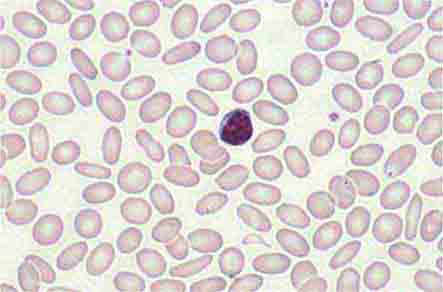
FIGURE 81e-14 Elliptocytosis. Small lymphocyte in center of field. Elliptical shape of red cells is related to weakened membrane structure, usually due to mutations in spectrin.
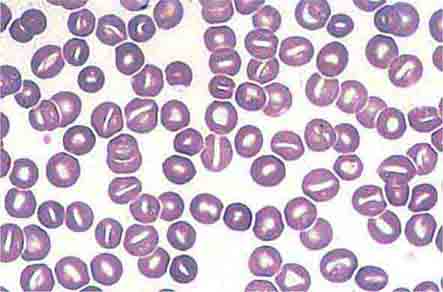
FIGURE 81e-15 Stomatocytosis. Red cells characterized by a wide transverse slit or stoma. This often is seen as an artifact in a dehydrated blood smear. These cells can be seen in hemolytic anemias and in conditions in which the red cell is overhydrated or dehydrated.
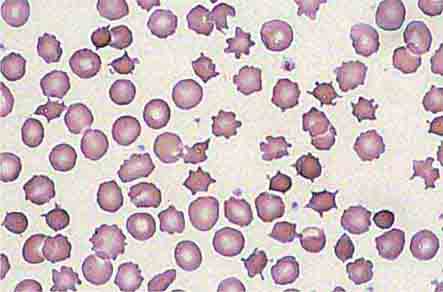
FIGURE 81e-16 Acanthocytosis. Spiculated red cells are of two types: acanthocytes are contracted dense cells with irregular membrane projections that vary in length and width; echinocytes have small, uniform, and evenly spaced membrane projections. Acanthocytes are present in severe liver disease, in patients with abetalipoproteinemia, and in rare patients with McLeod blood group. Echinocytes are found in patients with severe uremia, in glycolytic red cell enzyme defects, and in microangiopathic hemolytic anemia.
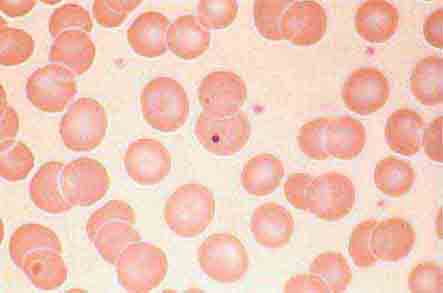
FIGURE 81e-17 Howell-Jolly bodies. Howell-Jolly bodies are tiny nuclear remnants that normally are removed by the spleen. They appear in the blood after splenectomy (defect in removal) and with maturation/dysplastic disorders (excess production).
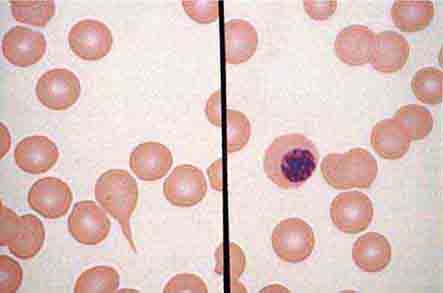
FIGURE 81e-18 Teardrop cells and nucleated red blood cells characteristic of myelofibrosis. A teardrop-shaped red blood cell (left panel) and a nucleated red blood cell (right panel) as typically seen with myelofibrosis and extramedullary hematopoiesis.
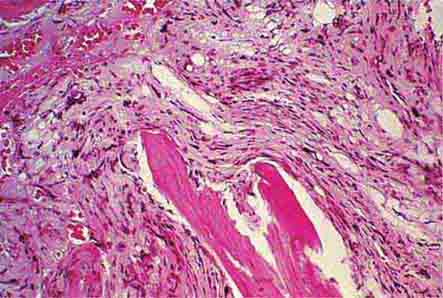
FIGURE 81e-19 Myelofibrosis of the bone marrow. Total replacement of marrow precursors and fat cells by a dense infiltrate of reticulin fibers and collagen (H&E stain).
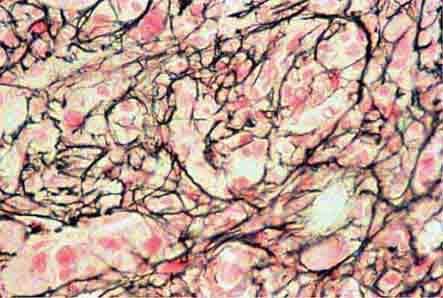
FIGURE 81e-20 Reticulin stain of marrow myelofibrosis. Silver stain of a myelofibrotic marrow showing an increase in reticulin fibers (black-staining threads).
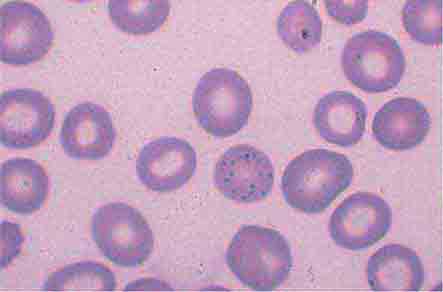
FIGURE 81e-21 Stippled red cell in lead poisoning. Mild hypochromia. Coarsely stippled red cell.
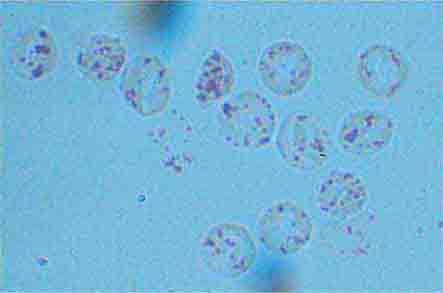
FIGURE 81e-22 Heinz bodies. Blood mixed with hypotonic solution of crystal violet. The stained material is precipitates of denatured hemoglobin within cells.
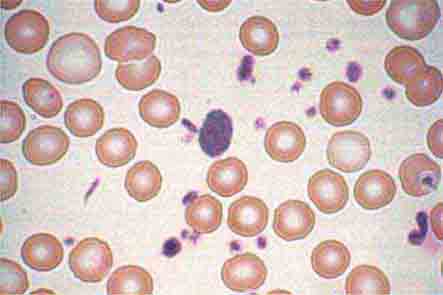
FIGURE 81e-23 Giant platelets. Giant platelets, together with a marked increase in the platelet count, are seen in myeloproliferative disorders, especially primary thrombocythemia.
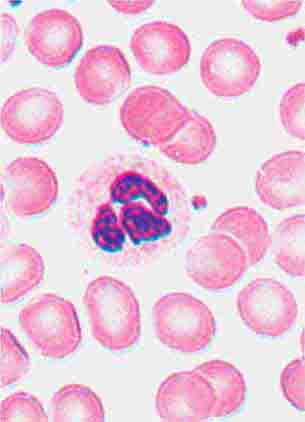
FIGURE 81e-24 Normal granulocytes. The normal granulocyte has a segmented nucleus with heavy, clumped chromatin; fine neutrophilic granules are dispersed throughout the cytoplasm.
FIGURE 81e-25 Normal monocytes. The film was prepared from the buffy coat of the blood from a normal donor. L, lymphocyte; M, monocyte; N, neutrophil.
FIGURE 81e-26 Normal eosinophils. The film was prepared from the buffy coat of the blood from a normal donor. E, eosinophil; L, lymphocyte; N, neutrophil.
FIGURE 81e-27 Normal basophil. The film was prepared from the buffy coat of the blood from a normal donor. B, basophil; L, lymphocyte.
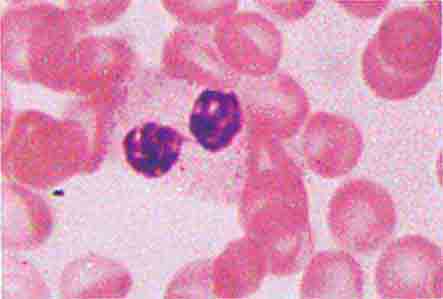
FIGURE 81e-28 Pelger-Hüet anomaly. In this benign disorder, the majority of granulocytes are bilobed. The nucleus frequently has a spectacle-like, or “pince-nez,” configuration.
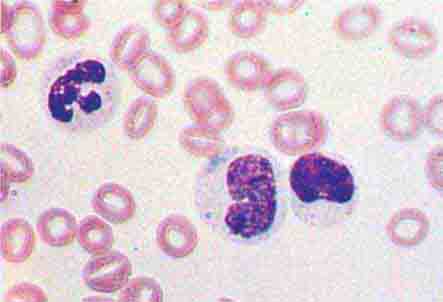
FIGURE 81e-29 Döhle body. Neutrophil band with Döhle body. The neutrophil with a sausage-shaped nucleus in the center of the field is a band form. Döhle bodies are discrete, blue-staining nongranular areas found in the periphery of the cytoplasm of the neutrophil in infections and other toxic states. They represent aggregates of rough endoplasmic reticulum.
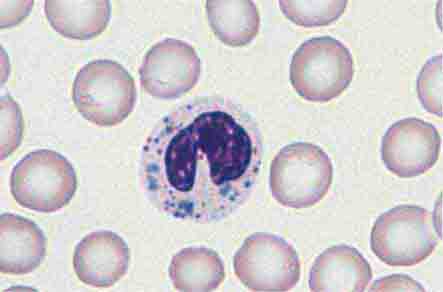
FIGURE 81e-30 Chédiak-Higashi disease. Note giant granules in neutrophil.
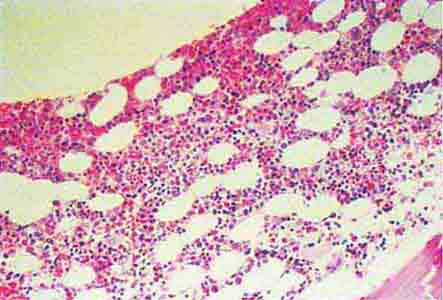
FIGURE 81e-31 Normal bone marrow. Low-power view of normal adult marrow (hematoxylin and eosin [H&E] stain), showing a mix of fat cells (clear areas) and hematopoietic cells. The percentage of the space that consists of hematopoietic cells is referred to as marrow cellularity. In adults, normal marrow cellularity is 35–40%. If demands for increased marrow production occur, cellularity may increase to meet the demand. As people age, the marrow cellularity decreases and the marrow fat increases. Patients >70 years old may have a 20–30% marrow cellularity.
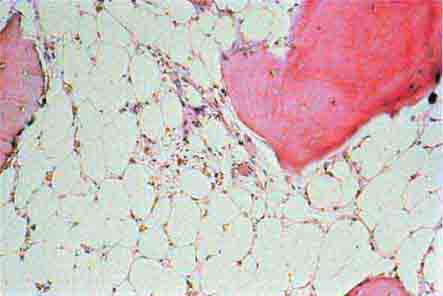
FIGURE 81e-32 Aplastic anemia bone marrow. Normal hematopoietic precursor cells are virtually absent, leaving behind fat cells, reticuloendothelial cells, and the underlying sinusoidal structure.
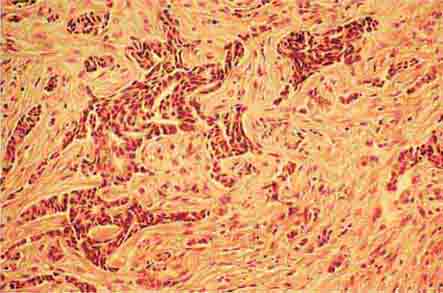
FIGURE 81e-33 Metastatic cancer in the bone marrow. Marrow biopsy specimen infiltrated with metastatic breast cancer and reactive fibrosis (H&E stain).
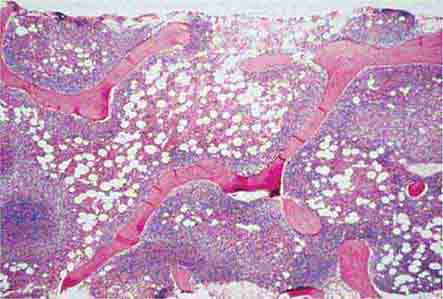
FIGURE 81e-34 Lymphoma in the bone marrow. Nodular (follicular) lymphoma infiltrate in a marrow biopsy specimen. Note the characteristic paratrabecular location of the lymphoma cells.
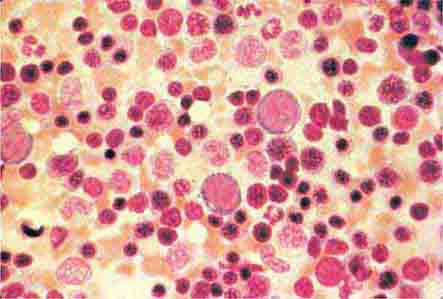
FIGURE 81e-35 Erythroid hyperplasia of the marrow. Marrow aspirate specimen with a myeloid/erythroid ratio (M/E ratio) of 1:1–2, typical for a patient with a hemolytic anemia or one recovering from blood loss.
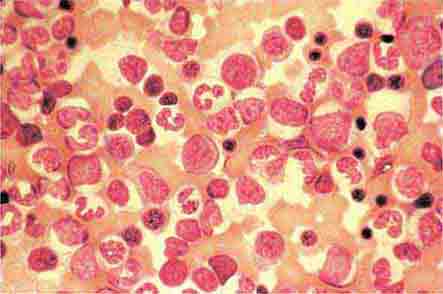
FIGURE 81e-36 Myeloid hyperplasia of the marrow. Marrow aspirate specimen showing a myeloid/erythroid ratio of ≥3:1, suggesting either a loss of red blood cell precursors or an expansion of myeloid elements.
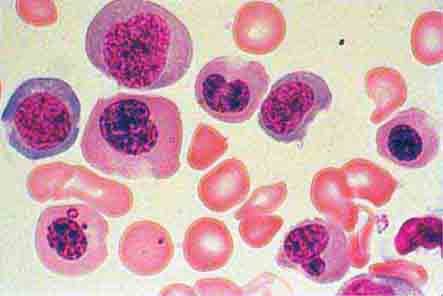
FIGURE 81e-37 Megaloblastic erythropoiesis. High-power view of megaloblastic red blood cell precursors from a patient with a macrocytic anemia. Maturation is delayed, with late normoblasts showing a more immature-appearing nucleus with a lattice-like pattern with normal cytoplasmic maturation.
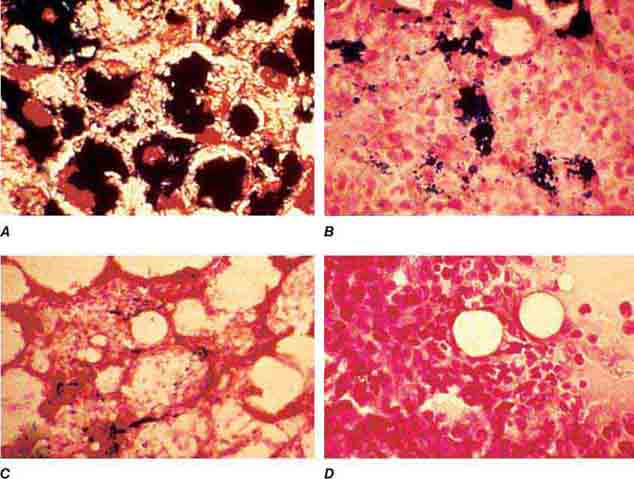
FIGURE 81e-38 Prussian blue staining of marrow iron stores. Iron stores can be graded on a scale of 0 to 4+. A. A marrow with excess iron stores (>4+); B. normal stores (2–3+); C. minimal stores (1+); and D. absent iron stores (0).
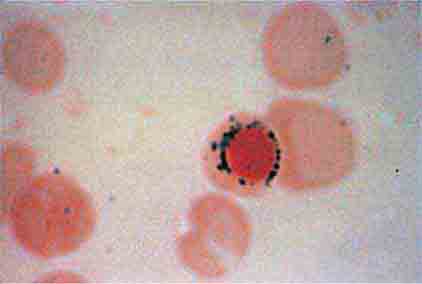
FIGURE 81e-39 Ringed sideroblast. An orthochromatic normoblast with a collar of blue granules (mitochondria encrusted with iron) surrounding the nucleus.
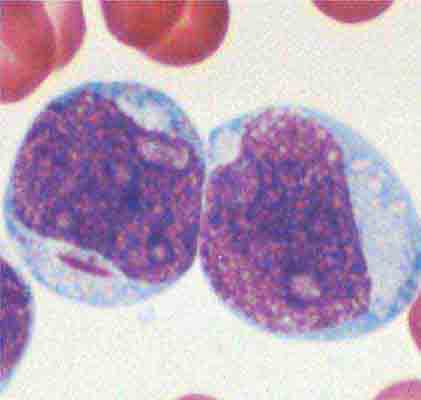
FIGURE 81e-40 Acute myeloid leukemia. Leukemic myeloblast with an Auer rod. Note two to four large, prominent nucleoli in each cell.
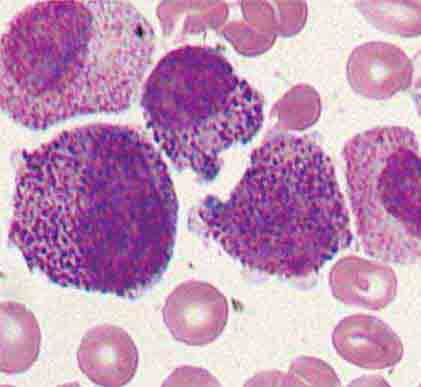
FIGURE 81e-41 Acute promyelocytic leukemia. Note prominent cytoplasmic granules in the leukemia cells.
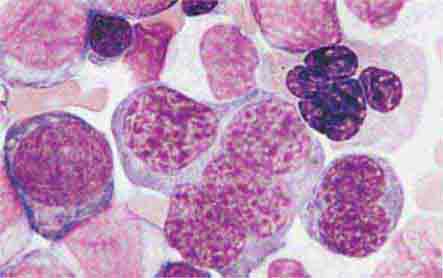
FIGURE 81e-42 Acute erythroleukemia. Note giant dysmorphic erythroblasts; two are binucleate, and one is multinucleate.
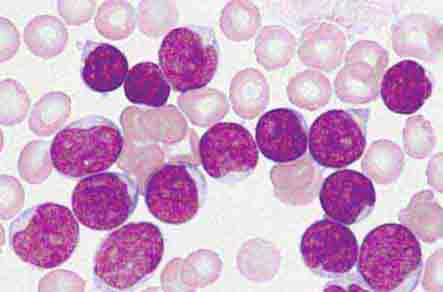
FIGURE 81e-43 Acute lymphoblastic leukemia.
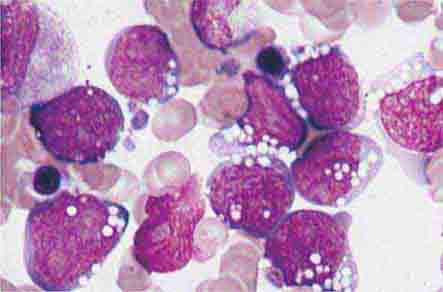
FIGURE 81e-44 Burkitt’s leukemia, acute lymphoblastic leukemia.
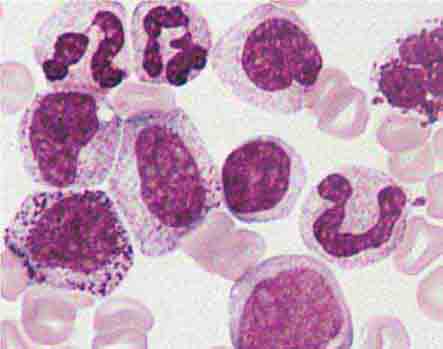
FIGURE 81e-45 Chronic myeloid leukemia in the peripheral blood.
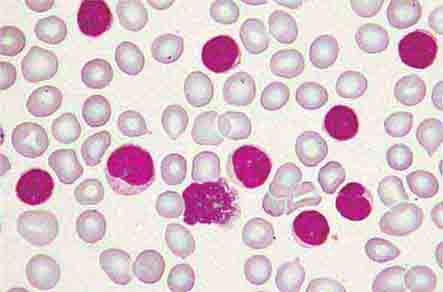
FIGURE 81e-46 Chronic lymphoid leukemia in the peripheral blood.
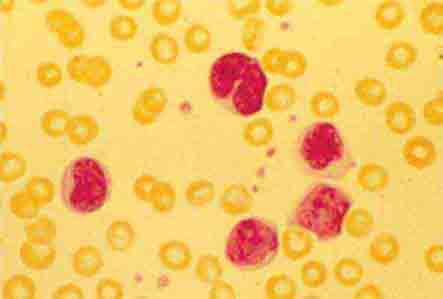
FIGURE 81e-47 Sézary’s syndrome. Lymphocytes with frequently convoluted nuclei (Sézary cells) in a patient with advanced mycosis fungoides.
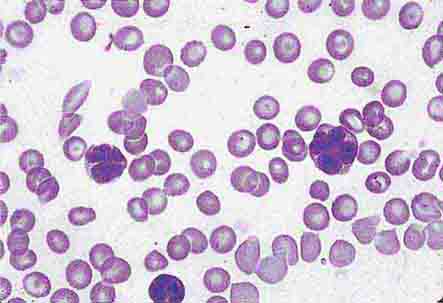
FIGURE 81e-48 Adult T cell leukemia. Peripheral blood smear showing leukemia cells with typical “flower-shaped” nucleus.
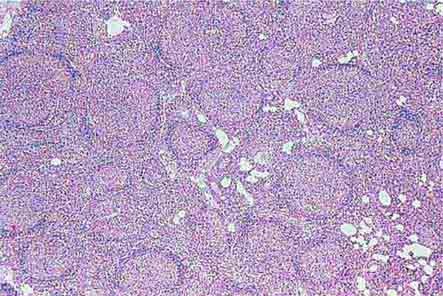
FIGURE 81e-49 Follicular lymphoma in a lymph node. The normal nodal architecture is effaced by nodular expansions of tumor cells. Nodules vary in size and contain predominantly small lymphocytes with cleaved nuclei along with variable numbers of larger cells with vesicular chromatin and prominent nucleoli.
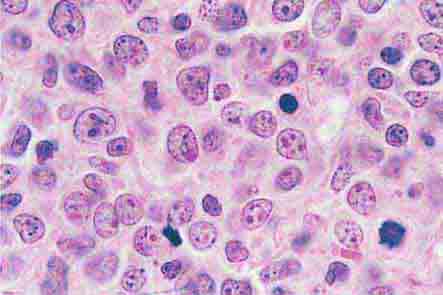
FIGURE 81e-50 Diffuse large B cell lymphoma in a lymph node. The neoplastic cells are heterogeneous but predominantly large cells with vesicular chromatin and prominent nucleoli.
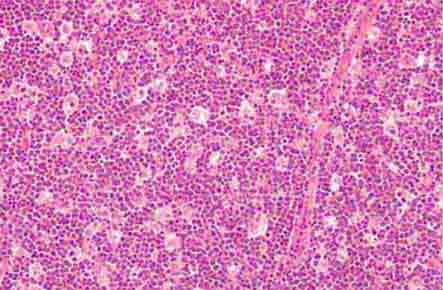
FIGURE 81e-51 Burkitt’s lymphoma in a lymph node. Burkitt’s lymphoma with starry-sky appearance. The lighter areas are macrophages attempting to clear dead cells.
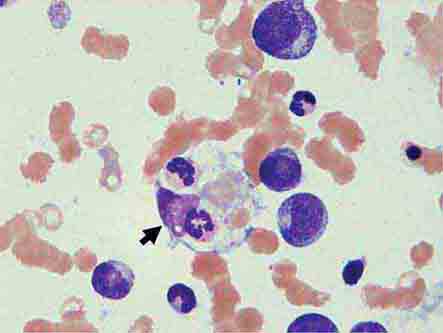
FIGURE 81e-52 Erythrophagocytosis accompanying aggressive lymphoma. The central macrophage is ingesting red cells, neutrophils, and platelets. (Courtesy of Dr. Kiyomi Tsukimori, Kyushu University, Fukuoka, Japan.)
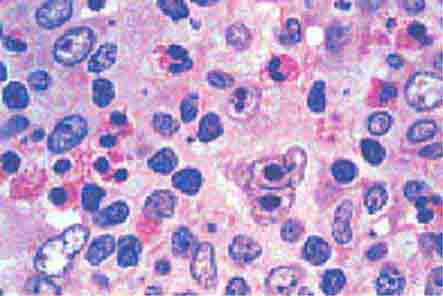
FIGURE 81e-53 Hodgkin’s disease. A Reed-Sternberg cell is present near the center of the field; a large cell with a bilobed nucleus and prominent nucleoli giving an “owl’s eyes” appearance. The majority of the cells are normal lymphocytes, neutrophils, and eosinophils that form a pleiomorphic cellular infiltrate.
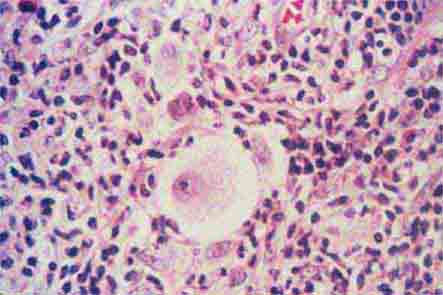
FIGURE 81e-54 Lacunar cell; Reed-Sternberg cell variant in nodular sclerosing Hodgkin’s disease. High-power view of single mononuclear lacunar cell with retracted cytoplasm in a patient with nodular sclerosing Hodgkin’s disease.
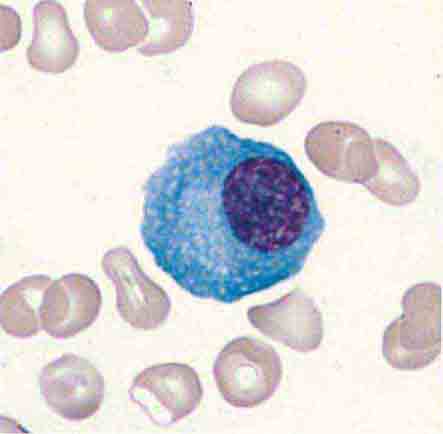
FIGURE 81e-55 Normal plasma cell.
FIGURE 81e-56 Multiple myeloma.

FIGURE 81e-57 Serum color in hemoglobinemia. The distinctive red coloration of plasma (hemoglobinemia) in a spun blood sample in a patient with intravascular hemolysis.
ACKNOWLEDGMENT
Figures in this e-chapter were borrowed from Williams Hematology, 7th edition, M Lichtman et al (eds). New York, McGraw-Hill, 2005; Hematology in General Practice, 4th edition, RS Hillman, KA Ault, New York, McGraw-Hill, 2005.
[/level-membership-for-internal-medicine-category][not-level-membership-for-internal-medicine-category]
FIGURE 76e-21 Non-Hodgkin’s lymphoma involving the skin, with typical violaceous, “plum-colored” nodules. (Courtesy of Jean Bolognia, MD; with permission.)
FIGURE 76e-22 Basal cell carcinoma, with central ulceration and a pearly, rolled, telangiectatic tumor border.
FIGURE 76e-23 Mycosis fungoides is a cutaneous T cell lymphoma. Plaque-stage lesions are seen in this patient.
FIGURE 76e-24 Metastatic carcinoma to the skin is characterized by inflammatory, often ulcerated dermal nodules.
FIGURE 76e-25 Keratoacanthoma is a low-grade squamous cell carcinoma that presents as an exophytic nodule with central keratinous debris.
FIGURE 76e-26 Squamous cell carcinoma is seen here as a hyperkeratotic, crusted, and somewhat eroded plaque on the lower lip. Sun-exposed skin of the head, neck, hands, and arms are other typical sites of involvement.
FIGURE 76e-27 Actinic keratoses consist of hyperkeratotic erythematous papules and patches on sun-exposed skin. They arise in middle-aged to older adults and have some potential for malignant transformation. (Courtesy of Robert Swerlick, MD; with permission.)
MELANOMA AND BENIGN PIGMENTED LESIONS
(Figs. 76e-28 to 76e-33) As the prognosis of melanoma is related primarily to the microscopic depth of invasion, and as early detection with surgical treatment can be curative in a high percentage of patients, it is essential that all clinicians acquire some facility in evaluating pigmented lesions. Three clinicopathologic subtypes of melanoma—superficial spreading, lentigo maligna, and acral lentiginous melanoma—typically display features noted in the “ABCD rule”: asymmetry (one half of the lesion varies from the other half); border irregularity (the circumferential border exhibits an irregular, sometimes jagged appearance); color (there is uneven coloration and tone to the pigmented lesion, with various shades of brown, black, red, and white in different areas); and diameter (the diameter is typically >6 mm). The more uncommon subtype, nodular melanoma, may not manifest all these features but rather may present as a more symmetric, evenly pigmented, or amelanotic lesion. Dysplastic (atypical) melanocytic nevi may occur as solitary or multiple lesions as well as in the setting of familial melanoma. These nevi display some degree of asymmetry, border irregularity, and color variation. Ordinary nevi may be acquired or congenital and are quite common.
FIGURE 76e-28 Nevi are benign proliferations of nevomelanocytes characterized by regularly shaped hyperpigmented macules or papules of a uniform color.
FIGURE 76e-29 Dysplastic nevi are irregularly pigmented and shaped nevomelanocytic lesions that may be associated with familial melanoma.
FIGURE 76e-30 Superficial spreading melanoma, the most common type of malignant melanoma, is characterized by color variegation (black, blue, brown, pink, and white) and irregular borders.
FIGURE 76e-31 Lentigo maligna melanoma occurs on sun-exposed skin as a large, hyperpigmented macule or plaque with irregular borders and variable pigmentation. (Courtesy of Alvin Solomon, MD; with permission.)
FIGURE 76e-32 Nodular melanoma most commonly manifests as a rapidly growing, often ulcerated or crusted black nodule. (Courtesy of S. Wright Caughman, MD; with permission.)
FIGURE 76e-33 Acral lentiginous melanoma is more common among blacks, Asians, and Hispanics and occurs as an enlarging hyperpigmented macule or plaque on the palms or soles. Lateral pigment diffusion is present.
INFECTIOUS DISEASE AND THE SKIN
(Figs. 76e-34 to 76e-58) One of the roles of the skin is to function as a barrier from the outside world. In this capacity, exposure to infectious agents occurs, and bacterial, viral, fungal, and parasitic infections may result. In addition, the skin may be secondarily involved and provides diagnostic clues to systemic infections such as meningococcemia, Rocky Mountain spotted fever, Lyme disease, and septic emboli. Most sexually transmitted bacterial and viral diseases exhibit cutaneous involvement; examples include primary and secondary syphilis, chancroid, genital herpes simplex, and condyloma acuminatum.
FIGURE 76e-34 Erysipelas is a streptococcal infection of the superficial dermis and consists of well-demarcated, erythematous, edematous, warm plaques.
FIGURE 76e-35 Varicella, with numerous lesions in various stages of evolution: vesicles on an erythematous base, umbilicated vesicles, and crusts. (Courtesy of Robert Hartman, MD; with permission.)
FIGURE 76e-36 Herpes zoster is seen in this HIV-infected patient as hemorrhagic vesicles and pustules on an erythematous base in a dermatomal distribution. (Courtesy of Robert Swerlick, MD; with permission.)
FIGURE 76e-37 Impetigo contagiosa is a superficial streptococcal or Staphylococcus aureus infection consisting of honey-colored crusts and erythematous weeping erosions. Bullous lesions are occasionally seen.
FIGURE 76e-38 Tender vesicles and erosions in the mouth of a patient with hand-foot-and-mouth disease. (Courtesy of Stephen D. Gellis, MD; with permission.)
FIGURE 76e-39 Lacy reticular rash of erythema infectiosum (fifth disease).
FIGURE 76e-40 Molluscum contagiosum is a cutaneous poxvirus infection characterized by multiple umbilicated flesh-colored or hypopigmented papules. (Courtesy of Yale Resident’s Slide Collection; with permission.)
FIGURE 76e-41 Oral hairy leukoplakia often presents as white plaques on the lateral tongue and is associated with Epstein-Barr virus infection. (From K Wolff et al: Fitzpatrick’s Color Atlas & Synopsis of Clinical Dermatology, 5th ed. New York, McGraw-Hill, 2005. www.accessmedicine.com.)
FIGURE 76e-42 Fulminant meningococcemia, with extensive angular purpuric patches. (Courtesy of Stephen D. Gellis, MD; with permission.)
FIGURE 76e-43 Rocky Mountain spotted fever, with pinpoint petechial lesions on the palm and volar aspect of the wrist. (Courtesy of Robert Swerlick, MD; with permission.)
FIGURE 76e-44 Erythema migrans, the early cutaneous manifestation of Lyme disease, is characterized by erythematous annular patches, often with a central erythematous papule at the tick-bite site. (Courtesy of Yale Resident’s Slide Collection; with permission.)
FIGURE 76e-45 Primary syphilis, with a firm, nontender chancre. (Courtesy of Gregory Cox, MD; with permission.)
FIGURE 76e-46 Secondary syphilis commonly affects the palms and soles, with scaling, firm, red-brown papules. (Courtesy of Alvin Solomon, MD; with permission.)
FIGURE 76e-47 Condylomata lata are moist, somewhat verrucous inter-triginous plaques seen in secondary syphilis. (Courtesy of Yale Resident’s Slide Collection; with permission.)
FIGURE 76e-48 Secondary syphilis, with the characteristic papulosquamous truncal eruption.
FIGURE 76e-49 A. Tinea corporis is a superficial fungal infection, seen here as an erythematous annular scaly plaque with central clearing. B. A common presentation of chronic dermatophyte infection involves the feet (tinea pedis), hands (tinea manum), and nails (tinea unguium).
FIGURE 76e-50 Scabies, with typical scaling erythematous papules and few linear burrows.
FIGURE 76e-51 Skin lesions caused by Chironex fleckeri sting. (Courtesy of V. Pranava Murthy, MD; with permission.)
FIGURE 76e-52 Chancroid, with characteristic penile ulcers and associated left inguinal adenitis (bubo).
FIGURE 76e-53 Condylomata acuminata are lesions induced by human papillomavirus and in this patient are seen as multiple verrucous papules coalescing into plaques. (Courtesy of S. Wright Caughman, MD; with permission.)
FIGURE 76e-54 A patient with features of polar lepromatous leprosy: multiple nodular skin lesions, particularly of the forehead, and loss of eyebrows. (Courtesy of Robert Gelber, MD; with permission.)
FIGURE 76e-55 Skin lesions of neutropenic patients. A. Hemorrhagic papules on the foot of a patient undergoing treatment for multiple myeloma. Biopsy and culture demonstrated Aspergillosis species. B. Eroded nodule on the hard palate of a patient undergoing chemotherapy. Biopsy and culture demonstrated Mucor species. C. Ecthyma gangrenosum in a neutropenic patient with Pseudomonas aeruginosa bacteremia.
FIGURE 76e-56 Septic emboli, with hemorrhage and infarction due to acute Staphylococcus aureus endocarditis. (Courtesy of L. Baden, MD; with permission.)
FIGURE 76e-57 Vegetations (arrows) due to viridans streptococcal endocarditis involving the mitral valve. (Courtesy of AW Karchmer, MD; with permission.)
FIGURE 76e-58 Disseminated gonococcemia in the skin is seen as hemorrhagic papules and pustules with purpuric centers in an acral distribution. (Courtesy of Daniel M. Musher, MD; with permission.)
IMMUNOLOGICALLY MEDIATED SKIN DISEASE
(Figs. 76e-59 to 76e-70) Immunologically mediated skin disease may be largely localized to skin and mucous membranes and manifest with blisters and erosions such as pemphigus, pemphigoid, and dermatitis herpetiformis. In diseases such as systemic lupus erythematosus, dermatomyositis, and vasculitis, skin manifestations are often only one element of a widespread process.
FIGURE 76e-59 Lupus erythematosus. A. Systemic lupus erythematosus, with prominent, scaly malar erythema. Involvement of other sun-exposed sites is also common. B. Acute lupus erythematosus on the upper chest, with brightly erythematous and slightly edematous coalescence of papules and plaques. (B: Courtesy of Robert Swerlick, MD; with permission.)
FIGURE 76e-60 Discoid lupus erythematosus. Atrophic, depigmented plaques and patches surrounded by hyperpigmentation and erythema in association with scarring and alopecia are characteristic of this cutaneous form of lupus.
FIGURE 76e-61 Dermatomyositis. Periorbital violaceous erythema characterizes the classic heliotrope rash. (Courtesy of James Krell, MD; with permission.)
FIGURE 76e-62 Scleroderma characterized by typical expressionless, mask-like facies.
FIGURE 76e-63 Scleroderma, with acral sclerosis and focal digital ulcers.
FIGURE 76e-64 Dermatomyositis often involves the hands as erythematous flat-topped papules over the knuckles (Gottron’s sign) and periungual telangiectasias.
FIGURE 76e-65 Erythema multiforme is characterized by multiple erythematous plaques with a target or iris morphology and usually represents a hypersensitivity reaction to drugs or infections (especially herpes simplex virus). (Courtesy of Yale Resident’s Slide Collection; with permission.)
FIGURE 76e-66 Dermatitis herpetiformis, manifested by pruritic, grouped vesicles in a typical location. The vesicles are often excoriated and may also occur on the knees, buttocks, elbows, and posterior scalp.
FIGURE 76e-67 Pemphigus vulgaris. A. Eroded bullae on the back. B. The oral mucosa is almost invariably involved, sometimes with erosions on the gingiva, buccal mucosa, palate, posterior pharynx, or tongue. (B: Courtesy of Robert Swerlick, MD; with permission.)
FIGURE 76e-68 Erythema nodosum is a panniculitis characterized by tender deep-seated nodules and plaques, usually located on the lower extremities. (Courtesy of Robert Swerlick, MD; with permission.)
FIGURE 76e-69 Vasculitis. Palpable purpuric papules on the lower legs are seen in this patient with cutaneous small-vessel vasculitis. (Courtesy of Robert Swerlick, MD; with permission.)
FIGURE 76e-70 Bullous pemphigoid, with tense vesicles and bullae on an erythematous, urticarial base. (Courtesy of Yale Resident’s Slide Collection; with permission.)
SKIN MANIFESTATIONS OF INTERNAL DISEASE
(Figs. 76e-71 to 76e-78) While many systemic diseases also have cutaneous manifestations, there are well-recognized dermatologic markers of internal disease, some of which are shown in this section. Many of these dermatologic markers may precede, accompany, or follow diagnosis of systemic disease. Acanthosis nigricans is a prototypical dermatologic process that often occurs in association with underlying systemic abnormalities, most commonly obesity and insulin resistance. It may also be associated with other endocrine disorders and several rare genetic syndromes. Malignant acanthosis nigricans may occur in association with several malignancies, especially adenocarcinoma of the gastrointestinal tract, lung, and breast. Other markers of internal disease in this section include pretibial myxedema, which is associated with thyroid disease, and Sweet syndrome, which may be associated with hematologic malignancies, solid tumors, infections, or inflammatory bowel disease. The skin is also involved in many systemic inflammatory diseases such as sarcoidosis, rheumatoid arthritis, and lupus erythematosus.
FIGURE 76e-71 Acanthosis nigricans, with typical hyperpigmented plaques on a velvet-like, verrucous surface on the neck.
FIGURE 76e-72 Pretibial myxedema manifesting as waxy, infiltrated plaques in a patient with Graves’ disease.
FIGURE 76e-73 Erythematous, indurated plaque of Sweet syndrome, with a pseudovesicular border. (Courtesy of Robert Swerlick, MD, with permission.)
FIGURE 76e-74 Bilateral rheumatoid nodules of the upper extremities. (Courtesy of Robert Swerlick, MD; with permission.)
FIGURE 76e-75 Neurofibromatosis, with numerous flesh-colored cutaneous neurofibromas.
FIGURE 76e-76 Coumarin necrosis. Shown is cutaneous and subcutaneous necrosis of a breast. Other fatty areas, such as buttocks and thighs, are also common sites of involvement. (Courtesy of Kim Yancey, MD; with permission.)
FIGURE 76e-77 Sarcoid. A. Infiltrated papules and plaques of variable color are seen in a typical paranasal and periorbital location. B. Infiltrated, hyperpigmented, and slightly erythematous coalescent papules and plaques on the upper arm. (B: Courtesy of Robert Swerlick, MD; with permission.)
FIGURE 76e-78 Pyoderma gangrenosum on the dorsal aspect of both hands. Multiple necrotic ulcers are surrounded by a violaceous and undermined border. (Courtesy of Robert Swerlick, MD; with permission.)
SECTION 10 |
HEMATOLOGIC ALTERATIONS |
77 |
Anemia and Polycythemia |
HEMATOPOIESIS AND THE PHYSIOLOGIC BASIS OF RED CELL PRODUCTION
Hematopoiesis is the process by which the formed elements of blood are produced. The process is regulated through a series of steps beginning with the hematopoietic stem cell. Stem cells are capable of producing red cells, all classes of granulocytes, monocytes, platelets, and the cells of the immune system. The precise molecular mechanism—either intrinsic to the stem cell itself or through the action of extrinsic factors—by which the stem cell becomes committed to a given lineage is not fully defined. However, experiments in mice suggest that erythroid cells come from a common erythroid/megakaryocyte progenitor that does not develop in the absence of expression of the GATA-1 and FOG-1 (friend of GATA-1) transcription factors (Chap. 89e). Following lineage commitment, hematopoietic progenitor and precursor cells come increasingly under the regulatory influence of growth factors and hormones. For red cell production, erythropoietin (EPO) is the primary regulatory hormone. EPO is required for the maintenance of committed erythroid progenitor cells that, in the absence of the hormone, undergo programmed cell death (apoptosis). The regulated process of red cell production is erythropoiesis, and its key elements are illustrated in Fig. 77-1.
FIGURE 77-1 The physiologic regulation of red cell production by tissue oxygen tension. Hb, hemoglobin.
In the bone marrow, the first morphologically recognizable erythroid precursor is the pronormoblast. This cell can undergo four to five cell divisions, which result in the production of 16–32 mature red cells. With increased EPO production, or the administration of EPO as a drug, early progenitor cell numbers are amplified and, in turn, give rise to increased numbers of erythrocytes. The regulation of EPO production itself is linked to tissue oxygenation.
In mammals, O2 is transported to tissues bound to the hemoglobin contained within circulating red cells. The mature red cell is 8 μm in diameter, anucleate, discoid in shape, and extremely pliable in order to traverse the microcirculation successfully; its membrane integrity is maintained by the intracellular generation of ATP. Normal red cell production results in the daily replacement of 0.8–1% of all circulating red cells in the body, since the average red cell lives 100–120 days. The organ responsible for red cell production is called the erythron. The erythron is a dynamic organ made up of a rapidly proliferating pool of marrow erythroid precursor cells and a large mass of mature circulating red blood cells. The size of the red cell mass reflects the balance of red cell production and destruction. The physiologic basis of red cell production and destruction provides an understanding of the mechanisms that can lead to anemia.
The physiologic regulator of red cell production, the glycoprotein hormone EPO, is produced and released by peritubular capillary lining cells within the kidney. These cells are highly specialized epithelial-like cells. A small amount of EPO is produced by hepatocytes. The fundamental stimulus for EPO production is the availability of O2 for tissue metabolic needs. Key to EPO gene regulation is hypoxia-inducible factor (HIF)-1α. In the presence of O2, HIF-1α is hydroxylated at a key proline, allowing HIF-1α to be ubiquitinated and degraded via the proteasome pathway. If O2 becomes limiting, this critical hydroxylation step does not occur, allowing HIF-1α to partner with other proteins, translocate to the nucleus, and upregulate the expression of the EPO gene, among others.
Impaired O2 delivery to the kidney can result from a decreased red cell mass (anemia), impaired O2 loading of the hemoglobin molecule or a high O2 affinity mutant hemoglobin (hypoxemia), or, rarely, impaired blood flow to the kidney (renal artery stenosis). EPO governs the day-to-day production of red cells, and ambient levels of the hormone can be measured in the plasma by sensitive immunoassays—the normal level being 10–25 U/L. When the hemoglobin concentration falls below 100–120 g/L (10–12 g/dL), plasma EPO levels increase in proportion to the severity of the anemia (Fig. 77-2). In circulation, EPO has a half-clearance time of 6–9 h. EPO acts by binding to specific receptors on the surface of marrow erythroid precursors, inducing them to proliferate and to mature. With EPO stimulation, red cell production can increase four- to fivefold within a 1- to 2-week period, but only in the presence of adequate nutrients, especially iron. The functional capacity of the erythron, therefore, requires normal renal production of EPO, a functioning erythroid marrow, and an adequate supply of substrates for hemoglobin synthesis. A defect in any of these key components can lead to anemia. Generally, anemia is recognized in the laboratory when a patient’s hemoglobin level or hematocrit is reduced below an expected value (the normal range). The likelihood and severity of anemia are defined based on the deviation of the patient’s hemoglobin/hematocrit from values expected for age- and sex-matched normal subjects. The hemoglobin concentration in adults has a Gaussian distribution. The mean hematocrit value for adult males is 47% (standard deviation, ±7%) and that for adult females is 42% (±5%). Any single hematocrit or hemoglobin value carries with it a likelihood of associated anemia. Thus, a hematocrit of <39% in an adult male or <35% in an adult female has only about a 25% chance of being normal. Hematocrit levels are less useful than hemoglobin levels in assessing anemia because they are calculated rather than measured directly. Suspected low hemoglobin or hematocrit values are more easily interpreted if previous values for the same patient are known for comparison. The World Health Organization (WHO) defines anemia as a hemoglobin level <130 g/L (13 g/dL) in men and <120 g/L (12 g/dL) in women.
FIGURE 77-2 Erythropoietin (EPO) levels in response to anemia. When the hemoglobin level falls to 120 g/L (12 g/dL), plasma EPO levels increase logarithmically. In the presence of chronic kidney disease or chronic inflammation, EPO levels are typically lower than expected for the degree of anemia. As individuals age, the level of EPO needed to sustain normal hemoglobin levels appears to increase. (From RS Hillman et al: Hematology in Clinical Practice, 5th ed. New York, McGraw-Hill, 2010.)
The critical elements of erythropoiesis—EPO production, iron availability, the proliferative capacity of the bone marrow, and effective maturation of red cell precursors—are used for the initial classification of anemia (see below).
ANEMIA
CLINICAL PRESENTATION OF ANEMIA
Signs and Symptoms Anemia is most often recognized by abnormal screening laboratory tests. Patients less commonly present with advanced anemia and its attendant signs and symptoms. Acute anemia is due to blood loss or hemolysis. If blood loss is mild, enhanced O2 delivery is achieved through changes in the O2–hemoglobin dissociation curve mediated by a decreased pH or increased CO2 (Bohr effect). With acute blood loss, hypovolemia dominates the clinical picture, and the hematocrit and hemoglobin levels do not reflect the volume of blood lost. Signs of vascular instability appear with acute losses of 10–15% of the total blood volume. In such patients, the issue is not anemia but hypotension and decreased organ perfusion. When >30% of the blood volume is lost suddenly, patients are unable to compensate with the usual mechanisms of vascular contraction and changes in regional blood flow. The patient prefers to remain supine and will show postural hypotension and tachycardia. If the volume of blood lost is >40% (i.e., >2 L in the average-sized adult), signs of hypovolemic shock including confusion, dyspnea, diaphoresis, hypotension, and tachycardia appear (Chap. 129). Such patients have significant deficits in vital organ perfusion and require immediate volume replacement.
With acute hemolysis, the signs and symptoms depend on the mechanism that leads to red cell destruction. Intravascular hemolysis with release of free hemoglobin may be associated with acute back pain, free hemoglobin in the plasma and urine, and renal failure. Symptoms associated with more chronic or progressive anemia depend on the age of the patient and the adequacy of blood supply to critical organs. Symptoms associated with moderate anemia include fatigue, loss of stamina, breathlessness, and tachycardia (particularly with physical exertion). However, because of the intrinsic compensatory mechanisms that govern the O2–hemoglobin dissociation curve, the gradual onset of anemia—particularly in young patients—may not be associated with signs or symptoms until the anemia is severe (hemoglobin <70–80 g/L [7–8 g/dL]). When anemia develops over a period of days or weeks, the total blood volume is normal to slightly increased, and changes in cardiac output and regional blood flow help compensate for the overall loss in O2-carrying capacity. Changes in the position of the O2–hemoglobin dissociation curve account for some of the compensatory response to anemia. With chronic anemia, intracellular levels of 2,3-bisphosphoglycerate rise, shifting the dissociation curve to the right and facilitating O2 unloading. This compensatory mechanism can only maintain normal tissue O2 delivery in the face of a 20–30 g/L (2–3 g/dL) deficit in hemoglobin concentration. Finally, further protection of O2 delivery to vital organs is achieved by the shunting of blood away from organs that are relatively rich in blood supply, particularly the kidney, gut, and skin.
Certain disorders are commonly associated with anemia. Chronic inflammatory states (e.g., infection, rheumatoid arthritis, cancer) are associated with mild to moderate anemia, whereas lymphoproliferative disorders, such as chronic lymphocytic leukemia and certain other B cell neoplasms, may be associated with autoimmune hemolysis.
APPROACH TO THE PATIENT:
Anemia
The evaluation of the patient with anemia requires a careful history and physical examination. Nutritional history related to drugs or alcohol intake and family history of anemia should always be assessed. Certain geographic backgrounds and ethnic origins are associated with an increased likelihood of an inherited disorder of the hemoglobin molecule or intermediary metabolism. Glucose-6-phosphate dehydrogenase (G6PD) deficiency and certain hemoglobinopathies are seen more commonly in those of Middle Eastern or African origin, including African Americans who have a high frequency of G6PD deficiency. Other information that may be useful includes exposure to certain toxic agents or drugs and symptoms related to other disorders commonly associated with anemia. These include symptoms and signs such as bleeding, fatigue, malaise, fever, weight loss, night sweats, and other systemic symptoms. Clues to the mechanisms of anemia may be provided on physical examination by findings of infection, blood in the stool, lymphadenopathy, splenomegaly, or petechiae. Splenomegaly and lymphadenopathy suggest an underlying lymphoproliferative disease, whereas petechiae suggest platelet dysfunction. Past laboratory measurements are helpful to determine a time of onset.
In the anemic patient, physical examination may demonstrate a forceful heartbeat, strong peripheral pulses, and a systolic “flow” murmur. The skin and mucous membranes may be pale if the hemoglobin is <80–100 g/L (8–10 g/dL). This part of the physical examination should focus on areas where vessels are close to the surface such as the mucous membranes, nail beds, and palmar creases. If the palmar creases are lighter in color than the surrounding skin when the hand is hyperextended, the hemoglobin level is usually <80 g/L (8 g/dL).
LABORATORY EVALUATION
Table 77-1 lists the tests used in the initial workup of anemia. A routine complete blood count (CBC) is required as part of the evaluation and includes the hemoglobin, hematocrit, and red cell indices: the mean cell volume (MCV) in femtoliters, mean cell hemoglobin (MCH) in picograms per cell, and mean concentration of hemoglobin per volume of red cells (MCHC) in grams per liter (non-SI: grams per deciliter). The red cell indices are calculated as shown in Table 77-2, and the normal variations in the hemoglobin and hematocrit with age are shown in Table 77-3. A number of physiologic factors affect the CBC, including age, sex, pregnancy, smoking, and altitude. High-normal hemoglobin values may be seen in men and women who live at altitude or smoke heavily. Hemoglobin elevations due to smoking reflect normal compensation due to the displacement of O2 by CO in hemoglobin binding. Other important information is provided by the reticulocyte count and measurements of iron supply including serum iron, total iron-binding capacity (TIBC; an indirect measure of serum transferrin), and serum ferritin. Marked alterations in the red cell indices usually reflect disorders of maturation or iron deficiency. A careful evaluation of the peripheral blood smear is important, and clinical laboratories often provide a description of both the red and white cells, a white cell differential count, and the platelet count. In patients with severe anemia and abnormalities in red blood cell morphology and/or low reticulocyte counts, a bone marrow aspirate or biopsy can assist in the diagnosis. Other tests of value in the diagnosis of specific anemias are discussed in chapters on specific disease states.
|
LABORATORY TESTS IN ANEMIA DIAGNOSIS |
aM/E ratio, ratio of myeloid to erythroid precursors.
|
RED BLOOD CELL INDICES |
|
CHANGES IN NORMAL HEMOGLOBIN/HEMATOCRIT VALUES WITH AGE, SEX, AND PREGNANCY |
The components of the CBC also help in the classification of anemia. Microcytosis is reflected by a lower than normal MCV (<80), whereas high values (>100) reflect macrocytosis. The MCH and MCHC reflect defects in hemoglobin synthesis (hypochromia). Automated cell counters describe the red cell volume distribution width (RDW). The MCV (representing the peak of the distribution curve) is insensitive to the appearance of small populations of macrocytes or microcytes. An experienced laboratory technician will be able to identify minor populations of large or small cells or hypochromic cells before the red cell indices change.
Peripheral Blood Smear The peripheral blood smear provides important information about defects in red cell production (Chap. 81e). As a complement to the red cell indices, the blood smear also reveals variations in cell size (anisocytosis) and shape (poikilocytosis). The degree of anisocytosis usually correlates with increases in the RDW or the range of cell sizes. Poikilocytosis suggests a defect in the maturation of red cell precursors in the bone marrow or fragmentation of circulating red cells. The blood smear may also reveal polychromasia—red cells that are slightly larger than normal and grayish blue in color on the Wright-Giemsa stain. These cells are reticulocytes that have been prematurely released from the bone marrow, and their color represents residual amounts of ribosomal RNA. These cells appear in circulation in response to EPO stimulation or to architectural damage of the bone marrow (fibrosis, infiltration of the marrow by malignant cells, etc.) that results in their disordered release from the marrow. The appearance of nucleated red cells, Howell-Jolly bodies, target cells, sickle cells, and others may provide clues to specific disorders (Figs. 77-3 to 77-11).
FIGURE 77-3 Normal blood smear (Wright stain). High-power field showing normal red cells, a neutrophil, and a few platelets. (From RS Hillman et al: Hematology in Clinical Practice, 5th ed. New York, McGraw-Hill, 2010.)
FIGURE 77-4 Severe iron-deficiency anemia. Microcytic and hypochromic red cells smaller than the nucleus of a lymphocyte associated with marked variation in size (anisocytosis) and shape (poikilocytosis). (From RS Hillman et al: Hematology in Clinical Practice, 5th ed. New York, McGraw-Hill, 2010.)
FIGURE 77-5 Macrocytosis. Red cells are larger than a small lymphocyte and well hemoglobinized. Often macrocytes are oval shaped (macro-ovalocytes).
FIGURE 77-6 Howell-Jolly bodies. In the absence of a functional spleen, nuclear remnants are not culled from the red cells and remain as small homogeneously staining blue inclusions on Wright stain. (From RS Hillman et al: Hematology in Clinical Practice, 5th ed. New York, McGraw-Hill, 2010.)
FIGURE 77-7 Red cell changes in myelofibrosis. The left panel shows a teardrop-shaped cell. The right panel shows a nucleated red cell. These forms can be seen in myelofibrosis.
FIGURE 77-8 Target cells. Target cells have a bull’s-eye appearance and are seen in thalassemia and in liver disease. (From RS Hillman et al: Hematology in Clinical Practice, 5th ed. New York, McGraw-Hill, 2010.)
FIGURE 77-9 Red cell fragmentation. Red cells may become fragmented in the presence of foreign bodies in the circulation, such as mechanical heart valves, or in the setting of thermal injury. (From RS Hillman et al: Hematology in Clinical Practice, 5th ed. New York, McGraw-Hill, 2010.)
FIGURE 77-10 Uremia. The red cells in uremia may acquire numerous regularly spaced, small, spiny projections. Such cells, called burr cells or echinocytes, are readily distinguishable from irregularly spiculated acanthocytes shown in Fig. 77-11.
FIGURE 77-11 Spur cells. Spur cells are recognized as distorted red cells containing several irregularly distributed thornlike projections. Cells with this morphologic abnormality are also called acanthocytes. (From RS Hillman et al: Hematology in Clinical Practice, 5th ed. New York, McGraw-Hill, 2010.)
Reticulocyte Count An accurate reticulocyte count is key to the initial classification of anemia. Reticulocytes are red cells that have been recently released from the bone marrow. They are identified by staining with a supravital dye that precipitates the ribosomal RNA (Fig. 77-12). These precipitates appear as blue or black punctate spots and can be counted manually or, currently, by fluorescent emission of dyes that bind to RNA. This residual RNA is metabolized over the first 24–36 h of the reticulocyte’s life span in circulation. Normally, the reticulocyte count ranges from 1 to 2% and reflects the daily replacement of 0.8–1.0% of the circulating red cell population. A corrected reticulocyte count provides a reliable measure of effective red cell production.
FIGURE 77-12 Reticulocytes. Methylene blue stain demonstrates residual RNA in newly made red cells. (From RS Hillman et al: Hematology in Clinical Practice, 5th ed. New York, McGraw-Hill, 2010.)
In the initial classification of anemia, the patient’s reticulocyte count is compared with the expected reticulocyte response. In general, if the EPO and erythroid marrow responses to moderate anemia [hemoglobin <100 g/L (10 g/dL)] are intact, the red cell production rate increases to two to three times normal within 10 days following the onset of anemia. In the face of established anemia, a reticulocyte response less than two to three times normal indicates an inadequate marrow response.
To use the reticulocyte count to estimate marrow response, two corrections are necessary. The first correction adjusts the reticulocyte count based on the reduced number of circulating red cells. With anemia, the percentage of reticulocytes may be increased while the absolute number is unchanged. To correct for this effect, the reticulocyte percentage is multiplied by the ratio of the patient’s hemoglobin or hematocrit to the expected hemoglobin/hematocrit for the age and sex of the patient (Table 77-4). This provides an estimate of the reticulocyte count corrected for anemia. To convert the corrected reticulocyte count to an index of marrow production, a further correction is required, depending on whether some of the reticulocytes in circulation have been released from the marrow prematurely. For this second correction, the peripheral blood smear is examined to see if there are polychromatophilic macrocytes present.
|
CALCULATION OF RETICULOCYTE PRODUCTION INDEX |
Correction #2 for Longer Life of Prematurely Released Reticulocytes in the Blood:
This correction produces the reticulocyte production index.
In a person whose reticulocyte count is 9%, hemoglobin 7.5 gm/dL, and hematocrit 23%, the reticulocyte production index
These cells, representing prematurely released reticulocytes, are referred to as “shift” cells, and the relationship between the degree of shift and the necessary shift correction factor is shown in Fig. 77-13. The correction is necessary because these prematurely released cells survive as reticulocytes in circulation for >1 day, thereby providing a falsely high estimate of daily red cell production. If polychromasia is increased, the reticulocyte count, already corrected for anemia, should be divided again by 2 to account for the prolonged reticulocyte maturation time. The second correction factor varies from 1 to 3 depending on the severity of anemia. In general, a correction of 2 is simply used. An appropriate correction is shown in Table 77-4. If polychromatophilic cells are not seen on the blood smear, the second correction is not required. The now doubly corrected reticulocyte count is the reticulocyte production index, and it provides an estimate of marrow production relative to normal. In many hospital laboratories, the reticulocyte count is reported not only as a percentage but also in absolute numbers. If so, no correction for dilution is required. A summary of the appropriate marrow response to varying degrees of anemia is shown in Table 77-5.
FIGURE 77-13 Correction of the reticulocyte count. To use the reticulocyte count as an indicator of effective red cell production, the reticulocyte percentage must be corrected based on the level of anemia and the circulating life span of the reticulocytes. Erythroid cells take ~4.5 days to mature. At a normal hemoglobin, reticulocytes are released to the circulation with ~1 day left as reticulocytes. However, with different levels of anemia, reticulocytes (and even earlier erythroid cells) may be released from the marrow prematurely. Most patients come to clinical attention with hematocrits in the mid-20s, and thus a correction factor of 2 is commonly used because the observed reticulocytes will live for 2 days in the circulation before losing their RNA.
|
NORMAL MARROW RESPONSE TO ANEMIA |
Premature release of reticulocytes is normally due to increased EPO stimulation. However, if the integrity of the bone marrow release process is lost through tumor infiltration, fibrosis, or other disorders, the appearance of nucleated red cells or polychromatophilic macrocytes should still invoke the second reticulocyte correction. The shift correction should always be applied to a patient with anemia and a very high reticulocyte count to provide a true index of effective red cell production. Patients with severe chronic hemolytic anemia may increase red cell production as much as six- to sevenfold. This measure alone confirms the fact that the patient has an appropriate EPO response, a normally functioning bone marrow, and sufficient iron available to meet the demands for new red cell formation. If the reticulocyte production index is <2 in the face of established anemia, a defect in erythroid marrow proliferation or maturation must be present.
Tests of Iron Supply and Storage The laboratory measurements that reflect the availability of iron for hemoglobin synthesis include the serum iron, the TIBC, and the percent transferrin saturation. The percent transferrin saturation is derived by dividing the serum iron level (× 100) by the TIBC. The normal serum iron ranges from 9 to 27 μmol/L (50–150 μg/dL), whereas the normal TIBC is 54–64 μmol/L (300–360 μg/dL); the normal transferrin saturation ranges from 25 to 50%. A diurnal variation in the serum iron leads to a variation in the percent transferrin saturation. The serum ferritin is used to evaluate total body iron stores. Adult males have serum ferritin levels that average ~100 μg/L, corresponding to iron stores of ~1 g. Adult females have lower serum ferritin levels averaging 30 μg/L, reflecting lower iron stores (~300 mg). A serum ferritin level of 10–15 μg/L indicates depletion of body iron stores. However, ferritin is also an acute-phase reactant and, in the presence of acute or chronic inflammation, may rise several-fold above baseline levels. As a rule, a serum ferritin >200 μg/L means there is at least some iron in tissue stores.
Bone Marrow Examination A bone marrow aspirate and smear or a needle biopsy can be useful in the evaluation of some patients with anemia. In patients with hypoproliferative anemia and normal iron status, a bone marrow is indicated. Marrow examination can diagnose primary marrow disorders such as myelofibrosis, a red cell maturation defect, or an infiltrative disease (Figs. 77-14 to 77-16). The increase or decrease of one cell lineage (myeloid vs erythroid) compared to another is obtained by a differential count of nucleated cells in a bone marrow smear (the myeloid/erythroid [M/E] ratio). A patient with a hypoproliferative anemia (see below) and a reticulocyte production index <2 will demonstrate an M/E ratio of 2 or 3:1. In contrast, patients with hemolytic disease and a production index >3 will have an M/E ratio of at least 1:1. Maturation disorders are identified from the discrepancy between the M/E ratio and the reticulocyte production index (see below). Either the marrow smear or biopsy can be stained for the presence of iron stores or iron in developing red cells. The storage iron is in the form of ferritin or hemosiderin. On carefully prepared bone marrow smears, small ferritin granules can normally be seen under oil immersion in 20–40% of developing erythroblasts. Such cells are called sideroblasts.
FIGURE 77-14 Normal bone marrow. This is a low-power view of a section of a normal bone marrow biopsy stained with hematoxylin and eosin (H&E). Note that the nucleated cellular elements account for ~40–50% and the fat (clear areas) accounts for ~50–60% of the area. (From RS Hillman et al: Hematology in Clinical Practice, 5th ed. New York, McGraw-Hill, 2010.)
FIGURE 77-15 Erythroid hyperplasia. This marrow shows an increase in the fraction of cells in the erythroid lineage as might be seen when a normal marrow compensates for acute blood loss or hemolysis. The myeloid/erythroid (M/E) ratio is about 1:1. (From RS Hillman et al: Hematology in Clinical Practice, 5th ed. New York, McGraw-Hill, 2010.)
FIGURE 77-16 Myeloid hyperplasia. This marrow shows an increase in the fraction of cells in the myeloid or granulocytic lineage as might be seen in a normal marrow responding to infection. The myeloid/erythroid (M/E) ratio is >3:1. (From RS Hillman et al: Hematology in Clinical Practice, 5th ed. New York, McGraw-Hill, 2010.)
Additional laboratory tests may be of value in confirming specific diagnoses. For details of these tests and how they are applied in individual disorders, see Chaps. 126 to 130.
DEFINITION AND CLASSIFICATION OF ANEMIA
Initial Classification of Anemia The functional classification of anemia has three major categories. These are (1) marrow production defects (hypoproliferation), (2) red cell maturation defects (ineffective erythropoiesis), and (3) decreased red cell survival (blood loss/hemolysis). The classification is shown in Fig. 77-17. A hypoproliferative anemia is typically seen with a low reticulocyte production index together with little or no change in red cell morphology (a normocytic, normochromic anemia) (Chap. 126). Maturation disorders typically have a slight to moderately elevated reticulocyte production index that is accompanied by either macrocytic (Chap. 128) or microcytic (Chaps. 126, 127) red cell indices. Increased red blood cell destruction secondary to hemolysis results in an increase in the reticulocyte production index to at least three times normal (Chap. 129), provided sufficient iron is available. Hemorrhagic anemia does not typically result in production indices of more than 2.0–2.5 times normal because of the limitations placed on expansion of the erythroid marrow by iron availability.
FIGURE 77-17 The physiologic classification of anemia. CBC, complete blood count.
In the first branch point of the classification of anemia, a reticulocyte production index >2.5 indicates that hemolysis is most likely. A reticulocyte production index <2 indicates either a hypoproliferative anemia or maturation disorder. The latter two possibilities can often be distinguished by the red cell indices, by examination of the peripheral blood smear, or by a marrow examination. If the red cell indices are normal, the anemia is almost certainly hypoproliferative in nature. Maturation disorders are characterized by ineffective red cell production and a low reticulocyte production index. Bizarre red cell shapes—macrocytes or hypochromic microcytes—are seen on the peripheral blood smear. With a hypoproliferative anemia, no erythroid hyperplasia is noted in the marrow, whereas patients with ineffective red cell production have erythroid hyperplasia and an M/E ratio <1:1.
Hypoproliferative Anemias At least 75% of all cases of anemia are hypoproliferative in nature. A hypoproliferative anemia reflects absolute or relative marrow failure in which the erythroid marrow has not proliferated appropriately for the degree of anemia. The majority of hypoproliferative anemias are due to mild to moderate iron deficiency or inflammation. A hypoproliferative anemia can result from marrow damage, iron deficiency, or inadequate EPO stimulation. The last may reflect impaired renal function, suppression of EPO production by inflammatory cytokines such as interleukin 1, or reduced tissue needs for O2 from metabolic disease such as hypothyroidism. Only occasionally is the marrow unable to produce red cells at a normal rate, and this is most prevalent in patients with renal failure. With diabetes mellitus or myeloma, the EPO deficiency may be more marked than would be predicted by the degree of renal insufficiency. In general, hypoproliferative anemias are characterized by normocytic, normochromic red cells, although microcytic, hypochromic cells may be observed with mild iron deficiency or long-standing chronic inflammatory disease. The key laboratory tests in distinguishing between the various forms of hypoproliferative anemia include the serum iron and iron-binding capacity, evaluation of renal and thyroid function, a marrow biopsy or aspirate to detect marrow damage or infiltrative disease, and serum ferritin to assess iron stores. An iron stain of the marrow will determine the pattern of iron distribution. Patients with the anemia of acute or chronic inflammation show a distinctive pattern of serum iron (low), TIBC (normal or low), percent transferrin saturation (low), and serum ferritin (normal or high). These changes in iron values are brought about by hepcidin, the iron regulatory hormone that is produced by the liver and is increased in inflammation (Chap. 126). A distinct pattern of results is noted in mild to moderate iron deficiency (low serum iron, high TIBC, low percent transferrin saturation, low serum ferritin) (Chap. 126). Marrow damage by drugs, infiltrative disease such as leukemia or lymphoma, or marrow aplasia is diagnosed from the peripheral blood and bone marrow morphology. With infiltrative disease or fibrosis, a marrow biopsy is required.
Maturation Disorders The presence of anemia with an inappropriately low reticulocyte production index, macro- or microcytosis on smear, and abnormal red cell indices suggests a maturation disorder. Maturation disorders are divided into two categories: nuclear maturation defects, associated with macrocytosis, and cytoplasmic maturation defects, associated with microcytosis and hypochromia usually from defects in hemoglobin synthesis. The inappropriately low reticulocyte production index is a reflection of the ineffective erythropoiesis that results from the destruction within the marrow of developing erythroblasts. Bone marrow examination shows erythroid hyperplasia.
Nuclear maturation defects result from vitamin B12 or folic acid deficiency, drug damage, or myelodysplasia. Drugs that interfere with cellular DNA synthesis, such as methotrexate or alkylating agents, can produce a nuclear maturation defect. Alcohol, alone, is also capable of producing macrocytosis and a variable degree of anemia, but this is usually associated with folic acid deficiency. Measurements of folic acid and vitamin B12 are critical not only in identifying the specific vitamin deficiency but also because they reflect different pathogenetic mechanisms (Chap. 128).
Cytoplasmic maturation defects result from severe iron deficiency or abnormalities in globin or heme synthesis. Iron deficiency occupies an unusual position in the classification of anemia. If the iron-deficiency anemia is mild to moderate, erythroid marrow proliferation is blunted and the anemia is classified as hypoproliferative. However, if the anemia is severe and prolonged, the erythroid marrow will become hyperplastic despite the inadequate iron supply, and the anemia will be classified as ineffective erythropoiesis with a cytoplasmic maturation defect. In either case, an inappropriately low reticulocyte production index, microcytosis, and a classic pattern of iron values make the diagnosis clear and easily distinguish iron deficiency from other cytoplasmic maturation defects such as the thalassemias. Defects in heme synthesis, in contrast to globin synthesis, are less common and may be acquired or inherited (Chap. 430). Acquired abnormalities are usually associated with myelodysplasia, may lead to either a macro- or microcytic anemia, and are frequently associated with mitochondrial iron loading. In these cases, iron is taken up by the mitochondria of the developing erythroid cell but not incorporated into heme. The iron-encrusted mitochondria surround the nucleus of the erythroid cell, forming a ring. Based on the distinctive finding of so-called ringed sideroblasts on the marrow iron stain, patients are diagnosed as having a sideroblastic anemia—almost always reflecting myelodysplasia. Again, studies of iron parameters are helpful in the differential diagnosis of these patients.
Blood Loss/Hemolytic Anemia In contrast to anemias associated with an inappropriately low reticulocyte production index, hemolysis is associated with red cell production indices ≥2.5 times normal. The stimulated erythropoiesis is reflected in the blood smear by the appearance of increased numbers of polychromatophilic macrocytes. A marrow examination is rarely indicated if the reticulocyte production index is increased appropriately. The red cell indices are typically normocytic or slightly macrocytic, reflecting the increased number of reticulocytes. Acute blood loss is not associated with an increased reticulocyte production index because of the time required to increase EPO production and, subsequently, marrow proliferation. Subacute blood loss may be associated with modest reticulocytosis. Anemia from chronic blood loss presents more often as iron deficiency than with the picture of increased red cell production.
The evaluation of blood loss anemia is usually not difficult. Most problems arise when a patient presents with an increased red cell production index from an episode of acute blood loss that went unrecognized. The cause of the anemia and increased red cell production may not be obvious. The confirmation of a recovering state may require observations over a period of 2–3 weeks, during which the hemoglobin concentration will rise and the reticulocyte production index fall (Chap. 129).
Hemolytic disease, while dramatic, is among the least common forms of anemia. The ability to sustain a high reticulocyte production index reflects the ability of the erythroid marrow to compensate for hemolysis and, in the case of extravascular hemolysis, the efficient recycling of iron from the destroyed red cells to support red cell production. With intravascular hemolysis, such as paroxysmal nocturnal hemoglobinuria, the loss of iron may limit the marrow response. The level of response depends on the severity of the anemia and the nature of the underlying disease process.
Hemoglobinopathies, such as sickle cell disease and the thalassemias, present a mixed picture. The reticulocyte index may be high but is inappropriately low for the degree of marrow erythroid hyperplasia (Chap. 127).
Hemolytic anemias present in different ways. Some appear suddenly as an acute, self-limited episode of intravascular or extravascular hemolysis, a presentation pattern often seen in patients with autoimmune hemolysis or with inherited defects of the Embden-Meyerhof pathway or the glutathione reductase pathway. Patients with inherited disorders of the hemoglobin molecule or red cell membrane generally have a lifelong clinical history typical of the disease process. Those with chronic hemolytic disease, such as hereditary spherocytosis, may actually present not with anemia but with a complication stemming from the prolonged increase in red cell destruction such as symptomatic bilirubin gallstones or splenomegaly. Patients with chronic hemolysis are also susceptible to aplastic crises if an infectious process interrupts red cell production.
The differential diagnosis of an acute or chronic hemolytic event requires the careful integration of family history, the pattern of clinical presentation, and—whether the disease is congenital or acquired—careful examination of the peripheral blood smear. Precise diagnosis may require more specialized laboratory tests, such as hemoglobin electrophoresis or a screen for red cell enzymes. Acquired defects in red cell survival are often immunologically mediated and require a direct or indirect antiglobulin test or a cold agglutinin titer to detect the presence of hemolytic antibodies or complement-mediated red cell destruction (Chap. 129).
POLYCYTHEMIA
Polycythemia is defined as an increase in the hemoglobin above normal. This increase may be real or only apparent because of a decrease in plasma volume (spurious or relative polycythemia). The term erythrocytosis may be used interchangeably with polycythemia, but some draw a distinction between them: erythrocytosis implies documentation of increased red cell mass, whereas polycythemia refers to any increase in red cells. Often patients with polycythemia are detected through an incidental finding of elevated hemoglobin or hematocrit levels. Concern that the hemoglobin level may be abnormally high is usually triggered at 170 g/L (17 g/dL) for men and 150 g/L (15 g/dL) for women. Hematocrit levels >50% in men or >45% in women may be abnormal. Hematocrits >60% in men and >55% in women are almost invariably associated with an increased red cell mass. Given that the machine that quantitates red cell parameters actually measures hemoglobin concentrations and calculates hematocrits, hemoglobin levels may be a better index.
Features of the clinical history that are useful in the differential diagnosis include smoking history; current living at high altitude; or a history of congenital heart disease, sleep apnea, or chronic lung disease.
Patients with polycythemia may be asymptomatic or experience symptoms related to the increased red cell mass or the underlying disease process that leads to the increased red cell mass. The dominant symptoms from an increased red cell mass are related to hyperviscosity and thrombosis (both venous and arterial), because the blood viscosity increases logarithmically at hematocrits >55%. Manifestations range from digital ischemia to Budd-Chiari syndrome with hepatic vein thrombosis. Abdominal vessel thromboses are particularly common. Neurologic symptoms such as vertigo, tinnitus, headache, and visual disturbances may occur. Hypertension is often present. Patients with polycythemia vera may have aquagenic pruritus and symptoms related to hepatosplenomegaly. Patients may have easy bruising, epistaxis, or bleeding from the gastrointestinal tract. Peptic ulcer disease is common. Patients with hypoxemia may develop cyanosis on minimal exertion or have headache, impaired mental acuity, and fatigue.
The physical examination usually reveals a ruddy complexion. Splenomegaly favors polycythemia vera as the diagnosis (Chap. 131). The presence of cyanosis or evidence of a right-to-left shunt suggests congenital heart disease presenting in the adult, particularly tetralogy of Fallot or Eisenmenger’s syndrome (Chap. 236). Increased blood viscosity raises pulmonary artery pressure; hypoxemia can lead to increased pulmonary vascular resistance. Together, these factors can produce cor pulmonale.
Polycythemia can be spurious (related to a decrease in plasma volume; Gaisbock’s syndrome), primary, or secondary in origin. The secondary causes are all associated with increases in EPO levels: either a physiologically adapted appropriate elevation based on tissue hypoxia (lung disease, high altitude, CO poisoning, high-affinity hemoglobinopathy) or an abnormal overproduction (renal cysts, renal artery stenosis, tumors with ectopic EPO production). A rare familial form of polycythemia is associated with normal EPO levels but hyperresponsive EPO receptors due to mutations.
APPROACH TO THE PATIENT:
Polycythemia
As shown in Fig. 77-18, the first step is to document the presence of an increased red cell mass using the principle of isotope dilution by administering 51Cr-labeled autologous red blood cells to the patient and sampling blood radioactivity over a 2-h period. If the red cell mass is normal (<36 mL/kg in men, <32 mL/kg in women), the patient has spurious or relative polycythemia. If the red cell mass is increased (>36 mL/kg in men, >32 mL/kg in women), serum EPO levels should be measured. If EPO levels are low or unmeasurable, the patient most likely has polycythemia vera. A mutation in JAK2 (Val617Phe), a key member of the cytokine intracellular signaling pathway, can be found in 90–95% of patients with polycythemia vera. Many of those without this particular JAK2 mutation have mutations in exon 12. As a practical matter, few centers assess red cell mass in the setting of an increased hematocrit. The short workup is to measure EPO levels, check for JAK2 mutation, and perform an abdominal ultrasound to assess spleen size. Tests that support the diagnosis of polycythemia vera include elevated white blood cell count, increased absolute basophil count, and thrombocytosis.
FIGURE 77-18 An approach to the differential diagnosis of patients with an elevated hemoglobin (possible polycythemia). AV, atrioventricular; COPD, chronic obstructive pulmonary disease; CT, computed tomography; EPO, erythropoietin; hct, hematocrit; hgb, hemoglobin; IVP, intravenous pyelogram; RBC, red blood cell.
If serum EPO levels are elevated, one needs to distinguish whether the elevation is a physiologic response to hypoxia or related to autonomous EPO production. Patients with low arterial O2 saturation (<92%) should be further evaluated for the presence of heart or lung disease, if they are not living at high altitude. Patients with normal O2 saturation who are smokers may have elevated EPO levels because of CO displacement of O2. If carboxyhemoglobin (COHb) levels are high, the diagnosis is “smoker’s polycythemia.” Such patients should be urged to stop smoking. Those who cannot stop smoking require phlebotomy to control their polycythemia. Patients with normal O2 saturation who do not smoke either have an abnormal hemoglobin that does not deliver O2 to the tissues (evaluated by finding elevated O2–hemoglobin affinity) or have a source of EPO production that is not responding to the normal feedback inhibition. Further workup is dictated by the differential diagnosis of EPO-producing neoplasms. Hepatoma, uterine leiomyoma, and renal cancer or cysts are all detectable with abdominopelvic computed tomography scans. Cerebellar hemangiomas may produce EPO, but they present with localizing neurologic signs and symptoms rather than polycythemia-related symptoms.
78 |
Bleeding and Thrombosis |
The human hemostatic system provides a natural balance between procoagulant and anticoagulant forces. The procoagulant forces include platelet adhesion and aggregation and fibrin clot formation; anticoagulant forces include the natural inhibitors of coagulation and fibrinolysis. Under normal circumstances, hemostasis is regulated to promote blood flow; however, it is also prepared to clot blood rapidly to arrest blood flow and prevent exsanguination. After bleeding is successfully halted, the system remodels the damaged vessel to restore normal blood flow. The major components of the hemostatic system, which function in concert, are (1) platelets and other formed elements of blood, such as monocytes and red cells; (2) plasma proteins (the coagulation and fibrinolytic factors and inhibitors); and (3) the vessel wall.
STEPS OF NORMAL HEMOSTASIS
PLATELET PLUG FORMATION
On vascular injury, platelets adhere to the site of injury, usually the denuded vascular intimal surface. Platelet adhesion is mediated primarily by Von Willebrand factor (VWF), a large multimeric protein present in both plasma and the extracellular matrix of the subendothelial vessel wall, which serves as the primary “molecular glue,” providing sufficient strength to withstand the high levels of shear stress that would tend to detach them with the flow of blood. Platelet adhesion is also facilitated by direct binding to subendothelial collagen through specific platelet membrane collagen receptors.
Platelet adhesion results in subsequent platelet activation and aggregation. This process is enhanced and amplified by humoral mediators in plasma (e.g., epinephrine, thrombin); mediators released from activated platelets (e.g., adenosine diphosphate, serotonin); and vessel wall extracellular matrix constituents that come in contact with adherent platelets (e.g., collagen, VWF). Activated platelets undergo the release reaction, during which they secrete contents that further promote aggregation and inhibit the naturally anticoagulant endothelial cell factors. During platelet aggregation (platelet-platelet interaction), additional platelets are recruited from the circulation to the site of vascular injury, leading to the formation of an occlusive platelet thrombus. The platelet plug is anchored and stabilized by the developing fibrin mesh.
The platelet glycoprotein (Gp) IIb/IIIa (αIIbβ3) complex is the most abundant receptor on the platelet surface. Platelet activation converts the normally inactive Gp IIb/IIIa receptor into an active receptor, enabling binding to fibrinogen and VWF. Because the surface of each platelet has about 50,000 Gp IIb/IIIa–binding sites, numerous activated platelets recruited to the site of vascular injury can rapidly form an occlusive aggregate by means of a dense network of intercellular fibrinogen bridges. Because this receptor is the key mediator of platelet aggregation, it has become an effective target for antiplatelet therapy.
FIBRIN CLOT FORMATION
Plasma coagulation proteins (clotting factors) normally circulate in plasma in their inactive forms. The sequence of coagulation protein reactions that culminate in the formation of fibrin was originally described as a waterfall or a cascade. Two pathways of blood coagulation have been described in the past: the so-called extrinsic, or tissue factor, pathway and the so-called intrinsic, or contact activation, pathway. We now know that coagulation is normally initiated through tissue factor (TF) exposure and activation through the classic extrinsic pathway but with critically important amplification through elements of the classic intrinsic pathway, as illustrated in Fig. 78-1. These reactions take place on phospholipid surfaces, usually the activated platelet surface. Coagulation testing in the laboratory can reflect other influences due to the artificial nature of the in vitro systems used (see below).
FIGURE 78-1 Coagulation is initiated by tissue factor (TF) exposure, which, with factor (F) VIIa, activates FIX and FX, which in turn, with FVIII and FV as cofactors, respectively, results in thrombin formation and subsequent conversion of fibrinogen to fibrin. Thrombin activates FXI, FVIII, and FV, amplifying the coagulation signal. Once the TF/FVIIa/FXa complex is formed, tissue factor pathway inhibitor (TFPI) inhibits the TF/FVIIa pathway, making coagulation dependent on the amplification loop through FIX/FVIII. Coagulation requires calcium (not shown) and takes place on phospholipid surfaces, usually the activated platelet membrane.
[/not-level-membership-for-internal-medicine-category]
