[level-membership-for-internal-medicine-category]
289e |
Tumors and Trauma of the Heart |
TUMORS OF THE HEART
PRIMARY TUMORS
Primary tumors of the heart are rare. Approximately three-quarters are histologically benign, and the majority of these tumors are myxomas. Malignant tumors, almost all of which are sarcomas, account for 25% of primary cardiac tumors. All cardiac tumors, regardless of pathologic type, have the potential to cause life-threatening complications. Many tumors are now surgically curable; thus, early diagnosis is imperative.
Clinical Presentation Cardiac tumors may present with a wide array of cardiac and noncardiac manifestations. These manifestations depend in large part on the location and size of the tumor and are often nonspecific features of more common forms of heart disease, such as chest pain, syncope, heart failure, murmurs, arrhythmias, conduction disturbances, and pericardial effusion with or without tamponade. Additionally, embolic phenomena and constitutional symptoms may occur.
Myxoma Myxomas are the most common type of primary cardiac tumor in adults, accounting for one-third to one-half of all cases at postmortem examination, and about three-quarters of the tumors treated surgically. They occur at all ages, most commonly in the third through sixth decades, with a female predilection. Approximately 90% of myxomas are sporadic; the remainder are familial with autosomal dominant transmission. The familial variety often occurs as part of a syndrome complex (Carney complex) that includes (1) myxomas (cardiac, skin, and/or breast), (2) lentigines and/or pigmented nevi, and (3) endocrine overactivity (primary nodular adrenal cortical disease with or without Cushing’s syndrome, testicular tumors, and/or pituitary adenomas with gigantism or acromegaly). Certain constellations of findings have been referred to as the NAME syndrome (nevi, atrial myxoma, myxoid neurofibroma, and ephelides) or the LAMB syndrome (lentigines, atrial myxoma, and blue nevi), although these syndromes probably represent subsets of the Carney complex. The genetic basis of this complex has not been elucidated completely; however, patients frequently have inactivating mutations in the tumor-suppressor gene PRKAR1A, which encodes the protein kinase A type I-α regulatory subunit.
Pathologically, myxomas are gelatinous structures that consist of myxoma cells embedded in a stroma rich in glycosaminoglycans. Most are solitary, arise from the interatrial septum in the vicinity of the fossa ovalis (particularly the left atrium), and are often pedunculated on a fibrovascular stalk. In contrast to sporadic tumors, familial or syndromic tumors tend to occur in younger individuals, are often multiple, may be ventricular in location, and are more likely to recur after initial resection.
Myxomas commonly present with obstructive signs and symptoms. The most common clinical presentation mimics that of mitral valve disease: either stenosis owing to tumor prolapse into the mitral orifice or regurgitation resulting from tumor-induced valvular trauma. Ventricular myxomas may cause outflow obstruction similar to that caused by subaortic or subpulmonic stenosis. The symptoms and signs of myxoma may be sudden in onset or positional in nature, owing to the effects of gravity on tumor position. A characteristic low-pitched sound, a “tumor plop,” may be appreciated on auscultation during early or mid-diastole and is thought to result from the impact of the tumor against the mitral valve or ventricular wall. Myxomas also may present with peripheral or pulmonary emboli or with constitutional signs and symptoms, including fever, weight loss, cachexia, malaise, arthralgias, rash, digital clubbing, Raynaud’s phenomenon, hypergammaglobulinemia, anemia, polycythemia, leukocytosis, elevated erythrocyte sedimentation rate, thrombocytopenia, and thrombocytosis. These features account for the frequent misdiagnosis of patients with myxomas as having endocarditis, collagen vascular disease, or a paraneoplastic syndrome.
Two-dimensional transthoracic or omniplane transesophageal echocardiography is useful in the diagnosis of cardiac myxoma and allows assessment of tumor size and determination of the site of tumor attachment, both of which are important considerations in the planning of surgical excision (Fig. 289e-1). Computed tomography (CT) and magnetic resonance imaging (MRI) may provide important information regarding size, shape, composition, and surface characteristics of the tumor (Fig. 289e-2).
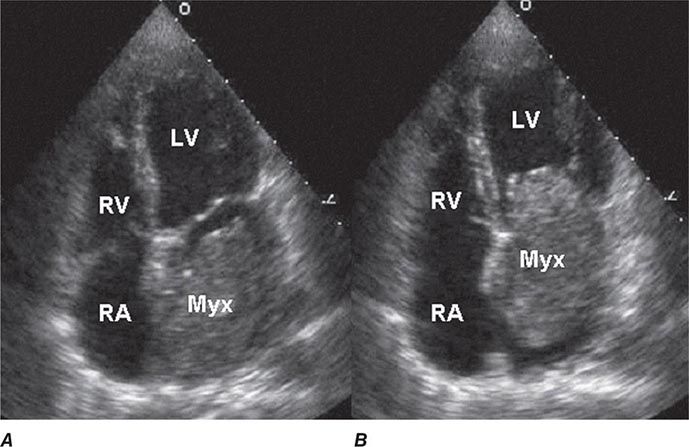
FIGURE 289e-1 Transthoracic echocardiogram demonstrating a large atrial myxoma. The myxoma (Myx) fills the entire left atrium in systole (A) and prolapses across the mitral valve and into the left ventricle (LV) during diastole (B). RA, right atrium; RV, right ventricle. (Courtesy of Dr. Michael Tsang; with permission.)
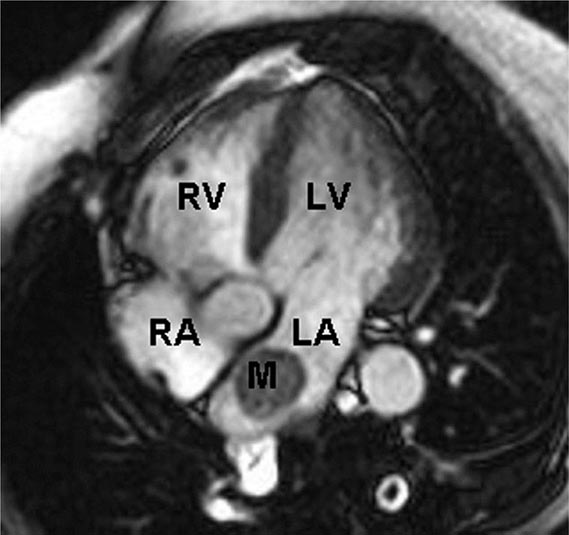
FIGURE 289e-2 Cardiac magnetic resonance imaging demonstrating a rounded mass (M) within the left atrium (LA). Pathologic evaluation at the time of surgery revealed it to be an atrial myxoma. LV, left ventricle; RA, right atrium; RV, right ventricle.
Although cardiac catheterization and angiography were previously performed routinely before tumor resection, they no longer are considered mandatory when adequate noninvasive information is available and other cardiac disorders (e.g., coronary artery disease) are not considered likely. Additionally, catheterization of the chamber from which the tumor arises carries the risk of tumor embolization. Because myxomas may be familial, echocardiographic screening of first-degree relatives is appropriate, particularly if the patient is young and has multiple tumors or evidence of myxoma syndrome.
Other Benign Tumors Cardiac lipomas, although relatively common, are usually incidental findings at postmortem examination; however, they may grow as large as 15 cm and may present with symptoms owing to mechanical interference with cardiac function, arrhythmias, or conduction disturbances or as an abnormality of the cardiac silhouette on chest x-ray. Papillary fibroelastomas are the most common tumors of the cardiac valves. Although usually clinically silent, they can cause valve dysfunction and may embolize distally, resulting in transient ischemic attacks, stroke, or myocardial infarction. In general, these tumors should be resected even when asymptomatic, although a more conservative approach may be considered for small, right-sided lesions. Rhabdomyomas and fibromas are the most common cardiac tumors in infants and children and usually occur in the ventricles, where they may produce mechanical obstruction to blood flow, thereby mimicking valvular stenosis, congestive heart failure (CHF), restrictive or hypertrophic cardiomyopathy, or pericardial constriction. Rhabdomyomas are probably hamartomatous growths, are multiple in 90% of cases, and are strongly associated with tuberous sclerosis. These tumors have a tendency to regress completely or partially; only tumors that cause obstruction require surgical resection. Fibromas are usually single, are often calcified, tend to grow and cause obstructive symptoms, and should be resected. Hemangiomas and mesotheliomas are generally small tumors, most often intramyocardial in location, and may cause atrioventricular (AV) conduction disturbances and even sudden death as a result of their propensity to develop in the region of the AV node. Other benign tumors arising from the heart include teratoma, chemodectoma, neurilemoma, granular cell myoblastoma, and bronchogenic cysts.
Sarcoma Almost all malignant primary cardiac tumors are sarcomas, which may be of several histologic types. In general, these tumors are characterized by rapid progression that culminates in the patient’s death within weeks to months from the time of presentation as a result of hemodynamic compromise, local invasion, or distant metastases. Sarcomas commonly involve the right side of the heart, are characterized by rapid growth, frequently invade the pericardial space, and may obstruct the cardiac chambers or venae cavae. Sarcomas also may occur on the left side of the heart and may be mistaken for myxomas.
TUMORS METASTATIC TO THE HEART
Tumors metastatic to the heart are much more common than primary tumors, and their incidence is likely to increase as the life expectancy of patients with various forms of malignant neoplasms is extended by more effective therapy. Although cardiac metastases may occur with any tumor type, the relative incidence is especially high in malignant melanoma and, to a somewhat lesser extent, leukemia and lymphoma. In absolute terms, the most common primary originating sites of cardiac metastases are carcinoma of the breast and lung, reflecting the high incidence of those cancers. Cardiac metastases almost always occur in the setting of widespread primary disease, and most often there is either primary or metastatic disease elsewhere in the thoracic cavity. Nevertheless, cardiac metastasis occasionally may be the initial presentation of an extrathoracic tumor.
Cardiac metastases may occur via hematogenous or lymphangitic spread or by direct tumor invasion. They generally manifest as small, firm nodules; diffuse infiltration also may occur, especially with sarcomas or hematologic neoplasms. The pericardium is most often involved, followed by myocardial involvement of any chamber and, rarely, by involvement of the endocardium or cardiac valves.
Cardiac metastases are clinically apparent only ~10% of the time, are usually not the cause of the patient’s presentation, and rarely are the cause of death. The vast majority occur in the setting of a previously recognized malignant neoplasm. As with primary cardiac tumors, the clinical presentation reflects more the location and size of the tumor than its histologic type. When symptomatic, cardiac metastases may result in a variety of clinical features, including dyspnea, acute pericarditis, cardiac tamponade, ectopic tachyarrhythmias, heart block, and CHF. Importantly, many of these signs and symptoms may also result from myocarditis, pericarditis, or cardiomyopathy induced by radiotherapy or chemotherapy.
Electrocardiographic (ECG) findings are nonspecific. On chest x-ray, the cardiac silhouette is most often normal but may be enlarged or exhibit a bizarre contour. Echocardiography is useful for identifying pericardial effusions and visualizing larger metastases, although CT and radionuclide imaging with gallium or thallium may define the tumor burden more clearly. Cardiac MRI offers superb image quality and plays a central role in the diagnostic evaluation of cardiac metastases and cardiac tumors in general. Pericardiocentesis may allow for a specific cytologic diagnosis in patients with malignant pericardial effusions. Angiography is rarely necessary but may delineate discrete lesions.
TRAUMATIC CARDIAC INJURY
Traumatic cardiac injury may be caused by either penetrating or nonpenetrating trauma. Penetrating injuries most often result from gunshot or knife wounds, and the site of entry is usually obvious. Nonpenetrating injuries most often occur during motor vehicle accidents, either from rapid deceleration or from impact of the chest against the steering wheel, and may be associated with significant cardiac injury even in the absence of external signs of thoracic trauma.
NONPENETRATING CARDIAC INJURY
Myocardial contusions are the most common form of nonpenetrating cardiac injury and may initially be overlooked in trauma patients as the clinical focus is directed toward other, more obvious injuries. Myocardial necrosis may occur as a direct result of the blunt injury or as a result of traumatic coronary laceration or thrombosis. The contused myocardium is pathologically similar to infarcted myocardium and may be associated with atrial or ventricular arrhythmias; conduction disturbances, including bundle branch block; or ECG abnormalities resembling those of infarction or pericarditis. Thus, it is important to consider contusion as a cause of otherwise unexplained ECG changes in a trauma patient. Serum creatine kinase, myocardial band (CK-MB) isoenzyme levels are increased in ~20% of patients who experience blunt chest trauma but may be falsely elevated in the presence of massive skeletal muscle injury. Cardiac troponin levels are more specific for identifying cardiac injury in this setting. Echocardiography is useful in detecting structural and functional sequelae of contusion, including wall motion abnormalities (most commonly involving the right ventricle, interventricular septum, or left ventricular apex), pericardial effusion, valvular dysfunction, and ventricular rupture.
Rupture of the cardiac valves or their supporting structures, most commonly of the tricuspid or mitral valve, leads to acute valvular incompetence. This complication is usually heralded by the development of a loud murmur, may be associated with rapidly progressive heart failure, and can be diagnosed by either transthoracic or transesophageal echocardiography.
The most serious consequence of nonpenetrating cardiac injury is myocardial rupture, which may result in hemopericardium and tamponade (free wall rupture) or intracardiac shunting (ventricular septal rupture). Although it generally is fatal, up to 40% of patients with cardiac rupture have been reported to survive long enough to reach a specialized trauma center. Hemopericardium also may result from traumatic rupture of a pericardial vessel or a coronary artery. Additionally, a pericardial effusion may develop weeks or even months after blunt chest trauma as a manifestation of the post–cardiac injury syndrome, which resembles the postpericardiotomy syndrome (Chap. 288).
Blunt, nonpenetrating, often innocent-appearing injuries to the chest may trigger ventricular fibrillation even in absence of overt signs of injury. This syndrome, referred to as commotio cordis, occurs most often in adolescents during sporting events (e.g., baseball, hockey, football, and lacrosse) and probably results from an impact to the chest wall overlying the heart during the susceptible phase of repolarization just before the peak of the T wave. Survival depends on prompt defibrillation. Sudden emotional or physical trauma, even in the absence of direct cardiac trauma, may precipitate a transient catecholamine-mediated cardiomyopathy referred to as tako-tsubo syndrome or the apical ballooning syndrome (Chap. 287).
Rupture or transection of the aorta, usually just above the aortic valve or at the site of the ligamentum arteriosum, is a common consequence of nonpenetrating chest trauma and is the most common vascular deceleration injury. The clinical presentation is similar to that of aortic dissection (Chap. 301); the arterial pressure and pulse amplitude may be increased in the upper extremities and decreased in the lower extremities, and chest x-ray may reveal mediastinal widening. Occasionally, aortic rupture is contained by the aortic adventitia, resulting in a false, or pseudo-, aneurysm that may be discovered months or years after the initial injury.
PENETRATING CARDIAC INJURY
Penetrating injuries of the heart produced by knife or bullet wounds usually result in rapid clinical deterioration and frequently in death as a result of hemopericardium/pericardial tamponade or massive hemorrhage. Nonetheless, up to half of such patients may survive long enough to reach a specialized trauma center if immediate resuscitation is performed. Prognosis in these patients relates to the mechanism of injury, their clinical condition at presentation, and the specific cardiac chamber(s) involved. Iatrogenic cardiac or coronary arterial perforation may complicate placement of central venous or intracardiac catheters, pacemaker leads, or intracoronary stents and is associated with a better prognosis than are other forms of penetrating cardiac trauma.
Traumatic rupture of a great vessel from penetrating injury is usually associated with hemothorax and, less often, hemopericardium. Local hematoma formation may compress major vessels and produce ischemic symptoms, and AV fistulas may develop, occasionally resulting in high-output CHF.
Occasionally, patients who survive penetrating cardiac injuries may subsequently present with a new cardiac murmur or CHF as a result of mitral regurgitation or an intracardiac shunt (i.e., ventricular or atrial septal defect, aortopulmonary fistula, or coronary AV fistula) that was undetected at the time of the initial injury or developed subsequently. Therefore, trauma patients should be examined carefully several weeks after the injury. If a mechanical complication is suspected, it can be confirmed by echocardiography or cardiac catheterization.
290e |
Cardiac Manifestations of Systemic Disease |
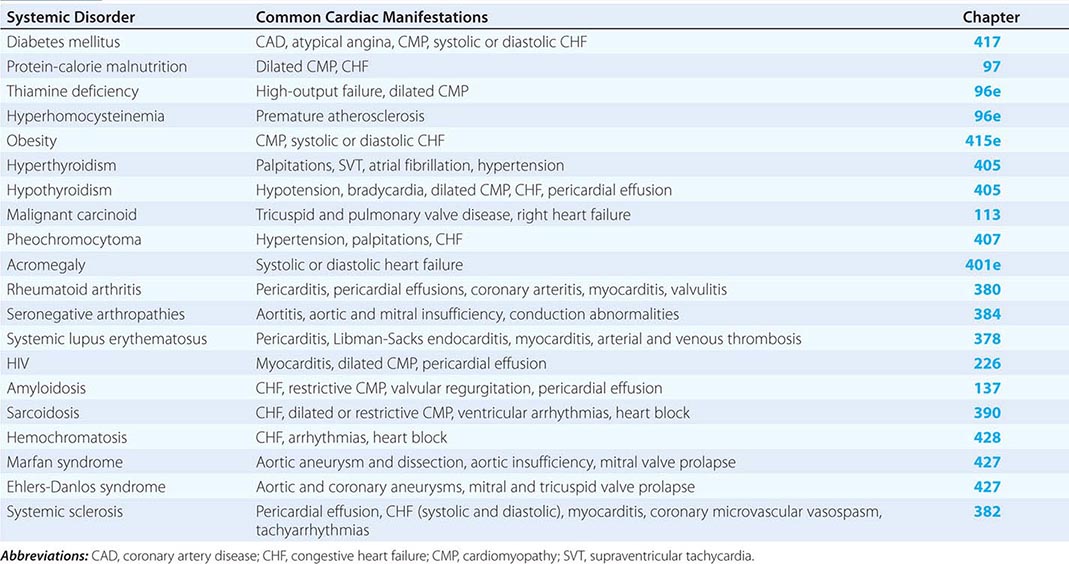
The common systemic disorders that have associated cardiac manifestations are summarized in Table 290e-1.
|
COMMON SYSTEMIC DISORDERS AND THEIR ASSOCIATED CARDIAC MANIFESTATIONS |
DIABETES MELLITUS
(See also Chap. 417) Diabetes mellitus, both insulin- and non-insulin-dependent, is an independent risk factor for coronary artery disease (CAD; Chap. 291e) and accounts for 14–50% of new cases of cardiovascular disease. Furthermore, CAD is the most common cause of death in adults with diabetes mellitus. In the diabetic population, the incidence of CAD relates to the duration of diabetes and the level of glycemic control, and its pathogenesis involves endothelial dysfunction, increased lipoprotein peroxidation, increased inflammation, a prothrombotic state, and associated metabolic abnormalities.
Compared to their nondiabetic counterparts, diabetic patients are more likely to have a myocardial infarction, have a greater burden of CAD, have larger infarct size, and have more postinfarct complications, including heart failure, shock, and death. Importantly, diabetic patients are more likely to have atypical ischemic symptoms; nausea, dyspnea, pulmonary edema, arrhythmias, heart block, or syncope may be their anginal equivalent. Additionally, “silent ischemia,” resulting from autonomic nervous system dysfunction, is more common in diabetic patients, accounting for up to 90% of their ischemic episodes. Thus, one must have a low threshold for suspecting CAD in diabetic patients. The treatment of diabetic patients with CAD must include aggressive risk factor management (Chap. 418). Considerations regarding pharmacologic therapy and revascularization strategies are similar in diabetic and nondiabetic patients except that diabetic patients have higher morbidity and mortality rates associated with revascularization, have an increased risk of restenosis after percutaneous coronary intervention (PCI), and have improved survival when treated with surgical bypass compared with PCI for multivessel CAD.
Patients with diabetes mellitus also may have abnormal left ventricular systolic and diastolic function, reflecting concomitant epicardial CAD and/or hypertension, coronary microvascular disease, endothelial dysfunction, ventricular hypertrophy, and autonomic dysfunction. Furthermore, the increase in intramyocardial lipid deposition (predominantly nonesterified fatty acids) that is characteristic of diabetic states may contribute to both systolic and diastolic dysfunction by impairing insulin signaling, reducing trans-sarcolemma calcium flux, and inducing myocyte apoptosis. A restrictive cardiomyopathy may be present with abnormal myocardial relaxation and elevated ventricular filling pressures. Histologically, interstitial fibrosis is seen, and intramural arteries may demonstrate intimal thickening, hyaline deposition, and inflammatory changes. Diabetic patients have an increased risk of developing clinical heart failure, which probably contributes to their excessive cardiovascular morbidity and mortality rates. There is some evidence that insulin therapy may ameliorate diabetes-related myocardial dysfunction.
MALNUTRITION AND VITAMIN DEFICIENCY
Malnutrition (See also Chap. 97) In patients whose intake of protein, calories, or both is severely deficient, the heart may become thin, pale, and hypokinetic with myofibrillar atrophy and interstitial edema. The systolic pressure and cardiac output fall, and the pulse pressure narrows. Generalized edema is common and relates to a variety of factors, including reduced serum oncotic pressure and myocardial dysfunction. Such profound states of protein and calorie malnutrition, termed kwashiorkor and marasmus, respectively, are most common in underdeveloped countries. However, significant nutritional heart disease also may occur in developed nations, particularly in patients with chronic diseases such as AIDS, patients with anorexia nervosa, and patients with severe cardiac failure in whom gastrointestinal hypoperfusion and venous congestion may lead to anorexia and malabsorption. Open-heart surgery poses increased risk in malnourished patients; such patients may benefit from preoperative hyperalimentation.
Thiamine Deficiency (Beriberi) (See also Chap. 96e) Generalized malnutrition often is accompanied by thiamine deficiency; however, this hypovitaminosis also may occur in the presence of an adequate protein and caloric intake, particularly in East Asia, where polished rice deficient in thiamine may be a major dietary component. In Western nations where the use of thiamine-enriched flour is widespread, clinical thiamine deficiency is limited primarily to alcoholics, food faddists, and patients receiving chemotherapy. Nonetheless, when thiamine stores are measured using the thiamine-pyrophosphate effect (TPPE), thiamine deficiency has been found in 20–90% of patients with chronic heart failure. This deficiency appears to result from both reduced dietary intake and a diuretic-induced increase in the urinary excretion of thiamine. The acute administration of thiamine to these patients increases the left ventricular ejection fraction and the excretion of salt and water.
Clinically, patients with thiamine deficiency usually have evidence of generalized malnutrition, peripheral neuropathy, glossitis, and anemia. The classic associated cardiovascular syndrome is characterized by high-output heart failure, tachycardia, and often elevated biventricular filling pressures. The major cause of the high-output state is vasomotor depression leading to reduced systemic vascular resistance, the precise mechanism of which is not understood. The cardiac examination may reveal a wide pulse pressure, tachycardia, a third heart sound, and an apical systolic murmur. The electrocardiogram (ECG) may reveal decreased voltage, a prolonged QT interval, and T-wave abnormalities. The chest x-ray generally reveals cardiomegaly and signs of congestive heart failure (CHF). The response to thiamine is often dramatic, with an increase in systemic vascular resistance, a decrease in cardiac output, clearing of pulmonary congestion, and a reduction in heart size often occurring in 12–48 h. Although the response to inotropes and diuretics may be poor before thiamine therapy, these agents may be important after thiamine repletion, since the left ventricle may not be able to handle the increased work load presented by the return of vascular tone.
Vitamin B6, B12, and Folate Deficiency (See also Chap. 96e) Vitamin B6, vitamin B12, and folate are cofactors in the metabolism of homocysteine. Their deficiency probably contributes to the majority of cases of hyperhomocysteinemia, a disorder associated with increased atherosclerotic risk. Supplementation of these vitamins has reduced the incidence of hyperhomocysteinemia in the United States; however, the clinical cardiovascular benefit of normalizing elevated homocysteine levels has not been proved.
OBESITY
(See also Chap. 415e) Obesity is associated with an increased prevalence of hypertension, glucose intolerance, atherosclerotic CAD, atrial fibrillation, obstructive sleep apnea, and pulmonary hypertension, and is associated with increased cardiovascular morbidity and mortality rates. In addition, obese patients have a distinct hemodynamic profile characterized by increased total and central blood volumes, increased cardiac output, and elevated left ventricular filling pressure. The elevated cardiac output appears to be required to support the metabolic demands of the excess adipose tissue. Left ventricular filling pressure is often at the upper limits of normal at rest and rises excessively with exercise, contributing to exertional dyspnea. In part as a result of chronic volume overload, eccentric cardiac hypertrophy with cardiac dilation and ventricular diastolic and/or systolic dysfunction may develop. In addition, altered levels of adipokines secreted by adipose tissue may contribute to adverse myocardial remodeling via direct effects on cardiac myocytes and other cells. Pathologically, there is left and, in some cases, right ventricular hypertrophy and generalized cardiac dilation. Pulmonary congestion, peripheral edema, and exercise intolerance may all ensue; however, the recognition of these findings may be difficult in massively obese patients.
Treatment with angiotensin-converting enzyme inhibitors, sodium restriction, and diuretics may be useful to control heart failure symptoms. Weight reduction, however, is the most effective therapy and results in reduction in blood volume and the return of cardiac output toward normal. However, rapid weight reduction may be dangerous, as cardiac arrhythmias and sudden death owing to electrolyte imbalance have been described.
THYROID DISEASE
(See also Chap. 405) Thyroid hormone exerts a major influence on the cardiovascular system by a number of direct and indirect mechanisms, and not surprisingly, cardiovascular effects are prominent in both hypo- and hyperthyroidism. Thyroid hormone causes increases in total-body metabolism and oxygen consumption that indirectly increase the cardiac workload. In addition, thyroid hormone exerts direct inotropic, chronotropic, and dromotropic effects that are similar to those seen with adrenergic stimulation (e.g., tachycardia, increased cardiac output); they are mediated at least partly by both transcriptional and nontranscriptional effects of thyroid hormone on myosin, calcium-activated ATPase, Na+-K+-ATPase, and myocardial β-adrenergic receptors.
Hyperthyroidism Common cardiovascular manifestations of hyperthyroidism include palpitations, systolic hypertension, and fatigue. Sinus tachycardia is present in ~40% of hyperthyroid patients, and atrial fibrillation is present in ~15%. Physical examination may reveal a hyperdynamic precordium, a widened pulse pressure, increases in the intensity of the first heart sound and the pulmonic component of the second heart sound, and a third heart sound. An increased incidence of mitral valve prolapse has been described in hyperthyroid patients, in which case a midsystolic murmur may be heard at the left sternal border with or without a midsystolic click. A systolic pleuropericardial friction rub (Means-Lerman scratch) may be heard at the left second intercostal space during expiration and is thought to result from the hyperdynamic cardiac motion.
Elderly patients with hyperthyroidism may present with only cardiovascular manifestations of thyrotoxicosis such as sinus tachycardia, atrial fibrillation, and hypertension, all of which may be resistant to therapy until the hyperthyroidism is controlled. Angina pectoris and CHF are unusual with hyperthyroidism unless there is coexistent heart disease; in such cases, symptoms often resolve with treatment of the hyperthyroidism.
Hypothyroidism Cardiac manifestations of hypothyroidism include a reduction in cardiac output, stroke volume, heart rate, systolic blood pressure, and pulse pressure. Pericardial effusions are present in about one-third of patients, rarely progress to tamponade, and probably result from increased capillary permeability. Other clinical signs include cardiomegaly, bradycardia, weak arterial pulses, distant heart sounds, and pleural effusions. Although the signs and symptoms of myxedema may mimic those of CHF, in the absence of other cardiac disease, myocardial failure is uncommon. The ECG generally reveals sinus bradycardia and low voltage and may show prolongation of the QT interval, decreased P-wave voltage, prolonged AV conduction time, intraventricular conduction disturbances, and nonspecific ST-T-wave abnormalities. Chest x-ray may show cardiomegaly, often with a “water bottle” configuration; pleural effusions; and, in some cases, evidence of CHF. Pathologically, the heart is pale and dilated and often demonstrates myofibrillar swelling, loss of striations, and interstitial fibrosis.
Patients with hypothyroidism frequently have elevations of cholesterol and triglycerides, resulting in premature atherosclerotic CAD. Before treatment with thyroid hormone, patients with hypothyroidism frequently do not have angina pectoris, presumably because of the low metabolic demands caused by their condition. However, angina and myocardial infarction may be precipitated during initiation of thyroid hormone replacement, especially in elderly patients with underlying heart disease. Therefore, replacement should be done with care, starting with low doses that are increased gradually.
MALIGNANT CARCINOID
(See also Chap. 113) Carcinoid tumors most often originate in the small bowel and elaborate a variety of vasoactive amines (e.g., serotonin), kinins, indoles, and prostaglandins that are believed to be responsible for the diarrhea, flushing, and labile blood pressure that characterize the carcinoid syndrome. Some 50% of patients with carcinoid syndrome have cardiac involvement, usually manifesting as abnormalities of the tricuspid or pulmonic valves. These patients invariably have hepatic metastases that allow vasoactive substances to circumvent hepatic metabolism. Left-sided cardiac involvement is rare and indicates either pulmonary carcinoid or an intracardiac shunt. Pathologically, carcinoid lesions are fibrous plaques that consist of smooth-muscle cells embedded in a stroma of glycosaminoglycans and collagen. They occur on the cardiac valves, where they cause valvular dysfunction, as well as on the endothelium of the cardiac chambers and great vessels.
Carcinoid heart disease most often presents as tricuspid regurgitation, pulmonic stenosis, or both. In some cases, a high cardiac output state may occur, presumably as a result of a decrease in systemic vascular resistance resulting from vasoactive substances released by the tumor. Treatment with somatostatin analogues (e.g., octreotide) or interferon α improves symptoms and survival in patients with carcinoid heart disease but does not appear to improve valvular abnormalities. Treatment with diuretics usually mitigates the symptoms of right heart failure; in some severely symptomatic patients, valve replacement is indicated. Coronary artery spasm, presumably due to a circulating vasoactive substance, may occur in patients with carcinoid syndrome.
PHEOCHROMOCYTOMA
(See also Chap. 407) In addition to causing labile or sustained hypertension, the high circulating levels of catecholamines resulting from a pheochromocytoma may cause direct myocardial injury. Focal myocardial necrosis and inflammatory cell infiltration are present in ~50% of patients who die with pheochromocytoma and may contribute to clinically significant left ventricular failure and pulmonary edema. In addition, associated hypertension results in left ventricular hypertrophy. Left ventricular dysfunction and CHF may resolve after removal of the tumor.
ACROMEGALY
(See also Chap. 401e) Exposure of the heart to excessive growth hormone may cause CHF as a result of high cardiac output, diastolic dysfunction owing to ventricular hypertrophy (with increased left ventricular chamber size or wall thickness), or global systolic dysfunction. Hypertension occurs in up to one-third of patients with acromegaly and is characterized by suppression of the renin-angiotensin-aldosterone axis and increases in total-body sodium and plasma volume. Some form of cardiac disease occurs in about one-third of patients with acromegaly and is associated with a doubling of the risk of cardiac death.
RHEUMATOID ARTHRITIS AND THE COLLAGEN VASCULAR DISEASES
Rheumatoid Arthritis (See also Chap. 380) Rheumatoid arthritis may be associated with inflammatory changes in any or all cardiac structures, although pericarditis is the most common clinical entity. Pericardial effusions are found on echocardiography in 10–50% of patients with rheumatoid arthritis, particularly those with subcutaneous nodules. Nonetheless, only a small fraction of these patients have symptomatic pericarditis, and when present, it usually follows a benign course, only occasionally progressing to cardiac tamponade or constrictive pericarditis. The pericardial fluid is generally exudative, with decreased concentrations of complement and glucose and elevated cholesterol. Coronary arteritis with intimal inflammation and edema is present in ~20% of cases but only rarely results in angina pectoris or myocardial infarction. Inflammation and granuloma formation may affect the cardiac valves, most often the mitral and aortic valves, and may cause clinically significant regurgitation owing to valve deformity. Myocarditis is uncommon and rarely results in cardiac dysfunction.
Treatment is directed at the underlying rheumatoid arthritis and may include glucocorticoids. Urgent pericardiocentesis should be performed in patients with tamponade, but pericardiectomy usually is required in cases of pericardial constriction.
Seronegative Arthropathies (See also Chap. 384) The seronegative arthropathies, including ankylosing spondylitis, reactive arthritis, psoriatic arthritis, and the arthritides associated with ulcerative colitis and regional enteritis, are all strongly associated with the HLA-B27 histocompatibility antigen and may be accompanied by a pancarditis and proximal aortitis. The aortic inflammation usually is limited to the aortic root but may extend to involve the aortic valve, mitral valve, and ventricular myocardium, resulting in aortic and mitral regurgitation, conduction abnormalities, and ventricular dysfunction. One-tenth of these patients have significant aortic insufficiency, and one-third have conduction disturbances; both are more common in patients with peripheral joint involvement and long-standing disease. Treatment with aortic valve replacement and permanent pacemaker implantation may be required. Occasionally, aortic regurgitation precedes the onset of arthritis, and therefore, the diagnosis of a seronegative arthritis should be considered in young males with isolated aortic regurgitation.
Systemic Lupus Erythematosus (SLE) (See also Chap. 378) A significant percentage of patients with SLE have cardiac involvement. Pericarditis is common, occurring in about two-thirds of patients, and generally follows a benign course, although rarely tamponade or constriction may result. The characteristic endocardial lesions of SLE are verrucous valvular abnormalities known as Libman-Sacks endocarditis. They most often are located on the left-sided cardiac valves, particularly on the ventricular surface of the posterior mitral leaflet, and are made up almost entirely of fibrin. These lesions may embolize or become infected but rarely cause hemodynamically important valvular regurgitation. Myocarditis generally parallels the activity of the disease and, although common histologically, seldom results in clinical heart failure unless associated with hypertension. Although arteritis of epicardial coronary arteries may occur, it rarely results in myocardial ischemia. There is, however, an increased incidence of coronary atherosclerosis that probably is related more to associated risk factors and glucocorticoid use than to SLE itself. Patients with the antiphospholipid antibody syndrome may have a higher incidence of cardiovascular abnormalities, including valvular regurgitation, venous and arterial thrombosis, premature stroke, myocardial infarction, pulmonary hypertension, and cardiomyopathy.
SECTION 5 |
CORONARY AND PERIPHERAL VASCULAR DISEASE |
291e |
The Pathogenesis, Prevention, and Treatment of Atherosclerosis |
PATHOGENESIS
Atherosclerosis remains the major cause of death and premature disability in developed societies. Moreover, current predictions estimate that by the year 2020 cardiovascular diseases, notably atherosclerosis, will become the leading global cause of total disease burden. Although many generalized or systemic risk factors predispose to its development, atherosclerosis affects various regions of the circulation preferentially and has distinct clinical manifestations that depend on the particular circulatory bed affected. Atherosclerosis of the coronary arteries commonly causes myocardial infarction (MI) (Chap. 295) and angina pectoris (Chap. 293). Atherosclerosis of the arteries supplying the central nervous system frequently provokes strokes and transient cerebral ischemia (Chap. 446). In the peripheral circulation, atherosclerosis causes intermittent claudication and gangrene and can jeopardize limb viability. Involvement of the splanchnic circulation can cause mesenteric ischemia. Atherosclerosis can affect the kidneys either directly (e.g., renal artery stenosis) or as a common site of atheroembolic disease (Chap. 301).
Even within a particular arterial bed, stenoses due to atherosclerosis tend to occur focally, typically in certain predisposed regions. In the coronary circulation, for example, the proximal left anterior descending coronary artery exhibits a particular predilection for developing atherosclerotic disease. Similarly, atherosclerosis preferentially affects the proximal portions of the renal arteries and, in the extracranial circulation to the brain, the carotid bifurcation. Indeed, atherosclerotic lesions often form at branch points of arteries, regions characterized disturbed hydrodynamics. Not all manifestations of atherosclerosis result from stenotic, occlusive disease. Ectasia and the development of aneurysmal disease, for example, frequently occur in the aorta (Chap. 301). In addition to focal, flow-limiting stenoses, nonocclusive intimal atherosclerosis also occurs diffusely in affected arteries, as shown by intravascular imaging and postmortem studies.
Atherogenesis in humans typically occurs over a period of many years, usually many decades. Growth of atherosclerotic plaques probably does not occur in a smooth, linear fashion but discontinuously, with periods of relative quiescence punctuated by periods of rapid evolution. After a generally prolonged “silent” period, atherosclerosis may become clinically manifest. The clinical expressions of atherosclerosis may be chronic, as in the development of stable, effort-induced angina pectoris or predictable and reproducible intermittent claudication. Alternatively, a dramatic acute clinical event such as MI, stroke, or sudden cardiac death may first herald the presence of atherosclerosis. Other individuals may never experience clinical manifestations of arterial disease despite the presence of widespread atherosclerosis demonstrated postmortem.
INITIATION OF ATHEROSCLEROSIS
An integrated view of experimental results in animals and studies of human atherosclerosis suggests that the “fatty streak” represents the initial lesion of atherosclerosis. These early lesions most often seem to arise from focal increases in the content of lipoproteins within regions of the intima. In particular, the fraction of lipoproteins related to low-density lipoprotein (LDL) that bear apolipoprotein B appear causally related to atherosclerosis. This accumulation of lipoprotein particles may not result simply from increased permeability, or “leakiness,” of the overlying endothelium (Fig. 291e-1). Rather, the lipoproteins may collect in the intima of arteries because they bind to constituents of the extracellular matrix, increasing the residence time of the lipid-rich particles within the arterial wall. Lipoproteins that accumulate in the extracellular space of the intima of arteries often associate with proteoglycans of the arterial extracellular matrix, an interaction that may slow the egress of these lipid-rich particles from the intima. Lipoprotein particles in the extracellular space of the intima, particularly those retained by binding to matrix macromolecules, may undergo oxidative modifications. Considerable evidence supports a pathogenic role for products of oxidized lipoproteins in atherogenesis. Lipoproteins sequestered from (plasma) antioxidants in the extracellular space of the intima become particularly susceptible to oxidative modification, giving rise to hydroperoxides, lysophospholipids, oxysterols, and aldehydic breakdown products of fatty acids and phospholipids. Modifications of the apoprotein moieties may include breaks in the peptide backbone as well as derivatization of certain amino acid residues. Local production of hypochlorous acid by myeloperoxidase associated with inflammatory cells within the plaque yields chlorinated species such as chlorotyrosyl moieties. Considerable evidence supports the presence of such oxidation products in atherosclerotic lesions.
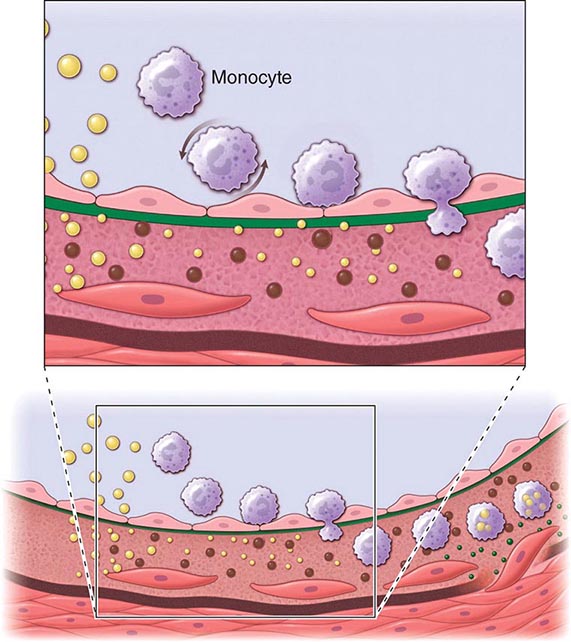
FIGURE 291e-1 Cross-sectional view of an artery depicting steps in development of an atheroma, from left to right. The upper panel shows a detail of the boxed area below. The endothelial monolayer overlying the intima contacts blood. Hypercholesterolemia promotes accumulation of low-density lipoprotein (LDL) particles (yellow spheres) in the intima. The lipoprotein particles often associate with constituents of the extracellular matrix, notably proteoglycans. Sequestration within the intima separates lipoproteins from some plasma antioxidants and favors oxidative modification. Such modified lipoprotein particles (darker spheres) may trigger a local inflammatory response that signals subsequent steps in lesion formation. The augmented expression of various adhesion molecules for leukocytes recruits monocytes to the site of a nascent arterial lesion.
Once adherent, some white blood cells migrate into the intima. The directed migration of leukocytes probably depends on chemoattractant factors, including modified lipoprotein particles themselves and chemoattractant cytokines (depicted by the smaller green spheres), such as the chemokine macrophage chemoattractant protein-1 produced by vascular wall cells in response to modified lipoproteins. Leukocytes in the evolving fatty streak can divide and exhibit augmented expression of receptors for modified lipoproteins (scavenger receptors). These mononuclear phagocytes ingest lipids and become foam cells, represented by a cytoplasm filled with lipid droplets. As the fatty streak evolves into a more complicated atherosclerotic lesion, smooth-muscle cells migrate from the media (bottom of lower panel hairline) through the internal elastic membrane (solid wavy line) and accumulate within the expanding intima, where they lay down extracellular matrix that forms the bulk of the advanced lesion (bottom panel, right side).
Leukocyte Recruitment Accumulation of leukocytes characterizes the formation of early atherosclerotic lesions (Fig. 291e-1). Thus, from its very inception, atherogenesis involves elements of inflammation, a process that now provides a unifying theme in the pathogenesis of this disease. The inflammatory cell types typically found in the evolving atheroma include monocyte-derived macrophages and dendritic cells, T and B lymphocytes, and mast cells. Hypercholesterolemia augments the portion of particularly proinflammatory monocytes in blood that preferentially enter the nascent atheroma in mice. A number of adhesion molecules or receptors for leukocytes expressed on the surface of the arterial endothelial cell probably participate in the recruitment of leukocytes to the nascent atheroma. Proinflammatory cytokines can augment the expression of leukocyte adhesion molecules.
Laminar shear forces such as those encountered in most regions of normal arteries also can suppress the expression of leukocyte adhesion molecules. Sites of predilection for atherosclerotic lesions (e.g., distal to flow dividers) often have low shear stress and/or disturbed flow. Ordered, pulsatile laminar shear of normal blood flow augments the production of nitric oxide by endothelial cells. This molecule, in addition to its vasodilator properties, can act at the low levels constitutively produced by arterial endothelium as a local anti-inflammatory autacoid, e.g., limiting local adhesion molecule expression. Exposure of endothelial cells to laminar shear stress increases the transcription of Krüppel-like factor 2 (KLF2), which augments the activity of numerous salutary endothelial functions including nitric oxide synthase. Laminar shear stress also stimulates endothelial cells to produce superoxide dismutase, an antioxidant enzyme. These examples indicate how hemodynamic forces may influence the cellular events that underlie atherosclerotic lesion initiation and potentially explain the favored localization of atherosclerotic lesions at sites that experience disturbed flow or low shear stress.
Once captured on the surface of the arterial endothelial cell by adhesion receptors, the leukocytes penetrate the endothelial layer and take up residence in the intima. In addition to products of modified lipoproteins, cytokines (protein mediators of inflammation) can regulate the expression of adhesion molecules involved in leukocyte recruitment. For example, interleukin 1 (IL-1) and tumor necrosis factor (TNF) induce or augment the expression of leukocyte adhesion molecules on endothelial cells. Because products of lipoprotein oxidation can induce cytokine release from vascular wall cells, this pathway may provide an additional link between arterial accumulation of lipoproteins and leukocyte recruitment. Chemoattractant cytokines appear to direct the migration of leukocytes into the arterial wall.
Foam-Cell Formation Once resident within the intima, the mononuclear phagocytes mature into macrophages and become lipid-laden foam cells, a conversion that requires the uptake of lipoprotein particles by receptor-mediated endocytosis. One might suppose that the “classic” LDL receptor mediates this lipid uptake; however, humans or animals lacking effective LDL receptors due to genetic alterations (e.g., familial hypercholesterolemia) have abundant arterial lesions and extraarterial xanthomata rich in macrophage-derived foam cells. In addition, the exogenous cholesterol suppresses expression of the LDL receptor; thus, the level of this cell-surface receptor for LDL decreases under conditions of cholesterol excess. Candidates for alternative receptors that can mediate lipid loading of foam cells include a number of macrophage “scavenger” receptors, which preferentially endocytose modified lipoproteins, and other receptors for oxidized LDL or very low-density lipoprotein (VLDL). Monocyte attachment to the endothelium, migration into the intima, and maturation to form lipid-laden macrophages thus represent key steps in the formation of the fatty streak, the precursor of fully formed atherosclerotic plaques.
ATHEROMA EVOLUTION AND COMPLICATIONS
Although the fatty streak commonly precedes the development of a more advanced atherosclerotic plaque, not all fatty streaks progress to form complex atheromata. By ingesting lipids from the extracellular space, the mononuclear phagocytes bearing such scavenger receptors may remove lipoproteins from the developing lesion. Some lipid-laden macrophages may leave the artery wall, exporting lipid in the process. Lipid accumulation, and hence the propensity to form an atheroma, ensues if the amount of lipid entering the artery wall exceeds that removed by mononuclear phagocytes or other pathways. Macrophages also proliferate in plaques in response to hematopoietic growth factors overexpressed in lesions, another aspect of the dynamic regulation and flux of cells during atherogenesis.
Export by phagocytes may constitute one response to local lipid overload in the evolving lesion. Another mechanism, reverse cholesterol transport mediated by high-density lipoproteins (HDLs), probably provides an independent pathway for lipid removal from atheroma. This transfer of cholesterol from the cell to the HDL particle involves specialized cell-surface molecules such as the ATP binding cassette (ABC) transporters. ABCA1, the gene mutated in Tangier disease, a condition characterized by very low HDL levels, transfers cholesterol from cells to nascent HDL particles and ABCG1 to mature HDL particles. “Reverse cholesterol transport” mediated by these ABC transporters allows HDL loaded with cholesterol to deliver it to hepatocytes by binding to scavenger receptor B1 or other receptors. The liver cell can metabolize the sterol to bile acids that can be excreted. Thus, macrophages may play a vital role in the dynamic economy of lipid accumulation in the arterial wall during atherogenesis.
Some lipid-laden foam cells within the expanding intimal lesion perish. Some foam cells may die as a result of programmed cell death, or apoptosis. This death of mononuclear phagocytes results in the formation of the lipid-rich center, often called the necrotic core, in established atherosclerotic plaques. Impaired clearance of dead foam cells (efferocytosis) in plaques may hasten lipid core formation. Macrophages loaded with modified lipoproteins may elaborate microparticles or exosomes (which may contain regulatory microRNAs), cytokines, and growth factors that can further signal some of the cellular events in lesion complication. Whereas accumulation of lipid-laden macrophages characterizes the fatty streak, buildup of fibrous tissue formed by extracellular matrix typifies the more advanced atherosclerotic lesion. The smooth-muscle cell synthesizes the bulk of the extracellular matrix of the complex atherosclerotic lesion. A number of growth factors or cytokines elaborated by mononuclear phagocytes can stimulate smooth-muscle cell proliferation and production of extracellular matrix. Cytokines found in the plaque, including IL-1 and TNF, can induce local production of growth factors, including forms of platelet-derived growth factor (PDGF), fibroblast growth factors, and others, which may contribute to plaque evolution and complication. Other cytokines, notably interferon γ (IFN-γ) derived from activated T cells within lesions, can limit the synthesis of interstitial forms of collagen by smooth-muscle cells. These examples illustrate how atherogenesis involves a complex mix of mediators that in the balance determines the characteristics of particular lesions.
The accumulation of smooth-muscle cells and their elaboration of extracellular matrix probably provide a critical transition, yielding a fibrofatty lesion in place of a simple accumulation of macrophage-derived foam cells. For example, PDGF elaborated by activated platelets, macrophages, and endothelial cells can stimulate the migration of smooth-muscle cells normally resident in the tunica media into the intima. Such growth factors and cytokines produced locally can stimulate the proliferation of resident smooth-muscle cells or resident stem cells in the intima as well as those that may migrate in from the media. Transforming growth factor β (TGF-β), among other mediators, potently stimulates interstitial collagen production by smooth-muscle cells. These mediators may arise not only from neighboring vascular cells or leukocytes (a “paracrine” pathway), but also, in some instances, from the same cell that responds to the factor (an “autocrine” pathway). Together, these alterations in smooth-muscle cells, signaled by these mediators acting at short distances, can hasten transformation of the fatty streak into a more fibrous smooth-muscle cell and extracellular matrix—rich lesion.
In addition to locally produced mediators, products of blood coagulation and thrombosis likely contribute to atheroma evolution and complication. This involvement justifies the use of the term atherothrombosis to convey the inextricable links between atherosclerosis and thrombosis. Fatty streak formation begins beneath a morphologically intact endothelium. In advanced fatty streaks, however, microscopic breaches in endothelial integrity may occur. Microthrombi rich in platelets can form at such sites of limited endothelial denudation, owing to exposure of the thrombogenic extracellular matrix of the underlying basement membrane. Activated platelets release numerous factors that can promote the fibrotic response, including PDGF and TGF-β. Thrombin not only generates fibrin during coagulation, but also stimulates protease-activated receptors that can signal smooth-muscle migration, proliferation, and extracellular matrix production. Many arterial mural microthrombi resolve without clinical manifestation by a process of local fibrinolysis, resorption, and endothelial repair, yet can lead to lesion progression by stimulating these profibrotic functions of smooth-muscle cells (Fig. 291e-2D).
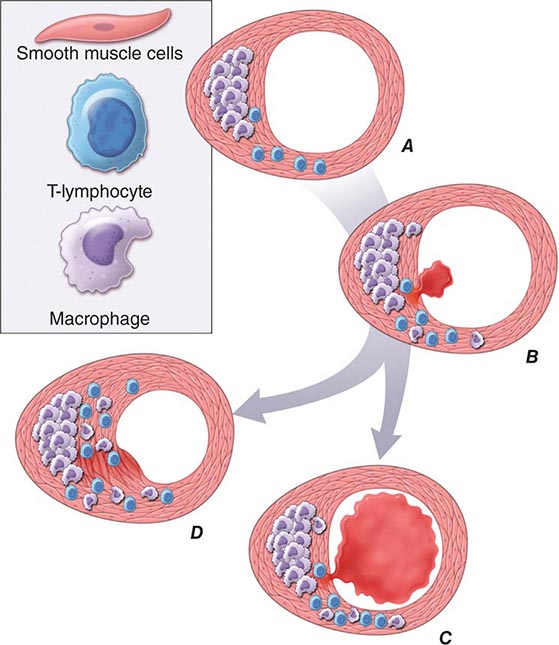
FIGURE 291e-2 Plaque rupture, thrombosis, and healing. A. Arterial remodeling during atherogenesis. During the initial part of the life history of an atheroma, growth is often outward, preserving the caliber of the lumen. This phenomenon of “compensatory enlargement” accounts in part for the tendency of coronary arteriography to underestimate the degree of atherosclerosis. B. Rupture of the plaque’s fibrous cap causes thrombosis. Physical disruption of the atherosclerotic plaque commonly causes arterial thrombosis by allowing blood coagulant factors to contact thrombogenic collagen found in the arterial extracellular matrix and tissue factor produced by macrophage-derived foam cells in the lipid core of lesions. In this manner, sites of plaque rupture form the nidus for thrombi. The normal artery wall has several fibrinolytic or antithrombotic mechanisms that tend to resist thrombosis and lyse clots that begin to form in situ. Such antithrombotic or thrombolytic molecules include thrombomodulin, tissue- and urokinase-type plasminogen activators, heparan sulfate proteoglycans, prostacyclin, and nitric oxide. C. When the clot overwhelms the endogenous fibrinolytic mechanisms, it may propagate and lead to arterial occlusion. The consequences of this occlusion depend on the degree of existing collateral vessels. In a patient with chronic multivessel occlusive coronary artery disease (CAD), collateral channels have often formed. In such circumstances, even a total arterial occlusion may not lead to myocardial infarction (MI), or it may produce an unexpectedly modest or a non-ST-segment elevation infarct because of collateral flow. In a patient with less advanced disease and without substantial stenotic lesions to provide a stimulus for collateral vessel formation, sudden plaque rupture and arterial occlusion commonly produces an ST-segment elevation infarction. These are the types of patients who may present with MI or sudden death as a first manifestation of coronary atherosclerosis. In some cases, the thrombus may lyse or organize into a mural thrombus without occluding the vessel. Such instances may be clinically silent. D. The subsequent thrombin-induced fibrosis and healing causes a fibroproliferative response that can lead to a more fibrous lesion that can produce an eccentric plaque that causes a hemodynamically significant stenosis. In this way, a nonocclusive mural thrombus, even if clinically silent or causing unstable angina rather than infarction, can provoke a healing response that can promote lesion fibrosis and luminal encroachment. Such a sequence of events may convert a “vulnerable” atheroma with a thin fibrous cap that is prone to rupture into a more “stable” fibrous plaque with a reinforced cap. Angioplasty of unstable coronary lesions may “stabilize” the lesions by a similar mechanism, producing a wound followed by healing.
Microvessels As atherosclerotic lesions advance, abundant plexi of microvessels develop in connection with the artery’s vasa vasorum. Newly developing microvascular networks may contribute to lesion complications in several ways. These blood vessels provide an abundant surface area for leukocyte trafficking and may serve as the portal for entry and exit of white blood cells from the established atheroma. Microvessels in the plaques may also furnish foci for intraplaque hemorrhage. Like the neovessels in the diabetic retina, microvessels in the atheroma may be friable and prone to rupture and can produce focal hemorrhage. Such a vascular leak can provoke thrombosis in situ, yielding local thrombin generation, which in turn can activate smooth-muscle and endothelial cells through ligation of protease-activated receptors. Atherosclerotic plaques often contain fibrin and hemosiderin, an indication that episodes of intraplaque hemorrhage contribute to plaque complications.
CALCIFICATION As they advance, atherosclerotic plaques also accumulate calcium. Microvesicles derived from lesional cells can stimulate calcification, and this process co-localizes with regions of heightened inflammation. Mineralization of the atherosclerotic plaque recapitulates many aspects of bone formation, including the regulatory participation of transcription factors such as Runx2.
Plaque Evolution Smooth-muscle cells and macrophages die in the atherosclerotic plaque. Indeed, complex atheromata often have a mostly fibrous character and lack the cellularity of less advanced lesions. This relative paucity of smooth-muscle cells in advanced atheromata may result from the predominance of cytostatic mediators such as TGF-β and IFN-γ (which can inhibit smooth-muscle cell proliferation) and also from smooth-muscle cell apoptosis. Thus, during the evolution of the atherosclerotic plaque, a complex and highly regulated balance between entry and egress of lipoproteins and leukocytes, cell proliferation and cell death, extracellular matrix production, and remodeling, as well as calcification and neovascularization, contribute to lesion formation. Many mediators related to atherogenic risk factors, including those derived from lipoproteins, cigarette smoking, and angiotensin II, provoke the production of proinflammatory cytokines and alter the behavior of the intrinsic vascular wall cells and infiltrating leukocytes that underlie the complex pathogenesis of these lesions. Thus, advances in vascular biology have led to increased understanding of the mechanisms that link risk factors to the pathogenesis of atherosclerosis and its complications.
PATHOPHYSIOLOGIC CONSEQUENCES OF ATHEROSCLEROSIS
Atherosclerotic lesions occur ubiquitously in Western societies, and the prevalence of this disease is on the rise globally. Most atheromata produce no symptoms, and many never cause clinical manifestations. Numerous patients with diffuse atherosclerosis may succumb to unrelated illnesses without ever having experienced a clinically significant manifestation of atherosclerosis. Arterial remodeling during atheroma formation accounts for some of this variability in the clinical expression of atherosclerotic disease (Fig. 291e-2A). During the initial phases of atheroma development, the plaque usually grows outward, in an abluminal direction. Vessels affected by atherogenesis tend to increase in diameter, a phenomenon known as compensatory enlargement, a type of vascular remodeling. The growing atheroma does not encroach on the arterial lumen until the burden of atherosclerotic plaque exceeds ~40% of the area encompassed by the internal elastic lamina. Thus, during much of its life history, an atheroma will not cause stenosis that can limit tissue perfusion.
Flow-limiting stenoses commonly form later in the history of the plaque. Many such plaques cause stable syndromes such as demand-induced angina pectoris or intermittent claudication in the extremities. In the coronary circulation and other circulations, even total vascular occlusion by an atheroma does not invariably lead to infarction. The hypoxic stimulus of repeated bouts of ischemia characteristically induces formation of collateral vessels in the myocardium, mitigating the consequences of an acute occlusion of an epicardial coronary artery. By contrast, many lesions that cause acute or unstable atherosclerotic syndromes, particularly in the coronary circulation, may arise from atherosclerotic plaques that do not produce a flow-limiting stenosis. Such lesions may produce only minimal luminal irregularities on traditional angiograms and often do not meet the traditional criteria for “significance” by arteriography. Thrombi arising from such nonocclusive stenoses may explain the frequency of MI as an initial manifestation of coronary artery disease (CAD) (in at least one-third of cases) in patients who report no prior history of angina pectoris, a syndrome usually caused by flow-limiting stenoses.
Plaque Instability and Rupture Postmortem studies afford considerable insight into the microanatomic substrate underlying the “instability” of plaques that do not cause critical stenoses. A superficial erosion of the endothelium or a frank plaque rupture or fissure usually produces the thrombus that causes episodes of unstable angina pectoris or the occlusive and relatively persistent thrombus that causes acute MI (Fig. 291e-2B). Rupture of the plaque’s fibrous cap (Fig. 291e-2C) permits contact between coagulation factors in the blood and highly thrombogenic tissue factor expressed by macrophage foam cells in the plaque’s lipid-rich core. If the ensuing thrombus is nonocclusive or transient, the episode of plaque disruption may not cause symptoms or may result in episodic ischemic symptoms such as rest angina. Occlusive thrombi that endure often cause acute MI, particularly in the absence of a well-developed collateral circulation that supplies the affected territory. Repetitive episodes of plaque disruption and healing provide one likely mechanism of transition of the fatty streak to a more complex fibrous lesion (Fig. 291e-2D). The healing process in arteries, as in skin wounds, involves the laying down of new extracellular matrix and fibrosis.
Not all atheromata exhibit the same propensity to rupture. Pathologic studies of culprit lesions that have caused acute MI reveal several characteristic features. Plaques that have caused thromboses tend to have thin fibrous caps, relatively large lipid cores, a high content of macrophages, outward remodeling, and spotty (rather than dense) calcification. Morphometric studies of such culprit lesions show that at sites of plaque rupture, macrophages and T lymphocytes predominate and contain relatively few smooth-muscle cells. The cells that concentrate at sites of plaque rupture bear markers of inflammatory activation. In addition, patients with active atherosclerosis and acute coronary syndromes display signs of disseminated inflammation. Inflammatory mediators regulate processes that govern the integrity of the plaque’s fibrous cap and, hence, its propensity to rupture. For example, the T cell–derived cytokine IFN-γ, which is found in atherosclerotic plaques, can inhibit growth and collagen synthesis of smooth-muscle cells, as noted above. Cytokines derived from activated macrophages and lesional T cells can boost production of proteolytic enzymes that can degrade the extracellular matrix of the plaque’s fibrous cap. Thus, inflammatory mediators can impair the collagen synthesis required for maintenance and repair of the fibrous cap and trigger degradation of extracellular matrix macromolecules, processes that weaken the plaque’s fibrous cap and enhance its susceptibility to rupture (so-called vulnerable plaques, Fig. 291e-3). In contrast to plaques with these features of vulnerability, those with a dense extracellular matrix and relatively thick fibrous cap without substantial tissue factor–rich lipid cores seem generally resistant to rupture and unlikely to provoke thrombosis.
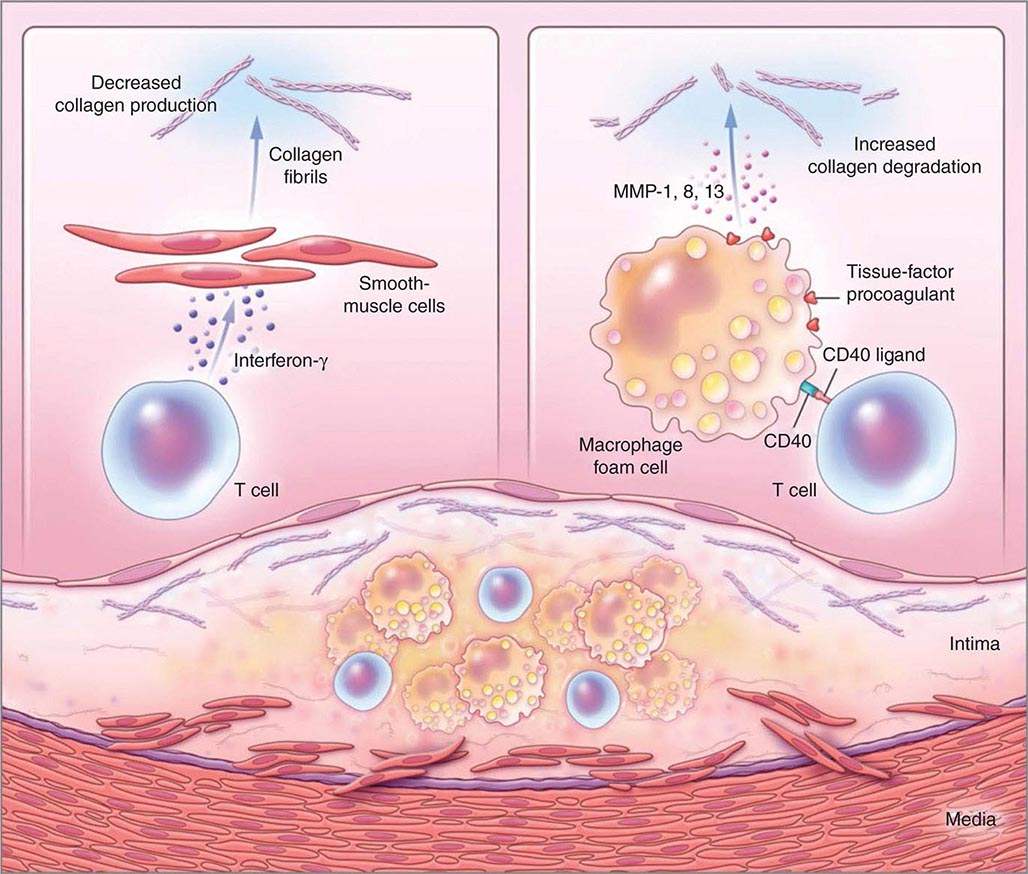
FIGURE 291e-3 Inflammatory pathways that predispose atherosclerotic plaques to rupture and provoke thrombosis. A cross-section of an atheromatous plaque at the bottom of the figure shows the central lipid core that contains macrophage foam cells (yellow) and T cells (blue). The intima and media also contain arterial smooth-muscle cells (red), which are the source of arterial collagen (depicted as triple helical coiled structures). Activated T cells (of the type 1 helper T cell subtype) secrete cytokine interferon γ, which inhibits the production of the new, interstitial collagen that is required to repair and maintain the plaque’s protective fibrous cap (upper left). The T cells can also activate the macrophages in the intimal lesion by expressing the inflammatory mediator CD40 ligand (CD154), which engages its cognate receptor (CD40) on the phagocyte. This inflammatory signalling causes overproduction of interstitial collagenases (matrix metalloproteinases [MMPs] 1, 8, and 13) that catalyze the initial rate-limiting step in collagen breakdown (top right). CD40 ligation also causes macrophages to overproduce tissue-factor procoagulant. Thus, inflammatory signalling puts the collagen in the plaque’s fibrous cap in double jeopardy—decreasing synthesis and increasing breakdown—rendering the cap susceptible to rupture. Inflammatory activation also boosts tissue-factor production, which triggers thrombus formation in the disrupted plaque. These mechanisms link inflammation in the plaque to the thrombotic complications of atherosclerosis, including the acute coronary syndromes. (Adapted from P Libby: N Engl J Med 368:2004, 2013.)
Functional features of the atheromatous plaque, in addition to its degree of luminal encroachment, influence the clinical manifestations of this disease. This enhanced understanding of plaque biology provides insight into the diverse ways in which atherosclerosis can present clinically and the reasons why the disease may remain silent or stable for prolonged periods, punctuated by acute complications at certain times. Increased understanding of atherogenesis provides new insight into the mechanisms linking it to the risk factors discussed below, indicates the ways in which current therapies may improve outcomes, and suggests new targets for future intervention.
PREVENTION AND TREATMENT
THE CONCEPT OF ATHEROSCLEROTIC RISK FACTORS
The systematic study of risk factors for atherosclerosis emerged from a coalescence of experimental results, as well as from cross-sectional and ultimately longitudinal studies in humans. The prospective, community-based Framingham Heart Study provided rigorous support for the concept that hypercholesterolemia, hypertension, and other factors correlate with cardiovascular risk. Similar observational studies performed worldwide bolstered the concept of “risk factors” for cardiovascular disease.
From a practical viewpoint, the cardiovascular risk factors that have emerged from such studies fall into two categories: those modifiable by lifestyle and/or pharmacotherapy, and those that are immutable, such as age and sex. The weight of evidence supporting various risk factors differs. For example, hypercholesterolemia and hypertension certainly predict coronary risk, but the magnitude of the contributions of other so-called nontraditional risk factors, such as levels of homocysteine, levels of lipoprotein (a) [Lp(a)], and infection, remains controversial. Moreover, some biomarkers that predict cardiovascular risk may not participate in the causal pathway for the disease or its complications. Genetic studies using genome-wide association (GWAS) approaches and Mendelian randomization approaches have helped to distinguish between risk markers and factors that contribute causally to the disease. For example, recent genetic studies suggest that C-reactive protein (CRP) does not itself mediate atherogenesis, despite its ability to predict risk, whereas Lp(a) and apolipoprotein C3 have emerged as a causal risk factor. Table 291e-1 lists a number of risk factors implicated in atherosclerosis. The sections below will consider some of these factors and approaches to their modification.
|
MAJOR RISK FACTORS FOR ATHEROSCLEROSIS |
aHDL cholesterol ≥1.6 mmol/L (≥60 mg/dL) has been viewed as a “negative” risk factor.
Abbreviations: BMI, body mass index; BP, blood pressure; CHD, coronary heart disease; HDL, high-density lipoprotein; LDL, low-density lipoprotein.
Lipid Disorders
Abnormalities in plasma lipoproteins and derangements in lipid metabolism rank among the most firmly established and best understood risk factors for atherosclerosis. Chapter 421 describes the lipoprotein classes and provides a detailed discussion of lipoprotein metabolism.
The American College of Cardiology and American Heart Association (ACC/AHA) promulgated new guidelines on risk assessment, lifestyle measures, and cholesterol management in 2013. The panels that produced these guidelines followed an evidence-based approach. the 2013 cholesterol guideline focused on 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase inhibitors (statins) rather than other classes of lipid-modulating drugs, including fibric acid derivatives, cholesterol absorption inhibitors such as ezetimibe, and niacin products. The guideline cites the lack of contemporary randomized clinical trial evidence that supports the efficacy of these nonstatin lipid-modifying agents in cardiovascular event reduction. The cholesterol guideline defined four statin benefit groups (Table 291e-2): (1) all individuals who have clinical atherosclerotic cardiovascular disease (ASCVD), therefore considered “secondary prevention”; (2) those with LDL cholesterol ≥190 mg/dL without a secondary cause such as a high intake of saturated or trans fats, various drugs, or certain diseases; (3) individuals with diabetes without established cardiovascular disease who are 40–75 years old and have LDL cholesterol of 70–189 mg/dL; and (4) those without established ASCVD without diabetes who are 40–75 years old and who have LDL cholesterol of 70–189 mg/dL and a calculated ASCVD risk ≥7.5%. An online risk calculator based on pooled cohorts was provided to aid clinicians and patients in calculating their risk (http://my.americanheart.org/professional/StatementsGuidelines/PreventionGuidelines/Prevention-Guidelines_UCM_457698_SubHomePage.jsp). Other validated risk calculators that incorporate family history of CAD and a marker of inflammation (high-sensitivity CRP [hsCRP]) that apply to U.S. women and men exist (http://www.reynoldsriskscore.org). Downloadable applications for risk calculation on handheld devices are readily available.
|
SUMMARY OF THE FOUR STATIN BENEFIT GROUPS DESCRIBED IN THE 2013 ACC/AHA GUIDELINE ON THE TREATMENT OF BLOOD CHOLESTEROL TO REDUCE ATHEROSCLEROTIC CARDIOVASCULAR RISK IN ADULTS |
Abbreviations: ACC/AHA, American College of Cardiology and American Heart Association; ASCVD, atherosclerotic cardiovascular disease; LDL-C, low-density lipoprotein cholesterol.
Source: Adapted from NJ Stone et al: 2013 ACC/AHA Guideline on the Treatment of Blood Cholesterol to Reduce Atherosclerotic Cardiovascular Risk in Adults. J Am Coll Cardiol 2013, doi: 10.1016/j.jacc.2013.11.002.
The 2013 guideline emphasized a patient-centered approach and recommended that clinicians and patients engage in a risk-benefit conversation before starting statin therapy and not rely solely on calculated risks or arbitrary category assignment. It further emphasizes that medications do not supplant a healthy lifestyle. The guideline also provides some practical suggestions regarding management of muscle symptoms attributed to statins, an issue of considerable concern to many patients and practitioners alike.
In a major departure from prior guidelines, the 2013 guideline eliminates LDL targets as goals of therapy. The panel did so because major clinical trials did not titrate therapy to a goal, but rather used fixed doses of statins. Instead, the new guideline suggests different intensities of statin therapy based on risk category (Fig. 291e-4).
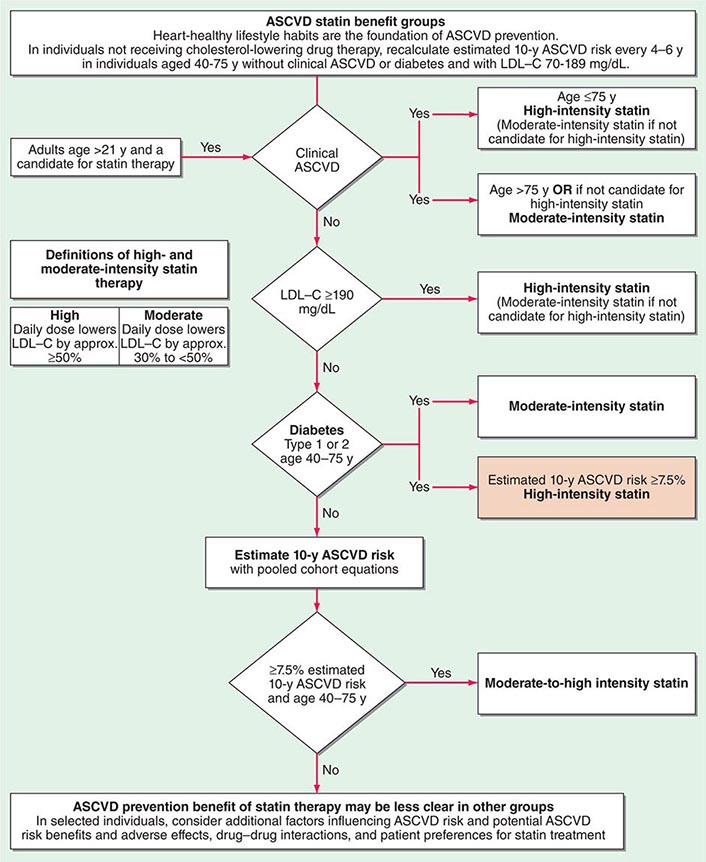
FIGURE 291e-4 Major recommendations for statin therapy for atherosclerotic cardiovascular disease (ASCVD) prevention. LDL-C, low-density lipoprotein cholesterol. (From NJ Stone et al: J Am Coll Cardiol, 2013, doi: 10.1016/j.jacc.2013.11.002.)
The 2013 guideline focus on statins reflects an extensive body of rigorous evidence that supports the effectiveness of this class of drugs in cardiovascular event reduction and an acceptable risk-benefit relationship (Fig. 291e-5). Moreover, because almost all statins are now available as generic statins medications, cost has become much less of an impediment to their use.
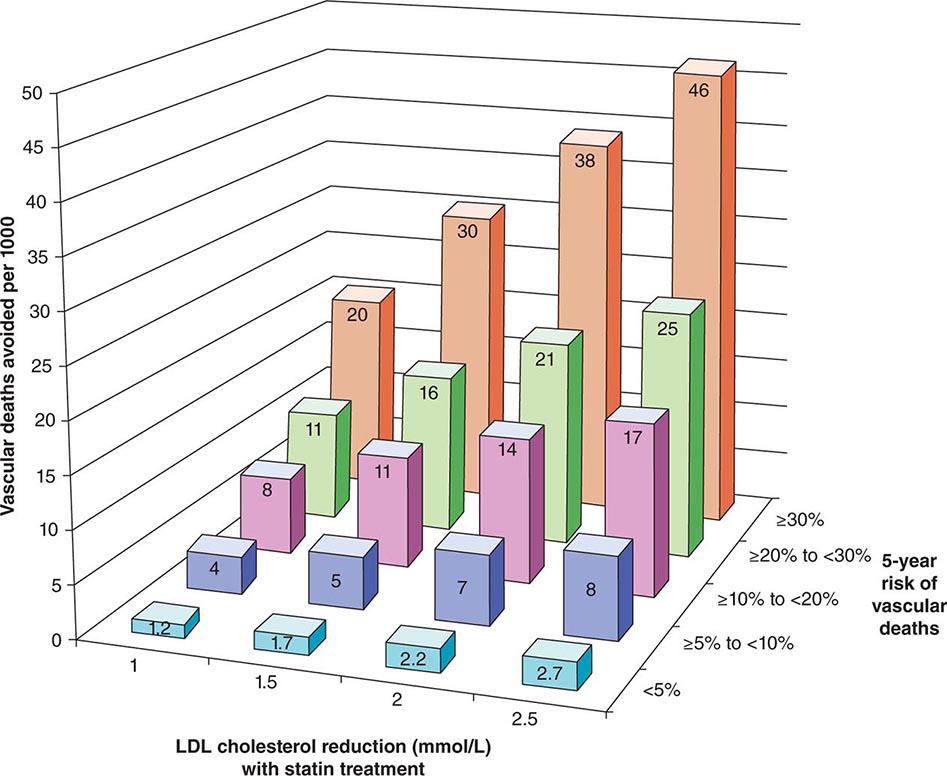
FIGURE 291e-5 The Cholesterol Treatment Trialists Collaboration meta-analyzed 27 randomized clinical trials evaluating statin therapy. They found profound decreases in both major vascular events and vascular death (not shown) proportional to the magnitude of low-density lipoprotein (LDL) cholesterol reduction achieved with statin treatment. This diagram shows the results of this meta-analysis for vascular death. (From Lancet 380:581, 2012.)
The clinical use of effective pharmacologic strategies for lowering LDL has reduced cardiovascular events markedly, but a considerable burden of residual risk remains even in patients treated with high-intensity statins. Hence, current studies are evaluating other avenues to address the residual burden of cardiovascular disease that persists despite statin treatment. Inhibitors of genetic studies identified proprotein convertase subtilisin kexin-like 9 (PCSK9) as a regulator of LDL levels associated with cardiovascular outcomes. Interaction of the LDL receptor with PCSK9 hastens the receptor’s degradation, and hence yields higher circulating LDL concentrations. Genetic variants that lower PCSK9 activity appear to protect against cardiovascular events. Monoclonal antibodies that neutralize PCSK9 lower LDL levels even in statin-treated patients and are currently under investigation as novel therapeutics to lower cardiovascular risk.
LDL-lowering therapies do not appear to exert their beneficial effect on cardiovascular events by causing a marked “regression” of stenoses. Studies of lipid lowering monitored by angiography or by intravascular imaging modalities have shown at best a modest reduction in coronary artery stenoses over the duration of study, despite abundant evidence of event reduction. These results suggest that the beneficial mechanism of lipid lowering by statins does not require a substantial reduction in the fixed stenoses. Rather, the benefit may derive from “stabilization” of atherosclerotic lesions without substantially decreased stenosis. Such stabilization of atherosclerotic lesions and the attendant decrease in coronary events may result from the egress of lipids or from favorably influencing aspects of the biology of atherogenesis discussed above. In addition, as sizable lesions may protrude abluminally rather than into the lumen due to complementary enlargement, shrinkage of such plaques may not be apparent on angiograms. The consistent benefit of statins may depend not only on their salutary effects on the lipid profile, but also on direct modulation of plaque biology independent of lipid lowering.
As the prevalence of metabolic syndrome and diabetes increases, many patients present with low concentrations of HDL (HDL cholesterol <1.0 mmol/L [<40 mg/dL]). A baseline measurement of HDL cholesterol indubitably correlates with future cardiovascular risk. Yet, the utility of therapies that raise HDL cholesterol levels in blood as effective interventions to reduce cardiovascular vascular events has come into question. Blood HDL levels vary inversely with those of triglycerides, and the independent role of HDL versus triglycerides as a cardiovascular risk factor remains unsettled. The 2013 guideline does not advocate any specific therapy for raising HDL. Indeed, multiple recent trials failed to show that raising HDL cholesterol levels improves cardiovascular outcomes, and recent genetic studies cast doubt on low HDL as a causal risk factor for atherosclerotic events. Weight loss and physical activity can raise HDL, and these lifestyle measures merit universal adoption (Table 291e-3). Nicotinic acid, particularly in combination with statins, can robustly raise HDL, but clinical trial data do not support the effectiveness of nicotinic acid in cardiovascular risk reduction. Agonists of nuclear receptors provide another potential avenue for raising HDL levels. Yet patients treated with peroxisome proliferator–activated receptors alpha and gamma (PPAR-α and -γ) agonists have not consistently shown improved cardiovascular outcomes, and at least some PPAR agonists have been associated with worsened cardiovascular outcomes. Other agents in clinical development raise HDL levels by inhibiting cholesteryl ester transfer protein (CETP). Two such agents have undergone large-scale clinical evaluation and have not shown efficacy in improving cardiovascular outcomes. Clinical studies currently under way will assess the effectiveness of two other CETP inhibitors that lack some of the adverse off-target actions encountered with the first agent tested.
|
HEART HEALTHY NUTRITION AND PHYSICAL ACTIVITY BEHAVIORS RECOMMENDED IN THE 2013 ACC/AHA GUIDELINE ON LIFESTYLE MANAGEMENT TO REDUCE CARDIOVASCULAR RISK |
Abbreviations: ACC/AHA, American College of Cardiology and American Heart Association; DASH, Dietary Approaches to Stop Hypertension; USDA, U.S. Department of Agriculture.
Source: Adapted from RH Eckel et al: 2013 AHA/ACC Guideline on Lifestyle Management to Reduce Cardiovascular Risk. J Am Coll Cardiol 2013, doi: 10.1016/j.jacc.2013.11.003.
The mechanism by which elevated LDL levels promote atherogenesis may involve oxidative modification. Yet, rigorous and well-controlled clinical trials have failed to demonstrate that antioxidant vitamin therapy improves coronary heart disease (CHD) outcomes. In regard to nontraditional risk factors including homocysteine and infection, large-scale clinical trials using vitamins to lower homocysteine or using antibiotics have not reduced cardiovascular events. Therefore, the current evidence base does not support the use of vitamins or antibiotics to lower cardiovascular risk.
Hypertension (See also Chap. 298) A wealth of epidemiologic data support a relationship between hypertension and atherosclerotic risk, and extensive clinical trial evidence has established that pharmacologic treatment of hypertension can reduce the risk of stroke, heart failure, and CHD events.
Diabetes Mellitus, Insulin Resistance, and the Metabolic Syndrome (See also Chap. 417) Most patients with diabetes mellitus die of atherosclerosis and its complications. Aging and rampant obesity underlie a current epidemic of type 2 diabetes mellitus. The abnormal lipoprotein profile associated with insulin resistance, known as diabetic dyslipidemia, accounts for part of the elevated cardiovascular risk in patients with type 2 diabetes. Although diabetic individuals often have LDL cholesterol levels near the average, the LDL particles tend to be smaller and denser and, therefore, more atherogenic. Other features of diabetic dyslipidemia include low HDL and elevated triglyceride levels. Hypertension also frequently accompanies obesity, insulin resistance, and dyslipidemia. This commonly encountered clinical cluster of risk factors has become known as the metabolic syndrome (Chap. 422). Despite legitimate concerns about whether clustered components confer more risk than the individual components, the metabolic syndrome concept may offer clinical utility.
Therapeutic objectives for intervention in these patients include addressing the underlying causes, including obesity and low physical activity, by initiating lifestyle measures (see below). Establishing that strict glycemic control reduces the risk of macrovascular complications of diabetes has proved much more elusive than the beneficial effects on microvascular complications such as retinopathy and renal disease. Indeed, “tight” glycemic control may increase adverse events in patients with type 2 diabetes, lending even greater importance to aggressive control of other aspects of risk in this patient population. In this regard, multiple clinical trials have demonstrated unequivocal benefit of statin therapy in diabetic patients over all ranges of LDL cholesterol levels (but not those with end-stage renal disease or advanced heart failure). Among the oral hypoglycemic agents, metformin possesses the best evidence base for cardiovascular event reduction. The novel oral hypoglycemic agents tested in sufficiently powered trials, the dipeptidyl peptidase-4 (DPP-4) inhibitors saxagliptin and alogliptin, did not show cardiovascular benefit. Indeed, saxagliptin was associated with a slight increase in heart failure. Diabetic populations appear to derive particular benefit from antihypertensive strategies that block the action of angiotensin II. Thus, the antihypertensive regimen for patients with the metabolic syndrome should include angiotensin-converting enzyme inhibitors or angiotensin receptor blockers when possible. Many of these individuals will require more than one antihypertensive agent to reach the 2013 goals for individuals 18 years of age or older with diabetes to achieve a systolic blood pressure of less than 140 mmHg and a diastolic blood pressure of less than 90 mmHg.
Male Sex/Postmenopausal State Decades of observational studies have verified excess coronary risk in men compared with premenopausal women. After menopause, however, coronary risk accelerates in women. Although observational and experimental studies have suggested that estrogen therapy reduces coronary risk, large-scale randomized clinical trials have not demonstrated a net benefit of estrogen with or without progestins on CHD outcomes. In the Heart and Estrogen/Progestin Replacement Study (HERS), postmenopausal female survivors of acute MI were randomized to an estrogen/progestin combination or to placebo. This study showed no overall reduction in recurrent coronary events in the active treatment arm. Indeed, early in the 5-year course of this trial, a trend occurred toward an increase in vascular events in the treated women. Extended follow-up of this cohort did not disclose an accrual of benefit in the treatment group. The Women’s Health Initiative (WHI) study arm, using a similar estrogen plus progesterone regimen, was halted due to a small but significant hazard of cardiovascular events, stroke, and breast cancer. The estrogen without progestin arm of WHI (conducted in women without a uterus) was stopped early due to an increase in strokes, and failed to afford protection from MI or CHD death during observation over 7 years. The excess cardiovascular events in these trials may result from an increase in thromboembolism (Chap. 413). Physicians should work with women to provide information and help weigh the small but evident CHD risk of estrogen ± progestin versus the benefits for postmenopausal symptoms and osteoporosis, taking personal preferences into account. Post hoc analyses of observational studies suggest that estrogen therapy in women younger than or closer to menopause than the women enrolled in WHI might confer cardiovascular benefit. Thus, the timing in relation to menopause or the age at which estrogen therapy begins may influence its risk/benefit balance.
The lack of efficacy of estrogen therapy in cardiovascular risk reduction highlights the need for redoubled attention to known modifiable risk factors in women. Meta-analysis supports the efficacy of statins to reduce cardiovascular events in women in primary prevention, as well as in those who have already experienced a cardiovascular event.
Dysregulated Coagulation or Fibrinolysis Thrombosis ultimately causes the gravest complications of atherosclerosis. The propensity to form thrombi and/or lyse clots once they form influences the manifestations of atherosclerosis. Thrombosis provoked by atheroma rupture and subsequent healing may promote plaque growth. Certain individual characteristics can influence thrombosis or fibrinolysis and have received attention as potential coronary risk factors. For example, fibrinogen levels correlate with coronary risk and provide information about coronary risk independent of the lipoprotein profile.
The stability of an arterial thrombus depends on the balance between fibrinolytic factors, such as plasmin, and inhibitors of the fibrinolytic system, such as plasminogen activator inhibitor 1 (PAI-1). Individuals with diabetes mellitus or the metabolic syndrome have elevated levels of PAI-1 in plasma, and this probably contributes to the increased risk of thrombotic events. Lp(a) (Chap. 421) may modulate fibrinolysis, and individuals with elevated Lp(a) levels have increased CHD risk.
Aspirin reduces CHD events in several contexts. Chapter 293 discusses aspirin therapy in stable ischemic heart disease, Chap. 294 reviews recommendations for aspirin treatment in acute coronary syndromes, and Chap. 446 describes aspirin’s role in preventing recurrent ischemic stroke. In primary prevention, pooled trial data show that low-dose aspirin treatment (81 mg/d to 325 mg on alternate days) can reduce the risk of a first MI in men. Although the Women’s Health Study (WHS) showed that aspirin (100 mg on alternate days) reduced strokes by 17%, it did not prevent MI in women. Current AHA guidelines recommend the use of low-dose aspirin (75–160 mg/d) for women with high cardiovascular risk (≥20% 10-year risk), for men with a ≥10% 10-year risk of CHD, and for all aspirin-tolerant patients with established cardiovascular disease who lack contraindications.
Inflammation An accumulation of clinical evidence shows that markers of inflammation correlate with coronary risk. For example, plasma levels of CRP, as measured by a high-sensitivity assay (hsCRP), prospectively predict the risk of MI. CRP levels also correlate with the outcome in patients with acute coronary syndromes. In contrast to several other novel risk factors, CRP adds predictive information to that derived from established risk factors, such as those included in the Framingham score (Fig. 291e-6). Mendelian randomization studies do not support a causal role for CRP in cardiovascular disease. Thus, CRP serves as a validated biomarker of risk, but probably not as a direct contributor to pathogenesis.
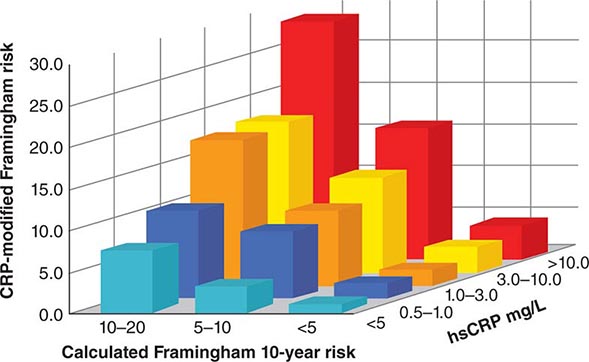
FIGURE 291e-6 C-reactive protein (CRP) level adds to the predictive value of the Framingham score. hsCRP, high-sensitivity measurement of CRP. (Adapted from PM Ridker et al: Circulation 109:2818, 2004.)
Elevations in acute-phase reactants such as fibrinogen and CRP reflect the overall inflammatory burden, not just vascular foci of inflammation. Visceral adipose tissue releases proinflammatory cytokines that drive CRP production and may represent a major extravascular stimulus to the elevation of inflammatory markers in obese and overweight individuals. Indeed, CRP levels rise with body mass index (BMI) or visceral adipose depot as assessed by imaging, and weight reduction lowers CRP levels. Infectious agents might also furnish inflammatory stimuli related to cardiovascular risk.
Statin therapy likely reduces cardiovascular events in part by muting the inflammatory aspects of the pathogenesis of atherosclerosis. For example, in statin trials conducted in both primary (JUPITER) and secondary (PROVE-IT/TIMI-22) prevention populations, prespecified analyses showed that those who achieved lower levels of both LDL and CRP had better clinical outcomes than did those who only reached the lower level of either the inflammatory marker or the atherogenic lipoprotein (Fig. 291e-7). The anti-inflammatory effect of statins appears independent of LDL lowering, because these two variables correlated very poorly in individual subjects in multiple clinical trials.
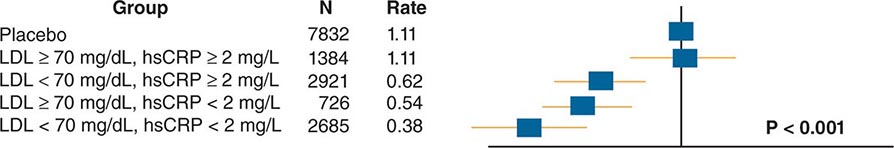
FIGURE 291e-7 Evidence from the JUPITER study that both low-density lipoprotein (LDL)-lowering and anti-inflammatory actions contribute to the benefit of statin therapy in primary prevention. See text for explanation. hsCRP, high-sensitivity measurement of C-reactive protein (CRP). (Adapted from PM Ridker et al: Lancet 373:1175, 2009.)
Lifestyle Modification The prevention of atherosclerosis presents a long-term challenge to all health care professionals and for public health policy. Both individual practitioners and organizations providing health care should strive to help patients optimize their risk factor profiles long before atherosclerotic disease becomes manifest. The current accumulation of cardiovascular risk in youth and in certain minority populations presents a particularly vexing concern from a public health perspective.
The ACC/AHA 2013 Guideline on Lifestyle Management to Reduce Cardiovascular Risk relied on rigorous evidentiary reviews. Few lifestyle interventions have undergone rigorous evaluation in randomized clinical trials. Therefore, these guidelines reflected judicious analysis of carefully selected observational studies and of intervention studies that relied primarily on biomarkers or surrogate endpoints rather than “hard” cardiovascular outcomes. Table 291e-3 summarizes the ACC/AHA lifestyle recommendations.
The care plan for all patients seen by internists should include measures to assess and minimize cardiovascular risk. Physicians must counsel patients about the health risks of tobacco use and provide guidance and resources regarding smoking cessation. Similarly, physicians should advise all patients about prudent dietary and physical activity habits for maintaining ideal body weight. Both National Institutes of Health (NIH) and AHA statements recommend at least 30 min of moderate-intensity physical activity per day. Obesity, particularly the male pattern of centripetal or visceral fat accumulation, can contribute to the elements of the “metabolic syndrome” cluster. Physicians should encourage their patients to take personal responsibility for behavior related to modifiable risk factors for the development of premature atherosclerotic disease. Conscientious counseling and patient education may forestall the need for pharmacologic measures intended to reduce coronary risk.
Issues in Risk Assessment A growing panel of markers of coronary risk presents a perplexing array to the practitioner. Markers measured in peripheral blood include size fractions of LDL particles and concentrations of homocysteine, Lp(a), fibrinogen, CRP, PAI-1, myeloperoxidase, lipoprotein-associated phospholipase A2, and imaging assessment of subclinical atherosclerosis, among many others. In general, such specialized tests add little to the information available from a careful history and physical examination combined with measurement of a plasma lipoprotein profile and fasting blood glucose. The hsCRP measurement may well prove an exception in view of its robustness in risk prediction, ease of reproducible and standardized measurement, relative stability in individuals over time, ability to add to the risk information disclosed by standard measurements such as the components of the Framingham risk score, and most importantly, the demonstration in a large-scale trial (JUPITER) that allocating therapy can reduce cardiovascular events in those deemed ineligible by traditional risk assessment criteria. The addition of information regarding a family history of premature atherosclerosis (a simply obtained indicator of genetic susceptibility), together with the inflammation marker hsCRP, permits correct reclassification of risk in individuals, especially those whose Framingham scores place them at intermediate risk.
Available data do not support the routine use of imaging studies to screen for subclinical disease (e.g., measurement of carotid intima-media thickness, coronary artery calcification, and use of computed tomographic coronary angiograms [CTA]). Inappropriate use of such imaging modalities may promote excessive alarm in asymptomatic individuals and prompt invasive diagnostic and therapeutic procedures of unproven value for both asymptomatic atherosclerosis and incidental findings. Widespread application of such modalities for screening should await proof that targeting therapies based on their application provides clinical benefit.
The 2013 ACC/AHA Guideline on the Assessment of Cardiovascular Risk recommends the use of newer risk markers if uncertainty persists after assessing quantitative risk using the pooled cohort calculator. The guideline states that family history, hsCRP, coronary artery calcium (CAC) score, or ankle-brachial index (ABI) may then be considered to inform treatment decision making. It discourages carotid intima-media thickness (CIMT) for routine measurement in clinical practice for risk assessment for a first ASCVD event. The guideline panel deemed the contribution to risk assessment for a first ASCVD event using apolipoprotein B (ApoB), chronic kidney disease, albuminuria, or cardiorespiratory fitness as uncertain at present.
Progress in human genetics holds considerable promise for risk prediction and for individualization of cardiovascular therapy. Many early reports identified single-nucleotide polymorphisms (SNPs) in candidate genes as predictors of cardiovascular risk. The validation of such genetic markers of risk and drug responsiveness in multiple populations often proved disappointing. The era of GWAS has led to discovery of sites of genetic variation that reproducibly indicate heightened cardiovascular risk (e.g., chromosome 9p21). The advent of technology that permits relatively rapid and inexpensive exome or whole-genome sequencing promises to identify new therapeutic targets, sharpen risk prediction, and deploy preventive or therapeutic measures in a more personalized manner. Despite this considerable promise, genetic scores for risk prediction have not yet demonstrated consistent improvement over algorithms that use traditional tools.
THE CHALLENGE OF IMPLEMENTATION: CHANGING PHYSICIAN AND PATIENT BEHAVIOR
Despite declining age-adjusted rates of coronary death, cardiovascular mortality worldwide is rising due to aging of the population and due to subsiding of communicable diseases and increased prevalence of risk factors in developing countries. Enormous challenges remain regarding translation of the current evidence base into practice. Physicians must learn how to help individuals adopt a healthy lifestyle in a culturally appropriate manner and to deploy their increasingly powerful pharmacologic tools most economically and effectively. The obstacles to implementation of current evidence-based prevention and treatment of atherosclerosis involve economics, education, physician awareness, and patient adherence to recommended regimens. Future goals in the treatment of atherosclerosis should include more widespread implementation of the current evidence-based guidelines regarding risk factor management and, when appropriate, drug therapy.
292e |
Atlas of Atherosclerosis |
Knowledge about the biology of human atherosclerosis and the risk factors for the disease has expanded considerably. The application of vascular biology to human atherosclerosis has revealed many new insights into the mechanisms that promote clinical events. The series of animated video presentations presented here illustrates some of the evolving information about risk factors for atherosclerosis and the pathophysiology of clinical events.
The importance of blood pressure as a risk factor for atherosclerosis and cardiovascular events has long been recognized. More recent clinical information has highlighted the importance of pulse pressure—the difference between the systolic pressure and minimum diastolic arterial pressure—as a prognostic indicator of cardiovascular risk. The video clip on pulse pressure explains the pathophysiology of this readily measured clinical variable.
Physicians possess a great deal of knowledge about the role of cholesterol in the prediction of atherosclerosis and its complications, but knowledge about the mechanism that links hypercholesterolemia to cardiovascular events has lagged the epidemiologic and observational findings. Low-density lipoprotein (LDL) provides an example of a well-understood cardiovascular risk factor. Several of the animations included in this series highlight the role of modified LDL as a trigger for inflammation and other aspects of the pathobiology of arterial plaques that lead to their aggravation and clinical events. Physicians have useful tools for modulating LDL, but other aspects of dyslipidemia are on the rise and provide a growing challenge to the practitioner. In particular, low levels of high-density lipoprotein (HDL) and elevated levels of triglycerides characterize the constellation of findings denoted by some as the “metabolic syndrome.” In the wake of increasing obesity worldwide, these features of the lipoprotein profile require renewed focus. Several of the animations in this collection discuss the concept of the metabolic syndrome and the role of lipid profile components other than LDL in atherogenesis.
The traditional approach to atherosclerosis focused on arterial stenoses as a cause of ischemia and cardiovascular events. Physicians now have effective revascularization modalities for addressing flow-limiting stenoses, but atherosclerotic plaques that do not cause stenoses nonetheless may precipitate clinical events, such as unstable angina and acute myocardial infarction. Thus, it is necessary to add to the traditional focus on stenosis an enlarged appreciation of the pathobiology of atherosclerosis that underlies many acute coronary syndromes. The animation on the development and complication of atherosclerotic plaque explains some of these emerging concepts in plaque activation as they apply to the precipitation of thrombotic complications of atherosclerosis.
ACKNOWLEDGMENTS
From Peter Libby, MD: Changes and Challenges in Cardiovascular Protection: A Special CME Activity for Physicians. Created under an unrestricted educational grant from Merck & Co., Inc. Copyright © 2002, Cardinal Health; used with permission.
VIDEO 292e-1 Pulse pressure. Considerable evidence suggests that pulse pressure serves as an important risk factor for future cardiovascular events. This video clip explains the derivation of pulse pressure and some of the pathophysiology that determines this parameter. (With permission from the Academy for Health Care Education.)
VIDEO 292e-2 Plaque instability. Most coronary thromboses result from a physical disruption of the atherosclerotic plaque. This animation explains some of the current concepts of the pathophysiology of atherosclerotic plaque disruption and how it triggers arterial thrombosis.
VIDEO 292e-3 Lipoprotein menagerie. The lipid profile confers important information regarding cardiovascular risk and the effects of therapies; understanding lipoprotein metabolism provides insight into the pathophysiology of arterial disease. This animation presents the rudiments of lipoprotein metabolism that are important in clinical medicine.
VIDEO 292e-4 Formation and complication of atherosclerotic plaques. Physicians now understand the generation of atherosclerotic plaques as a dynamic process involving an interchange between cells of the artery wall, inflammatory cells recruited from blood, and risk factors such as lipoproteins. This animation reviews current thinking about how risk factors alter the biology of the artery wall and can incite initiation and progression of atherosclerosis. It also discusses the importance of inflammation in these processes and portrays the role of inflammation in plaque disruption and thrombosis. Finally, this animation depicts the concept of stabilization of atherosclerotic plaques by interventions such as lipid lowering.
VIDEO 292e-5 Atherogenesis. This video clip highlights some of the current thinking about mechanisms of atherogenesis.
VIDEO 292e-6 Metabolic syndrome. A number of important cardiovascular risk factors tend to cluster in a pattern that has been described by some as the metabolic syndrome. Although controversy persists regarding whether cardiovascular risk due to these factors is additive or synergistic, their clinical importance is growing. This animation discusses some of the metabolic derangements that underlie the metabolic syndrome.
293 |
Ischemic Heart Disease |
Ischemic heart disease (IHD) is a condition in which there is an inadequate supply of blood and oxygen to a portion of the myocardium; it typically occurs when there is an imbalance between myocardial oxygen supply and demand. The most common cause of myocardial ischemia is atherosclerotic disease of an epicardial coronary artery (or arteries) sufficient to cause a regional reduction in myocardial blood flow and inadequate perfusion of the myocardium supplied by the involved coronary artery. Chapter 291e deals with the development and treatment of atherosclerosis. This chapter focuses on the chronic manifestations and treatment of IHD. The subsequent chapters address the acute phases of IHD.
EPIDEMIOLOGY AND GLOBAL TRENDS
![]() IHD causes more deaths and disability and incurs greater economic costs than any other illness in the developed world. IHD is the most common, serious, chronic, life-threatening illness in the United States, where 13 million persons have IHD, >6 million have angina pectoris, and >7 million have sustained a myocardial infarction. Genetic factors, a high-fat and energy-rich diet, smoking, and a sedentary lifestyle are associated with the emergence of IHD (Chap. 291e). In the United States and Western Europe, IHD is growing among low-income groups, but primary prevention has delayed the disease to later in life in all socioeconomic groups. Despite these sobering statistics, it is worth noting that epidemiologic data show a decline in the rate of deaths due to IHD, about half of which is attributable to treatments and half to prevention by risk factor modification.
IHD causes more deaths and disability and incurs greater economic costs than any other illness in the developed world. IHD is the most common, serious, chronic, life-threatening illness in the United States, where 13 million persons have IHD, >6 million have angina pectoris, and >7 million have sustained a myocardial infarction. Genetic factors, a high-fat and energy-rich diet, smoking, and a sedentary lifestyle are associated with the emergence of IHD (Chap. 291e). In the United States and Western Europe, IHD is growing among low-income groups, but primary prevention has delayed the disease to later in life in all socioeconomic groups. Despite these sobering statistics, it is worth noting that epidemiologic data show a decline in the rate of deaths due to IHD, about half of which is attributable to treatments and half to prevention by risk factor modification.
Obesity, insulin resistance, and type 2 diabetes mellitus are increasing and are powerful risk factors for IHD. These trends are occurring in the general context of population growth and as a result of the increase in the average age of the world’s population. With urbanization in countries with emerging economies and a growing middle class, elements of the energy-rich Western diet are being adopted. As a result, the prevalence of risk factors for IHD and the prevalence of IHD itself are both increasing rapidly, so that in analyses of the global burden of disease, there is a shift from communicable to noncommunicable diseases. Population subgroups that appear to be particularly affected are men in South Asian countries, especially India and the Middle East. In light of the projection of large increases in IHD throughout the world, IHD is likely to become the most common cause of death worldwide by 2020.
PATHOPHYSIOLOGY
Central to an understanding of the pathophysiology of myocardial ischemia is the concept of myocardial supply and demand. In normal conditions, for any given level of a demand for oxygen, the myocardium will control the supply of oxygen-rich blood to prevent underperfusion of myocytes and the subsequent development of ischemia and infarction. The major determinants of myocardial oxygen demand (MVO2) are heart rate, myocardial contractility, and myocardial wall tension (stress). An adequate supply of oxygen to the myocardium requires a satisfactory level of oxygen-carrying capacity of the blood (determined by the inspired level of oxygen, pulmonary function, and hemoglobin concentration and function) and an adequate level of coronary blood flow. Blood flows through the coronary arteries in a phasic fashion, with the majority occurring during diastole. About 75% of the total coronary resistance to flow occurs across three sets of arteries: (1) large epicardial arteries (Resistance 1 = R1), (2) prearteriolar vessels (R2), and (3) arteriolar and intramyocardial capillary vessels (R3). In the absence of significant flow-limiting atherosclerotic obstructions, R1 is trivial; the major determinant of coronary resistance is found in R2 and R3.
The normal coronary circulation is dominated and controlled by the heart’s requirements for oxygen. This need is met by the ability of the coronary vascular bed to vary its resistance (and, therefore, blood flow) considerably while the myocardium extracts a high and relatively fixed percentage of oxygen. Normally, intramyocardial resistance vessels demonstrate a great capacity for dilation (R2 and R3 decrease). For example, the changing oxygen needs of the heart with exercise and emotional stress affect coronary vascular resistance and in this manner regulate the supply of oxygen and substrate to the myocardium (metabolic regulation). The coronary resistance vessels also adapt to physiologic alterations in blood pressure to maintain coronary blood flow at levels appropriate to myocardial needs (autoregulation).
By reducing the lumen of the coronary arteries, atherosclerosis limits appropriate increases in perfusion when the demand for flow is augmented, as occurs during exertion or excitement. When the luminal reduction is severe, myocardial perfusion in the basal state is reduced. Coronary blood flow also can be limited by spasm (see Prinzmetal’s angina in Chap. 294), arterial thrombi, and, rarely, coronary emboli as well as by ostial narrowing due to aortitis. Congenital abnormalities such as the origin of the left anterior descending coronary artery from the pulmonary artery may cause myocardial ischemia and infarction in infancy, but this cause is very rare in adults.
Myocardial ischemia also can occur if myocardial oxygen demands are markedly increased and particularly when coronary blood flow may be limited, as occurs in severe left ventricular hypertrophy due to aortic stenosis. The latter can present with angina that is indistinguishable from that caused by coronary atherosclerosis largely owing to subendocardial ischemia (Chap. 283). A reduction in the oxygen-carrying capacity of the blood, as in extremely severe anemia or in the presence of carboxyhemoglobin, rarely causes myocardial ischemia by itself but may lower the threshold for ischemia in patients with moderate coronary obstruction.
Not infrequently, two or more causes of ischemia coexist in a patient, such as an increase in oxygen demand due to left ventricular hypertrophy secondary to hypertension and a reduction in oxygen supply secondary to coronary atherosclerosis and anemia. Abnormal constriction or failure of normal dilation of the coronary resistance vessels also can cause ischemia. When it causes angina, this condition is referred to as microvascular angina.
CORONARY ATHEROSCLEROSIS
Epicardial coronary arteries are the major site of atherosclerotic disease. The major risk factors for atherosclerosis (high levels of plasma low-density lipoprotein [LDL], low plasma high-density lipoprotein [HDL], cigarette smoking, hypertension, and diabetes mellitus [Chap. 291e]) disturb the normal functions of the vascular endothelium. These functions include local control of vascular tone, maintenance of an antithrombotic surface, and control of inflammatory cell adhesion and diapedesis. The loss of these defenses leads to inappropriate constriction, luminal thrombus formation, and abnormal interactions between blood cells, especially monocytes and platelets, and the activated vascular endothelium. Functional changes in the vascular milieu ultimately result in the subintimal collections of fat, smooth muscle cells, fibroblasts, and intercellular matrix that define the atherosclerotic plaque. Rather than viewing atherosclerosis strictly as a vascular problem, it is useful to consider it in the context of alterations in the nature of the circulating blood (hyperglycemia; increased concentrations of LDL cholesterol, tissue factor, fibrinogen, von Willebrand factor, coagulation factor VII, and platelet microparticles). The combination of a “vulnerable vessel” in a patient with “vulnerable blood” promotes a state of hypercoagulability and hypofibrinolysis. This is especially true in patients with diabetes mellitus.
Atherosclerosis develops at irregular rates in different segments of the epicardial coronary tree and leads eventually to segmental reductions in cross-sectional area, i.e., plaque formation. There is also a predilection for atherosclerotic plaques to develop at sites of increased turbulence in coronary flow, such as at branch points in the epicardial arteries. When a stenosis reduces the diameter of an epicardial artery by 50%, there is a limitation of the ability to increase flow to meet increased myocardial demand. When the diameter is reduced by ∼80%, blood flow at rest may be reduced, and further minor decreases in the stenotic orifice area can reduce coronary flow dramatically to cause myocardial ischemia at rest or with minimal stress.
Segmental atherosclerotic narrowing of epicardial coronary arteries is caused most commonly by the formation of a plaque, which is subject to rupture or erosion of the cap separating the plaque from the bloodstream. Upon exposure of the plaque contents to blood, two important and interrelated processes are set in motion: (1) platelets are activated and aggregate, and (2) the coagulation cascade is activated, leading to deposition of fibrin strands. A thrombus composed of platelet aggregates and fibrin strands traps red blood cells and can reduce coronary blood flow, leading to the clinical manifestations of myocardial ischemia.
The location of the obstruction influences the quantity of myocardium rendered ischemic and determines the severity of the clinical manifestations. Thus, critical obstructions in vessels, such as the left main coronary artery and the proximal left anterior descending coronary artery, are particularly hazardous. Chronic severe coronary narrowing and myocardial ischemia frequently are accompanied by the development of collateral vessels, especially when the narrowing develops gradually. When well developed, such vessels can by themselves provide sufficient blood flow to sustain the viability of the myocardium at rest but not during conditions of increased demand.
With progressive worsening of a stenosis in a proximal epicardial artery, the distal resistance vessels (when they function normally) dilate to reduce vascular resistance and maintain coronary blood flow. A pressure gradient develops across the proximal stenosis, and poststenotic pressure falls. When the resistance vessels are maximally dilated, myocardial blood flow becomes dependent on the pressure in the coronary artery distal to the obstruction. In these circumstances, ischemia, manifest clinically by angina or electrocardiographically by ST-segment deviation, can be precipitated by increases in myocardial oxygen demand caused by physical activity, emotional stress, and/or tachycardia. Changes in the caliber of the stenosed coronary artery due to physiologic vasomotion, loss of endothelial control of dilation (as occurs in atherosclerosis), pathologic spasm (Prinzmetal’s angina), or small platelet-rich plugs also can upset the critical balance between oxygen supply and demand and thereby precipitate myocardial ischemia.
EFFECTS OF ISCHEMIA
During episodes of inadequate perfusion caused by coronary atherosclerosis, myocardial tissue oxygen tension falls and may cause transient disturbances of the mechanical, biochemical, and electrical functions of the myocardium (Fig. 293-1). Coronary atherosclerosis is a focal process that usually causes nonuniform ischemia. During ischemia, regional disturbances of ventricular contractility cause segmental hypokinesia, akinesia, or, in severe cases, bulging (dyskinesia), which can reduce myocardial pump function.
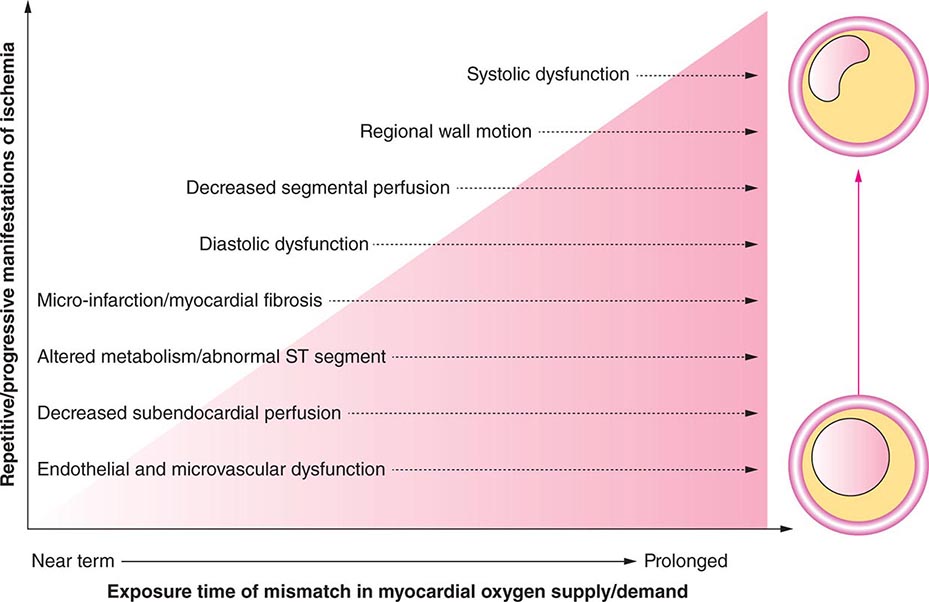
FIGURE 293-1 Cascade of mechanisms and manifestations of ischemia. (Modified from LJ Shaw et al: J Am Coll Cardiol 54:1561, 2009. Original figure illustration by Rob Flewell.)
The abrupt development of severe ischemia, as occurs with total or subtotal coronary occlusion, is associated with almost instantaneous failure of normal muscle relaxation and then contraction. The relatively poor perfusion of the subendocardium causes more intense ischemia of this portion of the wall (compared with the subepicardial region). Ischemia of large portions of the ventricle causes transient left ventricular failure, and if the papillary muscle apparatus is involved, mitral regurgitation can occur. When ischemia is transient, it may be associated with angina pectoris; when it is prolonged, it can lead to myocardial necrosis and scarring with or without the clinical picture of acute myocardial infarction (Chap. 295).
A wide range of abnormalities in cell metabolism, function, and structure underlie these mechanical disturbances during ischemia. The normal myocardium metabolizes fatty acids and glucose to carbon dioxide and water. With severe oxygen deprivation, fatty acids cannot be oxidized, and glucose is converted to lactate; intracellular pH is reduced, as are the myocardial stores of high-energy phosphates, i.e., ATP and creatine phosphate. Impaired cell membrane function leads to the leakage of potassium and the uptake of sodium by myocytes as well as an increase in cytosolic calcium. The severity and duration of the imbalance between myocardial oxygen supply and demand determine whether the damage is reversible (≤20 min for total occlusion in the absence of collaterals) or permanent, with subsequent myocardial necrosis (>20 min).
Ischemia also causes characteristic changes in the electrocardiogram (ECG) such as repolarization abnormalities, as evidenced by inversion of T waves and, when more severe, displacement of ST segments (Chap. 268). Transient T-wave inversion probably reflects nontransmural, intramyocardial ischemia; transient ST-segment depression often reflects patchy subendocardial ischemia; and ST-segment elevation is thought to be caused by more severe transmural ischemia. Another important consequence of myocardial ischemia is electrical instability, which may lead to isolated ventricular premature beats or even ventricular tachycardia or ventricular fibrillation (Chap. 277). Most patients who die suddenly from IHD do so as a result of ischemia-induced ventricular tachyarrhythmias (Chap. 327).
ASYMPTOMATIC VERSUS SYMPTOMATIC IHD
Although the prevalence is decreasing, postmortem studies of accident victims and military casualties in Western countries still show that coronary atherosclerosis can begin before age 20 and is present even among adults who were asymptomatic during life. Exercise stress tests in asymptomatic persons may show evidence of silent myocardial ischemia, i.e., exercise-induced ECG changes not accompanied by angina pectoris; coronary angiographic studies of such persons may reveal coronary artery plaques and previously unrecognized obstructions (Chap. 272). Postmortem examination of patients with such obstructions without a history of clinical manifestations of myocardial ischemia often shows macroscopic scars secondary to myocardial infarction in regions supplied by diseased coronary arteries, with or without collateral circulation. According to population studies, ∼25% of patients who survive acute myocardial infarction may not come to medical attention, and these patients have the same adverse prognosis as do those who present with the classic clinical picture of acute myocardial infarction (Chap. 295). Sudden death may be unheralded and is a common presenting manifestation of IHD (Chap. 327).
Patients with IHD also can present with cardiomegaly and heart failure secondary to ischemic damage of the left ventricular myocardium that may have caused no symptoms before the development of heart failure; this condition is referred to as ischemic cardiomyopathy. In contrast to the asymptomatic phase of IHD, the symptomatic phase is characterized by chest discomfort due to either angina pectoris or acute myocardial infarction (Chap. 295). Having entered the symptomatic phase, the patient may exhibit a stable or progressive course, revert to the asymptomatic stage, or die suddenly.
STABLE ANGINA PECTORIS
This episodic clinical syndrome is due to transient myocardial ischemia. Various diseases that cause myocardial ischemia and the numerous forms of discomfort with which it may be confused are discussed in Chap. 19. Males constitute ∼70% of all patients with angina pectoris and an even greater proportion of those less than 50 years of age. It is, however, important to note that angina pectoris in women is often atypical in presentation (see below).
HISTORY
The typical patient with angina is a man >50 years or a woman >60 years of age who complains of episodes of chest discomfort, usually described as heaviness, pressure, squeezing, smothering, or choking and only rarely as frank pain. When the patient is asked to localize the sensation, he or she typically places a hand over the sternum, sometimes with a clenched fist, to indicate a squeezing, central, substernal discomfort (Levine’s sign). Angina is usually crescendo-decrescendo in nature, typically lasts 2 to 5 min, and can radiate to either shoulder and to both arms (especially the ulnar surfaces of the forearm and hand). It also can arise in or radiate to the back, interscapular region, root of the neck, jaw, teeth, and epigastrium. Angina is rarely localized below the umbilicus or above the mandible. A useful finding in assessing a patient with chest discomfort is the fact that myocardial ischemic discomfort does not radiate to the trapezius muscles; that radiation pattern is more typical of pericarditis.
Although episodes of angina typically are caused by exertion (e.g., exercise, hurrying, or sexual activity) or emotion (e.g., stress, anger, fright, or frustration) and are relieved by rest, they also may occur at rest (Chap. 294) and while the patient is recumbent (angina decubitus). The patient may be awakened at night by typical chest discomfort and dyspnea. Nocturnal angina may be due to episodic tachycardia, diminished oxygenation as the respiratory pattern changes during sleep, or expansion of the intrathoracic blood volume that occurs with recumbency; the latter causes an increase in cardiac size (end-diastolic volume), wall tension, and myocardial oxygen demand that can lead to ischemia and transient left ventricular failure.
The threshold for the development of angina pectoris may vary by time of day and emotional state. Many patients report a fixed threshold for angina, which occurs predictably at a certain level of activity, such as climbing two flights of stairs at a normal pace. In these patients, coronary stenosis and myocardial oxygen supply are fixed, and ischemia is precipitated by an increase in myocardial oxygen demand; they are said to have stable exertional angina. In other patients, the threshold for angina may vary considerably within any particular day and from day to day. In such patients, variations in myocardial oxygen supply, most likely due to changes in coronary vasomotor tone, may play an important role in defining the pattern of angina. A patient may report symptoms upon minor exertion in the morning (a short walk or shaving) yet by midday be capable of much greater effort without symptoms. Angina may also be precipitated by unfamiliar tasks, a heavy meal, exposure to cold, or a combination of these factors.
Exertional angina typically is relieved in 1–5 min by slowing or ceasing activities and even more rapidly by rest and sublingual nitroglycerin (see below). Indeed, the diagnosis of angina should be suspect if it does not respond to the combination of these measures. The severity of angina can be conveniently summarized by the Canadian Cardiac Society functional classification (Table 293-1). Its impact on the patient’s functional capacity can be described by using the New York Heart Association functional classification (Table 293-1).
|
CARDIOVASCULAR DISEASE CLASSIFICATION CHART |
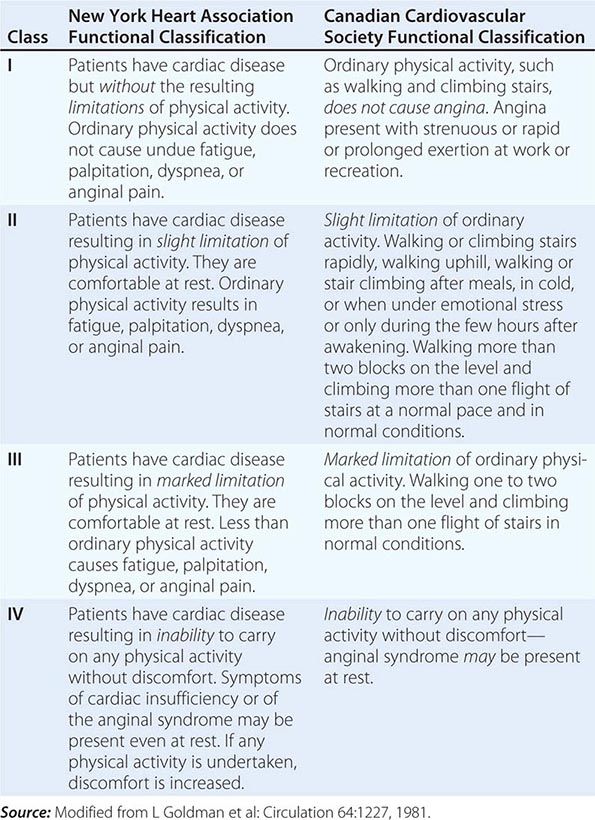
Sharp, fleeting chest pain or a prolonged, dull ache localized to the left submammary area is rarely due to myocardial ischemia. However, especially in women and diabetic patients, angina pectoris may be atypical in location and not strictly related to provoking factors. In addition, this symptom may exacerbate and remit over days, weeks, or months. Its occurrence can be seasonal, occurring more frequently in the winter in temperate climates. Anginal “equivalents” are symptoms of myocardial ischemia other than angina. They include dyspnea, nausea, fatigue, and faintness and are more common in the elderly and in diabetic patients.
Systematic questioning of a patient with suspected IHD is important to uncover the features of an unstable syndrome associated with increased risk, such as angina occurring with less exertion than in the past, occurring at rest, or awakening the patient from sleep. Since coronary atherosclerosis often is accompanied by similar lesions in other arteries, a patient with angina should be questioned and examined for peripheral arterial disease (intermittent claudication [Chap. 302]), stroke, or transient ischemic attacks (Chap. 446). It is also important to uncover a family history of premature IHD (<55 years in first-degree male relatives and <65 in female relatives) and the presence of diabetes mellitus, hyperlipidemia, hypertension, cigarette smoking, and other risk factors for coronary atherosclerosis (Chap. 291e).
The history of typical angina pectoris establishes the diagnosis of IHD until proven otherwise. The coexistence of advanced age, male sex, the postmenopausal state, and risk factors for atherosclerosis increase the likelihood of hemodynamically significant coronary disease. A particularly challenging problem is the evaluation and management of patients with persistent ischemic-type chest discomfort but no flow-limiting obstructions in their epicardial coronary arteries. This situation arises more often in women than in men. Potential etiologies include microvascular coronary disease (detectable on coronary reactivity testing in response to vasoactive agents such as intracoronary adenosine, acetylcholine, and nitroglycerin) and abnormal cardiac nociception. Treatment of microvascular coronary disease should focus on efforts to improve endothelial function, including nitrates, beta blockers, calcium antagonists, statins, and angiotensin-converting enzyme (ACE) inhibitors. Abnormal cardiac nociception is more difficult to manage and may be ameliorated in some cases by imipramine.
PHYSICAL EXAMINATION
The physical examination is often normal in patients with stable angina when they are asymptomatic. However, because of the increased likelihood of IHD in patients with diabetes and/or peripheral arterial disease, clinicians should search for evidence of atherosclerotic disease at other sites, such as an abdominal aortic aneurysm, carotid arterial bruits, and diminished arterial pulses in the lower extremities. The physical examination also should include a search for evidence of risk factors for atherosclerosis such as xanthelasmas and xanthomas (Chap. 291e). Evidence for peripheral arterial disease should be sought by evaluating the pulse contour at multiple locations and comparing the blood pressure between the arms and between the arms and the legs (ankle-brachial index). Examination of the fundi may reveal an increased light reflex and arteriovenous nicking as evidence of hypertension. There also may be signs of anemia, thyroid disease, and nicotine stains on the fingertips from cigarette smoking.
Palpation may reveal cardiac enlargement and abnormal contraction of the cardiac impulse (left ventricular dyskinesia). Auscultation can uncover arterial bruits, a third and/or fourth heart sound, and, if acute ischemia or previous infarction has impaired papillary muscle function, an apical systolic murmur due to mitral regurgitation. These auscultatory signs are best appreciated with the patient in the left lateral decubitus position. Aortic stenosis, aortic regurgitation (Chap. 283), pulmonary hypertension (Chap. 304), and hypertrophic cardiomyopathy (Chap. 287) must be excluded, since these disorders may cause angina in the absence of coronary atherosclerosis. Examination during an anginal attack is useful, since ischemia can cause transient left ventricular failure with the appearance of a third and/or fourth heart sound, a dyskinetic cardiac apex, mitral regurgitation, and even pulmonary edema. Tenderness of the chest wall, localization of the discomfort with a single fingertip on the chest, or reproduction of the pain with palpation of the chest makes it unlikely that the pain is caused by myocardial ischemia. A protuberant abdomen may indicate that the patient has the metabolic syndrome and is at increased risk for atherosclerosis.
LABORATORY EXAMINATION
Although the diagnosis of IHD can be made with a high degree of confidence from the history and physical examination, a number of simple laboratory tests can be helpful. The urine should be examined for evidence of diabetes mellitus and renal disease (including microalbuminuria) since these conditions accelerate atherosclerosis. Similarly, examination of the blood should include measurements of lipids (cholesterol—total, LDL, HDL—and triglycerides), glucose (hemoglobin A1C), creatinine, hematocrit, and, if indicated based on the physical examination, thyroid function. A chest x-ray is important as it may show the consequences of IHD, i.e., cardiac enlargement, ventricular aneurysm, or signs of heart failure. These signs can support the diagnosis of IHD and are important in assessing the degree of cardiac damage. Evidence exists that an elevated level of high-sensitivity C-reactive protein (CRP) (specifically, between 0 and 3 mg/dL) is an independent risk factor for IHD and may be useful in therapeutic decision making about the initiation of hypolipidemic treatment. The major benefit of high-sensitivity CRP is in reclassifying the risk of IHD in patients in the “intermediate” risk category on the basis of traditional risk factors.
ELECTROCARDIOGRAM
A 12-lead ECG recorded at rest may be normal in patients with typical angina pectoris, but there may also be signs of an old myocardial infarction (Chap. 268). Although repolarization abnormalities, i.e., ST-segment and T-wave changes, as well as left ventricular hypertrophy and disturbances of cardiac rhythm or intraventricular conduction are suggestive of IHD, they are nonspecific, since they also can occur in pericardial, myocardial, and valvular heart disease or, in the case of the former, transiently with anxiety, changes in posture, drugs, or esophageal disease. The presence of left ventricular hypertrophy (LVH) is a significant indication of increased risk of adverse outcomes from IHD. Of note, even though LVH and cardiac rhythm disturbances are nonspecific indicators of the development of IHD, they may be contributing factors to episodes of angina in patients in whom IHD has developed as a consequence of conventional risk factors. Dynamic ST-segment and T-wave changes that accompany episodes of angina pectoris and disappear thereafter are more specific.
STRESS TESTING
Electrocardiographic The most widely used test for both the diagnosis of IHD and the estimation of risk and prognosis involves recording the 12-lead ECG before, during, and after exercise, usually on a treadmill (Fig. 293-2). The test consists of a standardized incremental increase in external workload (Table 293-2) while symptoms, the ECG, and arm blood pressure are monitored. Exercise duration is usually symptom-limited, and the test is discontinued upon evidence of chest discomfort, severe shortness of breath, dizziness, severe fatigue, ST-segment depression >0.2 mV (2 mm), a fall in systolic blood pressure >10 mmHg, or the development of a ventricular tachyarrhythmia. This test is used to discover any limitation in exercise performance, detect typical ECG signs of myocardial ischemia, and establish their relationship to chest discomfort. The ischemic ST-segment response generally is defined as flat or downsloping depression of the ST segment >0.1 mV below baseline (i.e., the PR segment) and lasting longer than 0.08 s (Fig. 293-1). Upsloping or junctional ST-segment changes are not considered characteristic of ischemia and do not constitute a positive test. Although T-wave abnormalities, conduction disturbances, and ventricular arrhythmias that develop during exercise should be noted, they are also not diagnostic. Negative exercise tests in which the target heart rate (85% of maximal predicted heart rate for age and sex) is not achieved are considered nondiagnostic.
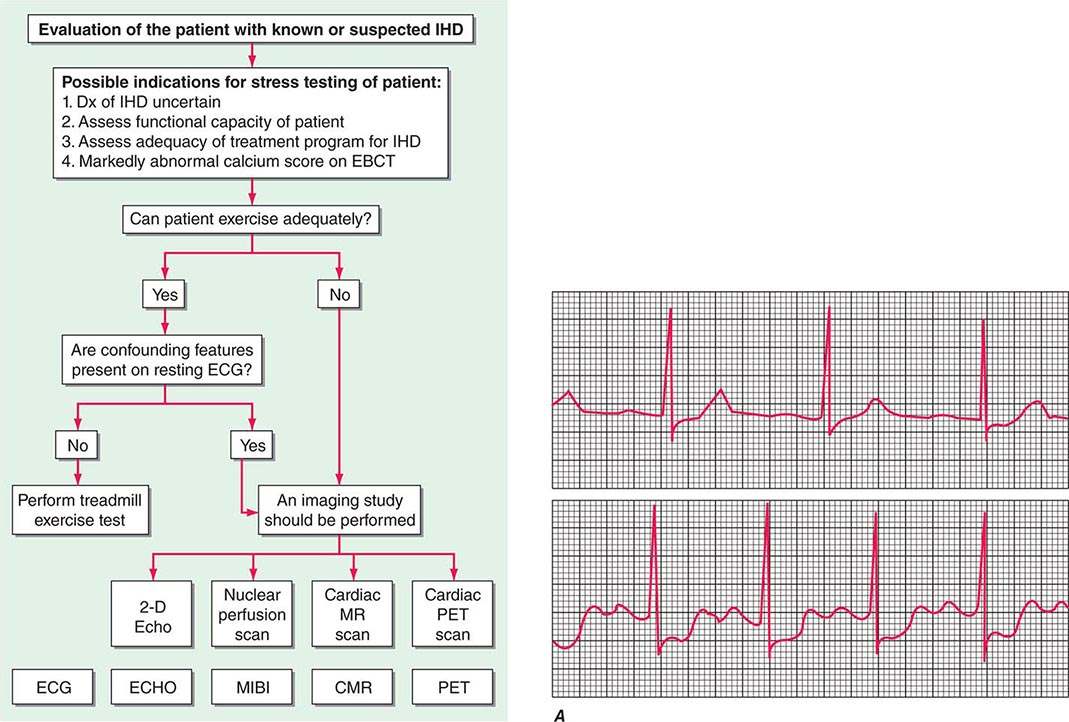
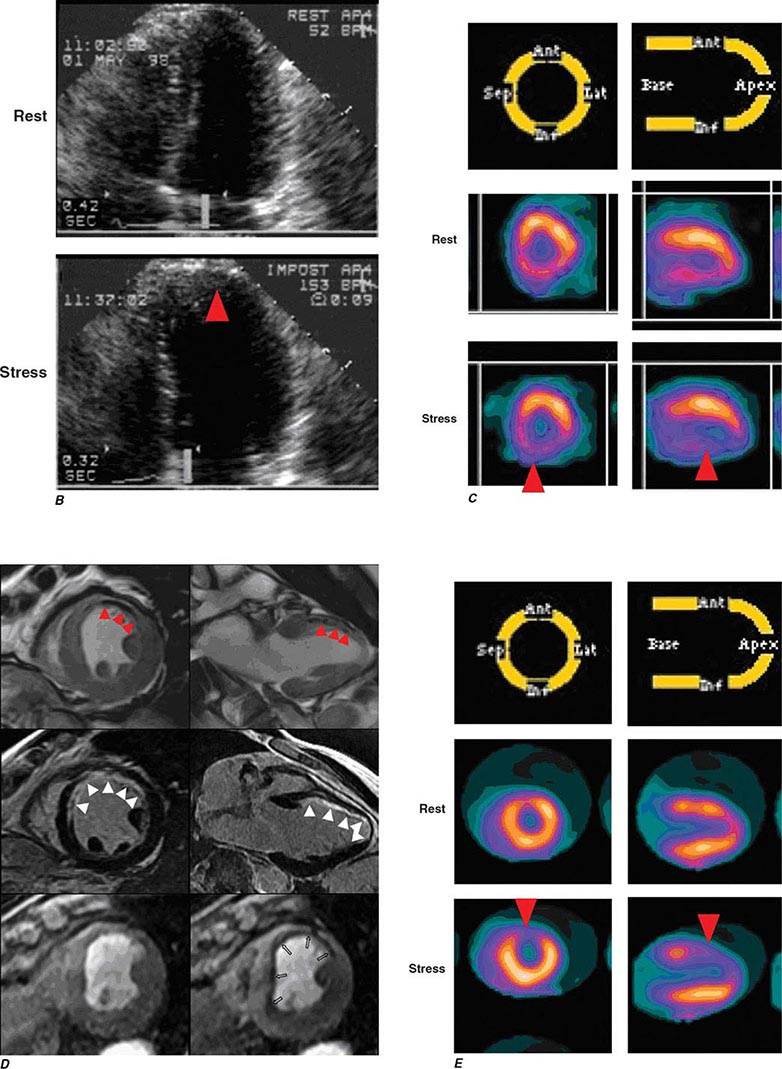
FIGURE 293-2 Evaluation of the patient with known or suspected ischemic heart disease. On the left of the figure is an algorithm for identifying patients who should be referred for stress testing and the decision pathway for determining whether a standard treadmill exercise with electrocardiogram (ECG) monitoring alone is adequate. A specialized imaging study is necessary if the patient cannot exercise adequately (pharmacologic challenge is given) or if there are confounding features on the resting ECG (symptom-limited treadmill exercise may be used to stress the coronary circulation). Panels B–E, continued on the next page, are examples of the data obtained with ECG monitoring and specialized imaging procedures. CMR, cardiac magnetic resonance; EBCT, electron beam computed tomography; ECHO, echocardiography; IHD, ischemic heart disease; MIBI, methoxyisobutyl isonitrite; MR, magnetic resonance; PET, positron emission tomography. A. Lead V4 at rest (top panel) and after 4.5 min of exercise (bottom panel). There is 3 mm (0.3 mV) of horizontal ST-segment depression, indicating a positive test for ischemia. (Modified from BR Chaitman, in E Braunwald et al [eds]: Heart Disease, 8th ed, Philadelphia, Saunders, 2008.) B. A 45-year-old avid jogger who began experiencing classic substernal chest pressure underwent an exercise echo study. With exercise the patient’s heart rate increased from 52 to 153 beats/min. The left ventricular chamber dilated with exercise, and the septal and apical portions became akinetic to dyskinetic (red arrow). These findings are strongly suggestive of a significant flow-limiting stenosis in the proximal left anterior descending artery, which was confirmed at coronary angiography. (Modified from SD Solomon, in E. Braunwald et al [eds]: Primary Cardiology, 2nd ed, Philadelphia, Saunders, 2003.) C. Stress and rest myocardial perfusion single-photon emission computed tomography images obtained with 99m-technetium sestamibi in a patient with chest pain and dyspnea on exertion. The images demonstrate a medium-size and severe stress perfusion defect involving the inferolateral and basal inferior walls, showing nearly complete reversibility, consistent with moderate ischemia in the right coronary artery territory (red arrows). (Images provided by Dr. Marcello Di Carli, Nuclear Medicine Division, Brigham and Women’s Hospital, Boston, MA.) D. A patient with a prior myocardial infarction presented with recurrent chest discomfort. On cardiac magnetic resonance (CMR) cine imaging, a large area of anterior akinesia was noted (marked by the arrows in the top left and right images, systolic frame only). This area of akinesia was matched by a larger extent of late gadolinium-DTPA enhancements consistent with a large transmural myocardial infarction (marked by arrows in the middle left and right images). Resting (bottom left) and adenosine vasodilating stress (bottom right) first-pass perfusion images revealed reversible perfusion abnormality that extended to the inferior septum. This patient was found to have an occluded proximal left anterior descending coronary artery with extensive collateral formation. This case illustrates the utility of different modalities in a CMR examination in characterizing ischemic and infarcted myocardium. DTPA, diethylenetriamine penta-acetic acid. (Images provided by Dr. Raymond Kwong, Cardiovascular Division, Brigham and Women’s Hospital, Boston, MA.) E. Stress and rest myocardial perfusion PET images obtained with rubidium-82 in a patient with chest pain on exertion. The images demonstrate a large and severe stress perfusion defect involving the mid and apical anterior, anterolateral, and anteroseptal walls and the left ventricular apex, showing complete reversibility, consistent with extensive and severe ischemia in the mid-left anterior descending coronary artery territory (red arrows). (Images provided by Dr. Marcello Di Carli, Nuclear Medicine Division, Brigham and Women’s Hospital, Boston, MA.)
|
RELATION OF METABOLIC EQUIVALENT TASKS (METs) TO STAGES IN VARIOUS TESTING PROTOCOLS |
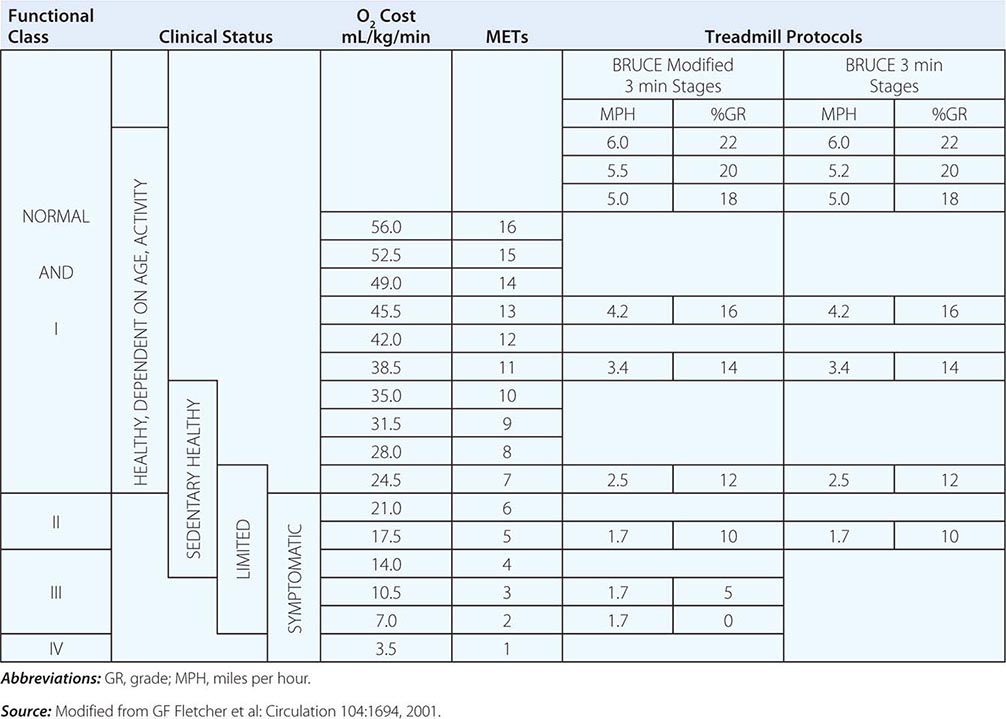
In interpreting ECG stress tests, the probability that coronary artery disease (CAD) exists in the patient or population under study (i.e., pretest probability) should be considered. Overall, false-positive or false-negative results occur in one-third of cases. However, a positive result on exercise indicates that the likelihood of CAD is 98% in males who are >50 years with a history of typical angina pectoris and who develop chest discomfort during the test. The likelihood decreases if the patient has atypical or no chest pain by history and/or during the test.
The incidence of false-positive tests is significantly increased in patients with low probabilities of IHD, such as asymptomatic men age <40 or premenopausal women with no risk factors for premature atherosclerosis. It is also increased in patients taking cardioactive drugs, such as digitalis and antiarrhythmic agents, and in those with intraventricular conduction disturbances, resting ST-segment and T-wave abnormalities, ventricular hypertrophy, or abnormal serum potassium levels. Obstructive disease limited to the circumflex coronary artery may result in a false-negative stress test since the lateral portion of the heart that this vessel supplies is not well represented on the surface 12-lead ECG. Since the overall sensitivity of exercise stress electrocardiography is only ∼75%, a negative result does not exclude CAD, although it makes the likelihood of three-vessel or left main CAD extremely unlikely.
A medical professional should be present throughout the exercise test. It is important to measure total duration of exercise, the times to the onset of ischemic ST-segment change and chest discomfort, the external work performed (generally expressed as the stage of exercise), and the internal cardiac work performed, i.e., by the heart rate–blood pressure product. The depth of the ST-segment depression and the time needed for recovery of these ECG changes are also important. Because the risks of exercise testing are small but real—estimated at one fatality and two nonfatal complications per 10,000 tests—equipment for resuscitation should be available. Modified (heart rate–limited rather than symptom-limited) exercise tests can be performed safely in patients as early as 6 days after uncomplicated myocardial infarction (Table 293-2). Contraindications to exercise stress testing include rest angina within 48 h, unstable rhythm, severe aortic stenosis, acute myocarditis, uncontrolled heart failure, severe pulmonary hypertension, and active infective endocarditis.
The normal response to graded exercise includes progressive increases in heart rate and blood pressure. Failure of the blood pressure to increase or an actual decrease with signs of ischemia during the test is an important adverse prognostic sign, since it may reflect ischemia-induced global left ventricular dysfunction. The development of angina and/or severe (>0.2 mV) ST-segment depression at a low workload, i.e., before completion of stage II of the Bruce protocol, and/or ST-segment depression that persists >5 min after the termination of exercise increases the specificity of the test and suggests severe IHD and a high risk of future adverse events.
Cardiac Imaging (See also Chap. 270e) When the resting ECG is abnormal (e.g., preexcitation syndrome, >1 mm of resting ST-segment depression, left bundle branch block, paced ventricular rhythm), information gained from an exercise test can be enhanced by stress myocardial radionuclide perfusion imaging after the intravenous administration of thallium-201 or 99m-technetium sestamibi during exercise (or with pharmacologic) stress. Contemporary data also suggest positron emission tomography (PET) imaging (with exercise or pharmacologic stress) using N-13 ammonia or rubidium-82 nuclide as another technique for assessing perfusion. Images obtained immediately after cessation of exercise to detect regional ischemia are compared with those obtained at rest to confirm reversible ischemia and regions of persistently absent uptake that signify infarction.
A sizable fraction of patients who need noninvasive stress testing to identify myocardial ischemia and increased risk of coronary events cannot exercise because of peripheral vascular or musculoskeletal disease, exertional dyspnea, or deconditioning. In these circumstances, an intravenous pharmacologic challenge is used in place of exercise. For example, dipyridamole or adenosine can be given to create a coronary “steal” by temporarily increasing flow in nondiseased segments of the coronary vasculature at the expense of diseased segments. Alternatively, a graded incremental infusion of dobutamine may be administered to increase MVO2. A variety of imaging options are available to accompany these pharmacologic stressors (Fig. 293-2). The development of a transient perfusion defect with a tracer such as thallium-201 or 99m-technetium sestamibi is used to detect myocardial ischemia.
Echocardiography is used to assess left ventricular function in patients with chronic stable angina and patients with a history of a prior myocardial infarction, pathologic Q waves, or clinical evidence of heart failure. Two-dimensional echocardiography can assess both global and regional wall motion abnormalities of the left ventricle that are transient when due to ischemia. Stress (exercise or dobutamine) echocardiography may cause the emergence of regions of akinesis or dyskinesis that are not present at rest. Stress echocardiography, like stress myocardial perfusion imaging, is more sensitive than exercise electrocardiography in the diagnosis of IHD. Cardiac magnetic resonance (CMR) stress testing is also evolving as an alternative to radionuclide, PET, or echocardiographic stress imaging. CMR stress testing performed with dobutamine infusion can be used to assess wall motion abnormalities accompanying ischemia, as well as myocardial perfusion. CMR can be used to provide more complete ventricular evaluation using multislice magnetic resonance imaging (MRI) studies.
Atherosclerotic plaques become progressively calcified over time, and coronary calcification in general increases with age. For this reason, methods for detecting coronary calcium have been developed as a measure of the presence of coronary atherosclerosis. These methods involve computed tomography (CT) applications that achieve rapid acquisition of images (electron beam [EBCT] and multidetector [MDCT] detection). Coronary calcium detected by these imaging techniques most commonly is quantified by using the Agatston score, which is based on the area and density of calcification. Although the diagnostic accuracy of this imaging method is high (sensitivity, 90–94%; specificity, 95–97%; negative predictive value, 93–99%), its prognostic utility has not been defined. Thus, its role in CT, EBCT, and MDCT scans for the detection and management of patients with IHD has not been clarified.
CORONARY ARTERIOGRAPHY
(See also Chap. 272) This diagnostic method outlines the lumina of the coronary arteries and can be used to detect or exclude serious coronary obstruction. However, coronary arteriography provides no information about the arterial wall, and severe atherosclerosis that does not encroach on the lumen may go undetected. Of note, atherosclerotic plaques characteristically are scattered throughout the coronary tree, tend to occur more frequently at branch points, and grow progressively in the intima and media of an epicardial coronary artery at first without encroaching on the lumen, causing an outward bulging of the artery—a process referred to as remodeling (Chap. 291e). Later in the course of the disease, further growth causes luminal narrowing.
Indications Coronary arteriography is indicated in (1) patients with chronic stable angina pectoris who are severely symptomatic despite medical therapy and are being considered for revascularization, i.e., a percutaneous coronary intervention (PCI) or coronary artery bypass grafting (CABG); (2) patients with troublesome symptoms that present diagnostic difficulties in whom there is a need to confirm or rule out the diagnosis of IHD; (3) patients with known or possible angina pectoris who have survived cardiac arrest; (4) patients with angina or evidence of ischemia on noninvasive testing with clinical or laboratory evidence of ventricular dysfunction; and (5) patients judged to be at high risk of sustaining coronary events based on signs of severe ischemia on noninvasive testing, regardless of the presence or severity of symptoms (see below).
Examples of other indications for coronary arteriography include the following:
1. Patients with chest discomfort suggestive of angina pectoris but a negative or nondiagnostic stress test who require a definitive diagnosis for guiding medical management, alleviating psychological stress, career or family planning, or insurance purposes.
2. Patients who have been admitted repeatedly to the hospital for a suspected acute coronary syndrome (Chaps. 294 and 295), but in whom this diagnosis has not been established and in whom the presence or absence of CAD should be determined.
3. Patients with careers that involve the safety of others (e.g., pilots, firefighters, police) who have questionable symptoms or suspicious or positive noninvasive tests and in whom there are reasonable doubts about the state of the coronary arteries.
4. Patients with aortic stenosis or hypertrophic cardiomyopathy and angina in whom the chest pain could be due to IHD.
5. Male patients >45 years and females >55 years who are to undergo a cardiac operation such as valve replacement or repair and who may or may not have clinical evidence of myocardial ischemia.
6. Patients after myocardial infarction, especially those who are at high risk after myocardial infarction because of the recurrence of angina or the presence of heart failure, frequent ventricular premature contractions, or signs of ischemia on the stress test.
7. Patients with angina pectoris, regardless of severity, in whom noninvasive testing indicates a high risk of coronary events (poor exercise performance or severe ischemia).
8. Patients in whom coronary spasm or another nonatherosclerotic cause of myocardial ischemia (e.g., coronary artery anomaly, Kawasaki disease) is suspected.
Noninvasive alternatives to diagnostic coronary arteriography include CT angiography and CMR angiography (Chap. 270e). Although these new imaging techniques can provide information about obstructive lesions in the epicardial coronary arteries, their exact role in clinical practice has not been rigorously defined. Important aspects of their use that should be noted include the substantially higher radiation exposure with CT angiography compared to conventional diagnostic arteriography and the limitations on CMR imposed by cardiac movement during the cardiac cycle, especially at high heart rates.
PROGNOSIS
The principal prognostic indicators in patients known to have IHD are age, the functional state of the left ventricle, the location(s) and severity of coronary artery narrowing, and the severity or activity of myocardial ischemia. Angina pectoris of recent onset, unstable angina (Chap. 294), early postmyocardial infarction angina, angina that is unresponsive or poorly responsive to medical therapy, and angina accompanied by symptoms of congestive heart failure all indicate an increased risk for adverse coronary events. The same is true for the physical signs of heart failure, episodes of pulmonary edema, transient third heart sounds, and mitral regurgitation and for echocardiographic or radioisotopic (or roentgenographic) evidence of cardiac enlargement and reduced (<0.40) ejection fraction.
Most important, any of the following signs during noninvasive testing indicates a high risk for coronary events: inability to exercise for 6 min, i.e., stage II (Bruce protocol) of the exercise test; a strongly positive exercise test showing onset of myocardial ischemia at low workloads (≥0.1 mV ST-segment depression before completion of stage II, ≥0.2 mV ST-segment depression at any stage, ST-segment depression for >5 min after the cessation of exercise, a decline in systolic pressure >10 mmHg during exercise, or the development of ventricular tachyarrhythmias during exercise); the development of large or multiple perfusion defects or increased lung uptake during stress radioisotope perfusion imaging; and a decrease in left ventricular ejection fraction during exercise on radionuclide ventriculography or during stress echocardiography. Conversely, patients who can complete stage III of the Bruce exercise protocol and have a normal stress perfusion scan or negative stress echocardiographic evaluation are at very low risk for future coronary events. The finding of frequent episodes of ST-segment deviation on ambulatory ECG monitoring (even in the absence of symptoms) is also an adverse prognostic finding.
On cardiac catheterization, elevations of left ventricular end-diastolic pressure and ventricular volume and reduced ejection fraction are the most important signs of left ventricular dysfunction and are associated with a poor prognosis. Patients with chest discomfort but normal left ventricular function and normal coronary arteries have an excellent prognosis. Obstructive lesions of the left main (>50% luminal diameter) or left anterior descending coronary artery proximal to the origin of the first septal artery are associated with a greater risk than are lesions of the right or left circumflex coronary artery because of the greater quantity of myocardium at risk. Atherosclerotic plaques in epicardial arteries with fissuring or filling defects indicate increased risk. These lesions go through phases of inflammatory cellular activity, degeneration, endothelial dysfunction, abnormal vasomotion, platelet aggregation, and fissuring or hemorrhage. These factors can temporarily worsen the stenosis and cause thrombosis and/or abnormal reactivity of the vessel wall, thus exacerbating the manifestations of ischemia. The recent onset of symptoms, the development of severe ischemia during stress testing (see above), and unstable angina pectoris (Chap. 294) all reflect episodes of rapid progression in coronary lesions.
With any degree of obstructive CAD, mortality is greatly increased when left ventricular function is impaired; conversely, at any level of left ventricular function, the prognosis is influenced importantly by the quantity of myocardium perfused by critically obstructed vessels. Therefore, it is essential to collect all the evidence substantiating past myocardial damage (evidence of myocardial infarction on ECG, echocardiography, radioisotope imaging, or left ventriculography), residual left ventricular function (ejection fraction and wall motion), and risk of future damage from coronary events (extent of coronary disease and severity of ischemia defined by noninvasive stress testing). The larger the quantity of established myocardial necrosis is, the less the heart is able to withstand additional damage and the poorer the prognosis is. Risk estimation must include age, presenting symptoms, all risk factors, signs of arterial disease, existing cardiac damage, and signs of impending damage (i.e., ischemia).
The greater the number and severity of risk factors for coronary atherosclerosis (advanced age [>75 years], hypertension, dyslipidemia, diabetes, morbid obesity, accompanying peripheral and/or cerebrovascular disease, previous myocardial infarction), the worse the prognosis of an angina patient. Evidence exists that elevated levels of CRP in the plasma, extensive coronary calcification on electron beam CT (see above), and increased carotid intimal thickening on ultrasound examination also indicate an increased risk of coronary events.
|
TREATMENT |
STABLE ANGINA PECTORIS |
Once the diagnosis of IHD has been made, each patient must be evaluated individually with respect to his or her level of understanding, expectations and goals, control of symptoms, and prevention of adverse clinical outcomes such as myocardial infarction and premature death. The degree of disability and the physical and emotional stress that precipitates angina must be recorded carefully to set treatment goals. The management plan should include the following components: (1) explanation of the problem and reassurance about the ability to formulate a treatment plan, (2) identification and treatment of aggravating conditions, (3) recommendations for adaptation of activity as needed, (4) treatment of risk factors that will decrease the occurrence of adverse coronary outcomes, (5) drug therapy for angina, and (6) consideration of revascularization.
EXPLANATION AND REASSURANCE
Patients with IHD need to understand their condition and realize that a long and productive life is possible even though they have angina pectoris or have experienced and recovered from an acute myocardial infarction. Offering results of clinical trials showing improved outcomes can be of great value in encouraging patients to resume or maintain activity and return to work. A planned program of rehabilitation can encourage patients to lose weight, improve exercise tolerance, and control risk factors with more confidence.
IDENTIFICATION AND TREATMENT OF AGGRAVATING CONDITIONS
A number of conditions may increase oxygen demand or decrease oxygen supply to the myocardium and may precipitate or exacerbate angina in patients with IHD. Left ventricular hypertrophy, aortic valve disease, and hypertrophic cardiomyopathy may cause or contribute to angina and should be excluded or treated. Obesity, hypertension, and hyperthyroidism should be treated aggressively to reduce the frequency and severity of anginal episodes. Decreased myocardial oxygen supply may be due to reduced oxygenation of the arterial blood (e.g., in pulmonary disease or, when carboxyhemoglobin is present, due to cigarette or cigar smoking) or decreased oxygen-carrying capacity (e.g., in anemia). Correction of these abnormalities, if present, may reduce or even eliminate angina pectoris.
ADAPTATION OF ACTIVITY
Myocardial ischemia is caused by a discrepancy between the demand of the heart muscle for oxygen and the ability of the coronary circulation to meet that demand. Most patients can be helped to understand this concept and utilize it in the rational programming of activity. Many tasks that ordinarily evoke angina may be accomplished without symptoms simply by reducing the speed at which they are performed. Patients must appreciate the diurnal variation in their tolerance of certain activities and should reduce their energy requirements in the morning, immediately after meals, and in cold or inclement weather. On occasion, it may be necessary to recommend a change in employment or residence to avoid physical stress.
Physical conditioning usually improves the exercise tolerance of patients with angina and has substantial psychological benefits. A regular program of isotonic exercise that is within the limits of the individual patient’s threshold for the development of angina pectoris and that does not exceed 80% of the heart rate associated with ischemia on exercise testing should be strongly encouraged. Based on the results of an exercise test, the number of metabolic equivalent tasks (METs) performed at the onset of ischemia can be estimated (Table 293-2) and a practical exercise prescription can be formulated to permit daily activities that will fall below the ischemic threshold (Table 293-3).
|
ENERGY REQUIREMENTS FOR SOME COMMON ACTIVITIES |
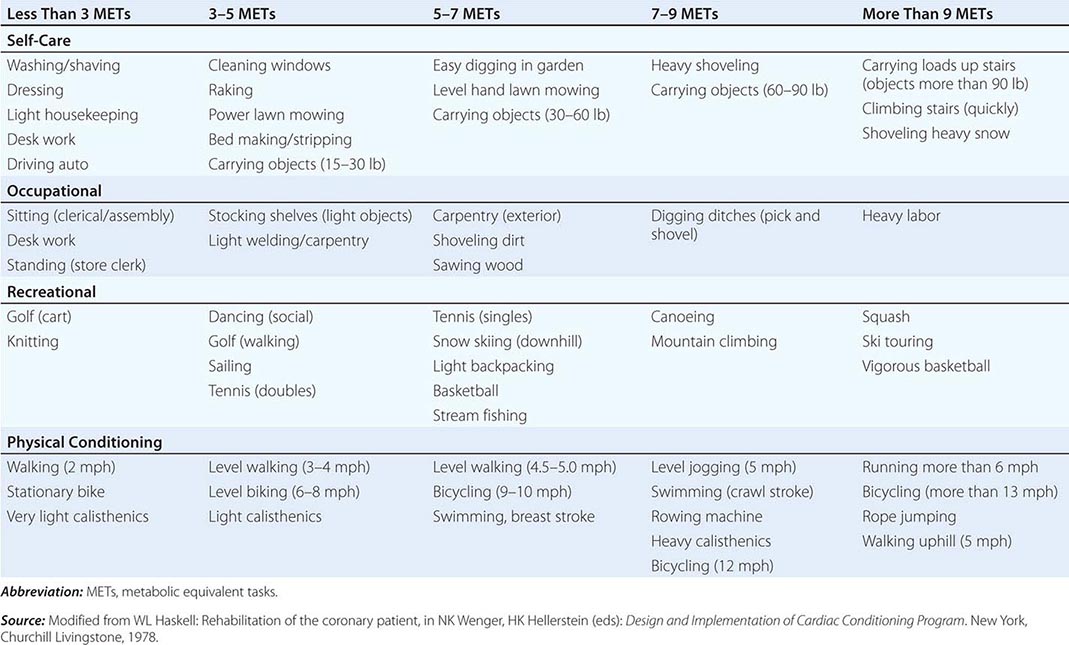
TREATMENT OF RISK FACTORS
A family history of premature IHD is an important indicator of increased risk and should trigger a search for treatable risk factors such as hyperlipidemia, hypertension, and diabetes mellitus. Obesity impairs the treatment of other risk factors and increases the risk of adverse coronary events. In addition, obesity often is accompanied by three other risk factors: diabetes mellitus, hypertension, and hyperlipidemia. The treatment of obesity and these accompanying risk factors is an important component of any management plan. A diet low in saturated and trans-unsaturated fatty acids and a reduced caloric intake to achieve optimal body weight are a cornerstone in the management of chronic IHD. It is especially important to emphasize weight loss and regular exercise in patients with the metabolic syndrome or overt diabetes mellitus.
Cigarette smoking accelerates coronary atherosclerosis in both sexes and at all ages and increases the risk of thrombosis, plaque instability, myocardial infarction, and death (Chap. 291e). In addition, by increasing myocardial oxygen needs and reducing oxygen supply, it aggravates angina. Smoking cessation studies have demonstrated important benefits with a significant decline in the occurrence of these adverse outcomes. The physician’s message must be clear and strong and supported by programs that achieve and monitor abstinence (Chap. 470). Hypertension (Chap. 298) is associated with an increased risk of adverse clinical events from coronary atherosclerosis as well as stroke. In addition, the left ventricular hypertrophy that results from sustained hypertension aggravates ischemia. There is evidence that long-term effective treatment of hypertension can decrease the occurrence of adverse coronary events.
Diabetes mellitus (Chap. 417) accelerates coronary and peripheral atherosclerosis and is frequently associated with dyslipidemias and increases in the risk of angina, myocardial infarction, and sudden coronary death. Aggressive control of the dyslipidemia (target LDL cholesterol <70 mg/dL) and hypertension (target blood pressure 120/80 mmHg) that are frequently found in diabetic patients is highly effective and therefore essential, as described below.
DYSLIPIDEMIA
The treatment of dyslipidemia is central in aiming for long-term relief from angina, reduced need for revascularization, and reduction in myocardial infarction and death. The control of lipids can be achieved by the combination of a diet low in saturated and trans-unsaturated fatty acids, exercise, and weight loss. Nearly always, HMG-CoA reductase inhibitors (statins) are required and can lower LDL cholesterol (25–50%), raise HDL cholesterol (5–9%), and lower triglycerides (5–30%). A powerful treatment effect of statins on atherosclerosis, IHD, and outcomes is seen regardless of the pretreatment LDL cholesterol level. Fibrates or niacin can be used to raise HDL cholesterol and lower triglycerides (Chaps. 291e and 421). Controlled trials with lipid-regulating regimens have shown equal proportional benefit for men, women, the elderly, diabetic patients, and smokers.
Compliance with the health-promoting behaviors listed above is generally very poor, and a conscientious physician must not underestimate the major effort required to meet this challenge. Many patients who are discharged from the hospital with proven coronary disease do not receive adequate treatment for dyslipidemia. In light of the proof that treating dyslipidemia brings major benefits, physicians need to establish treatment pathways, monitor compliance, and follow up regularly.
RISK REDUCTION IN WOMEN WITH IHD
The incidence of clinical IHD in premenopausal women is very low; however, after menopause, the atherogenic risk factors increase (e.g., increased LDL, reduced HDL) and the rate of clinical coronary events accelerates to the levels observed in men. Women have not given up cigarette smoking as effectively as have men. Diabetes mellitus, which is more common in women, greatly increases the occurrence of clinical IHD and amplifies the deleterious effects of hypertension, hyperlipidemia, and smoking. Cardiac catheterization and coronary revascularization are underused in women and are performed at a later and more severe stage of the disease than in men. When cholesterol lowering, beta blockers after myocardial infarction, and coronary artery bypass grafting are applied in the appropriate patient groups, women benefit to the same degree as men.
DRUG THERAPY
The commonly used drugs for the treatment of angina pectoris are summarized in Tables 293-4 through 293-6. Pharmacotherapy for IHD is designed to reduce the frequency of anginal episodes, myocardial infarction, and coronary death. There is a wealth of trial data to emphasize how important this medical management is when added to the health-promoting behaviors discussed above. To achieve maximum benefit from medical therapy for IHD, it is frequently necessary to combine agents from different classes and titrate the doses as guided by the individual profile of risk factors, symptoms, hemodynamic responses, and side effects.
|
NITRATE THERAPY IN PATIENTS WITH ISCHEMIC HEART DISEASE |
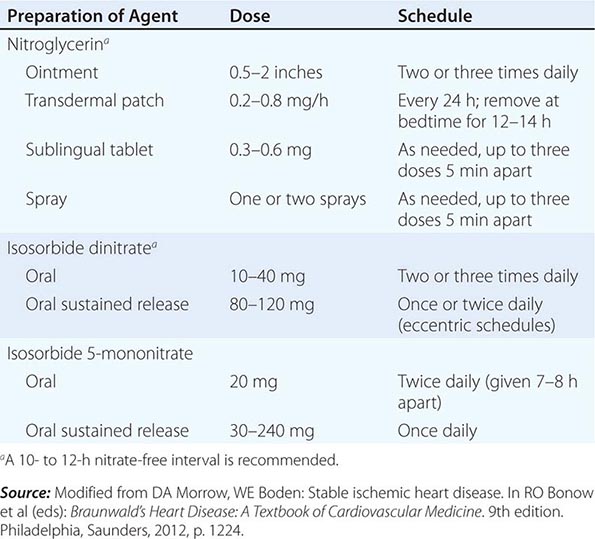
|
PROPERTIES OF BETA BLOCKERS IN CLINICAL USE FOR ISCHEMIC HEART DISEASE |
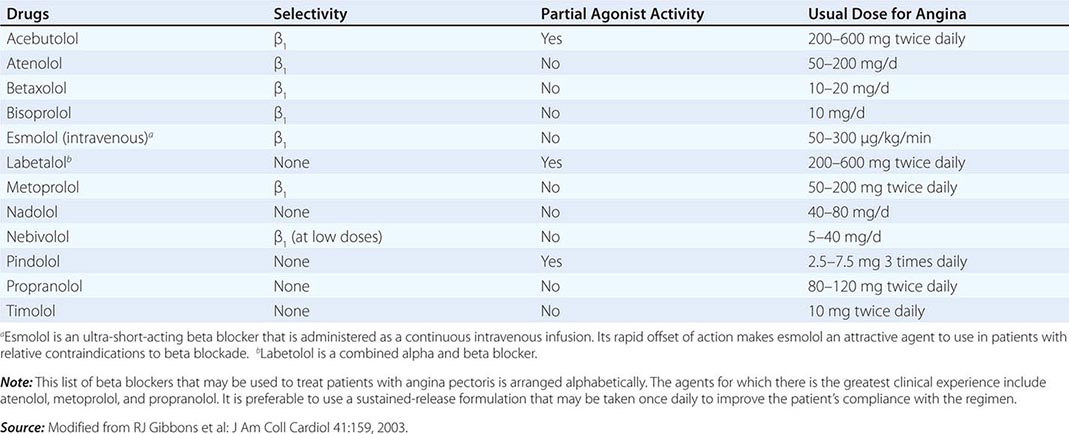
|
CALCIUM CHANNEL BLOCKERS IN CLINICAL USE FOR ISCHEMIC HEART DISEASE |
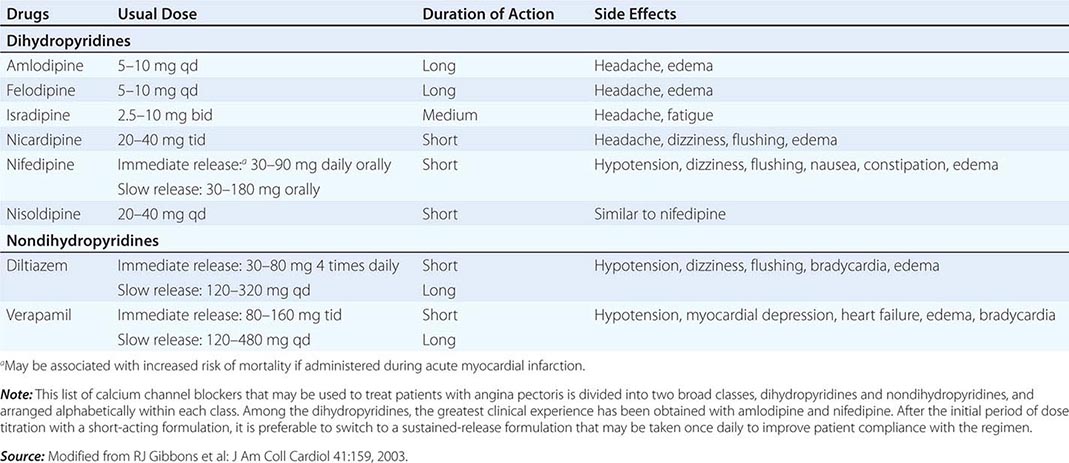
NITRATES
The organic nitrates are a valuable class of drugs in the management of angina pectoris (Table 293-4). Their major mechanisms of action include systemic venodilation with concomitant reduction in left ventricular end-diastolic volume and pressure, thereby reducing myocardial wall tension and oxygen requirements; dilation of epicardial coronary vessels; and increased blood flow in collateral vessels. When metabolized, organic nitrates release nitric oxide (NO) that binds to guanylyl cyclase in vascular smooth muscle cells, leading to an increase in cyclic guanosine monophosphate, which causes relaxation of vascular smooth muscle. Nitrates also exert antithrombotic activity by NO-dependent activation of platelet guanylyl cyclase, impairment of intraplatelet calcium flux, and platelet activation.
The absorption of these agents is most rapid and complete through the mucous membranes. For this reason, nitroglycerin is most commonly administered sublingually in tablets of 0.4 or 0.6 mg. Patients with angina should be instructed to take the medication both to relieve angina and also approximately 5 min before stress that is likely to induce an episode. The value of this prophylactic use of the drug cannot be overemphasized.
Nitrates improve exercise tolerance in patients with chronic angina and relieve ischemia in patients with unstable angina as well as patients with Prinzmetal’s variant angina (Chap. 294). A diary of angina and nitroglycerin use may be valuable for detecting changes in the frequency, severity, or threshold for discomfort that may signify the development of unstable angina pectoris and/or herald an impending myocardial infarction.
Long-Acting Nitrates None of the long-acting nitrates are as effective as sublingual nitroglycerin for the acute relief of angina. These organic nitrate preparations can be swallowed, chewed, or administered as a patch or paste by the transdermal route (Table 293-4). They can provide effective plasma levels for up to 24 h, but the therapeutic response is highly variable. Different preparations and/or administration during the daytime should be tried only to prevent discomfort while avoiding side effects such as headache and dizziness. Individual dose titration is important to prevent side effects. To minimize the effects of tolerance, the minimum effective dose should be used and a minimum of 8 h each day kept free of the drug to restore any useful response(s).
β-Adrenergic Blockers These drugs represent an important component of the pharmacologic treatment of angina pectoris (Table 293-5). They reduce myocardial oxygen demand by inhibiting the increases in heart rate, arterial pressure, and myocardial contractility caused by adrenergic activation. Beta blockade reduces these variables most strikingly during exercise but causes only small reductions at rest. Long-acting beta-blocking drugs or sustained-release formulations offer the advantage of once-daily dosing (Table 293-5). The therapeutic aims include relief of angina and ischemia. These drugs also can reduce mortality and reinfarction rates in patients after myocardial infarction and are moderately effective antihypertensive agents.
Relative contraindications include asthma and reversible airway obstruction in patients with chronic lung disease, atrioventricular conduction disturbances, severe bradycardia, Raynaud’s phenomenon, and a history of mental depression. Side effects include fatigue, reduced exercise tolerance, nightmares, impotence, cold extremities, intermittent claudication, bradycardia (sometimes severe), impaired atrioventricular conduction, left ventricular failure, bronchial asthma, worsening claudication, and intensification of the hypoglycemia produced by oral hypoglycemic agents and insulin. Reducing the dose or even discontinuation may be necessary if these side effects develop and persist. Since sudden discontinuation can intensify ischemia, the doses should be tapered over 2 weeks. Beta blockers with relative β1-receptor specificity such as metoprolol and atenolol may be preferable in patients with mild bronchial obstruction and insulin-requiring diabetes mellitus.
Calcium Channel Blockers Calcium channel blockers (Table 293-6) are coronary vasodilators that produce variable and dose-dependent reductions in myocardial oxygen demand, contractility, and arterial pressure. These combined pharmacologic effects are advantageous and make these agents as effective as beta blockers in the treatment of angina pectoris. They are indicated when beta blockers are contraindicated, poorly tolerated, or ineffective. Because of differences in the dose-response relationship on cardiac electrical activity between the dihydropyridine and nondihydropyridine calcium channel blockers, verapamil and diltiazem may produce symptomatic disturbances in cardiac conduction and bradyarrhythmias. They also exert negative inotropic actions and are more likely to aggravate left ventricular failure, particularly when used in patients with left ventricular dysfunction, especially if the patients are also receiving beta blockers. Although useful effects usually are achieved when calcium channel blockers are combined with beta blockers and nitrates, individual titration of the doses is essential with these combinations. Variant (Prinzmetal’s) angina responds particularly well to calcium channel blockers (especially members of the dihydropyridine class), supplemented when necessary by nitrates (Chap. 294).
Verapamil ordinarily should not be combined with beta blockers because of the combined adverse effects on heart rate and contractility. Diltiazem can be combined with beta blockers in patients with normal ventricular function and no conduction disturbances. Amlodipine and beta blockers have complementary actions on coronary blood supply and myocardial oxygen demands. Whereas the former decreases blood pressure and dilates coronary arteries, the latter slows heart rate and decreases contractility. Amlodipine and the other second-generation dihydropyridine calcium antagonists (nicardipine, isradipine, long-acting nifedipine, and felodipine) are potent vasodilators and are useful in the simultaneous treatment of angina and hypertension. Short-acting dihydropyridines should be avoided because of the risk of precipitating infarction, particularly in the absence of concomitant beta blocker therapy.
Choice Between Beta Blockers and Calcium Channel Blockers for Initial Therapy Since beta blockers have been shown to improve life expectancy after acute myocardial infarction (Chaps. 294 and 295) and calcium channel blockers have not, the former may also be preferable in patients with angina and a damaged left ventricle. However, calcium channel blockers are indicated in patients with the following: (1) inadequate responsiveness to the combination of beta blockers and nitrates; many of these patients do well with a combination of a beta blocker and a dihydropyridine calcium channel blocker; (2) adverse reactions to beta blockers such as depression, sexual disturbances, and fatigue; (3) angina and a history of asthma or chronic obstructive pulmonary disease; (4) sick-sinus syndrome or significant atrioventricular conduction disturbances; (5) Prinzmetal’s angina; or (6) symptomatic peripheral arterial disease.
Antiplatelet Drugs Aspirin is an irreversible inhibitor of platelet cyclooxygenase and thereby interferes with platelet activation. Chronic administration of 75–325 mg orally per day has been shown to reduce coronary events in asymptomatic adult men over age 50, patients with chronic stable angina, and patients who have or have survived unstable angina and myocardial infarction. There is a dose-dependent increase in bleeding when aspirin is used chronically. It is preferable to use an enteric-coated formulation in the range of 81–162 mg/d. Administration of this drug should be considered in all patients with IHD in the absence of gastrointestinal bleeding, allergy, or dyspepsia. Clopidogrel (300–600 mg loading and 75 mg/d) is an oral agent that blocks P2Y12 ADP receptor–mediated platelet aggregation. It provides benefits similar to those of aspirin in patients with stable chronic IHD and may be substituted for aspirin if aspirin causes the side effects listed above. Clopidogrel combined with aspirin reduces death and coronary ischemic events in patients with an acute coronary syndrome (Chap. 294) and also reduces the risk of thrombus formation in patients undergoing implantation of a stent in a coronary artery (Chap. 296e). Alternative antiplatelet agents that block the P2Y12 platelet receptor such as prasugrel and ticagrelor have been shown to be more effective than clopidogrel for prevention of ischemic events after placement of a stent for an acute coronary syndrome but are associated with an increased risk of bleeding. Although combined treatment with clopidogrel and aspirin for at least a year is recommended in patients with an acute coronary syndrome treated with implantation of a drug-eluting stent, studies have not shown any benefit from the routine addition of clopidogrel to aspirin in patients with chronic stable IHD.
OTHER THERAPIES
The angiotensin-converting enzyme (ACE) inhibitors are widely used in the treatment of survivors of myocardial infarction, patients with hypertension or chronic IHD including angina pectoris, and those at high risk of vascular diseases such as diabetes. The benefits of ACE inhibitors are most evident in IHD patients at increased risk, especially if diabetes mellitus or left ventricle dysfunction is present, and those who have not achieved adequate control of blood pressure and LDL cholesterol on beta blockers and statins. However, the routine administration of ACE inhibitors to IHD patients who have normal left ventricular function and have achieved blood pressure and LDL goals on other therapies does not reduce the incidence of events and therefore is not cost-effective.
Despite treatment with nitrates, beta blockers, or calcium channel blockers, some patients with IHD continue to experience angina, and additional medical therapy is now available to alleviate their symptoms. Ranolazine, a piperazine derivative, may be useful for patients with chronic angina despite standard medical therapy. Its antianginal action is believed to occur via inhibition of the late inward sodium current (INa). The benefits of INa inhibition include limitation of the Na overload of ischemic myocytes and prevention of Ca2+ overload via the Na+–Ca2+ exchanger. A dose of 500–1000 mg orally twice daily is usually well tolerated. Ranolazine is contraindicated in patients with hepatic impairment or with conditions or drugs associated with QTc prolongation and when drugs that inhibit the CYP3A metabolic system (e.g., ketoconazole, diltiazem, verapamil, macrolide antibiotics, HIV protease inhibitors, and large quantities of grapefruit juice) are being used.
Nonsteroidal anti-inflammatory drug (NSAID) use in patients with IHD may be associated with a small but finite increased risk of myocardial infarction and mortality. For this reason, they generally should be avoided in IHD patients. If they are required for symptom relief, it is advisable to coadminister aspirin and strive to use an NSAID associated with the lowest risk of cardiovascular events, in the lowest dose required, and for the shortest period of time.
Another class of agents opens ATP-sensitive potassium channels in myocytes, leading to a reduction of free intracellular calcium ions. The major drug in this class is nicorandil, which typically is administered orally in a dose of 20 mg twice daily for prevention of angina. (Nicorandil is not available for use in the United States but is used in several other countries.)
Angina and Heart Failure Transient left ventricular failure with angina can be controlled by the use of nitrates. For patients with established congestive heart failure, the increased left ventricular wall tension raises myocardial oxygen demand. Treatment of congestive heart failure with an ACE inhibitor, a diuretic, and digoxin (Chap. 279) reduces heart size, wall tension, and myocardial oxygen demand, which helps control angina and ischemia. If the symptoms and signs of heart failure are controlled, an effort should be made to use beta blockers not only for angina but because trials in heart failure have shown significant improvement in survival. A trial of the intravenous ultra-short-acting beta blocker esmolol may be useful to establish the safety of beta blockade in selected patients. Nocturnal angina often can be relieved by the treatment of heart failure.
The combination of congestive heart failure and angina in patients with IHD usually indicates a poor prognosis and warrants serious consideration of cardiac catheterization and coronary revascularization.
CORONARY REVASCULARIZATION
Clinical trials have confirmed that with the initial diagnosis of stable IHD, it is first appropriate to initiate a thorough medical regimen as described above. Revascularization should be considered in the presence of unstable phases of the disease, intractable symptoms, severe ischemia or high-risk coronary anatomy, diabetes, and impaired left ventricular (LV) function. Revascularization should be employed in conjunction with but not replace the continuing need to modify risk factors and assess medical therapy. An algorithm for integrating medical therapy and revascularization options in patients with IHD is shown in Fig. 293-3.
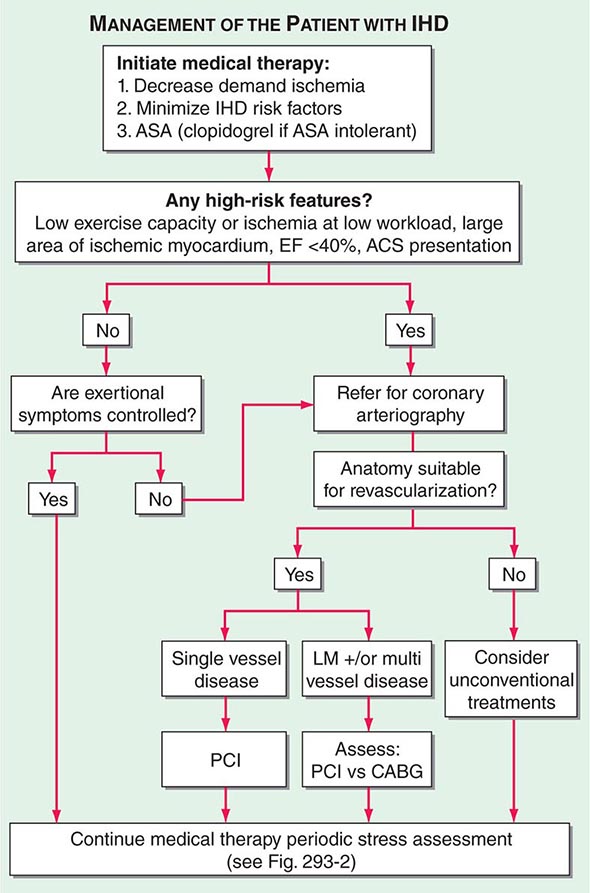
FIGURE 293-3 Algorithm for management of a patient with ischemic heart disease. All patients should receive the core elements of medical therapy as shown at the top of the algorithm. If high-risk features are present, as established by the clinical history, exercise test data, and imaging studies, the patient should be referred for coronary arteriography. Based on the number and location of the diseased vessels and their suitability for revascularization, the patient is treated with a percutaneous coronary intervention (PCI) or coronary artery bypass graft (CABG) surgery or should be considered for unconventional treatments. See text for further discussion. ACS, acute coronary syndrome; ASA, aspirin; EF, ejection fraction; IHD, ischemic heart disease; LM, left main.
PERCUTANEOUS CORONARY INTERVENTION
(See also Chap. 296e) Percutaneous coronary intervention (PCI) involving balloon dilatation usually accompanied by coronary stenting is widely used to achieve revascularization of the myocardium in patients with symptomatic IHD and suitable stenoses of epicardial coronary arteries. Whereas patients with stenosis of the left main coronary artery and those with three-vessel IHD (especially with diabetes and/or impaired LV function) who require revascularization are best treated with CABG, PCI is widely employed in patients with symptoms and evidence of ischemia due to stenoses of one or two vessels and even in selected patients with three-vessel disease (and, perhaps, in some patients with left main disease) and may offer many advantages over surgery.
Indications and Patient Selection The most common clinical indication for PCI is symptom-limiting angina pectoris, despite medical therapy, accompanied by evidence of ischemia during a stress test. PCI is more effective than medical therapy for the relief of angina. PCI improves outcomes in patients with unstable angina or when used early in the course of myocardial infarction with and without cardiogenic shock. However, in patients with stable exertional angina, clinical trials have confirmed that PCI does not reduce the occurrence of death or myocardial infarction compared to optimum medical therapy. PCI can be used to treat stenoses in native coronary arteries as well as in bypass grafts in patients who have recurrent angina after CABG.
Risks When coronary stenoses are discrete and symmetric, two and even three vessels can be treated in sequence. However, case selection is essential to avoid a prohibitive risk of complications, which are usually due to dissection or thrombosis with vessel occlusion, uncontrolled ischemia, and ventricular failure (Chap. 296e). Oral aspirin, a P2Y12 antagonist, and an antithrombin agent are given to reduce coronary thrombus formation. Left main coronary artery stenosis generally is regarded as a contraindication to PCI; such patients should be treated with CABG. In selected cases such as patients with prohibitive surgical risks, PCI of an unprotected left main can be considered, but such a procedure should be performed only by a highly skilled operator; importantly, there are regional differences in the use of this approach internationally.
Efficacy Primary success, i.e., adequate dilation (an increase in luminal diameter >20% to a residual diameter obstruction <50%) with relief of angina, is achieved in >95% of cases. Recurrent stenosis of the dilated vessels occurs in ∼20% of cases within 6 months of PCI with bare metal stents, and angina will recur within 6 months in 10% of cases. Restenosis is more common in patients with diabetes mellitus, arteries with small caliber, incomplete dilation of the stenosis, long stents, occluded vessels, obstructed vein grafts, dilation of the left anterior descending coronary artery, and stenoses containing thrombi. In diseased vein grafts, procedural success has been improved by the use of capture devices or filters that prevent embolization, ischemia, and infarction.
It is usual clinical practice to administer aspirin indefinitely and a P2Y12 antagonist for 1–3 months after the implantation of a bare metal stent. Although aspirin in combination with a thienopyridine may help prevent coronary thrombosis during and shortly after PCI with stenting, there is no evidence that these medications reduce the incidence of restenosis.
The use of drug-eluting stents that locally deliver antiproliferative drugs can reduce restenosis to less than 10%. Advances in PCI, especially the availability of drug-eluting stents, have vastly extended the use of this revascularization option in patients with IHD. Of note, however, the delayed endothelial healing in the region of a drug-eluting stent also extends the period during which the patient is at risk for subacute stent thrombosis. Current recommendations are to administer aspirin indefinitely and a P2Y12 antagonist daily for at least 1 year after implantation of a drug-eluting stent. When a situation arises in which temporary discontinuation of antiplatelet therapy is necessary, the clinical circumstances should be reviewed with the operator who performed the PCI and a coordinated plan should be established for minimizing the risk of late stent thrombus; central to this plan is the discontinuation of antiplatelet therapy for the shortest acceptable period. The risk of stent thrombosis is dependent on stent size and length, complexity of the lesions, age, diabetes, and technique. However, compliance with dual antiplatelet therapy and individual responsiveness to platelet inhibition are very important factors as well.
Successful PCI produces effective relief of angina in >95% of cases. The majority of patients with symptomatic IHD who require revascularization can be treated initially by PCI. Successful PCI is less invasive and expensive than CABG and permits savings in the initial cost of care. Successful PCI avoids the risk of stroke associated with CABG surgery, allows earlier return to work, and allows the resumption of an active life. However, the early health-related and economic benefit of PCI is reduced over time because of the greater need for follow-up and the increased need for repeat procedures. When directly compared in patients with diabetes or three-vessel or left main CAD, CABG was superior to PCI in preventing major adverse cardiac or cerebrovascular events over a 12-month follow-up.
CORONARY ARTERY BYPASS GRAFTING
Anastomosis of one or both of the internal mammary arteries or a radial artery to the coronary artery distal to the obstructive lesion is the preferred procedure. For additional obstructions that cannot be bypassed by an artery, a section of a vein (usually the saphenous) is used to form a connection between the aorta and the coronary artery distal to the obstructive lesion.
Although some indications for CABG are controversial, certain areas of agreement exist:
1. The operation is relatively safe, with mortality rates <1% in patients without serious comorbid disease and normal LV function and when the procedure is performed by an experienced surgical team.
2. Intraoperative and postoperative mortality rates increase with the severity of ventricular dysfunction, comorbidities, age >80 years, and lack of surgical experience. The effectiveness and risk of CABG vary widely depending on case selection and the skill and experience of the surgical team.
3. Occlusion of venous grafts is observed in 10–20% of patients during the first postoperative year and in approximately 2% per year during 5- to 7-year follow-up and 4% per year thereafter. Long-term patency rates are considerably higher for internal mammary and radial artery implantations than for saphenous vein grafts. In patients with left anterior descending coronary artery obstruction, survival is better when coronary bypass involves the internal mammary artery rather than a saphenous vein. Graft patency and outcomes are improved by meticulous treatment of risk factors, particularly dyslipidemia.
4. Angina is abolished or greatly reduced in ∼90% of patients after complete revascularization. Although this usually is associated with graft patency and restoration of blood flow, the pain may also have been alleviated as a result of infarction of the ischemic segment or a placebo effect. Within 3 years, angina recurs in about one-fourth of patients but is rarely severe.
5. Survival may be improved by operation in patients with stenosis of the left main coronary artery as well as in patients with three- or two-vessel disease with significant obstruction of the proximal left anterior descending coronary artery. The survival benefit is greater in patients with abnormal LV function (ejection fraction <50%). Survival may also be improved in the following patients: (a) patients with obstructive CAD who have survived sudden cardiac death or sustained ventricular tachycardia; (b) patients who have undergone previous CABG and have multiple saphenous vein graft stenoses, especially of a graft supplying the left anterior descending coronary artery; and (c) patients with recurrent stenosis after PCI and high-risk criteria on noninvasive testing.
6. Minimally invasive CABG through a small thoracotomy and/or off-pump surgery can reduce morbidity and shorten convalescence in suitable patients but does not appear to reduce significantly the risk of neurocognitive dysfunction postoperatively.
7. Among patients with type 2 diabetes mellitus and multivessel coronary disease, CABG surgery plus optimal medical therapy is superior to optimal medical therapy alone in preventing major cardiovascular events, a benefit mediated largely by a significant reduction in nonfatal myocardial infarction. The benefits of CABG are especially evident in diabetic patients treated with an insulin-sensitizing strategy as opposed to an insulin-providing strategy. CABG has also been shown to be superior to PCI (including the use of drug-eluting stents) in preventing death, myocardial infarction, and repeat revascularization in patients with diabetes mellitus and multivessel IHD.
Indications for CABG usually are based on the severity of symptoms, coronary anatomy, and ventricular function. The ideal candidate is male, <80 years of age, has no other complicating disease, and has troublesome or disabling angina that is not adequately controlled by medical therapy or does not tolerate medical therapy. The patient wishes to lead a more active life and has severe stenoses of two or three epicardial coronary arteries with objective evidence of myocardial ischemia as a cause of the chest discomfort. Great symptomatic benefit can be anticipated in such patients. Congestive heart failure and/or LV dysfunction, advanced age (>80 years), reoperation, urgent need for surgery, and the presence of diabetes mellitus are all associated with a higher perioperative mortality rate.
LV dysfunction can be due to noncontractile or hypocontractile segments that are viable but are chronically ischemic (hibernating myocardium). As a consequence of chronic reduction in myocardial blood flow, these segments downregulate their contractile function. They can be detected by using radionuclide scans of myocardial perfusion and metabolism, PET, cardiac MRI, or delayed scanning with thallium-201 or by improvement of regional functional impairment provoked by low-dose dobutamine. In such patients, revascularization improves myocardial blood flow, can return function, and can improve survival.
The Choice Between PCI and CABG All the clinical characteristics of each individual patient must be used to decide on the method of revascularization (e.g., LV function, diabetes, lesion complexity). A number of randomized clinical trials have compared PCI and CABG in patients with multivessel CAD who were suitable technically for both procedures. The redevelopment of angina requiring repeat coronary angiography and repeat revascularization is higher with PCI. This is a result of restenosis in the stented segment (a problem largely solved with drug-eluting stents) and the development of new stenoses in unstented portions of the coronary vasculature. It has been argued that PCI with stenting focuses on culprit lesions, whereas a bypass graft to the target vessel also provides a conduit around future culprit lesions proximal to the anastomosis of the graft to the native vessel (Fig. 293-4). By contrast, stroke rates are lower with PCI.
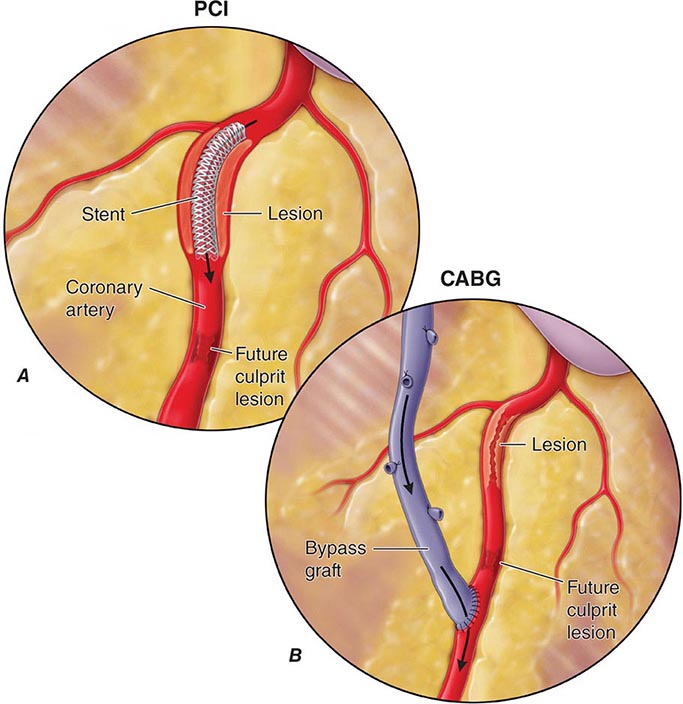
FIGURE 293-4 Difference in the approach to the lesion with percutaneous coronary intervention (PCI) and coronary artery bypass grafting (CABG). PCI is targeted at the “culprit” lesion or lesions, whereas CABG is directed at the epicardial vessel, including the culprit lesion or lesions and future culprits, proximal to the insertion of the vein graft, a difference that may account for the superiority of CABG, at least in the intermediate term, in patients with multivessel disease. (Reproduced from BJ Gersh, RL Frye: N Engl J Med 352:2235, 2005.)
Based on available evidence, it is now recommended that patients with an unacceptable level of angina despite optimal medical management be considered for coronary revascularization. Patients with single- or two-vessel disease with normal LV function and anatomically suitable lesions ordinarily are advised to undergo PCI (Chap. 296e). Patients with three-vessel disease (or two-vessel disease that includes the proximal left descending coronary artery) and impaired global LV function (LV ejection fraction <50%) or diabetes mellitus and those with left main CAD or other lesions unsuitable for catheter-based procedures should be considered for CABG as the initial method of revascularization. In light of the complexity of the decision making, it is desirable to have a multidisciplinary team, including a cardiologist and a cardiac surgeon in conjunction with the patient’s primary care physician, provide input along with ascertaining the patient’s preferences before committing to a particular revascularization option.
UNCONVENTIONAL TREATMENTS FOR IHD
On occasion clinicians will encounter a patient who has persistent disabling angina despite maximally tolerated medical therapy and for whom revascularization is not an option (e.g., small diffusely diseased vessels not amenable to stent implantation or acceptable targets for bypass grafting). In such situations, unconventional treatments should be considered.
Enhanced external counterpulsation utilizes pneumatic cuffs on the lower extremities to provide diastolic augmentation and systolic unloading of blood pressure to decrease cardiac work and oxygen consumption while enhancing coronary blood flow. Clinical trials have shown that regular application improves angina, exercise capacity, and regional myocardial perfusion. Experimental approaches such as gene and stem cell therapies are also under active study.
ASYMPTOMATIC (SILENT) ISCHEMIA
Obstructive CAD, acute myocardial infarction, and transient myocardial ischemia are frequently asymptomatic. During continuous ambulatory ECG monitoring, the majority of ambulatory patients with typical chronic stable angina are found to have objective evidence of myocardial ischemia (ST-segment depression) during episodes of chest discomfort while they are active outside the hospital. In addition, many of these patients also have more frequent episodes of asymptomatic ischemia. Frequent episodes of ischemia (symptomatic and asymptomatic) during daily life appear to be associated with an increased likelihood of adverse coronary events (death and myocardial infarction). In addition, patients with asymptomatic ischemia after a myocardial infarction are at greater risk for a second coronary event. The widespread use of exercise ECG during routine examinations has also identified some of these previously unrecognized patients with asymptomatic CAD. Longitudinal studies have demonstrated an increased incidence of coronary events in asymptomatic patients with positive exercise tests.
294 |
Non-ST-Segment Elevation Acute Coronary Syndrome (Non-ST-Segment Elevation Myocardial Infarction and Unstable Angina) |
Patients with ischemic heart disease fall into two large groups: patients with chronic coronary artery disease (CAD) who most commonly present with stable angina (Chap. 293) and patients with acute coronary syndromes (ACSs). These include patients with acute myocardial infarction with ST-segment elevation (STEMI) on their presenting electrocardiogram (Chap. 295) and those with non-ST-segment elevation acute coronary syndrome (NSTE-ACS). The latter include patients with non-ST-segment elevation myocardial infarction (NSTEMI), who, by definition, have evidence of myocyte necrosis, and those with unstable angina (UA), who do not. The relative incidence of NSTEMI compared to STEMI appears to be increasing (Fig. 294-1). Every year in the United States, approximately 1.1 million patients are admitted to hospitals with NSTE-ACS as compared with ∼300,000 patients with acute STEMI. Women comprise more than one-third of patients with NSTE-ACS, but less than one-fourth of patients with STEMI.
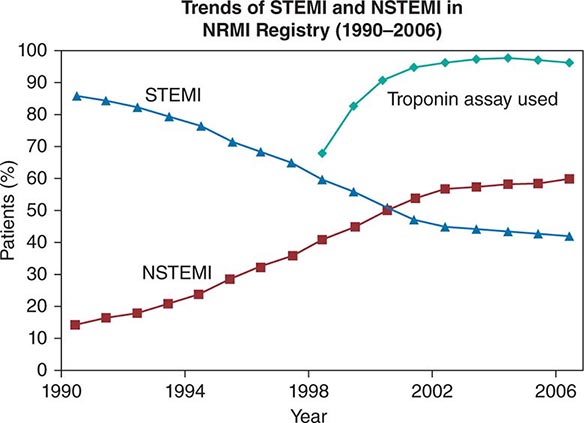
FIGURE 294-1 Trends of incidence of ST-segment elevation myocardial infarction (STEMI) and non-ST-segment elevation myocardial infarction (NSTEMI) and of frequency of use of troponin assay to diagnose acute myocardial infarction. NRMI, National Registry of Myocardial Infarction. (From N Arora, RG Brindis, CP Cannon: Acute coronary syndrome in North America, in Theroux P [ed]: Acute Coronary Syndromes, 2nd ed. Philadelphia: Elsevier, 2011.)
PATHOPHYSIOLOGY
NSTE-ACS is most commonly caused by an imbalance between oxygen supply and oxygen demand resulting from a partially occluding thrombus forming on a disrupted atherothrombotic coronary plaque or on eroded coronary artery endothelium. Severe ischemia or myocardial necrosis may occur consequent to the reduction of coronary blood flow caused by the thrombus and by downstream embolization of platelet aggregates and/or atherosclerotic debris. Other causes of NSTE-ACS include: (1) dynamic obstruction (e.g., coronary spasm, as in Prinzmetal’s variant angina [see “Prinzmetal’s Variant Angina” later]); (2) severe mechanical obstruction due to progressive coronary atherosclerosis; and (3) increased myocardial oxygen demand produced by conditions such as fever, tachycardia, and thyrotoxicosis in the presence of fixed epicardial coronary obstruction. More than one of these processes may be involved.
Among patients with NSTE-ACS studied at angiography, approximately 10% have stenosis of the left main coronary artery, 35% have three-vessel CAD, 20% have two-vessel disease, 20% have single-vessel disease, and 15% have no apparent critical epicardial coronary artery stenosis; some of the latter may have obstruction of the coronary microcirculation and/or spasm. The “culprit lesion” responsible for ischemia may show an eccentric stenosis with scalloped or overhanging edges and a narrow neck on coronary angiography. Optical coherence tomography (an invasive technique) and contrast-enhanced coronary computed tomographic angiography (CCTA), a noninvasive technique (Fig. 294-2), have shown that culprit lesions are composed of a lipid-rich core with a thin fibrous cap. Patients with NSTE-ACS frequently have multiple such plaques that are at risk of disruption (vulnerable plaques).
FIGURE 294-2 Coronary computed tomographic angiogram showing an obstructive plaque in the right coronary artery. (From PJ de Feyter, K Nieman. Multislice computed tomography in acute coronary syndromes, in Theroux P [ed]: Acute Coronary Syndromes, 2nd ed. Philadelphia: Elsevier, 2011.)
CLINICAL PRESENTATION
Diagnosis The diagnosis of NSTE-ACS is based largely on the clinical presentation. Typically, chest discomfort is severe and has at least one of three features: (1) it occurs at rest (or with minimal exertion), lasting >10 minutes; (2) it is of relatively recent onset (i.e., within the prior 2 weeks); and/or (3) it occurs with a crescendo pattern (i.e., distinctly more severe, prolonged, or frequent than previous episodes). The diagnosis of NSTEMI is established if a patient with these clinical features develops evidence of myocardial necrosis, as reflected in abnormally elevated levels of biomarkers of cardiac necrosis (see below).
History and Physical Examination The chest discomfort, often severe enough to be described as frank pain, is typically located in the substernal region or sometimes in the epigastrium, and radiates to the left arm, left shoulder, and/or neck. Anginal “equivalents” such as dyspnea, epigastric discomfort, nausea, or weakness may occur instead of chest pain and appear to be more frequent in women, the elderly, and patients with diabetes mellitus. The physical examination resembles that in patients with stable angina (Chap. 293) and may be unremarkable. If the patient has a large area of myocardial ischemia or a large NSTEMI, the physical findings can include diaphoresis; pale, cool skin; sinus tachycardia; a third and/or fourth heart sound; basilar rales; and, sometimes, hypotension.
Electrocardiogram ST-segment depression occurs in 20 to 25% of patients; it may be transient in patients without biomarker evidence of myocardial necrosis, but may be persistent for several days in NSTEMI. T-wave changes are common but are less specific signs of ischemia, unless they are new and deep T-wave inversions (≥0.3 mV).
Cardiac Biomarkers Patients with NSTEMI have elevated biomarkers of necrosis, such as cardiac troponin I or T, which are specific, sensitive, and the preferred markers of myocardial necrosis. The MB isoform of creatine kinase (CK-MB) is a less sensitive alternative. Elevated levels of these markers distinguish patients with NSTEMI from those with UA. There is a characteristic temporal rise and fall of the plasma concentration of these markers and a direct relationship between the degree of elevation and mortality (see Fig. 294-4B). However, in patients without a clear clinical history of myocardial ischemia, minor cardiac troponin (cTn) elevations have been reported and can be caused by congestive heart failure, myocarditis, or pulmonary embolism, or using high-sensitivity assays, they may occur in ostensibly normal subjects. Thus, in patients with an unclear history, small elevations of cTn, especially if they are persistent, may not be diagnostic of an ACS.
With more widespread measurement of troponin, especially using high-sensitivity assays, an increasing fraction of patients with NSTE-ACS are found to have NSTEMI, whereas the fraction of patients with UA is dwindling.
DIAGNOSTIC EVALUATION
In addition to the clinical examination, three major noninvasive tools are used in the evaluation of NSTEMI-ACS: the electrocardiogram (ECG), cardiac biomarkers, and stress testing. CCTA is an additional emerging option (Fig. 294-2). The goals are to: (1) recognize or exclude myocardial infarction (MI) using cardiac biomarkers, preferably cTn; (2) detect rest ischemia (using serial or continuous ECGs); and (3) detect significant coronary obstruction at rest with CCTA and myocardial ischemia using stress testing (Chap 270e).
Patients with a low likelihood of ischemia are usually managed with an emergency department–based critical pathway (which, in some institutions, is carried out in a “chest pain unit”) (Fig. 294-3). Evaluation of such patients includes clinical monitoring for recurrent ischemic discomfort and continuous monitoring of ECGs and cardiac markers, typically obtained at baseline and at 4–6 h and 12 h after presentation. If new elevations in cardiac markers or ST-T-wave changes on the ECG are noted, the patient should be admitted to the hospital. Patients who remain pain free with negative markers may proceed to stress testing to determine the presence of ischemia or CCTA to detect coronary luminal obstruction (Fig. 294-2).
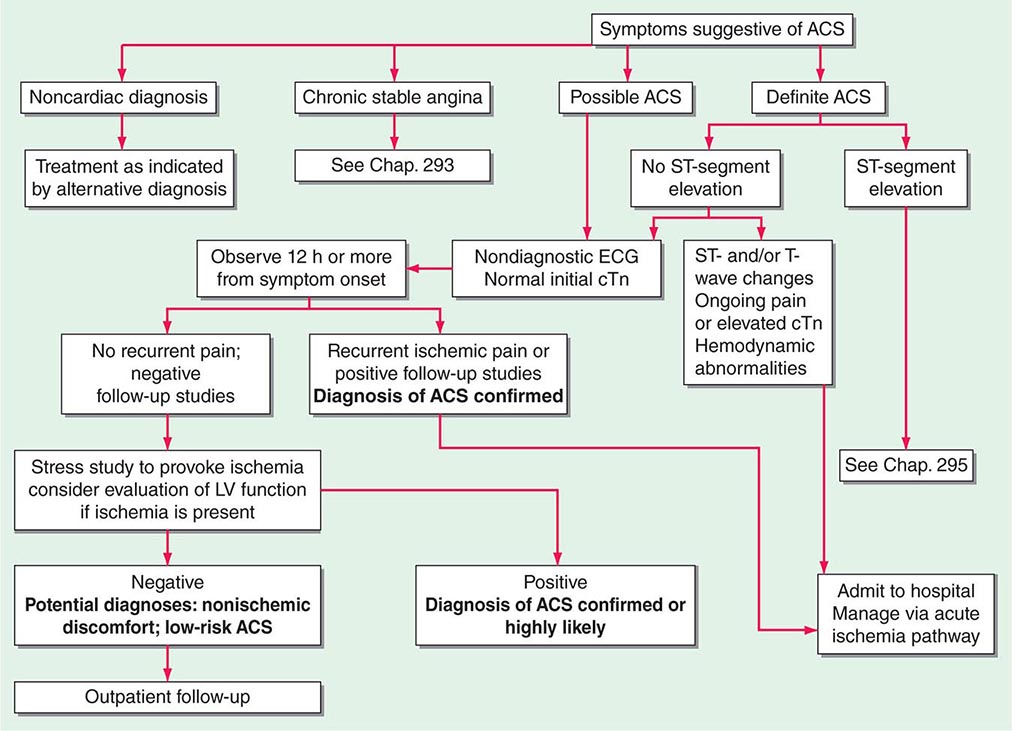
FIGURE 294-3 Algorithm for evaluation and management of patients with suspected acute coronary syndrome (ACS). Follow-up studies refer to ST deviations and elevation of troponin levels. cTn, cardiac troponin; ECG, electrocardiogram; LV, left ventricular. (Modified from JL Anderson et al: J Am Coll Cardiol 61:e179, 2013.)
RISK STRATIFICATION
Patients with documented NSTE-ACS exhibit a wide spectrum of early (30 days) risk of death, ranging from 1 to 10%, and a recurrent ACS rate of 5 to 15% during the first year. Assessment of risk can be accomplished by clinical risk scoring systems such as that developed from the Thrombolysis in Myocardial Infarction (TIMI) Trials, which includes seven independent risk factors (Fig. 294-4A). The presence of an abnormally elevated cTn is especially important, as is its peak level, which correlates with the extent of myocardial damage (Fig. 294-4B). Other risk factors include diabetes mellitus, left ventricular dysfunction, renal dysfunction, and elevated levels of B-type natriuretic peptides and C-reactive protein. Multimarker strategies are now gaining favor, both to define more fully the pathophysiologic mechanisms underlying a given patient’s presentation and to stratify the patient’s risk further. Patients with ACS without elevated levels of cTn (infrequently encountered with the new sensitive troponin assays) are considered to have UA and have a more favorable prognosis than those with cTn elevations (NSTEMI).
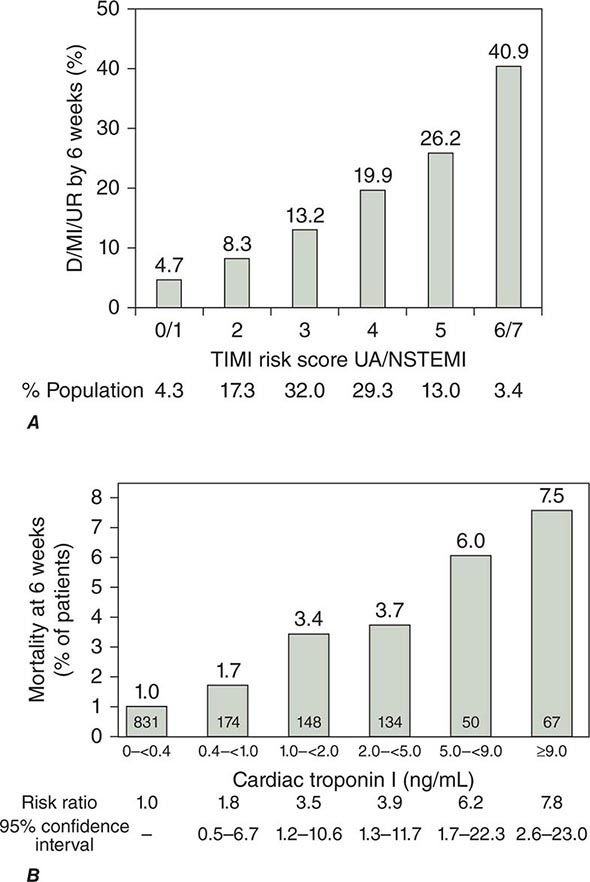
FIGURE 294-4 A. Death (D), myocardial infarction (MI), or need for urgent revascularization (UR) through 6 weeks by Thrombolysis in Myocardial Infarction (TIMI) Risk Score in the unfractionated heparin arm of the TIMI 11B trial. (From EM Antman et al: JAMA 284:835, 2000.) B. Mortality rate at 42 days by baseline cardiac troponin I levels in the TIMI 3B trial. (From EM Antman et al: N Engl J Med 335:1342, 1996.)
Early risk assessment is useful both in predicting the risk of recurrent cardiac events and in identifying patients who would derive the greatest benefit from an early invasive strategy. For example, in the TACTICS-TIMI 18 Trial, an early invasive strategy conferred a 40% reduction in recurrent cardiac events in patients with an elevated cTn level, whereas no benefit was observed in those without detectable troponin.
|
TREATMENT |
NON-ST-SEGMENT ELEVATION ACUTE CORONARY SYNDROME (NON-ST-SEGMENT ELEVATION MYOCARDIAL INFARCTION AND UNSTABLE ANGINA) |
MEDICAL TREATMENT
Patients should be placed at bed rest with continuous ECG monitoring for ST-segment deviation and cardiac arrhythmias. Ambulation is permitted if the patient shows no recurrence of ischemia (symptoms or ECG changes) and does not develop an elevation of a biomarker of necrosis for 12–24 h. Medical therapy involves simultaneous anti-ischemic and antithrombotic treatments and consideration of coronary revascularization.
ANTI-ISCHEMIC TREATMENT (TABLE 294-1)
|
DRUGS COMMONLY USED IN INTENSIVE MEDICAL MANAGEMENT OF PATIENTS WITH UNSTABLE ANGINA AND NON-ST-SEGMENT ELEVATION MYOCARDIAL INFARCTION |
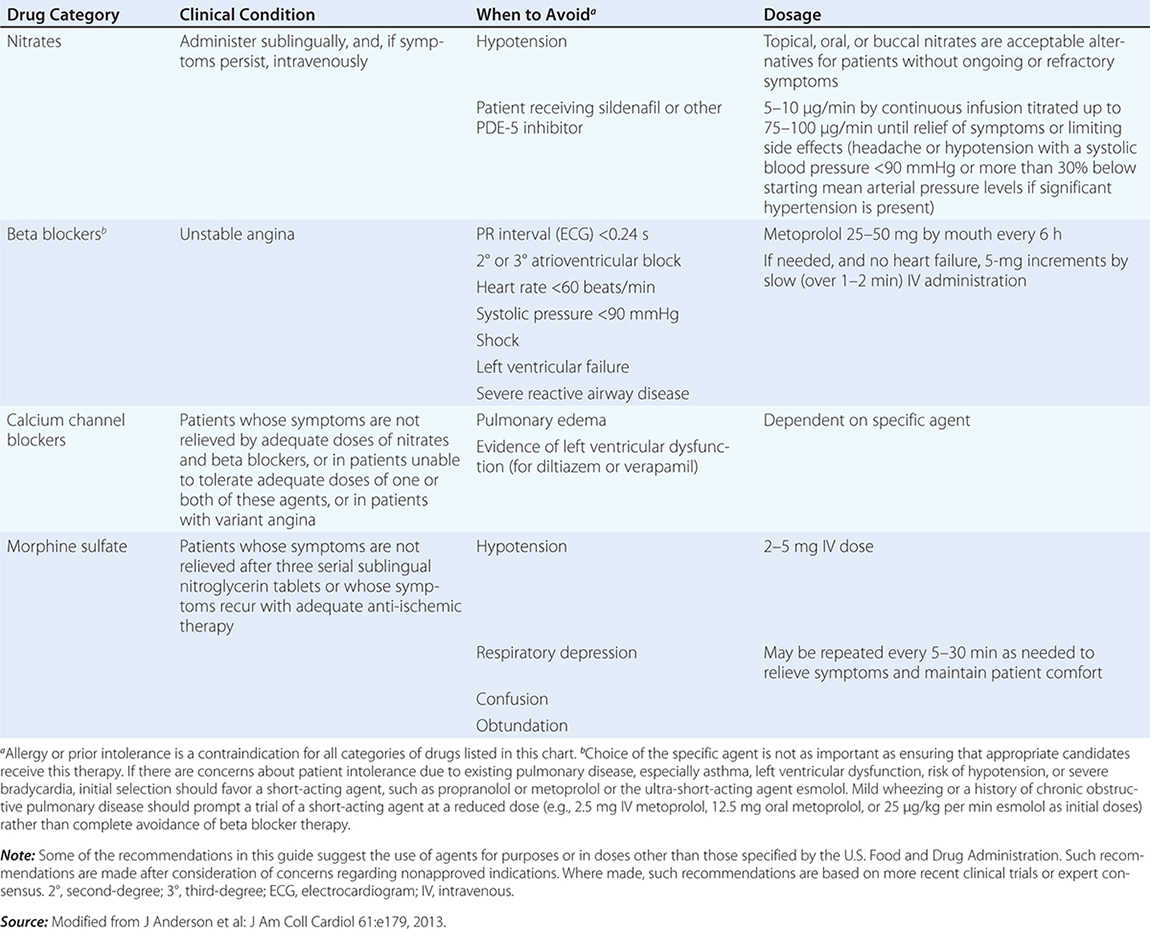
To provide relief and prevention of recurrence of chest pain, initial treatment should include bed rest, nitrates, beta adrenergic blockers, and inhaled oxygen in the presence of hypoxemia.
Nitrates These should first be given sublingually or by buccal spray (0.3–0.6 mg) if the patient is experiencing ischemic pain. If pain persists after three doses given 5 min apart, intravenous nitroglycerin (5–10 μg/min using nonabsorbing tubing) is recommended. The rate of the infusion may be increased by 10 μg/min every 3–5 min until symptoms are relieved, systolic arterial pressure falls to <100 mmHg, or the dose reaches 200 μg/min. Topical or oral nitrates (Chap. 293) can be used when the pain has resolved, or they may replace intravenous nitroglycerin when the patient has been pain-free for 12–24 h. The only absolute contraindications to the use of nitrates are hypotension or the use of sildenafil or other phosphodiesterase-5 inhibitors within the previous 24–48 h.
Beta Adrenergic Blockers and Other Agents Beta blockers are the other mainstay of anti-ischemic treatment. They may be started by the intravenous route in patients with severe ischemia, but this is contraindicated in the presence of heart failure. Ordinarily, oral beta blockade targeted to a heart rate of 50–60 beats/min is recommended. Heart rate–slowing calcium channel blockers, e.g., verapamil or diltiazem, are recommended for patients who have persistent symptoms or ECG signs of ischemia after treatment with full-dose nitrates and beta blockers and in patients with contraindications to either class of these agents. Additional medical therapy includes angiotensin-converting enzyme (ACE) inhibitors or, if these are not tolerated, angiotensin receptor blockers. Early administration of intensive HMG-CoA reductase inhibitors (statins), such as atorvastatin 80 mg/d, prior to percutaneous coronary intervention (PCI), and continued thereafter, has been shown to reduce complications of the procedure and recurrences of ACS.
ANTITHROMBOTIC THERAPY (TABLE 294-2)
|
CLINICAL USE OF ANTITHROMBOTIC THERAPY |
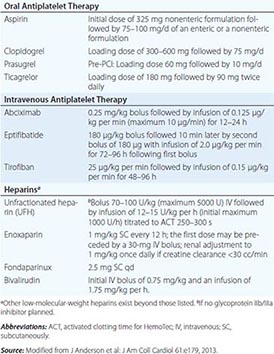
This is the second major cornerstone of treatment. There are two components of antithrombotic therapy: antiplatelet drugs and anticoagulants.
Antiplatelet Drugs (See Chap. 143) Initial treatment should begin with the platelet cyclooxygenase inhibitor aspirin. The typical initial dose is 325 mg/d, with lower doses (75–100 mg/d) recommended thereafter. Contraindications are active bleeding or aspirin intolerance. “Aspirin resistance” has been noted in 2–8% of patients but frequently has been related to noncompliance.
In the absence of a high risk for bleeding, patients with NSTE-ACS, irrespective of whether an invasive or conservative strategy (see below) is selected, should receive a platelet P2Y12 receptor blocker to inhibit platelet activation. The thienopyridine clopidogrel is an inactive prodrug that is converted into an active metabolite that causes irreversible blockade of the platelet P2Y12 receptor. When added to aspirin, so-called dual antiplatelet therapy, it has been shown to confer a 20% relative reduction in cardiovascular death, MI, or stroke, compared to aspirin alone, but to be associated with a moderate (absolute 1%) increase in major bleeding.
Continued benefit of treatment with the combination of aspirin and clopidogrel has been observed both in patients treated conservatively and in those who underwent PCI. This regimen should continue for at least 1 year in patients with NSTE-ACS, especially those with a drug-eluting stent, to prevent stent thrombosis. Up to one-third of patients have an inadequate response to clopidogrel, and a substantial proportion of these cases are related to a genetic variant of the cytochrome P450 system. A variant of the 2C19 gene leads to reduced conversion of clopidogrel into its active metabolite, which, in turn, reduces platelet inhibition and is associated with increases in the incidence of adverse cardiovascular events. Alternate P2Y12 blockers, such as prasugrel or ticagrelor (see below) used with aspirin, should be considered in patients with NSTE-ACS who develop a coronary event while receiving clopidogrel and aspirin or who are hyporesponsive to clopidogrel as identified by platelet and/or genetic testing, although such testing is not yet widespread.
A second P2Y12 blocker, prasugrel, also a thienopyridine, achieves a more rapid onset and higher level of platelet inhibition than clopidogrel. It has been approved for ACS patients following angiography in whom PCI is planned. It should be administered at a loading dose of 60 mg followed by 10 mg/d for up to 15 months. The TRITON-TIMI 38 trial showed that relative to clopidogrel, prasugrel reduced the risk of cardiovascular death, MI, or stroke significantly, albeit with an increase in major bleeding. Stent thrombosis was reduced by half. This agent is contraindicated in patients with prior stroke or transient ischemic attack or at high risk for bleeding. It has not been found to be effective in patients treated by a conservative strategy (see below).
Ticagrelor is a novel, potent, reversible platelet P2Y12 inhibitor. It has been shown in the PLATO trial to reduce the risk of cardiovascular death, MI, or stroke compared with clopidogrel in ACS patients who are treated by either an invasive or a conservative strategy. This agent reduced mortality but increased the risk of bleeding not associated with coronary artery bypass grafting. After a loading dose of 180 mg, 90 mg bid is administered as maintenance.
Prior to the development of the oral P2Y12 receptor blockers, many trials had shown the benefit of intravenous glycoprotein IIb/IIIa inhibitors. Their benefit, however, has been small (i.e., only a 10% reduction in death or MI, with a significant increase in major bleeding). Two recent studies failed to show a benefit of routine early initiation of a drug in this class compared with their use only in patients who undergo PCI. The addition of these agents to aspirin and a P2Y12 inhibitor (i.e., triple antiplatelet therapy) should be reserved for unstable patients with recurrent rest pain, elevated cTn, and ECG changes, as well as those who have a coronary thrombus evident on angiography when they undergo PCI.
Anticoagulants (See Chap. 143) Four options are available for anticoagulant therapy to be added to antiplatelet agents: (1) unfractionated heparin (UFH), long the mainstay of therapy; (2) the low-molecular-weight heparin (LMWH), enoxaparin, which has been shown to be superior to UFH in reducing recurrent cardiac events, especially in patients managed by a conservative strategy but with some increase in bleeding; (3) bivalirudin, a direct thrombin inhibitor that is similar in efficacy to either UFH or LMWH but causes less bleeding and is used just prior to and/or during PCI; and (4) the indirect factor Xa inhibitor, fondaparinux, which is equivalent in efficacy to enoxaparin but appears to have a lower risk of major bleeding.
Excessive bleeding is the most important adverse effect of all antithrombotic agents, including both antiplatelet agents and anticoagulants. Therefore, attention must be directed to the doses of antithrombotic agents, accounting for body weight, creatinine clearance, and a previous history of excessive bleeding, as a means of reducing the risk of bleeding. Patients who have experienced a stroke are at higher risk of intracranial bleeding with potent antiplatelet agents and combinations of antithrombotic drugs.
INVASIVE VERSUS CONSERVATIVE STRATEGY
Multiple clinical trials have demonstrated the benefit of an early invasive strategy in high-risk patients (i.e., patients with multiple clinical risk factors, ST-segment deviation, and/or positive biomarkers) (Table 294-3). In this strategy, following treatment with anti-ischemic and antithrombotic agents, coronary arteriography is carried out within ∼48 h of presentation, followed by coronary revascularization (PCI or coronary artery bypass grafting), depending on the coronary anatomy. In low-risk patients, the outcomes from an invasive strategy are similar to those obtained from a conservative strategy. The latter consists of anti-ischemic and antithrombotic therapy followed by “watchful waiting,” in which the patient is closely observed and coronary arteriography is carried out only if rest pain or ST-segment changes recur, a biomarker of necrosis becomes positive, or there is evidence of severe ischemia on a stress test.
|
CLASS I RECOMMENDATIONS FOR USE OF AN EARLY INVASIVE STRATEGY IN PATIENTS WITH NON-ST-SEGMENT ELEVATION ACUTE CORONARY SYNDROMEa |
aAny one of the high-risk indicators. bSee Antman (JAMA 284:835, 2000).
Abbreviations: CABG, coronary artery bypass grafting; CHF, congestive heart failure; EF, ejection fraction; MR, mitral regurgitation; PCI, percutaneous coronary intervention; TIMI, Thrombolysis in Myocardial Infarction; TnI, troponin I; TnT, troponin T; VT, ventricular tachycardia.
Source: Modified from J Anderson et al: J Am Coll Cardiol 61:e179, 2013.
LONG-TERM MANAGEMENT
The time of hospital discharge is a “teachable moment” for the patient with NSTE-ACS, when the physician can review and optimize the medical regimen. Risk-factor modification is key, and the caregiver should discuss with the patient the importance of smoking cessation, achieving optimal weight, daily exercise, blood-pressure control, following an appropriate diet, control of hyperglycemia (in diabetic patients), and lipid management as recommended for patients with chronic stable angina (Chap. 293).
There is evidence of benefit with long-term therapy with five classes of drugs that are directed at different components of the atherothrombotic process. Beta blockers, statins (at a high dose, e.g., atorvastatin 80 mg/d), and ACE inhibitors or angiotensin receptor blockers are recommended for long-term plaque stabilization. Antiplatelet therapy, now recommended to be the combination of low-dose (75–100 mg/d) aspirin and a P2Y12 inhibitor (clopidogrel, prasugrel, or ticagrelor) for 1 year, with aspirin continued thereafter, prevents or reduces the severity of any thrombosis that would occur if a plaque were to rupture.
Registries have shown that women and racial minorities, as well as patients with NSTE-ACS at high risk, including the elderly and patients with diabetes or chronic kidney disease, are less likely to receive evidence-based pharmacologic and interventional therapies with resultant poorer clinical outcomes and quality of life. Special attention should be directed to these groups.
PRINZMETAL’S VARIANT ANGINA
In 1959 Prinzmetal et al. described a syndrome of severe ischemic pain that usually occurs at rest and is associated with transient ST-segment elevation. Prinzmetal’s variant angina (PVA) is caused by focal spasm of an epicardial coronary artery, leading to severe transient myocardial ischemia and occasionally infarction. The cause of the spasm is not well defined, but it may be related to hypercontractility of vascular smooth muscle due to adrenergic vasoconstrictors, leukotrienes, or serotonin. For reasons that are not clear, the prevalence of PVA has decreased substantially during the past few decades.
Clinical and Angiographic Manifestations Patients with PVA are generally younger and have fewer coronary risk factors (with the exception of cigarette smoking) than do patients with NSTE-ACS. Cardiac examination is usually unremarkable in the absence of ischemia. The clinical diagnosis of PVA is made by the detection of transient ST-segment elevation with rest pain. Many patients also exhibit multiple episodes of asymptomatic ST-segment elevation (silent ischemia). Small elevations of troponin may occur in patients with prolonged attacks.
Coronary angiography demonstrates transient coronary spasm as the diagnostic hallmark of PVA. Atherosclerotic plaques in at least one proximal coronary artery occur in about half of patients, and in these patients, spasm usually occurs within 1 cm of the plaque. Focal spasm is most common in the right coronary artery, and it may occur at one or more sites in one artery or in multiple arteries simultaneously. Hyperventilation or intracoronary acetylcholine has been used to provoke focal coronary stenosis on angiography or to provoke rest angina with ST-segment elevation to establish the diagnosis.
Prognosis Many patients with PVA pass through an acute, active phase, with frequent episodes of angina and cardiac events during the first 6 months after presentation. Survival at 5 years is excellent (∼90–95%). Patients with no or mild fixed coronary obstruction tend to experience a more benign course than do patients with associated severe obstructive lesions. Nonfatal MI occurs in up to 20% of patients by 5 years. Patients with PVA who develop serious arrhythmias during spontaneous episodes of pain are at a higher risk for sudden cardiac death. In most patients who survive an infarction or the initial 3- to 6-month period of frequent episodes, there is a tendency for symptoms and cardiac events to diminish over time.
295 |
ST-Segment Elevation Myocardial Infarction |
Acute myocardial infarction (AMI) is one of the most common diagnoses in hospitalized patients in industrialized countries. In the United States, approximately 525,000 patients experience a new AMI, and 190,000 experience a recurrent AMI each year. More than half of AMI-related deaths occur before the stricken individual reaches the hospital. The in-hospital mortality rate after admission for AMI has declined from 10% to about 6% over the past decade. The 1-year mortality rate after AMI is about 15%. Mortality is approximately fourfold higher in elderly patients (over age 75) as compared with younger patients.
When patients with prolonged ischemic discomfort at rest are first seen, the working clinical diagnosis is that they are suffering from an acute coronary syndrome (Fig. 295-1). The 12-lead electrocardiogram (ECG) is a pivotal diagnostic and triage tool because it is at the center of the decision pathway for management; it permits distinction of those patients presenting with ST-segment elevation from those presenting without ST-segment elevation. Serum cardiac biomarkers are obtained to distinguish unstable angina (UA) from non-ST-segment elevation myocardial infarction (NSTEMI) and to assess the magnitude of an ST-segment elevation myocardial infarction (STEMI). This chapter focuses on the evaluation and management of patients with STEMI, while Chap. 294 discusses UA/NSTEMI.
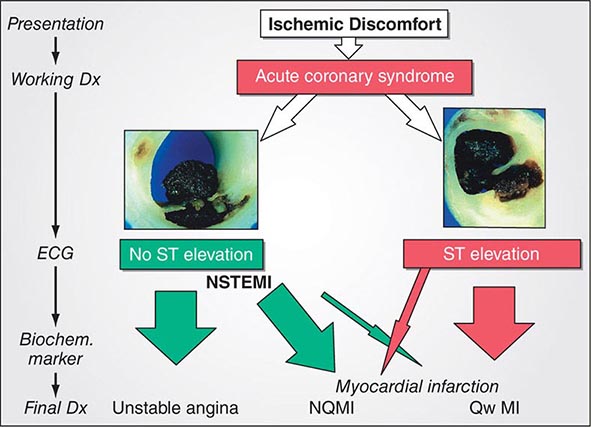
FIGURE 295-1 Acute coronary syndromes. Following disruption of a vulnerable plaque, patients experience ischemic discomfort resulting from a reduction of flow through the affected epicardial coronary artery. The flow reduction may be caused by a completely occlusive thrombus (right) or subtotally occlusive thrombus (left). Patients with ischemic discomfort may present with or without ST-segment elevation. Of patients with ST-segment elevation, the majority (wide red arrow) ultimately develop a Q wave on the ECG (Qw MI), while a minority (thin red arrow) do not develop Q wave and, in older literature, were said to have sustained a non-Q-wave MI (NQMI). Patients who present without ST-segment elevation are suffering from either unstable angina or a non-ST-segment elevation MI (NSTEMI) (wide green arrows), a distinction that is ultimately made based on the presence or absence of a serum cardiac marker such as CK-MB or a cardiac troponin detected in the blood. The majority of patients presenting with NSTEMI do not develop a Q wave on the ECG; a minority develop a Qw MI (thin green arrow). Dx, diagnosis; ECG, electrocardiogram; MI, myocardial infarction. (Adapted from CW Hamm et al: Lancet 358:1533, 2001, and MJ Davies: Heart 83:361, 2000; with permission from the BMJ Publishing Group.)
PATHOPHYSIOLOGY: ROLE OF ACUTE PLAQUE RUPTURE
STEMI usually occurs when coronary blood flow decreases abruptly after a thrombotic occlusion of a coronary artery previously affected by atherosclerosis. Slowly developing, high-grade coronary artery stenoses do not typically precipitate STEMI because of the development of a rich collateral network over time. Instead, STEMI occurs when a coronary artery thrombus develops rapidly at a site of vascular injury. This injury is produced or facilitated by factors such as cigarette smoking, hypertension, and lipid accumulation. In most cases, STEMI occurs when the surface of an atherosclerotic plaque becomes disrupted (exposing its contents to the blood) and conditions (local or systemic) favor thrombogenesis. A mural thrombus forms at the site of plaque disruption, and the involved coronary artery becomes occluded. Histologic studies indicate that the coronary plaques prone to disruption are those with a rich lipid core and a thin fibrous cap (Chap. 291e). After an initial platelet monolayer forms at the site of the disrupted plaque, various agonists (collagen, ADP, epinephrine, serotonin) promote platelet activation. After agonist stimulation of platelets, thromboxane A2 (a potent local vasoconstrictor) is released, further platelet activation occurs, and potential resistance to fibrinolysis develops.
In addition to the generation of thromboxane A2, activation of platelets by agonists promotes a conformational change in the glycoprotein IIb/IIIa receptor (Chap. 140). Once converted to its functional state, this receptor develops a high affinity for soluble adhesive proteins (i.e., integrins) such as fibrinogen. Since fibrinogen is a multivalent molecule, it can bind to two different platelets simultaneously, resulting in platelet cross-linking and aggregation.
The coagulation cascade is activated on exposure of tissue factor in damaged endothelial cells at the site of the disrupted plaque. Factors VII and × are activated, ultimately leading to the conversion of prothrombin to thrombin, which then converts fibrinogen to fibrin (Chap. 141). Fluid-phase and clot-bound thrombin participates in an autoamplification reaction leading to further activation of the coagulation cascade. The culprit coronary artery eventually becomes occluded by a thrombus containing platelet aggregates and fibrin strands.
In rare cases, STEMI may be due to coronary artery occlusion caused by coronary emboli, congenital abnormalities, coronary spasm, and a wide variety of systemic—particularly inflammatory—diseases. The amount of myocardial damage caused by coronary occlusion depends on (1) the territory supplied by the affected vessel, (2) whether or not the vessel becomes totally occluded, (3) the duration of coronary occlusion, (4) the quantity of blood supplied by collateral vessels to the affected tissue, (5) the demand for oxygen of the myocardium whose blood supply has been suddenly limited, (6) endogenous factors that can produce early spontaneous lysis of the occlusive thrombus, and (7) the adequacy of myocardial perfusion in the infarct zone when flow is restored in the occluded epicardial coronary artery.
Patients at increased risk for developing STEMI include those with multiple coronary risk factors (Chap. 291e) and those with UA (Chap. 294). Less common underlying medical conditions predisposing patients to STEMI include hypercoagulability, collagen vascular disease, cocaine abuse, and intracardiac thrombi or masses that can produce coronary emboli.
There have been major advances in the management of STEMI with recognition that the “chain of survival” involves a highly integrated system starting with prehospital care and extending to early hospital management so as to provide expeditious implementation of a reperfusion strategy.
CLINICAL PRESENTATION
In up to one-half of cases, a precipitating factor appears to be present before STEMI, such as vigorous physical exercise, emotional stress, or a medical or surgical illness. Although STEMI may commence at any time of the day or night, circadian variations have been reported such that clusters are seen in the morning within a few hours of awakening.
Pain is the most common presenting complaint in patients with STEMI. The pain is deep and visceral; adjectives commonly used to describe it are heavy, squeezing, and crushing, although, occasionally, it is described as stabbing or burning (Chap. 19). It is similar in character to the discomfort of angina pectoris (Chap. 293) but commonly occurs at rest, is usually more severe, and lasts longer. Typically, the pain involves the central portion of the chest and/or the epigastrium, and, on occasion, it radiates to the arms. Less common sites of radiation include the abdomen, back, lower jaw, and neck. The frequent location of the pain beneath the xiphoid and epigastrium and the patients’ denial that they may be suffering a heart attack are chiefly responsible for the common mistaken impression of indigestion. The pain of STEMI may radiate as high as the occipital area but not below the umbilicus. It is often accompanied by weakness, sweating, nausea, vomiting, anxiety, and a sense of impending doom. The pain may commence when the patient is at rest, but when it begins during a period of exertion, it does not usually subside with cessation of activity, in contrast to angina pectoris.
The pain of STEMI can simulate pain from acute pericarditis (Chap. 288), pulmonary embolism (Chap. 300), acute aortic dissection (Chap. 301), costochondritis, and gastrointestinal disorders. These conditions should therefore be considered in the differential diagnosis. Radiation of discomfort to the trapezius is not seen in patients with STEMI and may be a useful distinguishing feature that suggests pericarditis is the correct diagnosis. However, pain is not uniformly present in patients with STEMI. The proportion of painless STEMIs is greater in patients with diabetes mellitus, and it increases with age. In the elderly, STEMI may present as sudden-onset breathlessness, which may progress to pulmonary edema. Other less common presentations, with or without pain, include sudden loss of consciousness, a confusional state, a sensation of profound weakness, the appearance of an arrhythmia, evidence of peripheral embolism, or merely an unexplained drop in arterial pressure.
PHYSICAL FINDINGS
Most patients are anxious and restless, attempting unsuccessfully to relieve the pain by moving about in bed, altering their position, and stretching. Pallor associated with perspiration and coolness of the extremities occurs commonly. The combination of substernal chest pain persisting for >30 min and diaphoresis strongly suggests STEMI. Although many patients have a normal pulse rate and blood pressure within the first hour of STEMI, about one-fourth of patients with anterior infarction have manifestations of sympathetic nervous system hyperactivity (tachycardia and/or hypertension), and up to one-half with inferior infarction show evidence of parasympathetic hyperactivity (bradycardia and/or hypotension).
The precordium is usually quiet, and the apical impulse may be difficult to palpate. In patients with anterior wall infarction, an abnormal systolic pulsation caused by dyskinetic bulging of infarcted myocardium may develop in the periapical area within the first days of the illness and then may resolve. Other physical signs of ventricular dysfunction include fourth and third heart sounds, decreased intensity of the first heart sound, and paradoxical splitting of the second heart sound (Chap. 267). A transient midsystolic or late systolic apical systolic murmur due to dysfunction of the mitral valve apparatus may be present. A pericardial friction rub may be heard in patients with transmural STEMI at some time in the course of the disease, if they are examined frequently. The carotid pulse is often decreased in volume, reflecting reduced stroke volume. Temperature elevations up to 38°C may be observed during the first week after STEMI. The arterial pressure is variable; in most patients with transmural infarction, systolic pressure declines by approximately 10–15 mmHg from the preinfarction state.
LABORATORY FINDINGS
STEMI progresses through the following temporal stages: (1) acute (first few hours–7 days), (2) healing (7–28 days), and (3) healed (≥29 days). When evaluating the results of diagnostic tests for STEMI, the temporal phase of the infarction must be considered. The laboratory tests of value in confirming the diagnosis may be divided into four groups: (1) ECG, (2) serum cardiac biomarkers, (3) cardiac imaging, and (4) nonspecific indices of tissue necrosis and inflammation.
ELECTROCARDIOGRAM
The electrocardiographic manifestations of STEMI are described in Chap. 268. During the initial stage, total occlusion of an epicardial coronary artery produces ST-segment elevation. Most patients initially presenting with ST-segment elevation ultimately evolve Q waves on the ECG. However, Q waves in the leads overlying the infarct zone may vary in magnitude and even appear only transiently, depending on the reperfusion status of the ischemic myocardium and restoration of transmembrane potentials over time. A small proportion of patients initially presenting with ST-segment elevation will not develop Q waves when the obstructing thrombus is not totally occlusive, obstruction is transient, or if a rich collateral network is present. Among patients presenting with ischemic discomfort but without ST-segment elevation, if a serum cardiac biomarker of necrosis (see below) is detected, the diagnosis of NSTEMI is ultimately made (Fig. 295-1). A minority of patients who present initially without ST-segment elevation may develop a Q-wave MI. Previously, it was believed that transmural myocardial infarction (MI) is present if the ECG demonstrates Q waves or loss of R waves, and nontransmural MI may be present if the ECG shows only transient ST-segment and T-wave changes. However, electrocardiographic-pathologic correlations are far from perfect and terms such as Q-wave MI, non-Q-wave MI, transmural MI, and nontransmural MI, have been replaced by STEMI and NSTEMI (Fig. 295-1). Contemporary studies using magnetic resonance imaging (MRI) suggest that the development of a Q wave on the ECG is more dependent on the volume of infarcted tissue rather than the transmurality of infarction.
SERUM CARDIAC BIOMARKERS
Certain proteins, called serum cardiac biomarkers, are released from necrotic heart muscle after STEMI. The rate of liberation of specific proteins differs depending on their intracellular location, their molecular weight, and the local blood and lymphatic flow. Cardiac biomarkers become detectable in the peripheral blood once the capacity of the cardiac lymphatics to clear the interstitium of the infarct zone is exceeded and spillover into the venous circulation occurs. The temporal pattern of protein release is of diagnostic importance. The criteria for AMI require a rise and/or fall in cardiac biomarker values with at least one value above the 99th percentile of the upper reference limit for normal individuals
Cardiac-specific troponin T (cTnT) and cardiac-specific troponin I (cTnI) have amino-acid sequences different from those of the skeletal muscle forms of these proteins. These differences permitted the development of quantitative assays for cTnT and cTnI with highly specific monoclonal antibodies. Since cTnT and cTnI are not normally detectable in the blood of healthy individuals but may increase after STEMI to levels many times higher than the upper reference limit (the highest value seen in 99% of a reference population not suffering from MI), the measurement of cTnT or cTnI is of considerable diagnostic usefulness, and they are now the preferred biochemical markers for MI (Fig. 295-2). With improvements in the assays for the cardiac-specific troponins, it is now possible to detect concentrations <1 ng/L in patients without ischemic-type chest discomfort. The cardiac troponins are particularly valuable when there is clinical suspicion of either skeletal muscle injury or a small MI that may be below the detection limit for creatine phosphokinase (CK) and its MB isoenzyme (CK-MB) measurements, and they are, therefore, of particular value in distinguishing UA from NSTEMI. In practical terms, the high-sensitivity troponin assays are of less immediate value in patients with STEMI. Contemporary urgent reperfusion strategies necessitate making a decision (based largely on a combination of clinical and ECG findings) before the results of blood tests have returned from the laboratory. Levels of cTnI and cTnT may remain elevated for 7–10 days after STEMI.
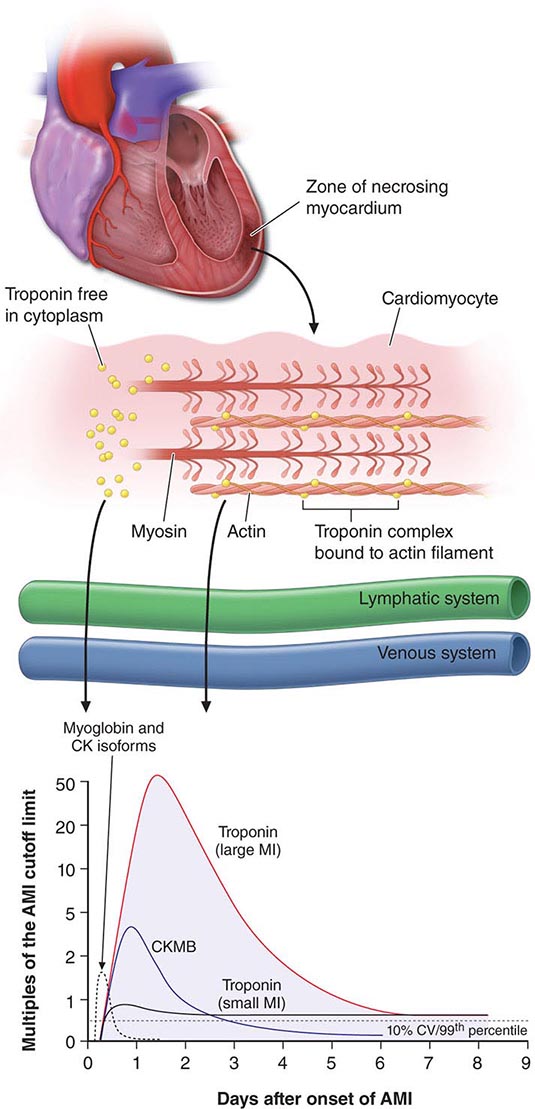
FIGURE 295-2 The zone of necrosing myocardium is shown at the top of the figure, followed in the middle portion of the figure by a diagram of a cardiomyocyte that is in the process of releasing biomarkers. The biomarkers that are released into the interstitium are first cleared by lymphatics followed subsequently by spillover into the venous system. After disruption of the sarcolemmal membrane of the cardiomyocyte, the cytoplasmic pool of biomarkers is released first (left-most arrow in bottom portion of figure). Markers such as myoglobin and CK isoforms are rapidly released, and blood levels rise quickly above the cutoff limit; this is then followed by a more protracted release of biomarkers from the disintegrating myofilaments that may continue for several days. Cardiac troponin levels rise to about 20 to 50 times the upper reference limit (the 99th percentile of values in a reference control group) in patients who have a “classic” acute myocardial infarction (MI) and sustain sufficient myocardial necrosis to result in abnormally elevated levels of the MB fraction of creatine kinase (CK-MB). Clinicians can now diagnose episodes of microinfarction by sensitive assays that detect cardiac troponin elevations above the upper reference limit, even though CK-MB levels may still be in the normal reference range (not shown). CV, coefficient of variation. (Modified from EM Antman: Decision making with cardiac troponin tests. N Engl J Med 346:2079, 2002 and AS Jaffe, L Babiun, FS Apple: Biomarkers in acute cardiac disease: The present and the future. J Am Coll Cardiol 48:1, 2006.)
CK rises within 4–8 h and generally returns to normal by 48–72 h (Fig. 295-2). An important drawback of total CK measurement is its lack of specificity for STEMI, as CK may be elevated with skeletal muscle disease or trauma, including intramuscular injection. The MB isoenzyme of CK has the advantage over total CK that it is not present in significant concentrations in extracardiac tissue and, therefore, is considerably more specific. However, cardiac surgery, myocarditis, and electrical cardioversion often result in elevated serum levels of the MB isoenzyme. A ratio (relative index) of CK-MB mass to CK activity ≥2.5 suggests but is not diagnostic of a myocardial rather than a skeletal muscle source for the CK-MB elevation.
Many hospitals are using cTnT or cTnI rather than CK-MB as the routine serum cardiac marker for diagnosis of STEMI, although any of these analytes remain clinically acceptable. It is not cost-effective to measure both a cardiac-specific troponin and CK-MB at all time points in every patient.
While it has long been recognized that the total quantity of protein released correlates with the size of the infarct, the peak protein concentration correlates only weakly with infarct size. Recanalization of a coronary artery occlusion (either spontaneously or by mechanical or pharmacologic means) in the early hours of STEMI causes earlier peaking of biomarker measurements (Fig. 295-2) because of a rapid washout from the interstitium of the infarct zone, quickly overwhelming lymphatic clearance of the proteins.
The nonspecific reaction to myocardial injury is associated with polymorphonuclear leukocytosis, which appears within a few hours after the onset of pain and persists for 3–7 days; the white blood cell count often reaches levels of 12,000–15,000/μL. The erythrocyte sedimentation rate rises more slowly than the white blood cell count, peaking during the first week and sometimes remaining elevated for 1 or 2 weeks.
CARDIAC IMAGING
Abnormalities of wall motion on two-dimensional echocardiography (Chap. 270e) are almost universally present. Although acute STEMI cannot be distinguished from an old myocardial scar or from acute severe ischemia by echocardiography, the ease and safety of the procedure make its use appealing as a screening tool in the Emergency Department setting. When the ECG is not diagnostic of STEMI, early detection of the presence or absence of wall motion abnormalities by echocardiography can aid in management decisions, such as whether the patient should receive reperfusion therapy (e.g., fibrinolysis or a percutaneous coronary intervention [PCI]). Echocardiographic estimation of left ventricular (LV) function is useful prognostically; detection of reduced function serves as an indication for therapy with an inhibitor of the renin-angiotensin-aldosterone system. Echocardiography may also identify the presence of right ventricular (RV) infarction, ventricular aneurysm, pericardial effusion, and LV thrombus. In addition, Doppler echocardiography is useful in the detection and quantitation of a ventricular septal defect and mitral regurgitation, two serious complications of STEMI.
[/level-membership-for-internal-medicine-category][not-level-membership-for-internal-medicine-category]
289e |
Tumors and Trauma of the Heart |
TUMORS OF THE HEART
PRIMARY TUMORS
Primary tumors of the heart are rare. Approximately three-quarters are histologically benign, and the majority of these tumors are myxomas. Malignant tumors, almost all of which are sarcomas, account for 25% of primary cardiac tumors. All cardiac tumors, regardless of pathologic type, have the potential to cause life-threatening complications. Many tumors are now surgically curable; thus, early diagnosis is imperative.
Clinical Presentation Cardiac tumors may present with a wide array of cardiac and noncardiac manifestations. These manifestations depend in large part on the location and size of the tumor and are often nonspecific features of more common forms of heart disease, such as chest pain, syncope, heart failure, murmurs, arrhythmias, conduction disturbances, and pericardial effusion with or without tamponade. Additionally, embolic phenomena and constitutional symptoms may occur.
Myxoma Myxomas are the most common type of primary cardiac tumor in adults, accounting for one-third to one-half of all cases at postmortem examination, and about three-quarters of the tumors treated surgically. They occur at all ages, most commonly in the third through sixth decades, with a female predilection. Approximately 90% of myxomas are sporadic; the remainder are familial with autosomal dominant transmission. The familial variety often occurs as part of a syndrome complex (Carney complex) that includes (1) myxomas (cardiac, skin, and/or breast), (2) lentigines and/or pigmented nevi, and (3) endocrine overactivity (primary nodular adrenal cortical disease with or without Cushing’s syndrome, testicular tumors, and/or pituitary adenomas with gigantism or acromegaly). Certain constellations of findings have been referred to as the NAME syndrome (nevi, atrial myxoma, myxoid neurofibroma, and ephelides) or the LAMB syndrome (lentigines, atrial myxoma, and blue nevi), although these syndromes probably represent subsets of the Carney complex. The genetic basis of this complex has not been elucidated completely; however, patients frequently have inactivating mutations in the tumor-suppressor gene PRKAR1A, which encodes the protein kinase A type I-α regulatory subunit.
Pathologically, myxomas are gelatinous structures that consist of myxoma cells embedded in a stroma rich in glycosaminoglycans. Most are solitary, arise from the interatrial septum in the vicinity of the fossa ovalis (particularly the left atrium), and are often pedunculated on a fibrovascular stalk. In contrast to sporadic tumors, familial or syndromic tumors tend to occur in younger individuals, are often multiple, may be ventricular in location, and are more likely to recur after initial resection.
Myxomas commonly present with obstructive signs and symptoms. The most common clinical presentation mimics that of mitral valve disease: either stenosis owing to tumor prolapse into the mitral orifice or regurgitation resulting from tumor-induced valvular trauma. Ventricular myxomas may cause outflow obstruction similar to that caused by subaortic or subpulmonic stenosis. The symptoms and signs of myxoma may be sudden in onset or positional in nature, owing to the effects of gravity on tumor position. A characteristic low-pitched sound, a “tumor plop,” may be appreciated on auscultation during early or mid-diastole and is thought to result from the impact of the tumor against the mitral valve or ventricular wall. Myxomas also may present with peripheral or pulmonary emboli or with constitutional signs and symptoms, including fever, weight loss, cachexia, malaise, arthralgias, rash, digital clubbing, Raynaud’s phenomenon, hypergammaglobulinemia, anemia, polycythemia, leukocytosis, elevated erythrocyte sedimentation rate, thrombocytopenia, and thrombocytosis. These features account for the frequent misdiagnosis of patients with myxomas as having endocarditis, collagen vascular disease, or a paraneoplastic syndrome.
Two-dimensional transthoracic or omniplane transesophageal echocardiography is useful in the diagnosis of cardiac myxoma and allows assessment of tumor size and determination of the site of tumor attachment, both of which are important considerations in the planning of surgical excision (Fig. 289e-1). Computed tomography (CT) and magnetic resonance imaging (MRI) may provide important information regarding size, shape, composition, and surface characteristics of the tumor (Fig. 289e-2).
FIGURE 289e-1 Transthoracic echocardiogram demonstrating a large atrial myxoma. The myxoma (Myx) fills the entire left atrium in systole (A) and prolapses across the mitral valve and into the left ventricle (LV) during diastole (B). RA, right atrium; RV, right ventricle. (Courtesy of Dr. Michael Tsang; with permission.)
FIGURE 289e-2 Cardiac magnetic resonance imaging demonstrating a rounded mass (M) within the left atrium (LA). Pathologic evaluation at the time of surgery revealed it to be an atrial myxoma. LV, left ventricle; RA, right atrium; RV, right ventricle.
Although cardiac catheterization and angiography were previously performed routinely before tumor resection, they no longer are considered mandatory when adequate noninvasive information is available and other cardiac disorders (e.g., coronary artery disease) are not considered likely. Additionally, catheterization of the chamber from which the tumor arises carries the risk of tumor embolization. Because myxomas may be familial, echocardiographic screening of first-degree relatives is appropriate, particularly if the patient is young and has multiple tumors or evidence of myxoma syndrome.
Other Benign Tumors Cardiac lipomas, although relatively common, are usually incidental findings at postmortem examination; however, they may grow as large as 15 cm and may present with symptoms owing to mechanical interference with cardiac function, arrhythmias, or conduction disturbances or as an abnormality of the cardiac silhouette on chest x-ray. Papillary fibroelastomas are the most common tumors of the cardiac valves. Although usually clinically silent, they can cause valve dysfunction and may embolize distally, resulting in transient ischemic attacks, stroke, or myocardial infarction. In general, these tumors should be resected even when asymptomatic, although a more conservative approach may be considered for small, right-sided lesions. Rhabdomyomas and fibromas are the most common cardiac tumors in infants and children and usually occur in the ventricles, where they may produce mechanical obstruction to blood flow, thereby mimicking valvular stenosis, congestive heart failure (CHF), restrictive or hypertrophic cardiomyopathy, or pericardial constriction. Rhabdomyomas are probably hamartomatous growths, are multiple in 90% of cases, and are strongly associated with tuberous sclerosis. These tumors have a tendency to regress completely or partially; only tumors that cause obstruction require surgical resection. Fibromas are usually single, are often calcified, tend to grow and cause obstructive symptoms, and should be resected. Hemangiomas and mesotheliomas are generally small tumors, most often intramyocardial in location, and may cause atrioventricular (AV) conduction disturbances and even sudden death as a result of their propensity to develop in the region of the AV node. Other benign tumors arising from the heart include teratoma, chemodectoma, neurilemoma, granular cell myoblastoma, and bronchogenic cysts.
Sarcoma Almost all malignant primary cardiac tumors are sarcomas, which may be of several histologic types. In general, these tumors are characterized by rapid progression that culminates in the patient’s death within weeks to months from the time of presentation as a result of hemodynamic compromise, local invasion, or distant metastases. Sarcomas commonly involve the right side of the heart, are characterized by rapid growth, frequently invade the pericardial space, and may obstruct the cardiac chambers or venae cavae. Sarcomas also may occur on the left side of the heart and may be mistaken for myxomas.
TUMORS METASTATIC TO THE HEART
Tumors metastatic to the heart are much more common than primary tumors, and their incidence is likely to increase as the life expectancy of patients with various forms of malignant neoplasms is extended by more effective therapy. Although cardiac metastases may occur with any tumor type, the relative incidence is especially high in malignant melanoma and, to a somewhat lesser extent, leukemia and lymphoma. In absolute terms, the most common primary originating sites of cardiac metastases are carcinoma of the breast and lung, reflecting the high incidence of those cancers. Cardiac metastases almost always occur in the setting of widespread primary disease, and most often there is either primary or metastatic disease elsewhere in the thoracic cavity. Nevertheless, cardiac metastasis occasionally may be the initial presentation of an extrathoracic tumor.
Cardiac metastases may occur via hematogenous or lymphangitic spread or by direct tumor invasion. They generally manifest as small, firm nodules; diffuse infiltration also may occur, especially with sarcomas or hematologic neoplasms. The pericardium is most often involved, followed by myocardial involvement of any chamber and, rarely, by involvement of the endocardium or cardiac valves.
Cardiac metastases are clinically apparent only ~10% of the time, are usually not the cause of the patient’s presentation, and rarely are the cause of death. The vast majority occur in the setting of a previously recognized malignant neoplasm. As with primary cardiac tumors, the clinical presentation reflects more the location and size of the tumor than its histologic type. When symptomatic, cardiac metastases may result in a variety of clinical features, including dyspnea, acute pericarditis, cardiac tamponade, ectopic tachyarrhythmias, heart block, and CHF. Importantly, many of these signs and symptoms may also result from myocarditis, pericarditis, or cardiomyopathy induced by radiotherapy or chemotherapy.
Electrocardiographic (ECG) findings are nonspecific. On chest x-ray, the cardiac silhouette is most often normal but may be enlarged or exhibit a bizarre contour. Echocardiography is useful for identifying pericardial effusions and visualizing larger metastases, although CT and radionuclide imaging with gallium or thallium may define the tumor burden more clearly. Cardiac MRI offers superb image quality and plays a central role in the diagnostic evaluation of cardiac metastases and cardiac tumors in general. Pericardiocentesis may allow for a specific cytologic diagnosis in patients with malignant pericardial effusions. Angiography is rarely necessary but may delineate discrete lesions.
TRAUMATIC CARDIAC INJURY
Traumatic cardiac injury may be caused by either penetrating or nonpenetrating trauma. Penetrating injuries most often result from gunshot or knife wounds, and the site of entry is usually obvious. Nonpenetrating injuries most often occur during motor vehicle accidents, either from rapid deceleration or from impact of the chest against the steering wheel, and may be associated with significant cardiac injury even in the absence of external signs of thoracic trauma.
NONPENETRATING CARDIAC INJURY
Myocardial contusions are the most common form of nonpenetrating cardiac injury and may initially be overlooked in trauma patients as the clinical focus is directed toward other, more obvious injuries. Myocardial necrosis may occur as a direct result of the blunt injury or as a result of traumatic coronary laceration or thrombosis. The contused myocardium is pathologically similar to infarcted myocardium and may be associated with atrial or ventricular arrhythmias; conduction disturbances, including bundle branch block; or ECG abnormalities resembling those of infarction or pericarditis. Thus, it is important to consider contusion as a cause of otherwise unexplained ECG changes in a trauma patient. Serum creatine kinase, myocardial band (CK-MB) isoenzyme levels are increased in ~20% of patients who experience blunt chest trauma but may be falsely elevated in the presence of massive skeletal muscle injury. Cardiac troponin levels are more specific for identifying cardiac injury in this setting. Echocardiography is useful in detecting structural and functional sequelae of contusion, including wall motion abnormalities (most commonly involving the right ventricle, interventricular septum, or left ventricular apex), pericardial effusion, valvular dysfunction, and ventricular rupture.
Rupture of the cardiac valves or their supporting structures, most commonly of the tricuspid or mitral valve, leads to acute valvular incompetence. This complication is usually heralded by the development of a loud murmur, may be associated with rapidly progressive heart failure, and can be diagnosed by either transthoracic or transesophageal echocardiography.
The most serious consequence of nonpenetrating cardiac injury is myocardial rupture, which may result in hemopericardium and tamponade (free wall rupture) or intracardiac shunting (ventricular septal rupture). Although it generally is fatal, up to 40% of patients with cardiac rupture have been reported to survive long enough to reach a specialized trauma center. Hemopericardium also may result from traumatic rupture of a pericardial vessel or a coronary artery. Additionally, a pericardial effusion may develop weeks or even months after blunt chest trauma as a manifestation of the post–cardiac injury syndrome, which resembles the postpericardiotomy syndrome (Chap. 288).
Blunt, nonpenetrating, often innocent-appearing injuries to the chest may trigger ventricular fibrillation even in absence of overt signs of injury. This syndrome, referred to as commotio cordis, occurs most often in adolescents during sporting events (e.g., baseball, hockey, football, and lacrosse) and probably results from an impact to the chest wall overlying the heart during the susceptible phase of repolarization just before the peak of the T wave. Survival depends on prompt defibrillation. Sudden emotional or physical trauma, even in the absence of direct cardiac trauma, may precipitate a transient catecholamine-mediated cardiomyopathy referred to as tako-tsubo syndrome or the apical ballooning syndrome (Chap. 287).
Rupture or transection of the aorta, usually just above the aortic valve or at the site of the ligamentum arteriosum, is a common consequence of nonpenetrating chest trauma and is the most common vascular deceleration injury. The clinical presentation is similar to that of aortic dissection (Chap. 301); the arterial pressure and pulse amplitude may be increased in the upper extremities and decreased in the lower extremities, and chest x-ray may reveal mediastinal widening. Occasionally, aortic rupture is contained by the aortic adventitia, resulting in a false, or pseudo-, aneurysm that may be discovered months or years after the initial injury.
PENETRATING CARDIAC INJURY
Penetrating injuries of the heart produced by knife or bullet wounds usually result in rapid clinical deterioration and frequently in death as a result of hemopericardium/pericardial tamponade or massive hemorrhage. Nonetheless, up to half of such patients may survive long enough to reach a specialized trauma center if immediate resuscitation is performed. Prognosis in these patients relates to the mechanism of injury, their clinical condition at presentation, and the specific cardiac chamber(s) involved. Iatrogenic cardiac or coronary arterial perforation may complicate placement of central venous or intracardiac catheters, pacemaker leads, or intracoronary stents and is associated with a better prognosis than are other forms of penetrating cardiac trauma.
Traumatic rupture of a great vessel from penetrating injury is usually associated with hemothorax and, less often, hemopericardium. Local hematoma formation may compress major vessels and produce ischemic symptoms, and AV fistulas may develop, occasionally resulting in high-output CHF.
Occasionally, patients who survive penetrating cardiac injuries may subsequently present with a new cardiac murmur or CHF as a result of mitral regurgitation or an intracardiac shunt (i.e., ventricular or atrial septal defect, aortopulmonary fistula, or coronary AV fistula) that was undetected at the time of the initial injury or developed subsequently. Therefore, trauma patients should be examined carefully several weeks after the injury. If a mechanical complication is suspected, it can be confirmed by echocardiography or cardiac catheterization.
290e |
Cardiac Manifestations of Systemic Disease |
The common systemic disorders that have associated cardiac manifestations are summarized in Table 290e-1.
|
COMMON SYSTEMIC DISORDERS AND THEIR ASSOCIATED CARDIAC MANIFESTATIONS |
DIABETES MELLITUS
(See also Chap. 417) Diabetes mellitus, both insulin- and non-insulin-dependent, is an independent risk factor for coronary artery disease (CAD; Chap. 291e) and accounts for 14–50% of new cases of cardiovascular disease. Furthermore, CAD is the most common cause of death in adults with diabetes mellitus. In the diabetic population, the incidence of CAD relates to the duration of diabetes and the level of glycemic control, and its pathogenesis involves endothelial dysfunction, increased lipoprotein peroxidation, increased inflammation, a prothrombotic state, and associated metabolic abnormalities.
Compared to their nondiabetic counterparts, diabetic patients are more likely to have a myocardial infarction, have a greater burden of CAD, have larger infarct size, and have more postinfarct complications, including heart failure, shock, and death. Importantly, diabetic patients are more likely to have atypical ischemic symptoms; nausea, dyspnea, pulmonary edema, arrhythmias, heart block, or syncope may be their anginal equivalent. Additionally, “silent ischemia,” resulting from autonomic nervous system dysfunction, is more common in diabetic patients, accounting for up to 90% of their ischemic episodes. Thus, one must have a low threshold for suspecting CAD in diabetic patients. The treatment of diabetic patients with CAD must include aggressive risk factor management (Chap. 418). Considerations regarding pharmacologic therapy and revascularization strategies are similar in diabetic and nondiabetic patients except that diabetic patients have higher morbidity and mortality rates associated with revascularization, have an increased risk of restenosis after percutaneous coronary intervention (PCI), and have improved survival when treated with surgical bypass compared with PCI for multivessel CAD.
Patients with diabetes mellitus also may have abnormal left ventricular systolic and diastolic function, reflecting concomitant epicardial CAD and/or hypertension, coronary microvascular disease, endothelial dysfunction, ventricular hypertrophy, and autonomic dysfunction. Furthermore, the increase in intramyocardial lipid deposition (predominantly nonesterified fatty acids) that is characteristic of diabetic states may contribute to both systolic and diastolic dysfunction by impairing insulin signaling, reducing trans-sarcolemma calcium flux, and inducing myocyte apoptosis. A restrictive cardiomyopathy may be present with abnormal myocardial relaxation and elevated ventricular filling pressures. Histologically, interstitial fibrosis is seen, and intramural arteries may demonstrate intimal thickening, hyaline deposition, and inflammatory changes. Diabetic patients have an increased risk of developing clinical heart failure, which probably contributes to their excessive cardiovascular morbidity and mortality rates. There is some evidence that insulin therapy may ameliorate diabetes-related myocardial dysfunction.
MALNUTRITION AND VITAMIN DEFICIENCY
Malnutrition (See also Chap. 97) In patients whose intake of protein, calories, or both is severely deficient, the heart may become thin, pale, and hypokinetic with myofibrillar atrophy and interstitial edema. The systolic pressure and cardiac output fall, and the pulse pressure narrows. Generalized edema is common and relates to a variety of factors, including reduced serum oncotic pressure and myocardial dysfunction. Such profound states of protein and calorie malnutrition, termed kwashiorkor and marasmus, respectively, are most common in underdeveloped countries. However, significant nutritional heart disease also may occur in developed nations, particularly in patients with chronic diseases such as AIDS, patients with anorexia nervosa, and patients with severe cardiac failure in whom gastrointestinal hypoperfusion and venous congestion may lead to anorexia and malabsorption. Open-heart surgery poses increased risk in malnourished patients; such patients may benefit from preoperative hyperalimentation.
Thiamine Deficiency (Beriberi) (See also Chap. 96e) Generalized malnutrition often is accompanied by thiamine deficiency; however, this hypovitaminosis also may occur in the presence of an adequate protein and caloric intake, particularly in East Asia, where polished rice deficient in thiamine may be a major dietary component. In Western nations where the use of thiamine-enriched flour is widespread, clinical thiamine deficiency is limited primarily to alcoholics, food faddists, and patients receiving chemotherapy. Nonetheless, when thiamine stores are measured using the thiamine-pyrophosphate effect (TPPE), thiamine deficiency has been found in 20–90% of patients with chronic heart failure. This deficiency appears to result from both reduced dietary intake and a diuretic-induced increase in the urinary excretion of thiamine. The acute administration of thiamine to these patients increases the left ventricular ejection fraction and the excretion of salt and water.
Clinically, patients with thiamine deficiency usually have evidence of generalized malnutrition, peripheral neuropathy, glossitis, and anemia. The classic associated cardiovascular syndrome is characterized by high-output heart failure, tachycardia, and often elevated biventricular filling pressures. The major cause of the high-output state is vasomotor depression leading to reduced systemic vascular resistance, the precise mechanism of which is not understood. The cardiac examination may reveal a wide pulse pressure, tachycardia, a third heart sound, and an apical systolic murmur. The electrocardiogram (ECG) may reveal decreased voltage, a prolonged QT interval, and T-wave abnormalities. The chest x-ray generally reveals cardiomegaly and signs of congestive heart failure (CHF). The response to thiamine is often dramatic, with an increase in systemic vascular resistance, a decrease in cardiac output, clearing of pulmonary congestion, and a reduction in heart size often occurring in 12–48 h. Although the response to inotropes and diuretics may be poor before thiamine therapy, these agents may be important after thiamine repletion, since the left ventricle may not be able to handle the increased work load presented by the return of vascular tone.
Vitamin B6, B12, and Folate Deficiency (See also Chap. 96e) Vitamin B6, vitamin B12, and folate are cofactors in the metabolism of homocysteine. Their deficiency probably contributes to the majority of cases of hyperhomocysteinemia, a disorder associated with increased atherosclerotic risk. Supplementation of these vitamins has reduced the incidence of hyperhomocysteinemia in the United States; however, the clinical cardiovascular benefit of normalizing elevated homocysteine levels has not been proved.
OBESITY
(See also Chap. 415e) Obesity is associated with an increased prevalence of hypertension, glucose intolerance, atherosclerotic CAD, atrial fibrillation, obstructive sleep apnea, and pulmonary hypertension, and is associated with increased cardiovascular morbidity and mortality rates. In addition, obese patients have a distinct hemodynamic profile characterized by increased total and central blood volumes, increased cardiac output, and elevated left ventricular filling pressure. The elevated cardiac output appears to be required to support the metabolic demands of the excess adipose tissue. Left ventricular filling pressure is often at the upper limits of normal at rest and rises excessively with exercise, contributing to exertional dyspnea. In part as a result of chronic volume overload, eccentric cardiac hypertrophy with cardiac dilation and ventricular diastolic and/or systolic dysfunction may develop. In addition, altered levels of adipokines secreted by adipose tissue may contribute to adverse myocardial remodeling via direct effects on cardiac myocytes and other cells. Pathologically, there is left and, in some cases, right ventricular hypertrophy and generalized cardiac dilation. Pulmonary congestion, peripheral edema, and exercise intolerance may all ensue; however, the recognition of these findings may be difficult in massively obese patients.
Treatment with angiotensin-converting enzyme inhibitors, sodium restriction, and diuretics may be useful to control heart failure symptoms. Weight reduction, however, is the most effective therapy and results in reduction in blood volume and the return of cardiac output toward normal. However, rapid weight reduction may be dangerous, as cardiac arrhythmias and sudden death owing to electrolyte imbalance have been described.
THYROID DISEASE
(See also Chap. 405) Thyroid hormone exerts a major influence on the cardiovascular system by a number of direct and indirect mechanisms, and not surprisingly, cardiovascular effects are prominent in both hypo- and hyperthyroidism. Thyroid hormone causes increases in total-body metabolism and oxygen consumption that indirectly increase the cardiac workload. In addition, thyroid hormone exerts direct inotropic, chronotropic, and dromotropic effects that are similar to those seen with adrenergic stimulation (e.g., tachycardia, increased cardiac output); they are mediated at least partly by both transcriptional and nontranscriptional effects of thyroid hormone on myosin, calcium-activated ATPase, Na+-K+-ATPase, and myocardial β-adrenergic receptors.
Hyperthyroidism Common cardiovascular manifestations of hyperthyroidism include palpitations, systolic hypertension, and fatigue. Sinus tachycardia is present in ~40% of hyperthyroid patients, and atrial fibrillation is present in ~15%. Physical examination may reveal a hyperdynamic precordium, a widened pulse pressure, increases in the intensity of the first heart sound and the pulmonic component of the second heart sound, and a third heart sound. An increased incidence of mitral valve prolapse has been described in hyperthyroid patients, in which case a midsystolic murmur may be heard at the left sternal border with or without a midsystolic click. A systolic pleuropericardial friction rub (Means-Lerman scratch) may be heard at the left second intercostal space during expiration and is thought to result from the hyperdynamic cardiac motion.
Elderly patients with hyperthyroidism may present with only cardiovascular manifestations of thyrotoxicosis such as sinus tachycardia, atrial fibrillation, and hypertension, all of which may be resistant to therapy until the hyperthyroidism is controlled. Angina pectoris and CHF are unusual with hyperthyroidism unless there is coexistent heart disease; in such cases, symptoms often resolve with treatment of the hyperthyroidism.
Hypothyroidism Cardiac manifestations of hypothyroidism include a reduction in cardiac output, stroke volume, heart rate, systolic blood pressure, and pulse pressure. Pericardial effusions are present in about one-third of patients, rarely progress to tamponade, and probably result from increased capillary permeability. Other clinical signs include cardiomegaly, bradycardia, weak arterial pulses, distant heart sounds, and pleural effusions. Although the signs and symptoms of myxedema may mimic those of CHF, in the absence of other cardiac disease, myocardial failure is uncommon. The ECG generally reveals sinus bradycardia and low voltage and may show prolongation of the QT interval, decreased P-wave voltage, prolonged AV conduction time, intraventricular conduction disturbances, and nonspecific ST-T-wave abnormalities. Chest x-ray may show cardiomegaly, often with a “water bottle” configuration; pleural effusions; and, in some cases, evidence of CHF. Pathologically, the heart is pale and dilated and often demonstrates myofibrillar swelling, loss of striations, and interstitial fibrosis.
Patients with hypothyroidism frequently have elevations of cholesterol and triglycerides, resulting in premature atherosclerotic CAD. Before treatment with thyroid hormone, patients with hypothyroidism frequently do not have angina pectoris, presumably because of the low metabolic demands caused by their condition. However, angina and myocardial infarction may be precipitated during initiation of thyroid hormone replacement, especially in elderly patients with underlying heart disease. Therefore, replacement should be done with care, starting with low doses that are increased gradually.
MALIGNANT CARCINOID
(See also Chap. 113) Carcinoid tumors most often originate in the small bowel and elaborate a variety of vasoactive amines (e.g., serotonin), kinins, indoles, and prostaglandins that are believed to be responsible for the diarrhea, flushing, and labile blood pressure that characterize the carcinoid syndrome. Some 50% of patients with carcinoid syndrome have cardiac involvement, usually manifesting as abnormalities of the tricuspid or pulmonic valves. These patients invariably have hepatic metastases that allow vasoactive substances to circumvent hepatic metabolism. Left-sided cardiac involvement is rare and indicates either pulmonary carcinoid or an intracardiac shunt. Pathologically, carcinoid lesions are fibrous plaques that consist of smooth-muscle cells embedded in a stroma of glycosaminoglycans and collagen. They occur on the cardiac valves, where they cause valvular dysfunction, as well as on the endothelium of the cardiac chambers and great vessels.
Carcinoid heart disease most often presents as tricuspid regurgitation, pulmonic stenosis, or both. In some cases, a high cardiac output state may occur, presumably as a result of a decrease in systemic vascular resistance resulting from vasoactive substances released by the tumor. Treatment with somatostatin analogues (e.g., octreotide) or interferon α improves symptoms and survival in patients with carcinoid heart disease but does not appear to improve valvular abnormalities. Treatment with diuretics usually mitigates the symptoms of right heart failure; in some severely symptomatic patients, valve replacement is indicated. Coronary artery spasm, presumably due to a circulating vasoactive substance, may occur in patients with carcinoid syndrome.
PHEOCHROMOCYTOMA
(See also Chap. 407) In addition to causing labile or sustained hypertension, the high circulating levels of catecholamines resulting from a pheochromocytoma may cause direct myocardial injury. Focal myocardial necrosis and inflammatory cell infiltration are present in ~50% of patients who die with pheochromocytoma and may contribute to clinically significant left ventricular failure and pulmonary edema. In addition, associated hypertension results in left ventricular hypertrophy. Left ventricular dysfunction and CHF may resolve after removal of the tumor.
ACROMEGALY
(See also Chap. 401e) Exposure of the heart to excessive growth hormone may cause CHF as a result of high cardiac output, diastolic dysfunction owing to ventricular hypertrophy (with increased left ventricular chamber size or wall thickness), or global systolic dysfunction. Hypertension occurs in up to one-third of patients with acromegaly and is characterized by suppression of the renin-angiotensin-aldosterone axis and increases in total-body sodium and plasma volume. Some form of cardiac disease occurs in about one-third of patients with acromegaly and is associated with a doubling of the risk of cardiac death.
RHEUMATOID ARTHRITIS AND THE COLLAGEN VASCULAR DISEASES
Rheumatoid Arthritis (See also Chap. 380) Rheumatoid arthritis may be associated with inflammatory changes in any or all cardiac structures, although pericarditis is the most common clinical entity. Pericardial effusions are found on echocardiography in 10–50% of patients with rheumatoid arthritis, particularly those with subcutaneous nodules. Nonetheless, only a small fraction of these patients have symptomatic pericarditis, and when present, it usually follows a benign course, only occasionally progressing to cardiac tamponade or constrictive pericarditis. The pericardial fluid is generally exudative, with decreased concentrations of complement and glucose and elevated cholesterol. Coronary arteritis with intimal inflammation and edema is present in ~20% of cases but only rarely results in angina pectoris or myocardial infarction. Inflammation and granuloma formation may affect the cardiac valves, most often the mitral and aortic valves, and may cause clinically significant regurgitation owing to valve deformity. Myocarditis is uncommon and rarely results in cardiac dysfunction.
Treatment is directed at the underlying rheumatoid arthritis and may include glucocorticoids. Urgent pericardiocentesis should be performed in patients with tamponade, but pericardiectomy usually is required in cases of pericardial constriction.
Seronegative Arthropathies (See also Chap. 384) The seronegative arthropathies, including ankylosing spondylitis, reactive arthritis, psoriatic arthritis, and the arthritides associated with ulcerative colitis and regional enteritis, are all strongly associated with the HLA-B27 histocompatibility antigen and may be accompanied by a pancarditis and proximal aortitis. The aortic inflammation usually is limited to the aortic root but may extend to involve the aortic valve, mitral valve, and ventricular myocardium, resulting in aortic and mitral regurgitation, conduction abnormalities, and ventricular dysfunction. One-tenth of these patients have significant aortic insufficiency, and one-third have conduction disturbances; both are more common in patients with peripheral joint involvement and long-standing disease. Treatment with aortic valve replacement and permanent pacemaker implantation may be required. Occasionally, aortic regurgitation precedes the onset of arthritis, and therefore, the diagnosis of a seronegative arthritis should be considered in young males with isolated aortic regurgitation.
Systemic Lupus Erythematosus (SLE) (See also Chap. 378) A significant percentage of patients with SLE have cardiac involvement. Pericarditis is common, occurring in about two-thirds of patients, and generally follows a benign course, although rarely tamponade or constriction may result. The characteristic endocardial lesions of SLE are verrucous valvular abnormalities known as Libman-Sacks endocarditis. They most often are located on the left-sided cardiac valves, particularly on the ventricular surface of the posterior mitral leaflet, and are made up almost entirely of fibrin. These lesions may embolize or become infected but rarely cause hemodynamically important valvular regurgitation. Myocarditis generally parallels the activity of the disease and, although common histologically, seldom results in clinical heart failure unless associated with hypertension. Although arteritis of epicardial coronary arteries may occur, it rarely results in myocardial ischemia. There is, however, an increased incidence of coronary atherosclerosis that probably is related more to associated risk factors and glucocorticoid use than to SLE itself. Patients with the antiphospholipid antibody syndrome may have a higher incidence of cardiovascular abnormalities, including valvular regurgitation, venous and arterial thrombosis, premature stroke, myocardial infarction, pulmonary hypertension, and cardiomyopathy.
SECTION 5 |
CORONARY AND PERIPHERAL VASCULAR DISEASE |
291e |
The Pathogenesis, Prevention, and Treatment of Atherosclerosis |
PATHOGENESIS
Atherosclerosis remains the major cause of death and premature disability in developed societies. Moreover, current predictions estimate that by the year 2020 cardiovascular diseases, notably atherosclerosis, will become the leading global cause of total disease burden. Although many generalized or systemic risk factors predispose to its development, atherosclerosis affects various regions of the circulation preferentially and has distinct clinical manifestations that depend on the particular circulatory bed affected. Atherosclerosis of the coronary arteries commonly causes myocardial infarction (MI) (Chap. 295) and angina pectoris (Chap. 293). Atherosclerosis of the arteries supplying the central nervous system frequently provokes strokes and transient cerebral ischemia (Chap. 446). In the peripheral circulation, atherosclerosis causes intermittent claudication and gangrene and can jeopardize limb viability. Involvement of the splanchnic circulation can cause mesenteric ischemia. Atherosclerosis can affect the kidneys either directly (e.g., renal artery stenosis) or as a common site of atheroembolic disease (Chap. 301).
Even within a particular arterial bed, stenoses due to atherosclerosis tend to occur focally, typically in certain predisposed regions. In the coronary circulation, for example, the proximal left anterior descending coronary artery exhibits a particular predilection for developing atherosclerotic disease. Similarly, atherosclerosis preferentially affects the proximal portions of the renal arteries and, in the extracranial circulation to the brain, the carotid bifurcation. Indeed, atherosclerotic lesions often form at branch points of arteries, regions characterized disturbed hydrodynamics. Not all manifestations of atherosclerosis result from stenotic, occlusive disease. Ectasia and the development of aneurysmal disease, for example, frequently occur in the aorta (Chap. 301). In addition to focal, flow-limiting stenoses, nonocclusive intimal atherosclerosis also occurs diffusely in affected arteries, as shown by intravascular imaging and postmortem studies.
Atherogenesis in humans typically occurs over a period of many years, usually many decades. Growth of atherosclerotic plaques probably does not occur in a smooth, linear fashion but discontinuously, with periods of relative quiescence punctuated by periods of rapid evolution. After a generally prolonged “silent” period, atherosclerosis may become clinically manifest. The clinical expressions of atherosclerosis may be chronic, as in the development of stable, effort-induced angina pectoris or predictable and reproducible intermittent claudication. Alternatively, a dramatic acute clinical event such as MI, stroke, or sudden cardiac death may first herald the presence of atherosclerosis. Other individuals may never experience clinical manifestations of arterial disease despite the presence of widespread atherosclerosis demonstrated postmortem.
INITIATION OF ATHEROSCLEROSIS
An integrated view of experimental results in animals and studies of human atherosclerosis suggests that the “fatty streak” represents the initial lesion of atherosclerosis. These early lesions most often seem to arise from focal increases in the content of lipoproteins within regions of the intima. In particular, the fraction of lipoproteins related to low-density lipoprotein (LDL) that bear apolipoprotein B appear causally related to atherosclerosis. This accumulation of lipoprotein particles may not result simply from increased permeability, or “leakiness,” of the overlying endothelium (Fig. 291e-1). Rather, the lipoproteins may collect in the intima of arteries because they bind to constituents of the extracellular matrix, increasing the residence time of the lipid-rich particles within the arterial wall. Lipoproteins that accumulate in the extracellular space of the intima of arteries often associate with proteoglycans of the arterial extracellular matrix, an interaction that may slow the egress of these lipid-rich particles from the intima. Lipoprotein particles in the extracellular space of the intima, particularly those retained by binding to matrix macromolecules, may undergo oxidative modifications. Considerable evidence supports a pathogenic role for products of oxidized lipoproteins in atherogenesis. Lipoproteins sequestered from (plasma) antioxidants in the extracellular space of the intima become particularly susceptible to oxidative modification, giving rise to hydroperoxides, lysophospholipids, oxysterols, and aldehydic breakdown products of fatty acids and phospholipids. Modifications of the apoprotein moieties may include breaks in the peptide backbone as well as derivatization of certain amino acid residues. Local production of hypochlorous acid by myeloperoxidase associated with inflammatory cells within the plaque yields chlorinated species such as chlorotyrosyl moieties. Considerable evidence supports the presence of such oxidation products in atherosclerotic lesions.
FIGURE 291e-1 Cross-sectional view of an artery depicting steps in development of an atheroma, from left to right. The upper panel shows a detail of the boxed area below. The endothelial monolayer overlying the intima contacts blood. Hypercholesterolemia promotes accumulation of low-density lipoprotein (LDL) particles (yellow spheres) in the intima. The lipoprotein particles often associate with constituents of the extracellular matrix, notably proteoglycans. Sequestration within the intima separates lipoproteins from some plasma antioxidants and favors oxidative modification. Such modified lipoprotein particles (darker spheres) may trigger a local inflammatory response that signals subsequent steps in lesion formation. The augmented expression of various adhesion molecules for leukocytes recruits monocytes to the site of a nascent arterial lesion.
Once adherent, some white blood cells migrate into the intima. The directed migration of leukocytes probably depends on chemoattractant factors, including modified lipoprotein particles themselves and chemoattractant cytokines (depicted by the smaller green spheres), such as the chemokine macrophage chemoattractant protein-1 produced by vascular wall cells in response to modified lipoproteins. Leukocytes in the evolving fatty streak can divide and exhibit augmented expression of receptors for modified lipoproteins (scavenger receptors). These mononuclear phagocytes ingest lipids and become foam cells, represented by a cytoplasm filled with lipid droplets. As the fatty streak evolves into a more complicated atherosclerotic lesion, smooth-muscle cells migrate from the media (bottom of lower panel hairline) through the internal elastic membrane (solid wavy line) and accumulate within the expanding intima, where they lay down extracellular matrix that forms the bulk of the advanced lesion (bottom panel, right side).
Leukocyte Recruitment Accumulation of leukocytes characterizes the formation of early atherosclerotic lesions (Fig. 291e-1). Thus, from its very inception, atherogenesis involves elements of inflammation, a process that now provides a unifying theme in the pathogenesis of this disease. The inflammatory cell types typically found in the evolving atheroma include monocyte-derived macrophages and dendritic cells, T and B lymphocytes, and mast cells. Hypercholesterolemia augments the portion of particularly proinflammatory monocytes in blood that preferentially enter the nascent atheroma in mice. A number of adhesion molecules or receptors for leukocytes expressed on the surface of the arterial endothelial cell probably participate in the recruitment of leukocytes to the nascent atheroma. Proinflammatory cytokines can augment the expression of leukocyte adhesion molecules.
Laminar shear forces such as those encountered in most regions of normal arteries also can suppress the expression of leukocyte adhesion molecules. Sites of predilection for atherosclerotic lesions (e.g., distal to flow dividers) often have low shear stress and/or disturbed flow. Ordered, pulsatile laminar shear of normal blood flow augments the production of nitric oxide by endothelial cells. This molecule, in addition to its vasodilator properties, can act at the low levels constitutively produced by arterial endothelium as a local anti-inflammatory autacoid, e.g., limiting local adhesion molecule expression. Exposure of endothelial cells to laminar shear stress increases the transcription of Krüppel-like factor 2 (KLF2), which augments the activity of numerous salutary endothelial functions including nitric oxide synthase. Laminar shear stress also stimulates endothelial cells to produce superoxide dismutase, an antioxidant enzyme. These examples indicate how hemodynamic forces may influence the cellular events that underlie atherosclerotic lesion initiation and potentially explain the favored localization of atherosclerotic lesions at sites that experience disturbed flow or low shear stress.
Once captured on the surface of the arterial endothelial cell by adhesion receptors, the leukocytes penetrate the endothelial layer and take up residence in the intima. In addition to products of modified lipoproteins, cytokines (protein mediators of inflammation) can regulate the expression of adhesion molecules involved in leukocyte recruitment. For example, interleukin 1 (IL-1) and tumor necrosis factor (TNF) induce or augment the expression of leukocyte adhesion molecules on endothelial cells. Because products of lipoprotein oxidation can induce cytokine release from vascular wall cells, this pathway may provide an additional link between arterial accumulation of lipoproteins and leukocyte recruitment. Chemoattractant cytokines appear to direct the migration of leukocytes into the arterial wall.
Foam-Cell Formation Once resident within the intima, the mononuclear phagocytes mature into macrophages and become lipid-laden foam cells, a conversion that requires the uptake of lipoprotein particles by receptor-mediated endocytosis. One might suppose that the “classic” LDL receptor mediates this lipid uptake; however, humans or animals lacking effective LDL receptors due to genetic alterations (e.g., familial hypercholesterolemia) have abundant arterial lesions and extraarterial xanthomata rich in macrophage-derived foam cells. In addition, the exogenous cholesterol suppresses expression of the LDL receptor; thus, the level of this cell-surface receptor for LDL decreases under conditions of cholesterol excess. Candidates for alternative receptors that can mediate lipid loading of foam cells include a number of macrophage “scavenger” receptors, which preferentially endocytose modified lipoproteins, and other receptors for oxidized LDL or very low-density lipoprotein (VLDL). Monocyte attachment to the endothelium, migration into the intima, and maturation to form lipid-laden macrophages thus represent key steps in the formation of the fatty streak, the precursor of fully formed atherosclerotic plaques.
ATHEROMA EVOLUTION AND COMPLICATIONS
Although the fatty streak commonly precedes the development of a more advanced atherosclerotic plaque, not all fatty streaks progress to form complex atheromata. By ingesting lipids from the extracellular space, the mononuclear phagocytes bearing such scavenger receptors may remove lipoproteins from the developing lesion. Some lipid-laden macrophages may leave the artery wall, exporting lipid in the process. Lipid accumulation, and hence the propensity to form an atheroma, ensues if the amount of lipid entering the artery wall exceeds that removed by mononuclear phagocytes or other pathways. Macrophages also proliferate in plaques in response to hematopoietic growth factors overexpressed in lesions, another aspect of the dynamic regulation and flux of cells during atherogenesis.
Export by phagocytes may constitute one response to local lipid overload in the evolving lesion. Another mechanism, reverse cholesterol transport mediated by high-density lipoproteins (HDLs), probably provides an independent pathway for lipid removal from atheroma. This transfer of cholesterol from the cell to the HDL particle involves specialized cell-surface molecules such as the ATP binding cassette (ABC) transporters. ABCA1, the gene mutated in Tangier disease, a condition characterized by very low HDL levels, transfers cholesterol from cells to nascent HDL particles and ABCG1 to mature HDL particles. “Reverse cholesterol transport” mediated by these ABC transporters allows HDL loaded with cholesterol to deliver it to hepatocytes by binding to scavenger receptor B1 or other receptors. The liver cell can metabolize the sterol to bile acids that can be excreted. Thus, macrophages may play a vital role in the dynamic economy of lipid accumulation in the arterial wall during atherogenesis.
Some lipid-laden foam cells within the expanding intimal lesion perish. Some foam cells may die as a result of programmed cell death, or apoptosis. This death of mononuclear phagocytes results in the formation of the lipid-rich center, often called the necrotic core, in established atherosclerotic plaques. Impaired clearance of dead foam cells (efferocytosis) in plaques may hasten lipid core formation. Macrophages loaded with modified lipoproteins may elaborate microparticles or exosomes (which may contain regulatory microRNAs), cytokines, and growth factors that can further signal some of the cellular events in lesion complication. Whereas accumulation of lipid-laden macrophages characterizes the fatty streak, buildup of fibrous tissue formed by extracellular matrix typifies the more advanced atherosclerotic lesion. The smooth-muscle cell synthesizes the bulk of the extracellular matrix of the complex atherosclerotic lesion. A number of growth factors or cytokines elaborated by mononuclear phagocytes can stimulate smooth-muscle cell proliferation and production of extracellular matrix. Cytokines found in the plaque, including IL-1 and TNF, can induce local production of growth factors, including forms of platelet-derived growth factor (PDGF), fibroblast growth factors, and others, which may contribute to plaque evolution and complication. Other cytokines, notably interferon γ (IFN-γ) derived from activated T cells within lesions, can limit the synthesis of interstitial forms of collagen by smooth-muscle cells. These examples illustrate how atherogenesis involves a complex mix of mediators that in the balance determines the characteristics of particular lesions.
The accumulation of smooth-muscle cells and their elaboration of extracellular matrix probably provide a critical transition, yielding a fibrofatty lesion in place of a simple accumulation of macrophage-derived foam cells. For example, PDGF elaborated by activated platelets, macrophages, and endothelial cells can stimulate the migration of smooth-muscle cells normally resident in the tunica media into the intima. Such growth factors and cytokines produced locally can stimulate the proliferation of resident smooth-muscle cells or resident stem cells in the intima as well as those that may migrate in from the media. Transforming growth factor β (TGF-β), among other mediators, potently stimulates interstitial collagen production by smooth-muscle cells. These mediators may arise not only from neighboring vascular cells or leukocytes (a “paracrine” pathway), but also, in some instances, from the same cell that responds to the factor (an “autocrine” pathway). Together, these alterations in smooth-muscle cells, signaled by these mediators acting at short distances, can hasten transformation of the fatty streak into a more fibrous smooth-muscle cell and extracellular matrix—rich lesion.
In addition to locally produced mediators, products of blood coagulation and thrombosis likely contribute to atheroma evolution and complication. This involvement justifies the use of the term atherothrombosis to convey the inextricable links between atherosclerosis and thrombosis. Fatty streak formation begins beneath a morphologically intact endothelium. In advanced fatty streaks, however, microscopic breaches in endothelial integrity may occur. Microthrombi rich in platelets can form at such sites of limited endothelial denudation, owing to exposure of the thrombogenic extracellular matrix of the underlying basement membrane. Activated platelets release numerous factors that can promote the fibrotic response, including PDGF and TGF-β. Thrombin not only generates fibrin during coagulation, but also stimulates protease-activated receptors that can signal smooth-muscle migration, proliferation, and extracellular matrix production. Many arterial mural microthrombi resolve without clinical manifestation by a process of local fibrinolysis, resorption, and endothelial repair, yet can lead to lesion progression by stimulating these profibrotic functions of smooth-muscle cells (Fig. 291e-2D).
FIGURE 291e-2 Plaque rupture, thrombosis, and healing. A. Arterial remodeling during atherogenesis. During the initial part of the life history of an atheroma, growth is often outward, preserving the caliber of the lumen. This phenomenon of “compensatory enlargement” accounts in part for the tendency of coronary arteriography to underestimate the degree of atherosclerosis. B. Rupture of the plaque’s fibrous cap causes thrombosis. Physical disruption of the atherosclerotic plaque commonly causes arterial thrombosis by allowing blood coagulant factors to contact thrombogenic collagen found in the arterial extracellular matrix and tissue factor produced by macrophage-derived foam cells in the lipid core of lesions. In this manner, sites of plaque rupture form the nidus for thrombi. The normal artery wall has several fibrinolytic or antithrombotic mechanisms that tend to resist thrombosis and lyse clots that begin to form in situ. Such antithrombotic or thrombolytic molecules include thrombomodulin, tissue- and urokinase-type plasminogen activators, heparan sulfate proteoglycans, prostacyclin, and nitric oxide. C. When the clot overwhelms the endogenous fibrinolytic mechanisms, it may propagate and lead to arterial occlusion. The consequences of this occlusion depend on the degree of existing collateral vessels. In a patient with chronic multivessel occlusive coronary artery disease (CAD), collateral channels have often formed. In such circumstances, even a total arterial occlusion may not lead to myocardial infarction (MI), or it may produce an unexpectedly modest or a non-ST-segment elevation infarct because of collateral flow. In a patient with less advanced disease and without substantial stenotic lesions to provide a stimulus for collateral vessel formation, sudden plaque rupture and arterial occlusion commonly produces an ST-segment elevation infarction. These are the types of patients who may present with MI or sudden death as a first manifestation of coronary atherosclerosis. In some cases, the thrombus may lyse or organize into a mural thrombus without occluding the vessel. Such instances may be clinically silent. D. The subsequent thrombin-induced fibrosis and healing causes a fibroproliferative response that can lead to a more fibrous lesion that can produce an eccentric plaque that causes a hemodynamically significant stenosis. In this way, a nonocclusive mural thrombus, even if clinically silent or causing unstable angina rather than infarction, can provoke a healing response that can promote lesion fibrosis and luminal encroachment. Such a sequence of events may convert a “vulnerable” atheroma with a thin fibrous cap that is prone to rupture into a more “stable” fibrous plaque with a reinforced cap. Angioplasty of unstable coronary lesions may “stabilize” the lesions by a similar mechanism, producing a wound followed by healing.
Microvessels As atherosclerotic lesions advance, abundant plexi of microvessels develop in connection with the artery’s vasa vasorum. Newly developing microvascular networks may contribute to lesion complications in several ways. These blood vessels provide an abundant surface area for leukocyte trafficking and may serve as the portal for entry and exit of white blood cells from the established atheroma. Microvessels in the plaques may also furnish foci for intraplaque hemorrhage. Like the neovessels in the diabetic retina, microvessels in the atheroma may be friable and prone to rupture and can produce focal hemorrhage. Such a vascular leak can provoke thrombosis in situ, yielding local thrombin generation, which in turn can activate smooth-muscle and endothelial cells through ligation of protease-activated receptors. Atherosclerotic plaques often contain fibrin and hemosiderin, an indication that episodes of intraplaque hemorrhage contribute to plaque complications.
CALCIFICATION As they advance, atherosclerotic plaques also accumulate calcium. Microvesicles derived from lesional cells can stimulate calcification, and this process co-localizes with regions of heightened inflammation. Mineralization of the atherosclerotic plaque recapitulates many aspects of bone formation, including the regulatory participation of transcription factors such as Runx2.
Plaque Evolution Smooth-muscle cells and macrophages die in the atherosclerotic plaque. Indeed, complex atheromata often have a mostly fibrous character and lack the cellularity of less advanced lesions. This relative paucity of smooth-muscle cells in advanced atheromata may result from the predominance of cytostatic mediators such as TGF-β and IFN-γ (which can inhibit smooth-muscle cell proliferation) and also from smooth-muscle cell apoptosis. Thus, during the evolution of the atherosclerotic plaque, a complex and highly regulated balance between entry and egress of lipoproteins and leukocytes, cell proliferation and cell death, extracellular matrix production, and remodeling, as well as calcification and neovascularization, contribute to lesion formation. Many mediators related to atherogenic risk factors, including those derived from lipoproteins, cigarette smoking, and angiotensin II, provoke the production of proinflammatory cytokines and alter the behavior of the intrinsic vascular wall cells and infiltrating leukocytes that underlie the complex pathogenesis of these lesions. Thus, advances in vascular biology have led to increased understanding of the mechanisms that link risk factors to the pathogenesis of atherosclerosis and its complications.
PATHOPHYSIOLOGIC CONSEQUENCES OF ATHEROSCLEROSIS
Atherosclerotic lesions occur ubiquitously in Western societies, and the prevalence of this disease is on the rise globally. Most atheromata produce no symptoms, and many never cause clinical manifestations. Numerous patients with diffuse atherosclerosis may succumb to unrelated illnesses without ever having experienced a clinically significant manifestation of atherosclerosis. Arterial remodeling during atheroma formation accounts for some of this variability in the clinical expression of atherosclerotic disease (Fig. 291e-2A). During the initial phases of atheroma development, the plaque usually grows outward, in an abluminal direction. Vessels affected by atherogenesis tend to increase in diameter, a phenomenon known as compensatory enlargement, a type of vascular remodeling. The growing atheroma does not encroach on the arterial lumen until the burden of atherosclerotic plaque exceeds ~40% of the area encompassed by the internal elastic lamina. Thus, during much of its life history, an atheroma will not cause stenosis that can limit tissue perfusion.
Flow-limiting stenoses commonly form later in the history of the plaque. Many such plaques cause stable syndromes such as demand-induced angina pectoris or intermittent claudication in the extremities. In the coronary circulation and other circulations, even total vascular occlusion by an atheroma does not invariably lead to infarction. The hypoxic stimulus of repeated bouts of ischemia characteristically induces formation of collateral vessels in the myocardium, mitigating the consequences of an acute occlusion of an epicardial coronary artery. By contrast, many lesions that cause acute or unstable atherosclerotic syndromes, particularly in the coronary circulation, may arise from atherosclerotic plaques that do not produce a flow-limiting stenosis. Such lesions may produce only minimal luminal irregularities on traditional angiograms and often do not meet the traditional criteria for “significance” by arteriography. Thrombi arising from such nonocclusive stenoses may explain the frequency of MI as an initial manifestation of coronary artery disease (CAD) (in at least one-third of cases) in patients who report no prior history of angina pectoris, a syndrome usually caused by flow-limiting stenoses.
Plaque Instability and Rupture Postmortem studies afford considerable insight into the microanatomic substrate underlying the “instability” of plaques that do not cause critical stenoses. A superficial erosion of the endothelium or a frank plaque rupture or fissure usually produces the thrombus that causes episodes of unstable angina pectoris or the occlusive and relatively persistent thrombus that causes acute MI (Fig. 291e-2B). Rupture of the plaque’s fibrous cap (Fig. 291e-2C) permits contact between coagulation factors in the blood and highly thrombogenic tissue factor expressed by macrophage foam cells in the plaque’s lipid-rich core. If the ensuing thrombus is nonocclusive or transient, the episode of plaque disruption may not cause symptoms or may result in episodic ischemic symptoms such as rest angina. Occlusive thrombi that endure often cause acute MI, particularly in the absence of a well-developed collateral circulation that supplies the affected territory. Repetitive episodes of plaque disruption and healing provide one likely mechanism of transition of the fatty streak to a more complex fibrous lesion (Fig. 291e-2D). The healing process in arteries, as in skin wounds, involves the laying down of new extracellular matrix and fibrosis.
Not all atheromata exhibit the same propensity to rupture. Pathologic studies of culprit lesions that have caused acute MI reveal several characteristic features. Plaques that have caused thromboses tend to have thin fibrous caps, relatively large lipid cores, a high content of macrophages, outward remodeling, and spotty (rather than dense) calcification. Morphometric studies of such culprit lesions show that at sites of plaque rupture, macrophages and T lymphocytes predominate and contain relatively few smooth-muscle cells. The cells that concentrate at sites of plaque rupture bear markers of inflammatory activation. In addition, patients with active atherosclerosis and acute coronary syndromes display signs of disseminated inflammation. Inflammatory mediators regulate processes that govern the integrity of the plaque’s fibrous cap and, hence, its propensity to rupture. For example, the T cell–derived cytokine IFN-γ, which is found in atherosclerotic plaques, can inhibit growth and collagen synthesis of smooth-muscle cells, as noted above. Cytokines derived from activated macrophages and lesional T cells can boost production of proteolytic enzymes that can degrade the extracellular matrix of the plaque’s fibrous cap. Thus, inflammatory mediators can impair the collagen synthesis required for maintenance and repair of the fibrous cap and trigger degradation of extracellular matrix macromolecules, processes that weaken the plaque’s fibrous cap and enhance its susceptibility to rupture (so-called vulnerable plaques, Fig. 291e-3). In contrast to plaques with these features of vulnerability, those with a dense extracellular matrix and relatively thick fibrous cap without substantial tissue factor–rich lipid cores seem generally resistant to rupture and unlikely to provoke thrombosis.
FIGURE 291e-3 Inflammatory pathways that predispose atherosclerotic plaques to rupture and provoke thrombosis. A cross-section of an atheromatous plaque at the bottom of the figure shows the central lipid core that contains macrophage foam cells (yellow) and T cells (blue). The intima and media also contain arterial smooth-muscle cells (red), which are the source of arterial collagen (depicted as triple helical coiled structures). Activated T cells (of the type 1 helper T cell subtype) secrete cytokine interferon γ, which inhibits the production of the new, interstitial collagen that is required to repair and maintain the plaque’s protective fibrous cap (upper left). The T cells can also activate the macrophages in the intimal lesion by expressing the inflammatory mediator CD40 ligand (CD154), which engages its cognate receptor (CD40) on the phagocyte. This inflammatory signalling causes overproduction of interstitial collagenases (matrix metalloproteinases [MMPs] 1, 8, and 13) that catalyze the initial rate-limiting step in collagen breakdown (top right). CD40 ligation also causes macrophages to overproduce tissue-factor procoagulant. Thus, inflammatory signalling puts the collagen in the plaque’s fibrous cap in double jeopardy—decreasing synthesis and increasing breakdown—rendering the cap susceptible to rupture. Inflammatory activation also boosts tissue-factor production, which triggers thrombus formation in the disrupted plaque. These mechanisms link inflammation in the plaque to the thrombotic complications of atherosclerosis, including the acute coronary syndromes. (Adapted from P Libby: N Engl J Med 368:2004, 2013.)
Functional features of the atheromatous plaque, in addition to its degree of luminal encroachment, influence the clinical manifestations of this disease. This enhanced understanding of plaque biology provides insight into the diverse ways in which atherosclerosis can present clinically and the reasons why the disease may remain silent or stable for prolonged periods, punctuated by acute complications at certain times. Increased understanding of atherogenesis provides new insight into the mechanisms linking it to the risk factors discussed below, indicates the ways in which current therapies may improve outcomes, and suggests new targets for future intervention.
PREVENTION AND TREATMENT
THE CONCEPT OF ATHEROSCLEROTIC RISK FACTORS
The systematic study of risk factors for atherosclerosis emerged from a coalescence of experimental results, as well as from cross-sectional and ultimately longitudinal studies in humans. The prospective, community-based Framingham Heart Study provided rigorous support for the concept that hypercholesterolemia, hypertension, and other factors correlate with cardiovascular risk. Similar observational studies performed worldwide bolstered the concept of “risk factors” for cardiovascular disease.
From a practical viewpoint, the cardiovascular risk factors that have emerged from such studies fall into two categories: those modifiable by lifestyle and/or pharmacotherapy, and those that are immutable, such as age and sex. The weight of evidence supporting various risk factors differs. For example, hypercholesterolemia and hypertension certainly predict coronary risk, but the magnitude of the contributions of other so-called nontraditional risk factors, such as levels of homocysteine, levels of lipoprotein (a) [Lp(a)], and infection, remains controversial. Moreover, some biomarkers that predict cardiovascular risk may not participate in the causal pathway for the disease or its complications. Genetic studies using genome-wide association (GWAS) approaches and Mendelian randomization approaches have helped to distinguish between risk markers and factors that contribute causally to the disease. For example, recent genetic studies suggest that C-reactive protein (CRP) does not itself mediate atherogenesis, despite its ability to predict risk, whereas Lp(a) and apolipoprotein C3 have emerged as a causal risk factor. Table 291e-1 lists a number of risk factors implicated in atherosclerosis. The sections below will consider some of these factors and approaches to their modification.
|
MAJOR RISK FACTORS FOR ATHEROSCLEROSIS |
aHDL cholesterol ≥1.6 mmol/L (≥60 mg/dL) has been viewed as a “negative” risk factor.
Abbreviations: BMI, body mass index; BP, blood pressure; CHD, coronary heart disease; HDL, high-density lipoprotein; LDL, low-density lipoprotein.
Lipid Disorders
Abnormalities in plasma lipoproteins and derangements in lipid metabolism rank among the most firmly established and best understood risk factors for atherosclerosis. Chapter 421 describes the lipoprotein classes and provides a detailed discussion of lipoprotein metabolism.
The American College of Cardiology and American Heart Association (ACC/AHA) promulgated new guidelines on risk assessment, lifestyle measures, and cholesterol management in 2013. The panels that produced these guidelines followed an evidence-based approach. the 2013 cholesterol guideline focused on 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase inhibitors (statins) rather than other classes of lipid-modulating drugs, including fibric acid derivatives, cholesterol absorption inhibitors such as ezetimibe, and niacin products. The guideline cites the lack of contemporary randomized clinical trial evidence that supports the efficacy of these nonstatin lipid-modifying agents in cardiovascular event reduction. The cholesterol guideline defined four statin benefit groups (Table 291e-2): (1) all individuals who have clinical atherosclerotic cardiovascular disease (ASCVD), therefore considered “secondary prevention”; (2) those with LDL cholesterol ≥190 mg/dL without a secondary cause such as a high intake of saturated or trans fats, various drugs, or certain diseases; (3) individuals with diabetes without established cardiovascular disease who are 40–75 years old and have LDL cholesterol of 70–189 mg/dL; and (4) those without established ASCVD without diabetes who are 40–75 years old and who have LDL cholesterol of 70–189 mg/dL and a calculated ASCVD risk ≥7.5%. An online risk calculator based on pooled cohorts was provided to aid clinicians and patients in calculating their risk (http://my.americanheart.org/professional/StatementsGuidelines/PreventionGuidelines/Prevention-Guidelines_UCM_457698_SubHomePage.jsp). Other validated risk calculators that incorporate family history of CAD and a marker of inflammation (high-sensitivity CRP [hsCRP]) that apply to U.S. women and men exist (http://www.reynoldsriskscore.org). Downloadable applications for risk calculation on handheld devices are readily available.
|
SUMMARY OF THE FOUR STATIN BENEFIT GROUPS DESCRIBED IN THE 2013 ACC/AHA GUIDELINE ON THE TREATMENT OF BLOOD CHOLESTEROL TO REDUCE ATHEROSCLEROTIC CARDIOVASCULAR RISK IN ADULTS |
Abbreviations: ACC/AHA, American College of Cardiology and American Heart Association; ASCVD, atherosclerotic cardiovascular disease; LDL-C, low-density lipoprotein cholesterol.
Source: Adapted from NJ Stone et al: 2013 ACC/AHA Guideline on the Treatment of Blood Cholesterol to Reduce Atherosclerotic Cardiovascular Risk in Adults. J Am Coll Cardiol 2013, doi: 10.1016/j.jacc.2013.11.002.
The 2013 guideline emphasized a patient-centered approach and recommended that clinicians and patients engage in a risk-benefit conversation before starting statin therapy and not rely solely on calculated risks or arbitrary category assignment. It further emphasizes that medications do not supplant a healthy lifestyle. The guideline also provides some practical suggestions regarding management of muscle symptoms attributed to statins, an issue of considerable concern to many patients and practitioners alike.
In a major departure from prior guidelines, the 2013 guideline eliminates LDL targets as goals of therapy. The panel did so because major clinical trials did not titrate therapy to a goal, but rather used fixed doses of statins. Instead, the new guideline suggests different intensities of statin therapy based on risk category (Fig. 291e-4).
FIGURE 291e-4 Major recommendations for statin therapy for atherosclerotic cardiovascular disease (ASCVD) prevention. LDL-C, low-density lipoprotein cholesterol. (From NJ Stone et al: J Am Coll Cardiol, 2013, doi: 10.1016/j.jacc.2013.11.002.)
The 2013 guideline focus on statins reflects an extensive body of rigorous evidence that supports the effectiveness of this class of drugs in cardiovascular event reduction and an acceptable risk-benefit relationship (Fig. 291e-5). Moreover, because almost all statins are now available as generic statins medications, cost has become much less of an impediment to their use.
FIGURE 291e-5 The Cholesterol Treatment Trialists Collaboration meta-analyzed 27 randomized clinical trials evaluating statin therapy. They found profound decreases in both major vascular events and vascular death (not shown) proportional to the magnitude of low-density lipoprotein (LDL) cholesterol reduction achieved with statin treatment. This diagram shows the results of this meta-analysis for vascular death. (From Lancet 380:581, 2012.)
The clinical use of effective pharmacologic strategies for lowering LDL has reduced cardiovascular events markedly, but a considerable burden of residual risk remains even in patients treated with high-intensity statins. Hence, current studies are evaluating other avenues to address the residual burden of cardiovascular disease that persists despite statin treatment. Inhibitors of genetic studies identified proprotein convertase subtilisin kexin-like 9 (PCSK9) as a regulator of LDL levels associated with cardiovascular outcomes. Interaction of the LDL receptor with PCSK9 hastens the receptor’s degradation, and hence yields higher circulating LDL concentrations. Genetic variants that lower PCSK9 activity appear to protect against cardiovascular events. Monoclonal antibodies that neutralize PCSK9 lower LDL levels even in statin-treated patients and are currently under investigation as novel therapeutics to lower cardiovascular risk.
LDL-lowering therapies do not appear to exert their beneficial effect on cardiovascular events by causing a marked “regression” of stenoses. Studies of lipid lowering monitored by angiography or by intravascular imaging modalities have shown at best a modest reduction in coronary artery stenoses over the duration of study, despite abundant evidence of event reduction. These results suggest that the beneficial mechanism of lipid lowering by statins does not require a substantial reduction in the fixed stenoses. Rather, the benefit may derive from “stabilization” of atherosclerotic lesions without substantially decreased stenosis. Such stabilization of atherosclerotic lesions and the attendant decrease in coronary events may result from the egress of lipids or from favorably influencing aspects of the biology of atherogenesis discussed above. In addition, as sizable lesions may protrude abluminally rather than into the lumen due to complementary enlargement, shrinkage of such plaques may not be apparent on angiograms. The consistent benefit of statins may depend not only on their salutary effects on the lipid profile, but also on direct modulation of plaque biology independent of lipid lowering.
[/not-level-membership-for-internal-medicine-category]
