[level-membership-for-surgery-category]Chapter 5
Atherosclerosis
Christopher D. Owens
Vascular surgeons care for a diverse set of clinical manifestations related to atherosclerosis—from symptomatic transient cerebral ischemic attacks to aortoiliac occlusions to aortic aneurysm repairs in patients with advanced coronary heart disease (CHD). However, despite the wide range of manifestations, culprit lesions are more alike than different. Accumulation of large amounts of cholesterol ester in the arterial wall and formation of complex advanced plaque are common to all these lesions. Although their formation insidiously spans decades, atherosclerotic lesions can reach a clinical horizon within minutes and manifest as catastrophic myocardial infarction (MI), stroke, or limb ischemia. Far from being a simple cholesterol deposition problem, plaque formation and evolution are now recognized as much more dynamic in nature. Rather than following a progressive degenerative process, individual plaque formations may progress and regress or undergo growth spurts. Inflammatory changes within the fibrous cap may render them vulnerable to rupture or erosion and ulceration. An understanding of each of these processes helps the vascular surgeon in developing evidence-based treatment strategies for each of the clinical syndromes of atherosclerosis, which together constitute a massive public health problem.
Atherosclerotic-related cardiovascular disease (CVD) is the leading cause of death in every region of the world except sub-Saharan Africa, and it has now eclipsed infectious disease in most underserved nations.1 However, during the last two decades, CHD age-specific death rates fell by greater than 40% in high-income countries.2 The prolonged survival has translated into growing disease prevalence and a staggering financial burden. It is estimated that 80,700,000 Americans have CVD with an estimated annual cost of treatment of $448.5 billion.3 Globally, 202 million people have peripheral arterial disease (PAD), which disproportionately affects blacks and individuals living in low to middle-income countries.1,3 Particularly troubling is the high prevalence of cardiovascular risk factors in children and young adults.4,5 A sedentary lifestyle, abdominal obesity, and poor diets contribute to dyslipidemia and high blood pressure. Autopsy studies in children and young adults demonstrate a link between these risk factors and early lesions.4
This chapter outlines the existing theories of the pathophysiology of atherosclerosis and the relationship to traditional and emerging risk factors. It also describes our current conceptual understanding of the fundamental biology of atherosclerotic plaque.
Normal Artery Structure and Function
A detailed presentation of normal arterial structure can be found elsewhere in this text (see Chapters 3 and 4); however, the relevant anatomy is presented here. Histologically, the arterial wall consists of three distinct layers: the intima (endothelium and subendothelial space), the media (smooth muscle cells [SMCs]), and the adventitia (vasa vasorum and connective tissue elements). The intima consists of the endothelium, its basement membrane, and the subendothelial space, which extends to the internal elastic laminae. The endothelium is a single layer of cells lining the inner surface of all blood vessels.
Endothelium
The endothelium is an organ with autocrine, paracrine, and endocrine functions that serve to regulate blood vessel tone and thrombogenicity. The endothelium synthesizes an array of factors that regulate vascular tone (e.g., nitric oxide [NO], hyperpolarizing factors, prostaglandins, endothelin, angiotensin II) and interactions with blood elements (e.g., adhesion molecules, thrombomodulin, plasminogen activators), which all work in balance to maintain vascular homeostasis.
Endothelial dysfunction is regarded as the earliest manifestation of vessel injury and is present before histologic evidence of atherosclerosis.6 Hence functional testing of the endothelium is an important tool for investigators studying atherosclerosis. Endothelial function can be detected in vivo by invasive and noninvasive techniques. Catheter-directed intraarterial administration of acetylcholine has been used to detect the presence of endothelium-dependent vasodilatation in the coronary and peripheral vasculature. Paradoxical contraction with low doses of acetylcholine is regarded as positive for endothelial dysfunction in the coronary circulation. An acute increase in blood flow can also stimulate endothelial relaxation. Flow-mediated, endothelium-dependent vasodilatation can be measured noninvasively by high-resolution ultrasound techniques.7 The primary mediator of flow mediated vasodilation is NO.8 Administration of nitroglycerin can detect the ability of vascular smooth muscle to relax in the presence of exogenous NO and thus detects endothelial-independent relaxation.
Subendothelial Space
The subendothelial space is particularly relevant in atherosclerosis because it is the location where atherogenic particles are retained and modified so that they can be taken up by macrophages and SMCs.9 In terms of volume, the major component of the extracellular intimal matrix is proteoglycans, which form a three-dimensional network that fills 60% of the space.9 The remaining mass of this compartment is filled with type IV collagen of the basement membrane, type I and III collagen randomly dispersed in this matrix, and occasional elastin fibers. Extending from the proteoglycans are sulfated glycosaminoglycan chains, which serve to interact with positive residues on apolipoproteins. Large versican-like proteoglycans containing chondroitin sulfate readily interacts with apolipoprotein B-100 (apoB-100), the apolipoprotein associated with low-density lipoprotein (LDL).10 The stoichiometry is such that 1 µg of versican can bind to 50 to 100 µg of LDL at physiologic ionic strength, thus suggesting that several lipoprotein particles are retained on each proteoglycan molecule.10,11
Smooth Muscle Cells
In children, very few SMCs or macrophages are present in the subendothelial space; however, with age and lesion formation, their concentration increases. In autopsy studies, abundant SMCs are found in the coronary arteries in atherosclerosis-prone areas, but they are relatively sparse in areas resistant to atherosclerosis.12 Whereas human medial SMCs predominantly express proteins involved in the contractile function of the cell, those found in the intima express lower levels of these proteins, have a higher proliferative index, and exhibit greater synthetic capacity for extracellular matrix, proteases, and inflammatory cytokines.13,14 These “synthetic” SMCs migrate more readily and can produce up to 25 to 46 times more collagen than “contractile” SMCs can. Thus SMCs directly contribute to intimal thickening as a result of increased proteoglycan production, inflammatory cell recruitment, and retention of atherogenic particles within the subendothelial space.
Vasa Vasorum
The vasa vasorum may contribute to atherosclerosis as well. First-order vasa vasorum run longitudinally to the lumen of the host vessel, whereas second-order vasa vasorum are arranged circumferentially. As plaque volume increases, there is an increase in the vasa vasorum visible on histologic section. Whether this is a response to plaque growth or contributes to plaque growth is unknown. Although the development of vasa vasorum in plaque involves angiogenesis, it is unclear what signals this process. Atherosclerotic vasa vasorum can proliferate and thereby lead to extensive neovascularization directed toward lipid-rich atheroma. The neovessels of plaque may provide an additional portal for trafficking leukocytes to promote the inflammatory process or become friable and contribute to intraplaque hemorrhage.15
Atherosclerotic Lesions
Lesions of the arterial wall have been divided into six types based on their histopathologic features.16,17 However, although useful for comparing pathologic specimens, the classification system has limited clinical practicality. The assumption of a relentless, linear progression of the disease has recently been challenged, and though this assumption is true under experimental conditions in some animal models,18,19 it is not necessarily the case in humans.20,21
Autopsy studies from the Bogalusa Heart Study and the Pathobiological Determinants of Atherosclerosis in Youth study demonstrate that atherosclerotic lesions form in early childhood and increase with age and that a significant percentage of them are raised.4,22–24 In an intravascular ultrasound (IVUS) study of heart donors, 17% of individuals younger than 20 years had evidence of atherosclerosis.25 Notably, both the Bogalusa Heart Study and the Pathobiological Determinants of Atherosclerosis in Youth study emphasize that the number and severity of early lesions are directly related to known cardiovascular risk factors, thus suggesting that a paradigm shift is needed to address risk factors in childhood rather than pharmacologic therapy later in the disease process.
Diffuse intimal thickening has been identified in the atherosclerotic-prone areas of coronary arteries as early as 36 weeks of gestation.26 Although the fatty streak—dominated by lipid-filled macrophages—is itself benign, it is the precursor of the more clinically relevant late lesion. The fatty streak is the first lesion visible to the naked eye, and because of its positive Sudan staining, it is referred to as sudanophilic. Its yellow color is attributed to lipid in the form of cholesterol and cholesterol esters within macrophages and SMCs. From the aforementioned studies, it is clear that atherosclerosis begins early in life in all races, is associated with risk factors, and occurs in anatomic locales susceptible to the formation of more advanced fibrous plaque.
Advanced lesions, or fibrous plaque, are characterized histologically by the amount of extracellular lipid and fibrous connective tissue seen. They are whitish in gross appearance and are elevated so that they protrude into the lumen. These fibroatheromas are prone to produce clinical sequelae by erosion of the surface endothelial cells, rupture of the fibrous cap, erosion of a calcium nodule, or intraplaque hemorrhage (Fig. 5-1).16,27 Plaques differ in consistency and may be relatively soft and friable or densely sclerotic and calcific. Likewise, some have well-formed fibrous caps, whereas others are covered by a narrow zone of loose connective tissue or by endothelium alone.
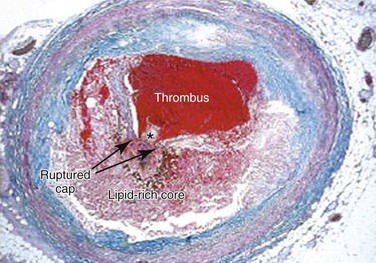
Figure 5-1 Thrombotic complication of a fibroatheroma seen on a trichrome-stained (thrombus and intraplaque hemorrhage stain red; collagen stains blue) cross-section of a human coronary artery. The fibrous cap has ruptured (area between the arrows), and the highly thrombotic lipid core is exposed to circulating blood with the subsequent production of acute occlusive thrombosis.
The necrotic core usually occupies the deeper central regions of the plaque and contains amorphous lipid and cholesterol crystals. The term atheroma is derived from the Greek word athere, meaning “porridge-like gruel.” SMCs and inflammatory cells are located adjacent to the necrotic core and at the shoulders of the plaque, where it is most susceptible to rupture.
The fibrous caps contain varied levels of SMCs adjacent to the collagen and basement membrane. These cells have reduced proliferative ability and may be thought of as senescent. However, the fibrous cap of ruptured plaque is often infiltrated with foam cells, which are largely of macrophage origin28 and thus indicative of active inflammation and vulnerability to rupture. A priori identification of these so-called thin-cap fibroatheromas is an active area of cardiovascular imaging research.29
Theories of Pathogenesis
Each of the well-regarded theories of atherosclerosis discussed in this section attempts to explain the underlying pathogenesis of atherosclerotic plaque. From the time initially proposed, each theory has undergone a steady evolution as our methodology of scientific inquiry has advanced from histologic descriptors to powerful molecular mechanisms. Each theory, held accountable over time, must incorporate new scientific data within a clinical context of risk factors and biomarkers to result in a model of atherosclerotic lesion formation.
Lipid Hypothesis
Cholesterol has been one of the most studied molecules in biomedical research. Since it was first isolated from gallstones in 1784, organic chemists and biochemists, physiologists and physicists, epidemiologists, and other medical scientists have devoted significant portions of their careers to its study and have collectively produced a body of work culminating in 13 Nobel Prizes.30 The modern era of cholesterol research began in St. Petersburg, Russia, at the turn of the 20th century, when Nikolai Anitschkow produced vascular lesions in rabbits by feeding them purified cholesterol dissolved in sunflower oil.31,32 These lesions closely resembled those seen in human atherosclerosis. He determined that the earliest lesions consisted of foam cells that were Sudan positive and contained birefringent crystals and hypothesized that they were caused by elevated serum cholesterol. He also noted a distinctive pattern consisting of location of the lesions near arterial branch points and concluded that this was probably determined by hemodynamic factors. Nevertheless, this work was criticized because the levels of cholesterol produced were too high and could not be experimentally reproduced in more conventional animal models such as rats and dogs.31,32
Cholesterol is absolutely insoluble in water, and the early work done in Russia provided no clues as to how it was transported to the arterial wall and formed lesions. The discovery that cholesterol was associated with proteins that allowed it to be transported in the aqueous environment led to investigations into lipoproteins. The first investigation in which the lipoprotein content in whole serum was accurately quantified involved analytic ultracentrifugation and was led by John Gofman of the Donner Laboratory at Berkley.33 Lipoprotein fractions were isolated and characterized by their densities and floatation characteristics.33 Of importance, this group noted that it was not simply the total cholesterol that was important but the species of lipoprotein contained within the cholesterol.34
Cholesterol is transported in the aqueous environment by esterification of the sterol of long-chain fatty acids and packaging of these esters with the hydrophobic cores of plasma lipoproteins. With its polar hydroxyl group esterified, cholesterol remains sequestered within this core, which is essentially an oil droplet composed of cholesteryl esters and triglycerides, solubilized by a surface monolayer of phospholipid and unesterified cholesterol and stabilized by protein. In persons who are fasting, lipids circulate in plasma as lipoprotein particles that are defined on the basis of their density as very-low-density lipoprotein (VLDL), intermediate-density lipoprotein (IDL), LDL, and high-density lipoprotein (HDL). The protein of the VLDL, IDL, and LDL molecule is apoB-100, which has β-electrophoretic mobility.
The major milestone in the lipoprotein field was discovery of the defective gene associated with familial hypercholesterolemia by Goldstein and Brown. In fibroblasts cultured from normal human subjects, the activity of 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase, the rate-controlling enzyme in cholesterol biosynthesis, is regulated by the content of LDL (but not HDL) in the culture medium. Goldstein and Brown found that 3-hydroxy-3-methylglutaryl coenzyme A reductase was not sensitive to normal feedback regulation by LDL in cultured skin fibroblasts from homozygous familial hypercholesterolemia patients.35,36 They then determined that the defect was related to deficient binding of LDL to the cells of patients with familial hypercholesterolemia and that suppression of 3-hydroxy-3-methylglutaryl coenzyme A reductase activity was related to the amount of LDL bound.37 Therefore these patients had overproduction of cholesterol because they lacked the appropriate receptor. They demonstrated that cellular uptake of LDL absolutely requires the LDL receptor, without which the LDL cholesterol concentration builds up to 800 to 1000 mg/dL (20.72 to 25.90 mmol/L). Recognition of the importance of this work resulted in Goldstein and Brown sharing the Nobel Prize in Physiology and Medicine in 1985.
Epidemiologic evidence from numerous studies, including the Japanese migration studies38,39 and the Framingham Heart Study,40 demonstrated the association between cholesterol and incident CHD. Other studies showed that diets rich in unsaturated fats resulted in lowered cholesterol and reduced cardiovascular events.41
Widespread acceptance of the causative role of cholesterol began with publication of the Coronary Primary Prevention Trial, sponsored by the National Institutes of Health.42,43 This trial demonstrated that reducing cholesterol with the bile acid–binding resin cholestyramine would reduce cardiovascular events. A decade later, this trial was reinforced with powerful data emerging from the statin era.44 Although the primacy of the “cholesterol hypothesis” has been called into question, there is no doubt that lipids play a critical role in the pathogenesis of atherosclerosis.
Response-to-Injury Hypothesis
The basis of this hypothesis lies in the similarities between atherosclerosis and the response of arteries to experimental injury. Although the origins of the hypothesis date back to Virchow, the modern version of the hypothesis builds on earlier work by Duguid (1949),45 Poole and Florey (1958),46 and French (1966)47 and was formally advanced in 1973 by Ross and Glomset,48 who emphasized the importance of injury to the endothelium as a seminal event initiating the development of atherosclerosis lesions. Even though the definition of the term injury has been modified over the last three decades,49 all response-to-injury hypotheses have emphasized the primacy of the endothelium in stimulating the cascade of events leading to lesion formation.48–53
Endothelial injury may result from mechanical disruption, exposure to toxic or infectious agents, or endogenous inflammatory signals. Injury to the endothelium allows adhesion of platelets and an influx of LDL and other serum factors into the subendothelial space. Platelets release their alpha granules and stimulate migration of SMCs into the intima, where they proliferate and form a thickened neointima responsible for narrowing of the arterial lumen. Restoration of a healthy endothelial cell layer abates the process. During work on this hypothesis, Ross et al performed experiments leading to the discovery of platelet-derived growth factor (PDGF).54,55
Others counter that it is not injury to the endothelium that is the initiating event but rather retention of inflammatory lipids in the subendothelial space, which renders a particular area susceptible to atherosclerosis. By noting that LDL accumulates in the intima within 2 hours after a bolus infusion56 and that the accumulation occurs before the formation of fatty streaks,57,58 they concluded that retention of LDL is the initiating event of atherosclerosis. The so-called response-to-retention theory purports that retained apoB–containing lipoproteins stimulate a macrophage- and T cell–dominated inflammatory response in the arterial wall.59,60
Monoclonal Hypothesis
During the same year that Ross and Glomset were formulating the response-to-injury hypothesis of atherosclerosis, another group of investigators at the same institution (University of Washington) were writing their own hypothesis of atherosclerosis. This hypothesis suggests that each lesion of atherosclerosis is derived from a single SMC that serves as a precursor for the clonal expansion of proliferating SMCs.61 The hypothesis put forth by Benditt and Benditt uses the concept that in every female cell there is only one active X chromosome and the progeny of that cell will express the same X chromosome as the parent cell. Glucose-6-phosphate dehydrogenase (G6PD) has two isoforms that can be separated by electrophoresis. Its gene is located on the X chromosome and can therefore be used to identify the progeny of a parent cell. This approach was used to determine that uterine leiomyomas are composed of cells with the same active X chromosome, whereas adjacent normal myometrium was composed of a mixture of cells containing both G6PD isoforms and, therefore, that both X chromosomes were active.62
The Benditts examined a series of atherosclerotic plaque from four black females and compared them with adjacent normal areas of arterial wall. They determined that SMCs from the plaque contained only one G6PD isoform, whereas adjacent control areas contained a mixture of isoforms. This allowed them to conclude that each lesion is a clonal outgrowth derived from a single precursor SMC located in the intima. It is noteworthy that SMCs within individual human neonatal intimal thickenings are monoclonal in origin, whereas cells from the subjacent media are polyclonal.
Others challenged the hypothesis by noting that identification of a single enzyme phenotype does not necessarily imply clonal origin.63,64 Regardless, the work was importantly heuristic. It hinted toward the role that modern molecular biology would play in unraveling the genetic basis of atherosclerosis.
Atherosclerosis as a Chronic Inflammatory Disease
The original version of the response-to-injury hypothesis of atherosclerosis initially proposed that endothelial denudation was the first step in atherosclerosis.48 Subsequent versions of the hypothesis proposed that an endothelium chronically bathed in serum with high concentrations of LDL or exposed to other cardiovascular risk factors would render susceptible areas of the endothelium dysfunctional or activated.52 Indeed, an intact endothelium may be a necessary factor for lesion progression, and it is now clear that developing atheromas are covered by an intact endothelium throughout most stages of lesion progression.17,49,65–67 In humans, only the most advanced ulcerated lesions are focally devoid of endothelium. The injury results in increased adhesiveness and an increase in permeability of the endothelium to inflammatory cells (Fig. 5-2).
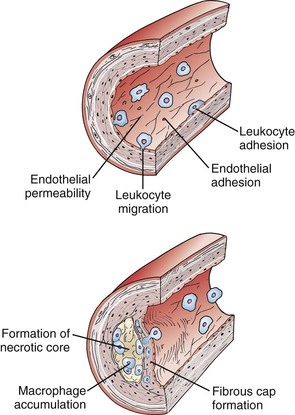
Figure 5-2 Initiation and progression of atherosclerotic plaque. Cardiovascular risk factors, hemodynamic forces, toxins, and infectious agents interact with the vessel at the level of the endothelium to produce injury, resulting in decreased nitric oxide (NO) production and increased permeability. Once injured, the endothelium increases the expression of leukocyte adhesion molecules such as vascular cell adhesion molecule-1, intracellular adhesion molecule-1, and P- and E-selectin, which increases the adherence of macrophages and other leukocytes. Permeability of the endothelium also increases and permits entry of leukocytes and lipoproteins into the subendothelial space. Chemokines and cytokines such as monocyte chemotactic protein-1 (MCP-1) and interleukin-8 (IL-8) further enhance the recruitment of leukocytes and smooth muscle cells (SMCs) into the subendothelial space. Lipoproteins retained in the subendothelial space are biochemically modified such that they can be taken up by macrophages and SMCs to form foam cells. Foam cells at the central-most position of the developing atheroma become necrotic and form the central lipid core, whereas the shoulder regions contain SMCs, macrophages, and other leukocytes. Platelet-derived growth factor (PDGF) and transforming growth factor-β (TGF-β) stimulate SMC migration and collagen formation in the subendothelial space, as well as formation of the fibrous cap.
Atherosclerosis is now recognized as an inflammatory disease, and components of the innate and adaptive immune system are involved in every step of the atherosclerotic process. Much of our modern understanding comes from examination of human pathology specimens and transgenic animals. Genetic deletion of apolipoprotein E (apoE−/−) or the LDL receptor (ldlr−/−), which produces mice with severe hypercholesterolemia and atherosclerotic lesions with features of mature human atheroma, has become a cornerstone in atherosclerosis research laboratories.68–70 The importance of the LDL receptor in cholesterol regulation was noted by Goldstein and Brown when studying patients with familial hypercholesterolemia. Apolipoprotein E suppresses atherosclerosis, and apoE−/−– mice have very low amounts of pre-β HDL and their plasma is poor at promoting the efflux of cholesterol from lipid-laden macrophages.71 Crossbreeding these mice with other strains carrying null mutations in immunologically relevant genes produces a robust research tool to dissect out the contribution of individual components of immune pathways in atherosclerosis. For example, some of the earliest approaches using compound mutant mice involved the global loss of the entire adaptive immune system. Rag1−/− or Rag2−/− mice lack the V(D)J recombinase required to form lymphocyte antigen receptor genes and hence have a complete loss of B and T cells. In apoE−/− mice, a regular chow diet produced plasma cholesterol levels between 390 and 470 mg/dL. Double knockout apoE−/−/Rag1−/− animals had a 40% reduction in aortic atherosclerotic lesion reduction compared with immunocompetent animals and provided an important piece of evidence of the role cellular immunity plays in the pathogenesis of atherosclerosis.72
LDL Retention
Trafficking of circulating LDL into and out of the subendothelial space is probably a function of concentration gradients, as well as endothelial permeability.9,73 Normal intima does not retain LDL particles, thus suggesting that most particles return to plasma or are degraded in situ. However, subendothelial retention of apoB-100–containing lipoproteins is an early event in atherosclerosis.73 Factors favoring net retention include a balance of uptake and degradation of LDL by macrophages, egress of LDL-containing macrophages into the circulation, and complex interaction with proteoglycans such that the LDL particles are sufficiently modified to maintain the gradient. Once bound to the matrix, the lipoprotein is modified so that it becomes oxidized.9,74 Oxidized LDL, unlike native LDL, is chemotactic to monocytes and rapidly taken up by macrophages to form foam cells by scavenger receptors.
Monocytes
As early as 1958, Poole and Florey noted that macrophages adhere to the surface of endothelial cells overlying atheroma.46 In studies of hypercholesterolemic monkeys, monocytes are seen attached to the endothelium within 12 days of initiation of an atherogenic diet.19 It is now apparent that monocytes play a very early role in atheroma formation.75,76 Endothelium, which is rendered activated, expresses adhesion molecules that interact with circulating leukocytes, principally monocytes and lymphocytes. E- and P-selectins slow monocytes, mediate rolling, and loosely tether them to the endothelium.77 More permanent fixation is due to members of the immunoglobulin superfamily—vascular cell adhesion molecule-1 (VCAM-1)78 and intercellular adhesion molecule-1 (ICAM-1)—which are soon upregulated and firmly fix the leukocytes to the wall. The importance of the selectins, VCAM-1, and ICAM-1 in the pathogenesis of atherosclerosis is evident in either ICAM-1 or VCAM-1 mutant animals exhibiting far less atherosclerosis than do wild type controls.79–81 The adherent leukocytes are stimulated to migrate into the subendothelial space by a number of chemokines, including MCP-1 and oxidized LDL.82–84
Macrophages
Once within the subendothelial space, monocytes undergo a series of phenotypic modulations and become resident tissue macrophages that take up oxidized LDL via the scavenger receptor A (SR-A), as well as CD36.85 One of the key signals for macrophage activation, macrophage colony-stimulating factor, can enhance scavenger receptor expression and promote replication of macrophages and their production of proinflammatory cytokines. Experiments in mutant mice deficient in macrophage colony-stimulating factor have shown the importance of this factor in atheroma formation.86,87 Although most foam cells are macrophage in origin, SMCs also take up lipids via scavenger receptors and CD36.
In mice, proinflammatory subsets of macrophages called M1-macrophages, induced by hyperlipidemia, produce the inflammatory cytokines IL-1β and TNF-α. The IL-1 gene family encodes three major proteins. The first two, IL-1α and IL-1β, exert proinflammatory effects by binding to the IL-1 receptor type 1. The third is IL-1 receptor antagonist (IL-1Ra), an endogenous inhibitor that competitively blocks the binding of IL-1α and IL-1β to the IL-1 receptor. Direct evidence for the proatherogenic role of IL-1 was obtained in experiments in which apoE−/− mice received subcutaneous injection of recombinant IL-1 receptor antagonist. Mice injected with IL-1 receptor antagonist displayed a marked reduction in atherosclerotic lesion size. Similarly IL-18, previously called interferon-γ (IFN-γ)–inducing factor, is a Th1-promoting cytokine and therefore has the capacity to promote inflammation through the innate and the adaptive immune pathways.88,89 Knockout of IL-18 in atherosclerosis-prone animals reduces aortic atherosclerotic lesion size.
Macrophages also contribute to thrombosis in several pivotal ways. As discussed later, atherosclerotic lesions that have ruptured characteristically contain abundant macrophages underlying a thin and collagen-poor fibrous cap. Specific collagenases MMP-1, MMP-8, and MMP-13, co-localizes with macrophages in human atheroma. Rupture of the fibrous cap may be a balance between collagen synthesis by SMCs and collagen breakdown by interstitial (matrix metalloproteinases) collagenases generated by activated macrophages. Studies in collagenase-resistant mutant “knockin” mice crossbred with apoE−/− mice demonstrated increased intimal collagen and SMC content.90 Similarly, in compound mutant apolipoprotein E–null mice crossed with MMP-13/collagenase-3−/− mice have increased fibrillar collagen that is thicker and more aligned in aortic atherosclerosis.91 Hence interstitial collagenases produced by activated macrophages greatly influence the structure and integrity of the fibrous cap overlying atherosclerotic plaques. Macrophages and SMCs within atherosclerotic plaques also overexpress the potent procoagulant tissue factor in response to C-reactive protein or CD40 ligand. Hence inflammation links atherosclerosis and thrombosis, leading some to refer to it as atherothrombosis.
Lymphocytes and Adaptive Immunity
The finding in the early 1980s that macrophages expressed major histocompatibility class II antigens needed for antigen presentation to CD4+ T cells suggested that adaptive immunity was involved in the atherosclerotic process.92 T lymphocytes, which account for as much as 10% to 20% of the leukocyte population, are found most abundantly in the shoulder and fibrous cap region of the atheroma. In advanced lesions, these T cells display markers of chronic activation and produce the prototypical Th1 cytokine, (IFN-γ) which further stimulates expression of class II major histocompatibility antigens in SMCs and macrophages. The crossbreeding of severe combined immunodeficiency (scid)/scid mice that lack T and B cells with apoE−/− mice produces offspring that are both hypercholesterolemic and immunodeficient. Similar to what was found with apoE−/−/Rag2−/− double mutants, when lesions in these mice were compared with those in the immunocompetent apoE−/− animals, a dramatic 70% reduction of lesion size was observed. However, transfer of oxidized LDL-reactive T cells to apoE−/−/scid/scid mice is more efficient at lesion acceleration than the transfer of T cells with no specificity to a plaque-derived antigen, demonstrating the importance of antigen presentation in the pathogenesis of atherosclerosis.93
Because of the participation of all components of the innate and adaptive immune system, atherosclerosis resembles other inflammatory and autoimmune diseases such as rheumatoid arthritis and type 1 diabetes mellitus. Potential endogenous autoantigens to activate T cells include LDL or heat shock protein 60.93,94 Cellular and humeral immune responses are mounted toward these antigens in humans and mice, and protective immunization strategies in mice have provided encouraging results.
In mice, functionally distinct T cell subsets appear to exist in atheroma. Natural killer T cells appear to accelerate atherosclerosis when recognizing lipid antigens presented through CD1 molecules.95 Conversely, regulatory T cells may suppress Th1 effector cells through production of antiinflammatory cytokines such as IL-10 and transforming growth factor-β (TGF-β).96 T cells, although far fewer in number than macrophages, likely serve as key regulators of the concerted immune response.
Mast cells are located at the shoulder and more central area of the cap, where they may participate in rupture or erosion of the cap.97 Once recruited to the subendothelial space in the intima, the white blood cells can perpetuate and amplify the ongoing inflammatory response that led to their recruitment.98 The CD40 receptor and CD40 ligand are expressed by several inflammatory cells, including macrophages and B and T lymphocytes.99 It is thought that this system contributes to leukocyte adhesion, matrix degradation, and cytokine-induced inflammation. Interruption of the CD40 signaling pathway reduces progression of atherosclerosis in experimental models.
Smooth Muscle Cells
Proinflammatory mediators can stimulate the migration of SMCs from the tunica media into the intima. Growth factors produced locally provide a paracrine stimulus for SMC proliferation and activation. Activated SMCs appear capable of producing growth factors (e.g., PDGFs, fibroblast growth factors) that can stimulate their own proliferation and that of their neighbors in an autocrine and paracrine fashion.100 TGF-β stimulates the production of matrix and collagen in the subendothelial space. As the plaque matures, necrotic foam cells contribute to the central lipid core, whereas collagen contributes to the overlying fibrous cap of a mature fibroatheroma.
Calcification
Bone morphogenetic protein-2 (BMP-2), a member of the TGF-β family, and inorganic phosphate induce the osteochondrogenic phenotype in SMCs.101 BMP-2, in turn, is produced by endothelial cells exposed to hypoxia, reactive oxygen species, turbulent flow, high pressure, or inflammation.101 Atherosclerotic calcification proceeds through a process similar to chondrogenesis, whereby cartilaginous metaplasia precedes osteoblast induction. This is distinct from medial artery calcification, which proceeds through a process similar to intramembranous bone formation. The latter is common in patients with diabetes or chronic kidney disease. The reason that some plaques undergo calcific changes whereas others do not is not clear.
Thus the inflammatory reaction stimulated by modified LDL and white blood cells in the subendothelial space provides a nidus for the subsequent events leading to the formation of mature fibrocalcific atherosclerotic plaque. Notably, this process, which probably begins in childhood, is silent until sufficient arterial stenosis exists or until a catastrophic plaque-destabilizing event occurs and produces a clinical symptom.
Putting It All Together: The Inflammasome
Clinically, cholesterol is an established causative risk factor for atherosclerosis. That cholesterol leads to vascular inflammation by activating the immune system remains an attractive hypothesis, albeit one that requires more studies before it can be confirmed or rejected in humans. Recent evidence has shown that crystalline cholesterol is present early in the formation of atherosclerotic lesions and coincides with the presence of immune cells.102,103 Cells of innate immunity are equipped with receptors that detect pathogen-associated molecular patterns and endogenous danger signals. Once these receptors are activated, monocytes and macrophages secrete inflammatory cytokines such as IL-1β into the arterial wall. In the last 10 years, considerable efforts in several laboratories have established a role for the interleukin 1 family in complex diseases, including atherosclerosis, diabetes mellitus, rheumatoid arthritis, and gout. As discussed earlier, IL-1β has several layers of regulation and is synthesized as pro-IL-1β and requires activated caspase-1 to cleave pro-IL-1β and IL-18 into their active and secreted form. Caspase-1 activation requires a second signal mediated through a complex of intracellular proteins known as inflammasomes. The nucleotide-binding domain leucine-rich (NLR) family, pyrin domain–containing 3 (NLRP3) inflammasome has been most extensively studied. When NLRP3 receptors are activated, they oligomerize and recruit caspase-1 through the adapter protein ASC (apoptosis-associated specklike protein containing a caspase recruitment domain) and autocatalytically activate caspase-1, which cleaves pro-IL-1β into its mature forms.104 NLRP3 inflammasome activation historically included pore-forming toxins, extracellular adenosine triphosphate, viral DNA, and gout-associated uric acid crystals. However, recent evidence demonstrates that cholesterol crystals can also activate the inflammasome and therefore increase secretion of the potent proinflammatory cytokines IL-1β and IL-18. These data argue that cholesterol crystals are not solely an inert byproduct within the wall of the artery but an active contributor to atherosclerosis.102,103
Moreover, hypercholesterolemic ldlr−/− mice reconstituted with bone marrow from mice deficient in NLRP3, ASC, or IL-1β have significantly reduced aortic lesion sizes compared with those reconstituted with wild-type bone marrow.102 These mice showed significantly lowered levels of IL-1β and IL-18. The implication of caspase-1 was evaluated recently in atherosclerosis as a pathogenic enzyme because this deficiency decreases atherosclerosis in apoE-deficient mice.105 Hence within the vascular wall, lipids and inflammation appear to be inextricably linked.
C-Reactive Protein
The clinical detection of inflammation within the vascular wall is limited to the measurement of spillover circulating biomarkers of the inflammatory process. Many prospective studies have established an association between biomarkers of inflammation and first and recurrent cardiovascular events. For example, studies have noted that the circulating soluble ICAM-1 and VCAM-1 are elevated in the plasma of patients before the development of peripheral arterial disease.106 Similarly, the inflammatory cytokines IL-6 and IL-1β, as well as acute-phase reactants C-reactive protein, fibrinogen, and serum amyloid A, have all been associated with peripheral arterial disease and its progression.107,108 Among the many inflammatory biomarkers evaluated, high-sensitivity C-reactive protein (hsCRP) has emerged to be the leading biomarker for clinical application. This is because CRP is a very stable analyte over time, has a relatively long half-life, has no diurnal variation, and requires no special processing for sampling.109 In addition, very low (<0.5 mg/L) and very high (>10 mg/L) values of hsCRP both provide important prognostic information on cardiovascular risk.110,111
Currently, more than 20 large prospective studies across North America and Europe demonstrate the relationship between the baseline inflammatory state, as measured by the hsCRP test, and the risk for future cardiovascular events. Furthermore, in 28,000 apparently healthy women, hsCRP was a stronger predictor of cardiovascular events (including coronary events, stroke events, and cardiovascular death) than were LDL cholesterol, total cholesterol, or other markers of inflammation and thrombosis.112,113 Prediction models (Reynolds Risk Score Project: www.reynoldsriskscore.org) for future cardiovascular risk demonstrate that hsCRP enters the model just behind high blood pressure but ahead of smoking and total and LDL cholesterol.114 The predictive value of hsCRP for cardiovascular risk is similar in the United States and Europe, thus demonstrating consistency in different populations.115 hsCRP is predictive in both primary and secondary prevention. In the Pravastatin or Atorvastatin Evaluation and Infection Therapy—Thrombolysis in Myocardial Infarction 22 (PROVE IT-TIMI 22) study, patients with acute coronary syndromes and the fewest subsequent events were those who reached LDL cholesterol levels of less than 70 mg/dL (1.81 mmol/L) and those who reduced their CRP levels to less than 2 mg/L (19.1 mmol/L).116,117 Accordingly, hsCRP is at least as robust a biomarker for predicting future cardiovascular events as are traditional cholesterol-based biomarkers (Fig. 5-3).
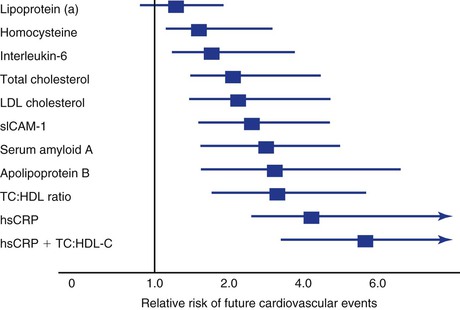
Figure 5-3 Comparison of lipid and nonlipid risk factors for cardiovascular disease (CVD) in 28,000 women enrolled in a nested case-control study from the Women’s Health Study cohort of postmenopausal, apparently healthy women. CRP adds to the total predictive value of cholesterol. Note that lipoprotein (a) and homocysteine are relatively weak predictors of CVD. HDL, High-density lipoprotein; hsCRP, high-sensitivity C-reactive protein; LDL, low-density lipoprotein; sICAM-1, soluble intercellular adhesion molecule-1; TC, total cholesterol.
The recently completed Justification for the Use of Statins in Primary Prevention: An Intervention Trial Evaluating Rosuvastatin (JUPITER) adds further circumstantial evidence of the role inflammation plays in atherosclerosis. The primary objective of JUPITER was to determine whether treatment with rosuvastatin would reduce the rate of first major cardiovascular event in patients with normal cholesterol levels but high levels of inflammation.118 The inclusion criteria of the trial stipulated that patients have a median hsCRP greater than 2.0 mg/L (median 4.2 mg/L) and LDL levels less than 130 mg/dL (median, 108 mg/L). The trial was terminated early because rosuvastatin resulted in a 44% reduction in the composite primary endpoint of MI, stroke, hospitalization for unstable angina, arterial revascularization, and cardiovascular death. Of particular interest to the surgical community was the 46% reduction in arterial revascularization and 48% reduction in stroke in patients allocated to rosuvastatin. The latter is particularly intriguing given the favorable data of statins in patients with carotid stenosis.119–121 Thus JUPITER determined that individuals with elevated levels of inflammatory biomarkers are at high vascular risk even when other risk factors are acceptable. Further, individuals at increased risk due to inflammation benefit from therapy they otherwise would not have received.122 Whether or not reducing inflammation with a drug that does not also modify lipids is an intriguing hypothesis that is currently being tested.123
Localization of Atherosclerosis
The propensity of atherosclerosis to occur at specific sites in the arterial tree has been observed since the time of Anitschkow’s cholesterol-fed rabbits.121 Other investigators noted increased incorporation of 3H-thymidine into endothelial cells at branch sites in animals not exposed to injury; this was thought to be due to hemodynamic consequences.124–126 Insull and colleagues found that atherosclerosis was predominantly located at intercostal branch points of the human thoracic aorta.127 However, it is not entirely clear why, given a constellation of risk factors to which the entire arterial tree is subjected, atherosclerosis should occur at these specific sites.
Only recently have the molecular mechanisms underlying this phenomenon become elucidated. Areas of well-developed laminar shear stress are relatively resistant to atheroma formation, whereas areas of turbulent or low shear stress (such as the carotid bifurcation) are more susceptible to atherosclerosis. Shear stress response elements occur in the promoter regions of a number of atheroprotective genes. eNOS is one such gene. Laminar shear increases eNOS activity and thus NO production, which renders endothelial cells more thromboresistant and results in less adhesion molecule expression and decreased SMC migration. NO reduces VCAM-1 gene expression through a novel pathway involving the inhibition of nuclear factor κB (NF-κB). Superoxide dismutase is expressed at higher shear stress and may reduce oxidative stress by catabolizing the highly reactive superoxide anion O2−. Thus areas of high laminar shear stress have antiinflammatory and antioxidant properties and exhibit less adhesion to circulating leukocytes.
Branch points, bifurcations, and major curvatures disrupt laminar flow and cause boundary layer separation, flow reversal, and shifting stagnation points. Such areas are characterized by increased particle contact time with the luminal surface, which may favor lipid deposition. Hemodynamic forces may predispose specific areas of the arterial tree to atherosclerosis when the appropriate risk factors are present. Areas of arterial branch points demonstrate elevated NF-κB activity.128 VCAM-1, an NF-κB–dependent protein, is expressed on endothelium at arterial branch points even in normal cholesterolemic animals. Disrupted laminar flow may promote a phenotypic transition in arterial cells favoring a proliferative state. NF-κB binds to several areas in the cyclin D1 promotor and stimulates G1-to-S transition.129 Alternatively, laminar shear stress induces the cyclin-dependent kinase inhibitor p21 and suppress the G0/G1-to-S phase transition.130
Hence fluid mechanical forces influence endothelial gene expression through certain response elements sensitive to shear stress. Endothelial dysfunction, characterized by decreased NO production, promotes vessel wall entry and modification of circulating LDL. Therefore it is possible that the differential regulation of endothelial genes by distinct flow profiles allows certain areas of the vasculature to more effectively resist the influence of risk factors such as hyperlipidemia and diabetes.
Progression/Regression of Plaques
DePalma and others, in an important series of experiments, noted progression and regression of atherosclerosis in canines and subhuman primates by serial laparotomy and autopsy examinations.131–134 The observation that plaque reduction correlated with a reduction in serum cholesterol paved the way for cholesterol-lowering trials in laboratory animals with bile acid sequestrants to test the hypothesis that reduced serum cholesterol would reduce plaque burden.133,134 The success of these early animal trials engendered considerable optimism that atherosclerosis was not universally progressive and eventually leads to lumen occlusion. Hence our current understanding of atherosclerotic plaque emphasizes a dynamic evolution.
Serial angiographic studies of human coronary arteries demonstrate periods of intermittent growth spurts followed by relative quiescence.21,135 What may account for the nonuniform progression of these atherosclerotic lesions? One prevailing theory suggests that most plaque disruptions with in situ thrombus formation do not always proceed to total occlusion and clinical sequelae but instead are clinically silent (Fig. 5-4). Though unnoticed by the patient or clinician, they are far from benign. The local nonocclusive platelet thrombus induces a healing response with local inflammatory cytokine and growth factor production. TGF-β and PDGF stimulate SMC collagen production and migration. Thrombin production accelerates SMC migration and proliferation, and fibrin stimulates wound contraction and progressive luminal narrowing.136,137 Healed fibrous cap ruptures can be detected microscopically by the identification of breaks in the fibrous cap with a surrounding repair reaction consisting of a proteoglycan-rich mass or collagen-rich scar.138 Thus in contrast to slow steady plaque growth in which compensatory enlargement of the vessel may protect from luminal encroachment, acute plaque rupture with nonocclusive thrombosis may signal a cascade of events leading to a fibrous atheroma and constrictive remodeling. Whether this mechanism is operative in peripheral vessels (e.g., femoropopliteal arteries) remains to be determined.
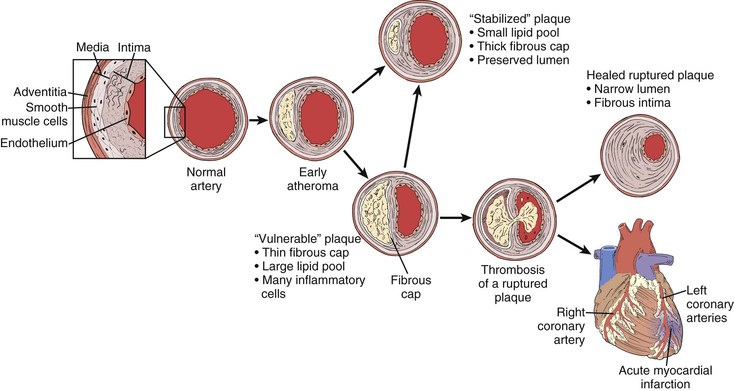
Figure 5-4 Life cycle of human atherosclerotic plaque. Rather than relentless progressive enlargement, human atherosclerotic plaque may undergo periods of progression, regression, and growth spurts. As the plaque extrudes into the lumen, the vessel undergoes compensatory enlargement, which preserves the lumen. The so-called vulnerable plaque consists of a relatively large lipid core and a thin (<100 mm) fibrous cap. Thrombotic complications can occur as a result of cap rupture, superficial endothelial erosion, intraplaque hemorrhage, or erosion of a calcified nodule in which circulating blood elements come in contact with the thrombogenic lipid core. The fate of such an event may be manifested as a myocardial infarction, transient ischemic attack, or in situ thrombosis of a lower extremity artery. Far more commonly, however, they result in a nonocclusive thrombus inciting a healing response. These healed ruptures result in a fibrous plaque with a narrowed lumen.
Angiographic studies are limited to imaging silhouettes of the vessel lumen and vastly underestimate the true burden of atherosclerosis and its diffuse nature.139 The use of cross-sectional images by IVUS reveals that segments of arteries that appear normal by angiography may nonetheless harbor substantial atherosclerotic disease.140 Through morphometric studies of nonhuman primates and detailed autopsy studies of human coronary arteries, we understand that vessels undergo compensatory enlargement (remodeling) and recognize that lumen encroachment is a relatively late occurrence in the evolution of atherosclerotic plaque.141,142 IVUS studies have demonstrated that this phenomenon is not limited to the coronary arteries but has also been documented in the femoral and carotid territories, thus indicating that this is a universal response to atherosclerosis. Studies of human femoral arteries have demonstrated that progression of atherosclerosis involves changes in plaque and vessel volume that result in a change in lumen volume.143
More recently, IVUS has been used to document plaque progression/regression in human coronary arteries with intensive statin treatment.144,145 The Reversal of Atherosclerosis with Aggressive Lipid Lowering (REVERSAL) trial measured the rate of disease progression in patients treated with either 40 mg of pravastatin or 80 mg of atorvastatin. LDL was reduced to 79 mg/dL (2.05 mmol/L) in the atorvastatin arm versus 110 mg/dL (2.85 mmol/L) in the pravastatin arm, and CRP was reduced 36.4% versus 5.2%. This resulted in a significant change in the primary endpoint of total atheroma progression of −0.4% in the atorvastatin arm versus 2.7% in the pravastatin arm.145 In an open-label trial known as A Study to Evaluate the Effect of Rosuvastatin on Intravascular Ultrasound–Derived Coronary Atheroma Burden (ASTEROID), 80 mg of rosuvastatin resulted in a change in atheroma volume of −0.98% over a 24-month treatment period. Collectively, these trials suggest that atherosclerotic plaque, far from being fixed, is a dynamic and mutable lesion that may regress in response to intensive hypolipidemic therapy. These trials also reflect the clinical benefit seen in the intensive treatment arms in trials such as PROVE IT-TIMI 22 and provide further rationale for recommendations of reducing LDL cholesterol below 100 mg/dL (2.59 mmol/L).146 Further effort is needed to determine whether intensive statin therapy can reduce the progression of atherosclerotic disease in other vascular territories, such as the femoral arteries.
Thrombotic Complications of Atherosclerosis
Much of the practice in vascular surgery is driven by the detection of flow-limiting stenosis and applying percutaneous or surgical therapies to restore flow and alleviate the resultant downstream ischemia. However, a significant amount of resources are expended in caring for the acute thrombotic events complicating atherosclerosis, including not only MI but also stroke and in situ thrombosis of the peripheral circulation. These catastrophic clinical syndromes often occur suddenly and without warning. Recent work, primarily in the coronary circulation, has substantially improved our understanding of these events. Less is known about the pathogenesis of thrombotic occlusions in the periphery, but in the carotid arteries, intraplaque hemorrhage is commonly seen in excised endarterectomy specimens from symptomatic patients.147
In the coronary circulation, various lines of evidence reveal that generally, the culprit lesions are not necessarily those associated with the tightest stenosis.148–150 This is not to say that high-grade stenosis does not undergo thrombotic complications; on a per-lesion basis, the individual probability of a high-grade lesion is higher than the probability of a less severe lesion.151 However, because noncritical stenoses outnumber lesions producing critical stenosis, the total probability of a thrombotic complication attributable to the noncritical stenosis is higher. It must also be remembered that these assessments are based on standard planar angiography and do not take into account compensatory enlargement. Thus low-grade stenosis does not necessarily equate with smaller lesions. Other variables such as inflammation account for plaque rupture.
It is thought that physical disruption of the atherosclerotic plaque rather than critical stenosis can commonly precipitate arterial thrombosis. Four mechanisms of plaque disruption may cause thrombosis or rapid plaque expansion.27 Each may be operative in different vascular territories or with a different constellation of risk factors. For example, complete fracture of the plaque’s fibrous cap causes most cases of fatal coronary thrombosis.152 Plaque rupture is probably caused by both mechanical and biologic factors.15,28 Physical forces acting on the shoulder region of the plaque, where circumferential wall stress and cap fatigue are the greatest, render this area particularly vulnerable, especially when the central lipid core accounts for greater than 40% of the total lesion area and the cap is relatively thin, less than 100 µm.153 Inflammation within the plaque may destabilize the cap and potentiate injury by hemodynamic factors. Macrophages and mast cells degrade extracellular matrix by phagocytosis and secretion of proteolytic enzymes.154 SMCs and macrophages, when stimulated by inflammatory cytokines, produce matrix metalloproteinases (MMPs), including collagenases, elastases, gelatinases, and stromelysins, that degrade the matrix of the fibrous cap and result in thinning and weakening.155–160 It is now apparent that endothelial cells, SMCs, and macrophages all increase the expression of CD40 ligand and its receptor under the direction of the inflammatory cytokines. Ligation of CD40 in turn upregulates the production of MMPs, inflammatory cytokines, and tissue factor, thus emphasizing the autocrine and paracrine nature of the local inflammatory response of the fibrous cap.157–160
Once the plaque is fractured, blood is exposed to the underlying thrombogenic substrate and thrombosis ensues. Tissue factor initiates the extrinsic clotting cascade and is a major regulator of coagulation and thrombosis. Thrombus formation and platelet adhesion create further stenosis and thrombotic occlusion. As previously mentioned, most thrombotic complications of plaque lead to progression of stenosis rather than occlusion of the artery. It is likely that cardiovascular risk factors serve individually or collectively to increase the systemic procoagulant activity of blood and affect the outcome. Cigarette smoking, hyperglycemia, and elevated LDL cholesterol all increase blood thrombogenicity.161 These same risk factors are characterized by endothelial abnormalities such as increased generation of superoxide anion and decreased endothelium-derived NO.7,162–164 Thus risk factors are linked to progression of atherosclerosis and to its thrombotic complications at susceptible areas in the vascular tree through abnormalities in blood coagulation, as well as endothelial function.
Whereas complete fracture of the fibrous cap may be the most common cause underlying coronary thrombosis, other mechanisms may be more important in the periphery. Intraplaque hemorrhage can transform an asymptomatic carotid plaque into a symptomatic lesion and produce transient ischemic attacks or stroke. Neovascularization and proliferation of the vasa vasorum, as in diabetic retinopathy, may produce a local microvascular network that is fragile and friable. Intraplaque hemorrhage can cause rapid plaque expansion and thinning or disruption of the cap. Superficial plaque erosion producing in situ thrombus without plaque rupture is prevalent in patients with diabetes and in women.27,165 Apoptosis of endothelial cells may promote superficial erosion, and various inflammatory stimuli may promote apoptosis.166 In particular, macrophage-derived myeloperoxidase may be operative in that it promotes both tissue factor generation and endothelial cell apoptosis, thus linking superficial erosion and in situ thrombosis.167 Finally, erosion through the intima of a calcified nodule represents another less common form of atherosclerotic thrombosis that may be relevant in both the coronary and peripheral circulations.
Distinct artery-dependent patterns of atherosclerosis probably account for differences in the pathogenesis of thrombotic-related complications.168 Both coronary and carotid atherosclerotic plaques appear to be laden with foam cells and to have large lipid cores; plaque rupture or intraplaque hemorrhage is common. In the femoral artery, plaque is more commonly fibrous without extensive foam cells. Here, it is likely that superficial plaque erosion and erosion of a calcific nodule more commonly lead to thrombosis.
Identification of Vulnerable Lesions
How does one predict a priori which patients may be more likely to progress from an asymptomatic carotid lesion to one with symptoms or who may develop an MI following noncardiac vascular surgery or the progression of a Hunter’s canal stenosis? After all, many patients do perfectly fine with stable 60% carotid bifurcation disease or stable claudication. Understanding of the in vivo structural, functional, and biologic aspects of an atherosclerotic lesion may identify plaques that are “vulnerable” to either progression or symptomatic transformation.
Identification of such vulnerable lesions has been an area of intense recent work. As discussed previously, IVUS evaluation determines the true extent of the size of the atherosclerotic plaque as opposed to what is visible on angiography, the latter of which dramatically underestimates the true dimensions of the lesion. IVUS can detect the degree to which an artery has remodeled in response to a plaque. For instance, it has been determined that excessive expansive remodeling may result in a thin vulnerable plaque.169
Impaired endothelial vasodilator function is seen in patients with cardiovascular risk factors, and cardiovascular risk factors predict CHD and stroke. Brachial artery flow mediated vasodilation has been used as a surrogate for the more relevant coronary circulation.170,171 Impaired flow mediated vasodilation is seen in patients with inflammation, as well as in those with classic risk factors,172,173 and it is often present before symptoms of peripheral or coronary arterial disease are evident.6 Impaired flow mediated vasodilation has been demonstrated to predict adverse events in patients undergoing vascular surgery.174–176 Hence the classic Framingham risk factors, as well as inflammation, appear to impair endothelial function and promote atherosclerosis.
Finally, molecular imaging is on the immediate horizon to help identify vulnerable plaque. For example, plaque with active inflammation may be identified by extensive accumulation of macrophages. Successful detection of plaque inflammation with magnetic resonance imaging was possible with gadolinium-loaded micelles coupled with antibodies to the scavenger receptor.177 Alternatively, computed tomography may be used with a novel iodinated nanoparticulate contrast agent to detect inflammation in atherosclerotic plaque.178 Contrast-enhanced ultrasound may also be used by attaching VCAM-1 to microbubbles and identifying areas of active adhesion molecule expression.179
Selected Key References
Collins T, Cybulsky MI. NF-κB: pivotal mediator or innocent bystander in atherogenesis? J Clin Invest. 2001;107:255–264.
A comprehensive review of the cellular molecular signals involved in inflammation..
Cook NR, Buring JE, Ridker PM. The effect of including C-reactive protein in cardiovascular risk prediction models for women. Ann Intern Med. 2006;145:21–29.
Demonstrates that the plasma concentration of high-sensitivity C-reactive protein adds predictive value beyond traditional Framingham risk factors to the 10-year cardiovascular risk in women. The risk can be calculated at www.reynoldsriskscore.org..
DePalma RG, Hubay CA, Insull W Jr, Robinson AV, Hartman PH. Progression and regression of experimental atherosclerosis. Surg Gynecol Obstet. 1970;131:633–647.
Duewell P, Kono H, Rayner KJ, Sirois CM, Vladimer G, Bauernfeind FG, Abela GS, Franchi L, Nuñez G, Schnurr M, Espevik T, Lien E, Fitzgerald KA, Rock KL, Moore KJ, Wright SD, Hornung V, Latz E. NLRP3 Inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature. 2010;464:1357–1361.
Provides important mechanistic insight to the link between cholesterol and inflammation..
Glagov S, Weisenberg E, Zarins CK, Stankunavicius R, Kolettis GJ. Compensatory enlargement of human atherosclerotic coronary arteries. N Engl J Med. 1987;316:1371–1375.
One of the most frequently cited papers related to arterial remodeling..
Goldstein JL, Brown MS. Familial hypercholesterolemia: identification of a defect in the regulation of 3-hydroxy-3-methylglutaryl coenzyme A reductase activity associated with overproduction of cholesterol. Proc Natl Acad Sci U S A. 1973;70:2804–2808.
Ross R. Atherosclerosis—an inflammatory disease. N Engl J Med. 1999;340:115–126.
Steinberg D. Thematic review series: the pathogenesis of atherosclerosis. An interpretive history of the cholesterol controversy: Part I. J Lipid Res. 2004;45:1583–1593.
The reference list can be found on the companion Expert Consult website at www.expertconsult.com.
References
1. Bonow RO, et al. World Heart Day 2002: The international burden of cardiovascular disease: responding to the emerging global epidemic. Circulation. 2002;106:1602–1605.
2. Ford ES, et al. Explaining the decrease in U.S. deaths from coronary disease, 1980-2000. N Engl J Med. 2007;356:2388–2398.
3. Rosamond W, et al. Heart disease and stroke statistics—2008 update: a report from the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation. 2008;117:e25–e146.
4. McGill HC Jr, et al. Preventing heart disease in the 21st century: implications of the Pathobiological Determinants of Atherosclerosis in Youth (PDAY) study. Circulation. 2008;117:1216–1227.
5. Hayman LL, et al. Primary prevention of cardiovascular disease in nursing practice: focus on children and youth: a scientific statement from the American Heart Association Committee on Atherosclerosis, Hypertension, and Obesity in Youth of the Council on Cardiovascular Disease in the Young, Council on Cardiovascular Nursing, Council on Epidemiology and Prevention, and Council on Nutrition, Physical Activity, and Metabolism. Circulation. 2007;116:344–357.
6. Lieberman EH, et al. Flow-induced vasodilation of the human brachial artery is impaired in patients <40 years of age with coronary artery disease. Am J Cardiol. 1996;78:1210–1214.
7. Celermajer DS, et al. Non-invasive detection of endothelial dysfunction in children and adults at risk of atherosclerosis. Lancet. 1992;340:1111–1115.
8. Joannides R, et al. Nitric oxide is responsible for flow-dependent dilatation of human peripheral conduit arteries in vivo. Circulation. 1995;91:1314–1319.
9. Camejo G, et al. Association of apo B lipoproteins with arterial proteoglycans: pathological significance and molecular basis. Atherosclerosis. 1998;139:205–222.
10. Camejo G, et al. Binding of low density lipoproteins by proteoglycans synthesized by proliferating and quiescent human arterial smooth muscle cells. J Biol Chem. 1993;268:14131–14137.
11. Pentikainen MO, et al. The proteoglycan decorin links low density lipoproteins with collagen type I. J Biol Chem. 1997;272:7633–7638.
12. Doran AC, et al. Role of smooth muscle cells in the initiation and early progression of atherosclerosis. Arterioscler Thromb Vasc Biol. 2008;28:812–819.
13. Owens GK, et al. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol Rev. 2004;84:767–801.
14. Ang AH, et al. Collagen synthesis by cultured rabbit aortic smooth-muscle cells. Alteration with phenotype. Biochem J. 1990;265:461–469.
15. Fuster V, et al. Atherothrombosis and high-risk plaque: Part I: evolving concepts. J Am Coll Cardiol. 2005;46:937–954.
16. Stary HC, et al. A definition of advanced types of atherosclerotic lesions and a histological classification of atherosclerosis. A report from the Committee on Vascular Lesions of the Council on Arteriosclerosis, American Heart Association. Arterioscler Thromb Vasc Biol. 1995;15:1512–1531.
17. Stary HC, et al. A definition of initial, fatty streak, and intermediate lesions of atherosclerosis. A report from the Committee on Vascular Lesions of the Council on Arteriosclerosis, American Heart Association. Circulation. 1994;89:2462–2478.
18. Faggiotto A, et al. Studies of hypercholesterolemia in the nonhuman primate. II. Fatty streak conversion to fibrous plaque. Arteriosclerosis. 1984;4:341–356.
19. Faggiotto A, et al. Studies of hypercholesterolemia in the nonhuman primate. I. Changes that lead to fatty streak formation. Arteriosclerosis. 1984;4:423–440.
20. Bruschke AV, et al. The dynamics of progression of coronary atherosclerosis studied in 168 medically treated patients who underwent coronary arteriography three times. Am Heart J. 1989;117:296–305.
21. Yokoya K, et al. Process of progression of coronary artery lesions from mild or moderate stenosis to moderate or severe stenosis: a study based on four serial coronary arteriograms per year. Circulation. 1999;100:903–909.
22. Newman WP 3rd, et al. Relation of serum lipoprotein levels and systolic blood pressure to early atherosclerosis. The Bogalusa Heart Study. N Engl J Med. 1986;314:138–144.
23. Berenson GS, et al. Association between multiple cardiovascular risk factors and atherosclerosis in children and young adults. The Bogalusa Heart Study. N Engl J Med. 1998;338:1650–1656.
24. McGill HC Jr, et al. Effects of nonlipid risk factors on atherosclerosis in youth with a favorable lipoprotein profile. Circulation. 2001;103:1546–1550.
25. Tuzcu EM, et al. High prevalence of coronary atherosclerosis in asymptomatic teenagers and young adults: evidence from intravascular ultrasound. Circulation. 2001;103:2705–2710.
26. Nakashima Y, et al. Distributions of diffuse intimal thickening in human arteries: preferential expression in atherosclerosis-prone arteries from an early age. Virchows Arch. 2002;441:279–288.
27. Libby P, et al. Pathophysiology of coronary artery disease. Circulation. 2005;111:3481–3488.
28. van der Wal AC, et al. Site of intimal rupture or erosion of thrombosed coronary atherosclerotic plaques is characterized by an inflammatory process irrespective of the dominant plaque morphology. Circulation. 1994;89:36–44.
29. Hong MK, et al. A three-vessel virtual histology intravascular ultrasound analysis of frequency and distribution of thin-cap fibroatheromas in patients with acute coronary syndrome or stable angina pectoris. Am J Cardiol. 2008;101:568–572.
30. Brown MS, et al. A receptor-mediated pathway for cholesterol homeostasis. Science. 1986;232:34–47.
31. Steinberg D. Thematic review series: the pathogenesis of atherosclerosis. An interpretive history of the cholesterol controversy: Part I. J Lipid Res. 2004;45:1583–1593.
32. Steinberg D. Thematic review series: the pathogenesis of atherosclerosis. An interpretive history of the cholesterol controversy: Part II: the early evidence linking hypercholesterolemia to coronary disease in humans. J Lipid Res. 2005;46:179–190.
33. Gofman JW, et al. Ultracentrifugal studies of lipoproteins of human serum. J Biol Chem. 1949;179:973–979.
34. Gofman JW, et al. Atherosclerosis, lipoproteins, and coronary artery disease. Wisc Med J. 1952;51:687–689.
35. Goldstein JL, et al. Familial hypercholesterolemia: identification of a defect in the regulation of 3-hydroxy-3-methylglutaryl coenzyme A reductase activity associated with overproduction of cholesterol. Proc Natl Acad Sci U S A. 1973;70:2804–2808.
36. Brown MS, et al. Regulation of 3-hydroxy-3-methylglutaryl coenzyme A reductase activity in cultured human fibroblasts. Comparison of cells from a normal subject and from a patient with homozygous familial hypercholesterolemia. J Biol Chem. 1974;249:789–796.
37. Brown MS, et al. Familial hypercholesterolemia: defective binding of lipoproteins to cultured fibroblasts associated with impaired regulation of 3-hydroxy-3-methylglutaryl coenzyme A reductase activity. Proc Natl Acad Sci U S A. 1974;71:788–792.
38. Robertson TL, et al. Epidemiologic studies of coronary heart disease and stroke in Japanese men living in Japan, Hawaii, and California. Coronary heart disease risk factors in Japan and Hawaii. Am J Cardiol. 1977;39:244–249.
39. Robertson TL, et al. Epidemiologic studies of coronary heart disease and stroke in Japanese men living in Japan, Hawaii, and California. Incidence of myocardial infarction and death from coronary heart disease. Am J Cardiol. 1977;39:239–243.
40. Kannel WB, et al. Factors of risk in the development of coronary heart disease—six-year follow-up experience. The Framingham Study. Ann Intern Med. 1961;55:33–50.
41. Dayton S, et al. Controlled trial of a diet high in unsaturated fat for prevention of atherosclerotic complications. Lancet. 1968;2:1060–1062.
42. The Lipid Research Clinics Coronary Primary Prevention Trial results. II. The relationship of reduction in incidence of coronary heart disease to cholesterol lowering. JAMA. 1984;251:365–374.
44. Scandinavian Simvastatin Survival Study Group. Randomised trial of cholesterol lowering in 4444 patients with coronary heart disease: the Scandinavian Simvastatin Survival Study (4S). Lancet. 1994;344:1383–1389.
45. Duguid JB. Pathogenesis of atherosclerosis. Lancet. 1949;2:925–927.
46. Poole JC, et al. Changes in the endothelium of the aorta and the behaviour of macrophages in experimental atheroma of rabbits. J Pathol Bacteriol. 1958;75:245–251.
47. French JE. Atherosclerosis in relation to the structure and function of the arterial intima, with special reference to the endothelium. Int Rev Exp Pathol. 1966;5:253–353.
48. Ross R, et al. Atherosclerosis and the arterial smooth muscle cell: proliferation of smooth muscle is a key event in the genesis of the lesions of atherosclerosis. Science. 1973;180:1332–1339.
49. Ross R. The pathogenesis of atherosclerosis: a perspective for the 1990s. Nature. 1993;362:801–809.
50. Ross R, et al. The pathogenesis of atherosclerosis (second of two parts). N Engl J Med. 1976;295:420–425.
51. Ross R, et al. The pathogenesis of atherosclerosis (first of two parts). N Engl J Med. 1976;295:369–377.
52. Ross R. The pathogenesis of atherosclerosis—an update. N Engl J Med. 1986;314:488–500.
53. Ross R. Atherosclerosis—an inflammatory disease. N Engl J Med. 1999;340:115–126.
54. Ross R, et al. A platelet-dependent serum factor that stimulates the proliferation of arterial smooth muscle cells in vitro. Proc Natl Acad Sci U S A. 1974;71:1207–1210.
55. Ross R, et al. The biology of platelet-derived growth factor. Cell. 1986;46:155–169.
56. Nievelstein PF, et al. Lipid accumulation in rabbit aortic intima 2 hours after bolus infusion of low density lipoprotein. A deep-etch and immunolocalization study of ultrarapidly frozen tissue. Arterioscler Thromb. 1991;11:1795–1805.
57. Schwenke DC, et al. Initiation of atherosclerotic lesions in cholesterol-fed rabbits. II. Selective retention of LDL vs. selective increases in LDL permeability in susceptible sites of arteries. Arteriosclerosis. 1989;9:908–918.
58. Schwenke DC, et al. Initiation of atherosclerotic lesions in cholesterol-fed rabbits. I. Focal increases in arterial LDL concentration precede development of fatty streak lesions. Arteriosclerosis. 1989;9:895–907.
59. Tabas I, et al. Subendothelial lipoprotein retention as the initiating process in atherosclerosis: update and therapeutic implications. Circulation. 2007;116:1832–1844.
60. Williams KJ, et al. The response-to-retention hypothesis of early atherogenesis. Arterioscler Thromb Vasc Biol. 1995;15:551–561.
61. Benditt EP, et al. Evidence for a monoclonal origin of human atherosclerotic plaques. Proc Natl Acad Sci U S A. 1973;70:1753–1756.
62. Linder D, et al. Glucose-6-phosphate dehydrogenase mosaicism: utilization as a cell marker in the study of leiomyomas. Science. 1965;150:67–69.
63. Fialkow PJ. Use of genetic markers to study cellular origin and development of tumors in human females. Adv Cancer Res. 1972;15:191–226.
64. Fialkow PJ. The origin and development of human tumors studied with cell markers. N Engl J Med. 1974;291:26–35.
65. Stary HC. Changes in components and structure of atherosclerotic lesions developing from childhood to middle age in coronary arteries. Basic Res Cardiol. 1994;89(Suppl 1):17–32.
66. Stary HC, et al. A definition of initial, fatty streak, and intermediate lesions of atherosclerosis. A report from the Committee on Vascular Lesions of the Council on Arteriosclerosis, American Heart Association. Arterioscler Thromb. 1994;14:840–856.
67. Taylor KE, et al. Preservation and structural adaptation of endothelium over experimental foam cell lesions. Quantitative ultrastructural study. Arteriosclerosis. 1989;9:881–894.
68. Plump AS, et al. Severe hypercholesterolemia and atherosclerosis in apolipoprotein E-deficient mice created by homologous recombination in ES cells. Cell. 1992;71(2):343–353.
69. Ishibashi S, et al. Hypercholesterolemia in low density lipoprotein receptor knockout mice and its reversal by adenovirus-mediated gene delivery. J Clin Invest. 1993;92(2):883–893.
70. Ishibashi S, et al. The two-receptor model of lipoprotein clearance: tests of the hypothesis in “knockout” mice lacking the low density lipoprotein receptor, apolipoprotein E, or both proteins. Proc Natl Acad Sci U S A. 1994;91(10):4431–4435.
71. Raffai RL. Apolipoprotein E regulation of myeloid cell plasticity in atherosclerosis. Curr Opin Lipidol. 2012;23(5):471–478.
72. Dansky HM, et al. T and B lymphocytes play a minor role in atherosclerotic plaque formation in the apolipoprotein E-deficient mouse. Proc Natl Acad Sci U S A. 1997;94(9):4642–4646.
73. Skalen K, et al. Subendothelial retention of atherogenic lipoproteins in early atherosclerosis. Nature. 2002;417:750–754.
74. Yla-Herttuala S, et al. Evidence for the presence of oxidatively modified low density lipoprotein in atherosclerotic lesions of rabbit and man. J Clin Invest. 1989;84:1086–1095.
75. Gerrity RG. The role of the monocyte in atherogenesis: II. Migration of foam cells from atherosclerotic lesions. Am J Pathol. 1981;103:191–200.
76. Gerrity RG. The role of the monocyte in atherogenesis: I. Transition of blood-borne monocytes into foam cells in fatty lesions. Am J Pathol. 1981;103:181–190.
77. Burger PC, et al. Platelet P-selectin facilitates atherosclerotic lesion development. Blood. 2003;101:2661–2666.
78. Cybulsky MI, et al. Endothelial expression of a mononuclear leukocyte adhesion molecule during atherogenesis. Science. 1991;251:788–791.
79. Dong ZM, et al. The combined role of P- and E-selectins in atherosclerosis. J Clin Invest. 1998;102(1):145–152.
80. Bourdillon MC, et al. ICAM-1 deficiency reduces atherosclerotic lesions in double-knockout mice (ApoE(-/-)/ICAM-1(-/-)) fed a fat or a chow diet. Arterioscler Thromb Vasc Biol. 2000;20(12):2630–2635.
81. Dansky HM, et al. Adhesion of monocytes to arterial endothelium and initiation of atherosclerosis are critically dependent on vascular cell adhesion molecule-1 gene dosage. Arterioscler Thromb Vasc Biol. 2001;21(10):1662–1667.
82. Gu L, et al. Absence of monocyte chemoattractant protein-1 reduces atherosclerosis in low density lipoprotein receptor–deficient mice. Mol Cell. 1998;2:275–281.
83. Holvoet P, et al. Oxidized low-density lipoprotein correlates positively with toll-like receptor 2 and interferon regulatory factor-1 and inversely with superoxide dismutase-1 expression: studies in hypercholesterolemic swine and THP-1 cells. Arterioscler Thromb Vasc Biol. 2006;26:1558–1565.
84. Quehenberger O. Thematic review series: the immune system and atherogenesis. Molecular mechanisms regulating monocyte recruitment in atherosclerosis. J Lipid Res. 2005;46:1582–1590.
85. Endemann G, et al. CD36 is a receptor for oxidized low density lipoprotein. J Biol Chem. 1993;268:11811–11816.
86. Smith JD, et al. Decreased atherosclerosis in mice deficient in both macrophage colony-stimulating factor (op) and apolipoprotein E. Proc Natl Acad Sci U S A. 1995;92:8264–8268.
87. Rajavashisth T, et al. Heterozygous osteopetrotic (op) mutation reduces atherosclerosis in LDL receptor–deficient mice. J Clin Invest. 1998;101:2702–2710.
88. Okamura H, et al. Cloning of a new cytokine that induces IFN-gamma production by T cells. Nature. 1995;378(6552):88–91.
89. Ushio S, et al. Cloning of the cDNA for human IFN-gamma-inducing factor, expression in Escherichia coli, and studies on the biologic activities of the protein. J Immunol. 1996;156(11):4274–4279.
90. Fukumoto Y, et al. Genetically determined resistance to collagenase action augments interstitial collagen accumulation in atherosclerotic plaques. Circulation. 2004;110(14):1953–1959.
92. Jonasson L, et al. Expression of class II transplantation antigen on vascular smooth muscle cells in human atherosclerosis. J Clin Invest. 1985;76(1):125–131.
93. Zhou X, et al. Adoptive transfer of CD4+ T cells reactive to modified low-density lipoprotein aggravates atherosclerosis. Arterioscler Thromb Vasc Biol. 2006;26(4):864–870.
94. Palinski W, et al. Low density lipoprotein undergoes oxidative modification in vivo. Proc Natl Acad Sci U S A. 1989;86(4):1372–1376.
95. Tupin E, et al. CD1d-dependent activation of NKT cells aggravates atherosclerosis. J Exp Med. 2004;199(3):417–422.
96. Ait-Oufella H, et al. Natural regulatory T cells control the development of atherosclerosis in mice. Nat Med. 2006;12(2):178–180.
97. Kovanen PT, et al. Infiltrates of activated mast cells at the site of coronary atheromatous erosion or rupture in myocardial infarction. Circulation. 1995;92:1084–1088.
98. Sun J, et al. Mast cells promote atherosclerosis by releasing proinflammatory cytokines. Nat Med. 2007;13:719–724.
99. Schonbeck U, et al. CD40 signaling and plaque instability. Circ Res. 2001;89:1092–1103.
100. Geng YJ, et al. Progression of atheroma: a struggle between death and procreation. Arterioscler Thromb Vasc Biol. 2002;22:1370–1380.
101. Johnson KA, et al. Transglutaminase 2 is central to induction of the arterial calcification program by smooth muscle cells. Circ Res. 2008;102:529–537.
102. Duewell P, et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature. 2010;464(7293):1357–1361.
103. Rajamaki K, et al. Cholesterol crystals activate the NLRP3 inflammasome in human macrophages: a novel link between cholesterol metabolism and inflammation. PLoS One. 2010;5(7):e11765.
104. Martinon F, et al. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell. 2002;10(2):417–426.
105. Gage J, et al. Caspase-1 deficiency decreases atherosclerosis in apolipoprotein E-null mice. Can J Cardiol. 2012;28(2):222–229.
106. Pradhan AD, et al. Symptomatic peripheral arterial disease in women: nontraditional biomarkers of elevated risk. Circulation. 2008;117(6):823–831.
107. McDermott MM, et al. Patterns of inflammation associated with peripheral arterial disease: the InCHIANTI study. Am Heart J. 2005;150(2):276–281.
108. Tzoulaki I, et al. C-reactive protein, interleukin-6, and soluble adhesion molecules as predictors of progressive peripheral atherosclerosis in the general population: Edinburgh Artery Study. Circulation. 2005;112(7):976–983.
109. Kaptoge S, et al. C-reactive protein concentration and risk of coronary heart disease, stroke, and mortality: an individual participant meta-analysis. Lancet. 2010;375(9709):132–140.
110. Ridker PM. C-reactive protein and the prediction of cardiovascular events among those at intermediate risk: moving an inflammatory hypothesis toward consensus. J Am Coll Cardiol. 2007;49(21):2129–2138.
111. Ridker PM, et al. Clinical usefulness of very high and very low levels of C-reactive protein across the full range of Framingham Risk Scores. Circulation. 2004;109(16):1955–1959.
112. Ridker PM, et al. Comparison of C-reactive protein and low-density lipoprotein cholesterol levels in the prediction of first cardiovascular events. N Engl J Med. 2002;347:1557–1565.
113. Ridker PM, et al. C-reactive protein and other markers of inflammation in the prediction of cardiovascular disease in women. N Engl J Med. 2000;342:836–843.
114. Cook NR, et al. The effect of including C-reactive protein in cardiovascular risk prediction models for women. Ann Intern Med. 2006;45:21–29.
115. Boekholdt SM, et al. C-reactive protein levels and coronary artery disease incidence and mortality in apparently healthy men and women: the EPIC-Norfolk prospective population study 1993-2003. Atherosclerosis. 2006;187:415–422.
116. Ridker PM, et al. C-reactive protein levels and outcomes after statin therapy. N Engl J Med. 2005;352:20–28.
117. Morrow DA, et al. Clinical relevance of C-reactive protein during follow-up of patients with acute coronary syndromes in the Aggrastat-to-Zocor Trial. Circulation. 2006;114:281–288.
118. Ridker PM, et al. Rosuvastatin to prevent vascular events in men and women with elevated C-reactive protein. N Engl J Med. 2008;359(21):2195–2207.
119. Amarenco P, et al. Results of the Stroke Prevention by Aggressive Reduction in Cholesterol Levels (SPARCL) trial by stroke subtypes. Stroke. 2009;40(4):1405–1409.
120. Amarenco P, et al. High-dose atorvastatin after stroke or transient ischemic attack. N Engl J Med. 2006;355(6):549–559.
121. Kennedy J, et al. Statins are associated with better outcomes after carotid endarterectomy in symptomatic patients. Stroke. 2005;36(10):2072–2076.
122. Owens CD. Statins and other agents for vascular inflammation. J Vasc Surg. 2012;56(6):1799–1806.
123. Ridker PM. Moving beyond JUPITER: Will inhibiting inflammation reduce vascular event rates? Curr Atheroscler Rep. 2013;15(1):295.
124. Schwartz SM, et al. Cell replication in the aortic endothelium: a new method for study of the problem. Lab Invest. 1973;28:699–707.
125. Wright HP. Mitosis patterns in aortic endothelium. Atherosclerosis. 1972;15:93–100.
126. Spaet TH, et al. Mitotic activity of rabbit blood vessels. Proc Soc Exp Biol Med. 1967;125:1197–1201.
127. Insull W Jr, et al. Cholesterol, triglyceride, and phospholipid content of intima, media, and atherosclerotic fatty streak in human thoracic aorta. J Clin Invest. 1966;45:513–523.
128. Collins T, et al. NF-κB: Pivotal mediator or innocent bystander in atherogenesis? J Clin Invest. 2001;107:255–264.
129. Guttridge DC, et al. NF-κB controls cell growth and differentiation through transcriptional regulation of cyclin D1. Mol Cell Biol. 1999;19:5785–5799.
130. Akimoto S, et al. Laminar shear stress inhibits vascular endothelial cell proliferation by inducing cyclin-dependent kinase inhibitor p21 (Sdi1/Cip1/Waf1). Circ Res. 2000;86:185–190.
131. DePalma RG, et al. Progression and regression of experimental atherosclerosis. Surg Gynecol Obstet. 1970;131:633–647.
132. DePalma RG, et al. Animal models for the study of progression and regression of atherosclerosis. Surgery. 1972;72:268–278.
133. DePalma RG, et al. Atherosclerotic plaque regression in rhesus monkeys induced by bile acid sequestrant. Exp Mol Pathol. 1979;31:423–439.
134. DePalma RG, et al. Regression of atherosclerotic plaques in rhesus monkeys. Angiographic, morphologic, and angiochemical changes. Arch Surg. 1980;115:1268–1278.
135. Lichtlen PR, et al. Anatomical progression of coronary artery disease in humans as seen by prospective, repeated, quantitated coronary angiography. Relation to clinical events and risk factors. The INTACT Study Group. Circulation. 1992;86:828–838.
136. Asada Y, et al. Fibrin-rich and platelet-rich thrombus formation on neointima: recombinant tissue factor pathway inhibitor prevents fibrin formation and neointimal development following repeated balloon injury of rabbit aorta. Thromb Haemost. 1998;80:506–511.
137. Courtman DW, et al. Sequential injury of the rabbit abdominal aorta induces intramural coagulation and luminal narrowing independent of intimal mass: extrinsic pathway inhibition eliminates luminal narrowing. Circ Res. 1998;82:996–1006.
138. Burke AP, et al. Healed plaque ruptures and sudden coronary death: evidence that subclinical rupture has a role in plaque progression. Circulation. 2001;103:934–940.
140. Mintz GS, et al. American College of Cardiology Clinical Expert Consensus Document on Standards for Acquisition, Measurement and Reporting of Intravascular Ultrasound Studies (IVUS). A report of the American College of Cardiology Task Force on Clinical Expert Consensus Documents. J Am Coll Cardiol. 2001;37:1478–1492.
141. Clarkson TB, et al. Remodeling of coronary arteries in human and nonhuman primates. JAMA. 1994;271:289–294.
142. Glagov S, et al. Compensatory enlargement of human atherosclerotic coronary arteries. N Engl J Med. 1987;316:1371–1375.
143. Hagenaars T, et al. Progression of atherosclerosis at one-year follow-up seen with volumetric intravascular ultrasound in femoropopliteal arteries. Am J Cardiol. 2000;85:226–231.
144. Nissen SE, et al. Effect of very high-intensity statin therapy on regression of coronary atherosclerosis: the ASTEROID trial. JAMA. 2006;295:1556–1565.
145. Nissen SE, et al. Effect of intensive compared with moderate lipid-lowering therapy on progression of coronary atherosclerosis: a randomized controlled trial. JAMA. 2004;291:1071–1080.
146. Cannon CP, et al. Intensive versus moderate lipid lowering with statins after acute coronary syndromes. N Engl J Med. 2004;350:1495–1504.
147. Montauban van Swijndregt AD, et al. Cerebral ischemic disease and morphometric analyses of carotid plaques. Ann Vasc Surg. 1999;13:468–474.
148. Little WC, et al. Can coronary angiography predict the site of a subsequent myocardial infarction in patients with mild-to-moderate coronary artery disease? Circulation. 1988;78:1157–1166.
149. Fuster V, et al. Insights into the pathogenesis of acute ischemic syndromes. Circulation. 1988;77:1213–1220.
150. Ambrose JA, et al. Angiographic progression of coronary artery disease and the development of myocardial infarction. J Am Coll Cardiol. 1988;12:56–62.
151. Falk E. Plaque rupture with severe pre-existing stenosis precipitating coronary thrombosis. Characteristics of coronary atherosclerotic plaques underlying fatal occlusive thrombi. Br Heart J. 1983;50:127–134.
152. Davies MJ, et al. The pathological basis and microanatomy of occlusive thrombus formation in human coronary arteries. Philos Trans R Soc Lond B Biol Sci. 1981;294:225–229.
153. Maseri A, et al. Is there a vulnerable plaque? Circulation. 2003;107:2068–2071.
154. Kaartinen M, et al. Accumulation of activated mast cells in the shoulder region of human coronary atheroma, the predilection site of atheromatous rupture. Circulation. 1994;90:1669–1678.
155. Shah PK, et al. Matrix metalloproteinase hypothesis of plaque rupture: players keep piling up but questions remain. Circulation. 2001;104:1878–1880.
156. Shah PK, et al. Human monocyte-derived macrophages induce collagen breakdown in fibrous caps of atherosclerotic plaques. Potential role of matrix-degrading metalloproteinases and implications for plaque rupture. Circulation. 1995;92:1565–1569.
157. Mach F, et al. Activation of monocyte/macrophage functions related to acute atheroma complication by ligation of CD40: induction of collagenase, stromelysin, and tissue factor. Circulation. 1997;96:396–399.
158. Mach F, et al. Functional CD40 ligand is expressed on human vascular endothelial cells, smooth muscle cells, and macrophages: implications for CD40-CD40 ligand signaling in atherosclerosis. Proc Natl Acad Sci U S A. 1997;94:1931–1936.
159. Schonbeck U, et al. Ligation of CD40 activates interleukin 1β-converting enzyme (caspase-1) activity in vascular smooth muscle and endothelial cells and promotes elaboration of active interleukin 1β. J Biol Chem. 1997;272:19569–19574.
160. Schonbeck U, et al. Regulation of matrix metalloproteinase expression in human vascular smooth muscle cells by T lymphocytes: a role for CD40 signaling in plaque rupture? Circ Res. 1997;81:448–454.
161. Edelberg JM, et al. Regulation of vascular bed–specific prothrombotic potential. Circ Res. 2001;89:117–124.
162. Ohara Y, et al. Hypercholesterolemia increases endothelial superoxide anion production. J Clin Invest. 1993;91:2546–2551.
163. Tesfamariam B, et al. Free radicals mediate endothelial cell dysfunction caused by elevated glucose. Am J Physiol. 1992;263:H321–H326.
164. Creager MA, et al. Impaired vasodilation of forearm resistance vessels in hypercholesterolemic humans. J Clin Invest. 1990;86:228–234.
165. Farb A, et al. Coronary plaque erosion without rupture into a lipid core. A frequent cause of coronary thrombosis in sudden coronary death. Circulation. 1996;93:1354–1363.
166. Sugiyama S, et al. Hypochlorous acid, a macrophage product, induces endothelial apoptosis and tissue factor expression: involvement of myeloperoxidase-mediated oxidant in plaque erosion and thrombogenesis. Arterioscler Thromb Vasc Biol. 2004;24:1309–1314.
167. Hazen SL. Myeloperoxidase and plaque vulnerability. Arterioscler Thromb Vasc Biol. 2004;24:1143–1146.
168. Dalager S, et al. Artery-related differences in atherosclerosis expression: implications for atherogenesis and dynamics in intima-media thickness. Stroke. 2007;38:2698–2705.
169. Chatzizisis YS, et al. Prediction of the localization of high-risk coronary atherosclerotic plaques on the basis of low endothelial shear stress: an intravascular ultrasound and histopathology natural history study. Circulation. 2008;117:993–1002.
170. Corretti MC, et al. Guidelines for the ultrasound assessment of endothelial-dependent flow-mediated vasodilation of the brachial artery: a report of the International Brachial Artery Reactivity Task Force. J Am Coll Cardiol. 2002;39:257–265.
171. Fichtlscherer S, et al. Elevated C-reactive protein levels and impaired endothelial vasoreactivity in patients with coronary artery disease. Circulation. 2000;102:1000–1006.
172. Brevetti G, et al. Endothelial dysfunction in peripheral arterial disease is related to increase in plasma markers of inflammation and severity of peripheral circulatory impairment but not to classic risk factors and atherosclerotic burden. J Vasc Surg. 2003;38:374–379.
173. Cleland SJ, et al. Endothelial dysfunction as a possible link between C-reactive protein levels and cardiovascular disease. Clin Sci (Lond). 2000;98:531–535.
174. Brevetti G, et al. Endothelial dysfunction and cardiovascular risk prediction in peripheral arterial disease: additive value of flow-mediated dilation to ankle-brachial pressure index. Circulation. 2003;108:2093–2098.
175. Gokce N, et al. Risk stratification for postoperative cardiovascular events via noninvasive assessment of endothelial function: a prospective study. Circulation. 2002;105:1567–1572.
176. Gokce N, et al. Predictive value of noninvasively determined endothelial dysfunction for long-term cardiovascular events in patients with peripheral vascular disease. J Am Coll Cardiol. 2003;41:1769–1775.
177. Amirbekian V, et al. Detecting and assessing macrophages in vivo to evaluate atherosclerosis noninvasively using molecular MRI. Proc Natl Acad Sci U S A. 2007;104:961–966.
178. Hyafil F, et al. Noninvasive detection of macrophages using a nanoparticulate contrast agent for computed tomography. Nat Med. 2007;13:636–641.
179. Kaufmann BA, et al. Molecular imaging of inflammation in atherosclerosis with targeted ultrasound detection of vascular cell adhesion molecule-1. Circulation. 2007;116:276–284.
[/level-membership-for-surgery-category][not-level-membership-for-surgery-category]Chapter 5
Atherosclerosis
Christopher D. Owens
Vascular surgeons care for a diverse set of clinical manifestations related to atherosclerosis—from symptomatic transient cerebral ischemic attacks to aortoiliac occlusions to aortic aneurysm repairs in patients with advanced coronary heart disease (CHD). However, despite the wide range of manifestations, culprit lesions are more alike than different. Accumulation of large amounts of cholesterol ester in the arterial wall and formation of complex advanced plaque are common to all these lesions. Although their formation insidiously spans decades, atherosclerotic lesions can reach a clinical horizon within minutes and manifest as catastrophic myocardial infarction (MI), stroke, or limb ischemia. Far from being a simple cholesterol deposition problem, plaque formation and evolution are now recognized as much more dynamic in nature. Rather than following a progressive degenerative process, individual plaque formations may progress and regress or undergo growth spurts. Inflammatory changes within the fibrous cap may render them vulnerable to rupture or erosion and ulceration. An understanding of each of these processes helps the vascular surgeon in developing evidence-based treatment strategies for each of the clinical syndromes of atherosclerosis, which together constitute a massive public health problem.
Atherosclerotic-related cardiovascular disease (CVD) is the leading cause of death in every region of the world except sub-Saharan Africa, and it has now eclipsed infectious disease in most underserved nations.1 However, during the last two decades, CHD age-specific death rates fell by greater than 40% in high-income countries.2 The prolonged survival has translated into growing disease prevalence and a staggering financial burden. It is estimated that 80,700,000 Americans have CVD with an estimated annual cost of treatment of $448.5 billion.3 Globally, 202 million people have peripheral arterial disease (PAD), which disproportionately affects blacks and individuals living in low to middle-income countries.1,3 Particularly troubling is the high prevalence of cardiovascular risk factors in children and young adults.4,5 A sedentary lifestyle, abdominal obesity, and poor diets contribute to dyslipidemia and high blood pressure. Autopsy studies in children and young adults demonstrate a link between these risk factors and early lesions.4
This chapter outlines the existing theories of the pathophysiology of atherosclerosis and the relationship to traditional and emerging risk factors. It also describes our current conceptual understanding of the fundamental biology of atherosclerotic plaque.
Normal Artery Structure and Function
A detailed presentation of normal arterial structure can be found elsewhere in this text (see Chapters 3 and 4); however, the relevant anatomy is presented here. Histologically, the arterial wall consists of three distinct layers: the intima (endothelium and subendothelial space), the media (smooth muscle cells [SMCs]), and the adventitia (vasa vasorum and connective tissue elements). The intima consists of the endothelium, its basement membrane, and the subendothelial space, which extends to the internal elastic laminae. The endothelium is a single layer of cells lining the inner surface of all blood vessels.
Endothelium
The endothelium is an organ with autocrine, paracrine, and endocrine functions that serve to regulate blood vessel tone and thrombogenicity. The endothelium synthesizes an array of factors that regulate vascular tone (e.g., nitric oxide [NO], hyperpolarizing factors, prostaglandins, endothelin, angiotensin II) and interactions with blood elements (e.g., adhesion molecules, thrombomodulin, plasminogen activators), which all work in balance to maintain vascular homeostasis.
Endothelial dysfunction is regarded as the earliest manifestation of vessel injury and is present before histologic evidence of atherosclerosis.6 Hence functional testing of the endothelium is an important tool for investigators studying atherosclerosis. Endothelial function can be detected in vivo by invasive and noninvasive techniques. Catheter-directed intraarterial administration of acetylcholine has been used to detect the presence of endothelium-dependent vasodilatation in the coronary and peripheral vasculature. Paradoxical contraction with low doses of acetylcholine is regarded as positive for endothelial dysfunction in the coronary circulation. An acute increase in blood flow can also stimulate endothelial relaxation. Flow-mediated, endothelium-dependent vasodilatation can be measured noninvasively by high-resolution ultrasound techniques.7 The primary mediator of flow mediated vasodilation is NO.8 Administration of nitroglycerin can detect the ability of vascular smooth muscle to relax in the presence of exogenous NO and thus detects endothelial-independent relaxation.
Subendothelial Space
The subendothelial space is particularly relevant in atherosclerosis because it is the location where atherogenic particles are retained and modified so that they can be taken up by macrophages and SMCs.9 In terms of volume, the major component of the extracellular intimal matrix is proteoglycans, which form a three-dimensional network that fills 60% of the space.9 The remaining mass of this compartment is filled with type IV collagen of the basement membrane, type I and III collagen randomly dispersed in this matrix, and occasional elastin fibers. Extending from the proteoglycans are sulfated glycosaminoglycan chains, which serve to interact with positive residues on apolipoproteins. Large versican-like proteoglycans containing chondroitin sulfate readily interacts with apolipoprotein B-100 (apoB-100), the apolipoprotein associated with low-density lipoprotein (LDL).10 The stoichiometry is such that 1 µg of versican can bind to 50 to 100 µg of LDL at physiologic ionic strength, thus suggesting that several lipoprotein particles are retained on each proteoglycan molecule.10,11
Smooth Muscle Cells
In children, very few SMCs or macrophages are present in the subendothelial space; however, with age and lesion formation, their concentration increases. In autopsy studies, abundant SMCs are found in the coronary arteries in atherosclerosis-prone areas, but they are relatively sparse in areas resistant to atherosclerosis.12 Whereas human medial SMCs predominantly express proteins involved in the contractile function of the cell, those found in the intima express lower levels of these proteins, have a higher proliferative index, and exhibit greater synthetic capacity for extracellular matrix, proteases, and inflammatory cytokines.13,14 These “synthetic” SMCs migrate more readily and can produce up to 25 to 46 times more collagen than “contractile” SMCs can. Thus SMCs directly contribute to intimal thickening as a result of increased proteoglycan production, inflammatory cell recruitment, and retention of atherogenic particles within the subendothelial space.
Vasa Vasorum
The vasa vasorum may contribute to atherosclerosis as well. First-order vasa vasorum run longitudinally to the lumen of the host vessel, whereas second-order vasa vasorum are arranged circumferentially. As plaque volume increases, there is an increase in the vasa vasorum visible on histologic section. Whether this is a response to plaque growth or contributes to plaque growth is unknown. Although the development of vasa vasorum in plaque involves angiogenesis, it is unclear what signals this process. Atherosclerotic vasa vasorum can proliferate and thereby lead to extensive neovascularization directed toward lipid-rich atheroma. The neovessels of plaque may provide an additional portal for trafficking leukocytes to promote the inflammatory process or become friable and contribute to intraplaque hemorrhage.15
Atherosclerotic Lesions
Lesions of the arterial wall have been divided into six types based on their histopathologic features.16,17 However, although useful for comparing pathologic specimens, the classification system has limited clinical practicality. The assumption of a relentless, linear progression of the disease has recently been challenged, and though this assumption is true under experimental conditions in some animal models,18,19 it is not necessarily the case in humans.20,21
Autopsy studies from the Bogalusa Heart Study and the Pathobiological Determinants of Atherosclerosis in Youth study demonstrate that atherosclerotic lesions form in early childhood and increase with age and that a significant percentage of them are raised.4,22–24 In an intravascular ultrasound (IVUS) study of heart donors, 17% of individuals younger than 20 years had evidence of atherosclerosis.25 Notably, both the Bogalusa Heart Study and the Pathobiological Determinants of Atherosclerosis in Youth study emphasize that the number and severity of early lesions are directly related to known cardiovascular risk factors, thus suggesting that a paradigm shift is needed to address risk factors in childhood rather than pharmacologic therapy later in the disease process.
Diffuse intimal thickening has been identified in the atherosclerotic-prone areas of coronary arteries as early as 36 weeks of gestation.26 Although the fatty streak—dominated by lipid-filled macrophages—is itself benign, it is the precursor of the more clinically relevant late lesion. The fatty streak is the first lesion visible to the naked eye, and because of its positive Sudan staining, it is referred to as sudanophilic. Its yellow color is attributed to lipid in the form of cholesterol and cholesterol esters within macrophages and SMCs. From the aforementioned studies, it is clear that atherosclerosis begins early in life in all races, is associated with risk factors, and occurs in anatomic locales susceptible to the formation of more advanced fibrous plaque.
Advanced lesions, or fibrous plaque, are characterized histologically by the amount of extracellular lipid and fibrous connective tissue seen. They are whitish in gross appearance and are elevated so that they protrude into the lumen. These fibroatheromas are prone to produce clinical sequelae by erosion of the surface endothelial cells, rupture of the fibrous cap, erosion of a calcium nodule, or intraplaque hemorrhage (Fig. 5-1).16,27 Plaques differ in consistency and may be relatively soft and friable or densely sclerotic and calcific. Likewise, some have well-formed fibrous caps, whereas others are covered by a narrow zone of loose connective tissue or by endothelium alone.
Figure 5-1 Thrombotic complication of a fibroatheroma seen on a trichrome-stained (thrombus and intraplaque hemorrhage stain red; collagen stains blue) cross-section of a human coronary artery. The fibrous cap has ruptured (area between the arrows), and the highly thrombotic lipid core is exposed to circulating blood with the subsequent production of acute occlusive thrombosis.
The necrotic core usually occupies the deeper central regions of the plaque and contains amorphous lipid and cholesterol crystals. The term atheroma is derived from the Greek word athere, meaning “porridge-like gruel.” SMCs and inflammatory cells are located adjacent to the necrotic core and at the shoulders of the plaque, where it is most susceptible to rupture.
The fibrous caps contain varied levels of SMCs adjacent to the collagen and basement membrane. These cells have reduced proliferative ability and may be thought of as senescent. However, the fibrous cap of ruptured plaque is often infiltrated with foam cells, which are largely of macrophage origin28 and thus indicative of active inflammation and vulnerability to rupture. A priori identification of these so-called thin-cap fibroatheromas is an active area of cardiovascular imaging research.29
Theories of Pathogenesis
Each of the well-regarded theories of atherosclerosis discussed in this section attempts to explain the underlying pathogenesis of atherosclerotic plaque. From the time initially proposed, each theory has undergone a steady evolution as our methodology of scientific inquiry has advanced from histologic descriptors to powerful molecular mechanisms. Each theory, held accountable over time, must incorporate new scientific data within a clinical context of risk factors and biomarkers to result in a model of atherosclerotic lesion formation.
Lipid Hypothesis
Cholesterol has been one of the most studied molecules in biomedical research. Since it was first isolated from gallstones in 1784, organic chemists and biochemists, physiologists and physicists, epidemiologists, and other medical scientists have devoted significant portions of their careers to its study and have collectively produced a body of work culminating in 13 Nobel Prizes.30 The modern era of cholesterol research began in St. Petersburg, Russia, at the turn of the 20th century, when Nikolai Anitschkow produced vascular lesions in rabbits by feeding them purified cholesterol dissolved in sunflower oil.31,32 These lesions closely resembled those seen in human atherosclerosis. He determined that the earliest lesions consisted of foam cells that were Sudan positive and contained birefringent crystals and hypothesized that they were caused by elevated serum cholesterol. He also noted a distinctive pattern consisting of location of the lesions near arterial branch points and concluded that this was probably determined by hemodynamic factors. Nevertheless, this work was criticized because the levels of cholesterol produced were too high and could not be experimentally reproduced in more conventional animal models such as rats and dogs.31,32
Cholesterol is absolutely insoluble in water, and the early work done in Russia provided no clues as to how it was transported to the arterial wall and formed lesions. The discovery that cholesterol was associated with proteins that allowed it to be transported in the aqueous environment led to investigations into lipoproteins. The first investigation in which the lipoprotein content in whole serum was accurately quantified involved analytic ultracentrifugation and was led by John Gofman of the Donner Laboratory at Berkley.33 Lipoprotein fractions were isolated and characterized by their densities and floatation characteristics.33 Of importance, this group noted that it was not simply the total cholesterol that was important but the species of lipoprotein contained within the cholesterol.34
Cholesterol is transported in the aqueous environment by esterification of the sterol of long-chain fatty acids and packaging of these esters with the hydrophobic cores of plasma lipoproteins. With its polar hydroxyl group esterified, cholesterol remains sequestered within this core, which is essentially an oil droplet composed of cholesteryl esters and triglycerides, solubilized by a surface monolayer of phospholipid and unesterified cholesterol and stabilized by protein. In persons who are fasting, lipids circulate in plasma as lipoprotein particles that are defined on the basis of their density as very-low-density lipoprotein (VLDL), intermediate-density lipoprotein (IDL), LDL, and high-density lipoprotein (HDL). The protein of the VLDL, IDL, and LDL molecule is apoB-100, which has β-electrophoretic mobility.
The major milestone in the lipoprotein field was discovery of the defective gene associated with familial hypercholesterolemia by Goldstein and Brown. In fibroblasts cultured from normal human subjects, the activity of 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase, the rate-controlling enzyme in cholesterol biosynthesis, is regulated by the content of LDL (but not HDL) in the culture medium. Goldstein and Brown found that 3-hydroxy-3-methylglutaryl coenzyme A reductase was not sensitive to normal feedback regulation by LDL in cultured skin fibroblasts from homozygous familial hypercholesterolemia patients.35,36 They then determined that the defect was related to deficient binding of LDL to the cells of patients with familial hypercholesterolemia and that suppression of 3-hydroxy-3-methylglutaryl coenzyme A reductase activity was related to the amount of LDL bound.37 Therefore these patients had overproduction of cholesterol because they lacked the appropriate receptor. They demonstrated that cellular uptake of LDL absolutely requires the LDL receptor, without which the LDL cholesterol concentration builds up to 800 to 1000 mg/dL (20.72 to 25.90 mmol/L). Recognition of the importance of this work resulted in Goldstein and Brown sharing the Nobel Prize in Physiology and Medicine in 1985.
Epidemiologic evidence from numerous studies, including the Japanese migration studies38,39 and the Framingham Heart Study,40 demonstrated the association between cholesterol and incident CHD. Other studies showed that diets rich in unsaturated fats resulted in lowered cholesterol and reduced cardiovascular events.41
Widespread acceptance of the causative role of cholesterol began with publication of the Coronary Primary Prevention Trial, sponsored by the National Institutes of Health.42,43 This trial demonstrated that reducing cholesterol with the bile acid–binding resin cholestyramine would reduce cardiovascular events. A decade later, this trial was reinforced with powerful data emerging from the statin era.44 Although the primacy of the “cholesterol hypothesis” has been called into question, there is no doubt that lipids play a critical role in the pathogenesis of atherosclerosis.
Response-to-Injury Hypothesis
The basis of this hypothesis lies in the similarities between atherosclerosis and the response of arteries to experimental injury. Although the origins of the hypothesis date back to Virchow, the modern version of the hypothesis builds on earlier work by Duguid (1949),45 Poole and Florey (1958),46 and French (1966)47 and was formally advanced in 1973 by Ross and Glomset,48 who emphasized the importance of injury to the endothelium as a seminal event initiating the development of atherosclerosis lesions. Even though the definition of the term injury has been modified over the last three decades,49 all response-to-injury hypotheses have emphasized the primacy of the endothelium in stimulating the cascade of events leading to lesion formation.48–53
Endothelial injury may result from mechanical disruption, exposure to toxic or infectious agents, or endogenous inflammatory signals. Injury to the endothelium allows adhesion of platelets and an influx of LDL and other serum factors into the subendothelial space. Platelets release their alpha granules and stimulate migration of SMCs into the intima, where they proliferate and form a thickened neointima responsible for narrowing of the arterial lumen. Restoration of a healthy endothelial cell layer abates the process. During work on this hypothesis, Ross et al performed experiments leading to the discovery of platelet-derived growth factor (PDGF).54,55
Others counter that it is not injury to the endothelium that is the initiating event but rather retention of inflammatory lipids in the subendothelial space, which renders a particular area susceptible to atherosclerosis. By noting that LDL accumulates in the intima within 2 hours after a bolus infusion56 and that the accumulation occurs before the formation of fatty streaks,57,58 they concluded that retention of LDL is the initiating event of atherosclerosis. The so-called response-to-retention theory purports that retained apoB–containing lipoproteins stimulate a macrophage- and T cell–dominated inflammatory response in the arterial wall.59,60
Monoclonal Hypothesis
During the same year that Ross and Glomset were formulating the response-to-injury hypothesis of atherosclerosis, another group of investigators at the same institution (University of Washington) were writing their own hypothesis of atherosclerosis. This hypothesis suggests that each lesion of atherosclerosis is derived from a single SMC that serves as a precursor for the clonal expansion of proliferating SMCs.61 The hypothesis put forth by Benditt and Benditt uses the concept that in every female cell there is only one active X chromosome and the progeny of that cell will express the same X chromosome as the parent cell. Glucose-6-phosphate dehydrogenase (G6PD) has two isoforms that can be separated by electrophoresis. Its gene is located on the X chromosome and can therefore be used to identify the progeny of a parent cell. This approach was used to determine that uterine leiomyomas are composed of cells with the same active X chromosome, whereas adjacent normal myometrium was composed of a mixture of cells containing both G6PD isoforms and, therefore, that both X chromosomes were active.62
The Benditts examined a series of atherosclerotic plaque from four black females and compared them with adjacent normal areas of arterial wall. They determined that SMCs from the plaque contained only one G6PD isoform, whereas adjacent control areas contained a mixture of isoforms. This allowed them to conclude that each lesion is a clonal outgrowth derived from a single precursor SMC located in the intima. It is noteworthy that SMCs within individual human neonatal intimal thickenings are monoclonal in origin, whereas cells from the subjacent media are polyclonal.
Others challenged the hypothesis by noting that identification of a single enzyme phenotype does not necessarily imply clonal origin.63,64 Regardless, the work was importantly heuristic. It hinted toward the role that modern molecular biology would play in unraveling the genetic basis of atherosclerosis.
Atherosclerosis as a Chronic Inflammatory Disease
The original version of the response-to-injury hypothesis of atherosclerosis initially proposed that endothelial denudation was the first step in atherosclerosis.48 Subsequent versions of the hypothesis proposed that an endothelium chronically bathed in serum with high concentrations of LDL or exposed to other cardiovascular risk factors would render susceptible areas of the endothelium dysfunctional or activated.52 Indeed, an intact endothelium may be a necessary factor for lesion progression, and it is now clear that developing atheromas are covered by an intact endothelium throughout most stages of lesion progression.17,49,65–67 In humans, only the most advanced ulcerated lesions are focally devoid of endothelium. The injury results in increased adhesiveness and an increase in permeability of the endothelium to inflammatory cells (Fig. 5-2).
Figure 5-2 Initiation and progression of atherosclerotic plaque. Cardiovascular risk factors, hemodynamic forces, toxins, and infectious agents interact with the vessel at the level of the endothelium to produce injury, resulting in decreased nitric oxide (NO) production and increased permeability. Once injured, the endothelium increases the expression of leukocyte adhesion molecules such as vascular cell adhesion molecule-1, intracellular adhesion molecule-1, and P- and E-selectin, which increases the adherence of macrophages and other leukocytes. Permeability of the endothelium also increases and permits entry of leukocytes and lipoproteins into the subendothelial space. Chemokines and cytokines such as monocyte chemotactic protein-1 (MCP-1) and interleukin-8 (IL-8) further enhance the recruitment of leukocytes and smooth muscle cells (SMCs) into the subendothelial space. Lipoproteins retained in the subendothelial space are biochemically modified such that they can be taken up by macrophages and SMCs to form foam cells. Foam cells at the central-most position of the developing atheroma become necrotic and form the central lipid core, whereas the shoulder regions contain SMCs, macrophages, and other leukocytes. Platelet-derived growth factor (PDGF) and transforming growth factor-β (TGF-β) stimulate SMC migration and collagen formation in the subendothelial space, as well as formation of the fibrous cap.
Atherosclerosis is now recognized as an inflammatory disease, and components of the innate and adaptive immune system are involved in every step of the atherosclerotic process. Much of our modern understanding comes from examination of human pathology specimens and transgenic animals. Genetic deletion of apolipoprotein E (apoE−/−) or the LDL receptor (ldlr−/−), which produces mice with severe hypercholesterolemia and atherosclerotic lesions with features of mature human atheroma, has become a cornerstone in atherosclerosis research laboratories.68–70 The importance of the LDL receptor in cholesterol regulation was noted by Goldstein and Brown when studying patients with familial hypercholesterolemia. Apolipoprotein E suppresses atherosclerosis, and apoE−/−– mice have very low amounts of pre-β HDL and their plasma is poor at promoting the efflux of cholesterol from lipid-laden macrophages.71 Crossbreeding these mice with other strains carrying null mutations in immunologically relevant genes produces a robust research tool to dissect out the contribution of individual components of immune pathways in atherosclerosis. For example, some of the earliest approaches using compound mutant mice involved the global loss of the entire adaptive immune system. Rag1−/− or Rag2−/− mice lack the V(D)J recombinase required to form lymphocyte antigen receptor genes and hence have a complete loss of B and T cells. In apoE−/− mice, a regular chow diet produced plasma cholesterol levels between 390 and 470 mg/dL. Double knockout apoE−/−/Rag1−/− animals had a 40% reduction in aortic atherosclerotic lesion reduction compared with immunocompetent animals and provided an important piece of evidence of the role cellular immunity plays in the pathogenesis of atherosclerosis.72
LDL Retention
Trafficking of circulating LDL into and out of the subendothelial space is probably a function of concentration gradients, as well as endothelial permeability.9,73 Normal intima does not retain LDL particles, thus suggesting that most particles return to plasma or are degraded in situ. However, subendothelial retention of apoB-100–containing lipoproteins is an early event in atherosclerosis.73 Factors favoring net retention include a balance of uptake and degradation of LDL by macrophages, egress of LDL-containing macrophages into the circulation, and complex interaction with proteoglycans such that the LDL particles are sufficiently modified to maintain the gradient. Once bound to the matrix, the lipoprotein is modified so that it becomes oxidized.9,74 Oxidized LDL, unlike native LDL, is chemotactic to monocytes and rapidly taken up by macrophages to form foam cells by scavenger receptors.
Monocytes
As early as 1958, Poole and Florey noted that macrophages adhere to the surface of endothelial cells overlying atheroma.46 In studies of hypercholesterolemic monkeys, monocytes are seen attached to the endothelium within 12 days of initiation of an atherogenic diet.19 It is now apparent that monocytes play a very early role in atheroma formation.75,76 Endothelium, which is rendered activated, expresses adhesion molecules that interact with circulating leukocytes, principally monocytes and lymphocytes. E- and P-selectins slow monocytes, mediate rolling, and loosely tether them to the endothelium.77 More permanent fixation is due to members of the immunoglobulin superfamily—vascular cell adhesion molecule-1 (VCAM-1)78 and intercellular adhesion molecule-1 (ICAM-1)—which are soon upregulated and firmly fix the leukocytes to the wall. The importance of the selectins, VCAM-1, and ICAM-1 in the pathogenesis of atherosclerosis is evident in either ICAM-1 or VCAM-1 mutant animals exhibiting far less atherosclerosis than do wild type controls.79–81 The adherent leukocytes are stimulated to migrate into the subendothelial space by a number of chemokines, including MCP-1 and oxidized LDL.82–84
Macrophages
Once within the subendothelial space, monocytes undergo a series of phenotypic modulations and become resident tissue macrophages that take up oxidized LDL via the scavenger receptor A (SR-A), as well as CD36.85 One of the key signals for macrophage activation, macrophage colony-stimulating factor, can enhance scavenger receptor expression and promote replication of macrophages and their production of proinflammatory cytokines. Experiments in mutant mice deficient in macrophage colony-stimulating factor have shown the importance of this factor in atheroma formation.86,87 Although most foam cells are macrophage in origin, SMCs also take up lipids via scavenger receptors and CD36.
In mice, proinflammatory subsets of macrophages called M1-macrophages, induced by hyperlipidemia, produce the inflammatory cytokines IL-1β and TNF-α. The IL-1 gene family encodes three major proteins. The first two, IL-1α and IL-1β, exert proinflammatory effects by binding to the IL-1 receptor type 1. The third is IL-1 receptor antagonist (IL-1Ra), an endogenous inhibitor that competitively blocks the binding of IL-1α and IL-1β to the IL-1 receptor. Direct evidence for the proatherogenic role of IL-1 was obtained in experiments in which apoE−/− mice received subcutaneous injection of recombinant IL-1 receptor antagonist. Mice injected with IL-1 receptor antagonist displayed a marked reduction in atherosclerotic lesion size. Similarly IL-18, previously called interferon-γ (IFN-γ)–inducing factor, is a Th1-promoting cytokine and therefore has the capacity to promote inflammation through the innate and the adaptive immune pathways.88,89 Knockout of IL-18 in atherosclerosis-prone animals reduces aortic atherosclerotic lesion size.
Macrophages also contribute to thrombosis in several pivotal ways. As discussed later, atherosclerotic lesions that have ruptured characteristically contain abundant macrophages underlying a thin and collagen-poor fibrous cap. Specific collagenases MMP-1, MMP-8, and MMP-13, co-localizes with macrophages in human atheroma. Rupture of the fibrous cap may be a balance between collagen synthesis by SMCs and collagen breakdown by interstitial (matrix metalloproteinases) collagenases generated by activated macrophages. Studies in collagenase-resistant mutant “knockin” mice crossbred with apoE−/− mice demonstrated increased intimal collagen and SMC content.90 Similarly, in compound mutant apolipoprotein E–null mice crossed with MMP-13/collagenase-3−/− mice have increased fibrillar collagen that is thicker and more aligned in aortic atherosclerosis.91 Hence interstitial collagenases produced by activated macrophages greatly influence the structure and integrity of the fibrous cap overlying atherosclerotic plaques. Macrophages and SMCs within atherosclerotic plaques also overexpress the potent procoagulant tissue factor in response to C-reactive protein or CD40 ligand. Hence inflammation links atherosclerosis and thrombosis, leading some to refer to it as atherothrombosis.
Lymphocytes and Adaptive Immunity
The finding in the early 1980s that macrophages expressed major histocompatibility class II antigens needed for antigen presentation to CD4+ T cells suggested that adaptive immunity was involved in the atherosclerotic process.92 T lymphocytes, which account for as much as 10% to 20% of the leukocyte population, are found most abundantly in the shoulder and fibrous cap region of the atheroma. In advanced lesions, these T cells display markers of chronic activation and produce the prototypical Th1 cytokine, (IFN-γ) which further stimulates expression of class II major histocompatibility antigens in SMCs and macrophages. The crossbreeding of severe combined immunodeficiency (scid)/scid mice that lack T and B cells with apoE−/− mice produces offspring that are both hypercholesterolemic and immunodeficient. Similar to what was found with apoE−/−/Rag2−/− double mutants, when lesions in these mice were compared with those in the immunocompetent apoE−/− animals, a dramatic 70% reduction of lesion size was observed. However, transfer of oxidized LDL-reactive T cells to apoE−/−/scid/scid mice is more efficient at lesion acceleration than the transfer of T cells with no specificity to a plaque-derived antigen, demonstrating the importance of antigen presentation in the pathogenesis of atherosclerosis.93
Because of the participation of all components of the innate and adaptive immune system, atherosclerosis resembles other inflammatory and autoimmune diseases such as rheumatoid arthritis and type 1 diabetes mellitus. Potential endogenous autoantigens to activate T cells include LDL or heat shock protein 60.93,94 Cellular and humeral immune responses are mounted toward these antigens in humans and mice, and protective immunization strategies in mice have provided encouraging results.
In mice, functionally distinct T cell subsets appear to exist in atheroma. Natural killer T cells appear to accelerate atherosclerosis when recognizing lipid antigens presented through CD1 molecules.95
[/not-level-membership-for-surgery-category]
