[level-membership-for-internal-medicine-category]
Chapter 9 Atherosclerosis and dyslipidaemia
EPIDEMIOLOGY AND AETIOLOGY
The association between elevated serum cholesterol and coronary heart disease (CHD) was first reported in the 1930s, and subsequent large epidemiologic studies confirmed the strong relationship between serum cholesterol and CHD.1–3 Current predictions estimate that by the year 2020 cardiovascular diseases, notably atherosclerosis, will become the leading global cause of total disease burden.4 Lipoprotein disorders or dyslipidaemias are common and are an important risk factor for coronary artery disease and all types of atherosclerotic vascular disease.5 Cardiovascular risk was found to be positively associated with low-density lipoprotein (LDL-C) levels and inversely with high density lipoprotein (HDL-C) levels.6 The relationship between fasting serum triglyceride (TG) and cardiovascular risk has been confounded by the inverse association between TG and HDL-C, as well as by the association of TG with other risk factors such as diabetes mellitus and body mass (see Table 9.1).6 Triglycerides alone are now considered an important independent predictor of cardiovascular risk.1
| CAUSE | EFFECT ON LIPID PROFILE |
|---|---|
Source: Therapeutic Guidelines (Cardiovascular).17
Atherosclerosis affects various regions of the circulation and will present with unique clinical symptoms depending on the particular vascular bed affected. In coronary arteries it causes myocardial infarction and angina, whereas stroke and transient cerebral ischaemia can occur when it is present in the arteries of the nervous system.2,7 In the peripheral circulation it can result in intermittent claudication and gangrene, which can jeopardise limb viability.4
Atherogenesis or the pathogenesis of atherosclerosis usually occurs over decades in a discontinuous fashion with relative periods of quiescence punctuated with periods of rapid evolution.3 The eventual clinical manifestation may be chronic, as is the case in angina, or acute, such as a heart attack or stroke or sudden death. Regardless, individuals may live with atherosclerosis and experience no symptoms or reduced life expectancy and morbidity.
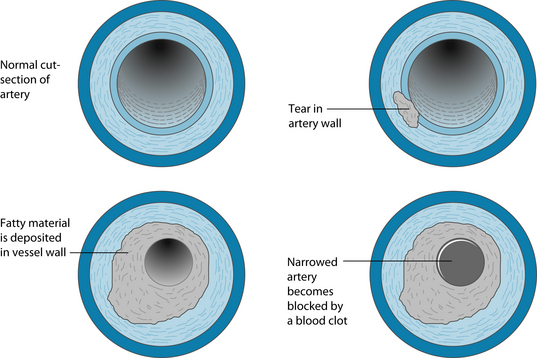
The ‘fatty streak’ represents the initial lesion, usually due to focal increases in the density of lipoproteins within the region of the intima of a blood vessel; hence the reason for vigilance in maintaining low levels of these particles in the bloodstream.8 The lipoproteins often associate with glycosaminoglycans in the arterial extracellular matrix and become more ‘fixed’ in the vasculature. These lipoprotein macromolecules undergo oxidative modifications, giving rise to hydroperoxides, lysophospholipids, oxysterols and aldehydic breakdown products.4 Constituents of oxidatively modified LDL-C can augment expression of leukocyte adhesion molecules, recruiting leukocytes and macrophages into the area, which undergoes inflammation.9
Areas of inflammation produce altered laminar flow in the arteries affected, and reduce the production of nitric oxide, which is locally both vasodilatory and anti-inflammatory. The inflammatory process eventually penetrates or tears the endothelial layer with the products of lipoprotein oxidation, inducing cytokines and tumour necrosis factor alpha (TNF-α) released from the vascular cell walls.4 Once inside the walls of the artery mononuclear phagocytes mature into macrophages and become foam cells.
The cytokines and TNF-α produce growth factors that induce the smooth muscle to produce local growth factors such as platelet-derived growth factor and fibroblast growth factors.9 This fibro-fatty material recruits more smooth muscle cells into the intima from the tunica media layer of the vessel. At this point transforming growth factor beta (TGF-β), amongst other mediators, stimulates collagen production by the smooth muscle cells transforming the fatty streak into a more fibrous smooth-muscle cell and extracellular-rich lesion.4 In addition to this process products of blood coagulation and thrombosis contribute to atheroma evolution, which justifies the alternative term for the process, ‘atherothrombosis’ (see Figure 9.1).
Unfortunately the prevalence of traditional risk factors is almost as high in those without cardiovascular disease as in subsequently affected individuals.1 Recently, several novel key biomarkers have been implicated in the pathology of atherosclerosis. There are several, but the more commonly researched include C-reactive protein (CRP), homocysteine (Hcy) and indicators of oxidative stress.8,10
C-reactive protein (CRP)
CRP is a calcium-binding pentameric protein consisting of five identical, non-covalently linked 23-kDa subunits.1 It is present in trace amounts in humans and appears to have been highly conserved over hundreds of millions of years. It is synthesised primarily by hepatocytes in response to activation of several cytokines, such as interleukins 1 and 6, and TNF-α.1 In healthy people in the absence of active inflammatory states, CRP levels are usually below 1 mg/L. CRP is expressed in atherosclerotic plaque and may enhance expression of local adhesion molecules, increase expression of endothelial plasminogen activation inhibitor 1, reduce endothelial nitric oxide bioactivity and alter low-density lipoprotein (LDL) cholesterol by macrophages.10
Homocysteine (Hcy)
The homocysteine theory of arteriosclerosis attributes one of the underlying causes of vascular disease to elevation of blood homocysteine concentrations as the result of dietary, genetic, metabolic, hormonal or toxic factors.11 Dietary deficiency of vitamin B6 and folic acid and absorptive deficiency of vitamin B12, which result from traditional food processing or abnormal absorption of B vitamins, are important factors in causing elevations in blood homocysteine.11,10 It is believed that homocysteinylation of lipoproteins increases their atherogenicity and hence their cytotoxic effects on endothelial tissue.12 It is hypothesised that oxidative stress and methylative modification is the mechanism by which Hcy is pro-atherosclerotic.13
Oxidative stress
Endothelial dysfunction is the initial step in the pathogenesis of atherosclerosis.14,15 Nitric oxide (NO) plays an important role in the regulation of vascular tone and the suppression of smooth muscle cell proliferation. An increase in oxidative stress inactivates NO, impairing endothelium-dependent vasodilation and creating endothelial dysfunction.16 Dyslipidaemia may be primary or secondary. The secondary causes are shown in Table 9.1, and should be looked for in patients and treated appropriately. If secondary causes have been excluded, primary dyslipidaemia due to genetic and environmental (particularly dietary) factors is diagnosed.17
RISK FACTORS
The general risk factors applicable to atherosclerosis relate to lifestyle choices and genetics. Overall estimates of the heritability of myocardial infarction, one of the major clinical outcomes of atherosclerosis, have varied widely, ranging from very little to greater than 50%.18,19 It is important to note that heritability varies from population to population. In populations in which the environment is relatively homogeneous, for example in Iceland, genetics will tend to predominate, whereas in populations exhibiting large environmental differences, for example in Los Angeles, genetics are relatively less important.1 The known risk factors for atherosclerosis generally exhibit very significant heritability estimates as well (see Figure 9.2). Smoking and alcohol consumption as well as a diet rich in refined sugars and saturated fats will adversely influence the condition (see the box below).20 Hypertension, diabetes and obesity have a pronounced negative influence on the disease.1 Exercise is also a significant factor.3 In women the protective effects of oestrogen are greatly diminished in menopause.1 There are several medical conditions that drastically increase the risk associated with this condition, and all of these may cause altered blood fat profiles as secondary to their primary condition.
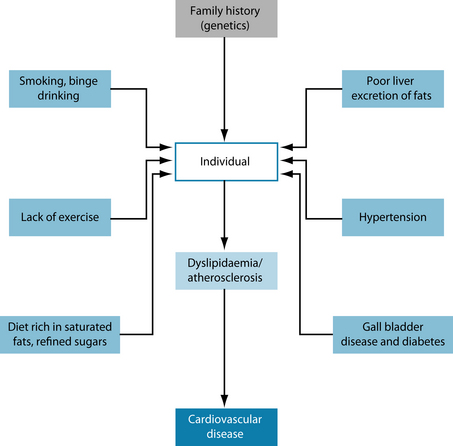
The influence of environmental factors on lipoprotein metabolism and cardiovascular disease has been dramatically revealed by comparisons of different populations.3 Studies have shown that intrauterine under-nutrition is associated with obesity and other detrimental metabolic characteristics in adulthood.1 In particular under-nutrition
GENETIC AND ENVIRONMENTAL RISK FACTORS FOR CARDIOVASCULAR DISEASE1
was associated with a premature onset of neonatal leptin surge, which appears to be involved in the formation of the energy-regulation circuits in the hypothalamus that affect metabolism in adults.1
CONVENTIONAL TREATMENT
The assessment of dyslipidaemia involves the measurement of fasting total cholesterol (TC), high density lipoprotein (HDL-C), low-density lipoprotein (LDL-C) and triglyceride (TG).10 Fasting usually takes place from midnight prior to the blood test.
Dyslipidaemia is treated in cases of:
Source: Therapeutic Guidelines (Cardiovascular).17
The question that needs to be addressed in relation to dyslipidaemia is: who should in fact be treated and when? Tables 9.3 and 9.4 list the current Australian medical protocols. The aim of treatment is to reduce the progression of atherosclerosis and improve survival rates of individuals. An even more important aim is to prevent myocardial infarct (heart attack) and stroke in patients with established cardiovascular disease. A reduction in triglyceride levels should prevent the occurrence of pancreatitis in susceptible individuals as well. Common pharmacological interventions are listed in Table 9.5.
Table 9.3 Patients at increased risk and warranting therapy, based on their fasting lipid levels
| PATIENT CHARACTERISTICS | LIPID LEVEL (FASTING) |
|---|---|
| Diabetes mellitus, but the patient is not in the ‘very high risk’ category (see below). | Total cholesterol > 5.5 mmol/L |
| Aboriginal or Torres Strait Islander Hypertension | |
| HDL cholesterol < 1 mmol/L | Total cholesterol > 6.5 mmol/L |
| Familial hypercholesterolaemia identified by:
Family history of coronary heart disease: |
If aged 18 years or less at treatment initiation:
If aged more than 18 years at treatment initiation: |
| Patients not already identified who are: | Total cholesterol > 7.5 mmol/L or triglyceride > 4 mmol/L |
| Patients not already identified | Total cholesterol > 9 mmol/L or triglyceride > 8 mmol/L |
Source: Therapeutic Guidelines (Cardiovascular).17
Table 9.4 Patients in the ‘very high risk’ category warranting therapy regardless of their fasting lipid levels
| The following patients are in the ‘very high risk’ category, regardless of their fasting lipid levels: |
Source: Therapeutic Guidelines (Cardiovascular).17
Table 9.5 Pharmacological treatment of dyslipidaemia
| DRUG | BENEFIT | DISADVANTAGE |
|---|---|---|
| Statin | Reduces LDL-C by 30–50%. Best tolerated | Reduces coenzyme Q10 |
| Bile-acid binding resin | Reduces LDL-C by 15–25% | Poorly tolerated, may worsen hypertriglyceridaemia |
| Nicotinic acid | Reduces LDL-C by 15–30%, triglycerides by 25–40%. Increases HDL-C by 20–35% | Causes severe flushing, which limits use |
| Ezetimibe | Reduces LDL-C by 18%. Well tolerated | Generally not used alone |
| Fibrate | Reduces LDL-C by 18%,∗ and triglycerides by 40–80%. Increases HDL-C by 10–30% | ∗Can increase LDL-C in some instances. |
Source: Taken from Australian Medicines Handbook.45
KEY TREATMENT PROTOCOLS
Plasma total homocysteine (Hcy) has been associated with cardiovascular risk in multiple large-scale epidemiological studies, and it has been considered as an independent risk factor for atherosclerosis.21 Evidence indicates that although mild hyper-Hcy may be regarded as a minor risk factor for CHD in low-risk patients, it can play a role in triggering new events in patients with known CHD, also by interacting with the
‘classical’ CV risk factors.21 The administration of vitamins B6 (300 mg/day) and B12 (250 μg/day) and folic acid (10 mg/day) over a 120-day period reduced plasma Hcy levels by 24%; this in turn resulted in a 28.5% reduction in coronary risk.22 In a study of 23 patients with angiographically proven coronary artery disease, daily dosing with a multivitamin containing 0.65 mg folic acid, 150 mg alpha-tocopherol, 150 mg ascorbic acid, 12.5 mg beta-carotene and 0.4 mg B12 produced a 28% reduction in Hcy and a decrease in in vitro LDL oxidation of 39%.23 However, in patients with stable coronary artery disease, Hcy-lowering therapy with B vitamins does not affect levels of inflammatory markers (CRP and soluble CD4 ligands CD40L) associated with atherogenesis.24 After two months treatment with oral folic acid in 16 patients with insulin-dependent diabetes, plasma Hcy fell by 25%, however there was no difference in endothelial function or markers of oxidant stress.25 There was however both a reduction in Hcy levels and an increase in endothelial function in a study of 60 patients receiving either 5 mg of folic acid or 600 mg of N-acetylcysteine.26 Of interest is a small study in which 12 patients with coronary artery disease underwent therapy with high dose (10 mg daily) folic acid therapy. Whereas resting myocardial blood flow remained unchanged, stress myocardial blood flow and coronary flow reserve increased significantly along with reduction in Hcy.27 Phenolic compounds from extra virgin olive oil (hydroxytyrosol, homovanillyl alcohol, caffeic acid and ferulic acid) in vivo significantly reduced homocysteine-induced endothelial dysfunction.28
C-reactive protein (CRP)
C-reactive protein (CRP) is an inflammatory protein that may play a role in the pathogenesis of atherosclerosis. Evidence now suggests that a single nucleotide polymorphism that results in the CRP-757C allele may be responsible for an increase in the intima thickness in the carotid arteries of cardiovascular patients.29 Loss of body fat has a positive correlation with lower CRP levels. A study of 33 obese subjects over 20 weeks showed a 35% reduction in CRP levels only after significant loss of body fat; this reduction was maintained for the duration of the study.30 In epidemiological trials high fibre intakes have consistently been associated with reduction in cardiovascular disease risk and CRP levels. A data synthesis of seven clinical trials reported significantly lower CRP concentrations in six of them in the vicinity of 25–54% through increased dietary fibre consumption with dosages ranging between 3.3 and 7.8 g/MJ.31 Inflammatory reactions in coronary plaques play an important role in the pathogenesis of acute atherothrombotic events. The association of serum CRP with the incidence of first major coronary heart disease (CHD) event was studied in 936 men.32 The results indicated that there was a prognostic significance to raised CRP and CHD events.32 Antioxidants may play a role in reducing levels of CRP. Vitamin C supplementation at 515 mg daily reduced CRP levels by 24%.33 Alpha-tocopherol (vitamin E) supplementation (especially at higher doses) in human subjects and animal models has been shown to be both antioxidant and anti-inflammatory, decreasing C-reactive protein (CRP) and the release of pro-inflammatory cytokines such as chemokines IL-8 and PAI-1.34
Oxidative stress
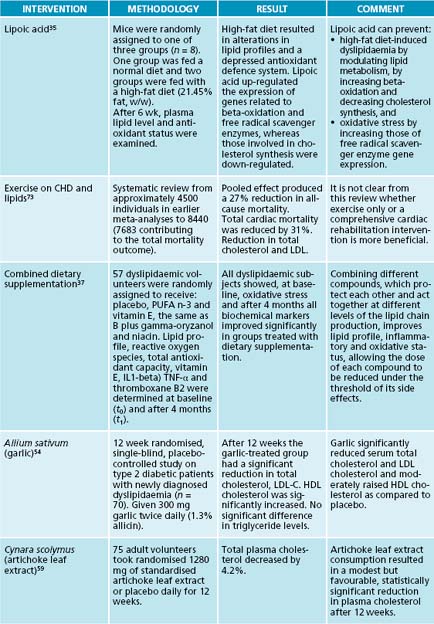
Oxidative stress plays a pivotal role in atherogenesis, and some antioxidants, such as lipoic acid, can have an effect on attenuating dyslipidaemia. Supplementation with lipoic acid induced decreases in lipid peroxidation, plasma cholesterol, triacylglycerols and low-density lipoprotein cholesterol, and an increase in high-density lipoprotein in mice fed a high fat diet.35 Similarly, rabbits on a high-cholesterol diet had significantly lower levels of LDL-C and total cholesterol when given 4.2 mg/kg of lipoic acid daily.36 It is posited that lipoic acid reduces oxidative stress by increasing free radical scavenger enzyme expression in the cell.35 A combination of omega-3 fatty acids, vitamin E, gamma oryzanol and niacin produced significant improvements in lipid profiles in a study amongst 57 participants.37 In mice vitamin E has a modest effect at best on experimental atherosclerosis, and only in situations of severe vitamin E deficiency.38 However, the benefits of vitamin E may be due to its effects on smooth muscle proliferation. Cell culture studies have shown that alpha-tocopherol brings about inhibition of smooth muscle cell proliferation. This takes place via inhibition of protein kinase C activity. Alpha-tocopherol also inhibits LDL-induced smooth muscle cell proliferation and protein kinase C activity.39 Forty-four patients undergoing outpatient treatment for atherosclerosis with simvastatin 10 mg were administered 90 mg of coenzyme Q10 for 12 weeks. The positive effects of coenzyme Q10 were particularly expressed in relation to the antiatherogenic fraction of cholesterol, which increased by 23%. The index of atherogenicity decreased by 27%. At the background of coenzyme Q10 treatment 30% reduction in plasma lipoperoxide levels occurred, demonstrating the potentially independent role of coenzyme Q10 in the positive modification of oxidative stress.40 A study of 21 patients using 60 mg of coenzyme Q10 alone demonstrated its potentially independent role in the positive modification of oxidative stress, antiatherogenic fraction of the lipid profile, atherogenic ratio and platelet aggregatability.41
Polygonum multiflorum stilbene-glycoside is a water-soluble fraction of Polygonum multiflorum, a traditional Chinese herbal medicines, and has protective effects on the cardiovascular system. Rabbits fed for 12 weeks on a high-cholesterol diet and Polygonum multiflorum stilbene-glycoside had a decrease in the atherosclerotic lesioned area of 43% when given 50 mg/kg, and 60% when given 100 mg/kg.42 This was possibly due to reductions in intercellular adhesion molecule-1 protein and the vascular endothelial growth factor.
Cocoa is rich in plant-derived flavonoids, which have a therapeutic benefit in atherosclerosis. In humans, flavonol-rich cocoa counteracts lipid peroxidation and therefore lowers the plasma level of F2-isoprostanes, markers of in vivo lipid peroxidation, and plasma levels of oxidised LDL in hypercholesterolaemic patients.43
Alteration of lipid profile with natural products
Nicotinic acid, or niacin, has established efficacy for the treatment of dyslipidaemia, but the clinical use of niacin has been limited by cutaneous flushing, a well-recognised associated adverse effect. Flushing has been cited as the main reason for the discontinuation of niacin therapy, estimated at rates as high as 25–40%.44 It has good LDL- and triglyceride-lowering effects and produces a marked increase in HDL (typically reduces LDL by 15–30% and triglycerides by 25–40%, and increases HDL by 20–35%).45 Niacin promotes angiographic regression when used in combination with other drugs that lower LDL cholesterol and can reduce cardiovascular risk in patients with coronary heart disease.46 The Therapeutic Guidelines17 recommend daily dosing of 500 mg slowly increasing to 3 g daily.
Policosanol is a mixture of higher primary aliphatic alcohols purified from sugarcane wax with cholesterol-lowering effects proven in patients with type II hypercholesterolaemia and dyslipidaemia due to type 2 diabetes mellitus.47 A once-daily dose of 5 mg has been shown to reduce LDL-C by 32.6% after 12 weeks as well as improve lipid ratios in a cohort of adolescents.47 The effects of policosanol were compared to those of lovastatin in patients with dyslipidaemia and type 2 diabetes in a randomised double-blind study. A daily dose of 10 mg of policosanol for 8 weeks lowered LDL-C by 29.9%, TC by 21.1% and TGs by 13.6%.48 Policosanol (10 mg/day) was slightly more effective than lovastatin (20 mg/day) in reducing the LDL-C/HDL-C and total cholesterol/HDL-C ratios, in increasing HDL-C levels and in preventing LDL oxidation.48 However a double-blind, randomised, placebo-controlled trial amongst 70 patients contradicted previous evidence. The addition of 10 mg policosanol daily to a dietetic regimen failed to produce any significant change in BMI, TC, HDL-C, LDL-C and TG plasma levels.49 In a double-blind, randomised, placebo-controlled trial of 68 patients with primary hypercholesterolaemia, a daily dose of policosanol 20 mg over 8 weeks did not produce any significant change in the parameters mentioned above.50
Allium sativum (garlic) and many of its preparations have been widely recognised as effective in the prevention and treatment of atherosclerosis and other risk factors for cardiovascular disease. Studies have suggested a multitude of physiological effects, including inhibition of platelet activity and increased levels of antioxidant enzymes.51 Garlic has been shown to inhibit the enzymes involved in lipid synthesis, decrease platelet aggregation, prevent lipid peroxidation of oxidised erythrocytes and LDL, increase antioxidant status, and inhibit angiotensin-converting enzyme.52 A small study involving the administration of 600 mg fish oil with 500 mg garlic oil daily concluded that the total cholesterol, low-density lipoprotein, serum triglyceride, very low-density lipoprotein and the total cholesterol:high-density lipoprotein ratio decreased by 20%, 21%, 37%, 36.7% and 23.4%, respectively, and the high-density lipoprotein increased by 5.1% after 60 days of supplementation.53 A single-blind, placebo-controlled study concluded after 6 weeks of garlic supplementation that TC, LDL-C and TG all decreased by 12.1%, 17.3% and 6.3% respectively, while HDL-C rose by 15.7%. In newly diagnosed dyslipidaemia in type 2 diabetics, 600 mg of garlic daily resulted in 12.03% reduction in total cholesterol, 17.99% reduction in LDL-C as well as an 8.81% increase in HDL-C levels.54 Long-term results, however, may not be as promising. In its evidence report on garlic, the Agency for Health Care Research and Quality reviewed 37 randomised trials and found that garlic preparations lowered total cholesterol by small amounts in the short term, but no reduction was observed at 6 months.51
The principal essential fatty acids in fish oil are eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA).55 Fish oil has been reported to have anti-inflammatory and immunosuppressive effects, and EPA competitively inhibits synthesis of thromboxane A2. Both EPA and DHA interfere with prostaglandin synthesis in platelets and blood vessels, resulting in decreased platelet activity.55
The beneficial effect of dietary fish oil on plasma triglycerides is thought to be related to the high quantities of omega-3 polyunsaturated fatty acids found in many types of fish.17 In an attempt to further explain the mechanism of action of fish oil, a study of a fish oil-rich diet administered to apolipoprotein E-deficient mice resulted in reduced adhesion molecule expression in atherosclerotic lesions.56 Because these molecules are involved in lesion progression, the effect of fish oil may explain the observed decrease in atherogenesis. There is now considerable evidence that omega-3 fatty acid treatment at the prescription strength of 4 g/day effectively and safely lowers triglyceride levels and increases low-density lipoprotein size, as well as affecting high-density lipoprotein metabolism.57 Daily doses of > 6 capsules are required to reduce triglyceride concentrations for brands where a 1 g capsule contains 300 mg of omega-3 fatty acids (180 mg eicosapentaenoic acid and 120 mg docosahexaenoic acid).45
Cocoa butter, a fat derived from cocoa plants and found predominantly in dark chocolate, contains an average of 33% monounsaturated oleic acid and 33% stearic acid.43 In hypertensive patients, daily consumption of 100 g flavonoid-rich chocolate over 2 weeks led to a significant 12% reduction of serum total and LDL cholesterol levels. Moreover, cocoa appears to inhibit LDL oxidation.43
Promoting liver choleresis
Bile acids and their salts are essential in the management of cholesterol levels. They are synthesised from cholesterol in the liver. There are three groups: primary (cholic and chenodeoxycholic acids and their salts); secondary, made from the action of bacteria on primary bile salts (deoxycholate and lithocholate); and tertiary, the result of modifications of secondary bile salts (sulfate ester of lithocholate, ursodeoxycholate and 7-β-epimer of chenodeoxycholate).58 Saponins have been implicated in interference with the absorption of cholesterol, bile acids and fats and have shown potential in the reduction of blood cholesterol levels.58 Their cholesterol-lowering effect may also be linked to their binding of bile salts and increasing their faecal excretion.
Choleretics stimulate bile production by hepatocytes and most have effective cholagogue properties as well.58 One such compound is Cynara scolymus. A study of 75 normal and moderately hypercholesterolaemic volunteers reduced plasma cholesterol
by 4.2% with daily intake of C. scolymus of 1280 mg daily for 12 weeks.59 In 2004 an in vitro study of an artichoke leaf extract increased the activity of an endothelial nitric oxide synthase (eNOS) promoter.60 The compounds thought responsible were the flavonoids luteolin and cynaroside, with the activity suspected to occur through an increase in eNOS gene transcription.60 Apart from this choleretic activity C. scolymus has long been known for its antioxidant ability. The antioxidant activity of the herbal medicine was confirmed by its in vitro inhibition of LDL oxidation in rats. It also raised levels of glutathione peroxidase activity.61 A methanolic extract of C. scolymus was found to suppress serum triglyceride elevation in olive loaded mice, with the components responsible identified as sesquiterpenes (cynaropicrin, aguerin B and grosheimin) together with new sesquiterpene glycosides (cynarascolosides A, B and C).62 Curcuma comosa is an indigenous plant of Thailand, and has been traditionally and widely used as an anti-inflammatory agent for the treatment of postpartum uterine bleeding and uterine inflammation.63 The administration of an ethyl acetate extract to hypercholesterolaemic hamsters (0–500 mg/kg for 7 days) resulted in a decrease in both plasma triglyceride and cholesterol levels in a dose-dependent manner.64 Plasma HDL-C was increased and LDL-C was reduced; there is also a choleretic effect.64
INTEGRATIVE MEDICAL CONSIDERATIONS
Current evidence tentatively supports yoga, acupuncture and traditional Chinese medicine in the treatment of cardiovascular disease. Yoga retarded progression and increased regression of coronary atherosclerosis in patients with severe coronary artery disease.65 Acupuncture and moxibustion improved the plaque of carotid atherosclerosis, so as to alleviate and prevent the development of ischaemic cerebrovascular disease.66 A study of the press needle technique repeated 200 times up to four times a day showed a reduction in triglycerides, homocysteine and CRP.67 A small study (40 patients) showed the return to normal levels of plasma endothelin, nitric oxide and raised apoprotein after treatment with collateral circulation in a limb affected by atherosclerosis increased significantly as well.68
Example treatment
Prescription
As a minimum all patients should be given dietary advice. This includes enriching their diet with a good source of fibre, both soluble and insoluble, between 10 and 60 g daily.69,70 Smoking should be ceased,71,33 and alcohol intake should be reduced to a moderate level (1–2 glasses of wine daily) at a minimum.72 Binge drinking, particularly with beer and spirits, should be discouraged. The optimal level of exercise is the equivalent of 30 minutes daily of an intensity at 65–80% peak oxygen consumption.73,74 Cynara scolymus is beneficial as an antioxidant and choleretic.59,61 It reduced the oxidative effects on plasma lipids at the same time as reducing their levels.62 The effect on nitric oxide adds a vasodilator dimension to its actions.60 Allium sativum is beneficial for both its anti-thrombotic activity and its ability to modify lipid components, in raising HDL-C levels and reducing both TG and LDL-C fractions in the blood.54,52 Crataegus oxyacantha possesses many cardiovascular benefits, particularly the ability to reduce resting diastolic blood pressure and heart rate as well as potent antioxidant activity. Coenzyme Q10 has a
positive effect in reducing oxidative stress as well as other antiatherogenic effects.40,41 Policosanol has a more definite therapeutic application, particularly in reducing the more harmful blood fat fractions such as LDL-C, TG and total cholesterol (TC).47,48 Fish oil is prescribed to modify plasma lipid profiles to increase HDL-C levels while reducing LDL-C and TG fractions at the same time.57,45 Its ability to reduce platelet aggregation makes it particularly good for patients at risk of infarction and stroke. The liquids available to practitioners may contain almost 2.8 g of omega-3 fatty acids per 5 mL; this makes it easy to use doses of approximately 6 g (equivalent to 6 × 1000 mg capsules) daily. At this level profound changes may occur to blood lipid profiles.
Dietary and lifestyle modification
Irrespective of whether an individual is in a high-risk category, dietary treatment should be the first line of therapy for dyslipidaemia. Naturally most protocols are centred on fat reduction and an increase in fibre intake. The National Cholesterol Education Program Adult Treatment Panel III (ATP III)75 recommends a low saturated fat diet with nutritional supplementation (fish oil, oat bran or plant sterol). These diets generally lowered total cholesterol TC (7–18%) and LDL-C (7–15%) concentrations respectively.75 Intake of dietary fibre lowers plasma lipids. Soluble fibre lowers serum TC and LDL-C without significant alteration in serum HDL-C and TG.70 Subjects taking 45 g of oats daily as part of a low caloric diet for 6 weeks had significant reductions in both total cholesterol and LDL-C.69 A trial in 2008 compared a low-fat diet with a very low carbohydrate diet. Each diet provided around 1500 kcal daily and around 20–30 g protein. The diets differed in the amount of carbohydrate, being 12% for the low-carbohydrate diet and 56% for the low fat diet. The low-carbohydrate diet was higher in fats, particularly omega-6 fatty acids. The very low carbohydrate diet resulted in profound alterations in fatty acid composition and reduced inflammation compared to a low-fat diet, as well as reducing atherosclerosis.76
Lifestyle advice includes alcohol and smoking reduction. The importance of smoking as a risk factor for coronary heart disease is beyond doubt. Free radicals in cigarette smoke cause oxidative damage to proteins, DNA and lipids, contributing to the pathobiology of atherosclerosis, heart disease and cancer. As an exercise to estimate the magnitude of risk reduction, a review of 20 independent studies was undertaken.71 Cessation of smoking was associated with a substantial reduction in risk of all-cause mortality among patients with CHD of 36%.71 Both active and passive smoking raise plasma levels of CRP.33 This causes oxidative damage to vascular endothelium via raised plasma fats. Light to moderate alcohol intake is known to have cardioprotective properties; however, the magnitude of protection depends on other factors and may be confined to some subsets of the population.72 This cardioprotective effect seems to be larger among middle-aged and elderly adults than young adults. The levels of alcohol at which the risk of CHD is lowest and the levels of alcohol at which the risk of CHD exceeds the risk among abstainers are lower for women than for men. There is evidence to suggest that wine may have more beneficial effects than beer and distilled spirits, with drinking patterns producing greater risk reduction through steady versus binge drinking.72
At the patient’s age, a vigorous exercise program is recommended to be based on her physical ability (after medical consultation). Exercise is well known to have positive effects on lipid levels. A 2002 study attempted to ascertain the level of exercise required to produce optimal benefits. A level of exercise equivalent to jogging 32.0 km per week at 65 to 80% of peak oxygen consumption resulted in greater improvements than did the lower amounts of exercise, which were the equivalent of jogging
19.2 km a week at 65 to 80% of peak oxygen consumption and the equivalent of walking 19.2 km per week at 40 to 55% of peak oxygen consumption. All three interventions were superior to the control condition in all 11 lipid variables.74 In the National Cholesterol Education Program mentioned above, exercise intervention increased HDL-C levels (5–14%) and lowered TG levels (4–18%) respectively.75 A meta-analysis of studies on the effect of exercise on identifiable risk factors in 8440 patients indicated a pooled effect estimate for total mortality reduction of 27%.73 As well as this, there was a significant net reduction in total cholesterol of –0.57 mmol/L and LDL-C –0.51 mmol/L respectively.73
McCully K.S. Homocysteine, vitamins and vascular disease prevention. Am J Clin Nutr. 2007;86(5):1563S-1568S.
Therapeutic guidelines, cardiovascular (version 5). Online. Available: http://etg.hcn.net.au/. Accessed 23 April 2009.
Topol E.J., et al. Textbook of cardiovascular medicine, 3rd edn. Lippincott Williams & Wilkins, 2007.
1. Topol E.J., editor. Textbook of cardiovascular medicine, 3rd edn, Philadelphia: Lippincott Williams & Wilkins, 2007.
2. Peck M.D., Ai A.L. Cardiac conditions. J Gerontol Soc Work. 2008;50(Suppl 1):13-44.
3. Vanuzzo D., et al. [Cardiovascular risk and cardiometabolic risk: an epidemiological evaluation]. G Ital Cardiol (Rome). 2008;9(4 Suppl 1):6S-17S.
4. Harrison’s principles of internal medicine. Online [cited 2009, June 2]. Available: http://www.accessmedicine.com/content.aspx?aID=2872140&searchStr=atherosclerosis.
5. Arora S., Nicholls S.J. Atherosclerotic plaque reduction: blood pressure, dyslipidemia, atherothrombosis. Drugs Today (Barc). 2008;44(9):711-718.
6. Rizzo M., et al. Atherogenic dyslipidemia and oxidative stress: a new look. Transl Res. 2009;153(5):217-223.
7. Easton J.D., et al. Definition and evaluation of transient ischemic attack: a scientific statement for healthcare professionals from the American Heart Association/American Stroke Association Stroke Council; Council on Cardiovascular Surgery and Anesthesia; Council on Cardiovascular Radiology and Intervention; Council on Cardiovascular Nursing; and the Interdisciplinary Council on Peripheral Vascular Disease. The American Academy of Neurology affirms the value of this statement as an educational tool for neurologists. Stroke. 2009;40(6):2276-2293.
8. Sobenin I.A., et al. [Reduction of cardiovascular risk in primary prophylaxy of coronary heart disease]. Klin Med (Mosk). 2005;83(4):52-55.
9. Rodriguez G., et al. [Role of inflammation in atherogenesis]. Invest Clin. 2009;50(1):109-129.
10. Liapis C.D., et al. What a vascular surgeon should know and do about atherosclerotic risk factors. J Vasc Surg. 2009;49(5):1348-1354.
11. McCully K.S. Homocysteine, vitamins and vascular disease prevention. Am J Clin Nutr. 2007;86(5):1563S-1568S.
12. Ferretti G., et al. Homocysteinylation of low-density lipoproteins (LDL) from subjects with Type 1 diabetes: effect on oxidative damage of human endothelial cells. Diabetic Med. 2006;23(7):808-813.
13. Su J., et al. A comparative study on pathogenic effects of homocysteine and cysteine on atherosclerosis. Wei Sheng Yan Jiu. 2009;38(1):43-46.
14. Harrison D.G., Gongora M.C. Oxidative stress and hypertension. Med Clin North Am. 2009;93(3):621-635.
15. Roberts C.K., Sindhu K.K. Oxidative stress and metabolic syndrome. Life Sci. 2009;84(21–22):705-712.
16. Higashi Y., et al. Endothelial function and oxidative stress in cardiovascular diseases. Circ J. 2009;73(3):411-418. [Epub 2009, Feb 4]
17. Therapeutic guidelines, cardiovascular (version 5). Online. Available: http://etg.hcn.net.au/. Accessed 23 April 2009.
18. Cottart C.H., et al. [Biology of arterial ageing and arteriosclerosis.]. C R Biol. 2009;332(5):433-447.
19. Roy H., et al. Molecular genetics of atherosclerosis. Hum Genet. 2009;125(5–6):467-491.
20. Walker C., Reamy B.V. Diets for cardiovascular disease prevention: what is the evidence? Am Fam Physician. 2009;79(7):571-578.
21. Cesari M., et al. Homocysteine-lowering treatment in coronary heart disease. Curr Med Chem Cardiovasc Hematol Agents. 2005;3(4):289-295.
22. Garces P.A., et al. [Lowering plasma homocysteine with vitamins B6, B12, and folic acid. Effect on lipids concentration in patients with secondary hyperlipoproteinemia type IV, with and without Lovastatin treatment]. Arch Latinoam Nutr. 2006;56(1):36-42.
23. Bunout D., et al. Effects of supplementation with folic acid and antioxidant vitamins on homocysteine levels and LDL oxidation in coronary patients. Nutrition. 2000;16(2):107-110.
24. Bleie O., et al. Homocysteine-lowering therapy does not affect inflammatory markers of atherosclerosis in patients with stable coronary artery disease. J Intern Med. 2007;262(2):244-253.
25. Wotherspoon F., et al. The effect of oral folic acid upon plasma homocysteine, endothelial function and oxidative stress in patients with type 1 diabetes and microalbuminuria. Int J Clin Pract. 2008;62(4):569-574.
26. Yilmaz H., et al. Effects of folic acid and N-acetylcysteine on plasma homocysteine levels and endothelial function in patients with coronary artery disease. Acta Cardiol. 2007;62(6):579-585.
27. Graf S., et al. 13N-ammonia rest/stress PET: folic acid improves global coronary vasoreactivity in coronary artery disease patients with normal or elevated homocysteine levels. Nuklearmedizin. 2006;45(6):248-253.
28. Manna C., et al. Olive oil phenolic compounds inhibit homocysteine-induced endothelial cell adhesion regardless of their different antioxidant activity. J Agric Food Chem. 2009;57(9):3478-3482.
29. Ben-Assayag E., et al. Association of the -757T>C polymorphism in the CRP gene with circulating C-reactive protein levels and carotid atherosclerosis. Thromb Res. 2009;24(4):458-462.
30. Belza A., et al. Effect of diet-induced energy deficit and body fat reduction on high-sensitive CRP and other inflammatory markers in obese subjects. Int J Obes (Lond). 2009;33(4):456-464.
31. North C.J., et al. The effects of dietary fibre on C-reactive protein, an inflammation marker predicting cardiovascular disease. Eur J Clin Nutr. 2009;63(8):921-933.
32. Koenig W., et al. C-reactive protein, a sensitive marker of inflammation, predicts future risk of coronary heart disease in initially healthy middle-aged men: results from the MONICA (Monitoring Trends and Determinants in Cardiovascular Disease) Augsburg Cohort Study, 1984 to 1992. Circulation. 1999;99(2):237-242.
33. Block G., et al. Plasma C-reactive protein concentrations in active and passive smokers: influence of antioxidant supplementation. J Am Coll Nutr. 2004;23(2):141-147.
34. Singh U., Devaraj S., Vitamin E. inflammation and atherosclerosis. Vitam Horm. 2007;76:519-549.
35. Yang R.L., et al. Lipoic acid prevents high-fat diet-induced dyslipidemia and oxidative stress: a microarray analysis. Nutrition. 2008;24(6):582-588.
36. Amom Z., et al. Lipid lowering effect of antioxidant alpha-lipoic acid in experimental atherosclerosis. J Clin Biochem Nutr. 2008;43(2):88-94.
37. Accinni R., et al. Effects of combined dietary supplementation on oxidative and inflammatory status in dyslipidemic subjects. Nutr Metab Cardiovasc Dis. 2006;16(2):121-127.
38. Suarna C., et al. Protective effect of vitamin E supplements on experimental atherosclerosis is modest and depends on preexisting vitamin E deficiency. Free Radic Biol Med. 2006;41(5):722-730.
39. Kartal Ozer N., et al. Molecular mechanisms of cholesterol or homocysteine effect in the development of atherosclerosis: role of vitamin E. Biofactors. 2003;19(1–2):63-70.
40. Chapidze G.E., et al. [Combination treatment with coenzyme Q10 and simvastatin in patients with coronary atherosclerosis]. Kardiologiia. 2006;46(8):11-13.
41. Chapidze G., et al. Prevention of coronary atherosclerosis by the use of combination therapy with antioxidant coenzyme Q10 and statins. Georgian Med News. 2005;118:20-25.
42. Yang P.Y., et al. Reduction of atherosclerosis in cholesterol-fed rabbits and decrease of expressions of intracellular adhesion molecule-1 and vascular endothelial growth factor in foam cells by a water-soluble fraction of Polygonum multiflorum. J Pharmacol Sci. 2005;99(3):294-300.
43. Corti R., et al. Cocoa and cardiovascular health. Circulation. 2009;119:1433-1441.
44. Davidson M. Niacin use and cutaneous flushing: mechanisms and strategies for prevention. Am J Cardiol. 2008;101(8 Suppl. 1):S14-S19.
45. Australian medicines handbook. Adelaide: Australian Medicines Handbook Pty Ltd; 2009.
46. Berra K. Clinical update on the use of niacin for the treatment of dyslipidemia. J Am Acad Nurse Pract. 2004;16(12):526-534.
47. Castano G., et al. A randomized, double-blind, placebo-controlled study of the efficacy and tolerability of policosanol in adolescents with type II hypercholesterolemia. Curr Ther Res Clin Exp. 2002;63(4):286-303.
48. Castano G., et al. Effects of policosanol and lovastatin on lipid profile and lipid peroxidation in patients with dyslipidemia associated with type 2 diabetes mellitus. Int J Clin Pharmacol Res. 2002;22(3–4):89-99.
49. Francini-Pesenti F., et al. Sugar cane policosanol failed to lower plasma cholesterol in primitive, diet-resistant hypercholesterolemia: a double blind, controlled study. Complement Ther Med. 2008;16(2):61-65.
50. Francini-Pesenti F., et al. Effect of sugar cane policosanol on lipid profile in primary hypercholesterolemia. Phytother Res. 2008;22(3):318-322.
51. Mansoor G.A. Herbs and alternative therapies in the hypertension clinic. Am J Hypertens. 2001;14:971-975.
52. Rahman K., Lowe G.M. Garlic and cardiovascular disease: a critical review. J Nutr. 2006;136(3 Suppl):736S-740S.
53. Jeyaraj S., et al. Effect of combined supplementation of fish oil with garlic pearls on the serum lipid profile in hypercholesterolemic subjects. Indian Heart J. 2005;57(4):327-331.
54. Ashraf R., et al. Effects of garlic on dyslipidemia in patients with type 2 diabetes mellitus. J Ayub Med Coll Abbottabad. 2005;17(3):60-64.
55. Tassoni D., et al. The role of eicosanoids in the brain. Asia Pac J Clin Nutr. 2008;17(Suppl 1):220-228.
56. Casos K., et al. Atherosclerosis prevention by a fish oil-rich diet in apoE(–/–) mice is associated with a reduction of endothelial adhesion molecules. Atherosclerosis. 2008;201(2):306-317.
57. Goldberg R.B., Sabharwal A.K. Fish oil in the treatment of dyslipidemia. Curr Opin Endocrinol Diabetes Obes. 2008;15(2):167-174.
58. Mills S., Bone K. Principles and practice of phytotherapy. St Louis: Churchill Livingstone; 2000.
59. Bundy R., et al. Artichoke leaf extract (Cynara scolymus) reduces plasma cholesterol in otherwise healthy hypercholesterolemic adults: a randomized, double blind placebo controlled trial. Phytomedicine. 2008;15(9):668-675.
60. Li H., et al. Flavonoids from artichoke (Cynara scolymus L.) up-regulate endothelial-type nitric-oxide synthase gene expression in human endothelial cells. J Pharmacol Exp Ther. 2004;310(3):926-932.
61. Jimenez-Escrig A., et al. In vitro antioxidant activities of edible artichoke (Cynara scolymus L.) and effect on biomarkers of antioxidants in rats. J Agric Food Chem. 2003;51(18):5540-5545.
62. Shimoda H., et al. Anti-hyperlipidemic sesquiterpenes and new sesquiterpene glycosides from the leaves of artichoke (Cynara scolymus L.): structure requirement and mode of action. Bioorg Med Chem Lett. 2003;13(2):223-228.
63. Jantaratnotai N., et al. Inhibitory effect of Curcuma comosa on NO production and cytokine expression in LPS-activated microglia. Life Sci. 2006;78(6):571-577.
64. Piyachaturawat P., et al. Reduction of plasma cholesterol by Curcuma comosa extract in hypercholesterolaemic hamsters. J Ethnopharmacol. 1999;66(2):199-204.
65. Manchanda S.C., et al. Retardation of coronary atherosclerosis with yoga lifestyle intervention. J Assoc Physicians India. 2000;48(7):687-694.
66. Wang W.Z., et al. [Clinical study on acupuncture and moxibustion for treatment of plaque of carotid atherosclerosis]. Zhongguo Zhen Jiu. 2005;25(5):312-314.
67. Omura Y., et al. Anatomical relationship between traditional acupuncture point ST 36 and Omura’s ST 36 (True ST 36) with their therapeutic effects: 1) inhibition of cancer cell division by markedly lowering cancer cell telomere while increasing normal cell telomere, 2) improving circulatory disturbances, with reduction of abnormal increase in high triglyceride, L-homocystein, CRP, or cardiac troponin I, T in blood by the stimulation of Omura’s ST 36–Part 1. Acupunct Electrother Res. 2007;32(1–2):31-70.
68. Wang C., et al. [Clinical study on treatment of atherosclerosis obliterans by integrated traditional Chinese and Western medicine]. Zhongguo Zhong Xi Yi Jie He Za Zhi. 2000;20(11):828-830.
69. Saltzman E., et al. An oat-containing hypocaloric diet reduces systolic blood pressure and improves lipid profile beyond effects of weight loss in men and women. J Nutr. 2001;131(5):1465-1470.
70. Mia M.A., et al. Dietary fibre and coronary heart disease. Mymensingh Med J. 2002;11(2):133-135.
71. Critchley J.A., Capewell S. Smoking cessation for the secondary prevention of coronary heart disease. Cochrane Database Syst Rev. (4):2003;. CD003041
72. Tolstrup J., Gronbaek M. Alcohol and atherosclerosis: recent insights. Curr Atheroscler Rep. 2007;9(2):116-124.
73. Jolliffe J., et al. Exercise-based rehabilitation for coronary heart disease. Cochrane Database Syst Rev. (1):2001;. CD003316
74. Kraus W.E., et al. Effects of the amount and intensity of exercise on plasma lipoproteins. N Engl J Med. 2002;347(19):1483-1492.
75. Varady K.A., Jones P.J. Combination diet and exercise interventions for the treatment of dyslipidemia: an effective preliminary strategy to lower cholesterol levels? J Nutr. 2005;135(8):1829-1835.
76. Forsythe C.E., et al. Comparison of low fat and low carbohydrate diets on circulating fatty acid composition and markers of inflammation. Lipids. 2008;1:65-77.
[/level-membership-for-internal-medicine-category][not-level-membership-for-internal-medicine-category]
Chapter 9 Atherosclerosis and dyslipidaemia
EPIDEMIOLOGY AND AETIOLOGY
The association between elevated serum cholesterol and coronary heart disease (CHD) was first reported in the 1930s, and subsequent large epidemiologic studies confirmed the strong relationship between serum cholesterol and CHD.1–3 Current predictions estimate that by the year 2020 cardiovascular diseases, notably atherosclerosis, will become the leading global cause of total disease burden.4 Lipoprotein disorders or dyslipidaemias are common and are an important risk factor for coronary artery disease and all types of atherosclerotic vascular disease.5 Cardiovascular risk was found to be positively associated with low-density lipoprotein (LDL-C) levels and inversely with high density lipoprotein (HDL-C) levels.6 The relationship between fasting serum triglyceride (TG) and cardiovascular risk has been confounded by the inverse association between TG and HDL-C, as well as by the association of TG with other risk factors such as diabetes mellitus and body mass (see Table 9.1).6 Triglycerides alone are now considered an important independent predictor of cardiovascular risk.1
| CAUSE | EFFECT ON LIPID PROFILE |
|---|---|
Source: Therapeutic Guidelines (Cardiovascular).17
Atherosclerosis affects various regions of the circulation and will present with unique clinical symptoms depending on the particular vascular bed affected. In coronary arteries it causes myocardial infarction and angina, whereas stroke and transient cerebral ischaemia can occur when it is present in the arteries of the nervous system.2,7 In the peripheral circulation it can result in intermittent claudication and gangrene, which can jeopardise limb viability.4
Atherogenesis or the pathogenesis of atherosclerosis usually occurs over decades in a discontinuous fashion with relative periods of quiescence punctuated with periods of rapid evolution.3 The eventual clinical manifestation may be chronic, as is the case in angina, or acute, such as a heart attack or stroke or sudden death. Regardless, individuals may live with atherosclerosis and experience no symptoms or reduced life expectancy and morbidity.
The ‘fatty streak’ represents the initial lesion, usually due to focal increases in the density of lipoproteins within the region of the intima of a blood vessel; hence the reason for vigilance in maintaining low levels of these particles in the bloodstream.8 The lipoproteins often associate with glycosaminoglycans in the arterial extracellular matrix and become more ‘fixed’ in the vasculature. These lipoprotein macromolecules undergo oxidative modifications, giving rise to hydroperoxides, lysophospholipids, oxysterols and aldehydic breakdown products.4 Constituents of oxidatively modified LDL-C can augment expression of leukocyte adhesion molecules, recruiting leukocytes and macrophages into the area, which undergoes inflammation.9
Areas of inflammation produce altered laminar flow in the arteries affected, and reduce the production of nitric oxide, which is locally both vasodilatory and anti-inflammatory. The inflammatory process eventually penetrates or tears the endothelial layer with the products of lipoprotein oxidation, inducing cytokines and tumour necrosis factor alpha (TNF-α) released from the vascular cell walls.4 Once inside the walls of the artery mononuclear phagocytes mature into macrophages and become foam cells.
The cytokines and TNF-α produce growth factors that induce the smooth muscle to produce local growth factors such as platelet-derived growth factor and fibroblast growth factors.9 This fibro-fatty material recruits more smooth muscle cells into the intima from the tunica media layer of the vessel. At this point transforming growth factor beta (TGF-β), amongst other mediators, stimulates collagen production by the smooth muscle cells transforming the fatty streak into a more fibrous smooth-muscle cell and extracellular-rich lesion.4 In addition to this process products of blood coagulation and thrombosis contribute to atheroma evolution, which justifies the alternative term for the process, ‘atherothrombosis’ (see Figure 9.1).
Unfortunately the prevalence of traditional risk factors is almost as high in those without cardiovascular disease as in subsequently affected individuals.1 Recently, several novel key biomarkers have been implicated in the pathology of atherosclerosis. There are several, but the more commonly researched include C-reactive protein (CRP), homocysteine (Hcy) and indicators of oxidative stress.8,10
C-reactive protein (CRP)
CRP is a calcium-binding pentameric protein consisting of five identical, non-covalently linked 23-kDa subunits.1 It is present in trace amounts in humans and appears to have been highly conserved over hundreds of millions of years. It is synthesised primarily by hepatocytes in response to activation of several cytokines, such as interleukins 1 and 6, and TNF-α.1 In healthy people in the absence of active inflammatory states, CRP levels are usually below 1 mg/L. CRP is expressed in atherosclerotic plaque and may enhance expression of local adhesion molecules, increase expression of endothelial plasminogen activation inhibitor 1, reduce endothelial nitric oxide bioactivity and alter low-density lipoprotein (LDL) cholesterol by macrophages.10
Homocysteine (Hcy)
The homocysteine theory of arteriosclerosis attributes one of the underlying causes of vascular disease to elevation of blood homocysteine concentrations as the result of dietary, genetic, metabolic, hormonal or toxic factors.11 Dietary deficiency of vitamin B6 and folic acid and absorptive deficiency of vitamin B12, which result from traditional food processing or abnormal absorption of B vitamins, are important factors in causing elevations in blood homocysteine.11,10 It is believed that homocysteinylation of lipoproteins increases their atherogenicity and hence their cytotoxic effects on endothelial tissue.12 It is hypothesised that oxidative stress and methylative modification is the mechanism by which Hcy is pro-atherosclerotic.13
Oxidative stress
Endothelial dysfunction is the initial step in the pathogenesis of atherosclerosis.14,15 Nitric oxide (NO) plays an important role in the regulation of vascular tone and the suppression of smooth muscle cell proliferation. An increase in oxidative stress inactivates NO, impairing endothelium-dependent vasodilation and creating endothelial dysfunction.16 Dyslipidaemia may be primary or secondary. The secondary causes are shown in Table 9.1, and should be looked for in patients and treated appropriately. If secondary causes have been excluded, primary dyslipidaemia due to genetic and environmental (particularly dietary) factors is diagnosed.17
RISK FACTORS
The general risk factors applicable to atherosclerosis relate to lifestyle choices and genetics. Overall estimates of the heritability of myocardial infarction, one of the major clinical outcomes of atherosclerosis, have varied widely, ranging from very little to greater than 50%.18,19 It is important to note that heritability varies from population to population. In populations in which the environment is relatively homogeneous, for example in Iceland, genetics will tend to predominate, whereas in populations exhibiting large environmental differences, for example in Los Angeles, genetics are relatively less important.1 The known risk factors for atherosclerosis generally exhibit very significant heritability estimates as well (see Figure 9.2). Smoking and alcohol consumption as well as a diet rich in refined sugars and saturated fats will adversely influence the condition (see the box below).20 Hypertension, diabetes and obesity have a pronounced negative influence on the disease.1 Exercise is also a significant factor.3 In women the protective effects of oestrogen are greatly diminished in menopause.1 There are several medical conditions that drastically increase the risk associated with this condition, and all of these may cause altered blood fat profiles as secondary to their primary condition.
The influence of environmental factors on lipoprotein metabolism and cardiovascular disease has been dramatically revealed by comparisons of different populations.3 Studies have shown that intrauterine under-nutrition is associated with obesity and other detrimental metabolic characteristics in adulthood.1 In particular under-nutrition
GENETIC AND ENVIRONMENTAL RISK FACTORS FOR CARDIOVASCULAR DISEASE1
was associated with a premature onset of neonatal leptin surge, which appears to be involved in the formation of the energy-regulation circuits in the hypothalamus that affect metabolism in adults.1
CONVENTIONAL TREATMENT
The assessment of dyslipidaemia involves the measurement of fasting total cholesterol (TC), high density lipoprotein (HDL-C), low-density lipoprotein (LDL-C) and triglyceride (TG).10 Fasting usually takes place from midnight prior to the blood test.
Dyslipidaemia is treated in cases of:
Source: Therapeutic Guidelines (Cardiovascular).17
The question that needs to be addressed in relation to dyslipidaemia is: who should in fact be treated and when? Tables 9.3 and 9.4 list the current Australian medical protocols. The aim of treatment is to reduce the progression of atherosclerosis and improve survival rates of individuals. An even more important aim is to prevent myocardial infarct (heart attack) and stroke in patients with established cardiovascular disease. A reduction in triglyceride levels should prevent the occurrence of pancreatitis in susceptible individuals as well. Common pharmacological interventions are listed in Table 9.5.
Table 9.3 Patients at increased risk and warranting therapy, based on their fasting lipid levels
| PATIENT CHARACTERISTICS | LIPID LEVEL (FASTING) |
|---|---|
| Diabetes mellitus, but the patient is not in the ‘very high risk’ category (see below). | Total cholesterol > 5.5 mmol/L |
| Aboriginal or Torres Strait Islander Hypertension | |
| HDL cholesterol < 1 mmol/L | Total cholesterol > 6.5 mmol/L |
| Familial hypercholesterolaemia identified by:
Family history of coronary heart disease: |
If aged 18 years or less at treatment initiation:
If aged more than 18 years at treatment initiation: |
| Patients not already identified who are: | Total cholesterol > 7.5 mmol/L or triglyceride > 4 mmol/L |
| Patients not already identified | Total cholesterol > 9 mmol/L or triglyceride > 8 mmol/L |
Source: Therapeutic Guidelines (Cardiovascular).17
Table 9.4 Patients in the ‘very high risk’ category warranting therapy regardless of their fasting lipid levels
| The following patients are in the ‘very high risk’ category, regardless of their fasting lipid levels: |
Source: Therapeutic Guidelines (Cardiovascular).17
Table 9.5 Pharmacological treatment of dyslipidaemia
| DRUG | BENEFIT | DISADVANTAGE |
|---|---|---|
| Statin | Reduces LDL-C by 30–50%. Best tolerated | Reduces coenzyme Q10 |
| Bile-acid binding resin | Reduces LDL-C by 15–25% | Poorly tolerated, may worsen hypertriglyceridaemia |
| Nicotinic acid | Reduces LDL-C by 15–30%, triglycerides by 25–40%. Increases HDL-C by 20–35% | Causes severe flushing, which limits use |
| Ezetimibe | Reduces LDL-C by 18%. Well tolerated | Generally not used alone |
| Fibrate | Reduces LDL-C by 18%,∗ and triglycerides by 40–80%. Increases HDL-C by 10–30% | ∗Can increase LDL-C in some instances. |
Source: Taken from Australian Medicines Handbook.45
KEY TREATMENT PROTOCOLS
Plasma total homocysteine (Hcy) has been associated with cardiovascular risk in multiple large-scale epidemiological studies, and it has been considered as an independent risk factor for atherosclerosis.21 Evidence indicates that although mild hyper-Hcy may be regarded as a minor risk factor for CHD in low-risk patients, it can play a role in triggering new events in patients with known CHD, also by interacting with the
‘classical’ CV risk factors.21 The administration of vitamins B6 (300 mg/day) and B12 (250 μg/day) and folic acid (10 mg/day) over a 120-day period reduced plasma Hcy levels by 24%; this in turn resulted in a 28.5% reduction in coronary risk.22 In a study of 23 patients with angiographically proven coronary artery disease, daily dosing with a multivitamin containing 0.65 mg folic acid, 150 mg alpha-tocopherol, 150 mg ascorbic acid, 12.5 mg beta-carotene and 0.4 mg B12 produced a 28% reduction in Hcy and a decrease in in vitro LDL oxidation of 39%.23
[/not-level-membership-for-internal-medicine-category]
