[level-membership-for-neurology-category]
CHAPTER 52 ASSESSMENT AND MANAGEMENT PRINCIPLES
By virtue of their capacity to divert the functionality of neural networks into clinically overt discharges, epileptic seizures represent a fascinating window onto brain functions and also the source of a virtually infinite variety of ictal manifestations. The epilepsies encompass a large variety of syndromes reflecting a multitude of brain lesions as well as gene and protein dysfunction that results in neuronal hyperexcitability. An ever-increasing understanding, described in an exponentially growing number of dedicated textbooks, translates into a capacity for more precise diagnosis and optimization of syndrome-dependent management, compounded by the flurry of antiepileptic drugs made available for seizure treatment in the last two decades.
DIAGNOSIS STEP 1: DEFINING THE SEIZURE TYPE(S)
Investigating the Seizure Episode
Validating the Core Features of Epileptic Seizures
“Desperately Seeking” a Witness to the Seizure(s)
If a seizure is primarily characterized by a lack of responsiveness, particular attention should be paid to the presence of automatisms, which, though often noticed, are infrequently reported spontaneously by witnesses. The diagnostic value of oroalimentary automatisms has been mentioned, the same is true for manual, pedal, and verbal automatisms, which all strongly suggest an epileptic origin for seizures. These automatic activities tend to imitate seemingly natural or purposeful gestures or speech, although they usually appear meaningless or inappropriate during a seizure. They must be distinguished from elementary motor activity leading to posture, change in muscle tone, clonic jerk, or a scream. In the 1989 classification of seizures and epilepsies, automatisms are specifically associated with complex partial seizures. In fact, as previously described, subtle automatisms may also occur during simple partial seizures and, at times, during absence seizures.
Classifying the Seizure Episode
 The episode was primarily marked by a complete loss of consciousness with a fall if the patient was standing and may have progressed to “convulsions.”
The episode was primarily marked by a complete loss of consciousness with a fall if the patient was standing and may have progressed to “convulsions.”

Seizures Associated With Complete Loss of Consciousness, a Fall, and Convulsive Features
Differential Diagnoses
 Syncope can result from various pathophysiological mechanisms with a common endpoint being a decrease in cerebral perfusion responsible for acute cerebral and brainstem dysfunction. Vasovagal syncope is the most frequent and can be mistaken for GTCS. It typically occurs in adolescents and young adults following emotionally salient stimuli (pain, sight of blood during venous puncture, warm and enclosed atmosphere) or after standing motionless for prolonged periods (related to progressive venous blood sequestration in the lower limbs). Thus, vasovagal syncope usually occurs in the standing position and often aborts if a patient lies down in the early phase of an attack. More rarely, vasovagal syncope will occur in a seated patient at the end of a meal, triggered by digestion-induced splanchnic blood sequestration. Prodromes include vertigo, visual and auditory disturbances, nausea, sweating, and the feeling of an imminent fainting or death. Intense pallor and general hypotonia follow, resulting in a progressive nontraumatic fall and loss of consciousness, with eyes closed or rolled upward. At times, syncope might progress to brief axial hypertonia associated with a few irregular clonic limb movements (fewer than six), “convulsive syncope.” Urination and biting of the tip of the tongue can occur, but normal consciousness is restored much more rapidly than in GTCS. Thus, detailed analysis of all signs and symptoms usually results in a clear distinction of syncope from GTCS, even in the presence of clonic movements, urination, and tongue biting. When necessary, a tilt test can be used to confirm the diagnosis of vasovagal syncope. Cardiogenic syncope represents a less frequent form of attack, observed in older patients with cardiovascular pathology but no other precipitating factors. It is characterized by a more abrupt loss of consciousness and fall.
Syncope can result from various pathophysiological mechanisms with a common endpoint being a decrease in cerebral perfusion responsible for acute cerebral and brainstem dysfunction. Vasovagal syncope is the most frequent and can be mistaken for GTCS. It typically occurs in adolescents and young adults following emotionally salient stimuli (pain, sight of blood during venous puncture, warm and enclosed atmosphere) or after standing motionless for prolonged periods (related to progressive venous blood sequestration in the lower limbs). Thus, vasovagal syncope usually occurs in the standing position and often aborts if a patient lies down in the early phase of an attack. More rarely, vasovagal syncope will occur in a seated patient at the end of a meal, triggered by digestion-induced splanchnic blood sequestration. Prodromes include vertigo, visual and auditory disturbances, nausea, sweating, and the feeling of an imminent fainting or death. Intense pallor and general hypotonia follow, resulting in a progressive nontraumatic fall and loss of consciousness, with eyes closed or rolled upward. At times, syncope might progress to brief axial hypertonia associated with a few irregular clonic limb movements (fewer than six), “convulsive syncope.” Urination and biting of the tip of the tongue can occur, but normal consciousness is restored much more rapidly than in GTCS. Thus, detailed analysis of all signs and symptoms usually results in a clear distinction of syncope from GTCS, even in the presence of clonic movements, urination, and tongue biting. When necessary, a tilt test can be used to confirm the diagnosis of vasovagal syncope. Cardiogenic syncope represents a less frequent form of attack, observed in older patients with cardiovascular pathology but no other precipitating factors. It is characterized by a more abrupt loss of consciousness and fall.

Seizures Primarily Characterized by an Impairment of Consciousness









The large variety of ictal complex partial seizure manifestations cannot be detailed herein, nor the distinctive features of temporal, frontal, parietal, occipital, or insular seizures. These descriptions that are of relevance for localization of seizure onset zones in epilepsy surgery candidates are addressed in Chapters 54.
Differential Diagnosis (Psychogenic Nonepileptic Seizures, Parasomnia)
 Psychogenic nonepileptic seizures can resemble dialeptic seizures especially in patients who have suffered or observed such seizures in others. Automatisms are very unlikely to occur in psychogenic nonepileptic seizures, but tremor-like focal limb movements can be associated with apparent impairment of consciousness. Eye closure with active resistance to opening remains a very suggestive sign of psychogenic nonepileptic seizures in this form of nonepileptic seizure.
Psychogenic nonepileptic seizures can resemble dialeptic seizures especially in patients who have suffered or observed such seizures in others. Automatisms are very unlikely to occur in psychogenic nonepileptic seizures, but tremor-like focal limb movements can be associated with apparent impairment of consciousness. Eye closure with active resistance to opening remains a very suggestive sign of psychogenic nonepileptic seizures in this form of nonepileptic seizure.
Seizures Primarily Characterized by Abnormal Sensations with Preserved Consciousness
Differential Diagnoses of Simple Partial Seizures
 Presyncopal states of vasovagal origin have been described in a previous section. The distinction from epileptic seizures relies on the presence of specific precipitating factors; progressive onset, intense pallor, and a combination of sensory and vegetative symptoms. Whereas many of these symptoms can occur together during seizures, the specific combination of vertigo, visual and auditory disturbances, nausea, sweating, and a feeling of imminent fainting or death is very unlikely to result from an epileptic fit.
Presyncopal states of vasovagal origin have been described in a previous section. The distinction from epileptic seizures relies on the presence of specific precipitating factors; progressive onset, intense pallor, and a combination of sensory and vegetative symptoms. Whereas many of these symptoms can occur together during seizures, the specific combination of vertigo, visual and auditory disturbances, nausea, sweating, and a feeling of imminent fainting or death is very unlikely to result from an epileptic fit.




Generalized myoclonic seizures are described in this section because they are associated with fully preserved consciousness. However, due to their benign features, this type of seizure almost never leads to medical attention or to spontaneous complaint by patients interviewed after a first GTCS. They are primarily encountered in juvenile myoclonic epilepsy and characterized by single or repetitive brief bilateral and symmetrical jerks of proximal limb segments, typically the shoulders, leading to the release of held objects (most likely those used during washing and breakfast due to their morning time of occurrence). Falls may occur when the lower limbs are affected.
Laboratory Investigations of Seizure Episodes
Electroencephalography
 The presence of interictal epileptiform electroencephalographic abnormalities (spikes, sharp waves, spikes and waves) favors an epileptic origin of a seizure but does not exclude a nonepileptic attack, because a few percent of normal individuals demonstrate incidental electroencephalographic findings of these types. Conversely, interictal electroencephalograms often prove normal in epileptic patients, of whom 15% will consistently fail to show epileptiform abnormalities on repeat investigation.
The presence of interictal epileptiform electroencephalographic abnormalities (spikes, sharp waves, spikes and waves) favors an epileptic origin of a seizure but does not exclude a nonepileptic attack, because a few percent of normal individuals demonstrate incidental electroencephalographic findings of these types. Conversely, interictal electroencephalograms often prove normal in epileptic patients, of whom 15% will consistently fail to show epileptiform abnormalities on repeat investigation.
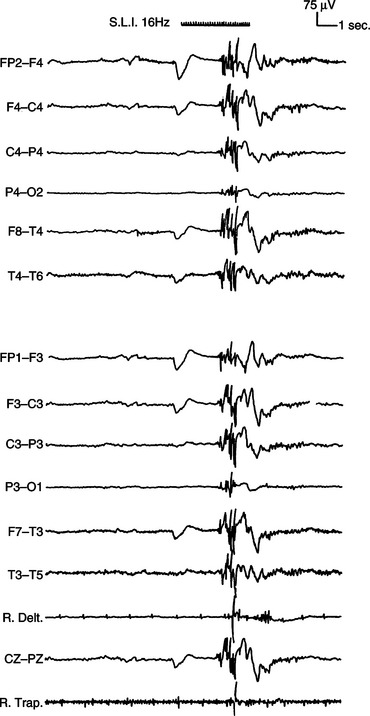
In contrast to these limitations a postictal electroencephalogram has the advantage of being more readily obtained than an ictal electroencephalogram, while still demonstrating abnormalities such as postictal slow waves that are directly related to a preceding seizure (Fig. 52-1). When present, such slow waves strongly support an epileptic origin for attacks and may differentiate between generalized and focal seizures depending on the scalp distribution.
DIAGNOSIS STEP 2: IDENTIFYING THE EPILEPTIC SYNDROME
Information Needed to Identify an Epileptic Syndrome (in Addition to Seizure Type)
Past History
Severe head trauma results in post-traumatic epilepsy in 0.5% to 5% of cases. One should distinguish early post-traumatic seizures occurring during the first week from post-traumatic epilepsy that develops later, although the former is a risk factor for the latter. The delay between head trauma and the onset of post-traumatic epilepsy is less than 1 year in 60% of cases, less than 3 years in 80%, and less than 10 years in 95%. The severity of head trauma is assessed by the presence of the following criteria: penetrating open trauma (associated with a 30% risk of developing post-traumatic epilepsy), intracranial hematoma, any type of computed tomography (CT) scan or MRI-detectable brain lesion, prolonged neurological deficit, coma or post-traumatic amnesia greater than 24 hours, and depressed skull fracture.
Age at Onset
Age at onset is a major diagnostic criterion in the vast majority of idiopathic or generalized epilepsy syndromes. The main exceptions are cryptogenic and symptomatic partial epilepsies, which can occur at virtually any age. Table 52-1 provides an overview of the age-dependent syndromes by increasing age at onset.
TABLE 52-1 Overview of the Age-Dependent Syndromes by Increasing Age at Onset
Electroencephalography
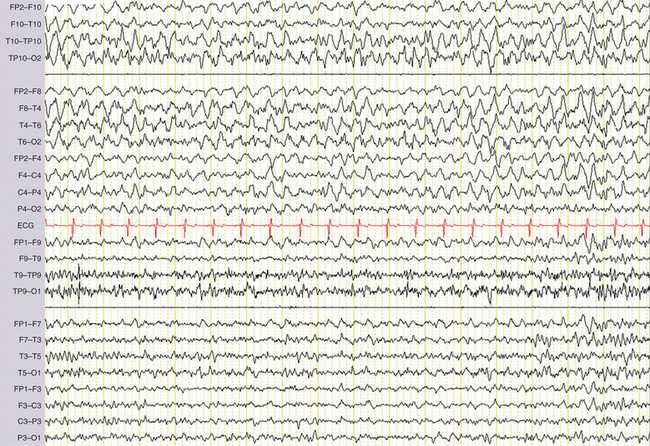
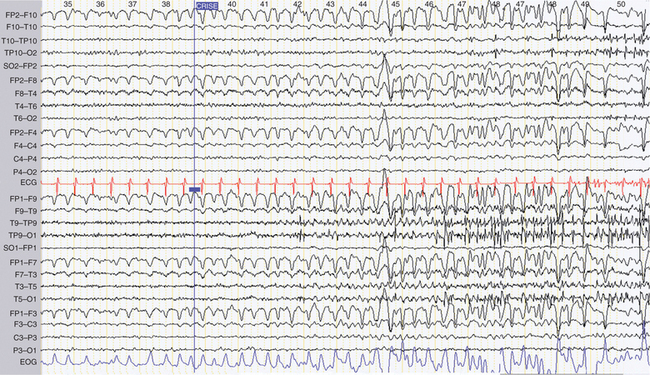
Standard electroencephalography typically lasts 20 minutes, including periods of hyperventilation and intermittent photic stimulation. Hyperventilation favors the occurrence of all types of interictal electroencephalographic abnormalities and can precipitate absences as well as partial seizures. It also elicits physiological slow waves, at times of very high amplitude, in children and young adults. Intermittent photic stimulation can also be responsible for physiological photomyogenic electroencephalographic responses, which should be distinguished from the abnormal photoparoxysmal response, which is characterized by generalized spike-and-wave or polyspike-and-wave discharges that can culminate in a clinically overt seizure (Fig. 52-2). Photoparoxysmal response is observed mainly in juvenile myoclonic epilepsy and in the rare, purely photosensitive epilepsies. Photoparoxysmal response can also be seen in nonepileptic patients, especially in family members of juvenile myoclonic epilepsy probands.
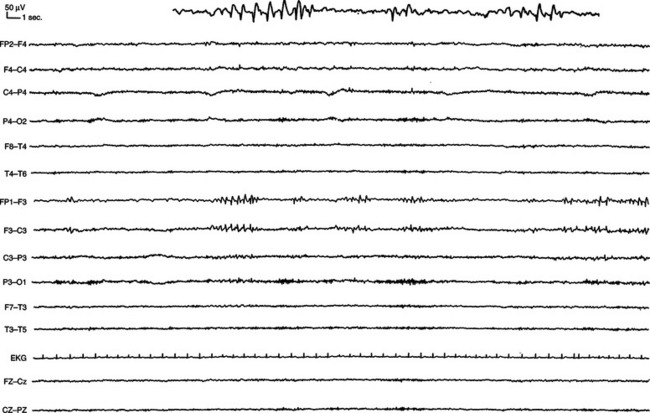
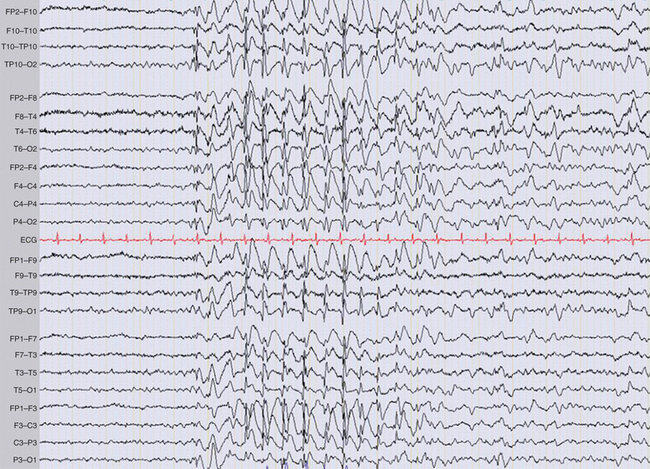
Patients with epilepsy can show epileptiform abnormalities, including focal or generalized, symmetrical or asymmetrical spikes (duration less than 80 milliseconds), sharp waves (duration, 80 to 200 milliseconds), polyspike, spike-and-wave, and polyspike-and-wave complexes that can be isolated or assembled into rhythmic bursts or discharges of various durations, amplitudes, and frequencies (Fig. 52-3). When lasting more than a few second(s), these rhythmic epileptiform patterns constitute ictal discharges. Bilateral symmetrical generalized spike-and-wave discharges with a frequency that is equal to or higher than 3 Hz are characteristic of typical absence seizures and more generally of idiopathic generalized epilepsies (Fig. 52-4). Conversely, bilateral asymmetrical generalized spike-and-wave discharges with a frequency that is equal to or lower than 2.5 Hz are seen in atypical absence seizures and cryptogenic or symptomatic generalized epilepsies (Fig. 52-5). Generalized tonic or tonic-clonic seizures are characterized by a diffuse flattening of the electroencephalogram, reflecting a very high frequency discharge of low amplitude. Various types of focal ictal discharges are seen at the onset of partial seizures, including rhythmic spikes, sharp waves, or slow waves, as well as low-amplitude fast-activity discharges (Fig. 52-6). Patients with epilepsy can also show nonepileptiform, and thus less specific electroencephalographic findings, such as intermittent slow waves. Permanently abnormal background activity or a slow wave focus that is poorly or not reactive to external stimuli suggests an underlying encephalopathy or brain lesion.
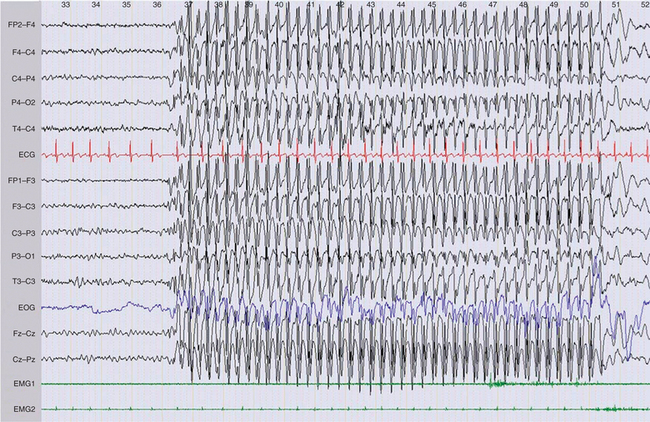
Figure 52-3 Generalized 3-Hz spike and wave discharge during an absence seizure in childhood absence epilepsy.
Neuroimaging
 Although CT scanning is much less sensitive than MRI in the identification of epileptogenic brain lesions such as hippocampal sclerosis, malformations of cortical development, cavernous angiomas, and low-grade tumors, it has two important applications:
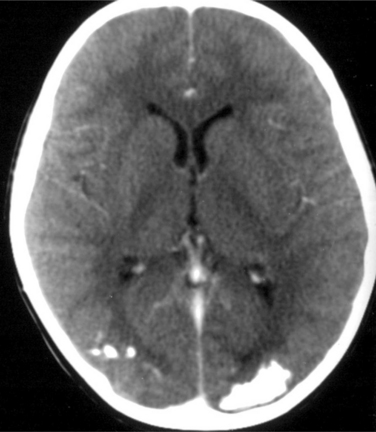
Although CT scanning is much less sensitive than MRI in the identification of epileptogenic brain lesions such as hippocampal sclerosis, malformations of cortical development, cavernous angiomas, and low-grade tumors, it has two important applications:In addition, CT scanning may be more sensitive than MRI in detecting calcifications, such as those observed around the ventricles in tuberous sclerosis, or the occipital calcifications associated with celiac disease (Fig. 52-7).
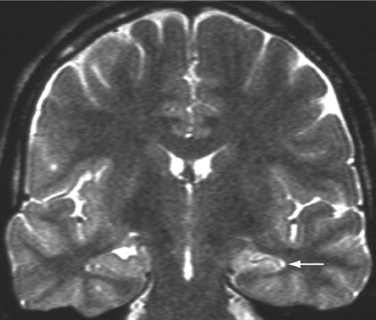
 MRI is the gold standard for neuroimaging of the nonidiopathic epilepsies. It should be performed at 1.5 T or higher, by a radiologist familiar with the special sequences and slices orientations for optimal investigation of epileptic disorders. A standard epilepsy protocol should typically include a three-dimensional 1-mm3 resolution T1-weighted sequence as well as T2-weighted and FLAIR images in the axial and coronal planes, the latter perpendicular to the long axis of the hippocampus. Provided such a protocol, MRI can detect abnormalities in up to 80% of patients with temporal lobe epilepsy and approximately two thirds of patients with nonidiopathic epilepsies. The most frequently encountered epileptogenic brain lesions are hippocampal sclerosis (Fig. 52-8), gliotic scars following various cerebral insults, low-grade tumors, and, in particular, dysembryoplasic neuroepithelial tumors, gangliogliomas, oligodendrogliomas, cavernous angiomas, and malformations of cortical development, including cortical dysplasia, heterotopia, and polymicrogyria.
MRI is the gold standard for neuroimaging of the nonidiopathic epilepsies. It should be performed at 1.5 T or higher, by a radiologist familiar with the special sequences and slices orientations for optimal investigation of epileptic disorders. A standard epilepsy protocol should typically include a three-dimensional 1-mm3 resolution T1-weighted sequence as well as T2-weighted and FLAIR images in the axial and coronal planes, the latter perpendicular to the long axis of the hippocampus. Provided such a protocol, MRI can detect abnormalities in up to 80% of patients with temporal lobe epilepsy and approximately two thirds of patients with nonidiopathic epilepsies. The most frequently encountered epileptogenic brain lesions are hippocampal sclerosis (Fig. 52-8), gliotic scars following various cerebral insults, low-grade tumors, and, in particular, dysembryoplasic neuroepithelial tumors, gangliogliomas, oligodendrogliomas, cavernous angiomas, and malformations of cortical development, including cortical dysplasia, heterotopia, and polymicrogyria.
A Systematic Approach to the Classification of Epileptic Syndromes
Idiopathic Epilepsies of Late Childhood and Adolescence
The idiopathic epilepsies, in particular, childhood absence epilepsy, juvenile myoclonic epilepsy, and benign childhood epilepsy with centrotemporal spikes,1 represent the most prevalent epileptic syndromes of childhood and adolescence. They should be identifiable by any physician involved in the care of patients with epilepsy, inasmuch as specific treatments exist for these syndromes.
Benign childhood epilepsy with centrotemporal spikes is the most frequent epileptic syndrome of childhood with an age at onset ranging between 3 and 13 years. Seizures are typically sleep-related and characterized by tonic-clonic contraction of one side of the face and ipsilateral tongue and pharyngeal-laryngeal muscles, ictal anarthria, and profuse dribbling at times preceded by somatosensory symptoms in the same body parts, and preserved consciousness. Secondary involvement of the ipsilateral upper limb can also be seen, as well as secondary generalization. Interictal electroencephalography shows typical high-amplitude, slow biphasic centrotemporal spikes, with a characteristic tangential dipolar distribution (Fig. 52-9). Spikes greatly increase in frequency and often become bilateral during sleep. Antiepileptic drug treatment can be avoided if seizures are brief and appear only during sleep.
Nonidiopathic Partial Epilepsies
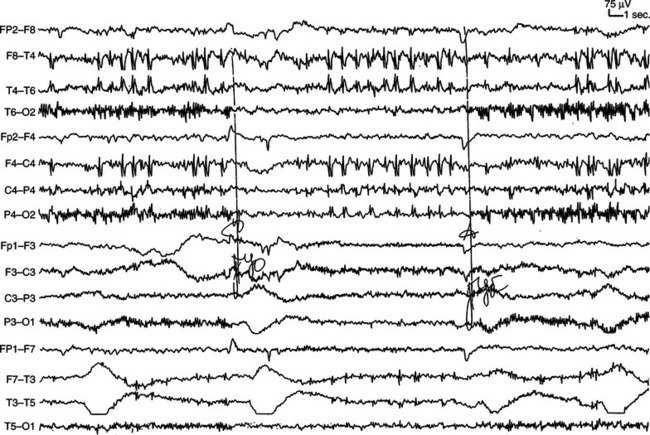
Nonidiopathic partial epilepsies represent between 50% and 60% of all epilepsies and occur across the entire life span. The seizure type varies greatly as a function of the cortical regions involved by an ictal discharge and often provides information about the most likely side and lobe of onset. Such information is useful in interpretation of the relationship between an MRI-detected abnormality and associated seizures or can help in detection of subtle brain lesions or malformations of cortical development. The multiple forms of nonidiopathic partial epilepsies need to be distinguished because specific management issues arise in this frequent disorder. For instance, mesial temporal lobe epilepsy with MRI changes of hippocampal sclerosis is characterized by a high rate of pharmacoresistance that is estimated to lie between 60% and 90% and shows an excellent response to epilepsy surgery in 80% of operated patients (Fig. 52-9). Despite these figures, the majority of patients with mesial temporal lobe epilepsy are referred for presurgical evaluation after an average delay of 15 to 20 years, primarily because the epileptic syndrome has not been properly assessed.
Occasional Seizures
Occasional seizures must be distinguished from all of the above conditions as they usually do not require antiepileptic treatment but rather control of a provocative agent or situation.2 The latter includes intake of excessive alcohol; of illicit drugs such as cocaine, codeine, amphetamines, and phencyclidine; and of many psychotropic drugs including antipsychotics, antidepressants, and lithium. Abrupt abstinence of alcohol, benzodiazepines, and barbiturates in chronic abusers can also precipitate a seizure. In contrast to alcohol-related seizures, alcohol-related epilepsy is characterized by relapsing partial or generalized seizures appearing in a chronic alcohol-abuser, independent of acute alcohol toxicity or sudden abstinence. It usually requires antiepileptic drug treatment.
Nonketotic hyperglycemia is one particular such disorder that can complicate preexisting diabetes but also often occurs in patients without prior history of hyperglycemia. This specific metabolic disorder is responsible for reflex motor seizures triggered by active or passive posture or movements that can progress to epilepsia partialis continua. Biochemical testing shows hyperglycemia of varying severity without associated ketosis. MRI is normal. Antiepileptic treatments are ineffective, whereas normalization of glycemia by insulin rapidly leads to seizure control.
Progressive Myoclonic Epilepsies
Progressive myoclonic epilepsies include a variety of rare genetic seizure disorders associated with progressive neurological impairment, primarily characterized by action myoclonus and cerebellar dysfunction.3 They are thus of special interest to neurologists despite their rarity. Progressive myoclonic epilepsies at an early stage are often mistakenly diagnosed as juvenile myoclonic epilepsy, due to seemingly comparable age and seizure types at onset. However, a detailed analysis of the myoclonic jerks and neurological examination differentiate the two forms of myoclonic epilepsy, allowing anticipation of the far more severe prognosis of progressive myoclonic epilepsies. Various types of seizures can occur in these disorders, including generalized myoclonic seizures but also GTCS, atypical absences, and partial seizures that predominantly originate in the occipital lobe. However, the main clinical feature of progressive myoclonic epilepsy is the presence of action myoclonus that needs to be actively searched for. This movement disorder progressively worsens over years, together with the development of cerebellar dysfunction and cognitive impairment of varying severity, depending on the underlying etiology. Five diseases account for the great majority of progressive myoclonic epilepsies: Unverricht-Lundborg disease, myoclonic epilepsy with ragged red fibers, adult neuronal ceroid-lipofuscinosis (Kufs’ disease), Lafora’s disease, and sialidosis.
Autoimmune and Endocrinological Seizure Disorders
Celiac disease is associated with epilepsy in 3.5% to 5.5% of cases. Seizures typically originate in the occipital lobe, where bilateral calcifications are often observed (see Fig. 52-8). It is to be distinguished from Sturge-Weber syndrome, where the calcified occipital pial angiomata are usually unilateral. Epilepsy is often the presenting manifestation of this disorder, before the occurrence of intestinal symptoms. Diagnosis is confirmed by the detection of IgA endomysial antibodies. A gluten-free diet is then required, although its impact on epilepsy remains controversial.
Several other rare forms of autoimmune limbic encephalitis have been described based on the demonstration of specific antibodies, including anti-Hu, which is found in paraneoplastic syndromes and those directed toward voltage-gated potassium channels, or the more recently discovered anti-neuropil antibodies. Recognizing these conditions has important therapeutic consequences as they can respond dramatically to corticosteroids.4
CONCLUSION
ACKNOWLEDGMENTS
We wish to thank Renzo Guerrini and Richard Frackowiak for their help on this chapter.
1 Callenbach PM, van den Maagdenberg AM, Frants RR, et al. Clinical and genetic aspects of idiopathic epilepsies in childhood. Eur J Paediatr Neurol. 2005;9:91-103.
2 Delanty N, Vaughan CJ, French JA. Medical causes of seizures. Lancet. 1998;352:383-390.
3 Shahwan A, Farrell M, Delanty N. Progressive myoclonic epilepsies: a review of genetic and therapeutic aspects. Lancet Neurol. 2005;4:239-248.
4 Lang B, Dale RC, Vincent A. New autoantibody mediated disorders of the central nervous system. Curr Opin Neurol. 2003;16:351-357.
[/level-membership-for-neurology-category][not-level-membership-for-neurology-category]
CHAPTER 52 ASSESSMENT AND MANAGEMENT PRINCIPLES
By virtue of their capacity to divert the functionality of neural networks into clinically overt discharges, epileptic seizures represent a fascinating window onto brain functions and also the source of a virtually infinite variety of ictal manifestations. The epilepsies encompass a large variety of syndromes reflecting a multitude of brain lesions as well as gene and protein dysfunction that results in neuronal hyperexcitability. An ever-increasing understanding, described in an exponentially growing number of dedicated textbooks, translates into a capacity for more precise diagnosis and optimization of syndrome-dependent management, compounded by the flurry of antiepileptic drugs made available for seizure treatment in the last two decades.
DIAGNOSIS STEP 1: DEFINING THE SEIZURE TYPE(S)
Investigating the Seizure Episode
Validating the Core Features of Epileptic Seizures
“Desperately Seeking” a Witness to the Seizure(s)
If a seizure is primarily characterized by a lack of responsiveness, particular attention should be paid to the presence of automatisms, which, though often noticed, are infrequently reported spontaneously by witnesses. The diagnostic value of oroalimentary automatisms has been mentioned, the same is true for manual, pedal, and verbal automatisms, which all strongly suggest an epileptic origin for seizures. These automatic activities tend to imitate seemingly natural or purposeful gestures or speech, although they usually appear meaningless or inappropriate during a seizure. They must be distinguished from elementary motor activity leading to posture, change in muscle tone, clonic jerk, or a scream. In the 1989 classification of seizures and epilepsies, automatisms are specifically associated with complex partial seizures. In fact, as previously described, subtle automatisms may also occur during simple partial seizures and, at times, during absence seizures.
Classifying the Seizure Episode
The episode was primarily marked by a complete loss of consciousness with a fall if the patient was standing and may have progressed to “convulsions.”Seizures Associated With Complete Loss of Consciousness, a Fall, and Convulsive Features
Differential Diagnoses
Syncope can result from various pathophysiological mechanisms with a common endpoint being a decrease in cerebral perfusion responsible for acute cerebral and brainstem dysfunction. Vasovagal syncope is the most frequent and can be mistaken for GTCS. It typically occurs in adolescents and young adults following emotionally salient stimuli (pain, sight of blood during venous puncture, warm and enclosed atmosphere) or after standing motionless for prolonged periods (related to progressive venous blood sequestration in the lower limbs). Thus, vasovagal syncope usually occurs in the standing position and often aborts if a patient lies down in the early phase of an attack. More rarely, vasovagal syncope will occur in a seated patient at the end of a meal, triggered by digestion-induced splanchnic blood sequestration. Prodromes include vertigo, visual and auditory disturbances, nausea, sweating, and the feeling of an imminent fainting or death. Intense pallor and general hypotonia follow, resulting in a progressive nontraumatic fall and loss of consciousness, with eyes closed or rolled upward. At times, syncope might progress to brief axial hypertonia associated with a few irregular clonic limb movements (fewer than six), “convulsive syncope.” Urination and biting of the tip of the tongue can occur, but normal consciousness is restored much more rapidly than in GTCS. Thus, detailed analysis of all signs and symptoms usually results in a clear distinction of syncope from GTCS, even in the presence of clonic movements, urination, and tongue biting. When necessary, a tilt test can be used to confirm the diagnosis of vasovagal syncope. Cardiogenic syncope represents a less frequent form of attack, observed in older patients with cardiovascular pathology but no other precipitating factors. It is characterized by a more abrupt loss of consciousness and fall.