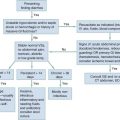
Aspirin and Nonsteroidal Agents
Aspirin
Unfortunately, the severity of this poisoning may be underestimated because of the lack of familiarity with the clinical picture. Salicylate toxicity can cause metabolic acidosis, seizure, hyperthermia, pulmonary edema, cerebral edema, renal failure, and death. Morbidity and mortality are increased by delayed diagnosis in elderly patients with chronic medical problems and in young patients diagnosed with an acute illness.1
Principles of Disease
Salts of salicylic acid are rapidly absorbed intact from the gastrointestinal tract, with appreciable serum concentrations occurring within 30 minutes. Two thirds of a therapeutic dose is absorbed in 1 hour, and peak levels occur in 2 to 4 hours. Large ingestions frequently delay gastric emptying, and ingestions of enteric capsules may cause a prolonged absorption with rising serum levels for 12 hours or more.2
In the intestinal wall, liver, and red blood cells, aspirin is hydrolyzed to free salicylic acid, which reversibly binds to albumin (which contains a genetically determined variable number of salicylate-binding sites). In the liver, salicylate is conjugated with glucuronic acid and glycine (Fig. 149-1). A small fraction is hydroxylated. Free salicylate and its conjugates are eliminated by renal excretion. At therapeutic salicylate concentrations, elimination follows first-order kinetics, and excretion is proportional to salicylate concentration. When serum salicylate concentrations are greater than 30 mg/dL, however, elimination follows zero-order kinetics, and the metabolic rate is constant. The metabolic pathways become saturated, and the pH-sensitive urinary excretion of salicylic acid determines the half-life, which becomes prolonged and may approach 15 to 30 hours with toxic doses.3
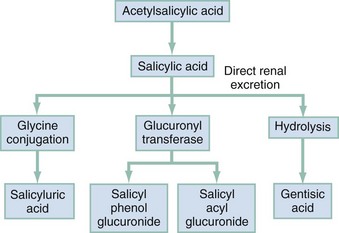
Figure 149-1 Aspirin metabolism.
Pathophysiology
Acid-Base Disturbances and Metabolic Effects.: Salicylate stimulates the medullary respiratory center early and increases the sensitivity of the respiratory center to pH and carbon dioxide partial pressure (PCO2). Hyperventilation develops early, then subsequently becomes a compensatory mechanism to the metabolic acidosis. Prolonged high serum concentrations eventually depress the respiratory center. Respiratory alkalosis is compensated by the buffering of the hemoglobin-oxyhemoglobin system, the exchange of intracellular hydrogen ions for extracellular cations, and the urinary excretion of bicarbonate. Loss of bicarbonate decreases buffering capacity and intensifies the metabolic acidosis.4,5
Toxicity results primarily from salicylate interference with aerobic metabolism by uncoupling of mitochondrial oxidative phosphorylation. Inhibition of the Krebs dehydrogenase cycle increases production of pyruvic acid and increases conversion to lactic acid. Increased lipid metabolism increases production of ketone bodies. Metabolic rate, temperature, tissue carbon dioxide, and oxygen consumption are increased. Tissue glycolysis predisposes to hypoglycemia. (Hepatic gluconeogenesis and release of epinephrine may cause the less common hyperglycemia.) Inefficiency of anaerobic metabolism results in less energy being used to create adenosine triphosphate, and energy is released as heat, causing the hyperthermia frequently seen in salicylate poisoning.4
Only nonionized particles can cross the lipophilic cell membrane and accumulate in the brain and other tissues. Because ASA has a low pKa,3,5 the majority of salicylate is ionized, and little salicylate enters tissues at the physiologic pH of 7.4. However, as pH decreases, more particles become un-ionized and cross the cell membrane and blood-brain barrier, markedly increasing the movement of salicylate into the tissues and central nervous system (CNS).4,5
Fluid and Electrolyte Abnormalities.: Significant potassium loss in salicylate toxicity is caused by vomiting, secondary to stimulation of the medullary chemoreceptor trigger zone; increased renal excretion of sodium, bicarbonate, and potassium as a compensatory response to the respiratory alkalosis; salicylate-induced increased permeability of the renal tubules with further loss of potassium; intracellular accumulation of sodium and water; and inhibition of the active transport system, secondary to uncoupling of oxidative phosphorylation. The net result is rapid depletion of potassium stores.4
A salicylate-induced decrease in renal blood flow or direct nephrotoxicity may cause acute nonoliguric renal failure. Salicylate-induced secretion of inappropriate antidiuretic hormone may also affect renal function.6
Pulmonary and Cerebral Edema.: The exact mechanism by which salicylates increase alveolar capillary membrane permeability is unknown. Theories include inhibition of prostacyclin, changes in platelet-vessel interaction, and neurogenic influences. In adults, the risk factors for salicylate-induced pulmonary edema include age older than 30 years, cigarette smoking, chronic salicylate ingestion, metabolic acidosis, neurologic symptoms, and serum salicylate concentration greater than 40 mg/dL. Risk factors in children include high serum salicylate levels, large anion gap, decreased serum potassium concentration, and low PCO2.7
Chronic Ingestion Physiology.: Physiologic changes of aging predispose elderly patients to toxicity from chronic therapeutic ingestion. Decreased liver blood flow rates decrease biotransformation of salicylate, and decreased renal function decreases salicylate clearance. Chronic ingestion allows salicylates more time to pass through the blood-brain barrier. In addition, chronic ingestion of aspirin decreases albumin binding, increasing the free salicylate that can enter the cell. This causes significant clinical illness with a relatively low serum salicylate concentration. A patient with chronic salicylate toxicity and a serum concentration of 40 mg/dL may be more ill than a patient with an acute ingestion and serum concentration of 80 mg/dL.8
Clinical Features
Shortness of breath and altered sensorium are caused by pulmonary and cerebral edema, respectively. Noncardiac pulmonary edema may be more common in children than scattered case reports suggest.9 Failure to recognize salicylate toxicity as the cause of pulmonary edema increases the likelihood of morbidity and mortality in these patients.
Diagnostic Strategies
Low pH and bicarbonate level portend severe disease. The pH begins to drop when the patient is unable to compensate for the acidosis. Lactic acid accumulates, and serum bicarbonate is consumed. When pH is less than 7.4 and both PCO2 and bicarbonate level are low, the patient begins to decompensate hemodynamically. In the intubated patient or the acidotic patient with low PCO2 and bicarbonate level, hemodialysis should be undertaken.10
Differential Considerations
The symptoms of salicylism (hyperthermia, altered mental status or coma, pulmonary edema, and shock) mimic sepsis and the symptoms of many other diseases (Box 149-1). This is especially true with chronic ingestion; serum salicylate concentration is relatively low, and the severity of the poisoning is not recognized.11 Death is caused by CNS depression and cardiovascular collapse.
Management
Specific treatment of salicylate toxicity has two main objectives: (1) to correct fluid deficits and acid-base abnormalities and (2) to increase excretion (Box 149-2). Electrolyte values are helpful to guide replacement and to assess renal function necessary to excrete salicylates. Serum salicylate levels should be repeated until they are decreasing to measure the effectiveness of treatment and to guide the decision for dialysis.
Activated Charcoal
Although activated charcoal (AC), in a single dose or multiple doses, somewhat reduces salicylate absorption and has been recommended in the past, there is no evidence of outcome benefit. Whereas there is no evidence that AC in general decreases toxicity or alters therapy or course of the illness, this has not been specifically examined for salicylates.12 In chronic salicylate poisoning, because the presentation is long after absorption, AC would not be expected to be of any benefit.
Urine Alkalinization
Urine alkalinization is difficult to achieve because the excretion of salicylic acid in the urine decreases urine pH. In addition, potassium depletion must be corrected to attain an alkaline urine. Alkaline urine should not be produced at the cost of systemic alkalemia. Forced diuresis does not significantly increase salicylate excretion and may potentiate cerebral and pulmonary edema. Salicylate clearance varies in direct proportion to flow rate but increases exponentially with pH.13
Hemodialysis
Hemodialysis is indicated for patients with the following: serum salicylate levels greater than 100 mg/dL in acute and 50 mg/dL in chronic salicylate poisoning; altered mental status, including coma; seizure; endotracheal intubation other than for coingestants; renal or hepatic failure; pulmonary edema; severe acid-base imbalance; rapidly rising serum salicylate level; and failure to respond to more conservative treatment. Exchange transfusion can be considered in young infants or unusual cases of congenital salicylism.14
Pregnancy
Greater salicylate concentration on the fetal side of the placenta and relative fetal acidemia contribute to fetal distress from maternal salicylate poisoning. Salicylate poisoning during pregnancy is associated with fetal demise, and delivery of the distressed fetus should be considered if the fetus is viable.15
Nonsteroidal Anti-Inflammatory Drugs
Principles of Disease
Traditional NSAIDs are nonselective and inhibit both COX-1 and COX-2. Newer, specific COX-2 inhibitors (e.g., celecoxib, rofecoxib, parecoxib, meloxicam) have fewer side effects than traditional NSAIDs while maintaining analgesic and anti-inflammatory efficacy. However, COX-2 inhibitors permit unopposed thromboxane A2 production by platelets, thereby potentiating platelet aggregation, thrombosis, and vasoconstriction.16 Therapeutic use of rofecoxib, particularly at higher doses, was associated with increased risk of myocardial infarction and stroke, and it has subsequently been withdrawn from the market.17,18
Pharmacokinetics
The NSAIDs are highly bound to plasma proteins, mainly albumin, and therefore have small volumes of distribution (0.10-0.17 L/kg). They are eliminated by hepatic biotransformation, principally oxidation and glucuronidation, with the metabolites being excreted in the urine. Metabolites are inactive, except for those of sulindac, nabumetone (inactive parent drugs metabolized to active agent in vivo), and phenylbutazone. Plasma half-lives are short (1-4 hours), except for naproxen (12-15 hours), oxaprozin (25-50 hours), piroxicam (45 hours), and phenylbutazone (50-100 hours). Elimination half-lives are not substantially prolonged in overdose.19
Clinical Features
Most NSAID overdoses, even with large amounts, are asymptomatic or cause only minor CNS or gastrointestinal disturbances. Significant toxicity has not been reported after overdose of the newer COX-2 inhibitors. Of all isolated celecoxib exposures reported to the Texas poison control centers during a 5-year period, no more than minor effects were reported and then in only 12% of cases.20
Ibuprofen is the most common NSAID ingested in overdose and is representative of the propionic acid derivatives. Despite rare case reports of coma, seizure, hypotension, hypothermia, upper gastrointestinal tract bleeding, acute renal failure, and metabolic acidosis, most ibuprofen overdoses follow a benign, rapidly self-limited course. About 50% of adults and 7% of children have symptoms. Symptomatic overdose occurs only after ingestion of at least 100 mg/kg, and all those who develop symptoms do so within 4 hours of ingestion. Life-threatening symptoms or signs are rare, and most toxicity is a mild gastrointestinal or CNS disturbance that resolves in 24 hours.21–25 Less common clinical effects include mild metabolic acidosis, muscle fasciculations, mydriasis, chills, diaphoresis, hyperventilation, mildly elevated systolic blood pressure, asymptomatic bradycardia, hypotension, dyspnea, tinnitus, and rash.
Renal dysfunction is seen only after massive acute overdose and in association with a period of relative hypovolemia with hypotension.21–26 It is reversible and usually responds to supportive measures, but rare deaths have been reported.27 Serum ibuprofen concentrations do not predict toxicity.21
Overdose with mefenamic acid, a fenamate, is associated with a relatively high incidence of seizures, which occur 2 to 7 hours after ingestion.28 Rapid recovery is the rule with supportive care and intravenous benzodiazepines. Serum mefenamic acid concentrations correlate with seizures but do not aid in acute management.
Phenylbutazone, a pyrazolone, is now rarely used because of its association with aplastic anemia and agranulocytosis. Although phenylbutazone overdose is rare, it is much more severe than overdose with other NSAIDs.29 Mild poisoning consists of nausea, abdominal pain, and drowsiness. Severely poisoned patients have early onset of abdominal pain, nausea, vomiting, hematemesis, diarrhea, restlessness, dizziness, coma, convulsions, hyperpyrexia, electrolyte disturbances, hyperventilation, alkalosis or acidosis, respiratory arrest, hypotension, cyanosis, edema, electrocardiographic abnormalities, or cardiac arrest. Late sequelae of severe poisoning (2-7 days) include renal, hepatic, and hematologic dysfunction.29,30 The clinical course is prolonged compared with that of other NSAID poisoning, reflecting the prolonged elimination half-lives of phenylbutazone and its principal metabolite, oxyphenbutazone.30
Management
There is no evidence supporting the use of gastric emptying or AC for NSAID overdose. All patients with nontrivial overdoses should be observed until 4 hours after ingestion and until symptoms are noted to be mild or improving. Hypotension, if it occurs, is managed with intravenous crystalloid solution. Although it is rarely indicated and not studied, extracorporeal membrane oxygenation has successfully managed refractory hypotension after massive ibuprofen overdose.30
Because of high protein binding and rapid metabolism, urine alkalinization, hemodialysis, or hemoperfusion is not clinically useful. Enhanced elimination with plasmapheresis has been attempted in severe phenylbutazone poisoning.31
Disposition
Key Concepts
 ASA intoxication, especially chronic salicylism, should be considered in the differential diagnosis of altered mental status in the elderly.
ASA intoxication, especially chronic salicylism, should be considered in the differential diagnosis of altered mental status in the elderly.
 Potassium stores are rapidly depleted in patients with salicylate intoxication.
Potassium stores are rapidly depleted in patients with salicylate intoxication.
 The acute toxic dose of ASA is 300 mg/kg, and 500 mg/kg is potentially lethal.
The acute toxic dose of ASA is 300 mg/kg, and 500 mg/kg is potentially lethal.
 Acidosis signifies severe salicylism as unbound salicylate is moving into the cell.
Acidosis signifies severe salicylism as unbound salicylate is moving into the cell.




References
1. Chapman, BJ, Proudfoot, AT. Adult salicylate poisoning: Deaths and outcome in patients with high plasma salicylate concentrations. Q J Med. 1989;72:699.
2. Drummond, R, Kadri, N, St-Cyr, J. Delayed salicylate toxicity following enteric-coated acetylsalicylic acid overdose: A case report and review of the literature. CJEM. 2001;3:44.
3. Roberts, LJ, Morrow, JD. Analgesic-antipyretic and anti-inflammatory agents and drugs employed in the treatment of gout. In: Hardman J, Limbird L, eds. Goodman and Gilman’s The Pharmacological Basis of Therapeutics. New York: McGraw Hill; 2001:687–729.
4. O’Malley, G. Emergency department management of the salicylate-poisoned patient. Emerg Med Clin North Am. 2007;25:333.
5. Dromgoole, S, Furst, D. Salicylates. In: Evans WE, Schentag JJ, Jusko WJ, eds. Applied Pharmacokinetics: Principles of Therapeutic Drug Monitoring. Vancouver, Wash: Applied Therapeutics, 1992.
6. Pemeger, TV, Whelton, PK. Risk of kidney failure associated with the use of acetaminophen, aspirin, and nonsteroidal anti-inflammatory drugs. N Engl J Med. 1994;331:1675.
7. Gonzolea, E, et al. Recurrent ARDS in a 39-year-old woman with migraine headaches. Chest. 1998;114:919.
8. Gaudreault, P, Temple, AR, Lovejoy, FH. The relative severity of acute versus chronic salicylate poisoning in children: A clinical comparison. Pediatrics. 1982;70:566.
9. Fisher, CJ, Albertson, TE, Foulke, GE. Salicylate-induced pulmonary edema: Clinical characteristics in children. Am J Emerg Med. 1985;3:33.
10. Greenberg, MI, Henderson, RG, Hoffman, M. Deleterious effects of endotracheal intubation in salicylate poisoning. Ann Emerg Med. 2003;4:231.
11. Lamesh, RA. Accidental chronic salicylate intoxication in an elderly patient: Major morbidity despite early recognition. Vet Hum Toxicol. 1993;35:34.
12. Chyka, PA, Seger, D, Krenzelok, EP, Vale, JA, American Academy of Clinical Toxicology; European Association of Poisons Centres and Clinical Toxicologists. Position paper: Single-dose activated charcoal. Clin Toxicol (Phila). 2005;43:1.
13. Proudfoot, A, Krenzelok, EP, Brent, J, Vale, JA. Does urinary alkalinization increase salicylate elimination? If so, Why? Toxicol Rev. 2003;3:29.
14. Lund, B, Seifert, SA, Mayersohn, M. Efficacy of sustained low-efficiency dialysis in the treatment of salicylate toxicity. Nephrol Dial Transplant. 2005;7:1483.
15. Palatnick, W, Tenenbein, M. Aspirin poisoning during pregnancy: Increased fetal sensitivity. Am J Perinatol. 1998;15:39.
16. Wright, JM. The double-edged sword of COX-2 selective NSAIDs. CMAJ. 2002;167:1131.
17. Graham, D, et al. Risk of myocardial infarction and sudden cardiac death in patients treated with cyclo-oxygenase 2 selective and non-selective non-steroidal anti-inflammatory drugs: Nested case-control study. Lancet. 2005;365:475.
18. Levesque, L, Brophy, J, Zhang, B. The risk for myocardial infarction with cyclooxygenase 2 inhibitors: A population study of elderly adults. Ann Intern Med. 2005;142:481.
19. Hall, AH, et al. Ibuprofen overdose: 126 cases. Ann Emerg Med. 1986;15:1308.
20. Forrester, MB. Celecoxib exposures reported to Texas poison control centers from 1999 to 2004. Hum Exp Toxicol. 2006;25:261.
21. McElwee, NE, et al. A prospective, population-based study of acute ibuprofen overdose: Complications are rare and routine serum levels not warranted. Ann Emerg Med. 1990;19:652.
22. Perry, SJ, Streete, PJ, Volans, GN. Ibuprofen overdose: The first two years of over-the-counter sales. Hum Toxicol. 1987;6:173.
23. Halpern, SM, Fitzpatrick, R, Volans, GN. Ibuprofen toxicity: A review of adverse reactions and overdose. Adverse Drug React Toxicol Rev. 1993;12:107.
24. Hall, AH, et al. Ibuprofen overdose in adults. J Toxicol Clin Toxicol. 1992;30:23.
25. Le, HT, Bosse, GM, Tsai, Y. Ibuprofen overdose complicated by renal failure, adult respiratory distress syndrome, and metabolic acidosis. J Toxicol Clin Toxicol. 1994;32:315.
26. Perazella, MA, Buller, GK. Can ibuprofen cause acute renal failure in a normal individual? A case of acute overdose. Am J Kidney Dis. 1991;18:600.
27. Holubek, W, et al. A report of two deaths from massive ibuprofen ingestion. J Med Toxicol. 2007;3:52.
28. Balali-Mood, M, et al. Mefenamic acid overdosage. Lancet. 1981;1:1354.
29. Court, H, Volans, GN. Poisoning after overdose with non-steroidal anti-inflammatory drugs. Adverse Drug React Acute Poisoning Rev. 1984;3:1.
30. Marciniak, KE, et al. Massive ibuprofen overdose requiring extracorporeal membrane oxygenation for cardiovascular support. Pediatr Crit Care Med. 2007;8:180.
31. Virji, MA, Venkataraman, S, Lower, DR, Rao, KN. Role of laboratory in the management of phenylbutazone poisoning. J Toxicol Clin Toxicol. 2003;41:1013.