PART 12: Critical Care Medicine
|
RESPIRATORY CRITICAL CARE |
321 |
Approach to the Patient with Critical Illness |
The care of critically ill patients requires a thorough understanding of pathophysiology and centers initially on the resuscitation of patients at the extremes of physiologic deterioration. This resuscitation is often fast-paced and occurs early, without a detailed awareness of the patient’s chronic medical problems. While physiologic stabilization is taking place, intensivists attempt to gather important background medical information to supplement the real-time assessment of the patient’s current physiologic conditions. Numerous tools are available to assist intensivists in the accurate assessment of pathophysiology and management of incipient organ failure, offering a window of opportunity for diagnosing and treating underlying disease(s) in a stabilized patient. Indeed, the use of invasive interventions such as mechanical ventilation and renal replacement therapy is commonplace in the intensive care unit (ICU). An appreciation of the risks and benefits of such aggressive and often invasive interventions is vital to ensure an optimal outcome. Nonetheless, intensivists must recognize when a patient’s chances for recovery are remote or nonexistent and must counsel and comfort dying patients and their significant others. Critical care physicians often must redirect the goals of care from resuscitation and cure to comfort when the resolution of an underlying illness is not possible.
ASSESSMENT OF ILLNESS SEVERITY
In the ICU, illnesses are frequently categorized by degree of severity. Numerous severity-of-illness (SOI) scoring systems have been developed and validated over the past three decades. Although these scoring systems have been validated as tools to assess populations of critically ill patients, their utility in predicting individual patient outcomes is not clear. SOI scoring systems are important for defining populations of critically ill patients. Such systematic scoring allows effective comparison of groups of patients enrolled in clinical trials. In verifying a purported benefit of therapy, investigators must be confident that different groups involved in a clinical trial have similar illness severities. SOI scores are also useful in guiding hospital administrative policies, directing the allocation of resources such as nursing and ancillary care and assisting in assessments of quality of ICU care over time. Scoring system validations are based on the premise that age, chronic medical illnesses, and derangements from normal physiology are associated with increased mortality rates. All existing SOI scoring systems are derived from patients who have already been admitted to the ICU.
SOI scoring systems cannot be used to predict survival in individual patients. No established scoring systems that purport to direct clinicians’ decision-making regarding criteria for admission to an ICU are available, although such models are being developed. Thus the use of SOI scoring systems to direct therapy and clinical decision-making cannot be recommended at present. Instead, these tools should be used as a source of important data to complement clinical bedside decision-making.
The most commonly utilized scoring systems are the APACHE (Acute Physiology and Chronic Health Evaluation) and the SAPS (Simplified Acute Physiology Score) systems.
THE APACHE II SCORING SYSTEM
The APACHE II system is the most commonly used SOI scoring system in North America. Age, type of ICU admission (after elective surgery vs. nonsurgical or after emergency surgery), chronic health problems, and 12 physiologic variables (the worst values for each in the first 24 h after ICU admission) are used to derive a score. The predicted hospital mortality rate is derived from a formula that takes into account the APACHE II score, the need for emergency surgery, and a weighted, disease-specific diagnostic category (Table 321–1). The relationship between APACHE II score and mortality risk is illustrated in Fig. 321-1. Updated versions of the APACHE scoring system (APACHE III and APACHE IV) have been published.
|
CALCULATION OF ACUTE PHYSIOLOGY AND CHRONIC HEALTH EVALUATION II (APACHE II) SCOREa |
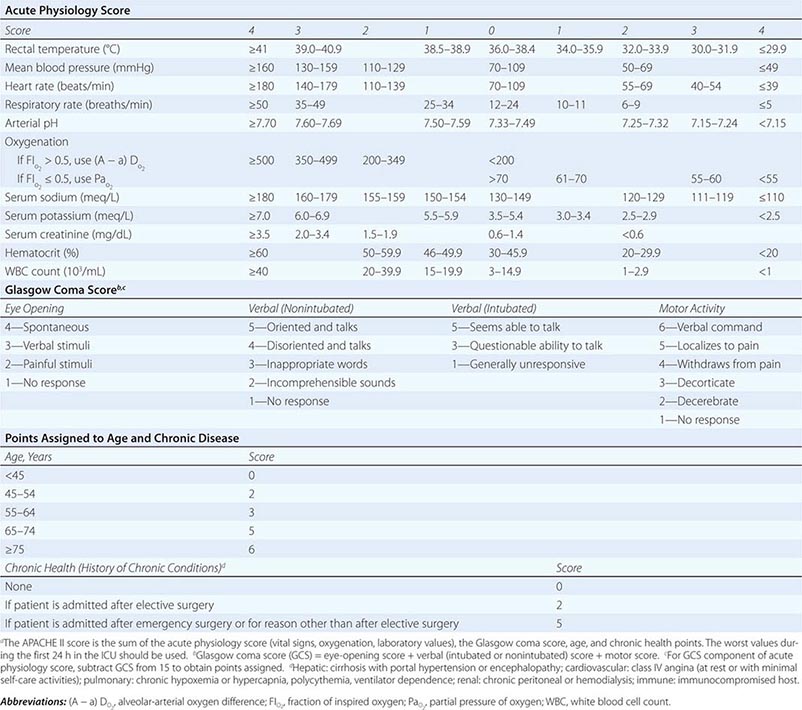
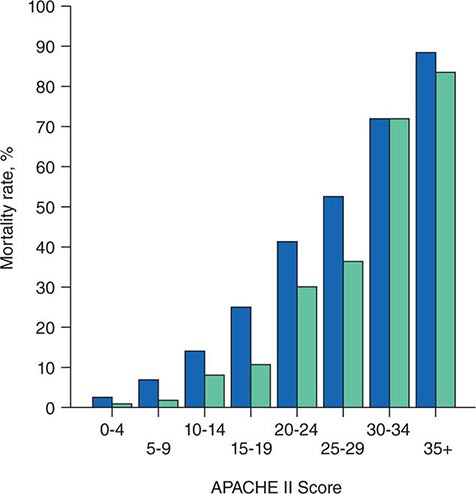
FIGURE 321-1 APACHE II survival curve. Blue, nonoperative; green, postoperative.
THE SAPS SCORING SYSTEM
The SAPS II score, used more frequently in Europe than in the United States, was derived in a manner similar to the APACHE score. This score is not disease specific but rather incorporates three underlying disease variables: AIDS, metastatic cancer, and hematologic malignancy. SAPS 3, which utilizes a 1-h rather than a 24-h window for measuring physiologic derangement scores, was developed in 2005.
SHOCK
See also Chap. 324.
INITIAL EVALUATION
Shock, a common condition necessitating ICU admission or occurring in the course of critical care, is defined by the presence of multisystem end-organ hypoperfusion. Clinical indicators include reduced mean arterial pressure (MAP), tachycardia, tachypnea, cool skin and extremities, acute altered mental status, and oliguria. Hypotension is usually, though not always, present. The end result of multiorgan hypoperfusion is tissue hypoxia, often with accompanying lactic acidosis. Since the MAP is the product of cardiac output and systemic vascular resistance (SVR), reductions in blood pressure can be caused by decreases in cardiac output and/or SVR. Accordingly, once shock is contemplated, the initial evaluation of a hypotensive patient should include an early bedside assessment of the adequacy of cardiac output (Fig. 321-2). Clinical evidence of diminished cardiac output includes a narrow pulse pressure—a marker that correlates with stroke volume—and cool extremities with delayed capillary refill. Signs of increased cardiac output include a widened pulse pressure (particularly with a reduced diastolic pressure), warm extremities with bounding pulses, and rapid capillary refill. If a hypotensive patient has clinical signs of increased cardiac output, it can be inferred that the reduced blood pressure is from decreased SVR.
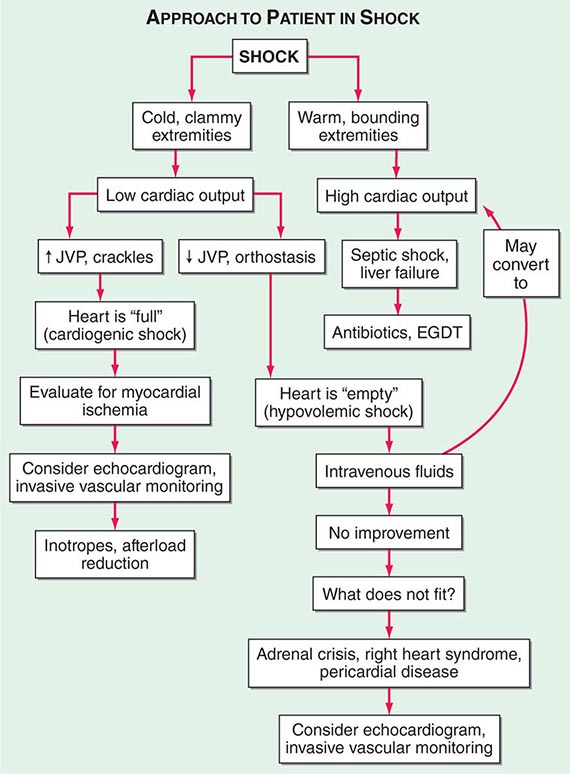
FIGURE 321-2 Approach to the patient in shock. EGDT, early goal-directed therapy; JVP, jugular venous pulse.
In hypotensive patients with signs of reduced cardiac output, an assessment of intravascular volume status is appropriate. A hypotensive patient with decreased intravascular volume status may have a history suggesting hemorrhage or other volume losses (e.g., vomiting, diarrhea, polyuria). Although evidence of a reduced jugular venous pressure (JVP) is often sought, static measures of right atrial pressure do not predict fluid responsiveness reliably; the change in right atrial pressure as a function of spontaneous respiration is a better predictor of fluid responsiveness (Fig. 321-3). Patients with fluid-responsive (i.e., hypovolemic) shock also may manifest large changes in pulse pressure as a function of respiration during mechanical ventilation (Fig. 321-4). A hypotensive patient with increased intravascular volume and cardiac dysfunction may have S3 and/or S4 gallops on examination, increased JVP, extremity edema, and crackles on lung auscultation. The chest x-ray may show cardiomegaly, widening of the vascular pedicle, Kerley B lines, and pulmonary edema. Chest pain and electrocardiographic changes consistent with ischemia may be noted (Chap. 326).
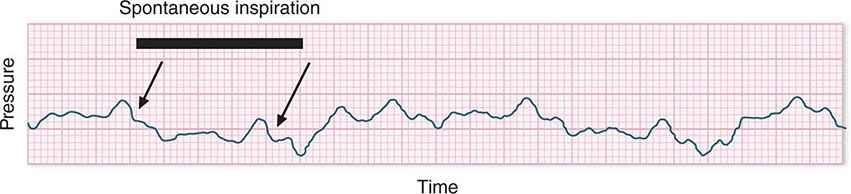
FIGURE 321-3 Right atrial pressure change during spontaneous respiration in a patient with shock whose cardiac output will increase in response to intravenous fluid administration. The right atrial pressure decreases from 7 mmHg to 4 mmHg. The horizontal bar marks the time of spontaneous inspiration.
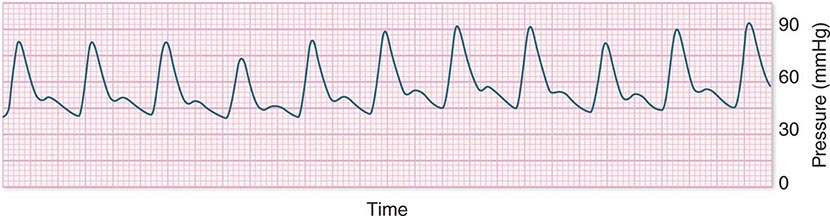
FIGURE 321-4 Pulse pressure change during mechanical ventilation in a patient with shock whose cardiac output will increase in response to intravenous fluid administration. The pulse pressure (systolic minus diastolic blood pressure) changes during mechanical ventilation in a patient with septic shock.
In hypotensive patients with clinical evidence of increased cardiac output, a search for causes of decreased SVR is appropriate. The most common cause of high-cardiac-output hypotension is sepsis (Chap. 325). Other causes include liver failure, severe pancreatitis, burns and other trauma that elicit the systemic inflammatory response syndrome (SIRS), anaphylaxis, thyrotoxicosis, and peripheral arteriovenous shunts.
In summary, the most common categories of shock are hypovolemic, cardiogenic, and high-cardiac-output with decreased SVR (high-output hypotension). Certainly more than one category can occur simultaneously (e.g., hypovolemic and septic shock).
The initial assessment of a patient in shock should take only a few minutes. It is important that aggressive resuscitation is instituted on the basis of the initial assessment, particularly since early resuscitation from septic and cardiogenic shock may improve survival (see below). If the initial bedside assessment yields equivocal or confounding data, more objective assessments such as echocardiography and/or invasive vascular monitoring may be useful. The goal of early resuscitation is to reestablish adequate tissue perfusion and thus to prevent or minimize end-organ injury.
MECHANICAL VENTILATORY SUPPORT
(See also Chap. 323) During the initial resuscitation of patients in shock, principles of advanced cardiac life support should be followed. As such patients may be obtunded and unable to protect the airway, an early assessment of the airway is mandatory. Early intubation and mechanical ventilation often are required. Reasons for the institution of endotracheal intubation and mechanical ventilation include acute hypoxemic respiratory failure and ventilatory failure, which frequently accompany shock. Acute hypoxemic respiratory failure may occur in patients with cardiogenic shock and pulmonary edema (Chap. 326) as well as in those who are in septic shock with pneumonia or acute respiratory distress syndrome (ARDS) (Chaps. 322 and 325). Ventilatory failure often occurs as a consequence of an increased load on the respiratory system in the form of acute metabolic (often lactic) acidosis or decreased lung compliance due to pulmonary edema. Inadequate perfusion to respiratory muscles in the setting of shock may be another reason for early intubation and mechanical ventilation. Normally, the respiratory muscles receive a very small percentage of the cardiac output. However, in patients who are in shock with respiratory distress, the percentage of cardiac output dedicated to respiratory muscles may increase by tenfold or more. Lactic acid production from inefficient respiratory muscle activity presents an additional ventilatory load.
Mechanical ventilation may relieve the work of breathing and allow redistribution of a limited cardiac output to other vital organs. Patients demonstrate respiratory distress by an inability to speak full sentences, accessory use of respiratory muscles, paradoxical abdominal muscle activity, extreme tachypnea (>40 breaths/min), and decreasing respiratory rate despite an increasing drive to breathe. When patients with shock are treated with mechanical ventilation, a major goal is for the ventilator to assume all or the majority of the work of breathing, facilitating a state of minimal respiratory muscle work. With the institution of mechanical ventilation for shock, further declines in MAP are frequently seen. The reasons include impeded venous return from positive-pressure ventilation, reduced endogenous catecholamine secretion once the stress associated with respiratory failure abates, and the actions of drugs used to facilitate endotracheal intubation (e.g., propofol, opiates). Accordingly, hypotension should be anticipated during endotracheal intubation. Because many of these patients may be fluid responsive, IV volume administration should be considered. Figure 321-2 summarizes the diagnosis and treatment of different types of shock For further discussion of individual forms of shock, see Chaps. 324, 325, and 326.
RESPIRATORY FAILURE
Respiratory failure is one of the most common reasons for ICU admission. In some ICUs, ≥75% of patients require mechanical ventilation during their stay. Respiratory failure can be categorized mechanistically on the basis of pathophysiologic derangements in respiratory function.
TYPE I: ACUTE HYPOXEMIC RESPIRATORY FAILURE
This type of respiratory failure occurs with alveolar flooding and subsequent intrapulmonary shunt physiology. Alveolar flooding may be a consequence of pulmonary edema, pneumonia, or alveolar hemorrhage. Pulmonary edema can be further categorized as occurring due to elevated pulmonary microvascular pressures, as seen in heart failure and intravascular volume overload or ARDS (“low-pressure pulmonary edema,” Chap. 322). This syndrome is defined by acute onset (≤1 week) of bilateral opacities on chest imaging that are not fully explained by cardiac failure or fluid overload and of shunt physiology requiring positive end-expiratory pressure (PEEP). Type I respiratory failure occurs in clinical settings such as sepsis, gastric aspiration, pneumonia, near-drowning, multiple blood transfusions, and pancreatitis. The mortality rate among patients with ARDS was traditionally very high (50–70%), although changes in patient care have led to mortality rates closer to 30% (see below).
For many years, physicians have suspected that mechanical ventilation of patients with ARDS may propagate lung injury. Cyclical collapse and reopening of alveoli may be partly responsible for this adverse effect. As seen in Fig. 321-5, the pressure-volume relationship of the lung in ARDS is not linear. Alveoli may collapse at very low lung volumes. Animal studies have suggested that stretching and overdistention of injured alveoli during mechanical ventilation can further injure the lung. Concern over this alveolar overdistention, termed ventilator-induced “volutrauma,” led to a multicenter, randomized, prospective trial comparing traditional ventilator strategies for ARDS (large tidal volume: 12 mL/kg of ideal body weight) with a low tidal volume (6 mL/kg of ideal body weight). This study showed a dramatic reduction in mortality rate in the low-tidal-volume group from that in the high-tidal-volume group (31% versus 39.8%). In addition, a “fluid-conservative” management strategy (maintaining a low central venous pressure [CVP] or pulmonary capillary wedge pressure [PCWP]) is associated with fewer days of mechanical ventilation than a “fluid-liberal” strategy (maintaining a relatively high CVP or PCWP) in ARDS.
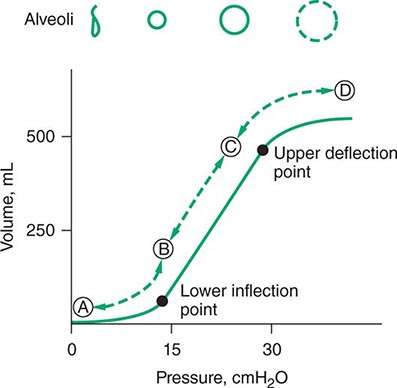
FIGURE 321-5 Pressure-volume relationship in the lungs of a patient with acute respiratory distress syndrome (ARDS). At the lower inflection point, collapsed alveoli begin to open and lung compliance changes. At the upper deflection point, alveoli become overdistended. The shape and size of alveoli are illustrated at the top of the figure.
TYPE II RESPIRATORY FAILURE
This type of respiratory failure is a consequence of alveolar hypoventilation and results from the inability to eliminate carbon dioxide effectively. Mechanisms are categorized by impaired central nervous system (CNS) drive to breathe, impaired strength with failure of neuromuscular function in the respiratory system, and increased load(s) on the respiratory system. Reasons for diminished CNS drive to breathe include drug overdose, brainstem injury, sleep-disordered breathing, and severe hypothyroidism. Reduced strength can be due to impaired neuromuscular transmission (e.g., myasthenia gravis, Guillain-Barré syndrome, amyotrophic lateral sclerosis) or respiratory muscle weakness (e.g., myopathy, electrolyte derangements, fatigue).
The overall load on the respiratory system can be subclassified into resistive loads (e.g., bronchospasm), loads due to reduced lung compliance (e.g., alveolar edema, atelectasis, intrinsic positive end-expiratory pressure [auto-PEEP]—see below), loads due to reduced chest wall compliance (e.g., pneumothorax, pleural effusion, abdominal distention), and loads due to increased minute ventilation requirements (e.g., pulmonary embolus with increased dead-space fraction, sepsis).
The mainstays of therapy for type II respiratory failure are directed at reversing the underlying cause(s) of ventilatory failure. Noninvasive positive-pressure ventilation with a tight-fitting facial or nasal mask, with avoidance of endotracheal intubation, often stabilizes these patients. This approach has been shown to be beneficial in treating patients with exacerbations of chronic obstructive pulmonary disease; it has been tested less extensively in other kinds of respiratory failure but may be attempted nonetheless in the absence of contraindications (hemodynamic instability, inability to protect the airway, respiratory arrest).
TYPE III RESPIRATORY FAILURE
This form of respiratory failure results from lung atelectasis. Because atelectasis occurs so commonly in the perioperative period, this form is also called perioperative respiratory failure. After general anesthesia, decreases in functional residual capacity lead to collapse of dependent lung units. Such atelectasis can be treated by frequent changes in position, chest physiotherapy, upright positioning, and control of incisional and/or abdominal pain. Noninvasive positive-pressure ventilation may also be used to reverse regional atelectasis.
TYPE IV RESPIRATORY FAILURE
This form results from hypoperfusion of respiratory muscles in patients in shock. Normally, respiratory muscles consume <5% of total cardiac output and oxygen delivery. Patients in shock often experience respiratory distress due to pulmonary edema (e.g., in cardiogenic shock), lactic acidosis, and anemia. In this setting, up to 40% of cardiac output may be distributed to the respiratory muscles. Intubation and mechanical ventilation can allow redistribution of the cardiac output away from the respiratory muscles and back to vital organs while the shock is treated.
CARE OF THE MECHANICALLY VENTILATED PATIENT
(See also Chap. 323) Whereas a thorough understanding of the pathophysiology of respiratory failure is essential for optimal patient care, recognition of a patient’s readiness to be liberated from mechanical ventilation is likewise important. Several studies have shown that daily spontaneous breathing trials can identify patients who are ready for extubation. Accordingly, all intubated, mechanically ventilated patients should undergo daily screening of respiratory function. If oxygenation is stable (i.e., PaO2/FIO2 [partial pressure of oxygen/fraction of inspired oxygen] >200 and PEEP ≤5 cmH2O), cough and airway reflexes are intact, and no vasopressor agents or sedatives are being administered, the patient has passed the screening test and should undergo a spontaneous breathing trial. This trial consists of a period of breathing through the endotracheal tube without ventilator support (both continuous positive airway pressure [CPAP] of 5 cmH2O and an open T-piece breathing system can be used) for 30–120 min. The spontaneous breathing trial is declared a failure and stopped if any of the following occur: (1) respiratory rate >35/min for >5 min, (2) O2 saturation <90%, (3) heart rate >140/min or a 20% increase or decrease from baseline, (4) systolic blood pressure <90 mmHg or >180 mmHg, or (5) increased anxiety or diaphoresis. If, at the end of the spontaneous breathing trial, none of the above events has occurred and the ratio of the respiratory rate and tidal volume in liters (f/VT) is <105, the patient can be extubated. Such protocol-driven approaches to patient care can have an important impact on the duration of mechanical ventilation and ICU stay. In spite of such a careful approach to liberation from mechanical ventilation, up to 10% of patients develop respiratory distress after extubation and may require resumption of mechanical ventilation. Many of these patients will require reintubation. The use of noninvasive ventilation in patients in whom extubation fails may be associated with worse outcomes than are obtained with immediate reintubation.
Mechanically ventilated patients frequently require sedatives and analgesics. Opiates are the mainstay of therapy for pain control in mechanically ventilated patients. After adequate pain control has been ensured, additional indications for sedation include anxiolysis; treatment of subjective dyspnea; psychosis; facilitation of nursing care; reduction of autonomic hyperactivity, which may precipitate myocardial ischemia; and reduction of total O2 consumption (VO2).
Neuromuscular blocking agents are occasionally needed to facilitate mechanical ventilation in patients with profound ventilator dyssynchrony despite optimal sedation, particularly in the setting of severe ARDS. Use of these agents may result in prolonged weakness—a myopathy known as the postparalytic syndrome. For this reason, neuromuscular blocking agents typically are used as a last resort when aggressive sedation fails to achieve patient-ventilator synchrony. Because neuromuscular blocking agents result in pharmacologic paralysis without altering mental status, sedative-induced amnesia is mandatory when these agents are administered.
Amnesia can be achieved reliably with benzodiazepines such as lorazepam and midazolam as well as the the IV anesthetic agent propofol. Outside the setting of pharmacologic paralysis, few data support the idea that amnesia is mandatory in all patients who require intubation and mechanical ventilation. Since many of these critically patients have impaired hepatic and renal function, sedatives and opiates may accumulate when given for prolonged periods. A nursing protocol–driven approach to sedation of mechanically ventilated patients or daily interruption of sedative infusions paired with daily spontaneous breathing trials has been shown to prevent excessive drug accumulation and shorten the duration of both mechanical ventilation and ICU stay.
MULTIORGAN SYSTEM FAILURE
Multiorgan system failure, which is commonly associated with critical illness, is defined by the simultaneous presence of physiologic dysfunction and/or failure of two or more organs. Typically, this syndrome occurs in the setting of severe sepsis, shock of any kind, severe inflammatory conditions such as pancreatitis, and trauma. The fact that multiorgan system failure occurs commonly in the ICU is a testament to our current ability to stabilize and support single-organ failure. The ability to support single-organ failure aggressively (e.g., by mechanical ventilation or by renal replacement therapy) has reduced rates of early mortality in critical illness. As a result, it is uncommon for critically ill patients to die in the initial stages of resuscitation. Instead, many patients succumb to critical illness later in the ICU stay, after the initial presenting problem has been stabilized.
Although there is debate regarding specific definitions of organ failure, several general principles governing the syndrome of multiorgan system failure apply. First, organ failure, no matter how it is defined, must persist beyond 24 h. Second, mortality risk increases with the accrual of failing organs. Third, the prognosis worsens with increased duration of organ failure. These observations remain true across various critical care settings (e.g., medical versus surgical). SIRS is a common basis for multiorgan system failure. Although infection is a common cause of SIRS, “sterile” triggers such as pancreatitis, trauma, and burns often are invoked to explain multiorgan system failure.
MONITORING IN THE ICU
Because respiratory failure and circulatory failure are common in critically ill patients, monitoring of the respiratory and cardiovascular systems is undertaken frequently. Evaluation of respiratory gas exchange is routine in critical illness. The “gold standard” remains arterial blood-gas analysis, in which pH, PaO2, partial pressure of carbon dioxide (PCO2), and O2 saturation are measured directly. With arterial blood-gas analysis, the two main functions of the lung—oxygenation of arterial blood and elimination of CO2—can be assessed directly. In fact, the blood pH, which has a profound effect on the drive to breathe, can be assessed only by such sampling. Although sampling of arterial blood is generally safe, it may be painful and cannot provide continuous information. In light of these limitations, noninvasive monitoring of respiratory function is often employed.
PULSE OXIMETRY
The most commonly utilized noninvasive technique for monitoring respiratory function, pulse oximetry takes advantage of differences in the absorptive properties of oxygenated and deoxygenated hemoglobin. At wavelengths of 660 nm, oxyhemoglobin reflects light more effectively than does deoxyhemoglobin, whereas the reverse is true in the infrared spectrum (940 nm). A pulse oximeter passes both wavelengths of light through a perfused digit such as a finger, and the relative intensity of light transmission at these two wavelengths is recorded. From this information, the relative percentage of oxyhemoglobin is derived. Since arterial pulsations produce phasic changes in the intensity of transmitted light, the pulse oximeter is designed to detect only light of alternating intensity. This feature allows distinction of arterial and venous blood O2 saturations.
RESPIRATORY SYSTEM MECHANICS
Respiratory system mechanics can be measured in patients during mechanical ventilation (Chap. 323). When volume-controlled modes of mechanical ventilation are used, accompanying airway pressures can easily be measured as long as the patient is passive. The peak airway pressure is determined by two variables: airway resistance and respiratory system compliance. At the end of inspiration, inspiratory flow can be stopped transiently. This end-inspiratory pause (plateau pressure) is a static measurement, affected only by respiratory system compliance and not by airway resistance. Therefore, during volume-controlled ventilation, the difference between the peak (airway resistance + respiratory system compliance) and plateau (respiratory system compliance only) airway pressures provides a quantitative assessment of airway resistance. Accordingly, during volume-controlled ventilation, patients with increases in airway resistance typically have increased peak airway pressures as well as abnormally high gradients between peak and plateau airway pressures (typically >15 cmH2O) at an inspiratory flow rate of 1 L/sec. The compliance of the respiratory system is defined by the change in pressure of the respiratory system per unit change in volume.
The respiratory system can be divided into two components: the lungs and the chest wall. Normally, respiratory system compliance is ~100 mL/cmH2O. Pathophysiologic processes such as pleural effusions, pneumothorax, and increased abdominal girth all reduce chest wall compliance. Lung compliance may be reduced by pneumonia, pulmonary edema, interstitial lung disease, or auto-PEEP. Accordingly, patients with abnormalities in compliance of the respiratory system (lungs and/or chest wall) typically have elevated peak and plateau airway pressures but a normal gradient between these two pressures. Auto-PEEP occurs when there is insufficient time for emptying of alveoli before the next inspiratory cycle. Since the alveoli have not decompressed completely, alveolar pressure remains positive at the end of exhalation (functional residual capacity). This phenomenon results most commonly from critical narrowing of distal airways in disease processes such as asthma and COPD. Auto-PEEP with resulting alveolar overdistention may result in diminished lung compliance, reflected by abnormally increased plateau airway pressures. Modern mechanical ventilators allow breath-to-breath display of pressure and flow, permitting detection of problems such as patient-ventilator dyssynchrony, airflow obstruction, and auto-PEEP (Fig. 321–6).
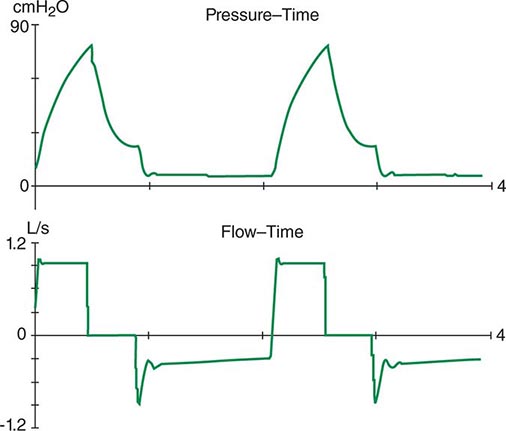
FIGURE 321-6 Increased airway resistance with auto-PEEP. The top waveform (airway pressure vs. time) shows a large difference between the peak airway pressure (80 cmH2O) and the plateau airway pressure (20 cmH2O). The bottom waveform (flow vs. time) demonstrates airflow throughout expiration (reflected by the flow tracing on the negative portion of the abscissa) that persists up to the next inspiratory effort.
CIRCULATORY STATUS
Oxygen delivery (QO2) is a function of cardiac output and the content of O2 in the arterial blood (CaO2). The CaO2 is determined by the hemoglobin concentration, the arterial hemoglobin saturation, and dissolved O2 not bound to hemoglobin. For normal adults:
QO2 = 50 dL/min × (1.39 × 15 g/dL [hemoglobin concentration] × 1.0 [hemoglobin % saturation] + 0.0031 × 100 [PaO2])
= 50 dL/min (cardiac output) × 21.6 mL O2 per dL blood (CaO2)
= 1058 mL O2 per min
It is apparent that nearly all of the O2 delivered to tissues is bound to hemoglobin and that the dissolved O2 (PaO2) contributes very little to O2 content in arterial blood or to O2 delivery. Normally, the content of O2 in mixed venous blood (C![]() O2) is 15.76 mL/dL since the mixed venous blood is 75% saturated. Therefore, the normal tissue extraction ratio for O2 is CaO2 – C
O2) is 15.76 mL/dL since the mixed venous blood is 75% saturated. Therefore, the normal tissue extraction ratio for O2 is CaO2 – C![]() O2/CaO2 ([21.16–15.76]/21.16) or ~25%. A pulmonary artery catheter allows measurements of O2 delivery and the O2 extraction ratio.
O2/CaO2 ([21.16–15.76]/21.16) or ~25%. A pulmonary artery catheter allows measurements of O2 delivery and the O2 extraction ratio.
Information on the mixed venous O2 saturation allows assessment of global tissue perfusion. A reduced mixed venous O2 saturation may be caused by inadequate cardiac output, reduced hemoglobin concentration, and/or reduced arterial O2 saturation. An abnormally high VO2 may also lead to a reduced mixed venous O2 saturation if O2 delivery is not concomitantly increased. Abnormally increased VO2 in peripheral tissues may be caused by problems such as fever, agitation, shivering, and thyrotoxicosis.
The pulmonary artery catheter originally was designed as a tool to guide therapy for acute myocardial infarction but has been used in the ICU for evaluation and treatment of a variety of other conditions, such as ARDS, septic shock, congestive heart failure, and acute renal failure. This device has never been validated as a tool associated with reduction in morbidity and mortality rates. Indeed, despite numerous prospective studies, mortality or morbidity rate benefits associated with use of the pulmonary artery catheter have never been reported in any setting. Accordingly, it appears that routine pulmonary artery catheterization is not indicated as a means of monitoring and characterizing circulatory status in most critically ill patients.
Static measurements of circulatory parameters (e.g., CVP, PCWP) do not provide reliable information on the circulatory status of critically ill patients. In contrast, dynamic assessments measuring the impact of breathing on the circulation are more reliable predictors of responsiveness to IV fluid administration. A decrease in CVP of >1 mmHg during inspiration in a spontaneously breathing patient may predict an increase in cardiac output after IV fluid administration. Similarly, a changing pulse pressure during mechanical ventilation has been shown to predict an increase in cardiac output after IV fluid administration in patients with septic shock.
PREVENTION OF COMPLICATIONS OF CRITICAL ILLNESS
SEPSIS IN THE CRITICAL CARE UNIT
(See also Chap. 325) Sepsis, defined as the presence of SIRS in the setting of known or suspected infection, is a significant problem in the care of critically ill patients, who often progress to severe sepsis with the failure of one or more organs. Sepsis is the leading cause of death in noncoronary ICUs in the United States, with case rates expected to increase as the population ages and a higher percentage of people are vulnerable to infection.
NOSOCOMIAL INFECTIONS IN THE ICU
Many therapeutic interventions in the ICU are invasive and predispose patients to infectious complications. These interventions include endotracheal intubation, indwelling vascular catheters, transurethral bladder catheters, and other catheters placed into sterile body cavities (e.g., tube thoracostomy, percutaneous intraabdominal drainage catheterization). The longer such devices remain in place, the more prone to these infections patients become. For example, ventilator-associated pneumonia correlates strongly with the duration of intubation and mechanical ventilation. Therefore, an important aspect of preventive care is the timely removal of invasive devices as soon as they are no longer needed. Moreover, multidrug-resistant organisms are commonplace in the ICU.
Infection control is critical in the ICU. Care bundles, which include measures such as frequent hand washing, are effective but underutilized strategies. Other components of care bundles, such as protective isolation of patients colonized or infected by drug-resistant organisms, are also commonly used. Silver-coated endotracheal tubes reportedly reduce the incidence of ventilator-associated pneumonia. Studies evaluating multifaceted, evidence-based strategies to decrease catheter-related bloodstream infections have shown improved outcomes with strict adherence to measures such as hand washing, full-barrier precautions during catheter insertion, chlorhexidine skin preparation, avoidance of the femoral site, and timely catheter removal.
DEEP VENOUS THROMBOSIS (DVT)
(See also Chap. 300) All ICU patients are at high risk for this complication because of their predilection for immobility. Therefore, all should receive some form of prophylaxis against DVT. The most commonly employed forms of prophylaxis are subcutaneous low-dose heparin injections and sequential compression devices for the lower extremities. Observational studies report an alarming incidence of DVTs despite the use of these standard prophylactic regimens. Furthermore, heparin prophylaxis may result in heparin-induced thrombocytopenia, another nosocomial complication in critically ill patients.
Low-molecular-weight heparins such as enoxaparin are more effective than unfractionated heparin for DVT prophylaxis in high-risk patients (e.g., those undergoing orthopedic surgery) and are associated with a lower incidence of heparin-induced thrombocytopenia. Fondaparinux, a selective factor Xa inhibitor, is even more effective than enoxaparin in high-risk orthopedic patients.
STRESS ULCERS
Prophylaxis against stress ulcers is frequently administered in most ICUs; typically, histamine-2 antagonists or proton pump inhibitors are given. Available data suggest that high-risk patients, such as those with coagulopathy, shock, or respiratory failure requiring mechanical ventilation, benefit from such prophylactic treatment.
NUTRITION AND GLYCEMIC CONTROL
These are important issues that may be associated with respiratory failure, impaired wound healing, and dysfunctional immune response in critically ill patients. Early enteral feeding is reasonable, though no data are available to suggest that this treatment improves patient outcome per se. Certainly, enteral feeding, if possible, is preferred over parenteral nutrition, which is associated with numerous complications, including hyperglycemia, fatty liver, cholestasis, and sepsis. When parenteral feeding is necessary to supplement enteral nutrition, delaying this intervention until day 8 in the ICU results in better recovery and fewer ICU-related complications. Tight glucose control is an area of controversy in critical care. Although one study showed a significant mortality benefit when glucose levels were aggressively normalized in a large group of surgical ICU patients, more recent data for a large population of both medical and surgical ICU patients suggested that tight glucose control resulted in increased rates of mortality.
ICU-ACQUIRED WEAKNESS
ICU-acquired weakness occurs frequently in patients who survive critical illness, particularly those with SIRS and/or sepsis. Both neuropathies and myopathies have been described, most commonly after ~1 week in the ICU. The mechanisms behind ICU-acquired weakness syndromes are poorly understood. Intensive insulin therapy may reduce polyneuropathy in critical illness. Very early physical and occupational therapy in mechanically ventilated patients reportedly results in significant improvements in functional independence at hospital discharge as well as in reduced durations of mechanical ventilation and delirium.
ANEMIA
Studies have shown that most ICU patients are anemic as a result of chronic inflammation. Phlebotomy also contributes to ICU anemia. A large multicenter study involving patients in many different ICU settings challenged the conventional notion that a hemoglobin level of 100 g/L (10 g/dL) is needed in critically ill patients, with similar outcomes noted in those whose transfusion trigger was 7 g/dL. Red blood cell transfusion is associated with impairment of immune function and increased risk of infections as well as of ARDS and volume overload, all of which may explain the findings in this study. Recently, a conservative transfusion strategy enhanced survival among patients with active upper gastrointestinal hemorrhage.
ACUTE RENAL FAILURE
(See also Chap. 334) Acute renal failure occurs in a significant percentage of critically ill patients. The most common underlying etiology is acute tubular necrosis, usually precipitated by hypoperfusion and/or nephrotoxic agents. Currently, no pharmacologic agents are available for prevention of renal injury in critical illness. Studies have shown convincingly that low-dose dopamine is not effective in protecting the kidneys from acute injury.
NEUROLOGIC DYSFUNCTION IN CRITICALLY ILL PATIENTS
DELIRIUM
(See also Chaps. 34 and 328) This state is defined by (1) an acute onset of changes or fluctuations in mental status, (2) inattention, (3) disorganized thinking, and (4) an altered level of consciousness (i.e., a state other than alertness). Delirium is reported to occur in a wide range of mechanically ventilated ICU patients and can be detected by the Confusion Assessment Method (CAM)-ICU or the Intensive Care Delirium Screening Checklist. These tools are used to ask patients to answer simple questions and perform simple tasks and can be used readily at the bedside. The differential diagnosis of delirium in ICU patients is broad and includes infectious etiologies (including sepsis), medications (particularly sedatives and analgesics), drug withdrawal, metabolic/electrolyte derangements, intracranial pathology (e.g., stroke, intracranial hemorrhage), seizures, hypoxia, hypertensive crisis, shock, and vitamin deficiencies (particularly thiamine). Patients with ICU delirium have increases in length of hospital stay, time on mechanical ventilation, cognitive impairment at hospital discharge, and 6-month mortality rate. Interventions to reduce ICU delirium are limited. The sedative dexmedetomidine has been less strongly associated with ICU delirium than midazolam. In addition, as mentioned above, very early physical and occupational therapy in mechanically ventilated patients has been demonstrated to reduce delirium.
ANOXIC CEREBRAL INJURY
(See also Chap. 330) This condition is common after cardiac arrest and often results in severe and permanent brain injury in survivors. Active cooling of patients after cardiac arrest has been shown to improve neurologic outcomes. Therefore, patients who present to the ICU after circulatory arrest from ventricular fibrillation or pulseless ventricular tachycardia should be actively cooled to achieve a core body temperature of 32–34°C.
STROKE
(See also Chap. 446) Stroke is a common cause of neurologic critical illness. Hypertension must be managed carefully, since abrupt reductions in blood pressure may be associated with further brain ischemia and injury. Acute ischemic stroke treated with tissue plasminogen activator (tPA) has an improved neurologic outcome when treatment is given within 3 h of onset of symptoms. The mortality rate is not reduced when tPA is compared with placebo, despite the improved neurologic outcome. The risk of cerebral hemorrhage is significantly higher in patients given tPA. No benefit is seen when tPA therapy is given beyond 3 h after symptom onset. Heparin has not been convincingly shown to improve outcomes in patients with acute ischemic stroke. Decompressive craniectomy is a surgical procedure that relieves increased intracranial pressure in the setting of space-occupying brain lesions or brain swelling from stroke; available evidence suggests that this procedure may improve survival among select patients (≤55 years or age), albeit at a cost of increased disability for some.
SUBARACHNOID HEMORRHAGE
(See also Chap. 446) Subarachnoid hemorrhage may occur secondary to aneurysm rupture and is often complicated by cerebral vasospasm, re-bleeding, and hydrocephalus. Vasospasm can be detected by either transcranial Doppler assessment or cerebral angiography; it is typically treated with the calcium channel blocker nimodipine, aggressive IV fluid administration, and therapy aimed at increasing blood pressure, typically with vasoactive drugs such as phenylephrine. The IV fluids and vasoactive drugs (hypertensive hypervolemic therapy) are used to overcome the cerebral vasospasm. Early surgical clipping or endovascular coiling of aneurysms is advocated to prevent complications related to re-bleeding. Hydrocephalus, typically heralded by a decreased level of consciousness, may require ventriculostomy drainage.
STATUS EPILEPTICUS
(See also Chap. 445) Recurrent or relentless seizure activity is a medical emergency. Cessation of seizure activity is required to prevent irreversible neurologic injury. Lorazepam is the most effective benzodiazepine for treating status epilepticus and is the treatment of choice for controlling seizures acutely. Phenytoin or fosphenytoin should be given concomitantly since lorazepam has a short half-life. Other drugs, such as gabapentin, carbamazepine, and phenobarbital, should be reserved for patients with contraindications to phenytoin (e.g., allergy or pregnancy) or ongoing seizures despite phenytoin.
BRAIN DEATH
(See also Chap. 330) Although deaths of critically ill patients usually are attributable to irreversible cessation of circulatory and respiratory function, a diagnosis of death also may be established by irreversible cessation of all functions of the entire brain, including the brainstem, even if circulatory and respiratory functions remain intact on artificial life support. Such a diagnosis requires demonstration of the absence of cerebral function (no response to any external stimulus) and brainstem functions (e.g., unreactive pupils, lack of ocular movement in response to head turning or ice-water irrigation of ear canals, positive apnea test [no drive to breathe]). Absence of brain function must have an established cause and be permanent without possibility of recovery; a sedative effect, hypothermia, hypoxemia, neuromuscular paralysis, and severe hypotension must be ruled out. If there is uncertainty about the cause of coma, studies of cerebral blood flow and electroencephalography should be performed.
WITHHOLDING OR WITHDRAWING CARE
(See also Chap. 10) Withholding or withdrawal of care occurs commonly in the ICU setting. The Task Force on Ethics of the Society of Critical Care Medicine reported that it is ethically sound to withhold or withdraw care if a patient or the patient’s surrogate makes such a request or if the physician judges that the goals of therapy are not achievable. Since all medical treatments are justified by their expected benefits, the loss of such an expectation justifies the act of withdrawing or withholding such treatment; these two actions are judged to be fundamentally similar. An underlying stipulation derived from this report is that an informed patient should have his or her wishes respected with regard to life-sustaining therapy. Implicit in this stipulation is the need to ensure that patients are thoroughly and accurately informed regarding the plausibility and expected results of various therapies.
The act of informing patients and/or surrogate decision-makers is the responsibility of the physician and other health care providers. If a patient or surrogate desires therapy deemed futile by the treating physician, the physician is not obligated ethically to provide such treatment. Rather, arrangements may be made to transfer the patient’s care to another care provider. Whether the decision to withdraw life support should be initiated by the physician or left to surrogate decision-makers alone is not clear. One study reported that slightly more than half of surrogate decision-makers preferred to receive such a recommendation, whereas the rest did not. Critical care providers should meet regularly with patients and/or surrogates to discuss prognosis when the withholding or withdrawal of care is being considered. After a consensus among caregivers has been reached, this information should be relayed to the patient and/or surrogate decision-maker. If a decision to withhold or withdraw life-sustaining care for a patient has been made, aggressive attention to analgesia and anxiolysis is needed.
322 |
Acute Respiratory Distress Syndrome |
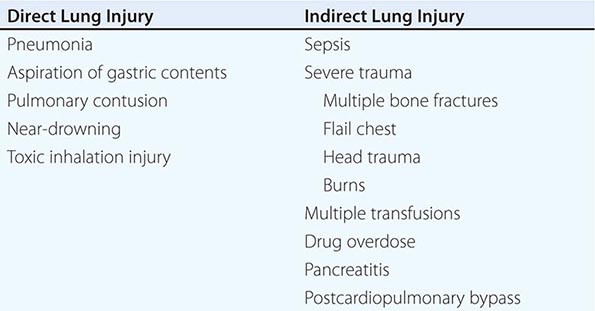
Acute respiratory distress syndrome (ARDS) is a clinical syndrome of severe dyspnea of rapid onset, hypoxemia, and diffuse pulmonary infiltrates leading to respiratory failure. ARDS is caused by diffuse lung injury from many underlying medical and surgical disorders. The lung injury may be direct, as occurs in toxic inhalation, or indirect, as occurs in sepsis (Table 322-1). The clinical features of ARDS are listed in Table 322-2. By expert consensus, ARDS is defined by three categories based on the degrees of hypoxemia (Table 322-2). These stages of mild, moderate, and severe ARDS are associated with mortality risk and with the duration of mechanical ventilation in survivors.
|
CLINICAL DISORDERS COMMONLY ASSOCIATED WITH ARDS |
|
DIAGNOSTIC CRITERIA FOR ARDS |
The annual incidence of ARDS is estimated to be as high as 60 cases/100,000 population. Approximately 10% of all intensive care unit (ICU) admissions involve patients with acute respiratory failure; ~20% of these patients meet the criteria for ARDS.
ETIOLOGY
While many medical and surgical illnesses have been associated with the development of ARDS, most cases (>80%) are caused by a relatively small number of clinical disorders: severe sepsis syndrome and/or bacterial pneumonia (~40–50%), trauma, multiple transfusions, aspiration of gastric contents, and drug overdose. Among patients with trauma, the most frequently reported surgical conditions in ARDS are pulmonary contusion, multiple bone fractures, and chest wall trauma/flail chest, whereas head trauma, near-drowning, toxic inhalation, and burns are rare causes. The risks of developing ARDS are increased in patients with more than one predisposing medical or surgical condition.
Several other clinical variables have been associated with the development of ARDS. These include older age, chronic alcohol abuse, metabolic acidosis, and severity of critical illness. Trauma patients with an Acute Physiology and Chronic Health Evaluation (APACHE) II score ≥16 (Chap. 321) have a 2.5-fold increased risk of developing ARDS, and those with a score >20 have an incidence of ARDS that is more than threefold greater than the incidence among those with APACHE II scores ≤9.
CLINICAL COURSE AND PATHOPHYSIOLOGY
The natural history of ARDS is marked by three phases—exudative, proliferative, and fibrotic—that each have characteristic clinical and pathologic features (Fig. 322-1).
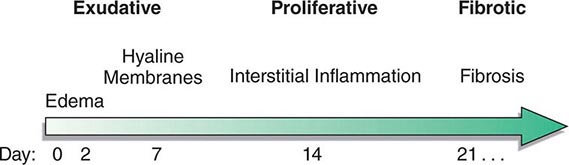
FIGURE 322-1 Diagram illustrating the time course for the development and resolution of ARDS. The exudative phase is notable for early alveolar edema and neutrophil-rich leukocytic infiltration of the lungs, with subsequent formation of hyaline membranes from diffuse alveolar damage. Within 7 days, a proliferative phase ensues with prominent interstitial inflammation and early fibrotic changes. Approximately 3 weeks after the initial pulmonary injury, most patients recover. However, some patients enter the fibrotic phase, with substantial fibrosis and bullae formation.
Exudative Phase In this phase (Fig. 322-2), alveolar capillary endothelial cells and type I pneumocytes (alve\olar epithelial cells) are injured, with consequent loss of the normally tight alveolar barrier to fluid and macromolecules. Edema fluid that is rich in protein accumulates in the interstitial and alveolar spaces. Significant concentrations of cytokines (e.g., interleukin 1, interleukin 8, and tumor necrosis factor α) and lipid mediators (e.g., leukotriene B4) are present in the lung in this acute phase. In response to proinflammatory mediators, leukocytes (especially neutrophils) traffic into the pulmonary interstitium and alveoli. In addition, condensed plasma proteins aggregate in the air spaces with cellular debris and dysfunctional pulmonary surfactant to form hyaline membrane whorls. Pulmonary vascular injury also occurs early in ARDS, with vascular obliteration by microthrombi and fibrocellular proliferation (Fig. 322-3).
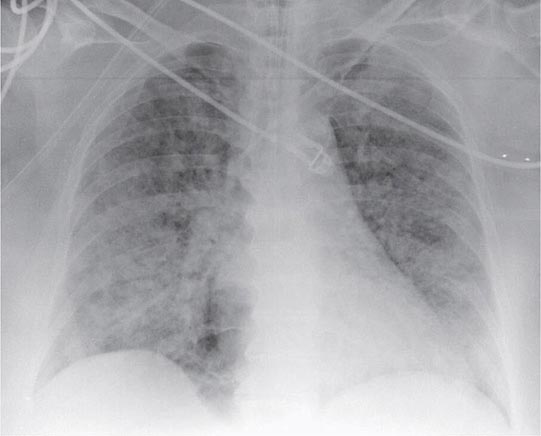
FIGURE 322-2 A representative anteroposterior chest x-ray in the exudative phase of ARDS shows diffuse interstitial and alveolar infiltrates that can be difficult to distinguish from left ventricular failure.
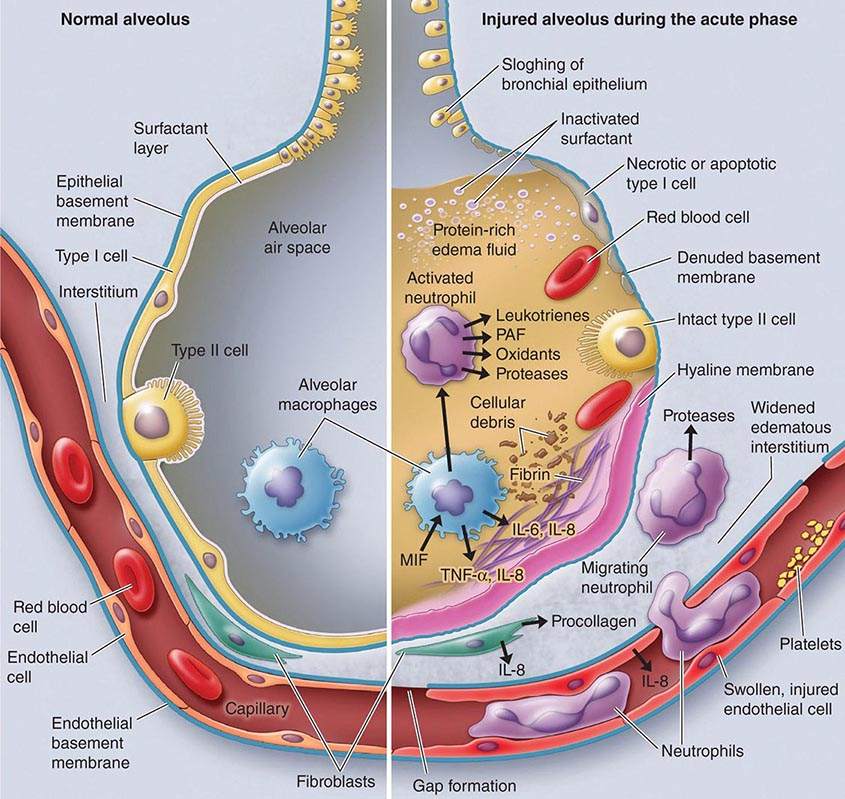
FIGURE 322-3 The normal alveolus (left) and the injured alveolus in the acute phase of acute lung injury and the acute respiratory distress syndrome (right). In the acute phase of the syndrome (right), there is sloughing of both the bronchial and alveolar epithelial cells, with the formation of protein-rich hyaline membranes on the denuded basement membrane. Neutrophils are shown adhering to the injured capillary endothelium and transmigrating through the interstitium into the air space, which is filled with protein-rich edema fluid. In the air space, an alveolar macrophage is secreting cytokines—i.e., interleukins 1, 6, 8, and 10 (IL-1, -6, -8, and -10) and tumor necrosis factor α (TNF-α)—that act locally to stimulate chemotaxis and activate neutrophils. Macrophages also secrete other cytokines, including IL-1, -6, and -10. IL-1 can also stimulate the production of extracellular matrix by fibroblasts. Neutrophils can release oxidants, proteases, leukotrienes, and other proinflammatory molecules, such as platelet-activating factor (PAF). A number of antiinflammatory mediators are also present in the alveolar milieu, including the IL-1-receptor antagonist, soluble TNF-α receptor, autoantibodies to IL-8, and cytokines such as IL-10 and IL-11 (not shown). The influx of protein-rich edema fluid into the alveolus has led to the inactivation of surfactant. MIF, macrophage inhibitory factor. (From LB Ware, MA Matthay: N Engl J Med 342:1334, 2000, with permission.)
Alveolar edema predominantly involves dependent portions of the lung, with diminished aeration and atelectasis. Collapse of large sections of dependent lung markedly decreases lung compliance. Consequently, intrapulmonary shunting and hypoxemia develop and the work of breathing increases, leading to dyspnea. The pathophysiologic alterations in alveolar spaces are exacerbated by microvascular occlusion that results in reductions in pulmonary arterial blood flow to ventilated portions of the lung (and thus in increased dead space) and in pulmonary hypertension. Thus, in addition to severe hypoxemia, hypercapnia secondary to an increase in pulmonary dead space is prominent in early ARDS.
The exudative phase encompasses the first 7 days of illness after exposure to a precipitating ARDS risk factor, with the patient experiencing the onset of respiratory symptoms. Although usually presenting within 12–36 h after the initial insult, symptoms can be delayed by 5–7 days. Dyspnea develops, with a sensation of rapid shallow breathing and an inability to get enough air. Tachypnea and increased work of breathing result frequently in respiratory fatigue and ultimately in respiratory failure. Laboratory values are generally nonspecific and are primarily indicative of underlying clinical disorders. The chest radiograph usually reveals alveolar and interstitial opacities involving at least three-quarters of the lung fields (Fig. 322-2). While characteristic for ARDS, these radiographic findings are not specific and can be indistinguishable from cardiogenic pulmonary edema (Chap. 326). Unlike the latter, however, the chest x-ray in ARDS rarely shows cardiomegaly, pleural effusions, or pulmonary vascular redistribution. Chest CT in ARDS reveals extensive heterogeneity of lung involvement (Fig. 322-4).
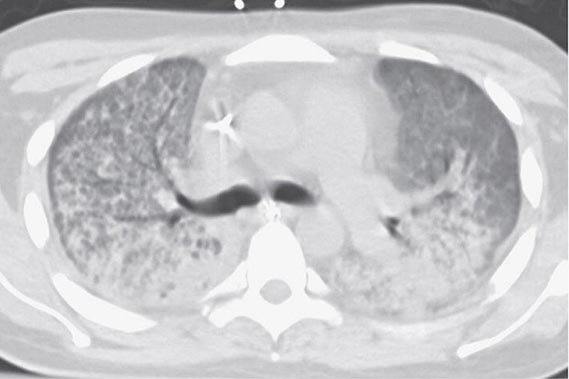
FIGURE 322-4 A representative CT scan of the chest during the exudative phase of ARDS, in which dependent alveolar edema and atelectasis predominate.
Because the early features of ARDS are nonspecific, alternative diagnoses must be considered. In the differential diagnosis of ARDS, the most common disorders are cardiogenic pulmonary edema, diffuse pneumonia, and alveolar hemorrhage. Less common diagnoses to consider include acute interstitial lung diseases (e.g., acute interstitial pneumonitis; Chap. 315), acute immunologic injury (e.g., hypersensitivity pneumonitis; Chap. 310), toxin injury (e.g., radiation pneumonitis; Chap. 263), and neurogenic pulmonary edema (Chap. 47e).
Proliferative Phase This phase of ARDS usually lasts from day 7 to day 21. Most patients recover rapidly and are liberated from mechanical ventilation during this phase. Despite this improvement, many patients still experience dyspnea, tachypnea, and hypoxemia. Some patients develop progressive lung injury and early changes of pulmonary fibrosis during the proliferative phase. Histologically, the first signs of resolution are often evident in this phase, with the initiation of lung repair, the organization of alveolar exudates, and a shift from a neutrophil- to a lymphocyte-predominant pulmonary infiltrate. As part of the reparative process, type II pneumocytes proliferate along alveolar basement membranes. These specialized epithelial cells synthesize new pulmonary surfactant and differentiate into type I pneumocytes.
Fibrotic Phase While many patients with ARDS recover lung function 3–4 weeks after the initial pulmonary injury, some enter a fibrotic phase that may require long-term support on mechanical ventilators and/or supplemental oxygen. Histologically, the alveolar edema and inflammatory exudates of earlier phases are now converted to extensive alveolar-duct and interstitial fibrosis. Marked disruption of acinar architecture leads to emphysema-like changes, with large bullae. Intimal fibroproliferation in the pulmonary microcirculation causes progressive vascular occlusion and pulmonary hypertension. The physiologic consequences include an increased risk of pneumothorax, reductions in lung compliance, and increased pulmonary dead space. Patients in this late phase experience a substantial burden of excess morbidity. Lung biopsy evidence for pulmonary fibrosis in any phase of ARDS is associated with increased mortality risk.
|
TREATMENT |
ACUTE RESPIRATORY DISTRESS SYNDROME |
GENERAL PRINCIPLES
Recent reductions in ARDS mortality rates are largely the result of general advances in the care of critically ill patients (Chap. 321). Thus, caring for these patients requires close attention to (1) the recognition and treatment of underlying medical and surgical disorders (e.g., sepsis, aspiration, trauma); (2) the minimization of procedures and their complications; (3) prophylaxis against venous thromboembolism, gastrointestinal bleeding, aspiration, excessive sedation, and central venous catheter infections; (4) prompt recognition of nosocomial infections; and (5) provision of adequate nutrition.
MANAGEMENT OF MECHANICAL VENTILATION
(See also Chap. 323) Patients meeting clinical criteria for ARDS frequently become fatigued from increased work of breathing and progressive hypoxemia, requiring mechanical ventilation for support.
Ventilator-Induced Lung Injury Despite its life-saving potential, mechanical ventilation can aggravate lung injury. Experimental models have demonstrated that ventilator-induced lung injury appears to require two processes: repeated alveolar overdistention and recurrent alveolar collapse. As is clearly evident from chest CT (Fig. 322-4), ARDS is a heterogeneous disorder, principally involving dependent portions of the lung with relative sparing of other regions. Because compliance differs in affected versus more “normal” areas of the lung, attempts to fully inflate the consolidated lung may lead to overdistention of and injury to the more normal areas. Ventilator-induced injury can be demonstrated in experimental models of acute lung injury, with high-tidal-volume (VT) ventilation resulting in additional, synergistic alveolar damage.
A large-scale, randomized controlled trial sponsored by the National Institutes of Health and conducted by the ARDS Network compared low VT ventilation (6 mL/kg of predicted body weight) to conventional VT ventilation (12 mL/kg predicted body weight). The mortality rate was significantly lower in the low VT patients (31%) than in the conventional VT patients (40%). This improvement in survival represents the most substantial ARDS-mortality benefit that has been demonstrated for any therapeutic intervention to date.
Prevention of Alveolar Collapse In ARDS, the presence of alveolar and interstitial fluid and the loss of surfactant can lead to a marked reduction of lung compliance. Without an increase in end-expiratory pressure, significant alveolar collapse can occur at end-expiration, with consequent impairment of oxygenation. In most clinical settings, positive end-expiratory pressure (PEEP) is empirically set to minimize FIO2 (inspired O2 percentage) and maximize PaO2 (arterial partial pressure of O2). On most modern mechanical ventilators, it is possible to construct a static pressure–volume curve for the respiratory system. The lower inflection point on the curve represents alveolar opening (or “recruitment”). The pressure at this point, usually 12–15 mmHg in ARDS, is a theoretical “optimal PEEP” for alveolar recruitment. Titration of the PEEP to the lower inflection point on the static pressure–volume curve has been hypothesized to keep the lung open, improving oxygenation and protecting against lung injury. Three large randomized trials have investigated the utility of PEEP-based strategies to keep the lung open. In all three trials, improvement in lung function was evident but overall mortality rates were not altered significantly. Until more data become available on the clinical utility of high PEEP, it is advisable to set PEEP to minimize FIO2 and optimize PaO2 (Chap. 323). Measurement of esophageal pressures to estimate transpulmonary pressure may help identify an optimal PEEP in some cases.
Oxygenation can also be improved by increasing mean airway pressure with inverse-ratio ventilation. In this technique, the inspiratory time (I) is lengthened so that it is longer than the expiratory time (E)—that is, I:E > 1:1. With diminished time to exhale, dynamic hyperinflation leads to increased end-expiratory pressure, similar to ventilator-prescribed PEEP. This mode of ventilation has the advantage of improving oxygenation with lower peak pressures than are required for conventional ventilation. Although inverse-ratio ventilation can improve oxygenation and can help reduce FIO2 to ≤0.60, thus avoiding possible oxygen toxicity, no benefit in ARDS mortality risk has been demonstrated. Recruitment maneuvers that transiently increase PEEP to “recruit” atelectatic lung can also increase oxygenation, but a mortality benefit has not been established.
In several randomized trials, mechanical ventilation in the prone position improved arterial oxygenation, but its effect on survival and other important clinical outcomes remains uncertain. Moreover, unless the critical-care team is experienced in “proning,” repositioning critically ill patients can be hazardous, leading to accidental endotracheal extubation, loss of central venous catheters, and orthopedic injury.
OTHER STRATEGIES IN MECHANICAL VENTILATION
Several additional mechanical-ventilation strategies that use specialized equipment have been tested in ARDS patients; most of these approaches have had mixed or disappointing results in adults. High-frequency ventilation (HFV) entails ventilating at extremely high respiratory rates (5–20 cycles per second) and low VTs (1–2 mL/kg). Use of partial liquid ventilation (PLV) with perfluorocarbon—an inert, high-density liquid that easily solubilizes oxygen and carbon dioxide—has yielded promising preliminary results, enhancing pulmonary function in patients with ARDS, but also has provided no survival benefit. Lung-replacement therapy with extracorporeal membrane oxygenation (ECMO), which provides a clear survival benefit in neonatal respiratory distress syndrome, may also have utility in selected adult patients with ARDS.
Data supporting the efficacy of “adjunctive” ventilator therapies (e.g., high PEEP, inverse ratio ventilation, recruitment maneuvers, prone positioning, HFV, ECMO, and PLV) remain incomplete. Accordingly, these modalities are reserved for use as rescue rather than primary therapies.
FLUID MANAGEMENT
(See also Chap. 321) Increased pulmonary vascular permeability leading to interstitial and alveolar edema fluid rich in protein is a central feature of ARDS. In addition, impaired vascular integrity augments the normal increase in extravascular lung water that occurs with increasing left atrial pressure. Maintaining a low left atrial filling pressure minimizes pulmonary edema and prevents further decrements in arterial oxygenation and lung compliance; improves pulmonary mechanics; shortens ICU stay and the duration of mechanical ventilation; and is associated with a lower mortality rate in both medical and surgical ICU patients. Thus, aggressive attempts to reduce left atrial filling pressures with fluid restriction and diuretics should be an important aspect of ARDS management, limited only by hypotension and hypoperfusion of critical organs such as the kidneys.
NEUROMUSCULAR BLOCKADE
In severe ARDS, sedation alone can be inadequate for the patient-ventilator synchrony required for lung-protective ventilation. This clinical problem was recently addressed in a multicenter, randomized, placebo-controlled trial of early neuromuscular blockade (with cisatracurium besylate) for 48 h. In severe ARDS, early neuromuscular blockade increased the rate of survival and ventilator-free days without increasing ICU-acquired paresis. These promising findings support the early administration of neuromuscular blockade if needed to facilitate mechanical ventilation in severe ARDS; however, these results must be replicated prior to their widespread application in clinical practice.
GLUCOCORTICOIDS
Many attempts have been made to treat both early and late ARDS with glucocorticoids, with the goal of reducing potentially deleterious pulmonary inflammation. Few studies have shown any benefit. Current evidence does not support the use of high-dose glucocorticoids in the care of ARDS patients.
OTHER THERAPIES
Clinical trials of surfactant replacement and multiple other medical therapies have proved disappointing. Inhaled nitric oxide and inhaled epoprostenol sodium can transiently improve oxygenation but do not improve survival or decrease time on mechanical ventilation.
RECOMMENDATIONS
Many clinical trials have been undertaken to improve the outcome of patients with ARDS; most have been unsuccessful in modifying the natural history. While results of large clinical trials must be judiciously applied to individual patients, evidence-based recommendations are summarized in Table 322-3, and an algorithm for the initial therapeutic goals and limits in ARDS management is provided in Fig. 322-5.
|
EVIDENCE-BASED RECOMMENDATIONS FOR ARDS THERAPIES |
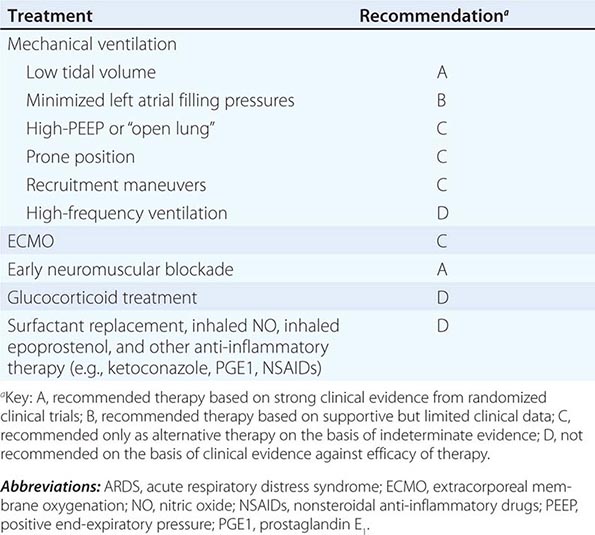
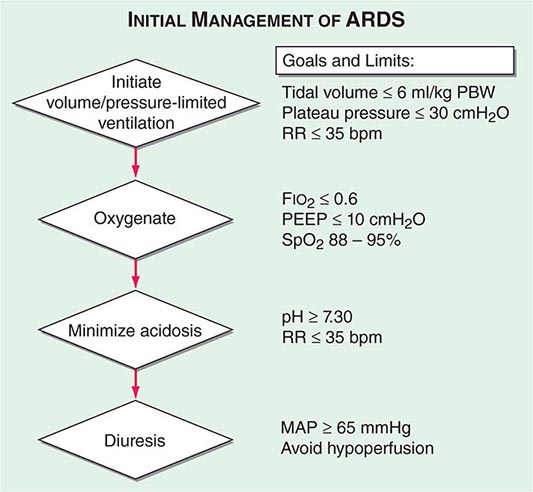
FIGURE 322-5 Algorithm for the initial management of ARDS. Clinical trials have provided evidence-based therapeutic goals for a stepwise approach to the early mechanical ventilation, oxygenation, and correction of acidosis and diuresis of critically ill patients with ARDS. FIO2, inspired O2 percentage; MAP, mean arterial pressure; PBW, predicted body weight; PEEP, positive end expiratory pressure; RR, respiratory rate; SpO2, arterial oxyhemoglobin saturation measured by pulse oximetry.
PROGNOSIS
Mortality Recent mortality estimates for ARDS range from 26% to 44%. There is substantial variability, but a trend toward improved ARDS outcomes appears evident. Of interest, mortality in ARDS is largely attributable to nonpulmonary causes, with sepsis and nonpulmonary organ failure accounting for >80% of deaths. Thus, improvement in survival is likely secondary to advances in the care of septic/infected patients and those with multiple organ failure (Chap. 321).
The major risk factors for ARDS mortality are nonpulmonary. Advanced age is an important risk factor. Patients >75 years of age have a substantially higher mortality risk (~60%) than those <45 (~20%). Moreover, patients >60 years of age with ARDS and sepsis have a threefold higher mortality risk than those <60. Other risk factors include preexisting organ dysfunction from chronic medical illness—in particular, chronic liver disease, cirrhosis, chronic alcohol abuse, chronic immunosuppression, sepsis, chronic renal disease, failure of any nonpulmonary organ, and increased APACHE III scores (Chap. 321). Patients with ARDS arising from direct lung injury (including pneumonia, pulmonary contusion, and aspiration; Table 322-1) are nearly twice as likely to die as those with indirect causes of lung injury, while surgical and trauma patients with ARDS—especially those without direct lung injury—have a higher survival rate than other ARDS patients.
An early (within 24 h of presentation) elevation in pulmonary dead space (>0.60) and severe arterial hypoxemia (PaO2/FIO2, <100 mmHg) predict increased mortality risk from ARDS; however, there is surprisingly little additional value in predicting ARDS mortality from other measures of the severity of lung injury, including the level of PEEP (≥10 cm H2O), respiratory system compliance (≤40 mL/cm H2O), the extent of alveolar infiltrates on chest radiography, and the corrected expired volume per minute (≥10 L/min).
Functional Recovery in ARDS Survivors While it is common for patients with ARDS to experience prolonged respiratory failure and remain dependent on mechanical ventilation for survival, it is a testament to the resolving powers of the lung that the majority of patients recover nearly normal lung function. Patients usually recover maximal lung function within 6 months. One year after endotracheal extubation, more than one-third of ARDS survivors have normal spirometry values and diffusion capacity. Most of the remaining patients have only mild abnormalities in pulmonary function. Unlike mortality risk, recovery of lung function is strongly associated with the extent of lung injury in early ARDS. Low static respiratory compliance, high levels of required PEEP, longer durations of mechanical ventilation, and high lung injury scores are all associated with less recovery of pulmonary function. Of note, when physical function is assessed 5 years after ARDS, exercise limitation and decreased physical quality of life are often documented despite normal or nearly normal pulmonary function. When caring for ARDS survivors, it is important to be aware of the potential for a substantial burden of psychological problems in patients and family caregivers, including significant rates of depression and posttraumatic stress disorder.
WEBSITES
ARDS Support Center for patient-oriented education: www.ards.org
NHLBI ARDS Clinical Trials information: www.ardsnet.org
ARDS Foundation: www.ardsusa.org
ACKNOWLEDGMENT
The authors acknowledge the contribution to this chapter by the previous author, Dr. Steven D. Shapiro.
323 |
Mechanical Ventilatory Support |
MECHANICAL VENTILATORY SUPPORT
Mechanical ventilation is used to assist or replace spontaneous breathing. It is implemented with special devices that can support ventilatory function and improve oxygenation through the application of high-oxygen-content gas and positive pressure. The primary indication for initiation of mechanical ventilation is respiratory failure, of which there are two basic types: (1) hypoxemic, which is present when arterial O2 saturation (SaO2) <90% occurs despite an increased inspired O2 fraction and usually results from ventilation-perfusion mismatch or shunt; and (2) hypercarbic, which is characterized by elevated arterial carbon dioxide partial pressure (PCO2) values (usually >50 mmHg) resulting from conditions that decrease minute ventilation or increase physiologic dead space such that alveolar ventilation is inadequate to meet metabolic demands. When respiratory failure is chronic, neither of the two types is obligatorily treated with mechanical ventilation, but when it is acute, mechanical ventilation may be lifesaving.
INDICATIONS
The most common reasons for instituting mechanical ventilation are acute respiratory failure with hypoxemia (acute respiratory distress syndrome, heart failure with pulmonary edema, pneumonia, sepsis, complications of surgery and trauma), which accounts for ~65% of all ventilated cases, and hypercarbic ventilatory failure—e.g., due to coma (15%), exacerbations of chronic obstructive pulmonary disease (COPD; 13%), and neuromuscular diseases (5%). The primary objectives of mechanical ventilation are to decrease the work of breathing, thus avoiding respiratory muscle fatigue, and to reverse life-threatening hypoxemia and progressive respiratory acidosis.
In some cases, mechanical ventilation is used as an adjunct to other forms of therapy. For example, it is used to reduce cerebral blood flow in patients with increased intracranial pressure. Mechanical ventilation also is used frequently in conjunction with endotracheal intubation for airway protection to prevent aspiration of gastric contents in otherwise unstable patients during gastric lavage for suspected drug overdose or during gastrointestinal endoscopy. In critically ill patients, intubation and mechanical ventilation may be indicated before the performance of essential diagnostic or therapeutic studies if it appears that respiratory failure may occur during those maneuvers.
TYPES OF MECHANICAL VENTILATION
There are two basic methods of mechanical ventilation: noninvasive ventilation (NIV) and invasive (or conventional mechanical) ventilation (MV).
Noninvasive Ventilation NIV has gained acceptance because it is effective in certain conditions, such as acute or chronic respiratory failure, and is associated with fewer complications—namely, pneumonia and tracheolaryngeal trauma. NIV usually is provided with a tight-fitting face mask or nasal mask similar to the masks traditionally used for treatment of sleep apnea. NIV has proved highly effective in patients with respiratory failure arising from acute exacerbations of chronic obstructive pulmonary disease. It is most frequently implemented as bilevel positive airway pressure ventilation or pressure-support ventilation. Both modes, which apply a preset positive pressure during inspiration and a lower pressure during expiration at the mask, are well tolerated by a conscious patient and optimize patient-ventilator synchrony. The major limitation to the widespread application of NIV has been patient intolerance: the tight-fitting mask required for NIV can cause both physical and psychological discomfort. In addition, NIV has had limited success in patients with acute hypoxemic respiratory failure, for whom endotracheal intubation and conventional MV remain the ventilatory method of choice.
The most important group of patients who benefit from a trial of NIV are those with exacerbations of COPD and respiratory acidosis (pH <7.35). Experience from several randomized trials has shown that, in patients with ventilatory failure characterized by blood pH levels between 7.25 and 7.35, NIV is associated with low failure rates (15–20%) and good outcomes (as judged by intubation rate, length of stay in intensive care, and—in some series—mortality rates). In more severely ill patients with a blood pH <7.25, the rate of NIV failure is inversely related to the severity of respiratory acidosis, with higher failure rates as the pH decreases. In patients with milder acidosis (pH >7.35), NIV is not better than conventional treatment that includes controlled oxygen delivery and pharmacotherapy for exacerbations of COPD (systemic glucocorticoids, bronchodilators, and, if needed, antibiotics).
Despite its benign outcomes, NIV is not useful in the majority of cases of respiratory failure and is contraindicated in patients with the conditions listed in Table 323-1. NIV can delay lifesaving ventilatory support in those cases and, in fact, can actually result in aspiration or hypoventilation. Once NIV is initiated, patients should be monitored; a reduction in respiratory frequency and a decrease in the use of accessory muscles (scalene, sternomastoid, and intercostals) are good clinical indicators of adequate therapeutic benefit. Arterial blood gases should be determined at least within hours of the initiation of therapy to ensure that NIV is having the desired effect. Lack of benefit within that time frame should alert the physician to the possible need for conventional MV.
|
CONTRAINDICATIONS FOR NONINVASIVE VENTILATION |
Conventional Mechanical Ventilation Conventional MV is implemented once a cuffed tube is inserted into the trachea to allow conditioned gas (warmed, oxygenated, and humidified) to be delivered to the airways and lungs at pressures above atmospheric pressure. Care should be taken during intubation to avoid brain-damaging hypoxia. In most cases, the administration of mild sedation may facilitate the procedure. Opiates and benzodiazepines are good choices but can have a deleterious effect on hemodynamics in patients with depressed cardiac function or low systemic vascular resistance. Morphine can promote histamine release from tissue mast cells and may worsen bronchospasm in patients with asthma; fentanyl, sufentanil, and alfentanil are acceptable alternatives. Ketamine may increase systemic arterial pressure and has been associated with hallucinatory responses. The shorter-acting agents etomidate and propofol have been used for both induction and maintenance of anesthesia in ventilated patients because they have fewer adverse hemodynamic effects, but both are significantly more expensive than older agents. Great care must be taken to avoid the use of neuromuscular paralysis during intubation of patients with renal failure, tumor lysis syndrome, crush injuries, medical conditions associated with elevated serum potassium levels, and muscular dystrophy syndromes; in particular, the use of agents whose mechanism of action includes depolarization at the neuromuscular junction, such as succinylcholine chloride, must be avoided.
PRINCIPLES OF MECHANICAL VENTILATION
Once the patient has been intubated, the basic goals of MV are to optimize oxygenation while avoiding ventilator-induced lung injury due to overstretch and collapse/re-recruitment. This concept, known as the “protective ventilatory strategy” (see below and Fig. 323-1) is supported by evidence linking high airway pressures and volumes and overstretching of the lung as well as collapse/re-recruitment to poor clinical outcomes (barotrauma and volume trauma). Although normalization of pH through elimination of CO2 is desirable, the risk of lung damage associated with the large volume and high pressures needed to achieve this goal has led to the acceptance of permissive hypercapnia. This condition is well tolerated when care is taken to avoid excess acidosis by pH buffering.
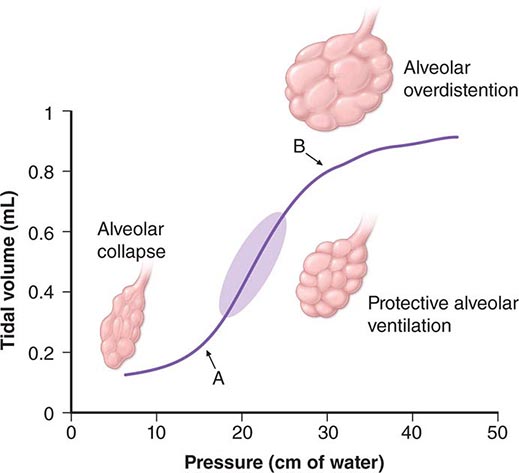
FIGURE 323-1 Hypothetical pressure-volume curve of the lung in a patient undergoing mechanical ventilation. Alveoli tend to close if the distending pressure falls below the lower inflection point (A), whereas they overstretch if the pressure within them is higher than that of the upper inflection point (B). Collapse and opening of ventilated alveoli are associated with poor outcomes in patients with acute respiratory failure. Protective ventilation (purple shaded area), using a lower tidal volume (6 mL/kg of ideal body weight) and maintaining positive end-expiratory pressure to prevent overstretching and collapse/opening of alveoli, has resulted in improved survival rates among patients receiving mechanical ventilatory support.
MODES OF VENTILATION
Mode refers to the manner in which ventilator breaths are triggered, cycled, and limited. The trigger, either an inspiratory effort or a time-based signal, defines what the ventilator senses to initiate an assisted breath. Cycle refers to the factors that determine the end of inspiration. For example, in volume-cycled ventilation, inspiration ends when a specific tidal volume is delivered. Other types of cycling include pressure cycling and time cycling. The limiting factors are operator-specified values, such as airway pressure, that are monitored by transducers internal to the ventilator circuit throughout the respiratory cycle; if the specified values are exceeded, inspiratory flow is terminated, and the ventilator circuit is vented to atmospheric pressure or the specified pressure at the end of expiration (positive end-expiratory pressure, or PEEP). Most patients are ventilated with assist-control ventilation, intermittent mandatory ventilation, or pressure-support ventilation, with the latter two modes often used simultaneously (Table 323-2).
|
CHARACTERISTICS OF THE MOST COMMONLY USED FORMS OF MECHANICAL VENTILATION |
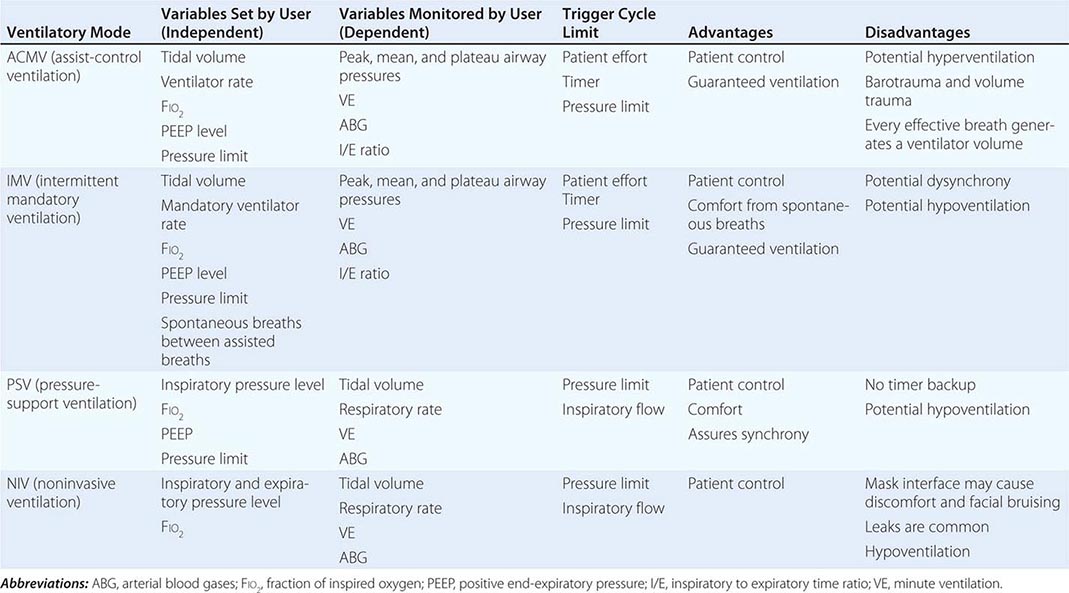
Assist-Control Ventilation (ACMV) ACMV is the most widely used mode of ventilation. In this mode, an inspiratory cycle is initiated either by the patient’s inspiratory effort or, if none is detected within a specified time window, by a timer signal within the ventilator. Every breath delivered, whether patient- or timer-triggered, consists of the operator-specified tidal volume. Ventilatory rate is determined either by the patient or by the operator-specified backup rate, whichever is of higher frequency. ACMV is commonly used for initiation of mechanical ventilation because it ensures a backup minute ventilation in the absence of an intact respiratory drive and allows for synchronization of the ventilator cycle with the patient’s inspiratory effort.
Problems can arise when ACMV is used in patients with tachypnea due to nonrespiratory or nonmetabolic factors, such as anxiety, pain, and airway irritation. Respiratory alkalemia may develop and trigger myoclonus or seizures. Dynamic hyperinflation leading to increased intrathoracic pressures (so-called auto-PEEP) may occur if the patient’s respiratory mechanics are such that inadequate time is available for complete exhalation between inspiratory cycles. Auto-PEEP can limit venous return, decrease cardiac output, and increase airway pressures, predisposing to barotrauma.
Intermittent Mandatory Ventilation (IMV) With this mode, the operator sets the number of mandatory breaths of fixed volume to be delivered by the ventilator; between those breaths, the patient can breathe spontaneously. In the most frequently used synchronized mode (SIMV), mandatory breaths are delivered in synchrony with the patient’s inspiratory efforts at a frequency determined by the operator. If the patient fails to initiate a breath, the ventilator delivers a fixed-tidal-volume breath and resets the internal timer for the next inspiratory cycle. SIMV differs from ACMV in that only a preset number of breaths are ventilator-assisted.
SIMV allows patients with an intact respiratory drive to exercise inspiratory muscles between assisted breaths; thus it is useful for both supporting and weaning intubated patients. SIMV may be difficult to use in patients with tachypnea because they may attempt to exhale during the ventilator-programmed inspiratory cycle. Consequently, the airway pressure may exceed the inspiratory pressure limit, the ventilator-assisted breath will be aborted, and minute volume may drop below that programmed by the operator. In this setting, if the tachypnea represents a response to respiratory or metabolic acidosis, a change in ACMV will increase minute ventilation and help normalize the pH while the underlying process is further evaluated and treated.
Pressure-Support Ventilation (PSV) This form of ventilation is patient-triggered, flow-cycled, and pressure-limited. It provides graded assistance and differs from the other two modes in that the operator sets the pressure level (rather than the volume) to augment every spontaneous respiratory effort. The level of pressure is adjusted by observing the patient’s respiratory frequency. During PSV, the inspiration is terminated when inspiratory airflow falls below a certain level; in most ventilators, this flow rate cannot be adjusted by the operator. With PSV, patients receive ventilator assistance only when the ventilator detects an inspiratory effort. PSV is often used in combination with SIMV to ensure volume-cycled backup for patients whose respiratory drive is depressed. PSV is well tolerated by most patients who are being weaned from MV; PSV parameters can be set to provide full ventilatory support and can be withdrawn to load the respiratory muscles gradually.
Other Modes of Ventilation There are other modes of ventilation, each with its own acronym and each with specific modifications of the manner and duration in which pressure is applied to the airway and lungs and of the interaction between the mechanical assistance provided by the ventilator and the patient’s respiratory effort. Although their use in acute respiratory failure is limited, the following modes have been used with varying levels of enthusiasm and adoption.
PRESSURE-CONTROL VENTILATION (PCV) This form of ventilation is time-triggered, time-cycled, and pressure-limited. A specified pressure is imposed at the airway opening throughout inspiration. Since the inspiratory pressure is specified by the operator, tidal volume and inspiratory flow rate are dependent, rather than independent, variables and are not operator-specified. PCV is the preferred mode of ventilation for patients in whom it is desirable to regulate peak airway pressures, such as those with preexisting barotrauma, and for post–thoracic surgery patients, in whom the shear forces across a fresh suture line should be limited. When PCV is used, minute ventilation is altered through changes in rate or in the pressure-control value, with consequent changes in tidal volume.
INVERSE-RATIO VENTILATION (IRV) This mode is a variant of PCV that incorporates the use of a prolonged inspiratory time with the appropriate shortening of the expiratory time. IRV has been used in patients with severe hypoxemic respiratory failure. This approach increases mean distending pressures without increasing peak airway pressures. It is thought to work in conjunction with PEEP to open collapsed alveoli and improve oxygenation. However, no clinical-trial data have shown that IRV improves outcomes.
CONTINUOUS POSITIVE AIRWAY PRESSURE (CPAP) CPAP is not a true support mode of ventilation because all ventilation occurs through the patient’s spontaneous efforts. The ventilator provides fresh gas to the breathing circuit with each inspiration and sets the circuit to a constant, operator-specified pressure. CPAP is used to assess extubation potential in patients who have been effectively weaned and who require little ventilatory support and in patients with intact respiratory system function who require an endotracheal tube for airway protection.
Nonconventional Ventilatory Strategies Several nonconventional strategies have been evaluated for their ability to improve oxygenation and reduce mortality rates in patients with advanced hypoxemic respiratory failure. These strategies include high-frequency oscillatory ventilation (HFOV), airway pressure release ventilation (APRV), extracorporeal membrane oxygenation (ECMO), and partial liquid ventilation (PLV) using perfluorocarbons. Although case reports and small uncontrolled cohort studies have shown benefit, randomized controlled trials have failed to demonstrate consistent improvements in outcome with most of these strategies. A recent randomized trial of ECMO documented positive outcomes, but the technique remains controversial because older studies failed to document positive results. Currently, these approaches should be thought of as “salvage” techniques and considered for patients with hypoxemia refractory to conventional therapy. Prone positioning of patients with refractory hypoxemia has also been explored because, in theory, lying prone should improve ventilation-perfusion matching. Several randomized trials in patients with acute lung injury did not demonstrate a survival advantage with prone positioning despite demonstration of a transient physiologic benefit. The administration of nitric oxide gas, which has bronchodilator and pulmonary vasodilator effects when delivered through the airways and improves arterial oxygenation in many patients with advanced hypoxemic respiratory failure, also failed to improve outcomes in these patients with acute lung injury.
The design of new ventilator modes reflect attempts to improve patient-ventilator synchrony—a major practical issue during MV—by allowing patients to trigger the ventilator with their own effort while also incorporating flow algorithms that terminate the cycles once certain preset criteria are reached; this approach has greatly improved patient comfort. New modes of ventilation that synchronize not only the timing but also the levels of assistance to match the patient’s effort have been developed. Proportional assist ventilation (PAV) and neurally adjusted ventilatory-assist ventilation (NAV) are two modes that are designed to deliver assisted breaths through algorithms incorporating not only pressure, volume, and time but also overall respiratory resistance as well as compliance (in the case of PAV) and neural activation of the diaphragm (in the case of NAV). Although these modes enhance patient-ventilator synchrony, their practical use in the everyday management of patients undergoing MV needs further study.
PROTECTIVE VENTILATORY STRATEGY
Whichever mode of MV is used in acute respiratory failure, the evidence from several important controlled trials indicates that a protective ventilation approach guided by the following principles (and summarized in Fig. 323-1) is safe and offers the best chance of a good outcome: (1) Set a target tidal volume close to 6 mL/kg of ideal body weight. (2) Prevent plateau pressure (static pressure in the airway at the end of inspiration) exceeding 30 cm H2O. (3) Use the lowest possible fraction of inspired oxygen (FIO2) to keep the SaO2 at ≥90%. (4) Adjust the PEEP to maintain alveolar patency while preventing overdistention and closure/reopening. With the application of these techniques, the mortality rate among patients with acute hypoxemic respiratory failure has decreased to ~30% from close to 50% a decade ago.
PATIENT MANAGEMENT
Once the patient has been stabilized with respect to gas exchange, definitive therapy for the underlying process responsible for respiratory failure is initiated. Subsequent modifications in ventilator therapy must be provided in parallel with changes in the patient’s clinical status. As improvement in respiratory function is noted, the first priority is to reduce the level of mechanical ventilatory support. Patients on full ventilatory support should be monitored frequently, with the goal of switching to a mode that allows for weaning as soon as possible. Protocols and guidelines that can be applied by paramedical personnel when physicians are not readily available have proved to be of value in shortening ventilator and intensive care unit (ICU) time, with very good outcomes. Patients whose condition continues to deteriorate after ventilatory support is initiated may require increased O2, PEEP, or one of the alternative modes of ventilation.
GENERAL SUPPORT DURING VENTILATION
Patients for whom mechanical ventilation has been initiated usually require sedation and analgesia to maintain an acceptable level of comfort. Often, this treatment consists of a combination of a benzodiazepine and an opiate administered intravenously. Medications commonly used for this purpose include lorazepam, midazolam, diazepam, morphine, and fentanyl. Oversedation must be avoided in the ICU because most (but not all) studies show that daily interruption of sedation in patients with improved ventilatory status results in a shorter time on the ventilator and a shorter ICU stay.
Immobilized patients receiving mechanical ventilatory support are at risk for deep venous thrombosis and decubitus ulcers. Venous thrombosis should be prevented with the use of subcutaneous heparin and/or pneumatic compression boots. Fractionated low-molecular-weight heparin appears to be equally effective for this purpose. To help prevent decubitus ulcers, frequent changes in body position and the use of soft mattress overlays and air mattresses are employed. Prophylaxis against diffuse gastrointestinal mucosal injury is indicated for patients undergoing MV. Histamine-receptor (H2-receptor) antagonists, antacids, and cytoprotective agents such as sucralfate have all been used for this purpose and appear to be effective. Nutritional support by enteral feeding through either a nasogastric or an orogastric tube should be initiated and maintained whenever possible. Delayed gastric emptying is common in critically ill patients taking sedative medications but often responds to promotility agents such as metoclopramide. Parenteral nutrition is an alternative to enteral nutrition in patients with severe gastrointestinal pathology who need prolonged MV.
COMPLICATIONS OF MECHANICAL VENTILATION
Endotracheal intubation and mechanical ventilation have direct and indirect effects on the lung and upper airways, the cardiovascular system, and the gastrointestinal system. Pulmonary complications include barotrauma, nosocomial pneumonia, oxygen toxicity, tracheal stenosis, and deconditioning of respiratory muscles. Barotrauma and volutrauma overdistend and disrupt lung tissue; may be clinically manifest by interstitial emphysema, pneumomediastinum, subcutaneous emphysema, or pneumothorax; and can result in the liberation of cytokines from overdistended tissues, further promoting tissue injury. Clinically significant pneumothorax requires tube thoracostomy. Intubated patients are at high risk for ventilator-associated pneumonia as a result of aspiration from the upper airways through small leaks around the endotracheal tube cuff; the most common organisms responsible for this condition are Pseudomonas aeruginosa, enteric gram-negative rods, and Staphylococcus aureus. Given the high associated mortality rates, early initiation of empirical antibiotics directed against likely pathogens is recommended. Hypotension resulting from elevated intrathoracic pressures with decreased venous return is almost always responsive to intravascular volume repletion. In patients who are judged to have respiratory failure on the basis of alveolar edema but in whom the cardiac or pulmonary origin of the edema is unclear, hemodynamic monitoring with a pulmonary arterial catheter may be of value in helping to clarify the cause of the edema. Gastrointestinal effects of positive-pressure ventilation include stress ulceration and mild to moderate cholestasis.
WEANING FROM MECHANICAL VENTILATION
The Decision to Wean It is important to consider discontinuation of mechanical ventilation once the underlying respiratory disease begins to reverse. Although the predictive capacities of multiple clinical and physiologic variables have been explored, the consensus from a ventilatory weaning task force cites the following conditions as indicating amenability to weaning: (1) Lung injury is stable or resolving. (2) Gas exchange is adequate, with low PEEP/FIO2 (<8 cmH2O) and FIO2(<0.5). (3) Hemodynamic variables are stable, and the patient is no longer receiving vasopressors). (4) The patient is capable of initiating spontaneous breaths. A “wean screen” based on these variables should be done at least daily. If the patient is deemed capable of beginning to wean, the recommendation is to perform a spontaneous breathing trial (SBT), whose value is supported by several randomized trials (Fig. 323-2). The SBT involves an integrated patient assessment during spontaneous breathing with little or no ventilatory support. The SBT is usually implemented with a T-piece using 1–5 cmH2O CPAP with 5–7 cmH2O or PSV from the ventilator to offset resistance from the endotracheal tube. Once it is determined that the patient can breathe spontaneously, a decision must be made about the removal of the artificial airway, which should be undertaken only when it is concluded that the patient has the ability to protect the airway, is able to cough and clear secretions, and is alert enough to follow commands. In addition, other factors must be taken into account, such as the possible difficulty of replacing the tube if that maneuver is required. If upper airway difficulty is suspected, an evaluation using a “cuff-leak” test (assessing the presence of air movement around a deflated endotracheal tube cuff) is supported by some internists. Despite all precautions, ~10–15% of extubated patients require reintubation. Several studies suggest that NIV can be used to obviate reintubation, particularly in patients with ventilatory failure secondary to COPD exacerbation; in this setting, earlier extubation with the use of prophylactic NIV has yielded good results. The use of NIV to facilitate weaning in respiratory failure of other etiologies is not currently indicated.
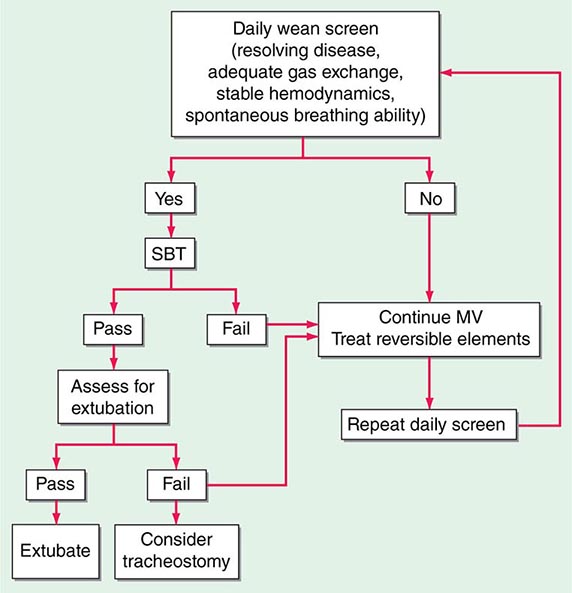
FIGURE 323-2 Flow chart to guide the daily approach to management of patients being considered for weaning off mechanical ventilation (MV). If attempts at extubation fail, a tracheostomy should be considered. SBT, spontaneous breathing trial.
Prolonged Mechanical Ventilation and Tracheostomy From 5% to 13% of patients undergoing MV will go on to require prolonged MV (>21 days). In these instances, critical care personnel must decide whether and when to perform a tracheostomy. This decision is individualized and is based on the risk and benefits of tracheostomy and prolonged intubation as well as the patient’s preferences and expected outcomes. A tracheostomy is thought to be more comfortable, to require less sedation, and to provide a more secure airway and may also reduce weaning time. However, tracheostomy carries the risk of complications, which occur in 5–40% of these procedures and include bleeding, cardiopulmonary arrest, hypoxia, structural damage, pneumothorax, pneumomediastinum, and wound infection. In patients with long-term tracheostomy, complex complications include tracheal stenosis, granulation, and erosion of the innominate artery. In general, if a patient needs MV for more than 10–14 days, a tracheostomy, planned under optimal conditions, is indicated. Whether it is completed at the bedside or as an operative procedure depends on local resources and experience. Some 5–10% of patients are deemed unable to wean in the ICU. These patients may benefit from transfer to special units where a multidisciplinary approach, including nutrition optimization, physical therapy with rehabilitation, and slower weaning methods (including SIMV with PSV), results in successful weaning rates of up to 30%. Unfortunately, close to 2% of ventilated patients may ultimately become dependent on ventilatory support to maintain life. Most of these patients remain in chronic care institutions, although some with strong social, economic, and family support may live a relatively fulfilling life with at-home ventilation.
SECTION 2 |
SHOCK AND CARDIAC ARREST |
324 |
Approach to the Patient with Shock |
Shock is the clinical syndrome that results from inadequate tissue perfusion. Irrespective of cause, the hypoperfusion-induced imbalance between the delivery of and requirements for oxygen and substrate leads to cellular dysfunction. The cellular injury created by the inadequate delivery of oxygen and substrates also induces the production and release of damage-associated molecular patterns (DAMPs or “danger signals”) and inflammatory mediators that further compromise perfusion through functional and structural changes within the microvasculature. This leads to a vicious cycle in which impaired perfusion is responsible for cellular injury that causes maldistribution of blood flow, further compromising cellular perfusion; the latter ultimately causes multiple organ failure (MOF) and, if the process is not interrupted, leads to death. The clinical manifestations of shock are also the result, in part, of autonomic neuroendocrine responses to hypoperfusion as well as the breakdown in organ function induced by severe cellular dysfunction (Fig. 324-1).
FIGURE 324-1 Shock-induced vicious cycle.
When very severe and/or persistent, inadequate oxygen delivery leads to irreversible cell injury, only rapid restoration of oxygen delivery can reverse the progression of the shock state. The fundamental approach to management, therefore, is to recognize overt and impending shock in a timely fashion and to intervene emergently to restore perfusion. Doing so often requires the expansion or reexpansion of intravascular blood volume. Control of any inciting pathologic process (e.g., continued hemorrhage, impairment of cardiac function, or infection) must occur simultaneously.
Clinical shock is usually accompanied by hypotension (i.e., a mean arterial pressure [MAP] <60 mmHg in previously normotensive persons). Multiple classification schemes have been developed in an attempt to synthesize the seemingly dissimilar processes leading to shock. Strict adherence to a classification scheme may be difficult from a clinical standpoint because of the frequent combination of two or more causes of shock in any individual patient, but the classification shown in Table 324-1 provides a useful reference point from which to discuss and further delineate the underlying processes.
|
CLASSIFICATION OF SHOCK |
PATHOGENESIS AND ORGAN RESPONSE
MICROCIRCULATION
Normally when cardiac output falls, systemic vascular resistance rises to maintain a level of systemic pressure that is adequate for perfusion of the heart and brain at the expense of other tissues such as muscle, skin, and especially the gastrointestinal (GI) tract. Systemic vascular resistance is determined primarily by the luminal diameter of arterioles. The metabolic rates of the heart and brain are high, and their stores of energy substrate are low. These organs are critically dependent on a continuous supply of oxygen and nutrients, and neither tolerates severe ischemia for more than brief periods (minutes). Autoregulation (i.e., the maintenance of blood flow over a wide range of perfusion pressures) is critical in sustaining cerebral and coronary perfusion despite significant hypotension. However, when MAP drops to ≤60 mmHg, blood flow to these organs falls, and their function deteriorates.
Arteriolar vascular smooth muscle has both α- and β-adrenergic receptors. The α1 receptors mediate vasoconstriction, while the β2 receptors mediate vasodilation. Efferent sympathetic fibers release norepinephrine, which acts primarily on α1 receptors as one of the most fundamental compensatory responses to reduced perfusion pressure. Other constrictor substances that are increased in most forms of shock include angiotensin II, vasopressin, endothelin 1, and thromboxane A2. Both norepinephrine and epinephrine are released by the adrenal medulla, and the concentrations of these catecholamines in the bloodstream rise. Circulating vasodilators in shock include prostacyclin (prostaglandin [PG] I2), nitric oxide (NO), and, importantly, products of local metabolism such as adenosine that match flow to the tissue’s metabolic needs. The balance between these various vasoconstrictors and vasodilators influences the microcirculation and determines local perfusion.
Transport to cells depends on microcirculatory flow; capillary permeability; the diffusion of oxygen, carbon dioxide, nutrients, and products of metabolism through the interstitium; and the exchange of these products across cell membranes. Impairment of the microcirculation that is central to the pathophysiologic responses in the late stages of all forms of shock results in the derangement of cellular metabolism that is ultimately responsible for organ failure.
The endogenous response to mild or moderate hypovolemia is an attempt at restitution of intravascular volume through alterations in hydrostatic pressure and osmolarity. Constriction of arterioles leads to reductions in both the capillary hydrostatic pressure and the number of capillary beds perfused, thereby limiting the capillary surface area across which filtration occurs. When filtration is reduced while intravascular oncotic pressure remains constant or rises, there is net reabsorption of fluid into the vascular bed, in accord with Starling’s law of capillary interstitial liquid exchange. Metabolic changes (including hyperglycemia and elevations in the products of glycolysis, lipolysis, and proteolysis) raise extracellular osmolarity, leading to an osmotic gradient that increases interstitial and intravascular volume at the expense of intracellular volume.
CELLULAR RESPONSES
Interstitial transport of nutrients is impaired in shock, leading to a decline in intracellular high-energy phosphate stores. Mitochondrial dysfunction and uncoupling of oxidative phosphorylation are the most likely causes for decreased amounts of adenosine triphosphate (ATP). As a consequence, there is an accumulation of hydrogen ions, lactate, reactive oxygen species, and other products of anaerobic metabolism. As shock progresses, these vasodilator metabolites override vasomotor tone, causing further hypotension and hypoperfusion. Dysfunction of cell membranes is thought to represent a common end-stage pathophysiologic pathway in the various forms of shock. Normal cellular transmembrane potential falls, and there is an associated increase in intracellular sodium and water, leading to cell swelling that interferes further with microvascular perfusion. In a preterminal event, homeostasis of calcium via membrane channels is lost with flooding of calcium into the cytosol and concomitant extracellular hypocalcemia. There is also evidence for a widespread but selective apoptotic (programmed cell death) loss of cells, contributing to organ and immune failure.
NEUROENDOCRINE RESPONSE
Hypovolemia, hypotension, and hypoxia are sensed by baroreceptors and chemoreceptors that contribute to an autonomic response that attempts to restore blood volume, maintain central perfusion, and mobilize metabolic substrates. Hypotension disinhibits the vasomotor center, resulting in increased adrenergic output and reduced vagal activity. Release of norepinephrine from adrenergic neurons induces significant peripheral and splanchnic vasoconstriction, a major contributor to the maintenance of central organ perfusion, while reduced vagal activity increases the heart rate and cardiac output. Loss of vagal activity is also recognized to upregulate the innate immune inflammatory response. The effects of circulating epinephrine released by the adrenal medulla in shock are largely metabolic, causing increased glycogenolysis and gluconeogenesis and reduced pancreatic insulin release. However, epinephrine also inhibits production and release of inflammatory mediators through stimulation of β-adrenergic receptors on innate immune cells.
Severe pain or other stresses cause the hypothalamic release of adrenocorticotropic hormone (ACTH). This stimulates cortisol secretion that contributes to decreased peripheral uptake of glucose and amino acids, enhances lipolysis, and increases gluconeogenesis. Increased pancreatic secretion of glucagon during stress accelerates hepatic gluconeogenesis and further elevates blood glucose concentration. These hormonal actions act synergistically to increase blood glucose for both selective tissue metabolism and the maintenance of blood volume. Many critically ill patients have recently been shown to exhibit low plasma cortisol levels and an impaired response to ACTH stimulation, which is linked to a decrease in survival. The importance of the cortisol response to stress is illustrated by the profound circulatory collapse that occurs in patients with adrenocortical insufficiency (Chap. 406).
Renin release is increased in response to adrenergic discharge and reduced perfusion of the juxtaglomerular apparatus in the kidney. Renin induces the formation of angiotensin I that is then converted to angiotensin II by the angiotensin converting enzyme; angiotensin II is an extremely potent vasoconstrictor and stimulator of aldosterone release by the adrenal cortex and of vasopressin by the posterior pituitary. Aldosterone contributes to the maintenance of intravascular volume by enhancing renal tubular reabsorption of sodium, resulting in the excretion of a low-volume, concentrated, sodium-free urine. Vasopressin has a direct action on vascular smooth muscle, contributing to vasoconstriction, and acts on the distal renal tubules to enhance water reabsorption.
CARDIOVASCULAR RESPONSE
Three variables—ventricular filling (preload), the resistance to ventricular ejection (afterload), and myocardial contractility—are paramount in controlling stroke volume (Chap. 265e). Cardiac output, the major determinant of tissue perfusion, is the product of stroke volume and heart rate. Hypovolemia leads to decreased ventricular preload that, in turn, reduces the stroke volume. An increase in heart rate is a useful but limited compensatory mechanism to maintain cardiac output. A shock-induced reduction in myocardial compliance is frequent, reducing ventricular end-diastolic volume and, hence, stroke volume at any given ventricular filling pressure. Restoration of intravascular volume may return stroke volume to normal but only at elevated filling pressures. Increased filling pressures stimulate release of brain natriuretic peptide (BNP) to secrete sodium and volume to relieve the pressure on the heart. Levels of BNP correlate with outcome following severe stress. In addition, sepsis, ischemia, myocardial infarction (MI), severe tissue trauma, hypothermia, general anesthesia, prolonged hypotension, and acidemia may all also impair myocardial contractility and reduce the stroke volume at any given ventricular end-diastolic volume. The resistance to ventricular ejection is significantly influenced by the systemic vascular resistance, which is elevated in most forms of shock. However, resistance is decreased in the early hyperdynamic stage of septic shock or neurogenic shock (Chap. 325), thereby initially allowing the cardiac output to be maintained or elevated.
The venous system contains nearly two-thirds of the total circulating blood volume, most in the small veins, and serves as a dynamic reservoir for autoinfusion of blood. Active venoconstriction as a consequence of α-adrenergic activity is an important compensatory mechanism for the maintenance of venous return and, therefore, of ventricular filling during shock. By contrast, venous dilation, as occurs in neurogenic shock, reduces ventricular filling and hence stroke volume and potentially cardiac output.
PULMONARY RESPONSE
The response of the pulmonary vascular bed to shock parallels that of the systemic vascular bed, and the relative increase in pulmonary vascular resistance, particularly in septic shock, may exceed that of the systemic vascular resistance, leading to right heart failure. Shock-induced tachypnea reduces tidal volume and increases both dead space and minute ventilation. Relative hypoxia and the subsequent tachypnea induce a respiratory alkalosis. Recumbency and involuntary restriction of ventilation secondary to pain reduce functional residual capacity and may lead to atelectasis. Shock and, in particular, resuscitation-induced reactive oxygen species (oxidant radical) generation are recognized as major causes of acute lung injury and subsequent acute respiratory distress syndrome (ARDS; Chap. 322). These disorders are characterized by noncardiogenic pulmonary edema secondary to diffuse pulmonary capillary endothelial and alveolar epithelial injury, hypoxemia, and bilateral diffuse pulmonary infiltrates. Hypoxemia results from perfusion of underventilated and nonventilated alveoli. Loss of surfactant and lung volume in combination with increased interstitial and alveolar edema reduces lung compliance. The work of breathing and the oxygen requirements of respiratory muscles increase.
RENAL RESPONSE
Acute kidney injury (Chap. 334), a serious complication of shock and hypoperfusion, occurs less frequently than heretofore because of early aggressive volume repletion. Acute tubular necrosis is now more frequently seen as a result of the interactions of shock, sepsis, the administration of nephrotoxic agents (such as aminoglycosides and angiographic contrast media), and rhabdomyolysis; the latter may be particularly severe in skeletal muscle trauma. The physiologic response of the kidney to hypoperfusion is to conserve salt and water. In addition to decreased renal blood flow, increased afferent arteriolar resistance accounts for diminished glomerular filtration rate (GFR) that, together with increased aldosterone and vasopressin, is responsible for reduced urine formation. Toxic injury causes necrosis of tubular epithelium and tubular obstruction by cellular debris with back leak of filtrate. The depletion of renal ATP stores that occurs with prolonged renal hypoperfusion contributes to subsequent impairment of renal function.
METABOLIC DERANGEMENTS
During shock, there is disruption of the normal cycles of carbohydrate, lipid, and protein metabolism. Through the citric acid cycle, alanine in conjunction with lactate, which is converted from pyruvate in the periphery in the presence of oxygen deprivation, enhances the hepatic production of glucose. With reduced availability of oxygen, the breakdown of glucose to pyruvate, and ultimately lactate, represents an inefficient cycling of substrate with minimal net energy production. An elevated plasma lactate/pyruvate ratio is preferable to lactate alone as a measure of anaerobic metabolism and reflects inadequate tissue perfusion. Decreased clearance of exogenous triglycerides coupled with increased hepatic lipogenesis causes a significant rise in serum triglyceride concentrations. There is increased protein catabolism as energy substrate, a negative nitrogen balance, and, if the process is prolonged, severe muscle wasting.
INFLAMMATORY RESPONSES
Activation of an extensive network of proinflammatory mediator pathways by the innate immune system plays a significant role in the progression of shock and contributes importantly to the development of multiple organ injury, multiple organ dysfunction (MOD), and MOF (Fig. 324-2). In those surviving the acute insult, there is a prolonged endogenous counterregulatory response to “turn off” or balance the excessive proinflammatory response. If balance is restored, the patient does well. If the response is excessive, adaptive immunity is suppressed and the patient is highly susceptible to secondary nosocomial infections, which may then drive the inflammatory response and lead to delayed MOF.
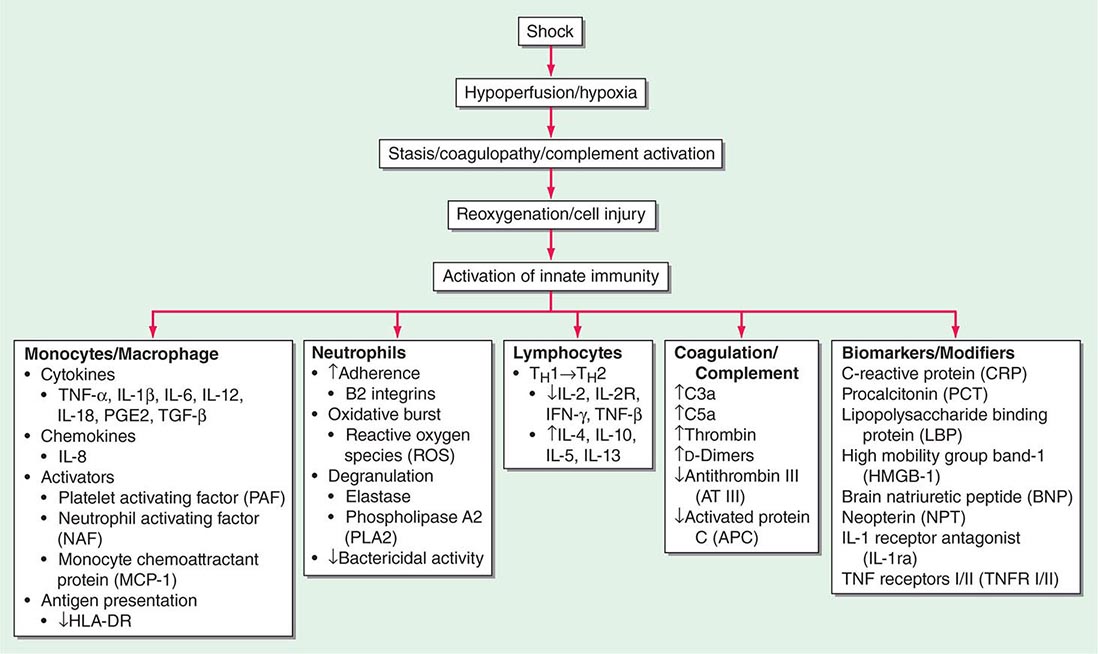
FIGURE 324-2 A schematic of the host immunoinflammatory response to shock. IFN, interferon; IL, interleukin; PG, prostaglandin; TGF, tumor growth factor; TNF, tumor necrosis factor.
Multiple humoral mediators are activated during shock and tissue injury. The complement cascade, activated through both the classic and alternate pathways, generates the anaphylatoxins C3a and C5a (Chap. 372e). Direct complement fixation to injured tissues can progress to the C5-C9 attack complex, causing further cell damage. Activation of the coagulation cascade (Chap. 141) causes microvascular thrombosis, with subsequent fibrinolysis leading to repeated episodes of ischemia and reperfusion. Components of the coagulation system (e.g., thrombin) are potent proinflammatory mediators that cause expression of adhesion molecules on endothelial cells and activation of neutrophils, leading to microvascular injury. Coagulation also activates the kallikrein-kininogen cascade, contributing to hypotension.
Eicosanoids are vasoactive and immunomodulatory products of arachidonic acid metabolism that include cyclooxygenase-derived prostaglandins (PGs) and thromboxane A2, as well as lipoxygenase-derived leukotrienes and lipoxins. Thromboxane A2 is a potent vasoconstrictor that contributes to the pulmonary hypertension and acute tubular necrosis of shock. PGI2 and PGE2 are potent vasodilators that enhance capillary permeability and edema formation. The cysteinyl leukotrienes LTC4 and LTD4 are pivotal mediators of the vascular sequelae of anaphylaxis, as well as of shock states resulting from sepsis or tissue injury. LTB4 is a potent neutrophil chemoattractant and secretagogue that stimulates the formation of reactive oxygen species. Platelet-activating factor, an ether-linked, arachidonyl-containing phospholipid mediator, causes pulmonary vasoconstriction, bronchoconstriction, systemic vasodilation, increased capillary permeability, and the priming of macrophages and neutrophils to produce enhanced levels of inflammatory mediators.
Tumor necrosis factor α (TNF-α), produced by activated macrophages, reproduces many components of the shock state, including hypotension, lactic acidosis, and respiratory failure. Interleukin 1β (IL-1β), originally defined as “endogenous pyrogen” and produced by tissue-fixed macrophages, is critical to the inflammatory response. Both are significantly elevated immediately following trauma and shock. IL-6, also produced predominantly by the macrophage, has a slightly delayed peak response but is the best single predictor of prolonged recovery and development of MOF following shock. Chemokines such as IL-8 are potent neutrophil chemoattractants and activators that upregulate adhesion molecules on the neutrophil to enhance aggregation, adherence, and damage to the vascular endothelium. While the endothelium normally produces low levels of NO, the inflammatory response stimulates the inducible isoform of NO synthase (iNOS), which is overexpressed and produces toxic nitroxyl-and oxygen-derived free radicals that contribute to the hyperdynamic cardiovascular response and tissue injury in sepsis.
Multiple inflammatory cells, including neutrophils, macrophages, and platelets, are major contributors to inflammation-induced injury. Margination of activated neutrophils in the microcirculation is a common pathologic finding in shock, causing secondary injury due to the release of toxic oxygen radicals, lipases (primarily PLA2), and proteases. Release of high levels of reactive oxygen intermediates/species (ROI/ROS) rapidly consumes endogenous essential antioxidants and generates diffuse oxygen radical damage. Newer efforts to control ischemia/reperfusion injury include treatment with carbon monoxide, hydrogen sulfide, or other agents to reduce oxidant stress. Tissue-fixed macrophages produce virtually all major mediators of the inflammatory response and orchestrate the progression and duration of the inflammatory response. A major source of activation of the monocyte/macrophage is through the highly conserved membrane toll-like receptors (TLRs) that recognize DAMPs, such as HMGB-1, and pathogen-associated molecular patterns (PAMPs), such as endotoxins released following tissue injury, and by pathogenic microbial organisms, respectively. TLRs also appear important in the chronic inflammation seen in Crohn’s disease, ulcerative colitis, and transplant rejection. The variability in individual responses is a genetic predisposition that, in part, is due to variants in genetic sequences affecting the function and production of various inflammatory mediators.
|
TREATMENT |
SHOCK |
MONITORING
Patients in shock require care in an intensive care unit (ICU). Careful and continuous assessment of the physiologic status is necessary. Arterial pressure through an indwelling line, pulse, and respiratory rate should be monitored continuously; a Foley catheter should be inserted to follow urine flow; and mental status should be assessed frequently. Sedated patients should be allowed to awaken (“drug holiday”) daily to assess their neurologic status and to shorten duration of ventilator support.
There is ongoing debate as to the indications for using the flow-directed pulmonary artery catheter (PAC; Swan-Ganz catheter) in the ICU. A recent Cochrane analysis showed that the use of a PAC did not alter mortality, length of stay, or cost for adult ICU patients. Most patients in the ICU can be safely managed without the use of a PAC. However, in shock with significant ongoing blood loss, fluid shifts, and underlying cardiac dysfunction, a PAC may be useful. The PAC is placed percutaneously via the subclavian or jugular vein through the central venous circulation and right heart into the pulmonary artery. There are ports both proximal in the right atrium and distal in the pulmonary artery to provide access for infusions and for cardiac output measurements. Right atrial and pulmonary artery pressures (PAPs) are measured, and the pulmonary capillary wedge pressure (PCWP) serves as an approximation of the left atrial pressure. Normal hemodynamic parameters and their derivation are summarized in Table 272-2 and Table 324-2.
|
NORMAL HEMODYNAMIC PARAMETERS |
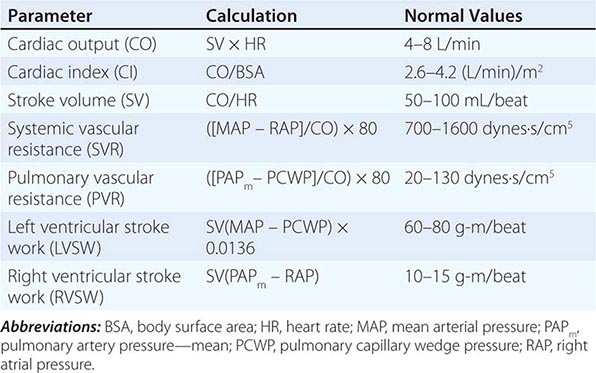
Cardiac output is determined by the thermodilution technique, and high-resolution thermistors can also be used to determine right ventricular end-diastolic volume to monitor further the response of the right heart to fluid resuscitation. A PAC with an oximeter port offers the additional advantage of online monitoring of the mixed venous oxygen saturation, an important index of overall tissue perfusion. Systemic and pulmonary vascular resistances are calculated as the ratio of the pressure drop across these vascular beds to the cardiac output (Chap. 272). Determinations of oxygen content in arterial and venous blood, together with cardiac output and hemoglobin concentration, allow calculation of oxygen delivery, oxygen consumption, and oxygen-extraction ratio (Table 324-3). The hemodynamic patterns associated with the various forms of shock are shown in Table 324-4.
|
OXYGEN TRANSPORT CALCULATIONS |
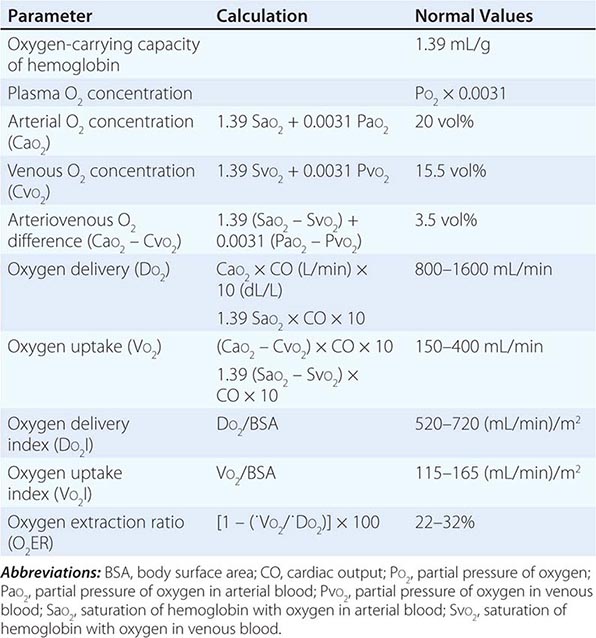
|
PHYSIOLOGIC CHARACTERISTICS OF THE VARIOUS FORMS OF SHOCK |
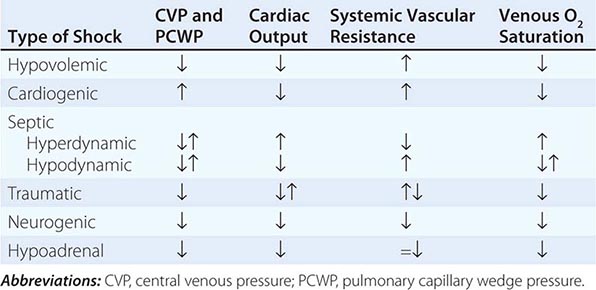
In resuscitation from shock, it is critical to restore tissue perfusion and optimize oxygen delivery, hemodynamics, and cardiac function rapidly. A reasonable goal of therapy is to achieve a normal mixed venous oxygen-saturation and arteriovenous oxygen-extraction ratio. To enhance oxygen delivery, red cell mass, arterial oxygen saturation, and cardiac output may be augmented singly or simultaneously. An increase in oxygen delivery not accompanied by an increase in oxygen consumption implies that oxygen availability is adequate and that oxygen consumption is not flow dependent. Conversely, an elevation of oxygen consumption with increased delivery implies that the oxygen supply was inadequate. However, cautious interpretation is required due to the link among increased oxygen delivery, cardiac work, and oxygen consumption. A reduction in systemic vascular resistance accompanying an increase in cardiac output indicates that compensatory vasoconstriction is reversing due to improved tissue perfusion. The determination of stepwise expansion of blood volume on cardiac performance allows identification of the optimum preload (Starling’s law). An algorithm for the resuscitation of the patient in shock is shown in Fig. 324-3.
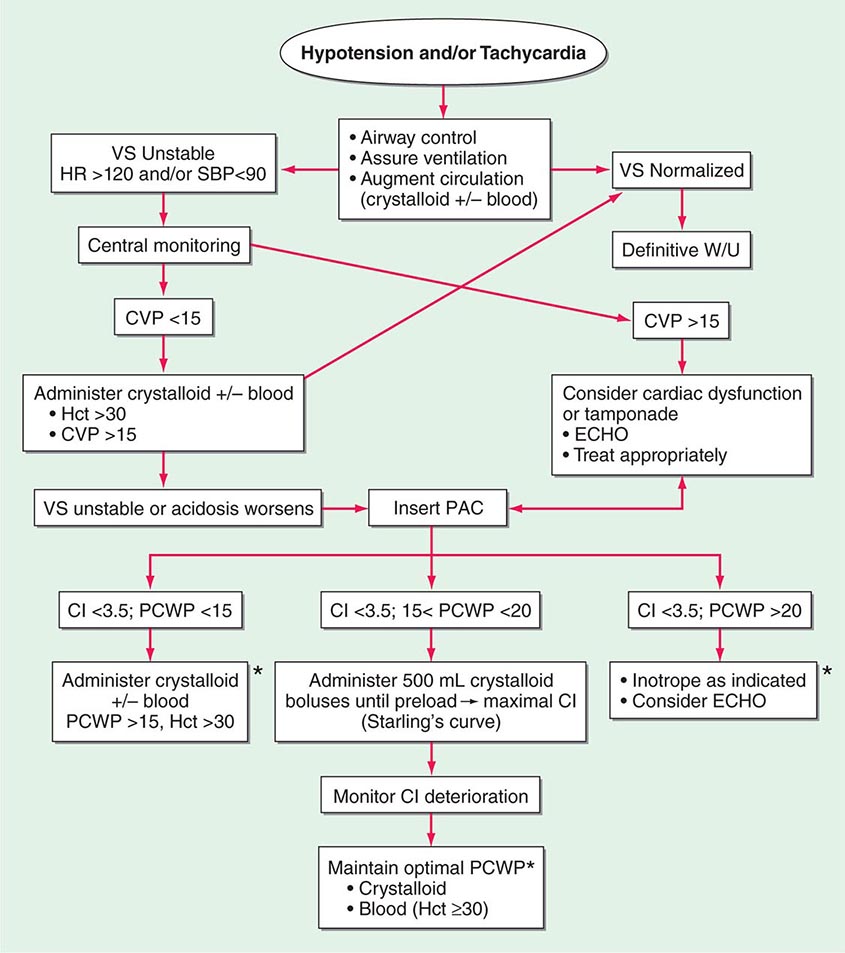
FIGURE 324-3 An algorithm for the resuscitation of the patient in shock. *Monitor SvO2, SVRI, and RVEDVI as additional markers of correction for perfusion and hypovolemia. Consider age-adjusted CI. CI, cardiac index in (L/min) per m2; CVP, central venous pressure; ECHO, echocardiogram; Hct, hematocrit; HR, heart rate; PAC, pulmonary artery catheter; PCWP, pulmonary capillary wedge pressure in mmHg; RVEDVI, right ventricular end-diastolic volume index; SBP, systolic blood pressure; SvO2, saturation of hemoglobin with O2 in venous blood; SVRI, systemic vascular resistance index; VS, vital signs; W/U, workup.
SPECIFIC FORMS OF SHOCK
HYPOVOLEMIC SHOCK
This most common form of shock results either from the loss of red blood cell mass and plasma from hemorrhage or from the loss of plasma volume alone due to extravascular fluid sequestration or GI, urinary, and insensible losses. The signs and symptoms of nonhemorrhagic hypovolemic shock are the same as those of hemorrhagic shock, although they may have a more insidious onset. The normal physiologic response to hypovolemia is to maintain perfusion of the brain and heart while attempting to restore an effective circulating blood volume. There is an increase in sympathetic activity, hyperventilation, collapse of venous capacitance vessels, release of stress hormones, and an attempt to replace the loss of intravascular volume through the recruitment of interstitial and intracellular fluid and by reduction of urine output.
Mild hypovolemia (≤20% of the blood volume) generates mild tachycardia but relatively few external signs, especially in a supine young patient (Table 324-5). With moderate hypovolemia (~20–40% of the blood volume), the patient becomes increasingly anxious and tachycardic; although normal blood pressure may be maintained in the supine position, there may be significant postural hypotension and tachycardia. If hypovolemia is severe (≥40% of the blood volume), the classic signs of shock appear; the blood pressure declines and becomes unstable even in the supine position, and the patient develops marked tachycardia, oliguria, and agitation or confusion. Perfusion of the central nervous system is well maintained until shock becomes severe. Hence, mental obtundation is an ominous clinical sign. The transition from mild to severe hypovolemic shock can be insidious or extremely rapid. If severe shock is not reversed rapidly, especially in elderly patients and those with comorbid illnesses, death is imminent. A very narrow time frame separates the derangements found in severe shock that can be reversed with aggressive resuscitation from those of progressive decompensation and irreversible cell injury.
|
HYPOVOLEMIC SHOCK |
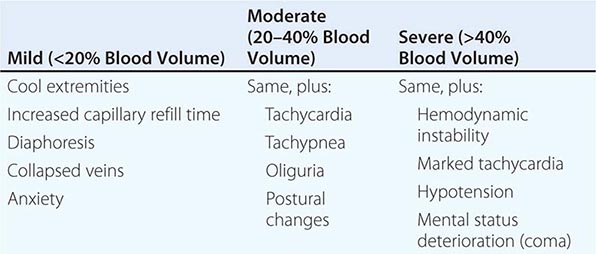
Diagnosis Hypovolemic shock is readily diagnosed when there are signs of hemodynamic instability and the source of volume loss is obvious. The diagnosis is more difficult when the source of blood loss is occult, as into the GI tract, or when plasma volume alone is depleted. Even after acute hemorrhage, hemoglobin and hematocrit values do not change until compensatory fluid shifts have occurred or exogenous fluid is administered. Thus, an initial normal hematocrit does not disprove the presence of significant blood loss. Plasma losses cause hemoconcentration, and free water loss leads to hypernatremia. These findings should suggest the presence of hypovolemia.
It is essential to distinguish between hypovolemic and cardiogenic shock (Chap. 326) because, although both may respond to volume initially, definitive therapy differs significantly. Both forms are associated with a reduced cardiac output and a compensatory sympathetic mediated response characterized by tachycardia and elevated systemic vascular resistance. However, the findings in cardiogenic shock of jugular venous distention, rales, and an S3 gallop distinguish it from hypovolemic shock and signify that ongoing volume expansion is undesirable and may cause further organ dysfunction.
TRAUMATIC SHOCK
Shock following trauma is, in large measure, due to hemorrhage. However, even when hemorrhage has been controlled, patients can continue to suffer loss of plasma volume into the interstitium of injured tissues. These fluid losses are compounded by injury-induced inflammatory responses, which contribute to the secondary microcirculatory injury. Proinflammatory mediators are induced by DAMPs released from injured tissue and are recognized by the highly conserved membrane receptors of the TLR family (see “Inflammatory Responses” above). These receptors on cells of the innate immune system, particularly the circulating monocyte, tissue-fixed macrophage, and dendritic cell, are potent activators of an excessive proinflammatory phenotype in response to cellular injury. This causes secondary tissue injury and maldistribution of blood flow, intensifying tissue ischemia and leading to multiple organ system failure. In addition, direct structural injury to the heart, chest, or head can also contribute to shock. For example, pericardial tamponade or tension pneumothorax impairs ventricular filling, whereas myocardial contusion depresses myocardial contractility.
CARDIOGENIC SHOCK
See Chap. 326.
COMPRESSIVE CARDIOGENIC SHOCK
With extrinsic compression, the heart and surrounding structures are less compliant, and therefore, normal filling pressures generate inadequate diastolic filling and stroke volume. Blood or fluid within the poorly distensible pericardial sac may cause tamponade (Chap. 288). Any cause of increased intrathoracic pressure, such as tension pneumothorax, herniation of abdominal viscera through a diaphragmatic hernia, or excessive positive-pressure ventilation to support pulmonary function, can also initiate compressive cardiogenic shock while simultaneously impeding venous return and preload. Although initially responsive to increased filling pressures produced by volume expansion, as compression increases, cardiogenic shock recurs. The window of opportunity gained by volume loading may be very brief until irreversible shock recurs. Diagnosis and intervention must occur urgently.
The diagnosis of compressive cardiogenic shock is most frequently based on clinical findings, the chest radiograph, and an echocardiogram. The diagnosis of compressive cardiac shock may be more difficult to establish in the setting of trauma when hypovolemia and cardiac compression are present simultaneously. The classic findings of pericardial tamponade include the triad of hypotension, neck vein distention, and muffled heart sounds (Chap. 288). Pulsus paradoxus (i.e., an inspiratory reduction in systolic pressure >10 mmHg) may also be noted. The diagnosis is confirmed by echocardiography, and treatment consists of immediate pericardiocentesis or the creation of an open subxiphoid pericardial window. A tension pneumothorax produces ipsilateral decreased breath sounds, tracheal deviation away from the affected thorax, and jugular venous distention. Radiographic findings include increased intrathoracic volume, depression of the diaphragm of the affected hemithorax, and shifting of the mediastinum to the contralateral side. Chest decompression must be carried out immediately and, ideally, should occur based on clinical findings rather than awaiting a chest radiograph. Release of air and restoration of normal cardiovascular dynamics are both diagnostic and therapeutic.
SEPTIC SHOCK
See Chap. 325.
NEUROGENIC SHOCK
Interruption of sympathetic vasomotor input after a high cervical spinal cord injury, inadvertent cephalad migration of spinal anesthesia, or devastating head injury may result in neurogenic shock. In addition to arteriolar dilation, venodilation causes pooling in the venous system, which decreases venous return and cardiac output. The extremities are often warm, in contrast to the usual sympathetic vasoconstriction-induced coolness in hypovolemic or cardiogenic shock. Treatment involves a simultaneous approach to the relative hypovolemia and to the loss of vasomotor tone. Excessive volumes of fluid may be required to restore normal hemodynamics if given alone. Once hemorrhage has been ruled out, norepinephrine or a pure α-adrenergic agent (phenylephrine) may be necessary to augment vascular resistance and maintain an adequate MAP.
HYPOADRENAL SHOCK
(See also Chap. 406) The normal host response to the stress of illness, operation, or trauma requires that the adrenal glands hypersecrete cortisol in excess of that normally required. Hypoadrenal shock occurs in settings in which unrecognized adrenal insufficiency complicates the host response to the stress induced by acute illness or major surgery. Adrenocortical insufficiency may occur as a consequence of the chronic administration of high doses of exogenous glucocorticoids. In addition, recent studies have shown that critical illness, including trauma and sepsis, may also induce a relative hypoadrenal state. Other, less common causes include adrenal insufficiency secondary to idiopathic atrophy, use of etomidate for intubation, tuberculosis, metastatic disease, bilateral hemorrhage, and amyloidosis. The shock produced by adrenal insufficiency is characterized by loss of homeostasis with reductions in systemic vascular resistance, hypovolemia, and reduced cardiac output. The diagnosis of adrenal insufficiency may be established by means of an ACTH stimulation test.
ADJUNCTIVE THERAPIES
The sympathomimetic amines dobutamine, dopamine, and norepinephrine are widely used in the treatment of all forms of shock. Dobutamine is inotropic with simultaneous afterload reduction, thus minimizing cardiac-oxygen consumption increases as cardiac output increases. Dopamine is an inotropic and chronotropic agent that also supports vascular resistance in those whose blood pressure will not tolerate peripheral vascular dilation. Norepinephrine primarily supports blood pressure through vasoconstriction and increases myocardial oxygen consumption while placing marginally perfused tissues, such as extremities and splanchnic organs, at risk for ischemia or necrosis, but it is also inotropic without significant chronotropy. Arginine-vasopressin (antidiuretic hormone) is being used increasingly to increase afterload and may better protect vital organ blood flow and prevent pathologic vasodilation.
REWARMING
Hypothermia is a frequent adverse consequence of massive volume resuscitation (Chap. 478e). The infusion of large volumes of refrigerated blood products and room temperature crystalloid solutions can rapidly drop core temperatures if fluid is not run through warming devices. Hypothermia may depress cardiac contractility and thereby further impair cardiac output and oxygen delivery/utilization. Hypothermia, particularly temperatures <35°C (<95°F), directly impairs the coagulation pathway, sometimes causing a significant coagulopathy. Rapid rewarming to >35°C (>95°F) significantly decreases the requirement for blood products and produces an improvement in cardiac function. The most effective method for rewarming is endovascular countercurrent warmers through femoral vein cannulation. This process does not require a pump and can rewarm a patient from 30° to 35°C (86° to 95°F) in 30–60 min.
325 |
Severe Sepsis and Septic Shock |
DEFINITIONS
(Table 325-1) Animals mount both local and systemic responses to microbes that traverse their epithelial barriers and enter underlying tissues. Fever or hypothermia, leukocytosis or leukopenia, tachypnea, and tachycardia are cardinal signs of the systemic response. To date, attempts to devise precise definitions for the harmful systemic reaction to infection (“sepsis”) have not resulted in a clinically useful level of specificity, in part because the systemic responses to infection, trauma, and other major stresses can be so similar. In general, when an infectious etiology is proven or strongly suspected and the response results in hypofunction of uninfected organs, the term sepsis (or severe sepsis) should be used. Septic shock refers to sepsis accompanied by hypotension that cannot be corrected by the infusion of fluids.
|
DEFINITIONS USED TO DESCRIBE THE CONDITION OF SEPTIC PATIENTS |

ETIOLOGY
![]() The systemic response to any class of microorganism can be harmful. Microbial invasion of the bloodstream is not essential because local inflammation can also elicit distant organ dysfunction and hypotension. In fact, blood cultures yield bacteria or fungi in only ~20–40% of cases of severe sepsis and 40–70% of cases of septic shock. In a prevalence study of 14,414 patients in intensive care units (ICUs) from 75 countries in 2007, 51% of patients were considered infected. Respiratory infection was most common (64%). Microbiologic results were positive in 70% of individuals considered infected; of the isolates, 62% were gram-negative bacteria (Pseudomonas species and Escherichia coli were most common), 47% were gram-positive bacteria (Staphylococcus aureus was most common), and 19% were fungi (Candida species). This distribution is similar to that reported a decade earlier from eight academic centers in the United States (Table 325-2). In patients whose blood cultures are negative, the etiologic agent is often established by culture or microscopic examination of infected material from a local site; specific identification of microbial DNA or RNA in blood or tissue samples is also used. In some case series, a majority of patients with a clinical picture of severe sepsis or septic shock have had negative microbiologic data.
The systemic response to any class of microorganism can be harmful. Microbial invasion of the bloodstream is not essential because local inflammation can also elicit distant organ dysfunction and hypotension. In fact, blood cultures yield bacteria or fungi in only ~20–40% of cases of severe sepsis and 40–70% of cases of septic shock. In a prevalence study of 14,414 patients in intensive care units (ICUs) from 75 countries in 2007, 51% of patients were considered infected. Respiratory infection was most common (64%). Microbiologic results were positive in 70% of individuals considered infected; of the isolates, 62% were gram-negative bacteria (Pseudomonas species and Escherichia coli were most common), 47% were gram-positive bacteria (Staphylococcus aureus was most common), and 19% were fungi (Candida species). This distribution is similar to that reported a decade earlier from eight academic centers in the United States (Table 325-2). In patients whose blood cultures are negative, the etiologic agent is often established by culture or microscopic examination of infected material from a local site; specific identification of microbial DNA or RNA in blood or tissue samples is also used. In some case series, a majority of patients with a clinical picture of severe sepsis or septic shock have had negative microbiologic data.
|
MICROORGANISMS INVOLVED IN EPISODES OF SEVERE SEPSIS AT EIGHT ACADEMIC MEDICAL CENTERS |
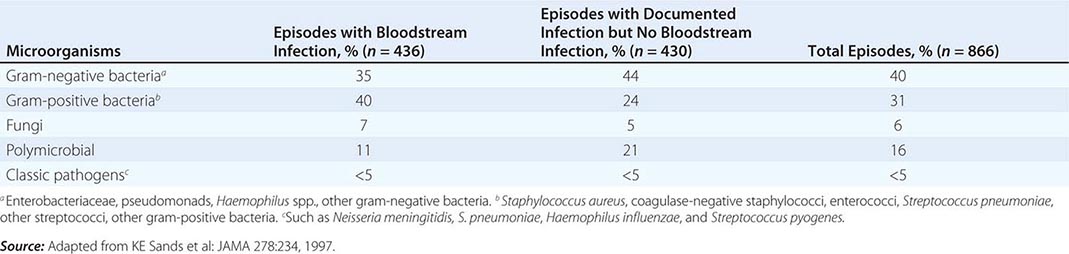
EPIDEMIOLOGY
Severe sepsis is a contributing factor in >200,000 deaths per year in the United States. The incidence of severe sepsis and septic shock has increased over the past 30 years, and the annual number of cases is now >750,000 (~3 per 1000 population). Approximately two-thirds of the cases occur in patients with significant underlying illness. Sepsis-related incidence and mortality rates increase with age and preexisting comorbidity. The rising incidence of severe sepsis in the United States has been attributable to the aging of the population, the increasing longevity of patients with chronic diseases, and the relatively high frequency with which sepsis has occurred in patients with AIDS. The widespread use of immunosuppressive drugs, indwelling catheters, and mechanical devices has also played a role. In the aforementioned international ICU prevalence study, the case–fatality rate among infected patients (33%) greatly exceeded that among uninfected patients (15%).
 Invasive bacterial infections are prominent causes of death around the world, particularly among young children. In sub-Saharan Africa, for example, careful screening for positive blood cultures found that community-acquired bacteremia accounted for at least one-fourth of deaths of children >1 year of age. Nontyphoidal Salmonella species, Streptococcus pneumoniae, Haemophilus influenzae, and E. coli were the most commonly isolated bacteria. Bacteremic children often had HIV infection or were severely malnourished.
Invasive bacterial infections are prominent causes of death around the world, particularly among young children. In sub-Saharan Africa, for example, careful screening for positive blood cultures found that community-acquired bacteremia accounted for at least one-fourth of deaths of children >1 year of age. Nontyphoidal Salmonella species, Streptococcus pneumoniae, Haemophilus influenzae, and E. coli were the most commonly isolated bacteria. Bacteremic children often had HIV infection or were severely malnourished.
PATHOPHYSIOLOGY
Sepsis is triggered most often by bacteria or fungi that do not ordinarily cause systemic disease in immunocompetent hosts (Table 325-2). To survive within the human body, these microbes often exploit acquired deficiencies in host defenses, indwelling catheters or other foreign matter, or obstructed fluid drainage conduits. Microbial pathogens, in contrast, can circumvent innate defenses because they (1) lack molecules that can be recognized by host receptors (see below) or (2) elaborate toxins or other virulence factors. In both cases, the body can mount a vigorous inflammatory reaction that results in sepsis or septic shock yet fails to kill the invaders. The septic response may also be induced by microbial exotoxins that act as superantigens (e.g., toxic shock syndrome toxin 1; Chap. 172) as well as by many pathogenic viruses.
Host Mechanisms for Sensing Microbes Animals have exquisitely sensitive mechanisms for recognizing and responding to certain highly conserved microbial molecules. Recognition of the lipid A moiety of lipopolysaccharide (LPS, also called endotoxin; Chap. 145e) is the best-studied example. A host protein (LPS-binding protein) binds lipid A and transfers the LPS to CD14 on the surfaces of monocytes, macrophages, and neutrophils. LPS then is passed to MD-2, a small receptor protein that is bound to Toll-like receptor (TLR) 4 to form a molecular complex that transduces the LPS recognition signal to the interior of the cell. This signal rapidly triggers the production and release of mediators, such as tumor necrosis factor (TNF; see below), that amplify the LPS signal and transmit it to other cells and tissues. Bacterial peptidoglycan and lipopeptides elicit responses in animals that are generally similar to those induced by LPS, although they interact with different TLRs. Having numerous TLR-based receptor complexes (10 different TLRs have been identified in humans) allows animals to recognize many conserved microbial molecules; others include lipopeptides (TLR2/1, TLR2/6), flagellin (TLR5), undermethylated DNA CpG sequences (TLR9), single-stranded RNA (TLR7, 8), and double-stranded RNA (TLR3). The ability of some TLRs to serve as receptors for host ligands (e.g., hyaluronans, heparan sulfate, saturated fatty acids, high-mobility group box 1) raises the possibility that they also play a role in producing noninfectious sepsis-like states. Other host pattern-recognition proteins that are important for sensing microbes include the intracellular NOD1 and NOD2 proteins, which recognize discrete fragments of bacterial peptidoglycan; the inflammasome, which senses some pathogens and produces interleukin (IL) 1β and IL-18; early complement components (principally in the alternative pathway); mannose-binding lectin and C-reactive protein, which activate the classic complement pathway; and Dectin-1 and complement receptor 3, which sense fungal β-glucan.
A host’s ability to recognize certain microbial molecules may influence both the potency of its own defenses and the pathogenesis of severe sepsis. For example, MD-2–TLR4 best senses LPS that has a bisphosphorylated, hexaacyl lipid A moiety (i.e., one with two phosphates and six fatty acyl chains). Most of the commensal aerobic and facultatively anaerobic gram-negative bacteria that trigger severe sepsis and shock (including E. coli, Klebsiella, and Enterobacter) make this lipid A structure. When they invade human hosts, often through breaks in an epithelial barrier, they are typically confined to the subepithelial tissue by a localized inflammatory response. Bacteremia, if it occurs, is intermittent and low grade because these bacteria are efficiently cleared from the bloodstream by TLR4-expressing Kupffer cells and splenic macrophages. These mucosal commensals seem to induce severe sepsis most often by triggering severe local tissue inflammation rather than by circulating within the bloodstream. One exception is Neisseria meningitidis. Its hexaacyl LPS seems to be shielded from host recognition by its polysaccharide capsule. This protection may allow meningococci to transit undetected from the nasopharyngeal mucosa into the bloodstream, where they can infect vascular endothelial cells and release large amounts of endotoxin and DNA. Host recognition of lipid A may nonetheless influence pathogenesis, as meningococci that produce pentaacyl LPS were isolated from the blood of patients with less severe coagulopathy than was found in patients whose isolates produced hexaacyl lipid A; underacylated N. meningitidis LPS has also been found in many isolates from patients with chronic meningococcemia. In contrast, gram-negative bacteria that make lipid A with fewer than six acyl chains (Yersinia pestis, Francisella tularensis, Vibrio vulnificus, Pseudomonas aeruginosa, and Burkholderia pseudomallei, among others) are poorly recognized by MD-2–TLR4. When these bacteria enter the body, they may initially induce relatively little inflammation. When they do trigger severe sepsis, it is often after they have multiplied to high density in tissues and blood. The importance of LPS recognition in disease pathogenesis was demonstrated by engineering of a virulent strain of Y. pestis that makes tetraacyl LPS at 37°C to produce hexaacyl LPS; unlike its virulent parent, the mutant strain stimulated local inflammation and was rapidly cleared from tissues. These findings were subsequently replicated in F. tularensis. For at least one large class of microbes—gram-negative aerobic bacteria—the pathogenesis of sepsis thus depends, at least in part, on whether the bacterium’s major signal molecule, LPS, can be sensed by the host.
Local and Systemic Host Responses to Invading Microbes Recognition of microbial molecules by tissue phagocytes triggers the production and/or release of numerous host molecules (cytokines, chemokines, prostanoids, leukotrienes, and others) that increase blood flow to the infected tissue (rubor), enhance the permeability of local blood vessels (tumor), recruit neutrophils and other cells to the site of infection (calor), and elicit pain (dolor). These reactions are familiar elements of local inflammation, the body’s frontline innate immune mechanism for eliminating microbial invaders. Systemic responses are activated by neural and/or humoral communication with the hypothalamus and brainstem; these responses enhance local defenses by increasing blood flow to the infected area, augmenting the number of circulating neutrophils, and elevating blood levels of numerous molecules (such as the microbial recognition proteins discussed above) that have anti-infective functions.
CYTOKINES AND OTHER MEDIATORS Cytokines can exert endocrine, paracrine, and autocrine effects (Chap. 372e). TNF-α stimulates leukocytes and vascular endothelial cells to release other cytokines (as well as additional TNF-α), to express cell-surface molecules that enhance neutrophil endothelial adhesion at sites of infection, and to increase prostaglandin and leukotriene production. Whereas blood levels of TNF-α are not elevated in individuals with localized infections, they increase in most patients with severe sepsis or septic shock. Moreover, IV infusion of TNF-α can elicit fever, tachycardia, hypotension, and other responses. In animals, larger doses of TNF-α induce shock and death.
Although TNF-α is a central mediator, it is only one of many proinflammatory molecules that contribute to innate host defense. Chemokines, most prominently IL-8 and IL-17, attract circulating neutrophils to the infection site. IL-1β exhibits many of the same activities as TNF-α. TNF-α, IL-1β, interferon γ, IL-12, IL-17, and other proinflammatory cytokines probably interact synergistically with one another and with additional mediators. The nonlinearity and multiplicity of these interactions have made it difficult to interpret the roles played by individual mediators in both tissues and blood.
COAGULATION FACTORS Intravascular thrombosis, a hallmark of the local inflammatory response, may help wall off invading microbes and prevent infection and inflammation from spreading to other tissues. IL-6 and other mediators promote intravascular coagulation initially by inducing blood monocytes and vascular endothelial cells to express tissue factor (Chap. 78). When tissue factor is expressed on cell surfaces, it binds to factor VIIa to form an active complex that can convert factors × and IX to their enzymatically active forms. The result is activation of both extrinsic and intrinsic clotting pathways, culminating in the generation of fibrin. Clotting is also favored by impaired function of the protein C–protein S inhibitory pathway and depletion of antithrombin and proteins C and S, whereas fibrinolysis is reduced by increases in plasma levels of plasminogen activator inhibitor 1. Thus, there may be a striking propensity toward intravascular fibrin deposition, thrombosis, and bleeding; this propensity has been most apparent in patients with intravascular endothelial infections such as meningococcemia (Chap. 180). Evidence points to tissue factor–expressing microparticles derived from leukocytes as a potential trigger for intravascular coagulation. The contact system is activated during sepsis but contributes more to the development of hypotension than to that of disseminated intravascular coagulation (DIC).
Neutrophil extracellular traps (NETs) are produced when neutrophils, stimulated by microbial agonists or IL-8, release granule proteins and chromatin to form an extracellular fibrillar matrix. NETs kill bacteria and fungi with antimicrobial granule proteins (e.g., elastase) and histones. It has been reported that NETs can form within hepatic sinusoids in animals injected with large amounts of LPS, and platelets can induce NET formation without killing neutrophils. A role played by NETs in organ hypofunction during sepsis has been proposed but not established.
CONTROL MECHANISMS Elaborate control mechanisms operate within both local sites of inflammation and the systemic compartment.
Local control mechanisms Host recognition of invading microbes within subepithelial tissues typically ignites immune responses that rapidly kill the invaders and then subside to allow tissue recovery. The forces that put out the fire and clean up the battleground include molecules that neutralize or inactivate microbial signals. Among these molecules are intracellular factors (e.g., suppressor of cytokine signaling 3 and IL-1 receptor–associated kinase 3) that diminish the production of proinflammatory mediators by neutrophils and macrophages; anti-inflammatory cytokines (IL-10, IL-4); and molecules derived from essential polyunsaturated fatty acids (lipoxins, resolvins, and protectins) that promote tissue restoration. Enzymatic inactivation of microbial signal molecules (e.g., LPS) may be required to restore homeostasis; a leukocyte enzyme, acyloxyacyl hydrolase, has been shown to prevent prolonged inflammation in mice by inactivating LPS.
Systemic control mechanisms The signaling apparatus that links microbial recognition to cellular responses in tissues is less active in the blood. For example, whereas LPS-binding protein plays a role in recognizing LPS, in plasma it also prevents LPS signaling by transferring LPS molecules into plasma lipoprotein particles that sequester the lipid A moiety so that it cannot interact with cells. At the high concentrations found in blood, LPS-binding protein also inhibits monocyte responses to LPS, and the soluble (circulating) form of CD14 strips off LPS that has bound to monocyte surfaces.
Systemic responses to infection also diminish cellular responses to microbial molecules. Circulating levels of cortisol and anti-inflammatory cytokines (e.g., IL-6 and IL-10) increase even in patients with minor infections. Glucocorticoids inhibit cytokine synthesis by monocytes in vitro; the increase in blood cortisol levels that occurs early in the systemic response presumably plays a similarly inhibitory role. Epinephrine inhibits the TNF-α response to endotoxin infusion in humans while augmenting and accelerating the release of IL-10; prostaglandin E2 has a similar “reprogramming” effect on the responses of circulating monocytes to LPS and other bacterial agonists. Cortisol, epinephrine, IL-10, and C-reactive protein reduce the ability of neutrophils to attach to vascular endothelium, favoring their demargination and thus contributing to leukocytosis while preventing neutrophil-endothelial adhesion in uninflamed organs. Studies in rodents have found that macrophage cytokine synthesis is inhibited by acetylcholine that is produced by choline acetyltransferase–secreting CD4+ T cells in response to stimulation by norepinephrine, whereas acetylcholine-producing B cells reduce neutrophil infiltration into tissues. Several lines of evidence thus suggest that the body’s neuroendocrine responses to injury and infection normally prevent inflammation within organs distant from a site of infection. There is also evidence that these responses may be immunosuppressive.
IL-6 plays important roles in the systemic compartment. Released by many different cell types, IL-6 is an important stimulus to the hypothalamic-pituitary-adrenal axis, is the major procoagulant cytokine, and is a principal inducer of the acute-phase response, which increases the blood concentrations of numerous molecules that have anti-infective, procoagulant, or anti-inflammatory actions. Blood levels of IL-1 receptor antagonist often greatly exceed those of circulating IL-1β, for example, and this excess may inhibit the binding of IL-1β to its receptors. High levels of soluble TNF receptors neutralize TNF-α that enters the circulation. Other acute-phase proteins are protease inhibitors or antioxidants; these may neutralize potentially harmful molecules released from neutrophils and other inflammatory cells. Increased hepatic production of hepcidin (stimulated largely by IL-6) promotes the sequestration of iron in hepatocytes, intestinal epithelial cells, and erythrocytes; this effect reduces iron acquisition by invading microbes while contributing to the normocytic, normochromic anemia associated with inflammation.
It may thus be said that both local and systemic responses to infectious agents benefit the host in important ways. Most of these responses and the molecules responsible for them have been highly conserved during animal evolution and therefore may be adaptive. Elucidating how they become maladaptive and contribute to lethality remains a major challenge for sepsis research.
Organ Dysfunction and Shock As the body’s responses to infection intensify, the mixture of circulating cytokines and other molecules becomes very complex: elevated blood levels of more than 60 molecules have been found in patients with septic shock. Although high concentrations of both pro- and anti-inflammatory molecules are found, the net mediator balance in the plasma of these extremely sick patients seems to be anti-inflammatory. For example, blood leukocytes from patients with severe sepsis are often hyporesponsive to agonists such as LPS. In patients with severe sepsis, persistence of leukocyte hyporesponsiveness has been associated with an increased risk of dying; at this time, the most predictive biomarker is a decrease in the expression of HLA-DR (class II) molecules on the surfaces of circulating monocytes, a response that seems to be induced by cortisol and/or IL-10. Apoptotic death of B cells, follicular dendritic cells, and CD4+ T lymphocytes also may contribute significantly to the immunosuppressive state.
ENDOTHELIAL INJURY Given the vascular endothelium’s important roles in regulating vascular tone, vascular permeability, and coagulation, many investigators have favored widespread vascular endothelial injury as the major mechanism for multiorgan dysfunction. In keeping with this idea, one study found high numbers of vascular endothelial cells in the peripheral blood of septic patients. Leukocyte-derived mediators and platelet-leukocyte-fibrin thrombi may contribute to vascular injury, but the vascular endothelium also seems to play an active role. Stimuli such as TNF-α induce vascular endothelial cells to produce and release cytokines, procoagulant molecules, platelet-activating factor, nitric oxide, and other mediators. In addition, regulated cell-adhesion molecules promote the adherence of neutrophils to endothelial cells. Although these responses can attract phagocytes to infected sites and activate their antimicrobial arsenals, endothelial cell activation can also promote increased vascular permeability, microvascular thrombosis, DIC, and hypotension.
Tissue oxygenation may decrease as the number of functional capillaries is reduced by luminal obstruction due to swollen endothelial cells, decreased deformability of circulating erythrocytes, leukocyte-platelet-fibrin thrombi, or compression by edema fluid. On the other hand, studies using orthogonal polarization spectral imaging of the microcirculation in the tongue found that sepsis-associated derangements in capillary flow could be reversed by applying acetylcholine to the surface of the tongue or by giving nitroprusside intravenously; these observations suggest a neuroendocrine basis for the loss of capillary filling. Oxygen utilization by tissues may also be impaired by changes (possibly induced by nitric oxide) that decrease oxidative phosphorylation and ATP production while increasing glycolysis. The local accumulation of lactic acid, a consequence of increased glycolysis, may decrease extracellular pH and contribute to the slowdown in cellular metabolism that occurs within affected tissues.
Remarkably, poorly functioning “septic” organs usually appear normal at autopsy. There is typically very little necrosis or thrombosis, and apoptosis is largely confined to lymphoid organs and the gastrointestinal tract. Moreover, organ function usually returns to normal if patients recover. These points suggest that organ dysfunction during severe sepsis has a basis that is principally biochemical, not structural.
SEPTIC SHOCK The hallmark of septic shock is a decrease in peripheral vascular resistance that occurs despite increased levels of vasopressor catecholamines. Before this vasodilatory phase, many patients experience a period during which oxygen delivery to tissues is compromised by myocardial depression, hypovolemia, and other factors. During this “hypodynamic” period, the blood lactate concentration is elevated and central venous oxygen saturation is low. Fluid administration is usually followed by the hyperdynamic vasodilatory phase, during which cardiac output is normal (or even high) and oxygen consumption declines despite adequate oxygen delivery. The blood lactate level may be normal or increased, and normalization of central venous oxygen saturation may reflect improved oxygen delivery, decreased oxygen uptake by tissues, or left-to-right shunting.
Prominent hypotensive molecules include nitric oxide, β-endorphin, bradykinin, platelet-activating factor, and prostacyclin. Agents that inhibit the synthesis or action of each of these mediators can prevent or reverse endotoxic shock in animals. However, in clinical trials, neither a platelet-activating factor receptor antagonist nor a bradykinin antagonist improved survival rates among patients with septic shock, and a nitric oxide synthase inhibitor, L-NG-methylarginine HCl, actually increased the mortality rate.
Severe Sepsis: A Single Pathogenesis? In some cases, circulating bacteria and their products almost certainly elicit multiorgan dysfunction and hypotension by directly stimulating inflammatory responses within the vasculature. In patients with fulminant meningococcemia, for example, mortality rates have correlated directly with blood levels of endotoxin and bacterial DNA and with the occurrence of DIC (Chap. 180). In most patients infected with other gram-negative bacteria, in contrast, circulating bacteria or bacterial molecules may reflect uncontrolled infection at a local tissue site and have little or no direct impact on distant organs; in these patients, inflammatory mediators or neural signals arising from the local site seem to be the key triggers for severe sepsis and septic shock. In a large series of patients with positive blood cultures, the risk of developing severe sepsis was strongly related to the site of primary infection: bacteremia arising from a pulmonary or abdominal source was eightfold more likely to be associated with severe sepsis than was bacteremic urinary tract infection, even after the investigators controlled for age, the kind of bacteria isolated from the blood, and other factors. A third pathogenesis may be represented by severe sepsis due to superantigen-producing S. aureus or Streptococcus pyogenes; the T cell activation induced by these toxins produces a cytokine profile that differs substantially from that elicited by gram-negative bacterial infection. Further evidence for different pathogenetic pathways has come from observations that the pattern of mRNA expression in peripheral-blood leukocytes from children with sepsis is different for gram-positive, gram-negative, and viral pathogens.
The pathogenesis of severe sepsis thus may differ according to the infecting microbe, the ability of the host’s innate defense mechanisms to sense and respond to it, the site of the primary infection, the presence or absence of immune defects, and the prior physiologic status of the host. Genetic factors are probably important as well, yet despite much study very few allelic polymorphisms have been associated with sepsis severity in more than one or two analyses. Further studies in this area are needed.
CLINICAL MANIFESTATIONS
The manifestations of the septic response are superimposed on the symptoms and signs of the patient’s underlying illness and primary infection. The rate at which severe sepsis develops may differ from patient to patient, and there are striking individual variations in presentation. For example, some patients with sepsis are normo- or hypothermic; the absence of fever is most common in neonates, in elderly patients, and in persons with uremia or alcoholism.
Hyperventilation, producing respiratory alkalosis, is often an early sign of the septic response. Disorientation, confusion, and other manifestations of encephalopathy may also develop early on, particularly in the elderly and in individuals with preexisting neurologic impairment. Focal neurologic signs are uncommon, although preexisting focal deficits may become more prominent.
Hypotension and DIC predispose to acrocyanosis and ischemic necrosis of peripheral tissues, most commonly the digits. Cellulitis, pustules, bullae, or hemorrhagic lesions may develop when hematogenous bacteria or fungi seed the skin or underlying soft tissue. Bacterial toxins may also be distributed hematogenously and elicit diffuse cutaneous reactions. On occasion, skin lesions may suggest specific pathogens. When sepsis is accompanied by cutaneous petechiae or purpura, infection with N. meningitidis (or, less commonly, H. influenzae) should be suspected (see Fig. 25e-42); in a patient who has been bitten by a tick while in an endemic area, petechial lesions also suggest Rocky Mountain spotted fever (see Fig. 211-1). A cutaneous lesion seen almost exclusively in neutropenic patients is ecthyma gangrenosum, often caused by P. aeruginosa. This bullous lesion surrounded by edema undergoes central hemorrhage and necrosis (see Fig. 189-1). Histopathologic examination shows bacteria in and around the wall of a small vessel, with little or no neutrophilic response. Hemorrhagic or bullous lesions in a septic patient who has recently eaten raw oysters suggest V. vulnificus bacteremia, whereas such lesions in a patient who has recently sustained a dog bite may indicate bloodstream infection due to Capnocytophaga canimorsus or Capnocytophaga cynodegmi. Generalized erythroderma in a septic patient suggests the toxic shock syndrome due to S. aureus or S. pyogenes.
Gastrointestinal manifestations such as nausea, vomiting, diarrhea, and ileus may suggest acute gastroenteritis. Stress ulceration can lead to upper gastrointestinal bleeding. Cholestatic jaundice, with elevated levels of serum bilirubin (mostly conjugated) and alkaline phosphatase, may precede other signs of sepsis. Hepatocellular or canalicular dysfunction appears to underlie most cases, and the results of hepatic function tests return to normal with resolution of the infection. Prolonged or severe hypotension may induce acute hepatic injury or ischemic bowel necrosis.
Many tissues may be unable to extract oxygen normally from the blood, so that anaerobic metabolism occurs despite near-normal mixed venous oxygen saturation. Blood lactate levels rise early because of increased glycolysis as well as impaired clearance of the resulting lactate and pyruvate by the liver and kidneys. The blood glucose concentration often increases, particularly in patients with diabetes, although impaired gluconeogenesis and excessive insulin release on occasion produce hypoglycemia. The cytokine-driven acute-phase response inhibits the synthesis of transthyretin while enhancing the production of C-reactive protein, fibrinogen, and complement components. Protein catabolism is often markedly accelerated. Serum albumin levels decline as a result of decreased hepatic synthesis and the movement of albumin into interstitial spaces.
MAJOR COMPLICATIONS
Cardiopulmonary Complications Ventilation-perfusion mismatching produces a fall in arterial Po2 early in the course. Increasing alveolar epithelial injury and capillary permeability result in increased pulmonary water content, which decreases pulmonary compliance and interferes with oxygen exchange. In the absence of pneumonia or heart failure, progressive diffuse pulmonary infiltrates and arterial hypoxemia occurring within 1 week of a known insult indicate the development of mild acute respiratory distress syndrome (ARDS) (200 mmHg < PaO2/FIO2 ≤ 300 mmHg), moderate ARDS (100 mmHg < PaO2/FIO2 ≤ 200 mmHg), or severe ARDS (PaO2/FIO2 ≤100 mmHg). Acute lung injury or ARDS develops in ~50% of patients with severe sepsis or septic shock. Respiratory muscle fatigue can exacerbate hypoxemia and hypercapnia. An elevated pulmonary capillary wedge pressure (>18 mmHg) suggests fluid volume overload or cardiac failure rather than ARDS. Pneumonia caused by viruses or by Pneumocystis may be clinically indistinguishable from ARDS.
Sepsis-induced hypotension (see “Septic Shock,” above) usually results initially from a generalized maldistribution of blood flow and blood volume and from hypovolemia that is due, at least in part, to diffuse capillary leakage of intravascular fluid. Other factors that may decrease effective intravascular volume include dehydration from antecedent disease or insensible fluid losses, vomiting or diarrhea, and polyuria. During early septic shock, systemic vascular resistance is usually elevated and cardiac output may be low. After fluid repletion, in contrast, cardiac output typically increases and systemic vascular resistance falls. Indeed, normal or increased cardiac output and decreased systemic vascular resistance distinguish septic shock from cardiogenic, extracardiac obstructive, and hypovolemic shock; other processes that can produce this combination include anaphylaxis, beriberi, cirrhosis, and overdoses of nitroprusside or narcotics.
Depression of myocardial function, manifested as increased end-diastolic and systolic ventricular volumes with a decreased ejection fraction, develops within 24 h in most patients with severe sepsis. Cardiac output is maintained despite the low ejection fraction because ventricular dilation permits a normal stroke volume. In survivors, myocardial function returns to normal over several days. Although myocardial dysfunction may contribute to hypotension, refractory hypotension is usually due to low systemic vascular resistance, and death most often results from refractory shock or the failure of multiple organs rather than from cardiac dysfunction per se.
Adrenal Insufficiency The diagnosis of adrenal insufficiency may be very difficult in critically ill patients. Whereas a plasma cortisol level of ≤15 μg/mL (≤10 μg/mL if the serum albumin concentration is <2.5 mg/dL) indicates adrenal insufficiency (inadequate production of cortisol), many experts now feel that the adrenocorticotropic hormone (CoSyntropin®) stimulation test is not useful for detecting less profound degrees of corticosteroid deficiency in patients who are critically ill. The concept of critical illness–related corticosteroid insufficiency (CIRCI) was proposed to encompass the different mechanisms that may produce corticosteroid activity that is inadequate for the severity of a patient’s illness. Although CIRCI may result from structural damage to the adrenal gland, it is more commonly due to reversible dysfunction of the hypothalamic-pituitary axis or to tissue corticosteroid resistance resulting from abnormalities of the glucocorticoid receptor or increased conversion of cortisol to cortisone. The major clinical manifestation of CIRCI is hypotension that is refractory to fluid replacement and requires pressor therapy. Some classic features of adrenal insufficiency, such as hyponatremia and hyperkalemia, are usually absent; others, such as eosinophilia and modest hypoglycemia, may sometimes be found. Specific etiologies include fulminant N. meningitidis bacteremia, disseminated tuberculosis, AIDS (with cytomegalovirus, Mycobacterium avium-intracellulare, or Histoplasma capsulatum disease), or the prior use of drugs that diminish glucocorticoid production, such as glucocorticoids, megestrol, etomidate, or ketoconazole.
Renal Complications Oliguria, azotemia, proteinuria, and nonspecific urinary casts are frequently found. Many patients are inappropriately polyuric; hyperglycemia may exacerbate this tendency. Most renal failure is due to acute tubular necrosis induced by hypovolemia, arterial hypotension, or toxic drugs, although some patients also have glomerulonephritis, renal cortical necrosis, or interstitial nephritis. Drug-induced renal damage may greatly complicate therapy, particularly when hypotensive patients are given aminoglycoside antibiotics. Nosocomial sepsis following acute renal injury is associated with a high mortality rate.
Coagulopathy Although thrombocytopenia occurs in 10–30% of patients, the underlying mechanisms are not understood. Platelet counts are usually very low (<50,000/μL) in patients with DIC; these low counts may reflect diffuse endothelial injury or microvascular thrombosis, yet thrombi have only infrequently been found on biopsy of septic organs.
Neurologic Complications Delirium (acute encephalopathy) is often an early manifestation of sepsis. Depending on the diagnostic criteria used, it occurs in 10–70% of septic patients at some point during the hospital course. When the septic illness lasts for weeks or months, “critical illness” polyneuropathy may prevent weaning from ventilatory support and produce distal motor weakness. Electrophysiologic studies are diagnostic. Guillain-Barré syndrome, metabolic disturbances, and toxin activity must be ruled out. Recent studies have documented long-term cognitive loss in survivors of severe sepsis.
Immunosuppression Patients with severe sepsis often become profoundly immunosuppressed. Manifestations include loss of delayed-type hypersensitivity reactions to common antigens, failure to control the primary infection, and increased risk for secondary infections (e.g., by opportunists such as Stenotrophomonas maltophilia, Acinetobacter calcoaceticus-baumannii, and Candida albicans). Approximately one-third of patients experience reactivation of herpes simplex virus, varicella-zoster virus, or cytomegalovirus infections; the latter are thought to contribute to adverse outcomes in some instances.
LABORATORY FINDINGS
Abnormalities that occur early in the septic response may include leukocytosis with a left shift, thrombocytopenia, hyperbilirubinemia, and proteinuria. Leukopenia may develop. The neutrophils may contain toxic granulations, Döhle bodies, or cytoplasmic vacuoles. As the septic response becomes more severe, thrombocytopenia worsens (often with prolonged thrombin time, decreased fibrinogen, and the presence of D-dimers, suggesting DIC), azotemia and hyperbilirubinemia become more prominent, and levels of aminotransferases rise. Active hemolysis suggests clostridial bacteremia, malaria, a drug reaction, or DIC; in the case of DIC, microangiopathic changes may be seen on a blood smear.
During early sepsis, hyperventilation induces respiratory alkalosis. With respiratory muscle fatigue and the accumulation of lactate, metabolic acidosis (with increased anion gap) typically supervenes. Evaluation of arterial blood gases reveals hypoxemia that is initially correctable with supplemental oxygen but whose later refractoriness to 100% oxygen inhalation indicates right-to-left shunting. The chest radiograph may be normal or may show evidence of underlying pneumonia, volume overload, or the diffuse infiltrates of ARDS. The electrocardiogram may show only sinus tachycardia or nonspecific ST–T wave abnormalities.
Most diabetic patients with sepsis develop hyperglycemia. Severe infection may precipitate diabetic ketoacidosis that may exacerbate hypotension (Chap. 417). Hypoglycemia occurs rarely and may indicate adrenal insufficiency. The serum albumin level declines as sepsis continues. Hypocalcemia is rare.
DIAGNOSIS
There is no specific diagnostic test for sepsis. Diagnostically sensitive findings in a patient with suspected or proven infection include fever or hypothermia, tachypnea, tachycardia, and leukocytosis or leukopenia (Table 325-1); acutely altered mental status, thrombocytopenia, an elevated blood lactate level, respiratory alkalosis, or hypotension also should suggest the diagnosis. The systemic response can be quite variable, however. In one study, 36% of patients with severe sepsis had a normal temperature, 40% had a normal respiratory rate, 10% had a normal pulse rate, and 33% had normal white blood cell counts. Moreover, the systemic responses of uninfected patients with other conditions may be similar to those characteristic of sepsis. Examples include pancreatitis, burns, trauma, adrenal insufficiency, pulmonary embolism, dissecting or ruptured aortic aneurysm, myocardial infarction, occult hemorrhage, cardiac tamponade, postcardiopulmonary bypass syndrome, anaphylaxis, tumor-associated lactic acidosis, and drug overdose.
Definitive etiologic diagnosis requires identification of the causative microorganism from blood or a local site of infection. At least two blood samples should be obtained (from two different venipuncture sites) for culture; in a patient with an indwelling catheter, one sample should be collected from each lumen of the catheter and another via venipuncture. In many cases, blood cultures are negative; this result can reflect prior antibiotic administration, the presence of slow-growing or fastidious organisms, or the absence of microbial invasion of the bloodstream. In these cases, Gram’s staining and culture of material from the primary site of infection or from infected cutaneous lesions may help establish the microbial etiology. Identification of microbial DNA in peripheral blood or tissue samples by polymerase chain reaction may also be definitive. The skin and mucosae should be examined carefully and repeatedly for lesions that might yield diagnostic information. With overwhelming bacteremia (e.g., pneumococcal sepsis in splenectomized individuals; fulminant meningococcemia; or infection with V. vulnificus, B. pseudomallei, or Y. pestis), microorganisms are sometimes visible on buffy coat smears of peripheral blood.
|
TREATMENT |
SEVERE SEPSIS AND SEPTIC SHOCK |
Patients in whom sepsis is suspected must be managed expeditiously. This task is best accomplished by personnel who are experienced in the care of the critically ill. Successful management requires urgent measures to treat the infection, to provide hemodynamic and respiratory support, and to remove or drain infected tissues. These measures should be initiated within 1 h of the patient’s presentation with severe sepsis or septic shock. Rapid assessment and diagnosis are therefore essential.
ANTIMICROBIAL AGENTS
Antimicrobial chemotherapy should be started as soon as samples of blood and other relevant sites have been obtained for culture. A large retrospective review of patients who developed septic shock found that the interval between the onset of hypotension and the administration of appropriate antimicrobial chemotherapy was the major determinant of outcome; a delay of as little as 1 h was associated with lower survival rates. Use of “inappropriate” antibiotics, defined on the basis of local microbial susceptibilities and published guidelines for empirical therapy (see below), was associated with fivefold lower survival rates, even among patients with negative cultures.
It is therefore very important to promptly initiate empirical antimicrobial therapy that is effective against both gram-positive and gram-negative bacteria (Table 325-3). Maximal recommended doses of antimicrobial drugs should be given intravenously, with adjustment for impaired renal function when necessary. Available information about patterns of antimicrobial susceptibility among bacterial isolates from the community, the hospital, and the patient should be taken into account. When culture results become available, the regimen can often be simplified because a single antimicrobial agent is usually adequate for the treatment of a known pathogen. Meta-analyses have concluded that, with one exception, combination antimicrobial therapy is not superior to monotherapy for treating gram-negative bacteremia; the exception is that aminoglycoside monotherapy for P. aeruginosa bacteremia is less effective than the combination of an aminoglycoside with an antipseudomonal β-lactam agent. Empirical antifungal therapy should be strongly considered if the septic patient is already receiving broad-spectrum antibiotics or parenteral nutrition, has been neutropenic for ≥5 days, has had a long-term central venous catheter in place, or has been hospitalized in an ICU for a prolonged period. The chosen antimicrobial regimen should be reconsidered daily in order to provide maximal efficacy with minimal resistance, toxicity, and cost.
|
INITIAL ANTIMICROBIAL THERAPY FOR SEVERE SEPSIS WITH NO OBVIOUS SOURCE IN ADULTS WITH NORMAL RENAL FUNCTION |

Most patients require antimicrobial therapy for at least 1 week. The duration of treatment is typically influenced by factors such as the site of tissue infection, the adequacy of surgical drainage, the patient’s underlying disease, and the antimicrobial susceptibility of the microbial isolate(s). The absence of an identified microbial pathogen is not necessarily an indication for discontinuing antimicrobial therapy because “appropriate” antimicrobial regimens seem to be beneficial in both culture-negative and culture-positive cases.
REMOVAL OF THE SOURCE OF INFECTION
Removal or drainage of a focal source of infection is essential. In one series, a focus of ongoing infection was found in ~80% of surgical ICU patients who died of severe sepsis or septic shock. Sites of occult infection should be sought carefully, particularly in the lungs, abdomen, and urinary tract. Indwelling IV or arterial catheters should be removed and the tip rolled over a blood agar plate for quantitative culture; after antibiotic therapy has been initiated, a new catheter should be inserted at a different site. Foley and drainage catheters should be replaced. The possibility of paranasal sinusitis (often caused by gram-negative bacteria) should be considered if the patient has undergone nasal intubation or has an indwelling nasogastric or feeding tube. Even in patients without abnormalities on chest radiographs, computed tomography (CT) of the chest may identify unsuspected parenchymal, mediastinal, or pleural disease. In the neutropenic patient, cutaneous sites of tenderness and erythema, particularly in the perianal region, must be carefully sought. In patients with sacral or ischial decubitus ulcers, it is important to exclude pelvic or other soft tissue pus collections with CT or magnetic resonance imaging (MRI). In patients with severe sepsis arising from the urinary tract, sonography or CT should be used to rule out ureteral obstruction, perinephric abscess, and renal abscess. Sonographic or CT imaging of the upper abdomen may disclose evidence of cholecystitis, bile duct dilation, and pus collections in the liver, subphrenic space, or spleen.
HEMODYNAMIC, RESPIRATORY, AND METABOLIC SUPPORT
The primary goals are to restore adequate oxygen and substrate delivery to the tissues as quickly as possible and to improve tissue oxygen utilization and cellular metabolism. Adequate organ perfusion is thus essential. Circulatory adequacy is assessed by measurement of arterial blood pressure and monitoring of parameters such as mentation, urine output, and skin perfusion. Indirect indices of oxygen delivery and consumption, such as central venous oxygen saturation, may also be useful. Initial management of hypotension should include the administration of IV fluids, typically beginning with 1–2 L of normal saline over 1–2 h. To avoid pulmonary edema, the central venous pressure should be maintained at 8–12 cmH2O. The urine output rate should be kept at >0.5 mL/kg per hour by continuing fluid administration; a diuretic such as furosemide may be used if needed. In about one-third of patients, hypotension and organ hypoperfusion respond to fluid resuscitation; a reasonable goal is to maintain a mean arterial blood pressure of >65 mmHg (systolic pressure >90 mmHg). If these guidelines cannot be met by volume infusion, vasopressor therapy is indicated (Chap. 326). Titrated doses of norepinephrine should be administered through a central catheter. If myocardial dysfunction produces elevated cardiac filling pressures and low cardiac output, inotropic therapy with dobutamine is recommended. Dopamine is rarely used.
In patients with septic shock, plasma vasopressin levels increase transiently but then decrease dramatically. Early studies found that vasopressin infusion can reverse septic shock in some patients, reducing or eliminating the need for catecholamine pressors. Although vasopressin may benefit patients who require less norepinephrine, its role in the treatment of septic shock seems to be a minor one overall.
CIRCI (see “Adrenal Insufficiency,” above) should be strongly considered in patients who develop hypotension that does not respond to fluid replacement therapy. Hydrocortisone (50 mg IV every 6 h) should be given; if clinical improvement occurs over 24–48 h, most experts would continue hydrocortisone therapy for 5–7 days before slowly tapering and discontinuing it. Meta-analyses of recent clinical trials have concluded that hydrocortisone therapy hastens recovery from sepsis-induced hypotension without increasing long-term survival.
Ventilator therapy is indicated for progressive hypoxemia, hypercapnia, neurologic deterioration, or respiratory muscle failure. Sustained tachypnea (respiratory rate, >30 breaths/min) is frequently a harbinger of impending respiratory collapse; mechanical ventilation is often initiated to ensure adequate oxygenation, to divert blood from the muscles of respiration, to prevent aspiration of oropharyngeal contents, and to reduce the cardiac afterload. The results of recent studies favor the use of low tidal volumes (6 mL/kg of ideal body weight, or as low as 4 mL/kg if the plateau pressure exceeds 30 cmH2O). Patients undergoing mechanical ventilation require careful sedation, with daily interruptions; elevation of the head of the bed helps to prevent nosocomial pneumonia. Stress-ulcer prophylaxis with a histamine H2-receptor antagonist may decrease the risk of gastrointestinal hemorrhage in ventilated patients.
Erythrocyte transfusion is generally recommended when the blood hemoglobin level decreases to ≤7 g/dL, with a target level of 9 g/dL in adults. Erythropoietin is not used to treat sepsis-related anemia. Bicarbonate is sometimes administered for severe metabolic acidosis (arterial pH <7.2), but there is little evidence that it improves either hemodynamics or the response to vasopressor hormones. DIC, if complicated by major bleeding, should be treated with transfusion of fresh-frozen plasma and platelets. Successful treatment of the underlying infection is essential to reverse both acidosis and DIC. Patients who are hypercatabolic and have acute renal failure may benefit greatly from intermittent hemodialysis or continuous veno-venous hemofiltration.
GENERAL SUPPORT
In patients with prolonged severe sepsis (i.e., that lasting more than 2 or 3 days), nutritional supplementation may reduce the impact of protein hypercatabolism; the available evidence favors the enteral delivery route. Prophylactic heparinization to prevent deep venous thrombosis is indicated for patients who do not have active bleeding or coagulopathy; when heparin is contraindicated, compression stockings or an intermittent compression device should be used. Recovery is also assisted by prevention of skin breakdown, nosocomial infections, and stress ulcers.
The role of tight control of the blood glucose concentration in recovery from critical illness has been addressed in numerous controlled trials. Meta-analyses of these trials have concluded that use of insulin to lower blood glucose levels to 100–120 mg/dL is potentially harmful and does not improve survival rates. Most experts now recommend using insulin only if it is needed to maintain the blood glucose concentration below ~180 mg/dL. Patients receiving intravenous insulin must be monitored frequently (every 1–2 h) for hypoglycemia.
OTHER MEASURES
Despite aggressive management, many patients with severe sepsis or septic shock die. Numerous interventions have been tested for their ability to improve survival rates among patients with severe sepsis. The list includes endotoxin-neutralizing proteins, inhibitors of cyclooxygenase or nitric oxide synthase, anticoagulants, polyclonal immunoglobulins, glucocorticoids, a phospholipid emulsion, and antagonists to TNF-α, IL-1, platelet-activating factor, and bradykinin. Unfortunately, none of these agents has improved rates of survival among patients with severe sepsis/septic shock in more than one large-scale, randomized, placebo-controlled clinical trial. Many factors have contributed to this lack of reproducibility, including (1) heterogeneity of the patient populations studied, the primary infection sites, the preexisting illnesses, and the inciting microbes; and (2) the nature of the “standard” therapy also used. A dramatic example of this problem was seen in a trial of tissue factor pathway inhibitor. Whereas the drug appeared to improve survival rates after 722 patients had been studied (p = .006), it did not do so in the next 1032 patients, and the overall result was negative. This inconsistency argues that the results of a clinical trial may not apply to individual patients, even within a carefully selected patient population. It also suggests that, at a minimum, a sepsis intervention should show a significant survival benefit in more than one placebo-controlled, randomized clinical trial before it is accepted as routine clinical practice. In one attempt to reduce patient heterogeneity in clinical trials, experts have called for changes that would restrict these trials to patients who have similar underlying diseases (e.g., major trauma) and inciting infections (e.g., pneumonia). Other investigators have proposed using specific biomarkers, such as IL-6 levels in blood or the expression of HLA-DR on peripheral-blood monocytes, to identify the patients most likely to benefit from certain interventions.
Recombinant activated protein C (aPC) was the first immunomodulatory drug to be approved by the U.S. Food and Drug Administration (FDA) for the treatment of patients with severe sepsis or septic shock. Approval was based on the results of a single randomized controlled trial in which the drug was given within 24 h of the patient’s first sepsis-related organ dysfunction; the 28-day survival rate was significantly higher among aPC recipients who were very sick (APACHE II score, ≥25) before infusion of the protein than among placebo-treated controls. Subsequent trials failed to show a benefit of aPC treatment in patients who were less sick (APACHE II score, <25) or in children, and, a decade after its licensure by the FDA, the drug was withdrawn from the market when a European trial failed to confirm its efficacy in adults with sepsis. Agents in ongoing or planned clinical trials include intravenous immunoglobulin, a polymyxin B hemofiltration column, and granulocyte-macrophage colony-stimulating factor, which has been reported to restore monocyte immunocompetence in patients with sepsis-associated immunosuppression.
A careful retrospective analysis found that the apparent efficacy of all sepsis therapeutics studied to date has been greatest among the patients at greatest risk of dying before treatment; conversely, use of many of these drugs has been associated with increased mortality rates among patients who are less ill. It is possible that neutralizing one of many different mediators may help patients who are very sick, whereas disrupting the mediator balance may be harmful to patients whose adaptive defense mechanisms are working well. This analysis suggests that if more aggressive early resuscitation improves survival rates among sicker patients, it will become more difficult to obtain additional benefit from other therapies; that is, if an intervention improves patients’ risk status, moving them into a “less severe illness” category, it will be harder to show that adding another agent to the therapeutic regimen is beneficial.
THE SURVIVING SEPSIS CAMPAIGN
An international consortium has advocated “bundling” of multiple therapeutic maneuvers into a unified algorithmic approach that will become the standard of care for severe sepsis. In theory, such a strategy would improve care by mandating measures that seem to bring maximal benefit, such as the rapid administration of appropriate antimicrobial therapy, fluids, and blood pressure support. Caution may be engendered by the fact that three of the key elements of the initial algorithm were eventually withdrawn for lack of evidence; moreover, the benefit of the current sepsis bundles has not been established in randomized controlled clinical trials.
PROGNOSIS
Approximately 20–35% of patients with severe sepsis and 40–60% of patients with septic shock die within 30 days. Others die within the ensuing 6 months. Late deaths often result from poorly controlled infection, immunosuppression, complications of intensive care, failure of multiple organs, or the patient’s underlying disease. Case–fatality rates are similar for culture-positive and culture-negative severe sepsis. Prognostic stratification systems such as APACHE II indicate that factoring in the patient’s age, underlying condition, and various physiologic variables can yield useful estimates of the risk of dying of severe sepsis. Age and prior health status are probably the most important risk factors (Fig. 325-1). In patients with no known preexisting morbidity, the case–fatality rate remains <10% until the fourth decade of life, after which it gradually increases to >35% in the very elderly. Death is significantly more likely in severely septic patients with preexisting illness. Septic shock is also a strong predictor of both short- and long-term mortality. Cognitive impairment may be significant in survivors, particularly those who are elderly.
FIGURE 325-1 Influence of age and prior health status on outcome of severe sepsis. With modern therapy, fewer than 10% of previously healthy young individuals (below 35 years of age) die with severe sepsis; the case–fatality rate then increases slowly through middle and old age. The most commonly identified etiologic agents in patients who die are Staphylococcus aureus, Streptococcus pyogenes, Streptococcus pneumoniae, and Neisseria meningitidis. Individuals with preexisting comorbidities are at greater risk of dying of severe sepsis at any age. The etiologic agents in these cases are likely to be S. aureus, Pseudomonas aeruginosa, various Enterobacteriaceae, enterococci, or fungi. (Adapted from DC Angus et al: Crit Care Med 29:1303, 2001.)
PREVENTION
Prevention offers the best opportunity to reduce morbidity and mortality from severe sepsis. In developed countries, most episodes of severe sepsis and septic shock are complications of nosocomial infections. These cases might be prevented by reducing the number of invasive procedures undertaken, by limiting the use (and duration of use) of indwelling vascular and bladder catheters, by reducing the incidence and duration of profound neutropenia (<500 neutrophils/μL), and by more aggressively treating localized nosocomial infections. Indiscriminate use of antimicrobial agents and glucocorticoids should be avoided, and optimal infection-control measures (Chap. 168) should be used. Studies indicate that 50–70% of patients who develop nosocomial severe sepsis or septic shock have experienced a less severe stage of the septic response on at least one previous day in the hospital. Research is needed to identify patients at increased risk and to develop adjunctive agents that can modulate the septic response before organ dysfunction or hypotension occurs.
326 |
Cardiogenic Shock and Pulmonary Edema |
Cardiogenic shock and pulmonary edema are life-threatening conditions that should be treated as medical emergencies. The most common joint etiology is severe left ventricular (LV) dysfunction that leads to pulmonary congestion and/or systemic hypoperfusion (Fig. 326-1). The pathophysiology of pulmonary edema and shock is discussed in Chaps. 47e and 324, respectively.
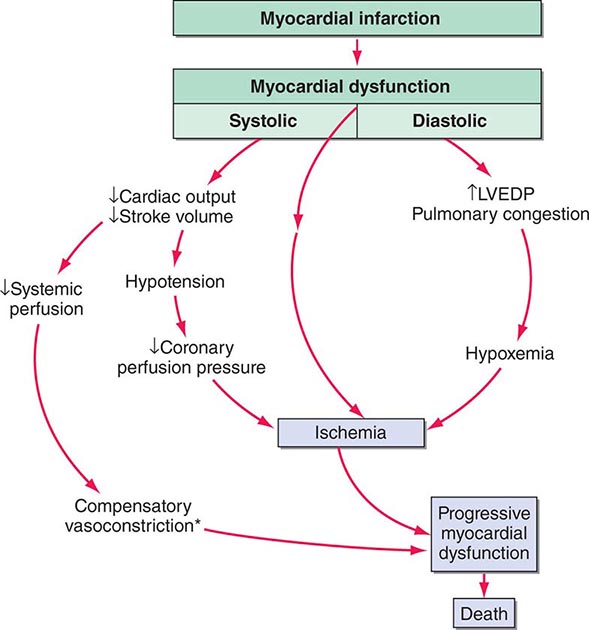
FIGURE 326-1 Pathophysiology of cardiogenic shock. Systolic and diastolic myocardial dysfunction results in a reduction in cardiac output and often pulmonary congestion. Systemic and coronary hypoperfusion occur, resulting in progressive ischemia. Although a number of compensatory mechanisms are activated in an attempt to support the circulation, these compensatory mechanisms may become maladaptive and produce a worsening of hemodynamics. *Release of inflammatory cytokines after myocardial infarction may lead to inducible nitric oxide expression, excess nitric oxide, and inappropriate vasodilation. This causes further reduction in systemic and coronary perfusion. A vicious spiral of progressive myocardial dysfunction occurs that ultimately results in death if it is not interrupted. LVEDP, left ventricular end-diastolic pressure. (From SM Hollenberg et al: Ann Intern Med 131:47, 1999.)
CARDIOGENIC SHOCK
Cardiogenic shock (CS) is characterized by systemic hypoperfusion due to severe depression of the cardiac index (<2.2 [L/min]/m2) and sustained systolic arterial hypotension (<90 mmHg) despite an elevated filling pressure (pulmonary capillary wedge pressure [PCWP] >18 mmHg). It is associated with in-hospital mortality rates >50%. The major causes of CS are listed in Table 326-1. Circulatory failure based on cardiac dysfunction may be caused by primary myocardial failure, most commonly secondary to acute myocardial infarction (MI) (Chap. 295), and less frequently by cardiomyopathy or myocarditis (Chap. 287), cardiac tamponade (Chap. 288), or critical valvular heart disease (Chap. 283).
|
ETIOLOGIES OF CARDIOGENIC SHOCK (CS)a AND CARDIOGENIC PULMONARY EDEMA |
aThe etiologies of CS are listed. Most of these can cause pulmonary edema instead of shock or pulmonary edema with CS. bThese cause CS but not pulmonary edema.
Abbreviations: LV, left ventricular; MI, myocardial infarction; MR, mitral regurgitation; RV, right ventricular; VSR, ventricular septal rupture.
Incidence The rate of CS complicating acute MI was 20% in the 1960s, stayed at ~8% for >20 years, but decreased to 5–7% in the first decade of this millennium largely due to increasing use of early reperfusion therapy for acute MI. Shock is more common with ST elevation MI (STEMI) than with non-ST elevation MI (Chap. 295).
LV failure accounts for ~80% of cases of CS complicating acute MI. Acute severe mitral regurgitation (MR), ventricular septal rupture (VSR), predominant right ventricular (RV) failure, and free wall rupture or tamponade account for the remainder.
Pathophysiology CS is characterized by a vicious circle in which depression of myocardial contractility, usually due to ischemia, results in reduced cardiac output and arterial blood pressure (BP), which result in hypoperfusion of the myocardium and further ischemia and depression of cardiac output (Fig. 326-1). Systolic myocardial dysfunction reduces stroke volume and, together with diastolic dysfunction, leads to elevated LV end-diastolic pressure and PCWP as well as to pulmonary congestion. Reduced coronary perfusion leads to worsening ischemia and progressive myocardial dysfunction and a rapid downward spiral, which, if uninterrupted, is often fatal. A systemic inflammatory response syndrome may accompany large infarctions and shock. Inflammatory cytokines, inducible nitric oxide synthase, and excess nitric oxide and peroxynitrite may contribute to the genesis of CS as they do to that of other forms of shock (Chap. 324). Lactic acidosis and hypoxemia from CS contribute to the vicious circle by worsening myocardial ischemia and hypotension. Severe acidosis reduces the efficacy of endogenous and exogenously administered catecholamines. Refractory sustained ventricular or atrial tachyarrhythmias can cause or exacerbate CS.
Patient Profile Older age, female sex, prior MI, diabetes, anterior MI location, and extensive coronary artery stenoses are associated with an increased risk of CS complicating MI. Shock associated with a first inferior MI should prompt a search for a mechanical cause. CS may rarely occur in the absence of significant stenosis, as seen in LV apical ballooning/Takotsubo’s cardiomyopathy.
Timing Shock is present on admission in only one-quarter of patients who develop CS complicating MI; one-quarter develop it rapidly thereafter, within 6 h of MI onset. Another quarter develop shock later on the first day. Subsequent onset of CS may be due to reinfarction, marked infarct expansion, or a mechanical complication.
Diagnosis Due to the unstable condition of these patients, supportive therapy must be initiated simultaneously with diagnostic evaluation (Fig. 326-2). A focused history and physical examination should be performed, blood specimens sent to the laboratory, and an electrocardiogram (ECG) and chest x-ray obtained.
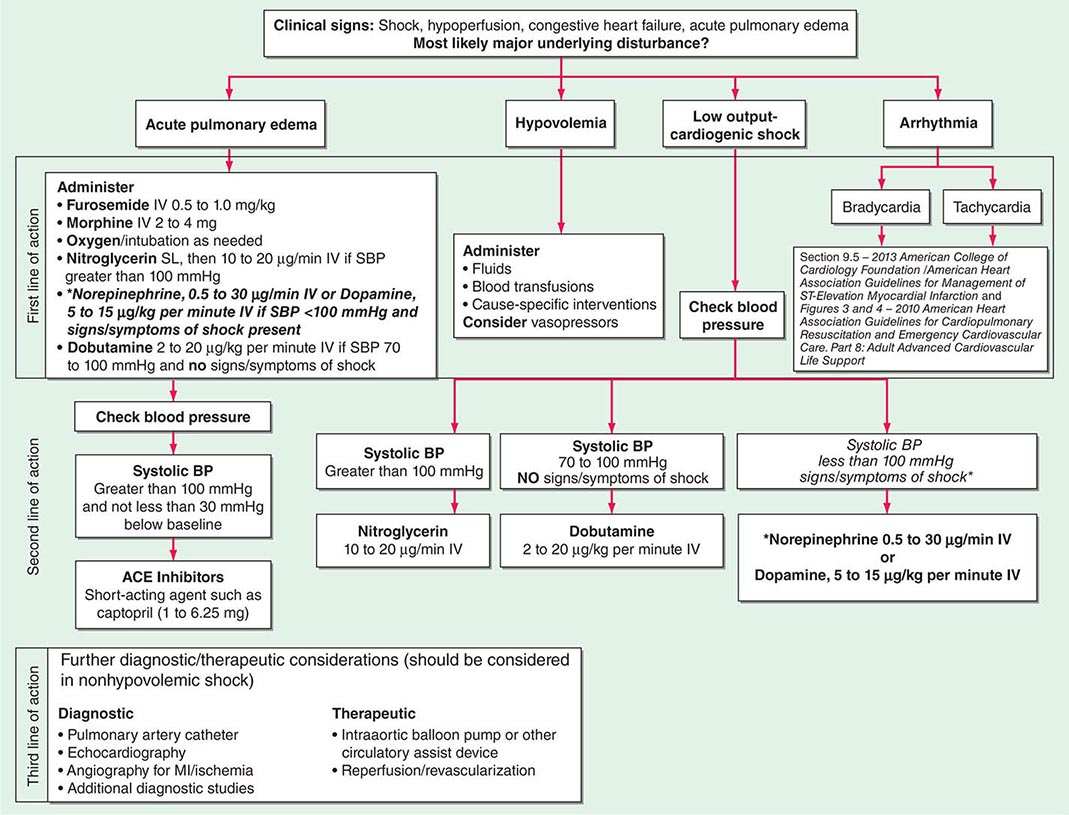
FIGURE 326-2 The emergency management of patients with cardiogenic shock, acute pulmonary edema, or both is outlined. *Furosemide: <0.5 mg/kg for new-onset acute pulmonary edema without hypervolemia; 1 mg/kg for acute on chronic volume overload, renal insufficiency. †For management of bradycardia and tachycardia, see Chaps. 274 and 276. Additional information can also be found in Section 9.5 of the 2013 American College of Cardiology Foundation/American Heart Association Guidelines for Management of ST-Elevation Myocardial Infarction and Figures 3 and 4 of the 2010 American Heart Association Guidelines for Cardiopulmonary Resuscitation and Emergency Cardiovascular Care. Part 8: Adult Advanced Cardiovascular Life Support. *Indicates modification from published guidelines. ACE, angiotensin-converting enzyme; BP, blood pressure; MI, myocardial infarction. (Modified from Guidelines 2000 for Cardiopulmonary Resuscitation and Emergency Cardiovascular Care. Part 7: The era of reperfusion: Section 1: Acute coronary syndromes [acute myocardial infarction]. The American Heart Association in collaboration with the International Liaison Committee on Resuscitation. Circulation 102:I172, 2000.)
Echocardiography is an invaluable diagnostic tool in patients with suspected CS.
CLINICAL FINDINGS Most patients have dyspnea and appear pale, apprehensive, and diaphoretic, and mental status may be altered. The pulse is typically weak and rapid, often in the range of 90–110 beats/min, or severe bradycardia due to high-grade heart block may be present. Systolic BP is reduced (<90 mmHg or ≥30 mmHg below baseline) with a narrow pulse pressure (<30 mmHg), but occasionally BP may be maintained by very high systemic vascular resistance. Tachypnea, Cheyne-Stokes respirations, and jugular venous distention may be present. There is typically a weak apical pulse and soft S1, and an S3 gallop may be audible. Acute, severe MR and VSR usually are associated with characteristic systolic murmurs (Chap. 295). Rales are audible in most patients with LV failure. Oliguria is common.
LABORATORY FINDINGS The white blood cell count is typically elevated with a left shift. Renal function is initially unchanged, but blood urea nitrogen and creatinine rise progressively. Hepatic transaminases may be markedly elevated due to liver hypoperfusion. The lactic acid level is elevated. Arterial blood gases usually demonstrate hypoxemia and anion gap metabolic acidosis, which may be compensated by respiratory alkalosis. Cardiac markers, creatine phosphokinase and its MB fraction, and troponins I and T are typically markedly elevated.
ELECTROCARDIOGRAM In CS due to acute MI with LV failure, Q waves and/or >2-mm ST elevation in multiple leads or left bundle branch block are usually present. More than one-half of all infarcts associated with shock are anterior. Global ischemia due to severe left main stenosis usually is accompanied by severe (e.g., >3 mm) ST depressions in multiple leads.
CHEST ROENTGENOGRAM The chest x-ray typically shows pulmonary vascular congestion and often pulmonary edema, but these findings may be absent in up to a third of patients. The heart size is usually normal when CS results from a first MI but is enlarged when it occurs in a patient with a previous MI.
ECHOCARDIOGRAM A two-dimensional echocardiogram with color-flow Doppler (Chap. 270e) should be obtained promptly in patients with suspected CS to help define its etiology. Doppler mapping demonstrates a left-to-right shunt in patients with VSR and the severity of MR when the latter is present. Proximal aortic dissection with aortic regurgitation or tamponade may be visualized, or evidence for pulmonary embolism may be obtained (Chap. 300).
PULMONARY ARTERY CATHETERIZATION The use of pulmonary artery (Swan-Ganz) catheters in patients with established or suspected CS is controversial (Chaps. 272 and 321). Their use is generally recommended for measurement of filling pressures and cardiac output to confirm the diagnosis and to optimize the use of IV fluids, inotropic agents, and vasopressors in persistent shock (Table 326-2). O2 saturation measurement from right atrial, RV, and pulmonary arterial blood samples can rule out a left-to-right shunt. In CS, low mixed venous O2 saturations and elevated arteriovenous (AV) O2 differences reflect low cardiac index and high fractional O2 extraction. However, when sepsis accompanies CS, AV O2 differences may not be elevated (Chap. 324). The PCWP is elevated. Use of sympathomimetic amines may return these measurements and the systemic BP to normal. Systemic vascular resistance may be low, normal, or elevated in CS. Equalization of right- and left-sided filling pressures (right atrial and PCWP) suggests cardiac tamponade as the cause of CS (Chap. 288).
|
HEMODYNAMIC PATTERNSa |
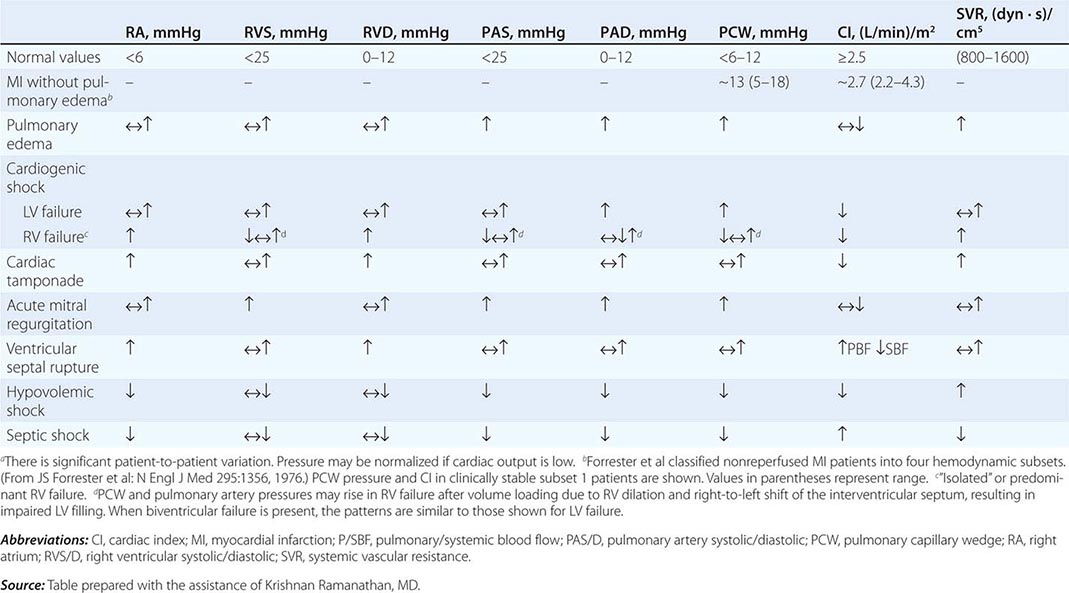
LEFT HEART CATHETERIZATION AND CORONARY ANGIOGRAPHY Measurement of LV pressure and definition of the coronary anatomy provide useful information and are indicated in most patients with CS complicating MI. Cardiac catheterization should be performed when there is a plan and capability for immediate coronary intervention (see below) or when a definitive diagnosis has not been made by other tests.
Prognosis Within this high-risk condition, there is a wide range of expected death rates based on age, severity of hemodynamic abnormalities, severity of the clinical manifestations of hypoperfusion, and the performance of early revascularization.
SHOCK SECONDARY TO RIGHT VENTRICULAR INFARCTION
Although transient hypotension is common in patients with RV infarction and inferior MI (Chap. 295), persistent CS due to RV failure accounts for only 3% of CS complicating MI. The salient features of RV shock are absence of pulmonary congestion, high right atrial pressure (which may be seen only after volume loading), RV dilation and dysfunction, only mildly or moderately depressed LV function, and predominance of single-vessel proximal right coronary artery occlusion. Management includes IV fluid administration to optimize right atrial pressure (10–15 mmHg); avoidance of excess fluids, which cause a shift of the interventricular septum into the LV; sympathomimetic amines; the early reestablishment of infarct-artery flow; and assist devices.
MITRAL REGURGITATION
(See also Chap. 295) Acute severe MR due to papillary muscle dysfunction and/or rupture may complicate MI and result in CS and/or pulmonary edema. This complication most often occurs on the first day, with a second peak several days later. The diagnosis is confirmed by echo-Doppler. Rapid stabilization with IABP is recommended, with administration of dobutamine as needed to raise cardiac output. Reducing the load against which the LV pumps (afterload) reduces the volume of regurgitant flow of blood into the left atrium. Mitral valve surgery is the definitive therapy and should be performed early in the course in suitable candidates.
VENTRICULAR SEPTAL RUPTURE
(See also Chap. 295) Echo-Doppler demonstrates shunting of blood from the left to the right ventricle and may visualize the opening in the interventricular septum. Timing and management are similar to those for MR with IABP support and surgical correction for suitable candidates.
FREE WALL RUPTURE
Myocardial rupture is a dramatic complication of STEMI that is most likely to occur during the first week after the onset of symptoms; its frequency increases with the age of the patient. The clinical presentation typically is a sudden loss of pulse, blood pressure, and consciousness but sinus rhythm on ECG (pulseless electrical activity) due to cardiac tamponade (Chap. 288). Free wall rupture may also result in CS due to subacute tamponade when the pericardium temporarily seals the rupture sites. Definitive surgical repair is required.
ACUTE FULMINANT MYOCARDITIS
(See also Chap. 287) Myocarditis can mimic acute MI with ST deviation or bundle branch block on the ECG and marked elevation of cardiac markers. Acute myocarditis causes CS in a small proportion of cases. These patients are typically younger than those with CS due to acute MI and often do not have typical ischemic chest pain. Echocardiography usually shows global LV dysfunction. Initial management is the same as for CS complicating acute MI (Fig. 326-2) but does not involve coronary revascularization. Endomyocardial biopsy is recommended to determine the diagnosis and need for immunosuppressives for entities such as giant cell myocarditis. Refractory CS can be managed with assist devices with or without ECMO.
PULMONARY EDEMA
The etiologies and pathophysiology of pulmonary edema are discussed in Chap. 47e.
Diagnosis Acute pulmonary edema usually presents with the rapid onset of dyspnea at rest, tachypnea, tachycardia, and severe hypoxemia. Crackles and wheezing due to alveolar flooding and airway compression from peribronchial cuffing may be audible. Release of endogenous catecholamines often causes hypertension.
It is often difficult to distinguish between cardiogenic and noncardiogenic causes of acute pulmonary edema. Echocardiography may identify systolic and diastolic ventricular dysfunction and valvular lesions. Electrocardiographic ST elevation and evolving Q waves are usually diagnostic of acute MI and should prompt immediate institution of MI protocols and coronary artery reperfusion therapy (Chap. 295). Brain natriuretic peptide levels, when substantially elevated, support heart failure as the etiology of acute dyspnea with pulmonary edema (Chap. 279).
The use of a Swan-Ganz catheter permits measurement of PCWP and helps differentiate high-pressure (cardiogenic) from normal-pressure (noncardiogenic) causes of pulmonary edema. Pulmonary artery catheterization is indicated when the etiology of the pulmonary edema is uncertain, when edema is refractory to therapy, or when it is accompanied by hypotension. Data derived from use of a catheter often alter the treatment plan, but no impact on mortality rates has been demonstrated.




