PART 16: Endocrinology and Metabolism
SECTION 1 |
ENDOCRINOLOGY |
399 |
Approach to the Patient with Endocrine Disorders |
The management of endocrine disorders requires a broad understanding of intermediary metabolism, reproductive physiology, bone metabolism, and growth. Accordingly, the practice of endocrinology is intimately linked to a conceptual framework for understanding hormone secretion, hormone action, and principles of feedback control (Chap. 400e). The endocrine system is evaluated primarily by measuring hormone concentrations, arming the clinician with valuable diagnostic information. Most disorders of the endocrine system are amenable to effective treatment once the correct diagnosis is determined. Endocrine deficiency disorders are treated with physiologic hormone replacement; hormone excess conditions, which usually are caused by benign glandular adenomas, are managed by removing tumors surgically or reducing hormone levels medically.
SCOPE OF ENDOCRINOLOGY
The specialty of endocrinology encompasses the study of glands and the hormones they produce. The term endocrine was coined by Starling to contrast the actions of hormones secreted internally (endocrine) with those secreted externally (exocrine) or into a lumen, such as the gastrointestinal tract. The term hormone, derived from a Greek phrase meaning “to set in motion,” aptly describes the dynamic actions of hormones as they elicit cellular responses and regulate physiologic processes through feedback mechanisms.
Unlike many other specialties in medicine, it is not possible to define endocrinology strictly along anatomic lines. The classic endocrine glands—pituitary, thyroid, parathyroid, pancreatic islets, adrenals, and gonads—communicate broadly with other organs through the nervous system, hormones, cytokines, and growth factors. In addition to its traditional synaptic functions, the brain produces a vast array of peptide hormones, and this has led to the discipline of neuroendocrinology. Through the production of hypothalamic releasing factors, the central nervous system (CNS) exerts a major regulatory influence over pituitary hormone secretion (Chap. 401e). The peripheral nervous system stimulates the adrenal medulla. The immune and endocrine systems are also intimately intertwined. The adrenal hormone cortisol is a powerful immunosuppressant. Cytokines and interleukins (ILs) have profound effects on the functions of the pituitary, adrenal, thyroid, and gonads. Common endocrine diseases such as autoimmune thyroid disease and type 1 diabetes mellitus are caused by dysregulation of immune surveillance and tolerance. Less common diseases such as polyglandular failure, Addison’s disease, and lymphocytic hypophysitis also have an immunologic basis.
The interdigitation of endocrinology with physiologic processes in other specialties sometimes blurs the role of hormones. For example, hormones play an important role in maintenance of blood pressure, intravascular volume, and peripheral resistance in the cardiovascular system. Vasoactive substances such as catecholamines, angiotensin II, endothelin, and nitric oxide are involved in dynamic changes of vascular tone in addition to their multiple roles in other tissues. The heart is the principal source of atrial natriuretic peptide, which acts in classic endocrine fashion to induce natriuresis at a distant target organ (the kidney). Erythropoietin, a traditional circulating hormone, is made in the kidney and stimulates erythropoiesis in bone marrow (Chap. 77). The kidney is also integrally involved in the renin-angiotensin axis (Chap. 406) and is a primary target of several hormones, including parathyroid hormone (PTH), mineralocorticoids, and vasopressin. The gastrointestinal tract produces a surprising number of peptide hormones, such as cholecystokinin, ghrelin, gastrin, secretin, and vasoactive intestinal peptide, among many others. Carcinoid and islet tumors can secrete excessive amounts of these hormones, leading to specific clinical syndromes (Chap. 113). Many of these gastrointestinal hormones are also produced in the CNS, where their functions are poorly understood. Adipose tissue produces leptin, which acts centrally to control appetite, along with adiponectin, resistin, and other hormones that regulate metabolism. As hormones such as inhibin, ghrelin, and leptin are discovered, they become integrated into the science and practice of medicine on the basis of their functional roles rather than their tissues of origin.
Characterization of hormone receptors frequently reveals unexpected relationships to factors in nonendocrine disciplines. The growth hormone (GH) and leptin receptors, for example, are members of the cytokine receptor family. The G protein–coupled receptors (GPCRs), which mediate the actions of many peptide hormones, are used in numerous physiologic processes, including vision, smell, and neurotransmission.
PATHOLOGIC MECHANISMS OF ENDOCRINE DISEASE
Endocrine diseases can be divided into three major types of conditions: (1) hormone excess, (2) hormone deficiency, and (3) hormone resistance (Table 399-1).
|
CAUSES OF ENDOCRINE DYSFUNCTION |
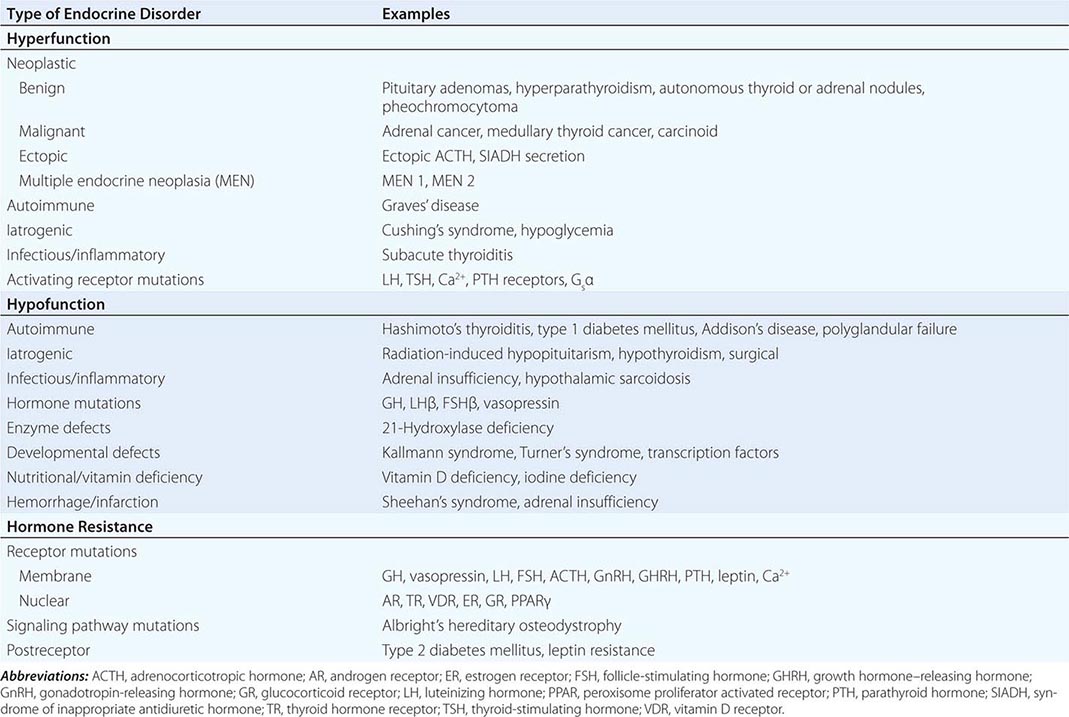
CAUSES OF HORMONE EXCESS
Syndromes of hormone excess can be caused by neoplastic growth of endocrine cells, autoimmune disorders, and excess hormone administration. Benign endocrine tumors, including parathyroid, pituitary, and adrenal adenomas, often retain the capacity to produce hormones, perhaps reflecting the fact that these tumors are relatively well differentiated. Many endocrine tumors exhibit subtle defects in their “set points” for feedback regulation. For example, in Cushing’s disease, impaired feedback inhibition of adrenocorticotropic hormone (ACTH) secretion is associated with autonomous function. However, the tumor cells are not completely resistant to feedback, as evidenced by ACTH suppression by higher doses of dexamethasone (e.g., high-dose dexamethasone test) (Chap. 406). Similar set point defects are also typical of parathyroid adenomas and autonomously functioning thyroid nodules.
The molecular basis of some endocrine tumors, such as the multiple endocrine neoplasia (MEN) syndromes (MEN 1, 2A, 2B), have provided important insights into tumorigenesis (Chap. 408). MEN 1 is characterized primarily by the triad of parathyroid, pancreatic islet, and pituitary tumors. MEN 2 predisposes to medullary thyroid carcinoma, pheochromocytoma, and hyperparathyroidism. The MEN1 gene, located on chromosome 11q13, encodes a putative tumor-suppressor gene, menin. Analogous to the paradigm first described for retinoblastoma, the affected individual inherits a mutant copy of the MEN1 gene, and tumorigenesis ensues after a somatic “second hit” leads to loss of function of the normal MEN1 gene (through deletion or point mutations).
In contrast to inactivation of a tumor-suppressor gene, as occurs in MEN 1 and most other inherited cancer syndromes, MEN 2 is caused by activating mutations in a single allele. In this case, activating mutations of the RET protooncogene, which encodes a receptor tyrosine kinase, leads to thyroid C cell hyperplasia in childhood before the development of medullary thyroid carcinoma. Elucidation of this pathogenic mechanism has allowed early genetic screening for RET mutations in individuals at risk for MEN 2, permitting identification of those who may benefit from prophylactic thyroidectomy and biochemical screening for pheochromocytoma and hyperparathyroidism.
Mutations that activate hormone receptor signaling have been identified in several GPCRs. For example, activating mutations of the luteinizing hormone (LH) receptor cause a dominantly transmitted form of male-limited precocious puberty, reflecting premature stimulation of testosterone synthesis in Leydig cells (Chap. 411). Activating mutations in these GPCRs are located predominantly in the transmembrane domains and induce receptor coupling to Gsα even in the absence of hormone. Consequently, adenylate cyclase is activated, and cyclic adenosine monophosphate (AMP) levels increase in a manner that mimics hormone action. A similar phenomenon results from activating mutations in Gsα. When these mutations occur early in development, they cause McCune-Albright syndrome. When they occur only in somatotropes, the activating Gsα mutations cause GH-secreting tumors and acromegaly (Chap. 403).
In autoimmune Graves’ disease, antibody interactions with the thyroid-stimulating hormone (TSH) receptor mimic TSH action, leading to hormone overproduction (Chap. 405). Analogous to the effects of activating mutations of the TSH receptor, these stimulating autoantibodies induce conformational changes that release the receptor from a constrained state, thereby triggering receptor coupling to G proteins.
CAUSES OF HORMONE DEFICIENCY
Most examples of hormone deficiency states can be attributed to glandular destruction caused by autoimmunity, surgery, infection, inflammation, infarction, hemorrhage, or tumor infiltration (Table 399-1). Autoimmune damage to the thyroid gland (Hashimoto’s thyroiditis) and pancreatic islet β cells (type 1 diabetes mellitus) is a prevalent cause of endocrine disease. Mutations in a number of hormones, hormone receptors, transcription factors, enzymes, and channels can also lead to hormone deficiencies.
HORMONE RESISTANCE
Most severe hormone resistance syndromes are due to inherited defects in membrane receptors, nuclear receptors, or the pathways that transduce receptor signals. These disorders are characterized by defective hormone action despite the presence of increased hormone levels. In complete androgen resistance, for example, mutations in the androgen receptor result in a female phenotypic appearance in genetic (XY) males, even though LH and testosterone levels are increased (Chap. 408). In addition to these relatively rare genetic disorders, more common acquired forms of functional hormone resistance include insulin resistance in type 2 diabetes mellitus, leptin resistance in obesity, and GH resistance in catabolic states. The pathogenesis of functional resistance involves receptor downregulation and postreceptor desensitization of signaling pathways; functional forms of resistance are generally reversible.
CLINICAL EVALUATION OF ENDOCRINE DISORDERS
Because most glands are relatively inaccessible, the physical examination usually focuses on the manifestations of hormone excess or deficiency as well as direct examination of palpable glands, such as the thyroid and gonads. For these reasons, it is important to evaluate patients in the context of their presenting symptoms, review of systems, family and social history, and exposure to medications that may affect the endocrine system. Astute clinical skills are required to detect subtle symptoms and signs suggestive of underlying endocrine disease. For example, a patient with Cushing’s syndrome may manifest specific findings, such as central fat redistribution, striae, and proximal muscle weakness, in addition to features seen commonly in the general population, such as obesity, plethora, hypertension, and glucose intolerance. Similarly, the insidious onset of hypothyroidism—with mental slowing, fatigue, dry skin, and other features—can be difficult to distinguish from similar, nonspecific findings in the general population. Clinical judgment that is based on knowledge of disease prevalence and pathophysiology is required to decide when to embark on more extensive evaluation of these disorders. Laboratory testing plays an essential role in endocrinology by allowing quantitative assessment of hormone levels and dynamics. Radiologic imaging tests such as computed tomography (CT) scan, magnetic resonance imaging (MRI), thyroid scan, and ultrasound are also used for the diagnosis of endocrine disorders. However, these tests generally are employed only after a hormonal abnormality has been established by biochemical testing.
HORMONE MEASUREMENTS AND ENDOCRINE TESTING
Immunoassays are the most important diagnostic tool in endocrinology, as they allow sensitive, specific, and quantitative determination of steady-state and dynamic changes in hormone concentrations. Immunoassays use antibodies to detect specific hormones. For many peptide hormones, these measurements are now configured to use two different antibodies to increase binding affinity and specificity. There are many variations of these assays; a common format involves using one antibody to capture the antigen (hormone) onto an immobilized surface and a second antibody, coupled to a chemiluminescent (immunochemiluminescent assay [ICMA]) or radioactive (immunoradiometric assay [IRMA]) signal, to detect the antigen. These assays are sensitive enough to detect plasma hormone concentrations in the picomolar to nanomolar range, and they can readily distinguish structurally related proteins, such as PTH from PTH-related peptide (PTHrP). A variety of other techniques are used to measure specific hormones, including mass spectroscopy, various forms of chromatography, and enzymatic methods; bioassays are now rarely used.
Most hormone measurements are based on plasma or serum samples. However, urinary hormone determinations remain useful for the evaluation of some conditions. Urinary collections over 24 h provide an integrated assessment of the production of a hormone or metabolite, many of which vary during the day. It is important to assure complete collections of 24-h urine samples; simultaneous measurement of creatinine provides an internal control for the adequacy of collection and can be used to normalize some hormone measurements. A 24-h urine free cortisol measurement largely reflects the amount of unbound cortisol, thus providing a reasonable index of biologically available hormone. Other commonly used urine determinations include 17-hydroxycorticosteroids, 17-ketosteroids, vanillylmandelic acid, metanephrine, catecholamines, 5-hydroxyindoleacetic acid, and calcium.
The value of quantitative hormone measurements lies in their correct interpretation in a clinical context. The normal range for most hormones is relatively broad, often varying by a factor of two- to tenfold. The normal ranges for many hormones are sex- and age-specific. Thus, using the correct normative database is an essential part of interpreting hormone tests. The pulsatile nature of hormones and factors that can affect their secretion, such as sleep, meals, and medications, must also be considered. Cortisol values increase fivefold between midnight and dawn; reproductive hormone levels vary dramatically during the female menstrual cycle.
For many endocrine systems, much information can be gained from basal hormone testing, particularly when different components of an endocrine axis are assessed simultaneously. For example, low testosterone and elevated LH levels suggest a primary gonadal problem, whereas a hypothalamic-pituitary disorder is likely if both LH and testosterone are low. Because TSH is a sensitive indicator of thyroid function, it is generally recommended as a first-line test for thyroid disorders. An elevated TSH level is almost always the result of primary hypothyroidism, whereas a low TSH is most often caused by thyrotoxicosis. These predictions can be confirmed by determining the free thyroxine level. In the less common circumstance when free thyroxine and TSH are both low, it is important to consider secondary hypopituitarism caused by hypothalamic-pituitary disease. Elevated calcium and PTH levels suggest hyperparathyroidism, whereas PTH is suppressed in hypercalcemia caused by malignancy or granulomatous diseases. A suppressed ACTH in the setting of hypercortisolemia, or increased urine free cortisol, is seen with hyperfunctioning adrenal adenomas.
It is not uncommon, however, for baseline hormone levels associated with pathologic endocrine conditions to overlap with the normal range. In this circumstance, dynamic testing is useful to separate the two groups further. There are a multitude of dynamic endocrine tests, but all are based on principles of feedback regulation, and most responses can be rationalized based on principles that govern the regulation of endocrine axes. Suppression tests are used in the setting of suspected endocrine hyperfunction. An example is the dexamethasone suppression test used to evaluate Cushing’s syndrome (Chaps. 403 and 406). Stimulation tests generally are used to assess endocrine hypofunction. The ACTH stimulation test, for example, is used to assess the adrenal gland response in patients with suspected adrenal insufficiency. Other stimulation tests use hypothalamic-releasing factors such as corticotropin-releasing hormone (CRH) and growth hormone–releasing hormone (GHRH) to evaluate pituitary hormone reserve (Chap. 403). Insulin-induced hypoglycemia also evokes pituitary ACTH and GH responses. Stimulation tests based on reduction or inhibition of endogenous hormones are now used infrequently. Examples include metyrapone inhibition of cortisol synthesis and clomiphene inhibition of estrogen feedback.
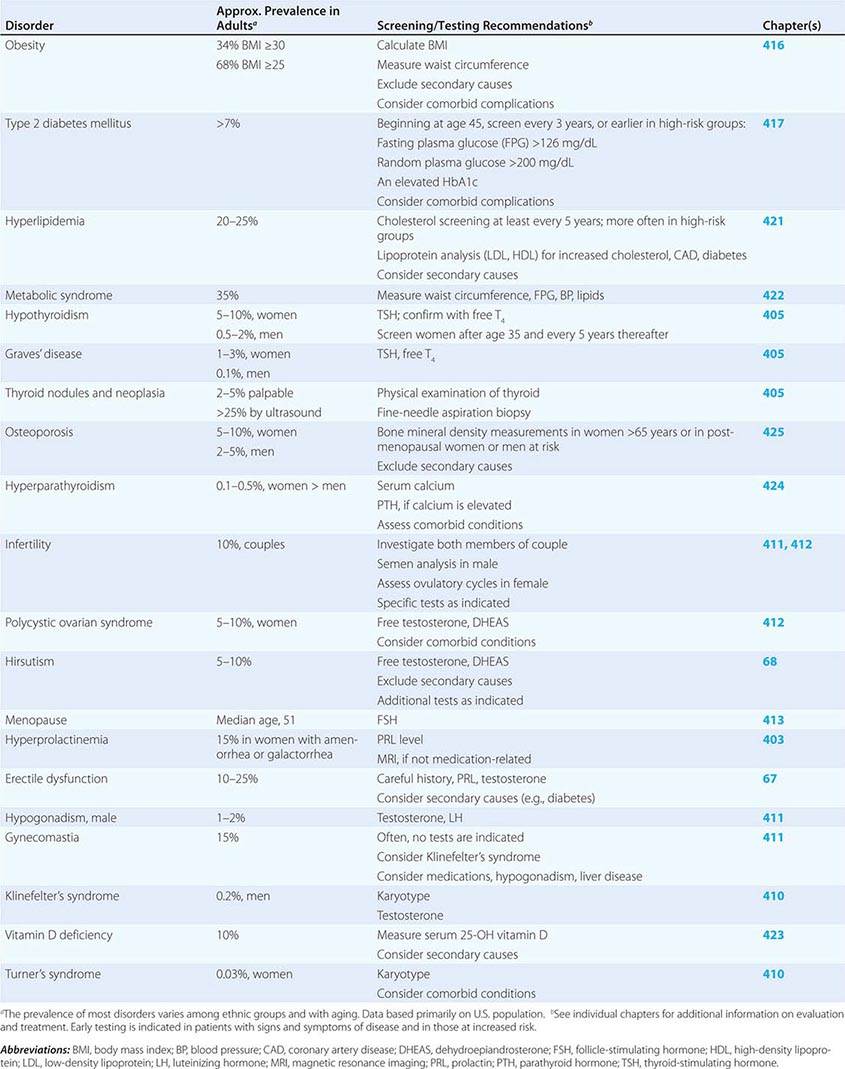
SCREENING AND ASSESSMENT OF COMMON ENDOCRINE DISORDERS
Many endocrine disorders are prevalent in the adult population (Table 399-2) and can be diagnosed and managed by general internists, family practitioners, or other primary health care providers. The high prevalence and clinical impact of certain endocrine diseases justifies vigilance for features of these disorders during routine physical examinations; laboratory screening is indicated in selected high-risk populations.
|
EXAMPLES OF PREVALENT ENDOCRINE AND METABOLIC DISORDERS IN THE ADULT |
400e |
Mechanisms of Hormone Action |
CLASSES OF HORMONES
Hormones can be divided into five major types: (1) amino acid derivatives such as dopamine, catecholamine, and thyroid hormone; (2) small neuropeptides such as gonadotropin-releasing hormone (GnRH), thyrotropin-releasing hormone (TRH), somatostatin, and vasopressin; (3) large proteins such as insulin, luteinizing hormone (LH), and parathyroid hormone (PTH); (4) steroid hormones such as cortisol and estrogen that are synthesized from cholesterol-based precursors; and (5) vitamin derivatives such as retinoids (vitamin A) and vitamin D. A variety of peptide growth factors, most of which act locally, share actions with hormones. As a rule, amino acid derivatives and peptide hormones interact with cell-surface membrane receptors. Steroids, thyroid hormones, vitamin D, and retinoids are lipid-soluble and interact with intracellular nuclear receptors, although many also interact with membrane receptors or intracellular signaling proteins as well.
HORMONE AND RECEPTOR FAMILIES
Hormones and receptors can be grouped into families, reflecting structural similarities and evolutionary origins (Table 400e-1). The evolution of these families generates diverse but highly selective pathways of hormone action. Recognition of these relationships has proven useful for extrapolating information gleaned from one hormone or receptor to other family members.
|
EXAMPLES OF MEMBRANE RECEPTOR FAMILIES AND SIGNALING PATHWAYS |
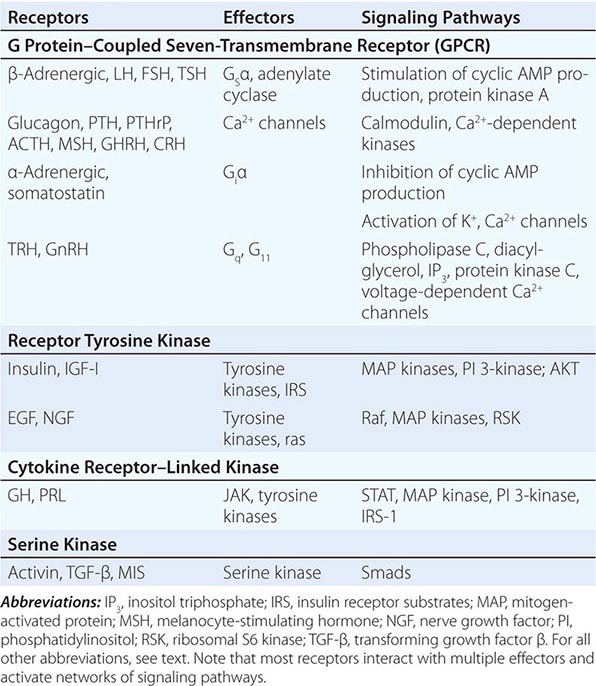
The glycoprotein hormone family, consisting of thyroid-stimulating hormone (TSH), follicle-stimulating hormone (FSH), LH, and human chorionic gonadotropin (hCG), illustrates many features of related hormones. The glycoprotein hormones are heterodimers that share the α subunit in common; the β subunits are distinct and confer specific biologic actions. The overall three-dimensional architecture of the β subunits is similar, reflecting the locations of conserved disulfide bonds that restrain protein conformation. The cloning of the β-subunit genes from multiple species suggests that this family arose from a common ancestral gene, probably by gene duplication and subsequent divergence to evolve new biologic functions.
As hormone families enlarge and diverge, their receptors must co-evolve to derive new biologic functions. Related G protein–coupled receptors (GPCRs), for example, have evolved for each of the glycoprotein hormones. These receptors are structurally similar, and each is coupled predominantly to the Gsα signaling pathway. However, there is minimal overlap of hormone binding. For example, TSH binds with high specificity to the TSH receptor but interacts minimally with the LH or FSH receptors. Nonetheless, there can be subtle physiologic consequences of hormone cross-reactivity with other receptors. Very high levels of hCG during pregnancy stimulate the TSH receptor and increase thyroid hormone levels, resulting in a compensatory decrease in TSH.
Insulin and insulin-like growth factor I (IGF-I) and IGF-II have structural similarities that are most apparent when precursor forms of the proteins are compared. In contrast to the high degree of specificity seen with the glycoprotein hormones, there is moderate cross-talk among the members of the insulin/IGF family. High concentrations of an IGF-II precursor produced by certain tumors (e.g., sarcomas) can cause hypoglycemia, partly because of binding to insulin and IGF-I receptors (Chap. 424). High concentrations of insulin also bind to the IGF-I receptor, perhaps accounting for some of the clinical manifestations seen in conditions with chronic hyperinsulinemia.
Another important example of receptor cross-talk is seen with PTH and parathyroid hormone–related peptide (PTHrP) (Chap. 424). PTH is produced by the parathyroid glands, whereas PTHrP is expressed at high levels during development and by a variety of tumors (Chap. 121). These hormones have amino acid sequence similarity, particularly in their amino-terminal regions. Both hormones bind to a single PTH receptor that is expressed in bone and kidney. Hypercalcemia and hypophosphatemia therefore may result from excessive production of either hormone, making it difficult to distinguish hyperparathyroidism from hypercalcemia of malignancy solely on the basis of serum chemistries. However, sensitive and specific assays for PTH and PTHrP now allow these disorders to be distinguished more readily.
Based on their specificities for DNA binding sites, the nuclear receptor family can be subdivided into type 1 receptors (glucocorticoid receptor, mineralocorticoid receptor, androgen receptor, estrogen receptor, progesterone receptor) that bind steroids and type 2 receptors (thyroid hormone receptor, vitamin D receptor, retinoic acid receptor, peroxisome proliferator activated receptor) that bind thyroid hormone, vitamin D, retinoic acid, or lipid derivatives. Certain functional domains in nuclear receptors, such as the zinc finger DNA-binding domains, are highly conserved. However, selective amino acid differences within this domain confer DNA sequence specificity. The hormone-binding domains are more variable, providing great diversity in the array of small molecules that bind to different nuclear receptors. With few exceptions, hormone binding is highly specific for a single type of nuclear receptor. One exception involves the glucocorticoid and mineralocorticoid receptors. Because the mineralocorticoid receptor also binds glucocorticoids with high affinity, an enzyme (11β-hydroxysteroid dehydrogenase) in renal tubular cells inactivates glucocorticoids, allowing selective responses to mineralocorticoids such as aldosterone. However, when very high glucocorticoid concentrations occur, as in Cushing’s syndrome, the glucocorticoid degradation pathway becomes saturated, allowing excessive cortisol levels to exert mineralocorticoid effects (sodium retention, potassium wasting). This phenomenon is particularly pronounced in ectopic adrenocorticotropic hormone (ACTH) syndromes (Chap. 406). Another example of relaxed nuclear receptor specificity involves the estrogen receptor, which can bind an array of compounds, some of which have little apparent structural similarity to the high-affinity ligand estradiol. This feature of the estrogen receptor makes it susceptible to activation by “environmental estrogens” such as resveratrol, octylphenol, and many other aromatic hydrocarbons. However, this lack of specificity provides an opportunity to synthesize a remarkable series of clinically useful antagonists (e.g., tamoxifen) and selective estrogen response modulators (SERMs) such as raloxifene. These compounds generate distinct conformations that alter receptor interactions with components of the transcription machinery (see below), thereby conferring their unique actions.
HORMONE SYNTHESIS AND PROCESSING
The synthesis of peptide hormones and their receptors occurs through a classic pathway of gene expression: transcription → mRNA → protein → posttranslational protein processing → intracellular sorting, followed by membrane integration or secretion (Chap. 82).
Many hormones are embedded within larger precursor polypeptides that are proteolytically processed to yield the biologically active hormone. Examples include proopiomelanocortin (POMC) → ACTH; proglucagon → glucagon; proinsulin → insulin; and pro-PTH → PTH, among others. In many cases, such as POMC and proglucagon, these precursors generate multiple biologically active peptides. It is provocative that hormone precursors are typically inactive, presumably adding an additional level of regulatory control. Prohormone conversion occurs not only for peptide hormones but also for certain steroids (testosterone → dihydrotestosterone) and thyroid hormone (T4 → T3).
Peptide precursor processing is intimately linked to intracellular sorting pathways that transport proteins to appropriate vesicles and enzymes, resulting in specific cleavage steps, followed by protein folding and translocation to secretory vesicles. Hormones destined for secretion are translocated across the endoplasmic reticulum under the guidance of an amino-terminal signal sequence that subsequently is cleaved. Cell-surface receptors are inserted into the membrane via short segments of hydrophobic amino acids that remain embedded within the lipid bilayer. During translocation through the Golgi and endoplasmic reticulum, hormones and receptors are subject to a variety of posttranslational modifications, such as glycosylation and phosphorylation, which can alter protein conformation, modify circulating half-life, and alter biologic activity.
Synthesis of most steroid hormones is based on modifications of the precursor, cholesterol. Multiple regulated enzymatic steps are required for the synthesis of testosterone (Chap. 411), estradiol (Chap. 412), cortisol (Chap. 406), and vitamin D (Chap. 423). This large number of synthetic steps predisposes to multiple genetic and acquired disorders of steroidogenesis.
Endocrine genes contain regulatory DNA elements similar to those found in many other genes, but their exquisite control by hormones reflects the presence of specific hormone response elements. For example, the TSH genes are repressed directly by thyroid hormones acting through the thyroid hormone receptor (TR), a member of the nuclear receptor family. Steroidogenic enzyme gene expression requires specific transcription factors, such as steroidogenic factor-1 (SF-1), acting in conjunction with signals transmitted by trophic hormones (e.g., ACTH or LH). For some hormones, substantial regulation occurs at the level of translational efficiency. Insulin biosynthesis, although it requires ongoing gene transcription, is regulated primarily at the translational and secretory levels in response to elevated levels of glucose or amino acids.
HORMONE SECRETION, TRANSPORT, AND DEGRADATION
The level of a hormone is determined by its rate of secretion and its circulating half-life. After protein processing, peptide hormones (e.g., GnRH, insulin, growth hormone [GH]) are stored in secretory granules. As these granules mature, they are poised beneath the plasma membrane for imminent release into the circulation. In most instances, the stimulus for hormone secretion is a releasing factor or neural signal that induces rapid changes in intracellular calcium concentrations, leading to secretory granule fusion with the plasma membrane and release of its contents into the extracellular environment and bloodstream. Steroid hormones, in contrast, diffuse into the circulation as they are synthesized. Thus, their secretory rates are closely aligned with rates of synthesis. For example, ACTH and LH induce steroidogenesis by stimulating the activity of the steroidogenic acute regulatory (StAR) protein (transports cholesterol into the mitochondrion) along with other rate-limiting steps (e.g., cholesterol side-chain cleavage enzyme, CYP11A1) in the steroidogenic pathway.
Hormone transport and degradation dictate the rapidity with which a hormonal signal decays. Some hormone signals are evanescent (e.g., somatostatin), whereas others are longer-lived (e.g., TSH). Because somatostatin exerts effects in virtually every tissue, a short half-life allows its concentrations and actions to be controlled locally. Structural modifications that impair somatostatin degradation have been useful for generating long-acting therapeutic analogues such as octreotide (Chap. 403). In contrast, the actions of TSH are highly specific for the thyroid gland. Its prolonged half-life accounts for relatively constant serum levels even though TSH is secreted in discrete pulses.
An understanding of circulating hormone half-life is important for achieving physiologic hormone replacement, as the frequency of dosing and the time required to reach steady state are intimately linked to rates of hormone decay. T4, for example, has a circulating half-life of 7 days. Consequently, >1 month is required to reach a new steady state, and single daily doses are sufficient to achieve constant hormone levels. T3, in contrast, has a half-life of 1 day. Its administration is associated with more dynamic serum levels, and it must be administered two to three times per day. Similarly, synthetic glucocorticoids vary widely in their half-lives; those with longer half-lives (e.g., dexamethasone) are associated with greater suppression of the hypothalamic-pituitary-adrenal (HPA) axis. Most protein hormones (e.g., ACTH, GH, prolactin [PRL], PTH, LH) have relatively short half-lives (<20 min), leading to sharp peaks of secretion and decay. The only accurate way to profile the pulse frequency and amplitude of these hormones is to measure levels in frequently sampled blood (every 10 min or less) over long durations (8–24 h). Because this is not practical in a clinical setting, an alternative strategy is to pool three to four samples drawn at about 30-min intervals, or interpret the results in the context of a relatively wide normal range. Rapid hormone decay is useful in certain clinical settings. For example, the short half-life of PTH allows the use of intraoperative PTH determinations to confirm successful removal of an adenoma. This is particularly valuable diagnostically when there is a possibility of multicentric disease or parathyroid hyperplasia, as occurs with multiple endocrine neoplasia (MEN) or renal insufficiency.
Many hormones circulate in association with serum-binding proteins. Examples include (1) T4 and T3 binding to thyroxine-binding globulin (TBG), albumin, and thyroxine-binding prealbumin (TBPA); (2) cortisol binding to cortisol-binding globulin (CBG); (3) androgen and estrogen binding to sex hormone–binding globulin (SHBG); (4) IGF-I and -II binding to multiple IGF-binding proteins (IGFBPs); (5) GH interactions with GH-binding protein (GHBP), a circulating fragment of the GH receptor extracellular domain; and (6) activin binding to follistatin. These interactions provide a hormonal reservoir, prevent otherwise rapid degradation of unbound hormones, restrict hormone access to certain sites (e.g., IGFBPs), and modulate the unbound, or “free,” hormone concentrations. Although a variety of binding protein abnormalities have been identified, most have little clinical consequence aside from creating diagnostic problems. For example, TBG deficiency can reduce total thyroid hormone levels greatly but the free concentrations of T4 and T3 remain normal. Liver disease and certain medications can also influence binding protein levels (e.g., estrogen increases TBG) or cause displacement of hormones from binding proteins (e.g., salsalate displaces T4 from TBG). In general, only unbound hormone is available to interact with receptors and thus elicit a biologic response. Short-term perturbations in binding proteins change the free hormone concentration, which in turn induces compensatory adaptations through feedback loops. SHBG changes in women are an exception to this self-correcting mechanism. When SHBG decreases because of insulin resistance or androgen excess, the unbound testosterone concentration is increased, potentially leading to hirsutism (Chap. 68). The increased unbound testosterone level does not result in an adequate compensatory feedback correction because estrogen, not testosterone, is the primary regulator of the reproductive axis.
An additional exception to the unbound hormone hypothesis involves megalin, a member of the low-density lipoprotein (LDL) receptor family that serves as an endocytotic receptor for carrier-bound vitamins A and D and SHBG-bound androgens and estrogens. After internalization, the carrier proteins are degraded in lysosomes and release their bound ligands within the cells. Membrane transporters have also been identified for thyroid hormones.
Hormone degradation can be an important mechanism for regulating concentrations locally. As noted above, 11β-hydroxysteroid dehydrogenase inactivates glucocorticoids in renal tubular cells, preventing actions through the mineralocorticoid receptor. Thyroid hormone deiodinases convert T4 to T3 and can inactivate T3. During development, degradation of retinoic acid by Cyp26b1 prevents primordial germ cells in the male from entering meiosis, as occurs in the female ovary.
HORMONE ACTION THROUGH RECEPTORS
Receptors for hormones are divided into two major classes: membrane and nuclear. Membrane receptors primarily bind peptide hormones and catecholamines. Nuclear receptors bind small molecules that can diffuse across the cell membrane, such as steroids and vitamin D. Certain general principles apply to hormone-receptor interactions regardless of the class of receptor. Hormones bind to receptors with specificity and an affinity that generally coincides with the dynamic range of circulating hormone concentrations. Low concentrations of free hormone (usually 10–12 to 10–9 M) rapidly associate and dissociate from receptors in a bimolecular reaction such that the occupancy of the receptor at any given moment is a function of hormone concentration and the receptor’s affinity for the hormone. Receptor numbers vary greatly in different target tissues, providing one of the major determinants of specific tissue responses to circulating hormones. For example, ACTH receptors are located almost exclusively in the adrenal cortex, and FSH receptors are found predominantly in the gonads. In contrast, insulin and TRs are widely distributed, reflecting the need for metabolic responses in all tissues.
MEMBRANE RECEPTORS
Membrane receptors for hormones can be divided into several major groups: (1) seven transmembrane GPCRs, (2) tyrosine kinase receptors, (3) cytokine receptors, and (4) serine kinase receptors (Fig. 400e-1). The seven transmembrane GPCR family binds a remarkable array of hormones, including large proteins (e.g., LH, PTH), small peptides (e.g., TRH, somatostatin), catecholamines (epinephrine, dopamine), and even minerals (e.g., calcium). The extracellular domains of GPCRs vary widely in size and are the major binding site for large hormones. The transmembrane-spanning regions are composed of hydrophobic α-helical domains that traverse the lipid bilayer. Like some channels, these domains are thought to circularize and form a hydrophobic pocket into which certain small ligands fit. Hormone binding induces conformational changes in these domains, transducing structural changes to the intracellular domain, which is a docking site for G proteins.
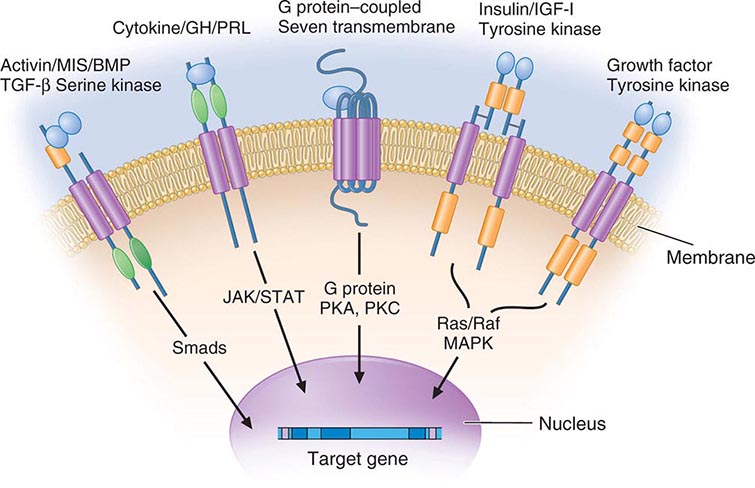
FIGURE 400e-1 Membrane receptor signaling. MAPK, mitogen-activated protein kinase; PKA, C, protein kinase A, C; TGF, transforming growth factor. For other abbreviations, see text.
The large family of G proteins, so named because they bind guanine nucleotides (guanosine triphosphate [GTP], guanosine diphosphate [GDP]), provides great diversity for coupling receptors to different signaling pathways. G proteins form a heterotrimeric complex that is composed of various α and βγ subunits. The α subunit contains the guanine nucleotide–binding site and hydrolyzes GTP → GDP. The βγ subunits are tightly associated and modulate the activity of the α subunit as well as mediating their own effector signaling pathways. G protein activity is regulated by a cycle that involves GTP hydrolysis and dynamic interactions between the α and αβ subunits. Hormone binding to the receptor induces GDP dissociation, allowing Gα to bind GTP and dissociate from the αβ complex. Under these conditions, the Gα subunit is activated and mediates signal transduction through various enzymes, such as adenylate cyclase and phospholipase C. GTP hydrolysis to GDP allows reassociation with the βγ subunits and restores the inactive state. As described below, a variety of endocrinopathies result from G protein mutations or from mutations in receptors that modify their interactions with G proteins. G proteins interact with other cellular proteins, including kinases, channels, G protein–coupled receptor kinases (GRKs), and arrestins, that mediate signaling as well as receptor desensitization and recycling.
The tyrosine kinase receptors transduce signals for insulin and a variety of growth factors, such as IGF-I, epidermal growth factor (EGF), nerve growth factor, platelet-derived growth factor, and fibroblast growth factor. The cysteine-rich extracellular ligand-binding domains contain growth factor binding sites. After ligand binding, this class of receptors undergoes autophosphorylation, inducing interactions with intracellular adaptor proteins such as Shc and insulin receptor substrates (IRS). In the case of the insulin receptor, multiple kinases are activated, including the Raf-Ras-MAPK and the Akt/protein kinase B pathways. The tyrosine kinase receptors play a prominent role in cell growth and differentiation as well as in intermediary metabolism.
The GH and PRL receptors belong to the cytokine receptor family. Analogous to the tyrosine kinase receptors, ligand binding induces receptor interaction with intracellular kinases—the Janus kinases (JAKs), which phosphorylate members of the signal transduction and activators of transcription (STAT) family—as well as with other signaling pathways (Ras, PI3-K, MAPK). The activated STAT proteins translocate to the nucleus and stimulate expression of target genes.
The serine kinase receptors mediate the actions of activins, transforming growth factor β, müllerian-inhibiting substance (MIS, also known as anti-müllerian hormone, AMH), and bone morphogenic proteins (BMPs). This family of receptors (consisting of type I and II subunits) signals through proteins termed smads (fusion of terms for Caenorhabditis elegans sma + mammalian mad). Like the STAT proteins, the smads serve a dual role of transducing the receptor signal and acting as transcription factors. The pleomorphic actions of these growth factors dictate that they act primarily in a local (paracrine or autocrine) manner. Binding proteins such as follistatin (which binds activin and other members of this family) function to inactivate the growth factors and restrict their distribution.
NUCLEAR RECEPTORS
The family of nuclear receptors has grown to nearly 100 members, many of which are still classified as orphan receptors because their ligands, if they exist, have not been identified (Fig. 400e-2). Otherwise, most nuclear receptors are classified on the basis of their ligands. Although all nuclear receptors ultimately act to increase or decrease gene transcription, some (e.g., glucocorticoid receptor) reside primarily in the cytoplasm, whereas others (e.g., TR) are located in the nucleus. After ligand binding, the cytoplasmically localized receptors translocate to the nucleus. There is growing evidence that certain nuclear receptors (e.g., glucocorticoid, estrogen) can also act at the membrane or in the cytoplasm to activate or repress signal transduction pathways, providing a mechanism for cross-talk between membrane and nuclear receptors.
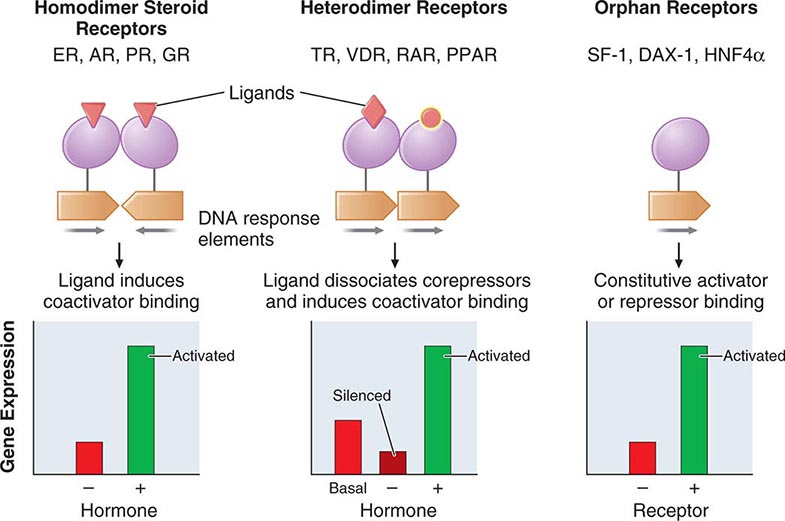
FIGURE 400e-2 Nuclear receptor signaling. AR, androgen receptor; DAX, dosage-sensitive sex-reversal, adrenal hypoplasia congenita, X-chromosome; ER, estrogen receptor; GR, glucocorticoid receptor; HNF4α, hepatic nuclear factor 4α; PPAR, peroxisome proliferator activated receptor; PR, progesterone receptor; RAR, retinoic acid receptor; SF-1, steroidogenic factor-1; TR, thyroid hormone receptor; VDR, vitamin D receptor.
The structures of nuclear receptors have been studied extensively, including by x-ray crystallography. The DNA binding domain, consisting of two zinc fingers, contacts specific DNA recognition sequences in target genes. Most nuclear receptors bind to DNA as dimers. Consequently, each monomer recognizes an individual DNA motif, referred to as a “half-site.” The steroid receptors, including the glucocorticoid, estrogen, progesterone, and androgen receptors, bind to DNA as homodimers. Consistent with this twofold symmetry, their DNA recognition half-sites are palindromic. The thyroid, retinoid, peroxisome proliferator activated, and vitamin D receptors bind to DNA preferentially as heterodimers in combination with retinoid × receptors (RXRs). Their DNA half-sites are typically arranged as direct repeats.
The carboxy-terminal hormone-binding domain mediates transcriptional control. For type II receptors such as TR and retinoic acid receptor (RAR), co-repressor proteins bind to the receptor in the absence of ligand and silence gene transcription. Hormone binding induces conformational changes, triggering the release of co-repressors and inducing the recruitment of coactivators that stimulate transcription. Thus, these receptors are capable of mediating dramatic changes in the level of gene activity. Certain disease states are associated with defective regulation of these events. For example, mutations in the TR prevent co-repressor dissociation, resulting in an autosomal dominant form of hormone resistance (Chap. 405). In promyelocytic leukemia, fusion of RARα to other nuclear proteins causes aberrant gene silencing that prevents normal cellular differentiation. Treatment with retinoic acid reverses this repression and allows cellular differentiation and apoptosis to occur. Most type 1 steroid receptors interact weakly with co-repressors, but ligand binding still induces interactions with an array of coactivators. X-ray crystallography shows that various SERMs induce distinct estrogen receptor conformations. The tissue-specific responses caused by these agents in breast, bone, and uterus appear to reflect distinct interactions with coactivators. The receptor-coactivator complex stimulates gene transcription by several pathways, including (1) recruitment of enzymes (histone acetyl transferases) that modify chromatin structure, (2) interactions with additional transcription factors on the target gene, and (3) direct interactions with components of the general transcription apparatus to enhance the rate of RNA polymerase II–mediated transcription. Studies of nuclear receptor-mediated transcription show that these are dynamic events that involve relatively rapid (e.g., 30–60 min) cycling of transcription complexes on any specific target gene.
FUNCTIONS OF HORMONES
The functions of individual hormones are described in detail in subsequent chapters. Nevertheless, it is useful to illustrate how most biologic responses require integration of several different hormone pathways. The physiologic functions of hormones can be divided into three general areas: (1) growth and differentiation, (2) maintenance of homeostasis, and (3) reproduction.
GROWTH
Multiple hormones and nutritional factors mediate the complex phenomenon of growth (Chap. 401e). Short stature may be caused by GH deficiency, hypothyroidism, Cushing’s syndrome, precocious puberty, malnutrition, chronic illness, or genetic abnormalities that affect the epiphyseal growth plates (e.g., FGFR3 and SHOX mutations). Many factors (GH, IGF-I, thyroid hormones) stimulate growth, whereas others (sex steroids) lead to epiphyseal closure. Understanding these hormonal interactions is important in the diagnosis and management of growth disorders. For example, delaying exposure to high levels of sex steroids may enhance the efficacy of GH treatment.
MAINTENANCE OF HOMEOSTASIS
Although virtually all hormones affect homeostasis, the most important among them are the following:
1. Thyroid hormone—controls about 25% of basal metabolism in most tissues
2. Cortisol—exerts a permissive action for many hormones in addition to its own direct effects
3. PTH—regulates calcium and phosphorus levels
4. Vasopressin—regulates serum osmolality by controlling renal free-water clearance
5. Mineralocorticoids—control vascular volume and serum electrolyte (Na+, K+) concentrations
6. Insulin—maintains euglycemia in the fed and fasted states
The defense against hypoglycemia is an impressive example of integrated hormone action (Chap. 420). In response to the fasting state and falling blood glucose, insulin secretion is suppressed, resulting in decreased glucose uptake and enhanced glycogenolysis, lipolysis, proteolysis, and gluconeogenesis to mobilize fuel sources. If hypoglycemia develops (usually from insulin administration or sulfonylureas), an orchestrated counterregulatory response occurs—glucagon and epinephrine rapidly stimulate glycogenolysis and gluconeogenesis, whereas GH and cortisol act over several hours to raise glucose levels and antagonize insulin action.
Although free-water clearance is controlled primarily by vasopressin, cortisol and thyroid hormone are also important for facilitating renal tubular responses to vasopressin (Chap. 404). PTH and vitamin D function in an interdependent manner to control calcium metabolism (Chap. 423). PTH stimulates renal synthesis of 1,25-dihydroxyvitamin D, which increases calcium absorption in the gastrointestinal tract and enhances PTH action in bone. Increased calcium, along with vitamin D, feeds back to suppress PTH, thus maintaining calcium balance.
Depending on the severity of a specific stress and whether it is acute or chronic, multiple endocrine and cytokine pathways are activated to mount an appropriate physiologic response. In severe acute stress such as trauma or shock, the sympathetic nervous system is activated and catecholamines are released, leading to increased cardiac output and a primed musculoskeletal system. Catecholamines also increase mean blood pressure and stimulate glucose production. Multiple stress-induced pathways converge on the hypothalamus, stimulating several hormones, including vasopressin and corticotropin-releasing hormone (CRH). These hormones, in addition to cytokines (tumor necrosis factor α, interleukin [IL] 2, IL-6) increase ACTH and GH production. ACTH stimulates the adrenal gland, increasing cortisol, which in turn helps sustain blood pressure and dampen the inflammatory response. Increased vasopressin acts to conserve free water.
REPRODUCTION
The stages of reproduction include (1) sex determination during fetal development (Chap. 410); (2) sexual maturation during puberty (Chaps. 411 and 412); (3) conception, pregnancy, lactation, and child rearing (Chap. 412); and (4) cessation of reproductive capability at menopause (Chap. 413). Each of these stages involves an orchestrated interplay of multiple hormones, a phenomenon well illustrated by the dynamic hormonal changes that occur during each 28-day menstrual cycle. In the early follicular phase, pulsatile secretion of LH and FSH stimulates the progressive maturation of the ovarian follicle. This results in gradually increasing estrogen and progesterone levels, leading to enhanced pituitary sensitivity to GnRH, which, when combined with accelerated GnRH secretion, triggers the LH surge and rupture of the mature follicle. Inhibin, a protein produced by the granulosa cells, enhances follicular growth and feeds back to the pituitary to selectively suppress FSH without affecting LH. Growth factors such as EGF and IGF-I modulate follicular responsiveness to gonadotropins. Vascular endothelial growth factor and prostaglandins play a role in follicle vascularization and rupture.
During pregnancy, the increased production of prolactin, in combination with placentally derived steroids (e.g., estrogen and progesterone), prepares the breast for lactation. Estrogens induce the production of progesterone receptors, allowing for increased responsiveness to progesterone. In addition to these and other hormones involved in lactation, the nervous system and oxytocin mediate the suckling response and milk release.
HORMONAL FEEDBACK REGULATORY SYSTEMS
Feedback control, both negative and positive, is a fundamental feature of endocrine systems. Each of the major hypothalamic-pituitary-hormone axes is governed by negative feedback, a process that maintains hormone levels within a relatively narrow range (Chap. 401e). Examples of hypothalamic-pituitary negative feedback include (1) thyroid hormones on the TRH-TSH axis, (2) cortisol on the CRH-ACTH axis, (3) gonadal steroids on the GnRH-LH/FSH axis, and (4) IGF-I on the growth hormone–releasing hormone (GHRH)-GH axis (Fig. 400e-3). These regulatory loops include both positive (e.g., TRH, TSH) and negative (e.g., T4, T3) components, allowing for exquisite control of hormone levels. As an example, a small reduction of thyroid hormone triggers a rapid increase of TRH and TSH secretion, resulting in thyroid gland stimulation and increased thyroid hormone production. When thyroid hormone reaches a normal level, it feeds back to suppress TRH and TSH, and a new steady state is attained. Feedback regulation also occurs for endocrine systems that do not involve the pituitary gland, such as calcium feedback on PTH, glucose inhibition of insulin secretion, and leptin feedback on the hypothalamus. An understanding of feedback regulation provides important insights into endocrine testing paradigms (see below).
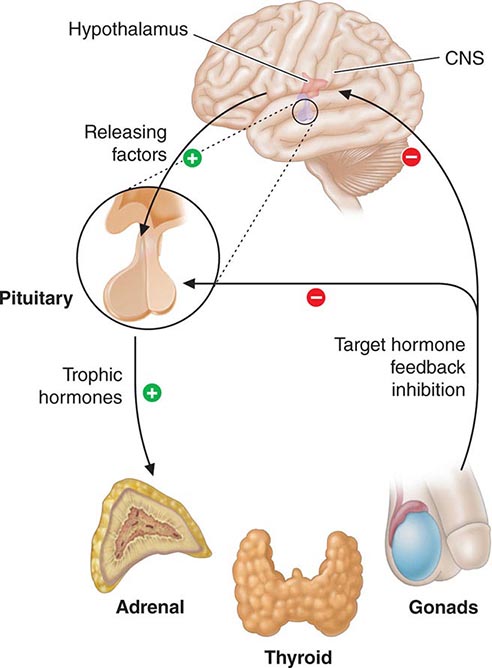
FIGURE 400e-3 Feedback regulation of endocrine axes. CNS, central nervous system.
Positive feedback control also occurs but is not well understood. The primary example is estrogen-mediated stimulation of the midcycle LH surge. Although chronic low levels of estrogen are inhibitory, gradually rising estrogen levels stimulate LH secretion. This effect, which is illustrative of an endocrine rhythm (see below), involves activation of the hypothalamic GnRH pulse generator. In addition, estrogen-primed gonadotropes are extraordinarily sensitive to GnRH, leading to amplification of LH release.
PARACRINE AND AUTOCRINE CONTROL
The previously mentioned examples of feedback control involve classic endocrine pathways in which hormones are released by one gland and act on a distant target gland. However, local regulatory systems, often involving growth factors, are increasingly recognized. Paracrine regulation refers to factors released by one cell that act on an adjacent cell in the same tissue. For example, somatostatin secretion by pancreatic islet δ cells inhibits insulin secretion from nearby β cells. Autocrine regulation describes the action of a factor on the same cell from which it is produced. IGF-I acts on many cells that produce it, including chondrocytes, breast epithelium, and gonadal cells. Unlike endocrine actions, paracrine and autocrine control are difficult to document because local growth factor concentrations cannot be measured readily.
Anatomic relationships of glandular systems also greatly influence hormonal exposure: the physical organization of islet cells enhances their intercellular communication; the portal vasculature of the hypothalamic-pituitary system exposes the pituitary to high concentrations of hypothalamic releasing factors; testicular seminiferous tubules gain exposure to high testosterone levels produced by the interdigitated Leydig cells; the pancreas receives nutrient information and local exposure to peptide hormones (incretins) from the gastrointestinal tract; and the liver is the proximal target of insulin action because of portal drainage from the pancreas.
HORMONAL RHYTHMS
The feedback regulatory systems described above are superimposed on hormonal rhythms that are used for adaptation to the environment. Seasonal changes, the daily occurrence of the light-dark cycle, sleep, meals, and stress are examples of the many environmental events that affect hormonal rhythms. The menstrual cycle is repeated on average every 28 days, reflecting the time required to follicular maturation and ovulation (Chap. 412). Essentially all pituitary hormone rhythms are entrained to sleep and to the circadian cycle, generating reproducible patterns that are repeated approximately every 24 h. The HPA axis, for example, exhibits characteristic peaks of ACTH and cortisol production in the early morning, with a nadir during the night. Recognition of these rhythms is important for endocrine testing and treatment. Patients with Cushing’s syndrome characteristically exhibit increased midnight cortisol levels compared with normal individuals (Chap. 406). In contrast, morning cortisol levels are similar in these groups, as cortisol is normally high at this time of day in normal individuals. The HPA axis is more susceptible to suppression by glucocorticoids administered at night as they blunt the early-morning rise of ACTH. Understanding these rhythms allows glucocorticoid replacement that mimics diurnal production by administering larger doses in the morning than in the afternoon. Disrupted sleep rhythms can alter hormonal regulation. For example, sleep deprivation causes mild insulin resistance, food craving, and hypertension, which are reversible, at least in the short term. Emerging evidence indicates that circadian clock pathways not only regulate sleep-wake cycles but also play important roles in virtually every cell type. For example, tissue-specific deletion of clock genes alters rhythms and levels of gene expression, as well as metabolic responses in liver, adipose, and other tissues.
Other endocrine rhythms occur on a more rapid time scale. Many peptide hormones are secreted in discrete bursts every few hours. LH and FSH secretion are exquisitely sensitive to GnRH pulse frequency. Intermittent pulses of GnRH are required to maintain pituitary sensitivity, whereas continuous exposure to GnRH causes pituitary gonadotrope desensitization. This feature of the hypothalamic-pituitary-gonadotrope axis forms the basis for using long-acting GnRH agonists to treat central precocious puberty or to decrease testosterone levels in the management of prostate cancer. It is important to be aware of the pulsatile nature of hormone secretion and the rhythmic patterns of hormone production in relating serum hormone measurements to normal values. For some hormones, integrated markers have been developed to circumvent hormonal fluctuations. Examples include 24-h urine collections for cortisol, IGF-I as a biologic marker of GH action, and HbA1c as an index of long-term (weeks to months) blood glucose control.
Often, one must interpret endocrine data only in the context of other hormones. For example, PTH levels typically are assessed in combination with serum calcium concentrations. A high serum calcium level in association with elevated PTH is suggestive of hyperparathyroidism, whereas a suppressed PTH in this situation is more likely to be caused by hypercalcemia of malignancy or other causes of hypercalcemia. Similarly, TSH should be elevated when T4 and T3 concentrations are low, reflecting reduced feedback inhibition. When this is not the case, it is important to consider secondary hypothyroidism, which is caused by a defect at the level of the pituitary.
401e |
Anterior Pituitary: Physiology of Pituitary Hormones |
The anterior pituitary often is referred to as the “master gland” because, together with the hypothalamus, it orchestrates the complex regulatory functions of many other endocrine glands. The anterior pituitary gland produces six major hormones: (1) prolactin (PRL), (2) growth hormone (GH), (3) adrenocorticotropic hormone (ACTH), (4) luteinizing hormone (LH), (5) follicle-stimulating hormone (FSH), and (6) thyroid-stimulating hormone (TSH) (Table 401e-1). Pituitary hormones are secreted in a pulsatile manner, reflecting stimulation by an array of specific hypothalamic releasing factors. Each of these pituitary hormones elicits specific responses in peripheral target tissues. The hormonal products of those peripheral glands, in turn, exert feedback control at the level of the hypothalamus and pituitary to modulate pituitary function (Fig. 401e-1). Pituitary tumors cause characteristic hormone excess syndromes. Hormone deficiency may be inherited or acquired. Fortunately, there are efficacious treatments for many pituitary hormone excess and deficiency syndromes. Nonetheless, these diagnoses are often elusive; this emphasizes the importance of recognizing subtle clinical manifestations and performing the correct laboratory diagnostic tests. For discussion of disorders of the posterior pituitary, or neurohypophysis, see Chap. 404.
|
ANTERIOR PITUITARY HORMONE EXPRESSION AND REGULATION |
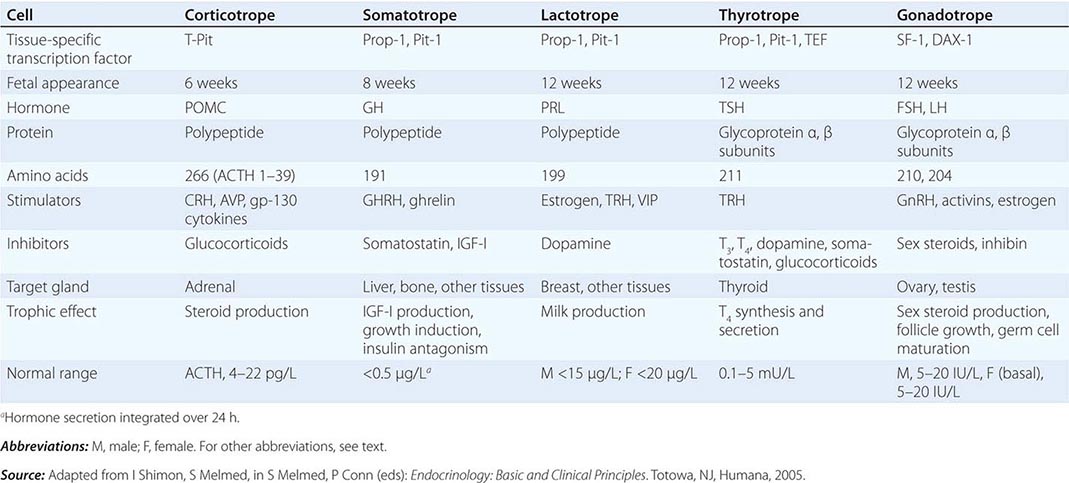
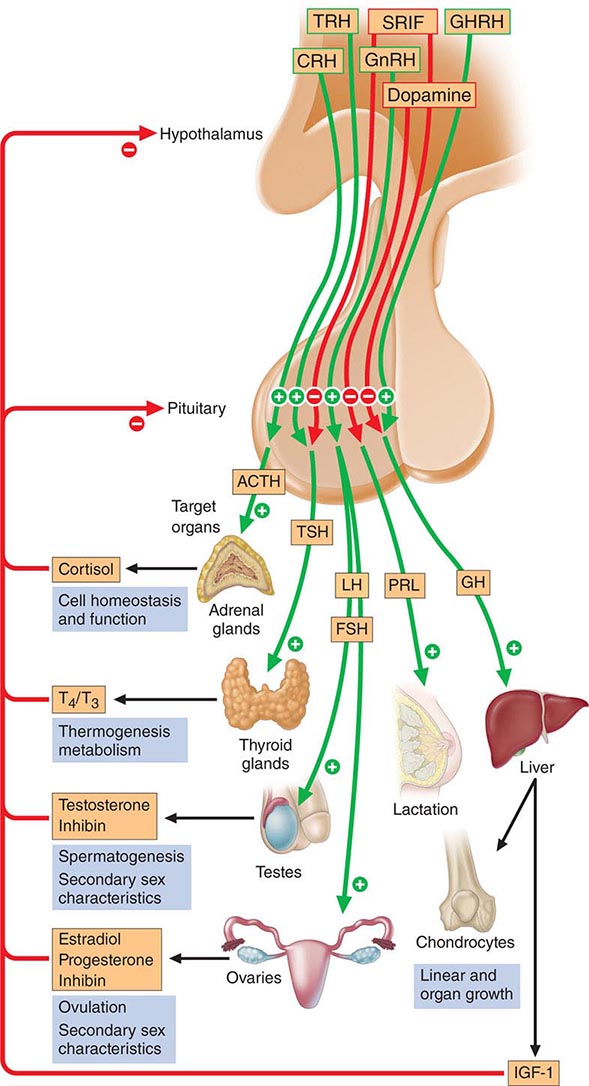
FIGURE 401e-1 Diagram of pituitary axes. Hypothalamic hormones regulate anterior pituitary trophic hormones that in turn determine target gland secretion. Peripheral hormones feed back to regulate hypothalamic and pituitary hormones. For abbreviations, see text.
ANATOMY AND DEVELOPMENT
ANATOMY
The pituitary gland weighs ~600 mg and is located within the sella turcica ventral to the diaphragma sella; it consists of anatomically and functionally distinct anterior and posterior lobes. The bony sella is contiguous to vascular and neurologic structures, including the cavernous sinuses, cranial nerves, and optic chiasm. Thus, expanding intrasellar pathologic processes may have significant central mass effects in addition to their endocrinologic impact.
Hypothalamic neural cells synthesize specific releasing and inhibiting hormones that are secreted directly into the portal vessels of the pituitary stalk. Blood supply of the pituitary gland comes from the superior and inferior hypophyseal arteries (Fig. 401e-2). The hypothalamic-pituitary portal plexus provides the major blood source for the anterior pituitary, allowing reliable transmission of hypothalamic peptide pulses without significant systemic dilution; consequently, pituitary cells are exposed to releasing or inhibiting factors and in turn release their hormones as discrete pulses into the systemic circulation (Fig. 401e-3).
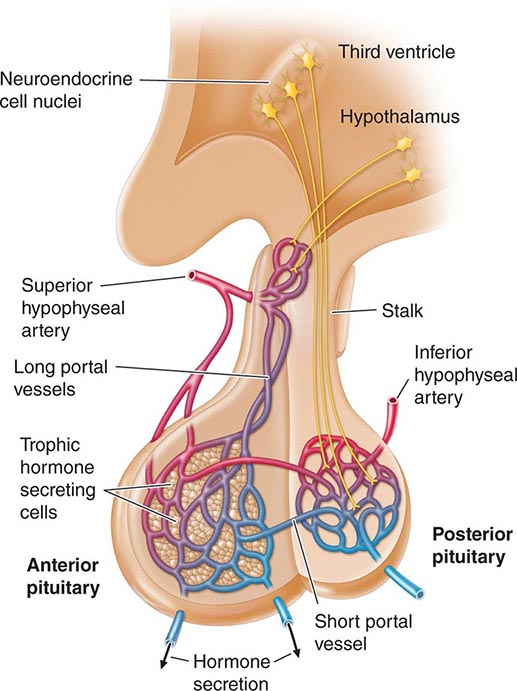
FIGURE 401e-2 Diagram of hypothalamic-pituitary vasculature. The hypothalamic nuclei produce hormones that traverse the portal system and impinge on anterior pituitary cells to regulate pituitary hormone secretion. Posterior pituitary hormones are derived from direct neural extensions.
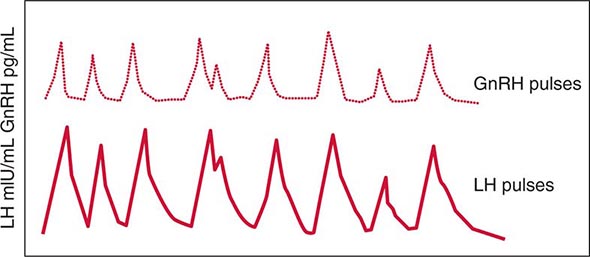
FIGURE 401e-3 Hypothalamic gonadotropin-releasing hormone (GnRH) pulses induce secretory pulses of luteinizing hormone (LH).
The posterior pituitary is supplied by the inferior hypophyseal arteries. In contrast to the anterior pituitary, the posterior lobe is directly innervated by hypothalamic neurons (supraopticohypophyseal and tuberohypophyseal nerve tracts) via the pituitary stalk (Chap. 404). Thus, posterior pituitary production of vasopressin (antidiuretic hormone [ADH]) and oxytocin is particularly sensitive to neuronal damage by lesions that affect the pituitary stalk or hypothalamus.
PITUITARY DEVELOPMENT
The embryonic differentiation and maturation of anterior pituitary cells have been elucidated in considerable detail. Pituitary development from Rathke’s pouch involves a complex interplay of lineage-specific transcription factors expressed in pluripotent precursor cells and gradients of locally produced growth factors (Table 401e-1). The transcription factor Prop-1 induces pituitary development of Pit-1-specific lineages as well as gonadotropes. The transcription factor Pit-1 determines cell-specific expression of GH, PRL, and TSH in somatotropes, lactotropes, and thyrotropes. Expression of high levels of estrogen receptors in cells that contain Pit-1 favors PRL expression, whereas thyrotrope embryonic factor (TEF) induces TSH expression. Pit-1 binds to GH, PRL, and TSH gene regulatory elements as well as to recognition sites on its own promoter, providing a mechanism for maintaining specific pituitary hormone phenotypic stability. Gonadotrope cell development is further defined by the cell-specific expression of the nuclear receptors steroidogenic factor (SF-1) and d osage-sensitive sex reversal, a drenal hypoplasia critical region, on chromosome X, gene 1 (DAX-1). Development of corticotrope cells, which express the proopiomelanocortin (POMC) gene, requires the T-Pit transcription factor. Abnormalities of pituitary development caused by mutations of Pit-1, Prop-1, SF-1, DAX-1, and T-Pit result in a rare, selective or combined pituitary hormone deficit syndromes.
ANTERIOR PITUITARY HORMONES
Each anterior pituitary hormone is under unique control, and each exhibits highly specific normal and dysregulated secretory characteristics.
PROLACTIN
Synthesis PRL consists of 198 amino acids and has a molecular mass of 21,500 kDa; it is weakly homologous to GH and human placental lactogen (hPL), reflecting the duplication and divergence of a common GH-PRL-hPL precursor gene. PRL is synthesized in lactotropes, which constitute about 20% of anterior pituitary cells. Lactotropes and somatotropes are derived from a common precursor cell that may give rise to a tumor that secretes both PRL and GH. Marked lactotrope cell hyperplasia develops during pregnancy and the first few months of lactation. These transient functional changes in the lactotrope population are induced by estrogen.
Secretion Normal adult serum PRL levels are about 10–25 μg/L in women and 10–20 μg/L in men. PRL secretion is pulsatile, with the highest secretory peaks occurring during rapid eye movement sleep. Peak serum PRL levels (up to 30 μg/L) occur between 4:00 and 6:00 A.M. The circulating half-life of PRL is about 50 min.
PRL is unique among the pituitary hormones in that the predominant central control mechanism is inhibitory, reflecting dopamine-mediated suppression of PRL release. This regulatory pathway accounts for the spontaneous PRL hypersecretion that occurs with pituitary stalk section, often a consequence of compressive mass lesions at the skull base. Pituitary dopamine type 2 (D2) receptors mediate inhibition of PRL synthesis and secretion. Targeted disruption (gene knockout) of the murine D2 receptor in mice results in hyperprolactinemia and lactotrope proliferation. As discussed below, dopamine agonists play a central role in the management of hyperprolactinemic disorders.
Thyrotropin-releasing hormone (TRH) (pyro Glu-His-Pro-NH2) is a hypothalamic tripeptide that elicits PRL release within 15–30 min after intravenous injection. The physiologic relevance of TRH for PRL regulation is unclear, and it appears primarily to regulate TSH (Chap. 405). Vasoactive intestinal peptide (VIP) also induces PRL release, whereas glucocorticoids and thyroid hormone weakly suppress PRL secretion.
Serum PRL levels rise transiently after exercise, meals, sexual intercourse, minor surgical procedures, general anesthesia, chest wall injury, acute myocardial infarction, and other forms of acute stress. PRL levels increase markedly (about tenfold) during pregnancy and decline rapidly within 2 weeks of parturition. If breast-feeding is initiated, basal PRL levels remain elevated; suckling stimulates transient reflex increases in PRL levels that last for about 30–45 min. Breast suckling activates neural afferent pathways in the hypothalamus that induce PRL release. With time, suckling-induced responses diminish and interfeeding PRL levels return to normal.
Action The PRL receptor is a member of the type I cytokine receptor family that also includes GH and interleukin (IL) 6 receptors. Ligand binding induces receptor dimerization and intracellular signaling by Janus kinase (JAK), which stimulates translocation of the signal transduction and activators of transcription (STAT) family to activate target genes. In the breast, the lobuloalveolar epithelium proliferates in response to PRL, placental lactogens, estrogen, progesterone, and local paracrine growth factors, including insulin-like growth factor I (IGF-I).
PRL acts to induce and maintain lactation, decrease reproductive function, and suppress sexual drive. These functions are geared toward ensuring that maternal lactation is sustained and not interrupted by pregnancy. PRL inhibits reproductive function by suppressing hypothalamic gonadotropin-releasing hormone (GnRH) and pituitary gonadotropin secretion and by impairing gonadal steroidogenesis in both women and men. In the ovary, PRL blocks folliculogenesis and inhibits granulosa cell aromatase activity, leading to hypoestrogenism and anovulation. PRL also has a luteolytic effect, generating a shortened, or inadequate, luteal phase of the menstrual cycle. In men, attenuated LH secretion leads to low testosterone levels and decreased spermatogenesis. These hormonal changes decrease libido and reduce fertility in patients with hyperprolactinemia.
GROWTH HORMONE
Synthesis GH is the most abundant anterior pituitary hormone, and GH-secreting somatotrope cells constitute up to 50% of the total anterior pituitary cell population. Mammosomatotrope cells, which coexpress PRL with GH, can be identified by using double immunostaining techniques. Somatotrope development and GH transcription are determined by expression of the cell-specific Pit-1 nuclear transcription factor. Five distinct genes encode GH and related proteins. The pituitary GH gene (hGH-N) produces two alternatively spliced products that give rise to 22-kDa GH (191 amino acids) and a less abundant 20-kDa GH molecule with similar biologic activity. Placental syncytiotrophoblast cells express a GH variant (hGH-V) gene; the related hormone human chorionic somatotropin (HCS) is expressed by distinct members of the gene cluster.
Secretion GH secretion is controlled by complex hypothalamic and peripheral factors. GH-releasing hormone (GHRH) is a 44-amino-acid hypothalamic peptide that stimulates GH synthesis and release. Ghrelin, an octanoylated gastric-derived peptide, and synthetic agonists of the GHS-R induce GHRH and also directly stimulate GH release. Somatostatin (somatotropin-release inhibiting factor [SRIF]) is synthesized in the medial preoptic area of the hypothalamus and inhibits GH secretion. GHRH is secreted in discrete spikes that elicit GH pulses, whereas SRIF sets basal GH secretory tone. SRIF also is expressed in many extrahypothalamic tissues, including the central nervous system (CNS), gastrointestinal tract, and pancreas, where it also acts to inhibit islet hormone secretion. IGF-I, the peripheral target hormone for GH, feeds back to inhibit GH; estrogen induces GH, whereas chronic glucocorticoid excess suppresses GH release.
Surface receptors on the somatotrope regulate GH synthesis and secretion. The GHRH receptor is a G protein–coupled receptor (GPCR) that signals through the intracellular cyclic AMP pathway to stimulate somatotrope cell proliferation as well as GH production. Inactivating mutations of the GHRH receptor cause profound dwarfism. A distinct surface receptor for ghrelin, the gastric-derived GH secretagogue, is expressed in both the hypothalamus and pituitary. Somatostatin binds to five distinct receptor subtypes (SSTR1 to SSTR5); SSTR2 and SSTR5 subtypes preferentially suppress GH (and TSH) secretion.
GH secretion is pulsatile, with highest peak levels occurring at night, generally correlating with sleep onset. GH secretory rates decline markedly with age so that hormone levels in middle age are about 15% of pubertal levels. These changes are paralleled by an age-related decline in lean muscle mass. GH secretion is also reduced in obese individuals, although IGF-I levels may not be suppressed, suggesting a change in the setpoint for feedback control. Elevated GH levels occur within an hour of deep sleep onset as well as after exercise, physical stress, and trauma and during sepsis. Integrated 24-h GH secretion is higher in women and is also enhanced by estrogen replacement likely reflective of increased peripheral GH-resistance. Using standard assays, random GH measurements are undetectable in ~50% of daytime samples obtained from healthy subjects and are also undetectable in most obese and elderly subjects. Thus, single random GH measurements do not distinguish patients with adult GH deficiency from normal persons.
GH secretion is profoundly influenced by nutritional factors. Using newer ultrasensitive GH assays with a sensitivity of 0.002 μg/L, a glucose load suppresses GH to <0.7 μg/L in women and to <0.07 μg/L in men. Increased GH pulse frequency and peak amplitudes occur with chronic malnutrition or prolonged fasting. GH is stimulated by intravenous L-arginine, dopamine, and apomorphine (a dopamine receptor agonist), as well as by α-adrenergic pathways. β-Adrenergic blockade induces basal GH and enhances GHRH- and insulin-evoked GH release.
Action The pattern of GH secretion may affect tissue responses. The higher GH pulsatility observed in men compared with the relatively continuous basal GH secretion in women may be an important biologic determinant of linear growth patterns and liver enzyme induction.
The 70-kDa peripheral GH receptor protein has structural homology with the cytokine/hematopoietic superfamily. A fragment of the receptor extracellular domain generates a soluble GH binding protein (GHBP) that interacts with GH in the circulation. The liver and cartilage contain the greatest number of GH receptors. GH binding to preformed receptor dimers is followed by internal rotation and subsequent signaling through the JAK/STAT pathway. Activated STAT proteins translocate to the nucleus, where they modulate expression of GH-regulated target genes. GH analogues that bind to the receptor but are incapable of mediating receptor signaling are potent antagonists of GH action. A GH receptor antagonist (pegvisomant) is approved for treatment of acromegaly.
GH induces protein synthesis and nitrogen retention and impairs glucose tolerance by antagonizing insulin action. GH also stimulates lipolysis, leading to increased circulating fatty acid levels, reduced omental fat mass, and enhanced lean body mass. GH promotes sodium, potassium, and water retention and elevates serum levels of inorganic phosphate. Linear bone growth occurs as a result of complex hormonal and growth factor actions, including those of IGF-I. GH stimulates epiphyseal prechondrocyte differentiation. These precursor cells produce IGF-I locally, and their proliferation is also responsive to the growth factor.
Insulin-Like Growth Factors Although GH exerts direct effects in target tissues, many of its physiologic effects are mediated indirectly through IGF-I, a potent growth and differentiation factor. The liver is the major source of circulating IGF-I. In peripheral tissues, IGF-I also exerts local paracrine actions that appear to be both dependent on and independent of GH. Thus, GH administration induces circulating IGF-I as well as stimulating local IGF-I production in multiple tissues.
Both IGF-I and IGF-II are bound to high-affinity circulating IGF-binding proteins (IGFBPs) that regulate IGF bioactivity. Levels of IGFBP3 are GH-dependent, and it serves as the major carrier protein for circulating IGF-I. GH deficiency and malnutrition usually are associated with low IGFBP3 levels. IGFBP1 and IGFBP2 regulate local tissue IGF action but do not bind appreciable amounts of circulating IGF-I.
Serum IGF-I concentrations are profoundly affected by physiologic factors. Levels increase during puberty, peak at 16 years, and subsequently decline by >80% during the aging process. IGF-I concentrations are higher in women than in men. Because GH is the major determinant of hepatic IGF-I synthesis, abnormalities of GH synthesis or action (e.g., pituitary failure, GHRH receptor defect, GH receptor defect or pharmacologic GH receptor blockade) reduce IGF-I levels. Hypocaloric states are associated with GH resistance; IGF-I levels are therefore low with cachexia, malnutrition, and sepsis. In acromegaly, IGF-I levels are invariably high and reflect a log-linear relationship with circulating GH concentrations.
IGF-I PHYSIOLOGY Injected IGF-I (100 μg/kg) induces hypoglycemia, and lower doses improve insulin sensitivity in patients with severe insulin resistance and diabetes. In cachectic subjects, IGF-I infusion (12 μg/kg per hour) enhances nitrogen retention and lowers cholesterol levels. Longer-term subcutaneous IGF-I injections enhance protein synthesis and are anabolic. Although bone formation markers are induced, bone turnover also may be stimulated by IGF-I. IGF-I has only been approved for use in patients with GH-resistance syndromes.
IGF-I side effects are dose-dependent, and overdose may result in hypoglycemia, hypotension, fluid retention, temporomandibular jaw pain, and increased intracranial pressure, all of which are reversible. Avascular femoral head necrosis has been reported. Chronic excess IGF-I administration presumably would result in features of acromegaly.
ADRENOCORTICOTROPIC HORMONE
(See also Chap. 406)
Synthesis ACTH-secreting corticotrope cells constitute about 20% of the pituitary cell population. ACTH (39 amino acids) is derived from the POMC precursor protein (266 amino acids) that also generates several other peptides, including β-lipotropin, β-endorphin, met-enkephalin, α-melanocyte-stimulating hormone (α-MSH), and corticotropin-like intermediate lobe protein (CLIP). The POMC gene is potently suppressed by glucocorticoids and induced by corticotropin-releasing hormone (CRH), arginine vasopressin (AVP), and proinflammatory cytokines, including IL-6, as well as leukemia inhibitory factor.
CRH, a 41-amino-acid hypothalamic peptide synthesized in the paraventricular nucleus as well as in higher brain centers, is the predominant stimulator of ACTH synthesis and release. The CRH receptor is a GPCR that is expressed on the corticotrope and signals to induce POMC transcription.
Secretion ACTH secretion is pulsatile and exhibits a characteristic circadian rhythm, peaking at about 6 A.M. and reaching a nadir about midnight. Adrenal glucocorticoid secretion, which is driven by ACTH, follows a parallel diurnal pattern. ACTH circadian rhythmicity is determined by variations in secretory pulse amplitude rather than changes in pulse frequency. Superimposed on this endogenous rhythm, ACTH levels are increased by physical and psychological stress, exercise, acute illness, and insulin-induced hypoglycemia.
Glucocorticoid-mediated negative regulation of the hypothalamic-pituitary-adrenal (HPA) axis occurs as a consequence of both hypothalamic CRH suppression and direct attenuation of pituitary POMC gene expression and ACTH release. In contrast, loss of cortisol feedback inhibition, as occurs in primary adrenal failure, results in extremely high ACTH levels.
Acute inflammatory or septic insults activate the HPA axis through the integrated actions of proinflammatory cytokines, bacterial toxins, and neural signals. The overlapping cascade of ACTH-inducing cytokines (tumor necrosis factor [TNF]; IL-1, -2, and -6; and leukemia inhibitory factor) activates hypothalamic CRH and AVP secretion, pituitary POMC gene expression, and local pituitary paracrine cytokine networks. The resulting cortisol elevation restrains the inflammatory response and enables host protection. Concomitantly, cytokine-mediated central glucocorticoid receptor resistance impairs glucocorticoid suppression of the HPA. Thus, the neuroendocrine stress response reflects the net result of highly integrated hypothalamic, intrapituitary, and peripheral hormone and cytokine signals acting to regulate cortisol secretion.
Action The major function of the HPA axis is to maintain metabolic homeostasis and mediate the neuroendocrine stress response. ACTH induces adrenocortical steroidogenesis by sustaining adrenal cell proliferation and function. The receptor for ACTH, designated melanocortin-2 receptor, is a GPCR that induces steroidogenesis by stimulating a cascade of steroidogenic enzymes (Chap. 406).
GONADOTROPINS: FSH AND LH
Synthesis and Secretion Gonadotrope cells constitute about 10% of anterior pituitary cells and produce two gonadotropin hormones—LH and FSH. Like TSH and hCG, LH and FSH are glycoprotein hormones that comprise α and β subunits. The α subunit is common to these glycoprotein hormones; specificity of hormone function is conferred by the β subunits, which are expressed by separate genes.
Gonadotropin synthesis and release are dynamically regulated. This is particularly true in women, in whom rapidly fluctuating gonadal steroid levels vary throughout the menstrual cycle. Hypothalamic GnRH, a 10-amino-acid peptide, regulates the synthesis and secretion of both LH and FSH. Brain kisspeptin, a product of the KISSI gene regulates hypothalamic GnRH release. GnRH is secreted in discrete pulses every 60–120 min, and the pulses in turn elicit LH and FSH pulses (Fig. 401e-3). The pulsatile mode of GnRH input is essential to its action; pulses prime gonadotrope responsiveness, whereas continuous GnRH exposure induces desensitization. Based on this phenomenon, long-acting GnRH agonists are used to suppress gonadotropin levels in children with precocious puberty and in men with prostate cancer (Chap. 115) and are used in some ovulation-induction protocols to reduce levels of endogenous gonadotropins (Chap. 412). Estrogens act at both the hypothalamus and the pituitary to modulate gonadotropin secretion. Chronic estrogen exposure is inhibitory, whereas rising estrogen levels, as occur during the preovulatory surge, exert positive feedback to increase gonadotropin pulse frequency and amplitude. Progesterone slows GnRH pulse frequency but enhances gonadotropin responses to GnRH. Testosterone feedback in men also occurs at the hypothalamic and pituitary levels and is mediated in part by its conversion to estrogens.
Although GnRH is the main regulator of LH and FSH secretion, FSH synthesis is also under separate control by the gonadal peptides inhibin and activin, which are members of the transforming growth factor β (TGF-β) family. Inhibin selectively suppresses FSH, whereas activin stimulates FSH synthesis (Chap. 412).
Action The gonadotropin hormones interact with their respective GPCRs expressed in the ovary and testis, evoking germ cell development and maturation and steroid hormone biosynthesis. In women, FSH regulates ovarian follicle development and stimulates ovarian estrogen production. LH mediates ovulation and maintenance of the corpus luteum. In men, LH induces Leydig cell testosterone synthesis and secretion, and FSH stimulates seminiferous tubule development and regulates spermatogenesis.
THYROID-STIMULATING HORMONE
Synthesis and Secretion TSH-secreting thyrotrope cells constitute 5% of the anterior pituitary cell population. TSH shares a common α subunit with LH and FSH but contains a specific TSH β subunit. TRH is a hypothalamic tripeptide (pyroglutamyl histidylprolinamide) that acts through a pituitary GPCR to stimulate TSH synthesis and secretion; it also stimulates the lactotrope cell to secrete PRL. TSH secretion is stimulated by TRH, whereas thyroid hormones, dopamine, somatostatin, and glucocorticoids suppress TSH by overriding TRH induction.
Thyrotrope cell proliferation and TSH secretion are both induced when negative feedback inhibition by thyroid hormones is removed. Thus, thyroid damage (including surgical thyroidectomy), radiation-induced hypothyroidism, chronic thyroiditis, and prolonged goitrogen exposure are associated with increased TSH levels. Long-standing untreated hypothyroidism can lead to elevated TSH levels as well as thyrotrope hyperplasia and pituitary enlargement, which may be evident on magnetic resonance imaging.
Action TSH is secreted in pulses, although the excursions are modest in comparison to other pituitary hormones because of the low amplitude of the pulses and the relatively long half-life of TSH. Consequently, single determinations of TSH suffice to precisely assess its circulating levels. TSH binds to a GPCR on thyroid follicular cells to stimulate thyroid hormone synthesis and release (Chap. 405).
402 |
Hypopituitarism |
Inadequate production of anterior pituitary hormones leads to features of hypopituitarism. Impaired production of one or more of the anterior pituitary trophic hormones can result from inherited disorders; more commonly, adult hypopituitarism is acquired and reflects the compressive mass effects of tumors or the consequences of local pituitary or hypothalamic traumatic, inflammatory, or vascular damage. These processes also may impair synthesis or secretion of hypothalamic hormones, with resultant pituitary failure (Table 402-1).
|
ETIOLOGY OF HYPOPITUITARISMa |
aTrophic hormone failure associated with pituitary compression or destruction usually occurs sequentially: growth hormone > follicle-stimulating hormone > luteinizing hormone > thyroid-stimulating hormone > adrenocorticotropic hormone. During childhood, growth retardation is often the presenting feature, and in adults, hypogonadism is the earliest symptom.
DEVELOPMENTAL AND GENETIC CAUSES OF HYPOPITUITARISM
Pituitary Dysplasia Pituitary dysplasia may result in aplastic, hypoplastic, or ectopic pituitary gland development. Because pituitary development follows midline cell migration from the nasopharyngeal Rathke’s pouch, midline craniofacial disorders may be associated with pituitary dysplasia. Acquired pituitary failure in the newborn also can be caused by birth trauma, including cranial hemorrhage, asphyxia, and breech delivery.
SEPTO-OPTIC DYSPLASIA Hypothalamic dysfunction and hypopituitarism may result from dysgenesis of the septum pellucidum or corpus callosum. Affected children have mutations in the HESX1 gene, which is involved in early development of the ventral prosencephalon. These children exhibit variable combinations of cleft palate, syndactyly, ear deformities, hypertelorism, optic nerve hypoplasia, micropenis, and anosmia. Pituitary dysfunction leads to diabetes insipidus, growth hormone (GH) deficiency and short stature, and, occasionally, thyroid-stimulating hormone (TSH) deficiency.
Tissue-Specific Factor Mutations Several pituitary cell–specific transcription factors, such as Pit-1 and Prop-1, are critical for determining the development and committed function of differentiated anterior pituitary cell lineages. Autosomal dominant or recessive Pit-1 mutations cause combined GH, prolactin (PRL), and TSH deficiencies. These patients usually present with growth failure and varying degrees of hypothyroidism. The pituitary may appear hypoplastic on magnetic resonance imaging (MRI).
Prop-1 is expressed early in pituitary development and appears to be required for Pit-1 function. Familial and sporadic PROP1 mutations result in combined GH, PRL, TSH, and gonadotropin deficiency. Over 80% of these patients have growth retardation; by adulthood, all are deficient in TSH and gonadotropins, and a small minority later develop adrenocorticotropic hormone (ACTH) deficiency. Because of gonadotropin deficiency, these individuals do not enter puberty spontaneously. In some cases, the pituitary gland appears enlarged on MRI. TPIT mutations result in ACTH deficiency associated with hypocortisolism.
Developmental Hypothalamic Dysfunction • KALLMANN SYNDROME Kallmann syndrome results from defective hypothalamic gonadotropin-releasing hormone (GnRH) synthesis and is associated with anosmia or hyposmia due to olfactory bulb agenesis or hypoplasia (Chap. 411). Classically, the syndrome may also be associated with color blindness, optic atrophy, nerve deafness, cleft palate, renal abnormalities, cryptorchidism, and neurologic abnormalities such as mirror movements. The initial genetic cause was identified in the X-linked KAL gene, mutations of which impair embryonic migration of GnRH neurons from the hypothalamic olfactory placode to the hypothalamus. Based on further studies, at least a dozen other genetic abnormalities, in addition to KAL mutations, have been found to cause isolated GnRH deficiency. Autosomal recessive (i.e., GPR54, KISS1) and dominant (i.e., FGFR1) modes of transmission have been described, and there is a growing list of genes associated with GnRH deficiency (GNRH1, PROK2, PROKR2, CH7, PCSK1, FGF8, NELF, WDR11, TAC3, TACR3). A fraction of patients have digenic mutations. Associated clinical features, in addition to GnRH deficiency, vary depending on the genetic cause. GnRH deficiency prevents progression through puberty. Males present with delayed puberty and pronounced hypogonadal features, including micropenis, probably the result of low testosterone levels during infancy. Females present with primary amenorrhea and failure of secondary sexual development.
Kallmann syndrome and other causes of congenital GnRH deficiency are characterized by low luteinizing hormone (LH) and follicle-stimulating hormone (FSH) levels and low concentrations of sex steroids (testosterone or estradiol). In sporadic cases of isolated gonadotropin deficiency, the diagnosis is often one of exclusion after other known causes of hypothalamic-pituitary dysfunction have been eliminated. Repetitive GnRH administration restores normal pituitary gonadotropin responses, pointing to a hypothalamic defect in these patients.
Long-term treatment of males with human chorionic gonadotropin (hCG) or testosterone restores pubertal development and secondary sex characteristics; women can be treated with cyclic estrogen and progestin. Fertility also may be restored by the administration of gonadotropins or by using a portable infusion pump to deliver subcutaneous, pulsatile GnRH.
BARDET-BIEDL SYNDROME This very rare genetically heterogeneous disorder is characterized by mental retardation, renal abnormalities, obesity, and hexadactyly, brachydactyly, or syndactyly. Central diabetes insipidus may or may not be associated. GnRH deficiency occurs in 75% of males and half of affected females. Retinal degeneration begins in early childhood, and most patients are blind by age 30. Numerous subtypes of Bardet-Biedl syndrome (BBS) have been identified, with genetic linkage to at least nine different loci. Several of the loci encode genes involved in basal body cilia function, and this may account for the diverse clinical manifestations.
LEPTIN AND LEPTIN RECEPTOR MUTATIONS Deficiencies of leptin or its receptor cause a broad spectrum of hypothalamic abnormalities, including hyperphagia, obesity, and central hypogonadism (Chap. 415e). Decreased GnRH production in these patients results in attenuated pituitary FSH and LH synthesis and release.
PRADER-WILLI SYNDROME This is a contiguous gene syndrome that results from deletion of the paternal copies of the imprinted SNRPN gene, the NECDIN gene, and possibly other genes on chromosome 15q. Prader-Willi syndrome is associated with hypogonadotropic hypogonadism, hyperphagia-obesity, chronic muscle hypotonia, mental retardation, and adult-onset diabetes mellitus (Chap. 83e). Multiple somatic defects also involve the skull, eyes, ears, hands, and feet. Diminished hypothalamic oxytocin- and vasopressin-producing nuclei have been reported. Deficient GnRH synthesis is suggested by the observation that chronic GnRH treatment restores pituitary LH and FSH release.
ACQUIRED HYPOPITUITARISM
Hypopituitarism may be caused by accidental or neurosurgical trauma; vascular events such as apoplexy; pituitary or hypothalamic neoplasms, craniopharyngioma, lymphoma, or metastatic tumors; inflammatory disease such as lymphocytic hypophysitis; infiltrative disorders such as sarcoidosis, hemochromatosis (Chap. 428), and tuberculosis; or irradiation.
Increasing evidence suggests that patients with brain injury, including contact sports trauma, subarachnoid hemorrhage, and irradiation, have transient hypopituitarism and require intermittent long-term endocrine follow-up, because permanent hypothalamic or pituitary dysfunction will develop in 25–40% of these patients.
Hypothalamic Infiltration Disorders These disorders—including sarcoidosis, histiocytosis X, amyloidosis, and hemochromatosis—frequently involve both hypothalamic and pituitary neuronal and neurochemical tracts. Consequently, diabetes insipidus occurs in half of patients with these disorders. Growth retardation is seen if attenuated GH secretion occurs before puberty. Hypogonadotropic hypogonadism and hyperprolactinemia are also common.
Inflammatory Lesions Pituitary damage and subsequent secretory dysfunction can be seen with chronic site infections such as tuberculosis, with opportunistic fungal infections associated with AIDS, and in tertiary syphilis. Other inflammatory processes, such as granulomas and sarcoidosis, may mimic the features of a pituitary adenoma. These lesions may cause extensive hypothalamic and pituitary damage, leading to trophic hormone deficiencies.
Cranial Irradiation Cranial irradiation may result in long-term hypothalamic and pituitary dysfunction, especially in children and adolescents, as they are more susceptible to damage after whole-brain or head and neck therapeutic irradiation. The development of hormonal abnormalities correlates strongly with irradiation dosage and the time interval after completion of radiotherapy. Up to two-thirds of patients ultimately develop hormone insufficiency after a median dose of 50 Gy (5000 rad) directed at the skull base. The development of hypopituitarism occurs over 5–15 years and usually reflects hypothalamic damage rather than primary destruction of pituitary cells. Although the pattern of hormone loss is variable, GH deficiency is most common, followed by gonadotropin and ACTH deficiency. When deficiency of one or more hormones is documented, the possibility of diminished reserve of other hormones is likely. Accordingly, anterior pituitary function should be continually evaluated over the long term in previously irradiated patients, and replacement therapy instituted when appropriate (see below).
Lymphocytic Hypophysitis This occurs most often in postpartum women; it usually presents with hyperprolactinemia and MRI evidence of a prominent pituitary mass that often resembles an adenoma, with mildly elevated PRL levels. Pituitary failure caused by diffuse lymphocytic infiltration may be transient or permanent but requires immediate evaluation and treatment. Rarely, isolated pituitary hormone deficiencies have been described, suggesting a selective autoimmune process targeted to specific cell types. Most patients manifest symptoms of progressive mass effects with headache and visual disturbance. The erythrocyte sedimentation rate often is elevated. Because the MRI image may be indistinguishable from that of a pituitary adenoma, hypophysitis should be considered in a postpartum woman with a newly diagnosed pituitary mass before an unnecessary surgical intervention is undertaken. The inflammatory process often resolves after several months of glucocorticoid treatment, and pituitary function may be restored, depending on the extent of damage.
Pituitary Apoplexy Acute intrapituitary hemorrhagic vascular events can cause substantial damage to the pituitary and surrounding sellar structures. Pituitary apoplexy may occur spontaneously in a preexisting adenoma; postpartum (Sheehan’s syndrome); or in association with diabetes, hypertension, sickle cell anemia, or acute shock. The hyperplastic enlargement of the pituitary, which occurs normally during pregnancy, increases the risk for hemorrhage and infarction. Apoplexy is an endocrine emergency that may result in severe hypoglycemia, hypotension and shock, central nervous system (CNS) hemorrhage, and death. Acute symptoms may include severe headache with signs of meningeal irritation, bilateral visual changes, ophthalmoplegia, and, in severe cases, cardiovascular collapse and loss of consciousness. Pituitary computed tomography (CT) or MRI may reveal signs of intratumoral or sellar hemorrhage, with pituitary stalk deviation and compression of pituitary tissue.
Patients with no evident visual loss or impaired consciousness can be observed and managed conservatively with high-dose glucocorticoids. Those with significant or progressive visual loss, cranial nerve palsy, or loss of consciousness require urgent surgical decompression. Visual recovery after sellar surgery is inversely correlated with the length of time after the acute event. Therefore, severe ophthalmoplegia or visual deficits are indications for early surgery. Hypopituitarism is common after apoplexy.
Empty Sella A partial or apparently totally empty sella is often an incidental MRI finding, and may be associated with intracranial hypertension. These patients usually have normal pituitary function, implying that the surrounding rim of pituitary tissue is fully functional. Hypopituitarism, however, may develop insidiously. Pituitary masses also may undergo clinically silent infarction and involution with development of a partial or totally empty sella by cerebrospinal fluid (CSF) filling the dural herniation. Rarely, small but functional pituitary adenomas may arise within the rim of normal pituitary tissue, and they are not always visible on MRI.
PRESENTATION AND DIAGNOSIS
The clinical manifestations of hypopituitarism depend on which hormones are lost and the extent of the hormone deficiency. GH deficiency causes growth disorders in children and leads to abnormal body composition in adults (see below). Gonadotropin deficiency causes menstrual disorders and infertility in women and decreased sexual function, infertility, and loss of secondary sexual characteristics in men. TSH and ACTH deficiency usually develop later in the course of pituitary failure. TSH deficiency causes growth retardation in children and features of hypothyroidism in children and adults. The secondary form of adrenal insufficiency caused by ACTH deficiency leads to hypocortisolism with relative preservation of mineralocorticoid production. PRL deficiency causes failure of lactation. When lesions involve the posterior pituitary, polyuria and polydipsia reflect loss of vasopressin secretion. In patients with long-standing pituitary damage, epidemiologic studies document an increased mortality rate, primarily from increased cardiovascular and cerebrovascular disease. Previous head or neck irradiation is also a determinant of increased mortality rates in patients with hypopituitarism, especially from cerebrovascular disease.
LABORATORY INVESTIGATION
Biochemical diagnosis of pituitary insufficiency is made by demonstrating low levels of respective pituitary trophic hormones in the setting of low levels of target hormones. For example, low free thyroxine in the setting of a low or inappropriately normal TSH level suggests secondary hypothyroidism. Similarly, a low testosterone level without elevation of gonadotropins suggests hypogonadotropic hypogonadism. Provocative tests may be required to assess pituitary reserve (Table 402-2). GH responses to insulin-induced hypoglycemia, arginine, L-dopa, growth hormone–releasing hormone (GHRH), or growth hormone–releasing peptides (GHRPs) can be used to assess GH reserve. Corticotropin-releasing hormone (CRH) administration induces ACTH release, and administration of synthetic ACTH (cosyntropin) evokes adrenal cortisol release as an indirect indicator of pituitary ACTH reserve (Chap. 406). ACTH reserve is most reliably assessed by measuring ACTH and cortisol levels during insulin-induced hypoglycemia. However, this test should be performed cautiously in patients with suspected adrenal insufficiency because of enhanced susceptibility to hypoglycemia and hypotension. Administering insulin to induce hypoglycemia is contraindicated in patients with active coronary artery disease or known seizure disorders.
|
TESTS OF PITUITARY SUFFICIENCY |
|
TREATMENT |
HYPOPITUITARISM |
Hormone replacement therapy, including glucocorticoids, thyroid hormone, sex steroids, growth hormone, and vasopressin, is usually safe and free of complications. Treatment regimens that mimic physiologic hormone production allow for maintenance of satisfactory clinical homeostasis. Effective dosage schedules are outlined in Table 402-3. Patients in need of glucocorticoid replacement require careful dose adjustments during stressful events such as acute illness, dental procedures, trauma, and acute hospitalization.
|
HORMONE REPLACEMENT THERAPY FOR ADULT HYPOPITUITARISMa |
DISORDERS OF GROWTH AND DEVELOPMENT
Skeletal Maturation and Somatic Growth The growth plate is dependent on a variety of hormonal stimuli, including GH, insulin-like growth factor (IGF) I, sex steroids, thyroid hormones, paracrine growth factors, and cytokines. The growth-promoting process also requires caloric energy, amino acids, vitamins, and trace metals and consumes about 10% of normal energy production. Malnutrition impairs chondrocyte activity, increases GH resistance, and reduces circulating IGF-I and IGFBP3 levels.
Linear bone growth rates are very high in infancy and are pituitary-dependent. Mean growth velocity is ~6 cm/year in later childhood and usually is maintained within a given range on a standardized percentile chart. Peak growth rates occur during midpuberty when bone age is 12 (girls) or 13 (boys). Secondary sexual development is associated with elevated sex steroids that cause progressive epiphyseal growth plate closure. Bone age is delayed in patients with all forms of true GH deficiency or GH receptor defects that result in attenuated GH action.
Short stature may occur as a result of constitutive intrinsic growth defects or because of acquired extrinsic factors that impair growth. In general, delayed bone age in a child with short stature is suggestive of a hormonal or systemic disorder, whereas normal bone age in a short child is more likely to be caused by a genetic cartilage dysplasia or growth plate disorder (Chap. 427).
GH Deficiency in Children • GH DEFICIENCY Isolated GH deficiency is characterized by short stature, micropenis, increased fat, high-pitched voice, and a propensity to hypoglycemia due to relatively unopposed insulin action. Familial modes of inheritance are seen in at least one-third of these individuals and may be autosomal dominant, recessive, or X-linked. About 10% of children with GH deficiency have mutations in the GH-N gene, including gene deletions and a wide range of point mutations. Mutations in transcription factors Pit-1 and Prop-1, which control somatotrope development, result in GH deficiency in combination with other pituitary hormone deficiencies, which may become manifest only in adulthood. The diagnosis of idiopathic GH deficiency (IGHD) should be made only after known molecular defects have been rigorously excluded.
GHRH RECEPTOR MUTATIONS Recessive mutations of the GHRH receptor gene in subjects with severe proportionate dwarfism are associated with low basal GH levels that cannot be stimulated by exogenous GHRH, GHRP, or insulin-induced hypoglycemia, as well as anterior pituitary hypoplasia The syndrome exemplifies the importance of the GHRH receptor for somatotrope cell proliferation and hormonal responsiveness.
GH INSENSITIVITY This is caused by defects of GH receptor structure or signaling. Homozygous or heterozygous mutations of the GH receptor are associated with partial or complete GH insensitivity and growth failure (Laron’s syndrome). The diagnosis is based on normal or high GH levels, with decreased circulating GH-binding protein (GHBP), and low IGF-I levels. Very rarely, defective IGF-I, IGF-I receptor, or IGF-I signaling defects are also encountered. STAT5B mutations result in both immunodeficiency as well as abrogated GH signaling, leading to short stature with normal or elevated GH levels and low IGF-I levels. Circulating GH receptor antibodies may rarely cause peripheral GH insensitivity.
NUTRITIONAL SHORT STATURE Caloric deprivation and malnutrition, uncontrolled diabetes, and chronic renal failure represent secondary causes of abrogated GH receptor function. These conditions also stimulate production of proinflammatory cytokines, which act to exacerbate the block of GH-mediated signal transduction. Children with these conditions typically exhibit features of acquired short stature with normal or elevated GH and low IGF-I levels.
PSYCHOSOCIAL SHORT STATURE Emotional and social deprivation lead to growth retardation accompanied by delayed speech, discordant hyperphagia, and an attenuated response to administered GH. A nurturing environment restores growth rates.
PRESENTATION AND DIAGNOSIS
Short stature is commonly encountered in clinical practice, and the decision to evaluate these children requires clinical judgment in association with auxologic data and family history. Short stature should be evaluated comprehensively if a patient’s height is >3 standard deviations (SD) below the mean for age or if the growth rate has decelerated. Skeletal maturation is best evaluated by measuring a radiologic bone age, which is based mainly on the degree of wrist bone growth plate fusion. Final height can be predicted using standardized scales (Bayley-Pinneau or Tanner-Whitehouse) or estimated by adding 6.5 cm (boys) or subtracting 6.5 cm (girls) from the midparental height.
LABORATORY INVESTIGATION
Because GH secretion is pulsatile, GH deficiency is best assessed by examining the response to provocative stimuli, including exercise, insulin-induced hypoglycemia, and other pharmacologic tests that normally increase GH to >7 μg/L in children. Random GH measurements do not distinguish normal children from those with true GH deficiency. Adequate adrenal and thyroid hormone replacement should be assured before testing. Age- and sex-matched IGF-I levels are not sufficiently sensitive or specific to make the diagnosis but can be useful to confirm GH deficiency. Pituitary MRI may reveal pituitary mass lesions or structural defects. Molecular analyses for known mutations should be undertaken when the cause of short stature remains cryptic, or when additional clinical features suggest a genetic cause.
|
TREATMENT |
DISORDERS OF GROWTH AND DEVELOPMENT |
Replacement therapy with recombinant GH (0.02–0.05 mg/kg per day SC) restores growth velocity in GH-deficient children to ~10 cm/year. If pituitary insufficiency is documented, other associated hormone deficits should be corrected, especially adrenal steroids. GH treatment is also moderately effective for accelerating growth rates in children with Turner’s syndrome and chronic renal failure.
In patients with GH insensitivity and growth retardation due to mutations of the GH receptor, treatment with IGF-I bypasses the dysfunctional GH receptor.
ADULT GH DEFICIENCY (AGHD)
This disorder usually is caused by acquired hypothalamic or pituitary somatotrope damage. Acquired pituitary hormone deficiency follows a typical pattern in which loss of adequate GH reserve foreshadows subsequent hormone deficits. The sequential order of hormone loss is usually GH → FSH/LH → TSH → ACTH. Patients previously diagnosed with childhood-onset GH deficiency should be retested as adults to affirm the diagnosis.
PRESENTATION AND DIAGNOSIS
The clinical features of AGHD include changes in body composition, lipid metabolism, and quality of life and cardiovascular dysfunction (Table 402-4). Body composition changes are common and include reduced lean body mass, increased fat mass with selective deposition of intraabdominal visceral fat, and increased waist-to-hip ratio. Hyperlipidemia, left ventricular dysfunction, hypertension, and increased plasma fibrinogen levels also may be present. Bone mineral content is reduced, with resultant increased fracture rates. Patients may experience social isolation, depression, and difficulty maintaining gainful employment. Adult hypopituitarism is associated with a threefold increase in cardiovascular mortality rates in comparison to age- and sex-matched controls, and this may be due to GH deficiency, as patients in these studies were replaced with other deficient pituitary hormones.
|
FEATURES OF ADULT GROWTH HORMONE DEFICIENCY |
Abbreviation: LDL, low-density lipoprotein. For other abbreviations, see text.
LABORATORY INVESTIGATION
AGHD is rare, and in light of the nonspecific nature of associated clinical symptoms, patients appropriate for testing should be selected carefully on the basis of well-defined criteria. With few exceptions, testing should be restricted to patients with the following predisposing factors: (1) pituitary surgery, (2) pituitary or hypothalamic tumor or granulomas, (3) history of cranial irradiation, (4) radiologic evidence of a pituitary lesion, (5) childhood requirement for GH replacement therapy, and rarely (6) unexplained low age- and sex-matched IGF-I levels. The transition of a GH-deficient adolescent to adulthood requires retesting to document subsequent adult GH deficiency. Up to 20% of patients previously treated for childhood-onset GH deficiency are found to be GH-sufficient on repeat testing as adults.
A significant proportion (~25%) of truly GH-deficient adults have low-normal IGF-I levels. Thus, as in the evaluation of GH deficiency in children, valid age- and sex-matched IGF-I measurements provide a useful index of therapeutic responses but are not sufficiently sensitive for diagnostic purposes. The most validated test to distinguish pituitary-sufficient patients from those with AGHD is insulin-induced (0.05–0.1 U/kg) hypoglycemia. After glucose reduction to ~40 mg/dL, most individuals experience neuroglycopenic symptoms (Chap. 420), and peak GH release occurs at 60 min and remains elevated for up to 2 h. About 90% of healthy adults exhibit GH responses >5 μg/L; AGHD is defined by a peak GH response to hypoglycemia of <3 μg/L. Although insulin-induced hypoglycemia is safe when performed under appropriate supervision, it is contraindicated in patients with diabetes, ischemic heart disease, cerebrovascular disease, or epilepsy and in elderly patients. Alternative stimulatory tests include intravenous arginine (30 g), GHRH (1 μg/kg), GHRP-6 (90 μg), and glucagon (1 mg). Combinations of these tests may evoke GH secretion in subjects who are not responsive to a single test.
|
TREATMENT |
ADULT GH DEFICIENCY |
Once the diagnosis of AGHD is unequivocally established, replacement of GH may be indicated. Contraindications to therapy include the presence of an active neoplasm, intracranial hypertension, and uncontrolled diabetes and retinopathy. The starting dose of 0.1–0.2 mg/d should be titrated (up to a maximum of 1.25 mg/d) to maintain IGF-I levels in the mid-normal range for age- and sex-matched controls (Fig. 402-1). Women require higher doses than men, and elderly patients require less GH. Long-term GH maintenance sustains normal IGF-I levels and is associated with persistent body composition changes (e.g., enhanced lean body mass and lower body fat). High-density lipoprotein cholesterol increases, but total cholesterol and insulin levels may not change significantly. Lumbar spine bone mineral density increases, but this response is gradual (>1 year). Many patients note significant improvement in quality of life when evaluated by standardized questionnaires. The effect of GH replacement on mortality rates in GH-deficient patients is currently the subject of long-term prospective investigation.
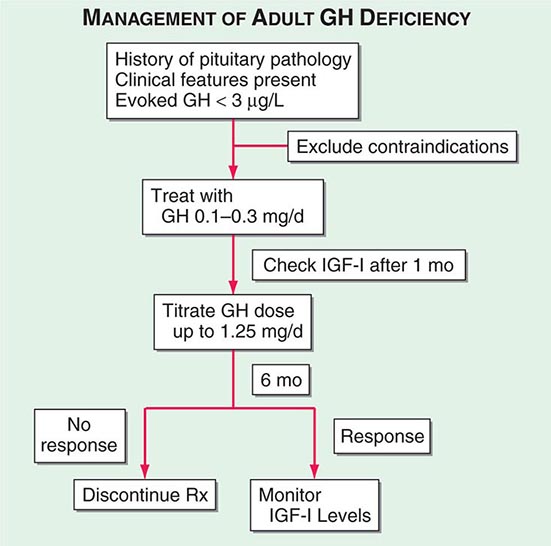
FIGURE 402-1 Management of adult growth hormone (GH) deficiency. IGF, insulin-like growth factor; Rx, Treatment.
About 30% of patients exhibit reversible dose-related fluid retention, joint pain, and carpal tunnel syndrome, and up to 40% exhibit myalgias and paresthesia. Patients receiving insulin require careful monitoring for dosing adjustments, as GH is a potent counterregulatory hormone for insulin action. Patients with type 2 diabetes mellitus initially develop further insulin resistance. However, glycemic control usually improves with the sustained loss of abdominal fat associated with long-term GH replacement. Headache, increased intracranial pressure, hypertension, and tinnitus occur rarely. Pituitary tumor regrowth and progression of skin lesions or other tumors are being assessed in long-term surveillance programs. To date, development of these potential side effects does not appear significant.
ACTH DEFICIENCY
PRESENTATION AND DIAGNOSIS
Secondary adrenal insufficiency occurs as a result of pituitary ACTH deficiency. It is characterized by fatigue, weakness, anorexia, nausea, vomiting, and, occasionally, hypoglycemia. In contrast to primary adrenal failure, hypocortisolism associated with pituitary failure usually is not accompanied by hyperpigmentation or mineralocorticoid deficiency.
ACTH deficiency is commonly due to glucocorticoid withdrawal after treatment-associated suppression of the hypothalamic-pituitary-adrenal (HPA) axis. Isolated ACTH deficiency may occur after surgical resection of an ACTH-secreting pituitary adenoma that has suppressed the HPA axis; this phenomenon is in fact suggestive of a surgical cure. The mass effects of other pituitary adenomas or sellar lesions may lead to ACTH deficiency, but usually in combination with other pituitary hormone deficiencies. Partial ACTH deficiency may be unmasked in the presence of an acute medical or surgical illness, when clinically significant hypocortisolism reflects diminished ACTH reserve. Rarely, TPIT or POMC mutations result in primary ACTH deficiency.
LABORATORY DIAGNOSIS
Inappropriately low ACTH levels in the setting of low cortisol levels are characteristic of diminished ACTH reserve. Low basal serum cortisol levels are associated with blunted cortisol responses to ACTH stimulation and impaired cortisol response to insulin-induced hypoglycemia, or testing with metyrapone or CRH. For a description of provocative ACTH tests, see Chap. 406.
|
TREATMENT |
ACTH DEFICIENCY |
Glucocorticoid replacement therapy improves most features of ACTH deficiency. The total daily dose of hydrocortisone replacement preferably should not exceed 25 mg daily, divided into two or three doses. Prednisone (5 mg each morning) is longer acting and has fewer mineralocorticoid effects than hydrocortisone. Some authorities advocate lower maintenance doses in an effort to avoid cushingoid side effects. Doses should be increased severalfold during periods of acute illness or stress.
GONADOTROPIN DEFICIENCY
Hypogonadism is the most common presenting feature of adult hypopituitarism even when other pituitary hormones are also deficient. It is often a harbinger of hypothalamic or pituitary lesions that impair GnRH production or delivery through the pituitary stalk. As noted below, hypogonadotropic hypogonadism is a common presenting feature of hyperprolactinemia.
A variety of inherited and acquired disorders are associated with isolated hypogonadotropic hypogonadism (IHH) (Chap. 411). Hypothalamic defects associated with GnRH deficiency include Kallmann syndrome and mutations in more than a dozen genes that regulate GnRH neuron migration, development, and function (see above). Mutations in GPR54, DAX1, kisspeptin, the GnRH receptor, and the LHβ or FSHβ subunit genes also cause pituitary gonadotropin deficiency. Acquired forms of GnRH deficiency leading to hypogonadotropism are seen in association with anorexia nervosa, stress, starvation, and extreme exercise but also may be idiopathic. Hypogonadotropic hypogonadism in these disorders is reversed by removal of the stressful stimulus or by caloric replenishment.
PRESENTATION AND DIAGNOSIS
In premenopausal women, hypogonadotropic hypogonadism presents as diminished ovarian function leading to oligomenorrhea or amenorrhea, infertility, decreased vaginal secretions, decreased libido, and breast atrophy. In hypogonadal adult men, secondary testicular failure is associated with decreased libido and potency, infertility, decreased muscle mass with weakness, reduced beard and body hair growth, soft testes, and characteristic fine facial wrinkles. Osteoporosis occurs in both untreated hypogonadal women and men.
LABORATORY INVESTIGATION
Central hypogonadism is associated with low or inappropriately normal serum gonadotropin levels in the setting of low sex hormone concentrations (testosterone in men, estradiol in women). Because gonadotropin secretion is pulsatile, valid assessments may require repeated measurements or the use of pooled serum samples. Men have reduced sperm counts.
Intravenous GnRH (100 μg) stimulates gonadotropes to secrete LH (which peaks within 30 min) and FSH (which plateaus during the ensuing 60 min). Normal responses vary according to menstrual cycle stage, age, and sex of the patient. Generally, LH levels increase about threefold, whereas FSH responses are less pronounced. In the setting of gonadotropin deficiency, a normal gonadotropin response to GnRH indicates intact pituitary gonadotrope function and suggests a hypothalamic abnormality. An absent response, however, does not reliably distinguish pituitary from hypothalamic causes of hypogonadism. For this reason, GnRH testing usually adds little to the information gained from baseline evaluation of the hypothalamic-pituitary-gonadotrope axis except in cases of isolated GnRH deficiency (e.g., Kallmann syndrome).
MRI examination of the sellar region and assessment of other pituitary functions usually are indicated in patients with documented central hypogonadism.
|
TREATMENT |
GONADOTROPIN DEFICIENCY |
In males, testosterone replacement is necessary to achieve and maintain normal growth and development of the external genitalia, secondary sex characteristics, male sexual behavior, and androgenic anabolic effects, including maintenance of muscle function and bone mass. Testosterone may be administered by intramuscular injections every 1–4 weeks or by using skin patches that are replaced daily (Chap. 411). Testosterone gels are also available. Gonadotropin injections (hCG or human menopausal gonadotropin [hMG]) over 12–18 months are used to restore fertility. Pulsatile GnRH therapy (25–150 ng/kg every 2 h), administered by a subcutaneous infusion pump, is also effective for treatment of hypothalamic hypogonadism when fertility is desired.
In premenopausal women, cyclical replacement of estrogen and progesterone maintains secondary sexual characteristics and integrity of genitourinary tract mucosa and prevents premature osteoporosis (Chap. 412). Gonadotropin therapy is used for ovulation induction. Follicular growth and maturation are initiated using hMG or recombinant FSH; hCG or human luteinizing hormone (hLH) is subsequently injected to induce ovulation. As in men, pulsatile GnRH therapy can be used to treat hypothalamic causes of gonadotropin deficiency.
DIABETES INSIPIDUS
See Chap. 404 for diagnosis and treatment of diabetes insipidus.
403 |
Anterior Pituitary Tumor Syndromes |
HYPOTHALAMIC, PITUITARY, AND OTHER SELLAR MASSES
EVALUATION OF SELLAR MASSES
Local Mass Effects Clinical manifestations of sellar lesions vary, depending on the anatomic location of the mass and the direction of its extension (Table 403-1). The dorsal sellar diaphragm presents the least resistance to soft tissue expansion from the sella; consequently, pituitary adenomas frequently extend in a suprasellar direction. Bony invasion may occur as well.
|
FEATURES OF SELLAR MASS LESIONSa |
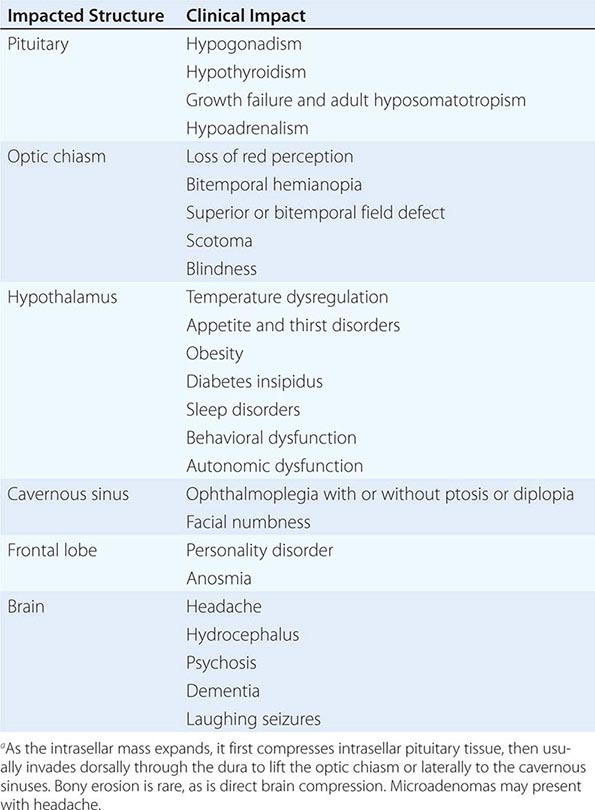
Headaches are common features of small intrasellar tumors, even with no demonstrable suprasellar extension. Because of the confined nature of the pituitary, small changes in intrasellar pressure stretch the dural plate; however, headache severity correlates poorly with adenoma size or extension.
Suprasellar extension can lead to visual loss by several mechanisms, the most common being compression of the optic chiasm, but rarely, direct invasion of the optic nerves or obstruction of cerebrospinal fluid (CSF) flow leading to secondary visual disturbances can occur. Pituitary stalk compression by a hormonally active or inactive intrasellar mass may compress the portal vessels, disrupting pituitary access to hypothalamic hormones and dopamine; this results in early hyperprolactinemia and later concurrent loss of other pituitary hormones. This “stalk section” phenomenon may also be caused by trauma, whiplash injury with posterior clinoid stalk compression, or skull base fractures. Lateral mass invasion may impinge on the cavernous sinus and compress its neural contents, leading to cranial nerve III, IV, and VI palsies as well as effects on the ophthalmic and maxillary branches of the fifth cranial nerve (Chap. 455). Patients may present with diplopia, ptosis, ophthalmoplegia, and decreased facial sensation, depending on the extent of neural damage. Extension into the sphenoid sinus indicates that the pituitary mass has eroded through the sellar floor. Aggressive tumors rarely invade the palate roof and cause nasopharyngeal obstruction, infection, and CSF leakage. Temporal and frontal lobe involvement may rarely lead to uncinate seizures, personality disorders, and anosmia. Direct hypothalamic encroachment by an invasive pituitary mass may cause important metabolic sequelae, including precocious puberty or hypogonadism, diabetes insipidus, sleep disturbances, dysthermia, and appetite disorders.
Magnetic Resonance Imaging Sagittal and coronal T1-weighted magnetic resonance imaging (MRI) before and after administration of gadolinium allows precise visualization of the pituitary gland with clear delineation of the hypothalamus, pituitary stalk, pituitary tissue and surrounding suprasellar cisterns, cavernous sinuses, sphenoid sinus, and optic chiasm. Pituitary gland height ranges from 6 mm in children to 8 mm in adults; during pregnancy and puberty, the height may reach 10–12 mm. The upper aspect of the adult pituitary is flat or slightly concave, but in adolescent and pregnant individuals, this surface may be convex, reflecting physiologic pituitary enlargement. The stalk should be midline and vertical. Computed tomography (CT) scan is reserved to define the extent of bony erosion or the presence of calcification.
Anterior pituitary gland soft tissue consistency is slightly heterogeneous on MRI, and signal intensity resembles that of brain matter on T1-weighted imaging (Fig. 403-1). Adenoma density is usually lower than that of surrounding normal tissue on T1-weighted imaging, and the signal intensity increases with T2-weighted images. The high phospholipid content of the posterior pituitary results in a “pituitary bright spot.”
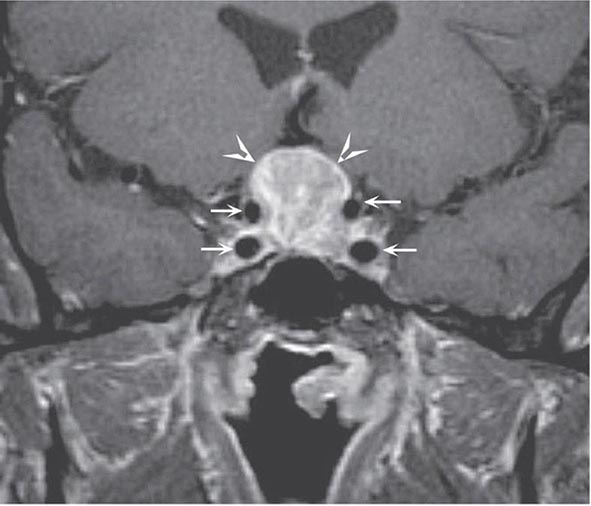
FIGURE 403-1 Pituitary adenoma. Coronal T1-weighted postcontrast magnetic resonance image shows a homogeneously enhancing mass (arrowheads) in the sella turcica and suprasellar region compatible with a pituitary adenoma; the small arrows outline the carotid arteries.
Sellar masses are encountered commonly as incidental findings on MRI, and most of them are pituitary adenomas (incidentalomas). In the absence of hormone hypersecretion, these small intrasellar lesions can be monitored safely with MRI, which is performed annually and then less often if there is no evidence of further growth. Resection should be considered for incidentally discovered larger macroadenomas, because about one-third become invasive or cause local pressure effects. If hormone hypersecretion is evident, specific therapies are indicated as described below. When larger masses (>1 cm) are encountered, they should also be distinguished from nonadenomatous lesions. Meningiomas often are associated with bony hyperostosis; craniopharyngiomas may be calcified and are usually hypodense, whereas gliomas are hyperdense on T2-weighted images.
Ophthalmologic Evaluation Because optic tracts may be contiguous to an expanding pituitary mass, reproducible visual field assessment using perimetry techniques should be performed on all patients with sellar mass lesions that impinge the optic chiasm (Chap. 39). Bitemporal hemianopia, often more pronounced superiorly, is observed classically. It occurs because nasal ganglion cell fibers, which cross in the optic chiasm, are especially vulnerable to compression of the ventral optic chiasm. Occasionally, homonymous hemianopia occurs from postchiasmal compression or monocular temporal field loss from prechiasmal compression. Invasion of the cavernous sinus can produce diplopia from ocular motor nerve palsy. Early diagnosis reduces the risk of optic atrophy, vision loss, or eye misalignment.
Laboratory Investigation The presenting clinical features of functional pituitary adenomas (e.g., acromegaly, prolactinomas, or Cushing’s syndrome) should guide the laboratory studies (Table 403-2). However, for a sellar mass with no obvious clinical features of hormone excess, laboratory studies are geared toward determining the nature of the tumor and assessing the possible presence of hypopituitarism. When a pituitary adenoma is suspected based on MRI, initial hormonal evaluation usually includes (1) basal prolactin (PRL); (2) insulin-like growth factor (IGF) I; (3) 24-h urinary free cortisol (UFC) and/or overnight oral dexamethasone (1 mg) suppression test; (4) α subunit, follicle-stimulating hormone (FSH), and luteinizing hormone (LH); and (5) thyroid function tests. Additional hormonal evaluation may be indicated based on the results of these tests. Pending more detailed assessment of hypopituitarism, a menstrual history, measurement of testosterone and 8 A.M. cortisol levels, and thyroid function tests usually identify patients with pituitary hormone deficiencies that require hormone replacement before further testing or surgery.
|
SCREENING TESTS FOR FUNCTIONAL PITUITARY ADENOMAS |
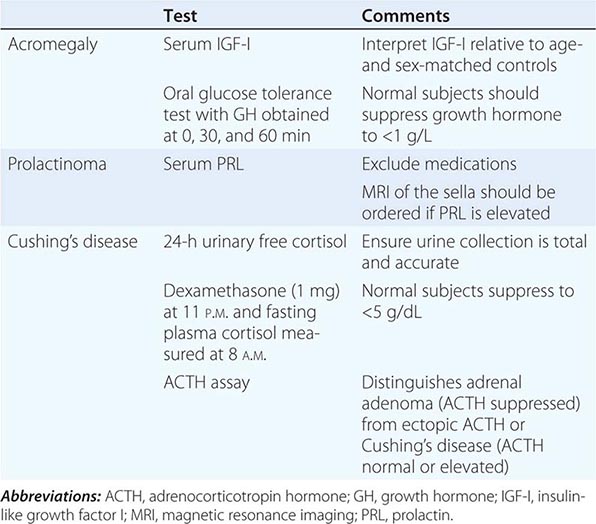
Histologic Evaluation Immunohistochemical staining of pituitary tumor specimens obtained at transsphenoidal surgery confirms clinical and laboratory studies and provides a histologic diagnosis when hormone studies are equivocal and in cases of clinically nonfunctioning tumors. Occasionally, ultrastructural assessment by electron microscopy is required for diagnosis.
|
TREATMENT |
HYPOTHALAMIC, PITUITARY, AND OTHER SELLAR MASSES |
OVERVIEW Successful management of sellar masses requires accurate diagnosis as well as selection of optimal therapeutic modalities. Most pituitary tumors are benign and slow-growing. Clinical features result from local mass effects and hormonal hyper- or hyposecretion syndromes caused directly by the adenoma or occurring as a consequence of treatment. Thus, lifelong management and follow-up are necessary for these patients.
MRI with gadolinium enhancement for pituitary visualization, new advances in transsphenoidal surgery and in stereotactic radiotherapy (including gamma-knife radiotherapy), and novel therapeutic agents have improved pituitary tumor management. The goals of pituitary tumor treatment include normalization of excess pituitary secretion, amelioration of symptoms and signs of hormonal hypersecretion syndromes, and shrinkage or ablation of large tumor masses with relief of adjacent structure compression. Residual anterior pituitary function should be preserved during treatment and sometimes can be restored by removing the tumor mass. Ideally, adenoma recurrence should be prevented.
TRANSSPHENOIDAL SURGERY Transsphenoidal rather than transfrontal resection is the desired surgical approach for pituitary tumors, except for the rare invasive suprasellar mass surrounding the frontal or middle fossa or the optic nerves or invading posteriorly behind the clivus. Intraoperative microscopy facilitates visual distinction between adenomatous and normal pituitary tissue as well as microdissection of small tumors that may not be visible by MRI (Fig. 403-2). Transsphenoidal surgery also avoids the cranial invasion and manipulation of brain tissue required by subfrontal surgical approaches. Endoscopic techniques with three-dimensional intraoperative localization have also improved visualization and access to tumor tissue. Individual surgical experience is a major determinant of outcome efficacy with these techniques.
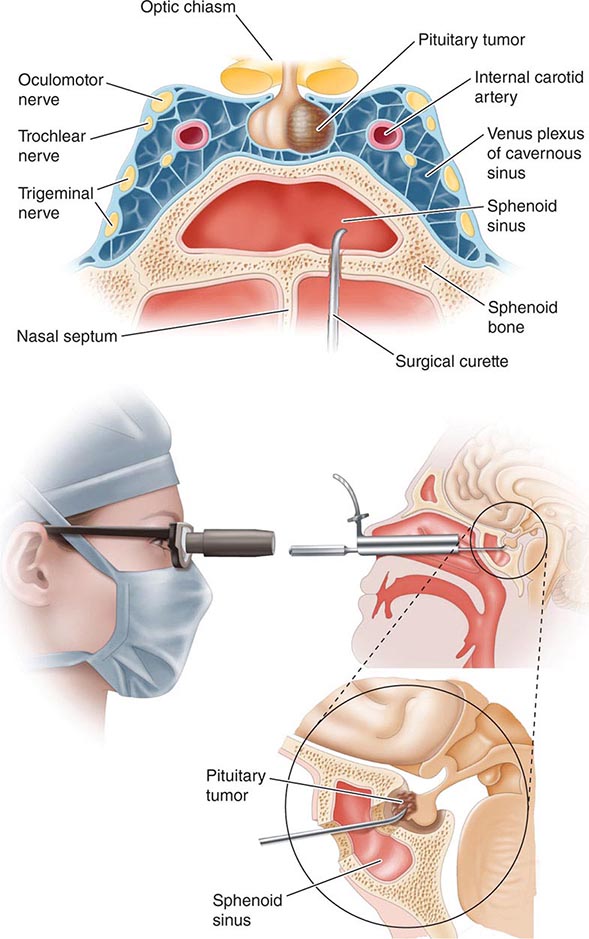
FIGURE 403-2 Transsphenoidal resection of pituitary mass via the endonasal approach. (Adapted from R Fahlbusch: Endocrinol Metab Clin 21:669, 1992.)
In addition to correction of hormonal hypersecretion, pituitary surgery is indicated for mass lesions that impinge on surrounding structures. Surgical decompression and resection are required for an expanding pituitary mass accompanied by persistent headache, progressive visual field defects, cranial nerve palsies, hydrocephalus, and, occasionally, intrapituitary hemorrhage and apoplexy. Transsphenoidal surgery sometimes is used for pituitary tissue biopsy to establish a histologic diagnosis. Whenever possible, the pituitary mass lesion should be selectively excised; normal pituitary tissue should be manipulated or resected only when critical for effective mass dissection. Nonselective hemihypophysectomy or total hypophysectomy may be indicated if no hypersecreting mass lesion is clearly discernible, multifocal lesions are present, or the remaining nontumorous pituitary tissue is obviously necrotic. This strategy, however, increases the likelihood of hypopituitarism and the need for lifelong hormone replacement.
Preoperative mass effects, including visual field defects and compromised pituitary function, may be reversed by surgery, particularly when the deficits are not long-standing. For large and invasive tumors, it is necessary to determine the optimal balance between maximal tumor resection and preservation of anterior pituitary function, especially for preserving growth and reproductive function in younger patients. Similarly, tumor invasion outside the sella is rarely amenable to surgical cure; the surgeon must judge the risk-versus-benefit ratio of extensive tumor resection.
Side Effects Tumor size, the degree of invasiveness, and experience of the surgeon largely determine the incidence of surgical complications. Operative mortality rate is about 1%. Transient diabetes insipidus and hypopituitarism occur in up to 20% of patients. Permanent diabetes insipidus, cranial nerve damage, nasal septal perforation, or visual disturbances may be encountered in up to 10% of patients. CSF leaks occur in 4% of patients. Less common complications include carotid artery injury, loss of vision, hypothalamic damage, and meningitis. Permanent side effects are rare after surgery for microadenomas.
RADIATION
Radiation is used either as a primary therapy for pituitary or parasellar masses or, more commonly, as an adjunct to surgery or medical therapy. Focused megavoltage irradiation is achieved by precise MRI localization, using a high-voltage linear accelerator and accurate isocentric rotational arcing. A major determinant of accurate irradiation is reproduction of the patient’s head position during multiple visits and maintenance of absolute head immobility. A total of <50 Gy (5000 rad) is given as 180-cGy (180-rad) fractions divided over about 6 weeks. Stereotactic radiosurgery delivers a large single high-energy dose from a cobalt-60 source (gamma knife), linear accelerator, or cyclotron. Long-term effects of gamma-knife surgery are unclear but appear to be similar to those encountered with conventional radiation. Proton beam therapy is available in some centers and provides concentrated radiation doses within a localized region.
The role of radiation therapy in pituitary tumor management depends on multiple factors, including the nature of the tumor, the age of the patient, and the availability of surgical and radiation expertise. Because of its relatively slow onset of action, radiation therapy is usually reserved for postsurgical management. As an adjuvant to surgery, radiation is used to treat residual tumor and in an attempt to prevent regrowth. Irradiation offers the only means for potentially ablating significant postoperative residual nonfunctioning tumor tissue. In contrast, PRL- and growth hormone (GH)-secreting tumor tissues are amenable to medical therapy.
Side Effects In the short term, radiation may cause transient nausea and weakness. Alopecia and loss of taste and smell may be more long-lasting. Failure of pituitary hormone synthesis is common in patients who have undergone head and neck or pituitary-directed irradiation. More than 50% of patients develop loss of GH, adrenocorticotropin hormone (ACTH), thyroid-stimulating hormone (TSH), and/or gonadotropin secretion within 10 years, usually due to hypothalamic damage. Lifelong follow-up with testing of anterior pituitary hormone reserve is therefore required after radiation treatment. Optic nerve damage with impaired vision due to optic neuritis is reported in about 2% of patients who undergo pituitary irradiation. Cranial nerve damage is uncommon now that radiation doses are ≤2 Gy (200 rad) at any one treatment session and the maximum dose is <50 Gy (5000 rad). The use of stereotactic radiotherapy may reduce damage to adjacent structures. Radiotherapy for pituitary tumors has been associated with adverse mortality rates, mainly from cerebrovascular disease. The cumulative risk of developing a secondary tumor after conventional radiation is 1.3% after 10 years and 1.9% after 20 years.
MEDICAL
Medical therapy for pituitary tumors is highly specific and depends on tumor type. For prolactinomas, dopamine agonists are the treatment of choice. For acromegaly, somatostatin analogues and GH receptor antagonists are indicated. For TSH-secreting tumors, somatostatin analogues and occasionally dopamine agonists are indicated. ACTH-secreting tumors and nonfunctioning tumors are generally not responsive to medications and require surgery and/or irradiation.
SELLAR MASSES
Sellar masses other than pituitary adenomas may arise from brain, hypothalamic, or pituitary tissues. Each exhibit features related to the lesion location but also unique to the specific etiology.
Hypothalamic Lesions Lesions involving the anterior and preoptic hypothalamic regions cause paradoxical vasoconstriction, tachycardia, and hyperthermia. Acute hyperthermia usually is due to a hemorrhagic insult, but poikilothermia may also occur. Central disorders of thermoregulation result from posterior hypothalamic damage. The periodic hypothermia syndrome is characterized by episodic attacks of rectal temperatures <30°C (86°F), sweating, vasodilation, vomiting, and bradycardia (Chap. 478e). Damage to the ventromedial hypothalamic nuclei by craniopharyngiomas, hypothalamic trauma, or inflammatory disorders may be associated with hyperphagia and obesity. This region appears to contain an energy-satiety center where melanocortin receptors are influenced by leptin, insulin, pro-opiomelanocortin (POMC) products, and gastrointestinal peptides (Chap. 415e). Polydipsia and hypodipsia are associated with damage to central osmoreceptors located in preoptic nuclei (Chap. 404). Slow-growing hypothalamic lesions can cause increased somnolence and disturbed sleep cycles as well as obesity, hypothermia, and emotional outbursts. Lesions of the central hypothalamus may stimulate sympathetic neurons, leading to elevated serum catecholamine and cortisol levels. These patients are predisposed to cardiac arrhythmias, hypertension, and gastric erosions.
Craniopharyngiomas are benign, suprasellar cystic masses that present with headaches, visual field deficits, and variable degrees of hypopituitarism. They are derived from Rathke’s pouch and arise near the pituitary stalk, commonly extending into the suprasellar cistern. Craniopharyngiomas are often large, cystic, and locally invasive. Many are partially calcified, exhibiting a characteristic appearance on skull x-ray and CT images. More than half of all patients present before age 20, usually with signs of increased intracranial pressure, including headache, vomiting, papilledema, and hydrocephalus. Associated symptoms include visual field abnormalities, personality changes and cognitive deterioration, cranial nerve damage, sleep difficulties, and weight gain. Hypopituitarism can be documented in about 90%, and diabetes insipidus occurs in about 10% of patients. About half of affected children present with growth retardation. MRI is generally superior to CT for evaluating cystic structure and tissue components of craniopharyngiomas. CT is useful to define calcifications and evaluate invasion into surrounding bony structures and sinuses.
Treatment usually involves transcranial or transsphenoidal surgical resection followed by postoperative radiation of residual tumor. Surgery alone is curative in less than half of patients because of recurrences due to adherence to vital structures or because of small tumor deposits in the hypothalamus or brain parenchyma. The goal of surgery is to remove as much tumor as possible without risking complications associated with efforts to remove firmly adherent or inaccessible tissue. In the absence of radiotherapy, about 75% of craniopharyngiomas recur, and 10-year survival is less than 50%. In patients with incomplete resection, radiotherapy improves 10-year survival to 70–90% but is associated with increased risk of secondary malignancies. Most patients require lifelong pituitary hormone replacement.
Developmental failure of Rathke’s pouch obliteration may lead to Rathke’s cysts, which are small (<5 mm) cysts entrapped by squamous epithelium and are found in about 20% of individuals at autopsy. Although Rathke’s cleft cysts do not usually grow and are often diagnosed incidentally, about a third present in adulthood with compressive symptoms, diabetes insipidus, and hyperprolactinemia due to stalk compression. Rarely, hydrocephalus develops. The diagnosis is suggested preoperatively by visualizing the cyst wall on MRI, which distinguishes these lesions from craniopharyngiomas. Cyst contents range from CSF-like fluid to mucoid material. Arachnoid cysts are rare and generate an MRI image that is isointense with CSF.
Sella chordomas usually present with bony clival erosion, local invasiveness, and, on occasion, calcification. Normal pituitary tissue may be visible on MRI, distinguishing chordomas from aggressive pituitary adenomas. Mucinous material may be obtained by fine-needle aspiration.
Meningiomas arising in the sellar region may be difficult to distinguish from nonfunctioning pituitary adenomas. Meningiomas typically enhance on MRI and may show evidence of calcification or bony erosion. Meningiomas may cause compressive symptoms.
Histiocytosis X includes a variety of syndromes associated with foci of eosinophilic granulomas. Diabetes insipidus, exophthalmos, and punched-out lytic bone lesions (Hand-Schüller-Christian disease) are associated with granulomatous lesions visible on MRI, as well as a characteristic axillary skin rash. Rarely, the pituitary stalk may be involved.
Pituitary metastases occur in ~3% of cancer patients. Bloodborne metastatic deposits are found almost exclusively in the posterior pituitary. Accordingly, diabetes insipidus can be a presenting feature of lung, gastrointestinal, breast, and other pituitary metastases. About half of pituitary metastases originate from breast cancer; about 25% of patients with metastatic breast cancer have such deposits. Rarely, pituitary stalk involvement results in anterior pituitary insufficiency. The MRI diagnosis of a metastatic lesion may be difficult to distinguish from an aggressive pituitary adenoma; the diagnosis may require histologic examination of excised tumor tissue. Primary or metastatic lymphoma, leukemias, and plasmacytomas also occur within the sella.
Hypothalamic hamartomas and gangliocytomas may arise from astrocytes, oligodendrocytes, and neurons with varying degrees of differentiation. These tumors may overexpress hypothalamic neuropeptides, including gonadotropin-releasing hormone (GnRH), growth hormone–releasing hormone (GHRH), and corticotropin-releasing hormone (CRH). With GnRH-producing tumors, children present with precocious puberty, psychomotor delay, and laughing-associated seizures. Medical treatment of GnRH-producing hamartomas with long-acting GnRH analogues effectively suppresses gonadotropin secretion and controls premature pubertal development. Rarely, hamartomas also are associated with craniofacial abnormalities; imperforate anus; cardiac, renal, and lung disorders; and pituitary failure as features of Pallister-Hall syndrome, which is caused by mutations in the carboxy terminus of the GLI3 gene. Hypothalamic hamartomas are often contiguous with the pituitary, and preoperative MRI diagnosis may not be possible. Histologic evidence of hypothalamic neurons in tissue resected at transsphenoidal surgery may be the first indication of a primary hypothalamic lesion.
Hypothalamic gliomas and optic gliomas occur mainly in childhood and usually present with visual loss. Adults have more aggressive tumors; about a third are associated with neurofibromatosis.
Brain germ cell tumors may arise within the sellar region. They include dysgerminomas, which frequently are associated with diabetes insipidus and visual loss. They rarely metastasize. Germinomas, embryonal carcinomas, teratomas, and choriocarcinomas may arise in the parasellar region and produce hCG. These germ cell tumors present with precocious puberty, diabetes insipidus, visual field defects, and thirst disorders. Many patients are GH-deficient with short stature.
PITUITARY ADENOMAS AND HYPERSECRETION SYNDROMES
Pituitary adenomas are the most common cause of pituitary hormone hypersecretion and hyposecretion syndromes in adults. They account for ~15% of all intracranial neoplasms and have been identified with a population prevalence of ~80/100,000. At autopsy, up to one-quarter of all pituitary glands harbor an unsuspected microadenoma (<10 mm diameter). Similarly, pituitary imaging detects small clinically inapparent pituitary lesions in at least 10% of individuals.
Pathogenesis Pituitary adenomas are benign neoplasms that arise from one of the five anterior pituitary cell types. The clinical and biochemical phenotypes of pituitary adenomas depend on the cell type from which they are derived. Thus, tumors arising from lactotrope (PRL), somatotrope (GH), corticotrope (ACTH), thyrotrope (TSH), or gonadotrope (LH, FSH) cells hypersecrete their respective hormones (Table 403-3). Plurihormonal tumors express various combinations of GH, PRL, TSH, ACTH, or the glycoprotein hormone α or β subunits. They may be diagnosed by careful immunocytochemistry or may manifest as clinical syndromes that combine features of these hormonal hypersecretory syndromes. Morphologically, these tumors may arise from a single polysecreting cell type or include cells with mixed function within the same tumor.
|
CLASSIFICATION OF PITUITARY ADENOMASa |
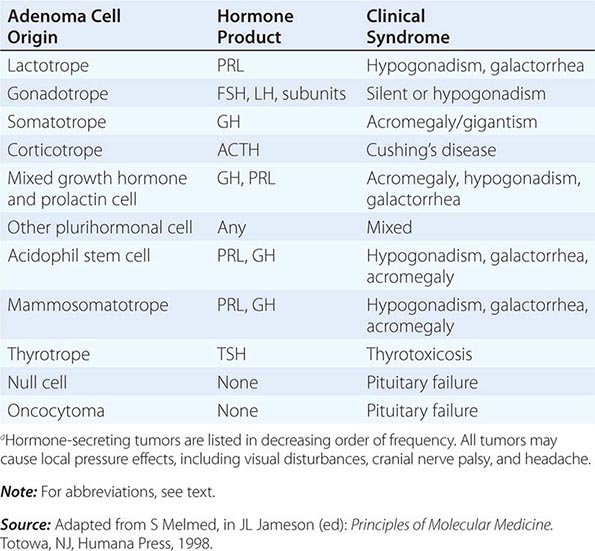
Hormonally active tumors are characterized by autonomous hormone secretion with diminished feedback responsiveness to physiologic inhibitory pathways. Hormone production does not always correlate with tumor size. Small hormone-secreting adenomas may cause significant clinical perturbations, whereas larger adenomas that produce less hormone may be clinically silent and remain undiagnosed (if no central compressive effects occur). About one-third of all adenomas are clinically nonfunctioning and produce no distinct clinical hypersecretory syndrome. Most of them arise from gonadotrope cells and may secrete small amounts of α- and β-glycoprotein hormone subunits or, very rarely, intact circulating gonadotropins. True pituitary carcinomas with documented extracranial metastases are exceedingly rare.
Almost all pituitary adenomas are monoclonal in origin, implying the acquisition of one or more somatic mutations that confer a selective growth advantage. Consistent with their clonal origin, complete surgical resection of small pituitary adenomas usually cures hormone hypersecretion. Nevertheless, hypothalamic hormones such as GHRH and CRH also enhance mitotic activity of their respective pituitary target cells in addition to their role in pituitary hormone regulation. Thus, patients who harbor rare abdominal or chest tumors that elaborate ectopic GHRH or CRH may present with somatotrope or corticotrope hyperplasia with GH or ACTH hypersecretion.
Several etiologic genetic events have been implicated in the development of pituitary tumors. The pathogenesis of sporadic forms of acromegaly has been particularly informative as a model of tumorigenesis. GHRH, after binding to its G protein–coupled somatotrope receptor, uses cyclic adenosine monophosphate (AMP) as a second messenger to stimulate GH secretion and somatotrope proliferation. A subset (~35%) of GH-secreting pituitary tumors contains sporadic mutations in Gsα (Arg 201 → Cys or His; Gln 227 → Arg). These mutations attenuate intrinsic GTPase activity, resulting in constitutive elevation of cyclic AMP, Pit-1 induction, and activation of cyclic AMP response element binding protein (CREB), thereby promoting somatotrope cell proliferation and GH secretion.
Characteristic loss of heterozygosity (LOH) in various chromosomes has been documented in large or invasive macroadenomas, suggesting the presence of putative tumor suppressor genes at these loci in up to 20% of sporadic pituitary tumors, including GH-, PRL-, and ACTH-producing adenomas and some nonfunctioning tumors. Lineage-specific cell cycle disruptions with elevated levels of CDK inhibitors are present in most of these adenomas.
Compelling evidence also favors growth factor promotion of pituitary tumor proliferation. Basic fibroblast growth factor (bFGF) is abundant in the pituitary and stimulates pituitary cell mitogenesis, whereas epithelial growth factor (EGF) receptor signaling induces both hormone synthesis and cell proliferation. Other factors involved in initiation and promotion of pituitary tumors include loss of negative-feedback inhibition (as seen with primary hypothyroidism or hypogonadism) and estrogen-mediated or paracrine angiogenesis. Growth characteristics and neoplastic behavior also may be influenced by several activated oncogenes, including RAS and pituitary tumor transforming gene (PTTG), or inactivation of growth suppressor genes, including MEG3.
Genetic Syndromes Associated with Pituitary Tumors Several familial syndromes are associated with pituitary tumors, and the genetic mechanisms for some of them have been unraveled (Table 403-4).
|
FAMILIAL PITUITARY TUMOR SYNDROMES |
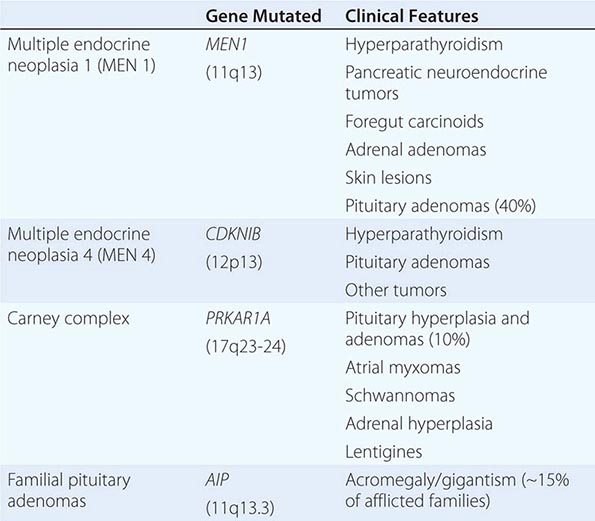
Multiple endocrine neoplasia (MEN) 1 is an autosomal dominant syndrome characterized primarily by a genetic predisposition to parathyroid, pancreatic islet, and pituitary adenomas (Chap. 408). MEN1 is caused by inactivating germline mutations in MENIN, a constitutively expressed tumor-suppressor gene located on chromosome 11q13. Loss of heterozygosity or a somatic mutation of the remaining normal MENIN allele leads to tumorigenesis. About half of affected patients develop prolactinomas; acromegaly and Cushing’s syndrome are less commonly encountered.
Carney’s syndrome is characterized by spotty skin pigmentation, myxomas, and endocrine tumors, including testicular, adrenal, and pituitary adenomas. Acromegaly occurs in about 20% of these patients. A subset of patients have mutations in the R1α regulatory subunit of protein kinase A (PRKAR1A).
McCune-Albright syndrome consists of polyostotic fibrous dysplasia, pigmented skin patches, and a variety of endocrine disorders, including acromegaly, adrenal adenomas, and autonomous ovarian function (Chap. 426e). Hormonal hypersecretion results from constitutive cyclic AMP production caused by inactivation of the GTPase activity of Gsα. The Gsα mutations occur postzygotically, leading to a mosaic pattern of mutant expression.
Familial acromegaly is a rare disorder in which family members may manifest either acromegaly or gigantism. A subset of families with a predisposition for familial pituitary tumors, especially acromegaly, have been found to harbor germline mutations in the AIP gene, which encodes the aryl hydrocarbon receptor interacting protein.
HYPERPROLACTINEMIA
Etiology Hyperprolactinemia is the most common pituitary hormone hypersecretion syndrome in both men and women. PRL-secreting pituitary adenomas (prolactinomas) are the most common cause of PRL levels >200 μg/L (see below). Less pronounced PRL elevation can also be seen with microprolactinomas but is more commonly caused by drugs, pituitary stalk compression, hypothyroidism, or renal failure (Table 403-5).
|
ETIOLOGY OF HYPERPROLACTINEMIA |
Note: Hyperprolactinemia >200 μg/L almost invariably is indicative of a prolactin-secreting pituitary adenoma. Physiologic causes, hypothyroidism, and drug-induced hyperprolactinemia should be excluded before extensive evaluation.
Pregnancy and lactation are the important physiologic causes of hyperprolactinemia. Sleep-associated hyperprolactinemia reverts to normal within an hour of awakening. Nipple stimulation and sexual orgasm also may increase PRL. Chest wall stimulation or trauma (including chest surgery and herpes zoster) invoke the reflex suckling arc with resultant hyperprolactinemia. Chronic renal failure elevates PRL by decreasing peripheral clearance. Primary hypothyroidism is associated with mild hyperprolactinemia, probably because of compensatory TRH secretion.
Lesions of the hypothalamic-pituitary region that disrupt hypothalamic dopamine synthesis, portal vessel delivery, or lactotrope responses are associated with hyperprolactinemia. Thus, hypothalamic tumors, cysts, infiltrative disorders, and radiation-induced damage cause elevated PRL levels, usually in the range of 30–100 μg/L. Plurihormonal adenomas (including GH and ACTH tumors) may hypersecrete PRL directly. Pituitary masses, including clinically nonfunctioning pituitary tumors, may compress the pituitary stalk to cause hyperprolactinemia.
Drug-induced inhibition or disruption of dopaminergic receptor function is a common cause of hyperprolactinemia (Table 403-5). Thus, antipsychotics and antidepressants are a relatively common cause of mild hyperprolactinemia. Most patients receiving risperidone have elevated prolactin levels, sometimes exceeding 200 μg/L. Methyldopa inhibits dopamine synthesis, and verapamil blocks dopamine release, also leading to hyperprolactinemia. Hormonal agents that induce PRL include estrogens and thyrotropin-releasing hormone (TRH).
Presentation and Diagnosis Amenorrhea, galactorrhea, and infertility are the hallmarks of hyperprolactinemia in women. If hyperprolactinemia develops before menarche, primary amenorrhea results. More commonly, hyperprolactinemia develops later in life and leads to oligomenorrhea and ultimately to amenorrhea. If hyperprolactinemia is sustained, vertebral bone mineral density can be reduced compared with age-matched controls, particularly when it is associated with pronounced hypoestrogenemia. Galactorrhea is present in up to 80% of hyperprolactinemic women. Although usually bilateral and spontaneous, it may be unilateral or expressed only manually. Patients also may complain of decreased libido, weight gain, and mild hirsutism.
In men with hyperprolactinemia, diminished libido, infertility, and visual loss (from optic nerve compression) are the usual presenting symptoms. Gonadotropin suppression leads to reduced testosterone, impotence, and oligospermia. True galactorrhea is uncommon in men with hyperprolactinemia. If the disorder is long-standing, secondary effects of hypogonadism are evident, including osteopenia, reduced muscle mass, and decreased beard growth.
The diagnosis of idiopathic hyperprolactinemia is made by exclusion of known causes of hyperprolactinemia in the setting of a normal pituitary MRI. Some of these patients may harbor small microadenomas below visible MRI sensitivity (~2 mm).
GALACTORRHEA
Galactorrhea, the inappropriate discharge of milk-containing fluid from the breast, is considered abnormal if it persists longer than 6 months after childbirth or discontinuation of breast-feeding. Postpartum galactorrhea associated with amenorrhea is a self-limiting disorder usually associated with moderately elevated PRL levels. Galactorrhea may occur spontaneously, or it may be elicited by nipple pressure. In both men and women, galactorrhea may vary in color and consistency (transparent, milky, or bloody) and arise either unilaterally or bilaterally. Mammography or ultrasound is indicated for bloody discharges (particularly from a single nipple), which may be caused by breast cancer. Galactorrhea is commonly associated with hyperprolactinemia caused by any of the conditions listed in Table 403-5. Acromegaly is associated with galactorrhea in about one-third of patients. Treatment of galactorrhea usually involves managing the underlying disorder (e.g., replacing T4 for hypothyroidism, discontinuing a medication, treating prolactinoma).
Laboratory Investigation Basal, fasting morning PRL levels (normally <20 μg/L) should be measured to assess hypersecretion. Both false-positive and false-negative results may be encountered. In patients with markedly elevated PRL levels (>1000 μg/L), reported results may be falsely lowered because of assay artifacts; sample dilution is required to measure these high values accurately. Falsely elevated values may be caused by aggregated forms of circulating PRL, which are usually biologically inactive (macroprolactinemia). Hypothyroidism should be excluded by measuring TSH and T4 levels.
|
TREATMENT |
HYPERPROLACTINEMIA |
Treatment of hyperprolactinemia depends on the cause of elevated PRL levels. Regardless of the etiology, however, treatment should be aimed at normalizing PRL levels to alleviate suppressive effects on gonadal function, halt galactorrhea, and preserve bone mineral density. Dopamine agonists are effective for most causes of hyperprolactinemia (see the treatment section for prolactinoma, below) regardless of the underlying cause.
If the patient is taking a medication known to cause hyperprolactinemia, the drug should be withdrawn, if possible. For psychiatric patients who require neuroleptic agents, supervised dose titration or the addition of a dopamine agonist can help restore normoprolactinemia and alleviate reproductive symptoms. However, dopamine agonists may worsen the underlying psychiatric condition, especially at high doses. Hyperprolactinemia usually resolves after adequate thyroid hormone replacement in hypothyroid patients or after renal transplantation in patients undergoing dialysis. Resection of hypothalamic or sellar mass lesions can reverse hyperprolactinemia caused by stalk compression and reduced dopamine tone. Granulomatous infiltrates occasionally respond to glucocorticoid administration. In patients with irreversible hypothalamic damage, no treatment may be warranted. In up to 30% of patients with hyperprolactinemia—usually without a visible pituitary microadenoma—the condition may resolve spontaneously.
PROLACTINOMA
Etiology and Prevalence Tumors arising from lactotrope cells account for about half of all functioning pituitary tumors, with a population prevalence of ~10/100,000 in men and ~30/100,000 in women. Mixed tumors that secrete combinations of GH and PRL, ACTH and PRL, and rarely TSH and PRL are also seen. These plurihormonal tumors are usually recognized by immunohistochemistry, sometimes without apparent clinical manifestations from the production of additional hormones. Microadenomas are classified as <1 cm in diameter and usually do not invade the parasellar region. Macroadenomas are >1 cm in diameter and may be locally invasive and impinge on adjacent structures. The female-to-male ratio for microprolactinomas is 20:1, whereas the sex ratio is near 1:1 for macroadenomas. Tumor size generally correlates directly with PRL concentrations; values >250 μg/L usually are associated with macroadenomas. Men tend to present with larger tumors than women, possibly because the features of male hypogonadism are less readily evident. PRL levels remain stable in most patients, reflecting the slow growth of these tumors. About 5% of microadenomas progress in the long term to macroadenomas.
Presentation and Diagnosis Women usually present with amenorrhea, infertility, and galactorrhea. If the tumor extends outside the sella, visual field defects or other mass effects may be seen. Men often present with impotence, loss of libido, infertility, or signs of central nervous system (CNS) compression, including headaches and visual defects. Assuming that physiologic and medication-induced causes of hyperprolactinemia are excluded (Table 403-5), the diagnosis of prolactinoma is likely with a PRL level >200 μg/L. PRL levels <100 μg/L may be caused by microadenomas, other sellar lesions that decrease dopamine inhibition, or nonneoplastic causes of hyperprolactinemia. For this reason, an MRI should be performed in all patients with hyperprolactinemia. It is important to remember that hyperprolactinemia caused secondarily by the mass effects of nonlactotrope lesions is also corrected by treatment with dopamine agonists despite failure to shrink the underlying mass. Consequently, PRL suppression by dopamine agonists does not necessarily indicate that the underlying lesion is a prolactinoma.
|
TREATMENT |
PROLACTINOMA |
Because microadenomas rarely progress to become macroadenomas, no treatment may be needed if patients are asymptomatic and fertility is not desired; these patients should be monitored by regular serial PRL measurements and MRI scans. For symptomatic microadenomas, therapeutic goals include control of hyperprolactinemia, reduction of tumor size, restoration of menses and fertility, and resolution of galactorrhea. Dopamine agonist doses should be titrated to achieve maximal PRL suppression and restoration of reproductive function (Fig. 403-3). A normalized PRL level does not ensure reduced tumor size. However, tumor shrinkage usually is not seen in those who do not respond with lowered PRL levels. For macroadenomas, formal visual field testing should be performed before initiating dopamine agonists. MRI and visual fields should be assessed at 6- to 12-month intervals until the mass shrinks and annually thereafter until maximum size reduction has occurred.
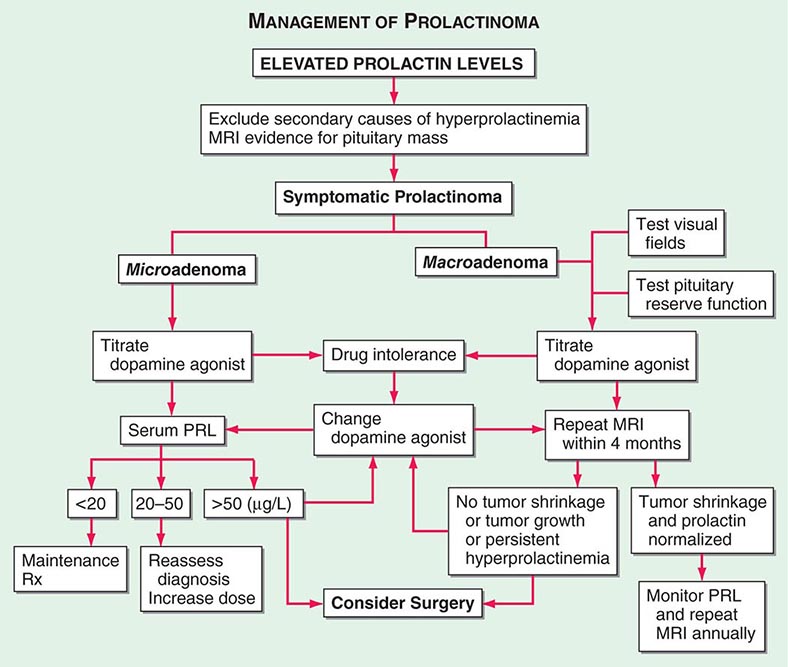
FIGURE 403-3 Management of prolactinoma. MRI, magnetic resonance imaging; PRL, prolactin.
MEDICAL
Oral dopamine agonists (cabergoline and bromocriptine) are the mainstay of therapy for patients with micro- or macroprolactinomas. Dopamine agonists suppress PRL secretion and synthesis as well as lactotrope cell proliferation. In patients with microadenomas who have achieved normoprolactinemia and significant reduction of tumor mass, the dopamine agonist may be withdrawn after 2 years. These patients should be monitored carefully for evidence of prolactinoma recurrence. About 20% of patients (especially males) are resistant to dopaminergic treatment; these adenomas may exhibit decreased D2 dopamine receptor numbers or a postreceptor defect. D2 receptor gene mutations in the pituitary have not been reported.
Cabergoline An ergoline derivative, cabergoline is a long-acting dopamine agonist with high D2 receptor affinity. The drug effectively suppresses PRL for >14 days after a single oral dose and induces prolactinoma shrinkage in most patients. Cabergoline (0.5–1.0 mg twice weekly) achieves normoprolactinemia and resumption of normal gonadal function in ~80% of patients with microadenomas; galactorrhea improves or resolves in 90% of patients. Cabergoline normalizes PRL and shrinks ~70% of macroprolactinomas. Mass effect symptoms, including headaches and visual disorders, usually improve dramatically within days after cabergoline initiation; improvement of sexual function requires several weeks of treatment but may occur before complete normalization of prolactin levels. After initial control of PRL levels has been achieved, cabergoline should be reduced to the lowest effective maintenance dose. In ~5% of treated patients harboring a microadenoma, hyperprolactinemia may resolve and not recur when dopamine agonists are discontinued after long-term treatment. Cabergoline also may be effective in patients resistant to bromocriptine. Adverse effects and drug intolerance are encountered less commonly than with bromocriptine.
BROMOCRIPTINE
The ergot alkaloid bromocriptine mesylate is a dopamine receptor agonist that suppresses prolactin secretion. Because it is short-acting, the drug is preferred when pregnancy is desired. In microadenomas, bromocriptine rapidly lowers serum prolactin levels to normal in up to 70% of patients, decreases tumor size, and restores gonadal function. In patients with macroadenomas, prolactin levels are also normalized in 70% of patients, and tumor mass shrinkage (≥50%) is achieved in most patients.
Therapy is initiated by administering a low bromocriptine dose (0.625–1.25 mg) at bedtime with a snack, followed by gradually increasing the dose. Most patients are controlled with a daily dose of ≤7.5 mg (2.5 mg tid).
SIDE EFFECTS
Side effects of dopamine agonists include constipation, nasal stuffiness, dry mouth, nightmares, insomnia, and vertigo; decreasing the dose usually alleviates these problems. Nausea, vomiting, and postural hypotension with faintness may occur in ~25% of patients after the initial dose. These symptoms may persist in some patients. In general, fewer side effects are reported with cabergoline. For the approximately 15% of patients who are intolerant of oral bromocriptine, cabergoline may be better tolerated. Intravaginal administration of bromocriptine is often efficacious in patients with intractable gastrointestinal side effects. Auditory hallucinations, delusions, and mood swings have been reported in up to 5% of patients and may be due to the dopamine agonist properties or to the lysergic acid derivative of the compounds. Rare reports of leukopenia, thrombocytopenia, pleural fibrosis, cardiac arrhythmias, and hepatitis have been described. Patients with Parkinson’s disease who receive at least 3 mg of cabergoline daily have been reported to be at risk for development of cardiac valve regurgitation. Studies analyzing over 500 prolactinoma patients receiving recommended doses of cabergoline (up to 2 mg weekly) have shown no evidence for an increased incidence of valvular disorders. Nevertheless, because no controlled prospective studies in pituitary tumor patients are available, it is prudent to perform echocardiograms before initiating standard-dose cabergoline therapy.
Surgery Indications for surgical adenoma debulking include dopamine resistance or intolerance and the presence of an invasive macroadenoma with compromised vision that fails to improve after drug treatment. Initial PRL normalization is achieved in about 70% of microprolactinomas after surgical resection, but only 30% of macroadenomas can be resected successfully. Follow-up studies have shown that hyperprolactinemia recurs in up to 20% of patients within the first year after surgery; long-term recurrence rates exceed 50% for macroadenomas. Radiotherapy for prolactinomas is reserved for patients with aggressive tumors that do not respond to maximally tolerated dopamine agonists and/or surgery.
PREGNANCY
The pituitary increases in size during pregnancy, reflecting the stimulatory effects of estrogen and perhaps other growth factors on pituitary vascularity and lactotrope cell hyperplasia. About 5% of microadenomas significantly increase in size, but 15–30% of macroadenomas grow during pregnancy. Bromocriptine has been used for more than 30 years to restore fertility in women with hyperprolactinemia, without evidence of teratogenic effects. Nonetheless, most authorities recommend strategies to minimize fetal exposure to the drug. For women taking bromocriptine who desire pregnancy, mechanical contraception should be used through three regular menstrual cycles to allow for conception timing. When pregnancy is confirmed, bromocriptine should be discontinued and PRL levels followed serially, especially if headaches or visual symptoms occur. For women harboring macroadenomas, regular visual field testing is recommended, and the drug should be reinstituted if tumor growth is apparent. Although pituitary MRI may be safe during pregnancy, this procedure should be reserved for symptomatic patients with severe headache and/or visual field defects. Surgical decompression may be indicated if vision is threatened. Although comprehensive data support the efficacy and relative safety of bromocriptine-facilitated fertility, patients should be advised of potential unknown deleterious effects and the risk of tumor growth during pregnancy. Because cabergoline is long-acting with a high D2-receptor affinity, it is not recommended for use in women when fertility is desired.
ACROMEGALY
Etiology GH hypersecretion is usually the result of a somatotrope adenoma but may rarely be caused by extrapituitary lesions (Table 403-6). In addition to the more common GH-secreting somatotrope adenomas, mixed mammosomatotrope tumors and acidophilic stem-cell adenomas secrete both GH and PRL. In patients with acidophilic stem-cell adenomas, features of hyperprolactinemia (hypogonadism and galactorrhea) predominate over the less clinically evident signs of acromegaly. Occasionally, mixed plurihormonal tumors are encountered that also secrete ACTH, the glycoprotein hormone α subunit, or TSH in addition to GH. Patients with partially empty sellas may present with GH hypersecretion due to a small GH-secreting adenoma within the compressed rim of pituitary tissue; some of these may reflect the spontaneous necrosis of tumors that were previously larger. GH-secreting tumors rarely arise from ectopic pituitary tissue remnants in the nasopharynx or midline sinuses.
|
CAUSES OF ACROMEGALY |
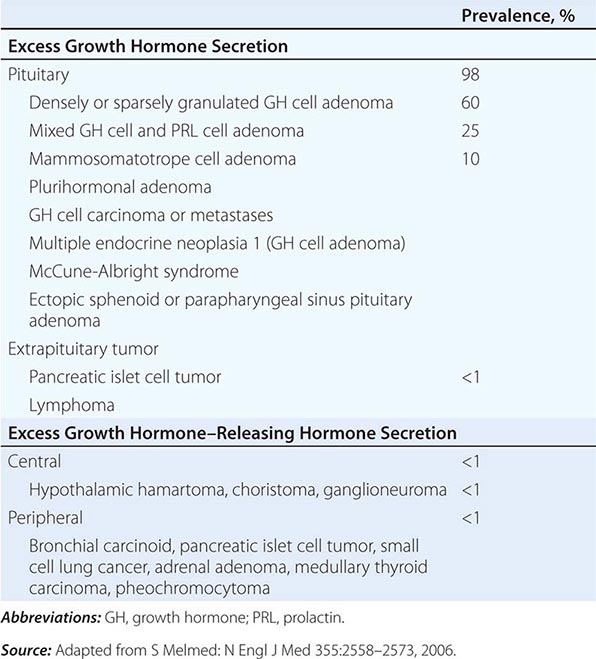
There are case reports of ectopic GH secretion by tumors of pancreatic, ovarian, lung, or hematopoietic origin. Rarely, excess GHRH production may cause acromegaly because of chronic stimulation of somatotropes. These patients present with classic features of acromegaly, elevated GH levels, pituitary enlargement on MRI, and pathologic characteristics of pituitary hyperplasia. The most common cause of GHRH-mediated acromegaly is a chest or abdominal carcinoid tumor. Although these tumors usually express positive GHRH immunoreactivity, clinical features of acromegaly are evident in only a minority of patients with carcinoid disease. Excessive GHRH also may be elaborated by hypothalamic tumors, usually choristomas or neuromas.
Presentation and Diagnosis Protean manifestations of GH and IGF-I hypersecretion are indolent and often are not clinically diagnosed for 10 years or more. Acral bony overgrowth results in frontal bossing, increased hand and foot size, mandibular enlargement with prognathism, and widened space between the lower incisor teeth. In children and adolescents, initiation of GH hypersecretion before epiphyseal long bone closure is associated with development of pituitary gigantism (Fig. 403-4). Soft tissue swelling results in increased heel pad thickness, increased shoe or glove size, ring tightening, characteristic coarse facial features, and a large fleshy nose. Other commonly encountered clinical features include hyperhidrosis, a deep and hollow-sounding voice, oily skin, arthropathy, kyphosis, carpal tunnel syndrome, proximal muscle weakness and fatigue, acanthosis nigricans, and skin tags. Generalized visceromegaly occurs, including cardiomegaly, macroglossia, and thyroid gland enlargement.
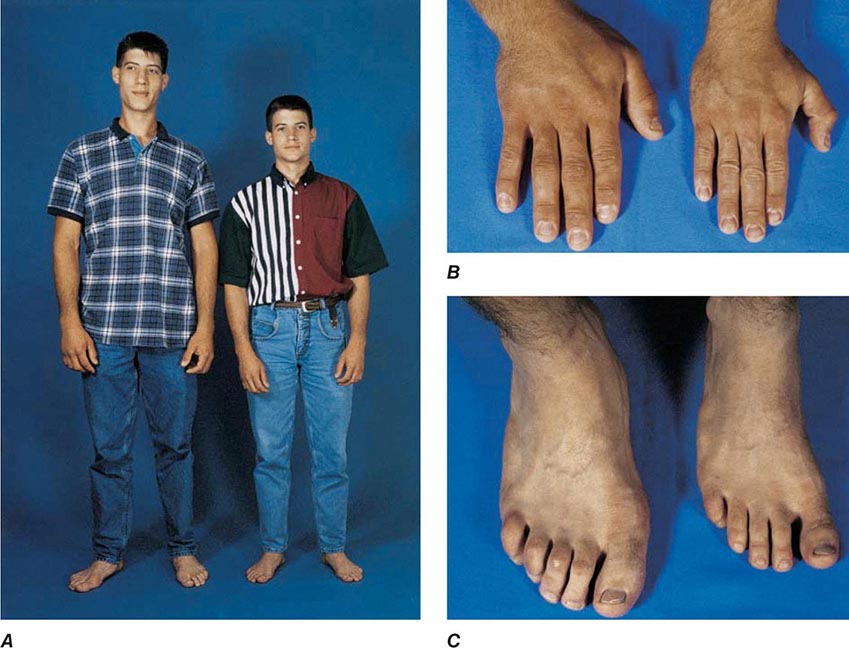
FIGURE 403-4 Features of acromegaly/gigantism. A 22-year-old man with gigantism due to excess growth hormone is shown to the left of his identical twin. The increased height and prognathism (A) and enlarged hand (B) and foot (C) of the affected twin are apparent. Their clinical features began to diverge at the age of approximately 13 years. (Reproduced from R Gagel, IE McCutcheon: N Engl J Med 324:524, 1999; with permission.)
The most significant clinical impact of GH excess occurs with respect to the cardiovascular system. Coronary heart disease, cardiomyopathy with arrhythmias, left ventricular hypertrophy, decreased diastolic function, and hypertension ultimately occur in most patients if untreated. Upper airway obstruction with sleep apnea occurs in more than 60% of patients and is associated with both soft tissue laryngeal airway obstruction and central sleep dysfunction. Diabetes mellitus develops in 25% of patients with acromegaly, and most patients are intolerant of a glucose load (as GH counteracts the action of insulin). Acromegaly is associated with an increased risk of colon polyps and mortality from colonic malignancy; polyps are diagnosed in up to one-third of patients. Overall mortality is increased about threefold and is due primarily to cardiovascular and cerebrovascular disorders and respiratory disease. Unless GH levels are controlled, survival is reduced by an average of 10 years compared with an age-matched control population.
Laboratory Investigation Age-matched serum IGF-I levels are elevated in acromegaly. Consequently, an IGF-I level provides a useful laboratory screening measure when clinical features raise the possibility of acromegaly. Due to the pulsatility of GH secretion, measurement of a single random GH level is not useful for the diagnosis or exclusion of acromegaly and does not correlate with disease severity. The diagnosis of acromegaly is confirmed by demonstrating the failure of GH suppression to <0.4 μg/L within 1–2 h of an oral glucose load (75 g). When newer ultrasensitive GH assays are used, normal nadir GH levels are even lower (<0.05 μg/L). About 20% of patients exhibit a paradoxical GH rise after glucose. PRL should be measured, as it is elevated in ~25% of patients with acromegaly. Thyroid function, gonadotropins, and sex steroids may be attenuated because of tumor mass effects. Because most patients will undergo surgery with glucocorticoid coverage, tests of ACTH reserve in asymptomatic patients are more efficiently deferred until after surgery.
|
TREATMENT |
ACROMEGALY |
The goal of treatment is to control GH and IGF-I hypersecretion, ablate or arrest tumor growth, ameliorate comorbidities, restore mortality rates to normal, and preserve pituitary function.
Surgical resection of GH-secreting adenomas is the initial treatment for most patients (Fig. 403-5). Somatostatin analogues are used as adjuvant treatment for preoperative shrinkage of large invasive macroadenomas, immediate relief of debilitating symptoms, and reduction of GH hypersecretion; in frail patients experiencing morbidity; and in patients who decline surgery or, when surgery fails, to achieve biochemical control. Irradiation or repeat surgery may be required for patients who cannot tolerate or do not respond to adjunctive medical therapy. The high rate of late hypopituitarism and the slow rate (5–15 years) of biochemical response are the main disadvantages of radiotherapy. Irradiation is also relatively ineffective in normalizing IGF-I levels. Stereotactic ablation of GH-secreting adenomas by gamma-knife radiotherapy is promising, but initial reports suggest that long-term results and side effects are similar to those observed with conventional radiation. Somatostatin analogues may be required while awaiting the full benefits of radiotherapy. Systemic co-morbid sequelae of acromegaly, including cardiovascular disease, diabetes, and arthritis, should be managed aggressively. Mandibular surgical repair may be indicated.
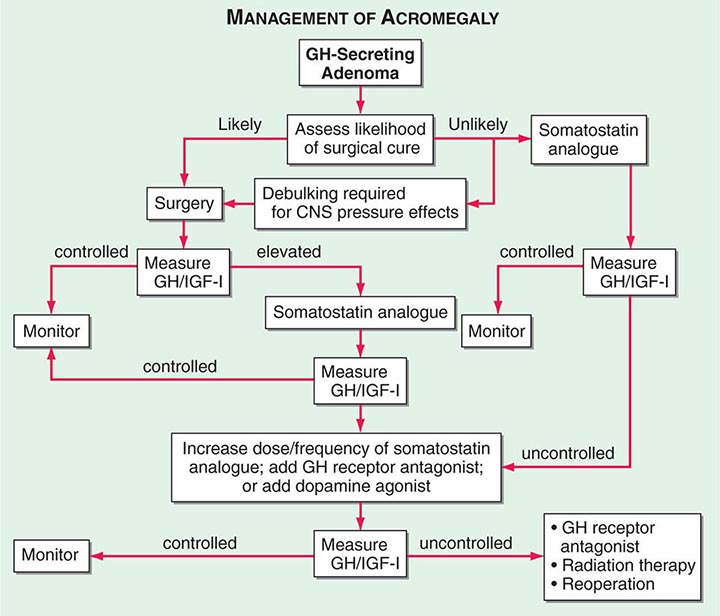
FIGURE 403-5 Management of acromegaly. GH, growth hormone; CNS, central nervous system; IGF, insulin-like growth factor. (Adapted from S Melmed et al: J Clin Endocrinol Metab 94:1509–1517, 2009; © The Endocrine Society.)
SURGERY
Transsphenoidal surgical resection by an experienced surgeon is the preferred primary treatment for both microadenomas (remission rate ~70%) and macroadenomas (<50% in remission). Soft tissue swelling improves immediately after tumor resection. GH levels return to normal within an hour, and IGF-I levels are normalized within 3–4 days. In ~10% of patients, acromegaly may recur several years after apparently successful surgery; hypopituitarism develops in up to 15% of patients after surgery.
SOMATOSTATIN ANALOGUES
Somatostatin analogues exert their therapeutic effects through SSTR2 and SSTR5 receptors, both of which are expressed by GH-secreting tumors. Octreotide acetate is an eight-amino-acid synthetic somatostatin analogue. In contrast to native somatostatin, the analogue is relatively resistant to plasma degradation. It has a 2-h serum half-life and possesses fortyfold greater potency than native somatostatin to suppress GH. Octreotide is administered by subcutaneous injection, beginning with 50 μg tid; the dose can be increased gradually up to 1500 μg/d. Fewer than 10% of patients do not respond to the analogue. Octreotide suppresses integrated GH levels and normalizes IGF-I levels in ~60% of treated patients.
The long-acting somatostatin depot formulations, octreotide and lanreotide, are the preferred medical treatment for patients with acromegaly. Sandostatin-LAR is a sustained-release, long-acting formulation of octreotide incorporated into microspheres that sustain drug levels for several weeks after intramuscular injection. GH suppression occurs for as long as 6 weeks after a 30-mg intramuscular injection; long-term monthly treatment sustains GH and IGF-I suppression and also reduces pituitary tumor size in ~50% of patients. Lanreotide autogel, a slow-release depot somatostatin preparation, is a cyclic somatostatin octapeptide analogue that suppresses GH and IGF-I hypersecretion after a 60-mg subcutaneous injection. Long-term (4–6 weeks) administration controls GH hypersecretion in about two-thirds of treated patients and improves patient compliance because of the long interval required between drug injections. Rapid relief of headache and soft tissue swelling occurs in ~75% of patients within days to weeks of somatostatin analogue initiation. Most patients report symptomatic improvement, including amelioration of headache, perspiration, obstructive apnea, and cardiac failure.
Side Effects Somatostatin analogues are well tolerated in most patients. Adverse effects are short-lived and mostly relate to drug-induced suppression of gastrointestinal motility and secretion. Transient nausea, abdominal discomfort, fat malabsorption, diarrhea, and flatulence occur in one-third of patients, and these symptoms usually remit within 2 weeks. Octreotide suppresses postprandial gallbladder contractility and delays gallbladder emptying; up to 30% of patients develop long-term echogenic sludge or asymptomatic cholesterol gallstones. Other side effects include mild glucose intolerance due to transient insulin suppression, asymptomatic bradycardia, hypothyroxinemia, and local injection site discomfort.
GH RECEPTOR ANTAGONIST
Pegvisomant antagonizes endogenous GH action by blocking peripheral GH binding to its receptor. Consequently, serum IGF-I levels are suppressed, reducing the deleterious effects of excess endogenous GH. Pegvisomant is administered by daily subcutaneous injection (10–20 mg) and normalizes IGF-I in ~70% of patients. GH levels, however, remain elevated as the drug does not target the pituitary adenoma. Side effects include reversible liver enzyme elevation, lipodystrophy, and injection site pain. Tumor size should be monitored by MRI.
Combined treatment with monthly somatostatin analogues and weekly or biweekly pegvisomant injections has been used effectively in resistant patients.
DOPAMINE AGONISTS
Bromocriptine and cabergoline may modestly suppress GH secretion in some patients. Very high doses of bromocriptine (≥20 mg/d) or cabergoline (0.5 mg/d) are usually required to achieve modest GH therapeutic efficacy. Combined treatment with octreotide and cabergoline may induce additive biochemical control compared with either drug alone.
RADIATION
External radiation therapy or high-energy stereotactic techniques are used as adjuvant therapy for acromegaly. An advantage of radiation is that patient compliance with long-term treatment is not required. Tumor mass is reduced, and GH levels are attenuated over time. However, 50% of patients require at least 8 years for GH levels to be suppressed to <5 μg/L; this level of GH reduction is achieved in about 90% of patients after 18 years but represents suboptimal GH suppression. Patients may require interim medical therapy for several years before attaining maximal radiation benefits. Most patients also experience hypothalamic-pituitary damage, leading to gonadotropin, ACTH, and/or TSH deficiency within 10 years of therapy.
In summary, surgery is the preferred primary treatment for GH-secreting microadenomas (Fig. 403-5). The high frequency of GH hypersecretion after macroadenoma resection usually necessitates adjuvant or primary medical therapy for these larger tumors. Patients unable to receive or respond to unimodal medical treatment may benefit from combined treatments, or can be offered radiation.
CUSHING’S SYNDROME (ACTH-PRODUCING ADENOMA)
(See also Chap. 406)
Etiology and Prevalence Pituitary corticotrope adenomas account for 70% of patients with endogenous causes of Cushing’s syndrome. However, it should be emphasized that iatrogenic hypercortisolism is the most common cause of cushingoid features. Ectopic tumor ACTH production, cortisol-producing adrenal adenomas, adrenal carcinoma, and adrenal hyperplasia account for the other causes; rarely, ectopic tumor CRH production is encountered.
ACTH-producing adenomas account for about 10–15% of all pituitary tumors. Because the clinical features of Cushing’s syndrome often lead to early diagnosis, most ACTH-producing pituitary tumors are relatively small microadenomas. However, macroadenomas also are seen and some ACTH-expressing adenomas are clinically silent. Cushing’s disease is 5–10 times more common in women than in men. These pituitary adenomas exhibit unrestrained ACTH secretion, with resultant hypercortisolemia. However, they retain partial suppressibility in the presence of high doses of administered glucocorticoids, providing the basis for dynamic testing to distinguish pituitary from nonpituitary causes of Cushing’s syndrome.
Presentation and Diagnosis The diagnosis of Cushing’s syndrome presents two great challenges: (1) to distinguish patients with pathologic cortisol excess from those with physiologic or other disturbances of cortisol production and (2) to determine the etiology of pathologic cortisol excess.
Typical features of chronic cortisol excess include thin skin, central obesity, hypertension, plethoric moon facies, purple striae and easy bruisability, glucose intolerance or diabetes mellitus, gonadal dysfunction, osteoporosis, proximal muscle weakness, signs of hyperandrogenism (acne, hirsutism), and psychological disturbances (depression, mania, and psychoses) (Table 403-7). Hematopoietic features of hypercortisolism include leukocytosis, lymphopenia, and eosinopenia. Immune suppression includes delayed hypersensitivity and infection propensity. These protean yet commonly encountered manifestations of hypercortisolism make it challenging to decide which patients mandate formal laboratory evaluation. Certain features make pathologic causes of hypercortisolism more likely; they include characteristic central redistribution of fat, thin skin with striae and bruising, and proximal muscle weakness. In children and young females, early osteoporosis may be particularly prominent. The primary cause of death is cardiovascular disease, but life-threatening infections and risk of suicide are also increased.
|
CLINICAL FEATURES OF CUSHING’S SYNDROME (ALL AGES) |
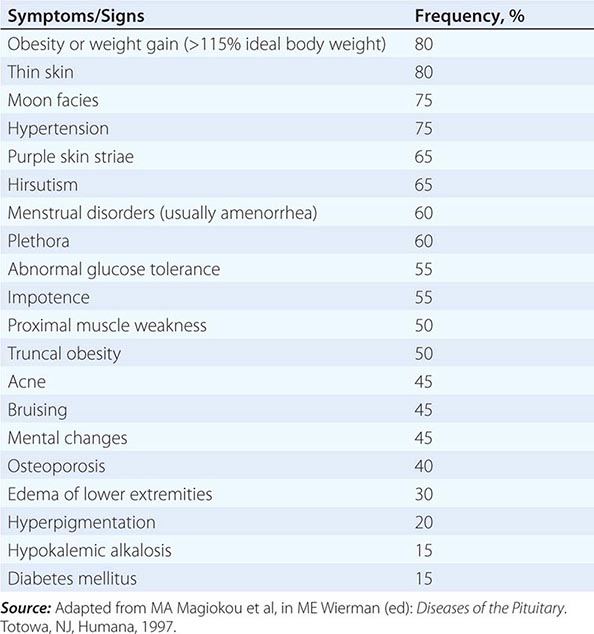
Rapid development of features of hypercortisolism associated with skin hyperpigmentation and severe myopathy suggests an ectopic tumor source of ACTH. Hypertension, hypokalemic alkalosis, glucose intolerance, and edema are also more pronounced in these patients. Serum potassium levels <3.3 mmol/L are evident in ~70% of patients with ectopic ACTH secretion but are seen in <10% of patients with pituitary-dependent Cushing’s syndrome.
Laboratory Investigation The diagnosis of Cushing’s syndrome is based on laboratory documentation of endogenous hypercortisolism. Measurement of 24-h urine free cortisol (UFC) is a precise and cost-effective screening test. Alternatively, the failure to suppress plasma cortisol after an overnight 1-mg dexamethasone suppression test can be used to identify patients with hypercortisolism. As nadir levels of cortisol occur at night, elevated midnight serum or salivary samples of cortisol are suggestive of Cushing’s syndrome. Basal plasma ACTH levels often distinguish patients with ACTH-independent (adrenal or exogenous glucocorticoid) from those with ACTH-dependent (pituitary, ectopic ACTH) Cushing’s syndrome. Mean basal ACTH levels are about eightfold higher in patients with ectopic ACTH secretion than in those with pituitary ACTH-secreting adenomas. However, extensive overlap of ACTH levels in these two disorders precludes using ACTH measurements to make the distinction. Preferably, dynamic testing based on differential sensitivity to glucocorticoid feedback or ACTH stimulation in response to CRH or cortisol reduction is used to distinguish ectopic from pituitary sources of excess ACTH (Table 403-8). Very rarely, circulating CRH levels are elevated, reflecting ectopic tumor-derived secretion of CRH and often ACTH. For further discussion of dynamic testing for Cushing’s syndrome, see Chap. 406.
|
DIFFERENTIAL DIAGNOSIS OF ACTH-DEPENDENT CUSHING’S SYNDROMEa |
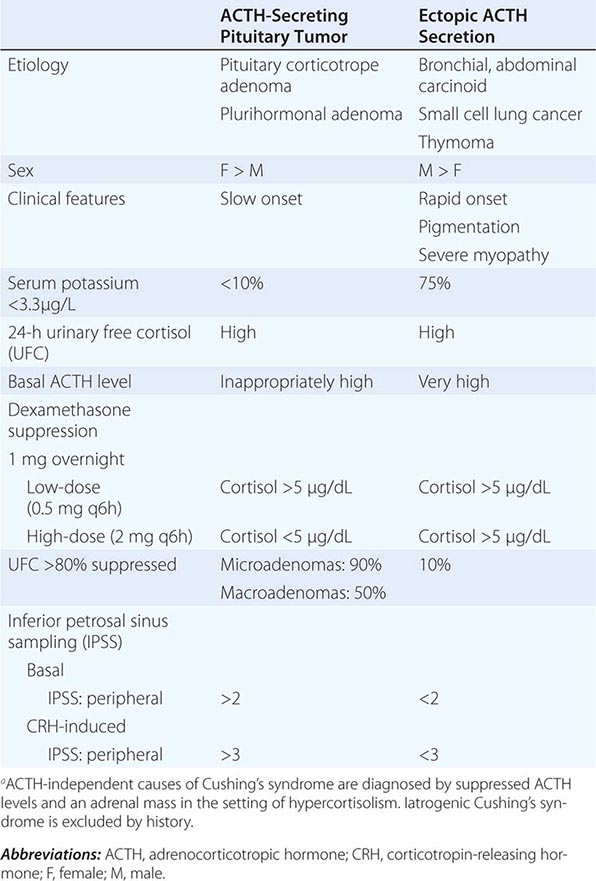
Most ACTH-secreting pituitary tumors are <5 mm in diameter, and about half are undetectable by sensitive MRI. The high prevalence of incidental pituitary microadenomas diminishes the ability to distinguish ACTH-secreting pituitary tumors accurately from nonsecreting incidentalomas.
Inferior Petrosal Venous Sampling Because pituitary MRI with gadolinium enhancement is insufficiently sensitive to detect small (<2 mm) pituitary ACTH-secreting adenomas, bilateral inferior petrosal sinus ACTH sampling before and after CRH administration may be required to distinguish these lesions from ectopic ACTH-secreting tumors that may have similar clinical and biochemical characteristics. Simultaneous assessment of ACTH in each inferior petrosal vein and in the diagnosis of peripheral circulation provides a strategy for confirming and localizing pituitary ACTH production. Sampling is performed at baseline and 2, 5, and 10 min after intravenous bovine CRH (1 μg/kg) injection. An increased ratio (>2) of inferior petrosal:peripheral vein ACTH confirms pituitary Cushing’s syndrome. After CRH injection, peak petrosal:peripheral ACTH ratios ≥3 confirm the presence of a pituitary ACTH-secreting tumor. The sensitivity of this test is >95%, with very rare false-positive results. False-negative results may be encountered in patients with aberrant venous drainage. Petrosal sinus catheterizations are technically difficult, and about 0.05% of patients develop neurovascular complications. The procedure should not be performed in patients with hypertension, in patients with known cerebrovascular disease, or in the presence of a well-visualized pituitary adenoma on MRI.
|
TREATMENT |
CUSHING’S SYNDROME |
Selective transsphenoidal resection is the treatment of choice for Cushing’s disease (Fig. 403-6). The remission rate for this procedure is ~80% for microadenomas but <50% for macroadenomas. However, surgery is rarely successful when the adenoma is not visible on MRI. After successful tumor resection, most patients experience a postoperative period of symptomatic ACTH deficiency that may last up to 12 months. This usually requires low-dose cortisol replacement, as patients experience both steroid withdrawal symptoms and have a suppressed hypothalamic-pituitary-adrenal axis. Biochemical recurrence occurs in approximately 5% of patients in whom surgery was initially successful.
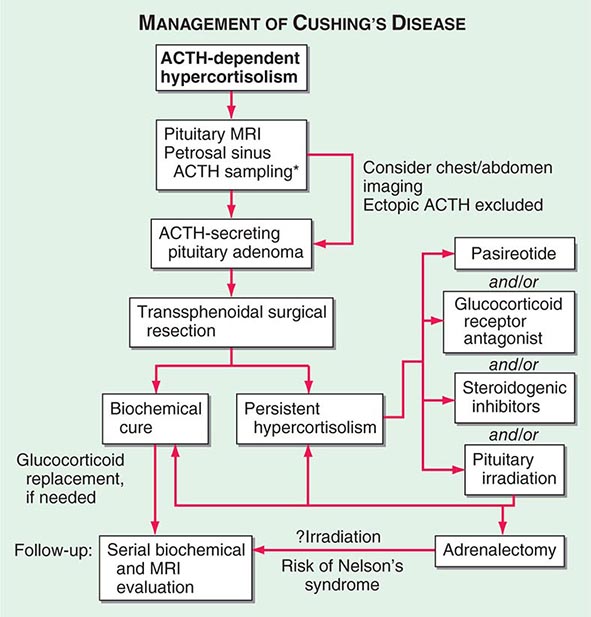
FIGURE 403-6 Management of Cushing’s syndrome. ACTH, adrenocorticotropin hormone; MRI, magnetic resonance imaging. *, Not usually required.
When initial surgery is unsuccessful, repeat surgery is sometimes indicated, particularly when a pituitary source for ACTH is well documented. In older patients, in whom issues of growth and fertility are less important, hemi- or total hypophysectomy may be necessary if a discrete pituitary adenoma is not recognized. Pituitary irradiation may be used after unsuccessful surgery, but it cures only about 15% of patients. Because the effects of radiation are slow and only partially effective in adults, steroidogenic inhibitors are used in combination with pituitary irradiation to block adrenal effects of persistently high ACTH levels.
Pasireotide (600 or 900 ug/day subcutaneously), a somatostatin analog with high affinity for SST5 > SST2 receptors, has been approved for treating patients with ACTH-secreting pituitary tumors when surgery is not an option or has been unsuccessful. In clinical trials, the drug lowered plasma ACTH levels, normalized 24-h urinary free cortisol levels in about 25% of patients, and resulted in up to 40% mean pituitary tumor shrinkage. Side effects include development of hyperglycemia and diabetes in about 70% of patients, likely due to suppressed pancreatic secretion of insulin and incretins. Because patients with hypercortisolism are insulin-resistant, hyperglycemia should be rigorously managed. Other side effects are similar to those encountered for somatostatin analogs and include transient abdominal discomfort, diarrhea, nausea, and gallstones (20% of patients). The drug requires consistent long-term administration.
Ketoconazole, an imidazole derivative antimycotic agent, inhibits several P450 enzymes and effectively lowers cortisol in most patients with Cushing’s disease when administered twice daily (600–1200 mg/d). Elevated hepatic transaminases, gynecomastia, impotence, gastrointestinal upset, and edema are common side effects.
Mifepristone (300–1200 mg/d), a glucocorticoid receptor antagonist, blocks peripheral cortisol action and is approved to treat hyperglycemia in Cushing’s disease. Because the drug does not target the pituitary tumor, both ACTH and cortisol levels remain elevated, thus obviating a reliable circulating biomarker. Side effects are largely due to general antagonism of other steroid hormones and include hypokalemia, endometrial hyperplasia, hypoadrenalism, and hypertension.
Metyrapone (2–4 g/d) inhibits 11β-hydroxylase activity and normalizes plasma cortisol in up to 75% of patients. Side effects include nausea and vomiting, rash, and exacerbation of acne or hirsutism. Mitotane (o,p ‘-DDD; 3–6 g/d orally in four divided doses) suppresses cortisol hypersecretion by inhibiting 11β-hydroxylase and cholesterol side-chain cleavage enzymes and by destroying adrenocortical cells. Side effects of mitotane include gastrointestinal symptoms, dizziness, gynecomastia, hyperlipidemia, skin rash, and hepatic enzyme elevation. It also may lead to hypoaldosteronism. Other agents include aminoglutethimide (250 mg tid), trilostane (200–1000 mg/d), cyproheptadine (24 mg/d), and IV etomidate (0.3 mg/kg per hour). Glucocorticoid insufficiency is a potential side effect of agents used to block steroidogenesis.
The use of steroidogenic inhibitors has decreased the need for bilateral adrenalectomy. Surgical removal of both adrenal glands corrects hypercortisolism but may be associated with significant morbidity rates and necessitates permanent glucocorticoid and mineralocorticoid replacement. Adrenalectomy in the setting of residual corticotrope adenoma tissue predisposes to the development of Nelson’s syndrome, a disorder characterized by rapid pituitary tumor enlargement and increased pigmentation secondary to high ACTH levels. Prophylactic radiation therapy may be indicated to prevent the development of Nelson’s syndrome after adrenalectomy.
NONFUNCTIONING AND GONADOTROPIN-PRODUCING PITUITARY ADENOMAS
Etiology and Prevalence Nonfunctioning pituitary adenomas include those that secrete little or no pituitary hormones as well as tumors that produce too little hormone to result in recognizable clinical features. They are the most common type of pituitary adenoma and are usually macroadenomas at the time of diagnosis because clinical features are not apparent until tumor mass effects occur. Based on immunohistochemistry, most clinically nonfunctioning adenomas can be shown to originate from gonadotrope cells. These tumors typically produce small amounts of intact gonadotropins (usually FSH) as well as uncombined α, LH β, and FSH β subunits. Tumor secretion may lead to elevated α and FSH β subunits and, rarely, to increased LH β subunit levels. Some adenomas express α subunits without FSH or LH. TRH administration often induces an atypical increase of tumor-derived gonadotropins or subunits.
Presentation and Diagnosis Clinically nonfunctioning tumors often present with optic chiasm pressure and other symptoms of local expansion or may be incidentally discovered on an MRI performed for another indication (incidentaloma). Rarely, menstruwal disturbances or ovarian hyperstimulation occur in women with large tumors that produce FSH and LH. More commonly, adenoma compression of the pituitary stalk or surrounding pituitary tissue leads to attenuated LH and features of hypogonadism. PRL levels are usually slightly increased, also because of stalk compression. It is important to distinguish this circumstance from true prolactinomas, as nonfunctioning tumors do not shrink in response to treatment with dopamine agonists.
Laboratory Investigation The goal of laboratory testing in clinically nonfunctioning tumors is to classify the type of the tumor, identify hormonal markers of tumor activity, and detect possible hypopituitarism. Free α subunit levels may be elevated in 10–15% of patients with nonfunctioning tumors. In female patients, peri- or postmenopausal basal FSH concentrations are difficult to distinguish from tumor-derived FSH elevation. Premenopausal women have cycling FSH levels, also preventing clear-cut diagnostic distinction from tumor-derived FSH. In men, gonadotropin-secreting tumors may be diagnosed because of slightly increased gonadotropins (FSH > LH) in the setting of a pituitary mass. Testosterone levels are usually low despite the normal or increased LH level, perhaps reflecting reduced LH bioactivity or the loss of normal LH pulsatility. Because this pattern of hormone test results is also seen in primary gonadal failure and, to some extent, with aging (Chap. 411), the finding of increased gonadotropins alone is insufficient for the diagnosis of a gonadotropin-secreting tumor. In the majority of patients with gonadotrope adenomas, TRH administration stimulates LH β subunit secretion; this response is not seen in normal individuals. GnRH testing, however, is not helpful for making the diagnosis. For nonfunctioning and gonadotropin-secreting tumors, the diagnosis usually rests on immunohistochemical analyses of surgically resected tumor tissue, as the mass effects of these tumors usually necessitate resection.
Although acromegaly or Cushing’s syndrome usually presents with unique clinical features, clinically inapparent (silent) somatotrope or corticotrope adenomas may only be diagnosed by immunostaining of resected tumor tissue. If PRL levels are <100 μg/L in a patient harboring a pituitary mass, a nonfunctioning adenoma causing pituitary stalk compression should be considered.
|
TREATMENT |
NONFUNCTIONING AND GONADOTROPIN-PRODUCING PITUITARY ADENOMAS |
Asymptomatic small nonfunctioning microadenomas adenomas with no threat to vision may be followed with regular MRI and visual field testing without immediate intervention. However, for macroadenomas, transsphenoidal surgery is indicated to reduce tumor size and relieve mass effects (Fig. 403-7). Although it is not usually possible to remove all adenoma tissue surgically, vision improves in 70% of patients with preoperative visual field defects. Preexisting hypopituitarism that results from tumor mass effects may improve or resolve completely. Beginning about 6 months postoperatively, MRI scans should be performed yearly to detect tumor regrowth. Within 5–6 years after successful surgical resection, ~15% of nonfunctioning tumors recur. When substantial tumor remains after transsphenoidal surgery, adjuvant radiotherapy may be indicated to prevent tumor regrowth. Radiotherapy may be deferred if no postoperative residual mass is evident. Nonfunctioning pituitary tumors respond poorly to dopamine agonist treatment and somatostatin analogues are largely ineffective for shrinking these tumors. The selective GnRH antagonist Nal-Glu GnRH suppresses FSH hypersecretion but has no effect on adenoma size.
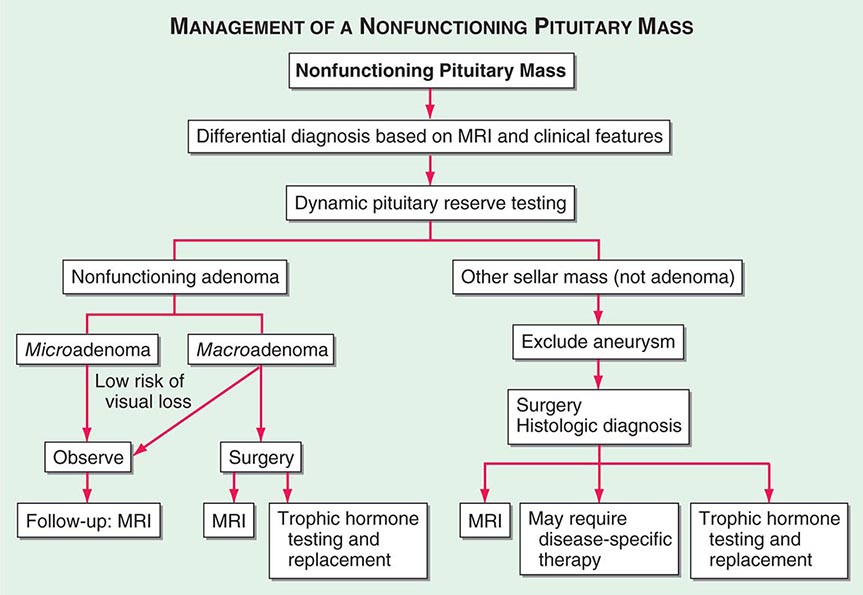
FIGURE 403-7 Management of a nonfunctioning pituitary mass. MRI, magnetic resonance imaging.
TSH-SECRETING ADENOMAS
TSH-producing macroadenomas are very rare but are often large and locally invasive when they occur. Patients usually present with thyroid goiter and hyperthyroidism, reflecting overproduction of TSH. Diagnosis is based on demonstrating elevated serum free T4 levels, inappropriately normal or high TSH secretion, and MRI evidence of a pituitary adenoma. Elevated uncombined α subunits are seen in many patients.
It is important to exclude other causes of inappropriate TSH secretion, such as resistance to thyroid hormone, an autosomal dominant disorder caused by mutations in the thyroid hormone β receptor (Chap. 405). The presence of a pituitary mass and elevated β subunit levels are suggestive of a TSH-secreting tumor. Dysalbuminemic hyperthyroxinemia syndromes, caused by mutations in serum thyroid hormone binding proteins, are also characterized by elevated thyroid hormone levels, but with normal rather than suppressed TSH levels. Moreover, free thyroid hormone levels are normal in these disorders, most of which are familial.
|
TREATMENT |
TSH-SECRETING ADENOMAS |
The initial therapeutic approach is to remove or debulk the tumor mass surgically, usually using a transsphenoidal approach. Total resection is not often achieved as most of these adenomas are large and locally invasive. Normal circulating thyroid hormone levels are achieved in about two-thirds of patients after surgery. Thyroid ablation or antithyroid drugs (methimazole and propylthiouracil) can be used to reduce thyroid hormone levels. Somatostatin analogue treatment effectively normalizes TSH and α subunit hypersecretion, shrinks the tumor mass in 50% of patients, and improves visual fields in 75% of patients; euthyroidism is restored in most patients. Because somatostatin analogues markedly suppress TSH, biochemical hypothyroidism often requires concomitant thyroid hormone replacement, which may also further control tumor growth.
404 |
Disorders of the Neurohypophysis |
The neurohypophysis, or posterior pituitary, is formed by axons that originate in large cell bodies in the supraoptic and paraventricular nuclei of the hypothalamus. It produces two hormones: (1) arginine vasopressin (AVP), also known as antidiuretic hormone, and (2) oxytocin. AVP acts on the renal tubules to reduce water loss by concentrating the urine. Oxytocin stimulates postpartum milk letdown in response to suckling. A deficiency of AVP secretion or action causes diabetes insipidus (DI), a syndrome characterized by the production of large amounts of dilute urine. Excessive or inappropriate AVP production impairs urinary water excretion and predisposes to hyponatremia if water intake is not reduced in parallel with urine output.
VASOPRESSIN
SYNTHESIS AND SECRETION
AVP is a nonapeptide composed of a six-member disulfide ring and a tripeptide tail (Fig. 404-1). It is synthesized via a polypeptide precursor that includes AVP, neurophysin, and copeptin, all encoded by a single gene on chromosome 20. After preliminary processing and folding, the precursor is packaged in neurosecretory vesicles, where it is transported down the axon; further processed to AVP, neurophysin, and copeptin; and stored in neurosecretory vesicles until released by exocytosis into peripheral blood.
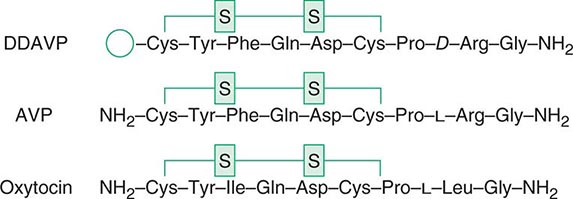
FIGURE 404-1 Primary structures of arginine vasopressin (AVP), oxytocin, and desmopressin (DDAVP).
AVP secretion is regulated primarily by the “effective” osmotic pressure of body fluids. This control is mediated by specialized hypothalamic cells known as osmoreceptors, which are extremely sensitive to small changes in the plasma concentration of sodium and its anions but normally are insensitive to other solutes such as urea and glucose. The osmoreceptors appear to include inhibitory as well as stimulatory components that function in concert to form a threshold, or set point, control system. Below this threshold, plasma AVP is suppressed to levels that permit the development of a maximum water diuresis. Above it, plasma AVP rises steeply in direct proportion to plasma osmolarity, quickly reaching levels sufficient to effect a maximum antidiuresis. The absolute levels of plasma osmolarity/sodium at which minimally and maximally effective levels of plasma AVP occur, vary appreciably from person to person, apparently due to genetic influences on the set and sensitivity of the system. However, the average threshold, or set point, for AVP release corresponds to a plasma osmolarity or sodium of about 280 mosmol/L or 135 meq/L, respectively; levels only 2–4% higher normally result in maximum antidiuresis.
Although it is relatively stable in a healthy adult, the set point of the osmoregulatory system can be lowered by pregnancy, the menstrual cycle, estrogen, and relatively large, acute reductions in blood pressure or volume. Those reductions are mediated largely by neuronal afferents that originate in transmural pressure receptors of the heart and large arteries and project via the vagus and glossopharyngeal nerves to the brainstem, from which postsynaptic projections ascend to the hypothalamus. These pathways maintain a tonic inhibitory tone that decreases when blood volume or pressure falls by >10–20%. This baroregulatory system is probably of minor importance in the physiology of AVP secretion because the hemodynamic changes required to affect it usually do not occur during normal activities. However, the baroregulatory system undoubtedly plays an important role in AVP secretion in patients with disorders that produce large, acute disturbances of hemodynamic function. However, the baroregulatory system undoubtedly plays an important role in AVP secretion in patients with disorders that produce large, acute disturbances of hemodynamic function.
AVP secretion also can be stimulated by nausea, acute hypoglycemia, glucocorticoid deficiency, smoking, and, possibly, hyperangiotensinemia. The emetic stimuli are extremely potent since they typically elicit immediate, 50- to 100-fold increases in plasma AVP even when the nausea is transient and is not associated with vomiting or other symptoms. They appear to act via the emetic center in the medulla and can be blocked completely by treatment with antiemetics such as fluphenazine. There is no evidence that pain or other noxious stresses have any effect on AVP unless they elicit a vasovagal reaction with its associated nausea and hypotension.
ACTION
The most important, if not the only, physiologic action of AVP is to reduce water excretion by promoting concentration of urine. This antidiuretic effect is achieved by increasing the hydroosmotic permeability of cells that line the distal tubule and medullary collecting ducts of the kidney (Fig. 404-2). In the absence of AVP, these cells are impermeable to water and reabsorb little, if any, of the relatively large volume of dilute filtrate that enters from the proximal nephron. The lack of reabsorption results in the excretion of very large volumes (as much as 0.2 mL/kg per min) of maximally dilute urine (specific gravity and osmolarity ~1.000 and 50 mosmol/L, respectively), a condition known as water diuresis. In the presence of AVP, these cells become selectively permeable to water, allowing the water to diffuse back down the osmotic gradient created by the hypertonic renal medulla. As a result, the dilute fluid passing through the tubules is concentrated and the rate of urine flow decreases. The magnitude of this effect varies in direct proportion to the plasma AVP concentration and the rate of solute excretion. At maximum levels of AVP and normal rates of solute excretion, it approximates a urine flow rate as low as 0.35 mL/min and a urine osmolarity as high as 1200 mosmol/L. This effect is reduced by a solute diuresis such as glucosuria in diabetes mellitus. Antidiuresis is mediated via binding to G protein–coupled V2 receptors on the serosal surface of the cell, activation of adenyl cyclase, and insertion into the luminal surface of water channels composed of a protein known as aquaporin 2 (AQP2). The V2 receptors and aquaporin 2 are encoded by genes on chromosomes Xq28 and 12q13, respectively.
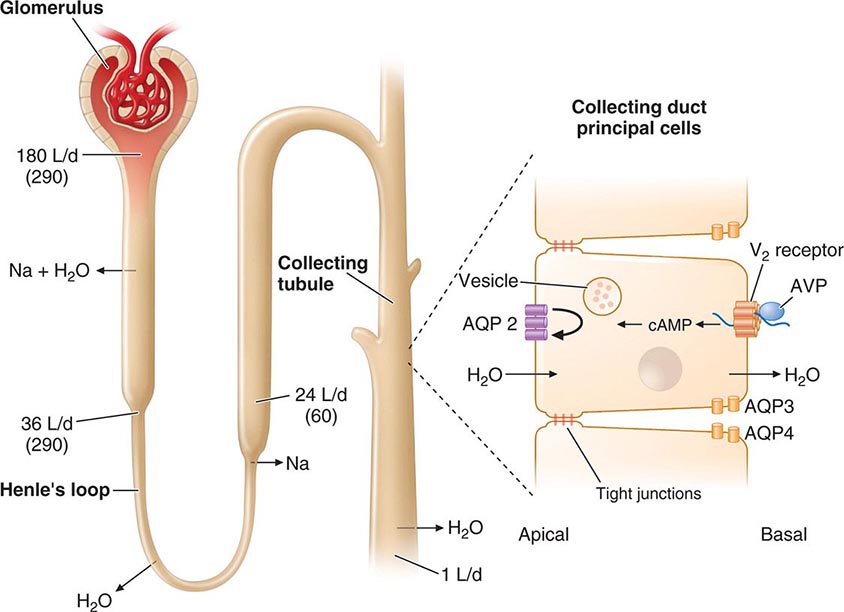
FIGURE 404-2 Antidiuretic effect of arginine vasopressin (AVP) in the regulation of urine volume. In a typical 70-kg adult, the kidney filters ~180 L/d of plasma. Of this, ~144 L (80%) is reabsorbed isosmotically in the proximal tubule and another 8 L (4–5%) is reabsorbed without solute in the descending limb of Henle’s loop. The remainder is diluted to an osmolarity of ~60 mmol/kg by selective reabsorption of sodium and chloride in the ascending limb. In the absence of AVP, the urine issuing from the loop passes largely unmodified through the distal tubules and collecting ducts, resulting in a maximum water diuresis. In the presence of AVP, solute-free water is reabsorbed osmotically through the principal cells of the collecting ducts, resulting in the excretion of a much smaller volume of concentrated urine. This antidiuretic effect is mediated via a G protein–coupled V2 receptor that increases intracellular cyclic AMP, thereby inducing translocation of aquaporin 2 (AQP 2) water channels into the apical membrane. The resultant increase in permeability permits an influx of water that diffuses out of the cell through AQP 3 and AQP 4 water channels on the basal-lateral surface. The net rate of flux across the cell is determined by the number of AQP 2 water channels in the apical membrane and the strength of the osmotic gradient between tubular fluid and the renal medulla. Tight junctions on the lateral surface of the cells serve to prevent unregulated water flow.
At high concentrations, AVP also causes contraction of smooth muscle in blood vessels in the skin and gastrointestinal tract, induces glycogenolysis in the liver, and potentiates adrenocorticotropic hormone (ACTH) release by corticotropin-releasing factor. These effects are mediated by V1a or V1b receptors that are coupled to phospholipase C. Their role, if any, in human physiology/pathophysiology is uncertain.
METABOLISM
AVP distributes rapidly into a space roughly equal to the extracellular fluid volume. It is cleared irreversibly with a half-life (t 1/2) of 10–30 min. Most AVP clearance is due to degradation in the liver and kidneys. During pregnancy, the metabolic clearance of AVP is increased three- to fourfold due to placental production of an N-terminal peptidase.
THIRST
Because AVP cannot reduce water loss below a certain minimum level obligated by urinary solute load and evaporation from skin and lungs, a mechanism for ensuring adequate intake is essential for preventing dehydration. This vital function is performed by the thirst mechanism. Like AVP, thirst is regulated primarily by an osmostat that is situated in the anteromedial hypothalamus and is able to detect very small changes in the plasma concentration of sodium and its anions. The thirst osmostat appears to be “set” about 3% higher than the AVP osmostat. This arrangement ensures that thirst, polydipsia, and dilution of body fluids do not occur until plasma osmolarity/sodium starts to exceed the defensive capacity of the antidiuretic mechanism.
OXYTOCIN
Oxytocin is also a nonapeptide that differs from AVP only at positions 3 and 8 (Fig. 404-1). However, it has relatively little antidiuretic effect and seems to act mainly on mammary ducts to facilitate milk letdown during nursing. It also may help initiate or facilitate labor by stimulating contraction of uterine smooth muscle, but it is not clear if this action is physiologic or necessary for normal delivery.
DEFICIENCIES OF AVP SECRETION AND ACTION
DIABETES INSIPIDUS
Clinical Characteristics A decrease of 75% or more in the secretion or action of AVP usually results in DI, a syndrome characterized by the production of abnormally large volumes of dilute urine. The 24-h urine volume exceeds 50 mL/kg body weight, and the osmolarity is less than 300 mosmol/L. The polyuria produces symptoms of urinary frequency, enuresis, and/or nocturia, which may disturb sleep and cause mild daytime fatigue or somnolence. It also results in a slight rise in plasma osmolarity that stimulates thirst and a commensurate increase in fluid intake (polydipsia). Overt clinical signs of dehydration are uncommon unless thirst and/or the compensatory increase of fluid intake are also impaired.
![]() Etiology A primary deficiency of AVP secretion usually results from agenesis or irreversible destruction of the neurohypophysis. It is referred to variously as neurohypophyseal DI, neurogenic DI, pituitary DI, cranial DI, or central DI. It can be caused by a variety of congenital, acquired, or genetic disorders, but in about one-half of all adult patients, it is idiopathic (Table 404-1). Pituitary DI caused by surgery in or around the neurohypophysis usually appears within 24 h. After a few days, it may transition to a 2- to 3-week period of inappropriate antidiuresis, after which the DI may or may not recur permanently. five genetic forms of pituitary DI are now known. By far, the most common is transmitted in an autosomal dominant mode and is caused by diverse mutations in the coding region of one allele of the AVP–neurophysin II (or AVP-NPII) gene. All the mutations alter one or more amino acids known to be critical for correct processing and/or folding of the prohormone, thus interfering with its trafficking through the endoplasmic reticulum. The misfolded mutant precursor accumulates and interferes with production of AVP by the normal allele, eventually destroying the magnocellular neurons in which it is produced. The AVP deficiency and DI are usually not present at birth but develop gradually over a period of several months to years, progressing from partial to severe at different rates depending on the mutation. Once established, the deficiency of AVP is permanent, but for unknown reasons, the DI occasionally improves or remits spontaneously in late middle age. The parvocellular neurons that make AVP and the magnocellular neurons that make oxytocin appear to be unaffected. There are also rare autosomal recessive forms of pituitary DI. One is due to an inactivating mutation in the AVP portion of the gene; another is due to a large deletion involving the majority of the AVP gene and regulatory sequences in the intergenic region. A third form is caused by mutations of the WFS 1 gene responsible for Wolfram’s syndrome (DI, diabetes mellitus, optic atrophy, and neural deafness [DIDMOAD]). An X-linked recessive form linked to a region on Xq28 has also been described.
Etiology A primary deficiency of AVP secretion usually results from agenesis or irreversible destruction of the neurohypophysis. It is referred to variously as neurohypophyseal DI, neurogenic DI, pituitary DI, cranial DI, or central DI. It can be caused by a variety of congenital, acquired, or genetic disorders, but in about one-half of all adult patients, it is idiopathic (Table 404-1). Pituitary DI caused by surgery in or around the neurohypophysis usually appears within 24 h. After a few days, it may transition to a 2- to 3-week period of inappropriate antidiuresis, after which the DI may or may not recur permanently. five genetic forms of pituitary DI are now known. By far, the most common is transmitted in an autosomal dominant mode and is caused by diverse mutations in the coding region of one allele of the AVP–neurophysin II (or AVP-NPII) gene. All the mutations alter one or more amino acids known to be critical for correct processing and/or folding of the prohormone, thus interfering with its trafficking through the endoplasmic reticulum. The misfolded mutant precursor accumulates and interferes with production of AVP by the normal allele, eventually destroying the magnocellular neurons in which it is produced. The AVP deficiency and DI are usually not present at birth but develop gradually over a period of several months to years, progressing from partial to severe at different rates depending on the mutation. Once established, the deficiency of AVP is permanent, but for unknown reasons, the DI occasionally improves or remits spontaneously in late middle age. The parvocellular neurons that make AVP and the magnocellular neurons that make oxytocin appear to be unaffected. There are also rare autosomal recessive forms of pituitary DI. One is due to an inactivating mutation in the AVP portion of the gene; another is due to a large deletion involving the majority of the AVP gene and regulatory sequences in the intergenic region. A third form is caused by mutations of the WFS 1 gene responsible for Wolfram’s syndrome (DI, diabetes mellitus, optic atrophy, and neural deafness [DIDMOAD]). An X-linked recessive form linked to a region on Xq28 has also been described.
|
CAUSES OF DIABETES INSIPIDUS |
A primary deficiency of plasma AVP also can result from increased metabolism by an N-terminal aminopeptidase produced by the placenta. It is referred to as gestational DI because the signs and symptoms manifest during pregnancy and usually remit several weeks after delivery.
Secondary deficiencies of AVP secretion result from inhibition by excessive intake of fluids. They are referred to as primary polydipsia and can be divided into three subcategories. One of them, dipsogenic DI, is characterized by inappropriate thirst caused by a reduction in the set of the osmoregulatory mechanism. It sometimes occurs in association with multifocal diseases of the brain such as neurosarcoid, tuberculous meningitis, and multiple sclerosis but is often idiopathic. The second subtype, psychogenic polydipsia, is not associated with thirst, and the polydipsia seems to be a feature of psychosis or obsessive compulsive disorder. The third subtype, iatrogenic polydipsia, results from recommendations to increase fluid intake for its presumed health benefits.
Primary deficiencies in the antidiuretic action of AVP result in nephrogenic DI. The causes can be genetic, acquired, or drug induced (Table 404-1). The most common genetic form is transmitted in a semirecessive X-linked manner. It is caused by mutations in the coding region of the V2 receptor gene that impair trafficking and/or ligand binding of the mutant receptor. There are also autosomal recessive or dominant forms of nephrogenic DI. They are caused by AQP2 gene mutations that result in complete or partial defects in trafficking and function of the water channels that mediate antidiuresis in the distal and collecting tubules of the kidney.
Secondary deficiencies in the antidiuretic response to AVP result from polyuria per se. They are caused by washout of the medullary concentration gradient and/or suppression of aquaporin function. They usually resolve 24–48 h after the polyuria is corrected but can complicate interpretation of some acute tests used for differential diagnosis.
Pathophysiology In pituitary, gestational, or nephrogenic DI, the polyuria results in a small (1–2%) decrease in body water and a commensurate increase in plasma osmolarity and sodium that stimulates thirst and a compensatory increase in water intake. As a result, hypernatremia and other overt physical or laboratory signs of dehydration do not develop unless the patient also has a defect in thirst or fails to increase fluid intake for some other reason.
In pituitary and nephrogenic DI, the severity of the defect in AVP secretion or action varies significantly from patient to patient. In some, the defect is so severe that it cannot be overcome by even an intense stimulus such as nausea or severe dehydration. In others, the defect in AVP secretion or action is incomplete, and a modest stimulus such as a few hours of fluid deprivation, smoking, or a vasovagal reaction can raise urine osmolarity as high as 800 mosmol/L. However, even when the defects are partial, the relation of urine osmolarity to plasma AVP in patients with nephrogenic DI (Fig. 404-3A) or of plasma AVP to plasma osmolarity and sodium in patients with pituitary DI (Fig. 404-3B) is subnormal.
FIGURE 404-3 Relationship of plasma AVP to urine osmolarity (A) and plasma osmolarity (B) before and during fluid deprivation–hypertonic saline infusion test in patients who are normal or have primary polydipsia (blue zones), pituitary diabetes insipidus (green zones), or nephrogenic diabetes insipidus (pink zones).
In primary polydipsia, the pathogenesis of the polydipsia and polyuria is the reverse of that in pituitary, nephrogenic, and gestational DI. In primary polydipsia, an abnormality in cognition or thirst causes excessive intake of fluids and an increase in body water that reduces plasma osmolarity/sodium, AVP secretion, and urinary concentration. Dilution of the urine, in turn, results in a compensatory increase in urinary free-water excretion that usually offsets the increase in intake and stabilizes plasma osmolarity/sodium at a level only 1–2% below basal. Thus, hyponatremia or clinically appreciable overhydration is uncommon unless the polydipsia is very severe or the compensatory water diuresis is impaired by a drug or disease that stimulates or mimics the antiduretic effect of endogenous AVP. A rise in plasma osmolarity and sodium produced by fluid deprivation or hypertonic saline infusion increases plasma AVP normally. However, the resultant increase in urine concentration is often subnormal because polyuria per se temporarily reduces the capacity of the kidney to concentrate the urine. Thus, the maximum level of urine osmolarity achieved during fluid deprivation is often indistinguishable from that in patients with partial pituitary or partial nephrogenic DI.
Differential Diagnosis When symptoms of urinary frequency, enuresis, nocturia, and/or persistent thirst are present in the absence of glucosuria, the possibility of DI should be evaluated by collecting a 24-h urine on ad libitum fluid intake. If the volume exceeds 50 mL/kg per day (3500 mL in a 70-kg male) and the osmolarity is below 300 mosmol/L, DI is confirmed and the patient should be evaluated further to determine the type in order to select the appropriate therapy.
The type of DI can sometimes be inferred from the clinical setting or medical history. Often, however, such information is lacking, ambiguous, or misleading, and other approaches to differential diagnosis are needed. If basal plasma osmolarity and sodium are within normal limits, the traditional approach is to determine the effect of fluid deprivation and injection of antidiuretic hormone on urine osmolarity. This approach suffices for differential diagnosis if fluid deprivation raises plasma osmolarity and sodium above the normal range without inducing concentration of the urine. In that event, primary polydipsia and partial defects in AVP secretion and action are excluded, and the effect on urine osmolarity of injecting 2 μg of the AVP analogue, desmopressin, indicates whether the patient has severe pituitary DI or severe nephrogenic DI. However, this approach is of little or no diagnostic value if fluid deprivation results in concentration of the urine because the increases in urine osmolarity achieved both before and after the injection of desmopressin are similar in patients with partial pituitary DI, partial nephrogenic DI, and primary polydipsia. These disorders can be differentiated by measuring plasma AVP during fluid deprivation and relating it to the concurrent level of plasma and urine osmolarity (Fig. 404-3). However, this approach does not always differentiate clearly between partial pituitary DI and primary polydipsia unless the measurement is made when plasma osmolarity and sodium are at or above the normal range. This level is difficult to achieve by fluid deprivation alone once urinary concentration occurs. Therefore it is usually necessary to give a short infusion of 3% saline condition (0.1 mL/kg body weight per minute for 60 to 90 minutes) and repeat the measurement of plasma AVP.
A simpler but equally reliable way to differentiate between pituitary DI, nephrogenic DI, and primary polydipsia is to measure basal plasma AVP to determine if a brain magnetic resonance imaging (MRI) is needed and sufficient for diagnosis (Fig. 404-4). If plasma AVP on ad libitum fluid intake is normal or elevated (>1 pg/mL) when measured by a sensitive and specific assay, both primary polydipsia and pituitary DI are excluded and the diagnosis of nephrogenic DI can be confirmed, if desired, by a 1- to 2-day outpatient trial of desmopressin therapy. If, however, basal plasma AVP is low or undetectable (<1 pg/mL), nephrogenic DI is very unlikely and MRI of the brain can be used to differentiate pituitary DI from primary polydipsia. In most healthy adults and children, the posterior pituitary emits a hyperintense signal visible in T1-weighted midsagittal images. This “bright spot” is almost always present in patients with primary polydipsia but is always absent or abnormally small in patients with pituitary DI, even if their AVP deficiency is partial. The MRI is also useful in searching for pathology responsible for pituitary DI or the dipsogenic form of primary polydipsia (Fig. 404-2). The principal caveat is that MRI is not reliable for differential diagnosis of DI in patients with empty sella because they typically lack a bright spot even when their AVP secretion and action are normal. MRI also cannot be used to differentiate pituitary from nephrogenic DI because many patients with nephrogenic DI also lack a posterior pituitary bright spot, probably because they have an abnormally high rate of AVP secretion and turnover.




