Chapter 9 Apoptosis Assessment
 Introduction
Introduction
A landmark of cellular self-destruction by apoptosis is the activation of nucleases and proteases that degrade the higher order chromatin structure of the DNA into fragments of 50 to 300 kilobases and subsequently into smaller DNA pieces of about 200 base-pairs in length. Activation of proteases, notably aspartate-specific cysteinyl proteases, referred to as caspases, is of primary relevance to apoptosis. Caspase-3 is considered to be the key mediator of apoptosis of mammalian cells, and its expression may be measured with immunohistochemical staining. Using fluorescent-labeled reagents, it is possible to tag the DNA break and identify the percentage of apoptotic cells with a high degree of accuracy.1–6
Measurable Features of Apoptosis
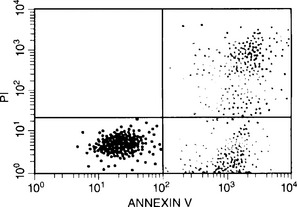
Apoptosis can also be characterized by changes in cell membrane structure. During apoptosis, the cell membrane’s phospholipid asymmetry changes—phosphatidylserine is exposed on the outer membrane, whereas membrane integrity is maintained. Annexin V specifically binds phosphatidylserine, whereas propidium iodide is a DNA-binding fluorochrome. When a cell population is exposed to both reagents, apoptotic cells stain positive for annexin V and negative for propidium iodide; necrotic cells stain positive for both, and live cells stain negative for both.3
This process of apoptosis and its analysis by flow cytometry are shown in Figures 9-1 and 9-2.
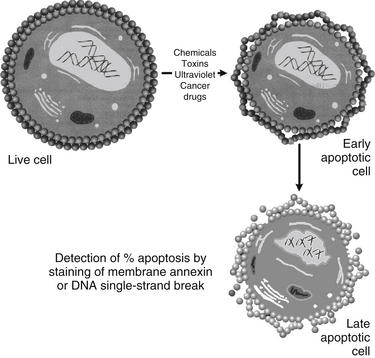
FIGURE 9-1 Detection of apoptosis using damaged membrane or DNA single-strand break and flow cytometry.
Another assessment of apoptosis involves ex vivo cell analysis. Specifically, the expression of active caspase-3 along with the Bcl-2/Bax ratio as markers of apoptosis can be measured. Immunohistochemical staining will reveal the expression of these apoptotic-related proteins, caspase-3 and cleaved caspase-3; the latter is indicative of apoptosis.7 Bcl-2 is anti-apoptotic gene product that exists in ratio to Bax and Bak, which are pro-apoptotic gene products. This ratio is indicative of the degree of apoptosis, with a decreased Bcl-2:Bax ratio indicative of apoptosis. Cells from Bax (−/−) and Bak(−/−) knockout animals do not respond to apoptosis inducers. In these cells, cytochrome C is not released from the mitochondrial membrane to initiate the caspase cascade.8 Thus, Bax and Bak are critical to apoptosis, and their expression in relation to Bcl-2 is highly correlative to apoptosis.
Different Stages of Apoptosis
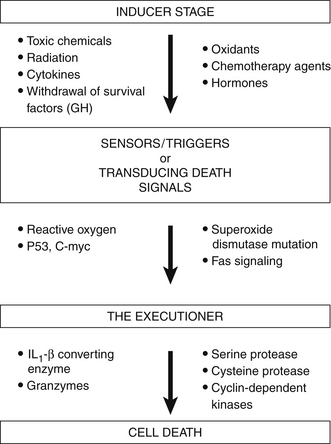
The process of apoptosis is divided into three different stages:
These stages of apoptosis are depicted in Figure 9-3. Induction represents the initial events that signal a cell so that apoptosis may begin. This induction phase may be induced by various physical agents, such as toxic chemicals, hypoxia, radiation, chemotherapy agents, hormones, and CD95 or Fas ligation. It has been proposed that the induction stage of apoptosis is prevented by many antioxidants (vitamin C, β-carotene, and vitamin E) and also by various biological response modifiers, including lentinan, thymic hormones, viral antigens, and cytokines.
Apoptosis is Induced by Chemicals to Control Malignancy
Many chemicals have the capacity to bind to DNA, form DNA adducts, or cause DNA single-strand breaks, possibly leading to cancer. However, the body is equipped with many factors, enzymes, suppressor genes, and cellular sensors, all with the capacity to prevent the consequences of this DNA damage by activating apoptosis-inducing signals.
An important prediction of the relevance of apoptosis to malignancy is that the rate of apoptosis versus mitosis should influence the behavior of a tumor. Recently, the relationship between the apoptotic and mitotic indexes in a tumor was demonstrated as predictive of outcome: a higher ratio of apoptosis to mitosis within the tumor correlated with positive prognosis. Further, it was found that this was not simply a function of cell death per se. Tumors with a high incidence of necrosis rather than apoptosis were correlated with poor prognosis. It therefore follows that treatments or conditions that favor apoptosis should have desirable effects, and that defects in the pathways leading to apoptosis are likely to play important roles in the process of oncogenesis.4,5
Many reactive chemicals and drugs such as acetaminophen, diquat, carbon tetrachloride, quinones, cyanide, polyhydroxyl polyether, methyl mercury, and organotin have been implicated in apoptosis (programmed cell death) and necrosis (toxic cell death).9–16
Most research on chemical induction of apoptosis is carried out with primary cultures of cell lines (e.g., neurons, thymocytes, carcinoma cells, leukemia cells, neuroblastoma, breast cancer cells, lymphoma); little has been published on the in vivo effects of chemicals on apoptotic cells in animal models and none in humans. Therefore, it was of interest to examine the effects of exposure to low levels of benzene, as well as through drinking water concentrations of up to 14 ppb on the apoptotic cell population, as well as to examine possible changes in the cell cycle progression.9
Evidence is sufficient for the carcinogenicity of benzene in humans; therefore, there is no safe level of exposure to this chemical or its metabolites. Published case reports, a case series, epidemiologic studies, and both cohort and case–control studies have shown statistically significant associations between leukemia and occupational exposure to benzene and benzene-containing solvents.17,18
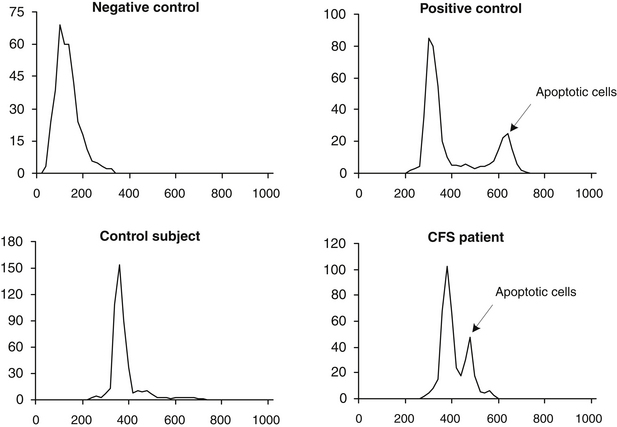
It has been indicated that possibly 800,000 persons are exposed to benzene from coke oven emissions at levels less than 0.1 ppm, and 5 million may be exposed to benzene from petroleum refinery emissions at levels of 0.1 to 1 ppm. Since then, numerous chemicals have been implicated in apoptosis (or programmed cell death), which arises from damage to DNA. One of the authors, Vojdani along with collaborators, hypothesized that in individuals with a certain genetic makeup, benzene or its metabolites act as haptens, which may induce programmed cell death. The study involved a group of 60 male and female subjects who were exposed to benzene-contaminated water (at concentrations up to 14 ppm for a period of 3 to 5 years).18a For comparison, a control group consisting of 30 healthy males and females with a similar age distribution and without a history of exposure to benzene were recruited. Peripheral blood lymphocytes of both groups were tested for percentage of apoptotic cell population, using flow cytometry. When exposed individuals were compared with the control group, statistically significant differences between each mean group were detected (27.5 ± 2.4 and 10 ± 2.6, respectively), indicating an increased rate of apoptosis in 86.6% of exposed individuals (P <0.0001; Mann-Whitney U-test). Flow cytometry analysis of apoptosis in a healthy control and a patient with chronic fatigue syndrome is shown in Figure 9-4.
 Clinical Applications
Clinical Applications
Apoptosis in Cancer
The failure of apoptosis in malignant cells in the context of irreparable DNA damage leads to tumor progression. Cancer therapies, namely, chemotherapy and radiation, control cancer by inflicting cell damage, which, in turn, triggers apoptosis. Unfortunately, >50% of all human cancers involve a mutation of p53, a central gene in apoptosis. p53 stimulates both the extrinsic death receptor pathway of apoptosis as well as the intrinsic mitochondrial pathway involving a decreased Bcl-2:Bax ratio. Thus, it is imperative to find therapies that promote apoptosis independent of p53. Promising therapies in this regard include curcumin,19 derived from Curcuma longa, and resveratrol,20 both of which are under investigative study for this application. Another promising cancer treatment involves the use of recombinant human apoptosis ligands to induce tumor necrosis factor-related apoptosis inducing ligand (TRAIL). These ligands induce apoptosis via TRAIL, a selective death receptor pathway in a broad range of cancer cell lines while sparing most normal cell types.21
Apoptosis in Autoimmune Diseases
In cancer, it is the tumor cells that forget to die; in autoimmunity, immune cells fail to die when they are supposed to. Virtually all tissues harbor apoptotic cells at one time or another. Damaged cells usually commit suicide for the greater good of the body; when this does not occur, disease may develop. Autoimmunity occurs when the antigen receptors on immune cells recognize specific antigens on healthy cells and cause the cells bearing those particular substances to die. Autoimmune disease results from perpetuated immune-mediated tissue destruction, and can involve immune cells that are resistant to apoptosis. Under normal conditions, the body allows a certain number of self-reactive lymphocytes to circulate. These cells normally do little harm, but they can become overactive through several processes. For instance, if these reactive lymphocytes recognize some foreign antigen such as microbes on food and haptenic chemicals, then exposure to that antigen causes them to become excited. If, due to molecular mimicry, these antigens are similar to normal tissues, the activated cells may expand their numbers and attack the healthy tissue, thus causing an autoimmune disease.1,22,23
Autoimmune reactions usually are self-limited—they disappear when the antigens that originally set them off are cleared away. In some instances, however, the autoreactive lymphocytes survive longer than they should and continue to induce apoptosis in normal cells. Some evidence in animals and humans has indicated that extended survival of autoreactive cells is implicated in at least two chronic autoimmune syndromes—systemic lupus erythematosus and rheumatoid arthritis. In other words, the lymphocytes undergo too little apoptosis, with the result that normal cells undergo too much.24,25
Apoptosis During Viral Infection
Epstein-Barr virus, which causes mononucleosis and has been linked to lymphomas in humans, uses a mechanism that has been seen in other viruses. Epstein-Barr virus produces substances that inhibit apoptosis. Papillomavirus, a major cause of cervical cancer, inactivates p53, a central mediator of apoptosis. Cowpox virus, a relative of which is used as the smallpox vaccine, is another virus that inhibits caspase activation and attendant apoptosis. Investigators interested in antiviral therapy are now exploring ways to block the activity of the antiapoptotic molecules manufactured by viruses.24
Apoptosis in Acquired Immunodeficiency Syndrome
Induction of apoptosis by viruses in healthy cells is believed to contribute to the immune deficiency found in patients with acquired immunodeficiency syndrome (AIDS). In these patients, infection with human immunodeficiency virus (HIV) causes T-helper cells to die. As T-helper cells gradually disappear, cytotoxic cells, such as natural killer cells, perish as well through apoptosis, because they cannot survive without the growth signals produced by T-helper cells. When the number of T cells dwindles, so does the body’s ability to fight infections, especially viral and parasitic infections. Researchers have shown that many more helper cells succumb in addition to those that are infected with HIV. It is also highly probable that a large number of the cells die through apoptosis. Apparently, Fas plays a crucial role in this process.
It is also possible that oxygen-free radicals trigger the suicide of virus-free T cells. These highly reactive substances are produced by inflammatory cells drawn to infected lymph nodes in HIV patients. Free radicals can damage DNA and membranes in cells. They will cause necrosis if they do extensive damage, but they can induce apoptosis if the damage is more subtle. In support of the free-radical theory, researchers have found that molecules capable of neutralizing free radicals prevent apoptosis in T cells obtained from AIDS patients.24,25
Therapies with antiapoptotic medication, such as Trolox, a water-soluble analog of vitamin E that prevents oxidative stress, and pyrrolidine dithiocarbamate, a potent inhibitor of nuclear factor-κB, are now the focus of AIDS and autoimmune disease studies.26,27
Additionally, protease inhibitors, which are the mainstay of HIV therapy, inhibit apoptosis in immune cells.28
Apoptosis in the Heart and Brain
Apoptosis also accounts for much of the pathology seen in such diseases as Alzheimer’s, Parkinson’s, Huntington’s, and amyotrophic lateral sclerosis (Lou Gehrig’s disease), which are marked by the loss of brain neurons. Elevated apoptosis in these neurologic diseases seems to be related to lack of production of the nerve growth factor and to free radical damage. It seems likely that a combination of such factors could cause many cells to destroy themselves. Manipulation of this process of cell killing may help in treating these neurologic diseases. Studies in animal models imply that long-term delivery of nerve growth factors could protect against programmed cell death in these conditions. Therefore, a greater understanding of the mechanisms involved in cell death should greatly enhance those important steps.22,26,29
 Conclusions
Conclusions
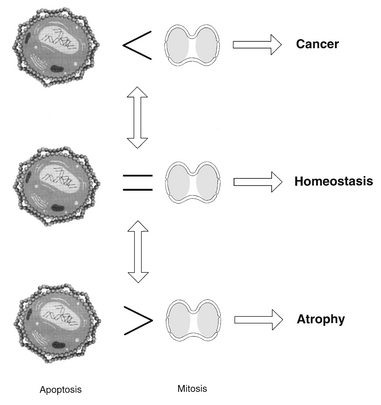
Apoptosis and cell proliferation play an important role in development, differentiation, homeostasis, and aging.2–6 The balance established between these two processes depends on various growth and death signals that are influenced by diet, nutrition, lifestyle, and other environmental factors. When the equilibrium between life and death is disrupted by aberrant signals (e.g., low levels of antioxidants in the blood or tissue cells), either tissue growth or atrophy occurs.
Under normal conditions with optimal nutritional factors, tissue homeostasis is sustained by balancing the effects of mitosis and apoptosis. The importance of this balance can clearly be seen when one of these processes becomes predominant (Figure 9-5). The apoptotic potential within each cell is critical for the health of the host. Apoptosis is an elegant response to overwhelming DNA damaging stress. This seemingly heroic sacrifice of self for the greater good underpins healthy living. Imbalance of apoptosis regulators, genetic mutations, and viral infections thwarts the healing impact of apoptosis. Finding ways to restore apoptotic balance is critical to health.
1. Wyllie A.H., Kerr J.F., Currie A.R. Cell death: the significance of apoptosis. Int Rev Cytol. 1980;68:251–306.
2. White E. Life, death and the pursuit of apoptosis. Genes Dev. 1996;10:1–15.
3. Jarvis W.D., Kolesnick R.N., Fornari F.A., et al. Induction of apoptotic DNA damage and cell death by activation of the sphingomyelin pathway. Proc Natl Acad Sci U S A. 1994;91:73–77.
4. Green D.R., Martin S.J. The killer and the executioner: how apoptosis controls malignancy. Curr Opin Immunol. 1995;7:694–703.
5. Arends M.J., McGregor A.H., Wyllie A.H. Apoptosis is inversely related to necrosis and determines net growth in tumors bearing constitutively expressed myc, ras and HPV oncogenes. J Pathol. 1994;144:1045–1057.
6. Marchetti P., Hirsch T., Zamzami M., et al. Mitochondrial permeability triggers lymphocyte apoptosis. J Immunol. 1996;157:4830–4836.
7. Amatya J.L., Takeshima Y., Shrestha L., et al. Evaluation of apoptosis and immunohistochemical expression of the apoptosis-related proteins in mesothelioma. Hiroshima J Med Sci. 2010;59(2):27–33.
8. Kandasamy K., Srinivasula S.M., Alnemri E.S., et al. Involvement of proapoptotic molecules Bax and Bak in tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)-induced mitochondrial disruption and apoptosis: differential regulation of cytochrome c and Smac/DIABLO release. Cancer Res. 2003;63(7):1712–1721.
9. Vojdani A., Mordechai E., Brautbar N. Abnormal apoptosis and cell cycle progression in humans exposed to methyl tertiary-butyl ether and benzene contaminating water. Human Exp Toxicol. 1997;16:485–494.
10. Walker P.R., Smith C., Youdale T., et al. Topoisomerase II-reactive chemotherapeutic drugs induce apoptosis in thymocytes. Cancer Res. 1991;51:1078–1085.
11. Brown D.B., Sun X.M., Cohen G.M. Dexamethasone-induced apoptosis involves cleavage of DNA to large fragments prior to internu-cleosomal fragmentation. J Biol Chem. 1993;268:3037–3039.
12. Reynolds E.S., Kanz M.F., Chicco P., Moslen M.T. 1.1-Dichloroethylene: an apoptotic hepatotoxin? Environ Health Perspect. 1984;57:313–320.
13. Aw T.Y., Nicotera P., Manzo L., Orrenius S. Tributyltin stimulates apoptosis in rat thymocytes. Arch Biochem Biophys. 1990;283:46–50.
14. Rossi A.D., Larsson O., Manzo L., et al. Modification of Ca2+ signaling by inorganic mercury in PC12 cells. FASEB. 1993;7:1507–1514.
15. Kunimoto M. Methyl mercury induces apoptosis of rat cerebellar neurons in primary culture. Biochem Biophys Res Commun. 1994;204:310–317.
16. Vivian B., Rossi A.D., Chow S.C., Nicotera P. Organotin compounds induce calcium overload and apoptosis in PC12 cells. Neurotoxicology. 1995;16:19–25.
17. Ledda-Columbano G.M., Coni P., Curto M., et al. Induction of two different modes of cell death, apoptosis and necrosis in rat liver after a single dose of thioacetamide. Am J Pathol. 1991;139:1099–1109.
18. ATSDR (Agency for Toxic Substances and Disease Registry). Toxicological profile for benzene, draft report. Atlanta, GA: Department of Health and Human Services Agency; 1987.
18a. Vojdani A., Mordechai E., Brautbar N. Abnormal apoptosis and cell cycle progression in humans exposed to methyl tertiary-butyl ether and benzene contaminating water. Hum Exp Toxicol. 1997 Sep;16(9):485–494.
19. Saha A., Kuzuhara T., Echigo N., Fujii A., et al. Apoptosis of human lung cancer cells by curcumin mediated through up-regulation of “growth arrest and DNA damage inducible genes 45 and 153.”. Biol Pharm Bull. 2010;33(8):1291–1299.
20. Fulda S., Debatin K.M. Sensitization for tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis by the chemopreventive agent resveratrol. Cancer Res. 2004;64(1):337–346.
21. Ashkenazi A., Holland P., Eckhardt S.G. Ligand-based targeting of apoptosis in cancer: the potential of recombinant human apoptosis ligand 2/tumor necrosis factor-related apoptosis-inducing ligand (rhApo2L/TRAIL). J Clin Oncol. 2008;26(21):3621–3630.
22. National Institute of Environmental Health Sciences. Sixth annual report on carcinogens. Benzene Case No. 71-43-2: 35. Research Triangle Park, NC: National Institute of Environmental Health Sciences; 1991.
23. Golstein P., Ojcius D.M., Ding-E Young J. Cell death mechanisms and the immune system. Immunol Rev. 1991;121:29–65.
24. Cohen J.J., Duke R.C., Fadok V.A., Sellins K.S. Apoptosis and programmed cell death in immunity. Ann Rev Immunol. 1992;10:267–293.
25. Duke R.C., Ojcius D.M., Ding-E Young J. Cell suicide in health and disease. Sci Am. 1996;275:80–87.
26. Martin S.J., Green D.R. Protease activation during apoptosis: death by a thousand cuts. Cell. 1995;82:349–352.
27. Forrest V.J., Kang Y., McClain D.E., et al. Oxidative stress-induced apoptosis prevented by Trolox. Free Radic Biol Med. 1994;16:675–684.
28. Rizza S.A., Badley A.D. HIV protease inhibitors impact on apoptosis. Med Chem. 2008;4(1):75–79.
29. Schreck R., Meier B., Mannel D.N., et al. Dithiocarbamates as potent inhibitors of nuclear factor kB activation in intact cells. J Exp Med. 1992;175:1181–1194.
