[level-membership-for-hematology-oncology-and-palliative-medicine-category]26
Aplastic anaemia
Classification and aetiology
Aplastic anaemia (AA) may be part of a congenital syndrome, be secondary to well-defined insults to the bone marrow, or arise apparently spontaneously with no identifiable cause. A simple classification is shown in Table 26.1. The most common congenital disorder is Fanconi’s anaemia. Affected children suffer from defective DNA repair and the aplasia often coexists with skeletal deformities, skin pigmentation (Fig 26.1) and renal abnormalities. To date, fifteen genes (termed FANC) have been identified. Dyskeratosis congenita, another form of constitutional aplasia, is distinguished by a later onset, nail dystrophy, leukoplakia of mucosal surfaces and a high incidence of epithelial tumours. There is defective telomere maintenance and patients usually have very short telomeres. This is also observed in 10–15% of patients with acquired AA.
Table 26.1
Classification of aplastic anaemia
| 1. Idiopathic AA | |
| 2. Congenital AA | Fanconi’s anaemia |
| Dyskeratosis congenita | |
| 3. Secondary AA | Drugs – idiosyncratic or dose-related |
| Chemicals | |
| Ionising radiation | |
| Infection |
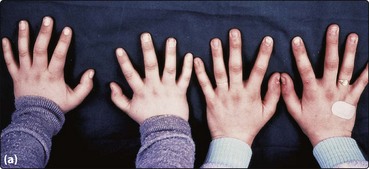
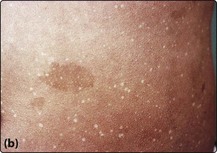
Fig 26.1 Fanconi’s anaemia.
(a) Digital abnormalities in brothers with the syndrome. (b) Skin pigmentation.
Infections known to predispose to AA include viral hepatitis and parvovirus infection. Exposure to chemicals, drugs and radiation can damage stem cells. Drugs may depress haematopoiesis idiosyncratically or predictably (Table 26.2). In roughly two-thirds of patients, no cause is apparent and AA is termed ‘idiopathic’. Improved haematopoiesis following immunosuppression (see below) suggests that in at least some cases the abnormal stem cell compartment is further compromised by poorly defined immune phenomena.
Table 26.2
Drugs associated with aplastic anaemia1
| Predictable | Cytotoxic agents |
| Idiosyncratic | Chloramphenicol |
| Sulfonamides | |
| Phenylbutazone | |
| Indometacin | |
| Gold salts | |
| Penicillamine | |
| Carbamazepine | |
| Phenytoin | |
| Carbimazole |
1This is a selective list of more commonly implicated agents.
Diagnosis
There are really two questions. Is the pancytopenia due to aplastic anaemia? Is this idiopathic AA or aplasia secondary to an identifiable cause (Table 26.3)?
Table 26.3
Marrow failure or infiltration








Hypersplenism



A reasonable sequence of investigations is as follows:
2 Bone marrow aspirate and trephine
This is the key diagnostic test. The marrow aspirate can be highly suggestive of aplasia with grossly hypocellular particles but a trephine biopsy is necessary to confirm the diagnosis and quantify the degree of hypocellularity (Fig 26.2). Aplasia may be patchy and if the trephine is surprisingly cellular in the context of the blood count, then further samples should be obtained. In practice the only likely confusion is with hypocellular myelodysplastic syndrome (see p. 50) or an atypical presentation of acute leukaemia, the latter particularly in childhood.
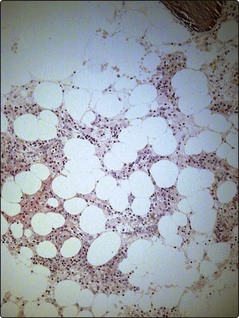
Measurement of severity
This is crucial as the severity defined from peripheral blood and bone marrow measurements predicts the response to treatment and survival (Table 26.4). The median survival of untreated severe AA is 3–6 months with only 20% of patients surviving longer than 1 year.
Table 26.4
AA is defined as non-severe (NSAA) unless it is:
1. Severe (SAA). Requires two of three peripheral blood criteria:
 Neutrophils less than 0.5 × 109/L
Neutrophils less than 0.5 × 109/L
 Platelets less than 20 × 109/L
Platelets less than 20 × 109/L
 Reticulocytes less than 20 × 109/L
Reticulocytes less than 20 × 109/L
 Less than 25% haematopoietic cells, or 25–50% haematopoietic cells and less than 30% cellularity
Less than 25% haematopoietic cells, or 25–50% haematopoietic cells and less than 30% cellularity
2. Very severe (VSAA). Requires a neutrophil count less than 0.2 × 109/L in the presence of at least one other peripheral blood criterion and the bone marrow features given for SAA
Management
Restoring normal haematopoiesis
There are two major options: immunosuppression and stem cell transplantation.
Immunosuppression or SCT?
Younger patients (less than 30 years) with SAA and a matched sibling donor should be transplanted. In VSAA (see Table 26.4) in this age group, the lack of a family donor should prompt a search for an HLA-matched unrelated donor. In patients 30–40 years with SAA, sibling SCT and immunosuppression produce similar survivals over 2–5 years. However, a significant proportion of patients receiving immunosuppression alone, approximately 25% at 10 years, evolve to clonal marrow diseases such as paroxysmal nocturnal haemoglobinuria, myelodysplastic syndrome and acute myeloid leukaemia. Thus in this group matched sibling SCT probably gives a better long-term prognosis. In older patients with SAA, and patients with non-severe AA, immunosuppression is generally the treatment of choice.






[/level-membership-for-hematology-oncology-and-palliative-medicine-category][not-level-membership-for-hematology-oncology-and-palliative-medicine-category]26
Aplastic anaemia
Classification and aetiology
Aplastic anaemia (AA) may be part of a congenital syndrome, be secondary to well-defined insults to the bone marrow, or arise apparently spontaneously with no identifiable cause. A simple classification is shown in Table 26.1. The most common congenital disorder is Fanconi’s anaemia. Affected children suffer from defective DNA repair and the aplasia often coexists with skeletal deformities, skin pigmentation (Fig 26.1) and renal abnormalities. To date, fifteen genes (termed FANC) have been identified. Dyskeratosis congenita, another form of constitutional aplasia, is distinguished by a later onset, nail dystrophy, leukoplakia of mucosal surfaces and a high incidence of epithelial tumours. There is defective telomere maintenance and patients usually have very short telomeres. This is also observed in 10–15% of patients with acquired AA.
Table 26.1
Classification of aplastic anaemia
| 1. Idiopathic AA | |
| 2. Congenital AA | Fanconi’s anaemia |
| Dyskeratosis congenita | |
| 3. Secondary AA | Drugs – idiosyncratic or dose-related |
| Chemicals | |
| Ionising radiation | |
| Infection |
Fig 26.1 Fanconi’s anaemia.
(a) Digital abnormalities in brothers with the syndrome. (b) Skin pigmentation.
Infections known to predispose to AA include viral hepatitis and parvovirus infection. Exposure to chemicals, drugs and radiation can damage stem cells. Drugs may depress haematopoiesis idiosyncratically or predictably (Table 26.2). In roughly two-thirds of patients, no cause is apparent and AA is termed ‘idiopathic’. Improved haematopoiesis following immunosuppression (see below) suggests that in at least some cases the abnormal stem cell compartment is further compromised by poorly defined immune phenomena.
Table 26.2
Drugs associated with aplastic anaemia1
| Predictable | Cytotoxic agents |
| Idiosyncratic | Chloramphenicol |
| Sulfonamides | |
| Phenylbutazone | |
| Indometacin | |
| Gold salts | |
| Penicillamine | |
| Carbamazepine | |
| Phenytoin | |
| Carbimazole |
1This is a selective list of more commonly implicated agents.
