33e |
Video Library of Gait Disorders |
Problems with gait and balance are major causes of falls, accidents, and resulting disability, especially in later life, and are often harbingers of neurologic disease. Early diagnosis is essential, especially for treatable conditions, because it may permit the institution of prophylactic measures to prevent dangerous falls and also to reverse or ameliorate the underlying cause. In this video, examples of gait disorders due to Parkinson’s disease, other extrapyramidal disorders, and ataxias, as well as other common gait disorders, are presented.
34 |
Confusion and Delirium |
Confusion, a mental and behavioral state of reduced comprehension, coherence, and capacity to reason, is one of the most common problems encountered in medicine, accounting for a large number of emergency department visits, hospital admissions, and inpatient consultations. Delirium, a term used to describe an acute confusional state, remains a major cause of morbidity and mortality, costing over $150 billion dollars yearly in health care costs in the United States alone. Despite increased efforts targeting awareness of this condition, delirium often goes unrecognized in the face of evidence that it is usually the cognitive manifestation of serious underlying medical or neurologic illness.
CLINICAL FEATURES OF DELIRIUM
A multitude of terms are used to describe patients with delirium, including encephalopathy, acute brain failure, acute confusional state, and postoperative or intensive care unit (ICU) psychosis. Delirium has many clinical manifestations, but is defined as a relatively acute decline in cognition that fluctuates over hours or days. The hallmark of delirium is a deficit of attention, although all cognitive domains—including memory, executive function, visuospatial tasks, and language—are variably involved. Associated symptoms that may be present in some cases include altered sleep-wake cycles, perceptual disturbances such as hallucinations or delusions, affect changes, and autonomic findings that include heart rate and blood pressure instability.
Delirium is a clinical diagnosis that is made only at the bedside. Two subtypes have been described—hyperactive and hypoactive—based on differential psychomotor features. The cognitive syndrome associated with severe alcohol withdrawal (i.e., “delirium tremens”) remains the classic example of the hyperactive subtype, featuring prominent hallucinations, agitation, and hyperarousal, often accompanied by life-threatening autonomic instability. In striking contrast is the hypoactive subtype, exemplified by benzodiazepine intoxication, in which patients are withdrawn and quiet, with prominent apathy and psychomotor slowing.
This dichotomy between subtypes of delirium is a useful construct, but patients often fall somewhere along a spectrum between the hyperactive and hypoactive extremes, sometimes fluctuating from one to the other. Therefore, clinicians must recognize this broad range of presentations of delirium to identify all patients with this potentially reversible cognitive disturbance. Hyperactive patients are often easily recognized by their characteristic severe agitation, tremor, hallucinations, and autonomic instability. Patients who are quietly hypoactive are more often overlooked on the medical wards and in the ICU.
The reversibility of delirium is emphasized because many etiologies, such as systemic infection and medication effects, can be treated easily. The long-term cognitive effects of delirium remain largely unknown. Some episodes of delirium continue for weeks, months, or even years. The persistence of delirium in some patients and its high recurrence rate may be due to inadequate initial treatment of the underlying etiology. In other instances, delirium appears to cause permanent neuronal damage and cognitive decline. Even if an episode of delirium completely resolves, there may be lingering effects of the disorder; a patient’s recall of events after delirium varies widely, ranging from complete amnesia to repeated re-experiencing of the frightening period of confusion, similar to what is seen in patients with posttraumatic stress disorder.
RISK FACTORS
An effective primary prevention strategy for delirium begins with identification of patients at high risk for this disorder, including those preparing for elective surgery or being admitted to the hospital. Although no single validated scoring system has been widely accepted as a screen for asymptomatic patients, there are multiple well-established risk factors for delirium.
The two most consistently identified risks are older age and baseline cognitive dysfunction. Individuals who are over age 65 or exhibit low scores on standardized tests of cognition develop delirium upon hospitalization at a rate approaching 50%. Whether age and baseline cognitive dysfunction are truly independent risk factors is uncertain. Other predisposing factors include sensory deprivation, such as preexisting hearing and visual impairment, as well as indices for poor overall health, including baseline immobility, malnutrition, and underlying medical or neurologic illness.
In-hospital risks for delirium include the use of bladder catheterization, physical restraints, sleep and sensory deprivation, and the addition of three or more new medications. Avoiding such risks remains a key component of delirium prevention as well as treatment. Surgical and anesthetic risk factors for the development of postoperative delirium include specific procedures such as those involving cardiopulmonary bypass, inadequate or excessive treatment of pain in the immediate postoperative period, and perhaps specific agents such as inhalational anesthetics.
The relationship between delirium and dementia (Chap. 448) is complicated by significant overlap between the two conditions, and it is not always simple to distinguish between them. Dementia and preexisting cognitive dysfunction serve as major risk factors for delirium, and at least two-thirds of cases of delirium occur in patients with coexisting underlying dementia. A form of dementia with parkinsonism, termed dementia with Lewy bodies, is characterized by a fluctuating course, prominent visual hallucinations, parkinsonism, and an attentional deficit that clinically resembles hyperactive delirium; patients with this condition are particularly vulnerable to delirium. Delirium in the elderly often reflects an insult to the brain that is vulnerable due to an underlying neurodegenerative condition. Therefore, the development of delirium sometimes heralds the onset of a previously unrecognized brain disorder.
EPIDEMIOLOGY
Delirium is common, but its reported incidence has varied widely with the criteria used to define this disorder. Estimates of delirium in hospitalized patients range from 18 to 64%, with higher rates reported for elderly patients and patients undergoing hip surgery. Older patients in the ICU have especially high rates of delirium that approach 75%. The condition is not recognized in up to one-third of delirious inpatients, and the diagnosis is especially problematic in the ICU environment, where cognitive dysfunction is often difficult to appreciate in the setting of serious systemic illness and sedation. Delirium in the ICU should be viewed as an important manifestation of organ dysfunction not unlike liver, kidney, or heart failure. Outside the acute hospital setting, delirium occurs in nearly one-quarter of patients in nursing homes and in 50 to 80% of those at the end of life. These estimates emphasize the remarkably high frequency of this cognitive syndrome in older patients, a population expected to grow in the upcoming decades.
Until recently, an episode of delirium was viewed as a transient condition that carried a benign prognosis. It is now recognized as a disorder with a substantial morbidity rate and increased mortality rate and often represents the first manifestation of a serious underlying illness. Recent estimates of in-hospital mortality rates among delirious patients have ranged from 25 to 33%, a rate similar to that of patients with sepsis. Patients with an in-hospital episode of delirium have a fivefold higher mortality rate in the months after their illness compared with age-matched nondelirious hospitalized patients. Delirious hospitalized patients have a longer length of stay, are more likely to be discharged to a nursing home, and are more likely to experience subsequent episodes of delirium and cognitive decline; as a result, this condition has enormous economic implications.
PATHOGENESIS
The pathogenesis and anatomy of delirium are incompletely understood. The attentional deficit that serves as the neuropsychological hallmark of delirium has a diffuse localization within the brainstem, thalamus, prefrontal cortex, and parietal lobes. Rarely, focal lesions such as ischemic strokes have led to delirium in otherwise healthy persons; right parietal and medial dorsal thalamic lesions have been reported most commonly, pointing to the importance of these areas to delirium pathogenesis. In most cases, delirium results from widespread disturbances in cortical and subcortical regions rather than a focal neuroanatomic cause. Electroencephalogram (EEG) data in persons with delirium usually show symmetric slowing, a nonspecific finding that supports diffuse cerebral dysfunction.
Multiple neurotransmitter abnormalities, proinflammatory factors, and specific genes likely play a role in the pathogenesis of delirium. Deficiency of acetylcholine may play a key role, and medications with anticholinergic properties also can precipitate delirium. Dementia patients are susceptible to episodes of delirium, and those with Alzheimer’s pathology and dementia with Lewy bodies or Parkinson’s disease dementia are known to have a chronic cholinergic deficiency state due to degeneration of acetylcholine-producing neurons in the basal forebrain. Additionally, other neurotransmitters are also likely to be involved in this diffuse cerebral disorder. For example, increases in dopamine can also lead to delirium. Patients with Parkinson’s disease treated with dopaminergic medications can develop a delirium-like state that features visual hallucinations, fluctuations, and confusion.
Not all individuals exposed to the same insult will develop signs of delirium. A low dose of an anticholinergic medication may have no cognitive effects on a healthy young adult but produce a florid delirium in an elderly person with known underlying dementia, although even healthy young persons develop delirium with very high doses of anticholinergic medications. This concept of delirium developing as the result of an insult in predisposed individuals is currently the most widely accepted pathogenic construct. Therefore, if a previously healthy individual with no known history of cognitive illness develops delirium in the setting of a relatively minor insult such as elective surgery or hospitalization, an unrecognized underlying neurologic illness such as a neurodegenerative disease, multiple previous strokes, or another diffuse cerebral cause should be considered. In this context, delirium can be viewed as a “stress test for the brain” whereby exposure to known inciting factors such as systemic infection and offending drugs can unmask a decreased cerebral reserve and herald a serious underlying and potentially treatable illness.
PREVENTION
In light of the high morbidity associated with delirium and the tremendously increased health care costs that accompany it, development of an effective strategy to prevent delirium in hospitalized patients is extremely important. Successful identification of high-risk patients is the first step, followed by initiation of appropriate interventions. Simple standardized protocols used to manage risk factors for delirium, including sleep-wake cycle reversal, immobility, visual impairment, hearing impairment, sleep deprivation, and dehydration, have been shown to be effective. Recent trials in the ICU have focused both on identifying sedatives, such as dexmedetomidine, that are less likely to lead to delirium in critically ill patients and on developing protocols for daily awakenings in which infusions of sedative medications are interrupted and the patient is reorientated by the staff. All hospitals and health care systems should work toward decreasing the incidence of delirium.
35 |
Dementia |
Dementia, a syndrome with many causes, affects >5 million people in the United States and results in a total annual health care cost between $157 and $215 billion. Dementia is defined as an acquired deterioration in cognitive abilities that impairs the successful performance of activities of daily living. Episodic memory, the ability to recall events specific in time and place, is the cognitive function most commonly lost; 10% of persons age >70 years and 20–40% of individuals age >85 years have clinically identifiable memory loss. In addition to memory, dementia may erode other mental faculties, including language, visuospatial, praxis, calculation, judgment, and problem-solving abilities. Neuropsychiatric and social deficits also arise in many dementia syndromes, manifesting as depression, apathy, anxiety, hallucinations, delusions, agitation, insomnia, sleep disturbances, compulsions, or disinhibition. The clinical course may be slowly progressive, as in Alzheimer’s disease (AD); static, as in anoxic encephalopathy; or may fluctuate from day to day or minute to minute, as in dementia with Lewy bodies. Most patients with AD, the most prevalent form of dementia, begin with episodic memory impairment, although in other dementias, such as frontotemporal dementia, memory loss is not typically a presenting feature. Focal cerebral disorders are discussed in Chap. 36 and illustrated in a video library in Chap. 37e; the pathogenesis of AD and related disorders is discussed in Chap. 448.
FUNCTIONAL ANATOMY OF THE DEMENTIAS
Dementia syndromes result from the disruption of specific large-scale neuronal networks; the location and severity of synaptic and neuronal loss combine to produce the clinical features (Chap. 36). Behavior, mood, and attention are modulated by ascending noradrenergic, serotonergic, and dopaminergic pathways, whereas cholinergic signaling is critical for attention and memory functions. The dementias differ in the relative neurotransmitter deficit profiles; accordingly, accurate diagnosis guides effective pharmacologic therapy.
AD begins in the entorhinal region of the medial temporal lobe, spreads to the hippocampus, and then moves to lateral and posterior temporal and parietal neocortex, eventually causing a more widespread degeneration. Vascular dementia is associated with focal damage in a variable patchwork of cortical and subcortical regions or white matter tracts that disconnect nodes within distributed networks. In keeping with its anatomy, AD typically presents with episodic memory loss accompanied later by aphasia or navigational problems. In contrast, dementias that begin in frontal or subcortical regions, such as frontotemporal dementia (FTD) or Huntington’s disease (HD), are less likely to begin with memory problems and more likely to present with difficulties with judgment, mood, executive control, movement, and behavior.
Lesions of frontal-striatal1 pathways produce specific and predictable effects on behavior. The dorsolateral prefrontal cortex has connections with a central band of the caudate nucleus. Lesions of either the caudate or dorsolateral prefrontal cortex, or their connecting white matter pathways, may result in executive dysfunction, manifesting as poor organization and planning, decreased cognitive flexibility, and impaired working memory. The lateral orbital frontal cortex connects with the ventromedial caudate, and lesions of this system cause impulsiveness, distractibility, and disinhibition. The anterior cingulate cortex and adjacent medial prefrontal cortex project to the nucleus accumbens, and interruption of this system produces apathy, poverty of speech, emotional blunting, or even akinetic mutism. All corticostriatal systems also include topographically organized projections through the globus pallidus and thalamus, and damage to these nodes can likewise reproduce the clinical syndrome of cortical or striatal injury.
THE CAUSES OF DEMENTIA
The single strongest risk factor for dementia is increasing age. The prevalence of disabling memory loss increases with each decade over age 50 and is usually associated with the microscopic changes of AD at autopsy. Yet some centenarians have intact memory function and no evidence of clinically significant dementia. Whether dementia is an inevitable consequence of normal human aging remains controversial.
The many causes of dementia are listed in Table 35-1. The frequency of each condition depends on the age group under study, access of the group to medical care, country of origin, and perhaps racial or ethnic background. AD is the most common cause of dementia in Western countries, accounting for more than half of all patients. Vascular disease is considered the second most frequent cause for dementia and is particularly common in elderly patients or populations with limited access to medical care, where vascular risk factors are undertreated. Often, vascular brain injury is mixed with neurodegenerative disorders, making it difficult, even for the neuropathologist, to estimate the contribution of cerebrovascular disease to the cognitive disorder in an individual patient. Dementias associated with Parkinson’s disease (PD) (Chap. 449) are common and may develop years after onset of a parkinsonian disorder, as seen with PD-related dementia (PDD), or can occur concurrently with or preceding the motor syndrome, as in dementia with Lewy bodies (DLB). In patients under the age of 65, FTD rivals AD as the most common cause of dementia. Chronic intoxications, including those resulting from alcohol and prescription drugs, are an important and often treatable cause of dementia. Other disorders listed in Table 35-1 are uncommon but important because many are reversible. The classification of dementing illnesses into reversible and irreversible disorders is a useful approach to differential diagnosis. When effective treatments for the neurodegenerative conditions emerge, this dichotomy will become obsolete.
|
DIFFERENTIAL DIAGNOSIS OF DEMENTIA |
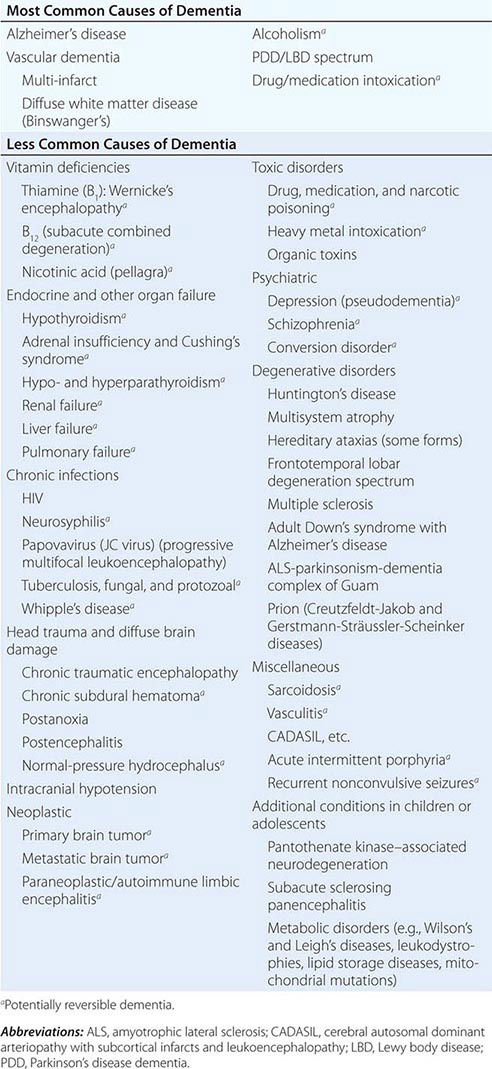
In a study of 1000 persons attending a memory disorders clinic, 19% had a potentially reversible cause of the cognitive impairment and 23% had a potentially reversible concomitant condition that may have contributed to the patient’s impairment. The three most common potentially reversible diagnoses were depression, normal pressure hydrocephalus (NPH), and alcohol dependence; medication side effects are also common and should be considered in every patient (Table 35-1).
Subtle cumulative decline in episodic memory is a common part of aging. This frustrating experience, often the source of jokes and humor, is referred to as benign forgetfulness of the elderly. Benign means that it is not so progressive or serious that it impairs reasonably successful and productive daily functioning, although the distinction between benign and more significant memory loss can be difficult to make. At age 85, the average person is able to learn and recall approximately one-half of the items (e.g., words on a list) that he or she could at age 18. A measurable cognitive problem that does not seriously disrupt daily activities is often referred to as mild cognitive impairment (MCI). Factors that predict progression from MCI to an AD dementia include a prominent memory deficit, family history of dementia, presence of an apolipoprotein ε4 (Apo ε4) allele, small hippocampal volumes, an AD-like signature of cortical atrophy, low cerebrospinal fluid Aβ, and elevated tau or evidence of brain amyloid deposition on positron emission tomography (PET) imaging.
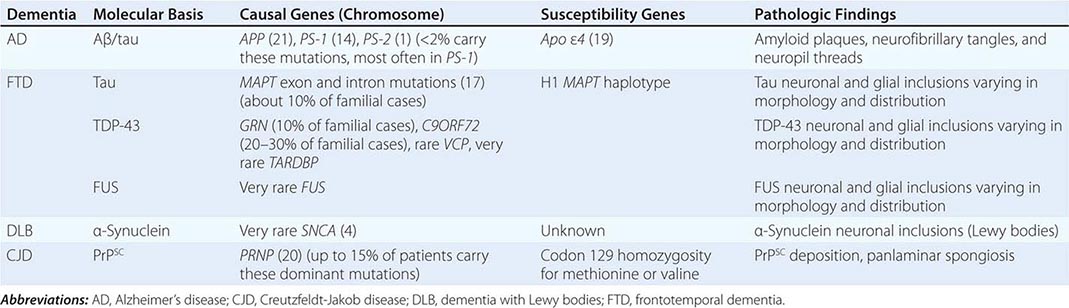
The major degenerative dementias include AD, DLB, FTD and related disorders, HD, and prion diseases, including Creutzfeldt-Jakob disease (CJD). These disorders are all associated with the abnormal aggregation of a specific protein: Aβ42 and tau in AD; α-synuclein in DLB; tau, TAR DNA-binding protein of 43 kDa (TDP-43), or fused in sarcoma (FUS) in FTD; huntingtin in HD; and misfolded prion protein (PrPsc) in CJD (Table 35-2).
|
THE MOLECULAR BASIS FOR DEGENERATIVE DEMENTIA |
APPROACH TO THE PATIENT:
Dementias
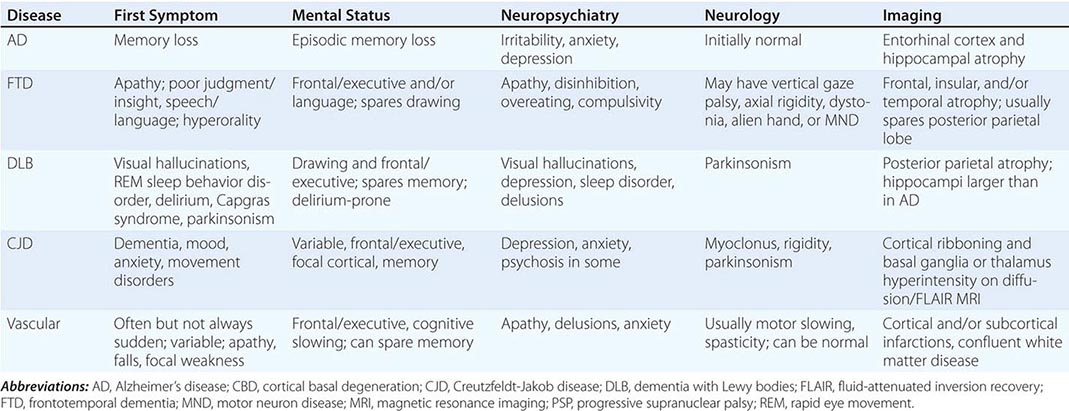
Three major issues should be kept at the forefront: (1) What is the best fit for a clinical diagnosis? (2) What component of the dementia syndrome is treatable or reversible? (3) Can the physician help to alleviate the burden on caregivers? A broad overview of the approach to dementia is shown in Table 35-3. The major degenerative dementias can usually be distinguished by the initial symptoms; neuropsychological, neuropsychiatric, and neurologic findings; and neuroimaging features (Table 35-4).
|
EVALUATION OF THE PATIENT WITH DEMENTIA |
|
CLINICAL DIFFERENTIATION OF THE MAJOR DEMENTIAS |
HISTORY
The history should concentrate on the onset, duration, and tempo of progression. An acute or subacute onset of confusion may be due to delirium (Chap. 34) and should trigger the search for intoxication, infection, or metabolic derangement. An elderly person with slowly progressive memory loss over several years is likely to suffer from AD. Nearly 75% of patients with AD begin with memory symptoms, but other early symptoms include difficulty with managing money, driving, shopping, following instructions, finding words, or navigating. Personality change, disinhibition, and weight gain or compulsive eating suggest FTD, not AD. FTD is also suggested by prominent apathy, compulsivity, loss of empathy for others, or progressive loss of speech fluency or single-word comprehension and by a relative sparing of memory and visuospatial abilities. The diagnosis of DLB is suggested by early visual hallucinations; parkinsonism; proneness to delirium or sensitivity to psychoactive medications; rapid eye movement (REM) behavior disorder (RBD; the loss of skeletal muscle paralysis during dreaming); or Capgras syndrome, the delusion that a familiar person has been replaced by an impostor.
A history of stroke with irregular stepwise progression suggests vascular dementia. Vascular dementia is also commonly seen in the setting of hypertension, atrial fibrillation, peripheral vascular disease, and diabetes. In patients suffering from cerebrovascular disease, it can be difficult to determine whether the dementia is due to AD, vascular disease, or a mixture of the two because many of the risk factors for vascular dementia, including diabetes, high cholesterol, elevated homocysteine, and low exercise, are also putative risk factors for AD. Moreover, many patients with a major vascular contribution to their dementia lack a history of stepwise decline. Rapid progression with motor rigidity and myoclonus suggests CJD (Chap. 453e). Seizures may indicate strokes or neoplasm but also occur in AD, particularly early-age-of-onset AD. Gait disturbance is common in vascular dementia, PD/DLB, or NPH. A history of high-risk sexual behaviors or intravenous drug use should trigger a search for central nervous system (CNS) infection, especially HIV or syphilis. A history of recurrent head trauma could indicate chronic subdural hematoma, chronic traumatic encephalopathy (a progressive dementia best characterized in contact sport athletes such as boxers and American football players), intracranial hypotension, or NPH. Subacute onset of severe amnesia and psychosis with mesial temporal T2/fluid-attenuated inversion recovery (FLAIR) hyperintensities on magnetic resonance imaging (MRI) should raise concern for paraneoplastic limbic encephalitis, especially in a long-term smoker or other patients at risk for cancer. Related autoimmune conditions, such as voltage-gated potassium channel (VGKC)- or N-methyl-D-aspartate (NMDA)-receptor antibody-mediated encephalopathy, can present with a similar tempo and imaging signature with or without characteristic motor manifestations such as myokymia (anti-VGKC) and faciobrachial dystonic seizures (anti-NMDA). Alcohol abuse creates risk for malnutrition and thiamine deficiency. Veganism, bowel irradiation, an autoimmune diathesis, a remote history of gastric surgery, and chronic antihistamine therapy for dyspepsia or gastroesophageal reflux predispose to B12 deficiency. Certain occupations, such as working in a battery or chemical factory, might indicate heavy metal intoxication. Careful review of medication intake, especially for sedatives and analgesics, may raise the issue of chronic drug intoxication. An autosomal dominant family history is found in HD and in familial forms of AD, FTD, DLB, or prion disorders. A history of mood disorders, the recent death of a loved one, or depressive signs, such as insomnia or weight loss, raise the possibility of depression-related cognitive impairments.
PHYSICAL AND NEUROLOGIC EXAMINATION
A thorough general and neurologic examination is essential to document dementia, to look for other signs of nervous system involvement, and to search for clues suggesting a systemic disease that might be responsible for the cognitive disorder. Typical AD spares motor systems until later in the course. In contrast, FTD patients often develop axial rigidity, supranuclear gaze palsy, or a motor neuron disease reminiscent of amyotrophic lateral sclerosis (ALS). In DLB, the initial symptoms may include the new onset of a parkinsonian syndrome (resting tremor, cogwheel rigidity, bradykinesia, festinating gait), but DLB often starts with visual hallucinations or dementia. Symptoms referable to the lower brainstem (RBD, gastrointestinal or autonomic problems) may arise years or even decades before parkinsonism or dementia. Corticobasal syndrome (CBS) features asymmetric akinesia and rigidity, dystonia, myoclonus, alien limb phenomena, pyramidal signs, and prefrontal deficits such as nonfluent aphasia with or without motor speech impairment, executive dysfunction, apraxia, or a behavioral disorder. Progressive supranuclear palsy (PSP) is associated with unexplained falls, axial rigidity, dysphagia, and vertical gaze deficits. CJD is suggested by the presence of diffuse rigidity, an akinetic-mute state, and prominent, often startle-sensitive myoclonus.
Hemiparesis or other focal neurologic deficits suggest vascular dementia or brain tumor. Dementia with a myelopathy and peripheral neuropathy suggests vitamin B12 deficiency. Peripheral neuropathy could also indicate another vitamin deficiency, heavy metal intoxication, thyroid dysfunction, Lyme disease, or vasculitis. Dry, cool skin, hair loss, and bradycardia suggest hypothyroidism. Fluctuating confusion associated with repetitive stereotyped movements may indicate ongoing limbic, temporal, or frontal seizures. In the elderly, hearing impairment or visual loss may produce confusion and disorientation misinterpreted as dementia. Profound bilateral sensorineural hearing loss in a younger patient with short stature or myopathy, however, should raise concern for a mitochondrial disorder.
COGNITIVE AND NEUROPSYCHIATRIC EXAMINATION
Brief screening tools such as the Mini-Mental State Examination (MMSE), the Montreal Cognitive Assessment (MOCA), and Cognistat can be used to capture dementia and follow progression. None of these tests is highly sensitive to early-stage dementia or discriminates between dementia syndromes. The MMSE is a 30 point test of cognitive function, with each correct answer being scored as 1 point. It includes tests in the areas of: orientation (e.g., identify season/date/month/year/floor/hospital/town/state/country); registration (e.g., name and restate 3 objects); recall (e.g., remember the same three objects 5 minutes later); and language (e.g., name pencil and watch; repeat “no if’s and’s or but’s”; follow a 3-step command; obey a written command; and write a sentence and copy a design). In most patients with MCI and some with clinically apparent AD, bedside screening tests may be normal, and a more challenging and comprehensive set of neuropsychological tests will be required. When the etiology for the dementia syndrome remains in doubt, a specially tailored evaluation should be performed that includes tasks of working and episodic memory, executive function, language, and visuospatial and perceptual abilities. In AD, the early deficits involve episodic memory, category generation (“name as many animals as you can in 1 minute”), and visuoconstructive ability. Usually deficits in verbal or visual episodic memory are the first neuropsychological abnormalities detected, and tasks that require the patient to recall a long list of words or a series of pictures after a predetermined delay will demonstrate deficits in most patients. In FTD, the earliest deficits on cognitive testing involve executive control or language (speech or naming) function, but some patients lack either finding despite profound social-emotional deficits. PDD or DLB patients have more severe deficits in visuospatial function but do better on episodic memory tasks than patients with AD. Patients with vascular dementia often demonstrate a mixture of executive control and visuospatial deficits, with prominent psychomotor slowing. In delirium, the most prominent deficits involve attention, working memory, and executive function, making the assessment of other cognitive domains challenging and often uninformative.
A functional assessment should also be performed to help the physician determine the day-to-day impact of the disorder on the patient’s memory, community affairs, hobbies, judgment, dressing, and eating. Knowledge of the patient’s functional abilities will help the clinician and the family to organize a therapeutic approach.
Neuropsychiatric assessment is important for diagnosis, prognosis, and treatment. In the early stages of AD, mild depressive features, social withdrawal, and irritability or anxiety are the most prominent psychiatric changes, but patients often maintain core social graces into the middle or late stages, when delusions, agitation, and sleep disturbance may emerge. In FTD, dramatic personality change with apathy, overeating, compulsions, disinhibition, euphoria, and loss of empathy are early and common. DLB is associated with visual hallucinations, delusions related to person or place identity, RBD, and excessive daytime sleepiness. Dramatic fluctuations occur not only in cognition but also in arousal. Vascular dementia can present with psychiatric symptoms such as depression, anxiety, delusions, disinhibition, or apathy.
LABORATORY TESTS
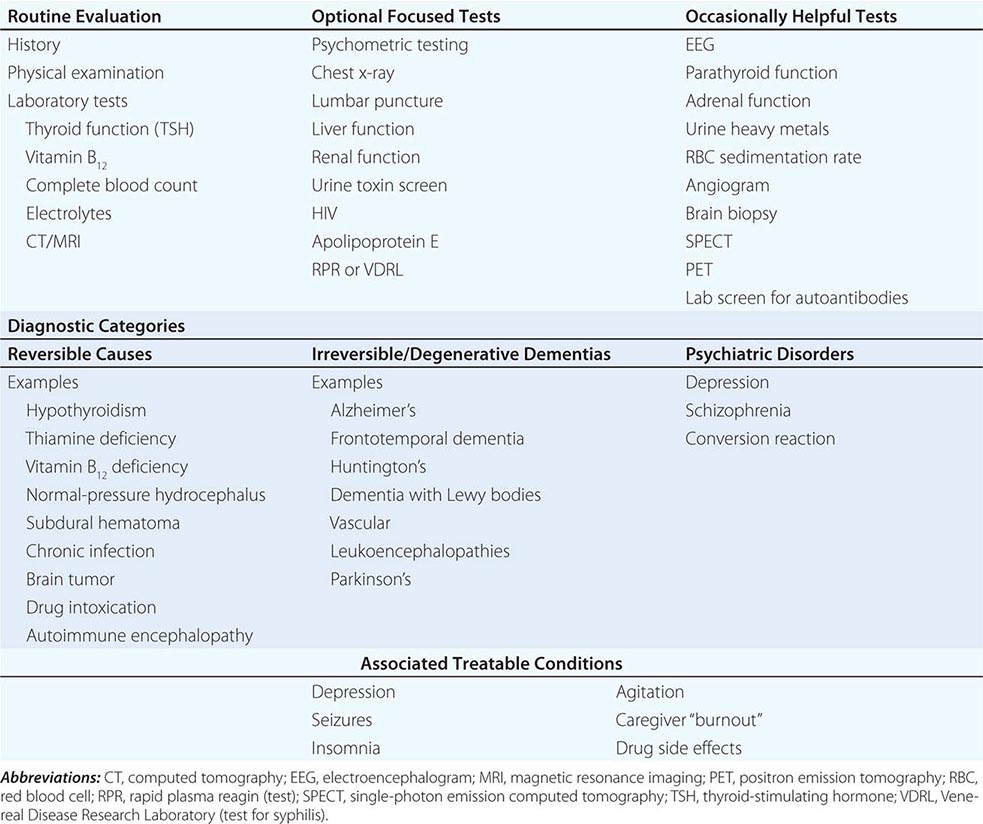
The choice of laboratory tests in the evaluation of dementia is complex and should be tailored to the individual patient. The physician must take measures to avoid missing a reversible or treatable cause, yet no single treatable etiology is common; thus, a screen must use multiple tests, each of which has a low yield. Cost/benefit ratios are difficult to assess, and many laboratory screening algorithms for dementia discourage multiple tests. Nevertheless, even a test with only a 1–2% positive rate is worth undertaking if the alternative is missing a treatable cause of dementia. Table 35-3 lists most screening tests for dementia. The American Academy of Neurology recommends the routine measurement of a complete blood count, electrolytes, renal and thyroid function, a vitamin B12 level, and a neuroimaging study (computed tomography [CT] or MRI).
Neuroimaging studies, especially MRI, help to rule out primary and metastatic neoplasms, locate areas of infarction or inflammation, detect subdural hematomas, and suggest NPH or diffuse white matter disease. They also help to establish a regional pattern of atrophy. Support for the diagnosis of AD includes hippocampal atrophy in addition to posterior-predominant cortical atrophy (Fig. 35-1). Focal frontal, insular, and/or anterior temporal atrophy (Fig. 35-1). Focal frontal, insular, and/or anterior temporal atrophy suggests FTD (Chap. 448). DLB often features less prominent atrophy, with greater involvement of amygdala than hippocampus. In CJD, magnetic resonance (MR) diffusion-weighted imaging reveals restricted diffusion within the cortical ribbon and basal ganglia in most patients. Extensive white matter abnormalities correlate with a vascular etiology (Fig. 35-2). Communicating hydrocephalus with vertex effacement (crowding of dorsal convexity gyri/sulci), gaping Sylvian fissures despite minimal cortical atrophy, and additional features shown in Fig. 35-3 suggest NPH. Single-photon emission computed tomography (SPECT) and PET scanning show temporal-parietal hypoperfusion or hypometabolism in AD and frontotemporal deficits in FTD, but these changes often reflect atrophy and can therefore be detected with MRI alone in many patients. Recently, amyloid imaging has shown promise for the diagnosis of AD, and Pittsburgh Compound-B (PiB) (not available outside of research settings) and 18F-AV-45 (florbetapir; approved by the U.S. Food and Drug Administration in 2013) are reliable radioligands for detecting brain amyloid associated with amyloid angiopathy or neuritic plaques of AD (Fig. 35-4). Because these abnormalities can be seen in cognitively normal older persons, however (~25% of individuals at age 65), amyloid imaging may also detect preclinical or incidental AD in patients lacking an AD-like dementia syndrome. Currently, the main clinical value of amyloid imaging is to exclude AD as the likely cause of dementia in patients who have negative scans. Once disease-modifying therapies become available, use of these biomarkers may help to identify treatment candidates before irreversible brain injury has occurred. In the meantime, the significance of detecting brain amyloid in an asymptomatic elder remains a topic of vigorous investigation. Similarly, MRI perfusion and structural/functional connectivity methods are being explored as potential treatment-monitoring strategies.
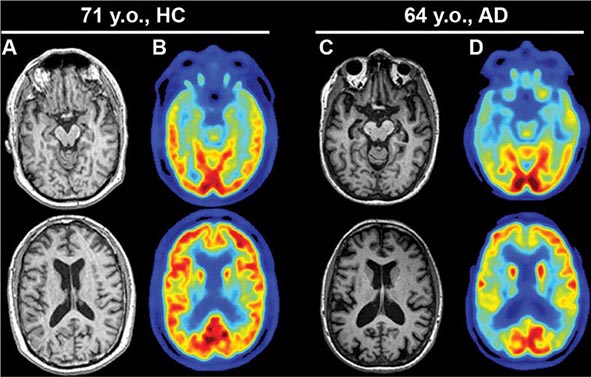
FIGURE 35-1 Alzheimer’s disease (AD). Axial T1-weighted magnetic resonance images of a healthy 71-year-old (A) and a 64-year-old with AD (C). Note the reduction in medial temporal lobe volume in the patient with AD. Fluorodeoxyglucose positron emission tomography scans of the same individuals (B and D) demonstrate reduced glucose metabolism in the posterior temporoparietal regions bilaterally in AD, a typical finding in this condition. HC, healthy control. (Images courtesy of Gil Rabinovici, University of California, San Francisco and William Jagust, University of California, Berkeley.)
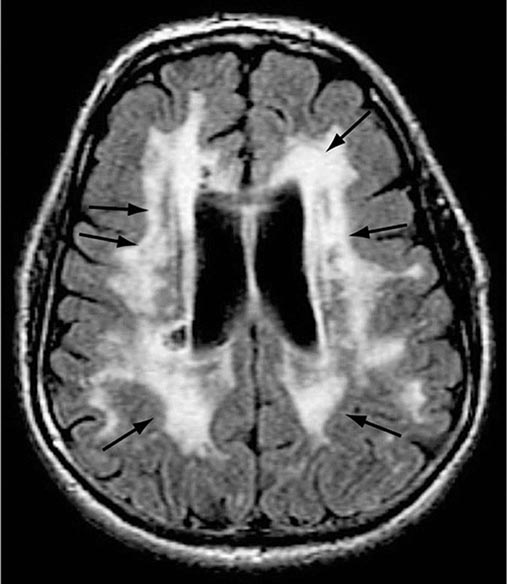
FIGURE 35-2 Diffuse white matter disease. Axial fluid-attenuated inversion recovery (FLAIR) magnetic resonance image through the lateral ventricles reveals multiple areas of hyperintensity (arrows) involving the periventricular white matter as well as the corona radiata and striatum. Although seen in some individuals with normal cognition, this appearance is more pronounced in patients with dementia of a vascular etiology.
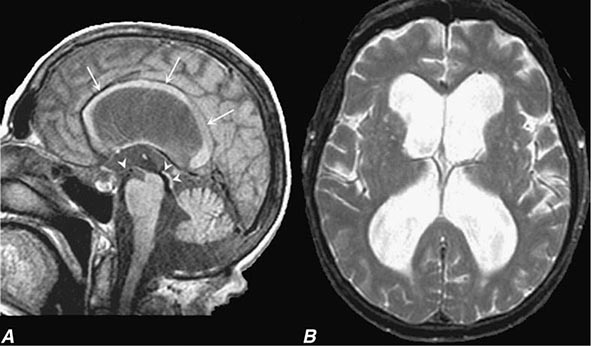
FIGURE 35-3 Normal-pressure hydrocephalus. A. Sagittal T1-weighted magnetic resonance image (MRI) demonstrates dilation of the lateral ventricle and stretching of the corpus callosum (arrows), depression of the floor of the third ventricle (single arrowhead), and enlargement of the aqueduct (double arrowheads). Note the diffuse dilation of the lateral, third, and fourth ventricles with a patent aqueduct, typical of communicating hydrocephalus. B. Axial T2-weighted MRIs demonstrate dilation of the lateral ventricles. This patient underwent successful ventriculoperitoneal shunting.
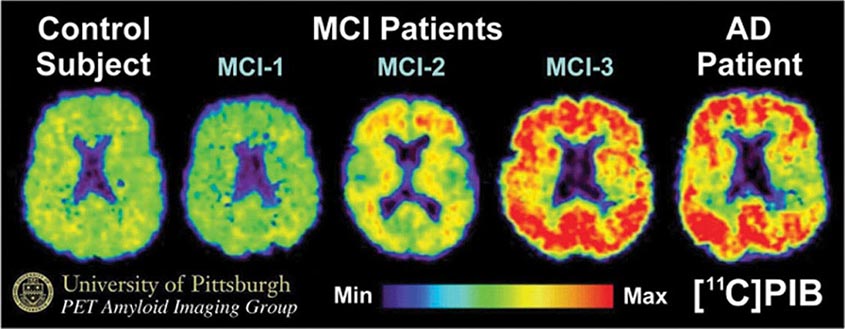
FIGURE 35-4 Positron emission tomography (PET) images obtained with the amyloid-imaging agent Pittsburgh Compound-B ([11C]PIB) in a normal control (left); three different patients with mild cognitive impairment (MCI; center); and a patient with mild Alzheimer’s disease (AD; right). Some MCI patients have control-like levels of amyloid, some have AD-like levels of amyloid, and some have intermediate levels. (Images courtesy of William Klunk and Chester Mathis, University of Pittsburgh.)
Lumbar puncture need not be done routinely in the evaluation of dementia, but it is indicated when CNS infection or inflammation are credible diagnostic possibilities. Cerebrospinal fluid (CSF) levels of Aβ42 and tau proteins show differing patterns with the various dementias, and the presence of low Aβ42 and mildly elevated CSF tau is highly suggestive of AD. The routine use of lumbar puncture in the diagnosis of dementia is debated, but the sensitivity and specificity of AD diagnostic measures are not yet high enough to warrant routine use. Formal psychometric testing helps to document the severity of cognitive disturbance, suggest psychogenic causes, and provide a more formal method for following the disease course. Electroencephalogram (EEG) is not routinely used but can help to suggest CJD (repetitive bursts of diffuse high-amplitude sharp waves, or “periodic complexes”) or an underlying nonconvulsive seizure disorder (epileptiform discharges). Brain biopsy (including meninges) is not advised except to diagnose vasculitis, potentially treatable neoplasms, or unusual infections when the diagnosis is uncertain. Systemic disorders with CNS manifestations, such as sarcoidosis, can usually be confirmed through biopsy of lymph node or solid organ rather than brain. MR angiography should be considered when cerebral vasculitis or cerebral venous thrombosis is a possible cause of the dementia.
1The striatum comprises the caudate/putamen.
36 |
Aphasia, Memory Loss, and Other Focal Cerebral Disorders |
The cerebral cortex of the human brain contains approximately 20 billion neurons spread over an area of 2.5 m2. The primary sensory and motor areas constitute 10% of the cerebral cortex. The rest is subsumed by modality-selective, heteromodal, paralimbic, and limbic areas collectively known as the association cortex (Fig. 36-1). The association cortex mediates the integrative processes that subserve cognition, emotion, and comportment. A systematic testing of these mental functions is necessary for the effective clinical assessment of the association cortex and its diseases. According to current thinking, there are no centers for “hearing words,” “perceiving space,” or “storing memories.” Cognitive and behavioral functions (domains) are coordinated by intersecting large-scale neural networks that contain interconnected cortical and subcortical components. Five anatomically defined large-scale networks are most relevant to clinical practice: (1) a perisylvian network for language, (2) a parietofrontal network for spatial orientation, (3) an occipitotemporal network for face and object recognition, (4) a limbic network for retentive memory, and (5) a prefrontal network for the executive control of cognition and comportment.
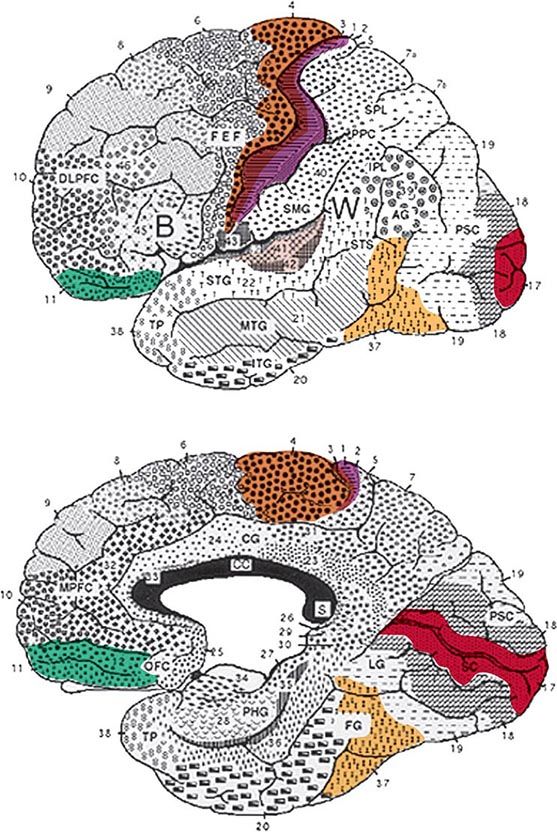
FIGURE 36-1 Lateral (top) and medial (bottom) views of the cerebral hemispheres. The numbers refer to the Brodmann cytoarchitectonic designations. Area 17 corresponds to the primary visual cortex, 41–42 to the primary auditory cortex, 1–3 to the primary somatosensory cortex, and 4 to the primary motor cortex. The rest of the cerebral cortex contains association areas. AG, angular gyrus; B, Broca’s area; CC, corpus callosum; CG, cingulate gyrus; DLPFC, dorsolateral prefrontal cortex; FEF, frontal eye fields (premotor cortex); FG, fusiform gyrus; IPL, inferior parietal lobule; ITG, inferior temporal gyrus; LG, lingual gyrus; MPFC, medial prefrontal cortex; MTG, middle temporal gyrus; OFC, orbitofrontal cortex; PHG, parahippocampal gyrus; PPC, posterior parietal cortex; PSC, peristriate cortex; SC, striate cortex; SMG, supramarginal gyrus; SPL, superior parietal lobule; STG, superior temporal gyrus; STS, superior temporal sulcus; TP, temporopolar cortex; W, Wernicke’s area.
THE LEFT PERISYLVIAN NETWORK FOR APHASIAS
The areas that are critical for language make up a distributed network located along the perisylvian region of the left hemisphere. One hub, located in the inferior frontal gyrus, is known as Broca’s area. Damage to this region impairs phonology, fluency, and the grammatical structure of sentences. The location of a second hub, known as Wernicke’s area, is less clearly settled but is traditionally thought to include the posterior parts of the temporal lobe. Cerebrovascular accidents that damage this area interfere with the ability to understand spoken or written sentences as well as the ability to express thoughts through meaningful words and statements. These two hubs are interconnected with each other and with surrounding parts of the frontal, parietal, and temporal lobes. Damage to this network gives rise to language impairments known as aphasia. Aphasia should be diagnosed only when there are deficits in the formal aspects of language, such as word finding, word choice, comprehension, spelling, or grammar. Dysarthria and mutism do not by themselves lead to a diagnosis of aphasia. In approximately 90% of right-handers and 60% of left-handers, aphasia occurs only after lesions of the left hemisphere.
CLINICAL EXAMINATION
The clinical examination of language should include the assessment of naming, spontaneous speech, comprehension, repetition, reading, and writing. A deficit of naming (anomia) is the single most common finding in aphasic patients. When asked to name a common object, the patient may fail to come up with the appropriate word, may provide a circumlocutious description of the object (“the thing for writing”), or may come up with the wrong word (paraphasia). If the patient offers an incorrect but related word (“pen” for “pencil”), the naming error is known as a semantic paraphasia; if the word approximates the correct answer but is phonetically inaccurate (“plentil” for “pencil”), it is known as a phonemic paraphasia. In most anomias, the patient cannot retrieve the appropriate name when shown an object but can point to the appropriate object when the name is provided by the examiner. This is known as a one-way (or retrieval-based) naming deficit. A two-way (comprehension-based) naming deficit exists if the patient can neither provide nor recognize the correct name. Spontaneous speech is described as “fluent” if it maintains appropriate output volume, phrase length, and melody or as “nonfluent” if it is sparse and halting and average utterance length is below four words. The examiner also should note the integrity of grammar as manifested by word order (syntax), tenses, suffixes, prefixes, plurals, and possessives. Comprehension can be tested by assessing the patient’s ability to follow conversation, asking yes-no questions (“Can a dog fly?” “Does it snow in summer?”), asking the patient to point to appropriate objects (“Where is the source of illumination in this room?”), or asking for verbal definitions of single words. Repetition is assessed by asking the patient to repeat single words, short sentences, or strings of words such as “No ifs, ands, or buts.” The testing of repetition with tongue twisters such as “hippopotamus” and “Irish constabulary” provides a better assessment of dysarthria and palilalia than of aphasia. It is important to make sure that the number of words does not exceed the patient’s attention span. Otherwise, the failure of repetition becomes a reflection of the narrowed attention span (working memory) rather than an indication of an aphasic deficit. Reading should be assessed for deficits in reading aloud as well as comprehension. Alexia describes an inability to either read aloud or comprehend single words and simple sentences; agraphia (or dysgraphia) is used to describe an acquired deficit in spelling.
Aphasias can arise acutely in cerebrovascular accidents (CVAs) or gradually in neurodegenerative diseases. The syndromes listed in Table 36-1 are most applicable to the former group, where gray matter and white matter at the lesion site are abruptly and jointly destroyed. Progressive neurodegenerative diseases can have cellular, laminar, and regional specificity, giving rise to a different set of aphasias that will be described separately. The syndromes outlined below are idealizations and rarely occur in pure form.
|
CLINICAL FEATURES OF APHASIAS AND RELATED CONDITIONS COMMONLY SEEN IN CEREBROVASCULAR ACCIDENTS |
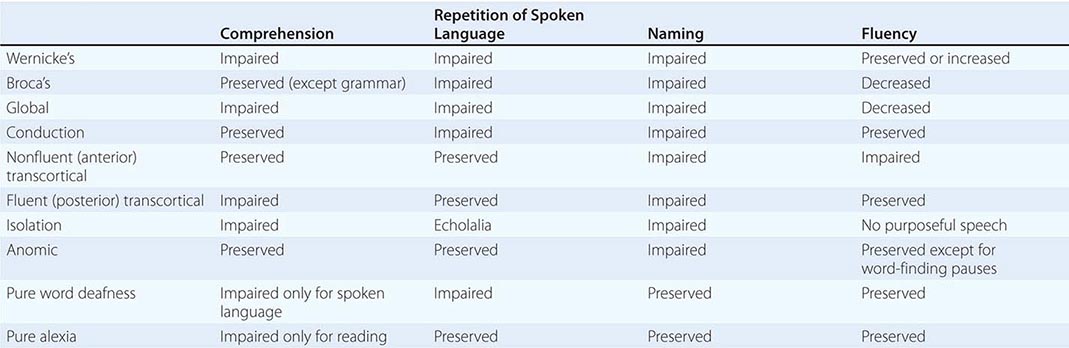
Wernicke’s Aphasia Comprehension is impaired for spoken and written words and sentences. Language output is fluent but is highly paraphasic and circumlocutious. Paraphasic errors may lead to strings of neologisms, which lead to “jargon aphasia.” Speech contains few substantive nouns. The output is therefore voluminous but uninformative. For example, a patient attempts to describe how his wife accidentally threw away something important, perhaps his dentures: “We don’t need it anymore, she says. And with it when that was downstairs was my teeth-tick … a … den … dentith … my dentist. And they happened to be in that bag … see? …Where my two … two little pieces of dentist that I use … that I … all gone. If she throws the whole thing away … visit some friends of hers and she can’t throw them away.”
Gestures and pantomime do not improve communication. The patient may not realize that his or her language is incomprehensible and may appear angry and impatient when the examiner fails to decipher the meaning of a severely paraphasic statement. In some patients this type of aphasia can be associated with severe agitation and paranoia. The ability to follow commands aimed at axial musculature may be preserved. The dissociation between the failure to understand simple questions (“What is your name?”) in a patient who rapidly closes his or her eyes, sits up, or rolls over when asked to do so is characteristic of Wernicke’s aphasia and helps differentiate it from deafness, psychiatric disease, or malingering. Patients with Wernicke’s aphasia cannot express their thoughts in meaning-appropriate words and cannot decode the meaning of words in any modality of input. This aphasia therefore has expressive as well as receptive components. Repetition, naming, reading, and writing also are impaired.
The lesion site most commonly associated with Wernicke’s aphasia is the posterior portion of the language network. An embolus to the inferior division of the middle cerebral artery, to the posterior temporal or angular branches in particular, is the most common etiology (Chap. 446). Intracerebral hemorrhage, head trauma, and neoplasm are other causes of Wernicke’s aphasia. A coexisting right hemianopia or superior quadrantanopia is common, and mild right nasolabial flattening may be found, but otherwise, the examination is often unrevealing. The paraphasic, neologistic speech in an agitated patient with an otherwise unremarkable neurologic examination may lead to the suspicion of a primary psychiatric disorder such as schizophrenia or mania, but the other components characteristic of acquired aphasia and the absence of prior psychiatric disease usually settle the issue. Prognosis for recovery of language function is guarded.
Broca’s Aphasia Speech is nonfluent, labored, interrupted by many word-finding pauses, and usually dysarthric. It is impoverished in function words but enriched in meaning-appropriate nouns. Abnormal word order and the inappropriate deployment of bound morphemes (word endings used to denote tenses, possessives, or plurals) lead to a characteristic agrammatism. Speech is telegraphic and pithy but quite informative. In the following passage, a patient with Broca’s aphasia describes his medical history: “I see … the dotor, dotor sent me … Bosson. Go to hospital. Dotor … kept me beside. Two, tee days, doctor send me home.”
Output may be reduced to a grunt or single word (“yes” or “no”), which is emitted with different intonations in an attempt to express approval or disapproval. In addition to fluency, naming and repetition are impaired. Comprehension of spoken language is intact except for syntactically difficult sentences with a passive voice structure or embedded clauses, indicating that Broca’s aphasia is not just an “expressive” or “motor” disorder and that it also may involve a comprehension deficit in decoding syntax. Patients with Broca’s aphasia can be tearful, easily frustrated, and profoundly depressed. Insight into their condition is preserved, in contrast to Wernicke’s aphasia. Even when spontaneous speech is severely dysarthric, the patient may be able to display a relatively normal articulation of words when singing. This dissociation has been used to develop specific therapeutic approaches (melodic intonation therapy) for Broca’s aphasia. Additional neurologic deficits include right facial weakness, hemiparesis or hemiplegia, and a buccofacial apraxia characterized by an inability to carry out motor commands involving oropharyngeal and facial musculature (e.g., patients are unable to demonstrate how to blow out a match or suck through a straw). The cause is most often infarction of Broca’s area (the inferior frontal convolution; “B” in Fig. 36-1) and surrounding anterior perisylvian and insular cortex due to occlusion of the superior division of the middle cerebral artery (Chap. 446). Mass lesions, including tumor, intracerebral hemorrhage, and abscess, also may be responsible. When the cause of Broca’s aphasia is stroke, recovery of language function generally peaks within 2 to 6 months, after which time further progress is limited. Speech therapy is more successful than in Wernicke’s aphasia.
Conduction Aphasia Speech output is fluent but contains many phonemic paraphasias, comprehension of spoken language is intact, and repetition is severely impaired. Naming elicits phonemic paraphasias, and spelling is impaired. Reading aloud is impaired, but reading comprehension is preserved. The lesion sites spare the functionality of Broca’s and Wernicke’s areas but may induce a disconnection between the two. Occasionally, a transient Wernicke’s aphasia may rapidly resolve into a conduction aphasia. The paraphasic output in conduction aphasia interferes with the ability to express meaning, but this deficit is not nearly as severe as the one displayed by patients with Wernicke’s aphasia. Associated neurologic signs in conduction aphasia vary according to the primary lesion site.
Transcortical Aphasias: Fluent and Nonfluent Clinical features of fluent (posterior) transcortical aphasia are similar to those of Wernicke’s aphasia, but repetition is intact. The lesion site disconnects the intact core of the language network from other temporoparietal association areas. Associated neurologic findings may include hemianopia. Cerebrovascular lesions (e.g., infarctions in the posterior watershed zone) and neoplasms that involve the temporoparietal cortex posterior to Wernicke’s area are common causes. The features of nonfluent (anterior) transcortical aphasia are similar to those of Broca’s aphasia, but repetition is intact and agrammatism is less pronounced. The neurologic examination may be otherwise intact, but a right hemiparesis also can exist. The lesion site disconnects the intact language network from prefrontal areas of the brain and usually involves the anterior watershed zone between anterior and middle cerebral artery territories or the supplementary motor cortex in the territory of the anterior cerebral artery.
Global and Isolation Aphasias Global aphasia represents the combined dysfunction of Broca’s and Wernicke’s areas and usually results from strokes that involve the entire middle cerebral artery distribution in the left hemisphere. Speech output is nonfluent, and comprehension of language is severely impaired. Related signs include right hemiplegia, hemisensory loss, and homonymous hemianopia. Isolation aphasia represents a combination of the two transcortical aphasias. Comprehension is severely impaired, and there is no purposeful speech output. The patient may parrot fragments of heard conversations (echolalia), indicating that the neural mechanisms for repetition are at least partially intact. This condition represents the pathologic function of the language network when it is isolated from other regions of the brain. Broca’s and Wernicke’s areas tend to be spared, but there is damage to the surrounding frontal, parietal, and temporal cortex. Lesions are patchy and can be associated with anoxia, carbon monoxide poisoning, or complete watershed zone infarctions.
Anomic Aphasia This form of aphasia may be considered the “minimal dysfunction” syndrome of the language network. Articulation, comprehension, and repetition are intact, but confrontation naming, word finding, and spelling are impaired. Word-finding pauses are uncommon, so language output is fluent but paraphasic, circumlocutious, and uninformative. The lesion sites can be anywhere within the left hemisphere language network, including the middle and inferior temporal gyri. Anomic aphasia is the single most common language disturbance seen in head trauma, metabolic encephalopathy, and Alzheimer’s disease.
Pure Word Deafness The most common causes are either bilateral or left-sided middle cerebral artery (MCA) strokes affecting the superior temporal gyrus. The net effect of the underlying lesion is to interrupt the flow of information from the auditory association cortex to the language network. Patients have no difficulty understanding written language and can express themselves well in spoken or written language. They have no difficulty interpreting and reacting to environmental sounds since primary auditory cortex and auditory association areas of the right hemisphere are spared. Because auditory information cannot be conveyed to the language network, however, it cannot be decoded into neural word representations, and the patient reacts to speech as if it were in an alien tongue that cannot be deciphered. Patients cannot repeat spoken language but have no difficulty naming objects. In time, patients with pure word deafness teach themselves lipreading and may appear to have improved. There may be no additional neurologic findings, but agitated paranoid reactions are common in the acute stages. Cerebrovascular lesions are the most common cause.
Pure Alexia Without Agraphia This is the visual equivalent of pure word deafness. The lesions (usually a combination of damage to the left occipital cortex and to a posterior sector of the corpus callosum—the splenium) interrupt the flow of visual input into the language network. There is usually a right hemianopia, but the core language network remains unaffected. The patient can understand and produce spoken language, name objects in the left visual hemifield, repeat, and write. However, the patient acts as if illiterate when asked to read even the simplest sentence because the visual information from the written words (presented to the intact left visual hemifield) cannot reach the language network. Objects in the left hemifield may be named accurately because they activate nonvisual associations in the right hemisphere, which in turn can access the language network through transcallosal pathways anterior to the splenium. Patients with this syndrome also may lose the ability to name colors, although they can match colors. This is known as a color anomia. The most common etiology of pure alexia is a vascular lesion in the territory of the posterior cerebral artery or an infiltrating neoplasm in the left occipital cortex that involves the optic radiations as well as the crossing fibers of the splenium. Because the posterior cerebral artery also supplies medial temporal components of the limbic system, a patient with pure alexia also may experience an amnesia, but this is usually transient because the limbic lesion is unilateral.
Apraxia and Aphemia Apraxia designates a complex motor deficit that cannot be attributed to pyramidal, extrapyramidal, cerebellar, or sensory dysfunction and that does not arise from the patient’s failure to understand the nature of the task. Apraxia of speech is used to designate articulatory abnormalities in the duration, fluidity, and stress of syllables that make up words. Intoning the words may improve articulation. It can arise with CVAs in the posterior part of Broca’s area or in the course of frontotemporal lobar degeneration (FTLD) with tauopathy. Aphemia is a severe form of acute speech apraxia that presents with severely impaired fluency (often mutism). Recovery is the rule and involves an intermediate stage of hoarse whispering. Writing, reading, and comprehension are intact, and so this is not a true aphasic syndrome. CVAs in parts of Broca’s area or subcortical lesions that undercut its connections with other parts of the brain may be present. Occasionally, the lesion site is on the medial aspects of the frontal lobes and may involve the supplementary motor cortex of the left hemisphere. Ideomotor apraxia is diagnosed when commands to perform a specific motor act (“cough,” “blow out a match”) or pantomime the use of a common tool (a comb, hammer, straw, or toothbrush) in the absence of the real object cannot be followed. The patient’s ability to comprehend the command is ascertained by demonstrating multiple movements and establishing that the correct one can be recognized. Some patients with this type of apraxia can imitate the appropriate movement (when it is demonstrated by the examiner) and show no impairment when handed the real object, indicating that the sensorimotor mechanisms necessary for the movement are intact. Some forms of ideomotor apraxia represent a disconnection of the language network from pyramidal motor systems so that commands to execute complex movements are understood but cannot be conveyed to the appropriate motor areas. Buccofacial apraxia involves apraxic deficits in movements of the face and mouth. Limb apraxia encompasses apraxic deficits in movements of the arms and legs. Ideomotor apraxia almost always is caused by lesions in the left hemisphere and is commonly associated with aphasic syndromes, especially Broca’s aphasia and conduction aphasia. Because the handling of real objects is not impaired, ideomotor apraxia by itself causes no major limitation of daily living activities. Patients with lesions of the anterior corpus callosum can display ideomotor apraxia confined to the left side of the body, a sign known as sympathetic dyspraxia. A severe form of sympathetic dyspraxia, known as the alien hand syndrome, is characterized by additional features of motor disinhibition on the left hand. Ideational apraxia refers to a deficit in the sequencing of goal-directed movements in patients who have no difficulty executing the individual components of the sequence. For example, when the patient is asked to pick up a pen and write, the sequence of uncapping the pen, placing the cap at the opposite end, turning the point toward the writing surface, and writing may be disrupted, and the patient may be seen trying to write with the wrong end of the pen or even with the removed cap. These motor sequencing problems usually are seen in the context of confusional states and dementias rather than focal lesions associated with aphasic conditions. Limb-kinetic apraxia involves clumsiness in the use of tools or objects that cannot be attributed to sensory, pyramidal, extrapyramidal, or cerebellar dysfunction. This condition can emerge in the context of focal premotor cortex lesions or corticobasal degeneration.
Gerstmann’s Syndrome The combination of acalculia (impairment of simple arithmetic), dysgraphia (impaired writing), finger anomia (an inability to name individual fingers such as the index and thumb), and right-left confusion (an inability to tell whether a hand, foot, or arm of the patient or examiner is on the right or left side of the body) is known as Gerstmann’s syndrome. In making this diagnosis, it is important to establish that the finger and left-right naming deficits are not part of a more generalized anomia and that the patient is not otherwise aphasic. When Gerstmann’s syndrome arises acutely and in isolation, it is commonly associated with damage to the inferior parietal lobule (especially the angular gyrus) in the left hemisphere.
Pragmatics and Prosody Pragmatics refers to aspects of language that communicate attitude, affect, and the figurative rather than literal aspects of a message (e.g., “green thumb” does not refer to the actual color of the finger). One component of pragmatics, prosody, refers to variations of melodic stress and intonation that influence attitude and the inferential aspect of verbal messages. For example, the two statements “He is clever.” and “He is clever?” contain an identical word choice and syntax but convey vastly different messages because of differences in the intonation with which the statements are uttered. Damage to right hemisphere regions corresponding to Broca’s area impairs the ability to introduce meaning-appropriate prosody into spoken language. The patient produces grammatically correct language with accurate word choice, but the statements are uttered in a monotone that interferes with the ability to convey the intended stress and affect. Patients with this type of aprosodia give the mistaken impression of being depressed or indifferent. Other aspects of pragmatics, especially the ability to infer the figurative aspect of a message, become impaired by damage to the right hemisphere or frontal lobes.
Subcortical Aphasia Damage to subcortical components of the language network (e.g., the striatum and thalamus of the left hemisphere) also can lead to aphasia. The resulting syndromes contain combinations of deficits in the various aspects of language but rarely fit the specific patterns described in Table 36-1. In a patient with a CVA, an anomic aphasia accompanied by dysarthria or a fluent aphasia with hemiparesis should raise the suspicion of a subcortical lesion site.
Progressive Aphasias Aphasias caused by major cerebrovascular accidents start suddenly and display maximal deficits at the onset. These are the “classic” aphasias described above. Aphasias caused by neurodegenerative diseases have an insidious onset and relentless progression. The neuropathology can be selective not only for gray matter but also for specific layers and cell types. The clinico-anatomic patterns are therefore different from those described in Table 36-1.
CLINICAL PRESENTATION AND DIAGNOSIS OF PRIMARY PROGRESSIVE APHASIA (PPA) Several neurodegenerative syndromes, such as typical Alzheimer-type (amnestic) and frontal-type (behavioral) dementias, can also undermine language as the disease progresses. In these cases, the aphasia is an ancillary component of the overall syndrome. When a neurodegenerative language disorder arises in relative isolation and becomes the primary concern that brings the patient to medical attention, a diagnosis of PPA is made.
LANGUAGE IN PPA The impairments of language in PPA have slightly different patterns from those seen in CVA-caused aphasias. Three major subtypes of PPA can be recognized. The agrammatic variant is characterized by consistently low fluency and impaired grammar but intact word comprehension. It most closely resembles Broca’s aphasia or anterior transcortical aphasia but usually lacks the right hemiparesis or dysarthria and has more profound impairments of grammar. Peak sites of neuronal loss (gray matter atrophy) include the left inferior frontal gyrus where Broca’s area is located. The neuropathology is usually an FTLD with tauopathy but can also be an atypical form of Alzheimer’s disease (AD) pathology. The semantic variant is characterized by preserved fluency and syntax but poor single-word comprehension and profound two-way naming impairments. This kind of aphasia is not seen with CVAs. It differs from Wernicke’s aphasia or posterior transcortical aphasia because speech is usually informative, repetition is intact, and comprehension of conversation is relatively preserved, as long as the meaning is not too dependent on words that the patient fails to understand. Peak atrophy sites are located in the left anterior temporal lobe, indicating that this part of the brain plays a critical role in the comprehension of words, especially words that denote concrete objects. The neuropathology is frequently an FTLD with abnormal precipitates of the 43-kDa transactive response DNA-binding protein TDP-43. The logopenic variant is characterized by preserved syntax and comprehension but frequent and severe word-finding pauses, anomia, circumlocutions, and simplifications during spontaneous speech. Peak atrophy sites are located in the temporoparietal junction and posterior temporal lobe, partially overlapping with traditional location of Wernicke’s area. However, the comprehension impairment of Wernicke’s aphasia is absent, perhaps because the underlying white matter, frequently damaged by cerebrovascular accidents, remains relatively intact in PPA. In contrast to Broca’s aphasia or agrammatic PPA, the interruption of fluency is variable so that speech may appear entirely normal if the patient is allowed to engage in small talk. Logopenic PPA resembles the anomic aphasia of Table 36-1 but usually has longer and more frequent word-finding pauses. Patients may also have poor phrase and word repetition, in which case the aphasia resembles the conduction aphasia in Table 36-1. Of all PPA subtypes, this is the one most commonly associated with the pathology of AD, but FTLD can also be the cause. In addition to these three major subtypes, PPA can also present in the form of pure word deafness or Gerstmann’s syndrome.
THE PARIETOFRONTAL NETWORK FOR NEGLECT AND RELATED CONDITIONS
Adaptive spatial orientation is subserved by a large-scale network containing three major cortical components. The cingulate cortex provides access to a motivational mapping of the extrapersonal space, the posterior parietal cortex to a sensorimotor representation of salient extrapersonal events, and the frontal eye fields to motor strategies for attentional behaviors (Fig. 36-2). Subcortical components of this network include the striatum and the thalamus. Damage to this network can undermine the distribution of attention within the extrapersonal space, giving rise to hemispatial neglect, simultanagnosia and object finding failures. The integration of egocentric (self-centered) with allocentric (object-centered) coordinates can also be disrupted, giving rise to impairments in route finding, the ability to avoid obstacles, and the ability to dress.
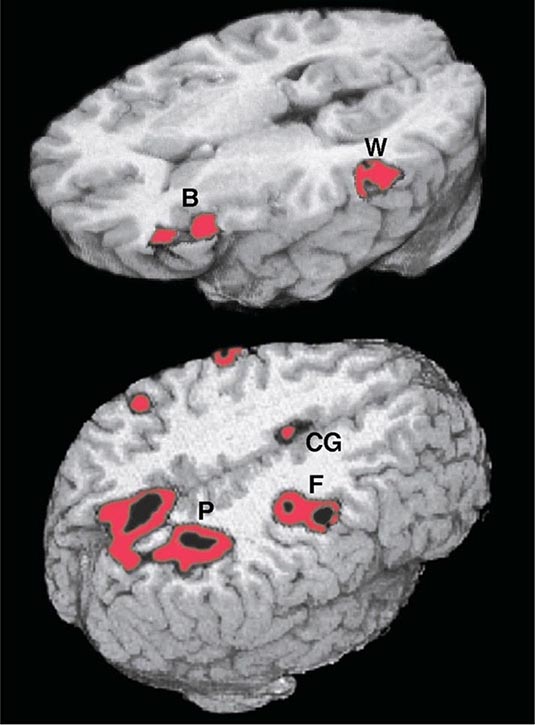
FIGURE 36-2 Functional magnetic resonance imaging of language and spatial attention in neurologically intact subjects. The red and black areas show regions of task-related significant activation. (Top) The subjects were asked to determine if two words were synonymous. This language task led to the simultaneous activation of the two epicenters of the language network, Broca’s area (B) and Wernicke’s area (W). The activations are exclusively in the left hemisphere. (Bottom) The subjects were asked to shift spatial attention to a peripheral target. This task led to the simultaneous activation of the three epicenters of the attentional network: the posterior parietal cortex (P), the frontal eye fields (F), and the cingulate gyrus (CG). The activations are predominantly in the right hemisphere. (Courtesy of Darren Gitelman, MD; with permission.)
HEMISPATIAL NEGLECT
Contralesional hemispatial neglect represents one outcome of damage to the cortical or subcortical components of this network. The traditional view that hemispatial neglect always denotes a parietal lobe lesion is inaccurate. According to one model of spatial cognition, the right hemisphere directs attention within the entire extrapersonal space, whereas the left hemisphere directs attention mostly within the contralateral right hemispace. Consequently, left hemisphere lesions do not give rise to much contralesional neglect because the global attentional mechanisms of the right hemisphere can compensate for the loss of the contralaterally directed attentional functions of the left hemisphere. Right hemisphere lesions, however, give rise to severe contralesional left hemispatial neglect because the unaffected left hemisphere does not contain ipsilateral attentional mechanisms. This model is consistent with clinical experience, which shows that contralesional neglect is more common, more severe, and longer lasting after damage to the right hemisphere than after damage to the left hemisphere. Severe neglect for the right hemispace is rare, even in left-handers with left hemisphere lesions.
Clinical Examination Patients with severe neglect may fail to dress, shave, or groom the left side of the body; fail to eat food placed on the left side of the tray; and fail to read the left half of sentences. When asked to copy a simple line drawing, the patient fails to copy detail on the left, and when the patient is asked to write, there is a tendency to leave an unusually wide margin on the left. Two bedside tests that are useful in assessing neglect are simultaneous bilateral stimulation and visual target cancellation. In the former, the examiner provides either unilateral or simultaneous bilateral stimulation in the visual, auditory, and tactile modalities. After right hemisphere injury, patients who have no difficulty detecting unilateral stimuli on either side experience the bilaterally presented stimulus as coming only from the right. This phenomenon is known as extinction and is a manifestation of the sensory-representational aspect of hemispatial neglect. In the target detection task, targets (e.g., A’s) are interspersed with foils (e.g., other letters of the alphabet) on a 21.5- to 28.0-cm (8.5 to 11 in.) sheet of paper, and the patient is asked to circle all the targets. A failure to detect targets on the left is a manifestation of the exploratory (motor) deficit in hemispatial neglect (Fig. 36-3A). Hemianopia is not by itself sufficient to cause the target detection failure because the patient is free to turn the head and eyes to the left. Target detection failures therefore reflect a distortion of spatial attention, not just of sensory input. Some patients with neglect also may deny the existence of hemiparesis and may even deny ownership of the paralyzed limb, a condition known as anosognosia.
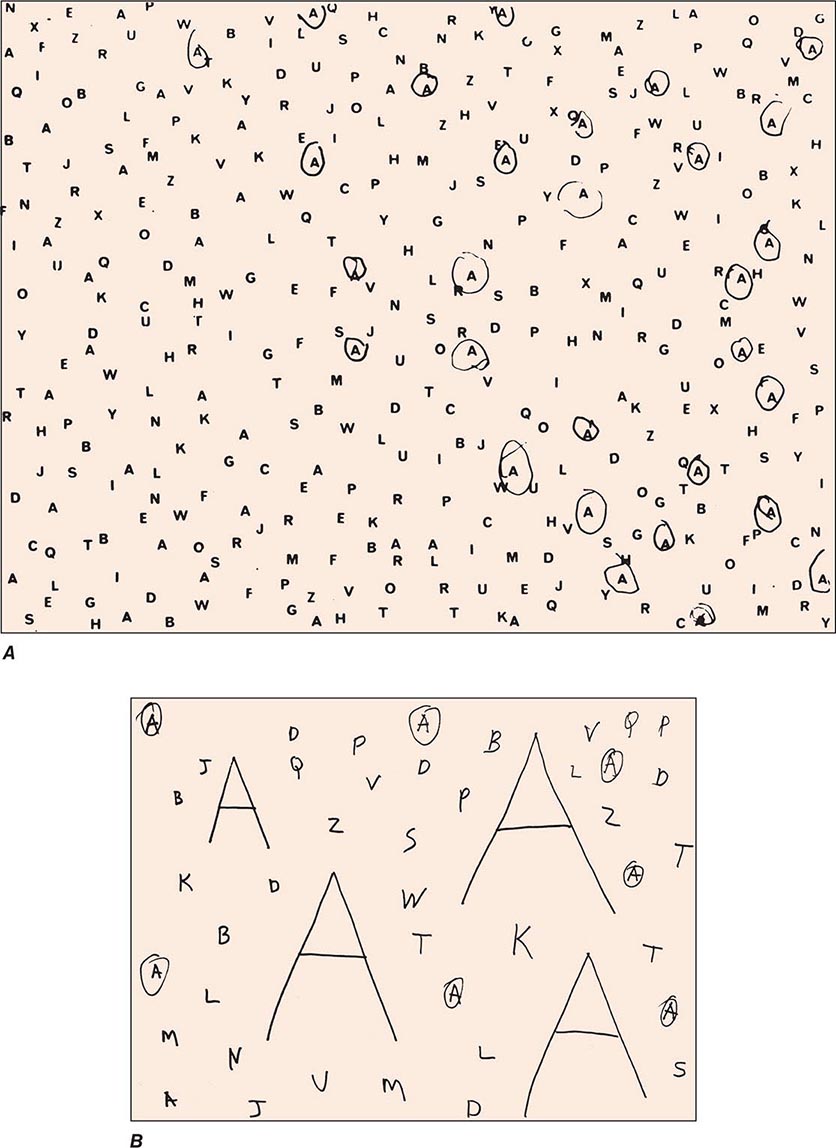
FIGURE 36-3 A. A 47-year-old man with a large frontoparietal lesion in the right hemisphere was asked to circle all the A’s. Only targets on the right are circled. This is a manifestation of left hemispatial neglect. B. A 70-year-old woman with a 2-year history of degenerative dementia was able to circle most of the small targets but ignored the larger ones. This is a manifestation of simultanagnosia.
BÁLINT’S SYNDROME, SIMULTANAGNOSIA, DRESSING APRAXIA, CONSTRUCTION APRAXIA, AND ROUTE FINDING
Bilateral involvement of the network for spatial attention, especially its parietal components, leads to a state of severe spatial disorientation known as Bálint’s syndrome. Bálint’s syndrome involves deficits in the orderly visuomotor scanning of the environment (oculomotor apraxia), accurate manual reaching toward visual targets (optic ataxia), and the ability to integrate visual information in the center of gaze with more peripheral information (simultanagnosia). A patient with simultanagnosia “misses the forest for the trees.” For example, a patient who is shown a table lamp and asked to name the object may look at its circular base and call it an ashtray. Some patients with simultanagnosia report that objects they look at may vanish suddenly, probably indicating an inability to look back at the original point of gaze after brief saccadic displacements. Movement and distracting stimuli greatly exacerbate the difficulties of visual perception. Simultanagnosia can occur without the other two components of Bálint’s syndrome.
A modification of the letter cancellation task described above can be used for the bedside diagnosis of simultanagnosia. In this modification, some of the targets (e.g., A’s) are made to be much larger than the others (7.5 to 10 cm vs 2.5 cm [3 to 4 in. vs 1 in.] in height), and all targets are embedded among foils. Patients with simultanagnosia display a counterintuitive but characteristic tendency to miss the larger targets (Fig. 36-3B). This occurs because the information needed for the identification of the larger targets cannot be confined to the immediate line of gaze and requires the integration of visual information across multiple fixation points. The greater difficulty in the detection of the larger targets also indicates that poor acuity is not responsible for the impairment of visual function and that the problem is central rather than peripheral. The test shown in Fig. 36-3B is not by itself sufficient to diagnose simultanagnosia because some patients with a frontal network syndrome may omit the large letters, perhaps because they lack the mental flexibility needed to realize that the two types of targets are symbolically identical despite being superficially different.
Bilateral parietal lesions can impair the integration of egocentric with allocentric spatial coordinates. One manifestation is dressing apraxia. A patient with this condition is unable to align the body axis with the axis of the garment and can be seen struggling as he or she holds a coat from its bottom or extends his or her arm into a fold of the garment rather than into its sleeve. Lesions that involve the posterior parietal cortex also lead to severe difficulties in copying simple line drawings. This is known as a construction apraxia and is much more severe if the lesion is in the right hemisphere. In some patients with right hemisphere lesions, the drawing difficulties are confined to the left side of the figure and represent a manifestation of hemispatial neglect; in others, there is a more universal deficit in reproducing contours and three-dimensional perspective. Impairments of route finding can be included in this group of disorders, which reflect an inability to orient the self with respect to external objects and landmarks.
Causes of Spatial Disorientation Cerebrovascular lesions and neoplasms in the right hemisphere are common causes of hemispatial neglect. Depending on the site of the lesion, a patient with neglect also may have hemiparesis, hemihypesthesia, and hemianopia on the left, but these are not invariant findings. The majority of these patients display considerable improvement of hemispatial neglect, usually within the first several weeks. Bálint’s syndrome, dressing apraxia, and route finding impairments are more likely to result from bilateral dorsal parietal lesions; common settings for acute onset include watershed infarction between the middle and posterior cerebral artery territories, hypoglycemia, and sagittal sinus thrombosis.
A progressive form of spatial disorientation, known as the posterior cortical atrophy syndrome, most commonly represents a variant of AD with unusual concentrations of neurofibrillary degeneration in the parieto-occipital cortex and the superior colliculus. The patient displays a progressive hemispatial neglect or Bálint’s syndrome, usually accompanied by dressing and construction apraxia. The corticobasal syndrome, which can be caused by AD or FTLD pathology, can also lead to a progressive left hemineglect syndrome. Both syndromes can impair route finding.
THE OCCIPITOTEMPORAL NETWORK FOR FACE AND OBJECT RECOGNITION
A patient with prosopagnosia cannot recognize familiar faces, including, sometimes, the reflection of his or her own face in the mirror. This is not a perceptual deficit because prosopagnosic patients easily can tell whether two faces are identical. Furthermore, a prosopagnosic patient who cannot recognize a familiar face by visual inspection alone can use auditory cues to reach appropriate recognition if allowed to listen to the person’s voice. The deficit in prosopagnosia is therefore modality-specific and reflects the existence of a lesion that prevents the activation of otherwise intact multimodal templates by relevant visual input. Prosopagnosic patients characteristically have no difficulty with the generic identification of a face as a face or a car as a car, but may not recognize the identity of an individual face or the make of an individual car. This reflects a visual recognition deficit for proprietary features that characterize individual members of an object class. When recognition problems become more generalized and extend to the generic identification of common objects, the condition is known as visual object agnosia. A patient with anomia cannot name the object but can describe its use. In contrast, a patient with visual agnosia is unable either to name a visually presented object or to describe its use. Face and object recognition disorders also can result from the simultanagnosia of Bálint’s syndrome, in which case they are known as apperceptive agnosias as opposed to the associative agnosias that result from inferior temporal lobe lesions.
CAUSES
The characteristic lesions in prosopagnosia and visual object agnosia of acute onset consist of bilateral infarctions in the territory of the posterior cerebral arteries. Associated deficits can include visual field defects (especially superior quadrantanopias) and a centrally based color blindness known as achromatopsia. Rarely, the responsible lesion is unilateral. In such cases, prosopagnosia is associated with lesions in the right hemisphere, and object agnosia with lesions in the left. Degenerative diseases of anterior and inferior temporal cortex can cause progressive associative prosopagnosia and object agnosia. The combination of progressive associative agnosia and a fluent aphasia is known as semantic dementia. Patients with semantic dementia fail to recognize faces and objects and cannot understand the meaning of words denoting objects. This needs to be differentiated from the semantic type of PPA where there is severe impairment in understanding words that denote objects and in naming faces and objects but a relative preservation of face and object recognition.
LIMBIC NETWORK FOR MEMORY AND AMNESIA
Limbic and paralimbic areas (such as the hippocampus, amygdala, and entorhinal cortex), the anterior and medial nuclei of the thalamus, the medial and basal parts of the striatum, and the hypothalamus collectively constitute a distributed network known as the limbic system. The behavioral affiliations of this network include the coordination of emotion, motivation, autonomic tone, and endocrine function. An additional area of specialization for the limbic network and the one that is of most relevance to clinical practice is that of declarative (explicit) memory for recent episodes and experiences. A disturbance in this function is known as an amnestic state. In the absence of deficits in motivation, attention, language, or visuospatial function, the clinical diagnosis of a persistent global amnestic state is always associated with bilateral damage to the limbic network, usually within the hippocampo-entorhinal complex or the thalamus. Damage to the limbic network does not necessarily destroy memories but interferes with their conscious recall in coherent form. The individual fragments of information remain preserved despite the limbic lesions and can sustain what is known as implicit memory. For example, patients with amnestic states can acquire new motor or perceptual skills even though they may have no conscious knowledge of the experiences that led to the acquisition of these skills.
The memory disturbance in the amnestic state is multimodal and includes retrograde and anterograde components. The retrograde amnesia involves an inability to recall experiences that occurred before the onset of the amnestic state. Relatively recent events are more vulnerable to retrograde amnesia than are more remote and more extensively consolidated events. A patient who comes to the emergency room complaining that he cannot remember his or her identity but can remember the events of the previous day almost certainly does not have a neurologic cause of memory disturbance. The second and most important component of the amnestic state is the anterograde amnesia, which indicates an inability to store, retain, and recall new knowledge. Patients with amnestic states cannot remember what they ate a few hours ago or the details of an important event they may have experienced in the recent past. In the acute stages, there also may be a tendency to fill in memory gaps with inaccurate, fabricated, and often implausible information. This is known as confabulation. Patients with the amnestic syndrome forget that they forget and tend to deny the existence of a memory problem when questioned. Confabulation is more common in cases where the underlying lesion also interferes with parts of the frontal network, as in the case of the Wernicke-Korsakoff syndrome or traumatic head injury.
CLINICAL EXAMINATION
A patient with an amnestic state is almost always disoriented, especially to time, and has little knowledge of current news. The anterograde component of an amnestic state can be tested with a list of four to five words read aloud by the examiner up to five times or until the patient can immediately repeat the entire list without an intervening delay. The next phase of the recall occurs after a period of 5 to 10 min during which the patient is engaged in other tasks. Amnestic patients fail this phase of the task and may even forget that they were given a list of words to remember. Accurate recognition of the words by multiple choice in a patient who cannot recall them indicates a less severe memory disturbance that affects mostly the retrieval stage of memory. The retrograde component of an amnesia can be assessed with questions related to autobiographical or historic events. The anterograde component of amnestic states is usually much more prominent than the retrograde component. In rare instances, occasionally associated with temporal lobe epilepsy or herpes simplex encephalitis, the retrograde component may dominate. Confusional states caused by toxic-metabolic encephalopathies and some types of frontal lobe damage lead to secondary memory impairments, especially at the stages of encoding and retrieval, even in the absence of limbic lesions. This sort of memory impairment can be differentiated from the amnestic state by the presence of additional impairments in the attention-related tasks described below in the section on the frontal lobes.
CAUSES, INCLUDING ALZHEIMER’S DISEASE
Neurologic diseases that give rise to an amnestic state include tumors (of the sphenoid wing, posterior corpus callosum, thalamus, or medial temporal lobe), infarctions (in the territories of the anterior or posterior cerebral arteries), head trauma, herpes simplex encephalitis, Wernicke-Korsakoff encephalopathy, paraneoplastic limbic encephalitis, and degenerative dementias such as AD and Pick’s disease. The one common denominator of all these diseases is the presence of bilateral lesions within one or more components in the limbic network. Occasionally, unilateral left-sided hippocampal lesions can give rise to an amnestic state, but the memory disorder tends to be transient. Depending on the nature and distribution of the underlying neurologic disease, the patient also may have visual field deficits, eye movement limitations, or cerebellar findings. AD and its prodromal state of mild cognitive impairment (MCI) are the most common causes of progressive memory impairments. The predilection of the entorhinal cortex and hippocampus for early neurofibrillary degeneration by typical AD pathology is responsible for the initially selective impairment of episodic memory. In time, additional impairments in language, attention, and visuospatial skills emerge as the neurofibrillary degeneration spreads to additional neocortical areas.
Transient global amnesia is a distinctive syndrome usually seen in late middle age. Patients become acutely disoriented and repeatedly ask who they are, where they are, and what they are doing. The spell is characterized by anterograde amnesia (inability to retain new information) and a retrograde amnesia for relatively recent events that occurred before the onset. The syndrome usually resolves within 24 to 48 h and is followed by the filling in of the period affected by the retrograde amnesia, although there is persistent loss of memory for the events that occurred during the ictus. Recurrences are noted in approximately 20% of patients. Migraine, temporal lobe seizures, and perfusion abnormalities in the posterior cerebral territory have been postulated as causes of transient global amnesia. The absence of associated neurologic findings occasionally may lead to the incorrect diagnosis of a psychiatric disorder.
THE PREFRONTAL NETWORK FOR EXECUTIVE FUNCTION AND BEHAVIOR
The frontal lobes can be subdivided into motor-premotor, dorsolateral prefrontal, medial prefrontal, and orbitofrontal components. The terms frontal lobe syndrome and prefrontal cortex refer only to the last three of these four components. These are the parts of the cerebral cortex that show the greatest phylogenetic expansion in primates, especially in humans. The dorsolateral prefrontal, medial prefrontal, and orbitofrontal areas, along with the subcortical structures with which they are interconnected (i.e., the head of the caudate and the dorsomedial nucleus of the thalamus), collectively make up a large-scale network that coordinates exceedingly complex aspects of human cognition and behavior.
The prefrontal network plays an important role in behaviors that require multitasking and the integration of thought with emotion. Cognitive operations impaired by prefrontal cortex lesions often are referred to as “executive functions.” The most common clinical manifestations of damage to the prefrontal network take the form of two relatively distinct syndromes. In the frontal abulic syndrome, the patient shows a loss of initiative, creativity, and curiosity and displays a pervasive emotional blandness, apathy, and lack of empathy. In the frontal disinhibition syndrome, the patient becomes socially disinhibited and shows severe impairments of judgment, insight, foresight, and the ability to mind rules of conduct. The dissociation between intact intellectual function and a total lack of even rudimentary common sense is striking. Despite the preservation of all essential memory functions, the patient cannot learn from experience and continues to display inappropriate behaviors without appearing to feel emotional pain, guilt, or regret when those behaviors repeatedly lead to disastrous consequences. The impairments may emerge only in real-life situations when behavior is under minimal external control and may not be apparent within the structured environment of the medical office. Testing judgment by asking patients what they would do if they detected a fire in a theater or found a stamped and addressed envelope on the road is not very informative because patients who answer these questions wisely in the office may still act very foolishly in real-life settings. The physician must therefore be prepared to make a diagnosis of frontal lobe disease based on historic information alone even when the mental state is quite intact in the office examination.
CLINICAL EXAMINATION
The emergence of developmentally primitive reflexes, also known as frontal release signs, such as grasping (elicited by stroking the palm) and sucking (elicited by stroking the lips) are seen primarily in patients with large structural lesions that extend into the premotor components of the frontal lobes or in the context of metabolic encephalopathies. The vast majority of patients with prefrontal lesions and frontal lobe behavioral syndromes do not display these reflexes. Damage to the frontal lobe disrupts a variety of attention-related functions, including working memory (the transient online holding and manipulation of information), concentration span, the scanning and retrieval of stored information, the inhibition of immediate but inappropriate responses, and mental flexibility. Digit span (which should be seven forward and five reverse) is decreased, reflecting poor working memory; the recitation of the months of the year in reverse order (which should take less than 15 s) is slowed as another indication of poor working memory; and the fluency in producing words starting with the letter a, f, or s that can be generated in 1 min (normally ≥12 per letter) is diminished even in nonaphasic patients, indicating an impairment in the ability to search and retrieve information from long-term stores. In “go–no go” tasks (where the instruction is to raise the finger upon hearing one tap but keep it still upon hearing two taps), the patient shows a characteristic inability to inhibit the response to the “no go” stimulus. Mental flexibility (tested by the ability to shift from one criterion to another in sorting or matching tasks) is impoverished; distractibility by irrelevant stimuli is increased; and there is a pronounced tendency for impersistence and perseveration. The ability for abstracting similarities and interpreting proverbs is also undermined.
The attentional deficits disrupt the orderly registration and retrieval of new information and lead to secondary memory deficits. The distinction of the underlying neural mechanisms is illustrated by the observation that severely amnestic patients who cannot remember events that occurred a few minutes ago may have intact if not superior working memory capacity as shown in tests of digit span.
CAUSES: TRAUMA, NEOPLASM, AND FRONTOTEMPORAL DEMENTIA
The abulic syndrome tends to be associated with damage in dorsolateral or dorsomedial prefrontal cortex, and the disinhibition syndrome with damage in orbitofrontal or ventromedial cortex. These syndromes tend to arise almost exclusively after bilateral lesions. Unilateral lesions confined to the prefrontal cortex may remain silent until the pathology spreads to the other side; this explains why thromboembolic CVA is an unusual cause of the frontal lobe syndrome. Common settings for frontal lobe syndromes include head trauma, ruptured aneurysms, hydrocephalus, tumors (including metastases, glioblastoma, and falx or olfactory groove meningiomas), and focal degenerative diseases. A major clinical form of FTLD known as the behavioral variant of frontotemporal dementia (bvFTD) causes a progressive frontal lobe syndrome. The behavioral changes can range from apathy to shoplifting, compulsive gambling, sexual indiscretions, remarkable lack of common sense, new ritualistic behaviors, and alterations in dietary preferences, usually leading to increased taste for sweets or rigid attachment to specific food items. In many patients with AD, neurofibrillary degeneration eventually spreads to prefrontal cortex and gives rise to components of the frontal lobe syndrome, but almost always on a background of severe memory impairment. Rarely, the bvFTD syndrome can arise in isolation in the context of an atypical form of AD pathology.
Lesions in the caudate nucleus or in the dorsomedial nucleus of the thalamus (subcortical components of the prefrontal network) also can produce a frontal lobe syndrome. This is one reason why the changes in mental state associated with degenerative basal ganglia diseases such as Parkinson’s disease and Huntington’s disease display components of the frontal lobe syndrome. Bilateral multifocal lesions of the cerebral hemispheres, none of which are individually large enough to cause specific cognitive deficits such as aphasia and neglect, can collectively interfere with the connectivity and therefore integrating (executive) function of the prefrontal cortex. A frontal lobe syndrome is therefore the single most common behavioral profile associated with a variety of bilateral multifocal brain diseases, including metabolic encephalopathy, multiple sclerosis, and vitamin B12 deficiency, among others. Many patients with the clinical diagnosis of a frontal lobe syndrome tend to have lesions that do not involve prefrontal cortex but involve either the subcortical components of the prefrontal network or its connections with other parts of the brain. To avoid making a diagnosis of “frontal lobe syndrome” in a patient with no evidence of frontal cortex disease, it is advisable to use the diagnostic term frontal network syndrome, with the understanding that the responsible lesions can lie anywhere within this distributed network. A patient with frontal lobe disease raises potential dilemmas in differential diagnosis: the abulia and blandness may be misinterpreted as depression, and the disinhibition as idiopathic mania or acting out. Appropriate intervention may be delayed while a treatable tumor keeps expanding.
CARING FOR PATIENTS WITH DEFICITS OF HIGHER CEREBRAL FUNCTION
Brain damage may cause a dissociation between feeling states and their expression so that a patient who may superficially appear jocular could still be suffering from an underlying depression that needs to be treated. If neuroleptics become absolutely necessary for the control of agitation, atypical neuroleptics are preferable because of their lower extrapyramidal side effects. Treatment with neuroleptics in elderly patients with dementia requires weighing the potential benefits against the potentially serious side effects.
Spontaneous improvement of cognitive deficits due to acute neurologic lesions is common. It is most rapid in the first few weeks but may continue for up to 2 years, especially in young individuals with single brain lesions. Some of the initial deficits appear to arise from remote dysfunction (diaschisis) in parts of the brain that are interconnected with the site of initial injury. Improvement in these patients may reflect, at least in part, a normalization of the remote dysfunction. Other mechanisms may involve functional reorganization in surviving neurons adjacent to the injury or the compensatory use of homologous structures, e.g., the right superior temporal gyrus with recovery from Wernicke’s aphasia. Cognitive rehabilitation procedures have been used in the treatment of higher cortical deficits. There are few controlled studies, but some show a benefit of rehabilitation in the recovery from hemispatial neglect and aphasia. Determining driving competence is challenging, especially in the early stages of dementing diseases. The diagnosis of a neurodegenerative disease is not by itself sufficient for asking the patient to stop driving. An on-the-road driving test and reports from family members may help time decisions related to this very important activity.
Some of the deficits described in this chapter are so complex that they may bewilder not only the patient and family but also the physician. It is imperative to carry out a systematic clinical evaluation to characterize the nature of the deficits and explain them in lay terms to the patient and family. An enlightened approach to patients with damage to the cerebral cortex requires an understanding of the principles that link neural networks to higher cerebral functions in health and disease.
37e |
Primary Progressive Aphasia, Memory Loss, and Other Focal Cerebral Disorders |
Language and memory are essential human functions. For the experienced clinician, the recognition of different types of language and memory disturbances often provides essential clues to the anatomic localization and diagnosis of neurologic disorders. This video illustrates classic disorders of language and speech (including the aphasias), memory (the amnesias), and other disorders of cognition that are commonly encountered in clinical practice.
38 |
Sleep Disorders |
Disturbed sleep is among the most frequent health complaints that physicians encounter. More than one-half of adults in the United States experience at least intermittent sleep disturbance, and only 30% of adult Americans report consistently obtaining a sufficient amount of sleep. The Institute of Medicine has estimated that 50–70 million Americans suffer from a chronic disorder of sleep and wakefulness, which can adversely affect daytime functioning as well as physical and mental health. Over the last 20 years, the field of sleep medicine has emerged as a distinct specialty in response to the impact of sleep disorders and sleep deficiency on overall health.
PHYSIOLOGY OF SLEEP AND WAKEFULNESS
Given the opportunity, most healthy young adults will sleep 7–8 h per night, although the timing, duration, and internal structure of sleep vary among individuals. In the United States, adults tend to have one consolidated sleep episode each night, although in some cultures sleep may be divided into a mid-afternoon nap and a shortened night sleep. This pattern changes considerably over the life span, as infants and young children sleep considerably more than older people.
The stages of human sleep are defined on the basis of characteristic patterns in the electroencephalogram (EEG), the electrooculogram (EOG—a measure of eye-movement activity), and the surface electromyogram (EMG) measured on the chin, neck, and legs. The continuous recording of these electrophysiologic parameters to define sleep and wakefulness is termed polysomnography.
Polysomnographic profiles define two basic states of sleep: (1) rapid eye movement (REM) sleep and (2) non–rapid eye movement (NREM) sleep. NREM sleep is further subdivided into three stages: N1, N2, and N3, characterized by increasing arousal threshold and slowing of the cortical EEG. REM sleep is characterized by a low-amplitude, mixed-frequency EEG similar to that of NREM stage N1 sleep. The EOG shows bursts of rapid eye movements similar to those seen during eyes-open wakefulness. EMG activity is absent in nearly all skeletal muscles, reflecting the brainstem-mediated muscle atonia that is characteristic of REM sleep.
ORGANIZATION OF HUMAN SLEEP
Normal nocturnal sleep in adults displays a consistent organization from night to night (Fig. 38-1). After sleep onset, sleep usually progresses through NREM stages N1–N3 sleep within 45–60 min. NREM stage N3 sleep (also known as slow-wave sleep) predominates in the first third of the night and comprises 15–25% of total nocturnal sleep time in young adults. Sleep deprivation increases the rapidity of sleep onset and both the intensity and amount of slow-wave sleep.
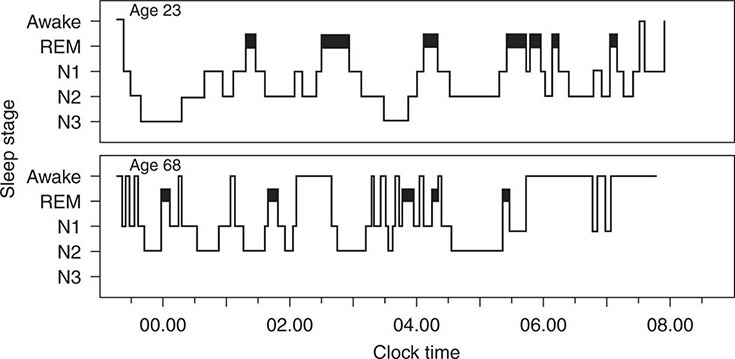
FIGURE 38-1 Wake-sleep architecture. Alternating stages of wakefulness, the three stages of NREM sleep (N1–N3), and REM sleep (solid bars) occur over the course of the night for representative young and older adult men. Characteristic features of sleep in older people include reduction of N3 slow-wave sleep, frequent spontaneous awakenings, early sleep onset, and early morning awakening. NREM, non–rapid eye movement; REM, rapid eye movement. (From the Division of Sleep and Circadian Disorders, Brigham and Women’s Hospital.)
The first REM sleep episode usually occurs in the second hour of sleep. NREM and REM sleep alternate through the night with an average period of 90–110 min (the “ultradian” sleep cycle). Overall, in a healthy young adult, REM sleep constitutes 20–25% of total sleep, and NREM stages N1 and N2 constitute 50–60%.
Age has a profound impact on sleep state organization (Fig. 38-1). N3 sleep is most intense and prominent during childhood, decreasing with puberty and across the second and third decades of life. N3 sleep declines during adulthood to the point where it may be completely absent in older adults. The remaining NREM sleep becomes more fragmented, with many more frequent awakenings from NREM sleep. It is the increased frequency of awakenings, rather than a decreased ability to fall back asleep, that accounts for the increased wakefulness during the sleep episode in older people. While REM sleep may account for 50% of total sleep time in infancy, the percentage falls off sharply over the first postnatal year as a mature REM-NREM cycle develops; thereafter, REM sleep occupies about 25% of total sleep time.
Sleep deprivation degrades cognitive performance, particularly on tests that require continual vigilance. Paradoxically, older people are less vulnerable to the neurobehavioral performance impairment induced by acute sleep deprivation than young adults, maintaining their reaction time and sustaining vigilance with fewer lapses of attention. However, it is more difficult for older adults to obtain recovery sleep after staying awake all night, as the ability to sleep during the daytime declines with age.
After sleep deprivation, NREM sleep is generally recovered first, followed by REM sleep. However, because REM sleep tends to be most prominent in the second half of the night, sleep truncation (e.g., by an alarm clock) results in selective REM sleep deprivation. This may increase REM sleep pressure to the point where the first REM sleep may occur much earlier in the nightly sleep episode. Because several disorders (see below) also cause sleep fragmentation, it is important that the patient have sufficient sleep opportunity (at least 8 h per night) for several nights prior to a diagnostic polysomnogram.
There is growing evidence that sleep deficiency in humans may cause glucose intolerance and contribute to the development of diabetes, obesity, and the metabolic syndrome, as well as impaired immune responses, accelerated atherosclerosis, and increased risk of cardiac disease and stroke. For these reasons, the Institute of Medicine declared sleep deficiency and sleep disorders “an unmet public health problem.”
WAKE AND SLEEP ARE REGULATED BY BRAIN CIRCUITS
Two principal neural systems govern the expression of the sleep and wakefulness. The ascending arousal system, illustrated in green in Fig. 38-2, consists of clusters of nerve cells extending from the upper pons to the hypothalamus and basal forebrain that activate the cerebral cortex, thalamus (which is necessary to relay sensory information to the cortex), and other forebrain regions. The ascending arousal neurons use monoamines (norepinephrine, dopamine, serotonin, and histamine), glutamate, or acetylcholine as neurotransmitters to activate their target neurons. Additional arousal-promoting neurons in the hypothalamus use the peptide neurotransmitter orexin (also known as hypocretin, shown in blue) to reinforce activity in the other arousal cell groups.
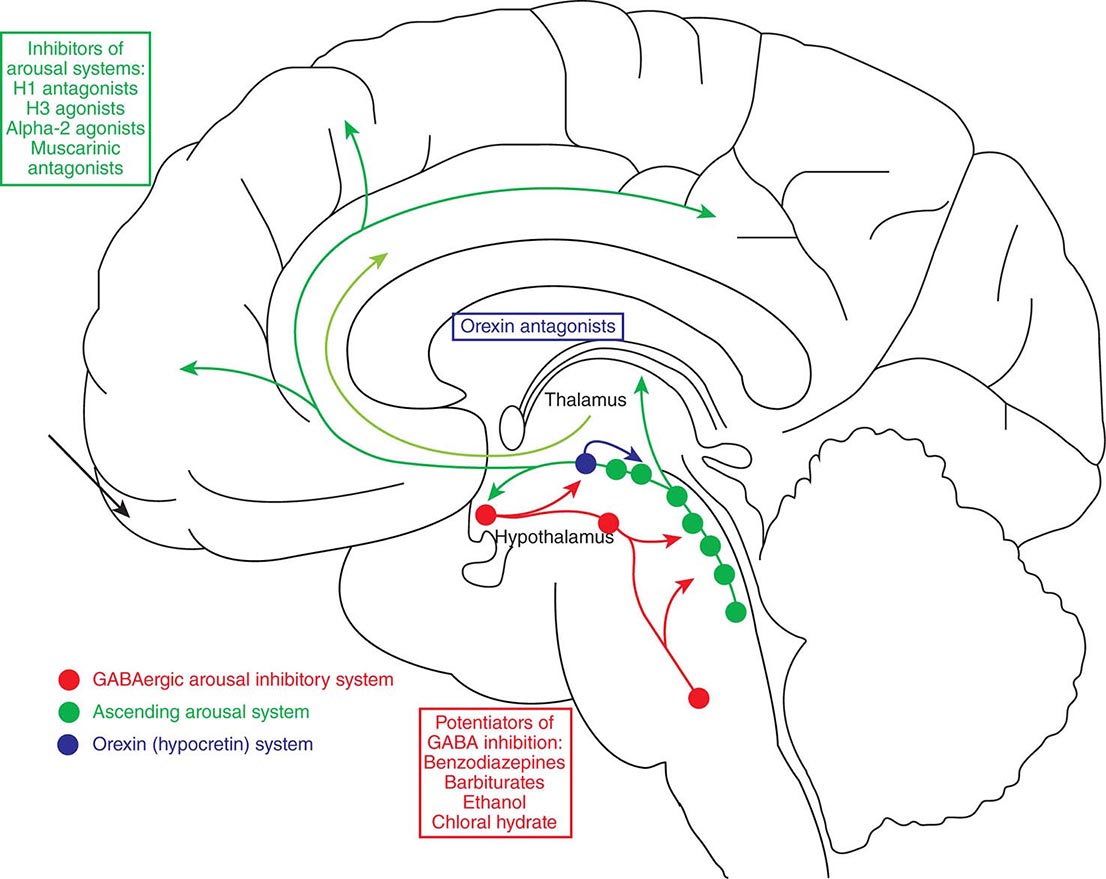
FIGURE 38-2 Relationship of drugs for insomnia with wake-sleep systems. The arousal system in the brain (green) includes monoaminergic, glutamatergic, and cholinergic neurons in the brainstem that activate neurons in the hypothalamus, thalamus, basal forebrain, and cerebral cortex. Orexin neurons (blue) in the hypothalamus, which are lost in narcolepsy, reinforce and stabilize arousal by activating other components of the arousal system. The sleep-promoting system (red) consists of GABAergic neurons in the preoptic area, lateral hypothalamus, and brainstem that inhibit the components of the arousal system, thus allowing sleep to occur. Drugs used to treat insomnia include those that block the effects of arousal system neurotransmitters (green and blue) and those that enhance the effects of γ-aminobutyric acid (GABA) produced by the sleep system (red).
Damage to the arousal system at the level of the rostral pons and lower midbrain causes coma, indicating that the ascending arousal influence from this level is critical in maintaining wakefulness. Damage to the hypothalamic branch of the arousal system causes profound sleepiness, but usually not coma. Specific loss of the orexin neurons produces the sleep disorder narcolepsy (see below).
The arousal system is turned off during sleep by inhibitory inputs from cell groups in the sleep-promoting system, shown in Fig. 38-2 in red. These neurons in the preoptic area, lateral hypothalamus, and pons use γ-aminobutyric acid (GABA) to inhibit the arousal system. Many sleep-promoting neurons are themselves inhibited by inputs from the arousal system. This mutual inhibition between the arousal- and sleep-promoting systems forms a neural circuit akin to what electrical engineers call a “flip-flop switch.” A switch of this type tends to promote rapid transitions between the on (wake) and off (sleep) states, while avoiding intermediate states. The relatively rapid transitions between waking and sleeping states, as seen in the EEG of humans and animals, is consistent with this model.
Neurons in the ventrolateral preoptic nucleus, one of the key sleep-promoting sites, are lost during normal human aging, correlating with reduced ability to maintain sleep (sleep fragmentation). The ventrolateral preoptic neurons are also injured in Alzheimer’s disease, which may in part account for the poor sleep quality in those patients.
Transitions between NREM and REM sleep appear to be governed by a similar switch in the brainstem. GABAergic REM-Off neurons have been identified in the lower midbrain that inhibit REM-On neurons in the upper pons. The REM-On group contains both GABAergic neurons that inhibit the REM-Off group (thus satisfying the conditions for a REM flip-flop switch) as well as glutamatergic neurons that project widely in the central nervous system (CNS) to cause the key phenomena associated with REM sleep. REM-On neurons that project to the medulla and spinal cord activate inhibitory (GABA and glycine-containing) interneurons, which in turn hyperpolarize the motor neurons, producing the atonia of REM sleep. REM-On neurons that project to the forebrain may be important in producing dreams.
The REM sleep switch receives cholinergic input, which favors transitions to REM sleep, and monoaminergic (norepinephrine and serotonin) input that prevents REM sleep. As a result, drugs that increase monoamine tone (e.g., serotonin or norepinephrine reuptake inhibitors) tend to reduce the amount of REM sleep. Damage to the neurons that promote REM sleep atonia can produce REM sleep behavior disorder, a condition in which patients act out their dreams (see below).
SLEEP-WAKE CYCLES ARE DRIVEN BY HOMEOSTATIC, ALLOSTATIC, AND CIRCADIAN INPUTS
The gradual increase in sleep drive with prolonged wakefulness, followed by deeper slow-wave sleep and prolonged sleep episodes, demonstrates that there is a homeostatic mechanism that regulates sleep. The neurochemistry of sleep homeostasis is only partially understood, but with prolonged wakefulness, adenosine levels rise in parts of the brain. Adenosine may act through A1 receptors to directly inhibit many arousal-promoting brain regions. In addition, adenosine promotes sleep through A2a receptors; inhibition of these receptors by caffeine is one of the chief ways in which people fight sleepiness. Other humoral factors, such as prostaglandin D2, have also been implicated in this process. Both adenosine and prostaglandin D2 activate the sleep-promoting neurons in the ventrolateral preoptic nucleus.
Allostasis is the physiologic response to a threat that cannot be managed by homeostatic mechanisms (e.g., the presence of physical danger or psychological threat). These stress responses can severely impact the need for and ability to sleep. For example, insomnia is very common in patients with anxiety and other psychiatric disorders. Stress-induced insomnia is even more common, affecting most people at some time in their lives. Positron emission tomography (PET) studies in patients with chronic insomnia show hyperactivation of the components of the ascending arousal system, as well as their targets in the limbic system in the forebrain (e.g., cingulate cortex and amygdala). The limbic areas are not only targets for the arousal system, but they also send excitatory outputs back to the arousal system, which contributes to a vicious cycle of anxiety about wakefulness that makes it more difficult to sleep. Approaches to treating insomnia rely on drugs that either inhibit the output of the ascending arousal system (green and blue in Fig. 38-2) or potentiate the output of the sleep-promoting system (red in Fig. 38-2). However, behavioral approaches (cognitive behavioral therapy and sleep hygiene) that may reduce forebrain limbic activity at bedtime are often equally or more successful.
Sleep is also regulated by a strong circadian timing signal, driven by the suprachiasmatic nuclei (SCN) of the hypothalamus, as described below. The SCN sends outputs to key sites in the hypothalamus, which impose 24-h rhythms on a wide range of behaviors and body systems, including the wake-sleep cycle.
PHYSIOLOGY OF CIRCADIAN RHYTHMICITY
The wake-sleep cycle is the most evident of many 24-h rhythms in humans. Prominent daily variations also occur in endocrine, thermoregulatory, cardiac, pulmonary, renal, immune, gastrointestinal, and neurobehavioral functions. At the molecular level, endogenous circadian rhythmicity is driven by self-sustaining transcriptional/translational feedback loops. In evaluating daily rhythms in humans, it is important to distinguish between diurnal components passively evoked by periodic environmental or behavioral changes (e.g., the increase in blood pressure and heart rate that occurs upon assumption of the upright posture) and circadian rhythms actively driven by an endogenous oscillatory process (e.g., the circadian variations in adrenal cortisol and pineal melatonin secretion that persist across a variety of environmental and behavioral conditions).
While it is now recognized that most cells in the body have circadian clocks that regulate diverse physiologic processes, most of these disparate clocks are unable to maintain the synchronization with each other that is required to produce useful 24-h rhythms aligned with the external light-dark cycle. The neurons in the SCN are interconnected with one another in such a way as to produce a near-24-h synchronous rhythm of neural activity that is then transmitted to the rest of the body. Bilateral destruction of the SCN results in a loss of most endogenous circadian rhythms including wake-sleep behavior and rhythms in endocrine and metabolic systems. The genetically determined period of this endogenous neural oscillator, which averages ~24.15 h in humans, is normally synchronized to the 24-h period of the environmental light-dark cycle through direct input from intrinsically photosensitive ganglion cells in the retina to the SCN. Humans are exquisitely sensitive to the resetting effects of light, particularly the shorter wavelengths (~460–500 nm) of the visible spectrum. Small differences in circadian period contribute to variations in diurnal preference in young adults (with the circadian period shorter in those who typically go to bed and rise earlier compared to those who typically go to bed and wake up later), whereas changes in homeostatic sleep regulation may underlie the age-related tendency toward earlier sleep-wake timing.
The timing and internal architecture of sleep are directly coupled to the output of the endogenous circadian pacemaker. Paradoxically, the endogenous circadian rhythm for wake propensity peaks just before the habitual bedtime, whereas that of sleep propensity peaks near the habitual wake time. These rhythms are thus timed to oppose the rise of sleep tendency throughout the usual waking day and the decline of sleep propensity during the habitual sleep episode, respectively. Misalignment of the endogenous circadian pacemaker with the desired wake-sleep cycle can, therefore, induce insomnia, decreased alertness, and impaired performance evident in night-shift workers and airline travelers.
BEHAVIORAL AND PHYSIOLOGIC CORRELATES OF SLEEP STATES AND STAGES
Polysomnographic staging of sleep correlates with behavioral changes during specific states and stages. During the transitional state (stage N1) between wakefulness and deeper sleep, individuals may respond to faint auditory or visual signals. Formation of short-term memories is inhibited at the onset of NREM stage N1 sleep, which may explain why individuals aroused from that transitional sleep stage frequently lack situational awareness. After sleep deprivation, such transitions may intrude upon behavioral wakefulness notwithstanding attempts to remain continuously awake (see “Shift-Work Disorder,” below).
Awakenings from REM sleep are associated with recall of vivid dream imagery over 80% of the time, especially later in the night. Imagery may also be reported after NREM sleep interruptions. Certain disorders may occur during specific sleep stages and are described below under “Parasomnias.” These include sleepwalking, night terrors, and enuresis (bed wetting), which occur most commonly in children during deep (N3) NREM sleep, and REM sleep behavior disorder, which occurs mainly among older men who fail to maintain full atonia during REM sleep, and often call out, thrash around, or even act out entire dreams.
All major physiologic systems are influenced by sleep. Blood pressure and heart rate decrease during NREM sleep, particularly during N3 sleep. During REM sleep, bursts of eye movements are associated with large variations in both blood pressure and heart rate mediated by the autonomic nervous system. Cardiac dysrhythmias may occur selectively during REM sleep. Respiratory function also changes. In comparison to relaxed wakefulness, respiratory rate becomes slower but more regular during NREM sleep (especially N3 sleep) and becomes irregular during bursts of eye movements in REM sleep. Decreases in minute ventilation during NREM sleep are out of proportion to the decrease in metabolic rate, resulting in a slightly higher PCO2.
Endocrine function also varies with sleep. N3 sleep is associated with secretion of growth hormone in men, while sleep in general is associated with augmented secretion of prolactin in both men and women. Sleep has a complex effect on the secretion of luteinizing hormone (LH): during puberty, sleep is associated with increased LH secretion, whereas sleep in the postpubertal female inhibits LH secretion in the early follicular phase of the menstrual cycle. Sleep onset (and probably N3 sleep) is associated with inhibition of thyroid-stimulating hormone and of the adrenocorticotropic hormone–cortisol axis, an effect that is superimposed on the prominent circadian rhythms in the two systems.
The pineal hormone melatonin is secreted predominantly at night in both day- and night-active species, reflecting the direct modulation of pineal activity by a circuitous neural pathway that links the SCN to the sympathetic nervous system, which innervates the pineal gland. Melatonin secretion does not require sleep, but melatonin secretion is inhibited by ambient light, an effect mediated by the neural connection from the retina to the pineal gland via the SCN. Sleep efficiency is highest when the sleep episode coincides with endogenous melatonin secretion. Administration of exogenous melatonin can hasten sleep onset and increase sleep efficiency when administered at a time when endogenous melatonin levels are low, such as in the afternoon or evening or at the desired bedtime in patients with delayed sleep-wake phase disorder, but it does not increase sleep efficiency if administered when endogenous melatonin levels are elevated. This may explain why melatonin is often ineffective in the treatment of patients with primary insomnia.
Sleep is accompanied by alterations of thermoregulatory function. NREM sleep is associated with an increase in the firing of warm-responsive neurons in the preoptic area and a fall in body temperature; conversely, skin warming without increasing core body temperature has been found to increase NREM sleep. REM sleep is associated with reduced thermoregulatory responsiveness.
DISORDERS OF SLEEP AND WAKEFULNESS
EVALUATION OF DAYTIME SLEEPINESS
Up to 25% of the adult population has persistent daytime sleepiness that impairs an individual’s ability to perform optimally in school, at work, while driving, and in other conditions that require alertness. Sleepy students often have trouble staying alert and performing well in school, and sleepy adults struggle to stay awake and focused on their work. More than half of Americans have fallen asleep while driving. An estimated 1.2 million motor vehicle crashes per year are due to drowsy drivers, causing about 20% of all serious crash injuries and deaths. One needn’t fall asleep to have an accident, as the inattention and slowed responses of drowsy drivers are a major contributor. Reaction time is equally impaired by 24 h of sleep loss as by a blood alcohol concentration of 0.10 g/dL.
Identifying and quantifying sleepiness can be challenging. First, patients may describe themselves as “sleepy,” “fatigued,” or “tired,” and the meanings of these words may differ between patients. For clinical purposes, it is best to use the term “sleepiness” to describe a propensity to fall asleep; whereas “fatigue” is best used to describe a feeling of low physical or mental energy but without a tendency to actually sleep. Sleepiness is usually most evident when the patient is sedentary, whereas fatigue may interfere with more active pursuits. Sleepiness generally occurs with disorders that reduce the quality or quantity of sleep or that interfere with the neural mechanisms of arousal, whereas fatigue is more common in inflammatory disorders such as cancer, multiple sclerosis (Chap. 458), fibromyalgia (Chap. 396), chronic fatigue syndrome (Chap. 464e), or endocrine deficiencies such as hypothyroidism (Chap. 405) or Addison’s disease (Chap. 406). Second, sleepiness can affect judgment in a manner analogous to ethanol, such that patients may have limited insight into the condition and the extent of their functional impairment. Finally, patients may be reluctant to admit that sleepiness is a problem because they may have become unfamiliar with feeling fully alert and because sleepiness is sometimes viewed pejoratively as reflecting poor motivation or bad sleep habits.
Table 38-1 outlines the diagnostic and therapeutic approach to the patient with a complaint of excessive daytime sleepiness.
|
EVALUATION OF THE PATIENT WITH EXCESSIVE DAYTIME SLEEPINESS |
To determine the extent and impact of sleepiness on daytime function, it is helpful to ask patients about the occurrence of sleep episodes during normal waking hours, both intentional and unintentional. Specific areas to be addressed include the occurrence of inadvertent sleep episodes while driving or in other safety-related settings, sleepiness while at work or school (and the relationship of sleepiness to work and school performance), and the effect of sleepiness on social and family life. Standardized questionnaires such as the Epworth Sleepiness Scale are often used clinically to measure sleepiness.
Eliciting a history of daytime sleepiness is usually adequate, but objective quantification is sometimes necessary. The MSLT measures a patient’s propensity to sleep under quiet conditions. The test is performed after an overnight polysomnogram to establish that the patient has had an adequate amount of good-quality nighttime sleep. The MSLT consists of five 20-min nap opportunities every 2 h across the day. The patient is instructed to try to fall asleep, and the major endpoints are the average latency to sleep and the occurrence of REM sleep during the naps. An average sleep latency across the naps of less than 8 min is considered objective evidence of excessive daytime sleepiness. REM sleep normally occurs only during the nighttime sleep episode, and the occurrence of REM sleep in two or more of the MSLT naps provides support for the diagnosis of narcolepsy.
For the safety of the individual and the general public, physicians have a responsibility to help manage issues around driving in patients with sleepiness. Legal reporting requirements vary from state to state, but at a minimum, physicians should inform sleepy patients about their increased risk of having an accident and advise such patients not to drive a motor vehicle until the sleepiness has been treated effectively. This discussion is especially important for professional drivers, and it should be documented in the patient’s medical record.
INSUFFICIENT SLEEP
Insufficient sleep is probably the most common cause of excessive daytime sleepiness. The average adult needs 7.5–8 h of sleep, but on weeknights, the average U.S. adult gets only 6.75 h of sleep. Only 30% of the U.S. adult population reports consistently obtaining sufficient sleep. Insufficient sleep is especially common among shift workers, individuals working multiple jobs, and people in lower socioeconomic groups. Most teenagers need ≥9 h of sleep, but many fail to get enough sleep because of circadian phase delay, or social pressures to stay up late coupled with early school start times. Late evening light exposure, television viewing, video-gaming, social media, texting, and smartphone use often delay bedtimes despite the fixed, early wake times required for work or school. As is typical with any disorder that causes sleepiness, individuals with chronically insufficient sleep may feel inattentive, irritable, unmotivated, and depressed, and have difficulty with school, work, and driving. Individuals differ in their optimal amount of sleep, and it can be helpful to ask how much sleep the patient obtains on a quiet vacation when he or she can sleep without restrictions. Some patients may think that a short amount of sleep is normal or advantageous, and they may not appreciate their biological need for more sleep, especially if coffee and other stimulants mask the sleepiness. A 2-week sleep log documenting the timing of sleep and daily level of alertness is diagnostically useful and provides helpful feedback for the patient. Extending sleep to the optimal amount on a regular basis can resolve the sleepiness and other symptoms. As with any lifestyle change, extending sleep requires commitment and adjustments, but the improvements in daytime alertness make this change worthwhile.
SLEEP APNEA SYNDROMES
Respiratory dysfunction during sleep is a common, serious cause of excessive daytime sleepiness as well as of disturbed nocturnal sleep. At least 24% of middle-aged men and 9% of middle-aged women in the United States have a reduction or cessation of breathing dozens or more times each night during sleep, with 9% of men and 4% of women doing so more than a hundred times per night. These episodes may be due to an occlusion of the airway (obstructive sleep apnea), absence of respiratory effort (central sleep apnea), or a combination of these factors (mixed sleep apnea). Failure to recognize and treat these conditions appropriately may lead to impairment of daytime alertness, increased risk of sleep-related motor vehicle crashes, depression, hypertension, myocardial infarction, diabetes, stroke, and increased mortality. Sleep apnea is particularly prevalent in overweight men and in the elderly, yet it is estimated to go undiagnosed in most affected individuals. This is unfortunate because several effective treatments are available. Readers are referred to Chap. 319 for a comprehensive review of the diagnosis and treatment of patients with sleep apnea.
NARCOLEPSY
Narcolepsy is characterized by difficulty sustaining wakefulness, poor regulation of REM sleep, and disturbed nocturnal sleep. All patients with narcolepsy have excessive daytime sleepiness. This sleepiness is often severe, but in some, it is mild. In contrast to patients with disrupted sleep (e.g., sleep apnea), people with narcolepsy usually feel well rested upon awakening and then feel tired throughout much of the day. In addition, they often experience symptoms related to an intrusion of REM sleep characteristics. REM sleep is characterized by dreaming and muscle paralysis, and people with narcolepsy can have: (1) sudden muscle weakness without a loss of consciousness, which is usually triggered by strong emotions (cataplexy; Video 38-1); (2) dream-like hallucinations at sleep onset (hypnagogic hallucinations) or upon awakening (hypnopompic hallucinations); and (3) muscle paralysis upon awakening (sleep paralysis). With severe cataplexy, an individual may be laughing at a joke and then suddenly collapse to the ground, immobile but awake for 1–2 min. With milder episodes, patients may have mild weakness of the face or neck. Narcolepsy is one of the more common causes of chronic sleepiness and affects about 1 in 2000 people in the United States. Narcolepsy typically begins between age 10 and 20; once established, the disease persists for life.
VIDEO 38-1 A typical episode of severe cataplexy. The patient is joking and then falls to the ground with an abrupt loss of muscle tone. The electromyogram recordings (four lower traces on the right) show reductions in muscle activity during the period of paralysis. The electroencephalogram (top two traces) shows wakefulness throughout the episode. (Video courtesy of Giuseppe Plazzi, University of Bologna.)
Narcolepsy is caused by loss of the hypothalamic neurons that produce the orexin neuropeptides (also known as hypocretins). Research in mice and dogs first demonstrated that a loss of orexin signaling due to null mutations of either the orexin neuropeptides or one of the orexin receptors causes sleepiness and cataplexy nearly identical to that seen in people with narcolepsy. Although genetic mutations rarely cause human narcolepsy, researchers soon discovered that patients with narcolepsy had very low or undetectable levels of orexins in their cerebrospinal fluid, and autopsy studies showed a nearly complete loss of the orexin-producing neurons in the hypothalamus. The orexins normally promote long episodes of wakefulness and suppress REM sleep, and thus, loss of orexin signaling results in frequent intrusions of sleep during the usual waking episode, with REM sleep and fragments of REM sleep at any time of day (Fig. 38-3).
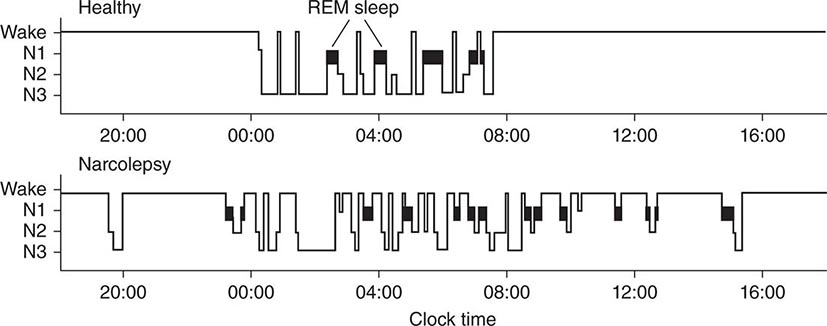
FIGURE 38-3 Polysomnographic recordings of a healthy individual and a patient with narcolepsy. The individual with narcolepsy enters rapid eye movement (REM) sleep quickly at night and has moderately fragmented sleep. During the day, the healthy subject stays awake from 8:00 AM until midnight, but the patient with narcolepsy dozes off frequently, with many daytime naps that include REM sleep.
Extensive evidence suggests that an autoimmune process likely causes this selective loss of the orexin-producing neurons. Certain human leukocyte antigens (HLAs) can increase the risk of autoimmune disorders (Chap. 373e), and narcolepsy has the strongest known HLA association. HLA DQB1*06:02 is found in about 90% of people with narcolepsy, whereas it occurs in only 12–25% of the general population. Researchers now hypothesize that in people with DQB1*06:02, an immune response against influenza, Streptococcus, or other infections may also damage the orexin-producing neurons through a process of molecular mimicry. This mechanism may account for the 8- to 12-fold increase in new cases of narcolepsy among children in Europe who received a particular brand of H1N1 influenza A vaccine (Pandemrix).
On rare occasions, narcolepsy can occur with neurologic disorders such as tumors or strokes that directly damage the orexin-producing neurons in the hypothalamus or their projections.
Diagnosis Narcolepsy is most commonly diagnosed by the history of chronic sleepiness plus cataplexy or other symptoms. Many disorders can cause feelings of weakness, but with true cataplexy, patients will describe definite functional weakness (e.g., slurred speech, dropping a cup, slumping into a chair) that has consistent emotional triggers such as heartfelt mirth when laughing at a great joke, happy surprise at unexpectedly seeing a friend, or intense anger. Cataplexy occurs in about half of all narcolepsy patients and is diagnostically very helpful because it occurs in almost no other disorder. In contrast, occasional hypnagogic hallucinations and sleep paralysis occur in about 20% of the general population, and these symptoms are not as diagnostically specific.
When narcolepsy is suspected, the diagnosis should be firmly established with a polysomnogram followed by an MSLT. The polysomnogram helps rule out other possible causes of sleepiness such as sleep apnea, and the MSLT provides essential, objective evidence of sleepiness plus REM sleep dysregulation. Across the five naps of the MSLT, most patients with narcolepsy will fall asleep in less than 8 min on average, and they will have episodes of REM sleep in at least two of the naps. Abnormal regulation of REM sleep is also manifested by the appearance of REM sleep within 15 min of sleep onset at night, which is rare in healthy individuals sleeping at their habitual bedtime. Stimulants should be stopped 1 week before the MSLT and antidepressants should be stopped 3 weeks prior, because these medications can affect the MSLT. In addition, patients should be encouraged to obtain a fully adequate amount of sleep each night for the week prior to the test to eliminate any effects of insufficient sleep.
EVALUATION OF INSOMNIA
Insomnia is the complaint of poor sleep and usually presents as difficulty initiating or maintaining sleep. People with insomnia are dissatisfied with their sleep and feel that it impairs their ability to function well in work, school, and social situations. Affected individuals often experience fatigue, decreased mood, irritability, malaise, and cognitive impairment.
Chronic insomnia, lasting more than 3 months, occurs in about 10% of adults and is more common in women, older adults, people of lower socioeconomic status, and individuals with medical, psychiatric, and substance abuse disorders. Acute or short-term insomnia affects over 30% of adults and is often precipitated by stressful life events such as a major illness or loss, change of occupation, medications, and substance abuse. If the acute insomnia triggers maladaptive behaviors such as increased nocturnal light exposure, frequently checking the clock, or attempting to sleep more by napping, it can lead to chronic insomnia.
Most insomnia begins in adulthood, but many patients may be predisposed and report easily disturbed sleep predating the insomnia, suggesting that their sleep is lighter than usual. Clinical studies and animal models indicate that insomnia is associated with activation during sleep of brain areas normally active only during wakefulness. The polysomnogram is rarely used in the evaluation of insomnia, as it typically confirms the patient’s subjective report of long latency to sleep and numerous awakenings but usually adds little new information. Many patients with insomnia have increased fast (beta) activity in the EEG during sleep; this fast activity is normally present only during wakefulness, which may explain why some patients report feeling awake for much of the night. The MSLT is rarely used in the evaluation of insomnia because, despite their feelings of low energy, most people with insomnia do not easily fall asleep during the day, and on the MSLT, their average sleep latencies are usually longer than normal.
Many factors can contribute to insomnia, and obtaining a careful history is essential so one can select therapies targeting the underlying factors. The assessment should focus on identifying predisposing, precipitating, and perpetuating factors.
Psychophysiologic Factors Many patients with insomnia have negative expectations and conditioned arousal that interfere with sleep. These individuals may worry about their insomnia during the day and have increasing anxiety as bedtime approaches if they anticipate a poor night of sleep. While attempting to sleep, they may frequently check the clock, which only heightens anxiety and frustration. They may find it easier to sleep in a new environment rather than their bedroom, as it lacks the negative associations.
Inadequate Sleep Hygiene Patients with insomnia sometimes develop counterproductive behaviors that contribute to their insomnia. These can include daytime napping that reduces sleep drive at night; an irregular sleep-wake schedule that disrupts their circadian rhythms; use of wake-promoting substances (e.g., caffeine, tobacco) too close to bedtime; engaging in alerting or stressful activities close to bedtime (e.g., arguing with a partner, work-related emailing and texting while in bed, sleeping with a smartphone or tablet at the bedside); and routinely using the bedroom for activities other than sleep or sex (e.g., TV, work), so the bedroom becomes associated with arousing or stressful feelings.
Psychiatric Conditions About 80% of patients with psychiatric disorders have sleep complaints, and about half of all chronic insomnia occurs in association with a psychiatric disorder. Depression is classically associated with early morning awakening, but it can also interfere with the onset and maintenance of sleep. Mania and hypomania can disrupt sleep and often are associated with substantial reductions in the total amount of sleep. Anxiety disorders can lead to racing thoughts and rumination that interfere with sleep and can be very problematic if the patient’s mind becomes active midway through the night. Panic attacks can occur during sleep and need to be distinguished from other parasomnias. Insomnia is common in schizophrenia and other psychoses, often resulting in fragmented sleep, less deep NREM sleep, and sometimes reversal of the day-night sleep pattern.
Medications and Drugs of Abuse A wide variety of psychoactive drugs can interfere with sleep. Caffeine, which has a half-life of 6–9 h, can disrupt sleep for up to 8–14 h, depending on the dose, variations in metabolism, and an individual’s caffeine sensitivity. Insomnia can also result from use of prescription medications too close to bedtime (e.g., theophylline, stimulants, antidepressants, glucocorticoids). Conversely, withdrawal of sedating medications such as alcohol, narcotics, or benzodiazepines can cause insomnia. Alcohol taken just before bed can shorten sleep latency, but it often produces rebound insomnia 2–3 h later as it wears off. This same problem with sleep maintenance can occur with short-acting benzodiazepines such as alprazolam.
Medical Conditions A large number of medical conditions disrupt sleep. Pain from rheumatologic disorders or a painful neuropathy commonly disrupts sleep. Some patients may sleep poorly because of respiratory conditions such as asthma, chronic obstructive pulmonary disease, cystic fibrosis, congestive heart failure, or restrictive lung disease, and some of these disorders are worse at night in bed due to circadian variations in airway resistance and postural changes that can result in paroxysmal nocturnal dyspnea. Many women experience poor sleep with the hormonal changes of menopause. Gastroesophageal reflux is also a common cause of difficulty sleeping.
Neurologic Disorders Dementia (Chap. 35) is often associated with poor sleep, probably due to a variety of factors, including napping during the day, altered circadian rhythms, and perhaps a weakened output of the brain’s sleep-promoting mechanisms. In fact, insomnia and nighttime wandering are some of the most common causes for institutionalization of patients with dementia, because they place a larger burden on caregivers. Conversely, in cognitively intact elderly men, fragmented sleep and poor sleep quality are associated with subsequent cognitive decline. Patients with Parkinson’s disease may sleep poorly due to rigidity, dementia, and other factors. Fatal familial insomnia is a very rare neurodegenerative condition caused by mutations in the prion protein gene, and although insomnia is a common early symptom, most patients present with other obvious neurologic signs such dementia, myoclonus, dysarthria, or autonomic dysfunction.
TREATMENTINSOMNIA
Treatment of insomnia improves quality of life and can promote long-term health. With improved sleep, patients often report less daytime fatigue, improved cognition, and more energy. Treating the insomnia can also improve the comorbid disease. For example, management of insomnia at the time of diagnosis of major depression often improves the response to antidepressants and reduces the risk of relapse. Sleep loss can heighten the perception of pain, so a similar approach is warranted in acute and chronic pain management.
The treatment plan should target all putative contributing factors: establish good sleep hygiene, treat medical disorders, use behavioral therapies for anxiety and negative conditioning, and use pharmacotherapy and/or psychotherapy for psychiatric disorders. Behavioral therapies should be the first-line treatment, followed by judicious use of sleep-promoting medications if needed.
TREATMENT OF MEDICAL AND PSYCHIATRIC DISEASE
If the history suggests that a medical or psychiatric disease contributes to the insomnia, then it should be addressed by, for example, treating the pain, improving breathing, and switching or adjusting the timing of medications.
IMPROVE SLEEP HYGIENE
Attention should be paid to improving sleep hygiene and avoiding counterproductive, arousing behaviors before bedtime. Patients should establish a regular bedtime and wake time, even on weekends, to help synchronize their circadian rhythms and sleep patterns. The amount of time allocated for sleep should not be more than their actual total amount of sleep. In the 30 min before bedtime, patients should establish a relaxing “wind-down” routine that can include a warm bath, listening to music, meditation, or other relaxation techniques. The bedroom should be off-limits to computers, televisions, radios, smartphones, videogames, and tablets. Once in bed, patients should try to avoid thinking about anything stressful or arousing such as problems with relationships or work. If they cannot fall asleep within 20 min, it often helps to get out of bed and read or listen to relaxing music in dim light as a form of distraction from any anxiety, but artificial light, including light from a television, cell phone, or computer, should be avoided, because light itself suppresses melatonin secretion and is arousing.
Table 38-2 outlines some of the key aspects of good sleep hygiene to improve insomnia.
|
METHODS TO IMPROVE SLEEP HYGIENE IN INSOMNIA PATIENTS |

COGNITIVE BEHAVIORAL THERAPY (CBT)
CBT uses a combination of the techniques above plus additional methods to improve insomnia. A trained therapist may use cognitive psychology techniques to reduce excessive worrying about sleep and to reframe faulty beliefs about the insomnia and its daytime consequences. The therapist may also teach the patient relaxation techniques, such as progressive muscle relaxation or meditation, to reduce autonomic arousal, intrusive thoughts, and anxiety.
MEDICATIONS FOR INSOMNIA
If insomnia persists after treatment of these contributing factors, pharmacotherapy is often used on a nightly or intermittent basis. A variety of sedatives can improve sleep.
Antihistamines, such as diphenhydramine, are the primary active ingredient in most over-the-counter sleep aids. These may be of benefit when used intermittently, but often produce rapid tolerance and can produce anticholinergic side effects such as dry mouth and constipation, which limit their use, particularly in the elderly.
Benzodiazepine receptor agonists (BzRAs) are an effective and well-tolerated class of medications for insomnia. BzRAs bind to the GABAA receptor and potentiate the postsynaptic response to GABA. GABAA receptors are found throughout the brain, and BzRAs may globally reduce neural activity and may enhance the activity of specific sleep-promoting GABAergic pathways. Classic BzRAs include lorazepam, triazolam, and clonazepam, whereas newer agents such as zolpidem and zaleplon have more selective affinity for the α1 subunit of the GABAA receptor.
Specific BzRAs are often chosen based on the desired duration of action. The most commonly prescribed agents in this family are zaleplon (5–20 mg), with a half-life of 1–2 h; zolpidem (5–10 mg) and triazolam (0.125–0.25 mg), with half-lives of 2–4 h; eszopiclone (1–3 mg), with a half-life of 5–8 h; and temazepam (15–30 mg), with a half-life of 8–20 h. Generally, side effects are minimal when the dose is kept low and the serum concentration is minimized during the waking hours (by using the shortest-acting effective agent). For chronic insomnia, intermittent use is recommended, unless the consequences of untreated insomnia outweigh concerns regarding chronic use.
The heterocyclic antidepressants (trazodone, amitriptyline,2 and doxepin) are the most commonly prescribed alternatives to BzRAs due to their lack of abuse potential and lower cost. Trazodone (25–100 mg) is used more commonly than the tricyclic antidepressants, because it has a much shorter half-life (5–9 h) and less anticholinergic activity.
Medications for insomnia are now among the most commonly prescribed medications, but they should be used cautiously. All sedatives increase the risk of injurious falls and confusion in the elderly, and therefore if needed, these medications should be used at the lowest effective dose. Morning sedation can interfere with driving and judgment, and when selecting a medication, one should consider the duration of action. Benzodiazepines carry a risk of addiction and abuse, especially in patients with a history of alcohol or sedative abuse. Like alcohol, some sleep-promoting medications can worsen sleep apnea. Sedatives can also produce complex behaviors during sleep, such as sleep walking and sleep eating, although this seems more likely at higher doses.
RESTLESS LEGS SYNDROME
Patients with restless legs syndrome (RLS) report an irresistible urge to move the legs. Many patients report a creepy-crawly or unpleasant deep ache within the thighs or calves, and those with more severe RLS may have discomfort in the arms as well. For most patients with RLS, these dysesthesias and restlessness are much worse in the evening and first half of the night. The symptoms appear with inactivity and can make sitting still in an airplane or when watching a movie a miserable experience. The sensations are temporarily relieved by movement, stretching, or massage. This nocturnal discomfort usually interferes with sleep, and patients may report daytime sleepiness as a consequence. RLS is very common, affecting 5–10% of adults and is more common in women and older adults.
A variety of factors can cause RLS. Iron deficiency is the most common treatable cause, and iron replacement should be considered if the ferritin level is less than 50 ng/mL. RLS can also occur with peripheral neuropathies and uremia and can be worsened by pregnancy, caffeine, alcohol, antidepressants, lithium, neuroleptics, and antihistamines. Genetic factors contribute to RLS, and polymorphisms in a variety of genes (BTBD9, MEIS1, MAP2K5/LBXCOR, and PTPRD) have been linked to RLS, although as yet, the mechanism through which they cause RLS remains unknown. Roughly one-third of patients (particularly those with an early age of onset) have multiple affected family members.
RLS is treated by addressing the underlying cause such as iron deficiency if present. Otherwise, treatment is symptomatic, and dopamine agonists are used most frequently. Agonists of dopamine D2/3 receptors such as pramipexole (0.25–0.5 mg q7PM) or ropinirole (0.5–4 mg q7PM) are considered first-line agents. Augmentation is a worsening of RLS such that symptoms begin earlier in the day and can spread to other body regions, and it can occur in about 25% of patients taking dopamine agonists. Other possible side effects of dopamine agonists include nausea, morning sedation, and increases in rewarding behavior such as gambling and sex. Opioids, benzodiazepines, pregabalin, and gabapentin may also be of therapeutic value. Most patients with restless legs also experience periodic limb movement disorder, although the reverse is not the case.
PERIODIC LIMB MOVEMENT DISORDER
Periodic limb movement disorder (PLMD) involves rhythmic twitches of the legs that disrupt sleep. The movements resemble a triple flexion reflex with extensions of the great toe and dorsiflexion of the foot for 0.5 to 5.0 s, which recur every 20–40 s during NREM sleep, in episodes lasting from minutes to hours. PLMD is diagnosed by a polysomnogram that includes recordings of the anterior tibialis and sometimes other muscles. The EEG shows that the movements of PLMD frequently cause brief arousals that disrupt sleep and can cause insomnia and daytime sleepiness. PLMD can be caused by the same factors that cause RLS (see above), and the frequency of leg movements improves with the same medications as used for RLS, including dopamine agonists. Recent genetic studies identified polymorphisms associated with RLS/PLMD, suggesting that they may have a common pathophysiology.
PARASOMNIAS
Parasomnias are abnormal behaviors or experiences that arise from or occur during sleep. A variety of parasomnias can occur during NREM sleep, from brief confusional arousals to sleepwalking and night terrors. The presenting complaint is usually related to the behavior itself, but the parasomnias can disturb sleep continuity or lead to mild impairments in daytime alertness. Two main parasomnias occur in REM sleep: REM sleep behavior disorder (RBD) and nightmares.
Sleepwalking (Somnambulism) Patients affected by this disorder carry out automatic motor activities that range from simple to complex. Individuals may walk, urinate inappropriately, eat, exit the house, or drive a car with minimal awareness. Full arousal may be difficult, and occasional individuals may respond to attempted awakening with agitation or violence. Sleepwalking arises from NREM stage N3 sleep, usually in the first few hours of the night, and the EEG usually shows the slow cortical activity of deep NREM sleep even when the patient is moving about. Sleepwalking is most common in children and adolescents, when these sleep stages are most robust. About 15% of children have occasional sleepwalking, and it persists in about 1% of adults. Episodes are usually isolated but may be recurrent in 1–6% of patients. The cause is unknown, although it has a familial basis in roughly one-third of cases. Sleepwalking can be worsened by insufficient sleep, which subsequently causes an increase in deep NREM sleep; alcohol; and stress. These should be addressed if present. Small studies have shown some efficacy of antidepressants and benzodiazepines; relaxation techniques and hypnosis can also be helpful. Patients and their families should improve home safety (e.g., replace glass doors, remove low tables to avoid tripping) to minimize the chance of injury if sleepwalking occurs.
Sleep Terrors This disorder occurs primarily in young children during the first few hours of sleep during NREM stage N3 sleep. The child often sits up during sleep and screams, exhibiting autonomic arousal with sweating, tachycardia, large pupils, and hyperventilation. The individual may be difficult to arouse and rarely recalls the episode on awakening in the morning. Treatment usually consists of reassuring the parents that the condition is self-limited and benign, and like sleepwalking, it may improve by avoiding insufficient sleep.
Sleep Bruxism Bruxism is an involuntary, forceful grinding of teeth during sleep that affects 10–20% of the population. The patient is usually unaware of the problem. The typical age of onset is 17–20 years, and spontaneous remission usually occurs by age 40. Sex distribution appears to be equal. In many cases, the diagnosis is made during dental examination, damage is minor, and no treatment is indicated. In more severe cases, treatment with a tooth guard is necessary to prevent tooth injury. Stress management or, in some cases, biofeedback can be useful when bruxism is a manifestation of psychological stress. There are anecdotal reports of benefit with benzodiazepines.
Sleep Enuresis Bedwetting, like sleepwalking and night terrors, is another parasomnia that occurs during sleep in the young. Before age 5 or 6 years, nocturnal enuresis should be considered a normal feature of development. The condition usually improves spontaneously by puberty, has a prevalence in late adolescence of 1–3%, and is rare in adulthood. Treatment consists of bladder training exercises and behavioral therapy. Symptomatic pharmacotherapy is usually accomplished in adults with desmopressin (0.2 mg qhs), oxybutynin chloride (5 mg qhs), or imipramine (10–25 mg qhs). Important causes of nocturnal enuresis in patients who were previously continent for 6–12 months include urinary tract infections or malformations, cauda equina lesions, emotional disturbances, epilepsy, sleep apnea, and certain medications.
REM Sleep Behavior Disorder (RBD) RBD (Video 38-2) is distinct from other parasomnias in that it occurs during REM sleep. The patient or the bed partner usually reports agitated or violent behavior during sleep, and upon awakening, the patient can often report a dream that accompanied the movements. During normal REM sleep, nearly all skeletal muscles are paralyzed, but in patients with RBD, the polysomnogram often shows limb movements during REM sleep, lasting for seconds to minutes. The movements can be dramatic, and it is not uncommon for the patient or the bed partner to be injured.
Video 38-2 Typical aggressive movements in rapid eye movement (REM) sleep behavior disorder. (Video courtesy of Dr. Carlos Schenck, University of Minnesota Medical School.)
RBD primarily afflicts older men, and most either have or will develop a neurodegenerative disorder. In longitudinal studies of RBD, half of the patients developed a synucleinopathy such as Parkinson’s disease (Chap. 449) or dementia with Lewy bodies (Chap. 448), or occasionally multiple system atrophy (Chap. 454), within 12 years, and over 80% developed a synucleinopathy by 20 years. RBD can occur in patients taking antidepressants, and in some, these medications may unmask this early indicator of neurodegeneration. Synucleinopathies probably cause neuronal loss in brainstem regions that regulate muscle atonia during REM sleep, and loss of these neurons permits movements to break through during REM sleep. RBD also occurs in about 30% of patients with narcolepsy, but the underlying cause is probably different, as they seem to be at no increased risk of a neurodegenerative disorder.
Many patients with RBD have sustained improvement with clonazepam (0.5–2.0 mg qhs).3 Melatonin at doses up to 9 mg nightly may also prevent attacks.
CIRCADIAN RHYTHM SLEEP DISORDERS
A subset of patients presenting with either insomnia or hypersomnia may have a disorder of sleep timing rather than sleep generation. Disorders of sleep timing can be either organic (i.e., due to an abnormality of circadian pacemaker[s]) or environmental/behavioral (i.e., due to a disruption of environmental synchronizers). Effective therapies aim to entrain the circadian rhythm of sleep propensity to an appropriate phase.
Delayed Sleep-Wake Phase Disorder Delayed sleep-wake phase disorder (DSWPD) is characterized by: (1) reported sleep onset and wake times intractably later than desired; (2) actual sleep times at nearly the same clock hours daily; and (3) if conducted at the habitual delayed sleep time, essentially normal sleep on polysomnography (except for delayed sleep onset). Patients with DSWPD exhibit an abnormally delayed endogenous circadian phase, which can be assessed by measuring, in a dimly lit environment, the onset of secretion of the endogenous circadian rhythm of pineal melatonin in either the blood or saliva, as light suppresses melatonin secretion. Dim-light melatonin onset (DLMO) in DSWPD patients typically occurs later in the evening than normal, which is about 8:00–9:00 PM (i.e., about 1–2 h before habitual bedtime). Patients tend to be young adults. The delayed circadian phase could be due to: (1) an abnormally long, genetically determined intrinsic period of the endogenous circadian pacemaker; (2) reduced phase-advancing capacity of the pacemaker; (3) slower rate of buildup of homeostatic sleep drive during wakefulness; or (4) an irregular prior sleep-wake schedule, characterized by frequent nights when the patient chooses to remain awake while exposed to artificial light well past midnight (for personal, social, school, or work reasons). In most cases, it is difficult to distinguish among these factors, as patients with either a behaviorally induced or biologically driven circadian phase delay may both exhibit a similar circadian phase delay in DLMO, making it difficult for both to fall asleep at the desired hour. DSWPD is a self-perpetuating condition that can persist for years and may not respond to attempts to reestablish normal bedtime hours. Treatment methods involving phototherapy with blue-enriched light during the morning hours and/or melatonin administration in the evening hours show promise in these patients, although the relapse rate is high. Patients with this circadian rhythm sleep disorder can be distinguished from those who have sleep-onset insomnia because DSWPD patients show late onset of dim-light melatonin secretion.
Advanced Sleep-Wake Phase Disorder Advanced sleep-wake phase disorder (ASWPD) is the converse of DSWPD. Most commonly, this syndrome occurs in older people, 15% of whom report that they cannot sleep past 5:00 AM, with twice that number complaining that they wake up too early at least several times per week. Patients with ASWPD are sleepy during the evening hours, even in social settings. Sleep-wake timing in ASWPD patients can interfere with a normal social life. Patients with this circadian rhythm sleep disorder can be distinguished from those who have early wakening due to insomnia because ASWPD patients show early onset of dim-light melatonin secretion.
In addition to age-related ASWPD, an early-onset familial variant of this condition has also been reported. In two families in which ASWPD was inherited in an autosomal dominant pattern, the syndrome was due to missense mutations in a circadian clock component (in the casein kinase binding domain of PER2 in one family, and in casein kinase I delta in the other) that altered the circadian period. Patients with ASWPD may benefit from bright-light and/or blue enriched phototherapy during the evening hours to reset the circadian pacemaker to a later hour.
Non-24-h Sleep-Wake Rhythm Disorder Non-24-h sleep-wake rhythm disorder (N24SWRD) can occur when the primary synchronizing input (i.e., the light-dark cycle) from the environment to the circadian pacemaker is compromised (as occurs in many blind people with no light perception) or when the maximal phase-advancing capacity of the circadian pacemaker cannot accommodate the difference between the 24-h geophysical day and the intrinsic period of the patient’s circadian pacemaker, resulting in loss of entrainment to the 24-h day. Rarely, self-selected exposure to artificial light may, in some sighted patients, inadvertently entrain the circadian pacemaker to a >24-h schedule. Affected patients with N24SWRD have difficulty maintaining a stable phase relationship between the output of the pacemaker and the 24-h day. Such patients typically present with an incremental pattern of successive delays in sleep propensity, progressing in and out of phase with local time. When the N24SWRD patient’s endogenous circadian rhythms are out of phase with the local environment, nighttime insomnia coexists with excessive daytime sleepiness. Conversely, when the endogenous circadian rhythms are in phase with the local environment, symptoms remit. The interval between symptomatic phases may last several weeks to several months in N24SWRD, depending on the period of the underlying nonentrained rhythm and the 24-h day. Nightly low-dose (0.5 mg) melatonin administration may improve sleep and, in some cases, induce synchronization of the circadian pacemaker.
Shift-Work Disorder More than 7 million workers in the United States regularly work at night, either on a permanent or rotating schedule. Many more begin the commute to work or school between 4:00 AM and 7:00 AM, requiring them to commute and then work during the time of day that they would otherwise be asleep. In addition, each week, millions of “day” workers and students elect to remain awake at night or awaken very early in the morning to work or study to meet work or school deadlines, drive long distances, compete in sporting events, or participate in recreational activities. Such schedules can result in both sleep loss and misalignment of circadian rhythms with respect to the sleep-wake cycle.
The circadian timing system usually fails to adapt successfully to the inverted schedules required by overnight work or the phase advance required by early morning (4:00 AM to 7:00 AM) start times. This leads to a misalignment between the desired work-rest schedule and the output of the pacemaker and to disturbed daytime sleep in most individuals. Excessive work hours (per day or per week), insufficient time off between consecutive days of work or school, and transmeridian travel may be contributing factors. Sleep deficiency, increased length of time awake prior to work, and misalignment of circadian phase produce decreased alertness and performance, increased reaction time, and increased risk of performance lapses, thereby resulting in greater safety hazards among night workers and other sleep-deprived individuals. Sleep disturbance nearly doubles the risk of a fatal work accident. Long-term night shift workers have higher rates of breast, colorectal, and prostate cancer and of cardiac, gastrointestinal, and reproductive disorders. The World Health Organization has added night-shift work to its list of probable carcinogens.
Sleep onset begins in local brain regions before gradually sweeping over the entire brain as sensory thresholds rise and consciousness is lost. A sleepy individual struggling to remain awake may attempt to continue performing routine and familiar motor tasks during the transition state between wakefulness and stage N1 sleep, while unable to adequately process sensory input from the environment. Motor vehicle operators who fail to heed the warning signs of sleepiness are especially vulnerable to sleep-related accidents, as sleep processes can intrude involuntarily upon the waking brain, causing catastrophic consequences. Such sleep-related attentional failures typically last only seconds but are known on occasion to persist for longer durations. There is a significant increase in the risk of sleep-related, fatal-to-the-driver highway crashes in the early morning and late afternoon hours, coincident with bimodal peaks in the daily rhythm of sleep tendency.
Resident physicians constitute another group of workers at greater risk for accidents and other adverse consequences of lack of sleep and misalignment of the circadian rhythm. Recurrent scheduling of resident physicians to work shifts of ≥24 consecutive hours impairs psychomotor performance to a degree that is comparable to alcohol intoxication, doubles the risk of attentional failures among intensive care unit resident physicians working at night, and significantly increases the risk of serious medical errors in intensive care units, including a fivefold increase in the risk of serious diagnostic mistakes. Some 20% of hospital resident physicians report making a fatigue-related mistake that injured a patient, and 5% admit making a fatigue-related mistake that resulted in the death of a patient. Moreover, working for >24 consecutive hours increases the risk of percutaneous injuries and more than doubles the risk of motor vehicle crashes on the commute home. For these reasons, in 2008, the Institute of Medicine concluded that the practice of scheduling resident physicians to work for more than 16 consecutive hours without sleep is hazardous for both resident physicians and their patients.
From 5 to 15% of individuals scheduled to work at night or in the early morning hours have much greater-than-average difficulties remaining awake during night work and sleeping during the day; these individuals are diagnosed with chronic and severe shift-work disorder (SWD). Patients with this disorder have a level of excessive sleepiness during work at night or in the early morning and insomnia during day sleep that the physician judges to be clinically significant; the condition is associated with an increased risk of sleep-related accidents and with some of the illnesses associated with night-shift work. Patients with chronic and severe SWD are profoundly sleepy at work. In fact, their sleep latencies during night work average just 2 min, comparable to mean daytime sleep latency durations of patients with narcolepsy or severe sleep apnea.
Jet Lag Disorder Each year, more than 60 million people fly from one time zone to another, often resulting in excessive daytime sleepiness, sleep-onset insomnia, and frequent arousals from sleep, particularly in the latter half of the night. The syndrome is transient, typically lasting 2–14 d depending on the number of time zones crossed, the direction of travel, and the traveler’s age and phase-shifting capacity. Travelers who spend more time outdoors at their destination reportedly adapt more quickly than those who remain in hotel rooms, presumably due to brighter (outdoor) light exposure. Avoidance of antecedent sleep loss and obtaining naps on the afternoon prior to overnight travel can reduce the difficulties associated with extended wakefulness. Laboratory studies suggest that low doses of melatonin can enhance sleep efficiency, but only if taken when endogenous melatonin concentrations are low (i.e., during the biologic daytime).
In addition to jet lag associated with travel across time zones, many patients report a behavioral pattern that has been termed social jet lag, in which bedtimes and wake times on weekends or days off occur 4–8 h later than during the week. Such recurrent displacement of the timing of the sleep-wake cycle is common in adolescents and young adults and is associated with sleep-onset insomnia, poorer academic performance, increased risk of depressive symptoms, and excessive daytime sleepiness.
MEDICAL IMPLICATIONS OF CIRCADIAN RHYTHMICITY
Prominent circadian variations have been reported in the incidence of acute myocardial infarction, sudden cardiac death, and stroke, the leading causes of death in the United States. Platelet aggregability is increased in the early morning hours, coincident with the peak incidence of these cardiovascular events. Recurrent circadian disruption combined with chronic sleep deficiency, such as occurs during night-shift work, is associated with increased plasma glucose concentrations after a meal due to inadequate pancreatic insulin secretion. Night shift workers with elevated fasting glucose have an increased risk of progressing to diabetes. Blood pressure of night workers with sleep apnea is higher than that of day workers. A better understanding of the possible role of circadian rhythmicity in the acute destabilization of a chronic condition such as atherosclerotic disease could improve the understanding of its pathophysiology.
Diagnostic and therapeutic procedures may also be affected by the time of day at which data are collected. Examples include blood pressure, body temperature, the dexamethasone suppression test, and plasma cortisol levels. The timing of chemotherapy administration has been reported to have an effect on the outcome of treatment. In addition, both the toxicity and effectiveness of drugs can vary with time of day. For example, more than a fivefold difference has been observed in mortality rates following administration of toxic agents to experimental animals at different times of day. Anesthetic agents are particularly sensitive to time-of-day effects. Finally, the physician must be aware of the public health risks associated with the ever-increasing demands made by the 24/7 schedules in our round-the-clock society.
ACKNOWLEDGMENT
John W. Winkelman, MD, PhD and Gary S. Richardson, MD contributed to this chapter in the prior edition and some material from that chapter has been retained here.
1No antidepressant has been approved by the U.S. Food and Drug Administration (FDA) for treating narcolepsy.
2Trazodone and amitriptyline have not been approved by the FDA for treating insomnia.
3No medications have been approved by the FDA for the treatment of RBD.
SECTION 4 |
DISORDERS OF EYES, EARS, NOSE, AND THROAT |
39 |
Disorders of the Eye |
THE HUMAN VISUAL SYSTEM
The visual system provides a supremely efficient means for the rapid assimilation of information from the environment to aid in the guidance of behavior. The act of seeing begins with the capture of images focused by the cornea and lens on a light-sensitive membrane in the back of the eye called the retina. The retina is actually part of the brain, banished to the periphery to serve as a transducer for the conversion of patterns of light energy into neuronal signals. Light is absorbed by pigment in two types of photoreceptors: rods and cones. In the human retina there are 100 million rods and 5 million cones. The rods operate in dim (scotopic) illumination. The cones function under daylight (photopic) conditions. The cone system is specialized for color perception and high spatial resolution. The majority of cones are within the macula, the portion of the retina that serves the central 10° of vision. In the middle of the macula a small pit termed the fovea, packed exclusively with cones, provides the best visual acuity.
Photoreceptors hyperpolarize in response to light, activating bipolar, amacrine, and horizontal cells in the inner nuclear layer. After processing of photoreceptor responses by this complex retinal circuit, the flow of sensory information ultimately converges on a final common pathway: the ganglion cells. These cells translate the visual image impinging on the retina into a continuously varying barrage of action potentials that propagates along the primary optic pathway to visual centers within the brain. There are a million ganglion cells in each retina and hence a million fibers in each optic nerve.
Ganglion cell axons sweep along the inner surface of the retina in the nerve fiber layer, exit the eye at the optic disc, and travel through the optic nerve, optic chiasm, and optic tract to reach targets in the brain. The majority of fibers synapse on cells in the lateral geniculate body, a thalamic relay station. Cells in the lateral geniculate body project in turn to the primary visual cortex. This afferent retinogeniculocortical sensory pathway provides the neural substrate for visual perception. Although the lateral geniculate body is the main target of the retina, separate classes of ganglion cells project to other subcortical visual nuclei involved in different functions. Ganglion cells that mediate pupillary constriction and circadian rhythms are light sensitive owing to a novel visual pigment, melanopsin. Pupil responses are mediated by input to the pretectal olivary nuclei in the midbrain. The pretectal nuclei send their output to the Edinger-Westphal nuclei, which in turn provide parasympathetic innervation to the iris sphincter via an interneuron in the ciliary ganglion. Circadian rhythms are timed by a retinal projection to the suprachiasmatic nucleus. Visual orientation and eye movements are served by retinal input to the superior colliculus. Gaze stabilization and optokinetic reflexes are governed by a group of small retinal targets known collectively as the brainstem accessory optic system.
The eyes must be rotated constantly within their orbits to place and maintain targets of visual interest on the fovea. This activity, called foveation, or looking, is governed by an elaborate efferent motor system. Each eye is moved by six extraocular muscles that are supplied by cranial nerves from the oculomotor (III), trochlear (IV), and abducens (VI) nuclei. Activity in these ocular motor nuclei is coordinated by pontine and midbrain mechanisms for smooth pursuit, saccades, and gaze stabilization during head and body movements. Large regions of the frontal and parietooccipital cortex control these brainstem eye movement centers by providing descending supranuclear input.
CLINICAL ASSESSMENT OF VISUAL FUNCTION
REFRACTIVE STATE
In approaching a patient with reduced vision, the first step is to decide whether refractive error is responsible. In emmetropia, parallel rays from infinity are focused perfectly on the retina. Sadly, this condition is enjoyed by only a minority of the population. In myopia, the globe is too long, and light rays come to a focal point in front of the retina. Near objects can be seen clearly, but distant objects require a diverging lens in front of the eye. In hyperopia, the globe is too short, and hence a converging lens is used to supplement the refractive power of the eye. In astigmatism, the corneal surface is not perfectly spherical, necessitating a cylindrical corrective lens. As an alternative to eyeglasses or contact lenses, refractive error can be corrected by performing laser in situ keratomileusis (LASIK) or photorefractive keratectomy (PRK) to alter the curvature of the cornea.
With the onset of middle age, presbyopia develops as the lens within the eye becomes unable to increase its refractive power to accommodate on near objects. To compensate for presbyopia an emmetropic patient must use reading glasses. A patient already wearing glasses for distance correction usually switches to bifocals. The only exception is a myopic patient, who may achieve clear vision at near simply by removing glasses containing the distance prescription.
Refractive errors usually develop slowly and remain stable after adolescence, except in unusual circumstances. For example, the acute onset of diabetes mellitus can produce sudden myopia because of lens edema induced by hyperglycemia. Testing vision through a pinhole aperture is a useful way to screen quickly for refractive error. If visual acuity is better through a pinhole than it is with the unaided eye, the patient needs refraction to obtain best corrected visual acuity.
VISUAL ACUITY
The Snellen chart is used to test acuity at a distance of 6 m (20 ft). For convenience, a scale version of the Snellen chart called the Rosenbaum card is held at 36 cm (14 in.) from the patient (Fig. 39-1). All subjects should be able to read the 6/6 m (20/20 ft) line with each eye using their refractive correction, if any. Patients who need reading glasses because of presbyopia must wear them for accurate testing with the Rosenbaum card. If 6/6 (20/20) acuity is not present in each eye, the deficiency in vision must be explained. If it is worse than 6/240 (20/800), acuity should be recorded in terms of counting fingers, hand motions, light perception, or no light perception. Legal blindness is defined by the Internal Revenue Service as a best corrected acuity of 6/60 (20/200) or less in the better eye or a binocular visual field subtending 20° or less. For driving the laws vary by state, but most states require a corrected acuity of 6/12 (20/40) in at least one eye for unrestricted privileges. Patients with a homonymous hemianopia should not drive.

FIGURE 39-1 The Rosenbaum card is a miniature, scale version of the Snellen chart for testing visual acuity at near. When the visual acuity is recorded, the Snellen distance equivalent should bear a notation indicating that vision was tested at near, not at 6 m (20 ft), or else the Jaeger number system should be used to report the acuity.
PUPILS
The pupils should be tested individually in dim light with the patient fixating on a distant target. There is no need to check the near response if the pupils respond briskly to light, because isolated loss of constriction (miosis) to accommodation does not occur. For this reason, the ubiquitous abbreviation PERRLA (pupils equal, round, and reactive to light and accommodation) implies a wasted effort with the last step. However, it is important to test the near response if the light response is poor or absent. Light-near dissociation occurs with neurosyphilis (Argyll Robertson pupil), with lesions of the dorsal midbrain (Parinaud’s syndrome), and after aberrant regeneration (oculomotor nerve palsy, Adie’s tonic pupil).
An eye with no light perception has no pupillary response to direct light stimulation. If the retina or optic nerve is only partially injured, the direct pupillary response will be weaker than the consensual pupillary response evoked by shining a light into the healthy fellow eye. A relative afferent pupillary defect (Marcus Gunn pupil) can be elicited with the swinging flashlight test (Fig. 39-2). It is an extremely useful sign in retrobulbar optic neuritis and other optic nerve diseases, in which it may be the sole objective evidence for disease. In bilateral optic neuropathy, no afferent pupil defect is present if the optic nerves are affected equally.
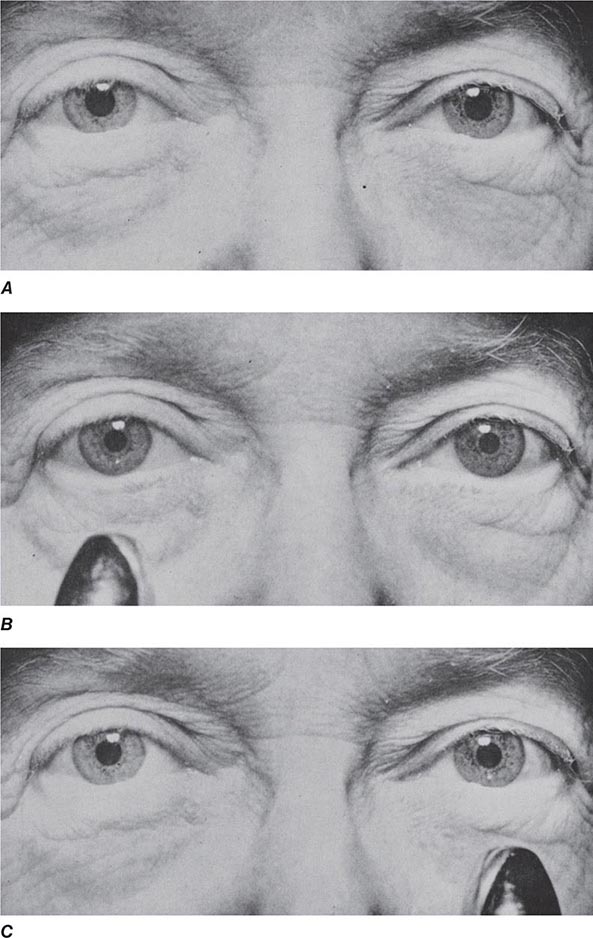
FIGURE 39-2 Demonstration of a relative afferent pupil defect (Marcus Gunn pupil) in the left eye, done with the patient fixating on a distant target. A. With dim background lighting, the pupils are equal and relatively large. B. Shining a flashlight into the right eye evokes equal, strong constriction of both pupils. C. Swinging the flashlight over to the damaged left eye causes dilation of both pupils, although they remain smaller than in A. Swinging the flashlight back over to the healthy right eye would result in symmetric constriction back to the appearance shown in B. Note that the pupils always remain equal; the damage to the left retina/optic nerve is revealed by weaker bilateral pupil constriction to a flashlight in the left eye compared with the right eye. (From P Levatin: Arch Ophthalmol 62:768, 1959. Copyright © 1959 American Medical Association. All rights reserved.)
Subtle inequality in pupil size, up to 0.5 mm, is a fairly common finding in normal persons. The diagnosis of essential or physiologic anisocoria is secure as long as the relative pupil asymmetry remains constant as ambient lighting varies. Anisocoria that increases in dim light indicates a sympathetic paresis of the iris dilator muscle. The triad of miosis with ipsilateral ptosis and anhidrosis constitutes Horner’s syndrome, although anhidrosis is an inconstant feature. Brainstem stroke, carotid dissection, and neoplasm impinging on the sympathetic chain occasionally are identified as the cause of Horner’s syndrome, but most cases are idiopathic.
Anisocoria that increases in bright light suggests a parasympathetic palsy. The first concern is an oculomotor nerve paresis. This possibility is excluded if the eye movements are full and the patient has no ptosis or diplopia. Acute pupillary dilation (mydriasis) can result from damage to the ciliary ganglion in the orbit. Common mechanisms are infection (herpes zoster, influenza), trauma (blunt, penetrating, surgical), and ischemia (diabetes, temporal arteritis). After denervation of the iris sphincter the pupil does not respond well to light, but the response to near is often relatively intact. When the near stimulus is removed, the pupil redilates very slowly compared with the normal pupil, hence the term tonic pupil. In Adie’s syndrome a tonic pupil is present, sometimes in conjunction with weak or absent tendon reflexes in the lower extremities. This benign disorder, which occurs predominantly in healthy young women, is assumed to represent a mild dysautonomia. Tonic pupils are also associated with Shy-Drager syndrome, segmental hypohidrosis, diabetes, and amyloidosis. Occasionally, a tonic pupil is discovered incidentally in an otherwise completely normal, asymptomatic individual. The diagnosis is confirmed by placing a drop of dilute (0.125%) pilocarpine into each eye. Denervation hypersensitivity produces pupillary constriction in a tonic pupil, whereas the normal pupil shows no response. Pharmacologic dilatation from accidental or deliberate instillation of anticholinergic agents (atropine, scopolamine drops) into the eye also can produce pupillary mydriasis. In this situation, normal strength (1%) pilocarpine causes no constriction.
Both pupils are affected equally by systemic medications. They are small with narcotic use (morphine, heroin) and large with anticholinergics (scopolamine). Parasympathetic agents (pilocarpine, demecarium bromide) used to treat glaucoma produce miosis. In any patient with an unexplained pupillary abnormality, a slit-lamp examination is helpful to exclude surgical trauma to the iris, an occult foreign body, perforating injury, intraocular inflammation, adhesions (synechia), angle-closure glaucoma, and iris sphincter rupture from blunt trauma.
EYE MOVEMENTS AND ALIGNMENT
Eye movements are tested by asking the patient, with both eyes open, to pursue a small target such as a penlight into the cardinal fields of gaze. Normal ocular versions are smooth, symmetric, full, and maintained in all directions without nystagmus. Saccades, or quick refixation eye movements, are assessed by having the patient look back and forth between two stationary targets. The eyes should move rapidly and accurately in a single jump to their target. Ocular alignment can be judged by holding a penlight directly in front of the patient at about 1 m. If the eyes are straight, the corneal light reflex will be centered in the middle of each pupil. To test eye alignment more precisely, the cover test is useful. The patient is instructed to look at a small fixation target in the distance. One eye is covered suddenly while the second eye is observed. If the second eye shifts to fixate on the target, it was misaligned. If it does not move, the first eye is uncovered and the test is repeated on the second eye. If neither eye moves the eyes are aligned orthotropically. If the eyes are orthotropic in primary gaze but the patient complains of diplopia, the cover test should be performed with the head tilted or turned in whatever direction elicits diplopia. With practice, the examiner can detect an ocular deviation (heterotropia) as small as 1–2° with the cover test. In a patient with vertical diplopia, a small deviation can be difficult to detect and easy to dismiss. The magnitude of the deviation can be measured by placing a prism in front of the misaligned eye to determine the power required to neutralize the fixation shift evoked by covering the other eye. Temporary press-on plastic Fresnel prisms, prism eyeglasses, or eye muscle surgery can be used to restore binocular alignment.
STEREOPSIS
Stereoacuity is determined by presenting targets with retinal disparity separately to each eye by using polarized images. The most popular office tests measure a range of thresholds from 800–40 seconds of arc. Normal stereoacuity is 40 seconds of arc. If a patient achieves this level of stereoacuity, one is assured that the eyes are aligned orthotropically and that vision is intact in each eye. Random dot stereograms have no monocular depth cues and provide an excellent screening test for strabismus and amblyopia in children.
COLOR VISION
The retina contains three classes of cones, with visual pigments of differing peak spectral sensitivity: red (560 nm), green (530 nm), and blue (430 nm). The red and green cone pigments are encoded on the × chromosome, and the blue cone pigment on chromosome 7. Mutations of the blue cone pigment are exceedingly rare. Mutations of the red and green pigments cause congenital X-linked color blindness in 8% of males. Affected individuals are not truly color blind; rather, they differ from normal subjects in the way they perceive color and how they combine primary monochromatic lights to match a particular color. Anomalous trichromats have three cone types, but a mutation in one cone pigment (usually red or green) causes a shift in peak spectral sensitivity, altering the proportion of primary colors required to achieve a color match. Dichromats have only two cone types and therefore will accept a color match based on only two primary colors. Anomalous trichromats and dichromats have 6/6 (20/20) visual acuity, but their hue discrimination is impaired. Ishihara color plates can be used to detect red-green color blindness. The test plates contain a hidden number that is visible only to subjects with color confusion from red-green color blindness. Because color blindness is almost exclusively X-linked, it is worth screening only male children.
The Ishihara plates often are used to detect acquired defects in color vision, although they are intended as a screening test for congenital color blindness. Acquired defects in color vision frequently result from disease of the macula or optic nerve. For example, patients with a history of optic neuritis often complain of color desaturation long after their visual acuity has returned to normal. Color blindness also can result from bilateral strokes involving the ventral portion of the occipital lobe (cerebral achromatopsia). Such patients can perceive only shades of gray and also may have difficulty recognizing faces (prosopagnosia). Infarcts of the dominant occipital lobe sometimes give rise to color anomia. Affected patients can discriminate colors but cannot name them.
VISUAL FIELDS
Vision can be impaired by damage to the visual system anywhere from the eyes to the occipital lobes. One can localize the site of the lesion with considerable accuracy by mapping the visual field deficit by finger confrontation and then correlating it with the topographic anatomy of the visual pathway (Fig. 39-3). Quantitative visual field mapping is performed by computer-driven perimeters that present a target of variable intensity at fixed positions in the visual field (Fig. 39-3A). By generating an automated printout of light thresholds, these static perimeters provide a sensitive means of detecting scotomas in the visual field. They are exceedingly useful for serial assessment of visual function in chronic diseases such as glaucoma and pseudotumor cerebri.
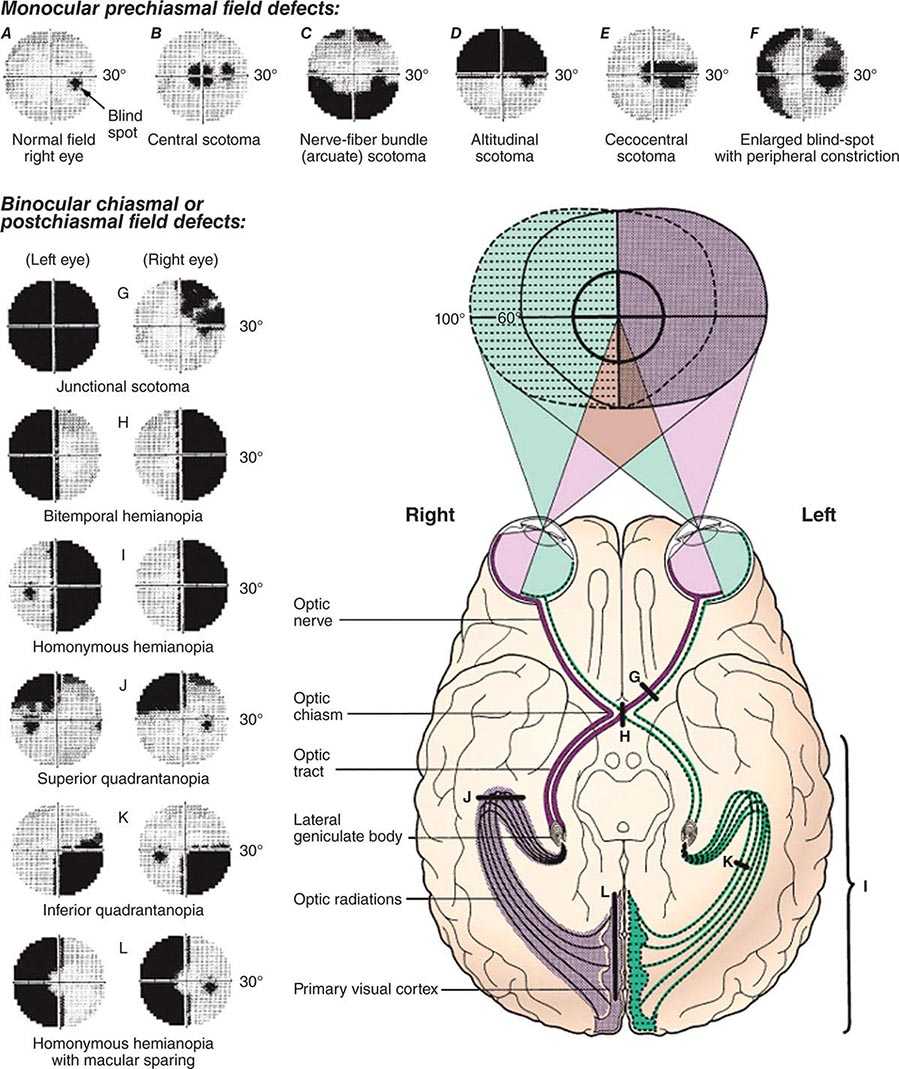
FIGURE 39-3 Ventral view of the brain, correlating patterns of visual field loss with the sites of lesions in the visual pathway. The visual fields overlap partially, creating 120° of central binocular field flanked by a 40° monocular crescent on either side. The visual field maps in this figure were done with a computer-driven perimeter (Humphrey Instruments, Carl Zeiss, Inc.). It plots the retinal sensitivity to light in the central 30° by using a gray scale format. Areas of visual field loss are shown in black. The examples of common monocular, prechiasmal field defects are all shown for the right eye. By convention, the visual fields are always recorded with the left eye’s field on the left and the right eye’s field on the right, just as the patient sees the world.
The crux of visual field analysis is to decide whether a lesion is before, at, or behind the optic chiasm. If a scotoma is confined to one eye, it must be due to a lesion anterior to the chiasm, involving either the optic nerve or the retina. Retinal lesions produce scotomas that correspond optically to their location in the fundus. For example, a superior-nasal retinal detachment results in an inferior-temporal field cut. Damage to the macula causes a central scotoma (Fig. 39-3B).
Optic nerve disease produces characteristic patterns of visual field loss. Glaucoma selectively destroys axons that enter the superotemporal or inferotemporal poles of the optic disc, resulting in arcuate scotomas shaped like a Turkish scimitar, which emanate from the blind spot and curve around fixation to end flat against the horizontal meridian (Fig. 39-3C). This type of field defect mirrors the arrangement of the nerve fiber layer in the temporal retina. Arcuate or nerve fiber layer scotomas also result from optic neuritis, ischemic optic neuropathy, optic disc drusen, and branch retinal artery or vein occlusion.
Damage to the entire upper or lower pole of the optic disc causes an altitudinal field cut that follows the horizontal meridian (Fig. 39-3D). This pattern of visual field loss is typical of ischemic optic neuropathy but also results from retinal vascular occlusion, advanced glaucoma, and optic neuritis.
About half the fibers in the optic nerve originate from ganglion cells serving the macula. Damage to papillomacular fibers causes a cecocentral scotoma that encompasses the blind spot and macula (Fig. 39-3E). If the damage is irreversible, pallor eventually appears in the temporal portion of the optic disc. Temporal pallor from a cecocentral scotoma may develop in optic neuritis, nutritional optic neuropathy, toxic optic neuropathy, Leber’s hereditary optic neuropathy, Kjer’s dominant optic atrophy, and compressive optic neuropathy. It is worth mentioning that the temporal side of the optic disc is slightly paler than the nasal side in most normal individuals. Therefore, it sometimes can be difficult to decide whether the temporal pallor visible on fundus examination represents a pathologic change. Pallor of the nasal rim of the optic disc is a less equivocal sign of optic atrophy.
At the optic chiasm, fibers from nasal ganglion cells decussate into the contralateral optic tract. Crossed fibers are damaged more by compression than are uncrossed fibers. As a result, mass lesions of the sellar region cause a temporal hemianopia in each eye. Tumors anterior to the optic chiasm, such as meningiomas of the tuberculum sella, produce a junctional scotoma characterized by an optic neuropathy in one eye and a superior-temporal field cut in the other eye (Fig. 39-3G). More symmetric compression of the optic chiasm by a pituitary adenoma (see Fig. 403-1), meningioma, craniopharyngioma, glioma, or aneurysm results in a bitemporal hemianopia (Fig. 39-3H). The insidious development of a bitemporal hemianopia often goes unnoticed by the patient and will escape detection by the physician unless each eye is tested separately.
It is difficult to localize a postchiasmal lesion accurately, because injury anywhere in the optic tract, lateral geniculate body, optic radiations, or visual cortex can produce a homonymous hemianopia (i.e., a temporal hemifield defect in the contralateral eye and a matching nasal hemifield defect in the ipsilateral eye) (Fig. 39-3I). A unilateral postchiasmal lesion leaves the visual acuity in each eye unaffected, although the patient may read the letters on only the left or right half of the eye chart. Lesions of the optic radiations tend to cause poorly matched or incongruous field defects in each eye. Damage to the optic radiations in the temporal lobe (Meyer’s loop) produces a superior quadrantic homonymous hemianopia (Fig. 39-3J), whereas injury to the optic radiations in the parietal lobe results in an inferior quadrantic homonymous hemianopia (Fig. 39-3K). Lesions of the primary visual cortex give rise to dense, congruous hemianopic field defects. Occlusion of the posterior cerebral artery supplying the occipital lobe is a common cause of total homonymous hemianopia. Some patients with hemianopia after occipital stroke have macular sparing, because the macular representation at the tip of the occipital lobe is supplied by collaterals from the middle cerebral artery (Fig. 39-3L). Destruction of both occipital lobes produces cortical blindness. This condition can be distinguished from bilateral prechiasmal visual loss by noting that the pupil responses and optic fundi remain normal.
DISORDERS
RED OR PAINFUL EYE
Corneal Abrasions Corneal abrasions are seen best by placing a drop of fluorescein in the eye and looking with the slit lamp, using a cobalt-blue light. A penlight with a blue filter will suffice if a slit lamp is not available. Damage to the corneal epithelium is revealed by yellow fluorescence of the exposed basement membrane underlying the epithelium. It is important to check for foreign bodies. To search the conjunctival fornices, the lower lid should be pulled down and the upper lid everted. A foreign body can be removed with a moistened cotton-tipped applicator after a drop of a topical anesthetic such as proparacaine has been placed in the eye. Alternatively, it may be possible to flush the foreign body from the eye by irrigating copiously with saline or artificial tears. If the corneal epithelium has been abraded, antibiotic ointment and a patch should be applied to the eye. A drop of an intermediate-acting cycloplegic such as cyclopentolate hydrochloride 1% helps reduce pain by relaxing the ciliary body. The eye should be reexamined the next day. Minor abrasions may not require patching, antibiotics, or cycloplegia.
Subconjunctival Hemorrhage This results from rupture of small vessels bridging the potential space between the episclera and the conjunctiva. Blood dissecting into this space can produce a spectacular red eye, but vision is not affected and the hemorrhage resolves without treatment. Subconjunctival hemorrhage is usually spontaneous but can result from blunt trauma, eye rubbing, or vigorous coughing. Occasionally it is a clue to an underlying bleeding disorder.
Pinguecula Pinguecula is a small, raised conjunctival nodule at the temporal or nasal limbus. In adults such lesions are extremely common and have little significance unless they become inflamed (pingueculitis). They are more apt to occur in workers with frequent outdoor exposure. A pterygium resembles a pinguecula but has crossed the limbus to encroach on the corneal surface. Removal is justified when symptoms of irritation or blurring develop, but recurrence is a common problem.
Blepharitis This refers to inflammation of the eyelids. The most common form occurs in association with acne rosacea or seborrheic dermatitis. The eyelid margins usually are colonized heavily by staphylococci. Upon close inspection, they appear greasy, ulcerated, and crusted with scaling debris that clings to the lashes. Treatment consists of strict eyelid hygiene, using warm compresses and eyelash scrubs with baby shampoo. An external hordeolum (sty) is caused by staphylococcal infection of the superficial accessory glands of Zeis or Moll located in the eyelid margins. An internal hordeolum occurs after suppurative infection of the oil-secreting meibomian glands within the tarsal plate of the eyelid. Topical antibiotics such as bacitracin/polymyxin B ophthalmic ointment can be applied. Systemic antibiotics, usually tetracyclines or azithromycin, sometimes are necessary for treatment of meibomian gland inflammation (meibomitis) or chronic, severe blepharitis. A chalazion is a painless, chronic granulomatous inflammation of a meibomian gland that produces a pealike nodule within the eyelid. It can be incised and drained or injected with glucocorticoids. Basal cell, squamous cell, or meibomian gland carcinoma should be suspected with any nonhealing ulcerative lesion of the eyelids.
Dacryocystitis An inflammation of the lacrimal drainage system, dacryocystitis can produce epiphora (tearing) and ocular injection. Gentle pressure over the lacrimal sac evokes pain and reflux of mucus or pus from the tear puncta. Dacryocystitis usually occurs after obstruction of the lacrimal system. It is treated with topical and systemic antibiotics, followed by probing, silicone stent intubation, or surgery to reestablish patency. Entropion (inversion of the eyelid) or ectropion (sagging or eversion of the eyelid) can also lead to epiphora and ocular irritation.
Conjunctivitis Conjunctivitis is the most common cause of a red, irritated eye. Pain is minimal, and visual acuity is reduced only slightly. The most common viral etiology is adenovirus infection. It causes a watery discharge, a mild foreign-body sensation, and photophobia. Bacterial infection tends to produce a more mucopurulent exudate. Mild cases of infectious conjunctivitis usually are treated empirically with broad-spectrum topical ocular antibiotics such as sulfacetamide 10%, polymyxin-bacitracin, or a trimethoprim-polymyxin combination. Smears and cultures usually are reserved for severe, resistant, or recurrent cases of conjunctivitis. To prevent contagion, patients should be admonished to wash their hands frequently, not to touch their eyes, and to avoid direct contact with others.
Allergic Conjunctivitis This condition is extremely common and often is mistaken for infectious conjunctivitis. Itching, redness, and epiphora are typical. The palpebral conjunctiva may become hypertropic with giant excrescences called cobblestone papillae. Irritation from contact lenses or any chronic foreign body also can induce formation of cobblestone papillae. Atopic conjunctivitis occurs in subjects with atopic dermatitis or asthma. Symptoms caused by allergic conjunctivitis can be alleviated with cold compresses, topical vasoconstrictors, antihistamines, and mast cell stabilizers such as cromolyn sodium. Topical glucocorticoid solutions provide dramatic relief of immune-mediated forms of conjunctivitis, but their long-term use is ill advised because of the complications of glaucoma, cataract, and secondary infection. Topical nonsteroidal anti-inflammatory drugs (NSAIDs) (e.g., ketorolac tromethamine) are better alternatives.
Keratoconjunctivitis Sicca Also known as dry eye, this produces a burning foreign-body sensation, injection, and photophobia. In mild cases the eye appears surprisingly normal, but tear production measured by wetting of a filter paper (Schirmer strip) is deficient. A variety of systemic drugs, including antihistaminic, anticholinergic, and psychotropic medications, result in dry eye by reducing lacrimal secretion. Disorders that involve the lacrimal gland directly, such as sarcoidosis and Sjögren’s syndrome, also cause dry eye. Patients may develop dry eye after radiation therapy if the treatment field includes the orbits. Problems with ocular drying are also common after lesions affecting cranial nerve V or VII. Corneal anesthesia is particularly dangerous, because the absence of a normal blink reflex exposes the cornea to injury without pain to warn the patient. Dry eye is managed by frequent and liberal application of artificial tears and ocular lubricants. In severe cases the tear puncta can be plugged or cauterized to reduce lacrimal outflow.
Keratitis Keratitis is a threat to vision because of the risk of corneal clouding, scarring, and perforation. Worldwide, the two leading causes of blindness from keratitis are trachoma from chlamydial infection and vitamin A deficiency related to malnutrition. In the United States, contact lenses play a major role in corneal infection and ulceration. They should not be worn by anyone with an active eye infection. In evaluating the cornea, it is important to differentiate between a superficial infection (keratoconjunctivitis) and a deeper, more serious ulcerative process. The latter is accompanied by greater visual loss, pain, photophobia, redness, and discharge. Slit-lamp examination shows disruption of the corneal epithelium, a cloudy infiltrate or abscess in the stroma, and an inflammatory cellular reaction in the anterior chamber. In severe cases, pus settles at the bottom of the anterior chamber, giving rise to a hypopyon. Immediate empirical antibiotic therapy should be initiated after corneal scrapings are obtained for Gram’s stain, Giemsa stain, and cultures. Fortified topical antibiotics are most effective, supplemented with subconjunctival antibiotics as required. A fungal etiology should always be considered in a patient with keratitis. Fungal infection is common in warm humid climates, especially after penetration of the cornea by plant or vegetable material.
Herpes Simplex The herpesviruses are a major cause of blindness from keratitis. Most adults in the United States have serum antibodies to herpes simplex, indicating prior viral infection (Chap. 216). Primary ocular infection generally is caused by herpes simplex type 1 rather than type 2. It manifests as a unilateral follicular blepharoconjunctivitis that is easily confused with adenoviral conjunctivitis unless telltale vesicles appear on the periocular skin or conjunctiva. A dendritic pattern of corneal epithelial ulceration revealed by fluorescein staining is pathognomonic for herpes infection but is seen in only a minority of primary infections. Recurrent ocular infection arises from reactivation of the latent herpesvirus. Viral eruption in the corneal epithelium may result in the characteristic herpes dendrite. Involvement of the corneal stroma produces edema, vascularization, and iridocyclitis. Herpes keratitis is treated with topical antiviral agents, cycloplegics, and oral acyclovir. Topical glucocorticoids are effective in mitigating corneal scarring but must be used with extreme caution because of the danger of corneal melting and perforation. Topical glucocorticoids also carry the risk of prolonging infection and inducing glaucoma.
Herpes Zoster Herpes zoster from reactivation of latent varicella (chickenpox) virus causes a dermatomal pattern of painful vesicular dermatitis. Ocular symptoms can occur after zoster eruption in any branch of the trigeminal nerve but are particularly common when vesicles form on the nose, reflecting nasociliary (V1) nerve involvement (Hutchinson’s sign). Herpes zoster ophthalmicus produces corneal dendrites, which can be difficult to distinguish from those seen in herpes simplex. Stromal keratitis, anterior uveitis, raised intraocular pressure, ocular motor nerve palsies, acute retinal necrosis, and postherpetic scarring and neuralgia are other common sequelae. Herpes zoster ophthalmicus is treated with antiviral agents and cycloplegics. In severe cases, glucocorticoids may be added to prevent permanent visual loss from corneal scarring.
Episcleritis This is an inflammation of the episclera, a thin layer of connective tissue between the conjunctiva and the sclera. Episcleritis resembles conjunctivitis, but it is a more localized process and discharge is absent. Most cases of episcleritis are idiopathic, but some occur in the setting of an autoimmune disease. Scleritis refers to a deeper, more severe inflammatory process that frequently is associated with a connective tissue disease such as rheumatoid arthritis, lupus erythematosus, polyarteritis nodosa, granulomatosis with polyangiitis (Wegener’s), or relapsing polychondritis. The inflammation and thickening of the sclera can be diffuse or nodular. In anterior forms of scleritis, the globe assumes a violet hue and the patient complains of severe ocular tenderness and pain. With posterior scleritis, the pain and redness may be less marked, but there is often proptosis, choroidal effusion, reduced motility, and visual loss. Episcleritis and scleritis should be treated with NSAIDs. If these agents fail, topical or even systemic glucocorticoid therapy may be necessary, especially if an underlying autoimmune process is active.
Uveitis Involving the anterior structures of the eye, uveitis also is called iritis or iridocyclitis. The diagnosis requires slit-lamp examination to identify inflammatory cells floating in the aqueous humor or deposited on the corneal endothelium (keratic precipitates). Anterior uveitis develops in sarcoidosis, ankylosing spondylitis, juvenile rheumatoid arthritis, inflammatory bowel disease, psoriasis, reactive arthritis, and Behçet’s disease. It also is associated with herpes infections, syphilis, Lyme disease, onchocerciasis, tuberculosis, and leprosy. Although anterior uveitis can occur in conjunction with many diseases, no cause is found to explain the majority of cases. For this reason, laboratory evaluation usually is reserved for patients with recurrent or severe anterior uveitis. Treatment is aimed at reducing inflammation and scarring by judicious use of topical glucocorticoids. Dilatation of the pupil reduces pain and prevents the formation of synechiae.
Posterior Uveitis This is diagnosed by observing inflammation of the vitreous, retina, or choroid on fundus examination. It is more likely than anterior uveitis to be associated with an identifiable systemic disease. Some patients have panuveitis, or inflammation of both the anterior and posterior segments of the eye. Posterior uveitis is a manifestation of autoimmune diseases such as sarcoidosis, Behçet’s disease, Vogt-Koyanagi-Harada syndrome, and inflammatory bowel disease. It also accompanies diseases such as toxoplasmosis, onchocerciasis, cysticercosis, coccidioidomycosis, toxocariasis, and histoplasmosis; infections caused by organisms such as Candida, Pneumocystis carinii, Cryptococcus, Aspergillus, herpes, and cytomegalovirus (see Fig. 219-1); and other diseases, such as syphilis, Lyme disease, tuberculosis, cat-scratch disease, Whipple’s disease, and brucellosis. In multiple sclerosis, chronic inflammatory changes can develop in the extreme periphery of the retina (pars planitis or intermediate uveitis).
Acute Angle-Closure Glaucoma This is an unusual but frequently misdiagnosed cause of a red, painful eye. Asian populations have a particularly high risk of angle-closure glaucoma. Susceptible eyes have a shallow anterior chamber because the eye has either a short axial length (hyperopia) or a lens enlarged by the gradual development of cataract. When the pupil becomes mid-dilated, the peripheral iris blocks aqueous outflow via the anterior chamber angle and the intraocular pressure rises abruptly, producing pain, injection, corneal edema, obscurations, and blurred vision. In some patients, ocular symptoms are overshadowed by nausea, vomiting, or headache, prompting a fruitless workup for abdominal or neurologic disease. The diagnosis is made by measuring the intraocular pressure during an acute attack or by performing gonioscopy, a procedure that allows one to observe a narrow chamber angle with a mirrored contact lens. Acute angle closure is treated with acetazolamide (PO or IV), topical beta blockers, prostaglandin analogues, α2-adrenergic agonists, and pilocarpine to induce miosis. If these measures fail, a laser can be used to create a hole in the peripheral iris to relieve pupillary block. Many physicians are reluctant to dilate patients routinely for fundus examination because they fear precipitating an angle-closure glaucoma. The risk is actually remote and more than outweighed by the potential benefit to patients of discovering a hidden fundus lesion visible only through a fully dilated pupil. Moreover, a single attack of angle closure after pharmacologic dilatation rarely causes any permanent damage to the eye and serves as an inadvertent provocative test to identify patients with narrow angles who would benefit from prophylactic laser iridectomy.
Endophthalmitis This results from bacterial, viral, fungal, or parasitic infection of the internal structures of the eye. It usually is acquired by hematogenous seeding from a remote site. Chronically ill, diabetic, or immunosuppressed patients, especially those with a history of indwelling IV catheters or positive blood cultures, are at greatest risk for endogenous endophthalmitis. Although most patients have ocular pain and injection, visual loss is sometimes the only symptom. Septic emboli from a diseased heart valve or a dental abscess that lodge in the retinal circulation can give rise to endophthalmitis. White-centered retinal hemorrhages known as Roth’s spots (Fig. 39-4) are considered pathognomonic for subacute bacterial endocarditis, but they also appear in leukemia, diabetes, and many other conditions. Endophthalmitis also occurs as a complication of ocular surgery, especially glaucoma filtering, occasionally months or even years after the operation. An occult penetrating foreign body or unrecognized trauma to the globe should be considered in any patient with unexplained intraocular infection or inflammation.
FIGURE 39-4 Roth’s spot, cotton-wool spot, and retinal hemorrhages in a 48-year-old liver transplant patient with candidemia from immunosuppression.





