175 Antidepressant Drug Overdose
Major depressive disorder (MDD) is a common and extremely important disease. The most recent national survey found a prevalence of 6.6% during the preceding 12 months and estimated that 16.2% of Americans will experience MDD during their lifetime.1 The treatment of MDD underwent a major revolution in the 1950s and 1960s with the introduction of the tricyclic antidepressants (TCAs) and monoamine oxidase inhibitors (MAOIs). Subsequently, the development of the so-called selective serotonin reuptake inhibitors (SSRIs) and serotonin and norepinephrine reuptake inhibitors (SNRIs) allowed effective treatment of depression without most of the side effects and toxicity associated with the older classes of medications. This favorable side-effect profile has led to a fundamental shift away from the use of TCAs and MAOIs and has dramatically increased the number of patients taking antidepressant medications.
Since they are used to treat MDD, it is not surprising that antidepressants have always figured prominently on the list of drugs used during intentional self-poisonings. According to data published by the American Association of Poison Control Centers, antidepressants have been the third most commonly ingested class of medications, after analgesics and sedatives/hypnotics/antipsychotics, for the past 15 years.2–4 As the use of TCAs and MAOIs has declined, so have the number of fatalities associated with these overdoses. In 1998, antidepressants were associated with almost 20% of fatal drug ingestions, but by 2008, this number had dropped to 8%.2,3 Despite this dramatic decrease, antidepressants remain the third most common cause of fatal drug ingestions.2
 Classification
Classification
As shown in Table 175-1, the most commonly used classification scheme divides antidepressant medications into tricyclic antidepressants, monoamine oxidase inhibitors, selective serotonin reuptake inhibitors, serotonin and norepinephrine reuptake inhibitors, and a miscellaneous group of drugs referred to as atypical antidepressants.5–7 This classification is suboptimal from a pharmacologic standpoint because it mixes structural (TCA) and functional (e.g., SSRI, MAOI) drug characteristics. In addition, as discussed later, functional characteristics can vary markedly among the drugs in each category, and significant overlap can occur between categories. Nevertheless, this classification scheme does provide a framework for discussing the pharmacology, clinical manifestations, and management of antidepressant overdose.
| Generic Name | Brand Name |
|---|---|
| Tricyclic Antidepressants | |
| Amitriptyline | Elavil |
| Amoxapine | Asendin |
| Clomipramine | Anafranil |
| Desipramine | Norpramin |
| Doxepin | Adapin, Sinequan |
| Imipramine | Tofranil |
| Maprotiline | Ludiomil |
| Nortriptyline | Pamelor |
| Protriptyline | Vivactil |
| Trimipramine | Surmontil |
| Monoamine Oxidase Inhibitors | |
| Isocarboxazid | Marplan |
| Phenelzine | Nardil |
| Tranylcypromine | Parnate |
| Moclobemide | Manerix |
| Selective Serotonin Reuptake Inhibitors | |
| Citalopram | Celexa |
| Escitalopram | Lexapro |
| Fluoxetine | Prozac |
| Fluvoxamine | Luvox |
| Paroxetine | Paxil |
| Sertraline | Zoloft |
| Serotonin and Norepinephrine Reuptake Inhibitors | |
| Venlafaxine | Effexor |
| Desvenlafaxine | Pristiq |
| Duloxetine | Cymbalta |
| Milnacipran | Savella |
| Atypical Antidepressants | |
| Bupropion | Wellbutrin |
| Mirtazapine | Remeron |
| Reboxetine | Edronax |
| Nefazodone | Serzone |
| Trazodone | Desyrel |
 Pharmacology
Pharmacology
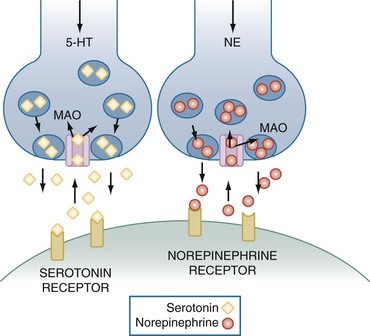
Before describing the pharmacology of the antidepressant drugs, it is important to review the release, reuptake, and metabolism of serotonin (5-hydroxytryptamine [5-HT]) and norepinephrine (NE), two monoamine neurotransmitters that are believed to play a major role in the pathogenesis of depression. As illustrated in Figure 175-1, 5-HT and NE are each synthesized by specific neurons and packaged into vesicles in the presynaptic nerve terminal. An action potential causes these vesicles to fuse with the nerve membrane, thereby releasing 5-HT or NE into the synaptic cleft. After release, these neurotransmitters bind to specific postsynaptic receptors. Seven serotonin receptor families (designated 5-HT1, 5-HT2, and so forth) have been identified, and many contain more than one receptor subtype (e.g., 5-HT1A, 5-HT1B).8 Each family and each receptor subtype appears to have specific functions and distributions throughout the body, although all are present in the central nervous system (CNS). NE binds to two major families of postsynaptic receptors termed α and β, and each has two major subtypes, referred to as α1, α2, β1, and β2. After release, the actions of 5-HT and NE are terminated primarily by active reuptake into the presynaptic neuron by amine-specific transporters. There, they are either repackaged into vesicles for future release or inactivated by the mitochondrial-bound enzyme, monoamine oxidase (MAO). MAO has the important role of inactivating a wide variety of monoamines and is found in the brain, gastrointestinal (GI) tract, and liver as well as other organs and tissues. There are two enzyme subtypes. MAO-A primarily functions to inactivate 5-HT, NE, and tyramine, whereas dopamine, phenylethylamine, tyramine, and tryptamine are the major substrates of MAO-B.8
Pharmacologic Actions
Most antidepressant medications act to increase the extraneuronal concentrations of serotonin and/or norepinephrine in the CNS. The TCAs, SSRIs, and SNRIs do this by inactivating specific transporters in the presynaptic neuron, thereby preventing the reuptake of these biogenic amines from the synaptic cleft. As shown in Table 175-2, the TCAs have a wide range of potencies and specificities for the 5-HT and NE transporters.5,9 For example, desipramine is the most potent inhibitor of NE reuptake, whereas clomipramine is the most effective serotonin reuptake blocker. The SSRIs, although much more specific, also demonstrate variable potency for transporter blockade.5,10–13 The SNRIs inhibit both 5-HT and NE reuptake, but with the exception of duloxetine have relatively low potency.5,14,15 At present, it is not clear that differences in drug selectivity translate into differences in efficacy, and differences in potency are largely eliminated through dosage adjustments.
TABLE 175-2 Potencies of Antidepressants for Blocking Neurotransmitter Reuptake
| Drug | Norepinephrine (NE) | Serotonin (5-HT) |
|---|---|---|
| Tricyclic Antidepressants | ||
| Desipramine | +++++ | ++ |
| Protriptyline | ++++ | ++ |
| Nortriptyline | +++ | + |
| Amoxapine | ++ | + |
| Doxepin | ++ | + |
| Clomipramine | + | +++++ |
| Imipramine | + | ++++ |
| Amitriptyline | + | ++ |
| Selective Serotonin Reuptake Inhibitors | ||
| Paroxetine | + | +++++ |
| Sertraline | ± | ++++ |
| Escitalopram | — | ++++ |
| Citalopram | — | +++ |
| Fluoxetine | ± | +++ |
| Fluvoxamine | — | ++ |
| Serotonin and Norepinephrine Reuptake Inhibitors | ||
| Duloxetine | +++ | +++++ |
| Venlafaxine | ± | + |
| Desvenlafaxine | + | ++ |
| Milnacipran | + | + |
Potency increases progressively from ± to +++++. —, no effect.
MAOIs prevent the breakdown of 5-HT and NE after reuptake has occurred.16 The antidepressant effect of these drugs requires the inhibition of MAO-A and is presumed to result from increased concentrations of 5-HT and NE in the brain. Most MAOIs, including isocarboxazid, phenelzine, and tranylcypromine, irreversibly inactivate both MAO-A and MAO-B. Recently, several new drugs which selectively and reversibly inactivate MAO-A have been developed. The most widely studied of these drugs, moclobemide, has been approved for use in several European countries but is not yet available in the United States.
The so-called atypical antidepressants act through a variety of different mechanisms.6,7,17 Bupropion primarily inhibits the reuptake of dopamine by blocking specific presynaptic transporters. Mirtazapine is a potent central α-adrenergic agonist that promotes the release of both serotonin and norepinephrine. It also acts as an antagonist at 5-HT2 and 5-HT3 receptors. Reboxetine is a selective inhibitor of NE reuptake. Nefazodone and trazodone act primarily by blocking 5-HT2A receptors.
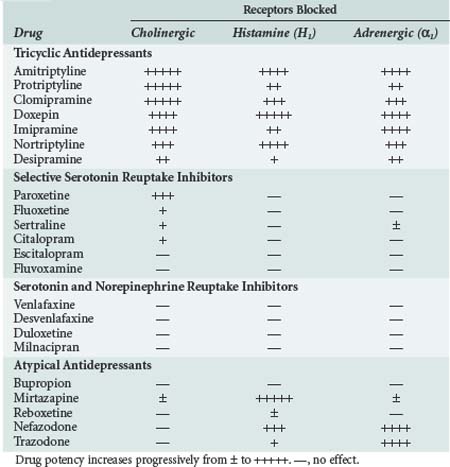
In addition to their therapeutic effects, many of the antidepressant drugs also have a variety of undesirable properties. As shown in Table 175-3, many of them block α1-adrenergic, cholinergic, and/or histamine (H1) receptors.5–15 The TCAs are the most potent antagonists of all three receptor types, although the atypical antidepressants, mirtazapine, nefazodone, and trazodone, also block H1 and/or α1-adrenergic receptors. In general, the SSRIs and SNRIs have little or no effect on these receptors. The TCAs also block fast inward sodium channels on myocardial cells, which is analogous to the effect of type I antiarrhythmic drugs.
Absorption, Distribution, Metabolism, and Excretion
In general, all antidepressants are well absorbed after oral administration, and peak plasma concentrations are usually achieved within several hours. Once absorbed, the TCAs in particular become tightly bound to plasma proteins and have a large volume of distribution. The MAOIs are metabolized primarily by hepatic acetylation, and the rate at which this process occurs varies widely among the population. Inactivation of the TCAs, SSRIs, SNRIs, and atypical antidepressants occurs largely via hepatic CYP450 enzymes, and the final byproduct is excreted in the urine. This means that coadministration of these drugs or use of another medication that inhibits CYP450 function may lead to significant drug toxicity.5,9,18
The duration of action of the antidepressants depends on the clearance rate of the parent compound as well as that of any active metabolites. Except for moclobemide, which is reversible and short acting, irreversible enzyme inactivation by the MAOIs causes their effects to last up to 2 weeks after these drugs have been ingested. In general, the other antidepressant drugs have half-lives in the range of 20 to 40 hours.5 Exceptions are fluoxetine, and its active metabolite norfluoxetine, which have half-lives of about 2 and 10 days, respectively; and venlafaxine and nefazodone, which have half-lives of approximately 5 and 3 hours, respectively.5 Because it takes approximately five half-lives for complete drug elimination to occur, most of the antidepressants can have prolonged effects after a toxic ingestion.
 Toxicology
Toxicology
The symptoms and signs that accompany an overdose, the severity and duration of toxicity, and even specific therapeutic strategies can be predicted based on a knowledge of the pharmacologic actions of each of the antidepressant drugs. Because of their potent antagonistic effects at cholinergic, adrenergic, and histaminic receptors and their ability to block sodium channels in the myocardium, TCAs are the most likely class of antidepressant drugs to cause major morbidity or death when taken in overdose.19 Not surprisingly, significant morbidity and mortality are very uncommon following ingestion of the SSRIs and SNRIs, which lack these properties.19 MAOIs and the atypical antidepressants have an intermediate toxicity profile.
Tricyclic Antidepressants
Clinical Features
The manifestations of TCA overdose are caused by the receptor and sodium channel blocking properties of these drugs.19,20 Patients typically present with symptoms and signs of an anticholinergic syndrome, or toxidrome, which may include mydriasis, dry mouth, slowed intestinal peristalsis or ileus, urinary retention, fever, flushing, sinus tachycardia, CNS depression that ranges from lethargy to coma, respiratory depression, and seizures. Blockade of α1-adrenergic receptors causes vasodilation, which decreases preload and vascular resistance and can lead to hypotension. Through their direct toxic effect on the myocardium, TCA overdose may slow depolarization and lead to prolongation of the QRS and QT intervals, heart block, and ventricular arrhythmias. Inhibition of the sodium current may also lead to decreases in myocardial contractility, stroke volume, and cardiac output. Hypotension can result from vasodilation, impaired contractility, or both. The life-threatening complications of TCA overdose, therefore, are ventricular arrhythmias, advanced heart block, shock, stupor and coma, respiratory depression, and recurrent generalized seizures.
Diagnosis and Initial Evaluation
The diagnosis of TCA overdose should be strongly suspected in any patient who presents with an anticholinergic toxidrome, especially if the electrocardiogram (ECG) demonstrates characteristic changes. Qualitative urine immunoassays for TCAs may be used to increase the level of suspicion, but they do not distinguish therapeutic from toxic ingestions, and they have relatively low specificity owing to cross-reactivity with other drugs including phenothiazines and diphenhydramine.21 Quantitative serum assays can be used to confirm a toxic ingestion, but long turnaround times often limit their clinical usefulness.
Since TCA overdose causes major morbidity and death in a relatively small proportion of patients, there has been a great deal of interest in identifying factors that can accurately predict major toxicity. Most studies have focused on ECG measurements and the serum drug concentration. Over the past 30 years, limb-lead QRS duration greater than 100 msec, a QTc greater than 430 msec, and a terminal 40-msec frontal plane QRS axis (T40) between 130 and 270 degrees all have been reported to predict future seizures, ventricular arrhythmias, and death, although the sensitivity and specificity of each has varied considerably.22–24 Similarly, an initial or maximum drug concentration greater than 1000 ng/mL has been found to have very good, fair, or poor prognostic value, depending on the study.22–26 A meta-analysis published in 2004 found that all four of these parameters have equally poor sensitivity and specificity for predicting seizures, ventricular arrhythmias, or death.27 On the other hand, QRS duration and serum concentration were found to have low negative likelihood ratios, indicating that these criteria can be used to predict the absence of future toxicity.27
Management
Prevention of Absorption
Activated charcoal is an inert, nonspecific adsorbent that irreversibly binds most drugs and toxins, including tricyclic antidepressants. Many studies in volunteers have shown that charcoal administration has a time-dependent effect on drug absorption. For example, after a single dose of nortriptyline, activated charcoal given at 30 minutes, 2 hours, or 4 hours reduced the peak serum concentration by 77%, 37%, and 19%, respectively.28 Despite its proven ability to reduce drug absorption, the efficacy of single-dose charcoal cannot be assessed because of the lack of satisfactorily designed clinical trials. The only prospective randomized placebo-controlled trial of single-dose charcoal in TCA-poisoned patients found no differences in clinical outcome or the rate of fall of drug concentrations.29 Aspiration appears to be the main complication of charcoal administration and is quite common. In their most recent position paper, the American Academy of Clinical Toxicology recommended that the administration of activated charcoal be considered only in patients who present within 1 hour of a potentially toxic ingestion.30
Although gastric lavage has been used in the initial management of most drug intoxications, there is virtually no evidence to support its use. In most poisoned patients, including those who have ingested TCAs, gastric lavage fails to significantly reduce drug absorption.31 Furthermore, several randomized trials comparing lavage plus activated charcoal with activated charcoal alone have failed to show an improvement in patient outcome.32–35 A number of serious risks of the procedure also have been well documented, including aspiration, cardiac arrhythmias, and esophageal perforation. Based on a thorough review of the literature, the American Academy of Clinical Toxicology has stated that “gastric lavage should not be employed routinely, if ever, in the management of poisoned patients.”36
Enhancement of Drug Elimination
Repeated doses of activated charcoal can increase drug clearance by interrupting enterohepatic circulation and by reducing the concentration of free drug in the intestinal lumen, thereby creating a diffusion gradient from the blood (a process referred to as gastrointestinal dialysis). Although multiple doses of activated charcoal increase the clearance of several drugs including carbamazepine, phenobarbital, and theophylline, studies examining TCA clearance in volunteer subjects have yielded inconclusive and often conflicting results, and no studies have examined this therapy in poisoned patients.37 For this reason, and because multiple-dose charcoal has been reported to cause intestinal obstruction, this therapy is not recommended for patients with TCA intoxication.37
Hemodialysis and charcoal hemoperfusion would be expected to be ineffective in removing TCAs and their active metabolites, because avid tissue and plasma protein binding leaves only a small fraction of free drug available for diffusion or adsorption. Although beneficial effects have been reported,38,39 based on these pharmacokinetic considerations, extracorporeal therapy is not recommended for patients with TCA poisoning.20
Sodium Bicarbonate
Several controlled trials in animals and case reports and case series in humans have demonstrated that administration of sodium bicarbonate is often effective in shortening the QRS interval, terminating ventricular arrhythmias, and increasing blood pressure after TCA overdose.40 Three potential mechanisms for these beneficial effects have been proposed.40 First, alkalinization of the serum increases protein binding, thereby reducing the concentration of free drug. Second, by causing drug ionization, alkalinization may reduce the affinity of TCAs for the myocardial sodium channel receptor. Third, an increase in the serum sodium concentration may overcome sodium channel blockade. This final mechanism may explain why hypertonic saline has been reported to reverse cardiac toxicity in some animal studies and in case reports in humans.41 It may also explain the observation that hyperventilation appears to be less effective than sodium bicarbonate administration.40 Based on this information, it is currently recommended that patients with evidence of cardiac toxicity (i.e., QRS or QT prolongation, ventricular arrhythmias, heart block, hypotension) receive sodium bicarbonate with the goal of achieving and maintaining an arterial pH of 7.50 to 7.55.42,43
Treatment of Specific Complications
Arrhythmias
Ventricular tachycardia and fibrillation accompanying TCA overdose are often refractory to drug therapy, and treatment should focus on the administration of sodium bicarbonate and the correction of acidemia, hypoxemia, and electrolyte abnormalities.43 Antiarrhythmic drugs categorized as class Ia (e.g., procainamide), Ic (e.g., flecainide, propafenone), and III (e.g., amiodarone, sotalol) are not only ineffective but also should be avoided because they, like the TCAs, can prolong depolarization. Case series have described the successful use of lidocaine,44 phenytoin,45,46 and magnesium sulfate47 in patients with refractory ventricular arrhythmias.
Hypotension
Because TCA-induced hypotension may result from vasodilation, impaired cardiac contractility, or both, right heart catheterization is often useful in determining the predominant cause and the most appropriate therapy. Arterial and venous dilation resulting from α1-adrenergic blockade cause a drop in systemic vascular resistance (SVR) and ventricular preload which are most effectively treated with volume resuscitation, followed if necessary by the use of one or more vasopressors. Norepinephrine may be more effective than dopamine in this setting,48 and high-dose glucagon has also been reported to be beneficial.49 On the other hand, impaired myocardial contractility leads to a fall in cardiac output and a compensatory rise in SVR and responds best to dobutamine and afterload reduction. Sodium bicarbonate administration may be effective in improving hypotension, regardless of the underlying cause.
Clinical Course and Monitoring
Patients with TCA overdose can become critically ill very rapidly, even when initial symptoms or signs are minimal.50 However, patients who develop major signs of toxicity (coma, seizures, respiratory depression, hypotension, ventricular arrhythmias) almost invariably do so within 6 hours of presentation, and almost all deaths occur within the first 16 hours.50 The maximum QRS duration also typically occurs within the first 6 hours22 and usually returns to normal within 12 to 18 hours.51 Patients rarely develop seizures or ventricular dysrhythmias after the QRS interval has decreased to less than 0.10 second.22,51 Based on this information, patients should be admitted to an intensive care unit (ICU) if they have signs of toxicity or QRS prolongation, or if they have been monitored for less than 6 hours in the emergency department. Patients should be transferred from the ICU only after their QRS interval has returned to normal.
Reuptake Inhibitors
Clinical Features
The SSRIs and SNRIs have a much more favorable side-effect profile than the TCAs, and overdoses are usually associated with little significant toxicity.19,52,53 The most common manifestations are lethargy, diaphoresis, nausea and vomiting, sinus tachycardia, and tremor.19 Seizures, cardiac conduction disturbances (including QRS and QTc prolongation), and atrial and ventricular arrhythmias are very uncommon but are most likely to occur after venlafaxine or citalopram ingestion.7,19,54,55 Mortality is also quite uncommon and in most reported cases has been associated with co-ingestion of other psychotropic agents, benzodiazepines, opiates, or alcohol.2,52,53
Another uncommon but potentially serious toxic manifestation of the SSRIs and SNRIs is a constellation of symptoms and signs referred to as serotonin syndrome.56 This disorder results from excessive stimulation of central and peripheral serotonin receptors and is characterized by the triad of altered mentation, autonomic dysfunction, and neuromuscular hyperactivity. Symptoms and signs range from mild to very severe and include delirium, diaphoresis, diarrhea, hyperthermia, tremor, hyperreflexia, clonus, and muscular rigidity. Laboratory findings are variable and nonspecific and may include leukocytosis and elevations of creatine phosphokinase and the hepatic transaminases. When it occurs, the serotonin syndrome usually develops within 6 hours following self-poisoning.56
Management
The treatment of SSRI and SNRI overdose is primarily supportive.53 As discussed earlier, gastric lavage is virtually never indicated, and single-dose activated charcoal should be considered only when patients present with major signs of toxicity within 1 hour of drug ingestion. Because major morbidity and mortality usually result from the effects of other medications, efforts must be made to identify all co-ingested substances.
Treatment of serotonin syndrome is also largely supportive, and usually the most important intervention is to identify and discontinue all serotonergic drugs.56 Serotonin syndrome usually has a benign course, and symptoms and signs typically resolve within 24 hours of onset. Occasionally, however, severe complications occur and require specific therapy; these include marked hyperthermia, rhabdomyolysis, disseminated intravascular coagulation, renal failure, and acute respiratory distress syndrome. Limited data suggest that the serotonin receptor (5-HT2A) antagonist, cyproheptadine, may be useful in severe cases.57
Monoamine Oxidase Inhibitors
The symptoms and signs that accompany MAOI overdose are believed to result primarily from a hyperadrenergic state produced by the inability to metabolize and inactivate NE in the central and peripheral nervous systems. Overdose with the irreversible MAOIs is commonly accompanied by life-threatening toxicity, and the mortality rate is similar to that of TCA ingestion.2,7,58 Clinical manifestations, which may be delayed for up to 24 hours, include mydriasis, flushing, diaphoresis, tachycardia, hypertension, hyperthermia, muscular rigidity, agitation, delirium, and seizures.59,60 Hypotension may occur later in the course, probably as the result of depletion of NE stores. Since MAOIs act to increase serotonin levels in the brain, overdose may also precipitate the serotonin syndrome.
Patients with MAOI overdose should receive activated charcoal if they present within 1 hour after drug ingestion.30 Severe hypertension is best controlled with sodium nitroprusside, and hypotension usually responds well to NE.56,58,59 Dopamine acts largely by releasing stored NE and should be avoided because it may either worsen the hyperadrenergic state or be ineffective due to endogenous NE depletion.59,60 Hyperthermia may be severe and require evaporative cooling techniques. Muscle rigidity usually responds to benzodiazepines but may require the use of neuromuscular blockade.56,59,60 Seizures typically respond to benzodiazepines, phenytoin, and phenobarbital.
Atypical Antidepressants
Relatively little is known about the consequences of overdose with the atypical antidepressants. It is recognized, however, that bupropion has the greatest toxicity.7,19 The most common manifestations of bupropion overdose are neurologic, and delirium and recurrent seizures are common. Cardiac complications including QRS and QTC prolongation and ventricular arrhythmias have been reported.7,61 Mirtazapine, reboxetine, nefazodone, and trazodone appear to produce relatively little toxicity following self-poisoning, and CNS depression is the most commonly reported effect.7,17,19,62–64
Key Points
Gillman PK. Tricyclic antidepressant pharmacology and therapeutic drug interactions updated. Br J Pharmacol. 2007;151:737-748.
Richelson E. Pharmacology of antidepressants. Mayo Clin Proc. 2001;76:511-527.
Krishnan KR. Revisiting monoamine oxidase inhibitors. J Clin Psychiatry. 2007;68(S8):35-41.
Woolf AD, Erdman AR, Nelson LS, et al. Tricyclic antidepressant poisoning: an evidence-based consensus guideline for out-of-hospital management. Clin Toxicol. 2007;45:203-233.
Blackman K, Brown SG, Wilkes GJ. Plasma alkalinization for tricyclic antidepressant toxicity: a systematic review. Emerg Med. 2001;13:204-210.
Boyer EW, Shannon M. The serotonin syndrome. N Engl J Med. 2005;352:1112-1120.
This is the most recent authoritative review of serotonin syndrome.
Buckley NA, Faunce TA. Atypical antidepressants in overdose. Drug Saf. 2003;26:539-551.
This is the best review of the pharmacology and toxicology of the atypical antidepressants.
1 Kessler RC, Berglund P, Demler O, et al. The epidemiology of major depressive disorder. JAMA. 2003;289:3095-3105.
2 Bronstein AC, Spyker DA, Cantilena LR, et al. 2008 annual report of the American Association of Poison Control Centers’ national poison data system. Clin Toxicol. 2009;47:911-1084.
3 Litovitz TL, Klein-Schwartz W, Caravati EM, et al. 1998 annual report of the American Association Of Poison Control Centers’ toxic exposure surveillance system. Am J Emerg Med. 1999;17:435.
4 Litovitz TL, Feldberg L, Soloway RA, et al. 1994 annual report of The American Association Of Poison Control Centers’ toxic exposure surveillance system. AM J Emerg Med. 1995;13:551.
5 Richelson E. Pharmacology of antidepressants. Mayo Clin Proc. 2001;76:511-527.
6 Reilly TH, Kirk MA. Atypical antipsychotics and newer antidepressants. Emerg Med Clin North Am. 2007;25:477-497.
7 Buckley NA, Faunce TA. Atypical antidepressants in overdose. Drug Saf. 2003;26:539-551.
8 Baldessarini RJ. Drugs acting on the central nervous system. In: Hardman JG, Limbird LE, editors. Goodman and Gilman’s The Pharmacologic Basis of Therapeutics. 10th ed. New York: McGraw-Hill; 2001:447-483.
9 Gillman PK. Tricyclic antidepressant pharmacology and therapeutic drug interactions updated. Br J Pharmacol. 2007;151:737-748.
10 Tang SW, Helmeste D. Paroxetine. Expert Opin Pharmacother. 2008;9:787-794.
11 Muijers RB, Plosker GL, Noble S. Spotlight on sertraline in the management of major depressive disorder in elderly patients. CNS Drugs. 2002;16:789-794.
12 Murdoch D, Keam SJ. Escitalopram. Drugs. 2005;65:2379-2404.
13 Perry R, Cassagnol M. Desvenlafaxine: A new serotonin-norepinephrine reuptake inhibitor for the treatment of adults with major depressive disorder. Clin Ther. 2009;31:1374-1404.
14 Frampton JE, Plosker GL. Duloxetine. CNS Drugs. 2007;21:581-609.
15 Pae CU, Marks DM, Shah M, et al. Milnacipran: Beyond a role of antidepressant. Clin Neuropharmacol. 2009;32:355-363.
16 Krishnan KR. Revisiting monoamine oxidase inhibitors. J Clin Psychiatry. 2007;68(S8):35-41.
17 Preskorn SH. Reboxetine: A norepinephrine selective reuptake pump inhibitor. J Psychiatric Pract. 2004;10:57-63.
18 Spina E, Santoro V, D’Arigo C. Clinically relevant pharmacokinetic drug interactions with second-generation antidepressants-an update. Clin Ther. 2008;30:1206-1227.
19 White NC, Litovitz T, Clancy C. Suicidal antidepressant overdoses: a comparative analysis by antidepressant type. J Med Toxicol. 2008;4:238-250.
20 Woolf AD, Erdman AR, Nelson LS, et al. Tricyclic antidepressant poisoning: an evidence-based consensus guideline for out-of-hospital management. Clin Toxicol. 2007;45:203-233.
21 Wu A, McKay C, Broussard L, et al. National Academy of Clinical Biochemistry laboratory medicine practice guidelines. Clin Chem. 2003;49:357-379.
22 Boehnert MT, Lovejoy FH. Value of the QRS duration versus the serum drug level in predicting seizures and ventricular arrhythmias after an acute overdose of tricyclic antidepressants. N Engl J Med. 1985;313:474-479.
23 Niemann JT, Bessen HA, Rothstein RJ, et al. Electrocardiographic criteria for tricyclic antidepressant cardiotoxicity. Am J Cardiol. 1986;57:1154-1159.
24 Wolfe TR, Caravati EM, Rollins DE. Terminal 40-ms frontal plane axis as a marker for tricyclic antidepressant overdose. Ann Emerg Med. 1989;18:348-351.
25 Bramble MG, Lishman AH, Purdon J. An analysis of plasma level and 24-hour ECG recordings in tricyclic antidepressant poisoning: implications for management. Q J Med. 1985;219:357-366.
26 Rudorfer MV. Cardiovascular changes and plasma drug levels after amitriptyline overdose. J Toxicol Clin Toxicol. 1982;19:67-78.
27 Bailey B, Buckley NA, Amre DK. A meta-analysis of prognostic indicators to predict seizures, arrhythmias, or death after tricyclic antidepressant overdose. J Toxicol Clin Toxicol. 2004;42:877-888.
28 Dawling S, Crome P, Braithwaite R. Effect of delayed administration of activated charcoal on nortriptyline absorption. Eur J Clin Pharmacol. 1978;14:445-447.
29 Crome P, Adams R, Ali C, et al. Activated charcoal in tricyclic antidepressant poisoning: pilot controlled clinical trial. Hum Toxicol. 1983;2:205-209.
30 Chyka PA, Seger D, Krenzelok EP, Vale JA, American Academy of Clinical Toxicology; European Association of Poison Centres and Clinical Toxicologists. Position paper: Single-dose activated charcoal. Clin Toxicol (Phila). 2005;43:61-87.
31 Watson WA, Leighton J, Guy J, et al. Recovery of cyclic antidepressants with gastric lavage. J Emerg Med. 1989;7:373-377.
32 Merigian KS, Woodard M, Hedges JR, et al. Prospective evaluation of gastric emptying in the self-poisoned patient. Am J Emerg Med. 1990;8:479-483.
33 Pond SM, Lewis-Driver DJ, Williams GM, et al. Gastric emptying in acute overdose: A prospective randomized controlled trial. Med J Aust. 1995;163:345-349.
34 Bosse GM, Barefoot JA, Pfeifer MP, et al. Comparison of three methods of gut decontamination in tricyclic antidepressant overdose. J Emerg Med. 1995;13:203-209.
35 Hulten BA, Adams R, Askenasi R, et al. Activated charcoal in tricyclic antidepressant poisoning. Hum Toxicol. 1988;7:307-310.
36 Vale JA, Kulig K, American Academy of Clinical Toxicology; European Association of Poison Centres and Clinical Toxicologists. Position paper: Gastric lavage. J Toxicol Clin Toxicol. 2004;42:933-943.
37 Vale JA, Krenzelok EP, Barceloux GD. Position statement and practice guidelines on the use of multidose activated charcoal in the treatment of acute poisoning. J Toxicol Clin Toxicol. 1999;37:731-751.
38 Ash SR, Levy H, Akmal M, et al. Treatment of severe tricyclic antidepressant overdose with extracorporeal sorbent detoxification. Adv Ren Replace Ther. 2002;9:31-41.
39 Bek K, Ozkaya O, Mutlu B, et al. Charcoal hemoperfusion in amitriptyline poisoning: experience in 20 children. Nephrology. 2008;13:193-197.
40 Blackman K, Brown SG, Wilkes GJ. Plasma alkalinization for tricyclic antidepressant toxicity: A systematic review. Emerg Med. 2001;13:204-210.
41 McKinney PE, Rasmussen R. Reversal of severe tricyclic antidepressant-induced cardiotoxicity with intravenous hypertonic saline solution. Ann Emerg Med. 2003;42:20-24.
42 Albertson TE, Dawson A, de Latorre F, et al. Tox-ACLS. Toxicologic-oriented advanced cardiac life support. Ann Emerg Med. 2001;37:S78-S90.
43 American Heart Association. Toxicology in ECC. Circulation. 2005;112:IV 126-32.
44 Langou RA, Van Dyke C, Tahan SR, et al. Cardiovascular manifestations of tricyclic antidepressant overdose. Am Heart J. 1980;100:458-464.
45 Boehnert M, Lovejoy F. The effect of phenytoin on cardiac conduction and ventricular arrhythmias in acute tricyclic antidepressant overdose. Vet Hum Toxicol. 1985;28:297.
46 Hagerman GA, Hanashiro PK. Reversal of tricyclic antidepressant-induced cardiac conduction abnormalities by phenytoin. Ann Emerg Med. 1981;10:82-86.
47 Citak A, Soysal D, Ucsel R, et al. Efficacy of long duration resuscitation and magnesium sulfate treatment in amitriptyline poisoning. Eur J Emerg Med. 2002;9:63-66.
48 Teba L, Schiebel F, Dedhia H, et al. Beneficial effect of norepinephrine in the treatment of circulatory shock caused by tricyclic antidepressant overdose. Am J Emerg Med. 1988;6:566-568.
49 Sensky PR, Olczak SA. High-dose intravenous glucagon in severe tricyclic poisoning. Postgrad Med J. 1999;75:611-612.
50 Callaham M, Kassal D. Epidemiology of fatal tricyclic antidepressant ingestion: Implications for management. Ann Emerg Med. 1985;14:29-37.
51 Shannon MW. Duration of QRS disturbances after severe tricyclic antidepressant intoxication. J Toxicol Clin Toxicol. 1992;30:377-386.
52 Whyte IM, Dawson AH, Buckley NA. Relative toxicity of venlafaxine and selective serotonin reuptake inhibitors in overdose compared to tricyclic antidepressants. Q J Med. 2003;96:369-374.
53 Nelson LS, Erdman AR, Booze LL, et al. Selective serotonin reuptake inhibitor poisoning: an evidence-based consensus guideline for out-of-hospital management. Clin Toxicol (Phila). 2007;45:315-332.
54 Catalano G, Catalano MC, Epstein MA, et al. QTc prolongation associated with citalopram overdose: A case report and literature review. Clin Neuropharmacol. 2001;24:158-162.
55 Brendel DH, Bodkin JA, Yang JM. Massive sertraline overdose. Ann Emerg Med. 2000;36:524-526.
56 Boyer EW, Shannon M. The serotonin syndrome. N Engl J Med. 2005;352:1112-1120.
57 Graudins A, Stearman A, Chan B. Treatment of the serotonin syndrome with cyproheptadine. J Emerg Med. 1998;16:615-619.
58 Buckley NA, McManus PR. Fatal toxicity of serotonergic and other antidepressant drugs: Analysis of United Kingdom mortality data. BMJ. 2002;325:1332-1333.
59 Linden CH, Rumack BH. Monoamine oxidase inhibitor overdose. Ann Emerg Med. 1984;13:1137-1144.
60 Thorp M, Toombs D, Harmon B. Monoamine oxidase inhibitor overdose. West J Med. 1997;166:275-277.
61 Harris CR, Gualtieri J, Stark G. Fatal bupropion overdose. J Toxicol Clin Toxicol. 1997;25:321-324.
62 Gaffney PN, Schuckman HA, Beeson MS. Nefazodone overdose. Ann Pharmacother. 1998;32:1249-1250.
63 Gamble DE, Peterson LG. Trazodone overdose. J Clin Psychiatry. 1986;47:544-546.
64 Bremner JD, Wingard P, Walshe TA. Safety of mirtazapine in overdose. J Clin Psychiatry. 1998;59:233-235.
