Stage Two: Coagulation
Coagulation is defined as production of fibrin, a thread-like protein that reinforces the platelet plug. Fibrin is produced by two convergent pathways (Fig. 44.2), referred to as the contact activation pathway (also known as the intrinsic pathway) and the tissue factor pathway (also known as the extrinsic pathway). The two pathways converge at factor Xa, after which they employ the same final series of reactions. In both pathways, each reaction in the sequence amplifies the reaction that follows. Hence, after this sequence is initiated, it becomes self-sustaining and self-reinforcing.
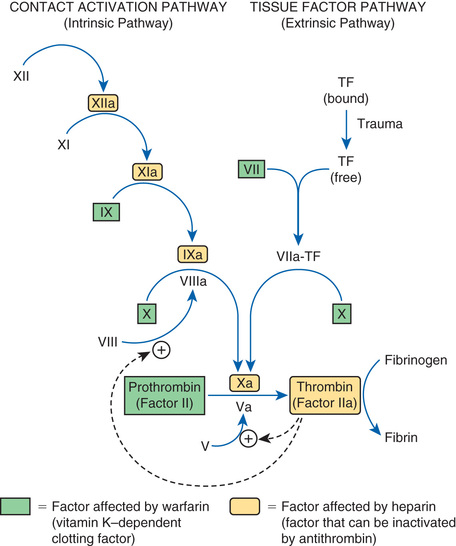
The tissue factor pathway is turned on by trauma to the vascular wall, which triggers release of tissue factor,1 also known as tissue thromboplastin. Tissue factor then combines with and thereby activates factor VII, which in turn activates factor X, which then catalyzes the conversion of prothrombin (factor II) into thrombin (factor IIa). Thrombin then does three things. First, it catalyzes the conversion of fibrinogen into fibrin. Second, it catalyzes the conversion of factor V into its active form (Va), a compound that greatly increases the activity of factor Xa, even though it has no direct catalytic activity of its own. Third, thrombin catalyzes the conversion of factor VIII into its active form (VIIIa), a compound that greatly increases the activity of factor IXa in the contact activation pathway.
The contact activation pathway is turned on when blood makes contact with collagen that has been exposed as a result of trauma to a blood vessel wall. Collagen contact stimulates conversion of factor XII into its active form, XIIa (see Fig. 44.2). Factor XIIa then activates factor XI, which activates factor IX, which activates factor X. After this, the contact activation pathway is the same as the tissue factor pathway. As noted, factor VIIIa, which is produced under the influence of thrombin, greatly increases the activity of factor IXa, even though it has no direct catalytic activity of its own.
Important to our understanding of anticoagulant drugs is the fact that four coagulation factors—factors VII, IX, X, and II (prothrombin)—require vitamin K for their synthesis. These factors appear in green boxes in Fig. 44.2. The significance of the vitamin K–dependent factors will become apparent when we discuss warfarin, an oral anticoagulant.
Keeping Hemostasis Under Control
To protect against widespread coagulation, the body must inactivate any clotting factors that stray from the site of vessel injury. Inactivation is accomplished with antithrombin, a protein that forms a complex with clotting factors and thereby inhibits their activity. The clotting factors that can be neutralized by antithrombin appear in yellow in Fig. 44.2. As we shall see, antithrombin is intimately involved in the action of heparin, an injectable anticoagulant drug.
Physiologic Removal of Clots
As healing of an injured vessel proceeds, removal of the clot is eventually necessary. The body accomplishes this with plasmin, an enzyme that degrades the fibrin meshwork of the clot. Plasmin is produced through the activation of its precursor, plasminogen. The fibrinolytic drugs (e.g., alteplase) act by promoting conversion of plasminogen into plasmin.
Thrombosis
A thrombus is a blood clot formed within a blood vessel or within the heart. Thrombosis (thrombus formation) reflects pathologic functioning of hemostatic mechanisms.
Arterial Thrombosis
Formation of an arterial thrombus begins with adhesion of platelets to the arterial wall. (Adhesion is stimulated by damage to the wall or rupture of an atherosclerotic plaque.) After adhesion, platelets release ADP and thromboxane A2 (TXA2), and thereby attract additional platelets to the evolving thrombus. With continued platelet aggregation, occlusion of the artery takes place. As blood flow comes to a stop, the coagulation cascade is initiated, causing the original plug to undergo reinforcement with fibrin. The consequence of an arterial thrombus is localized tissue injury owing to lack of perfusion.
Venous Thrombosis
Venous thrombi develop at sites where blood flow is slow. Stagnation of blood initiates the coagulation cascade, resulting in the production of fibrin, which enmeshes red blood cells and platelets to form the thrombus. The typical venous thrombus has a long tail that can break off to produce an embolus. Such emboli travel within the vascular system and become lodged at faraway sites, frequently the pulmonary arteries. Hence, unlike an arterial thrombus, whose harmful effects are localized, injury from a venous thrombus occurs secondary to embolization at a site distant from the original thrombus.
Overview of Drugs for Thromboembolic Disorders
The drugs considered fall into three major groups: (1) anticoagulants, (2) antiplatelet drugs, and (3) thrombolytic drugs, also known as fibrinolytic drugs. Anticoagulants (e.g., heparin, warfarin, dabigatran) disrupt the coagulation cascade and thereby suppress production of fibrin. Antiplatelet drugs (e.g., aspirin, clopidogrel) inhibit platelet aggregation. Thrombolytic drugs (e.g., alteplase) promote lysis of fibrin, causing dissolution of thrombi. Because these drugs are used only in a hospital setting, discussion of thrombolytics occurs in Chapter 89. Drugs that belong to these groups are shown in Table 44.1.
TABLE 44.1
Overview of Drugs for Thromboembolic Disorders
| Generic Name | Trade Name | Route | Action | Therapeutic Use |
| ANTICOAGULANTS | Anticoagulants decrease formation of fibrin | Used primarily to prevent thrombosis in veins and the atria of the heart | ||
| Vitamin K Antagonist | ||||
| Warfarin | Coumadin | PO | ||
| Heparin and Its Derivatives: Drugs That Activate Antithrombin | ||||
| Heparin (unfractionated) | SubQ, IV | |||
| LMW heparins | ||||
| Dalteparin | Fragmin | SubQ | ||
| Enoxaparin | Lovenox | SubQ | ||
| Fondaparinux | Arixtra | SubQ | ||
| Direct Thrombin Inhibitors | ||||
| Hirudin Analogs | ||||
| Desirudin | Iprivask | SubQ | ||
| Other Direct Thrombin Inhibitors | ||||
| Dabigatran | Pradaxa, Pradax  |
PO | ||
| Direct Factor Xa Inhibitors | ||||
| Rivaroxaban | Xarelto | PO | ||
| Apixaban | Eliquis | PO | ||
| Edoxaban | Savaysa | PO | ||
| ANTIPLATELET DRUGS | Antiplatelet drugs suppress platelet aggregation | Used primarily to prevent thrombosis in arteries | ||
| Cyclooxygenase Inhibitor | ||||
| Aspirin | PO | |||
| P2Y12 Adenosine Diphosphate Receptor Antagonists | ||||
| Clopidogrel | Plavix | PO | ||
| Prasugrel | Effient | PO | ||
| Ticagrelor | Brilinta | PO | ||
| Generic only | PO | |||
| Protease-Activated Receptor-1 (PAR-1) Antagonists | ||||
| Vorapaxar | Zontivity | PO | ||
| Other Antiplatelet Drugs | ||||
| Dipyridamole | Persantine | PO | ||
| Cilostazol | Pletal | PO | ||
Although the anticoagulants and the antiplatelet drugs both suppress thrombosis, they do so by different mechanisms. As a result, they differ in their effects and applications. The antiplatelet drugs are most effective at preventing arterial thrombosis, whereas anticoagulants are most effective against venous thrombosis.
Anticoagulants
By definition, anticoagulants are drugs that reduce formation of fibrin. Two basic mechanisms are involved. One anticoagulant—warfarin—inhibits the synthesis of clotting factors, including factor X and thrombin. All other anticoagulants inhibit the activity of clotting factors: either factor Xa, thrombin, or both.
Anticoagulants are in three pharmacologic classes—vitamin K antagonists, direct factor Xa inhibitors, and direct thrombin inhibitors (see Table 44.1).
Heparin and Its Derivatives: Drugs That Activate Antithrombin
All drugs in this group share the same mechanism of action. Specifically, they greatly enhance the activity of antithrombin, a protein that inactivates two major clotting factors: thrombin and factor Xa. In the absence of thrombin and factor Xa, production of fibrin is reduced, and hence clotting is suppressed.
Our discussion focuses on three preparations: unfractionated heparin, the low-molecular-weight (LMW) heparins, and fondaparinux. Although all three activate antithrombin, they do not have equal effects on thrombin and factor Xa. Specifically, heparin reduces the activity of thrombin and factor Xa more or less equally; the LMW heparins reduce the activity of factor Xa more than they reduce the activity of thrombin; and fondaparinux causes selective inhibition of factor Xa, having no effect on thrombin. Properties of the three preparations are shown in Table 44.2.
TABLE 44.2
Comparison of Drugs That Activate Antithrombin
| Property | Unfractionated Heparin | Low-Molecular-Weight Heparins | Fondaparinux |
| Molecular weight range | 3000–30,000 | 1000–9000 | 1728 |
| Mean molecular weight | 12,000–15,000 | 4000–5000 | 1728 |
| Mechanism of action | Activation of antithrombin, resulting in the inactivation of factor Xa and thrombin | Activation of antithrombin, resulting in preferential inactivation of factor Xa, plus some inactivation of thrombin | Activation of antithrombin, resulting in selective inactivation of factor Xa |
| Routes | IV, subQ | SubQ only | SubQ only |
| Nonspecific binding | Widespread | Minimal | Minimal |
| Laboratory monitoring | aPTT monitoring is essential | No aPTT monitoring required | No aPTT monitoring required |
| Dosage | Dosage must be adjusted on the basis of aPTT | Dosage is fixed | Dosage is fixed |
| Setting for use | Hospital | Hospital or home | Hospital or home |
| Cost | $3/day for heparin itself, but hospitalization and aPTT monitoring greatly increase the real cost | $35/day for LMW heparin (enoxaparin [Lovenox]), but home use and absence of aPTT monitoring greatly reduce the real cost | $59/day for fondaparinux, but home use and absence of aPTT monitoring greatly reduce the real cost |
Heparin (Unfractionated)
Heparin is a rapid-acting anticoagulant administered only by injection. Heparin differs from warfarin (an oral anticoagulant) in several respects, including mechanism, time course, indications, and management of overdose.
Chemistry
Heparin is not a single molecule, but rather a mixture of long polysaccharide chains, with molecular weights that range from 3000 to 30,000. The active region is a unique pentasaccharide (five-sugar) sequence found randomly along the chain. An important feature of heparin’s structure is the presence of many negatively charged groups. Because of these negative charges, heparin is highly polar and hence cannot readily cross membranes.
Mechanism of Anticoagulant Action
Heparin suppresses coagulation by helping antithrombin inactivate clotting factors, primarily thrombin and factor Xa. As shown in Fig. 44.3, binding of heparin to antithrombin produces a conformational change in antithrombin that greatly enhances its ability to inactivate both thrombin and factor Xa. However, the process of inactivating these two clotting factors is distinct. To inactivate thrombin, heparin must simultaneously bind with both thrombin and antithrombin, thereby forming a ternary complex. In contrast, to inactivate factor Xa, heparin binds only with antithrombin; heparin itself does not bind with factor Xa.
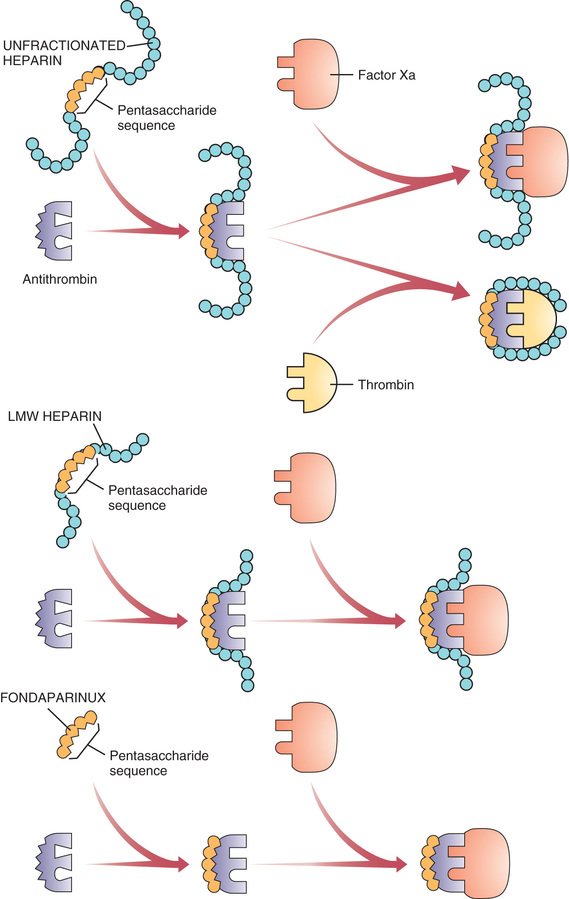
By activating antithrombin, and thereby promoting the inactivation of thrombin and factor Xa, heparin ultimately suppresses formation of fibrin. Because fibrin forms the framework of thrombi in veins, heparin is especially useful for prophylaxis of venous thrombosis. Because thrombin and factor Xa are inhibited as soon as they bind with the heparin-antithrombin complex, the anticoagulant effects of heparin develop quickly (within minutes of intravenous [IV] administration). This contrasts with warfarin, whose full effects are not seen for days.
Pharmacokinetics
Absorption and Distribution.
Because of its polarity and large size, heparin is unable to cross membranes, including those of the gastrointestinal (GI) tract. Consequently, heparin cannot be absorbed if given orally and therefore must be given by injection (IV or subcutaneous [subQ]). Because it cannot cross membranes, heparin does not traverse the placenta and does not enter breast milk.
Protein and Tissue Binding.
Heparin binds nonspecifically to plasma proteins, mononuclear cells, and endothelial cells. As a result, plasma levels of free heparin can be highly variable. Because of this variability, intensive monitoring is required (see later).
Metabolism and Excretion.
Heparin undergoes hepatic metabolism followed by renal excretion. Under normal conditions, the half-life is short (about 1.5 hours). However, in patients with hepatic or renal impairment, the half-life is increased.
Time Course.
Therapy is sometimes initiated with a bolus IV injection, and effects begin immediately. Duration of action is brief (hours) and varies with dosage. Effects are prolonged in patients with hepatic or renal impairment.
Therapeutic Uses
Heparin is a preferred anticoagulant for use during pregnancy (because it doesn’t cross the placenta) and in situations that require rapid onset of anticoagulant effects, including pulmonary embolism (PE) and massive deep vein thrombosis (DVT). In addition, heparin is used for patients undergoing open heart surgery and renal dialysis; during these procedures, heparin serves to prevent coagulation in devices of extracorporeal circulation (heart-lung machines, dialyzers). Low-dose therapy is used to prevent postoperative venous thrombosis. Heparin may also be useful for treating disseminated intravascular coagulation, a complex disorder in which fibrin clots form throughout the vascular system and in which bleeding tendencies may be present; bleeding can occur because massive fibrin production consumes available supplies of clotting factors. Heparin is also used as an adjunct to thrombolytic therapy of acute myocardial infarction (MI).
Adverse Effects
Hemorrhage.
All the drugs in this chapter increase the risk for patient bleeding. Bleeding develops in about 10% of patients and is the principal complication of treatment. Hemorrhage can occur at any site and may be fatal. Patients should be monitored closely for signs of blood loss. These include reduced blood pressure, increased heart rate, bruises, petechiae, hematomas, red or black stools, cloudy or discolored urine, pelvic pain (suggesting ovarian hemorrhage), headache or faintness (suggesting cerebral hemorrhage), and lumbar pain (suggesting adrenal hemorrhage). If bleeding develops, heparin should be withdrawn.
The risk for hemorrhage can be decreased in several ways. First, dosage should be carefully controlled so that the activated partial thromboplastin time (see later) does not exceed 2 times the control value. In addition, candidates for heparin therapy should be screened for risk factors (see “Warnings and Contraindications”). Finally, antiplatelet drugs (e.g., aspirin, clopidogrel) should be avoided.
 Black Box Warning: Spinal or Epidural Hematoma
Black Box Warning: Spinal or Epidural Hematoma
Heparin and all other anticoagulants pose a risk for spinal or epidural hematoma in patients undergoing spinal puncture or spinal or epidural anesthesia. Pressure on the spinal cord caused by the bleed can result in prolonged or permanent paralysis.
Risk for hematoma is increased by the following:
• Use of an indwelling epidural catheter
• Use of other anticoagulants (e.g., warfarin, dabigatran)
• Use of antiplatelet drugs (e.g., aspirin, clopidogrel)
• History of traumatic or repeated epidural or spinal puncture
• History of spinal deformity, spinal injury, or spinal surgery
Patients should be monitored for signs and symptoms of neurologic impairment. If impairment develops, immediate intervention is needed.
Heparin-Induced Thrombocytopenia (HIT).
This is a potentially fatal immune-mediated disorder characterized by reduced platelet counts (thrombocytopenia) and a seemingly paradoxical increase in thrombotic events. The underlying cause is development of antibodies against heparin–platelet protein complexes. These antibodies activate platelets and damage the vascular endothelium, thereby promoting both thrombosis and a rapid loss of circulating platelets. Thrombus formation poses a risk for DVT, PE, cerebral thrombosis, and MI. Ischemic injury secondary to thrombosis in the limbs may require amputation of an arm or leg. Coronary thrombosis can be fatal. The primary treatment for HIT is discontinuation of heparin and, if anticoagulation is still needed, substitution of a nonheparin anticoagulant (e.g., argatroban). The incidence of HIT is between 0.2% and 5% among patients who receive heparin for more than 4 days.
HIT should be suspected whenever platelet counts fall significantly or when thrombosis develops despite adequate anticoagulation. Accordingly, to reduce the risk for HIT, patients should be monitored for signs of thrombosis and for reductions in platelets. Platelet counts should be determined frequently (2–3 times a week) during the first 3 weeks of heparin use and monthly thereafter. If severe thrombocytopenia develops (platelet count below 100,000/mm3), heparin should be discontinued.
Hypersensitivity Reactions.
Because commercial heparin is extracted from animal tissues, these preparations may be contaminated with antigens that can promote allergy. Possible allergic responses include chills, fever, and urticaria. Anaphylactic reactions are rare.
Other Adverse Effects.
SubQ dosing may produce local irritation and hematoma. Vasospastic reactions that persist for several hours may develop after 1 or more weeks of treatment. Long-term, high-dose therapy may cause osteoporosis.
Warnings and Contraindications
Warnings.
Heparin must be used with extreme caution in all patients who have a high likelihood of bleeding. Among these are individuals with hemophilia, increased capillary permeability, dissecting aneurysm, peptic ulcer disease, severe hypertension, or threatened abortion. Heparin must also be used cautiously in patients with severe disease of the liver or kidneys.
Contraindications.
Heparin is contraindicated for patients with thrombocytopenia and uncontrollable bleeding. In addition, heparin should be avoided both during and immediately after surgery of the eye, brain, or spinal cord. Lumbar puncture and regional anesthesia are additional contraindications.
Drug Interactions
In heparin-treated patients, platelet aggregation is the major remaining defense against hemorrhage. Aspirin and other drugs that depress platelet function or affect coagulation will weaken this defense and hence must be employed with caution.
Laboratory Monitoring
The objective of anticoagulant therapy is to reduce blood coagulability to a level that is low enough to prevent thrombosis but not so low as to promote spontaneous bleeding. Because heparin levels can be highly variable, achieving this goal is difficult and requires careful control of dosage based on frequent tests of coagulation. The laboratory test employed most commonly is the activated partial thromboplastin time (aPTT). The normal value for the aPTT is 40 seconds. At therapeutic levels, heparin increases the aPTT by a factor of 1.5 to 2, making the aPTT 60 to 80 seconds. Because heparin has a rapid onset and brief duration, if an aPTT value falls outside the therapeutic range, coagulability can be quickly corrected through an adjustment in dosage: if the aPTT is too long (more than 80 seconds), the dosage should be lowered; conversely, if the aPTT is too short (less than 60 seconds), the dosage should be increased. Measurements of aPTTs should be made frequently (every 4–6 hours) during the initial phase of therapy. When an effective dosage has been established, measuring aPTT once a day will suffice.
Prescription and Preparations
Prescription.
Heparin is prescribed in units, not in milligrams. The heparin unit is an index of anticoagulant activity. Heparin dosage is titrated on the basis of laboratory monitoring, and hence dosage can be adjusted as needed based on test results.
Preparations.
Heparin sodium is supplied in single-dose vials; multidose vials; and unit-dose, preloaded syringes that have their own needles. Concentrations range from 1000 to 20,000 units/mL.
Dosage and Administration
General Considerations.
Heparin is administered by injection only. Two routes are employed: intravenous (either intermittent or continuous) and subcutaneous. Intramuscular injection causes hematoma and must not be done. Heparin is not administered orally because heparin is too large and too polar to permit intestinal absorption.
Dosage varies by indication. Postoperative prophylaxis of thrombosis, for example, requires relatively small doses. In other situations, such as open heart surgery, much larger doses are needed. The dosages given here are for “general anticoagulant therapy.” As a rule, the aPTT should be employed as a guideline for dosage titration; increases in the aPTT of 1.5- to 2-fold are therapeutic. Because heparin is formulated in widely varying concentrations, you must read the label carefully to ensure that dosing is correct.
PATIENT-CENTERED CARE ACROSS THE LIFE SPAN
Anticoagulants
| Life Stage | Patient Care Concerns |
| Infants | Heparin is commonly used in infants needing anticoagulation. Argatroban has been used successfully in infants with HIT. Warfarin is also administered to infants. |
| Children/adolescents | Many anticoagulants can be used safely in children, just in smaller doses. Side-effect profiles are similar to those of adults. |
| Pregnant women | Warfarin is classified in FDA Pregnancy Risk Category X and is contraindicated in pregnancy. LMW heparins and unfractionated heparin are commonly used in pregnancy. In pregnant women with HIT, argatroban is a safe alternative. |
| Breastfeeding women | Data are lacking regarding safety of these medications in breastfeeding. Warfarin and heparin are both safe to use. |
| Older adults | Atrial fibrillation becomes more common with age. In older adults, benefit must outweigh risk for bleeding secondary to falls, decreased renal function, or polypharmacy. |
Low-Molecular-Weight Heparins
Group Properties
LMW heparins are simply heparin preparations composed of molecules that are shorter than those found in unfractionated heparin. LMW heparins are as effective as unfractionated heparin and are easier to use because they can be given using a fixed dosage and don’t require aPTT monitoring. As a result, LMW heparins can be used at home, whereas unfractionated heparin must be given in a hospital when administering intravenously. Because of these advantages, LMW heparins are now considered first-line therapy for prevention and treatment of DVT. In the United States two LMW heparins are available: enoxaparin [Lovenox] and dalteparin [Fragmin]. Differences between LMW heparins and unfractionated heparin are shown in Table 44.2.
Production.
LMW heparins are made by depolymerizing unfractionated heparin (i.e., breaking unfractionated heparin into smaller pieces). Molecular weights in LMW preparations range between 1000 and 9000, with a mean of 4000 to 5000. In comparison, molecular weights in unfractionated heparin range between 3000 and 30,000, with a mean of 12,000 to 15,000.
Mechanism of Action.
Anticoagulant activity of LMW heparin is mediated by the same active pentasaccharide sequence that mediates anticoagulant action of unfractionated heparin. However, because LMW heparin molecules are short, they do not have quite the same effect as unfractionated heparin. Specifically, whereas unfractionated heparin is equally good at inactivating factor Xa and thrombin, LMW heparins preferentially inactivate factor Xa, being much less able to inactivate thrombin. Why the difference? To inactivate thrombin, a heparin chain must not only contain the pentasaccharide sequence that activates antithrombin but must also be long enough to provide a binding site for thrombin. This binding site is necessary because inactivation of thrombin requires simultaneous binding of thrombin with heparin and antithrombin (see Fig. 44.3). In contrast to unfractionated heparin chains, most (but not all) LMW heparin chains are too short to allow thrombin binding, and hence LMW heparins are less able to inactivate thrombin.
Therapeutic Use.
LMW heparins are approved for (1) prevention of DVT after abdominal surgery, hip replacement surgery, or knee replacement surgery; (2) treatment of established DVT, with or without PE; and (3) prevention of ischemic complications in patients with unstable angina, non–Q-wave MI, and ST-elevation MI (STEMI). In addition, these drugs have been used extensively off-label to prevent DVT after general surgery and in patients with multiple trauma and acute spinal injury. When used for prophylaxis or treatment of DVT, LMW heparins are at least as effective as unfractionated heparin, and possibly more effective.
Pharmacokinetics.
Compared with unfractionated heparin, LMW heparins have higher bioavailability and longer half-lives. Bioavailability is higher because LMW heparins do not undergo nonspecific binding to proteins and tissues and hence are more available for anticoagulant effects. Half-lives are prolonged (up to 6 times longer than that of unfractionated heparin) because LMW heparins undergo less binding to macrophages and hence undergo slower clearance by the liver. Because of increased bioavailability, plasma levels of LMW heparin are highly predictable. As a result, these drugs can be given using a fixed dosage, with no need for routine monitoring of coagulation. Because of their long half-lives, LMW heparins can be given just once or twice a day.
Administration, Dosing, and Monitoring.
All LMW heparins are administered by subQ injection. Dosage is sometimes based on body weight, depending on indication. Because plasma levels of LMW heparins are predictable for any given dose, these drugs can be employed using a fixed dosage without laboratory monitoring. This contrasts with unfractionated heparin, which requires dosage adjustments on the basis of aPTT measurements. Because LMW heparins have an extended half-life, dosing can be done once or twice daily. For prophylaxis of DVT, dosing is begun in the perioperative period and continued 5 to 10 days.
Adverse Effects and Interactions.
Bleeding is the major adverse effect. However, the incidence of bleeding complications is less than with unfractionated heparin. Despite the potential for bleeding, LMW heparins are considered safe for outpatient use. Like unfractionated heparin, LMW heparins can cause immune-mediated thrombocytopenia. As with unfractionated heparin, overdose with LMW heparins can be treated with protamine sulfate.
Like unfractionated heparin, LMW heparins can cause severe neurologic injury, including permanent paralysis, when given to patients undergoing spinal puncture or spinal or epidural anesthesia. The risk for serious harm is increased by concurrent use of antiplatelet drugs (e.g., aspirin, clopidogrel) or anticoagulants (e.g., warfarin, dabigatran). Patients should be monitored closely for signs of neurologic impairment.
Cost.
LMW heparins cost more than unfractionated heparin (e.g., about $63/day for dalteparin vs. $8/day for unfractionated heparin). However, because LMW heparins can be used at home and don’t require aPTT monitoring, the overall cost of treatment is lower than with unfractionated heparin.
Individual Preparations
In the United States two LMW heparins are available: enoxaparin and dalteparin. Additional LMW heparins are available in other countries. Each preparation is unique, so clinical experience with one may not apply fully to the other.
Enoxaparin.
Enoxaparin is approved for prevention of DVT after hip and knee replacement surgery or abdominal surgery in patients considered at high risk for thromboembolic complications (e.g., obese patients, those older than 40 years, and those with malignancy or a history of DVT or PE). The drug is also approved for preventing ischemic complications in patients with unstable angina, non–Q-wave MI, or STEMI.
Administration and Dosage.
Enoxaparin is administered by deep subQ injection. For patients with normal renal function (or moderate renal impairment), dosages are as follows:
• Prevention of DVT after hip or knee replacement surgery—30 mg every 12 hours starting 12 to 24 hours after surgery and continuing 7 to 10 days.
• Prevention of DVT after abdominal surgery—40 mg once daily, beginning 2 hours before surgery and continuing 7 to 10 days.
• Treatment of established DVT—1 mg/kg every 12 hours
• Patients with unstable angina or non–Q-wave MI—1 mg/kg every 12 hours (in conjunction with oral aspirin, 100–325 mg once daily) for 2 to 8 days.
• Patients with acute STEMI—30 mg/kg by IV bolus plus 1 mg/kg subQ, followed by 1 mg/kg subQ every 12 hours for up to 8 days.
For patients with severe renal impairment, dosage should be reduced.
Dalteparin.
Approved indications for dalteparin are prevention of DVT after hip replacement surgery or abdominal surgery in patients considered at high risk for thromboembolic complications, prevention of ischemic complications in patients with unstable angina or non–Q-wave MI, and management of symptomatic venous thromboembolism (VTE). Administration is by deep subQ injection. Dosages are as follows:
• Prevention of DVT after hip replacement surgery—2500 units 1 or 2 hours before surgery, 2500 units that evening (at least 6 hours after the first dose), and then 5000 units once daily for 5 to 10 days.
• Prevention of DVT after abdominal surgery—2500 units once daily for 5 to 10 days, starting 1 to 2 hours before surgery. In high-risk patients, this dose is increased to 5000 units once daily, starting the night before surgery.
• Patients with unstable angina or non–Q-wave MI—120 units/kg (but not more than 10,000 units total) every 12 hours for 5 to 8 days. Concurrent therapy with aspirin (75–165 mg/day) is required.
• Patients with symptomatic VTE—200 units/kg (but not more than 18,000 units total) once daily for 1 month, then 150 units/kg (but not more than 18,000 units total) once daily for months 2 through 6.
Fondaparinux
Actions
Fondaparinux [Arixtra] is a synthetic, subQ anticoagulant that enhances the activity of antithrombin, to cause selective inhibition of factor Xa. The result is reduced production of thrombin and hence reduced coagulation. Note that fondaparinux differs from the heparin preparations, which cause inactivation of thrombin as well as factor Xa.
Fondaparinux is closely related in structure and function to heparin and the LMW heparins. Structurally, fondaparinux is a pentasaccharide identical to the antithrombin-binding region of the heparins. Hence, like the heparins, fondaparinux is able to induce a conformational change in antithrombin, thereby increasing antithrombin’s activity—but only against factor Xa, not against thrombin. Why is fondaparinux selective for factor Xa? Because the drug is quite small—even smaller than the LMW heparins. As a result, it is too small to form a complex with both antithrombin and thrombin, and hence cannot reduce thrombin activity (see Fig. 44.3).
Fondaparinux has no effect on prothrombin time, aPTT, bleeding time, or platelet aggregation.
Therapeutic Use
Fondaparinux is approved for (1) preventing DVT after hip fracture surgery, hip replacement surgery, knee replacement surgery, or abdominal surgery; (2) treating acute PE (in conjunction with warfarin); and (3) treating acute DVT (in conjunction with warfarin). The drug is somewhat more effective than enoxaparin (an LMW heparin) at preventing DVT but may cause slightly more bleeding. Anticoagulation may persist for 2 to 4 days after the last dose. Fondaparinux is administered using a fixed dosage and does not require routine laboratory monitoring.
Pharmacokinetics
Fondaparinux is administered by subQ injection. Bioavailability is 100%. Plasma levels peak 2 hours after dosing. The drug is eliminated by the kidneys with a half-life of 17 to 21 hours. The half-life is increased in patients with renal impairment.
Adverse Effects
As with other anticoagulants, bleeding is the biggest concern. The risk is increased by advancing age and renal impairment. Fondaparinux should be used with caution in patients with moderate renal impairment, defined as creatinine clearance (CrCl) of 30 to 50 mL/minute, and avoided in patients with severe renal impairment, defined as CrCl below 30 mL/minute. The drug should also be avoided for prophylactic use in patients weighing less than 50 kg because low body weight increases bleeding risk. After surgery, at least 6 hours should elapse before starting fondaparinux. Aspirin and other drugs that interfere with hemostasis should be used with caution. In contrast to overdose with heparin or LMW heparins, overdose with fondaparinux cannot be treated with protamine sulfate.
Fondaparinux does not promote immune-mediated HIT, although it still can lower platelet counts. During clinical trials, thrombocytopenia developed in 3% of patients. Platelet counts should be monitored, and if they fall below 100,000/mm3, fondaparinux should be discontinued.
In patients undergoing anesthesia using an epidural or spinal catheter, fondaparinux (as well as other anticoagulants) can cause spinal or epidural hematoma, which can result in permanent paralysis. However, in clinical trials, when fondaparinux was administered no sooner than 2 hours after catheter removal, no hematomas were reported.
Preparations, Dosage, and Administration
Fondaparinux [Arixtra] is available in single-dose, prefilled syringes (2.5, 5, 7.5, and 10 mg). Dosing is done once a day by subQ injection.
For prevention of DVT, the recommended dosage is 2.5 mg once a day, starting 6 to 8 hours after surgery. The usual duration is 5 to 9 days.
For treatment of acute DVT or acute PE, dosage is based on body weight as follows: for patients under 50 kg, 5 mg once daily; for patients 50 to 100 kg, 7.5 mg once daily, and for patients over 100 kg, 10 mg once daily. The usual duration is 5 to 9 days when overlapping with warfarin.
Warfarin, a Vitamin K Antagonist
Warfarin [Coumadin, Jantoven], a vitamin K antagonist, is our oldest oral anticoagulant. The drug is similar to heparin in some respects and quite different in others. Like heparin, warfarin is used to prevent thrombosis. In contrast to heparin, warfarin has a delayed onset, which makes it inappropriate for emergencies. However, because it doesn’t require injection, warfarin is well suited for long-term prophylaxis. Like heparin, warfarin carries a significant risk for hemorrhage, which is amplified by the many drug interactions to which warfarin is subject.
History
The history of warfarin underscores its potential for harm. Warfarin was discovered after a farmer noticed that his cattle bled after eating spoiled clover silage. The causative agent was identified as bishydroxycoumarin (dicumarol). Research into derivatives of dicumarol led to the synthesis of warfarin. When warfarin was first developed, clinical use was ruled out owing to concerns about hemorrhage. So, instead of becoming a medicine, warfarin was used to kill rats. The drug proved especially effective in this application and remains one of our most widely used rodenticides. Clinical interest in warfarin was renewed after the report of a failed suicide attempt using huge doses of a warfarin-based rat poison. The clinical trials triggered by that event soon demonstrated that warfarin could be employed safely to treat humans.
Mechanism of Action
Warfarin suppresses coagulation by decreasing production of four clotting factors, namely, factors VII, IX, X, and prothrombin. These factors are known as vitamin K–dependent clotting factors because an active form of vitamin K is needed to make them. Warfarin works by inhibiting vitamin K epoxide reductase complex 1 (VKORC1), the enzyme needed to convert vitamin K to the required active form. Because of its mechanism, warfarin is referred to as a vitamin K antagonist, a term that is somewhat misleading because it implies antagonism of vitamin K actions, not antagonism of vitamin K activation. In therapeutic doses, warfarin reduces production of vitamin K–dependent clotting factors by 30% to 50%.
Pharmacokinetics
Absorption, Distribution, and Elimination
Warfarin is readily absorbed after oral dosing. When in the blood, about 99% of warfarin binds to albumin. Warfarin molecules that remain free (unbound) can readily cross membranes, including those of the placenta and milk-producing glands. Warfarin is inactivated in the liver, mainly by CYP2C9, the 2C9 isoenzyme of cytochrome P450. Metabolites are excreted in the urine and feces.
Time Course
Although warfarin acts quickly to inhibit clotting factor synthesis, noticeable anticoagulant effects are delayed because warfarin has no effect on clotting factors already in circulation. Hence, until these clotting factors decay, coagulation remains unaffected. Because decay of clotting factors occurs with a half-life of 6 hours to 2.5 days (depending on the clotting factor under consideration), initial responses may not be evident until 8 to 12 hours after the first dose. Peak effects take several days to develop.
After warfarin is discontinued, coagulation remains inhibited for 2 to 5 days because warfarin has a long half-life (1.5–2 days). Hence synthesis of new clotting factors remains suppressed, despite stopping dosing.
Therapeutic Uses
Overview of Uses
Warfarin is employed most frequently for long-term prophylaxis of thrombosis. Specific indications are (1) prevention of venous thrombosis and associated PE, (2) prevention of thromboembolism in patients with prosthetic heart valves, and (3) prevention of thrombosis in patients with atrial fibrillation. The drug has also been used to reduce the risk for recurrent transient ischemic attacks (TIAs) and recurrent MI. Because onset of effects is delayed, warfarin is not useful in emergencies. When rapid action is needed, anticoagulant therapy can be initiated with heparin.
Atrial Fibrillation
As discussed in Chapter 41, atrial fibrillation carries a high risk for stroke secondary to clot formation in the atrium. When people have atrial fibrillation, anticoagulant therapy is given long term to prevent clot formation. Until recently, warfarin was the only oral anticoagulant available and hence has been the reference standard for stroke prevention. However, four novel oral anticoagulants (NOACs)—dabigatran [Pradaxa, Pradax  ], apixaban [Eliquis], edoxaban [Savaysa], and rivaroxaban [Xarelto]—which are much easier to use than warfarin, are likely to replace warfarin as the treatment of choice for many patients.
], apixaban [Eliquis], edoxaban [Savaysa], and rivaroxaban [Xarelto]—which are much easier to use than warfarin, are likely to replace warfarin as the treatment of choice for many patients.
Monitoring Treatment
The anticoagulant effects of warfarin are evaluated by monitoring prothrombin time (PT)—a coagulation test that is especially sensitive to alterations in vitamin K–dependent factors. The average pretreatment value for PT is 12 seconds. Treatment with warfarin prolongs PT.
Traditionally, PT test results had been reported as a PT ratio, which is simply the ratio of the patient’s PT to a control PT. However, there is a serious problem with this form of reporting: test results can vary widely among laboratories. The underlying cause of variability is thromboplastin, a critical reagent employed in the PT test. To ensure that test results from different laboratories are comparable, results are now reported in terms of an international normalized ratio (INR). The INR is determined by multiplying the observed PT ratio by a correction factor specific to the particular thromboplastin preparation employed for the test.
The objective of treatment is to raise the INR to an appropriate value. Recommended INR ranges are shown in Table 44.3. As indicated, an INR of 2 to 3 is appropriate for most patients—although for some patients, the target INR is 2.5 to 3.5. If the INR is below the recommended range, warfarin dosage should be increased. Conversely, if the INR is above the recommended range, dosage should be reduced. Unfortunately, because warfarin has a delayed onset and prolonged duration of action, the INR cannot be altered quickly: after the dosage has been changed, it may take a week or more to reach the desired INR.
TABLE 44.3
Monitoring Warfarin Therapy: Recommended Ranges of Prothrombin Time–Derived Values
| Recommended Ranges | ||
| Condition Being Treated | Observed PT Ratio* | INR† |
| Acute myocardial infarction‡ | 1.3–1.5 | 2–3 |
| Atrial fibrillation‡ | 1.3–1.5 | 2–3 |
| Valvular heart disease‡ | 1.3–1.5 | 2–3 |
| Pulmonary embolism | 1.3–1.5 | 2–3 |
| Venous thrombosis§ | 1.3–1.5 | 2–3 |
| Tissue heart valves‡ | 1.3–1.5 | 2–3 |
| Mechanical heart valves | 1.5–2 | 3–4.5 |
| Systemic embolism | ||
| Prevention | 1.3–1.5 | 2–3 |
| Recurrent | 1.5–2 | 2–3 |
INR must be determined frequently during warfarin therapy. PT should be measured daily during the first 5 days of treatment, twice a week for the next 1 to 2 weeks, once a week for the next 1 to 2 months, and every 2 to 4 weeks thereafter. In addition, PT should be determined whenever a drug that interacts with warfarin is added to or deleted from the regimen.
INR can now be monitored at home. Several devices are available, including CoaguChek and the ProTime Microcoagulation System. These small, hand-held machines are easy to use, provide reliable results, and determine PT and INR values. In addition, the ProTime meter can be programmed by the prescriber with upper and lower INR values appropriate for the individual patient. When this is done, the meter will display either In Range, INR High, or INR Low, depending on the degree of anticoagulation. Home monitoring is more convenient than laboratory monitoring and gives patients a sense of empowerment. In addition, it improves anticoagulation control. In theory, home monitoring should help reduce bleeding (from excessive anticoagulation) and thrombosis (from insufficient anticoagulation). The CoaguChek meter costs about $1300, and the ProTime meter costs about $2700 to $3500. Each test costs about $10.
Adverse Effects
Hemorrhage
Bleeding is the major complication of warfarin therapy. Hemorrhage can occur at any site. Patients should be monitored closely for signs of bleeding (reduced blood pressure, increased heart rate, bruises, petechiae, hematomas, red or black stools, cloudy or discolored urine, pelvic pain, headache, and lumbar pain). If bleeding develops, warfarin should be discontinued. Severe overdose can be treated with vitamin K (see later). Patients should be encouraged to carry identification (e.g., Medic Alert bracelet) to inform emergency personnel of warfarin use. Of note, compared with warfarin, the newer oral anticoagulants—apixaban, rivaroxaban, and dabigatran—pose a significantly lower risk for serious bleeds.
Several measures can reduce the risk for bleeding. Candidates for treatment must be carefully screened for risk factors (see “Warnings and Contraindications”). INR must be measured frequently. A variety of drugs can potentiate warfarin’s effects (see later) and hence must be used with care.
Warfarin intensifies bleeding during surgery. Accordingly, surgeons must be informed of warfarin use. Patients anticipating elective procedures should discontinue warfarin several days before the appointment. If an emergency procedure must be performed, injection of vitamin K can help suppress bleeding.
Does warfarin increase bleeding during dental surgery? Yes, but not that much. Accordingly, most patients needn’t interrupt warfarin for dental procedures, including dental surgery. However, it is important that the INR be in the target range.
Fetal Hemorrhage and Teratogenesis From Use During Pregnancy
Warfarin can cross the placenta and affect the developing fetus. Fetal hemorrhage and death have occurred. In addition, warfarin can cause gross malformations, central nervous system defects, and optic atrophy. Accordingly, warfarin is classified in U.S. Food and Drug Administration (FDA) Pregnancy Risk Category X: the risks to the developing fetus outweigh any possible benefits of treatment. Women of childbearing age should be informed about the potential for teratogenesis and advised to postpone pregnancy. If pregnancy occurs, the possibility of termination should be discussed. If an anticoagulant is needed during pregnancy, heparin or LMW heparin, which do not cross the placenta, should be employed.
Use During Lactation
Warfarin enters breast milk. Women should be advised against breastfeeding.
Other Adverse Effects
Adverse effects other than hemorrhage are uncommon. Possible undesired responses include skin necrosis, alopecia, urticaria, dermatitis, fever, GI disturbances, and red-orange discoloration of urine, which must not be confused with hematuria. Long-term warfarin use (more than 12 months) may weaken bones and thereby increase the risk for fractures.
Drug Interactions
General Considerations
Warfarin is subject to a large number of clinically significant adverse interactions—perhaps more than any other drug. As a result of interactions, anticoagulant effects may be reduced to the point of permitting thrombosis, or they may be increased to the point of causing hemorrhage. Patients must be informed about the potential for hazardous interactions and instructed to avoid all drugs not specifically approved by the prescriber. This prohibition includes prescription drugs and over-the-counter products.
Interactions between warfarin and other drugs are shown in Table 44.4. As indicated, the interactants fall into three major categories: (1) drugs that increase anticoagulant effects, (2) drugs that promote bleeding, and (3) drugs that decrease anticoagulant effects. The major mechanisms by which anticoagulant effects can be increased are (1) displacement of warfarin from plasma albumin, (2) inhibition of the hepatic enzymes that degrade warfarin, and (3) decreased synthesis of clotting factors. The major mechanisms for decreasing anticoagulant effects are (1) acceleration of warfarin degradation through induction of hepatic drug-metabolizing enzymes, (2) increased synthesis of clotting factors, and (3) inhibition of warfarin absorption. Mechanisms by which drugs can promote bleeding, and thereby complicate anticoagulant therapy, include (1) inhibition of platelet aggregation, (2) inhibition of clotting factors, and (3) generation of GI ulcers.
TABLE 44.4
Interactions Between Warfarin and Other Drugs
| Drug Category | Mechanism of Interaction | Representative Interacting Drugs |
| Drugs that increase the effects of warfarin | Displacement of warfarin from albumin | |
| Inhibition of warfarin degradation | ||
| Decreased synthesis of clotting factors | Certain parenteral cephalosporins, including cefoperazone and cefamandole | |
| Drugs that promote bleeding | Inhibition of platelet aggregation | |
| Inhibition of clotting factors and/or thrombin | ||
| Promotion of ulcer formation | ||
| Drugs that decrease the effects of warfarin | Induction of drug-metabolizing enzymes | |
| Promotion of clotting factor synthesis | ||
| Reduction of warfarin absorption |
The existence of an interaction between warfarin and another drug does not absolutely preclude using the combination. The interaction does mean, however, that the combination must be used with due caution. The potential for harm is greatest when an interacting drug is being added to or withdrawn from the regimen. At these times, PT must be monitored and the dosage of warfarin adjusted to compensate for the effect of removing or adding an interacting drug.
Specific Interacting Drugs
Of the many drugs listed in Table 44.4, a few are especially likely to produce interactions of clinical significance. Four are discussed next.
Heparin.
The interaction of heparin with warfarin is obvious: being an anticoagulant itself, heparin directly increases the bleeding tendencies brought on by warfarin. Yet because onset of a therapeutic INR when starting warfarin therapy may take a few days, heparin is often administered alongside warfarin during this time. Combined therapy with heparin plus warfarin must be performed with care.
Aspirin.
Aspirin inhibits platelet aggregation. By blocking aggregation, aspirin can suppress formation of the platelet plug that initiates hemostasis. To make matters worse, aspirin can act directly on the GI tract to cause ulcers, thereby initiating bleeding. Therefore, when the antifibrin effects of warfarin are coupled with the antiplatelet and ulcerogenic effects of aspirin, the potential for hemorrhage is significant. Accordingly, patients should be warned specifically against using any product that contains aspirin, unless the provider has prescribed aspirin therapy. Drugs similar to aspirin (e.g., indomethacin, ibuprofen) should be avoided as well.
Nonaspirin Antiplatelet Drugs.
Like aspirin, other antiplatelet drugs can increase the risk for bleeding with warfarin. Accordingly, these drugs (e.g., clopidogrel, dipyridamole, ticlopidine, vorapaxar) should be used with caution.
Acetaminophen.
In the past, acetaminophen was considered safe for patients taking warfarin. In fact, acetaminophen was routinely recommended as an aspirin substitute for patients who needed a mild analgesic. Now, however, it appears that acetaminophen can increase the risk for bleeding: compared with nonusers of acetaminophen, those who take just 4 regular-strength tablets a day for a week are 10 times more likely to have a dangerously high INR. Unlike aspirin, which promotes bleeding by inhibiting platelet aggregation, acetaminophen is believed to inhibit warfarin degradation, thereby raising warfarin levels. At this time, the interaction between acetaminophen and warfarin has not been proved. Nonetheless, when the drugs are combined, the INR should be monitored closely.
Warnings and Contraindications
Like heparin, warfarin is contraindicated for patients with severe thrombocytopenia or uncontrollable bleeding and for patients undergoing lumbar puncture, regional anesthesia, or surgery of the eye, brain, or spinal cord. Also like heparin, warfarin must be used with extreme caution in patients at high risk for bleeding, including those with hemophilia, increased capillary permeability, dissecting aneurysm, GI ulcers, and severe hypertension, and in women anticipating abortion. In addition, warfarin is contraindicated in the presence of vitamin K deficiency, liver disease, and alcoholism—conditions that can disrupt hepatic synthesis of clotting factors. Warfarin is also contraindicated during pregnancy and lactation.
Vitamin K1 for Warfarin Overdose
The effects of warfarin overdose can be overcome with vitamin K1 (phytonadione). Vitamin K1 antagonizes warfarin’s actions and can thereby reverse warfarin-induced inhibition of clotting factor synthesis. (Vitamin K3—menadione—has no effect on warfarin action.)
As a rule, small doses—2.5 mg by oral administration (PO)—are preferred. Large doses (e.g., 10 mg PO) can cause prolonged resistance to warfarin, thereby hampering restoration of anticoagulation after bleeding is under control.
If vitamin K fails to control bleeding, levels of clotting factors can be raised quickly by infusing fresh whole blood, fresh-frozen plasma, or plasma concentrates of vitamin K–dependent clotting factors.
What About Dietary Vitamin K?
Like medicinal vitamin K, dietary vitamin K can reduce the anticoagulant effects of warfarin. Dietary sources include mayonnaise, canola oil, soybean oil, and green leafy vegetables. Patients do not need to avoid these foods but instead should keep intake of vitamin K constant. If vitamin K intake does increase, then warfarin dosage should be increased as well. Conversely, if vitamin K intake decreases, the warfarin dosage should decrease too.
Contrasts Between Warfarin and Heparin
Although heparin and warfarin are both anticoagulants, they differ in important ways (Table 44.5). Whereas warfarin is given orally, heparin is given by injection. Although both drugs decrease fibrin formation, they do so by different mechanisms: heparin inactivates thrombin and factor Xa, whereas warfarin inhibits synthesis of clotting factors. Heparin and warfarin differ with respect to time course of action: effects of heparin begin and fade rapidly, whereas effects of warfarin begin slowly but persist several days. Different tests are used to monitor therapy: changes in aPTT are used to monitor heparin treatment; changes in PT are used to monitor warfarin. Finally, these drugs differ with respect to management of overdose: protamine is given to counteract heparin; vitamin K1 is given to counteract warfarin.
TABLE 44.5
Contrasts Between Heparin and Warfarin
| Heparin | Warfarin | |
| Mechanism of action | Activates antithrombin, which then inactivates thrombin and factor Xa | Inhibits synthesis of vitamin K–dependent clotting factors, including prothrombin and factor X |
| Route | IV or subQ | PO |
| Onset | Rapid (minutes) | Slow (hours) |
| Duration | Brief (hours) | Prolonged (days) |
| Monitoring | aPTT | PT (INR)† |
| Antidote for overdose | Protamine | Vitamin K1 |
Dosage
Basic Considerations
Dosage requirements for warfarin vary widely among individuals, and hence dosage must be tailored to each patient. Traditionally, dosage adjustments have been done empirically (i.e., by trial and error). Dosing is usually begun at 2 to 5 mg/day. Maintenance dosages, which typically range from 2 to 10 mg/day, are determined by the target INR value. For most patients, dosage should be adjusted to produce an INR between 2 and 3.
Genetics and Dosage Adjustment
Patients with variant genes that code for VKORC1 and CYP2C9 are at increased risk for warfarin-induced bleeding and hence require reduced doses. As noted earlier, VKORC1 is the target enzyme that warfarin inhibits, and CYP2C9 is the enzyme that metabolizes warfarin. Variations in VKORC1 increase the enzyme’s sensitivity to inhibition by warfarin, and variations in CYP2C9 delay warfarin breakdown. With either variation, effects of warfarin are increased. To reduce the risk for bleeding, the FDA now recommends—but does not require—that patients undergo genetic testing for these variants. Dosage reductions based on this information can be determined using the calculator at www.warfarindosing.org.
Preparations
Warfarin sodium [Coumadin, Jantoven] is available in tablets (1, 2, 2.5, 3, 4, 5, 6, 7.5, and 10 mg) for oral use. In addition, warfarin is available in a formulation for parenteral dosing, which is not commonly done.
Direct Thrombin Inhibitors
The anticoagulants discussed in this section work by direct inhibition of thrombin. Hence they differ from the heparin-like anticoagulants, which inhibit thrombin indirectly (by enhancing the activity of antithrombin). One of the direct thrombin inhibitors—dabigatran—is administered orally; another—desirudin—is administered subcutaneously; and two others—bivalirudin and argatroban—are administered by continuous IV infusion. Only the subQ and PO drugs are suitable for outpatient use.
Dabigatran Etexilate
Dabigatran etexilate [Pradaxa, Pradax  ] is an oral prodrug that undergoes rapid conversion to dabigatran, a reversible, direct thrombin inhibitor. Compared with warfarin—our oldest oral anticoagulant—dabigatran has five major advantages: rapid onset; no need to monitor anticoagulation; few drug-food interactions; lower risk for major bleeding; and, because responses are predictable, the same dose can be used for all patients, regardless of age or weight. Contrasts between dabigatran and warfarin are shown in Table 44.6.
] is an oral prodrug that undergoes rapid conversion to dabigatran, a reversible, direct thrombin inhibitor. Compared with warfarin—our oldest oral anticoagulant—dabigatran has five major advantages: rapid onset; no need to monitor anticoagulation; few drug-food interactions; lower risk for major bleeding; and, because responses are predictable, the same dose can be used for all patients, regardless of age or weight. Contrasts between dabigatran and warfarin are shown in Table 44.6.
TABLE 44.6
Properties of Oral Anticoagulants
| Warfarin [Coumadin] | Rivaroxaban [Xarelto] | Apixaban [Eliquis] | Edoxaban [Savaysa] | Dabigatran Etexilate [Pradaxa, Pradax  ] ] |
|
| Mechanism | Decreased synthesis of vitamin K–dependent clotting factors | Inhibition of factor Xa | Inhibition of factor Xa | Inhibition of factor Xa | Direct inhibition of thrombin |
| INDICATIONS: | |||||
| Atrial fibrillation | Yes | Yes | Yes | Yes | Yes |
| Heart valve replacement | Yes | No | No | No | No |
| Knee or hip replacement | Yes | Yes | No | No | Yes* |
| Onset | Delayed (days) | Rapid (hours) | Rapid (hours) | Rapid (hours) | Rapid (hours) |
| Duration | Prolonged | Short | Short | Short | Short |
| Antidote available | Yes (oral/parenteral vitamin K) | No | No | No | No |
| Drug-food interactions | Many | Few | Few | Few | Few |
| INR testing needed | Yes | No | No | No | No |
| Dosage | Adjusted based on INR | Fixed | Fixed | Fixed | Fixed |
| Doses/day | One | One | Two | One | Two |
| Clinical experience | Extensive | Limited | Limited | Limited | Limited |
| Advantages, summary | Same as rivaroxaban | Same as rivaroxaban | Same as rivaroxaban | ||
| Disadvantages, summary | Same as rivaroxaban | Same as rivaroxaban | Same as rivaroxaban plus gastrointestinal disturbances are common | ||
Mechanism of Action
Dabigatran is a direct, reversible inhibitor of thrombin. The drug binds with and inhibits thrombin that is free in the blood as well as thrombin that is bound to clots. In contrast, heparin inhibits only free thrombin. By inhibiting thrombin, dabigatran (1) prevents the conversion of fibrinogen into fibrin and (2) prevents the activation of factor XIII and thereby prevents the conversion of soluble fibrin into insoluble fibrin.
Therapeutic Use
Atrial Fibrillation.
In the United States dabigatran was first approved for prevention of stroke and systemic embolism in patients with nonvalvular atrial fibrillation. Approval was based on the RE-LY trial, in which more than 18,000 patients were randomized to receive either dabigatran (110 or 150 mg twice daily) or warfarin (dosage adjusted to produce an INR of 2–3). At the lower dabigatran dose (110 mg twice daily), the incidence of bleeding with dabigatran was less than with warfarin, but protection against stroke was less, too. By contrast, at the higher dose (150 mg twice daily), the incidence of bleeding with dabigatran equaled that with warfarin, but the incidence of stroke or embolism was significantly lower. On the basis of these results, the FDA concluded that, for patients with atrial fibrillation, the benefit/risk profile of dabigatran was better at 150 mg twice daily than at 110 mg twice daily, and hence they approved the higher dose for these patients.
Knee or Hip Replacement.
Dabigatran is approved for prevention of VTE after knee or hip replacement surgery. The dosage is 220 mg once daily, after an initial dose of 110 mg.
DVT and PE Treatment.
In 2014, the FDA approved dabigatran for the treatment of DVT and PE in patients who have been treated with a parenteral anticoagulant for 5 to 10 days and to reduce the risk for recurrent DVT and PE in patients who have been previously treated. The dose for treatment is 150 mg twice daily.
Pharmacokinetics
Dabigatran etexilate is well absorbed from the GI tract, both in the presence and absence of food. (Food delays absorption but does not reduce the extent of absorption.) Plasma levels peak about 1 hour after dosing in the absence of food and 3 hours after dosing in the presence of food. In the blood, plasma esterases rapidly convert dabigatran etexilate to dabigatran, the drug’s active form. Protein binding in blood is low (about 35%). Dabigatran is not metabolized by hepatic enzymes. Elimination is primarily renal. The half-life is 13 hours in patients with normal renal function (CrCl 50 mL/min or higher) and increases to 18 hours in patients with moderate renal impairment (CrCl 30–50 mL/min).
Adverse Effects
Bleeding.
Like all other anticoagulants, dabigatran can cause bleeding. In the RE-LY trial, about 17% of patients taking 150 mg of dabigatran twice daily experienced bleeding of any intensity, and 3% experienced major bleeding. Patients who develop pathologic bleeding should stop taking the drug. Compared with warfarin, dabigatran is safer, posing a much lower risk for hemorrhagic stroke and other major bleeds.
Because dabigatran is not highly protein bound, dialysis can remove much of the drug (about 60% over 2–3 hours). Because dabigatran is eliminated primarily in the urine, maintaining adequate diuresis is important.
Owing to bleeding risk, dabigatran should be stopped before elective surgery. For patients with normal renal function (CrCl 50 mL/min or higher), dosing should stop 1 or 2 days before surgery. For patients with renal impairment (CrCl below 50 mL/min), dosing should stop 3 to 5 days before surgery.
GI Disturbances.
About 35% of patients experience dyspepsia (abdominal pain, bloating, nausea, vomiting) and/or gastritis-like symptoms (esophagitis, gastroesophageal reflux disease, gastric hemorrhage, erosive gastritis, hemorrhagic gastritis, GI ulcer). Symptoms of dyspepsia can be reduced by taking dabigatran with food and by using an acid-suppressing drug (proton pump inhibitor or histamine-2 receptor blocker). If these measures don’t help, patients may try a switch to warfarin, which carries a much lower risk for adverse GI effects.
Drug Interactions
Dabigatran is not metabolized by hepatic P450 enzymes, nor is it an inhibitor or inducer of these enzymes. Accordingly, dabigatran does not have metabolic interactions with other drugs.
Dabigatran etexilate is a substrate for intestinal P-glycoprotein, the transporter protein that can pump dabigatran and other drugs back into the intestine. Drugs that inhibit P-glycoprotein can increase dabigatran absorption and blood levels, and drugs that induce P-glycoprotein can decrease dabigatran absorption and blood levels. Combined use with a P-glycoprotein inhibitor (e.g., ketoconazole, amiodarone, verapamil, quinidine) could cause bleeding from excessive dabigatran levels, and hence these combinations should be avoided. Combined use with a P-glycoprotein inducer appears to be safe, even though it might reduce beneficial effects somewhat.
Bleeding risk is increased by other drugs that impair hemostasis.
Preparations, Dosage, Administration, and Storage
Preparations.
Dabigatran etexilate [Pradaxa] is available in three strengths: 75, 110, and 150-mg capsules.
Administration.
Dosing may be done with or without food. Patients should swallow the capsules intact. If the capsules are crushed, chewed, or opened, absorption will be increased by 75%, thereby posing a risk for bleeding.
Dosage for Atrial Fibrillation.
The usual dosage is 150 mg twice daily. If a dose is missed, it should be taken as soon as possible on the same day. However, if the missed dose cannot be taken at least 6 hours before the next scheduled dose, the missed dose should be skipped.
In patients with significant renal impairment (CrCl 15–30 mL/min), the dosage is 75 mg twice a day. For patients with greater renal impairment (CrCl below 15 mL/min), no dosing recommendation can be made.
Switching From Warfarin to Dabigatran.
Discontinue warfarin, wait until the INR falls below 2, and then start dabigatran.
Switching From Dabigatran to Warfarin.
Because onset of warfarin’s effects is delayed, warfarin should be started before stopping dabigatran, based on CrCl as follows:
• CrCl above 50 mL/min—start warfarin 3 days before stopping dabigatran.
• CrCl 31 to 50 mL—start warfarin 2 days before stopping dabigatran.
• CrCl 15 to 30 mL—start warfarin 1 day before stopping dabigatran.
Storage.
Dabigatran is unstable, especially when exposed to moisture. To maintain efficacy, the drug must be stored in the manufacturer-supplied bottle, which has a desiccant cap. Patients should open just one bottle at a time and should not distribute dabigatran to any other container, such as a weekly pill organizer. Current labeling says that, after the bottle is opened, dabigatran should be used within 30 days. However, recent evidence indicates that dabigatran capsules maintain efficacy for 4 months, provided they are stored in the original container—away from excessive moisture, heat, and cold—with the cap tightly closed after each use.
Hirudin Analogs
Desirudin
Desirudin [Iprivask] is a direct thrombin inhibitor given by subQ injection. Desirudin is indicated for prevention of DVT in patients undergoing elective hip replacement surgery. In clinical trials, patients experienced fewer thromboembolic events than those given unfractionated heparin or enoxaparin, an LMW heparin.
Desirudin is completely absorbed after subQ injection, achieving peak plasma levels in 1 to 3 hours. Elimination is primarily by renal excretion and partly by proteolytic cleavage. In patients with normal renal function, the elimination half-life is 2 to 3 hours. By contrast, in those with severe renal impairment, the half-life is greatly prolonged (up to 12 hours).
As with other anticoagulants, hemorrhage is the adverse effect of greatest concern. In clinical trials, the incidence of hemorrhage was 30% in the desirudin group compared with 33% in the enoxaparin group and 20% in the heparin group. Less serious effects include wound secretion, injection-site mass, anemia, nausea, and deep thrombophlebitis.
In patients undergoing spinal or epidural anesthesia, desirudin may cause spinal or epidural hematoma, which can result in long-term or even permanent paralysis. Hematoma risk is increased by use of other drugs that impair hemostasis (e.g., nonsteroidal antiinflammatory drugs [NSAIDs], antiplatelet drugs, warfarin, heparin). Patients should be monitored for signs of neurologic impairment and given immediate treatment if they develop.
Desirudin [Iprivask] is administered by deep subQ injection into the thigh or abdominal wall. For patients with normal renal function, the dosage is 15 mg every 12 hours, beginning 5 to 15 minutes before hip surgery (but after induction of regional block anesthesia, if used). For patients with moderate renal impairment (CrCl 30–50 mL/min), dosage is reduced to 5 mg every 12 hours. For those with severe renal impairment (CrCl below 30 mL/min), dosage is reduced to 1.7 mg every 12 hours. For all patients, the usual duration of treatment is 9 to 12 days.
Direct Factor Xa Inhibitors
Rivaroxaban
Actions and Uses
Rivaroxaban [Xarelto] is an oral anticoagulant that causes selective inhibition of factor Xa (activated factor X). Unlike fondaparinux, which acts indirectly (see earlier), rivaroxaban binds directly with the active center of factor Xa and thereby inhibits production of thrombin. Compared with warfarin, our oldest oral anticoagulant, rivaroxaban has several advantages: rapid onset, fixed dosage, lower bleeding risk, few drug interactions, and no need for INR monitoring. Rivaroxaban has three approved uses: (1) prevention of DVT and PE after total hip or knee replacement surgery, (2) prevention of stroke in patients with atrial fibrillation, and (3) treatment of DVT and PE unrelated to orthopedic surgery. Contrasts with warfarin are shown in Table 44.6.
Clinical Trials
Knee and Hip Replacement Patients.
In a series of trials known as RECORD (Regulation of Coagulation in Orthopedic Surgery to Prevent Deep Vein Thrombosis and Pulmonary Embolism), rivaroxaban was compared with enoxaparin (an LMW heparin) in patients who had undergone hip or knee replacement surgery. Patients who received rivaroxaban (10 mg once daily) were much less likely to experience DVT, VTE, PE, or death compared with patients who received enoxaparin (40 mg once daily or 30 mg twice daily). With both drugs, the incidence of major bleeding episodes was low (0.2%).
Nonvalvular Atrial Fibrillation Patients.
In a trial known as ROCKET AF, rivaroxaban was compared with warfarin for preventing stroke in patients with nonvalvular atrial fibrillation (i.e., patients with atrial fibrillation who do not have a prosthetic heart valve or hemodynamically significant valve disease). Rivaroxaban was at least as effective as warfarin and carried the same risk for major hemorrhagic events of all kinds—but had a lower risk for intracranial bleeds and fatal bleeds.
Pharmacokinetics
Rivaroxaban is administered orally, and bioavailability is high (80%–90%). Plasma levels peak 2 to 4 hours after dosing. Protein binding in blood is substantial (92%–95%). Rivaroxaban undergoes partial metabolism by CYP3A4 (the 3A4 isoenzyme of cytochrome P450) and is a substrate for P-glycoprotein, an efflux transporter that helps remove rivaroxaban from the body. Rivaroxaban is eliminated in the urine (36% as unchanged drug) and feces (7% as unchanged drug), with a half-life of 5 to 9 hours. In patients with renal impairment or hepatic impairment, rivaroxaban levels may accumulate.
Adverse Effects
Bleeding.
Bleeding is the most common adverse effect and can occur at any site. Patients have experienced epidural hematoma as well as major intracranial, retinal, adrenal, and GI bleeds. Some people have died. Bleeding risk is increased by other drugs that impede hemostasis. How does rivaroxaban compare with warfarin? The risk for hemorrhagic stroke and other major bleeds is significantly lower with rivaroxaban.
In the event of overdose, we have no specific antidote to reverse this drug’s anticoagulant effects. However, we can prevent further absorption of ingested rivaroxaban with activated charcoal. Treatment with several agents—recombinant factor VIIa, prothrombin complex concentrate (PCC), or activated PCC—can be considered. Preliminary studies of PCC have been promising, but more testing must be completed. Because rivaroxaban is highly protein bound, dialysis is unlikely to remove it from the blood.
Spinal or Epidural Hematoma.
Like all other anticoagulants, rivaroxaban poses a risk for spinal or epidural hematoma in patients undergoing spinal puncture or epidural anesthesia. Prolonged or permanent paralysis can result. Rivaroxaban should be discontinued at least 18 hours before removing an epidural catheter; after the catheter is out, another 6 hours should elapse before rivaroxaban is restarted. If a traumatic puncture occurs, rivaroxaban should be delayed for at least 24 hours. Anticoagulant-related spinal or epidural hematoma was discussed further earlier (see “Adverse Effects” under “Heparin”).
Drug Interactions.
Levels of rivaroxaban can be altered by drugs that inhibit or induce CYP3A4 and P-glycoprotein. Specifically, in patients with normal renal function, drugs that inhibit CYP3A4 strongly and also inhibit P-glycoprotein (e.g., ketoconazole, itraconazole, ritonavir) can raise rivaroxaban levels enough to increase the risk for bleeding. Similarly, in patients with renal impairment, drugs that inhibit CYP3A4 moderately and also inhibit P-glycoprotein (e.g., amiodarone, dronedarone, quinidine, diltiazem, verapamil, ranolazine, macrolide antibiotics) can raise rivaroxaban levels enough to increase the risk for bleeding. Conversely, drugs that induce CYP3A4 strongly and also induce P-glycoprotein (e.g., carbamazepine, phenytoin, rifampin, St. John’s wort) may reduce rivaroxaban levels enough to increase the risk for thrombotic events. Of note, rivaroxaban itself does not inhibit or induce cytochrome P450 enzymes or P-glycoprotein and hence is unlikely to alter the effects of other drugs.
Owing to the risk for bleeding, rivaroxaban should not be combined with other anticoagulants. Concurrent use with antiplatelet drugs and fibrinolytics should be done with caution.
Precautions
Renal Impairment.
Renal impairment can delay excretion of rivaroxaban and can thereby increase the risk for bleeding. Accordingly, rivaroxaban should be avoided in patients with severe renal impairment, indicated by a CrCl below 30 mL/minute. In patients with moderate renal impairment (CrCl 30–50 mL/min), rivaroxaban should be used with caution. If renal failure develops during treatment, rivaroxaban should be discontinued.
Hepatic Impairment.
In clinical trials, rivaroxaban levels and anticoagulation were excessive in patients with moderate hepatic impairment. Accordingly, in patients with moderate or severe hepatic impairment, rivaroxaban should not be used.
Pregnancy.
Rivaroxaban appears unsafe in pregnancy. The drug increases the risk for pregnancy-related hemorrhage and may have detrimental effects on the fetus. When pregnant rabbits were given high doses (10 mg/kg or more) during organogenesis, rivaroxaban increased fetal resorption, decreased fetal weight, and decreased the number of live fetuses. However, dosing of rats and rabbits early in pregnancy was not associated with gross fetal malformations. Rivaroxaban is classified in FDA Pregnancy Risk Category C and should be used only if the benefits are deemed to outweigh the risks to the mother and fetus.
Preparations, Dosage, and Administration
Rivaroxaban [Xarelto] is supplied in tablets (10, 15, and 20 mg). Whether dosing is done with food depends on the setting, as discussed later.
Prevention of DVT.
The recommended dosage is 10 mg once a day, with or without food, starting 6 to 10 hours after knee or hip replacement surgery. If a dose is missed, it should be taken as soon as possible, and the next dose should be taken as originally scheduled. Treatment duration is 12 days after knee replacement and 35 days after hip replacement.
Nonvalvular Atrial Fibrillation.
Dosing is done once a day with the evening meal. For patients with normal renal function, the dosage is 20 mg once daily, and for patients with moderate renal impairment, the dosage is 15 mg once daily. Patients with severe renal impairment should not use this drug.
Treatment of DVT/PE.
Dosing is started at 15 mg twice daily for the first 21 days, and then increased to 20 mg daily. Doses should be taken at approximately the same time each day.
Apixaban
Actions and Uses
Apixaban [Eliquis] is an additional oral anticoagulant that causes selective inhibition of factor Xa. Apixaban inhibits free and clot-bound factor Xa as well as prothrombinase activity. Apixaban has three approved uses: (1) prevention of stroke and systemic embolism in patients with nonvalvular atrial fibrillation, (2) treatment of DVT and PE, and (3) prophylaxis of DVT in patients undergoing hip or knee replacement.
Pharmacokinetics
Apixaban is administered orally, and bioavailability is moderate (~50%). Plasma levels peak 2 to 4 hours after dosing. Protein binding in blood is substantial (87%). Apixaban undergoes partial metabolism by CYP3A4. Apixaban is eliminated in the urine and feces, with a half-life of 12 hours after repeated dosing. In patients with renal impairment, apixaban levels may accumulate.
Adverse Effects
Bleeding.
As with rivaroxaban, bleeding is the most common adverse effect and can occur at any site. Bleeding risk is increased by other drugs that impede hemostasis. How does apixaban compare with warfarin? The risk for hemorrhagic stroke and other major bleeds is significantly lower with apixaban.
In the event of overdose, we have no specific antidote to reverse this drug’s anticoagulant effects. Treatment with several agents—recombinant factor VIIa, PCC, or activated PCC—can be considered, but testing has not been completed. Like rivaroxaban, apixaban is highly protein bound. Dialysis is unlikely to remove it from the blood.
Drug Interactions
Levels of rivaroxaban can be altered by drugs that inhibit or induce CYP3A4 and P-glycoprotein. Specifically, in patients with normal renal function, drugs that inhibit CYP3A4 strongly and also inhibit P-glycoprotein (e.g., ketoconazole, itraconazole, ritonavir) can raise apixaban levels enough to increase the risk for bleeding. Conversely, drugs that induce CYP3A4 strongly and also induce P-glycoprotein (e.g., carbamazepine, phenytoin, rifampin, St. John’s wort) may reduce apixaban levels enough to increase the risk for thrombotic events.
Precautions
Renal Impairment.
Renal impairment can delay excretion of apixaban, increasing the risk for bleeding. In patients with renal impairment, defined as a serum creatinine level greater than or equal to 1.5 mg/dL, apixaban dosing is decreased.
Pregnancy.
Studies of apixaban in pregnant patients are lacking. The drug may increase the risk for hemorrhage during pregnancy and delivery. Apixaban is classified in FDA Pregnancy Risk Category B.
Preparations, Dosage, and Administration
Apixaban [Eliquis] is supplied in tablets (2.5 and 5 mg). The recommended dose for most patients with atrial fibrillation is 5 mg taken orally twice daily. In patients with renal impairment, dosing is decreased to 2.5 mg twice daily. For the treatment of DVT, the dose is doubled to 10 mg twice daily. For prophylaxis after orthopedic surgery, the dose is only 2.5 mg twice daily.
Edoxaban
Edoxaban [Savaysa] is a newer oral anticoagulant that also causes selective inhibition of factor Xa. Edoxaban has two approved uses: (1) prevention of stroke and systemic embolism in patients with nonvalvular atrial fibrillation and (2) treatment of DVT or PE. Because it is a novel oral anticoagulant, like apixaban and rivaroxaban, edoxaban causes adverse effects and has drug interactions similar to these drugs.
Preparations, Dosage, and Administration
Edoxaban is available in 15-, 30-, and 60-mg tablets. The suggested dose for patients with atrial fibrillation is 60 mg orally daily. Treatment for DVT and PE is weight based. For patients <60 kg, the dose is 30 mg daily. For patients >60 kg, this dose is doubled to 60 mg daily. As with the other NOACs, doses should be decreased in patients with renal impairment.
Antiplatelet Drugs
Antiplatelet drugs suppress platelet aggregation. Because a platelet core constitutes the bulk of an arterial thrombus, the principal indication for the antiplatelet drugs is prevention of thrombosis in arteries. In contrast, the principal indication for anticoagulants (e.g., heparin, warfarin) is prevention of thrombosis in veins.
There are four major groups of antiplatelet drugs: aspirin (a “group” with one member), P2Y12 ADP receptor antagonists, PAR-1 antagonists, and GP IIb/IIIa receptor antagonists. As indicated in Fig. 44.1, aspirin and the P2Y12 ADP receptor antagonists affect only one pathway in platelet activation, and hence their antiplatelet effects are limited. In contrast, the GP IIb/IIIa antagonists block the final common step in platelet activation and hence have powerful antiplatelet effects. Properties of the major classes of antiplatelet drugs are shown in Table 44.7. Because GP IIb/IIIa antagonists are given only in the hospital setting, they are discussed further in Chapter 89.
TABLE 44.7
Properties of the Major Classes of Antiplatelet Drugs
| Aspirin, a Cyclooxygenase Inhibitor | P2Y12 ADP Receptor Blockers | PAR-1 Antagonists | |
| Representative drug | Aspirin | Clopidogrel [Plavix] | Vorapaxar [Zontivity] |
| Mechanism of antiplatelet action | Irreversibly inhibits cyclooxygenase and thereby blocks synthesis of thromboxane A2 | Irreversibly blocks receptors for ADP* | Reversibly blocks the PAR-1 expressed on platelets |
| Route | PO | PO | PO |
| Duration of effects | Effects persist 7–10 days after the last dose | Effects persist 7–10 days after the last dose* | Effects persist 7–10 days after the last dose |
| Cost | $3/month | $87/month | $320/month |
Aspirin
The basic pharmacology of aspirin is discussed in Chapter 55. Consideration here is limited to aspirin’s role in preventing arterial thrombosis.
Mechanism of Antiplatelet Action
Aspirin suppresses platelet aggregation by causing irreversible inhibition of cyclooxygenase, an enzyme required by platelets to synthesize TXA2. As noted, TXA2 is one of the factors that can promote platelet activation. In addition to activating platelets, TXA2 acts on vascular smooth muscle to promote vasoconstriction. Both actions promote hemostasis. By inhibiting cyclooxygenase, aspirin suppresses both TXA2-mediated vasoconstriction and platelet aggregation, thereby reducing the risk for arterial thrombosis. Because inhibition of cyclooxygenase by aspirin is irreversible, and because platelets lack the machinery to synthesize new cyclooxygenase, the effects of a single dose of aspirin persist for the life of the platelet (7–10 days).
In addition to inhibiting the synthesis of TXA2, aspirin can inhibit synthesis of prostacyclin by the blood vessel wall. Because prostacyclin has effects that are exactly opposite to those of TXA2—namely, suppression of platelet aggregation and promotion of vasodilation—suppression of prostacyclin synthesis can partially offset the beneficial effects of aspirin therapy. Fortunately, aspirin is able to inhibit synthesis of TXA2 at doses that are lower than those needed to inhibit synthesis of prostacyclin. Accordingly, if we keep the dosage of aspirin low (325 mg/day or less), we can minimize inhibition of prostacyclin production while maintaining inhibition of TXA2 production.
Indications for Antiplatelet Therapy
Antiplatelet therapy with aspirin has multiple indications of proven efficacy:
• Ischemic stroke (to reduce the risk for death and nonfatal stroke)
• TIAs (to reduce the risk for death and nonfatal stroke)
• Chronic stable angina (to reduce the risk for MI and sudden death)
• Unstable angina (to reduce the combined risk for death and nonfatal MI)
• Coronary stenting (to prevent reocclusion)
• Acute MI (to reduce the risk for vascular mortality)
• Previous MI (to reduce the combined risk for death and nonfatal MI)
• Primary prevention of MI (to prevent a first MI in men and in women aged 65 and older)
In all of these situations, prophylactic therapy with aspirin can reduce morbidity, and possibly mortality. Primary prevention of MI is discussed further next.
Primary Prevention of MI
In 2009, the U.S. Preventive Services Task Force (USPSTF) issued updated guidelines on the use of aspirin for primary prevention of MI. The USPSTF recommends the use of aspirin for men ages 45 to 79 years and women ages 55 to 79 years when the potential benefit of a reduction in MI outweighs the potential harm of an increase in GI hemorrhage. Cardiovascular risk is based on five factors—age, gender, cholesterol levels, blood pressure, and smoking status—and can be calculated using an online risk assessment tool, such as those at www.med-decisions.com. Although the optimal aspirin dosage for primary prevention is unknown, low doses (e.g., 81 mg/day) appear as effective as higher ones.
Adverse Effects
Even in low doses, aspirin increases the risk for GI bleeding and hemorrhagic stroke. Among middle-aged people taking aspirin for 5 years, the estimated rate of major GI bleeding episodes is 2 to 4 per 1000 patients, and the rate of hemorrhagic stroke is 0 to 2 episodes per 1000 patients. Use of enteric-coated or buffered aspirin may not reduce the risk for GI bleeding. Benefits of treatment must be weighed against bleeding risks. If GI bleeding occurs, adding a proton pump inhibitor (e.g., omeprazole [Prilosec]) to reduce gastric acidity can help.
Dosing
Dosage for preventing cardiovascular (CV) events should be low. Maximal inhibition of platelet cyclooxygenase, and hence maximal effects on platelet function, can be produced in a few days by taking 81 mg/day. Dosages higher than 81 mg/day offer no increase in benefits but do increase the risk for GI bleeding and stroke. Accordingly, for chronic therapy, a dosage of 81 mg/day is probably adequate. A higher dosage (e.g., 325 mg/day) is indicated for initial treatment of an acute event, such as MI, to establish full antiplatelet effects rapidly—after which 81 mg/day can be taken for maintenance.
P2Y12 Adenosine Diphosphate Receptor Antagonists
Drugs in this class block P2Y12 ADP receptors on the platelet surface, preventing ADP-stimulated aggregation (see Fig. 44.1). Three P2Y12 ADP receptor antagonists are available. Two of them—clopidogrel and prasugrel—cause irreversible receptor blockade, and the third—ticagrelor—causes reversible receptor blockade. Clopidogrel, prasugrel, and ticagrelor are used for secondary prevention of atherothrombotic events in patients with acute coronary syndrome (ACS), defined as unstable angina or MI. All three drugs are taken orally and can cause serious bleeding.
Clopidogrel
Clopidogrel [Plavix] is an oral antiplatelet drug with effects much like those of aspirin. The drug is taken to prevent stenosis of coronary stents and for secondary prevention of MI, ischemic stroke, and other vascular events.
Antiplatelet Actions
Clopidogrel blocks P2Y12 ADP receptors on platelets and thereby prevents ADP-stimulated platelet aggregation. As with aspirin, antiplatelet effects are irreversible and hence persist for the life of the platelet. Effects begin 2 hours after the first dose and plateau after 3 to 7 days of treatment. At the recommended dosage, platelet aggregation is inhibited by 40% to 60%. Platelet function and bleeding time return to baseline 7 to 10 days after the last dose.
Pharmacokinetics
Clopidogrel is rapidly absorbed from the GI tract, both in the presence and absence of food. Bioavailability is about 50%. Clopidogrel is a prodrug that undergoes metabolism to its active form, primarily by hepatic CYP2C19 (the 2C19 isoenzyme of cytochrome P450). People with variant forms of the CYP2C19 gene are poor metabolizers of clopidogrel and hence may not benefit adequately from the drug.
Therapeutic Use
Clopidogrel is used widely to prevent blockage of coronary artery stents and to reduce thrombotic events—MI, ischemic stroke, and vascular death—in patients with ACS and in those with atherosclerosis documented by recent MI, recent stroke, or established peripheral arterial disease. In patients with ACS, clopidogrel should always be combined with aspirin (75–325 mg once daily).
 Black Box Warning: Clopidogrel
Black Box Warning: Clopidogrel
Clopidogrel should not be used in poor metabolizers. Patients with variant forms of the CYP2C19 gene cannot reliably convert clopidogrel to its active form. When treated with standard dosages of clopidogrel, these poor metabolizers exhibit a higher rate of cardiovascular events compared with normal metabolizers.
Poor metabolizers can be identified by testing a blood or saliva sample for CYP2C19 variants or by simply measuring the platelet response to treatment. Unfortunately, even with this information, the course of action is not clear. Yes, we could give poor metabolizers higher doses—but doses that might be safe and effective have not been established. As an alternative, poor metabolizers could be treated with either prasugrel [Effient] or ticagrelor [Brilinta], two other P2Y12 ADP receptor antagonists discussed later.
Adverse Effects
Clopidogrel is generally well tolerated. Adverse effects are about the same as with aspirin. The most common complaints are abdominal pain, dyspepsia, diarrhea, and rash.
Bleeding.
Like all other antiplatelet drugs, clopidogrel poses a risk for serious bleeding. However, compared with aspirin, clopidogrel causes less GI bleeding (2% vs. 2.7%) and less intracranial hemorrhage (ICH) (0.4% vs. 0.5%). Owing to bleeding risk, clopidogrel should be discontinued 5 days before elective surgery. If possible, major bleeding should be managed without discontinuing clopidogrel because discontinuation would increase the risk of a thrombotic event.
Patients should be told about the risk for bleeding and warned that they may bruise or bleed more easily and that bleeding will take longer than usual to stop. Also, patients should be informed about signs of bleeding (e.g., blood in the urine, black tarry stools, vomitus that looks like coffee grounds) and instructed to contact the prescriber if these develop. Finally, patients who develop these symptoms should be warned not to stop clopidogrel until the prescriber says they should.
Thrombotic Thrombocytopenic Purpura (TTP).
Rarely, patients develop TTP, a potentially fatal condition characterized by thrombocytopenia, hemolytic anemia, neurologic symptoms, renal dysfunction, and fever. Most cases occur during the first 2 weeks of treatment. TTP is a serious disorder that requires urgent treatment, including plasmapheresis.
Drug Interactions
Drugs That Promote Bleeding.
Clopidogrel should be used with caution in patients taking other drugs that promote bleeding (e.g., heparin, warfarin, aspirin, and NSAIDs).
Proton Pump Inhibitors (PPIs).
Omeprazole [Prilosec, Losec  ] and other PPIs suppress secretion of gastric acid (see Chapter 62) and hence are often combined with clopidogrel to protect against GI bleeding. Unfortunately, PPIs may also reduce the antiplatelet effects of clopidogrel. PPIs inhibit CYP2C19, the enzyme that converts clopidogrel to its active form. Hence the dilemma: if clopidogrel is used alone, there is a significant risk for GI bleeding; however, if clopidogrel is combined with a PPI to reduce the risk for GI bleeding, antiplatelet effects may be reduced as well. After considering the available evidence, three organizations—the American College of Cardiology, the American Heart Association, and the American College of Gastroenterology—issued a consensus document on the problem. This document concludes that, although PPIs may reduce the antiplatelet effects of clopidogrel somewhat, there is no evidence that the reduction is large enough to be clinically relevant. Accordingly, for patients who have risk factors for GI bleeding (e.g., advanced age, use of NSAIDs or anticoagulants), the benefits of combining a PPI with clopidogrel probably outweigh any risk from reduced antiplatelet effects—and hence combining a PPI with clopidogrel is probably acceptable for these people. Conversely, for patients who lack risk factors for GI bleeding, combined use of clopidogrel with a PPI may reduce the benefits of clopidogrel without offering any meaningful GI protection—and hence combining a PPI with clopidogrel in these patients should probably be avoided. When a PPI is used with clopidogrel, pantoprazole [Protonix] would be a good choice because, compared with other PPIs, pantoprazole causes less inhibition of CYP2C19.
] and other PPIs suppress secretion of gastric acid (see Chapter 62) and hence are often combined with clopidogrel to protect against GI bleeding. Unfortunately, PPIs may also reduce the antiplatelet effects of clopidogrel. PPIs inhibit CYP2C19, the enzyme that converts clopidogrel to its active form. Hence the dilemma: if clopidogrel is used alone, there is a significant risk for GI bleeding; however, if clopidogrel is combined with a PPI to reduce the risk for GI bleeding, antiplatelet effects may be reduced as well. After considering the available evidence, three organizations—the American College of Cardiology, the American Heart Association, and the American College of Gastroenterology—issued a consensus document on the problem. This document concludes that, although PPIs may reduce the antiplatelet effects of clopidogrel somewhat, there is no evidence that the reduction is large enough to be clinically relevant. Accordingly, for patients who have risk factors for GI bleeding (e.g., advanced age, use of NSAIDs or anticoagulants), the benefits of combining a PPI with clopidogrel probably outweigh any risk from reduced antiplatelet effects—and hence combining a PPI with clopidogrel is probably acceptable for these people. Conversely, for patients who lack risk factors for GI bleeding, combined use of clopidogrel with a PPI may reduce the benefits of clopidogrel without offering any meaningful GI protection—and hence combining a PPI with clopidogrel in these patients should probably be avoided. When a PPI is used with clopidogrel, pantoprazole [Protonix] would be a good choice because, compared with other PPIs, pantoprazole causes less inhibition of CYP2C19.
CYP2C19 Inhibitors (Other Than PPIs).
Like the PPIs, several other drugs can inhibit CYP2C19. Among these are cimetidine, fluoxetine, fluvoxamine, fluconazole, ketoconazole, voriconazole, etravirine, felbamate, and ticlopidine. Because these drugs may reduce the antiplatelet effects of clopidogrel (by reducing its activation), use of alternative drugs is preferred.
Preparations, Dosage, and Administration
Clopidogrel [Plavix] is available in 75-mg tablets. The usual maintenance dosage is 75 mg once a day, taken with or without food. A 300-mg loading dose may be used for some patients. The optimal duration of treatment is unknown. Dosage needn’t be changed for older-adult patients or those with renal impairment. Patients being treated for ACS should take daily aspirin (75–325 mg). Clopidogrel should be withdrawn 5 days before elective surgery and then resumed as soon as possible.
Prasugrel
Actions and Uses
Prasugrel [Effient], a close relative of clopidogrel, is an oral antiplatelet drug approved for prevention of thrombotic events in patients with ACS. Like clopidogrel, prasugrel is a prodrug that undergoes conversion to an active metabolite, which then blocks P2Y12 ADP receptors on platelets, causing irreversible inhibition of platelet aggregation. Prasugrel is more effective than clopidogrel and has fewer drug interactions but causes more major bleeding.
Clinical Trial
In a trial known as TRITON-TIMI, prasugrel was compared directly with clopidogrel. The trial enrolled more than 13,000 patients with ACS who were scheduled for coronary angioplasty, also known as percutaneous coronary intervention. The goal with both drugs was to prevent thrombotic complications, including stent restenosis. Patients taking prasugrel experienced fewer thrombotic events but more major bleeding.
Pharmacokinetics
Prasugrel is rapidly absorbed after oral dosing, in both the presence and absence of food. Activation takes place in two steps. The process begins with hydrolysis by esterases in the intestine and ends with conversion to the active metabolite in the liver, primarily by CYP3A4 and CYP2B6, two isoenzymes of cytochrome P450. (Note that activation of prasugrel differs from activation of clopidogrel, which is mediated by CYP2C19.) The entire activation process is fast: plasma levels of the active metabolite peak about 30 minutes after dosing. Elimination of the active form is primarily by hepatic metabolism, followed by excretion in the urine and feces. The active metabolite has a half-life of 7 hours. In patients who weigh less than 60 kg, total exposure to the active metabolite is 30% to 40% higher than in heavier patients. Accordingly, these lighter patients may need a dosage reduction.
Adverse Effects
The principal adverse effect is bleeding, which occurs more often with prasugrel than with clopidogrel. According to results of TRITON-TIMI, among patients not undergoing coronary artery bypass surgery (CABG), the incidence of major bleeding was 2.4% with prasugrel versus 1.8% with clopidogrel, and the incidence of life-threatening bleeding was 0.4% versus 0.1%. Among patients who required CABG surgery, the incidence of major bleeding was greatly increased: 18.8% with prasugrel versus 2.7% with clopidogrel. Accordingly, if CABG surgery is anticipated, prasugrel should not be started. Prasugrel should be avoided by patients at increased risk for bleeding, including patients with active pathologic bleeding, patients older than 75 years, and patients with a history of TIAs or stroke. If possible, major bleeding should be managed without discontinuing prasugrel because discontinuation would increase the risk for a thrombotic event.
Rarely, patients experience hypersensitivity reactions, including potentially life-threatening angioedema. Onset may occur within hours of the first dose, or after 5 to 10 days of treatment.
Data from TRITON-TIMI suggest that prasugrel may increase the risk for cancer. Among patients using the drug, there was a 62% increase in the rate of new and worsening solid tumors. However, we don’t know of any plausible mechanism of tumor promotion. The FDA is monitoring for more cancer cases.
Drug Interactions
Other drugs that promote bleeding (e.g., warfarin, heparin, fibrinolytic drugs, chronic NSAIDs) will increase the risk for a serious bleed and hence should be used with great caution. PPIs, which may slow the activation of clopidogrel (by inhibiting CYP2C19), do not prevent the activation of prasugrel. Also, according to the prasugrel package insert, drugs that induce or inhibit CYP3A4 do not have a significant impact on prasugrel activity.
Preparations, Dosage, and Administration
Prasugrel [Effient] is supplied in 5- and 10-mg tablets for oral dosing, with or without food. Treatment consists of a 60-mg loading dose, followed by once-daily 10-mg maintenance doses. For patients who weigh less than 60 kg, maintenance doses may be reduced to 5 mg. All patients should take aspirin daily (80–325 mg).
Ticagrelor
Actions and Uses
Ticagrelor [Brilinta] is a P2Y12 ADP receptor antagonist indicated for prevention of thrombotic events in patients with ACS and to prevent further CV events in patients with history of MI. The drug inhibits platelet aggregation by blocking P2Y12 ADP receptors on the platelet surface. In contrast to clopidogrel and prasugrel, which cause irreversible receptor blockade, ticagrelor causes reversible blockade and hence the effects of ticagrelor wear off faster.
Clinical Trial
In a trial known as PLATO, ticagrelor was compared directly with clopidogrel in patients with recent-onset ACS (within the previous 24 hours). The trial randomized more than 18,000 patients to receive either ticagrelor (180 mg once followed by 90 mg twice daily) or clopidogrel (300 mg once followed by 75 mg once daily). All patients also took a daily aspirin. Compared with clopidogrel, ticagrelor produced a greater reduction in MI, stroke, stent restenosis, and CV death. Unfortunately, these advantages were offset by a greater risk for hemorrhagic events, including fatal intracranial bleeding.
Pharmacokinetics
Ticagrelor is administered by mouth, and food has little effect on absorption. Plasma levels peak 1.5 hours after dosing. Bioavailability is 36%. Unlike clopidogrel and prasugrel, which are prodrugs, ticagrelor is active as administered. In the liver, CYP3A4 converts much of each dose to an active metabolite. Later, the parent drug and active metabolite undergo inactivation by CYP3A4, followed by excretion in the feces (58%) and urine (26%). The elimination half-life is 7 hours for ticagrelor itself and 9 hours for the active metabolite.
Adverse Effects
The most common adverse effects are bleeding and dyspnea. Other adverse effects include headache, cough, dizziness, nausea, noncardiac chest pain, diarrhea, and bradycardia, including ventricular pauses.
Bleeding.
Like all other antiplatelet drugs, ticagrelor poses a risk for serious bleeding. In the PLATO study, serious non-CABG bleeding developed in 4.5% of patients taking ticagrelor, compared with 3.8% of patients taking clopidogrel. However, the incidence of CABG-related bleeding was the same with both drugs. Because of bleeding risk, ticagrelor should be discontinued 5 days before elective surgery and then resumed as soon as possible after the surgery is done.
Dyspnea.
In the PLATO study, dyspnea developed in 13.8% of patients taking ticagrelor, compared with 7.8% of patients taking clopidogrel. Dyspnea was usually mild to moderate, and often resolved despite continued drug use. Ticagrelor-related dyspnea does not require any specific intervention.
Ventricular Pauses.
Ticagrelor can cause ventricular pauses. In the PLATO trial, ventricular pauses were relatively common early in treatment (6% with ticagrelor vs. 3.5% with clopidogrel) but were much less common after 1 month (2.2% with ticagrelor vs. 1.6% with clopidogrel).
 Black Box Warning: Aspirin
Black Box Warning: Aspirin
Aspirin in low doses—75 to 100 mg/day—enhances the effects of ticagrelor. However, higher doses—more than 100 mg/day—actually reduce the benefits of ticagrelor. Accordingly, patients should be warned against taking more than 100 mg of aspirin a day.
Drug Interactions
Drugs That Promote Bleeding.
Anticoagulants (e.g., warfarin, heparin, dabigatran), fibrinolytics (e.g., alteplase, reteplase), and antiplatelet drugs (e.g., aspirin, abciximab) will increase the risk for a serious bleed and hence should be used with great caution.
Inhibitors and Inducers of CYP3A4.
Because ticagrelor and its active metabolite are eliminated by CYP3A4, drugs that induce CYP3A4 (e.g., rifampin, phenytoin, phenobarbital, carbamazepine) can reduce the therapeutic effects of both compounds, and drugs that inhibit CYP3A4 (e.g., ketoconazole, itraconazole, clarithromycin, telithromycin, ritonavir, saquinavir) can increase the risk for toxicity (by allowing both compounds to accumulate to dangerous levels).
Statins.
Ticagrelor inhibits CYP3A4 and can thereby increase levels of simvastatin and lovastatin. To avoid toxicity, dosages of these statins should not exceed 40 mg/day.
Digoxin.
Ticagrelor and its active metabolite can inhibit P-glycoprotein, a transport molecule that promotes renal, hepatic, and intestinal elimination of drugs (see Chapter 4). P-glycoprotein inhibition is of particular concern with digoxin, a heart drug with a low margin of safety. To avoid toxicity, digoxin levels should be checked during initial ticagrelor use and whenever ticagrelor dosage is changed.
Contraindications and Precautions
Ticagrelor is contraindicated for patients with active pathologic bleeding, a history of ICH, or severe hepatic impairment. In patients with moderate hepatic impairment, ticagrelor should be used with caution (because levels of ticagrelor itself and its active metabolite could become excessive, thereby increasing the risk for bleeding).
Preparations, Dosage, and Administration
Ticagrelor [Brilinta] is supplied in 90-mg tablets for dosing with or without food. After an initial loading dose (180 mg) combined with 325 mg of aspirin, patients take 90 mg twice daily. Daily aspirin (75–100 mg) should be continued as well. Doses for patients taking ticagrelor for secondary prevention of CV events should take 60 mg twice daily.
Protease-Activated Receptor-1 (PAR-1) Antagonists
Vorapaxar
Uses.
Vorapaxar [Zontivity] is approved for use in conjunction with aspirin and/or clopidogrel in the reduction of thrombotic CV events in patients with a history of MI or PAD. When used with other antiplatelet agents, vorapaxar reduces the rate of CV death, MI, stroke, and urgent coronary revascularization.
Mechanism of Action.
PAR-1 mediates the effects of thrombin, and these receptors are located on the surface of platelets. By reversibly antagonizing these receptors, vorapaxar inhibits thrombin-induced and thrombin receptor agonist peptide (TRAP)-induced platelet aggregation. Vorapaxar does not work on ADP receptors.
Pharmacokinetics.
Vorapaxar is well absorbed after oral administration. Antiplatelet effects begin within 1 hour. The drug undergoes extensive hepatic metabolism followed by excretion in the feces. Vorapaxar has a long half-life (8 days). Effects persist for 7 to 10 days after drug withdrawal (i.e., until new platelets have been synthesized).
Adverse Effects
Like all other antiplatelet drugs, vorapaxar poses a risk for serious bleeding. In the GUSTO study, serious non-CABG bleeding developed in 3% of patients taking vorapaxar. This is similar to the rates seen with ticagrelor in the PLATO trial.
Preparations, Dosage, and Administration.
Vorapaxar is available in 2.08-mg tablets. The recommended dosage is 1 tablet daily with or without food. Vorapaxar should be administered with clopidogrel or aspirin because it has not been studied alone in the prevention of thrombotic CV events.
Other Antiplatelet Drugs
Dipyridamole
Dipyridamole [Persantine] suppresses platelet aggregation, perhaps by increasing plasma levels of adenosine. The drug is approved only for prevention of thromboembolism after heart valve replacement. For this application, dipyridamole is always combined with warfarin. The recommended dosage is 75 to 100 mg 4 times a day. A fixed-dose combination of dipyridamole and aspirin is indicated for recurrent stroke (see later).
Dipyridamole Plus Aspirin
Actions and Use.
Dipyridamole combined with aspirin is available in a fixed-dose formulation sold as Aggrenox. The product is used to prevent recurrent ischemic stroke in patients who have had a previous stroke or TIA. Both drugs—aspirin and dipyridamole—suppress platelet aggregation. However, because they do so by different mechanisms, the combination is more effective than either drug alone.
Clinical Trial.
The benefit of combining aspirin and dipyridamole was demonstrated in the second European Stroke Prevention Study (ESPS-2), a randomized controlled trial that enrolled more than 6000 patients who had suffered a prior ischemic stroke or TIA. Some patients took aspirin alone (25 mg twice daily), some took dipyridamole alone (200 mg twice daily), some took both drugs, and some took placebo. After 24 months, the incidence of fatal or nonfatal ischemic stroke was reduced by 16% with dipyridamole alone, 18% with aspirin alone, and 37% with the combination. Unfortunately, ESPS-2 was tainted by scientific scandal (one investigator, who later resigned, was charged with creating and falsifying data). Although all fraudulent data were discarded before publication, some authorities remain skeptical of the results.
Adverse Effects.
The most common adverse effects of the combination are headache, dizziness, and GI disturbances (nausea, vomiting, diarrhea, abdominal pain, dyspepsia). Of course, bleeding is a concern: the product can cause hemorrhage (3.2% vs. 1.5% with placebo), nosebleed (2.4% vs. 1.5%), and purpura (1.4% vs. 0.4%). The aspirin in Aggrenox poses a risk for GI bleeding from peptic ulcers.
Preparations, Dosage, and Administration.
Aggrenox capsules contain 25 mg of aspirin and 200 mg of extended-release dipyridamole. The recommended dosage is 2 capsules a day—1 in the morning and 1 at night. The cost is about $317 a month, compared with $3 a month for aspirin alone. It is important to note that the daily dose of aspirin (50 mg) is lower than the dose recommended to prevent MI (at least 80 mg/day). Accordingly, supplemental aspirin may be needed.
Cilostazol
Actions and Therapeutic Use
Cilostazol [Pletal], a platelet inhibitor and vasodilator, is indicated for intermittent claudication. (Intermittent claudication is a syndrome characterized by pain, cramping, and weakness of the calf muscles brought on by walking and relieved by resting a few minutes. The underlying cause is atherosclerosis in the legs.) Cilostazol suppresses platelet aggregation by inhibiting phosphodiesterase type 3 (PDE-3) in platelets, and promotes vasodilation by inhibiting PDE-3 in blood vessels (primarily in the legs). Inhibition of platelet aggregation is greater than with aspirin, ticlopidine, or dipyridamole. Full effects take up to 12 weeks to develop but reverse quickly (within 48 hours) after drug withdrawal.
Adverse Effects
Cilostazol causes a variety of untoward effects. The most common is headache (34%). Others include diarrhea, abnormal stools, palpitations, dizziness, and peripheral edema.
Other drugs that inhibit PDE-3 have increased mortality in patients with heart failure. Whether cilostazol represents a risk is unknown. Nonetheless, heart failure is a contraindication to cilostazol use.
Drug and Food Interactions
Cilostazol is metabolized by hepatic CYP3A4, so cilostazol levels can be increased by CYP3A4 inhibitors (e.g., ketoconazole, itraconazole, erythromycin, fluoxetine, fluvoxamine, nefazodone, sertraline, and grapefruit juice). Metabolism of cilostazol can also be inhibited by omeprazole.
Preparations, Dosage, and Administration
Cilostazol [Pletal] is available in 50- and 100-mg tablets. The usual dosage is 100 mg twice daily, taken 30 minutes before or 2 hours after breakfast and the evening meal. Dosage should be reduced to 50 mg twice daily in patients taking omeprazole and drugs or foods that inhibit CYP3A4.

