CHAPTER 62 Anatomy, Histology, Embryology, Developmental Anomalies, and Pediatric Disorders of the Biliary Tract
EMBRYOLOGY OF THE LIVER AND BILIARY TRACT
The human liver is formed from two primordia (Fig. 62-1): the liver diverticulum and the septum transversum.1 Proximity of cardiac mesoderm, which expresses fibroblast growth factors (FGFs) 1, 2, and 8, and bone morphogenetic proteins cause the foregut endoderm to develop into the liver.2 Surrounding mesoderm and ectoderm participate in the hepatic specification of the endoderm, and many transcription factors, such as cJun, retinoblastoma gene, and nuclear factor κB, play important roles as regulators of liver embryogenesis.3 The liver diverticulum forms through proliferation of endodermal cells at the cranioventral junction of the yolk sac with the foregut and grows into the septum transversum in a cranioventral direction.4 The earliest marker of mammalian hepatic differentiation is the endodermal expression of albumin, transthyretin, and alpha fetoprotein. Cells that express these markers are called hepatoblasts, and they differentiate into hepatocytes and epithelial cells of the bile ducts. Signaling mediated by the stress-activated protein kinase (SAPK)/Jun N-terminal kinase (JNK) pathway promotes hepatoblast proliferation as well as survival.5 This early change occurs on the eighteenth day of gestation and corresponds to the 2.5-mm stage of the embryo. The signaling molecules that elicit embryonic induction of the liver from the mammalian gut endoderm or induction of other gut-derived organs are being defined. The homeobox gene Hhex is essential for proper hepatoblast differentiation and bile duct morphogenesis. Members of the GATA, FOXA, ONECUT1, and hepatocyte nuclear factor (HNF)3/forkhead transcription factor families are also required for the formation and differentiation of gut endoderm tissues.3,4 The septum transversum consists of mesenchymal cells and a capillary plexus formed by the branches of the two vitelline veins. At the 3- to 4-mm stage, between the third and fourth weeks of gestation, the growing diverticulum projects as an epithelial plug into the septum transversum.5 The homeodomain transcription factors Hex and Prox1, expressed in the anterior endoderm and hepatic diverticulum, are required for migration of hepatoblasts into the septum transversum that precedes liver growth and morphogenesis.6,7 Another homeodomain protein, Hlx, is necessary for hepatoblast proliferation. At the 5-mm stage, a solid cranial portion (hepatic) and a hollow caudal portion of the diverticulum can be clearly distinguished. The large hepatic portion differentiates into proliferating cords of hepatocytes and the intrahepatic bile ducts. HNF4a expression drives further hepatocyte differentiation and epithelial transformation into the characteristic sinusoidal architecture.8 The smaller, cystic portion, which initially is a cord of epithelial cells, forms the gallbladder, bile duct, and cystic duct through a process of elongation and recanalization.
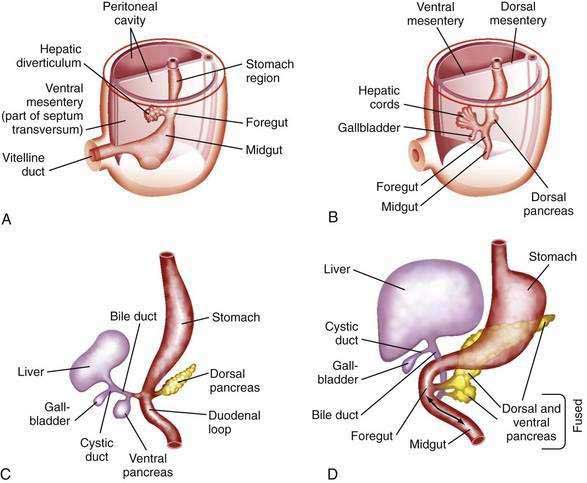
The intrahepatic bile ducts develop from primitive hepatocytes around branches of the portal vein. Cholangiocytes are associated with the basement membrane throughout bile duct development, suggesting that cholangiocytes receive morphogenic signals from components of the extracellular matrix including laminin and type IV collagen.9,10 A ring of hepatocytes in proximity to the portal vein branches first transforms into bile duct–type cells. A second layer of primitive hepatocytes is similarly transformed and produces a circular cleft around the portal vein that is lined on both sides by bile duct epithelial cells.11 This double-walled cylinder with a slit-like lumen, the ductal plate, can be detected at 9 weeks of gestation. Thus, the entire network of interlobular and intralobular bile ductules develops from the limiting plate. The transcription factors Hes1, HNF6, and HNF1β are required for gallbladder and bile duct development.6 The Notch and transforming growth factor-β (TGF-β) signaling pathways are activated in hepatoblasts surrounding the portal veins, allowing hepatoblasts to become cholangiocytes.5 In sections of the 10-mm embryo, many of the liver cords are traversed by double-walled canals that branch and morphologically are indistinguishable from bile capillaries of the adult. These structures differ from those of the adult in that they are bounded by six or more liver cells instead of two. The process of differentiation of bile ductular epithelial cells (cholangiocytes) from primitive hepatocytes has been documented in humans through the use of immunohistochemical staining with several anticytokeratin antibodies. During the phenotypic shift toward bile duct–type cells, hepatocytes first display increased reactivity for cytokeratins 8 and 18 and express cytokeratin 19 at 20 to 25 weeks of gestation.12 Cholangiocyte-mesenchymal cell interaction is important for the formation of bile ducts. During the transition from ductal plates to bile ducts, portal myofibroblasts significantly expand and surround newly formed bile ducts. Periportal connective tissue, corticosteroid hormones, and basal laminar components may play important roles in the differentiation of bile ducts. The ductal plate structure requires extensive remodeling through a process of reabsorption, possibly through apoptosis, to yield the characteristic anastomosing system of biliary channels that surround the portal vein. Proteins that appear to have a role in the promotion of apoptosis, specifically Fas antigen and c-myc, are consistently detected in primitive intrahepatic ductal cells.5 Lewis antigen, which is expressed in damaged and apoptotic cells, is also present. Bcl-2 protein, an inhibitor of apoptosis, is not found in early stages of intrahepatic bile duct cell development but becomes detectable later. Computed three-dimensional reconstruction of the developing ductal plate has shown that the ductal plate remodeling process starts at the porta hepatis at approximately 11 weeks of gestation and progresses toward the periphery of the liver.12 The process is in large part completed at term, but even at 40 weeks of gestation, some of the smallest portal vein branches may not be accompanied by an individual bile duct and may still be surrounded by a (discontinuous) ductal plate. In ductal plate malformation, which occurs in biliary disorders such as congenital hepatic fibrosis and Caroli’s disease (see later), insufficient reabsorption of ductal plates can result in the formation of large dilated segments of a primitive bile duct that surrounds the central portal vein.12
The gallbladder and extrahepatic bile ducts start to develop from hepatic endodermal cells and hepatoblasts immediately after formation of the liver primordium. Foxf1 is critical for mesenchymal epithelial cell induction of gallbladder morphogenesis.6 In embryos 5 to 6 mm in length, the original hepatic diverticulum differentiates cranially into proliferating hepatic cords and bile ducts and caudally into the gallbladder. The cystic portion of the liver diverticulum is hollow initially, but the lumen is filled as cells continually migrate into it. A study in 1994 showed that the primitive extrahepatic bile duct maintains continuity with the ductal plate, from which intrahepatic bile ducts are eventually formed.9,10 Contrary to long-held concepts of biliary development, no “solid stage” of endodermal occlusion of the bile duct lumen is found at any stage of gestation. At 16 mm, the cystic duct and proximal gallbladder are hollow, but the fundus of the gallbladder is still partially obstructed by remnants of the epithelial plug. The gallbladder is patent by the third month of gestation. Further development, until birth, consists primarily of continued growth. The characteristic folds of the gallbladder are formed toward the end of gestation and are moderately developed in the neonate. Bile secretion starts at the beginning of the fourth month of gestation; thereafter, the biliary system continuously contains bile, which is secreted into the gut and imparts a dark green color to the intestinal contents (meconium).
ANATOMY
BILE DUCTS
The adult human liver has more than 2 km of bile ductules and ducts. Quantitative computer-aided three-dimensional imaging has estimated the volume of the entire macroscopic duct system of human liver to be a mean of 20.4 cm.3,13 In these studies the mean internal surface of 398 cm2 is magnified approximately 5.5-fold by the presence of microvilli and cilia at the apical surface of cholangiocytes that play an important role in the regulation of cholangiocyte functions. These structures are far from being inert channels; they are capable of modifying biliary flow and composition significantly in response to hormones such as secretin.14,15 A general feature of bile ductules is their anatomic intimacy with portal blood and lymph vessels, which potentially allows selective exchange of materials between compartments. No major ultrastructural differences exist between cholangiocytes lining small and large bile ducts, but the functional properties of cholangiocytes are heterogeneous.15 For example, large, but not small, intrahepatic bile ducts are involved in secretin-regulated bile ductal secretion.16 Correspondingly, the secretin receptor and chloride-bicarbonate exchanger messenger ribonucleic acids (mRNAs) have been detected in large, but not small, intrahepatic bile duct units.15
Bile secretion begins at the level of the bile canaliculus, the smallest branch of the biliary tree.17 Its boundaries are formed by a specialized membrane of adjacent apical poles of liver cells. The canaliculi form a meshwork of polygonal channels between hepatocytes with many anastomotic interconnections.17 Bile then enters the small terminal channels (the canals of Hering), which have a basement membrane and are lined partly by hepatocytes and partly by cholangiocytes.13 The canals of Hering provide a conduit through which bile may traverse the limiting plate of hepatocytes to enter the larger perilobular or intralobular ducts.18,19 These smallest of biliary radicles are less than 15 to 20 µm in diameter with lumens surrounded by cuboidal epithelial cells. At the most proximal level, one or more fusiform-shaped ductular cells may share a canalicular lumen with a hepatocyte; gradually, the ductules become lined by two to four cuboidal epithelial cells as they approach the portal canal.17 Bile flows from the central lobular cells toward portal triads (from zone 3 to zone 1 of the liver acinus) (see Chapter 71). The terminal bile ductules are thought to proliferate as a result of chronic extrahepatic bile duct obstruction.19
The interlobular bile ducts form a richly anastomosing network that closely surrounds the branches of the portal vein.20–22 These bile ducts (Fig. 62-2) are initially 30 to 40 µm in diameter and are lined by a layer of cuboidal or columnar epithelium that displays a microvillar architecture on its luminal surface.17 The cells have a prominent Golgi apparatus and numerous vesicles that likely participate in the exchange of substances among cytoplasm, bile, and plasma through the processes of exocytosis and endocytosis.17 These ducts increase in caliber and possess smooth muscle fibers within their walls as they approach the hilum of the liver. The muscular component may provide the morphologic basis for the narrowing of the ducts at this level, as observed on cholangiography.22 Furthermore, as the ducts become progressively larger, the epithelium becomes thicker, and the surrounding layer of connective tissue grows thicker and contains many elastic fibers. These ducts anastomose further to form the large hilar, intrahepatic ducts, which are 1 to 1.5 mm in diameter and give rise to the main hepatic ducts.
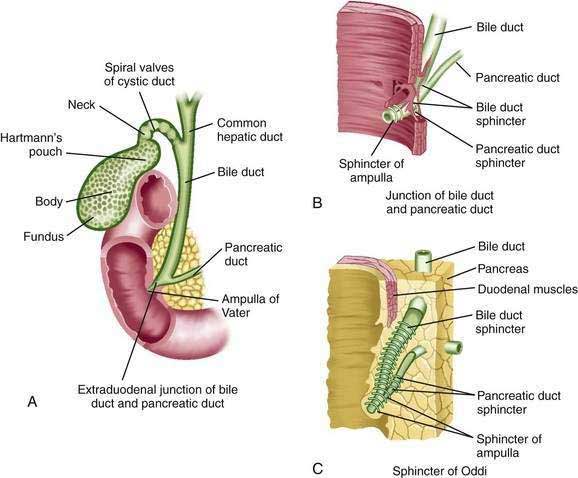
The common hepatic duct emerges from the porta hepatis after the union of the right and left hepatic ducts, each of which is 0.5 to 2.5 cm long (Fig. 62-3).23,24 The confluence of the right and left hepatic ducts is outside the liver in approximately 95% of cases; uncommonly, the ducts merge inside the liver, or the right and left hepatic ducts do not join until the cystic duct joins the right hepatic duct.24 As the hepatic ducts leave the porta hepatis, they lie within the two serous layers of the hepatoduodenal ligament. This sheath of fibrous tissue binds the hepatic ducts to the adjacent blood vessels. In the adult, the common hepatic duct is approximately 3 cm long and is joined by the cystic duct, usually at its right side, to form the bile duct (or common bile duct).24 However, the length and angle of junction of the cystic duct with the common hepatic duct are variable. The cystic duct enters the common hepatic duct directly in 70% of patients; alternatively, the cystic duct may run anterior or posterior to the bile duct and spiral around it before joining the bile duct on its medial side.23 The cystic duct may also course parallel to the common hepatic duct for 5 to 6 cm and enter it after running posterior to the first portion of the duodenum.
In humans, the large intrahepatic bile ducts at the hilum (1- to 1.5-mm diameter) have many irregular side branches and pouches (150- to 270-µm diameter) that are oriented in one plane, corresponding anatomically to the transverse fissure.17 Smaller pouches of the side branches are also found. Many side branches end as blind pouches, but others, particularly at the hilum, communicate with each other. At the bifurcation, side branches from several main bile ducts connect to form a plexus. The functional significance of these structures is not known. The blind pouches may serve to store or modify bile, whereas the biliary plexus provides anastomoses, which may allow exchange of material between the large bile ducts.
The anatomy of the hepatic hilum is particularly important to the surgeon. A plate of fibrous connective tissue in the hepatic hilum includes the umbilical plate that envelops the umbilical portion of the portal vein, the cystic plate in the gallbladder bed, and the Arantian plate that covers the ligamentum venosum.24 Histologic examination of the sagittal section of the hilar plate reveals abundant connective tissue, including neural fibers, lymphatic vessels, small capillaries, and small bile ducts. The bile ducts in the plate system correspond to the extrahepatic bile ducts, and their lengths are variable for every segment.24
Like the intestine, the cystic, common hepatic, and bile ducts possess mucosa, submucosa, and muscularis.22 The ducts are lined by a single layer of columnar epithelium. Mucus secreting tubular glands can be found at regular intervals in the submucosa, with openings to the surface of the mucosa. The bile duct is approximately 7 cm long, runs between layers of the lesser omentum, and lies anterior to the portal vein and to the right of the hepatic artery.24 The bile duct normally is 0.5 to 1.5 cm in diameter.19 The wall of the extrahepatic bile duct is supported by a layer of connective tissue with an admixture of occasional smooth muscle fibers. The smooth muscle component is conspicuous only at the neck of the gallbladder and at the lower end of the bile duct. The bile duct passes retroperitoneally behind the first portion of the duodenum in a notch on the back of the head of the pancreas and enters the second part of the duodenum. The duct then passes obliquely through the posterior medial aspect of the duodenal wall and joins the main pancreatic duct to form the ampulla of Vater (see Fig. 62-3).23 The mucous membrane bulge produced by the ampulla forms an eminence, the duodenal papilla. In approximately 10% to 15% of patients, the bile and pancreatic ducts open separately into the duodenum. The bile duct tapers to a diameter of 0.6 cm or less before its union with the pancreatic duct.24
As they course through the duodenal wall, the bile and pancreatic ducts are invested by a thickening of both the longitudinal and circular layers of smooth muscle (see Fig. 62-3) of the sphincter of Oddi.25 There is considerable variation in this structure, but it is usually composed of several parts: (1) the sphincter choledochus—circular muscle fibers that surround the intramural portion of the bile duct immediately before its junction with the pancreatic duct; (2) the sphincter pancreaticus, which is present in approximately one third of individuals and surrounds the intraduodenal portion of the pancreatic duct before its juncture with the ampulla; (3) the fasciculi longitudinales—longitudinal muscle bundles that span intervals between the bile and pancreatic ducts; and (4) the sphincter ampullae—longitudinal muscle fibers that surround a sparse layer of circular fibers around the ampulla of Vater.22 The sphincter choledochus constricts the lumen of the bile duct and thus prevents the flow of bile. Contraction of the fasciculi longitudinales shortens the length of the bile duct and thus promotes the flow of bile into the duodenum. The contraction of the sphincter ampullae shortens the ampulla and approximates the ampullary folds to prevent reflux of intestinal contents into the bile and pancreatic ducts. When both ducts end in the ampulla, however, contraction of the sphincter may cause reflux of bile into the pancreatic duct.25
The arterial supply of the bile ducts arises mainly from the right hepatic artery.20 An extraordinarily rich plexus of capillaries surrounds bile ducts as they pass through the portal tracts.20,26 Blood flowing through this peribiliary plexus empties into the hepatic sinusoids via the interlobular branches of the portal vein. The peribiliary plexus may modify biliary secretions through the bidirectional exchange of proteins, inorganic ions, and bile acids between blood and bile. Because blood flows in the direction (from the large toward the small ducts) opposite to that of bile flow, the peribiliary plexus presents a countercurrent stream of biliary-reabsorbed substances to hepatocytes.
An abundant anastomotic network of blood vessels from branches of the hepatic and gastroduodenal arteries supplies the bile duct.22,26 The supraduodenal portion of the duct is supplied by vessels running along its wall inferiorly from the retroduodenal artery and superiorly from the right hepatic artery. Injury to these blood vessels can result in bile duct stricturing.23
The lymphatic vessels of the hepatic, cystic, and proximal portions of the bile duct empty into glands at the hilum of the liver.22 Lymphatics draining from the lower portion of the bile duct drain into glands near the head of the pancreas.
GALLBLADDER
The gallbladder (see Fig. 62-3) is a storage reservoir that allows bile acids to be delivered in a high concentration and in a controlled manner to the duodenum for the solubilization of dietary lipid (see Chapter 64).22,27 It lies in a fossa on the undersurface of the right lobe of the liver.27 This distensible pear-shaped structure is 3 cm wide and 7 cm long in the adult and has a capacity of 30 to 50 mL.27 The gallbladder has a thin muscular layer with the smooth muscle cells largely oriented around the circumference of the gallbladder. The absorptive surface of the gallbladder is enhanced by numerous prominent folds. The gallbladder is covered anteriorly by an adventitia that is fused with the capsule of the liver. On its posterior aspect and at the apex, it is covered by the visceral peritoneum. The portions of the gallbladder are the fundus, body, infundibulum, and neck.22 The anterior portion of the fundus is located at the level of the right lateral border of the musculus rectus abdominis and the ninth costal cartilage. The posterior aspects of the fundus and body lie close to the transverse colon and duodenum, respectively. Thus, with perforation of the gallbladder, gallstones can readily penetrate these structures.27,28 The infundibulum is an area of tapering between the gallbladder body and neck. Hartmann’s pouch is a bulging of the inferior surface of the infundibulum that lies close to the neck of the gallbladder. Gallstones can become impacted in Hartmann’s pouch, thereby obstructing the cystic duct and producing cholecystitis.27 Extensive inflammation in Hartmann’s pouch can lead to obstruction of the adjacent common hepatic duct (Mirizzi’s syndrome).
The gallbladder is connected at its neck to the cystic duct, which empties into the bile duct (see Fig. 62-3).27 The cystic duct is approximately 4 cm long and maintains continuity with the surface columnar epithelium, lamina propria, muscularis, and serosa of the gallbladder. The mucous membrane of the gallbladder neck forms the spiral valve of Heister, which is involved in regulating flow into and out of the gallbladder.
The gallbladder is supplied by the cystic artery, which usually arises from the right hepatic artery.27,29 The artery divides into two branches near the neck of the gallbladder: a superficial branch that supplies the serosal surface and a deep branch that supplies the interior layers of the gallbladder wall. Variations in the origin and course of the cystic artery are common.27 Because the cystic artery is an end artery, the gallbladder is particularly susceptible to ischemic injury and necrosis that result from inflammation or interruption of hepatic arterial flow.
The cystic vein provides venous drainage from the gallbladder and cystic ducts and commonly empties into the portal vein and occasionally directly into the hepatic sinusoids.22,27 The lymph vessels of the gallbladder are connected with the lymph vessels of Glisson’s capsule. Subserosal and submucosal lymphatics empty into a lymph gland near the neck of the gallbladder.22 The sympathetic innervation of the gallbladder originates from the celiac axis and travels with branches of the hepatic artery and portal vein. Visceral pain is conducted through sympathetic fibers and is frequently referred to the right subcostal, epigastric, and right scapular regions. Branches of both vagus nerves provide parasympathetic innervation that likely contributes to the regulation of gallbladder motility.22
The gallbladder is lined by a mucosa that manifests multiple ridges and folds and is composed of a layer of columnar epithelial cells. The gallbladder wall consists of a mucosa, lamina propria, tunica muscularis, and serosa.30 The tunica muscularis is thick and invested with an interlocking array of longitudinal and spiral smooth muscle fibers. Tubuloalveolar glands are found in the region of the neck of the gallbladder and are involved in the production of mucus.27,30 The Rokitansky-Aschoff sinuses are invaginations of the surface epithelium that may extend through the muscularis.22 These structures can be a source of inflammation, most likely as a result of bacterial stasis and proliferation within the invaginations. The ducts of Luschka may be observed along the hepatic surface of the gallbladder and open directly into the intrahepatic bile ducts rather than into the gallbladder cavity. These structures are thought to represent a developmental anomaly, and when they are present in the gallbladder bed may be a source of a bile leak after cholecystectomy.27
CONGENITAL ANOMALIES OF THE EXTRAHEPATIC DUCTS
Accessory bile ducts are aberrant ducts that drain individual segments of the liver; they may drain directly into the gallbladder, cystic duct, right and left hepatic ducts, or bile duct.23,31 In rare cases, the right hepatic duct may connect to the gallbladder or cystic duct. These anomalies must be recognized on cholangiography in order to prevent inadvertent transection or ligation of bile ducts during surgery.
Complete duplication of the bile duct occurs rarely. In most cases, separate ducts drain the right and left hepatic lobes and open into the duodenum.23
Variation in the drainage and course of the cystic duct is common.23 Duplication of the cystic duct may also be encountered. The cystic duct is absent in most cases of agenesis of the gallbladder; rarely the duct alone may be absent, and the gallbladder empties directly into the common hepatic duct.
CONGENITAL ANOMALIES OF THE GALLBLADDER
A number of structural anomalies of the gallbladder have been described.23,31 Most of these defects are of no clinical importance, but occasionally the abnormal gallbladder may be a predisposing factor for bile stasis, inflammation, and formation of gallstones. Gallbladder disease in an anomalous or a malpositioned gallbladder may cause diagnostic confusion.
Agenesis of the gallbladder may be an isolated anomaly or occur in association with other congenital malformations.31 The abnormality has a frequency at autopsy of 0.04% to 0.13% and likely reflects a lack of development of the gallbladder bud or failure of the normal process of vacuolization. Incomplete vacuolization of the solid endodermal cord during development can result in congenital strictures of the gallbladder or cystic duct. Biliary atresia is commonly associated with an absent or atretic gallbladder. Hypoplasia of the gallbladder has been described, particularly in patients with cystic fibrosis. Ectopic tissues of foregut endodermal origin, including gastric, hepatic, adrenal, pancreatic, and thyroid tissues, may be found within the gallbladder wall.
A double gallbladder is another rare malformation, which occurs in approximately 1 to 5 per 10,000 persons in the general population.31,32 The two gallbladders may share a single cystic duct, forming a Y-shaped channel, or each may have a distinct cystic duct that enters the bile duct separately.23 Vesica fellae triplex, or triplication of the gallbladder, is another rare congenital anomaly.33 Multiple gallbladders are usually discovered because of cholelithiasis, sludge, cholecystitis, or neoplasia. Bilobed gallbladders and gallbladder diverticula are other rare anomalies. A single gallbladder may be divided by longitudinal septa into multiple chambers, probably secondary to incomplete vacuolization of the solid gallbladder bud during morphogenesis.32 Diverticula and septations of the gallbladder may promote bile stasis and gallstone formation.
Various malpositions of the gallbladder have been described.32 Rarely, the gallbladder lies under the left lobe of the liver, to the left of the falciform ligament. This defect likely results from migration of the embryonic bud from the hepatic diverticula to the left rather than to the right.23 Some researchers have proposed that the second gallbladder may develop independently from the left hepatic duct, with regression of the normal structure on the right. In other cases, a caudal bud that advances farther than the cranial bud may become buried within the cranial structure, creating an intrahepatic gallbladder. It is thought that if the caudal bud lags behind the movement of the cranial bud, a floating gallbladder results. In this setting, the gallbladder is covered completely with peritoneum and suspended from the undersurface of the liver by mesentery to the gallbladder or cystic duct; the gallbladder is abnormally mobile and prone to torsion. Rarely, gallbladders have been found in the abdominal wall, falciform ligament, and retroperitoneum.32
Several forms of “folded” gallbladders have been described. In one variant, the fundus appears to be bent, giving the appearance of a “Phrygian cap.”32 The gallbladder is usually located in a retroserosal position, and the anomaly is thought to result from aberrant folding of the gallbladder within the embryonic fossa. Aberrant folding of the fossa during the early stages of development can result in kinking between the body and the infundibulum of the gallbladder. Kinked gallbladders probably do not lead to clinical symptoms but may be a source of confusion in the interpretation of imaging studies.32
AN OVERVIEW OF DISORDERS OF THE BILIARY TRACT IN INFANTS AND CHILDREN
Cholestatic liver disease results from processes that interfere with either bile formation by hepatocytes or bile flow through the intrahepatic and extrahepatic biliary tree. A number of these disorders result from defective ontogenesis as well as from a failure of postnatal adaptation to the extrauterine environment. Table 62-1 provides a list of disorders that affect the biliary tract and occur in both infants and older children and that are discussed later in the chapter. There is a particular emphasis on neonatal cholangiopathies and the unique aspects of biliary disease in the older child. The general features of the many cholestatic liver diseases of the neonate are similar, and a central problem of pediatric hepatology is differentiating intrahepatic from extrahepatic cholestasis (Table 62-2).34 The treatment of metabolic or infective liver diseases and the surgical management of biliary anomalies require early diagnosis. Even when effective treatment is not possible, infants and children with progressive liver disease benefit from optimal nutritional support and medical management of chronic liver disease before they are referred for liver transplantation.
Table 62-1 Disorders of the Biliary Tract in Infants and Children
Modified from Balistreri WF. Neonatal cholestasis: Lessons from the past, issues for the future. Semin Liver Dis 1987; 7:61-6.
Table 62-2 Relative Frequencies of Various Forms of Neonatal Cholestasis
| DISORDER | FREQUENCY |
|---|---|
| Idiopathic neonatal hepatitis | 30-35 |
| Extrahepatic biliary atresia | 30 |
| α1-Antitrypsin deficiency | 7-10 |
| Intrahepatic cholestatic syndromes (Alagille syndrome, Byler’s disease, others) | 5-6 |
| Hepatitis (cytomegalovirus, rubella, herpes simplex virus, others) | 3-5 |
| Choledochal cyst | 2-4 |
| Bacterial sepsis | 2 |
| Endocrinopathy (hypothyroidism, panhypopituitarism) | ≈1 |
| Galactosemia | ≈1 |
| Inborn errors of bile acid metabolism | ≈1 |
| Other metabolic disorders | ≈1 |
Modified from Balistreri WF. Neonatal cholestasis: Lessons from the past, issues for the future. Semin Liver Dis 1987; 7:61.
Because of the immaturity of hepatobiliary function, the number of distinct disorders that exhibit cholestatic jaundice may be greater during the neonatal period than at any other time of life (see Table 62-1).35,36 Liver dysfunction in the infant, regardless of the cause, is commonly associated with bile secretory failure and cholestatic jaundice. Although cholestasis may be traced to the level of the hepatocyte or the biliary apparatus, in practice there may be considerable overlap among disorders with regard to the initial and subsequent sites of injury. For example, damage to the biliary epithelium often is a prominent feature of neonatal hepatitis that results from cytomegalovirus infection. Mechanical obstruction of the biliary tract invariably produces liver dysfunction and in the neonate may be associated with abnormalities of the liver parenchyma, such as giant cell transformation of hepatocytes. Whether giant cells, a frequent, nonspecific manifestation of neonatal liver injury, reflect the noxious effects of biliary obstruction or whether the hepatocytes and the biliary epithelium are damaged by a common agent during ontogenesis, such as a virus with tropism for both types of cells, is unknown. Furthermore, another common histologic variable that often accompanies neonatal cholestasis is bile ductular paucity or a diminution in the number of interlobular bile ducts.37 This finding may be of primary importance in patients with syndromic paucity of intrahepatic bile ducts but may also occur as an occasional feature of many other disorders, including idiopathic neonatal hepatitis, congenital cytomegalovirus infection, and α1-antitrypsin deficiency.38 Serial liver biopsies usually show a progressive decrease in the number of bile ductules per portal tract, with a variable amount of associated inflammation.
DIAGNOSIS
In most infants with cholestatic liver disease the condition appears during the first few weeks of life. Differentiating conjugated hyperbilirubinemia from the common unconjugated, physiologic hyperbilirubinemia of the neonate or the prolonged jaundice occasionally associated with breast-feeding is essential.39 The possibility of liver or biliary tract disease must be considered in any neonate older than 14 days with jaundice. The stools of a patient with well-established biliary atresia are acholic; however, early in the course of incomplete or evolving biliary obstruction, the stools may appear normal or only intermittently pigmented. Life-threatening but treatable disorders such as bacterial infection and a number of inborn errors of metabolism must be excluded. Furthermore, the success of surgical procedures in relieving the biliary obstruction of biliary atresia or a choledochal cyst depends on early diagnosis and surgery.
The approach to the evaluation of an infant with cholestatic liver disease is outlined in Table 62-3. The initial assessment should establish promptly whether cholestatic jaundice is present and assess the severity of liver dysfunction. A more detailed investigation may be required and should be guided by the clinical features of the case. All relevant diagnostic tests need not be performed in every patient. For example, ultrasonography may promptly establish a diagnosis of a choledochal cyst in a neonate with jaundice and thus obviate the need to exclude infectious and metabolic causes of liver disease. Numerous routine and specialized biochemical tests and imaging procedures have been proposed to distinguish intrahepatic from extrahepatic cholestasis in infants and thereby avoid unnecessary surgical exploration.39,40 Standard liver biochemical tests usually show variable elevations in serum direct bilirubin, aminotransferase, alkaline phosphatase, and lipid levels. Unfortunately, no single test has proved to have satisfactory discriminatory value, because at least 10% of infants with intrahepatic cholestasis have bile secretory failure sufficient to lead to an overlap in diagnostic test results with those suggestive of biliary atresia.41 The presence of bile pigment in stools is sometimes cited as evidence against biliary atresia, but coloration of feces with secretions and epithelial cells that have been shed by the cholestatic patient may be misleading.
Tests to Establish the Presence and Severity of Liver Disease
ALT, alanine aminotransferase; AST, aspartate aminotransferase; EBV, Epstein-Barr virus; HBsAg, hepatitis B surface antigen; MRCP, magnetic resonance cholangiopancreatography; STS, serologic test for syphilis; TORCH, toxoplasmosis, rubella, cytomegalovirus, herpesvirus.
Ultrasonography can be used to assess the size and echogenicity of the liver. Even in neonates, high-frequency, real-time ultrasonography usually can define the presence and size of the gallbladder, detect stones and sludge in the bile ducts and gallbladder, and demonstrate cystic or obstructive dilatation of the biliary system.42,43 Extrahepatic anomalies also may be identified. A triangular cord or bandlike periportal echogenicity (3 mm or greater in thickness), which represents a cone-shaped fibrotic mass cranial to the portal vein, appears to be a specific ultrasonographic finding in the early diagnosis of biliary atresia.42,43 The gallbladder “ghost” triad, defined as gallbladder length less than 1.9 cm, lack of smooth or complete echogenic mucosal lining with an indistinct wall, and irregular or lobular contour, has been proposed as additional criteria for biliary atresia.
Computed tomography provides information similar to that obtained by ultrasonography but is less suitable in patients younger than 2 years because of exposure to radiation, the paucity of intra-abdominal fat for contrast, and the need for heavy sedation or general anesthesia.44
Magnetic resonance cholangiopancreatography (MRCP), performed with T2-weighted turbo-spin echo sequences, is widely used to assess the biliary tract in all age groups. In a 1999 study, MRCP reliably demonstrated the bile duct and gallbladder in normal neonates. In some patients with biliary atresia, nonvisualization of the bile duct and demonstration of a small gallbladder have been characteristic MRCP findings.45 A more recent study found that MRCP is 82% accurate, 90% sensitive, and 77% specific for depicting extrahepatic biliary atresia. Contrary to previous reports, false-positive and false-negative findings occur with MRCP. Differentiation of severe intrahepatic cholestasis from biliary atresia may be difficult because the ability of MRCP to delineate the extrahepatic biliary tree depends on bile flow.46
The use of hepatobiliary scintigraphic imaging agents such as 99mTc iminodiacetic acid derivatives may be helpful in differentiating extrahepatic biliary atresia from other causes of neonatal jaundice.44 Unfortunately, a 1997 study showed that in 50% of patients who had a paucity of interlobular bile ducts but no extrahepatic obstruction, biliary excretion of radionuclide was absent.47 Twenty-five percent of patients who had idiopathic neonatal hepatitis also demonstrated no biliary excretion. Nevertheless, the modality remains useful for assessing cystic duct patency in patients with a hydropic gallbladder or cholelithiasis.
Percutaneous transhepatic cholangiopancreatography may be of value in visualizing the biliary tract in selected patients,48 but the technique is more difficult to perform in infants than in adults because the intrahepatic bile ducts are small and because most disorders that occur in infants do not result in dilatation of the biliary tree. Endoscopic retrograde cholangiopancreatography (ERCP) may be useful in evaluating children with extrahepatic biliary obstruction and has been performed successfully in a small number of cholestatic neonates.49 Considerable technical expertise is required of the operator to complete this procedure in infants. Most neonates require general anesthesia for a satisfactory examination.
Percutaneous liver biopsy is particularly valuable in evaluating cholestatic patients and can be undertaken in even the smallest infants with only sedation and local anesthesia.50 For example, a diagnosis of extrahepatic biliary atresia can be made on the basis of clinical and histologic criteria in 90% to 95% of patients. When doubt about the diagnosis persists, the patency of the biliary tree can be examined directly by a minilaparotomy and operative cholangiogram.
PEDIATRIC DISORDERS OF THE BILE DUCTS
BILIARY ATRESIA
Biliary atresia is characterized by the complete obstruction of bile flow as a result of the destruction or absence of all or a portion of the extrahepatic bile ducts.51 As part of the underlying disease process or as a result of biliary obstruction, concomitant injury and fibrosis of the intrahepatic bile ducts also occurs to a variable extent. The disorder occurs in 1 in 10,000 to 15,000 live births and accounts for approximately one third of cases of neonatal cholestatic jaundice (see Table 62-2). It is the most frequent cause of death from liver disease and reason for referral for liver transplantation in children (approximately 50% of all cases).52 The cause of biliary atresia is unknown. The disease is not inherited, and there have been several reports of dizygotic and monozygotic twins discordant for biliary atresia.53 In a study of 461 patients in France, seasonality, time clustering, and time-space clustering could not be demonstrated.54 Reports of familial cases have been rare; in most, a detailed histologic description of the extrahepatic biliary tree was not provided to exclude narrowing or hypoplasia of the bile duct associated with severe intrahepatic cholestasis. In the multistate case-controlled National Birth Defects Prevention Study conducted between 1997 and 2002, babies born to non-Hispanic black mothers were at greater risk than non-Hispanic white mothers. Conception during the spring and low dietary intakes of vitamin E, copper, phosphorus, and beta tocopherol were additional risk factors.55
Several mechanisms have been proposed to account for the progressive obliteration of the extrahepatic biliary tree.56 There is no evidence that biliary atresia results from a failure in morphogenesis or recanalization of the bile duct during embryonic development. Clinical features support the concept that in most cases, injury to the biliary tract occurs after birth. There is little support for an ischemic or toxic origin of extrahepatic bile duct injury.
Congenital infections with cytomegalovirus, rubella virus, human herpesvirus 6, and papillomavirus occasionally have been implicated.56 Reovirus type 3 has been implicated on the basis of the serologic evaluation of patients and immunolocalization of reovirus 3 antigens in a bile duct remnant of a patient with biliary atresia.57,58 The results of studies on the role of reovirus in biliary atresia have been contradictory. In a 1998 report, reovirus RNA was detected by reverse-transcriptase polymerase chain reaction methodology in hepatic or biliary tissues, or both, in 55% of patients who had biliary atresia and 78% of patients who had a choledochal cyst,59 compared with 21% of patients who had other hepatobiliary diseases and 12% of autopsy controls. Initial reports of the involvement of group C rotavirus in biliary atresia have not been confirmed.60
A significant increase in human leukocyte antigen (HLA) B12 has been found among patients with biliary atresia who had no associated anomalies.61 The HLA haplotypes A9, B5, A28, and B35 have been found more frequently. Oligonucleotide-based gene chip analysis of cRNA from livers of infants with biliary atresia has demonstrated a coordinated activation of genes involved in lymphocyte differentiation and inflammation.62 The finding of overexpression of osteopontin and γ-interferon indicates a potential role of type 1 T helper (Th1)–like cytokines in the pathogenesis. Biliary atresia is associated with oligoclonal expansions of CD4+ and CD8+ T cells within liver and extrahepatic bile duct remnant tissues, indicating the presence of activated T cells that react to specific antigenic stimulation.63 In a Rhesus rotavirus (RRV) murine model of biliary atresia, γ-interferon was particularly important in mediating bile duct injury.64 In other studies adoptive transfer of T cells from RRV-diseased mice into naïve syngeneic severe combined immunodeficient (SCID) recipients, at a time when viral infection could no longer be demonstrated, caused bile duct specific inflammation, possibly in response to bile duct autoantigens.65 Circulating markers of inflammation persist in biliary atresia and are largely unaffected by portoenterostomy (see later), with clear progressive elevation in both Th1 effectors interleukin (IL)-2 and interferon, some Th2 effectors (IL-4), as well as the macrophage marker (tumor necrosis factor-α [TNF-α]). Increased expression of soluble cell adhesion molecules, sICAM-1 and sVCAM-1, are also found and likely reflect ongoing recruitment of circulating inflammatory or immunocompetent cells into target tissues.66 Whether this immune response is induced by a viral infection or reflects a genetically programmed response to an infectious or environmental exposure remains unknown.
Extrahepatic anomalies occur in 10% to 25% of patients and include cardiovascular defects, polysplenia, malrotation, situs inversus, and bowel atresias.67,68 Some patients who have heterotaxia, including an infant with biliary atresia and polysplenia, have been found to have loss-of-function mutations in the CFC1 gene.69,70 This gene encodes a protein called CRYPTIC, which is involved in establishing the left-right axis during morphogenesis. In contrast, limited studies of infants with biliary atresia and heterotaxia have not found mutations in the INV gene, which is also involved in determining laterality.71 In a microarray analysis of liver tissue from infants with a so-called embryonic form of biliary atresia in which extrahepatic malformations and early onset of cholestatic jaundice occur, a unique pattern of expression of genes involved in chromatin integrity and function (Smarca-1, Rybp, and Hdac3) and overexpression of five imprinted genes (Igf2, Peg3, Peg10, Meg3, and IPW) was found, implying a failure to down-regulate embryonic gene programs that influence the development of the liver and other organs.72 Jagged1 (the gene defective in Alagille syndrome [see later]) missense mutations were identified in 9 of 102 patients with biliary atresia and were associated with a poor prognosis.73
Pathology
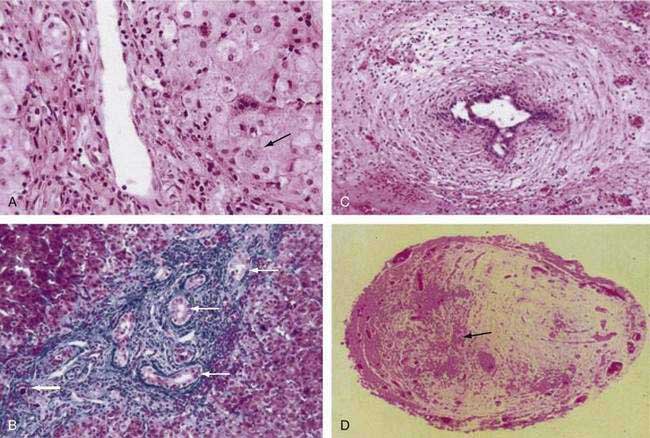
Histopathologic findings on initial liver biopsy specimens are of great importance in the management of patients with biliary atresia.51,52 Early in the course, hepatic architecture is generally preserved, with a variable degree of bile ductular proliferation, canalicular and cellular bile stasis, and portal tract edema and fibrosis (Fig. 62-4).52 The presence of bile plugs in portal triads is highly suggestive of large duct obstruction. Furthermore, bile ductules show varying injury to the biliary epithelium, including swelling, vacuolization, and even sloughing of cells into the lumen. Portal tracts may be infiltrated with inflammatory cells, and in approximately 25% of patients there may be giant cell transformation of hepatocytes to a degree observed more commonly in neonatal hepatitis. Bile ductules occasionally may assume a ductal plate configuration suggesting that the disease has interfered with the process of ductular remodeling that occurs during prenatal development.74 Biliary cirrhosis may be present initially or may evolve rapidly over the first months of life, with or without the successful restoration of bile flow.75
The morbid anatomic characteristics of the extrahepatic bile ducts in biliary atresia are highly variable. Kasai proposed a useful classification of the anatomic variants.76 Three main types have been defined on the basis of the site of the atresia. Type I is atresia of the bile duct with patent proximal ducts. Type II atresia involves the hepatic duct, with cystically dilated bile ducts at the porta hepatis. In type IIa atresia, the cystic and bile ducts are patent, whereas in type IIb atresia, these structures also are obliterated. These forms of biliary atresia have been referred to as “surgically correctable” but unfortunately account for less than 10% of all cases. Ninety percent or more of patients have type III atresia, involving obstruction of the common, hepatic, and cystic ducts, without cystically dilated hilar ducts. The entire perihilar area is in a cone of dense fibrous tissue. The gallbladder is involved to some extent in approximately 80% of patients. The type III variant has been characterized as noncorrectable, in that there are no patent hepatic or dilated hilar ducts that can be used for a biliary-enteric anastomosis.
Complete fibrous obliteration of at least a portion of the extrahepatic bile ducts is a consistent feature found on microscopic examination of the fibrous remnant.76 Other segments of the biliary tree may demonstrate lumens with varying degeneration of bile duct epithelial cells, inflammation, and fibrosis in the periductular tissues (see Fig. 62-4). In most patients, bile ducts within the liver that extend to the porta hepatis are patent during the first weeks of life but are destroyed progressively, presumably by the same process that damaged the extrahepatic ducts and by the effects of biliary obstruction. In more than 20% of patients, concentric tubular ductal structures similar to those observed in ductal plate malformations are found, indicating that the disease process interfered with the normal remodeling of the biliary tract.
Clinical Features
Most infants with biliary atresia are born at term after a normal pregnancy and have a normal birth weight.56 Female infants are affected more commonly than male infants. The perinatal course is typically unremarkable. Postnatal weight gain and development usually proceed normally. Jaundice is observed by the parents or the physician after the period of physiologic hyperbilirubinemia. Prolonged jaundice may be erroneously attributed to breastfeeding.77 The possibility of liver or biliary tract disease must be considered in any neonate older than 14 days with jaundice.78 The stools of a patient with well-established biliary atresia are acholic; however, early in the course the stools may appear normally pigmented or only intermittently pigmented.
The liver is typically enlarged with a firm edge palpable 2 to 6 cm below the right costal margin.52 The spleen is usually not enlarged early in the course but becomes enlarged as portal hypertension develops. Ascites and edema are not present initially, but coagulopathy may result from vitamin K deficiency.
Laboratory studies initially reveal evidence of cholestasis, with a serum bilirubin level of 6 to 12 mg/dL, at least 50% of which is conjugated.52 Serum aminotransferase and alkaline phosphatase levels are moderately elevated. Serum gamma glutamyl transpeptidase and 5′ nucleotidase levels are also elevated.
Treatment
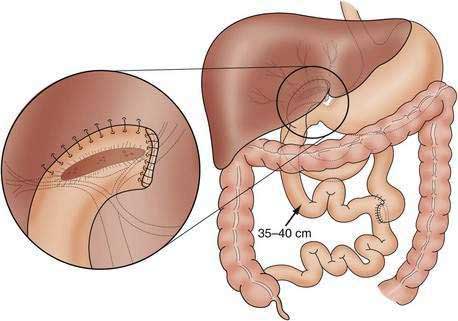
When the possibility of biliary atresia has been raised by clinical, pathologic, and imaging findings, exploratory laparotomy and operative cholangiography are necessary to document the site of obstruction and to direct attempts at surgical treatment.79–81 Sometimes, frozen sections of the transected porta hepatis are obtained to evaluate the presence and size of ductal remnants; however, the surgeon should avoid transection of the biliary tree, which may be patent but small as a result of biliary hypoplasia or markedly diminished bile flow associated with intrahepatic cholestasis. Patent proximal portions of the bile ducts or cystic structures in the porta hepatis allow conventional anastomosis with a segment of bowel in approximately 10% of patients. In most patients who have obliteration of the proximal extrahepatic biliary tree, the preferred surgical approach is the hepatoportoenterostomy procedure developed by Kasai (Fig. 62-5).82,83 The distal bile duct is transected, and the fibrous bile duct remnant is dissected to an area above the bifurcation of the portal vein.84 The dissection then progresses backward and laterally at this level, and the fibrous cone of tissue is transected flush with the liver surface, thereby exposing an area that may contain residual, microscopic bile ducts. The operation is completed by the anastomosis of a Roux-en-Y loop of jejunum around the bare edge of the transected tissue to provide a conduit for biliary drainage. A number of modifications of the enteric anastomosis, most involving exteriorization of the Roux-en-Y loop with diversion of the bile to the skin, have been used in an effort to decrease the high frequency of postoperative ascending cholangitis84; however, there may be severe fluid and electrolyte losses from the stoma and eventually massive bleeding from peristomal varices. There also is little evidence that the frequency of postoperative bacterial cholangitis is reduced through the use of these procedures.85 Many surgeons perform the original Kasai operation to prevent these complications and to facilitate liver transplantation, if required later. Multiple attempts at reexploration and revision of nonfunctional conduits should be avoided.85 Adjuvant therapy with glucocorticoids and ursodeoxycholic acid as a choleretic agent is widely prescribed postoperatively,86,87 but in a prospective, double-blind, randomized placebo-controlled trial, glucocorticoids did not reduce the need for liver transplantation after a Kasai portoenterostomy.88
Prognosis
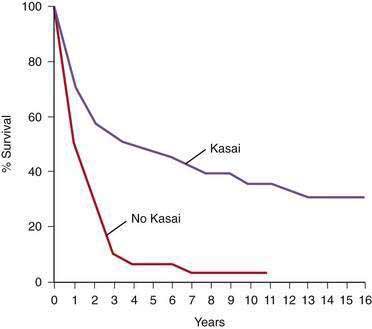
The prognosis of untreated biliary atresia is extremely poor; death from liver failure usually occurs within 2 years.89 Of 88 patients in the Biliary Atresia Registry (Surgical Section, American Academy of Pediatrics) who had either no surgery or a simple exploratory laparotomy, only 1 patient survived for more than 3 years. In the same series, follow-up data from numerous pediatric surgeons and practice settings in the United States disclosed a 5-year actuarial survival rate of 48% among 670 patients who had a Kasai operation (Fig. 62-6).90 Several large series from Europe and Japan have demonstrated similar or slightly better results.91–94 In a 2003 report from the Japanese Biliary Atresia Registry, 1381 patients had been enrolled since 1989.91 Jaundice resolved in 57% of patients after the Kasai operation, and the overall 5- and 10-year survival rates were 75.3% and 66.7%, respectively. At the time of the report, 57 of 108 patients had survived for 10 years without liver transplantation. In a series of all patients with biliary atresia identified in France over a period of 10 years (1986 to 1996), the overall survival rate of those treated with the Kasai operation and, if necessary, liver transplantation was 68%.92 The 10-year actuarial survival rate in patients with their native livers was 29%, a figure similar to the 31% compiled from 750 published cases by the authors. Therefore, children with biliary atresia derive long-term benefit from the hepatic portoenterostomy procedure, although most have some persisting liver dysfunction. Progressive biliary cirrhosis may result in death from hepatic failure or the need for liver transplantation despite an apparently successful restoration of bile flow.
Several factors have been found to contribute to the varying outcome after hepatic portoenterostomy. The age of the patient at the time of surgery is most critical.91,92,95 In several series, bile flow was reestablished in 80% to 90% of infants who were referred for surgery within 60 days of birth.91,96 Resolution of jaundice may still occur with diagnosis after 90 days of age, but long-term survival is compromised even in the era of liver transplantation.95 In a U.S. series, predictors of a poor outcome were white race, surgery at more than 60 days of age, cirrhosis on the initial liver biopsy specimen, totally nonpatent extrahepatic ducts, and absent ducts at the level of transection in the liver hilum.96 Independent prognostic factors for overall survival in the large French study were performance of the Kasai operation and age less than 45 days at surgery.92 Complete atresia of extrahepatic bile ducts and polysplenia syndrome were associated with a less favorable outcome. The experience of the surgical center was also important.92 A normal serum bilirubin level three months after surgery is predictive of long-term survival.97–99 Prehilar bile duct structures of at least 150 to 400 µm, particularly if lined with columnar epithelium, have not been consistently associated with a favorable prognosis.97,100 The quantity of the bile flow has been correlated with the total area of the biliary ductules identified in the excised porta hepatis specimen.101,102 The rate of progression of the underlying bile ductular and liver disease also limits survival.51,103 The disorder is not limited to the extrahepatic biliary tree and can be associated with progressive inflammation and destruction of the intrahepatic bile ducts and eventual cirrhosis.51 Recurring episodes of ascending bacterial cholangitis, which are most frequent during the first two years after surgery, can contribute to the ongoing bile duct injury and even lead to reobstruction.104 Cholangitis develops primarily in infants who have some degree of bile drainage, probably because of the access to ascending infection provided by patent bile ducts in the porta hepatis. Prophylactic oral antibiotics are often used to prevent recurrent cholangitis after a Kasai portoenterostomy, but controlled trials of this approach have not been done.105 Substantial hepatocyte injury, as indicated by lobular disarray and giant cell transformation on liver biopsy specimens, also has been associated with a poor outcome. The presence of the ductal plate malformation on liver biopsy specimens also predicts poor bile flow after hepatoportoenterostomy. Growth failure was associated with the need for transplantation or death by 24 months of age. Esophageal variceal hemorrhage alone is not an absolute indication for urgent liver transplantation in patients with good bile drainage and preserved liver synthetic function.106,107
Liver transplantation is essential in the management of children in whom portoenterostomy does not successfully restore bile flow, referral is late (probably at 120 days of age or later), and end-stage liver disease develops eventually despite bile drainage.92,108,109 Biliary atresia accounts for 40% to 50% of all liver transplants performed in children. The portoenterostomy is thought to make liver transplantation more difficult technically as a result of intra-abdominal adhesions and the various enteric conduits that are encountered110; however, with the use of reduced-size liver allografts and living-related donors, one-year survival rates of more than 90% can be expected.108,111,112
SPONTANEOUS PERFORATION OF THE BILE DUCT
Spontaneous perforation of the bile duct is a rare but distinct cholestatic disorder of infancy.113 The perforation usually occurs at the junction of the cystic and bile ducts. The cause is unknown, but there may be evidence of obstruction at the distal end of the bile duct secondary to stenosis or inspissated bile.114 Congenital weakness at the site of the perforation and injury produced by infection also have been suggested.
Clinical signs, including jaundice, acholic stools, dark urine, and ascites, typically occur during the first months of life.114 The infant also may experience vomiting and lack of weight gain. Progressive abdominal distention is a usual feature; bile staining of fluid within umbilical or inguinal hernias may be observed.
Mild to moderate conjugated hyperbilirubinemia with minimal elevation of serum aminotransferase levels is typical. Abdominal paracentesis reveals clear bile-stained ascitic fluid, which usually is sterile. Ultrasonography reveals ascites or loculated fluid in the right upper quadrant; the biliary tree is not dilated. Hepatobiliary scintigraphy demonstrates the free accumulation of isotope within the peritoneal cavity.114
Operative cholangiography is required to demonstrate the site of the perforation.115 Surgical treatment may involve simple drainage of the bilious ascites and repair of the site of the perforation.114–116 If the perforation is associated with obstruction of the bile duct, however, drainage via a cholecystojejunostomy may be required.
BILE PLUG SYNDROME
A plug of thick, inspissated bile and mucus also may cause obstruction of the bile duct.117,118 Otherwise healthy infants have been affected, but the condition occurs more commonly in sick, premature infants who cannot be fed and require prolonged parenteral nutrition. The pathogenesis may involve bile stasis, fasting, infection, and an increased bilirubin load. The cholestasis associated with massive hemolysis, or the inspissated bile syndrome, may have been a variant of the bile plug syndrome but is now infrequent with the advent of measures to prevent and treat Rh and ABO blood group incompatibilities. The clinical presentation may resemble that of biliary atresia. Ultrasonography may show dilated intrahepatic bile ducts. Exploratory laparotomy and operative cholangiography usually are required for diagnosis. Simple irrigation of the bile duct is curative.116
PRIMARY SCLEROSING CHOLANGITIS
Primary sclerosing cholangitis (PSC) is an uncommon, chronic, progressive disease of the biliary tract characterized by inflammation and fibrosis of the intrahepatic and extrahepatic biliary ductal systems leading eventually to biliary cirrhosis.119–121 Only aspects of PSC that are of particular importance to infants and children are discussed here (see Chapter 68 for a detailed discussion of PSC). PSC is a pathologic process that occurs in the absence of choledocholithiasis or a history of bile duct surgery. Sclerosing cholangitis may uncommonly present in the neonatal period; it may present later with features of autoimmunity (primary sclerosing cholangitis), often in association with inflammatory bowel disease; or it may occur with other disorders, including Langerhans cell histiocytosis, immunodeficiency, psoriasis, and cystic fibrosis. In adults, carcinoma of the bile ducts must also be excluded; however, this complication has not been reported in children. PSC is associated with inflammatory bowel disease (most often, ulcerative colitis) in 70% of adult patients, and in approximately 50% to 80% of children with the disorder.120,122 A male preponderance has been reported in some, but not all, large series of children with PSC. More than 200 cases of PSC have been reported in children, and most of these have occurred since the mid-1980s, presumably as a result of improvements in pediatric cholangiography.
The onset of PSC has been reported in the neonatal period; neonates accounted for 15 of 56 cases in a 1994 series of children with the disorder.122 Cholestatic jaundice and acholic stools were observed within the first two weeks of life. The presenting features were virtually identical to those of extrahepatic biliary atresia; however, percutaneous cholecystography disclosed a biliary system that was patent but exhibited rarefaction of segmental branches, stenosis, and focal dilatation of the intrahepatic bile ducts. The extrahepatic bile ducts were involved in six of eight patients. Jaundice subsided spontaneously within six months, but later in childhood all patients had clinical and biochemical features consistent with biliary cirrhosis and portal hypertension. In contrast with PSC in adults and older children, PSC in neonates has not been associated with intestinal disease.
Inflammatory bowel disease–associated PSC usually occurs in patients with ulcerative colitis, although cases have been reported in patients with Crohn’s disease.123 The bowel symptoms can precede, occur simultaneously with, or appear years after the diagnosis of PSC. As in adults, treatment of the bowel disease in infants, including colectomy, does not influence the progression of PSC. Celiac disease has also been associated with PSC.124
Lesions similar to those of PSC have been defined by cholangiography in Langerhans cell histiocytosis, but the process is caused by histiocytic infiltration and progressive scarring of portal tracts, with resulting distortion of intrahepatic bile ducts. Cholestasis can occur before the diagnosis of Langerhans cell histiocytosis has been established but most often is found later.125 Children with Langerhans cell histiocytosis may have involvement of multiple organs, with diabetes insipidus, bone lesions, skin lesions, lymphadenopathy, and exophthalmos. Chemotherapy does not affect the course of the biliary tract disease. Liver transplantation has been successful in several children who experienced progression to end-stage liver disease.126
In some children with a variety of immunodeficiencies, both cellular and humoral, sclerosing cholangitis appears to develop. Cryptosporidia and cytomegalovirus have been found concurrently in the biliary tract in some of these patients, as well as in adults with the acquired immunodeficiency syndrome (AIDS). 127,128 Treatment of the associated infection has no proven effect on the biliary tract disease.
There is no definitive diagnostic test for PSC; the diagnosis is based on a combination of biochemical, histologic, and radiologic data. Typically, adult patients exhibit fatigue, weight loss, pruritus, right upper quadrant pain, and intermittent jaundice. In children, the clinical presentation is more variable; the most common symptoms are abdominal pain, jaundice, and chronic diarrhea.120 Physical examination sometimes reveals hepatomegaly, which may be associated with splenomegaly, conjunctival icterus, and, rarely, ascites.
The serum alkaline phosphatase level is often elevated in patients with PSC, and serum aminotransferase levels may be mildly elevated129; however, in a 1995 series, 15 of 32 patients had a normal alkaline phosphatase level on presentation.130 Hyperbilirubinemia is seen in less than half of pediatric patients. Serum autoantibodies, including antinuclear antibodies and smooth muscle antibodies, may be found in some patients.129 Antineutrophil cytoplasmic antibodies (ANCAs) may be detected. On liver biopsy specimens, the histologic findings may be suggestive of PSC but usually are not diagnostic. Characteristic concentric periductal (“onion skin”) fibrosis may be present later in the course of the disease, but more often, only neoductular proliferation and fibrosis are found.131
Differentiating PSC from autoimmune hepatitis, particularly in the presence of circulating non–organ-specific autoantibodies and hepatic features on liver biopsy specimens, may be difficult. In 25% to 30% of cases an overlap syndrome may occur in children with both hepatic and cholestatic serum liver test results and with histologic features of autoimmune hepatitis and PSC.119 Serologic findings include the presence of antinuclear, smooth muscle, and anti–liver-kidney microsome type 1 (anti-LKM-1) antibodies and perinuclear ANCAs.132
The diagnosis of PSC is established by cholangiography.133 ERCP has been the method of choice for visualizing the intrahepatic and extrahepatic bile ducts122,134; however, a 2002 study of children demonstrated that MRCP was comparable to ERCP in correctly identifying changes of PSC in 13 cases and excluding abnormalities in 5.133 Irregularities of the intrahepatic and extrahepatic ducts can be found, including alternating strictures and areas of dilatation that produce a beaded appearance. Involvement of the intrahepatic bile ducts predominates in patients whose condition appears after the neonatal period. Occasionally, dominant strictures of the extrahepatic ducts or papillary stenosis is found. Small-duct PSC with a normal cholangiogram but histologic features of PSC rarely occurs in children.135
The prognosis of PSC in children is guarded.120 The clinical course of the disorder is variable but usually progressive. In a 1994 series of 56 children, the median survival time from onset of symptoms was approximately 10 years, similar to that reported in adults.122 In another study of 52 children, the median survival free of liver transplantation was 12.7 years.120 Analysis of survival factors at presentation indicates that older age, splenomegaly, and a prolonged prothrombin time predicted a poor outcome.130 The occurrence of jaundice after the neonatal period with a persisting serum bilirubin level of more than five times the upper normal value was also associated with a poor outcome. Hepatocellular carcinoma also may occur, but cholangiocarcinoma, an important complication of adult PSC, has not been reported in children.
The treatment of PSC in children is unsatisfactory.120,136 No published reports of controlled trials have demonstrated convincingly that any medical therapy improves histologic characteristics and prolongs survival. Uncontrolled experience has suggested some benefit for immunosuppressive therapy with prednisone and azathioprine in patients with the overlap syndrome.119 Ursodeoxycholic acid therapy in adults and in a limited number of children has led to an improvement in clinical symptoms and in liver test abnormalities, but a long-term benefit of treatment on survival has not been demonstrated.121 Liver transplantation is an important option for patients who experience progression to end-stage liver disease, and long-term results in children appear to be good137; however, recurrence of PSC after transplantation has been reported in children.131
CHOLEDOCHAL CYSTS
Incidence and Classification
Choledochal cysts are congenital anomalies of the biliary tract that are manifested by cystic dilatation of the extrahepatic and intrahepatic bile ducts.138,139 The incidence of choledochal cysts is 1 in 13,000 to 15,000 in Western countries and as high as 1 in 1000 in Japan.140 Choledochal cysts are not familial; female children are affected more commonly than male children. Cases have been described in utero and in older adult patients, but approximately two thirds of patients seek medical attention before the age of 10.
The classification proposed by Todani and colleagues (Fig. 62-7) are cited frequently.141,142 Several varieties of type I cysts, accounting for 80% to 90% of cases, exhibit segmental or diffuse fusiform dilatation of the bile duct. Type II cysts consist of a true choledochal diverticulum. Type III cysts consist of dilatation of the intraduodenal portion of the bile duct, or choledochocele. Type IV cysts may be subdivided into type IVa, or multiple intrahepatic and extrahepatic cysts, and type IVb, or multiple extrahepatic cysts. The type IVb variant either is uncommon or may overlap with type I. Whether type V, or Caroli’s disease, which consists of single or multiple dilatations of the intrahepatic ductal system, should be viewed as a form of choledochal cyst is unsettled.142,143
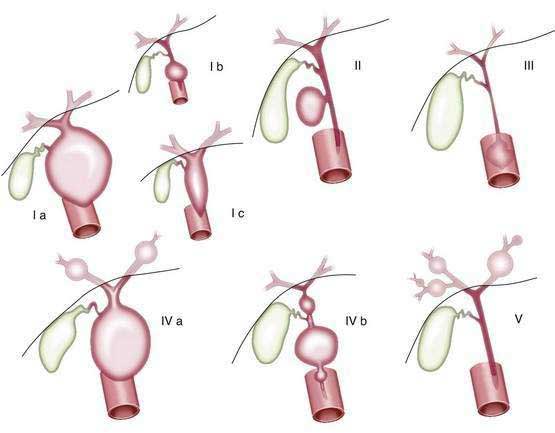
Figure 62-7. Classification of choledochal cysts according to Todani and colleagues.141 Ia, common type; Ib, segmental dilatation; Ic, diffuse dilatation; II, diverticulum; III, choledochocele; IVa, multiple cysts (intra- and extrahepatic); IVb, multiple cysts (extrahepatic); V, single or multiple dilatations of the intrahepatic ducts (Caroli’s disease).
(From Savader SJ, Benenati JF, Venbrux AC, et al. Choledochal cysts: Classification and cholangiographic appearance. AJR 1991; 156:327-31.)
Etiology
The cause of choledochal cysts has not been established.140 Congenital weakness of the bile duct wall, a primary abnormality of epithelial proliferation during embryologic ductal development, and congenital obstruction of bile ducts have been suggested. A relationship to other obstructive cholangiopathies, such as biliary atresia, has been proposed but not proved.144 Reovirus RNA has been detected by reverse-transcriptase polymerase chain reaction methodology in hepatic or biliary tissues of 78% of patients who have choledochal cysts.59 A high frequency (40%) of an anomalous union of the pancreatic and bile ducts, which may allow reflux of pancreatic secretions into the biliary tree, has been described.145 This process may result in progressive injury to the developing ductal system, with subsequent weakness and dilatation. Choledochal cysts have also been found in some patients with autosomal recessive polycystic renal disease.146
Pathology
The cysts are composed of a fibrous wall; there may be no epithelial lining or a low columnar epithelium.140 Mild chronic inflammation may be present. Complete, inflammatory obstruction of the terminal portion of the bile duct is common in infants who have a choledochal cyst.
Liver biopsy specimens in the affected neonate show typical features of large duct obstruction.140 Findings may mimic those observed in extrahepatic biliary atresia. Portal tract edema, bile ductular proliferation, and fibrosis may be prominent. A pattern of biliary cirrhosis may be observed in older patients with long-standing biliary obstruction. Carcinoma of the cyst wall may occur by adolescence.147,148
Clinical Features
The infantile form of choledochal cyst disease must be distinguished from other forms of hepatobiliary disease of the neonate, particularly biliary atresia.140 Disease often appears during the first months of life, and as many as 80% of patients have cholestatic jaundice and acholic stools.149 Vomiting, irritability, and failure to thrive may occur. Examination reveals hepatomegaly and in approximately one half of patients a palpable abdominal mass. In a series of 72 patients diagnosed postnatally, 50 (69%) exhibited jaundice that was associated with abdominal pain in 25 or with a palpable mass in 3; 13 (18%) had abdominal pain alone, and 2 (3%) had a palpable mass alone. In a 2008 series, adults were more likely to exhibit abdominal pain (97% versus 63%, P < 0.001), and children were more likely to experience jaundice (71% versus 25%, P = 0.001).148 In older patients, epigastric pain may result from pancreatitis. Intermittent jaundice and fever may result from recurrent episodes of cholangitis. The classic triad of abdominal pain, jaundice, and a palpable abdominal mass is observed in less than 20% of patients.148
Spontaneous perforation of a choledochal cyst may occur, particularly when bile flow is obstructed. Progressive hepatic injury can occur during the first months of life as a result of biliary obstruction caused by poor bile flow, sludge, protein plugs, and stones composed of fatty acids and calcium.150
Diagnosis
The diagnosis of a choledochal cyst is best established by ultrasonography (Fig. 62-8).44 In fact, several reports have demonstrated that antenatal ultrasonography can be used to detect a choledochal cyst in the fetus. Sequential ultrasonographic examinations have allowed the study of the evolution of choledochal cysts during pregnancy. In the older child, percutaneous transhepatic cholangiography or ERCP may help define the anatomic features of the cyst; its site of biliary origin, including an anomalous arrangement of the pancreaticobiliary junction; and the extent of both extrahepatic and intrahepatic disease, including the presence of intraductal strictures and calculi.151 MRCP is being used increasingly to evaluate the extent of the cyst and defects within the biliary tree and to detect an anomalous union of the pancreaticobiliary duct.152 MRCP was less effective than ERCP for detecting minor ductal abnormalities and small choledochoceles in adults.151
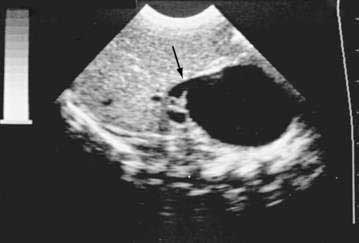
In practice, most pediatric surgeons rely on an operative cholangiogram to define the extent of intrahepatic and extrahepatic disease.140
Treatment
The preferred treatment is surgical excision of the cyst with reconstruction of the extrahepatic biliary tree.138,140 Biliary drainage is usually accomplished by a choledochojejunostomy with a Roux-en-Y anastomosis. Excision of the cyst reduces bile stasis and the risk of cholangitis and cholangiocarcinoma. Simple decompression and internal drainage should be done only when the complicated anatomic characteristics do not allow complete excision. Long-term follow-up is essential because recurrent cholangitis, lithiasis, anastomotic stricture, and pancreatitis may develop years after the initial surgery.139
CONGENITAL DILATATION OF THE INTRAHEPATIC BILE DUCTS
Nonobstructive saccular or fusiform dilatation of the intrahepatic bile ducts is a rare, congenital disorder.153,154 In the pure form, known as Caroli’s disease, dilatation is classically segmental and saccular and associated with stone formation and recurrent bacterial cholangitis. A more common type, Caroli’s syndrome, is associated with a portal tract lesion typical of congenital hepatic fibrosis (CHF).154 Dilatation of the extrahepatic bile ducts (choledochal cysts) also may be present. Renal disease occurs in both forms, renal tubular ectasia occurs with Caroli’s disease, and both conditions can be associated with autosomal recessive polycystic kidney disease (ARPKD) or, rarely, autosomal dominant polycystic kidney disease.155 Mutations in a polycystic kidney and hepatic disease 1 gene (PKHD1) have been identified in patients with ARPKD.156 The gene encodes a large protein (4074 amino acids) called fibrocystin to reflect the main resulting structural abnormalities in liver and kidney.157 The protein shares structural features with the hepatocyte growth factor receptor and appears to belong to a superfamily of proteins that are involved in the regulation of cell proliferation and of cellular adhesion and repulsion.158 Fibrocystin is localized to the primary cilia of renal epithelial cells and cholangiocytes, suggesting a link between ciliary dysfunction and cyst development.
Pathology
The intrahepatic cysts are in continuity with the biliary tract and lined by epithelium that may be ulcerated and hyperplastic.153 The cysts may contain inspissated bile, calculi, and purulent material.
Liver biopsy specimens may reveal normal tissue or features of acute or chronic cholangitis.159 Portal tract edema and fibrosis may be present. In cases associated with CHF, findings associated with the ductal plate malformation can be expected; the lumen of the portal bile duct forms an epithelium-lined circular cleft surrounding a central vascularized connective tissue core, or a series of bile duct lumens are arranged in a circle around a central fibrous tissue core.160
Clinical Features
Patients usually seek medical attention during childhood and adolescence because of hepatomegaly and abdominal pain.155,156 The disorder appears in the neonate as renal disease or cholestasis.156 The saccular or fusiform dilatation of bile ducts predisposes to stagnation of bile leading to the formation of biliary sludge and intraductal lithiasis. Fever and intermittent jaundice may occur during episodes of bacterial cholangitis. Hepatosplenomegaly is found in cases associated with CHF; affected patients may exhibit bleeding esophageal varices.153 The polycystic kidneys may be palpable.
Liver biochemical tests may have normal results or show mild to moderate elevations of serum bilirubin, alkaline phosphatase, and aminotransferase levels.154 Liver synthetic function is well preserved, but repeated episodes of infection and biliary obstruction within the cystic bile ducts eventually may lead to hepatic failure. The maximal concentrating capacity is the most frequently abnormal renal function test finding; variable elevations of blood urea nitrogen and serum creatinine levels reflect the severity of the underlying kidney disease.156
Diagnosis
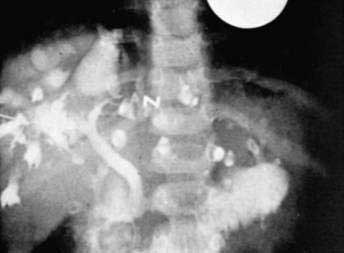
Ultrasonography, MRCP, and computed tomography are of great value in demonstrating the cystic dilatation of the intrahepatic bile ducts.161,162 Renal cysts or hyperechogenicity of papillae may be detected. Percutaneous or endoscopic cholangiography (Fig. 62-9) usually demonstrates a normal bile duct with segmental, saccular dilatations of the intrahepatic bile ducts.159 Rarely, the process may be limited to one lobe of the liver.
Prognosis and Treatment
The clinical course is often complicated by recurrent episodes of cholangitis156,159; sepsis and liver abscess may occur. The prognosis in the setting of persistent or recurrent infection is poor. Calculi frequently develop within the cystically dilated bile ducts and can complicate the treatment of cholangitis.163 Patients who have extensive hepatolithiasis may experience intractable abdominal pain. Removal of stones by surgery, endoscopy, or lithotripsy usually is not feasible.164 Hepatic resection is indicated for disease limited to a single lobe.165 Surgical drainage procedures generally are not effective and may complicate later liver transplantation. Therapy with ursodeoxycholic acid has been used successfully to dissolve intrahepatic stones.163 Cholangiocarcinoma may develop within the abnormal bile ducts.166 Portal hypertension and variceal bleeding may predominate in patients with CHF and Caroli’s disease.156 End-stage renal disease develops in some patients who have associated polycystic kidney disease. Liver transplantation is an option in patients who have extensive disease and frequent complications, including refractory cholangitis.167
NONSYNDROMIC PAUCITY OF THE INTERLOBULAR BILE DUCTS
A paucity of interlobular bile ducts may be an isolated and unexplained finding in infants and children with idiopathic cholestasis or a feature of a heterogeneous group of disorders that include congenital infections with rubella and cytomegalovirus and genetic disorders such as α1-antitrypsin deficiency and inborn errors of bile acid metabolism.168,169 Bile duct paucity has been observed in some cases of Williams and Noonan syndromes.170,171 Paucity of interlobular bile ducts has been defined as a ratio of the number of interlobular bile ducts to the number of portal tracts of less than 0.4.12,172 At least 10 portal tracts should be examined on a liver biopsy specimen to be confident that bile duct paucity is present. The structural abnormality has also been referred to as intrahepatic biliary atresia or intrahepatic biliary hypoplasia; however, these terms imply more insight into the pathogenesis of ductular paucity than currently prevails. Cases may arise from true biliary dysgenesis but more often result from active injury and loss of bile ducts.12,172 Bile duct paucity may occur without associated developmental anomalies and without a documented intrauterine infection or genetic disorder; however, this idiopathic form of nonsyndromic bile duct paucity is likely to be heterogeneous in cause with extremely variable clinical features and prognosis.169,173 Cholestasis typically develops early in infancy and may be associated with progressive liver disease.
SYNDROMIC PAUCITY OF THE INTERLOBULAR BILE DUCTS (ALAGILLE SYNDROME, OR ARTERIOHEPATIC DYSPLASIA)
Syndromic paucity of interlobular bile ducts (Alagille syndrome, or arteriohepatic dysplasia) is the most common form of familial intrahepatic cholestasis. This disorder is characterized by chronic cholestasis, a decreased number of interlobular bile ducts, and a variety of other congenital malformations.174
An autosomal dominant mode of transmission with incomplete penetrance and variable expressivity has been established from family studies.175 A partial deletion of the short arm of chromosome 20 was detected in some patients and led to the identification of the Alagille syndrome gene. Mutations in the jagged1 (JAG1) gene have been identified in approximately 94% of affected patients and include total gene deletions as well as protein truncating, splicing, and missense mutations.176 JAG1 encodes a ligand in the Notch signaling pathway that is involved in cell fate determination during development.177 Mutations in the gene encoding for the NOTCH2 receptor have been found in patients with Alagille syndrome who were negative for JAG1 mutations.178 There appears to be no phenotypic difference between patients with deletion of the entire JAG1 gene and those with intragenic mutations.179 The disorder may affect only one family member; such cases may represent spontaneous mutations of the JAG1 gene. Alternatively, it is possible that the variability in gene expression is so great that minimally affected family members are not diagnosed. A 1994 analysis of 33 families collected through 43 probands corroborated the autosomal dominant inheritance and concluded that the rate of penetrance is 94% and that 15% of cases are sporadic; however, expressivity was variable, and 26 persons (including 11 siblings) exhibited minor forms of the disease.180
Clinical Features
Chronic cholestasis of varying severity affects 95% of patients.181,182 Jaundice and clay-colored stools may be observed during the neonatal period and become apparent in most patients during the first 2 years of life. Intense pruritus may be present by 6 months of age.174 The liver and spleen are often enlarged. During the first years of life, xanthomata appear on the extensor surfaces of the fingers and in the creases of the palms and popliteal areas. Dysmorphic facies (Fig. 62-10) are usually recognized during infancy and become more characteristic with age.183 The forehead is typically broad, the eyes are deeply set and widely spaced, and the mandible is somewhat small and pointed, imparting a triangular appearance to the face. The malar eminence is flattened, and the ears are prominent. Extrahepatic anomalies have been described with this syndrome, but the phenotypic expression varies considerably. In a 1999 series of 92 patients, cholestasis occurred in 96%, cardiac murmur in 97%, butterfly vertebrae in 51%, posterior embryotoxon (mesodermal dysgenesis of the iris and cornea) in 78%, and characteristic facies in 96% of patients.182 Short stature is a regular feature but is only partially attributed to the severity of chronic cholestasis. Growth hormone insensitivity associated with elevated circulating levels of growth hormone–binding protein has been described in these patients.184 Mild to moderate mental retardation affects 15% to 20% of patients. Congenital heart disease occurs in most patients, and peripheral pulmonic stenosis is observed in approximately 90%.182,185 Systemic vascular malformations also may be present. Osseous abnormalities include a decreased bone age, variable shortening of the distal phalanges, and vertebral arch defects (e.g., butterfly vertebrae, hemivertebrae, and a decrease in the interpedicular distance). Ophthalmologic examination may reveal eye anomalies, including posterior embryotoxon, retinal pigmentation, and iris strands. Renal abnormalities and hypogonadism also have been described.182
Laboratory studies reveal an elevation of total serum bilirubin levels (usually 2 to 8 mg/dL) during infancy and intermittently later in life.182 Approximately 50% of the total serum bilirubin is conjugated. Serum alkaline phosphatase, gamma glutamyl transpeptidase, and 5′ nucleotidase levels may be extremely high and correlate somewhat with the degree of cholestasis. Serum aminotransferase levels are mildly to moderately increased. Serum cholesterol levels may be 200 mg/dL or higher, and serum triglyceride concentrations may range from 500 to 1000 mg/dL. Total serum bile acid concentrations are markedly elevated, but the bile acid profiles in serum, urine, and bile do not differ qualitatively from those seen in other cholestatic disorders.
Pathology
The hallmark of this condition is a paucity of interlobular bile ducts.173 Paucity may be defined as a significantly decreased ratio of the numbers of interlobular portal bile ducts to portal tracts (<0.4).172 The histologic features during the first months of life may overlap with those of neonatal hepatitis, in that there can be ballooning of hepatocytes, variable cholestasis, portal inflammation, and giant cell transformation. Often the number of interlobular bile ducts is not decreased on initial liver biopsy specimens, but bile duct injury consisting of cellular infiltration of portal triads contiguous to interlobular bile ducts, lymphocytic infiltration and pyknosis of biliary epithelium, and periductal fibrosis may be evident.168,186 Serial biopsy specimens from an individual patient may initially show bile duct proliferation, followed later in life by a paucity of bile ducts (Fig. 62-11).187 Paucity of interlobular bile ducts is usually apparent after 3 months. Mild periportal fibrosis also may be present, but progression to cirrhosis is uncommon. The extrahepatic bile ducts are patent but usually narrowed or hypoplastic. Ultrastructural studies have demonstrated the accumulation of bile pigment in the cytoplasm near lysosomes and vesicles of the outer convex space of the Golgi apparatus. The bile canaliculi most often appear to be structurally normal, but in some cases they may appear to be dilated with blunting and shortening of microvilli.186
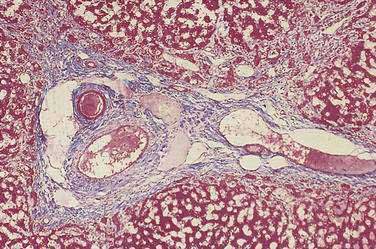
Pathogenesis
The mechanisms involved in the pathogenesis of bile duct paucity and cholestasis are unsettled. Also unknown is how the hepatobiliary disease relates to the multiplicity of congenital anomalies found in other organ systems. Mice homozygous for the Jag1 mutation die of hemorrhage early during embryogenesis and exhibit defects in remodeling of the embryonic and yolk sac vasculature.188 The strong JAG1 expression during human embryogenesis, both in the vascular system and in other mesenchymal and epithelial tissues, implicates abnormal angiogenesis in the pathogenesis of Alagille syndrome and particularly the paucity of interlobular bile ducts. In human embryos JAG1 is expressed in the distal cardiac outflow tract and pulmonary artery, major arteries, portal vein, optic vesicle, otocyst, branchial arches, metanephros, pancreas, and mesocardium; around the major bronchial branches; and in the neural tube.189 All these structures are affected in Alagille syndrome. Many of the JAG1 mutations generate premature termination codons, and many of these mutations produce a truncated protein that exerts a dominant-negative effect on Notch signaling.190 Although a vascular basis for the anomalies in Alagille syndrome seems possible, the precise mechanisms leading to bile duct paucity remain unknown. Notch signaling has an important role in the differentiation of biliary epithelial cells and is essential for their tubular formation during intrahepatic bile duct development.191 There is evidence that a lack of branching and elongation of bile ducts during postnatal liver growth contributes to peripheral bile duct paucity and cholestasis.192
Prognosis and Treatment
The clinical course is marked by varying degrees of cholestasis, sometimes worsened by intercurrent viral infections. Morbidity may result from pruritus, cutaneous xanthomata, and neuromuscular symptoms related to vitamin E deficiency. Treatment involves the provision of an adequate caloric intake, prevention or correction of fat-soluble vitamin deficiencies, and symptomatic measures to relieve pruritus. The long-term prognosis depends on the severity of the liver disease and associated malformations.185 Of 80 patients who had this disorder and who were followed by Alagille and associates, 21 patients died, but only 4 died as a result of liver disease.174 Partial external biliary diversion may be effective for treating severe pruritus and hypercholesterolemia in patients without cirrhosis who do not respond to medical therapy.193,194 In another series of 92 patients, the mortality rate was 17%.182 The factors that contributed significantly to mortality were hepatic disease or transplantation (25%), complex congenital heart disease (15%), and intracranial hemorrhage (25%). Hepatocellular carcinoma may occur.195 In a retrospective review of 268 patients, vascular anomalies such as intracranial aneurysms accounted for 34% of the mortality.196 On the basis of these studies, the 20-year predicted life expectancy is approximately 75% for all patients, 80% for those not requiring liver transplantation, and 60% for those who require liver transplantation. In a study of 168 patients with liver disease, actuarial survival rates with a native liver were 51% and 38% at 10 and 20 years, respectively, and overall survival rates were 68% and 62%, respectively.197 Neonatal cholestatic jaundice was associated with poorer survival with a native liver. Survival and candidacy for liver transplantation may be limited by the severity of associated cardiovascular anomalies.198 In a series of patients who underwent liver transplantation, a higher than expected mortality rate of 43% was attributed to cardiac disease or a previous Kasai procedure.199
MEDICAL MANAGEMENT OF CHRONIC CHOLESTASIS
Cholestatic liver disease in children adversely affects nutritional status, growth, and development, which all contribute to morbidity and mortality.200 In a child with chronic, and sometimes progressive, cholestatic liver disease, efforts should be directed to promoting growth and development and minimizing discomfort.201
Protein-energy malnutrition leading to growth failure is an inevitable consequence of chronic liver disease in 60% of children.201,202 Steatorrhea is common in children with cholestasis, as a result of impaired intraluminal lipolysis, solubilization, and intestinal absorption of long-chain triglycerides.203 Medium-chain triglycerides do not require solubilization by bile salts before intestinal absorption and thus can provide needed calories when administered orally in one of several commercial formulas or as an oil supplement.203
Significant morbid conditions resulting from fat-soluble vitamin deficiencies can be prevented in large part in cholestatic children.204 Because metabolic bone disease, manifesting as rickets and pathologic fractures, can result from vitamin D deficiency, vitamin D should be provided as D2 (5000 IU/day) or as 25-hydroxycholecalciferol (3 to 5 µg/kg/day). Supplements of elemental calcium (50 to 100 mg/kg/day) and phosphorus (25 to 50 mg/kg/day) also may be required. Bone mass can be reduced in cholestatic children even with normal serum 25-hydroxyvitamin D levels, possibly related to impaired insulin-like growth factor I production by the liver.205
Xerophthalmia, night blindness, and thickened skin have been reported in patients who have a vitamin A deficiency. Oral supplements of vitamin A, 5000 to 25,000 IU/day, should be administered.204
Vitamin K deficiency and associated coagulopathy may be treated initially with an oral water-soluble supplement administered in doses of 2.5 to 5 mg twice weekly to as much as 5 mg daily. Children who do not respond or who have significant bleeding require intramuscular injections of vitamin K.206
Chronic deficiency of vitamin E may produce a disabling, degenerative neuromuscular syndrome characterized by areflexia, ophthalmoplegia, cerebellar ataxia, peripheral neuropathy, and posterior column dysfunction.204 The onset can be observed within the first 2 years of life. Because serum vitamin E levels may be elevated spuriously in the presence of hyperlipidemia, the ratio of serum vitamin E to total serum lipids is most useful in monitoring the patient’s vitamin E status; deficiency in a child less than 12 years old, for example, is indicated by a ratio of less than 0.6. The child may not respond to massive doses of standard vitamin E preparations (150 to 200 IU/kg/day). Therapy with intramuscular dl-alpha-tocopherol (50 mg/day) or the water-soluble form of vitamin E, d-alpha-tocopherol polyethylene glycol-1000-succinate (15 to 25 IU/kg/day), is effective.204
Xanthomata and pruritus may cause substantial discomfort. Pruritus may be observed by three months of age.207 The success of most therapies for pruritus depends on the presence of patent bile ducts that allow bile acids and other biliary constituents to reach the gut lumen. Biliary diversion has been used as a successful alternative to relieve intractable pruritus in some patients with intrahepatic cholestasis.207,208 The antibiotic rifampin, through up-regulation of pathways for biotransformation and biliary excretion, and the choleretic bile acid ursodeoxycholic acid are used for the treatment of pruritus with varying degrees of success.209,210 Because of evidence that a component of pruritus may be of central neurogenic origin mediated by the opiate receptor system, opioid receptor antagonists such as naltrexone have been effective in some patients with severe pruritus unresponsive to other agents; however, side effects, withdrawal symptoms, and the lack of experience in children limit the general use of these medications.211 The nonabsorbable anion exchange resin cholestyramine may be used to bind bile acids, cholesterol, and presumably other potentially toxic agents in the intestinal lumen.207 This medication may lower serum lipid levels and bind the substances involved in the pathogenesis of pruritus. A dose of 0.25 to 0.5 g/kg/day is administered before breakfast or in divided doses before meals to relieve severe pruritus and xanthomata.207 Cholestyramine is relatively unpalatable, however, and carries modest risks for intestinal obstruction, caused by inspissation of the drug, and hyperchloremic acidosis. Colesevelam is a novel bile acid sequestrant that has superior bile acid–binding efficacy compared with cholestyramine and is taken in a more palatable tablet form.212 Its use in cholestatic liver disease has been limited. Pruritus also has been treated with exposure to ultraviolet B light.
PEDIATRIC DISORDERS OF THE GALLBLADDER
CHOLELITHIASIS
Cholelithiasis is uncommon in otherwise healthy children and usually occurs in patients who have a predisposing condition.213,214 An ultrasonographic survey of 1570 persons (ages 6 to 19 years) detected gallstones in only two female subjects, ages 13 and 18 years.215,216 None of the persons in the study population had undergone cholecystectomy. The overall prevalence of gallstone disease was 0.13% (0.27% in female subjects). Most cases come to light near the time of puberty, but gallstones have been reported at any age, including during fetal life. Pigmented gallstones predominate in infants and children.213 The conditions associated with an increased risk of cholelithiasis are listed in Table 62-4. An underlying cause of cholelithiasis can be identified in more than one half of children with calculous cholecystitis.
Table 62-4 Conditions Associated with Cholelithiasis in Children and Their Relative Frequencies According to Age*
| 0-12 MONTHS | 1-5 YEARS | 6-21 YEARS |
|---|---|---|
| Abdominal surgery | Hepatobiliary disease | Pregnancy |
| Total parenteral nutrition | Abdominal surgery | Obesity |
| Sepsis | Artificial heart valve | Hemolytic disease |
| Bronchopulmonary dysplasia | Malabsorption | Abdominal surgery |
| Hemolytic disease | Intestinal malabsorption | |
| Intestinal malabsorption | Hepatobiliary disease | |
| Necrotizing enterocolitis | Total parenteral nutrition | |
| Hepatobiliary disease |
* In approximate order of frequency.
Modified from Friesen CA, Roberts CC. Cholelithiasis: Clinical characteristics in children: Case analysis and literature review. Clin Pediatr (Phila) 1989; 28:294-8.
An in-depth discussion of the pathogenesis of gallstones can be found in Chapter 65; however, certain factors may assume greater importance during infancy and childhood.214,217 For example, an increased frequency of calculous cholecystitis is reported in sick premature infants, who often undergo a period of prolonged fasting without frequent stimulation of gallbladder contraction and who require periods of prolonged parenteral nutrition.217 Many of these patients have complicated medical courses that include frequent blood transfusions, episodes of sepsis, abdominal surgery, and use of diuretics and narcotic analgesics. Limited analyses of gallstones in such cases generally have shown the presence of mixed cholesterol-calcium bilirubinate stones.218 In the critically ill infant there may be a continuum from the common occurrence of an enlarged, distended gallbladder filled with sludge to the eventual development of cholelithiasis. As in adults, the incidence of gallstones is increased in children with disease or prior resection of the terminal ileum.218 In a 2007 series, 24% of 30 children with gallstones had calcium carbonate stones, previously considered rare.219
Black pigment gallstones occur commonly in patients who have chronic hemolytic disorders.220 These stones are composed predominantly of calcium bilirubinate, with substantial amounts of crystalline calcium carbonate and phosphate. In sickle cell disease, the risk of gallstones increases with age and occurs in at least 14% of children younger than 10 years and 36% of those between 10 and 20 years.221 The polymorphism in the promoter of the uridine diphosphate (UDP)-glucuronyl transferase 1A1 (UGT1A1) gene that underlies Gilbert’s syndrome, a chronic form of unconjugated hyperbilirubinemia (see Chapter 20), appears to be a major genetic risk factor, increasing the frequency and leading to an earlier age of onset of gallstones in patients with sickle cell disease.222
Obstructive jaundice in infants also may be caused by brown pigment cholelithiasis.223,224 Brown pigment stones are composed of varying proportions of calcium bilirubinate, calcium phosphate, calcium palmitate, cholesterol, and organic material. Unconjugated bilirubin accounts for a large percentage of the total bile biliary pigments. In several cases, bile has had high β-glucuronidase activity and on culture grew an abundant population of several bacteria. Pigment gallstones are postulated to have formed spontaneously in these infants, who had bacterial infections of the biliary tract.
Patients who have no identifiable cause of cholelithiasis are more likely to be female, older, and obese; have a family history of gallbladder disease; and have a greater likelihood of adult-like symptoms.218 Gallstones were detected in 10 of 493 obese children (2%; 8 girls, 2 boys).225 Cholesterol gallstones predominate in these patients. Insights into the pathogenesis of gallstones have been gained through careful studies of Pima Indians, who have an extraordinarily high prevalence of cholesterol gallstones. Highly saturated bile has not been detected among Pima Indians younger than age 13 years, but bile saturation increases significantly in both sexes during pubertal growth and development.226 In this population the sex-related difference in the size of the bile acid pool begins during puberty; young men show a significant rise in the size of the bile acid pool with age, whereas young women show only a slight rise. Because cholesterol gallstones are associated with smaller bile acid pools, the divergence in bile acid pool size between the two sexes also may account for the sex-related difference in the frequency of gallstones, which begins during adolescence.
Prolonged use of high-dose ceftriaxone, a third-generation cephalosporin, has been associated with the formation of calcium-ceftriaxone salt precipitates in the gallbladder. The process, also called biliary pseudolithiasis, is observed in 30% to 40% of children treated with the drug for severe infections.227 Patients may complain of abdominal pain and exhibit signs of intrahepatic cholestasis. Biliary sludge and gallbladder precipitates are found on ultrasonography.228 The problem generally resolves spontaneously with discontinuation of the drug.
Clinical Features
Most gallstones are found in the gallbladder.218 Children have a lower incidence of bile duct stones than adults. Most patients are asymptomatic; the gallstones are discovered either incidentally during the investigation of another problem or during screening because the patient has a condition associated with a high risk of cholelithiasis.214 Patients may complain of intermittent abdominal pain of variable severity; the pain may be localized to the right upper quadrant in older children but is generally poorly localized in infants.229 The physical examination findings are usually unremarkable. Tenderness in the right upper quadrant suggests cholecystitis, as occurs when a stone migrates to the neck of the gallbladder and obstructs the cystic duct. Infants may exhibit irritability, cholestatic jaundice, and acholic stools.214
Liver biochemical test results are usually normal.214 Plain films of the abdomen may reveal calculi, depending on the calcium content of the stone. Ultrasonography is considered the most sensitive and specific imaging technique for the demonstration of gallstones. Hepatobiliary scintigraphy is a valuable adjunct; failure to visualize the gallbladder provides evidence of acute cholecystitis (see later).
Treatment
Cholecystectomy remains the treatment of choice in patients who have symptoms or a nonfunctioning gallbladder.214 Laparoscopic cholecystectomy is done frequently in children and infants as young as 10 months.230 Operative cholangiography and exploration of the bile duct may be indicated on the basis of clinical imaging and operative findings. If choledocholithiasis is demonstrated prior to laparoscopic cholecystectomy in the older child and adolescent, then endoscopic sphincterotomy and stone extraction may be done first.
In asymptomatic patients without biochemical abnormalities (“silent gallstones”), management poses a more difficult problem. Epidemiologic studies and radiocarbon dating of gallstones in adults indicate a lag time of more than one decade between initial formation of a stone and development of symptoms.231 In patients who have underlying disorders such as hemolysis or ileal disease, cholecystectomy may be carried out at the same time as another surgical procedure. In addition, elective laparoscopic cholecystectomy is becoming the norm in children with chronic hemolytic anemias and asymptomatic cholelithiasis to prevent the potential complications of cholecystitis and choledocholithiasis.232 In cases associated with hepatic disease, severe obesity, or cystic fibrosis, the surgical risk of cholecystectomy may be substantial, and clinical judgment must be applied. In these cases, the patient should be counseled about the nature of the disease and the symptoms that may develop. Spontaneous resolution of cholelithiasis and even bile duct stones has been reported in infants. Because recurrence of lithiasis is rare in infants, cholecystectomy may not be required; however, patients with obstructive cholestasis are at risk for sepsis and cholangitis and should undergo surgery.214
Little experience has been reported in children with alternative therapies for gallstones such as medical dissolution with oral bile acid administration or shock-wave lithotripsy. Ursodeoxycholic acid therapy is of no value in the treatment of the predominantly pigment stones found in this age group. Furthermore, ursodeoxycholic acid failed to dissolve radiolucent gallstones in 10 children with cystic fibrosis.233
CALCULOUS CHOLECYSTITIS
Cholelithiasis may be associated with acute or chronic inflammation of the gallbladder (see also Chapter 65).214 Acute cholecystitis is often precipitated by impaction of a stone in the cystic duct. A progressive increase in pressure in the gallbladder secondary to fluid accumulation, the presence of stones, and the chemical irritant effects of bile acids can lead to progressive inflammation, congestion, and vascular compromise. Infarction, gangrene, and perforation can occur. Proliferation of bacteria within the obstructed gallbladder lumen can contribute to the process and lead to biliary sepsis.
Clinical Features
The acute onset of right upper quadrant pain is a constant feature of acute cholecystitis.218 The pain may be poorly localized in infants. Nausea and vomiting are frequent. Children have a higher frequency of jaundice (50%) than do adults. The patient may appear acutely ill with shallow respirations and may be febrile, particularly if bacterial infection is superimposed. Guarding of the abdomen is common, and palpation usually elicits tenderness in the right upper quadrant. Murphy’s sign may be present.
The onset of chronic cholecystitis is usually more indolent. The clinical course may be marked by recurrent episodes of upper abdominal discomfort. Older patients may experience intolerance to fatty foods. In one series, episodes of right upper quadrant pain developed in 64% of children with cholelithiasis and no cystic or bile duct obstruction and was most likely a consequence of chronic cholecystitis.234 Physical examination may yield negative findings or may disclose local tenderness over the gallbladder.
In acute cholecystitis, the white blood cell count is often elevated with a predominance of polymorphonuclear leukocytes.214 Serum bilirubin and alkaline phosphatase levels may be increased. Serum aminotransferase levels may be normal, but high elevations, suggestive of hepatocellular disease, can occur early with acute obstruction of the bile duct.
In patients with chronic cholecystitis, results of the complete blood count and liver biochemical tests are usually normal. In patients with an acute or chronic presentation, a plain film of the abdomen may demonstrate calcifications in the right upper quadrant.214 Abdominal ultrasonography is extremely useful in documenting the presence of stones in the gallbladder, may show thickening of the gallbladder wall, and may demonstrate dilatation of the biliary tract secondary to obstruction of the bile duct by a stone that has migrated from the gallbladder. MRCP may demonstrate similar findings but usually requires general anesthesia in infants and young children.235 Hepatobiliary scintigraphy rarely is necessary in the acutely ill patient but may be of value in demonstrating a malfunctioning gallbladder in patients with chronic cholecystitis.
Treatment
The acutely ill patient should be treated with intravenous fluids, analgesics, and broad-spectrum antibiotics. Cholecystectomy should be performed as soon as fluid deficits are corrected and infection is controlled.236 High-risk, acutely ill patients may benefit from percutaneous drainage via a transhepatic cholecystostomy. The results of surgery are excellent (see also Chapter 66). Care should be taken to exclude bile duct stones by operative cholangiography and, if necessary, exploration of the duct. Laparoscopic bile duct exploration for choledocholithiasis can be performed safely in children at the time of cholecystectomy and can clear all of the bile duct stones in most patients.237
Cholecystectomy is also the treatment of choice for patients with chronic calculous cholecystitis. Laparoscopic cholecystectomy is the preferred approach for most patients.238,239
ACALCULOUS CHOLECYSTITIS
Acalculous cholecystitis is an acute inflammation of the gallbladder without gallstones (see also Chapter 67).240 The disorder is uncommon in children but has been associated with infection or systemic illness. Pathogens have included streptococci (groups A and B); Leptospira interrogans; gram-negative organisms such as Salmonella and Shigella species and Escherichia coli; and parasitic infestations with Ascaris species or Giardia lamblia. In immunocompromised patients, pathogens such as Isospora belli and cytomegalovirus, Cryptosporidium, Aspergillus, and Candida species should be considered. Acalculous cholecystitis may follow abdominal trauma and has been observed in patients with systemic vasculitis, including polyarteritis nodosa, systemic lupus erythematosus, and mucocutaneous lymph node (Kawasaki’s) disease; however, in these conditions, gallbladder distention without inflammation also may occur. Congenital narrowing or inflammation of the cystic duct or external compression by enlarged lymph nodes has been associated with the disorder in children.
Clinical features of acute acalculous cholecystitis include right upper quadrant or epigastric pain, nausea, vomiting, fever, and occasionally jaundice.241 Right upper quadrant guarding and tenderness are present; a tender gallbladder is sometimes palpable. The findings may be less apparent in infants or critically ill patients because the presentation may be obscured by the underlying illness.
Laboratory evaluation may reveal elevated serum levels of alkaline phosphatase and conjugated bilirubin. Leukocytosis may occur. Ultrasonography discloses an enlarged, thick-walled gallbladder that may be distended with sludge but contains no calculi.241
Many patients respond to nonoperative management with nasogastric suction, intravenous fluids, and antibiotics, with resolution of clinical and imaging finding. Cholecystectomy will be required in cases associated with increasing gallbladder wall thickening and distension and with persistence of the nonshadowing echogenic materials or sludge in the gallbladder and pericholecystic fluid.240,241 The diagnosis is confirmed at laparotomy. The gallbladder is usually inflamed, and cultures of bile may yield positive results for the offending bacteria or contain parasites. The gallbladder may become gangrenous. Cholecystostomy drainage may be an alternative approach in critically ill patients.
Some children may present with chronic symptoms of right upper quadrant pain and nausea or vomiting.240 The white blood cell count and results of liver biochemical tests are usually normal. Most patients demonstrate abnormal gallbladder function on radionuclide hepatobiliary scanning.242 These patients generally have chronic inflammation in the gallbladder and require cholecystectomy.
ACUTE HYDROPS OF THE GALLBLADDER
Acute noncalculous, noninflammatory distention of the gallbladder may be observed in infants and children.243,244 The gallbladder is not acutely inflamed, and cultures of the bile are usually sterile. The absence of gallbladder inflammation and generally benign prognosis distinguish acute hydrops from acute acalculous cholecystitis. There may be a generalized mesenteric adenitis of lymph nodes near the cystic duct without mechanical compression. A temporal relationship to other infections, including scarlet fever and leptospirosis, has been observed in some cases.245 Acute hydrops also has been associated with Kawasaki’s disease and Henoch-Schönlein purpura.246 Like acalculous cholecystitis, the disorder can occur in children on prolonged parenteral nutrition. In some cases, a cause is not identified.
Acute hydrops is associated with the acute onset of cramping abdominal pain and often nausea and vomiting.246 Fever and jaundice may be present. The right upper quadrant is usually tender, and the distended gallbladder may be palpable.
The diagnosis of acute hydrops is confirmed in many patients at laparotomy.246 Cholecystectomy obviously is required if the gallbladder appears gangrenous. Pathologic examination of the gallbladder wall usually shows edema and mild inflammation. Cultures of the bile are usually sterile. These benign findings have led some surgeons to treat acute hydrops by a simple cholecystostomy instead of a cholecystectomy246; however, the treatment of gallbladder hydrops frequently is nonsurgical with a focus on supportive care and management of the intercurrent illness. In most patients, particularly in children on total parenteral nutrition in whom enteral feeding has been initiated, the process subsides spontaneously. Ultrasonography has been useful in establishing the diagnosis and following the spontaneous resolution of gallbladder distention. The prognosis is excellent. Gallbladder function can be expected to return to normal in most cases.246
GALLBLADDER DYSKINESIA
Gallbladder dyskinesia is recognized as a cause of chronic abdominal pain in children. The diagnosis is suggested by the presence of postprandial abdominal pain, the absence of cholelithiasis, and an abnormal ejection fraction on cholecystokinin-stimulated hepatobiliary scintigraphy. Pain relief after cholecystectomy has been variable in several reports.247 Gallbladder ejection fractions of less than 35% to 50% have sometimes been considered abnormal and an indication for surgery (see Chapter 63).
Gallbladder dyskinesia was the most common indication for surgery in 62 (58%) of the 107 children who underwent cholecystectomy in one series.248 In another published report of 51 children who underwent laparoscopic cholecystectomy for gallbladder dyskinesia after exclusion of more common gastrointestinal disorders, 27 of 38 (71%) patients available for follow-up experienced complete relief of symptoms.249 The presence of nausea, upper abdominal pain, and a gallbladder ejection fraction of less than 15% most reliably predicted benefit from cholecystectomy (positive predictive value of 93%). Histologic evidence of chronic cholecystitis was found in only 10 of 27 (41%) children with complete relief of symptoms and was not an independent predictor of a successful outcome. The presence of chronic inflammation in these patients suggests that they may have had a chronic acalculous cholecystitis rather than gallbladder dysmotility.
Adkins RBJr, Chapman WC, Reddy VS. Embryology, anatomy, and surgical applications of the extrahepatic biliary system. Surg Clin North Am. 2000;80:363-79. (Ref 23.)
Avisse C, Flament JB, Delattre JF. Ampulla of Vater. Anatomic, embryologic, and surgical aspects. Surg Clin North Am. 2000;80:201-12. (Ref 25.)
Bezerra JA, Balistreri WF. Cholestatic syndromes of infancy and childhood. Semin Gastrointest Dis. 2001;12:54-65. (Ref 39.)
Bogert PT, LaRusso NF. Cholangiocyte biology. Curr Opin Gastroenterol. 2007;23:299-305. (Ref 16.)
Davenport M. Biliary atresia. Semin Pediatr Surg. 2005;14:42-8. (Ref 85.)
Edil BH, Cameron JL, Reddy S, et al. Choledochal cyst disease in children and adults: A 30-year single-institution experience. J Am Coll Surg. 2008;206:1000-5. (Ref 148.)
Emerick KM, Rand EB, Goldmuntz E, et al. Features of Alagille syndrome in 92 patients: Frequency and relation to prognosis. Hepatology. 1999;29:822-9. (Ref 182.)
Feldstein AE, Perrault J, El-Youssif M, et al. Primary sclerosing cholangitis in children: A long-term follow-up study. Hepatology. 2003;38:210-17. (Ref 120.)
Imamoglu M, Sarihan H, Sari A, Ahmetoglu A. Acute acalculous cholecystitis in children: Diagnosis and treatment. J Pediatr Surg. 2002;37:36-9. (Ref 241.)
Kaechele V, Wabitsch M, Thiere D, et al. Prevalence of gallbladder stone disease in obese children and adolescents: Influence of the degree of obesity, sex, and pubertal development. J Pediatr Gastroenterol Nutr. 2006;42:66-70. (Ref 225.)
Kerkar N, Norton K, Suchy FJ. The hepatic fibrocystic diseases. Clin Liver Dis. 2006;10:55-71. (Ref 154.)
Lykavieris P, Hadchouel M, Chardot C, Bernard O. Outcome of liver disease in children with Alagille syndrome: A study of 163 patients. Gut. 2001;49:431-5. (Ref 197.)
Ng VL, Balistreri WF. Treatment options for chronic cholestasis in infancy and childhood. Curr Treat Options Gastroenterol. 2005;8:419-30. (Ref 200.)
Roskams TA, Theise ND, Balabaud C, et al. Nomenclature of the finer branches of the biliary tree: Canals, ductules, and ductular reactions in human livers. Hepatology. 2004;39:1739-45. (Ref 18.)
Shneider BL, Mazariegos GV. Biliary atresia: A transplant perspective. Liver Transpl. 2007;13:1482-95. (Ref 108.)
Sokol RJ, Mack C, Narkewicz MR, Karrer FM. Pathogenesis and outcome of biliary atresia: Current concepts. J Pediatr Gastroenterol Nutr. 2003;37:4-21. (Ref 52.)
Stringer MD, Taylor DR, Soloway RD. Gallstone composition: Are children different? J Pediatr. 2003;142:435-40. (Ref 216.)
Tanimizu N, Miyajima A. Molecular mechanism of liver development and regeneration. Int Rev Cytol. 2007;259:1-48. (Ref 5.)
1. Moore K, editor. The developing human: Clinically oriented embryology, 4th ed, Philadelphia: Saunders, 1988.
2. Calmont A, Wandzioch E, Tremblay KD, et al. An FGF response pathway that mediates hepatic gene induction in embryonic endoderm cells. Dev Cell. 2006;11:339-48.
3. Zaret KS. Genetic programming of liver and pancreas progenitors: Lessons for stem-cell differentiation. Nat Rev Genet. 2008;9:329-40.
4. Duncan SA. Mechanisms controlling early development of the liver. Mech Dev. 2003;120:19-33.
5. Tanimizu N, Miyajima A. Molecular mechanism of liver development and regeneration. Int Rev Cytol. 2007;259:1-48.
6. Costa RH, Kalinichenko VV, Holterman AX, Wang X. Transcription factors in liver development, differentiation, and regeneration. Hepatology. 2003;38:1331-47.
7. Bort R, Signore M, Tremblay K, et al. Hex homeobox gene controls the transition of the endoderm to a pseudostratified, cell emergent epithelium for liver bud development. Dev Biol. 2006;290:44-56.
8. Parviz F, Matullo C, Garrison WD, et al. Hepatocyte nuclear factor 4alpha controls the development of a hepatic epithelium and liver morphogenesis. Nat Genet. 2003;34:292-6.
9. Tan CE, Moscoso GJ. The developing human biliary system at the porta hepatis level between 11 and 25 weeks of gestation: A way to understanding biliary atresia. Part 2. Pathol Int. 1994;44:600-10.
10. Tan CE, Moscoso GJ. The developing human biliary system at the porta hepatis level between 29 days and 8 weeks of gestation: A way to understanding biliary atresia. Part 1. Pathol Int. 1994;44:587-99.
11. Faa G, Van Eyken P, Roskams T, et al. Expression of cytokeratin 20 in developing rat liver and in experimental models of ductular and oval cell proliferation. J Hepatol. 1998;29:628-33.
12. Desmet VJ. Ludwig symposium on biliary disorders—Part I. Pathogenesis of ductal plate abnormalities. Mayo Clin Proc. 1998;73:80-9.
13. Ludwig J, Ritman EL, LaRusso NF, et al. Anatomy of the human biliary system studied by quantitative computer-aided three-dimensional imaging techniques. Hepatology. 1998;27:893-9.
14. Alpini G, McGill JM, Larusso NF. The pathobiology of biliary epithelia. Hepatology. 2002;35:1256-68.
15. Marzioni M, Glaser SS, Francis H, et al. Functional heterogeneity of cholangiocytes. Semin Liver Dis. 2002;22:227-40.
16. Bogert PT, LaRusso NF. Cholangiocyte biology. Curr Opin Gastroenterol. 2007;23:299-305.
17. Jones AL, Schmucker DL, Renston RH, Murakami T. The architecture of bile secretion. A morphological perspective of physiology. Dig Dis Sci. 1980;25:609-29.
18. Roskams TA, Theise ND, Balabaud C, et al. Nomenclature of the finer branches of the biliary tree: Canals, ductules, and ductular reactions in human livers. Hepatology. 2004;39:1739-45.
19. Saxena R, Theise N. Canals of Hering: Recent insights and current knowledge. Semin Liver Dis. 2004;24:43-8.
20. Donato P, Coelho P, Rodrigues H, et al. Normal vascular and biliary hepatic anatomy: 3D demonstration by multidetector CT. Surg Radiol Anat. 2007;29:575-82.
21. Healey JEJr, Schroy PC. Anatomy of the biliary ducts within the human liver; analysis of the prevailing pattern of branchings and the major variations of the biliary ducts. AMA Arch Surg. 1953;66:599-616.
22. Jones A, Spring-Mills E. The liver and gallbladder. In: Weiss L, editor. Modern concepts of gastrointestinal histology. New York: Elsevier; 1983:738.
23. Adkins RBJr, Chapman WC, Reddy VS. Embryology, anatomy, and surgical applications of the extrahepatic biliary system. Surg Clin North Am. 2000;80:363-79.
24. Masunari H, Shimada H, Endo I, et al. Surgical anatomy of hepatic hilum with special reference of the plate system and extrahepatic duct. J Gastrointest Surg. 2008;12:1047-53.
25. Avisse C, Flament JB, Delattre JF. Ampulla of Vater. Anatomic, embryologic, and surgical aspects. Surg Clin North Am. 2000;80:201-12.
26. Takasaki S, Hano H. Three-dimensional observations of the human hepatic artery (arterial system in the liver). J Hepatol. 2001;34:455-66.
27. Frierson HFJr. The gross anatomy and histology of the gallbladder, extrahepatic bile ducts, Vaterian system, and minor papilla. Am J Surg Pathol. 1989;13:146-62.
28. Delattre JF, Avisse C, Flament JB. Anatomic basis of hepatic surgery. Surg Clin North Am. 2000;80:345-62.
29. Gordon KC. A comparative anatomical study of the distribution of the cystic artery in man and other species. J Anat. 1967;101:351-9.
30. Evett RD, Higgins JA, Brown ALJr. The fine structure of normal mucosa in human gall bladder. Gastroenterology. 1964;47:49-60.
31. Lamah M, Karanjia ND, Dickson GH. Anatomical variations of the extrahepatic biliary tree: Review of the world literature. Clin Anat. 2001;14:167-72.
32. Meilstrup JW, Hopper KD, Thieme GA. Imaging of gallbladder variants. AJR Am J Roentgenol. 1991;157:205-8.
33. Alicioglu B. An incidental case of triple gallbladder. World J Gastroenterol. 2007;13:2004-6.
34. Arya G, Balistreri WF. Pediatric liver disease in the United States: Epidemiology and impact. J Gastroenterol Hepatol. 2002;17:521-5.
35. Arrese M, Ananthananarayanan M, Suchy FJ. Hepatobiliary transport: molecular mechanisms of development and cholestasis. Pediatr Res. 1998;44:141-7.
36. Davenport M, Betalli P, D’Antiga L, et al. The spectrum of surgical jaundice in infancy. J Pediatr Surg. 2003;38:1471-9.
37. Suchy FJ. Clinical problems with developmental anomalies of the biliary tract. Semin Gastrointest Dis. 2003;14:156-64.
38. Birnbaum A, Suchy FJ. The intrahepatic cholangiopathies. Semin Liver Dis. 1998;18:263-9.
39. Bezerra JA, Balistreri WF. Cholestatic syndromes of infancy and childhood. Semin Gastrointest Dis. 2001;12:54-65.
40. Suchy FJ. Neonatal cholestasis. Pediatr Rev. 2004;25:388-96.
41. Rosenthal P. Assessing liver function and hyperbilirubinemia in the newborn. National Academy of Clinical Biochemistry. Clin Chem. 1997;43:228-34.
42. Park WH, Choi SO, Lee HJ. Technical innovation for noninvasive and early diagnosis of biliary atresia: The ultrasonographic “triangular cord” sign. J Hepatobiliary Pancreat Surg. 2001;8:337-41.
43. Kim MJ, Park YN, Han SJ, et al. Biliary atresia in neonates and infants: triangular area of high signal intensity in the porta hepatis at T2-weighted MR cholangiography with US and histopathologic correlation. Radiology. 2000;215:395-401.
44. Paltiel HJ. Imaging of neonatal cholestasis. Semin Ultrasound CT MR. 1994;15:290-305.
45. Jaw TS, Kuo YT, Liu GC, et al. MR cholangiography in the evaluation of neonatal cholestasis. Radiology. 1999;212:249-56.
46. Norton KI, Glass RB, Kogan D, et al. MR cholangiography in the evaluation of neonatal cholestasis: Initial results. Radiology. 2002;222:687-91.
47. Gilmour SM, Hershkop M, Reifen R, et al. Outcome of hepatobiliary scanning in neonatal hepatitis syndrome. J Nucl Med. 1997;38:1279-82.
48. Nwomeh BC, Caniano DA, Hogan M. Definitive exclusion of biliary atresia in infants with cholestatic jaundice: The role of percutaneous cholecysto-cholangiography. Pediatr Surg Int. 2007;23:845-9.
49. Iinuma Y, Narisawa R, Iwafuchi M, et al. The role of endoscopic retrograde cholangiopancreatography in infants with cholestasis. J Pediatr Surg. 2000;35:545-9.
50. Zerbini MC, Gallucci SD, Maezono R, et al. Liver biopsy in neonatal cholestasis: A review on statistical grounds. Mod Pathol. 1997;10:793-9.
51. Kahn E. Biliary atresia revisited. Pediatr Dev Pathol. 2004;7:109-24.
52. Sokol RJ, Mack C, Narkewicz MR, Karrer FM. Pathogenesis and outcome of biliary atresia: Current concepts. J Pediatr Gastroenterol Nutr. 2003;37:4-21.
53. Utterson EC, Shepherd RW, Sokol RJ, et al. Biliary atresia: Clinical profiles, risk factors, and outcomes of 755 patients listed for liver transplantation. J Pediatr. 2005;147:180-5.
54. Chardot C, Carton M, Spire-Bendelac N, et al. Epidemiology of biliary atresia in France: A national study 1986-96. J Hepatol. 1999;31:1006-13.
55. The NS, Honein MA, Caton AR, et al. Risk factors for isolated biliary atresia, National Birth Defects Prevention Study, 1997-2002. Am J Med Genet A. 2007;143A:2274-84.
56. Sokol RJ, Mack C. Etiopathogenesis of biliary atresia. Semin Liver Dis. 2001;21:517-24.
57. Glaser JH, Morecki R. Reovirus type 3 and neonatal cholestasis. Semin Liver Dis. 1987;7:100-7.
58. Morecki R, Glaser J. Reovirus 3 and neonatal biliary disease: Discussion of divergent results. Hepatology. 1989;10:515-17.
59. Tyler KL, Sokol RJ, Oberhaus SM, et al. Detection of reovirus RNA in hepatobiliary tissues from patients with extrahepatic biliary atresia and choledochal cysts. Hepatology. 1998;27:1475-82.
60. Riepenhoff-Talty M, Gouvea V, Evans MJ, et al. Detection of group C rotavirus in infants with extrahepatic biliary atresia. J Infect Dis. 1996;174:8-15.
61. Silveira TR, Salzano FM, Donaldson PT, et al. Association between HLA and extrahepatic biliary atresia. J Pediatr Gastroenterol Nutr. 1993;16:114-17.
62. Bezerra JA, Tiao G, Ryckman FC, et al. Genetic induction of proinflammatory immunity in children with biliary atresia. Lancet. 2002;360:1653-9.
63. Mack CL, Falta MT, Sullivan AK, et al. Oligoclonal expansions of CD4+ and CD8+ T-cells in the target organ of patients with biliary atresia. Gastroenterology. 2007;133:278-87.
64. Shivakumar P, Campbell KM, Sabla GE, et al. Obstruction of extrahepatic bile ducts by lymphocytes is regulated by IFN-gamma in experimental biliary atresia. J Clin Invest. 2004;114:322-9.
65. Mack CL, Tucker RM, Lu BR, et al. Cellular and humoral autoimmunity directed at bile duct epithelia in murine biliary atresia. Hepatology. 2006;44:1231-9.
66. Narayanaswamy B, Gonde C, Tredger JM, et al. Serial circulating markers of inflammation in biliary atresia—Evolution of the post-operative inflammatory process. Hepatology. 2007;46:180-7.
67. Davenport M, Savage M, Mowat AP, Howard ER. Biliary atresia splenic malformation syndrome: An etiologic and prognostic subgroup. Surgery. 1993;113:662-8.
68. Tanano H, Hasegawa T, Kawahara H, et al. Biliary atresia associated with congenital structural anomalies. J Pediatr Surg. 1999;34:1687-90.
69. Jacquemin E, Cresteil D, Raynaud N, Hadchouel M. CFCI gene mutation and biliary atresia with polysplenia syndrome. J Pediatr Gastroenterol Nutr. 2002;34:326-7.
70. Davit-Spraul A, Baussan C, Hermeziu B, et al. CFC1 gene involvement in biliary atresia with polysplenia syndrome. J Pediatr Gastroenterol Nutr. 2008;46:111-12.
71. Schon P, Tsuchiya K, Lenoir D, et al. Identification, genomic organization, chromosomal mapping and mutation analysis of the human INV gene, the ortholog of a murine gene implicated in left-right axis development and biliary atresia. Hum Genet. 2002;110:157-65.
72. Zhang DY, Sabla G, Shivakumar P, et al. Coordinate expression of regulatory genes differentiates embryonic and perinatal forms of biliary atresia. Hepatology. 2004;39:954-62.
73. Kohsaka T, Yuan ZR, Guo SX, et al. The significance of human jagged 1 mutations detected in severe cases of extrahepatic biliary atresia. Hepatology. 2002;36:904-12.
74. Low Y, Vijayan V, Tan CE. The prognostic value of ductal plate malformation and other histologic parameters in biliary atresia: An immunohistochemical study. J Pediatr. 2001;139:320-2.
75. Sokol RJ, Shepherd RW, Superina R, et al. Screening and outcomes in biliary atresia: Summary of a National Institutes of Health workshop. Hepatology. 2007;46:566-81.
76. Kasai M. Treatment of biliary atresia with special reference to hepatic porto-enterostomy and its modifications. Prog Pediatr Surg. 1974;6:5-52.
77. Wadhwani SI, Turmelle YP, Nagy R, et al. Prolonged neonatal jaundice and the diagnosis of biliary atresia: A single-center analysis of trends in age at diagnosis and outcomes. Pediatrics. 2008;121:e1438-40.
78. Hussein M, Howard ER, Mieli-Vergani G, Mowat AP. Jaundice at 14 days of age: Exclude biliary atresia. Arch Dis Child. 1991;66:1177-9.
79. Middlesworth W, Altman RP. Biliary atresia. Curr Opin Pediatr. 1997;9:265-9.
80. Ryckman FC, Alonso MH, Bucuvalas JC, Balistreri WF. Biliary atresia—Surgical management and treatment options as they relate to outcome. Liver Transpl Surg. 1998;4:S24-33.
81. Howard ER, Davenport M. The treatment of biliary atresia in Europe 1969-1995. Tohoku J Exp Med. 1997;181:75-83.
82. Ohi R. A history of the Kasai operation: Hepatic portoenterostomy for biliary atresia. World J Surg. 1988;12:871-4.
83. Kasai M, Mochizuki I, Ohkohchi N, Chiba T, Ohi R. Surgical limitation for biliary atresia: indication for liver transplantation. J Pediatr Surg. 1989;24:851-4.
84. Ohi R. Surgery for biliary atresia. Liver. 2001;21:175-82.
85. Davenport M. Biliary atresia. Semin Pediatr Surg. 2005;14:42-8.
86. Muraji T, Nio M, Ohhama Y, et al. Postoperative corticosteroid therapy for bile drainage in biliary atresia—A nationwide survey. J Pediatr Surg. 2004;39:1803-5.
87. Stringer MD, Davison SM, Rajwal SR, McClean P. Kasai portoenterostomy: 12-year experience with a novel adjuvant therapy regimen. J Pediatr Surg. 2007;42:1324-8.
88. Davenport M, Stringer MD, Tizzard SA, et al. Randomized, double-blind, placebo-controlled trial of corticosteroids after Kasai portoenterostomy for biliary atresia. Hepatology. 2007;46:1821-7.
89. Davenport M, Caponcelli E, Livesey E, et al. Surgical outcome in biliary atresia: Etiology affects the influence of age at surgery. Ann Surg. 2008;247:694-8.
90. Karrer FM, Lilly JR, Stewart BA, Hall RJ. Biliary atresia registry, 1976 to 1989. J Pediatr Surg. 1990;25:1076-80.
91. Nio M, Ohi R, Miyano T, et al. Five- and 10-year survival rates after surgery for biliary atresia: A report from the Japanese Biliary Atresia Registry. J Pediatr Surg. 2003;38:997-1000.
92. Chardot C, Carton M, Spire-Bendelac N, et al. Prognosis of biliary atresia in the era of liver transplantation: French national study from 1986 to 1996. Hepatology. 1999;30:606-11.
93. Chardot C, Serinet MO. Prognosis of biliary atresia: what can be further improved? J Pediatr. 2006;148:432-5.
94. Wildhaber BE, Majno P, Mayr J, et al. Biliary atresia: Swiss national study, 1994-2004. J Pediatr Gastroenterol Nutr. 2008;46:299-307.
95. Davenport M, Puricelli V, Farrant P, et al. The outcome of the older (≥100 days) infant with biliary atresia. J Pediatr Surg. 2004;39:575-81.
96. Karrer FM, Price MR, Bensard DD, et al. Long-term results with the Kasai operation for biliary atresia. Arch Surg. 1996;131:493-6.
97. Subramaniam R, Doig CM, Bowen J, Bruce J. Initial response to portoenterostomy determines long-term outcome in patients with biliary atresia. J Pediatr Surg. 2000;35:593-7.
98. Ohhama Y, Shinkai M, Fujita S, et al. Early prediction of long-term survival and the timing of liver transplantation after the Kasai operation. J Pediatr Surg. 2000;35:1031-4.
99. Shneider BL, Brown MB, Haber B, et al. A multicenter study of the outcome of biliary atresia in the United States, 1997 to 2000. J Pediatr. 2006;148:467-74.
100. Langenburg SE, Poulik J, Goretsky M, et al. Bile duct size does not predict success of portoenterostomy for biliary atresia. J Pediatr Surg. 2000;35:1006-7.
101. Schweizer P, Kirschner H, Schittenhelm C. Anatomy of the porta hepatis as a basis for extended hepatoporto-enterostomy for extrahepatic biliary atresia—A new surgical technique. Eur J Pediatr Surg. 2001;11:15-18.
102. Schweizer P, Kirschner HJ, Schittenhelm C. Anatomy of the porta hepatis (PH) as rational basis for the hepatoporto-enterostomy (HPE). Eur J Pediatr Surg. 1999;9:13-18.
103. Vazquez-Estevez J, Stewart B, Shikes RH, et al. Biliary atresia: early determination of prognosis. J Pediatr Surg. 1989;24:48-50.
104. Wu ET, Chen HL, Ni YH, et al. Bacterial cholangitis in patients with biliary atresia: Impact on short-term outcome. Pediatr Surg Int. 2001;17:390-5.
105. Bu LN, Chen HL, Chang CJ, et al. Prophylactic oral antibiotics in prevention of recurrent cholangitis after the Kasai portoenterostomy. J Pediatr Surg. 2003;38:590-3.
106. Miga D, Sokol RJ, Mackenzie T, et al. Survival after first esophageal variceal hemorrhage in patients with biliary atresia. J Pediatr. 2001;139:291-6.
107. van Heurn LW, Saing H, Tam PK. Portoenterostomy for biliary atresia: Long-term survival and prognosis after esophageal variceal bleeding. J Pediatr Surg. 2004;39:6-9.
108. Shneider BL, Mazariegos GV. Biliary atresia: A transplant perspective. Liver Transpl. 2007;13:1482-95.
109. Otte JB, de Ville de Goyet J, Reding R, et al. Sequential treatment of biliary atresia with Kasai portoenterostomy and liver transplantation: a review. Hepatology. 1994;20:41S-8S.
110. Sandler AD, Azarow KS, Superina RA. The impact of a previous Kasai procedure on liver transplantation for biliary atresia. J Pediatr Surg. 1997;32:416-19.
111. Miller CM, Gondolesi GE, Florman S, et al. One hundred nine living donor liver transplants in adults and children: A single-center experience. Ann Surg. 2001;234:301-11.
112. Diem HV, Evrard V, Vinh HT, et al. Pediatric liver transplantation for biliary atresia: Results of primary grafts in 328 recipients. Transplantation. 2003;75:1692-7.
113. Sahnoun L, Belghith M, Jouini R, et al. Spontaneous perforation of the extrahepatic bile duct in infancy: Report of two cases and literature review. Eur J Pediatr Surg. 2007;17:132-5.
114. Chardot C, Iskandarani F, De Dreuzy O, et al. Spontaneous perforation of the biliary tract in infancy: A series of 11 cases. Eur J Pediatr Surg. 1996;6:341-6.
115. Gauthier F, Chardot C, Branchereau S. [Role of the surgeon in the treatment of neuroblastoma in children]. Arch Pediatr. 1996;1:329s-30s.
116. Holland RM, Lilly JR. Surgical jaundice in infants: Other than biliary atresia. Semin Pediatr Surg. 1992;1:125-9.
117. Bernstein J, Braylan R, Brough AJ. Bile-plug syndrome: A correctable cause of obstructive jaundice in infants. Pediatrics. 1969;43:273-6.
118. Karrer FM, Hall RJ, Stewart BA, Lilly JR. Congenital biliary tract disease. Surg Clin North Am. 1990;70:1403-18.
119. Gregorio GV, Portmann B, Karani J, et al. Autoimmune hepatitis/sclerosing cholangitis overlap syndrome in childhood: A 16-year prospective study. Hepatology. 2001;33:544-53.
120. Feldstein AE, Perrault J, El-Youssif M, et al. Primary sclerosing cholangitis in children: A long-term follow-up study. Hepatology. 2003;38:210-17.
121. Maggs JR, Chapman RW. An update on primary sclerosing cholangitis. Curr Opin Gastroenterol. 2008;24:377-83.
122. Debray D, Pariente D, Urvoas E, et al. Sclerosing cholangitis in children. J Pediatr. 1994;124:49-56.
123. Stenhammar L, Hogberg L, Lewander P. Primary sclerosing cholangitis in childhood inflammatory bowel disease. Arch Dis Child. 1994;71:281-2.
124. Ludvigsson JF, Elfstrom P, Broome U, et al. Celiac disease and risk of liver disease: A general population-based study. Clin Gastroenterol Hepatol. 2007;5:63-9.
125. Squires RHJr, Weinberg AG, Zwiener RJ, Winick N. Langerhans’ cell histiocytosis presenting with hepatic dysfunction. J Pediatr Gastroenterol Nutr. 1993;16:190-3.
126. Braier J, Ciocca M, Latella A, et al. Cholestasis, sclerosing cholangitis, and liver transplantation in Langerhans cell histiocytosis. Med Pediatr Oncol. 2002;38:178-82.
127. Staines JD, Evans TJ. AIDS related cholangitis. Scott Med J. 1995;40:73.
128. Goddard EA, Mouton SC, Westwood AT, et al. Cryptosporidiosis of the gastrointestinal tract associated with sclerosing cholangitis in the absence of documented immunodeficiency: Cryptosporidium parvum and sclerosing cholangitis in an immunocompetent child. J Pediatr Gastroenterol Nutr. 2000;31:317-20.
129. Mieli-Vergani G, Vergani D. Sclerosing cholangitis in the paediatric patient. Best Pract Res Clin Gastroenterol. 2001;15:681-90.
130. Wilschanski M, Chait P, Wade JA, et al. Primary sclerosing cholangitis in 32 children: Clinical, laboratory, and radiographic features, with survival analysis. Hepatology. 1995;22:1415-22.
131. Batres LA, Russo P, Mathews M, et al. Primary sclerosing cholangitis in children: A histologic follow-up study. Pediatr Dev Pathol. 2005;8:568-76.
132. Terjung B, Bogsch F, Klein R, et al. Diagnostic accuracy of atypical p-ANCA in autoimmune hepatitis using ROC and multivariate regression analysis. Eur J Med Res. 2004;9:439-48.
133. Ferrara C, Valeri G, Salvolini L, Giovagnoni A. Magnetic resonance cholangiopancreatography in primary sclerosing cholangitis in children. Pediatr Radiol. 2002;32:413-17.
134. Meier PN. ERCP topics. Endoscopic retrograde cholangiopancreatography. Endoscopy. 2000;32:863-73.
135. Kaplan GG, Laupland KB, Butzner D, et al. The burden of large and small duct primary sclerosing cholangitis in adults and children: a population-based analysis. Am J Gastroenterol. 2007;102:1042-9.
136. Michaels A, Levy C. The medical management of primary sclerosing cholangitis. Medscape. J Med. 2008;10:61.
137. Liden H, Norrby J, Friman S, Olausson M. Liver transplantation for primary sclerosing cholangitis—A single-center experience. Transpl Int. 2000;1:S162-4.
138. O’Neill JAJr. Choledochal cyst. Curr Probl Surg. 1992;29:361-410.
139. Altman RP. Choledochal cyst. Semin Pediatr Surg. 1992;1:130-3.
140. Miyano T, Yamataka A. Choledochal cysts. Curr Opin Pediatr. 1997;9:283-8.
141. Todani T, Watanabe Y, Narusue M, et al. Congenital bile duct cysts: Classification, operative procedures, and review of thirty-seven cases including cancer arising from choledochal cyst. Am J Surg. 1977;134:263-9.
142. Savader SJ, Benenati JF, Venbrux AC, et al. Choledochal cysts: Classification and cholangiographic appearance. AJR Am J Roentgenol. 1991;156:327-31.
143. Todani T, Watanabe Y, Toki A, Morotomi Y. Classification of congenital biliary cystic disease: Special reference to type Ic and IVA cysts with primary ductal stricture. J Hepatobiliary Pancreat Surg. 2003;10:340-4.
144. Landing BH. Considerations of the pathogenesis of neonatal hepatitis, biliary atresia and choledochal cyst-the concept of infantile obstructive cholangiopathy. Prog Pediatr Surg. 1975;6:113-39.
145. Tashiro S, Imaizumi T, Ohkawa H, et al. Pancreaticobiliary maljunction: Retrospective and nationwide survey in Japan. J Hepatobiliary Pancreat Surg. 2003;10:345-51.
146. Goilav B, Norton KI, Satlin LM, et al. Predominant extrahepatic biliary disease in autosomal recessive polycystic kidney disease: A new association. Pediatr Transplant. 2006;10:294-8.
147. Watanabe Y, Toki A, Todani T. Bile duct cancer developed after cyst excision for choledochal cyst. J Hepatobiliary Pancreat Surg. 1999;6:207-12.
148. Edil BH, Cameron JL, Reddy S, et al. Choledochal cyst disease in children and adults: A 30-year single-institution experience. J Am Coll Surg. 2008;206:1000-5.
149. Todani T, Urushihara N, Morotomi Y, et al. Characteristics of choledochal cysts in neonates and early infants. Eur J Pediatr Surg. 1995;5:143-5.
150. Kaneko K, Ono Y, Tainaka T, Sumida W, Ando H. Fatty acid calcium stones in patients with pancreaticobiliary maljunction/choledochal cyst as another cause of obstructive symptoms besides protein plugs. J Pediatr Surg. 2008;43:564-7.
151. Shaffer E. Can MRCP replace ERCP in the diagnosis of congenital bile-duct cysts? Nat Clin Pract Gastroenterol Hepatol. 2006;3:76-7.
152. Fitoz S, Erden A, Boruban S. Magnetic resonance cholangiopancreatography of biliary system abnormalities in children. Clin Imaging. 2007;31:93-101.
153. Summerfield JA, Nagafuchi Y, Sherlock S, et al. Hepatobiliary fibropolycystic diseases. A clinical and histological review of 51 patients. J Hepatol. 1986;2:141-56.
154. Kerkar N, Norton K, Suchy FJ. The hepatic fibrocystic diseases. Clin Liver Dis. 2006;10:55-71.
155. Gunay-Aygun M, Avner ED, Bacallao RL, et al. Autosomal recessive polycystic kidney disease and congenital hepatic fibrosis: Summary statement of a first National Institutes of Health/Office of Rare Diseases conference. J Pediatr. 2006;149:159-64.
156. Guay-Woodford LM, Desmond RA. Autosomal recessive polycystic kidney disease: The clinical experience in North America. Pediatrics. 2003;111:1072-80.
157. Bergmann C, Senderek J, Kupper F, et al. PKHD1 mutations in autosomal recessive polycystic kidney disease (ARPKD). Hum Mutat. 2004;23:453-63.
158. Torres VE, Harris PC. Polycystic kidney disease: Genes, proteins, animal models, disease mechanisms and therapeutic opportunities. J Intern Med. 2007;261:17-31.
159. D’Agata ID, Jonas MM, Perez-Atayde AR, Guay-Woodford LM. Combined cystic disease of the liver and kidney. Semin Liver Dis. 1994;14:215-28.
160. Desmet VJ. What is congenital hepatic fibrosis? Histopathology. 1992;20:465-77.
161. Miller WJ, Sechtin AG, Campbell WL, Pieters PC. Imaging findings in Caroli’s disease. AJR Am J Roentgenol. 1995;165:333-7.
162. Guy F, Cognet F, Dranssart M, et al. Caroli’s disease: Magnetic resonance imaging features. Eur Radiol. 2002;12:2730-6.
163. Ros E, Navarro S, Bru C, et al. Ursodeoxycholic acid treatment of primary hepatolithiasis in Caroli’s syndrome. Lancet. 1993;342:404-6.
164. Bordas JM, Llach J, Gines A, et al. The role of therapeutic endoscopy associated with extracorporeal shock-wave lithotripsy and bile acid treatment in the management of Caroli’s disease. Endoscopy. 1999;31:280.
165. Kassahun WT, Kahn T, Wittekind C, et al. Caroli’s disease: liver resection and liver transplantation. Experience in 33 patients. Surgery. 2005;138:888-98.
166. Kasper HU, Stippel DL, Tox U, et al. [Primary cholangiocarcinoma in a case of Caroli’s disease: case report and literature review]. Pathologe. 2006;27:300-4.
167. Millwala F, Segev DL, Thuluvath PJ. Caroli’s disease and outcomes after liver transplantation. Liver Transpl. 2008;14:11-17.
168. Kahn E, Daum F, Markowitz J, et al. Nonsyndromatic paucity of interlobular bile ducts: Light and electron microscopic evaluation of sequential liver biopsies in early childhood. Hepatology. 1986;6:890-901.
169. Yehezkely-Schildkraut V, Munichor M, Mandel H, et al. Nonsyndromic paucity of interlobular bile ducts: report of 10 patients. J Pediatr Gastroenterol Nutr. 2003;37:546-9.
170. O’Reilly K, Ahmed SF, Murday V, McGrogan P. Biliary hypoplasia in Williams syndrome. Arch Dis Child. 2006;91:420-1.
171. Rasmussen KJ, Zador I, Zabel C, et al. Bile duct anomalies in a male child with Noonan syndrome: A case for ras and notch pathway synergism. Am J Med Genet A. 2008;146A:261-3.
172. Desmet VJ, van Eyken P, Roskams T. Histopathology of vanishing bile duct diseases. Adv Clin Path. 1998;2:87-99.
173. Hadchouel M. Paucity of interlobular bile ducts. Semin Diagn Pathol. 1992;9:24-30.
174. Alagille D, Estrada A, Hadchouel M, et al. Syndromic paucity of interlobular bile ducts (Alagille syndrome or arteriohepatic dysplasia): Review of 80 cases. J Pediatr. 1987;110:195-200.
175. Krantz ID, Piccoli DA, Spinner NB. Clinical and molecular genetics of Alagille syndrome. Curr Opin Pediatr. 1999;11:558-64.
176. Oda T, Elkahloun AG, Pike BL, et al. Mutations in the human Jagged1 gene are responsible for Alagille syndrome. Nat Genet. 1997;16:235-42.
177. Spinner NB. Alagille syndrome and the notch signaling pathway: New insights into human development. Gastroenterology. 1999;116:1257-60.
178. McDaniell R, Warthen DM, Sanchez-Lara PA, et al. NOTCH2 mutations cause Alagille syndrome, a heterogeneous disorder of the notch signaling pathway. Am J Hum Genet. 2006;79:169-73.
179. Spinner NB, Colliton RP, Crosnier C, et al. Jagged1 mutations in Alagille syndrome. Hum Mutat. 2001;17:18-33.
180. Dhorne-Pollet S, Deleuze JF, Hadchouel M, Bonaiti-Pellie C. Segregation analysis of Alagille syndrome. J Med Genet. 1994;31:453-7.
181. Alagille D. Alagille syndrome today. Clin Invest Med. 1996;19:325-30.
182. Emerick KM, Rand EB, Goldmuntz E, et al. Features of Alagille syndrome in 92 patients: Frequency and relation to prognosis. Hepatology. 1999;29:822-9.
183. Kamath BM, Loomes KM, Oakey RJ, et al. Facial features in Alagille syndrome: Specific or cholestasis facies? Am J Med Genet. 2002;112:163-70.
184. Bucuvalas JC, Horn JA, Carlsson L, et al. Growth hormone insensitivity associated with elevated circulating growth hormone-binding protein in children with Alagille syndrome and short stature. J Clin Endocrinol Metab. 1993;76:1477-82.
185. Goldmuntz E, Moore E, Spinner NB. The cardiovascular manifestations of Alagille syndrome and JAGI mutations. Methods Mol Med. 2006;126:217-31.
186. Kahn E. Paucity of interlobular bile ducts. Arteriohepatic dysplasia and nonsyndromic duct paucity. Perspect Pediatr Pathol. 1991;14:168-215.
187. Deutsch GH, Sokol RJ, Stathos TH, Knisely AS. Proliferation to paucity: evolution of bile duct abnormalities in a case of Alagille syndrome. Pediatr Dev Pathol. 2001;4:559-63.
188. Xue Y, Gao X, Lindsell CE, et al. Embryonic lethality and vascular defects in mice lacking the Notch ligand Jagged1. Hum Mol Genet. 1999;8:723-30.
189. McElhinney DB, Krantz ID, Bason L, et al. Analysis of cardiovascular phenotype and genotype-phenotype correlation in individuals with a JAG1 mutation and/or Alagille syndrome. Circulation. 2002;106:2567-74.
190. Boyer-Di Ponio J, Wright-Crosnier C, Groyer-Picard MT, et al. Biological function of mutant forms of JAGGED1 proteins in Alagille syndrome: Inhibitory effect on Notch signaling. Hum Mol Genet. 2007;16:2683-92.
191. Flynn DM, Nijjar S, Hubscher SG, et al. The role of Notch receptor expression in bile duct development and disease. J Pathol. 2004;204:55-64.
192. Libbrecht L, Spinner NB, Moore EC, et al. Peripheral bile duct paucity and cholestasis in the liver of a patient with Alagille syndrome: Further evidence supporting a lack of postnatal bile duct branching and elongation. Am J Surg Pathol. 2005;29:820-6.
193. Emerick KM, Whitington PF. Partial external biliary diversion for intractable pruritus and xanthomas in Alagille syndrome. Hepatology. 2002;35:1501-6.
194. Mattei P, von Allmen D, Piccoli D, Rand E. Relief of intractable pruritus in Alagille syndrome by partial external biliary diversion. J Pediatr Surg. 2006;41:104-7.
195. Kim B, Park SH, Yang HR, et al. Hepatocellular carcinoma occurring in Alagille syndrome. Pathol Res Pract. 2005;201:55-60.
196. Kamath BM, Spinner NB, Emerick KM, et al. Vascular anomalies in Alagille syndrome: A significant cause of morbidity and mortality. Circulation. 2004;109:1354-8.
197. Lykavieris P, Hadchouel M, Chardot C, Bernard O. Outcome of liver disease in children with Alagille syndrome: A study of 163 patients. Gut. 2001;49:431-5.
198. Englert C, Grabhorn E, Burdelski M, Ganschow R. Liver transplantation in children with Alagille syndrome: Indications and outcome. Pediatr Transplant. 2006;10:154-8.
199. Tzakis AG, Reyes J, Tepetes K, et al. Liver transplantation for Alagille’s syndrome. Arch Surg. 1993;128:337-9.
200. Ng VL, Balistreri WF. Treatment options for chronic cholestasis in infancy and childhood. Curr Treat Options Gastroenterol. 2005;8:419-30.
201. Squires RH. End-stage liver disease in children. Curr Treat Options Gastroenterol. 2001;4:409-21.
202. Sokol RJ, Stall C. Anthropometric evaluation of children with chronic liver disease. Am J Clin Nutr. 1990;52:203-8.
203. Ramaccioni V, Soriano HE, Arumugam R, Klish WJ. Nutritional aspects of chronic liver disease and liver transplantation in children. J Pediatr Gastroenterol Nutr. 2000;30:361-7.
204. Sokol RJ. Fat-soluble vitamins and their importance in patients with cholestatic liver diseases. Gastroenterol Clin North Am. 1994;23:673-705.
205. de Albuquerque Taveira AT, Fernandes MI, Galvao LC, et al. Impairment of bone mass development in children with chronic cholestatic liver disease. Clin Endocrinol (Oxf). 2007;66:518-23.
206. Mager DR, McGee PL, Furuya KN, Roberts EA. Prevalence of vitamin K deficiency in children with mild to moderate chronic liver disease. J Pediatr Gastroenterol Nutr. 2006;42:71-6.
207. Cohran VC, Heubi JE. Treatment of pediatric cholestatic liver disease. Curr Treat Options Gastroenterol. 2003;6:403-15.
208. Neimark E, Shneider B. Novel surgical and pharmacological approaches to chronic cholestasis in children: Partial external biliary diversion for intractable pruritus and xanthomas in Alagille syndrome. J Pediatr Gastroenterol Nutr. 2003;36:296-7.
209. Tandon P, Rowe BH, Vandermeer B, Bain VG. The efficacy and safety of bile Acid binding agents, opioid antagonists, or rifampin in the treatment of cholestasis-associated pruritus. Am J Gastroenterol. 2007;102:1528-36.
210. Marschall HU, Wagner M, Zollner G, et al. Complementary stimulation of hepatobiliary transport and detoxification systems by rifampicin and ursodeoxycholic acid in humans. Gastroenterology. 2005;129:476-85.
211. Bergasa NV. Update on the treatment of the pruritus of cholestasis. Clin Liver Dis. 2008;12:219-34.
212. Florentin M, Liberopoulos EN, Mikhailidis DP, Elisaf MS. Colesevelam hydrochloride in clinical practice: A new approach in the treatment of hypercholesterolaemia. Curr Med Res Opin. 2008;24:995-1009.
213. Friesen CA, Roberts CC. Cholelithiasis. Clinical characteristics in children. Case analysis and literature review. Clin Pediatr (Phila). 1989;28:294-8.
214. Rescorla FJ. Cholelithiasis, cholecystitis, and common bile duct stones. Curr Opin Pediatr. 1997;9:276-82.
215. Palasciano G, Portincasa P, Vinciguerra V, et al. Gallstone prevalence and gallbladder volume in children and adolescents: An epidemiological ultrasonographic survey and relationship to body mass index. Am J Gastroenterol. 1989;84:1378-82.
216. Stringer MD, Taylor DR, Soloway RD. Gallstone composition: Are children different? J Pediatr. 2003;142:435-40.
217. Wilcox DT, Casson D, Bowen J, et al. Cholelithiasis in early infancy. Pediatr Surg Int. 1997;12:198-9.
218. Holcomb GWJr, Holcomb GW3rd. Cholelithiasis in infants, children, and adolescents. Pediatr Rev. 1990;11:268-74.
219. Stringer MD, Soloway RD, Taylor DR, et al. Calcium carbonate gallstones in children. J Pediatr Surg. 2007;42:1677-82.
220. Alonso MH. Gall bladder abnormalities in children with sickle cell disease: Management with laparoscopic cholecystectomy. J Pediatr. 2004;145:580-1.
221. Walker TM, Hambleton IR, Serjeant GR. Gallstones in sickle cell disease: Observations from The Jamaican Cohort study. J Pediatr. 2000;136:80-5.
222. Chaar V, Keclard L, Diara JP, et al. Association of UGT1A1 polymorphism with prevalence and age at onset of cholelithiasis in sickle cell anemia. Haematologica. 2005;90:188-99.
223. Descos B, Bernard O, Brunelle F, et al. Pigment gallstones of the common bile duct in infancy. Hepatology. 1984;4:678-83.
224. Treem WR, Malet PF, Gourley GR, Hyams JS. Bile and stone analysis in two infants with brown pigment gallstones and infected bile. Gastroenterology. 1989;96:519-23.
225. Kaechele V, Wabitsch M, Thiere D, et al. Prevalence of gallbladder stone disease in obese children and adolescents: influence of the degree of obesity, sex, and pubertal development. J Pediatr Gastroenterol Nutr. 2006;42:66-70.
226. Bennion LJ, Knowler WC, Mott DM, et al. Development of lithogenic bile during puberty in Pima Indians. N Engl J Med. 1979;300:873-6.
227. Bor O, Dinleyici EC, Kebapci M, Aydogdu SD. Ceftriaxone-associated biliary sludge and pseudocholelithiasis during childhood: A prospective study. Pediatr Int. 2004;46:322-4.
228. Biner B, Oner N, Celtik C, et al. Ceftriaxone-associated biliary pseudolithiasis in children. J Clin Ultrasound. 2006;34:217-22.
229. Debray D, Pariente D, Gauthier F, et al. Cholelithiasis in infancy: A study of 40 cases. J Pediatr. 1993;122:385-91.
230. Siddiqui S, Newbrough S, Alterman D, et al. Efficacy of laparoscopic cholecystectomy in the pediatric population. J Pediatr Surg. 2008;43:109-13.
231. Mok HY, Druffel ER, Rampone WM. Chronology of cholelithiasis. Dating gallstones from atmospheric radiocarbon produced by nuclear bomb explosions. N Engl J Med. 1986;314:1075-7.
232. Curro G, Iapichino G, Lorenzini C, et al. Laparoscopic cholecystectomy in children with chronic hemolytic anemia. Is the outcome related to the timing of the procedure? Surg Endosc. 2006;20:252-5.
233. Colombo C, Bertolini E, Assaisso ML, et al. Failure of ursodeoxycholic acid to dissolve radiolucent gallstones in patients with cystic fibrosis. Acta Paediatr. 1993;82:562-5.
234. Reif S, Sloven DG, Lebenthal E. Gallstones in children. Characterization by age, etiology, and outcome. Am J Dis Child. 1991;145:105-8.
235. Metreweli C, So NM, Chu WC, Lam WW. Magnetic resonance cholangiography in children. Br J Radiol. 2004;77:1059-64.
236. Stevens KA, Chi A, Lucas LC, et al. Immediate laparoscopic cholecystectomy for acute cholecystitis: No need to wait. Am J Surg. 2006;192:756-61.
237. Bonnard A, Seguier-Lipszyc E, Liguory C, et al. Laparoscopic approach as primary treatment of common bile duct stones in children. J Pediatr Surg. 2005;40:1459-63.
238. Holcomb GW3rd, Morgan WM3rd, Neblett WW3rd, et al. Laparoscopic cholecystectomy in children: Lessons learned from the first 100 patients. J Pediatr Surg. 1999;34:1236-40.
239. St Peter SD, Keckler SJ, Nair A, et al. Laparoscopic cholecystectomy in the pediatric population. J Laparoendosc Adv Surg Tech A. 2008;18:127-30.
240. Tsakayannis DE, Kozakewich HP, Lillehei CW. Acalculous cholecystitis in children. J Pediatr Surg. 1996;31:127-30.
241. Imamoglu M, Sarihan H, Sari A, Ahmetoglu A. Acute acalculous cholecystitis in children: Diagnosis and treatment. J Pediatr Surg. 2002;37:36-9.
242. Bartel TB, Juweid ME, Ponto JA, Graham MM. Corn oil emulsion: A simple cholecystagogue for diagnosis of chronic acalculous cholecystitis. J Nucl Med. 2005;46:67-74.
243. Appleby GA, Forestier E, Starck CJ. Hydrops of the gallbladder in the neonatal period. Acta Paediatr Scand. 1981;70:117-20.
244. Suddleson EA, Reid B, Woolley MM, Takahashi M. Hydrops of the gallbladder associated with Kawasaki syndrome. J Pediatr Surg. 1987;22:956-9.
245. Barton LL, Luisiri A, Dawson JE. Hydrops of the gallbladder in childhood infections. Pediatr Infect Dis J. 1995;14:163-4.
246. Crankson S, Nazer H, Jacobsson B. Acute hydrops of the gallbladder in childhood. Eur J Pediatr. 1992;151:318-20.
247. Wood J, Holland AJ, Shun A, Martin HC. Biliary dyskinesia: is the problem with Oddi? Pediatr Surg Int. 2004;20:83-6.
248. Vegunta RK, Raso M, Pollock J, et al. Biliary dyskinesia: the most common indication for cholecystectomy in children. Surgery. 2005;138:726-31.
249. Carney DE, Kokoska ER, Grosfeld JL, et al. Predictors of successful outcome after cholecystectomy for biliary dyskinesia. J Pediatr Surg. 2004;39:813-16.