[level-membership-for-gastroenterology-and-hepatology-category]
CHAPTER 47 Anatomy, Histology, Embryology, and Developmental Anomalies of the Stomach and Duodenum
EMBRYOLOGY AND ANATOMY OF THE STOMACH
GENERAL CONSIDERATIONS
The stomach is recognizable in the fourth week of gestation as a dilation of the distal foregut (Fig. 47-1).1 As the stomach enlarges, the dorsal aspect grows more rapidly than the ventral aspect, thus forming the greater curvature. Additionally, during the enlargement process the stomach rotates 90 degrees around its longitudinal axis, orienting the greater curvature (the dorsal aspect) to the left and the lesser curvature (ventral aspect) to the right. The combined effects of rotation and ongoing differential growth result in the stomach lying transversely in the mid and left upper abdomen. The events also explain the vagal innervation of the stomach: the right vagus nerve innervating the posterior stomach wall (the primordial right side) and the left vagus nerve innervating the anterior wall (the primordial left side).
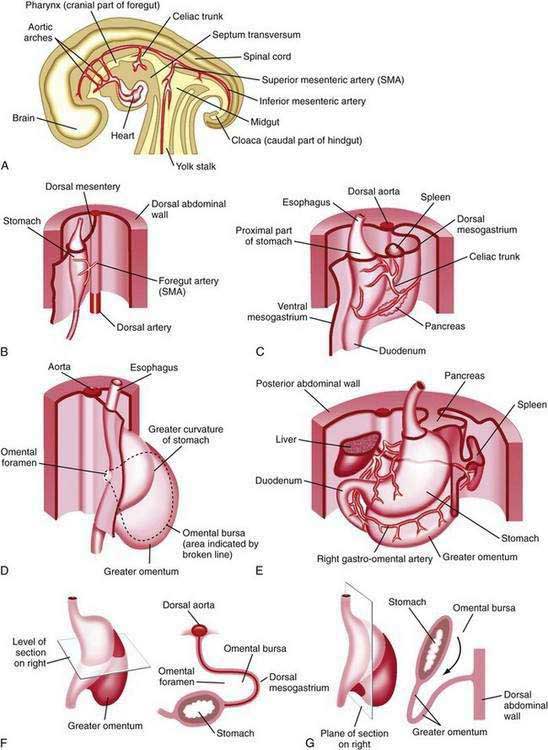
Figure 47-1. Development of the stomach and duodenum and formation of the omental bursa (lesser sac) and greater omentum. A, Median section of a 28-day embryo. B, Anterolateral view of a 28-day embryo. C, Embryo about 35 days old. D, Embryo about 40 days old. E, Embryo about 48 days old. F, Lateral view of the stomach and greater omentum of an embryo at about 52 days. The transverse section shows the omental foramen and omental bursa. G, Sagittal section showing the omental bursa and greater omentum. The embryology of the duodenum is discussed further in Chapters 55 and 96.
(From Moore KL, Persaud TVN. The developing human. 7th ed. Philadelphia: WB Saunders; 2003. p 258.)
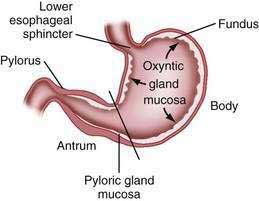
The stomach is divided into four regions that can be defined by anatomic or histologic landmarks (Fig. 47-2).2 Anatomically the cardia is a small ill-defined area of the stomach immediately adjacent to its junction with the esophagus. This region of the stomach has been the focus of intense investigation. Controversy exists as to the nature, location, extent, and even existence of cardiac mucosa. The fundus projects upward, above the cardia and gastroesophageal junction. This dome-shaped area of the stomach is its most superior portion and is in contact above with the left hemidiaphragm and to the left with the spleen. The body, or corpus, the largest portion of the stomach, is located immediately below and continuous with the fundus. The incisura angularis, a fixed, sharp indentation two thirds of the distance down the lesser curvature, marks the caudal aspect of the gastric body (Fig. 47-3). The gastric antrum extends from its indistinct border with the body to the junction of the pylorus with the duodenum. These gross anatomic landmarks correspond roughly with the mucosal histology because antral mucosa (pyloric gland mucosa) actually extends from an area on the lesser curvature somewhat above the incisura. The pylorus (pyloric channel) is a tubular structure joining the duodenum to the stomach and contains the palpable circular muscle, the pyloric sphincter. The pylorus is somewhat mobile owing to its enclosure between the peritoneum of the greater and lesser omenta but is generally located 2 cm to the right of midline at L1. Corresponding motor and secretory functions of these regions of the stomach are discussed in detail in Chapters 48 and 49.
TISSUE LAYERS OF THE STOMACH
The luminal surface of the gastric wall forms thick, longitudinally oriented folds, or rugae, that flatten with distention. Four layers make up the gastric wall: mucosa, submucosa, muscularis propria, and serosa. Mucosa lines the gastric lumen, appearing as a smooth, velvety blood-filled lining. The mucosa of the cardia, antrum, and pylorus is somewhat paler than that of the fundus and body. It is within the gastric mucosa that most of the functional secretory elements of the stomach are located (see Chapter 49). The submucosa, immediately deep to the mucosa, provides the dense connective tissue skeleton of collagen and elastin fibers. Lymphocytes, plasma cells, arterioles, venules, lymphatics, and the submucosal plexus are also contained within the submucosa. The third tissue layer, the muscularis propria, is a combination of three muscle layers: inner oblique, middle circular, and outer longitudinal. The inner oblique muscle fibers course over the gastric fundus, covering the anterior and posterior aspects of the stomach wall. The middle circular fibers encircle the body of the stomach, thickening distally to become the pyloric sphincter. The outer longitudinal muscle fibers course primarily along the greater and lesser curvatures of the stomach. The final layer of the stomach is the transparent serosa, a continuation of the visceral peritoneum.
MICROSCOPIC ANATOMY
The gastric mucosal surface is composed primarily of a simple layer of columnar epithelial cells 20 to 40 mm in height. These surface mucous cells (Fig. 47-4), which are similar throughout the stomach, contain basally located nuclei, prominent Golgi stacks, and dense cytoplasm with especially apically dense mucin-containing membrane-bound granules. The cells secrete mucus in granules that are released via exocytosis, apical expulsion, and cell exfoliation. The primary role of mucus, along with bicarbonate, is luminal cytoprotection from “the elements”: acid, pepsin, ingested substances, and pathogens. Cellular renewal time for a gastric surface mucous cell is approximately three days.
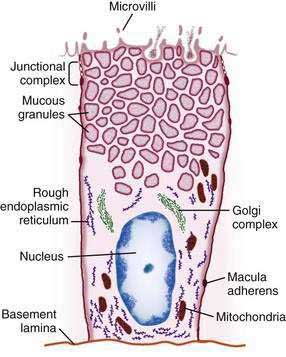
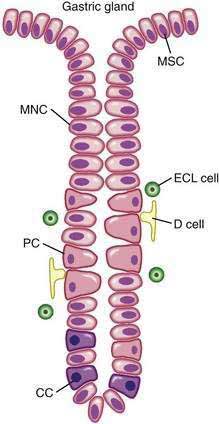
The surface epithelial lining is invaginated by gastric pits, or foveolae, that provide the gastric glands access to the gastric lumen, with a ratio of one pit to four or five gastric glands. The gastric glands of different anatomic regions of the stomach are lined with different types of specialized epithelial cells, allowing for differentiation of these regions by type of gastric gland (see Fig. 47-2). The first region, the cardia, is a small transition zone from esophageal squamous epithelium to gastric columnar epithelium. The cardia has been a controversial histologic area of discussion, with theories suggesting that its presence is pathologic. However, recent observations concluded that cardiac mucosa develops during gestation and is present at birth.3 The cardiac glands have a branched and tortuous configuration and are populated by mucous, endocrine, and undifferentiated cells. There is a gradual transition from cardiac glands to the second region, the acid-secreting segment of the stomach. This region encompasses the gastric fundus and body and contains the parietal (or oxyntic or fundic) glands. Parietal, chief (also known as peptic), endocrine, mucous neck, and undifferentiated cells compose the oxyntic glands. The final region, corresponding to the antrum and pylorus, contains the pyloric glands, composed of endocrine cells, including gastrin-producing G cells and mucous cells.
By far the most numerous and distinctive gastric glands are the oxyntic glands (Fig. 47-5), responsible for the secretion of acid, intrinsic factor, and most gastric enzymes. These fairly straight and simple tubular glands are closely associated in the areas of gastric fundus and body. A typical gland is subdivided into three areas: the isthmus (where surface mucous cells predominate), the neck (where parietal and mucous neck cells predominate), and the base (where chief cells predominate, along with some parietal and mucous neck cells). Endocrine cells, somatostatin-containing D cells, and histamine-secreting enterochromaffin-like (ECL) cells are scattered throughout the oxyntic epithelium.
The principal cell type of the oxyntic gland is the parietal cell (Fig. 47-6), responsible for the oxyntic mucosal secretion of 3 × 106 hydrogen ions per second, at a final hydrochloric acid (HCl) concentration of around 150 mmol/L. Parietal cells bulge into the lumina of the oxyntic glands and, as the primary hydrogen secretors, have ultrastructural characteristics different from other gastric cells: large mitochondria, microvilli lacking in glycocalyx, and a cytoplasmic canaliculi system in contact with the lumen. In the nonsecreting parietal cell, a cytoplasmic tubulovesicular system predominates and short microvilli line the apical canaliculus. In the secreting state, the tubulovesicular system disappears, leaving an extensive system of intracellular canaliculi containing long microvilli. Mitochondria occupy approximately 30% to 40% of the secreting parietal cell volume, providing energy required for acid secretion across apical microvilli (see Fig. 47-6). The so-called proton pump—the H+,K+-ATPase—resides in the apical microvillus membrane, as does carbonic anhydrase. The apical H+,K+-ATPase functions as the proton translocator in gastric acid secretion (see Chapter 49). Acid secretion begins within 5 to 10 minutes of stimulation. Additionally, parietal cells are the site of intrinsic factor secretion via membrane-associated vesicle transport.
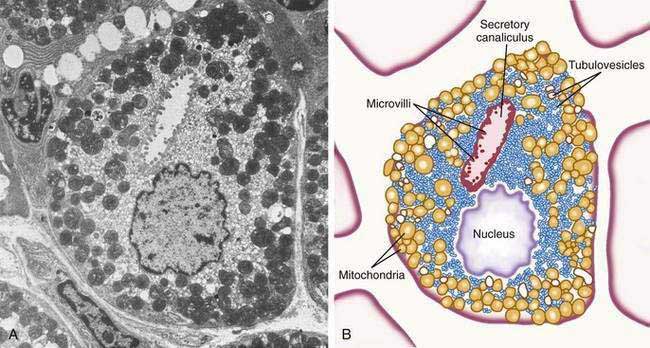
Figure 47-6. Parietal cell. A, Electron photomicrograph. B, Schematic.
(A and B, from Johnson LR. Gastrointestinal physiology. 6th ed. St Louis: Mosby; 2001. pp 78, 79.)
The final region of the stomach encompasses the antrum and pylorus and contains extensively coiled antral glands composed of endocrine and epithelial cells. The epithelial cells are predominantly mucous cells, and there are small numbers of pepsinogen II–secreting oxyntic cells. Although also small in number, gastrin-secreting (G) cells play a vital physiologic role and are the prototype of the open enteroendocrine cell. These cells, which occur either singly or in small clusters in the mid- to deep sections of antral glands (Fig. 47-7A), contain a basilar cytoplasm densely packed with gastrin-containing secretory granules (see Fig. 47-7B). Gastrin release is stimulated by gastric distention, vagal stimulation, dietary amino acids, and peptide, with rapid appearance of the hormone into the bloodstream in the postprandial period (see Chapter 49). The apical or luminal surface of the G cell is narrowed into small microvilli thought to contain receptors responsible for amino acid and peptide stimulation of gastrin release. Significant quantities of gastrin are also secreted into the gastric lumen; gastrin is a known gastric growth and differentiation factor, mediated through upregulation of heparin-binding epidermal-like growth factor (HB-EGF) in gastric parietal cells.4,5
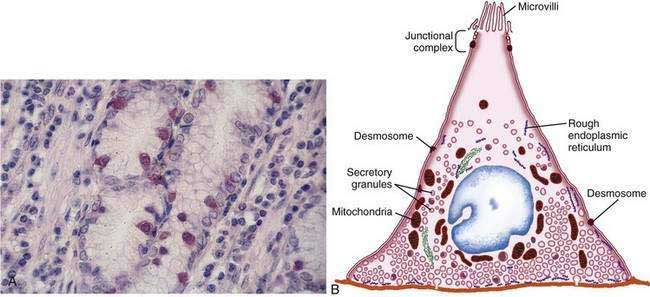
Antral enteroendocrine D cells found in close association with G cells manufacture somatostatin, a potent inhibitor of gastrin secretion. The D cells are also present in small numbers in oxyntic glands. Somatostatin is thought to inhibit acid secretion through paracrine (direct action on ECL and perhaps parietal cells or indirect action on G cells) or endocrine effects (direct action on parietal cells) (see Chapter 49 for more details).
CONGENITAL ANOMALIES OF THE STOMACH AND DUODENUM
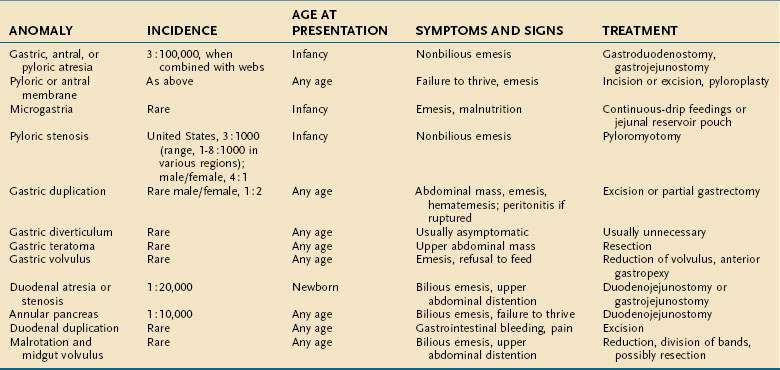
The congenital anomalies of the stomach and duodenum are summarized in Table 47-1.
GASTRIC ANOMALIES
Gastric Atresia
Pathogenesis
The cause of these lesions remains unknown, but the timing of a contrary developmental event may determine the type of atresia. For example, if there is fusion of redundant endoderm before eight weeks’ gestation (before muscle layer development), then discontinuity of gastric wall musculature would result in a segmental defect with or without a fibrous cord. On the other hand, if redundancy occurs after eight weeks’ gestation, when muscle layers are complete, a simple membrane develops. An alternative mechanism—focal ischemia at a critical time in development—has been proposed. Finally, failure of recanalization of the gastric lumen following temporary obstruction from mucosal proliferation has been proposed as a cause but is not a viable explanation because obstruction or recanalization does not occur in the stomach (unlike the esophagus and duodenum).6 Total epithelial detachment of gastric mucosa, associated with α6β4 integrin expression deficiency at the junction of epithelial cells and lamina propria, has been noted in a child with pyloric atresia.7
Genetic factors also are important. In addition to a familial form (autosomal recessive), gastric atresia is also associated with Down syndrome and junctional epidermolysis bullosa. In the case of epidermolysis bullosa–pyloric atresia–obstructive uropathy association, mutations in the α6 and β4 integrin subunits of the hemidesmosome have been noted.8,9 Junctional epidermolysis bullosa, also known as the JEB–pyloric atresia syndrome,10 is the only type that has been described.
Other associated anomalies are malrotation, atrial septal defect, absent gallbladder, tracheoesophageal fistula, vaginal atresia, and absent extrahepatic portal vein.11 In addition, gastric atresia may be associated with multiple intestinal atresias and immunodeficiency.12
Treatment
Following patient stabilization with fluids and gastric decompression, definitive treatment is surgical. Complete or incomplete antral membranes are treated by simple excision. Pyloric membranes require pyloroplasty. The presence of a concomitant duodenal atresia has been described (also known as windsock diaphragm), and its presence or absence is verified by passage of a catheter distally into the duodenum intraoperatively. Endoscopic therapy using a snare, papillotome, laser, or dilation via balloon also has been described. In cases involving atretic gap, gastroduodenostomy is considered curative. An alternative approach is pyloric sphincter reconstruction via longitudinal pyloromyotomy, followed by end-to-end anastomosis of cul-de-sacs of gastric and duodenal mucosa.13 Gastrojejunostomy is not recommended in children because of the risk of marginal ulcer.
Microgastria
Microgastria is an extremely rare congenital anomaly of the caudal part of the foregut. A small, tubular or saccular, incompletely rotated stomach is associated with a megaesophagus. Varying degrees of the anomaly occur owing to arrested development during the fifth week of gestation in differentiation of the greater curvature of the stomach; neither rotation nor fusiform dilation of the stomach occurs.14 A localized vascular insufficiency has been postulated to lead to the development of microgastria after the eighth week of gestation.15 The etiology is unknown.
Fortunately normal histology is preserved. Microgastria may occur as an isolated anomaly but more commonly is in association with duodenal atresia; nonrotation of the midgut; ileal duplication; hiatal hernia; asplenia; partial situs inversus; or renal, upper limb (microgastria–limb reduction anomaly), cardiac, pulmonary, skeletal, or spinal anomalies. In isolation, microgastria is not lethal, but other associated anomalies may be. It has been suggested that microgastria in association with limb reduction defects and central nervous system anomalies has a genetic basis, with an autosomal recessive pattern of inheritance.16 Chromosome analysis is normal.
Treatment
The medical management of microgastria includes frequent small-volume feedings or continuous-drip feedings into the stomach. An alternative is nocturnal drip feedings via jejunostomy to supplement oral intake. The surgical creation of a double-lumen Roux-en-Y pouch anastomosed to the greater curvature of the stomach has been described. This Hunt-Lawrence jejunal pouch has allowed normal growth and development and prevented reflux and dumping syndrome.17
Gastric Diverticulum (see also Chapter 23)
Clinical Features and Diagnosis
Most congenital gastric diverticula are asymptomatic and are incidental findings on radiography or endoscopy, or at autopsy (see Chapter 23). Size varies from 1 to 11 cm. Contrast radiography shows a rounded, well-delineated mobile pouch, often with an air-fluid level. Emptying of the diverticulum may be delayed. On endoscopy, the diverticulum is seen as a well-delineated opening; distention by the scope may reproduce symptoms. Unfortunately, both upper gastrointestinal radiologic studies and endoscopy may miss the diagnosis due to the typical location at the gastroesophageal junction. Symptoms may be epigastric or lower chest pain, indigestion, bleeding, or nonbilious emesis. The differential diagnosis includes an acquired gastric diverticulum found in association with pancreatitis, gastric outlet obstruction, trauma, ulcer disease, or malignancy. Hiatal hernia and hypertrophic gastric folds may mimic a diverticulum on contrast studies. Radiology cannot distinguish between congenital and acquired diverticula.
Gastric Duplication
Approximately 20% of all gastrointestinal duplications are gastric. Duplication of the stomach can occur in isolation, as a triplication (two gastric duplications in one individual), or with duplications of other structures in the gastrointestinal tract such as the esophagus or duodenum. Location is contiguous with the stomach, generally along the greater curvature or posterior wall and contains all layers of the gastric wall. Gastric and pancreatic mucosa lining the duplication are most clinically significant secondary to potential complications such as peptic ulcer disease and pancreatitis. Because the duplication rarely communicates with the stomach, a tubular, fusiform, or spherical cystic mass develops. Infrequently there may be a connection to the colon, pancreas, or pancreatic duplication; the connection may be the result of an acquired fistula from a penetrating peptic ulcer within the duplication. Several embryologic defects have been proposed as etiologies for duplications including errors in separation of notochord and endoderm, persistence of embryonic diverticula, and persistence of vacuoles within the epithelium of the primitive foregut.18 Most duplications occur in women (65%) and are detected during infancy or childhood (80%) with no familial tendency. Aside from concurrent duplications, vertebral anomalies are the second most commonly linked abnormality.19 Carcinomas arising in congenital duplications have been described in adults.
Clinical Features and Diagnosis
The clinical presentation of gastric duplication depends on factors such as size, location, and communicating structure (if any). Symptoms and signs vary and may include colic; abdominal mass; epigastric pain; failure to gain appropriate weight; vomiting; occult or frank upper or lower gastrointestinal bleeding secondary to peptic ulceration, the latter occurring via erosion into the colon; hematobilia via a communication with intrahepatic bile duct; respiratory distress or hemoptysis (perforated cyst fistulized to lung)20; pyloric obstruction; peritonitis secondary to rupture; pancreatitis; pancreatic pseudocyst; and acute abdomen. In early infancy symptoms may mimic those of hypertrophic pyloric stenosis. Diagnosis is suggested by an abdominal radiograph showing displacement and extrinsic compression of gastric lumen. Contrast radiography may demonstrate the duplication via a mass effect on the stomach (Fig. 47-8A) or the cyst may be imaged directly when there is communication with the gastrointestinal tract. Ultrasonography (see Fig. 47-8B) including prenatal ultrasonography,20 computed tomography (CT) scan (see Fig. 47-8C), magnetic resonance cholangiopancreatography (MRCP), Tc-99m pertechnetate, and endoscopy with endoscopic ultrasonography (EUS) may also demonstrate the lesion. Peristalsis identified by EUS in a juxtaenteric cyst is specific for a duplication cyst and may be considered as a diagnostic feature of this condition.21
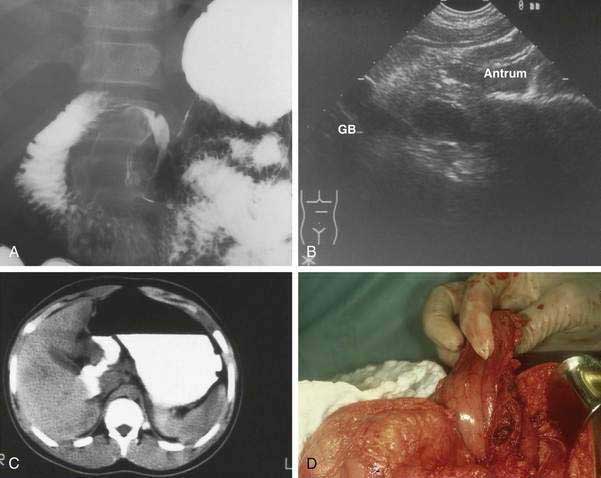
Treatment
Surgical excision is considered optimal therapy (see Fig. 47-8D). Laparoscopic resection has been described.22 When complete excision is not possible, as may be the case when cyst and viscus have a common muscle layer, debulking, cyst-gastrostomy, or partial gastrectomy may be necessary. Additionally, mucosectomy or mucosal surface ablation should be considered because the development of malignancy in enteric duplications has been documented in adults.23
Gastric Teratoma
Gastric teratomas are benign neoplasms of the stomach that occur almost exclusively in men. Gastric teratomas are rare, comprising only 1% of all childhood teratomas. These tumors may have their origins in pluripotential cells and contain all three embryonic germ cell layers. They are almost always diagnosed during infancy owing to their large size. Most are located along the greater curvature of the stomach and are extragastric, although intramural extension has been reported.24 The immature type (containing yolk cell tumor, germinoma, and embryonal carcinoma) may infiltrate regional structures—omentum, regional lymph nodes, left lobe of the liver—whereas the mature tissue form does not. In virtually all cases, gastric teratoma is an isolated finding and is not associated with other tumors or malformations.25
Clinical Features and Diagnosis
The typical patient is a male infant with an abdominal mass; mean age at presentation is 3.2 months.26 Vomiting may be present from intrinsic compression and gastrointestinal bleeding due to transmural growth and disruption of gastric mucosa. Polyhydramnios may be noted prenatally secondary to gastric obstruction by the mass. The newborn infant with a teratoma may be delivered prematurely or have respiratory distress on the basis of increased abdominal pressure. Delivery may be difficult, putting the infant at risk for injuries such as shoulder dystocia. Gastric teratoma associated with gastric perforation, mimicking meconium peritonitis, has been described.27
Noncontrast radiography demonstrates characteristic calcifications. Ultrasonography demonstrates solid and cystic areas, and CT or magnetic resonance imaging confirms the diagnosis and evaluates regional infiltration.26
Treatment
Tumor excision with primary gastric repair is the procedure of choice and is curative. Partial or total gastrectomy is required for intramural tumor extension. Malignant transformation to adenocarcinoma has been reported,28 as well as premalignant changes,29 and peritoneal gliomatosis has been observed. Fortunately even those cases with malignant histologic features or extension into adjacent tissues have an excellent prognosis.25 In the case of immature gastric teratomas, a serum alpha fetoprotein level may be useful, especially because of the possibility of recurrence or metastasis and the need for adjuvant chemotherapy.30
Infantile Hypertrophic Pyloric Stenosis
Infantile hypertrophic pyloric stenosis (IHPS) is a form of gastric outlet obstruction caused by hypertrophy of circular muscle surrounding the pyloric channel. Correction of IHPS is the most common abdominal operative procedure during the first 6 months of life. Because the muscular hypertrophy and obstruction tend to be an evolving process during the postnatal period, IHPS is arguably not a true congenital defect.31 The etiology of IHPS remains the subject of speculation. A localized lack of nitric oxide synthase, an enzyme associated with smooth muscle relaxation, or abnormal neuronal innervation associated with decreased muscle neurofilaments, nerve terminals, synaptic vesicle protein, and neural cell adhesion molecule32 has been implicated. However, anatomic studies cannot determine whether nitric oxide synthase deficiency is a primary or secondary event,33 and nitric oxide synthase deficiency is only notable in a subset of cases.34 Pacemaker cells that regulate gastrointestinal motility, the interstitial cell of Cajal, are observed only near the submucosa in IHPS instead of throughout the pylorus.35,36 Epidermal growth factor (EGF), EGF receptor, and heparin-binding EGF-like growth factor are markedly increased in smooth muscle cells in IHPS,37 but their triggers are unknown.
Familial clustering of IHPS is widely recognized, with autosomal dominant forms reported.38 Approximately 50% of identical twins are affected, leading credence to the roles of genetic and environmental factors. Male relatives of affected women are more likely to develop IHPS, such that siblings and offspring of affected women are more likely to develop IHPS than are relatives of affected men. Others at increased risk are first-born male infants, especially those with high birth weights or born to professional parents. IHPS also occurs in association with Turner’s syndrome, trisomy 18, Cornelia de Lange syndrome, esophageal atresia, Hirschsprung’s disease, phenylketonuria, and congenital rubella syndrome. Multiple reports have described an association between early macrolide exposure in infants, including exposure through breast milk,39 and the development of IHPS.40–42 In addition, a recent decline in the incidence of IHPS that parallels that of sudden infant death syndrome has been observed and coincides with the recommendation of supine infant sleeping position. This has led to the hypothesis that posture may contribute to the underlying etiology.43
Clinical Features and Diagnosis
When the presentation is typical and the olive palpated, no studies are necessary. However, in the minority of infants with projectile vomiting, definitive diagnosis requires radiologic studies. Noncontrast radiography demonstrates a distended stomach with paucity of gas beyond the stomach. Diagnosis is confirmed by abdominal ultrasonography of the pylorus, which has supplanted contrast radiography as the diagnostic study of choice for IHPS. Because dehydration may affect the pyloric ultrasound measurements, ensuring an adequate hydration status may be prudent before ultrasonographic evaluation.44 The length of the hypertrophied canal is variable and may range from as little as 14 mm to more than 20 mm. The numeric value for the lower limit of muscle thickness has varied in reports in the literature, ranging between 3 and 4.5 mm. This appears as a characteristic sonolucent “donut” (Fig. 47-9). Many consider the numeric value less important than the overall morphology of the canal and real-time observations. A negative ultrasonographic study hinges on an unequivocal diagnosis of a normal pyloric ring and a distensible antropyloric portion of the stomach.45 However, when the differential diagnosis includes IHPS, gastroesophageal reflux, or other upper gastrointestinal disorders, contrast radiography may be the appropriate first test. Contrast radiography must be performed carefully, and gastric contents should first be aspirated. The infant is given barium by nipple and imaged in a semiprone position. Characteristic findings include an elongated narrow pylorus with the appearance of a “double channel.” There is also indentation of the adjacent antrum and duodenum by the pyloric mass producing the so-called shoulders (Fig. 47-10). The most common abnormality that mimics IHPS is pylorospasm. Diagnosis of IHPS by endoscopy has been described in which the pylorus appears as a cauliflower-like narrowing, through which a 7.8-mm (external diameter) endoscope cannot be passed.46 However, another report on endoscopic diagnosis has refuted these claims.47 Endoscopy is also potentially beneficial to evaluate for eosinophilic gastroenteritis (see Chapter 27), which has been linked to pyloric stenosis.48
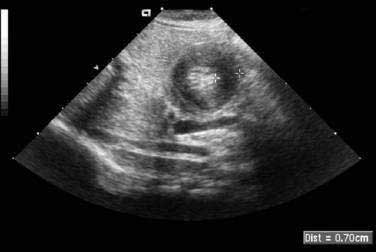
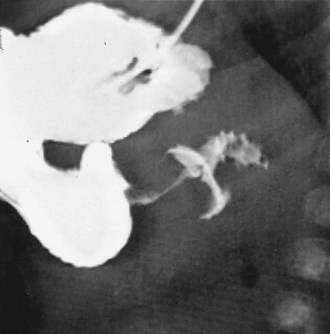
Treatment
The initial therapy for IHPS is fluid and electrolyte replacement to correct dehydration and hypochloremic metabolic alkalosis. Depending on severity, fluid and electrolyte repletion can usually be accomplished within 24 hours. Definitive therapy is the Ramstedt pyloromyotomy, which entails a longitudinal incision through the hypertrophied pyloric muscle down to the submucosa on the anterior surface of the pylorus. After spreading the muscle, the intact mucosa bulges through the incision to the level of the incised muscle. An alternative operation is the pyloric traumamyoplasty. With this procedure the pylorus is grasped with a Babcock clamp that disrupts the hypertrophied circular muscles in two places.49,50 Laparoscopic pyloromyotomy with its improved cosmetic results and reduced need for analgesics is becoming increasingly popular.51,52 Although infants may continue to vomit for the first few days postoperatively, persistent vomiting is suggestive of inadequate surgery.
Nonoperative therapy consists of the use of anticholinergic medications53 and paste-consistency feedings until such time that the muscle hypertrophy resolves.54 Because of the high failure rate, the prolonged recovery period (compared with surgery), and the low risk of pyloromyotomy, the nonoperative approach is rarely used in the United States.
The prognosis following surgery is excellent. The infant resumes normal growth and development. Although divergent gastric emptying rates have been observed many years following treatment of IHPS, gastric emptying by scintigraphy using radiolabeled liquids or solids was similar in patients treated surgically or conservatively versus controls.46
Adult Hypertrophic Pyloric Stenosis
Hypertrophic pyloric stenosis rarely occurs in adults. There are approximately 200 cases described in the literature. When HPS occurs in adults, its anatomic features are identical to the infantile type. In adults pyloric thickening is generally associated with peptic ulcer disease, hypertrophic gastopathy,55 or carcinoma. In a few cases, no etiology is determined; it is therefore unknown whether these are missed infantile cases or whether the hypertrophy occurred later in life. There is a family history of IHPS in some cases of adult HPS, thus again suggesting a role for genetic predisposition or missed infantile cases. In addition, 80% of adult HPS occur in men. The resected pylorus demonstrates normal mucosa and marked circumferential thickening of the muscularis propria.56 Microscopically there are variable degrees of inflammatory changes or edema, and degenerative changes in the ganglion cells of the myenteric plexus have been reported.55
Clinical Features and Diagnosis
Symptoms of adult HPS are similar to those observed in infancy: nausea, mild vomiting, early satiety, and epigastric pain, especially after eating. In contrast with the infantile form, the physical examination may not be helpful because the pyloric mass is difficult to palpate in adults. On contrast radiography, the elongated narrow pylorus is again apparent; gastric emptying is delayed, and the stomach may be dilated. Ultrasonography is the screening procedure of choice; it is generally considered abnormal if the pylorus is 1 cm or more thick with persistent elongation of more than 2 cm.55 Upper endoscopy is indicated to differentiate idiopathic HPS from carcinoma or chronic peptic ulcer disease.
Treatment
Traditionally, surgical pyloromyotomy or resection of the involved region has been considered the procedure of choice. Because of the risk of a small focus of carcinoma, surgical resection of the pylorus has been recommended. Endoscopic balloon dilation has also been efficacious in the management of HPS, but a high postprocedure recurrence rate—80% within the first 6 months—has been reported.57 Additionally, palliation of pyloric stenosis caused by gastric cancer using an endoscopically placed stent has been described.58
DUODENAL ANOMALIES
Duodenal Atresia and Stenosis
Duodenal atresia and stenosis are congenital defects characterized by complete and partial obstruction of the duodenum, respectively. Atresias occur in various anatomic configurations including a blind-ending pouch with no connection to the distal duodenum (least common), a pouch with a fibrous cord connecting to the distal duodenum, or a complete membrane obstructing the lumen (most common). Perforate membranes are also a cause of duodenal stenosis. All three lesions occur with greatest frequency near the ampulla of Vater, with most lesions (80%) occurring distal to this landmark. The overall incidence of the three anomalies combined is about 1 per 200,000 live births with a slight predilection for girls. The etiology of these lesions may relate to failure to recanalize the duodenal lumen by vacuolization at 8 to 10 weeks’ gestation. This is distinct from atresia or stenosis of the jejunum and ileum, which are caused by vascular accidents in utero.59 Duodenal stenosis has been observed in sonic hedgehog (shh)–mutant mice, thus adding to our understanding that mutations in signaling pathways may play a role in this malformation.60
In two series of more than 100 cases,61,62 more than 50% of affected patients had associated congenital defects including pancreatic defects; intestinal malrotation with congenital bands; esophageal atresia; Meckel’s diverticulum; imperforate anus; congenital heart disease; central nervous system lesions; renal anomalies; and, rarely, biliary tract anomalies. Trisomy 21 is strongly associated with duodenal atresia/stenosis/web in that anywhere from 25% to more than 50% of cases occur in infants and children with this chromosomal anomaly. Familial association is rare, although isolated case reports suggest a possible genetic association.63,64 A report of father and son with periampullary obstruction due to duodenal stenosis and annular pancreas (in the father) and segmental duodenal atresia (in the son) serves as a reminder that with increased survival of affected infants, a genetic basis may be realized in the future.65
Clinical Features and Diagnosis
The diagnosis of duodenal atresia may be suspected prenatally when ultrasonography demonstrates gastric and proximal duodenal dilation and polyhydramnios. Polyhydramnios is present in 33% to 50% of cases of duodenal atresia. The absence of gastric and proximal duodenal dilation in the presence of polyhydramnios does not exclude the diagnosis because intrauterine emesis may limit preobstructive dilation. High-frequency transvaginal transducers used in ultrasonography may overdiagnose intestinal dilation, so longer scanning is recommended once obstruction is suspected.66
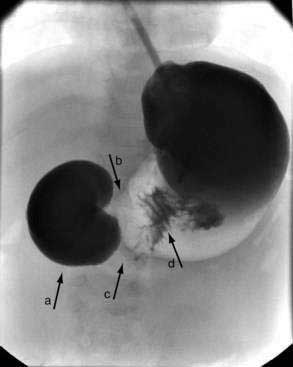
Noncontrast radiographs of the infant with duodenal obstruction classically demonstrate the presence of air in the stomach and in the first portion of the duodenum—the “double-bubble” sign (Fig. 47-11). The absence of air beyond the second bubble should be interpreted as probable duodenal atresia. Contrast radiography is generally effective in demonstrating atresias, stenosis, membranes, and other anomalies resulting in external compression of the duodenum (Fig. 47-12). In addition, normal or abnormal rotation and fixation of the bowel can be assessed. Competence of the ampullary sphincter has been noted to be compromised in few reported cases. Reflux of contrast medium through the ampulla poses risks of developing cholangitis and pancreatitis. Occasionally upper endoscopy is useful in diagnosing or defining a duodenal stenosis or membrane.
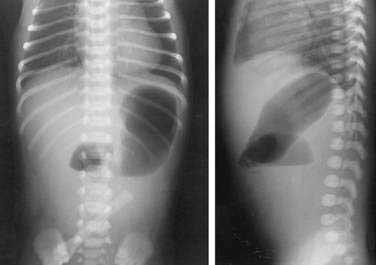
Figure 47-11. Anteroposterior and lateral noncontrast films of an infant with duodenal atresia demonstrate the “double-bubble” sign.
(Courtesy Marcia Pritchard, MD.)
Treatment
A newborn infant suspected of duodenal obstruction should have a nasogastric tube placed for decompression, and correction of fluid and electrolyte abnormalities should be instituted. The surgical approach in the past was duodenojejunostomy, but now duodenoduodenostomy is preferred.62 The operation has evolved from a side-to-side anastomosis to a proximal transverse to distal longitudinal (“diamond shaped”) anastomosis. Associated malrotation is corrected with a Ladd procedure. A catheter is passed into the distal duodenum to investigate for a second obstruction, which occurs in about 3% of cases. Membranes may be excised without anastomosis if the membrane was an isolated finding. Balloon dilation has been described for membranous duodenal stenosis.67,68 Endoscopic laser resection of membranes has been reported; unfortunately, subsequent scar formation has resulted in stenosis and the need for surgery.69 Late complications continue to plague patients even following surgical repair: motility problems, megaduodenum, gastroesophageal reflux unresponsive to medications, gastropathy, adhesions, and peptic ulcer disease occur months to years following primary repair.62
Approximately 12% of patients required revision or another intra-abdominal surgery over a 30-year follow-up period.70 Two teenagers have presented with choledochal cyst.61 Megaduodenum proximal to the obstruction, with abnormal peristalsis, is a common long-term issue, but most patients are asymptomatic. For symptomatic patients with megaduodenum, bowel plication may be indicated.71
Annular Pancreas
Annular pancreas is an unusual congenital malformation characterized by a thin ring of pancreatic tissue, most often encircling the second portion of the duodenum, contributing in variable degrees to obstruction (see also Chapter 55). The lesion may present in the neonatal period, in childhood, or adulthood. It is the most common congenital anomaly of the pancreas presenting in children. Some cases remain asymptomatic and are discovered as an incidental finding during endoscopic retrograde cholangiopancreatography (ERCP) or at autopsy. The anomalous tissue is histologically normal and contains a moderately sized pancreatic duct. The pancreatic tissue may penetrate the muscularis of the duodenal wall or remain distinct from the duodenum.
Several hypotheses exist regarding embryologic origin of annular pancreas. Evidence appears to favor Lecco’s 1910 hypothesis that the ventral pancreatic anlage becomes fixed to the duodenal wall before rotation during the fifth week of gestation. With subsequent growth and fusion of the dorsal and ventral anlagen, a partial (75%) or complete (25%) ring of pancreatic tissue is formed.6
Incidence of the disorder is approximately 1 in 100,000 live births, but this figure does not account for cases found during adulthood, during ERCP (when it is usually noted as an incidental finding), or at autopsy. The true incidence may be as high as 1 per 250 live births. In infancy the incidence is equal in male and female infants. In adulthood men outnumber women by 2 : 1. Infant and childhood cases are associated with other congenital anomalies in an estimated 40% to 70% of cases, including trisomy 21, duodenal atresia, cardiac defects, anorectal malformations, Meckel’s diverticulum, tracheoesophageal fistula, malrotation, genitourinary malformation, and situs inversus.72 In adults, compared with children, it is more common to have malrotation, duodenal web, Shatzki’s ring, duodenal atresia, tracheoesophageal fistula, and genitourinary abnormalities. Also adults are at increased risk of pancreatobiliary neoplasia. In 13 adults with annular pancreas, 6 had pancreatobiliary neoplasia, including 2 with adenocarcinoma of the pancrease, 2 with ampullary adenoma, and 1 with adenocarcinoma of the gallbladder.72 The finding of an annular pancreas in a woman and her child, as well as in two successive generations, suggests a possible hereditary link.73
Clinical Features and Diagnosis
Annular pancreas produces symptoms when tissue obstructs the duodenum or biliary tree. Controversy exists as to whether the annular pancreas actually plays a role in obstruction. The abnormally located pancreatic tissue is a visible indicator of an underlying duodenal abnormality that can range from minimal duodenal stenosis to atresia.74 Infants may present with high-grade obstructive symptoms and signs such as bilious emesis and upper abdominal distention indistinguishable from duodenal atresia or malrotation with midgut volvulus. During childhood intermittent bilious emesis and failure to thrive are common presenting symptoms, whereas during adulthood the most common symptom is abdominal pain. Other symptoms and signs in adults include nausea, vomiting, gastric outlet obstruction, pancreatitis, pancreatolithiasis, pancreas divisum, pancreatic mass, gastric or duodenal ulcer, or biliary obstruction resulting in jaundice. In the adult, development of symptoms peaks in the third to fifth decades.
Noncontrast radiologic studies of the infant may demonstrate the double-bubble sign identical to that seen in duodenal atresia (see Fig. 47-10). Contrast radiography should be done to ensure that the obstruction is not due to midgut volvulus, a surgical emergency. In adults, transabdominal ultrasound, EUS, CT, or magnetic resonance pancreatography may diagnose annular pancreas. ERCP may demonstrate ductular structures consistent with annular pancreas, but in some cases it may not be technically feasible owing to duodenal obstruction proximal to the major ampulla. Endoscopic ultrasonography is especially useful when prior gastric resection or duodenal obstruction precludes ERCP; in addition, a mass may be staged or undergo fine-needle aspiration at the time of EUS. The ability to evaluate for mass is a new consideration, given reports of ampullary carcinoma in association with annular pancreas; hence, jaundice should not be attributed to annular pancreas until carcinoma is ruled out.75 Magnetic resonance pancreatography, which allows spatial resolution of the entire pancreaticobiliary tree, can identify the annulus and the duct within that surrounds the duodenum. Finally, intraoperative diagnosis at laparotomy is not unusual.
Treatment
The preferred operative therapy for annular pancreas includes duodenoduodenostomy or duodenojejunostomy. Prognosis postoperatively is excellent with either, and postoperative deaths among infants are generally due to associated anomalies. Division or dissection of the pancreatic tissue is not recommended owing to the high risk of complications, including pancreatitis, pancreatic fistula, and incomplete relief of symptoms due to intrinsic duodenal narrowing. An annular pancreas identified at the time of organ procurement has been transplanted along with a long segment of duodenum with good results, so that annular pancreas can be considered suitable for transplantation.76
Duodenal Duplication Cysts
Duodenal duplication cysts are a rare anomaly, totaling only 7% of gastrointestinal duplications. Most commonly located posterior to the first or second portion of the duodenum, these spherical or tubular cysts generally do not communicate with the duodenal lumen but do share blood supply with the duodenum. Three histologic criteria for duodenal duplication cysts exist: gastrointestinal mucosa, a smooth muscle layer in the wall, and an association with the duodenal wall. The mucosa is typically duodenal, but in 15% of cases there is gastric mucosa, and very rarely, pancreatic tissue is found. Men and women are affected equally. Several embryologic theories have been postulated but none explain the diversity of anatomic varieties.77
Clinical Features and Diagnosis
Duplications may be clinically silent for years before presentation. Presenting signs and symptoms of these cysts are typically that of partial gastric outlet obstruction include vomiting, decreased oral intake, periumbilical tenderness, and abdominal distention. Conversely, an asymptomatic mass found on physical or radiologic examination may be noted first. If heterotopic gastric mucosa is present, bleeding or perforation may be the initial presenting sign. In the neonate duodenal obstruction due to a large duplication cyst has been reported. Infected duodenal duplication cyst has been noted as well.78 Pancreatitis may recur if the cyst compresses or is in communication with the pancreatic duct. Finally, jaundice and duodenojejunal intussusception resulting in small bowel obstruction have been described.79
Noncontrast, as well as contrast, radiography may demonstrate obstruction or compression effect, but in general, findings are nonspecific and only suggestive. Abdominal ultrasonography may show unilocular cystic structure with echogenic mucosa surrounded by thin hypoechoic halo of muscle layer.78 Peristaltic waves through the cyst may be evident on ultrasound. Antenatal ultrasound scans are more often identifying suspected cysts. CT may demonstrate an encapsulated, noncommunicating cyst posterior to the duodenum. On ERCP, a compressible periampullary mass may be seen.
Treatment
Surgical therapy should be individualized in accordance with the anatomy of the cyst. Because of potential complications, early neonatal resection, even for asymptomatic cysts, has been advocated.80 Operations that have been performed including local excision and cystojejunostomy. Mucosal stripping of the common muscular wall and resection coupled with removal of free walls has been recommended.81 This, however, may be complex because of the proximity of the cyst to the papilla and biliary-pancreatic confluence. Endoscopic drainage, as well as removal, has been successful in adult and pediatric cases. Invasive carcinoma has been reported in an adult with duodenal duplication cyst, so endoscopic drainage without resection may require reconsideration.
Carachi R, Azmy A. Foregut duplications. Pediatr Surg Int. 2002;18:371. (Ref 18.)
Çorapçioglu F, Ekingen G, Sarper N, Güvenç BH. Immature gastric teratoma of childhood: A case report and review of the literature. JPGN. 2004;39:292-4. (Ref 30.)
Fine J-D, Johnson LB, Weiner M, Suchindran C. Gastrointestinal complications of inherited epidermolysis bullosa: Cumulative experience of the National Epidermolysis Bullosa Registry. JPGN. 2008;46:147-58. (Ref 10.)
Gupta DK, Srinivas M, Dave S, et al. Gastric teratoma in children. Pediatr Surg Int. 2000;16:329. (Ref 26.)
Hellan M, Lee T, Lerner T. Diagnosis and therapy of primary hypertrophic pyloric stenosis in adults: Case report and review of literature. J Gastrointestinal Surg. 2006;10:265-9. (Ref 55.)
Johnson LR. Gastrointestinal physiology, 6th ed. St. Louis: Mosby; 2001. p 75. (Ref 2.)
MacMahon B. The continuing enigma of pyloric stenosis of infancy: A review. Epidemiology. 2006;17:195-201. (Ref 43.)
Merrot T, Anastasescu R, Pankevych T, et al. Duodenal duplications. Clinical characteristics, embryological hypotheses, histological findings, treatment. Eur J Pediatr Surg. 2006;16:18-23. (Ref 77.)
Moore KL, Persaud TVN. The developing human, 7th ed. Philadelphia: WB Saunders; 2003. p 258. (Ref 1.)
Naik-Mathuria B, Olutoye O. Foregut abnormalities. Surg Clin North Am. 2006;86:261-84. (Ref 50.)
Ng WT, Lee SY. Hypertophic pyloric stenosis, congenital or not congenital: A critical overview. Pediatr Surg Int. 2002;18:563. (Ref 31.)
Okoye BO, Parikh DH, Buick RG, et al. Pyloric atresia: Five new cases, a new association, and a review of the literature with guidelines. J Pediatr Surg. 2000;36:1242. (Ref 11.)
1. Moore KL, Persaud TVN. The developing human, 7th ed. Philadelphia: WB Saunders; 2003. p 258
2. Johnson LR. Gastrointestinal physiology, 6th ed. St. Louis: Mosby; 2001. p 75
3. DeHertogh G, Van Eyken P, Ectors N, et al. On the existence and location of cardiac mucosa: An autopsy study in embryos, fetuses, and infants. Gut. 2003;52:791.
4. Koh TJ, Chen D. Gastrin as a growth factor in the gastrointestinal tract. Regul Pept. 2000;93:37.
5. Sinclair NF, Raychowdhury R, Wang TC, et al. Gastrin regulates the heparin-binding epidermal-like growth factor promoter via a PKC/EGFR-dependent mechanism. Am J Physiol Gastrointest Liver Physiol. 2004;286:G992.
6. Skandalakis JE, Gray SW, Ricketts R. The stomach. In: Skandalakis JE, Gray SW, editors. Embryology for surgeons. 2nd ed. Baltimore: Williams & Wilkins; 1994:150.
7. Lachaux A, Bouvier R, Loras-Duclaux I, et al. Isolated deficient α6β4 integrin expression in the gut associated with intractable diarrhea. J Pediatr Gastroenterol Nutr. 1999;29:395.
8. Sonnenberg A, Calafat J, Janssen H, et al. Integrin α6/β4 complex is located in hemidesmosomes, suggesting a major role in epidermal cell-basement membrane adhesion. J Cell Biol. 1997;113:907.
9. Wallerstein R, Klein ML, Genieser N, et al. Epidermolysis bullosa, pyloric atresia, and obstructive uropathy: A report of two case reports with molecular correlation and clinical management. Pediatr Dermatol. 2000;17:286.
10. Fine J-D, Johnson LB, Weiner M, Suchindran C. Gastrointestinal complications of inherited epidermolysis bullosa: Cumulative experience of the National Epidermolysis Bullosa Registry. JPGN. 2008;46:147-58.
11. Okoye BO, Parikh DH, Buick RG, et al. Pyloric atresia: Five new cases, a new association, and a review of the literature with guidelines. J Pediatr Surg. 2000;36:1242.
12. Bass J. Pyloric atresia associated with multiple intestinal atresias and immune deficiency. J Pediatr Surg. 2002;37:941.
13. Dessant A, Iannuccelli M, Dore A, et al. Pyloric atresia: An attempt at anatomic pyloric sphincter reconstruction. J Pediatr Surg. 2000;35:1372.
14. Hernaiz Driever P, Gohlich-Ratmann G, Konig R, et al. Congenital microgastria, growth hormone deficiency, and diabetes insipidus. Eur J Pediatr. 1997;156:37.
15. Menon P, Rao HP, Cutinha BR, et al. Gastric augmentation in isolated congenital microgastria. J Pediatr Surg. 2003;38:E45.
16. al-Gazali LI, Bakir M, Dawodu A, et al. Recurrence of the severe form of microgastria-limb reduction defect in a consanguineous family. Clin Dysmorphol. 1999;8:253-8.
17. Kroes EJ, Festen C. Congenital microgastria: A case report and review of literature. Pediatr Surg Int. 1998;13:416.
18. Carachi R, Azmy A. Foregut duplications. Pediatr Surg Int. 2002;18:371.
19. Wieczorek RL, Seidman I, Ranson JH, et al. Congenital duplication of the stomach: case report and review of the english literature. Am J Gastroenterol. 1984;79:597-602.
20. Correia-Pinto J, Tavares ML, Monteiro J, et al. Prenatal diagnosis of abdominal enteric duplications. Prenat Diagn. 2000;20:163.
21. Bhatia V, Garg PK, Gupta SD, et al. Demonstration of peristalsis in gastric duplication cyst by EUS: Implications for diagnosis and symptomatology. Gastrointest Endosc. 2008;68:183-5.
22. Machado MAC, Santos VR, Martino RB, et al. Laparoscopic resection of gastric duplication. Surg Laparosc Endosc. 2003;13:268.
23. Kuraoka K, Nakayama H, Kagawa T, et al. Adenocarcinoma arising from a gastric duplication cyst with invasion to the stomach: A case report with literature review. J Clin Pathol. 2004;57:428-31.
24. Dunlap JP, James CA, Maxson RT, et al. Gastric teratoma with intramural extension. Pediatr Radiol. 1995;25:383.
25. Gore MD, Fernbach SK. Gastric teratoma. Radiology. 2002;225:497.
26. Gupta DK, Srinivas M, Dave S, et al. Gastric teratoma in children. Pediatr Surg Int. 2000;16:329.
27. Park WH, Choi S, Kim J. Congenital gastric teratoma with gastric perforation mimicking meconium peritonitis. J Pediatr Surg. 2002;37:E11.
28. Matsukuma S, Wada R, Daibou M, et al. Adenocarcinoma arising from gastric immature teratoma. Cancer. 1995;75:2663.
29. Bourke CJ, Mackay AJ, Payton D. Malignant gastric teratoma: Case report. Pediatr Surg Int. 1997;12:192.
30. Çorapçioglu F, Ekingen G, Sarper N, Güvenç BH. Immature gastric teratoma of childhood: A case report and review of the literature. JPGN. 2004;39:292-4.
31. Ng WT, Lee SY. Hypertophic pyloric stenosis, congenital or not congenital: A critical overview. Pediatr Surg Int. 2002;18:563.
32. Kobayashi H, O’Brian S, Puri P. Immunochemical characterization of neural cell adhesion molecule (NCAM), nitric oxide synthase, and neurofilament protein expression in pyloric muscle of patients with pyloric stenosis. J Pediatr Gastroenterol Nutr. 1995;20:319.
33. Abel RA. The ontogeny of the peptide innervation of the human pylorus with special reference to understanding the aetiology and pathogenesis of infantile hypertrophic pyloric stenosis. Ann R Coll Surg Engl. 2000;82:371.
34. Subramaniam R, Doig CM, Moore L. Nitric oxide synthase is absent in only a subset of cases of pyloric stenosis. J Pediatr Surg. 2001;36:616.
35. Vanderwinden JM, Liu H, de Laet MH, et al. Study of the interstitial cells of Cajal in infantile pyloric stenosis. Gastroenterology. 1996;111:279.
36. Vanderwinden JM, Rumessen JJ. Interstitial cells of Cajal in human gut and gastrointestinal disease. Microsc Res Tech. 1999;47:344.
37. Shima H, Ohshiro K, Puri P. Increased local synthesis of epidermal growth factors in infantile hypertrophic pyloric stenosis. Pediatr Res. 2000;47:201.
38. Holder-Espinasse M, Ahmad Z, Hamill J, et al. Familial syndromic duodenal atresia: Feingold syndrome. Eur J Pediatr Surg. 2004;14:112-16.
39. Sorensen HR, Skriver MV, Pedersen L, et al. Risk of infantile hypertrophic pyloric stenosis after maternal postnatal use of macrolides. Scand J Infect Dis. 2003;35:104.
40. Cooper WO, Griffin MR, Arbogast P, et al. Very early exposure to erythromycin and infantile hypertrophic pyloric stenosis. Arch Pediatr Adolesc Med. 2002;156:647.
41. Morrison W. Infantile hypertrophic pyloric stenosis in infants treated with azithromycin. Pediatr Infec Dis. 2007;26:186-8.
42. Maheshwai N. Are young infants treated with erythromycin at risk for developing hypertrophic pyloric stenosis? Arch Dis Child. 2007;92:271-3.
43. MacMahon B. The continuing enigma of pyloric stenosis of infancy: A review. Epidemiology. 2006;17:195-201.
44. Starinsky R, Klin B, Siman-Tov Y, et al. Does dehydration affect thickness of the pyloric muscle? An experimental study. Ultrasound Med Biol. 2001;28:421.
45. Hernanz-Schulman M. Infantile hypertrophic pyloric stenosis. Radiol. 2003;227:319-31.
46. De Baker A, Bove T, Vandenplas Y, et al. Contribution of endoscopy to early diagnosis of hypertrophic pyloric stenosis. J Pediatr Gastroenterol Nutr. 1994;18:78.
47. Michaud L, Gottrand F, Ategbo S, et al. Pitfalls of endoscopy for diagnosis of pyloric stenosis. J Pediatr Gastroenterol Nutr. 1995;21:483.
48. Khan S, Orenstein SR. Eosinophilic gastroenteritis masquerading as pyloric stenosis. Clin Pediatr. 2000;39:55.
49. Ordorica-Flores R, Leon-Villanueva V, Bracho-Blanchet E, et al. Infantile hypertrophic pyloric stenosis: A comparative study of pyloric trauma myoplasty and Fredet-Ramstedt pyloromyotomy. J Pediatr Surg. 2001;36:1000-3.
50. Naik-Mathuria B, Olutoye O. Foregut abnormalities. Surg Clin North Am. 2006;86:261-84.
51. Campbell BT, McLean K, Barnhart DC, et al. A comparison of laparoscopic and open pyloromyotomy at a teaching hospital. J Pediatr Surg. 2002;37:1068.
52. St. Peter SD, Ostlie DJ. Pyloric stenosis. From a retrospective analysis to a prospective clinical trial—The impact on surgical outcomes. Cur Opin in Pediatr. 2008;20:311-14.
53. Meissner PE, Engelmann G, Troeger J, et al. Conservative treatment of infantile hypertrophic pyloric stenosis with intravenous atropine sulfate does not replace pyloromyotomy. Pediatr Surg Int. 2006;22:1021-24.
54. Yamataka A, Tsukada K, Yokoyama-Laws Y, et al. Pyloromyotomy versus atropine sulfate for infantile hypertrophic pyloric stenosis. J Pediatr Surg. 2000;35:338.
55. Hellan M, Lee T, Lerner T. Diagnosis and therapy of primary hypertrophic pyloric stenosis in adults: Case report and review of literature. J Gastrointestinal Surg. 2006;10:265-9.
56. Graadt van Roggen JF, van Krieken JHJM. Adult hypertrophic pyloric stenosis: Case report and review. J Clin Pathol. 1998;51:479.
57. Kuwada SK, Alexander GL. Long-term outcome of endoscopic dilation of nonmalignant pyloric stenosis. Gastrointest Endosc. 1995;41:15.
58. Nakamura T, Kitagawa M, Takehira Y, et al. Palliation of pyloric stenosis caused by gastric cancer using an endoscopically placed covered ultraflex stent: Covered stent inside an occluded uncovered stent. Cardiovasc Intervent Radiol. 2000;23:315.
59. Hemming V, Rankin J. Small intestinal atresia in a defined population: Occurrence, prenatal diagnosis and survival. Prenat Diagn. 2007;27:1205-11.
60. Ramalho-Santos M, Melton DA, McMahon AP. Hedgehog signals regulate multiple aspects of gastrointestinal development. Development. 2000;127:2763.
61. Dalla Vecchia LK, Grosfeld JL, West KW, et al. Intestinal atresia and stenosis. Arch Surg. 1999;133:490.
62. Murshed R, Nicholls G, Spitz L. Intrinsic duodenal obstruction: Trends in management and outcome over 45 years (1951-1995) with relevance to prenatal counseling. Br J Obstet Gynaecol. 1999;106:1197.
63. Okti Poki H, Holland AJA, Pitkin J. Double bubble, double trouble. Pediatr Surg Int. 2005;21:428-31.
64. Holder-Espinasse M, Ahmad Z, Hamill J, et al. Familial syndromic duodenal atresia: Feingold syndrome. Eur J Pediatr Surg. 2004;14:112-16.
65. Mitchell CE, Marshall DG, Reid WD. Preampullary duodenal obstruction in a father and son. J Pediatr Surg. 1993;29:1582.
66. Zimmer EZ, Bronshtein M. Early diagnosis of duodenal atresia and possible sonographic pitfalls. Prenat Diag. 1996;16:564.
67. van Rijn RR, van Lienden KP, Fortuna TL, et al. Membranous duodenal stenosis: Initial experience with balloon dilatation in four children. Eur J Radiol. 2006;59:29-32.
68. Diamond IR, Hayes-Jordan A, Chait P, et al. A novel treatment of congenital duodenal stenosis: Image-guided treatment of congenital and acquired bowel strictures in children. J Laparoendosc Adv Surg Tech A. 2006;16:317-20.
69. Ladd AP, Madura JA. Congenital duodenal anomalies in the adult. Arch Surg. 2001;136:376.
70. Escobar MA, Ladd AP, Grosfeld JL, et al. Duodenal atresia and stenosis: Long term follow-up over 30 years. J Pediatr Surg. 2004;39:867.
71. Ein SH, Kim PCW, Miller HAB. The late nonfunctioning duodenal atresia repair-a second look. J Pediatr Surg. 2000;35:690.
72. Zyromski NJ, Sandoval JA, Pitt HA, et al. Annular pancreas: Dramatic differences between children and adults. J Am Coll Surg. 2008;2006:1019-27.
73. Paraskevas G, Papaziogas B, Lazaridis C, et al. Annular pancreas in adults: Embryological development, morphology and clinical significance. Surg Radiol Anat. 2001;23:437.
74. McCollum MO, Jamieson DH, Webber EM. Annular pancreas and duodenal stenosis. J Pediatr Surg. 2002;37:1776.
75. Benger JR, Thompson MH. Annular pancreas and obstructive jaundice. Am J Gastroenterol. 1997;92:713.
76. Romagnoli J, Papalois VE, Hakin NS. Transplantation of an annular pancreas with enteric drainage. Int Surg. 1998;83:36.
77. Merrot T, Anastasescu R, Pankevych T, et al. Duodenal duplications. Clinical characteristics, embryological hypotheses, histological findings, treatment. Eur J Pediatr Surg. 2006;16:18-23.
78. Oshima K, Suzuki N, Ikeda H, et al. Infected duodenal duplication with unusual clinical and radiological manifestations: A case report. Pediatr Radiol. 1998;28:518.
79. Zamir G, Gross E, Shmushkevich A, et al. Duodenal duplication cyst manifested by duodenojejunal intussusception and hyperbilirubinemia. J Pediatr Surg. 1999;34:1297.
80. Foley PT, Sithasanan N, McEwing R, et al. Enteric duplications presenting as antenatally detected abdominal cysts: Is delayed resection appropriate? J Pediatr Surg. 2006;38:1810-13.
81. Bergman KS, Jacir NN. Cystic duodenal duplication-staged management in a premature infant. J Pediatr Surg. 1993;28:1584.
[/level-membership-for-gastroenterology-and-hepatology-category][not-level-membership-for-gastroenterology-and-hepatology-category]
CHAPTER 47 Anatomy, Histology, Embryology, and Developmental Anomalies of the Stomach and Duodenum
EMBRYOLOGY AND ANATOMY OF THE STOMACH
GENERAL CONSIDERATIONS
The stomach is recognizable in the fourth week of gestation as a dilation of the distal foregut (Fig. 47-1).1 As the stomach enlarges, the dorsal aspect grows more rapidly than the ventral aspect, thus forming the greater curvature. Additionally, during the enlargement process the stomach rotates 90 degrees around its longitudinal axis, orienting the greater curvature (the dorsal aspect) to the left and the lesser curvature (ventral aspect) to the right. The combined effects of rotation and ongoing differential growth result in the stomach lying transversely in the mid and left upper abdomen. The events also explain the vagal innervation of the stomach: the right vagus nerve innervating the posterior stomach wall (the primordial right side) and the left vagus nerve innervating the anterior wall (the primordial left side).
Figure 47-1. Development of the stomach and duodenum and formation of the omental bursa (lesser sac) and greater omentum. A, Median section of a 28-day embryo. B, Anterolateral view of a 28-day embryo. C, Embryo about 35 days old. D, Embryo about 40 days old. E, Embryo about 48 days old. F, Lateral view of the stomach and greater omentum of an embryo at about 52 days. The transverse section shows the omental foramen and omental bursa. G, Sagittal section showing the omental bursa and greater omentum. The embryology of the duodenum is discussed further in Chapters 55 and 96.
(From Moore KL, Persaud TVN. The developing human. 7th ed. Philadelphia: WB Saunders; 2003. p 258.)
The stomach is divided into four regions that can be defined by anatomic or histologic landmarks (Fig. 47-2).2 Anatomically the cardia is a small ill-defined area of the stomach immediately adjacent to its junction with the esophagus. This region of the stomach has been the focus of intense investigation. Controversy exists as to the nature, location, extent, and even existence of cardiac mucosa. The fundus projects upward, above the cardia and gastroesophageal junction. This dome-shaped area of the stomach is its most superior portion and is in contact above with the left hemidiaphragm and to the left with the spleen. The body, or corpus, the largest portion of the stomach, is located immediately below and continuous with the fundus. The incisura angularis, a fixed, sharp indentation two thirds of the distance down the lesser curvature, marks the caudal aspect of the gastric body (Fig. 47-3). The gastric antrum extends from its indistinct border with the body to the junction of the pylorus with the duodenum. These gross anatomic landmarks correspond roughly with the mucosal histology because antral mucosa (pyloric gland mucosa) actually extends from an area on the lesser curvature somewhat above the incisura. The pylorus (pyloric channel) is a tubular structure joining the duodenum to the stomach and contains the palpable circular muscle, the pyloric sphincter. The pylorus is somewhat mobile owing to its enclosure between the peritoneum of the greater and lesser omenta but is generally located 2 cm to the right of midline at L1. Corresponding motor and secretory functions of these regions of the stomach are discussed in detail in Chapters 48 and 49.
TISSUE LAYERS OF THE STOMACH
The luminal surface of the gastric wall forms thick, longitudinally oriented folds, or rugae, that flatten with distention. Four layers make up the gastric wall: mucosa, submucosa, muscularis propria, and serosa. Mucosa lines the gastric lumen, appearing as a smooth, velvety blood-filled lining. The mucosa of the cardia, antrum, and pylorus is somewhat paler than that of the fundus and body. It is within the gastric mucosa that most of the functional secretory elements of the stomach are located (see Chapter 49). The submucosa, immediately deep to the mucosa, provides the dense connective tissue skeleton of collagen and elastin fibers. Lymphocytes, plasma cells, arterioles, venules, lymphatics, and the submucosal plexus are also contained within the submucosa. The third tissue layer, the muscularis propria, is a combination of three muscle layers: inner oblique, middle circular, and outer longitudinal. The inner oblique muscle fibers course over the gastric fundus, covering the anterior and posterior aspects of the stomach wall. The middle circular fibers encircle the body of the stomach, thickening distally to become the pyloric sphincter. The outer longitudinal muscle fibers course primarily along the greater and lesser curvatures of the stomach. The final layer of the stomach is the transparent serosa, a continuation of the visceral peritoneum.
MICROSCOPIC ANATOMY
The gastric mucosal surface is composed primarily of a simple layer of columnar epithelial cells 20 to 40 mm in height. These surface mucous cells (Fig. 47-4), which are similar throughout the stomach, contain basally located nuclei, prominent Golgi stacks, and dense cytoplasm with especially apically dense mucin-containing membrane-bound granules. The cells secrete mucus in granules that are released via exocytosis, apical expulsion, and cell exfoliation. The primary role of mucus, along with bicarbonate, is luminal cytoprotection from “the elements”: acid, pepsin, ingested substances, and pathogens. Cellular renewal time for a gastric surface mucous cell is approximately three days.
The surface epithelial lining is invaginated by gastric pits, or foveolae, that provide the gastric glands access to the gastric lumen, with a ratio of one pit to four or five gastric glands. The gastric glands of different anatomic regions of the stomach are lined with different types of specialized epithelial cells, allowing for differentiation of these regions by type of gastric gland (see Fig. 47-2). The first region, the cardia, is a small transition zone from esophageal squamous epithelium to gastric columnar epithelium. The cardia has been a controversial histologic area of discussion, with theories suggesting that its presence is pathologic. However, recent observations concluded that cardiac mucosa develops during gestation and is present at birth.3 The cardiac glands have a branched and tortuous configuration and are populated by mucous, endocrine, and undifferentiated cells. There is a gradual transition from cardiac glands to the second region, the acid-secreting segment of the stomach. This region encompasses the gastric fundus and body and contains the parietal (or oxyntic or fundic) glands. Parietal, chief (also known as peptic), endocrine, mucous neck, and undifferentiated cells compose the oxyntic glands. The final region, corresponding to the antrum and pylorus, contains the pyloric glands, composed of endocrine cells, including gastrin-producing G cells and mucous cells.
By far the most numerous and distinctive gastric glands are the oxyntic glands (Fig. 47-5), responsible for the secretion of acid, intrinsic factor, and most gastric enzymes. These fairly straight and simple tubular glands are closely associated in the areas of gastric fundus and body. A typical gland is subdivided into three areas: the isthmus (where surface mucous cells predominate), the neck (where parietal and mucous neck cells predominate), and the base (where chief cells predominate, along with some parietal and mucous neck cells). Endocrine cells, somatostatin-containing D cells, and histamine-secreting enterochromaffin-like (ECL) cells are scattered throughout the oxyntic epithelium.
The principal cell type of the oxyntic gland is the parietal cell (Fig. 47-6), responsible for the oxyntic mucosal secretion of 3 × 106 hydrogen ions per second, at a final hydrochloric acid (HCl) concentration of around 150 mmol/L. Parietal cells bulge into the lumina of the oxyntic glands and, as the primary hydrogen secretors, have ultrastructural characteristics different from other gastric cells: large mitochondria, microvilli lacking in glycocalyx, and a cytoplasmic canaliculi system in contact with the lumen. In the nonsecreting parietal cell, a cytoplasmic tubulovesicular system predominates and short microvilli line the apical canaliculus. In the secreting state, the tubulovesicular system disappears, leaving an extensive system of intracellular canaliculi containing long microvilli. Mitochondria occupy approximately 30% to 40% of the secreting parietal cell volume, providing energy required for acid secretion across apical microvilli (see Fig. 47-6). The so-called proton pump—the H+,K+-ATPase—resides in the apical microvillus membrane, as does carbonic anhydrase. The apical H+,K+-ATPase functions as the proton translocator in gastric acid secretion (see Chapter 49). Acid secretion begins within 5 to 10 minutes of stimulation. Additionally, parietal cells are the site of intrinsic factor secretion via membrane-associated vesicle transport.
Figure 47-6. Parietal cell. A, Electron photomicrograph. B, Schematic.
(A and B, from Johnson LR. Gastrointestinal physiology. 6th ed. St Louis: Mosby; 2001. pp 78, 79.)
The final region of the stomach encompasses the antrum and pylorus and contains extensively coiled antral glands composed of endocrine and epithelial cells. The epithelial cells are predominantly mucous cells, and there are small numbers of pepsinogen II–secreting oxyntic cells. Although also small in number, gastrin-secreting (G) cells play a vital physiologic role and are the prototype of the open enteroendocrine cell. These cells, which occur either singly or in small clusters in the mid- to deep sections of antral glands (Fig. 47-7A), contain a basilar cytoplasm densely packed with gastrin-containing secretory granules (see Fig. 47-7B). Gastrin release is stimulated by gastric distention, vagal stimulation, dietary amino acids, and peptide, with rapid appearance of the hormone into the bloodstream in the postprandial period (see Chapter 49). The apical or luminal surface of the G cell is narrowed into small microvilli thought to contain receptors responsible for amino acid and peptide stimulation of gastrin release. Significant quantities of gastrin are also secreted into the gastric lumen; gastrin is a known gastric growth and differentiation factor, mediated through upregulation of heparin-binding epidermal-like growth factor (HB-EGF) in gastric parietal cells.4,5
Antral enteroendocrine D cells found in close association with G cells manufacture somatostatin, a potent inhibitor of gastrin secretion. The D cells are also present in small numbers in oxyntic glands. Somatostatin is thought to inhibit acid secretion through paracrine (direct action on ECL and perhaps parietal cells or indirect action on G cells) or endocrine effects (direct action on parietal cells) (see Chapter 49 for more details).
