Analgesic Drugs
OPIOIDS
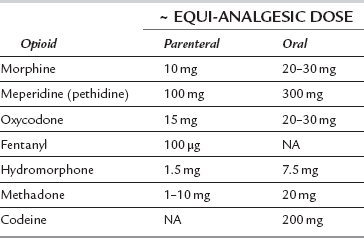
Dose conversion between different opioids is an inexact science, with equi-analgesic doses being based on relative potencies, often derived from studies that are not designed for dose equivalence calculation. Table 5.1 shows suggested equi-analgesic doses for some opioids.
Mechanism of Action
Opioid receptors were originally classified by pharmacological activity in animal preparations, and later by molecular sequence. The three main receptors were classified as μ (mu) or OP3, κ (kappa) or OP1 and δ (delta) or OP2. Another opioid-like receptor has been identified recently; it is termed the nociceptin orphanin FQ peptide receptor. Receptor nomenclature has changed several times in the last few years; the current International Union of Pharmacology (IUPHAR) classification is MOP (mu), KOP (kappa), DOP (delta) and NOP for the nociceptin orphanin FQ peptide receptor (Table 5.2).

Opioid receptors are distributed widely in both central and peripheral nervous systems. The different effects of currently available opioids are dependent on complex interactions at various receptors. There is a range of endogenous neuropeptide ligands active at these receptors (Table 5.2); they function as neurotransmitters, neuromodulators and neurohormones. The endogenous tetrapeptide endomorphins 1 and 2 are potent agonists acting specifically at the MOP receptor; they play a role in modulating inflammatory pain.
Pharmacodynamic Effects of Opioids
Analgesic Action



Opioid Structure
The structures of opioid analgesics are diverse, although for most opioids it is usually the laevorotatory (laevo) stereoisomer that is the active compound. The structures of some of the common agents are shown in Figures 5.1 and 5.2. Agents in current use include phenanthrenes (e.g. morphine – Fig. 5.1), phenylpiperidines (e.g. meperidine (pethidine), fentanyl – Fig. 5.2) and diphenylpropylamines (e.g. methadone, dextropropoxyphene). Structural modification affects agonist activity and alters physicochemical properties such as lipid solubility. A tertiary nitrogen is necessary for activity separated from a quaternary carbon by an ethylene chain. Chemical modifications that produce a quaternary nitrogen significantly reduce potency as a result of decreased CNS penetration. If the methyl group on the nitrogen is changed, antagonism of analgesia may be produced.
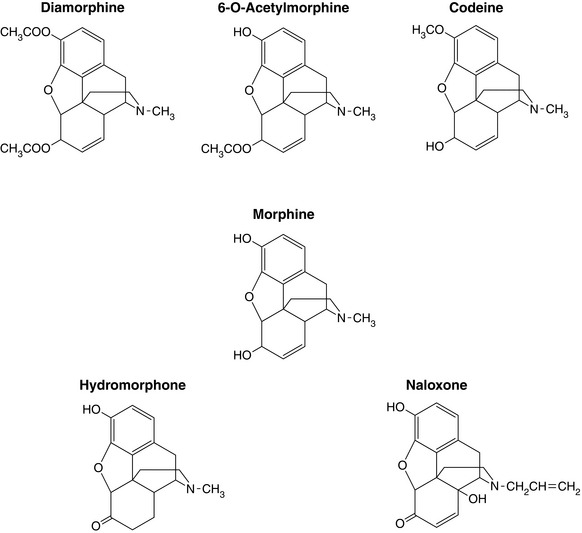
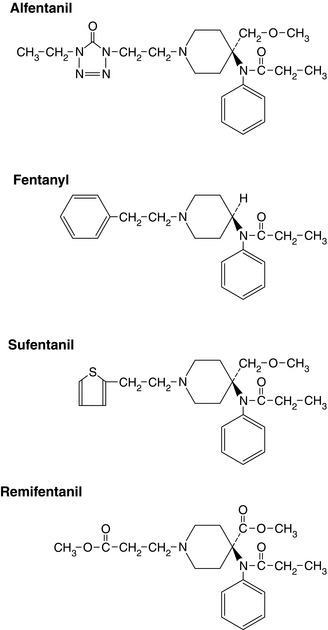
Other important positions for activity and metabolism, as seen on the morphine molecule (Fig. 5.1), include the C-3 phenol group (the distance of this from the nitrogen affects activity) and the C-6 alcohol group. Potency may be increased by hydroxylation of the C-3 phenol; oxidation of C-6 (e.g. hydromorphone); double acetylation at C-3 and C-6 (e.g. diamorphine); hydroxylation of C-14 and reducing the double bond at C-7/8. Further additions at the C-3 OH group reduce activity. A short-chain alkyl substitution is found in mixed agonist-antagonists, hydroxylation or bromination of C-14 produces full antagonists and removal or substitution of the methyl group reduces agonist activity.
Pharmacokinetics and Physicochemical Properties
After prolonged infusion, significant sequestration in fat stores and other body tissues occurs for highly lipid-soluble opioids. This is reflected in the ‘context-sensitive t½’ i.e. the time taken for the plasma concentration to reduce by 50% after an infusion designed to maintain constant plasma concentrations has been stopped (see Chapter 1). The context-sensitive t½ is increased after prolonged infusion for most opioids apart from remifentanil. For example, the elimination t½ for fentanyl after bolus administration is 3–5 h, but increases to 7–12 h after prolonged infusion.
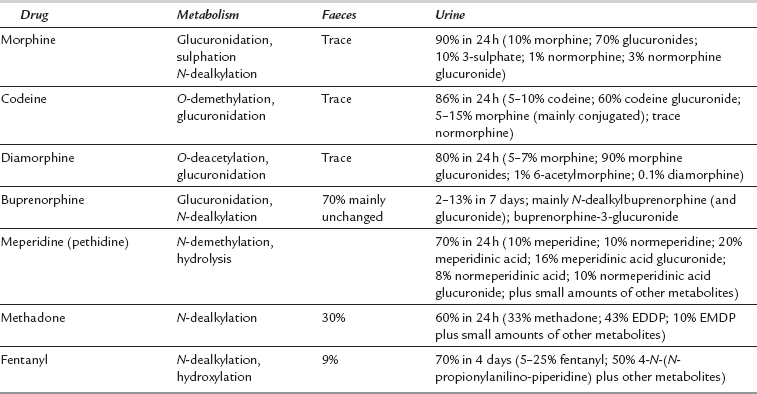
Most opioid metabolism occurs in the liver (phase I and II reactions) with the hydrophilic metabolites predominantly excreted renally, although a small amount may be excreted in the bile or unchanged in the urine. As a result, hepatic blood flow is one of the major determinants of plasma clearance. Metabolism of individual drugs is shown in Table 5.3. Enterohepatic recirculation may occur when water-soluble metabolites excreted in the gut may be metabolized by gut flora to the parent opioid and then reabsorbed. Lipid-soluble opioids may diffuse into the stomach, become ionized because of the low pH and then be reabsorbed in the small intestine; this results in a secondary peak in plasma concentration.
TABLE 5.3
Metabolism and Excretion of Some Opioids
A summary of physicochemical and pharmacokinetic properties of some opioids is shown in Table 5.4. Metabolism (including production of active metabolites), distribution between different tissues and elimination all interact within individual subjects to produce clinically important actions at receptor sites.
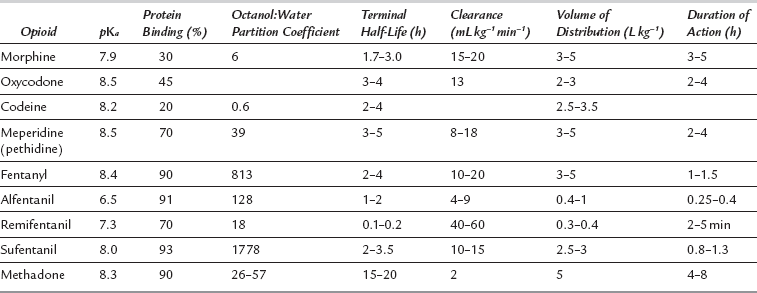
Factors affecting pharmacokinetics include:
 Age. Systemic dose is often calculated on body weight, although there is little evidence to support this in adult clinical practice. Age is often more important because of both pharmacokinetic and pharmacodynamic factors. Metabolism and volume of distribution are often reduced in the elderly, leading to increased free drug concentrations in the plasma. Hepatic blood flow may have declined by 40–50% by age 75 years, with reduced clearance of opioids. Increased CNS sensitivity to opioid effects is also found in the elderly.
Age. Systemic dose is often calculated on body weight, although there is little evidence to support this in adult clinical practice. Age is often more important because of both pharmacokinetic and pharmacodynamic factors. Metabolism and volume of distribution are often reduced in the elderly, leading to increased free drug concentrations in the plasma. Hepatic blood flow may have declined by 40–50% by age 75 years, with reduced clearance of opioids. Increased CNS sensitivity to opioid effects is also found in the elderly.




Routes of Administration
Opioids given parenterally have 100% bioavailability (see Chapter 1), although peak plasma concentrations may be affected by site of administration and haemodynamic status. Opioids may be given by many routes; variations between specific agents are discussed below. It is unclear how much cross-tolerance exists for different routes of administration, e.g. intravenous versus epidural.



MOP Agonists
Phenylpiperidine opioids (Fig. 5.2) are potent MOP receptor agonists with moderate (alfentanil) to high (sufentanil) lipid solubility and good diffusion through membranes. Both potency and time to reach the effect site vary considerably. In contrast to morphine, these agents do not cause histamine release. All except remifentanil may cause postoperative respiratory depression as a result of secondary peaks in plasma concentrations. This may be caused by release from body stores if large doses have been infused intra-operatively. Fentanyl, alfentanil and sufentanil are metabolized mainly in the liver to inactive metabolites. Very little is excreted unchanged in the urine (Table 5.3).
Morphine
Morphine is metabolized, at least in part, by microsomal UDP glucuronyl transferases (UDPGT) found in the liver, kidney and intestines. Several of these metabolites may have clinically significant effects (see below). Although morphine conjugation occurs in the liver, there is evidence that extrahepatic sites may also be important, e.g. kidney, gastrointestinal tract. The site of conjugation on the molecule also varies, leading to a variety of metabolites (Table 5.3). After glucuronidation, metabolites are excreted in urine or bile, dependent on molecular weight and polarity; more than 90% of morphine metabolites are excreted in the urine. The main metabolite in humans is morphine-3-glucuronide (60–80%) and this may have an excitatory effect via CNS actions not related to opioid receptor activation. Morphine-6-glucuronide (M-6-G) is active at the MOP receptor, producing analgesia and other MOP-related effects. It is significantly more potent than morphine. Therefore, M-6-G produces significant clinical effects despite only 10% of morphine being metabolized in this way. As it is renally excreted, it may accumulate in patients with impaired renal function, causing respiratory depression. Current evidence indicates that accumulation of morphine metabolites, especially M-6-G, becomes significant when creatinine clearance declines to 50 mL min–1 or less.
Diamorphine
Diamorphine is a prodrug; it is inactive at opioid receptors. However, it is converted rapidly to the active metabolites 6-monoacetylmorphine, morphine and M-6-G. Further metabolism is similar to that of morphine (Table 5.3) and similar problems may arise if there is renal impairment.
Partial Agonists
These agents have high affinity for the MOP receptor but limited efficacy (see Chapter 1). A ceiling effect is seen in the dose-response curve at less than the maximal analgesic effect of full MOP agonists. If given with a full MOP agonist, there may be a reduction in the maximal analgesic effect.
New Developments in Opioid Pharmacology
There is considerable interest in developing opioid analgesics with an improved side effect profile, and there are some promising recent developments in this area. It has been recognized that by using agents which act on more than one type of opioid receptor there may be beneficial effects (see Fig. 5.3) Some recently developed agents include:
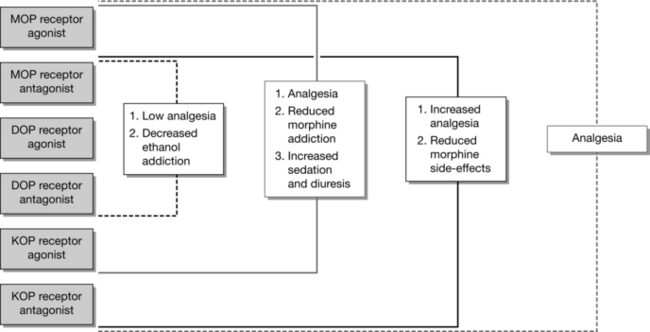
FIGURE 5.3 Schemtic representation of the potential clinical advantages of using bivalent ligands, with the various combinations of pharmacophores (antagonists and agonists). (Adapted from Dietis N, et al. British Journal of Anaesthesia 2009;103:38-49.)
 Oxycodone/Naloxone (Targinact®). This drug combination utilizes a fixed ratio of oxycodone and naloxone (2:1). The oral naloxone has a greater affinity for opioid receptors in the gut than oxycodone and therefore preferentially binds to these receptors. As a result, gastrointestinal side effects may be reduced, without any effect on the central analgesic effect of oxycodone.
Oxycodone/Naloxone (Targinact®). This drug combination utilizes a fixed ratio of oxycodone and naloxone (2:1). The oral naloxone has a greater affinity for opioid receptors in the gut than oxycodone and therefore preferentially binds to these receptors. As a result, gastrointestinal side effects may be reduced, without any effect on the central analgesic effect of oxycodone.


PARACETAMOL
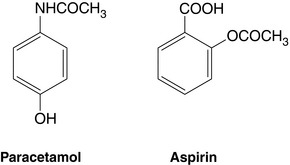
Paracetamol (acetaminophen) was first used in 1893 and is the only remaining p-aminophenol available in clinical practice. It is the active metabolite of the earlier, more toxic drugs acetanilide and phenacetin. Its structure is shown in Figure 5.4. Paracetamol is an effective analgesic and antipyretic but has no anti-inflammatory activity. In recommended doses, it is safe and has remarkably few side-effects.
NON-STEROIDAL ANTI-INFLAMMATORY DRUGS
The analgesic, anti-inflammatory and antipyretic effects of salicylates, derived from the bark of the willow tree, were described as early as 1763. Acetylsalicylic acid (aspirin) was first produced in 1853 (Fig. 5.4). More recently, many other NSAIDs have been developed with actions similar to aspirin. Perioperative analgesia using NSAIDs is free from many of the adverse effects of opioids, such as respiratory depression, sedation, nausea and vomiting and gastrointestinal stasis. NSAIDs have been shown to be effective analgesics in acute and chronic conditions, although significant contraindications and adverse effects limit their use.
Mechanism of Action
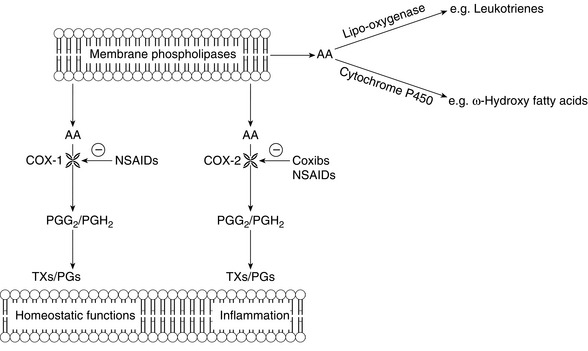
The mechanism of action of aspirin was discovered in the 1970s. It was shown to irreversibly inhibit the production of prostanoids (i.e. prostaglandins and thromboxanes) from arachidonic acid released from phospholipids in cell membranes (Fig. 5.5). The basal rate of prostaglandin production is low and regulated by tissue stimuli or trauma that activate phospholipases to release arachidonic acid. Prostaglandins are then produced by the enzyme prostaglandin endoperoxide synthase which has both cyclo-oxygenase and hydroperoxidase sites. At least two subtypes of cyclo-oxygenase enzyme have been identified in humans: COX-1 and COX-2. The prostanoids produced by COX-1 are functionally active in many areas, including the gastrointestinal tract, kidney, lung and cardiovascular systems. By contrast, the functional COX-2 enzyme is normally found less widely, e.g. brain, spinal cord, renal cortex, tracheal epithelium and vascular endothelium. However, COX-2 mRNA is widely distributed. In response to specific stimuli, especially those associated with inflammation, the expression of COX-2 isoenzyme is induced or upregulated, leading to increased local production of prostaglandins. A range of specific prostanoid receptors (e.g. EP1-4) are involved in peripheral sensitization associated with inflammation.
Pharmacodynamics
COX-2-SPECIFIC INHIBITORS
Mechanism of Action
The mechanism of action is similar to that of NSAIDs (Fig. 5.4). Both COX-1 and COX-2 enzymes have very similar active sites and catalytic properties, although COX-2 has a larger potential binding site because of a secondary internal pocket. This has allowed design of drugs to target predominantly COX-2. COX-2 is induced at sites of inflammation and trauma, producing prostaglandins, and these drugs, inhibit this process. However, COX-2 is an important constitutive enzyme in the CNS, including the spinal cord, and inhibition at this site is thought to be an important mechanism also.
Bell, R.F., Dahl, J.B., Moore, R.A., Kalso, E.A., Perioperative ketamine for acute postoperative pain. [Art. No.:CD004603]. Cochrane Database Syst. Rev. 2006;2006(1); doi:10.1002/14651858, CD004603.pub2
Christo, P.J. Opioid effectiveness and side effects in chronic pain. Anesthesiol. Clin. North America. 2003;21:699–713.
Colvin, L.A., Fallon, M.T. Opioid-induced hyperalgesia – a clinical challenge. Br. J. Anaesth. 2010;104:125–127.
Dietis, N., Guerrini, R., Calo, G., et al. Simultaneous targeting of multiple opioid receptors: a strategy to improve side-effect profile. Br. J. Anaesth. 2009;103:38–49.
Hinz, B., Brune, K. Paracetamol and cyclooxygenase inhibition: is there a cause for concern? Ann. Rheum. Dis. 2012;71(1):20–25.
Hocking, G., Cousins, M.J. Ketamine in chronic pain management: an evidence-based review. Anesth. Analg. 2003;97:1730–1739.
Mather, L.E. Trends in the pharmacology of opioids: implications for the pharmacotherapy of pain. Eur. J. Pain. 2001;5(Suppl. A):49–57.
McDonald, J., Lambert, D.G. Opioid receptors. Continuing Education in Anaesthesia, Critical Care and Pain. 2005;5:22–25.
Yaksh, T.L. Pharmacology and mechanisms of opioid analgesic activity. Acta Anaesthesiol. Scand. 1997;41:94–111.