452 |
Amyotrophic Lateral Sclerosis and Other Motor Neuron Diseases |
AMYOTROPHIC LATERAL SCLEROSIS
Amyotrophic lateral sclerosis (ALS) is the most common form of progressive motor neuron disease. It is a prime example of a neurodegenerative disease and is arguably the most devastating of the neurodegenerative disorders.
PATHOLOGY
The pathologic hallmark of motor neuron degenerative disorders is death of lower motor neurons (consisting of anterior horn cells in the spinal cord and their brainstem homologues innervating bulbar muscles) and upper, or corticospinal, motor neurons (originating in layer five of the motor cortex and descending via the pyramidal tract to synapse with lower motor neurons, either directly or indirectly via interneurons) (Chap. 30). Although at its onset ALS may involve selective loss of function of only upper or lower motor neurons, it ultimately causes progressive loss of both categories of motor neurons. Indeed, in the absence of clear involvement of both motor neuron types, the diagnosis of ALS is questionable. In a subset of cases, ALS arises concurrently with frontotemporal dementia (Chap. 448); in these instances, there is degeneration of frontotemporal cortical neurons and corresponding cortical atrophy.
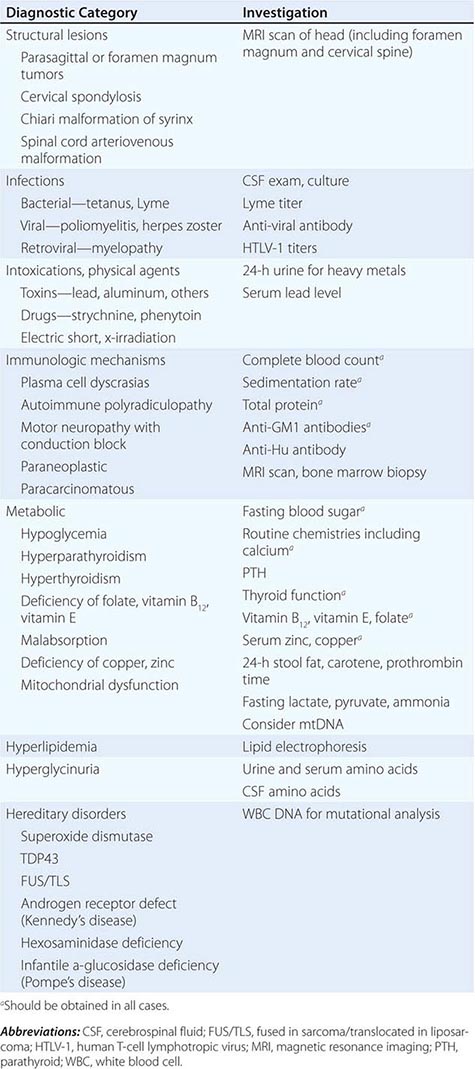
Other motor neuron diseases involve only particular subsets of motor neurons (Tables 452-1 and 452-2). Thus, in bulbar palsy and spinal muscular atrophy (SMA; also called progressive muscular atrophy), the lower motor neurons of brainstem and spinal cord, respectively, are most severely involved. By contrast, pseudobulbar palsy, primary lateral sclerosis (PLS), and familial spastic paraplegia (FSP) affect only upper motor neurons innervating the brainstem and spinal cord.
|
ETIOLOGY OF MOTOR NEURON DISORDERS |
|
SPORADIC MOTOR NEURON DISEASES |
In each of these diseases, the affected motor neurons undergo shrinkage, often with accumulation of the pigmented lipid (lipofuscin) that normally develops in these cells with advancing age. In ALS, the motor neuron cytoskeleton is typically affected early in the illness. Focal enlargements are frequent in proximal motor axons; ultrastructurally, these “spheroids” are composed of accumulations of neurofilaments and other proteins. Commonly in both sporadic and familial ALS, the affected neurons demonstrate ubiquitin-positive aggregates, typically associated with the protein TDP43 (see below). Also seen is proliferation of astroglia and microglia, the inevitable accompaniment of all degenerative processes in the central nervous system (CNS).
The death of the peripheral motor neurons in the brainstem and spinal cord leads to denervation and consequent atrophy of the corresponding muscle fibers. Histochemical and electrophysiologic evidence indicates that in the early phases of the illness denervated muscle can be reinnervated by sprouting of nearby distal motor nerve terminals, although reinnervation in this disease is considerably less extensive than in most other disorders affecting motor neurons (e.g., poliomyelitis, peripheral neuropathy). As denervation progresses, muscle atrophy is readily recognized in muscle biopsies and on clinical examination. This is the basis for the term amyotrophy. The loss of cortical motor neurons results in thinning of the corticospinal tracts that travel via the internal capsule (Fig. 452-1) and brainstem to the lateral and anterior white matter columns of the spinal cord. The loss of fibers in the lateral columns and resulting fibrillary gliosis impart a particular firmness (lateral sclerosis). A remarkable feature of the disease is the selectivity of neuronal cell death. By light microscopy, the entire sensory apparatus, the regulatory mechanisms for the control and coordination of movement, remains intact. Except in cases of frontotemporal dementia, the components of the brain required for cognitive processing are also preserved. However, immunostaining indicates that neurons bearing ubiquitin, a marker for degeneration, are also detected in nonmotor systems. Moreover, studies of glucose metabolism in the illness also indicate that there is neuronal dysfunction outside of the motor system. Within the motor system, there is some selectivity of involvement. Thus, motor neurons required for ocular motility remain unaffected, as do the parasympathetic neurons in the sacral spinal cord (the nucleus of Onufrowicz, or Onuf) that innervate the sphincters of the bowel and bladder.
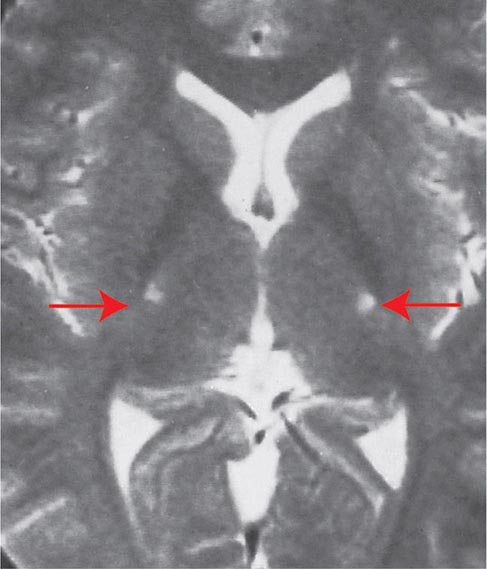
FIGURE 452-1 Amyotrophic lateral sclerosis. Axial T2-weighted magnetic resonance imaging (MRI) scan through the lateral ventricles of the brain reveals abnormal high signal intensity within the corticospinal tracts (arrows). This MRI feature represents an increase in water content in myelin tracts undergoing Wallerian degeneration secondary to cortical motor neuronal loss. This finding is commonly present in ALS, but can also be seen in AIDS-related encephalopathy, infarction, or other disease processes that produce corticospinal neuronal loss in a symmetric fashion.
CLINICAL MANIFESTATIONS
The manifestations of ALS are somewhat variable depending on whether corticospinal neurons or lower motor neurons in the brainstem and spinal cord are more prominently involved. With lower motor neuron dysfunction and early denervation, typically the first evidence of the disease is insidiously developing asymmetric weakness, usually first evident distally in one of the limbs. A detailed history often discloses recent development of cramping with volitional movements, typically in the early hours of the morning (e.g., while stretching in bed). Weakness caused by denervation is associated with progressive wasting and atrophy of muscles and, particularly early in the illness, spontaneous twitching of motor units, or fasciculations. In the hands, a preponderance of extensor over flexor weakness is common. When the initial denervation involves bulbar rather than limb muscles, the problem at onset is difficulty with chewing, swallowing, and movements of the face and tongue. Early involvement of the muscles of respiration may lead to death before the disease is far advanced elsewhere. With prominent corticospinal involvement, there is hyperactivity of the muscle-stretch reflexes (tendon jerks) and, often, spastic resistance to passive movements of the affected limbs. Patients with significant reflex hyperactivity complain of muscle stiffness often out of proportion to weakness. Degeneration of the corticobulbar projections innervating the brainstem results in dysarthria and exaggeration of the motor expressions of emotion. The latter leads to involuntary excess in weeping or laughing (pseudobulbar affect).
Virtually any muscle group may be the first to show signs of disease, but, as time passes, more and more muscles become involved until ultimately the disorder takes on a symmetric distribution in all regions. It is characteristic of ALS that, regardless of whether the initial disease involves upper or lower motor neurons, both will eventually be implicated. Even in the late stages of the illness, sensory, bowel and bladder, and cognitive functions are preserved. Even when there is severe brainstem disease, ocular motility is spared until the very late stages of the illness. As noted, in some cases (particularly those that are familial), ALS develops concurrently with frontotemporal dementia, characterized by early behavioral abnormalities with prominent behavioral features indicative of frontal lobe dysfunction.
A committee of the World Federation of Neurology has established diagnostic guidelines for ALS. Essential for the diagnosis is simultaneous upper and lower motor neuron involvement with progressive weakness and the exclusion of all alternative diagnoses. The disorder is ranked as “definite” ALS when three or four of the following are involved: bulbar, cervical, thoracic, and lumbosacral motor neurons. When two sites are involved, the diagnosis is “probable,” and when only one site is implicated, the diagnosis is “possible.” An exception is made for those who have progressive upper and lower motor neuron signs at only one site and a mutation in the gene encoding superoxide dismutase (SOD1; see below).
EPIDEMIOLOGY
The illness is relentlessly progressive, leading to death from respiratory paralysis; the median survival is from 3–5 years. There are very rare reports of stabilization or even regression of ALS. In most societies, there is an incidence of 1–3 per 100,000 and a prevalence of 3–5 per 100,000. It is striking that about 1 in 1000 adult deaths in North America and Western Europe (and probably elsewhere) are due to ALS; this finding predicts that some 300,000 individuals now alive in the United States will die of ALS. Several endemic foci of higher prevalence exist in the western Pacific (e.g., in specific regions of Guam or Papua New Guinea). In the United States and Europe, males are somewhat more frequently affected than females. Epidemiologic studies have incriminated risk factors for this disease including exposure to pesticides and insecticides, smoking, and, in one report, service in the military. Although ALS is overwhelmingly a sporadic disorder, some 5–10% of cases are inherited as an autosomal dominant trait.
FAMILIAL ALS
Several forms of selective motor neuron disease are inheritable (Table 452-3). Familial ALS (FALS) involves both corticospinal and lower motor neurons. Apart from its inheritance as an autosomal dominant trait, it is clinically indistinguishable from sporadic ALS. Genetic studies have identified mutations in multiple genes, including those encoding the protein C9orf 72 (open reading frame 72 on chromosome 9), cytosolic enzyme SOD1 (superoxide dismutase), the RNA binding proteins TDP43 (encoded by the TAR DNA binding protein gene), and FUS/TLS (fused in sarcoma/translocated in liposarcoma), as the most common causes of FALS. Mutations in C9orf72 account for ~45–50% of FALS and perhaps 4–5% of sporadic ALS cases. Mutations in SOD1 explain another 20% of cases of FALS, whereas TDP43 and FUS/TLS each represent about 5% of familial cases. It has recently been reported that ~1–2% of cases are caused by mutations in genes encoding the proteins optineuron and profilin-1 as well.
|
GENETIC MOTOR NEURON DISEASES |
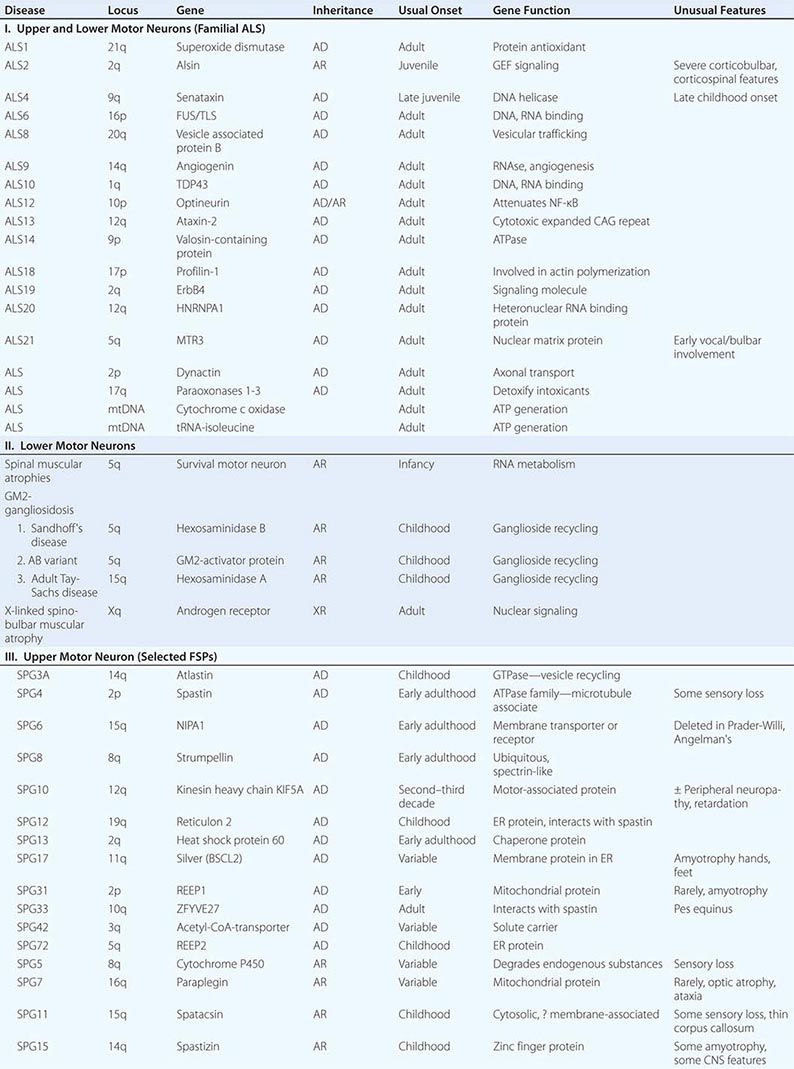
Rare mutations in other genes are also clearly implicated in ALS-like diseases. Thus, a familial, dominantly inherited motor disorder that in some individuals closely mimics the ALS phenotype arises from mutations in a gene that encodes a vesicle-binding protein. A predominantly lower motor neuron disease with early hoarseness due to laryngeal dysfunction has been ascribed to mutations in the gene encoding the cellular accessory motor protein dynactin. Mutations in senataxin, a helicase, cause an early adult-onset, slowly evolving ALS variant. Kennedy’s syndrome is an X-linked, adult-onset disorder that may mimic ALS, as described below.
Genetic analyses are also beginning to illuminate the pathogenesis of some childhood-onset motor neuron diseases. For example, a slowly disabling degenerative, predominantly upper motor neuron disease that starts in the first decade is caused by mutations in a gene that expresses a novel signaling molecule with properties of a guanine-exchange factor, termed alsin.
DIFFERENTIAL DIAGNOSIS
Because ALS is currently untreatable, it is imperative that potentially remediable causes of motor neuron dysfunction be excluded (Table 452-1). This is particularly true in cases that are atypical by virtue of (1) restriction to either upper or lower motor neurons, (2) involvement of neurons other than motor neurons, and (3) evidence of motor neuronal conduction block on electrophysiologic testing. Compression of the cervical spinal cord or cervicomedullary junction from tumors in the cervical regions or at the foramen magnum or from cervical spondylosis with osteophytes projecting into the vertebral canal can produce weakness, wasting, and fasciculations in the upper limbs and spasticity in the legs, closely resembling ALS. The absence of cranial nerve involvement may be helpful in differentiation, although some foramen magnum lesions may compress the twelfth cranial (hypoglossal) nerve, with resulting paralysis of the tongue. Absence of pain or of sensory changes, normal bowel and bladder function, normal roentgenographic studies of the spine, and normal cerebrospinal fluid (CSF) all favor ALS. Where doubt exists, magnetic resonance imaging (MRI) scans and contrast myelography should be performed to visualize the cervical spinal cord.
Another important entity in the differential diagnosis of ALS is multifocal motor neuropathy with conduction block (MMCB), discussed below. A diffuse, lower motor axonal neuropathy mimicking ALS sometimes evolves in association with hematopoietic disorders such as lymphoma or multiple myeloma. In this clinical setting, the presence of an M-component in serum should prompt consideration of a bone marrow biopsy. Lyme disease (Chap. 210) may also cause an axonal, lower motor neuropathy, although typically with intense proximal limb pain and a CSF pleocytosis.
Other treatable disorders that occasionally mimic ALS are chronic lead poisoning and thyrotoxicosis. These disorders may be suggested by the patient’s social or occupational history or by unusual clinical features. When the family history is positive, disorders involving the genes encoding C9orf72, cytosolic SOD1, TDP43, FUS/TLS, and adult hexosaminidase A or α-glucosidase deficiency must be excluded (Chap. 432e). These are readily identified by appropriate laboratory tests. Benign fasciculations are occasionally a source of concern because on inspection they resemble the fascicular twitchings that accompany motor neuron degeneration. The absence of weakness, atrophy, or denervation phenomena on electrophysiologic examination usually excludes ALS or other serious neurologic disease. Patients who have recovered from poliomyelitis may experience a delayed deterioration of motor neurons that presents clinically with progressive weakness, atrophy, and fasciculations. Its cause is unknown, but it is thought to reflect sublethal prior injury to motor neurons by poliovirus (Chap. 228).
Rarely, ALS develops concurrently with features indicative of more widespread neurodegeneration. Thus, one infrequently encounters otherwise typical ALS patients with a parkinsonian movement disorder or frontotemporal dementia, particularly in instances of C9orf72 mutations, which strongly suggests that the simultaneous occurrence of two disorders is a direct consequence of the gene mutation. As another example, prominent amyotrophy has been described as a dominantly inherited disorder in individuals with bizarre behavior and a movement disorder suggestive of parkinsonism; many such cases have now been ascribed to mutations that alter the expression of tau protein in brain (Chap. 448). In other cases, ALS develops simultaneously with a striking frontotemporal dementia. An ALS-like disorder has also been described in some individuals with chronic traumatic encephalopathy, associated with deposition of TDP43 and neurofibrillary tangles in motor neurons.
PATHOGENESIS
The cause of sporadic ALS is not well defined. Several mechanisms that impair motor neuron viability have been elucidated in mice and rats induced to develop motor neuron disease by SOD1 transgenes with ALS-associated mutations. One may loosely group the genetic causes of ALS into three categories. In one group, the primarily problem is inherent instability of the mutant proteins, with subsequent perturbations in protein degradation (SOD1, ubiquilin-1 and -2, p62). In the second, most rapidly growing category, the causative mutant genes perturb RNA processing, transport, and metabolism (C9orf73, TDP43, FUS). In the case of C9orf72, the molecular pathology is an expansion of an intronic hexanucleotide repeat (-GGGGCC-) beyond an upper normal of 30 repeats to hundreds or more repeats. As observed in other intronic repeat disorders such as myotonic dystrophy (Chap. 462e) and spinocerebellar atrophy type 8 (Chap. 450), data suggest that the expanded intronic repeats generate expanded RNA repeats that form intranuclear foci and confer toxicity by sequestering transcription factors or by undergoing noncanonical protein translation across all possible reading frames of the expanded RNA tracts. TDP43 and FUS are multifunctional proteins that bind RNA and DNA and shuttle between the nucleus and the cytoplasm, playing multiple roles in the control of cell proliferation, DNA repair and transcription, and gene translation, both in the cytoplasm and locally in dendritic spines in response to electrical activity. How mutations in FUS/TLS provoke motor neuron cell death is not clear, although this may represent loss of function of FUS/TLS in the nucleus or an acquired, toxic function of the mutant proteins in the cytosol. In the third group of ALS genes, the primary problem is defective axonal cytoskeleton and transport (dynactin, profilin-1). It is striking that variants in other genes (e.g., EphA4) influence survival in ALS but not ALS susceptibility. Beyond the upstream, primary defects, it is also evident that the ultimate neuronal cell death process is complex involving multiple cellular processes that accelerate cell death. These include but are not limited to excitotoxicity, impairment of axonal transport, oxidative stress, activation of endoplasmic reticulum stress and the unfolded protein response, and mitochondrial dysfunction.
Multiple recent studies have convincingly demonstrated that nonneuronal cells importantly influence the disease course, at least in ALS transgenic mice. A striking additional finding in neurodegenerative disorders is that miscreant proteins arising from gene defects in familial forms of these diseases are often implicated in sporadic forms of the same disorder. For example, germline mutations in the genes encoding β-amyloid and α-synuclein cause familial forms of Alzheimer’s and Parkinson’s diseases, and posttranslational, noninherited abnormalities in these proteins are also central to sporadic Alzheimer’s and Parkinson’s diseases. Analogously, recent reports propose that nonheritable, posttranslational modifications in SOD1 are pathogenic in sporadic ALS.
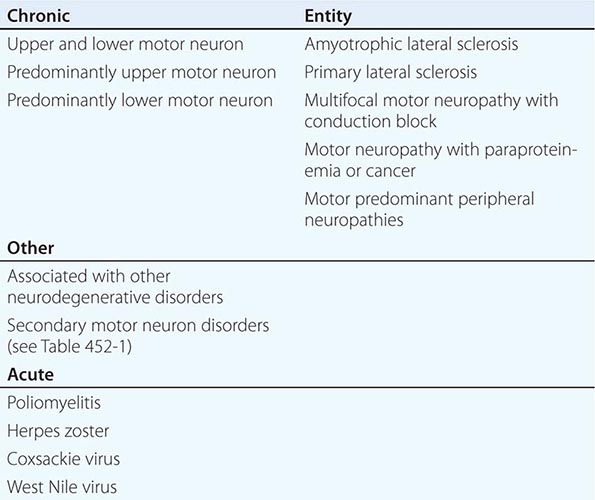
OTHER MOTOR NEURON DISEASES
SELECTED LOWER MOTOR NEURON DISORDERS
In these motor neuron diseases, the peripheral motor neurons are affected without evidence of involvement of the corticospinal motor system (Tables 452-1, 452-2, and 452-3).
X-Linked Spinobulbar Muscular Atrophy (Kennedy’s Disease) This is an X-linked lower motor neuron disorder in which progressive weakness and wasting of limb and bulbar muscles begins in males in mid-adult life and is conjoined with androgen insensitivity manifested by gynecomastia and reduced fertility (Chap. 411). In addition to gynecomastia, which may be subtle, two findings distinguishing this disorder from ALS are the absence of signs of pyramidal tract disease (spasticity) and the presence of a subtle sensory neuropathy in some patients. The underlying molecular defect is an expanded trinucleotide repeat (-CAG-) in the first exon of the androgen receptor gene on the × chromosome. DNA testing is available. An inverse correlation appears to exist between the number of -CAG- repeats and the age of onset of the disease.
Adult Tay-Sachs Disease Several reports have described adult-onset, predominantly lower motor neuropathies arising from deficiency of the enzymeβ-hexosaminidase (hex A). These tend to be distinguishable from ALS because they are very slowly progressive; dysarthria and radiographically evident cerebellar atrophy may be prominent. In rare cases, spasticity may also be present, although it is generally absent (Chap. 432e).
Spinal Muscular Atrophy The SMAs are a family of selective lower motor neuron diseases of early onset. Despite some phenotypic variability (largely in age of onset), the defect in the majority of families with SMA maps to a locus on chromosome 5 encoding a putative motor neuron survival protein (SMN, for survival motor neuron) that is important in the formation and trafficking of RNA complexes across the nuclear membrane. Neuropathologically these disorders are characterized by extensive loss of large motor neurons; muscle biopsy reveals evidence of denervation atrophy. Several clinical forms exist.
Infantile SMA (SMA I, Werdnig-Hoffmann disease) has the earliest onset and most rapidly fatal course. In some instances it is apparent even before birth, as indicated by decreased fetal movements late in the third trimester. Though alert, afflicted infants are weak and floppy (hypotonic) and lack muscle stretch reflexes. Death generally ensues within the first year of life. Chronic childhood SMA (SMA II) begins later in childhood and evolves with a more slowly progressive course. Juvenile SMA (SMA III, Kugelberg-Welander disease) manifests during late childhood and runs a slow, indolent course. Unlike most denervating diseases, in this chronic disorder, weakness is greatest in the proximal muscles; indeed, the pattern of clinical weakness can suggest a primary myopathy such as limb-girdle dystrophy. Electrophysiologic and muscle biopsy evidence of denervation distinguish SMA III from the myopathic syndromes. There is no primary therapy for SMA, although remarkable recent experimental data indicate that it may be possible to deliver the missing SMN gene to motor neurons using intravenously or intrathecally delivered adeno-associated viruses (e.g., AAV9) immediately after birth.
Multifocal Motor Neuropathy with Conduction Block In this disorder lower motor neuron function is regionally and chronically disrupted by remarkably focal blocks in conduction. Many cases have elevated serum titers of mono- and polyclonal antibodies to ganglioside GM1; it is hypothesized that the antibodies produce selective, focal, paranodal demyelination of motor neurons. MMCB is not typically associated with corticospinal signs. In contrast with ALS, MMCB may respond dramatically to therapy such as IV immunoglobulin or chemotherapy; thus, it is imperative that MMCB be excluded when considering a diagnosis of ALS.
Other Forms of Lower Motor Neuron Disease In individual families, other syndromes characterized by selective lower motor neuron dysfunction in an SMA-like pattern have been described. There are rare X-linked and autosomal dominant forms of apparent SMA. There is an ALS variant of juvenile onset, the Fazio-Londe syndrome, that involves mainly the musculature innervated by the brainstem. A component of lower motor neuron dysfunction is also found in degenerative disorders such as Machado-Joseph disease and the related olivopontocerebellar degenerations (Chap. 450).
SELECTED DISORDERS OF THE UPPER MOTOR NEURON
Primary Lateral Sclerosis This exceedingly rare disorder arises sporadically in adults in mid to late life. Clinically PLS is characterized by progressive spastic weakness of the limbs, preceded or followed by spastic dysarthria and dysphagia, indicating combined involvement of the corticospinal and corticobulbar tracts. Fasciculations, amyotrophy, and sensory changes are absent; neither electromyography nor muscle biopsy shows denervation. On neuropathologic examination, there is selective loss of the large pyramidal cells in the precentral gyrus and degeneration of the corticospinal and corticobulbar projections. The peripheral motor neurons and other neuronal systems are spared. The course of PLS is variable; although long-term survival is documented, the course may be as aggressive as in ALS, with ~3-year survival from onset to death. Early in its course, PLS raises the question of multiple sclerosis or other demyelinating diseases such as adrenoleukodystrophy as diagnostic considerations (Chap. 458). A myelopathy suggestive of PLS is infrequently seen with infection with the retrovirus human T cell lymphotropic virus 1 (HTLV-1) (Chap. 456). The clinical course and laboratory testing will distinguish these possibilities.
Familial Spastic Paraplegia In its pure form, FSP is usually transmitted as an autosomal trait; most adult-onset cases are dominantly inherited. Symptoms usually begin in the third or fourth decade, presenting as progressive spastic weakness beginning in the distal lower extremities; however, there are variants with onset so early that the differential diagnosis includes cerebral palsy. FSP typically has a long survival, presumably because respiratory function is spared. Late in the illness, there may be urinary urgency and incontinence and sometimes fecal incontinence; sexual function tends to be preserved.
In pure forms of FSP, the spastic leg weakness is often accompanied by posterior column (vibration and position) abnormalities and disturbance of bowel and bladder function. Some family members may have spasticity without clinical symptoms.
By contrast, particularly when recessively inherited, FSP may have complex or complicated forms in which altered corticospinal and dorsal column function is accompanied by significant involvement of other regions of the nervous system, including amyotrophy, mental retardation, optic atrophy, and sensory neuropathy.
Neuropathologically, in FSP, there is degeneration of the corticospinal tracts, which appear nearly normal in the brainstem but show increasing atrophy at more caudal levels in the spinal cord; in effect, the pathologic picture is of a dying-back or distal axonopathy of long neuronal fibers within the CNS.
Defects at numerous loci underlie both dominantly and recessively inherited forms of FSP (Table 452-3). More than 30 FSP genes have now been identified. The gene most commonly implicated in dominantly inherited FSP is spastin, which encodes a microtubule interacting protein. The most common childhood-onset dominant form arises from mutations in the atlastin gene. A kinesin heavy-chain protein implicated in microtubule motor function was found to be defective in a family with dominantly inherited FSP of variable-onset age.
An infantile-onset form of X-linked, recessive FSP arises from mutations in the gene for myelin proteolipid protein. This is an example of rather striking allelic variation, as most other mutations in the same gene cause not FSP but Pelizaeus-Merzbacher disease, a widespread disorder of CNS myelin. Another recessive variant is caused by defects in the paraplegin gene. Paraplegin has homology to metalloproteases that are important in mitochondrial function in yeast.
WEBSITES
Several websites provide valuable information on ALS including those offered by the Muscular Dystrophy Association (www.mdausa.org), the Amyotrophic Lateral Sclerosis Association (www.alsa.org), and the World Federation of Neurology and the Neuromuscular Unit at Washington University in St. Louis (www.neuro.wustl.edu).
453e |
Prion Diseases |
Prions are proteins that adopt an alternative conformation, which becomes self-propagating. Some prions cause degeneration of the central nervous system (CNS). Once relegated to causing a group of rare disorders of the CNS such as Creutzfeldt-Jakob disease (CJD), prions—as mounting evidence shows—also appear to play a key role in more common illnesses such as Alzheimer’s disease (AD) and Parkinson’s disease (PD). While CJD is caused by the accumulation of PrPSc, increasing data argue that Aβ prions cause AD, α-synuclein prions cause PD, and tau prions cause the frontotemporal dementias (FTDs). In this chapter, we confine our discussion to CJD, which typically presents with a rapidly progressive dementia as well as motor abnormalities. The illness is relentlessly progressive and generally causes death within 9 months of onset. Most CJD patients are between 50 and 75 years of age; however, patients as young as 17 and as old as 83 have been recorded.
CJD is one malady in a group of disorders caused by prions composed of the prion protein (PrP). PrP prions reproduce by binding to the normal, cellular isoform of the prion protein (PrPC) and stimulating conversion of PrPC into the disease-causing isoform PrPSc. PrPC is rich in α-helix and has little β-structure, whereas PrPSc has less α-helix and a high amount of β-structure (Fig. 453e-1). This α-to-β structural transition in PrP is the fundamental event underlying this group of prion diseases (Table 453e-1).
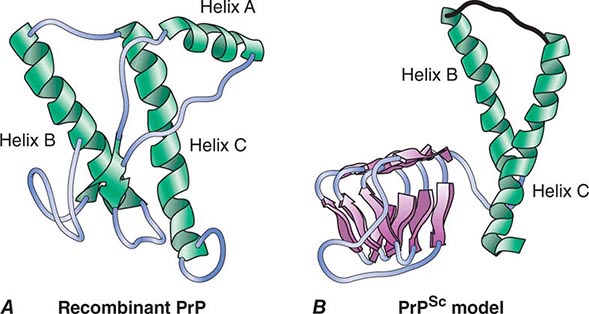
FIGURE 453e-1 Structures of prion proteins. A. NMR structure of Syrian hamster recombinant (rec) PrP(90–231). Presumably, the structure of the α-helical form of recPrP(90–231) resembles that of PrPC. recPrP(90–231) is viewed from the interface where PrPSc is thought to bind to PrPC. Shown are: α-helices A (residues 144–157), B (172–193), and C (200–227). Flat ribbons depict β-strands S1 (129–131) and S2 (161–163). B. Structural model of PrPSc. The 90–160 region has been modeled onto a β-helical architecture while the COOH terminal helices B and C are preserved as in PrPC.
|
GLOSSARY OF PRION TERMINOLOGY |

Four new concepts have emerged from studies of prions: (1) Prions are the only known transmissible pathogens that are devoid of nucleic acid; all other infectious agents possess genomes composed of either RNA or DNA that direct the synthesis of their progeny. (2) Prion diseases may be manifest as infectious, genetic, and sporadic disorders; no other group of illnesses with a single etiology presents with such a wide spectrum of clinical manifestations. (3) Prion diseases result from the accumulation of PrPSc, the conformation of which differs substantially from that of its precursor, PrPC. (4) Distinct strains of prions exhibit different biologic properties, which are epigenetically inherited. In other words, PrPSc can exist in a variety of different conformations, many of which seem to specify particular disease phenotypes.
How a specific conformation of a PrPSc molecule is imparted to PrPC during prion replication to produce nascent PrPSc with the same conformation is unknown. Additionally, it is unclear what factors determine where in the CNS a particular PrPSc molecule will be deposited.
SPECTRUM OF PRION DISEASES
The sporadic form of CJD is the most common prion disorder in humans. Sporadic CJD (sCJD) accounts for ~85% of all cases of human PrP prion disease, whereas inherited prion diseases account for 10–15% of all cases (Table 453e-2). Familial CJD (fCJD), Gerstmann-Sträussler-Scheinker (GSS) disease, and fatal familial insomnia (FFI) are all dominantly inherited prion diseases that are caused by mutations in the PrP gene.
|
THE PrP PRION DISEASES |
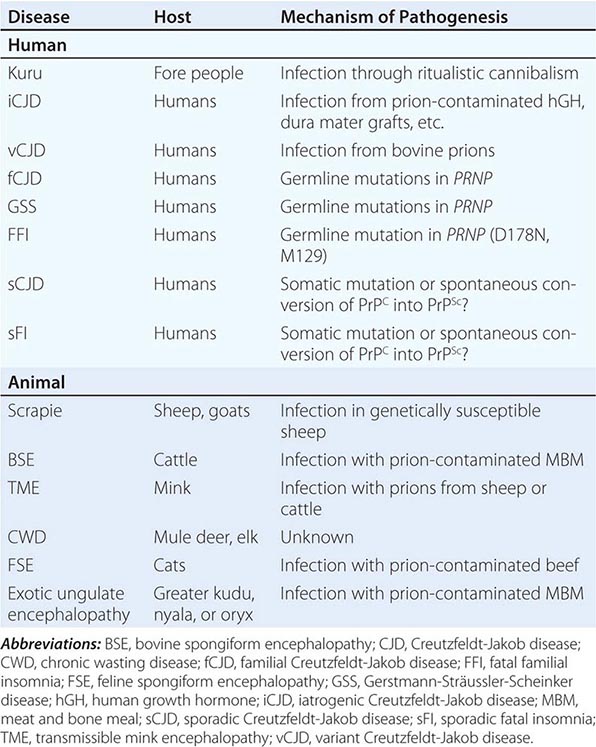
 Although infectious PrP prion diseases account for <1% of all cases and infection does not seem to play an important role in the natural history of these illnesses, the transmissibility of prions is an important biologic feature. Kuru of the Fore people of New Guinea is thought to have resulted from the consumption of brains from dead relatives during ritualistic cannibalism. With the cessation of ritualistic cannibalism in the late 1950s, kuru has nearly disappeared, with the exception of a few recent patients exhibiting incubation periods of >40 years. Iatrogenic CJD (iCJD) seems to be the result of the accidental inoculation of patients with prions. Variant CJD (vCJD) in teenagers and young adults in Europe is the result of exposure to tainted beef from cattle with bovine spongiform encephalopathy (BSE). Although occasional cases of iatrogenic CJD still occur, this form of CJD is currently on the decline due to public health measures aimed at preventing the spread of PrP prions.
Although infectious PrP prion diseases account for <1% of all cases and infection does not seem to play an important role in the natural history of these illnesses, the transmissibility of prions is an important biologic feature. Kuru of the Fore people of New Guinea is thought to have resulted from the consumption of brains from dead relatives during ritualistic cannibalism. With the cessation of ritualistic cannibalism in the late 1950s, kuru has nearly disappeared, with the exception of a few recent patients exhibiting incubation periods of >40 years. Iatrogenic CJD (iCJD) seems to be the result of the accidental inoculation of patients with prions. Variant CJD (vCJD) in teenagers and young adults in Europe is the result of exposure to tainted beef from cattle with bovine spongiform encephalopathy (BSE). Although occasional cases of iatrogenic CJD still occur, this form of CJD is currently on the decline due to public health measures aimed at preventing the spread of PrP prions.
Six diseases of animals are caused by prions (Table 453e-2). Scrapie of sheep and goats is the prototypic prion disease. Mink encephalopathy, BSE, feline spongiform encephalopathy, and exotic ungulate encephalopathy are all thought to occur after the consumption of prion-infected foodstuffs. The BSE epidemic emerged in Britain in the late 1980s and was shown to be due to industrial cannibalism. Whether BSE began as a sporadic case of BSE in a cow or started with scrapie in sheep is unknown. The origin of chronic wasting disease (CWD), a prion disease endemic in deer and elk in regions of North America, is uncertain. In contrast to other prion diseases, CWD is highly communicable. Feces from asymptomatic, infected cervids contain prions that are likely to be responsible for the spread of CWD.
EPIDEMIOLOGY
CJD is found throughout the world. The incidence of sCJD is approximately one case per million population, and thus it accounts for approximately 1 in every 10,000 deaths. Because sCJD is an age-dependent neurodegenerative disease, its incidence is expected to increase steadily as older segments of populations in developed and developing countries continue to expand. Although many geographic clusters of CJD have been reported, each has been shown to segregate with a PrP gene mutation. Attempts to identify common exposure to some etiologic agent have been unsuccessful for both the sporadic and familial cases. Ingestion of scrapie-infected sheep or goat meat as a cause of CJD in humans has not been demonstrated by epidemiologic studies, although speculation about this potential route of inoculation continues. Of particular interest are deer hunters who develop CJD, because up to 90% of culled deer in some game herds have been shown to harbor CWD prions. Whether prion disease in deer or elk has passed to cows, sheep, or directly to humans remains unknown. Studies with rodents demonstrate that oral infection with prions can occur, but the process is inefficient compared to intracerebral inoculation.
PATHOGENESIS
The human prion diseases were initially classified as neurodegenerative disorders of unknown etiology on the basis of pathologic changes being confined to the CNS. With the transmission of kuru and CJD to apes, investigators began to view these diseases as infectious CNS illnesses caused by slow viruses. Even though the familial nature of a subset of CJD cases was well described, the significance of this observation became more obscure with the transmission of CJD to animals. Eventually the meaning of heritable CJD became clear with the discovery of mutations in the PRNP gene of these patients. The prion concept explains how a disease can manifest as a heritable as well as an infectious illness. Moreover, the hallmark of all prion diseases, whether sporadic, dominantly inherited, or acquired by infection, is that they involve the aberrant metabolism of PrP.
A major feature that distinguishes prions from viruses is the finding that both PrP isoforms are encoded by a chromosomal gene. In humans, the PrP gene is designated PRNP and is located on the short arm of chromosome 20. Limited proteolysis of PrPSc produces a smaller, protease-resistant molecule of ~142 amino acids designated PrP 27-30; PrPC is completely hydrolyzed under the same conditions (Fig. 453e-2). In the presence of detergent, PrP 27-30 polymerizes into amyloid. Prion rods formed by limited proteolysis and detergent extraction are indistinguishable from the filaments that aggregate to form PrP amyloid plaques in the CNS. Both the rods and the PrP amyloid filaments found in brain tissue exhibit similar ultrastructural morphology and green-gold birefringence after staining with Congo red dye.
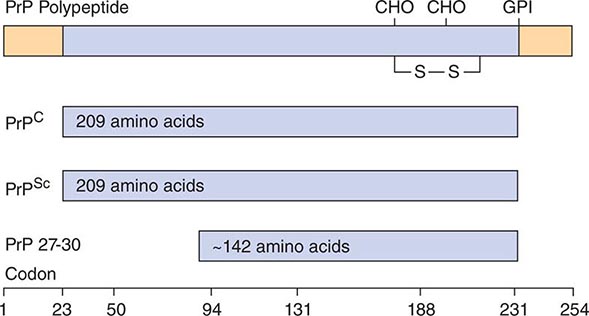
FIGURE 453e-2 Prion protein isoforms. Bar diagram of Syrian hamster PrP, which consists of 254 amino acids. After processing of the NH2 and COOH termini, both PrPC and PrPSc consist of 209 residues. After limited proteolysis, the NH2 terminus of PrPSc is truncated to form PrP 27–30 composed of ~142 amino acids. GPI, glycosylphosphatidyl inositol anchor attachment site; S—S, disulfide bond; CHO, N-linked sugars.
Prion Strains Distinct strains of prions exhibit different biologic properties, which are epigenetically heritable. The existence of prion strains raised the question of how heritable biologic information can be enciphered in a molecule other than nucleic acid. Various strains of prions have been defined by incubation times and the distribution of neuronal vacuolation. Subsequently, the patterns of PrPSc deposition were found to correlate with vacuolation profiles, and these patterns were also used to characterize prion strains.
Persuasive evidence that strain-specific information is enciphered in the tertiary structure of PrPSc comes from transmission of two different inherited human prion diseases to mice expressing a chimeric human-mouse PrP transgene. In FFI, the protease-resistant fragment of PrPSc after deglycosylation has a molecular mass of 19 kDa, whereas in fCJD and most sporadic prion diseases, it is 21 kDa (Table 453e-3). This difference in molecular mass was shown to be due to different sites of proteolytic cleavage at the NH2 termini of the two human PrPSc molecules, reflecting different tertiary structures. These distinct conformations were not unexpected because the amino acid sequences of the PrPs differ.
|
DISTINCT PRION STRAINS GENERATED IN HUMANS WITH INHERITED PRION DISEASES AND TRANSMITTED TO TRANSGENIC MICEa |
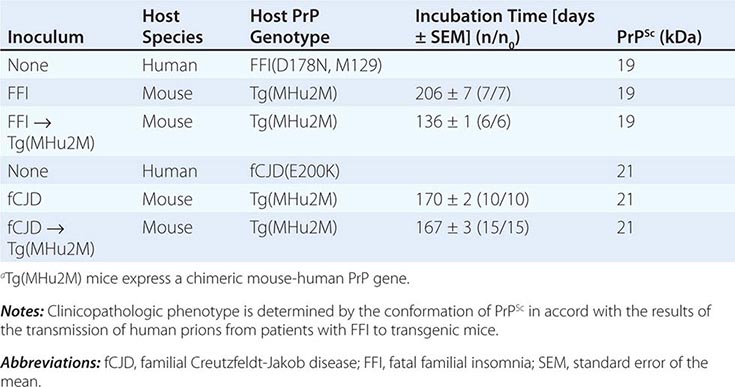
Extracts from the brains of patients with FFI transmitted disease into mice expressing a chimeric human-mouse PrP transgene and induced formation of the 19-kDa PrPSc, whereas brain extracts from FCJD and sCJD patients produced the 21-kDa PrPSc in mice expressing the same transgene. On second passage, these differences were maintained, demonstrating that chimeric PrPSc can exist in two different conformations based on the sizes of the protease-resistant fragments, even though the amino acid sequence of PrPSc is invariant.
This analysis was extended when patients with sporadic fatal insomnia (sFI) were identified. Although they did not carry a PRNP gene mutation, the patients demonstrated a clinical and pathologic phenotype that was indistinguishable from that of patients with FFI. Furthermore, 19-kDa PrPSc was found in their brains, and on passage of prion disease to mice expressing a chimeric human-mouse PrP transgene, 19-kDa PrPSc was also found. These findings indicate that the disease phenotype is dictated by the conformation of PrPSc and not the amino acid sequence. PrPSc acts as a template for the conversion of PrPC into nascent PrPSc. On the passage of prions into mice expressing a chimeric hamster-mouse PrP transgene, a change in the conformation of PrPSc was accompanied by the emergence of a new strain of prions.
Many new strains of prions were generated using recombinant (rec) PrP produced in bacteria; recPrP was polymerized into amyloid fibrils and inoculated into transgenic mice expressing high levels of wild-type mouse PrPC; approximately 500 days later, the mice died of prion disease. The incubation times of the “synthetic prions” in mice were dependent on the conditions used for polymerization of the amyloid fibrils. Highly stable amyloids gave rise to stable prions with long incubation times; low-stability amyloids led to prions with short incubation times. Amyloids of intermediate stability gave rise to prions with intermediate stabilities and intermediate incubation times. Such findings are consistent with earlier studies showing that the incubation times of synthetic and naturally occurring prions are directly proportional to the stability of the prion.
Species Barrier Studies on the role of the primary and tertiary structures of PrP in the transmission of prion disease have given new insights into the pathogenesis of these maladies. The amino acid sequence of PrP encodes the species of the prion, and the prion derives its PrPSc sequence from the last mammal in which it was passaged. While the primary structure of PrP is likely to be the most important or even sole determinant of the tertiary structure of PrPC, PrPSc seems to function as a template in determining the tertiary structure of nascent PrPSc molecules as they are formed from PrPC. In turn, prion diversity appears to be enciphered in the conformation of PrPSc, and thus prion strains seem to represent different conformers of PrPSc.
In general, transmission of PrP prion disease from one species to another is inefficient, in that not all intracerebrally inoculated animals develop disease, and those that fall ill do so only after long incubation times that can approach the natural life span of the animal. This “species barrier” to transmission is correlated with the degree of similarity between the amino acid sequences of PrPC in the inoculated host and of PrPSc in the prion inoculum. The importance of sequence similarity between the host and donor PrP argues that PrPC directly interacts with PrPSc in the prion conversion process.
SPORADIC AND INHERITED PrP PRION DISEASES
Several different scenarios might explain the initiation of sporadic prion disease: (1) A somatic mutation may be the cause and thus follow a path similar to that for germline mutations in inherited disease. In this situation, the mutant PrPSc must be capable of targeting wild-type PrPC, a process known to be possible for some mutations but less likely for others. (2) The activation energy barrier separating wild-type PrPC from PrPSc could be crossed on rare occasions when viewed in the context of a population. Most individuals would be spared, while presentations in the elderly with an incidence of ~1 per million would be seen. (3) PrPSc may be present at low levels in some normal cells, where it performs some important, as yet unknown, function. The level of PrPSc in such cells is hypothesized to be sufficiently low as to be not detected by routine bioassay. In some altered metabolic states, the cellular mechanisms for clearing PrPSc might become compromised, and the rate of PrPSc formation would then begin to exceed the capacity of the cell to clear it. The third possible mechanism is attractive because it suggests PrPSc is not simply a misfolded protein, as proposed for the first and second mechanisms, but that it is an alternatively folded molecule with a function. Moreover, the multitude of conformational states that PrPSc can adopt, as described above, raises the possibility that PrPSc or another prion-like protein might function in a process like short-term memory where information storage occurs in the absence of new protein synthesis.
More than 40 different mutations resulting in nonconservative substitutions in the human PRNP gene have been found to segregate with inherited human prion diseases. Missense mutations and expansions in the octapeptide repeat region of the gene are responsible for familial forms of prion disease. Five different mutations of the PRNP gene have been linked genetically to heritable prion disease.
Although phenotypes may vary dramatically within families, specific phenotypes tend to be observed with certain mutations. A clinical phenotype indistinguishable from typical sCJD is usually seen with substitutions at codons 180, 183, 200, 208, 210, and 232. Substitutions at codons 102, 105, 117, 198, and 217 are associated with the GSS variant of prion disease. The normal human PrP sequence contains five repeats of an eight-amino-acid sequence. Insertions from two to nine extra octarepeats frequently cause variable phenotypes ranging from a condition indistinguishable from sCJD to a slowly progressive dementing illness of many years in duration to an early-age-of-onset disorder that is similar to AD. A mutation at codon 178 resulting in substitution of asparagine for aspartic acid produces FFI if a methionine is encoded at the polymorphic residue 129 on the same allele. Typical CJD is seen if the D178N mutation occurs with a valine encoded at position 129 of the same allele.
HUMAN PRNP GENE POLYMORPHISMS
Polymorphisms influence the susceptibility to sporadic, inherited, and infectious forms of prion disease. The methionine/valine polymorphism at position 129 not only modulates the age of onset of some inherited prion diseases but can also determine the clinical phenotype. The finding that homozygosity at codon 129 predisposes to sCJD supports a model of prion production that favors PrP interactions between homologous proteins.
Substitution of the basic residue lysine at position 218 in mouse PrP produced dominant-negative inhibition of prion replication in transgenic mice. This same lysine at position 219 in human PrP has been found in 12% of the Japanese population, and this group appears to be resistant to prion disease. Dominant-negative inhibition of prion replication was also found with substitution of the basic residue arginine at position 171; sheep with arginine were resistant to scrapie prions but were susceptible to BSE prions that were inoculated intracerebrally.
INFECTIOUS PrP PRION DISEASES
IATROGENIC CJD
Accidental transmission of CJD to humans appears to have occurred with corneal transplantation, contaminated electroencephalogram (EEG) electrode implantation, and surgical procedures. Corneas from donors with unsuspected CJD have been transplanted to apparently healthy recipients who developed CJD after variable incubation periods. The same improperly decontaminated EEG electrodes that caused CJD in two young patients with intractable epilepsy caused CJD in a chimpanzee 18 months after their experimental implantation.
Surgical procedures may have resulted in accidental inoculation of patients with prions, presumably because some instrument or apparatus in the operating theater became contaminated when a CJD patient underwent surgery. Although the epidemiology of these studies is highly suggestive, no proof for such episodes exists.
Dura Mater Grafts More than 160 cases of CJD after implantation of dura mater grafts have been recorded. All of the grafts appear to have been acquired from a single manufacturer whose preparative procedures were inadequate to inactivate human prions. One case of CJD occurred after repair of an eardrum perforation with a pericardium graft.
Human Growth Hormone and Pituitary Gonadotropin Therapy The transmission of CJD prions from contaminated human growth hormone (hGH) preparations derived from human pituitaries has been responsible for fatal cerebellar disorders with dementia in >180 patients ranging in age from 10 to 41 years. These patients received injections of hGH every 2–4 days for 4–12 years. If it is thought that these patients developed CJD from injections of prion-contaminated hGH preparations, the possible incubation periods range from 4 to 30 years. Only recombinant hGH is now used therapeutically so that possible contamination with prions is no longer an issue. Four cases of CJD have occurred in women receiving human pituitary gonadotropin.
VARIANT CJD
The restricted geographic occurrence and chronology of vCJD raised the possibility that BSE prions had been transmitted to humans through the consumption of tainted beef. More than 190 cases of vCJD have occurred, with >90% of these in Britain. vCJD has also been reported in people either living in or originating from France, Ireland, Italy, Netherlands, Portugal, Spain, Saudi Arabia, United States, Canada, and Japan.
The steady decline in the number of vCJD cases over the past decade argues that there will not be a prion disease epidemic in Europe, similar to those seen for BSE and kuru. What is certain is that prion-tainted meat should be prevented from entering the human food supply.
The most compelling evidence that vCJD is caused by BSE prions was obtained from experiments in mice expressing the bovine PrP transgene. Both BSE and vCJD prions were efficiently transmitted to these transgenic mice and with similar incubation periods. In contrast to sCJD prions, vCJD prions did not transmit disease efficiently to mice expressing a chimeric human-mouse PrP transgene. Earlier studies with nontransgenic mice suggested that vCJD and BSE might be derived from the same source because both inocula transmitted disease with similar but very long incubation periods.
Attempts to determine the origin of BSE and vCJD prions have relied on passaging studies in mice, some of which are described above, as well as studies of the conformation and glycosylation of PrPSc. One scenario suggests that a particular conformation of bovine PrPSc was selected for heat resistance during the rendering process and was then reselected multiple times as cattle infected by ingesting prion-contaminated meat and bone meal (MBM) were slaughtered and their offal rendered into more MBM. Variant CJD cases have virtually disappeared with protection of the beef supply in Europe.
NEUROPATHOLOGY
Frequently the brains of patients with CJD have no recognizable abnormalities on gross examination. Patients who survive for several years have variable degrees of cerebral atrophy.
On light microscopy, the pathologic hallmarks of CJD are spongiform degeneration and astrocytic gliosis. The lack of an inflammatory response in CJD and other prion diseases is an important pathologic feature of these degenerative disorders. Spongiform degeneration is characterized by many 1- to 5-μm vacuoles in the neuropil between nerve cell bodies. Generally the spongiform changes occur in the cerebral cortex, putamen, caudate nucleus, thalamus, and molecular layer of the cerebellum. Astrocytic gliosis is a constant but nonspecific feature of prion diseases. Widespread proliferation of fibrous astrocytes is found throughout the gray matter of brains infected with CJD prions. Astrocytic processes filled with glial filaments form extensive networks.
Amyloid plaques have been found in ~10% of CJD cases. Purified CJD prions from humans and animals exhibit the ultrastructural and histochemical characteristics of amyloid when treated with detergents during limited proteolysis. In first passage from some human Japanese CJD cases, amyloid plaques have been found in mouse brains. These plaques stain with antibodies raised against PrP.
The amyloid plaques of GSS disease are morphologically distinct from those seen in kuru or scrapie. GSS plaques consist of a central dense core of amyloid surrounded by smaller globules of amyloid. Ultrastructurally, they consist of a radiating fibrillar network of amyloid fibrils, with scant or no neuritic degeneration. The plaques can be distributed throughout the brain but are most frequently found in the cerebellum. They are often located adjacent to blood vessels. Congophilic angiopathy has been noted in some cases of GSS disease.
In vCJD, a characteristic feature is the presence of “florid plaques.” These are composed of a central core of PrP amyloid, surrounded by vacuoles in a pattern suggesting petals on a flower.
CLINICAL FEATURES
Nonspecific prodromal symptoms occur in approximately a third of patients with CJD and may include fatigue, sleep disturbance, weight loss, headache, anxiety, vertigo, malaise, and ill-defined pain. Most patients with CJD present with deficits in higher cortical function. Similarly, psychiatric symptoms, such as depression, psychosis, and visual hallucinations, are often the defining features of the illness. These deficits almost always progress over weeks or months to a state of profound dementia characterized by memory loss, impaired judgment, and a decline in virtually all aspects of intellectual function. A few patients present with either visual impairment or cerebellar gait and coordination deficits. Frequently the cerebellar deficits are rapidly followed by progressive dementia. Visual problems often begin with blurred vision and diminished acuity, rapidly followed by dementia.
Other symptoms and signs include extrapyramidal dysfunction manifested as rigidity, masklike facies, or (less commonly) choreoathetoid movements; pyramidal signs (usually mild); seizures (usually major motor) and, less commonly, hypoesthesia; supranuclear gaze palsy; optic atrophy; and vegetative signs such as changes in weight, temperature, sweating, or menstruation.
Myoclonus Most patients (~90%) with CJD exhibit myoclonus that appears at various times throughout the illness. Unlike other involuntary movements, myoclonus persists during sleep. Startle myoclonus elicited by loud sounds or bright lights is frequent. It is important to stress that myoclonus is neither specific nor confined to CJD and tends to occur later in the course of CJD. Dementia with myoclonus can also be due to AD (Chap. 448), dementia with Lewy bodies (Chap. 448), corticobasal degeneration (Chap. 448) cryptococcal encephalitis (Chap. 239), or the myoclonic epilepsy disorder Unverricht-Lundborg disease (Chap. 445).
Clinical Course In documented cases of accidental transmission of CJD to humans, an incubation period of 1.5–2 years preceded the development of clinical disease. In other cases, incubation periods of up to 40 years have been suggested. Most patients with CJD live 6–12 months after the onset of clinical signs and symptoms, whereas some live for up to 5 years.
DIAGNOSIS
The constellation of dementia, myoclonus, and periodic electrical bursts in an afebrile 60-year-old patient generally indicates CJD. Clinical abnormalities in CJD are confined to the CNS. Fever, elevated sedimentation rate, leukocytosis in blood, or a pleocytosis in cerebrospinal fluid (CSF) should alert the physician to another etiology to explain the patient’s CNS dysfunction, although there are rare cases of CJD in which mild CSF pleocytosis is observed.
Variations in the typical course appear in inherited and transmitted forms of the disease. fCJD has an earlier mean age of onset than sCJD. In GSS disease, ataxia is usually a prominent and presenting feature, with dementia occurring late in the disease course. GSS disease presents earlier than CJD (mean age 43 years) and is typically more slowly progressive than CJD; death usually occurs within 5 years of onset. FFI is characterized by insomnia and dysautonomia; dementia occurs only in the terminal phase of the illness. Rare sporadic cases have been identified. vCJD has an unusual clinical course, with a prominent psychiatric prodrome that may include visual hallucinations and early ataxia, whereas frank dementia is usually a late sign of vCJD.
DIFFERENTIAL DIAGNOSIS
Many conditions mimic CJD. Dementia with Lewy bodies (Chap. 448) is the most common disorder to be mistaken for CJD. It can present in a subacute fashion with delirium, myoclonus, and extrapyramidal features. Other neurodegenerative disorders (Chap. 448) to consider include AD, FTD, corticobasal degeneration, progressive supranuclear palsy, ceroid lipofuscinosis, and myoclonic epilepsy with Lafora bodies (Chap. 445). The absence of abnormalities on diffusion-weighted and fluid-attenuated inversion recovery (FLAIR) magnetic resonance imaging (MRI) will almost always distinguish these dementing conditions from CJD.
Hashimoto’s encephalopathy, which presents as a subacute progressive encephalopathy with myoclonus and periodic triphasic complexes on the EEG, should be excluded in every case of suspected CJD. It is diagnosed by the finding of high titers of antithyroglobulin or antithyroid peroxidase (antimicrosomal) antibodies in the blood and improves with glucocorticoid therapy. Unlike CJD, fluctuations in severity typically occur in Hashimoto’s encephalopathy.
Intracranial vasculitides (Chap. 385) may produce nearly all of the symptoms and signs associated with CJD, sometimes without systemic abnormalities. Myoclonus is exceptional with cerebral vasculitis, but focal seizures may confuse the picture. Prominent headache, absence of myoclonus, stepwise change in deficits, abnormal CSF, and focal white matter changes on MRI or angiographic abnormalities all favor vasculitis.
Paraneoplastic conditions (Chap. 122), particularly limbic encephalitis and cortical encephalitis, can also mimic CJD. In many of these patients, dementia appears prior to the diagnosis of a tumor, and in some, no tumor is ever found. Detection of the paraneoplastic antibodies is often the only way to distinguish these cases from CJD.
Other diseases that can simulate CJD include neurosyphilis (Chap. 206), AIDS dementia complex (Chap. 226), progressive multifocal leukoencephalopathy (Chap. 164), subacute sclerosing panencephalitis, progressive rubella panencephalitis, herpes simplex encephalitis (Chap. 164), diffuse intracranial tumor (gliomatosis cerebri; Chap. 118), anoxic encephalopathy, dialysis dementia, uremia, hepatic encephalopathy, voltage-gated potassium channel (VGkC) autoimmune encephalopathy, and lithium or bismuth intoxication.
LABORATORY TESTS
The only specific diagnostic tests for CJD and other human prion diseases measure PrPSc. The most widely used method involves limited proteolysis that generates PrP 27-30, which is detected by immunoassay after denaturation. The conformation-dependent immunoassay (CDI) is based on immunoreactive epitopes that are exposed in PrPC but buried in PrPSc. In humans, the diagnosis of CJD can be established by brain biopsy if PrPSc is detected. If no attempt is made to measure PrPSc, but the constellation of pathologic changes frequently found in CJD is seen in a brain biopsy, then the diagnosis is reasonably secure (see “Neuropathology,” above). The high sensitivity and specificity of cortical ribboning and basal ganglia hyperintensity on FLAIR and diffusion-weighted MRI for the diagnosis of CJD have greatly diminished the need for brain biopsy in patients with suspected CJD. Because PrPSc is not uniformly distributed throughout the CNS, the apparent absence of PrPSc in a limited sample such as a biopsy does not rule out prion disease. At autopsy, sufficient brain samples should be taken for both PrPSc immunoassay, preferably by CDI, and immunohistochemistry of tissue sections.
To establish the diagnosis of either sCJD or familial prion disease, sequencing the PRNP gene must be performed. Finding the wild-type PRNP gene sequence permits the diagnosis of sCJD if there is no history to suggest infection from an exogenous source of prions. The identification of a mutation in the PRNP gene sequence that encodes a nonconservative amino acid substitution argues for familial prion disease.
CT may be normal or show cortical atrophy. MRI is valuable for distinguishing sCJD from most other conditions. On FLAIR sequences and diffusion-weighted imaging, ~90% of patients show increased intensity in the basal ganglia and cortical ribboning (Fig. 453e-3). This pattern is not seen with other neurodegenerative disorders but has been seen infrequently with viral encephalitis, paraneoplastic syndromes, or seizures. When the typical MRI pattern is present, in the proper clinical setting, diagnosis is facilitated. However, some cases of sCJD do not show this typical pattern, and other early diagnostic approaches are still needed.
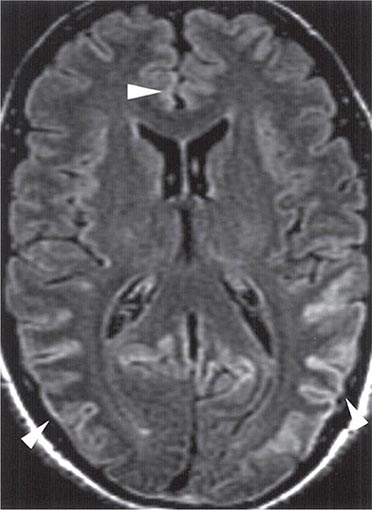
FIGURE 453e-3 T2-weighted (fluid-attenuated inversion recovery) magnetic resonance imaging showing hyperintensity in the cortex in a patient with sporadic CJD. This so-called “cortical ribboning” along with increased intensity in the basal ganglia on T2- or diffusion-weighted imaging can aid in the diagnosis of Creutzfeldt-Jakob disease.
CSF is nearly always normal but may show protein elevation and, rarely, mild pleocytosis. Although the stress protein 14-3-3 is elevated in the CSF of some patients with CJD, similar elevations of 14-3-3 are found in patients with other disorders; thus this elevation is not specific. Similarly, elevations of CSF neuron-specific enolase and tau occur in CJD but lack specificity for diagnosis.
The EEG is often useful in the diagnosis of CJD, although only approximately 60% of individuals show the typical pattern. During the early phase of CJD, the EEG is usually normal or shows only scattered theta activity. In most advanced cases, repetitive, high-voltage, triphasic, and polyphasic sharp discharges are seen, but in many cases their presence is transient. The presence of these stereotyped periodic bursts of <200 ms in duration, occurring every 1–2 s, makes the diagnosis of CJD very likely. These discharges are frequently but not always symmetric; there may be a one-sided predominance in amplitude. As CJD progresses, normal background rhythms become fragmentary and slower.
CARE OF CJD PATIENTS
Although CJD should not be considered either contagious or communicable, it is transmissible. The risk of accidental inoculation by aerosols is very small; nonetheless, procedures producing aerosols should be performed in certified biosafety cabinets. Biosafety level 2 practices, containment equipment, and facilities are recommended by the Centers for Disease Control and Prevention and the National Institutes of Health. The primary problem in caring for patients with CJD is the inadvertent infection of health care workers by needle and stab wounds. Electroencephalographic and electromyographic needles should not be reused after studies on patients with CJD have been performed.
There is no reason for pathologists or other morgue employees to resist performing autopsies on patients whose clinical diagnosis was CJD. Standard microbiologic practices outlined here, along with specific recommendations for decontamination, seem to be adequate precautions for the care of patients with CJD and the handling of infected specimens.
DECONTAMINATION OF CJD PRIONS
Prions are extremely resistant to common inactivation procedures, and there is some disagreement about the optimal conditions for sterilization. Some investigators recommend treating CJD-contaminated materials once with 1 N NaOH at room temperature, but we believe this procedure may be inadequate for sterilization. Autoclaving at 134°C for 5 h or treatment with 2 N NaOH for several hours is recommended for sterilization of prions. The term sterilization implies complete destruction of prions; any residual infectivity can be hazardous. Recent studies show that sCJD prions bound to stainless steel surfaces are resistant to inactivation by autoclaving at 134°C for 2 h; exposure of bound prions to an acidic detergent solution prior to autoclaving rendered prions susceptible to inactivation.
PREVENTION AND THERAPEUTICS
There is no known effective therapy for preventing or treating CJD. The finding that phenothiazines and acridines inhibit PrPSc formation in cultured cells led to clinical studies of quinacrine in CJD patients. Unfortunately, quinacrine failed to slow the rate of cognitive decline in CJD, possibly because therapeutic concentrations in the brain were not achieved. Although inhibition of the P-glycoprotein (Pgp) transport system resulted in substantially increased quinacrine levels in the brains of mice, the prion incubation times were not extended by treatment with the drug. Whether such an approach can be used to treat CJD remains to be established.
Like the acridines, anti-PrP antibodies have been shown to eliminate PrPSc from cultured cells. Additionally, such antibodies in mice, either administered by injection or produced from a transgene, have been shown to prevent prion disease when prions are introduced by a peripheral route, such as intraperitoneal inoculation. Unfortunately, the antibodies were ineffective in mice inoculated intracerebrally with prions. Several drugs, including pentosan polysulfate as well as porphyrin and phenylhydrazine derivatives, delay the onset of disease in animals inoculated intracerebrally with prions if the drugs are given intracerebrally beginning soon after inoculation.
DIFFERENT PRIONS CAUSING OTHER NEURODEGENERATIVE DISEASES
There is a rapidly expanding body of literature demonstrating that in addition to PrP, other proteins including amyloid beta (Aβ), tau, α-synuclein, and huntingtin can all becomes prions (Chap. 444e). Experimental studies have shown that transgenic mice expressing mutant amyloid precursor protein (APP) develop amyloid plaques containing fibrils composed of the Aβ peptide approximately a year after inoculation with synthetic Aβ peptides polymerized into amyloid fibrils or extracts prepared from the brains of patients with AD. Mutant tau aggregates in transgenic mice and cultured cells can trigger the aggregation of tau into fibrils that resemble those found in neurofibrillary tangles and Pick bodies. Such tangles have been found in AD, FTDs, Pick’s disease, and some cases of posttraumatic brain injury, all of which are likely to be caused by the prion isoforms of Aβ and/or tau.
In patients with advanced PD who received grafts of fetal substantia nigral neurons, Lewy bodies containing β-sheet–rich α-synuclein were identified in grafted cells approximately 10 years after transplantation, arguing for the axonal transport of misfolded α-synuclein crossing into grafted neurons, where it initiated aggregation of nascent α-synuclein into fibrils that coalesced into Lewy bodies. These findings combined with studies of multiple system atrophy (MSA) argue that the synucleinopathies are caused by prions. Brain homogenates from MSA patients injected into transgenic mice transmitted lethal neurodegeneration in approximately 3 months; moreover, recombinant synuclein injected into wild-type mice initiated the deposition of synuclein fibrils.
In summary, a wealth of evidence continues to accumulate arguing that proteins causing AD, PD, FTDs, ALS, and even HD acquire alternative conformations that become self-propagating. Each of these neurodegenerative diseases is caused by a different protein that undergoes a conformational transformation to become a prion. Prions explain many of the features that the neurodegenerative diseases have in common: (1) incidence increases with age, (2) steady progression over years, (3) spread from one region of the CNS to another, (4) protein deposits consisting of amyloid fibrils, and (5) late onset of inherited forms of the neurodegenerative diseases. Notably, amyloid plaques containing PrPSc are a nonobligatory feature of PrP prion disease in humans and animals. Furthermore, amyloid plaques in AD do not correlate with the level of dementia; however, the level of soluble (oligomeric) Aβ peptide does correlate with memory loss and other intellectual deficits.
454 |
Disorders of the Autonomic Nervous System |
The autonomic nervous system (ANS) innervates the entire neuraxis and influences all organ systems. It regulates blood pressure (BP), heart rate, sleep, and bladder and bowel function. It operates automatically; its full importance becomes recognized only when ANS function is compromised, resulting in dysautonomia. Hypothalamic disorders that cause disturbances in homeostasis are discussed in Chaps. 23 and 401e.
ANATOMIC ORGANIZATION
The activity of the ANS is regulated by central neurons responsive to diverse afferent inputs. After central integration of afferent information, autonomic outflow is adjusted to permit the functioning of the major organ systems in accordance with the needs of the whole organism. Connections between the cerebral cortex and the autonomic centers in the brainstem coordinate autonomic outflow with higher mental functions.
The preganglionic neurons of the parasympathetic nervous system leave the central nervous system (CNS) in the third, seventh, ninth, and tenth cranial nerves as well as the second and third sacral nerves, while the preganglionic neurons of the sympathetic nervous system exit the spinal cord between the first thoracic and the second lumbar segments (Fig. 454-1). These are thinly myelinated. The postganglionic neurons, located in ganglia outside the CNS, give rise to the postganglionic unmyelinated autonomic nerves that innervate organs and tissues throughout the body. Responses to sympathetic and parasympathetic stimulation are frequently antagonistic (Table 454-1), reflecting highly coordinated interactions within the CNS; the resultant changes in parasympathetic and sympathetic activity provide more precise control of autonomic responses than could be achieved by the modulation of a single system.
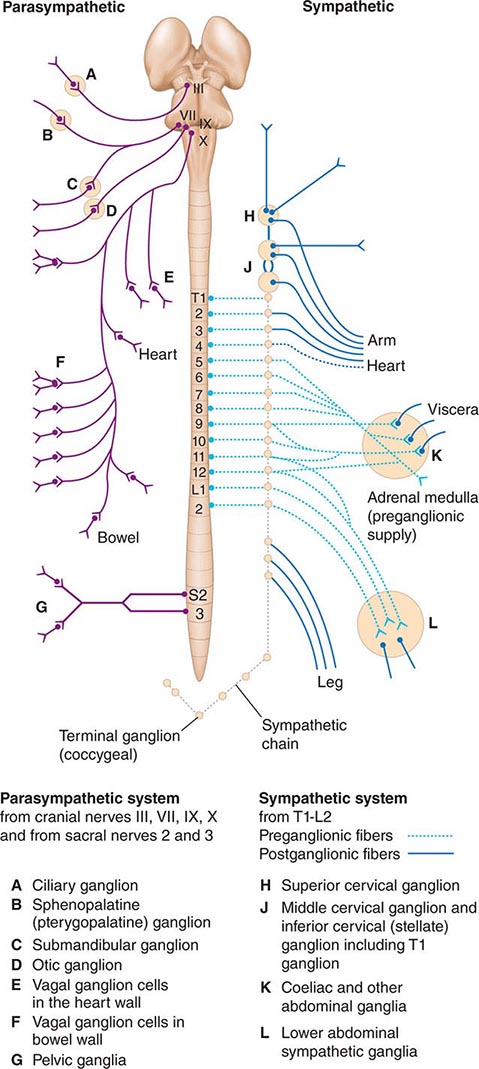
FIGURE 454-1 Schematic representation of the autonomic nervous system. (From M Moskowitz: Clin Endocrinol Metab 6:77, 1977.)
|
FUNCTIONAL CONSEQUENCES OF NORMAL ANS ACTIVATION |
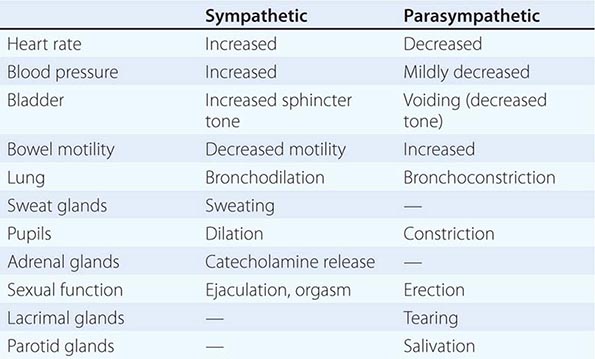
Acetylcholine (ACh) is the preganglionic neurotransmitter for both divisions of the ANS as well as the postganglionic neurotransmitter of the parasympathetic neurons; the preganglionic receptors are nicotinic, and the postganglionic are muscarinic in type. Norepinephrine (NE) is the neurotransmitter of the postganglionic sympathetic neurons, except for cholinergic neurons innervating the eccrine sweat glands.
CLINICAL EVALUATION
CLASSIFICATION
Disorders of the ANS may result from pathology of either the CNS or the peripheral nervous system (PNS) (Table 454-2). Signs and symptoms may result from interruption of the afferent limb, CNS processing centers, or efferent limb of reflex arcs controlling autonomic responses. For example, a lesion of the medulla produced by a posterior fossa tumor can impair BP responses to postural changes and result in orthostatic hypotension (OH). OH can also be caused by lesions of the spinal cord or peripheral vasomotor nerve fibers (e.g., diabetic autonomic neuropathy). Lesions of the efferent limb cause the most consistent and severe OH. The site of reflex interruption is usually established by the clinical context in which the dysautonomia arises, combined with judicious use of ANS testing and neuroimaging studies. The presence or absence of CNS signs, association with sensory or motor polyneuropathy, medical illnesses, medication use, and family history are often important considerations. Some syndromes do not fit easily into any classification scheme.
|
CLASSIFICATION OF CLINICAL AUTONOMIC DISORDERS |
Abbreviations: BP, blood pressure; CNS, central nervous system; HR, heart rate; POTS, postural orthostatic tachycardia syndrome.
SYMPTOMS OF AUTONOMIC DYSFUNCTION
Clinical manifestations can result from loss of function, overactivity, or dysregulation of autonomic circuits. Disorders of autonomic function should be considered in patients with unexplained OH, syncope, sleep dysfunction, altered sweating (hyperhidrosis or hypohidrosis), impotence, constipation or other gastrointestinal symptoms (bloating, nausea, vomiting of old food, diarrhea), or bladder disorders (urinary frequency, hesitancy, or incontinence). Symptoms may be widespread or regional in distribution. An autonomic history focuses on systemic functions (BP, heart rate, sleep, fever, sweating) and involvement of individual organ systems (pupils, bowel, bladder, sexual function). The autonomic symptom profile is a self-report questionnaire that can be used for formal assessment. It is also important to recognize the modulating effects of age. For example, OH typically produces lightheadedness in the young, whereas cognitive slowing is more common in the elderly. Specific symptoms of orthostatic intolerance are diverse (Table 454-3). Autonomic symptoms may vary dramatically, reflecting the dynamic nature of autonomic control over homeostatic function. For example, OH might be manifest only in the early morning, following a meal, with exercise, or with raised ambient temperature, depending on the regional vascular bed affected by the dysautonomia.
|
SYMPTOMS OF ORTHOSTATIC INTOLERANCE |
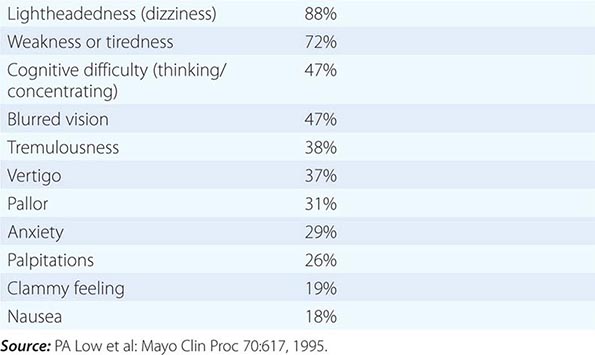
Early symptoms may be overlooked. Impotence, although not specific for autonomic failure, often heralds autonomic failure in men and may precede other symptoms by years (Chap. 67). A decrease in the frequency of spontaneous early morning erections may occur months before loss of nocturnal penile tumescence and development of total impotence. Bladder dysfunction may appear early in men and women, particularly in those with a CNS etiology. Cold feet may indicate increased peripheral vasomotor constriction. Brain and spinal cord disease above the level of the lumbar spine results first in urinary frequency and small bladder volumes and eventually in incontinence (upper motor neuron or spastic bladder). By contrast, PNS disease of autonomic nerve fibers results in large bladder volumes, urinary frequency, and overflow incontinence (lower motor neuron flaccid bladder). Measurement of bladder volume (postvoid residual) is a useful bedside test for distinguishing between upper and lower motor neuron bladder dysfunction in the early stages of dysautonomia. Gastrointestinal autonomic dysfunction typically presents as severe constipation. Diarrhea may develop (typically in diabetes mellitus) due to rapid transit of contents or uncoordinated small-bowel motor activity, or on an osmotic basis from bacterial overgrowth associated with small-bowel stasis. Impaired glandular secretory function may cause difficulty with food intake due to decreased salivation or eye irritation due to decreased lacrimation. Occasionally, temperature elevation and vasodilation can result from anhidrosis because sweating is normally important for heat dissipation (Chap. 23). Lack of sweating after a hot bath, during exercise, or on a hot day can suggest sudomotor failure.
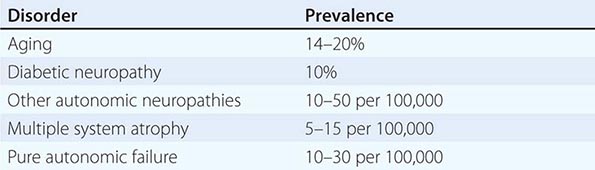
OH (also called orthostatic or postural hypotension) is perhaps the most disabling feature of autonomic dysfunction. The prevalence of OH is relatively high, especially when OH associated with aging and diabetes mellitus is included (Table 454-4). OH can cause a variety of symptoms, including dimming or loss of vision, lightheadedness, diaphoresis, diminished hearing, pallor, and weakness. Syncope results when the drop in BP impairs cerebral perfusion. Other manifestations of impaired baroreflexes are supine hypertension, a heart rate that is fixed regardless of posture, postprandial hypotension, and an excessively high nocturnal BP. Many patients with OH have a preceding diagnosis of hypertension or have concomitant supine hypertension, reflecting the great importance of baroreflexes in maintaining postural and supine normotension. The appearance of OH in patients receiving antihypertensive treatment may indicate overtreatment or the onset of an autonomic disorder. The most common causes of OH are not neurologic in origin; these must be distinguished from the neurogenic causes (Table 454-5). Neurocardiogenic and cardiac causes of syncope are considered in Chap. 27.
|
PREVALENCE OF ORTHOSTATIC HYPOTENSION IN DIFFERENT DISORDERS |
|
NONNEUROGENIC CAUSES OF ORTHOSTATIC HYPOTENSION |
|
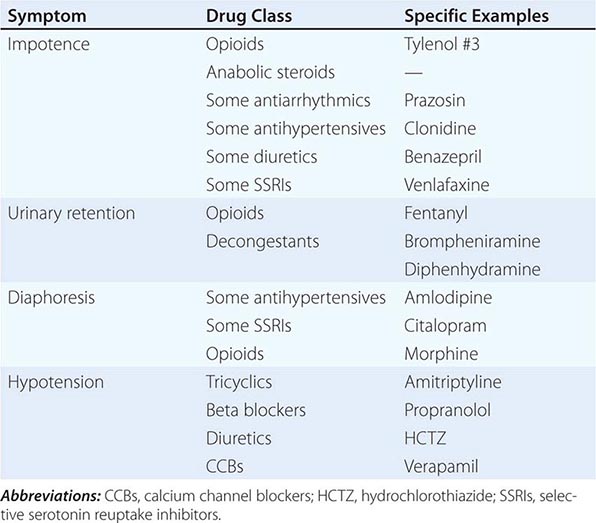
SOME DRUGS THAT AFFECT AUTONOMIC FUNCTION |
|
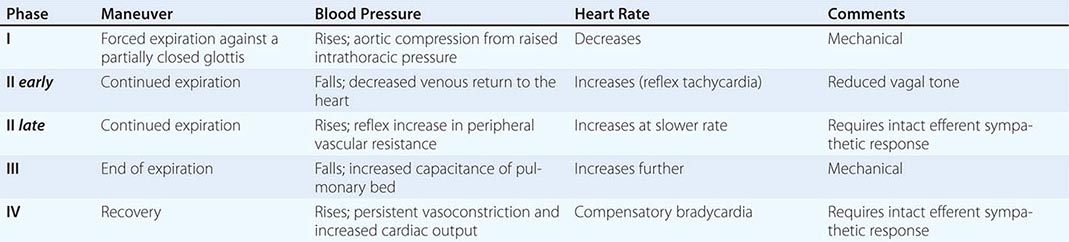
NORMAL BLOOD PRESSURE AND HEART RATE CHANGES DURING THE VALSALVA MANEUVER |
|
NEURAL PATHWAYS UNDERLYING SOME STANDARDIZED AUTONOMIC TESTS |
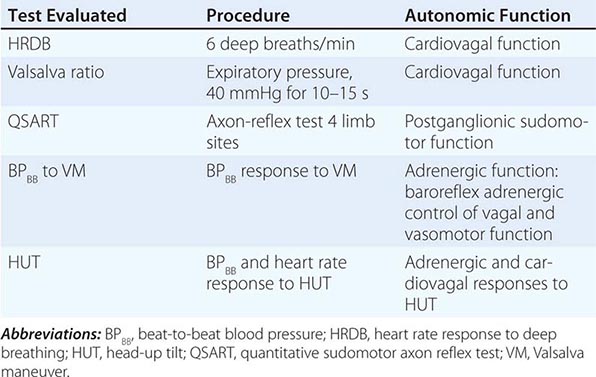
SPECIFIC SYNDROMES OF ANS DYSFUNCTION
MULTIPLE SYSTEM ATROPHY (CHAP. 449)
Multiple system atrophy (MSA) is an entity that comprises autonomic failure (OH or a neurogenic bladder) and either parkinsonism (MSA-p) or a cerebellar syndrome (MSA-c). MSA-p is the more common form; the parkinsonism is atypical in that it is usually unassociated with significant tremor or a response to levodopa. Symptomatic OH within 1 year of onset of parkinsonism predicts eventual development of MSA-p in 75% of patients. There is a very high frequency of impotence in men. Although autonomic abnormalities are common in advanced Parkinson’s disease (Chap. 449), the severity and distribution of autonomic failure are more severe and generalized in MSA. Brain magnetic resonance imaging (MRI) is a useful diagnostic adjunct: in MSA-p, iron deposition in the striatum may be evident as T2 hypointensity, and in MSA-c, cerebellar atrophy is present with a characteristic T2 hyperintense signal (“hot cross buns sign”) in the pons (Fig. 454-2). Cardiac postganglionic adrenergic innervation, measured by uptake of fluorodopamine on positron emission tomography, is markedly impaired in the dysautonomia of Parkinson’s disease (PD) but is usually normal in MSA. Neuropathologic changes include neuronal loss and gliosis in many CNS regions, including the brainstem, cerebellum, striatum, and intermediolateral cell column of the thoracolumbar spinal cord.
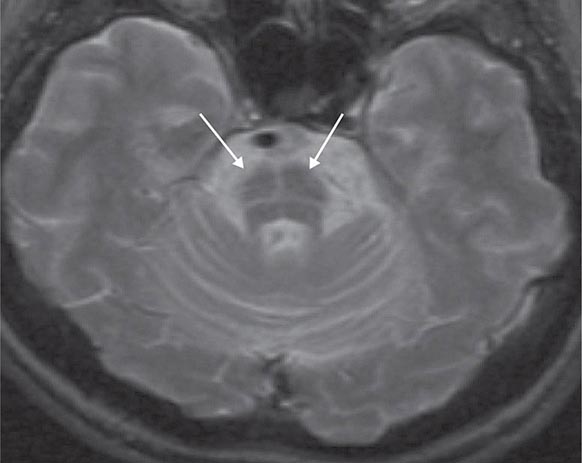
FIGURE 454-2 Multiple system atrophy, cerebellar type (MSA-c). Axial T2-weighted magnetic resonance image at the level of the pons shows a characteristic hyperintense signal, the “hot cross buns” sign. This appearance can also be seen in some spinocerebellar atrophies, as well as other neurodegenerative conditions affecting the brainstem.
MSA is uncommon, with a prevalence estimated at 2–5 per 100,000 individuals. Onset is typically in the mid-fifties, men are slightly more often affected than women, and most cases are sporadic. The diagnosis should be considered in adults over the age of 30 years who present with OH or urinary incontinence and either parkinsonism that is poorly responsive to dopamine replacement or a cerebellar syndrome. MSA generally progresses relentlessly to death 7–10 years after onset, but survival beyond 15 years has been reported. Factors that predict a worse prognosis include rapid progression of disability, bladder dysfunction, female gender, the MSA-p subtype, and an older age at onset. Attempts to slow the progression of MSA have thus far been unsuccessful, including trials of lithium, growth hormone, riluzole, rasagiline, minocycline, and a recent trail of rifampicin.
Management is symptomatic for neurogenic OH (see below), sleep disorders including laryngeal stridor, and gastrointestinal (GI) and urinary dysfunction. GI management includes frequent small meals, soft diet, stool softeners, and bulk agents. Gastroparesis is difficult to treat; metoclopramide stimulates gastric emptying but worsens parkinsonism by blocking central dopamine receptors. The peripheral dopamine (D2 and D3) receptor antagonist domperidone has been used patients with various GI conditions in many countries and is now available in the United States through the U.S. Food and Drug Administration’s (FDA) Expanded Access to Investigational Drugs program.
Autonomic dysfunction is also a common feature in dementia with Lewy bodies (Chap. 448); the severity is usually less than that found in MSA or PD. In multiple sclerosis (MS; Chap. 458), autonomic complications reflect the CNS location of MS involvement and generally worsen with disease duration and disability.
SPINAL CORD
Spinal cord lesions from any cause may result in focal autonomic deficits or autonomic hyperreflexia (e.g., spinal cord transection or hemisection) affecting bowel, bladder, sexual, temperature-regulation, or cardiovascular functions. Quadriparetic patients exhibit both supine hypertension and OH after upward tilting. Autonomic dysreflexia describes a dramatic increase in BP in patients with traumatic spinal cord lesions above the T6 level, often in response to stimulation of the bladder, skin, or muscles. A distended or obstructed bladder, suprapubic palpation, catheter insertion, and urinary infection are common triggers. Associated symptoms can include facial flushing, headache, hypertension, or piloerection. Potential complications include intracranial vasospasm or hemorrhage, cardiac arrhythmia, and death. Awareness of the syndrome, identifying the trigger, and careful monitoring of BP during procedures in patients with acute or chronic spinal cord injury are essential. In patients with supine hypertension, BP can be lowered by tilting the head upward or sitting the patient up. Vasodilator drugs may be used to treat acute elevations in BP. Clonidine can be used prophylactically to reduce the hypertension resulting from bladder stimulation. Dangerous increases or decreases in body temperature may result from an inability to experience the sensory accompaniments of heat or cold exposure or control peripheral vasoconstriction or sweating below the level of the spinal cord injury.
PERIPHERAL NERVE AND NEUROMUSCULAR JUNCTION DISORDERS
Peripheral neuropathies (Chap. 459) are the most common cause of chronic autonomic insufficiency. Polyneuropathies that affect small myelinated and unmyelinated fibers of the sympathetic and parasympathetic nerves commonly occur in diabetes mellitus, amyloidosis, chronic alcoholism, porphyria, and Guillain-Barré syndrome. Neuromuscular junction disorders with autonomic involvement include botulism and Lambert-Eaton syndrome (Chap. 461).
Diabetes Mellitus Autonomic neuropathy in patients with diabetes increases the mortality rate 1.5- to 3-fold, even after adjusting for other cardiovascular risk factors. Estimates of 5-year mortality risk among these patients range from 15 to 53%. Although many deaths are due to secondary vascular disease, there are patients who specifically suffer cardiac arrest due to autonomic neuropathy. The autonomic involvement is also predictive of other complications including renal disease, stroke, and sleep apnea. Diabetes mellitus is discussed in Chaps. 417–419.
Amyloidosis Autonomic neuropathy occurs in both sporadic and familial forms of amyloidosis (Chap. 137). The AL (immunoglobulin light chain) type is associated with primary amyloidosis or amyloidosis secondary to multiple myeloma. The ATTR type, with transthyretin as the primary protein component, is responsible for the most common form of inherited amyloidosis. Although patients usually present with a distal painful polyneuropathy accompanied by sensory loss, autonomic insufficiency can precede the development of the polyneuropathy or occur in isolation. The diagnosis can be made by protein electrophoresis of blood and urine, tissue biopsy (abdominal fat pad, rectal mucosa, or sural nerve) to search for amyloid deposits, and genetic testing for transthyretin mutations in familial cases. Treatment of familial cases with liver transplantation can be successful. The response of primary amyloidosis to melphalan and stem cell transplantation has been mixed. Death is usually due to cardiac or renal involvement. Postmortem studies reveal amyloid deposition in many organs, including two sites that contribute to autonomic failure: intraneural blood vessels and autonomic ganglia. Pathologic examination reveals a loss of both unmyelinated and myelinated nerve fibers.
Alcoholic Neuropathy Abnormalities in parasympathetic vagal and efferent sympathetic function are usually mild in alcoholic polyneuropathy. OH is usually due to brainstem involvement, rather than injury to the PNS. Impotence is a major problem, but concurrent gonadal hormone abnormalities may play a role in this symptom. Clinical symptoms of autonomic failure generally appear only when the stocking-glove polyneuropathy is severe, and there is usually coexisting Wernicke’s encephalopathy (Chap. 330). Autonomic involvement may contribute to the high mortality rates associated with alcoholism (Chap. 467).
Porphyria (Chap. 430) Autonomic dysfunction is most extensively documented in acute intermittent porphyria but can also occur with variegate porphyria and hereditary coproporphyria. Autonomic symptoms include tachycardia, sweating, urinary retention, abdominal pain, nausea and vomiting, insomnia, hypertension, and (less commonly) hypotension. Another prominent symptom is anxiety. Abnormal autonomic function can occur both during acute attacks and during remissions. Elevated catecholamine levels during acute attacks correlate with the degree of tachycardia and hypertension that is present.
Guillain-Barré Syndrome (Chap. 460) BP fluctuations and arrhythmias from autonomic instability can be severe. It is estimated that between 2 and 10% of patients with severe Guillain-Barré syndrome suffer fatal cardiovascular collapse. GI autonomic involvement, sphincter disturbances, abnormal sweating, and pupillary dysfunction can also occur. Demyelination has been described in the vagus and glossopharyngeal nerves, the sympathetic chain, and the white rami communicantes. Interestingly, the degree of autonomic involvement appears to be independent of the severity of motor or sensory neuropathy. Acute autonomic and sensory neuropathy is a variant that spares the motor system and presents with neurogenic OH and varying degrees of sensory loss. It is treated similarly to Guillain-Barré syndrome, but prognosis is less favorable, with persistent severe sensory deficits and variable degrees of OH in many patients.
Autoimmune Autonomic Ganglionopathy (AAG) This disorder presents with the subacute development of autonomic disturbances including OH, enteric neuropathy (gastroparesis, ileus, constipation/diarrhea), flaccid bladder, and cholinergic failure (e.g., loss of sweating, sicca complex, and a tonic pupil). A chronic form of AAG resembles pure autonomic failure (see below). Autoantibodies against the ganglionic ACh receptor (A3 AChR), which are present in approximately half of patients, are considered diagnostic of AAG. Pathology shows preferential involvement of small unmyelinated nerve fibers, with sparing of larger myelinated ones. Onset of the neuropathy follows a viral infection in approximately half of cases. Up to one-third of untreated patients experience significant functional improvement over time. Immunotherapies that have been reported to be helpful include plasmapheresis, intravenous immune globulin, glucocorticoids, azathioprine, rituximab, and mycophenolate mofetil. OH, gastroparesis, and sicca symptoms can be managed symptomatically.
AAG can also occur on a paraneoplastic basis, with adenocarcinoma or small-cell carcinoma of the lung, lymphoma, or thymoma being the most common (Chap. 122). In the paraneoplastic cases, distinctive additional features, such as cerebellar involvement or dementia, may be present (see Tables 122-1, 122-2, and 122-3). The neoplasm may be occult and possibly suppressed by the autoantibody.
Botulism Botulinum toxin binds presynaptically to cholinergic nerve terminals and, after uptake into the cytosol, blocks ACh release. This acute cholinergic neuropathy presents as motor paralysis and autonomic disturbances that include blurred vision, dry mouth, nausea, unreactive or sluggishly reactive pupils, constipation, and urinary retention (Chap. 178).
PURE AUTONOMIC FAILURE (PAF)
This sporadic syndrome consists of postural hypotension, impotence, bladder dysfunction, and impaired sweating. The disorder begins in midlife and occurs in women more often than men. The symptoms can be disabling, but the disease does not shorten life span. The clinical and pharmacologic characteristics suggest primary involvement of postganglionic sympathetic neurons. A severe reduction in the density of neurons within sympathetic ganglia results in low supine plasma NE levels and noradrenergic supersensitivity. Some patients who are initially labeled with this diagnosis subsequently go on to develop AAG or MSA. Skin biopsies can demonstrate phosphorylated α-synuclein inclusions in postganglionic sympathetic adrenergic and cholinergic nerve fibers from some individuals with PAF, distinguishing them from AAG and suggesting that PAF is a synucleinopathy; patients with PD also have α-synuclein inclusions in sympathetic nerve biopsies.
POSTURAL ORTHOSTATIC TACHYCARDIA SYNDROME (POTS)
This syndrome is characterized by symptomatic orthostatic intolerance without OH, accompanied by either an increase in heart rate to >120 beats/min or an increase of 30 beats/min with standing that subsides on sitting or lying down. Women are affected approximately five times more often than men, and most develop the syndrome between the ages of 15 and 50. Presyncopal symptoms (lightheadedness, weakness, blurred vision) combined with symptoms of autonomic overactivity (palpitations, tremulousness, nausea) are common. Recurrent unexplained episodes of dysautonomia and fatigue also occur. The pathogenesis is unclear, but there is increasing evidence for sympathetic denervation distally in the legs with preserved cardiovascular function. Hypovolemia, venous pooling, impaired brainstem regulation, or increased sympathetic activity may play a role. Optimal treatment is uncertain, but expansion of fluid volume with water, salt, and fludrocortisone can be helpful as initial interventions. If this approach is inadequate, then midodrine, pyridostigmine, phenobarbital, beta blockers, or clonidine can be tried. Reconditioning and a sustained exercise program are important adjuncts to treatment.
INHERITED DISORDERS
There are five known hereditary sensory and autonomic neuropathies (HSAN I–V). The most important autonomic variants are HSAN I and HSAN III. HSAN I is dominantly inherited and often presents as a distal small-fiber neuropathy (burning feet syndrome) associated with sensory loss and foot ulcers. The most common responsible gene, on chromosome 9q, is SPTLC1. SPTLC is an important enzyme in the regulation of ceramide. Cells from HSAN I patients with the mutation produce higher-than-normal levels of glucosyl ceramide, perhaps triggering apoptosis. HSAN III (Riley-Day syndrome; familial dysautonomia) is an autosomal recessive disorder of Ashkenazi Jewish children and adults and is much less prevalent than HSAN I. Decreased tearing, hyperhidrosis, reduced sensitivity to pain, areflexia, absent fungiform papillae on the tongue, and labile BP may be present. Episodic abdominal crises and fever are common. Pathologic examination of nerves reveals a loss of sympathetic, parasympathetic, and sensory neurons. The defective gene, IKBKAP, may prevent normal transcription of important molecules in neural development.
PRIMARY HYPERHIDROSIS
This syndrome presents with excess sweating of the palms of the hands and soles of the feet beginning in childhood or early adulthood. The condition tends to improve with age. The disorder affects 0.6–1.0% of the population. The etiology is unclear, but there may be a genetic component because 25% of patients have a positive family history. The condition can be socially embarrassing (e.g., shaking hands) or even disabling (e.g., inability to write without soiling the paper). Topical antiperspirants are occasionally helpful. More useful are potent anticholinergic drugs such as glycopyrrolate (1–2 mg PO tid). T2 ganglionectomy or sympathectomy is successful in >90% of patients with palmar hyperhidrosis. The advent of endoscopic transaxillary T2 sympathectomy has lowered the complication rate of the procedure. The most common postoperative complication is compensatory hyperhidrosis, which improves spontaneously over months. Other potential complications include recurrent hyperhidrosis (16%), Horner’s syndrome (<2%), gustatory sweating, wound infection, hemothorax, and intercostal neuralgia. Local injection of botulinum toxin has also been used to block cholinergic, postganglionic sympathetic fibers to sweat glands in patients with palmar hyperhidrosis. This approach is limited by the need for repetitive injections (the effect usually lasts 4 months before waning).
ACUTE SYMPATHETIC OVERACTIVITY SYNDROMES
The physician may be confronted occasionally with an acute state of sympathetic overactivity.
An autonomic storm is an acute state of sustained sympathetic surge that results in variable combinations of alterations in BP and heart rate, body temperature, respiration, and sweating. Causes of autonomic storm include brain and spinal cord injury, toxins and drugs, autonomic neuropathy, and chemodectomas (e.g., pheochromocytoma). Brain injury is the most common cause of autonomic storm and typically follows severe head trauma and postresuscitation encephalopathy anoxic-ischemic brain injury. Autonomic storm can also occur with other acute intracranial lesions such as hemorrhage, cerebral infarction, rapidly expanding tumors, subarachnoid hemorrhage, hydrocephalus, or (less commonly) an acute spinal cord lesion. The most consistent setting is that of an acute intracranial catastrophe of sufficient size and rapidity to produce a massive catecholaminergic surge. The surge can cause seizures, neurogenic pulmonary edema, and myocardial injury. Manifestations include fever, tachycardia, hypertension, tachypnea, hyperhidrosis, pupillary dilatation, and flushing. Lesions of the afferent limb of the baroreflex can result in milder recurrent autonomic storms; many of these follow neck irradiation.
Drugs and toxins may also be responsible, including sympathomimetics such as phenylpropanolamine, cocaine, amphetamines, and tricyclic antidepressants; tetanus; and, less often, botulinum toxin. Cocaine, including “crack,” can cause a hypertensive state with CNS hyperstimulation. Tricyclic overdose, such as from amitriptyline, can cause flushing, hypertension, tachycardia, fever, mydriasis, anhidrosis, and a toxic psychosis. The hyperadrenergic state associated with Guillain-Barré syndrome can produce a moderate autonomic storm. Pheochromocytoma presents with a paroxysmal or sustained hyperadrenergic state, headache, hyperhidrosis, palpitations, anxiety, tremulousness, and hypertension. Neuroleptic malignant syndrome refers to a syndrome of muscle rigidity, hyperthermia, and hypertension in psychotic patients treated with phenothiazines (Chap. 449). Management of autonomic storm includes ruling out other causes of autonomic instability, including malignant hyperthermia, porphyria, and seizures. Sepsis and encephalitis need to be excluded with appropriate studies. An electroencephalogram (EEG) should be done to search for seizure activity; MRI of the brain and spine is often necessary. The patient should be managed in an intensive care unit. Management with morphine sulphate (10 mg every 4 h) and labetalol (100–200 mg twice daily) may be helpful. Supportive treatment may need to be maintained for several weeks. For chronic and milder autonomic storm, propranolol and/or clonidine can be effective.
MISCELLANEOUS
Other conditions associated with autonomic failure include infections, malignancy, poisoning (organophosphates), and aging. Disorders of the hypothalamus can affect autonomic function and produce abnormalities in temperature control, satiety, sexual function, and circadian rhythms (Chap. 403).
REFLEX SYMPATHETIC DYSTROPHY AND CAUSALGIA
The failure to identify a primary role of the ANS in the pathogenesis of these disorders has resulted in a change of nomenclature. The terms complex regional pain syndrome (CRPS) types I and II are now used in place of reflex sympathetic dystrophy (RSD) and causalgia.
CRPS type I is a regional pain syndrome that often develops after tissue injury and most commonly affects one limb. Examples of associated injury include minor shoulder or limb trauma, fractures, myocardial infarction, or stroke. Allodynia (the perception of a nonpainful stimulus as painful), hyperpathia (an exaggerated pain response to a painful stimulus), and spontaneous pain occur. The symptoms are unrelated to the severity of the initial trauma and are not confined to the distribution of a single peripheral nerve. CRPS type II is a regional pain syndrome that develops after injury to a specific peripheral nerve, often a major nerve trunk. Spontaneous pain initially develops within the territory of the affected nerve but eventually may spread outside the nerve distribution.
Pain (usually burning or electrical in quality) is the primary clinical feature of CRPS. Vasomotor dysfunction, sudomotor abnormalities, or focal edema may occur alone or in combination but must be present for diagnosis. Limb pain syndromes that do not meet these criteria are best classified as “limb pain—not otherwise specified.” In CRPS, localized sweating (increased resting sweat output) and changes in blood flow may produce temperature differences between affected and unaffected limbs.
CRPS type I (RSD) has been classically divided into three clinical phases. Phase I consists of pain and swelling in the distal extremity occurring within weeks to 3 months after the precipitating event. The pain is diffuse, spontaneous, and either burning, throbbing, or aching in quality. The involved extremity is warm and edematous, and the joints are tender. Increased sweating and hair growth develop. In phase II (3–6 months after onset), thin, shiny, cool skin appears. After an additional 3–6 months (phase III), atrophy of the skin and subcutaneous tissue plus flexion contractures complete the clinical picture. Autonomic testing or bone scans are occasionally useful when the diagnosis is in doubt.
The natural history of typical CRPS may be more benign and more variable than previously recognized. A variety of surgical and medical treatments have been developed, with conflicting reports of efficacy. Clinical trials suggest that early mobilization with physical therapy or a brief course of glucocorticoids may be helpful for CRPS type I or II. Other medical treatments include the use of adrenergic blockers, nonsteroidal anti-inflammatory drugs, calcium channel blockers, phenytoin, opioids, and calcitonin. Stellate ganglion blockade is a commonly used invasive technique that often provides temporary pain relief, but the efficacy of repetitive blocks is uncertain.
455 |
Trigeminal Neuralgia, Bell’s Palsy, and Other Cranial Nerve Disorders |
Symptoms and signs of cranial nerve pathology are common in internal medicine. They often develop in the context of a widespread neurologic disturbance, and in such situations, cranial nerve involvement may represent the initial manifestation of the illness. In other disorders, involvement is largely restricted to one or several cranial nerves; these distinctive disorders are reviewed in this chapter. Disorders of ocular movement are discussed in Chap. 39, disorders of hearing in Chap. 43, and vertigo and disorders of vestibular function in Chap. 28.
FACIAL PAIN OR NUMBNESS
ANATOMIC CONSIDERATIONS
The trigeminal (fifth cranial) nerve supplies sensation to the skin of the face and anterior half of the head (Fig. 455-1). The motor part innervates the muscles involved in chewing (including masseters and pterygoids) as well as the tensor tympani of the middle ear (hearing especially for high-pitched tones). It is the largest of the cranial nerves. It exits in the lateral midpons and traverses the middle cranial fossa to the semilunar (gasserian, trigeminal) ganglion in Meckel’s cave, where the nerve divides into three divisions (ophthalmic [V1], maxillary [V2], and mandibular [V3]). V1 and V2 traverse the cavernous sinus to exit in the superior orbital fissure and foramen rotundum, located above and below the eye socket respectively; V3 exits through the foramen ovale. The trigeminal nerve is predominantly sensory, and motor innervation is exclusively carried in V3. The cornea is primarily innervated by V1, although an inferior crescent may be V2. Upon entering the pons, pain and temperature fibers descend ipsilaterally to the upper cervical spinal cord as the spinal tract of V, before synapsing with the spinal nucleus of V; this accounts for the facial numbness that can occur with spinal cord lesions above C2. In the brainstem, the spinal tract of V is also located adjacent to crossed ascending fibers of the spinothalamic tract, producing a “crossed” sensory loss for pain and temperature (ipsilateral face, contralateral arm/trunk/leg) with lesions of the lateral lower brainstem. CN V is also ensheathed by oligodendrocyte-derived, rather than Schwann cell–derived, myelin for up to 7 mm after it leaves the brainstem, unlike just a few millimeters for other cranial and spinal nerves; this may explain the high frequency of trigeminal neuralgia in multiple sclerosis (Chap. 458), a disorder of oligodendrocyte myelin.
FIGURE 455-1 The trigeminal nerve and its branches and sensory distribution on the face. The three major sensory divisions of the trigeminal nerve consist of the ophthalmic, maxillary, and mandibular nerves. (Adapted from Waxman SG: Clinical Neuroanatomy, 26th ed. http://www.accessmedicine.com. Copyright © The McGraw-Hill Companies, Inc. All rights reserved.)
TRIGEMINAL NEURALGIA (TIC DOULOUREUX)
Clinical Manifestations Trigeminal neuralgia is characterized by excruciating paroxysms of pain in the lips, gums, cheek, or chin and, very rarely, in the distribution of the ophthalmic division of the fifth nerve. The pain seldom lasts more than a few seconds or a minute or two but may be so intense that the patient winces, hence the term tic. The paroxysms, experienced as single jabs or clusters, tend to recur frequently, both day and night, for several weeks at a time. They may occur spontaneously or with movements of affected areas evoked by speaking, chewing, or smiling. Another characteristic feature is the presence of trigger zones, typically on the face, lips, or tongue, that provoke attacks; patients may report that tactile stimuli—e.g., washing the face, brushing the teeth, or exposure to a draft of air—generate excruciating pain. An essential feature of trigeminal neuralgia is that objective signs of sensory loss cannot be demonstrated on examination.
Trigeminal neuralgia is relatively common, with an estimated annual incidence of 4–8 per 100,000 individuals. Middle-aged and elderly persons are affected primarily, and ~60% of cases occur in women. Onset is typically sudden, and bouts tend to persist for weeks or months before remitting spontaneously. Remissions may be long-lasting, but in most patients, the disorder ultimately recurs.
Pathophysiology Symptoms result from ectopic generation of action potentials in pain-sensitive afferent fibers of the fifth cranial nerve root just before it enters the lateral surface of the pons. Compression or other pathology in the nerve leads to demyelination of large myelinated fibers that do not themselves carry pain sensation but become hyperexcitable and electrically coupled with smaller unmyelinated or poorly myelinated pain fibers in close proximity; this may explain why tactile stimuli, conveyed via the large myelinated fibers, can stimulate paroxysms of pain. Compression of the trigeminal nerve root by a blood vessel, most often the superior cerebellar artery or on occasion a tortuous vein, is now believed to be the source of trigeminal neuralgia in most patients. In cases of vascular compression, age-related brain sagging and increased vascular thickness and tortuosity may explain the prevalence of trigeminal neuralgia in later life.
Differential Diagnosis Trigeminal neuralgia must be distinguished from other causes of face and head pain (Chap. 21) and from pain arising from diseases of the jaw, teeth, or sinuses. Pain from migraine or cluster headache tends to be deep-seated and steady, unlike the superficial stabbing quality of trigeminal neuralgia; rarely, cluster headache is associated with trigeminal neuralgia, a syndrome known as cluster-tic. In temporal arteritis, superficial facial pain is present but is not typically shock-like, the patient frequently complains of myalgias and other systemic symptoms, and an elevated erythrocyte sedimentation rate (ESR) is usually present (Chap. 385). When trigeminal neuralgia develops in a young adult or is bilateral, multiple sclerosis (MS) is a key consideration, and in such cases, the cause is a demyelinating plaque near the root entry zone of the fifth nerve in the pons; often, evidence of facial sensory loss can be found on careful examination. Cases that are secondary to mass lesions—such as aneurysms, neurofibromas, acoustic schwannomas, or meningiomas—usually produce objective signs of sensory loss in the trigeminal nerve distribution (trigeminal neuropathy, see below).
Laboratory Evaluation An ESR is indicated if temporal arteritis is suspected. In typical cases of trigeminal neuralgia, neuroimaging studies are usually unnecessary but may be valuable if MS is a consideration or in assessing overlying vascular lesions in order to plan for decompression surgery.
TRIGEMINAL NEUROPATHY
A variety of diseases can affect the trigeminal nerve (Table 455-1). Most present with sensory loss on the face or with weakness of the jaw muscles. Deviation of the jaw on opening indicates weakness of the pterygoids on the side to which the jaw deviates. Some cases are due to Sjögren’s syndrome or a collagen-vascular disease such as systemic lupus erythematosus, scleroderma, or mixed connective tissue disease. Among infectious causes, herpes zoster (acute or postherpetic) and leprosy should be considered. Tumors of the middle cranial fossa (meningiomas), of the trigeminal nerve (schwannomas), or of the base of the skull (metastatic tumors) may cause a combination of motor and sensory signs. Lesions in the cavernous sinus can affect the first and second divisions of the trigeminal nerve, and lesions of the superior orbital fissure can affect the first (ophthalmic) division; the accompanying corneal anesthesia increases the risk of ulceration (neurokeratitis).
|
TRIGEMINAL NERVE DISORDERS |
Isolated sensory loss over the chin (mental neuropathy) can be the only manifestation of systemic malignancy. Rarely, an idiopathic form of trigeminal neuropathy is observed. It is characterized by numbness and paresthesias, sometimes bilaterally, with loss of sensation in the territory of the trigeminal nerve but without weakness of the jaw. Gradual recovery is the rule. Tonic spasm of the masticatory muscles, known as trismus, is symptomatic of tetanus (Chap. 177) or may occur in patients treated with phenothiazines.
FACIAL WEAKNESS
ANATOMIC CONSIDERATIONS
(Fig. 455-2) The seventh cranial nerve supplies all the muscles concerned with facial expression. The sensory component is small (the nervus intermedius); it conveys taste sensation from the anterior two-thirds of the tongue and probably cutaneous impulses from the anterior wall of the external auditory canal. The motor nucleus of the seventh nerve lies anterior and lateral to the abducens nucleus. After leaving the pons, the seventh nerve enters the internal auditory meatus with the acoustic nerve. The nerve continues its course in its own bony channel, the facial canal, and exits from the skull via the stylomastoid foramen. It then passes through the parotid gland and subdivides to supply the facial muscles.
FIGURE 455-2 The facial nerve. A, B, and C denote lesions of the facial nerve at the stylomastoid foramen, distal and proximal to the geniculate ganglion, respectively. Green lines indicate the parasympathetic fibers, red line indicates motor fibers, and purple lines indicate visceral afferent fibers (taste). (Adapted from MB Carpenter: Core Text of Neuroanatomy, 2nd ed. Williams & Wilkins, 1978.)
A complete interruption of the facial nerve at the stylomastoid foramen paralyzes all muscles of facial expression. The corner of the mouth droops, the creases and skinfolds are effaced, the forehead is unfurrowed, and the eyelids will not close. Upon attempted closure of the lids, the eye on the paralyzed side rolls upward (Bell’s phenomenon). The lower lid sags and falls away from the conjunctiva, permitting tears to spill over the cheek. Food collects between the teeth and lips, and saliva may dribble from the corner of the mouth. The patient complains of a heaviness or numbness in the face, but sensory loss is rarely demonstrable and taste is intact.
If the lesion is in the middle-ear portion, taste is lost over the anterior two-thirds of the tongue on the same side. If the nerve to the stapedius is interrupted, there is hyperacusis (sensitivity to loud sounds). Lesions in the internal auditory meatus may affect the adjacent auditory and vestibular nerves, causing deafness, tinnitus, or dizziness. Intrapontine lesions that paralyze the face usually affect the abducens nucleus as well, and often the corticospinal and sensory tracts.
If the peripheral facial paralysis has existed for some time and recovery of motor function is incomplete, a continuous diffuse contraction of facial muscles may appear. The palpebral fissure becomes narrowed, and the nasolabial fold deepens. Attempts to move one group of facial muscles may result in contraction of all (associated movements, or synkinesis). Facial spasms, initiated by movements of the face, may develop (hemifacial spasm). Anomalous regeneration of seventh nerve fibers may result in other troublesome phenomena. If fibers originally connected with the orbicularis oculi come to innervate the orbicularis oris, closure of the lids may cause a retraction of the mouth, or if fibers originally connected with muscles of the face later innervate the lacrimal gland, anomalous tearing (“crocodile tears”) may occur with any activity of the facial muscles, such as eating. Another facial synkinesia is triggered by jaw opening, causing closure of the eyelids on the side of the facial palsy (jaw-winking).
BELL’S PALSY
The most common form of facial paralysis is Bell’s palsy. The annual incidence of this idiopathic disorder is ~25 per 100,000 annually, or about 1 in 60 persons in a lifetime. Risk factors include pregnancy and diabetes mellitus.
Clinical Manifestations The onset of Bell’s palsy is fairly abrupt, with maximal weakness being attained by 48 h as a general rule. Pain behind the ear may precede the paralysis for a day or two. Taste sensation may be lost unilaterally, and hyperacusis may be present. In some cases, there is mild cerebrospinal fluid lymphocytosis. Magnetic resonance imaging (MRI) may reveal swelling and uniform enhancement of the geniculate ganglion and facial nerve and, in some cases, entrapment of the swollen nerve in the temporal bone. Approximately 80% of patients recover within a few weeks or months. Electromyography may be of some prognostic value; evidence of denervation after 10 days indicates there has been axonal degeneration, that there will be a long delay (3 months as a rule) before regeneration occurs, and that it may be incomplete. The presence of incomplete paralysis in the first week is the most favorable prognostic sign. Recurrences are reported in approximately 7% of cases.
Pathophysiology In acute Bell’s palsy, there is inflammation of the facial nerve with mononuclear cells, consistent with an infectious or immune cause. Herpes simplex virus (HSV) type 1 DNA was frequently detected in endoneurial fluid and posterior auricular muscle, suggesting that a reactivation of this virus in the geniculate ganglion may be responsible for most cases. Reactivation of varicella-zoster virus is associated with Bell’s palsy in up to one-third of cases and may represent the second most frequent cause. A variety of other viruses have also been implicated less commonly. An increased incidence of Bell’s palsy was also reported among recipients of inactivated intranasal influenza vaccine, and it was hypothesized that this could have resulted from the Escherichia coli enterotoxin used as adjuvant or reactivation of latent virus.
Differential Diagnosis There are many other causes of acute facial palsy that must be considered in the differential diagnosis of Bell’s palsy. Lyme disease can cause unilateral or bilateral facial palsies; in endemic areas, 10% or more of cases of facial palsy are likely due to infection with Borrelia burgdorferi (Chap. 210). The Ramsay Hunt syndrome, caused by reactivation of herpes zoster in the geniculate ganglion, consists of a severe facial palsy associated with a vesicular eruption in the external auditory canal and sometimes in the pharynx and other parts of the cranial integument; often the eighth cranial nerve is affected as well. Facial palsy that is often bilateral occurs in sarcoidosis (Chap. 390) and in Guillain-Barré syndrome (Chap. 460). Leprosy frequently involves the facial nerve, and facial neuropathy may also occur in diabetes mellitus, connective tissue diseases including Sjögren’s syndrome, and amyloidosis. The rare Melkersson-Rosenthal syndrome consists of recurrent facial paralysis; recurrent—and eventually permanent—facial (particularly labial) edema; and, less constantly, plication of the tongue. Its cause is unknown. Acoustic neuromas frequently involve the facial nerve by local compression. Infarcts, demyelinating lesions of MS, and tumors are the common pontine lesions that interrupt the facial nerve fibers; other signs of brainstem involvement are usually present. Tumors that invade the temporal bone (carotid body, cholesteatoma, dermoid) may produce a facial palsy, but the onset is insidious and the course progressive.
All these forms of nuclear or peripheral facial palsy must be distinguished from the supranuclear type. In the latter, the frontalis and orbicularis oculi muscles of the forehead are involved less than those of the lower part of the face, since the upper facial muscles are innervated by corticobulbar pathways from both motor cortices, whereas the lower facial muscles are innervated only by the opposite hemisphere. In supranuclear lesions, there may be a dissociation of emotional and voluntary facial movements, and often some degree of paralysis of the arm and leg or an aphasia (in dominant hemisphere lesions) is present.
Laboratory Evaluation The diagnosis of Bell’s palsy can usually be made clinically in patients with (1) a typical presentation, (2) no risk factors or preexisting symptoms for other causes of facial paralysis, (3) absence of cutaneous lesions of herpes zoster in the external ear canal, and (4) a normal neurologic examination with the exception of the facial nerve. Particular attention to the eighth cranial nerve, which courses near to the facial nerve in the pontomedullary junction and in the temporal bone, and to other cranial nerves is essential. In atypical or uncertain cases, an ESR, testing for diabetes mellitus, a Lyme titer, angiotensin-converting enzyme and chest imaging studies for possible sarcoidosis, a lumbar puncture for possible Guillain-Barré syndrome, or MRI scanning may be indicated. MRI often shows swelling and enhancement of the facial nerve in idiopathic Bell’s palsy (Fig. 455-3).
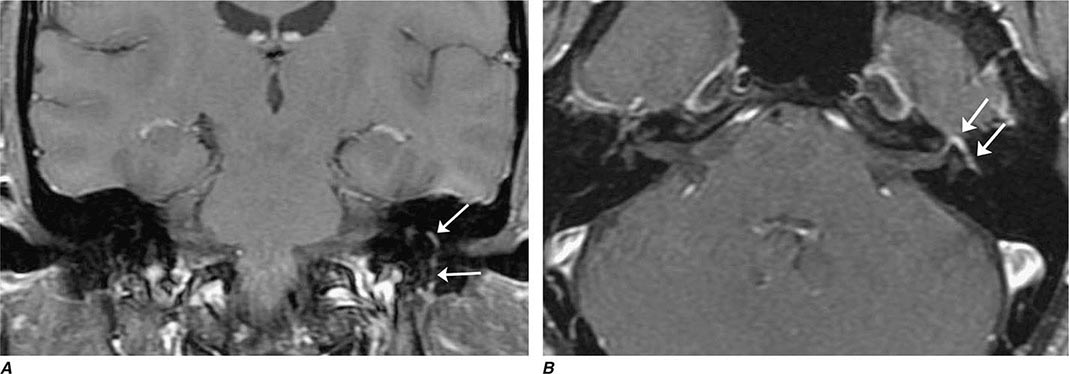
FIGURE 455-3 Axial and coronal T1-weighted images after gadolinium with fat suppression demonstrate diffuse smooth linear enhancement of the left facial nerve, involving the genu, tympanic, and mastoid segments within the temporal bone (arrows), without evidence of mass lesion. Although highly suggestive of Bell’s palsy, similar findings may be seen with other etiologies such as Lyme disease, sarcoidosis, and perineural malignant spread.
OTHER MOTOR DISORDERS OF THE FACE
Hemifacial spasm consists of painless irregular involuntary contractions on one side of the face. Most cases appear related to vascular compression of the exiting facial nerve in the pons. Other cases develop as a sequela to Bell’s palsy or are secondary to compression and/or demyelination of the nerve by tumor, infection, or MS. Mild cases can be treated with carbamazepine, gabapentin, or, if these drugs fail, baclofen. Local injections of botulinum toxin into affected muscles can relieve spasms for 3–4 months, and the injections can be repeated. Refractory cases due to vascular compression usually respond to surgical decompression of the facial nerve. Blepharospasm is an involuntary recurrent spasm of both eyelids that usually occurs in elderly persons as an isolated phenomenon or with varying degrees of spasm of other facial muscles. Severe, persistent cases of blepharospasm can be treated by local injection of botulinum toxin into the orbicularis oculi. Facial myokymia refers to a fine rippling activity of the facial muscles; it may be caused by MS or follow Guillain-Barré syndrome (Chap. 460).
Facial hemiatrophy occurs mainly in women and is characterized by a disappearance of fat in the dermal and subcutaneous tissues on one side of the face. It usually begins in adolescence or the early adult years and is slowly progressive. In its advanced form, the affected side of the face is gaunt, and the skin is thin, wrinkled, and brown. The facial hair may turn white and fall out, and the sebaceous glands become atrophic. Bilateral involvement may occur. A limited form of systemic sclerosis (scleroderma) may be the cause of some cases. Treatment is cosmetic, consisting of transplantation of skin and subcutaneous fat.
OTHER CRANIAL NERVE DISORDERS
GLOSSOPHARYNGEAL NEURALGIA
This form of neuralgia involves the ninth (glossopharyngeal) and sometimes portions of the tenth (vagus) cranial nerves. It resembles trigeminal neuralgia in many respects but is much less common. The pain is intense and paroxysmal; it originates on one side of the throat, approximately in the tonsillar fossa. In some cases, the pain is localized in the ear or may radiate from the throat to the ear because of involvement of the tympanic branch of the glossopharyngeal nerve. Spasms of pain may be initiated by swallowing or coughing. There is no demonstrable motor or sensory deficit; the glossopharyngeal nerve supplies taste sensation to the posterior third of the tongue and, together with the vagus nerve, sensation to the posterior pharynx. Cardiac symptoms—bradycardia or asystole, hypotension, and fainting—have been reported. Glossopharyngeal neuralgia can result from vascular compression, MS, or tumors, but many cases are idiopathic. Medical therapy is similar to that for trigeminal neuralgia, and carbamazepine is generally the first choice. If drug therapy is unsuccessful, surgical procedures—including microvascular decompression if vascular compression is evident—or rhizotomy of glossopharyngeal and vagal fibers in the jugular bulb is frequently successful.
Glossopharyngeal neuropathy in conjunction with vagus and accessory nerve palsies may occur with herpes zoster infection or with a tumor or aneurysm in the posterior fossa or in the jugular foramen. Hoarseness due to vocal cord paralysis, some difficulty in swallowing, deviation of the soft palate to the intact side, anesthesia of the posterior wall of the pharynx, and weakness of the upper part of the trapezius and sternocleidomastoid muscles make up the jugular foramen syndrome (Table 455-2).
|
CRANIAL NERVE SYNDROMES |
DYSPHAGIA AND DYSPHONIA
When the intracranial portion of one vagus (tenth cranial) nerve is interrupted, the soft palate droops ipsilaterally and does not rise in phonation. There is loss of the gag reflex on the affected side, as well as of the “curtain movement” of the lateral wall of the pharynx, whereby the faucial pillars move medially as the palate rises in saying “ah.” The voice is hoarse and slightly nasal, and the vocal cord lies immobile midway between abduction and adduction. Loss of sensation at the external auditory meatus and the posterior pinna may also be present.
The pharyngeal branches of both vagal nerves may be affected in diphtheria; the voice has a nasal quality, and regurgitation of liquids through the nose occurs during swallowing.
Injury to the vagus nerve in the carotid sheath can also occur with carotid dissection or following endarterectomy. The vagus nerve may be involved at the meningeal level by neoplastic and infectious processes and within the medulla by tumors, vascular lesions (e.g., the lateral medullary syndrome), and motor neuron disease. This nerve may be involved by infection with varicella zoster virus. Polymyositis and dermatomyositis, which cause hoarseness and dysphagia by direct involvement of laryngeal and pharyngeal muscles, may be confused with diseases of the vagus nerves. Dysphagia is also a symptom in some patients with myotonic dystrophy. Nonneurologic causes of dysphagia are discussed in Chap. 53.
The recurrent laryngeal nerves, especially the left, are most often damaged as a result of intrathoracic disease. Aneurysm of the aortic arch, an enlarged left atrium, and tumors of the mediastinum and bronchi are much more frequent causes of an isolated vocal cord palsy than are intracranial disorders. However, a substantial number of cases of recurrent laryngeal palsy remain idiopathic.
When confronted with a case of laryngeal palsy, the physician must attempt to determine the site of the lesion. If it is intramedullary, there are usually other signs, such as ipsilateral cerebellar dysfunction, loss of pain and temperature sensation over the ipsilateral face and contralateral arm and leg, and an ipsilateral Horner’s syndrome. If the lesion is extramedullary, the glossopharyngeal and spinal accessory nerves are frequently involved (jugular foramen syndrome). If it is extracranial in the posterior laterocondylar or retroparotid space, there may be a combination of ninth, tenth, eleventh, and twelfth cranial nerve palsies and a Horner’s syndrome (Table 455-2). If there is no sensory loss over the palate and pharynx and no palatal weakness or dysphagia, the lesion is below the origin of the pharyngeal branches, which leave the vagus nerve high in the cervical region; the usual site of disease is then the mediastinum.
NECK WEAKNESS
Isolated involvement of the accessory (eleventh cranial) nerve can occur anywhere along its route, resulting in partial or complete paralysis of the sternocleidomastoid and trapezius muscles. More commonly, involvement occurs in combination with deficits of the ninth and tenth cranial nerves in the jugular foramen or after exit from the skull (Table 455-2). An idiopathic form of accessory neuropathy, akin to Bell’s palsy, has been described, and it may be recurrent in some cases. Most but not all patients recover.
TONGUE PARALYSIS
The hypoglossal (twelfth cranial) nerve supplies the ipsilateral muscles of the tongue. The nucleus of the nerve or its fibers of exit may be involved by intramedullary lesions such as tumor, poliomyelitis, or most often motor neuron disease. Lesions of the basal meninges and the occipital bones (platybasia, invagination of occipital condyles, Paget’s disease) may compress the nerve in its extramedullary course or in the hypoglossal canal. Isolated lesions of unknown cause can occur. Atrophy and fasciculation of the tongue develop weeks to months after interruption of the nerve.
MULTIPLE CRANIAL NERVE PALSIES
Several cranial nerves may be affected by the same disease process. In this situation, the main clinical problem is to determine whether the lesion lies within the brainstem or outside it. Lesions that lie on the surface of the brainstem are characterized by involvement of adjacent cranial nerves (often occurring in succession) and late and rather slight involvement of the long sensory and motor pathways and segmental structures lying within the brainstem. The opposite is true of primary lesions within the brainstem. The extramedullary lesion is more likely to cause bone erosion or enlargement of the foramens of exit of cranial nerves. The intramedullary lesion involving cranial nerves often produces a crossed sensory or motor paralysis (cranial nerve signs on one side of the body and tract signs on the opposite side).
Involvement of multiple cranial nerves outside the brainstem is frequently the result of trauma, localized infections including varicella-zoster virus, infectious and noninfectious (especially carcinomatous) causes of meningitis (Chaps. 164 and 165), granulomatous diseases such as Wegener’s granulomatosis, Behçet’s disease, vascular disorders including those associated with diabetes, enlarging aneurysms, or locally infiltrating tumors. Among the tumors, nasopharyngeal cancers, lymphomas, neurofibromas, meningiomas, chordomas, cholesteatomas, carcinomas, and sarcomas have all been observed to involve a succession of lower cranial nerves. Owing to their anatomic relationships, the multiple cranial nerve palsies form a number of distinctive syndromes, listed in Table 455-2. Sarcoidosis is the cause of some cases of multiple cranial neuropathy; tuberculosis, the Chiari malformation, platybasia, and basilar invagination of the skull are additional causes. A purely motor disorder without atrophy always raises the question of myasthenia gravis (Chap. 461). As noted above, Guillain-Barré syndrome commonly affects the facial nerves bilaterally. In the Fisher variant of the Guillain-Barré syndrome, oculomotor paresis occurs with ataxia and areflexia in the limbs (Chap. 460). Wernicke’s encephalopathy can cause a severe ophthalmoplegia combined with other brainstem signs (Chap. 330).
The cavernous sinus syndrome (Fig. 455-4) is a distinctive and frequently life-threatening disorder. It often presents as orbital or facial pain; orbital swelling and chemosis due to occlusion of the ophthalmic veins; fever; oculomotor neuropathy affecting the third, fourth, and sixth cranial nerves; and trigeminal neuropathy affecting the ophthalmic (V1) and occasionally the maxillary (V2) divisions of the trigeminal nerve. Cavernous sinus thrombosis, often secondary to infection from orbital cellulitis (frequently Staphylococcus aureus), a cutaneous source on the face, or sinusitis (especially with mucormycosis in diabetic patients), is the most frequent cause; other etiologies include aneurysm of the carotid artery, a carotid-cavernous fistula (orbital bruit may be present), meningioma, nasopharyngeal carcinoma, other tumors, or an idiopathic granulomatous disorder (Tolosa-Hunt syndrome). The two cavernous sinuses directly communicate via intercavernous channels; thus, involvement on one side may extend to become bilateral. Early diagnosis is essential, especially when due to infection, and treatment depends on the underlying etiology.
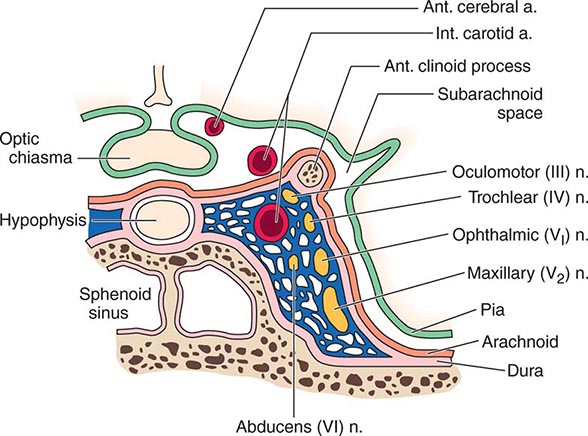
FIGURE 455-4 Anatomy of the cavernous sinus in coronal section, illustrating the location of the cranial nerves in relation to the vascular sinus, internal carotid artery (which loops anteriorly to the section), and surrounding structures.
In infectious cases, prompt administration of broad-spectrum antibiotics, drainage of any abscess cavities, and identification of the offending organism are essential. Anticoagulant therapy may benefit cases of primary thrombosis. Repair or occlusion of the carotid artery may be required for treatment of fistulas or aneurysms. The Tolosa-Hunt syndrome generally responds to glucocorticoids. A dramatic improvement in pain is usually evident within a few days; oral prednisone (60 mg daily) is usually continued for 2 weeks and then gradually tapered over a month, or longer if pain recurs. Occasionally an immunosuppressive medication, such as azathioprine or methotrexate, needs to be added to maintain an initial response to glucocorticoids.
An idiopathic form of multiple cranial nerve involvement on one or both sides of the face is occasionally seen. The syndrome consists of a subacute onset of boring facial pain, followed by paralysis of motor cranial nerves. The clinical features overlap those of the Tolosa-Hunt syndrome and appear to be due to idiopathic inflammation of the dura mater, which may be visualized by MRI. The syndrome is usually responsive to glucocorticoids.




