[level-membership-for-pediatrics-category]Chapter 128
Amenorrhea and Abnormalities of Puberty
Indications for evaluation of the adolescent pelvis usually are reports of abdominal pain, pelvic pain, or a mass. Many of the differential diagnoses for these complaints have been reviewed in Chapter 127. Other clinical symptoms resulting in imaging evaluation in female patients relate to abnormalities of development of secondary sexual characteristics of puberty. These changes may be seen earlier than normal (i.e., precocious puberty), may be delayed (i.e., delayed puberty), or may fail to develop (e.g., hypogonadism and sexual infantilism) in adolescents. The other key reason for adolescent gynecologic evaluation is amenorrhea (lack of menses), whether primary or secondary. Conditions causing pubertal delay also may cause primary or secondary amenorrhea.1,2
Puberty
Puberty among girls is the stage of development in between childhood and adulthood when activation of the hypothalamic-pituitary-ovarian-uterine axis produces maturation of the gonads, resulting in an increased production of sex hormones, development of secondary sex characteristics, a growth spurt, and development of reproductive capability. The earliest signs of puberty among girls are breast development (usually occurring between ages 8 and 13 years) and pubic hair growth (at age 8 to 14 years). These signs are followed by a growth spurt (at age 9.5 to 14.5 years), axillary hair development, and menarche (at age 10 to 16 years). Puberty is usually completed within about 4 years.1,3,4
Puberty among boys begins between 9 and 14 years of age and is completed in 3.5 to 4 years. It begins with testicular enlargement (usually occurring between age 9 and 13.5 years), followed by the appearance of pubic hair (at age 10 to 15 years), enlargement of the penis (at age 11 to 12.5 years), and development of axillary and facial hair, as well as a growth spurt (at age 10.5 to 16 years).1,3
Physiologic Changes at Puberty and the Normal Ovulatory Menstrual Cycle
Ordinarily, until the age of at least 8 years, an unknown “central restraining mechanism” prevents the pulsatile release of gonadotropin-releasing hormone (GnRH) from the arcuate nucleus of the hypothalamus. Pulsatile release of GnRH appears necessary for ovulation and development of the corpus luteum. In early puberty the pulsatile GnRH release is maximal only at night, but with time the typical adult pattern of continuous pulsatile GnRH secretion develops.1,4,5
With the earliest activation of GnRH, most girls undergo ovarian folliculogenesis without ovulation. Unopposed estrogen production leads to progressive uterine growth and endometrial proliferation. Breast budding, physiologic leukorrhea, and accelerated linear growth occur. As the hypothalamic-pituitary-ovarian-uterine axis matures over approximately a 2-year span, cycles with subnormal progesterone production and shortened intermenstrual intervals are replaced by normal corpus luteum function and fertile cycles.1,4,5
The typical ovulatory cycle has a 24- to 35-day intermenstrual interval and usually a premenstrual molimina. Longer intervals often are associated with anovulation. Improved nutrition and living conditions are thought to be responsible for the gradual decrease in mean menarchal age during the past century. In North America, the mean menarchal age currently is 12.4 years with a range of 9 to 17 years. Menarche usually occurs 2 to 5 years after breast bud development.1,5
Premature Thelarche and Adrenarche
Overview and Clinical Presentation: Premature thelarche and adrenarche are relatively common, self-limited variants of normal pubertal development in girls. Premature thelarche refers to premature breast development without other signs of precocious sexual maturation in girls younger than 8 years of age. It usually is seen between 1 and 4 years of age. A third of the cases resolve spontaneously. Premature adrenarche refers to the appearance of pubic and axillary hair without other signs of precocious sexual maturity. In both premature thelarche and premature adrenarche, bone age and patient height are normal to only slightly increased. The cause of premature thelarche or adrenarche is not certain. The levels of circulating sex hormones usually are normal. Increased end-organ sensitivity to normal levels of estrogen or androgen has been suggested as a possible cause.1,6,7
In boys, premature appearance of pubic and axillary hair without other signs or only minor signs of precocious puberty is a relatively common variant of normal development that may be due to an increase in circulating androgens from premature maturation of the adrenal glands of unknown cause. No penile enlargement is present, presumably because the levels of circulating androgens are not sufficiently elevated. The bone age and growth rate are slightly increased. The remainder of pubertal development occurs at a normal age.1,7
Precocious Puberty
Precocious puberty refers to the appearance of secondary sex characteristics before 8 years of age in girls and before 9 years of age in boys. Precocious puberty is divided into two main types: (1) complete, central, gonadotropin-dependent, or true precocious puberty and (2) incomplete, peripheral, gonadotropin-independent, pseudoprecocious puberty, or precocious pseudopuberty. Whereas the complete form is characteristically isosexual, with development of secondary sex characteristics that are appropriate for the patient’s gender, the incomplete form may be either isosexual or heterosexual. Incomplete heterosexual precocious puberty is manifested by signs of virilization in girls and by gynecomastia or other signs of feminization in boys.1,6,7
Complete or Central Isosexual Precocious Puberty
Overview and Pathophysiology: Complete or central isosexual precocious puberty results from premature activation of the hypothalamic-pituitary-gonadal complex with increased production of gonadotropic and sex hormones and an early onset of ovulation or spermatogenesis. Complete precocious puberty may be idiopathic or secondary to organic central nervous system (CNS) lesions.1,8,9
Etiology and Imaging: The cause of precocious puberty in girls is idiopathic in at least 80% of cases. About 20% of affected girls have a hypothalamic or pituitary lesion. Fewer than 10% of cases of true precocious puberty in boys have an idiopathic cause. A familial tendency to idiopathic early pubertal development is observed in some cases (i.e., constitutional or genetic precocious puberty).1,8,9
Possible causes of precocious puberty in either sex include intracranial tumors or cysts, hydrocephalus, sequelae of intracranial inflammatory processes or trauma, and other intracranial lesions that may activate the hypothalamus by pressure or invasion. Of the CNS neoplasms that may cause true precocious puberty, hamartoma of the tuber cinereum (Fig. 128-1) is the most common. This usually small CNS tumor is more common in boys than in girls, is generally benign, and is nonprogressive. The hypothalamic hamartoma secretes GnRH, and the resultant symptoms usually cannot be corrected surgically. The onset of puberty in patients with this lesion is usually at a younger age (2 years) than in patients with idiopathic precocious puberty. Other CNS neoplasms that may cause true precocious puberty in either sex usually are located in or near the hypothalamus and include hypothalamic or optic gliomas, astrocytoma, ependymoma, dysgerminoma, and prolactinoma. Suprasellar dysgerminoma (ectopic pinealoma) in boys also may cause incomplete precocious puberty through tumor secretion of human chorionic gonadotropin (hCG).1,9,10
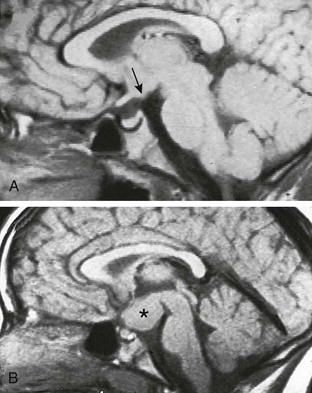
Figure 128-1 Precocious puberty caused by hamartoma of the tuber cinereum.
A and B, On midline sagittal T1-weighted magnetic resonance images of the brain, an arrow points to a small hamartoma in the brain of an 8-year-old and an asterisk indicates an unusually large hamartoma in a 7-year-old, both of whom had a long-standing history of precocious puberty.
True precocious puberty may be observed in some children with long-standing untreated hypothyroidism. The increased production of gonadotropic hormones and prolactin seen in these patients may be due to a hormonal overlap in the pituitary response to thyroid deficiency. Affected patients show little, if any, development of secondary sexual characteristics, especially pubic hair. All of these changes regress with treatment of the hypothyroidism.1,7
Incomplete (Pseudosexual) Precocious Puberty in Girls
Overview: Pseudosexual (or incomplete) precocious puberty in girls usually presents before 5 years of age. Excess circulating estrogens or related substances develop independent of stimulus by the hypothalamus and pituitary gland. These estrogens usually are produced by the ovaries or adrenal glands or may be from an exogenous source, such as food, parenteral or oral medications, or other substances.1,6,11
Etiology and Imaging: Gonadotropin levels are low, and the gonads remain immature. The most common ovarian source is an autonomous estrogen-secreting follicular (granulosa-thecal) cyst (Fig. 128-2). Other causes are estrogen-producing ovarian neoplasms, particularly granulosa cell (or granulosa-theca cell) tumors (Fig. 128-3) and rare estrogen-secreting adrenal neoplasms (adenomas or carcinomas).1,6,11
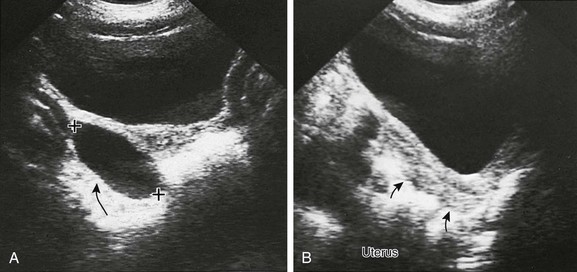
Figure 128-2 Precocious puberty due to an autonomous estrogen-secreting ovarian cyst.
A, A parasagittal sonogram of the right adnexa (arrow) of a 3-year-old with precocious puberty shows a large cyst (cursors). This cyst was proved to autonomously secrete estrogen. The left adnexa was normal. B, A midline longitudinal sonogram shows that the child’s uterus (arrows) is longer than normal for her age. It is not the tubular shape typical of childhood but appears almost pear shaped, with an echogenic central endometrial cavity indicating estrogenization.
McCune-Albright syndrome consists of polyostotic fibrous dysplasia, cutaneous café au lait spots, and precocious puberty. The syndrome is found predominantly in females. Autonomously functioning ovarian cysts are seen in some cases. In others, no anatomic cause is noted. McCune-Albright syndrome occasionally presents as complete or true precocious puberty. It has been suggested that the initially partial or incomplete precocious puberty in these patients becomes complete (gonadotropin dependent) as the result of an early maturation of the hypothalamic-pituitary-gonadal complex, which is caused by a long-standing exposure to estrogen.1,6
Virilizing Disorders (Heterosexual Precocious Puberty)
Overview and Clinical Presentation: Testosterone is produced in normal females by the adrenal gland (25%), by the ovary (25%), and by peripheral conversion of D4-androstenedione (50%), with only 1% typically free and biologically active. A form of pseudosexual precocity may occur in girls when androgen levels are excessive or end organs become excessively sensitive to normal amounts of circulating androgens. Either case may lead to heterosexual pseudoprecocious puberty as the girls undergo virilization and masculine secondary sexual characteristics develop. Clinical findings include increases in body and facial hair (hirsutism), acne, deepening of the voice, clitoromegaly, increased muscle mass, and temporal balding. Menstrual abnormalities are common among affected adolescents.1,11
Congenital Adrenal Hyperplasia
Overview and Imaging: Congenital adrenal hyperplasia is the most common form of hyperandrogenism. The increased production of androgenic substance is due in most cases to a deficiency of 21-hydroxylase and far less commonly to deficiencies in 11β-hydroxylase, 3β-hydroxysteroid dehydrogenase, or other enzymes involved in cortisol and/or aldosterone synthesis. Imaging of the adrenal gland will show enlargement (Fig. 128-4) that is usually bilateral and diffuse. The normal adrenal shape is maintained.1,12
Other Causes of Virilization in Girls
Overview, Etiology, and Pathophysiology: Adrenal adenomas or adrenal carcinomas may produce increased levels of androgens and virilization. Signs of Cushing syndrome may be seen as a result of associated tumor secretion of glucocorticoid hormones.1,12
Ovarian neoplasms may secrete androgens and cause virilization. These neoplasms include Sertoli-Leydig cell tumors, thecomas (luteoma—a virilizing dysgerminoma that contains theca cells), or gonadoblastomas. Exogenous exposure to androgens or androgen-like substances may cause virilization at all ages. Again, virilization at puberty may develop in some girls with rare anomalies of sex differentiation, such as 46,XY gonadal dysgenesis, even though they are phenotypically normal females at birth.1,13
Idiopathic hirsutism and polycystic ovarian disease also are considered to be among the virilizing disorders of females. Idiopathic hirsutism consists of an increase in body and facial hair (i.e., hirsutism or hypertrichosis) that occurs as the sole or predominant abnormality and is a relatively common problem in otherwise normal pubertal and postpubertal girls. Idiopathic hirsutism may be precipitated by any of the causes of virilization. It may be seen in families or as a result of polycystic ovary syndrome (PCOS) and the increased androgen production by the many small follicular cysts (see Chapter 127). The cause is usually idiopathic and believed to be a result of an altered response of the end organ (hair follicle) to normal levels of circulating androgens.1,14–16
Incomplete Isosexual (Pseudoprecocious) Puberty in Boys
Overview, Etiology, and Pathophysiology: Isosexual pseudoprecocity of sexual development in boys is due to an increase in circulating androgen or androgen-like substances either from production by adrenal glands or testes or from an exogenous source. Congenital adrenal hyperplasia (CAH) is the most common cause for excessive androgen production by the adrenal gland of either sex. Affected males are born with normal external genitalia, but if the CAH is untreated, signs of sexual precocity soon develop. Isosexual pseudoprecocious puberty in males, sometimes associated with signs of glucocorticoid excess, also can occur because of androgen-secreting neoplasms of the adrenal cortex (e.g., adenocarcinoma or benign adenoma). A rare cause of isosexual precocity in boys is an androgen-secreting Leydig cell tumor of the testis (see Chapter 126). Several extrapituitary hCG-secreting tumors also can result in an incomplete form of isosexual precocious puberty by stimulating testosterone production by the Leydig cells of the testis. These tumors include some hepatomas, hepatoblastomas, some teratomas or chorioepitheliomas of the mediastinum and retroperitoneum, and suprasellar germinoma or ectopic pinealoma. A familial form of gonadotropin-independent precocious puberty in boys is caused by premature maturation and sometimes hyperplasia of the Leydig cells of the testis, with production of testosterone. Exposure to exogenous androgens or the administration of hCG for undescended testes may be further causes of incomplete virilization in males.1,7,12
Adolescent Gynecomastia and Feminizing Disorders in Boys
Overview and Etiology: Mild breast development may occur transiently in adolescent boys between 13 and 15 years of age. It is usually bilateral, it is idiopathic, and it may be familial. Usually no hormonal abnormality is found. The gynecomastia generally regresses in 2 or 3 years, but in a few cases it may persist into adult life. Pathologic causes of gynecomastia in boys may be exposure to exogenous estrogens, an estrogen-secreting neoplasm of the testis or adrenal cortex, a prolactin-secreting neoplasm of the pituitary gland, Klinefelter syndrome, congenital bilateral anorchia, acquired testicular failure, and other conditions with some biochemical defect in testosterone production or androgen end-organ receptor.1,17
Clinical and Laboratory Evaluation of Precocious Puberty
Overview and Clinical Presentation: Physical examination can help determine the correct diagnosis. Physical examination should denote the type and degree of pubertal development as well as the size, shape, and firmness of the testes. In complete precocious puberty, both testes are enlarged; in partial sexual precocity they often are of normal size. Unilateral testicular enlargement suggests a testicular neoplasm. The abdomen should be evaluated for a flank or pelvic mass. The presence of café au lait spots should suggest the diagnosis of fibrous dysplasia or neurofibromatosis. Laboratory studies may include luteinizing hormone, follicle-stimulating hormone, and estradiol levels; gonadotropin response to GnRH; thyroid studies; and a vaginal smear in girls, as well as plasma and urinary testosterone levels.1,7,18
Imaging Workup of Precocious Puberty
Imaging: An anteroposterior film of the left hand, including carpal bones and the distal forearm, is used for bone age determination. Bone age often is advanced in patients with true precocious puberty and in patients with androgenic stimulation. The skeletal length often is increased early in patients with advanced bone age. However, if the precocious puberty is not treated, premature fusion of the epiphyses may develop and result in a decrease in the patient’s potential height.1,7
Abdominal ultrasound, with emphasis on the adrenals and the ovaries, has become the primary radiologic study in the initial evaluation of patients with precocious puberty. In girls with true precocious puberty, a pelvic ultrasound may show some bilateral enlargement of the ovaries and prominence of the uterus, and in rare cases a large estrogen-secreting ovarian cyst that has resulted in stimulation of the hypothalamic-pituitary complex. Small and multiple ovarian cysts/follicles may be seen in girls with true precocious puberty because of high gonadotropin levels. These cystic ovaries may look and be similar to those seen normally throughout childhood. Pseudoprecocity caused by the autonomous estrogen secretion of an ovarian cyst or tumor will be denoted by asymmetry of the ovaries of the child, with the autonomous mass being found in the larger ovary (see Fig. 128-2). The finding in prepubertal girls of an increased uterine size and a well-defined central endometrial echo indicates an increase in circulating estrogen from any cause. A skeletal survey is indicated when fibrous dysplasia is suspected.1,7,19
Patients with isosexual precocious puberty that is suspected to be of the complete or central type should have magnetic resonance imaging (MRI) of the brain with special attention to the tuber cinereum. Skull radiographs are of limited diagnostic value but may show intracranial calcifications, enlargement of the sella turcica, signs of increased intracranial pressure, or skull changes of fibrous dysplasia.1,7
Delayed or Absent Pubertal Development
Puberty is considered to be delayed and should be investigated in girls when secondary sex characteristics fail to appear by 13 years of age, which is considered two standard deviations beyond the norm. Evaluation for pubertal delays in boys is suggested when pubic and axillary hair or other external secondary characteristics, particularly enlargement of the testes and penis, fail to appear by age 14 years. Delayed puberty or failure of puberty to develop may be idiopathic (constitutional) or due to chronic systemic disorders, disorders of the hypothalamic-pituitary complex causing decreased gonadotropin secretion (i.e., secondary or hypogonadotropic hypogonadism), or primary disorders of the gonads with a secondary elevation of gonadotropic hormone secretion (i.e., primary or hypergonadotropic hypogonadism).1,20,21
Idiopathic (Constitutional) Pubertal Delay
Overview and Clinical Presentation: In some otherwise normal children, the onset of puberty is delayed for up to several years. When puberty eventually starts, it usually continues to completely normal secondary sexual development. These occurrences underline the fact that there may be constitutional or genetic causes for delay in the maturation of the hypothalamic-pituitary complex. Often, such children may experience a delay in bone age and height development. The ability to differentiate between constitutional pubertal delays in otherwise normal children and true disorders of the hypothalamic-pituitary axis may not be simple.1,21
Delayed Puberty in Chronic Systemic Disorders
Overview and Etiology: Delayed puberty may occur on a physiologic basis in patients with chronic diseases. Patients with other long-term debilitating processes, as well as anorexia nervosa, may have delays in pubertal development. Such delays also may be noted in children or adolescents who undergo prolonged and vigorous physical exertion, such as long-distance running or ballet. Puberty may develop or proceed normally in some of these patients after improvement or removal of the precipitating cause. However, any halt in the pubertal development of a patient is a cause for concern and an immediate endocrinologic workup.1,22,23
Hypogonadism Caused By Hypothalamic-Pituitary Disorders (Hypogonadotropic Hypogonadism)
Overview and Clinical Presentation: Several disorders of the hypothalamic-pituitary complex result in decreased gonadotropin production and, secondarily, a decrease in gonadal sex steroids. Impaired or absent gonadal function causes an absence of pubertal development. In girls, the uterus remains infantile, menses does not occur, and breast budding and other secondary sexual characteristics do not develop. In boys, the penis and scrotum are infantile and the testes remain immature and smaller than expected for age. Pubic and axillary hair is scanty or absent, the voice remains high pitched, and increased fat deposition often occurs, particularly at the hips, pelvis, abdomen, and breast. Delayed closure of the epiphyses with elongation of the limbs is observed unless an associated growth hormone deficiency is present.1,21,22
Etiology and Pathophysiology: Causative abnormalities include intracranial tumors such as craniopharyngioma, hypothalamic and optic gliomas, dysgerminoma, and other tumors of the pituitary gland and hypothalamus or adjacent areas. Other manifestations of pituitary insufficiency, such as diabetes insipidus and short stature, may be present. Primary or secondary hypogonadotropic hypogonadism also may be seen in patients with Langerhans cell histiocytosis and in patients with certain congenital midline defects of the face, the base of the skull, and the CNS (e.g., septo-optic dysplasia or holoprosencephaly), which may be associated with developmental anomalies of the hypothalamus and pituitary gland, resulting in hypogonadism.1,21
Functional causes of hypogonadism include idiopathic hypopituitarism, characterized by short stature resulting from growth hormone deficiency, and sometimes other findings associated with deficiencies of other pituitary hormones. Cases of isolated gonadotropic deficiency, sporadic or familial, may occur in association with anosmia or hyposmia and other abnormalities that characterize Kallmann syndrome. Isolated luteinizing hormone deficiency in males, which sometimes is familial, results in failure of pubertal development; it usually is associated with gynecomastia but with the preservation of spermatogenesis (i.e., fertile eunuch syndrome). Hypogonadism, which probably also is related to abnormalities of hypothalamic function, can be observed in Prader-Willi and Laurence-Moon-Biedl syndromes.1,21
Hypogonadism Caused By Gonadal Lesions (Hypergonadotropic Hypogonadism)
Overview, Etiology, and Pathophysiology: Certain congenital or acquired lesions of the gonads may result in hypogonadism and failure of pubertal development. Gonadal sex steroids are decreased, and as a result an increased secretion of gonadotropins by the pituitary occurs. Just as in hypogonadotropic hypogonadism, pubertal development is absent in girls or boys. The key gonadal lesions in girls include Turner syndrome, XX gonadal dysgenesis, XY gonadal dysgenesis, and a gonadal dysgenesis with galactosemia and immune oophoritis (often associated with Hashimoto thyroiditis, hypoparathyroidism, adrenal insufficiency, pernicious anemia, chronic active hepatitis, and candidiasis). Hypergonadotropic hypogonadism also can occur among patients with normal karyotypes who experience secondary ovarian failure either by infarction, as with cases of bilateral ovarian torsion, surgical removal of both ovaries, radiation therapy to the pelvis (usually of 800 cGy or greater), chemotherapy, or autoimmune oophoritis.1,21,24
Gonadal lesions in boys leading to hypogonadism include congenital bilateral anorchia with otherwise normal external genitalia, which is caused by resorption of the testes after the thirteenth week of gestation (vanishing testes and functional prepubertal castrate syndromes) and acquired testicular atrophy resulting from bilateral testicular torsion, surgical injury during bilateral orchiopexy, radiation, or other causes. Absence of puberty also is observed in some genetic males born with an entirely female phenotype, including those with XY gonadal dysgenesis, and some forms of male pseudohermaphroditism with impaired biosynthesis of active sex steroids as a result of an inherited enzymatic deficiency.1,25,26
Diagnostic and Historical Considerations
Overview, Clinical Presentation, and Imaging: Important information may be obtained from the patient’s family history, medical history, associated medical disorders, and physical examination. At the physical examination, special emphasis should be placed on pubertal staging (Tanner classification), assessment of general growth and maturation, detection of signs and symptoms of CNS disease, and systemic diseases or syndromes. Visual fields testing, gynecologic evaluation, and chromosomal analysis should be carried out if indicated. The hormonal studies include measurements of serum testosterone, estradiol, and gonadotropins; gonadotropic response to GnRH stimulation; and testosterone response to hCG stimulation. Measurements of circulating prolactin and growth hormone, as well as thyroid function tests, may be undertaken. Radiologic procedures include bone age determination, brain MRI, pelvic ultrasound to evaluate the size of the ovaries and uterus, and other studies as indicated.1,21
Analysis of Patients with Amenorrhea
Overview and Etiology: Causes of pubertal delay in girls may be similar to causes of primary or secondary amenorrhea. Primary amenorrhea is defined as a lack of menses by age 16 years. Secondary amenorrhea is defined as a cessation of menses at any point in time after menarche and before menopause.1,27,28
Primary amenorrhea has many causes and involves several organ systems (Box 128-1). It may be seen in adolescents with normal pubertal development, as well as those with delayed sexual development, delayed menarche with some pubertal development, and delayed menarche plus virilization. Many of its causes have already been reviewed, including hypogonadotropic hypogonadism, hypergonadotropic hypogonadism, pseudohermaphroditism, female adolescents with virilization, PCOS, genital obstruction (e.g., hematometra/hematometrocolpos), and uterine aplasia or hypoplasia. Secondary amenorrhea often is due physiologically to pregnancy and pathologically to PCOS.1,27,28
 What the Clinician Needs to Know
What the Clinician Needs to Know
• Bone age delayed, normal, or advanced
• Skeletal changes of polyostotic fibrous dysplasia (McCune-Albright syndrome)
• Presence of an intracranial lesion or congenital malformation on MRI/computed tomography that may activate the hypothalamus by pressure or invasion in the setting of central isosexual precocious puberty or result in decreased gonadotropin production in hypogonadotropic hypogonadism
• Presence of an ovarian lesion (cyst or neoplasm) on ultrasound in young girls with pseudosexual precocious puberty
• Presence of a testicular mass on ultrasound in pseudoprecocious puberty in boys
• Presence of enlarged adrenal glands (CAH) or adrenal mass in heterosexual precocious puberty
Argyropoulos, M, Kiortsis, D. MRI of the hypothalamic-pituitary axis in children. Pediatr Radiol. 2005;35:1045.
Cohen, HL, Bober, S, Bow, S. Imaging the pediatric pelvis: the normal and abnormal genital system and simulators of its diseases. Urol Radiol. 1992;14:273.
Garel, L, Dubois, J, Grignon, A, et al. US of the pediatric female pelvis: a clinical perspective. Radiographics. 2001;21(6):1393–1407.
Stranzinger, E, Strouse, PJ. Ultrasound of the pediatric female pelvis. Semin Ultrasound CT MR. 2008;29(2):98–113.
Warren, MP, Goodman, LR. Exercise-induced endocrine pathologies. J Endocrinol Invest. 2003;26(9):873–878.
References
1. Cohen, HL. Abnormalities of puberty and amenorrhea. In Slovis T, ed.: Caffey’s pediatric diagnostic imaging, 11th ed, Philadelphia: Elsevier, 2008.
2. Cohen, HL, Bober, S, Bow, S. Imaging the pediatric pelvis: the normal and abnormal genital system and simulators of its diseases. Urol Radiol. 1992;14:273.
3. The physiology of puberty. In Emans S, Goldstein D, eds.: Pediatric and adolescent gynecology, 3rd ed, Boston: Little Brown, 1990.
4. Lee, PA. Neuroendocrinology of puberty. Semin Reprod Endocrinol. 1988;6:13.
5. Lee, PA. Physiology of puberty. In: Becker K, Bilezkian J, Brenner W, Hung W, eds. Principles and practice of endocrinology and metabolism. Philadelphia: JB Lippincott, 1990.
6. Schwartz, ID, Root, AW. Puberty in girls: early, incomplete or precocious? Contemp Pediatr. 1990;7:147.
7. Wheeler, MD, Styne, DM. Diagnosis and management of precocious puberty. Pediatr Clin North Am. 1990;37:1255.
8. Bajpai, A, Sharma, J, Kabra, M, et al. Precocious puberty: clinical and endocrine profile and factors indicating neurogenic precocity in Indian children. J Pediatr Endocrinol Metab. 2002;15:1173.
9. Argyropoulos, M, Kiortsis, D. MRI of the hypothalamic-pituitary axis in children. Pediatr Radiol. 2005;35:1045.
10. Starceski, PJ. Hypothalamic hamartomas and sexual precocity. Am J Dis Child. 1990;144:225.
11. Rosenfield, RL. Hyperandrogenism in peripubertal girls. Pediatr Clin North Am. 1990;37:1333.
12. Kawashima, A, Sandler, C, Fishman, E, et al. Spectrum of CT findings in nonmalignant disease of the adrenal gland. Radiographics. 1998;18:393.
13. Larsen, W, Felmar, E, Wallace, M, et al. Sertoli-Leydig cell tumor of the ovary: a rare cause of amenorrhea. Obstet Gynecol. 1992;79:831.
14. Herter, L, Magalnaes, J, Spritzer, P. Association of ovarian volume and serum LH levels in adolescent patients with menstrual disorders and/or hirsutism. Braz J Med Biol Res. 1993;26:1041.
15. Battaglia, C, Artini, P, D’Ambrogio, G, et al. The role of color Doppler imaging in the diagnosis of polycystic ovary syndrome. Am J Obstet Gynecol. 1995;172:108.
16. Takahashi, K, Okada, M, Ozaki, T, et al. Transvaginal ultrasonographic morphology in polycystic ovarian syndrome. Gynecol Obstet Invest. 1995;39:201.
17. Mahoney, CP. Adolescent gynecomastia: differential diagnosis and management. Pediatr Clin North Am. 1990;37:1389.
18. The gonads: development and function of the reproductive system. In Ganong W, ed.: Review of medical physiology, 17th ed, Norwalk, CT: Appleton & Lange, 1995.
19. Cohen, HL, Eisenberg, P, Mandel, F, et al. Ovarian cysts are common in premenarchal girls: a sonographic study of 101 children 2–12 years old. AJR Am J Roentgenol. 1992;159:89.
20. Currarino, G, Wood, B, Majd, M. Abnormalities of the genital tract. In Silverman F, Kuhn J, eds.: Caffey’s pediatric x-ray diagnosis, 9th ed, St Louis: Mosby–Year Book, 1993.
21. Rosenfeld, RL. Clinical review 6: diagnosis and management of delayed puberty. J Clin Endocrinol Metab. 1990;70:559.
22. Emans, S. Amenorrhea in the adolescent. In Emans S, Goldstein D, eds.: Pediatric and adolescent gynecology, 5th ed, Philadelphia: Lippincott Williams and Wilkins, 2005.
23. Falsetti, L, Pasinetti, E, Mazzani, M, et al. Weight loss and menstrual cycle: clinical and endocrinological evaluation. Gynecol Endocrinol. 1992;6:49.
24. Tarani, L, Lampariello, S, Raguso, G, et al. Pregnancy in patients with Turner’s syndrome: six new cases and review of literature. Gynecol Endocrinol. 1998;12:83.
25. Shah, R, Woolley, M, Costin, G. Testicular feminization syndrome: the androgen insensitivity syndrome. J Pediatr Surg. 1992;27:757.
26. Lee, PA, O’Dea, L. Primary and secondary testicular insufficiency. Pediatr Clin North Am. 1990;37:1359.
27. Reid, R. Amenorrhea. In: Copeland L, ed. Textbook of gynecology. Philadelphia: WB Saunders, 1993.
28. Nidecker, A, Cohen, HL, Zinn, HL. Amenorrhea in the adolescent or young adult. In Bluth E, Benson C, Ralls P, et al, eds.: Ultrasound: a practical approach to clinical problems, 2nd ed, New York: Thieme, 2008.
[/level-membership-for-pediatrics-category][not-level-membership-for-pediatrics-category]Chapter 128
Amenorrhea and Abnormalities of Puberty
Indications for evaluation of the adolescent pelvis usually are reports of abdominal pain, pelvic pain, or a mass. Many of the differential diagnoses for these complaints have been reviewed in Chapter 127. Other clinical symptoms resulting in imaging evaluation in female patients relate to abnormalities of development of secondary sexual characteristics of puberty. These changes may be seen earlier than normal (i.e., precocious puberty), may be delayed (i.e., delayed puberty), or may fail to develop (e.g., hypogonadism and sexual infantilism) in adolescents. The other key reason for adolescent gynecologic evaluation is amenorrhea (lack of menses), whether primary or secondary. Conditions causing pubertal delay also may cause primary or secondary amenorrhea.1,2
Puberty
Puberty among girls is the stage of development in between childhood and adulthood when activation of the hypothalamic-pituitary-ovarian-uterine axis produces maturation of the gonads, resulting in an increased production of sex hormones, development of secondary sex characteristics, a growth spurt, and development of reproductive capability. The earliest signs of puberty among girls are breast development (usually occurring between ages 8 and 13 years) and pubic hair growth (at age 8 to 14 years). These signs are followed by a growth spurt (at age 9.5 to 14.5 years), axillary hair development, and menarche (at age 10 to 16 years). Puberty is usually completed within about 4 years.1,3,4
Puberty among boys begins between 9 and 14 years of age and is completed in 3.5 to 4 years. It begins with testicular enlargement (usually occurring between age 9 and 13.5 years), followed by the appearance of pubic hair (at age 10 to 15 years), enlargement of the penis (at age 11 to 12.5 years), and development of axillary and facial hair, as well as a growth spurt (at age 10.5 to 16 years).1,3
Physiologic Changes at Puberty and the Normal Ovulatory Menstrual Cycle
Ordinarily, until the age of at least 8 years, an unknown “central restraining mechanism” prevents the pulsatile release of gonadotropin-releasing hormone (GnRH) from the arcuate nucleus of the hypothalamus. Pulsatile release of GnRH appears necessary for ovulation and development of the corpus luteum. In early puberty the pulsatile GnRH release is maximal only at night, but with time the typical adult pattern of continuous pulsatile GnRH secretion develops.1,4,5
With the earliest activation of GnRH, most girls undergo ovarian folliculogenesis without ovulation. Unopposed estrogen production leads to progressive uterine growth and endometrial proliferation. Breast budding, physiologic leukorrhea, and accelerated linear growth occur. As the hypothalamic-pituitary-ovarian-uterine axis matures over approximately a 2-year span, cycles with subnormal progesterone production and shortened intermenstrual intervals are replaced by normal corpus luteum function and fertile cycles.1,4,5
The typical ovulatory cycle has a 24- to 35-day intermenstrual interval and usually a premenstrual molimina. Longer intervals often are associated with anovulation. Improved nutrition and living conditions are thought to be responsible for the gradual decrease in mean menarchal age during the past century. In North America, the mean menarchal age currently is 12.4 years with a range of 9 to 17 years. Menarche usually occurs 2 to 5 years after breast bud development.1,5
Premature Thelarche and Adrenarche
Overview and Clinical Presentation: Premature thelarche and adrenarche are relatively common, self-limited variants of normal pubertal development in girls. Premature thelarche refers to premature breast development without other signs of precocious sexual maturation in girls younger than 8 years of age. It usually is seen between 1 and 4 years of age. A third of the cases resolve spontaneously. Premature adrenarche refers to the appearance of pubic and axillary hair without other signs of precocious sexual maturity. In both premature thelarche and premature adrenarche, bone age and patient height are normal to only slightly increased. The cause of premature thelarche or adrenarche is not certain. The levels of circulating sex hormones usually are normal. Increased end-organ sensitivity to normal levels of estrogen or androgen has been suggested as a possible cause.1,6,7
In boys, premature appearance of pubic and axillary hair without other signs or only minor signs of precocious puberty is a relatively common variant of normal development that may be due to an increase in circulating androgens from premature maturation of the adrenal glands of unknown cause. No penile enlargement is present, presumably because the levels of circulating androgens are not sufficiently elevated. The bone age and growth rate are slightly increased. The remainder of pubertal development occurs at a normal age.1,7
Precocious Puberty
Precocious puberty refers to the appearance of secondary sex characteristics before 8 years of age in girls and before 9 years of age in boys. Precocious puberty is divided into two main types: (1) complete, central, gonadotropin-dependent, or true precocious puberty and (2) incomplete, peripheral, gonadotropin-independent, pseudoprecocious puberty, or precocious pseudopuberty. Whereas the complete form is characteristically isosexual, with development of secondary sex characteristics that are appropriate for the patient’s gender, the incomplete form may be either isosexual or heterosexual. Incomplete heterosexual precocious puberty is manifested by signs of virilization in girls and by gynecomastia or other signs of feminization in boys.1,6,7
Complete or Central Isosexual Precocious Puberty
Overview and Pathophysiology: Complete or central isosexual precocious puberty results from premature activation of the hypothalamic-pituitary-gonadal complex with increased production of gonadotropic and sex hormones and an early onset of ovulation or spermatogenesis. Complete precocious puberty may be idiopathic or secondary to organic central nervous system (CNS) lesions.1,8,9
Etiology and Imaging: The cause of precocious puberty in girls is idiopathic in at least 80% of cases. About 20% of affected girls have a hypothalamic or pituitary lesion. Fewer than 10% of cases of true precocious puberty in boys have an idiopathic cause. A familial tendency to idiopathic early pubertal development is observed in some cases (i.e., constitutional or genetic precocious puberty).1,8,9
Possible causes of precocious puberty in either sex include intracranial tumors or cysts, hydrocephalus, sequelae of intracranial inflammatory processes or trauma, and other intracranial lesions that may activate the hypothalamus by pressure or invasion. Of the CNS neoplasms that may cause true precocious puberty, hamartoma of the tuber cinereum (Fig. 128-1) is the most common. This usually small CNS tumor is more common in boys than in girls, is generally benign, and is nonprogressive. The hypothalamic hamartoma secretes GnRH, and the resultant symptoms usually cannot be corrected surgically. The onset of puberty in patients with this lesion is usually at a younger age (2 years) than in patients with idiopathic precocious puberty. Other CNS neoplasms that may cause true precocious puberty in either sex usually are located in or near the hypothalamus and include hypothalamic or optic gliomas, astrocytoma, ependymoma, dysgerminoma, and prolactinoma. Suprasellar dysgerminoma (ectopic pinealoma) in boys also may cause incomplete precocious puberty through tumor secretion of human chorionic gonadotropin (hCG).1,9,10
Figure 128-1 Precocious puberty caused by hamartoma of the tuber cinereum.
A and B, On midline sagittal T1-weighted magnetic resonance images of the brain, an arrow points to a small hamartoma in the brain of an 8-year-old and an asterisk indicates an unusually large hamartoma in a 7-year-old, both of whom had a long-standing history of precocious puberty.
True precocious puberty may be observed in some children with long-standing untreated hypothyroidism. The increased production of gonadotropic hormones and prolactin seen in these patients may be due to a hormonal overlap in the pituitary response to thyroid deficiency. Affected patients show little, if any, development of secondary sexual characteristics, especially pubic hair. All of these changes regress with treatment of the hypothyroidism.1,7
Incomplete (Pseudosexual) Precocious Puberty in Girls
Overview: Pseudosexual (or incomplete) precocious puberty in girls usually presents before 5 years of age. Excess circulating estrogens or related substances develop independent of stimulus by the hypothalamus and pituitary gland. These estrogens usually are produced by the ovaries or adrenal glands or may be from an exogenous source, such as food, parenteral or oral medications, or other substances.1,6,11
Etiology and Imaging: Gonadotropin levels are low, and the gonads remain immature. The most common ovarian source is an autonomous estrogen-secreting follicular (granulosa-thecal) cyst (Fig. 128-2). Other causes are estrogen-producing ovarian neoplasms, particularly granulosa cell (or granulosa-theca cell) tumors (Fig. 128-3) and rare estrogen-secreting adrenal neoplasms (adenomas or carcinomas).1,6,11
Figure 128-2 Precocious puberty due to an autonomous estrogen-secreting ovarian cyst.
A, A parasagittal sonogram of the right adnexa (arrow) of a 3-year-old with precocious puberty shows a large cyst (cursors). This cyst was proved to autonomously secrete estrogen. The left adnexa was normal. B, A midline longitudinal sonogram shows that the child’s uterus (arrows) is longer than normal for her age. It is not the tubular shape typical of childhood but appears almost pear shaped, with an echogenic central endometrial cavity indicating estrogenization.
McCune-Albright syndrome consists of polyostotic fibrous dysplasia, cutaneous café au lait spots, and precocious puberty. The syndrome is found predominantly in females. Autonomously functioning ovarian cysts are seen in some cases. In others, no anatomic cause is noted. McCune-Albright syndrome occasionally presents as complete or true precocious puberty. It has been suggested that the initially partial or incomplete precocious puberty in these patients becomes complete (gonadotropin dependent) as the result of an early maturation of the hypothalamic-pituitary-gonadal complex, which is caused by a long-standing exposure to estrogen.1,6
Virilizing Disorders (Heterosexual Precocious Puberty)
Overview and Clinical Presentation: Testosterone is produced in normal females by the adrenal gland (25%), by the ovary (25%), and by peripheral conversion of D4-androstenedione (50%), with only 1% typically free and biologically active. A form of pseudosexual precocity may occur in girls when androgen levels are excessive or end organs become excessively sensitive to normal amounts of circulating androgens. Either case may lead to heterosexual pseudoprecocious puberty as the girls undergo virilization and masculine secondary sexual characteristics develop. Clinical findings include increases in body and facial hair (hirsutism), acne, deepening of the voice, clitoromegaly, increased muscle mass, and temporal balding. Menstrual abnormalities are common among affected adolescents.1,11
Congenital Adrenal Hyperplasia
Overview and Imaging: Congenital adrenal hyperplasia is the most common form of hyperandrogenism. The increased production of androgenic substance is due in most cases to a deficiency of 21-hydroxylase and far less commonly to deficiencies in 11β-hydroxylase, 3β-hydroxysteroid dehydrogenase, or other enzymes involved in cortisol and/or aldosterone synthesis. Imaging of the adrenal gland will show enlargement (Fig. 128-4) that is usually bilateral and diffuse. The normal adrenal shape is maintained.1,12
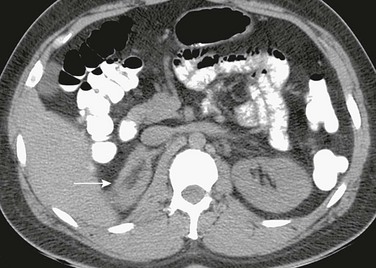
Figure 128-4 Congenital adrenal hyperplasia.
A computed tomography scan shows that the right adrenal gland (arrow
[/not-level-membership-for-pediatrics-category]
