447 |
Migraine and Other Primary Headache Disorders |
The general principles around headache as a cardinal symptom are covered elsewhere (Chap. 21); here we discuss disorders in which headache and associated features occur in the absence of any exogenous cause. The most common are migraine, tension-type headache, and the trigeminal autonomic cephalalgias, notably cluster headache; the complete list is summarized in Table 447-1.
|
PRIMARY HEADACHE DISORDERS, MODIFIED FROM INTERNATIONAL CLASSIFICATION OF HEADACHE DISORDERS-III-BETA (HEADACHE CLASSIFICATION COMMITTEE OF THE INTERNATIONAL HEADACHE SOCIETY, 2013) |

MIGRAINE
Migraine, the second most common cause of headache, and the most common headache-related, and indeed neurologic, cause of disability in the world, afflicts approximately 15% of women and 6% of men over a 1-year period. It is usually an episodic headache associated with certain features such as sensitivity to light, sound, or movement; nausea and vomiting often accompany the headache. A useful description of migraine is a recurring syndrome of headache associated with other symptoms of neurologic dysfunction in varying admixtures (Table 447-2). Migraine can often be recognized by its activators, referred to as triggers.
|
SYMPTOMS ACCOMPANYING SEVERE MIGRAINE ATTACKS IN 500 PATIENTS |
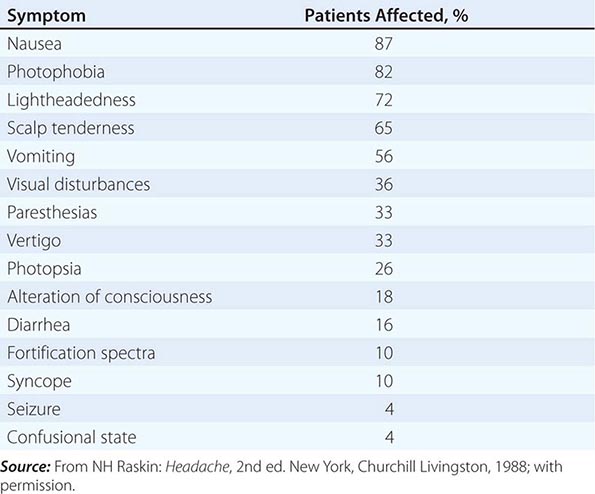
The brain of the migraineur is particularly sensitive to environmental and sensory stimuli; migraine-prone patients do not habituate easily to sensory stimuli. This sensitivity is amplified in females during the menstrual cycle. Headache can be initiated or amplified by various triggers, including glare, bright lights, sounds, or other afferent stimulation; hunger; let-down from stress; physical exertion; stormy weather or barometric pressure changes; hormonal fluctuations during menses; lack of or excess sleep; and alcohol or other chemical stimulation, such as with nitrates. Knowledge of a patient’s susceptibility to specific triggers can be useful in management strategies involving lifestyle adjustments.
Pathogenesis The sensory sensitivity that is characteristic of migraine is probably due to dysfunction of monoaminergic sensory control systems located in the brainstem and hypothalamus (Fig. 447-1).
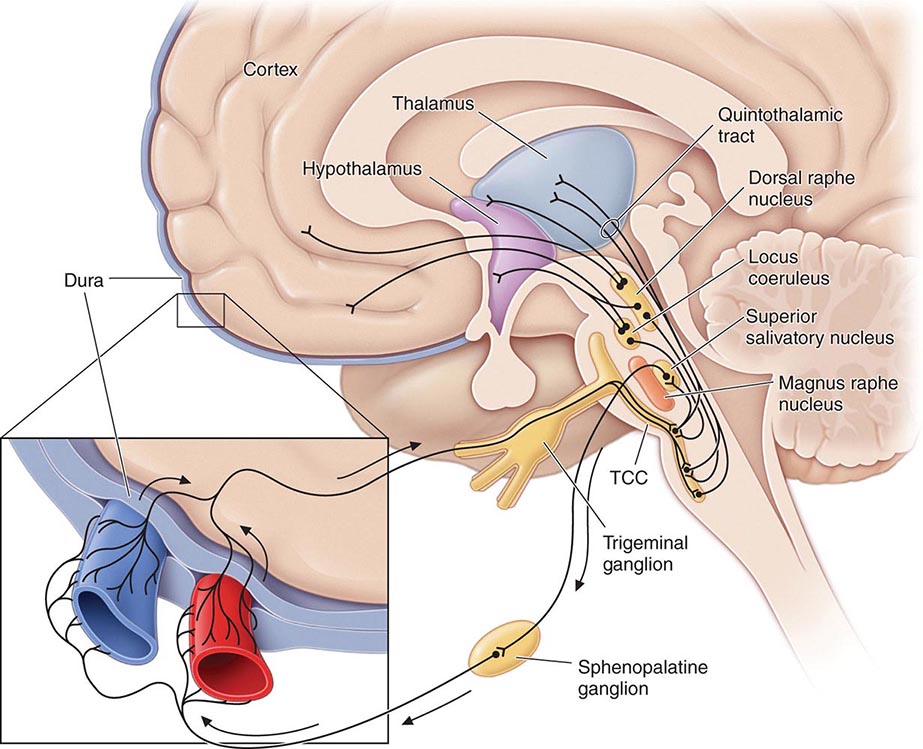
FIGURE 447-1 Brainstem pathways that modulate sensory input. The key pathway for pain in migraine is the trigeminovascular input from the meningeal vessels, which passes through the trigeminal ganglion and synapses on second-order neurons in the trigeminocervical complex (TCC). These neurons in turn project in the quintothalamic tract and, after decussating in the brainstem, synapse on neurons in the thalamus. Important modulation of the trigeminovascular nociceptive input comes from the dorsal raphe nucleus, locus coeruleus, and nucleus raphe magnus.
Activation of cells in the trigeminal nucleus results in the release of vasoactive neuropeptides, particularly calcitonin gene–related peptide (CGRP), at vascular terminations of the trigeminal nerve and within the trigeminal nucleus. CGRP receptor antagonists, gepants, have now been shown to be effective in the acute treatment of migraine, and monoclonal antibodies to CGRP have been shown effective in two early phase clinical trials. Centrally, the second-order trigeminal neurons cross the midline and project to ventrobasal and posterior nuclei of the thalamus for further processing. Additionally, there are projections to the periaqueductal gray and hypothalamus, from which reciprocal descending systems have established antinociceptive effects. Other brainstem regions likely to be involved in descending modulation of trigeminal pain include the nucleus locus coeruleus in the pons and the rostroventromedial medulla.
Pharmacologic and other data point to the involvement of the neurotransmitter 5-hydroxytryptamine (5-HT; also known as serotonin) in migraines. Approximately 60 years ago, methysergide was found to antagonize certain peripheral actions of 5-HT and was introduced as the first drug capable of preventing migraine attacks. The triptans were designed to stimulate selectively subpopulations of 5-HT receptors; at least 14 different 5-HT receptors exist in humans. The triptans are potent agonists of 5-HT1B and 5-HT1D receptors, and some are active at the 5-HT1F receptors; the latter’s exclusive agonists are called ditans. Triptans arrest nerve signaling in the nociceptive pathways of the trigeminovascular system, at least in the trigeminal nucleus caudalis and trigeminal sensory thalamus, in addition to cranial vasoconstriction, while ditans, now shown conclusively to be effective in acute migraine, act only at neural targets. An interesting range of neural targets is now being actively pursed for the acute and preventive management of migraine.
Data also support a role for dopamine in the pathophysiology of migraine. Most migraine symptoms can be induced by dopaminergic stimulation. Moreover, there is dopamine receptor hypersensitivity in migraineurs, as demonstrated by the induction of yawning, nausea, vomiting, hypotension, and other symptoms of a migraine attack by dopaminergic agonists at doses that do not affect nonmigraineurs. Dopamine receptor antagonists are effective therapeutic agents in migraine, especially when given parenterally or concurrently with other antimigraine agents. Moreover, hypothalamic activation, anterior to that seen in cluster headache, has now been shown in the premonitory phase of migraine using functional imaging, and this may hold a key to understanding some part of the role of dopamine in the disorder.
Migraine genes identified by studying families with familial hemiplegic migraine (FHM) reveal involvement of ion channels, suggesting that alterations in membrane excitability can predispose to migraine. Mutations involving the Cav2.1 (P/Q)–type voltage-gated calcium channel CACNA1A gene are now known to cause FHM 1; this mutation is responsible for about 50% of FHMs. Mutations in the Na+-K+ATPase ATP1A2 gene, designated FHM 2, are responsible for about 20% of FHMs. Mutations in the neuronal voltage-gated sodium channel SCN1A cause FHM 3. Functional neuroimaging has suggested that brainstem regions in migraine (Fig. 447-2) and the posterior hypothalamic gray matter region close to the human circadian pacemaker cells of the suprachiasmatic nucleus in cluster headache (Fig. 447-3) are good candidates for specific involvement in primary headache.
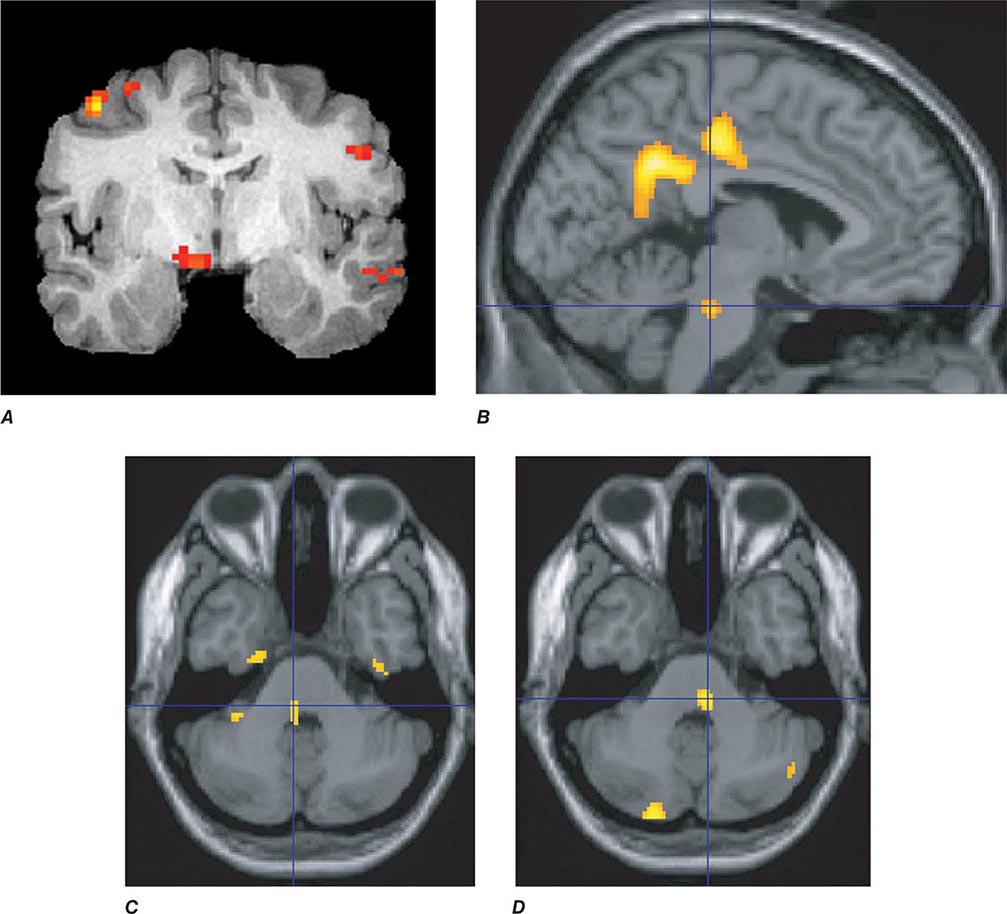
FIGURE 447-2 Positron emission tomography (PET) activation in migraine. Hypothalamic, dorsal midbrain, and dorsolateral pontine activation is seen in triggered attacks in the premonitory phase before pain, whereas in migraine attacks, dorsolateral pontine activation persists, as it does in chronic migraine (not shown). The dorsolateral pontine area, which includes the noradrenergic locus coeruleus, is fundamental to the expression of migraine. Moreover, lateralization of changes in this region of the brainstem correlates with lateralization of the head pain in hemicranial migraine; the scans shown in panels C and D are of patients with acute migraine headache on the right and left side, respectively. (Panel A from FH Maniyar et al: Brain 137:232, 2014; panel B from SK Afridi et al: Arch Neurol 2005;62:1270; Panels C and D from SK Afridi et al: Brain 128:932, 2005.)
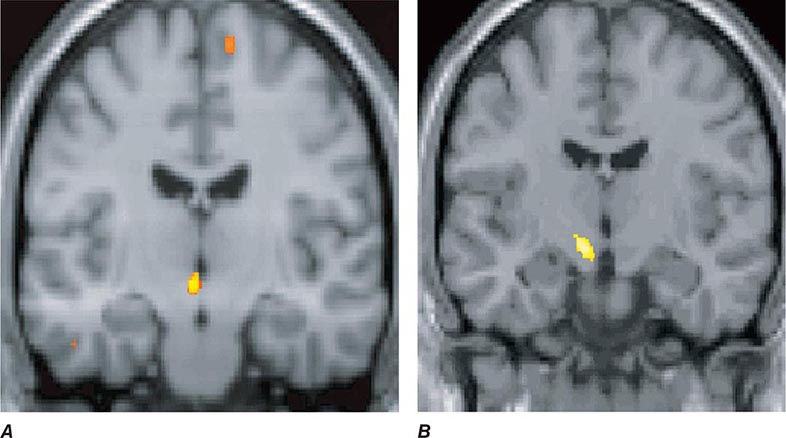
FIGURE 447-3 A. Posterior hypothalamic gray matter activation by positron emission tomography in a patient with acute cluster headache. (From A May et al: Lancet 352:275, 1998.) B. High-resolution T1-weighted magnetic resonance image obtained using voxel-based morphometry demonstrates increased gray matter activity, lateralized to the side of pain in a patient with cluster headache. (From A May et al: Nat Med 5:836, 1999.)
Diagnosis and Clinical Features Diagnostic criteria for migraine headache are listed in Table 447-3. A high index of suspicion is required to diagnose migraine: the migraine aura, consisting of visual disturbances with flashing lights or zigzag lines moving across the visual field or of other neurologic symptoms, is reported in only 20–25% of patients. A headache diary can often be helpful in making the diagnosis; this is also helpful in assessing disability and the frequency of treatment for acute attacks. Patients with episodes of migraine that occur daily or near-daily are considered to have chronic migraine (see “Chronic Daily Headache” in Chap. 21). Migraine must be differentiated from tension-type headache (discussed below), the most common primary headache syndrome seen in the population. Migraine has several forms that have been defined (Table 447-1): migraine with and without aura and chronic migraine, the latter occurring 15 days or more a month, as the most important. Migraine at its most basic level is headache with associated features, and tension-type headache is headache that is featureless. Most patients with disabling headache probably have migraine.
|
SIMPLIFIED DIAGNOSTIC CRITERIA FOR MIGRAINE |
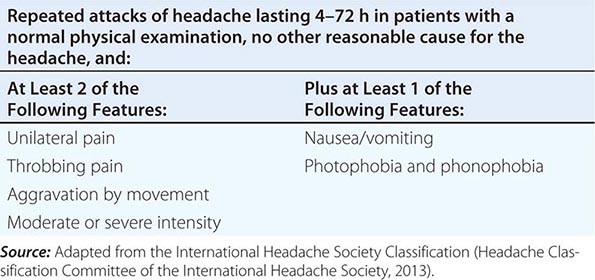
Patients with acephalgic migraine (typical aura without headache, 1.2.1.2 in Table 447-1) experience recurrent neurologic symptoms, often with nausea or vomiting, but with little or no headache. Vertigo can be prominent; it has been estimated that one-third of patients referred for vertigo or dizziness have a primary diagnosis of migraine. Migraine aura can have prominent brainstem symptoms, and the terms basilar artery and basilar-type migraine have now been replaced by migraine with brainstem aura (Table 447-1).
|
MIGRAINE HEADACHE |
Once a diagnosis of migraine has been established, it is important to assess the extent of a patient’s disease and disability. The Migraine Disability Assessment Score (MIDAS) is a well-validated, easy-to-use tool (Fig. 447-4).

FIGURE 447-4 The Migraine Disability Assessment Score (MIDAS) Questionnaire.
Patient education is an important aspect of migraine management. Information for patients is available at sites such as www.achenet.org, the website of the American Council for Headache Education (ACHE). It is helpful for patients to understand that migraine is an inherited tendency to headache; that migraine can be modified and controlled by lifestyle adjustments and medications, but it cannot be eradicated; and that, except in some occasions in women on oral estrogens or contraceptives, migraine is not associated with serious or life-threatening illnesses.
NONPHARMACOLOGIC MANAGEMENT
Migraine can often be managed to some degree by a variety of nonpharmacologic approaches. Most patients benefit by the identification and avoidance of specific headache triggers. A regulated lifestyle is helpful, including a healthy diet, regular exercise, regular sleep patterns, avoidance of excess caffeine and alcohol, and avoidance of acute changes in stress levels, being particularly wary of the let-down effect.
The measures that benefit a given individual should be used routinely because they provide a simple, cost-effective approach to migraine management. Patients with migraine do not encounter more stress than headache-free individuals; over-responsiveness to changes in stress appears to be the issue. Because the stresses of everyday living cannot be eliminated, lessening one’s response to stress by various techniques is helpful for many patients. These may include yoga, transcendental meditation, hypnosis, and conditioning techniques such as biofeedback. For most patients, this approach is, at best, an adjunct to pharmacotherapy. Nonpharmacologic measures are unlikely to prevent all migraine attacks. If these measures fail to prevent an attack, pharmacologic approaches are then needed to abort an attack.
ACUTE ATTACK THERAPIES FOR MIGRAINE
The mainstay of pharmacologic therapy is the judicious use of one or more of the many medicines that are effective in migraine (Table 447-4). The selection of the optimal regimen for a given patient depends on a number of factors, the most important of which is the severity of the attack. Mild migraine attacks can usually be managed by oral agents; the average efficacy rate is 50–70%. Severe migraine attacks may require parenteral therapy. Most drugs effective in the treatment of migraine are members of one of three major pharmacologic classes: nonsteroidal anti-inflammatory drugs, 5-HT1B/1D receptor agonists, and dopamine receptor antagonists.
|
TREATMENT OF ACUTE MIGRAINE |
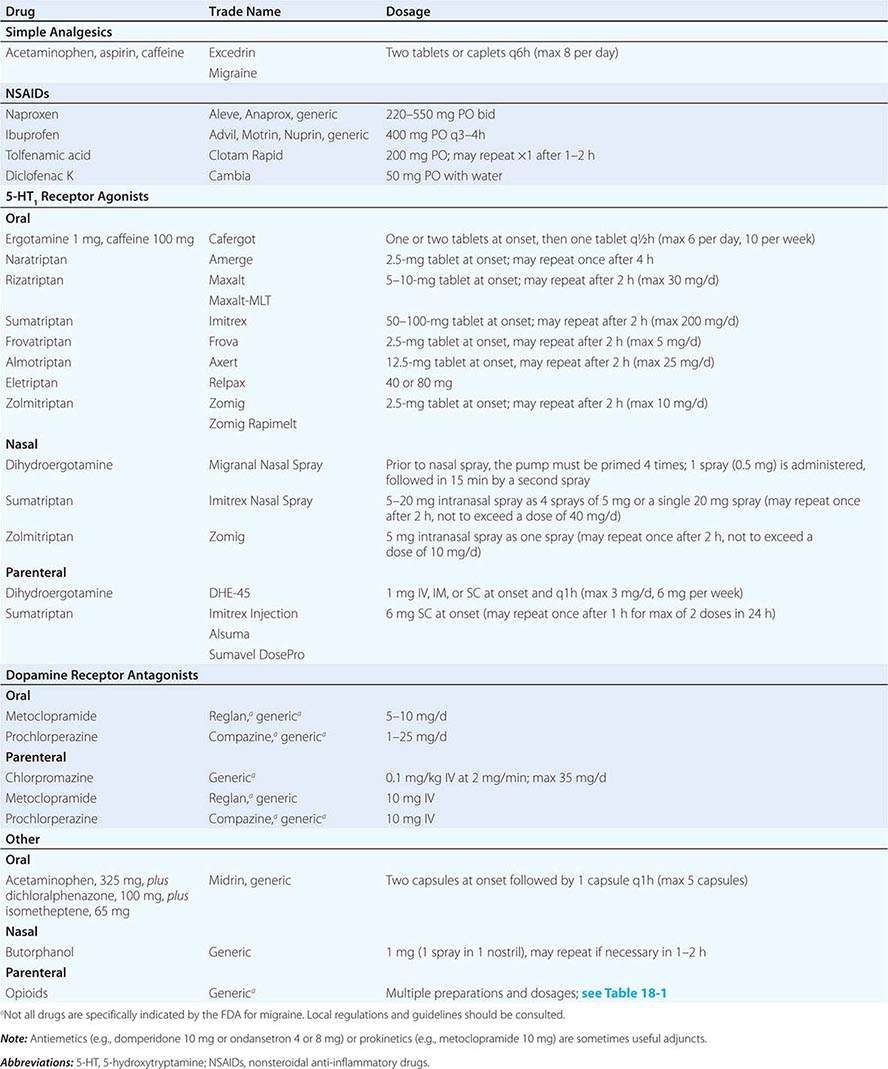
In general, an adequate dose of whichever agent is chosen should be used as soon as possible after the onset of an attack. If additional medication is required within 60 min because symptoms return or have not abated, the initial dose should be increased for subsequent attacks or a different class of drug tried as first-line treatment. Migraine therapy must be individualized; a standard approach for all patients is not possible. A therapeutic regimen may need to be constantly refined until one is identified that provides the patient with rapid, complete, and consistent relief with minimal side effects (Table 447-5).
|
CLINICAL STRATIFICATION OF ACUTE SPECIFIC MIGRAINE TREATMENTS |
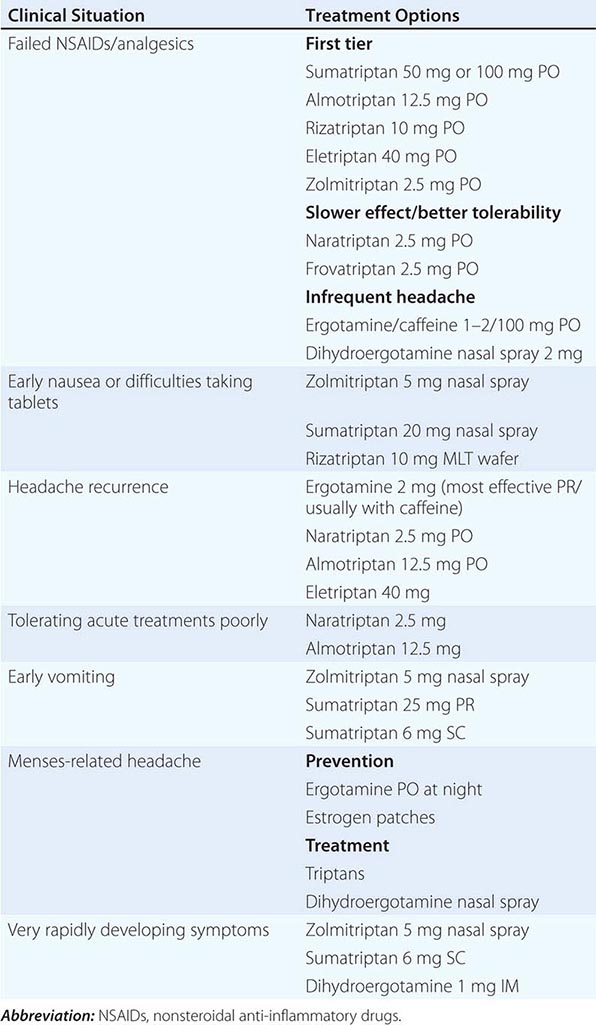
Nonsteroidal Anti-Inflammatory Drugs (NSAIDs) Both the severity and duration of a migraine attack can be reduced significantly by NSAIDs (Table 447-4). Indeed, many undiagnosed migraineurs self-treat with nonprescription NSAIDs. A general consensus is that NSAIDs are most effective when taken early in the migraine attack. However, the effectiveness of these agents in migraine is usually less than optimal in moderate or severe migraine attacks. The combination of acetaminophen, aspirin, and caffeine has been approved for use by the U.S. Food and Drug Administration (FDA) for the treatment of mild to moderate migraine. The combination of aspirin and metoclopramide has been shown to be comparable to a single dose of oral sumatriptan. Important side effects of NSAIDs include dyspepsia and gastrointestinal irritation.
5-HT1B/1D RECEPTOR AGONISTS
Oral Stimulation of 5-HT1B/1D receptors can stop an acute migraine attack. Ergotamine and dihydroergotamine are nonselective receptor agonists, whereas the triptans are selective 5-HT1B/1D receptor agonists. A variety of triptans, 5-HT1B/1D receptor agonists—sumatriptan, almotriptan, eletriptan, frovatriptan, naratriptan, rizatriptan, and zolmitriptan—are now available for the treatment of migraine.
Each drug in the triptan class has similar pharmacologic properties but varies slightly in terms of clinical efficacy. Rizatriptan and eletriptan are the most efficacious of the triptans currently available in the United States. Sumatriptan and zolmitriptan have similar rates of efficacy as well as time to onset, with an advantage of having multiple formulations, whereas almotriptan has a similar rate of efficacy to sumatriptan and is better tolerated, and frovatriptan and naratriptan are somewhat slower in onset and are better tolerated. Clinical efficacy appears to be related more to the tmax (time to peak plasma level) than to the potency, half-life, or bioavailability. This observation is consistent with a large body of data indicating that faster-acting analgesics are more effective than slower-acting agents.
Unfortunately, monotherapy with a selective oral 5-HT1B/1D receptor agonist does not result in rapid, consistent, and complete relief of migraine in all patients. Triptans are generally not effective in migraine with aura unless given after the aura is completed and the headache initiated. Side effects are common, although often mild and transient. Moreover, 5-HT1B/1D receptor agonists are contraindicated in individuals with a history of cardiovascular and cerebrovascular disease. Recurrence of headache, within usual time course of an attack, is another important limitation of triptan use and occurs at least occasionally in most patients. Evidence from randomized controlled trials show that coadministration of a longer-acting NSAID, naproxen 500 mg, with sumatriptan will augment the initial effect of sumatriptan and, importantly, reduce rates of headache recurrence.
Ergotamine preparations offer a nonselective means of stimulating 5-HT1 receptors. A nonnauseating dose of ergotamine should be sought because a dose that provokes nausea is too high and may intensify head pain. Except for a sublingual formulation of ergotamine, oral formulations of ergotamine also contain 100 mg caffeine (theoretically to enhance ergotamine absorption and possibly to add additional analgesic activity). The average oral ergotamine dose for a migraine attack is 2 mg. Because the clinical studies demonstrating the efficacy of ergotamine in migraine predated the clinical trial methodologies used with the triptans, it is difficult to assess the clinical efficacy of ergotamine versus the triptans. In general, ergotamine appears to have a much higher incidence of nausea than triptans but less headache recurrence.
Nasal Nasal formulations of dihydroergotamine (Migranal), zolmitriptan (Zomig nasal), or sumatriptan can be useful in patients requiring a nonoral route of administration. The nasal sprays result in substantial blood levels within 30–60 min. Although in theory nasal sprays might provide faster and more effective relief of a migraine attack than oral formulations, their reported efficacy is only approximately 50–60%. Studies with a new inhalational formulation of dihydroergotamine indicate that its absorption problems can be overcome to produce rapid onset of action with good tolerability.
Parenteral Administration of drugs by injection, such as dihydroergotamine and sumatriptan, is approved by the FDA for the rapid relief of a migraine attack. Peak plasma levels of dihydroergotamine are achieved 3 min after IV dosing, 30 min after IM dosing, and 45 min after SC dosing. If an attack has not already peaked, SC or IM administration of 1 mg of dihydroergotamine suffices for about 80–90% of patients. Sumatriptan, 4–6 mg SC, is effective in ~50–80% of patients, and can now be administered by a needle-free device.
DOPAMINE RECEPTOR ANTAGONISTS
Oral Oral dopamine receptor antagonists can be considered as adjunctive therapy in migraine. Drug absorption is impaired during migraine because of reduced gastrointestinal motility. Delayed absorption occurs even in the absence of nausea and is related to the severity of the attack and not its duration. Therefore, when oral NSAIDs and/or triptan agents fail, the addition of a dopamine receptor antagonist, such as metoclopramide 10 mg or domperidone 10 mg (not available in the United States), should be considered to enhance gastric absorption. In addition, dopamine receptor antagonists decrease nausea/vomiting and restore normal gastric motility.
Parenteral Dopamine receptor antagonists (e.g., chlorpromazine, prochlorperazine, metoclopramide) by injection can also provide significant acute relief of migraine; they can be used in combination with parenteral 5-HT1B/1D receptor agonists. A common IV protocol used for the treatment of severe migraine is the administration over 2 min of a mixture of 5 mg of prochlorperazine and 0.5 mg of dihydroergotamine.
OTHER MEDICATIONS FOR ACUTE MIGRAINE
Oral The combination of acetaminophen, dichloralphenazone, and isometheptene, one to two capsules, has been classified by the FDA as “possibly” effective in the treatment of migraine. Because the clinical studies demonstrating the efficacy of this combination analgesic in migraine predated the clinical trial methodologies used with the triptans, it is difficult to compare the efficacy of this sympathomimetic compound to other agents.
Nasal A nasal preparation of butorphanol is available for the treatment of acute pain. As with all opioids, the use of nasal butorphanol has little role in migraine treatment.
Parenteral Opioids are modestly effective in the acute treatment of migraine. For example, IV meperidine (50–100 mg) is given frequently in the emergency room. This regimen “works” in the sense that the pain of migraine is eliminated. However, this regimen is clearly suboptimal for patients with recurrent headache. Opioids do not treat the underlying headache mechanism; rather, they act to alter the pain sensation, and there is evidence their use may decrease the likelihood of a response to triptans in the future. Moreover, in patients taking oral opioids, such as oxycodone or hydrocodone, habituation or addiction can greatly confuse the treatment of migraine. Opioid craving and/or withdrawal can aggravate and accentuate migraine. Therefore, it is recommended that opioid use in migraine be limited to patients with severe, but infrequent, headaches that are unresponsive to other pharmacologic approaches or who have contraindications to other therapies.
MEDICATION-OVERUSE HEADACHE
Acute attack medications, particularly opioid or barbiturate-containing compound analgesics, have a propensity to aggravate headache frequency and induce a state of refractory daily or near-daily headache called medication-overuse headache. This condition is likely not a separate headache entity but a reaction of the migraine patient to a particular medicine. Migraine patients who have two or more headache days a week should be cautioned about frequent analgesic use (see “Chronic Daily Headache” in Chap. 21).
PREVENTIVE TREATMENTS FOR MIGRAINE
Patients with an increasing frequency of migraine attacks or with attacks that are either unresponsive or poorly responsive to abortive treatments are good candidates for preventive agents. In general, a preventive medication should be considered in the subset of patients with four or more attacks a month. Significant side effects are associated with the use of many of these agents; furthermore, determination of dose can be difficult because the recommended doses have been derived for conditions other than migraine. The mechanism of action of these drugs is unclear; it seems likely that the brain sensitivity that underlies migraine is modified. Patients are usually started on a low dose of a chosen treatment; the dose is then gradually increased, up to a reasonable maximum, to achieve clinical benefit.
Drugs that have the capacity to stabilize migraine are listed in Table 447-6. Drugs must be taken daily, and there is usually a lag of between 2 to 12 weeks before an effect is seen. The drugs that have been approved by the FDA for the prophylactic treatment of migraine include propranolol, timolol, sodium valproate, topiramate, and methysergide (not available). In addition, a number of other drugs appear to display prophylactic efficacy. This group includes amitriptyline, nortriptyline, flunarizine, phenelzine, gabapentin, and cyproheptadine. Placebo-controlled trials of onabotulinum toxin type A in episodic migraine were negative, whereas, overall, placebo-controlled trials in chronic migraine were positive. Phenelzine and methysergide are usually reserved for recalcitrant cases because of their serious potential side effects. Phenelzine is a monoamine oxidase inhibitor (MAOI); therefore, tyramine-containing foods, decongestants, and meperidine are contraindicated. Methysergide may cause retroperitoneal or cardiac valvular fibrosis when it is used for >6 months, and thus monitoring is required for patients using this drug; the risk of fibrosis is about 1:1500 and is likely to reverse after the drug is stopped.
|
PREVENTIVE TREATMENTS IN MIGRAINEa |
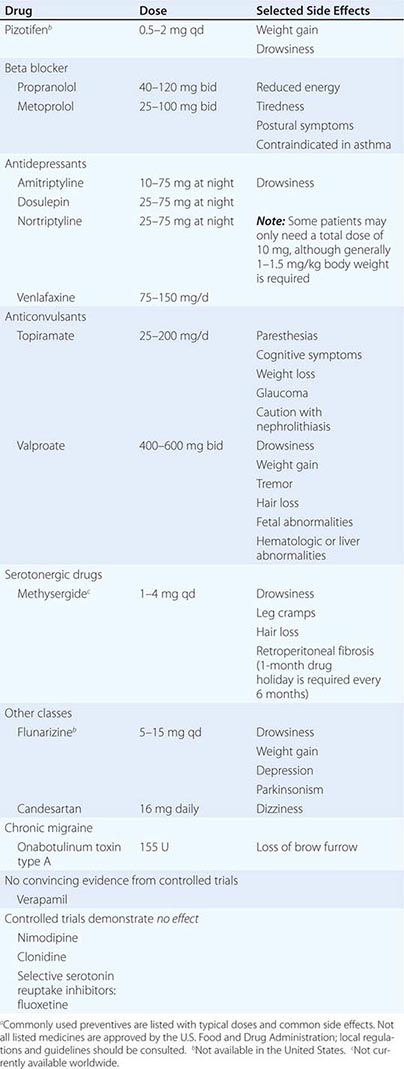
The probability of success with any one of the antimigraine drugs is 50–75%. Many patients are managed adequately with low-dose amitriptyline, propranolol, candesartan, topiramate, or valproate. If these agents fail or lead to unacceptable side effects, second-line agents such as methysergide or phenelzine can be used. Once effective stabilization is achieved, the drug is continued for ~6 months and then slowly tapered to assess the continued need. Many patients are able to discontinue medication and experience fewer and milder attacks for long periods, suggesting that these drugs may alter the natural history of migraine.
TENSION-TYPE HEADACHE
Clinical Features The term tension-type headache (TTH) is commonly used to describe a chronic head-pain syndrome characterized by bilateral tight, band-like discomfort. The pain typically builds slowly, fluctuates in severity, and may persist more or less continuously for many days. The headache may be episodic or chronic (present >15 days per month).
A useful clinical approach is to diagnose TTH in patients whose headaches are completely without accompanying features such as nausea, vomiting, photophobia, phonophobia, osmophobia, throbbing, and aggravation with movement. Such an approach neatly separates migraine, which has one or more of these features and is the main differential diagnosis, from TTH. The International Headache Society’s main definition of TTH allows an admixture of nausea, photophobia, or phonophobia in various combinations, although the appendix definition does not; this illustrates the difficulty in distinguishing these two clinical entities. In clinical practice, dichotomizing patients on the basis of the presence of associated features (migraine) and the absence of associated features (TTH) is highly recommended. Indeed patients whose headaches fit the TTH phenotype and who have migraine at other times, along with a family history of migraine, migrainous illnesses of childhood, or typical migraine triggers to their migraine attacks, may be biologically different from those who have TTH headache with none of the features. TTH may be infrequent (episodic) or occur on 15 days or more a month (chronic).
Pathophysiology The pathophysiology of TTH is incompletely understood. It seems likely that TTH is due to a primary disorder of central nervous system pain modulation alone, unlike migraine, which involves a more generalized disturbance of sensory modulation. Data suggest a genetic contribution to TTH, but this may not be a valid finding: given the current diagnostic criteria, the studies undoubtedly included many migraine patients. The name tension-type headache implies that pain is a product of nervous tension, but there is no clear evidence for tension as an etiology. Muscle contraction has been considered to be a feature that distinguishes TTH from migraine, but there appear to be no differences in contraction between the two headache types.
TRIGEMINAL AUTONOMIC CEPHALALGIAS, INCLUDING CLUSTER HEADACHE
The trigeminal autonomic cephalalgias (TACs) describe a grouping of primary headaches including cluster headache, paroxysmal hemicrania, SUNCT (short-lasting unilateral neuralgiform headache attacks with conjunctival injection and tearing)/SUNA (short-lasting unilateral neuralgiform headache attacks with cranial autonomic symptoms), and hemicrania continua (Table 447-1). TACs are characterized by relatively short-lasting attacks of head pain associated with cranial autonomic symptoms, such as lacrimation, conjunctival injection, or nasal congestion (Table 447-7). Pain is usually severe and may occur more than once a day. Because of the associated nasal congestion or rhinorrhea, patients are often misdiagnosed with “sinus headache” and treated with decongestants, which are ineffective.
|
CLINICAL FEATURES OF THE TRIGEMINAL AUTONOMIC CEPHALALGIAS |
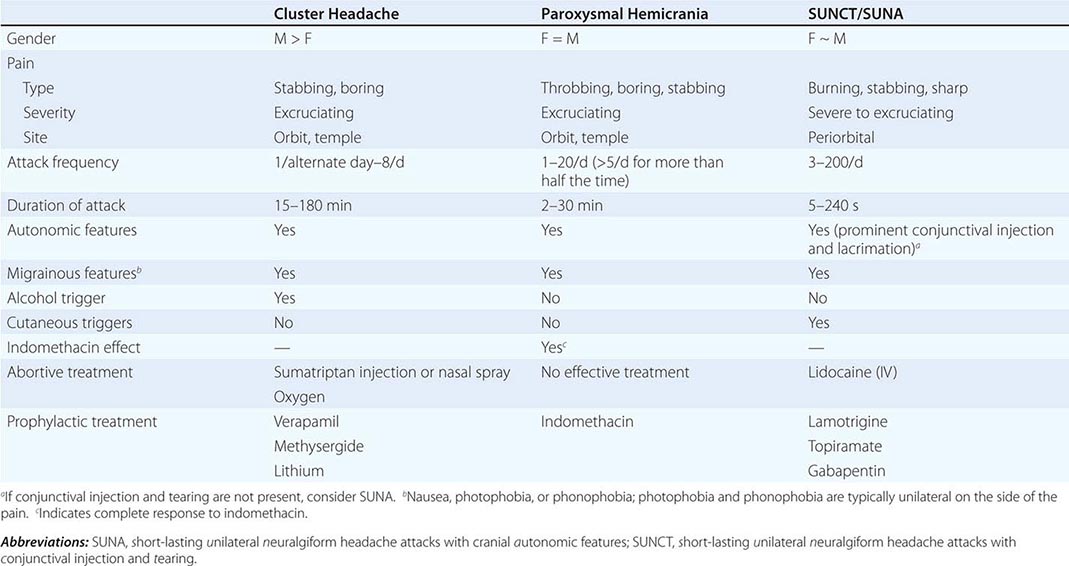
TACs must be differentiated from short-lasting headaches that do not have prominent cranial autonomic syndromes, notably trigeminal neuralgia, primary stabbing headache, and hypnic headache. The cycling pattern and length, frequency, and timing of attacks are useful in classifying patients. Patients with TACs should undergo pituitary imaging and pituitary function tests because there is an excess of TAC presentations in patients with pituitary tumor–related headache.
Cluster Headache Cluster headache is a relatively rare form of primary headache with a population frequency of approximately 0.1%. The pain is deep, usually retroorbital, often excruciating in intensity, nonfluctuating, and explosive in quality. A core feature of cluster headache is periodicity. At least one of the daily attacks of pain recurs at about the same hour each day for the duration of a cluster bout. The typical cluster headache patient has daily bouts of one to two attacks of relatively short-duration unilateral pain for 8 to 10 weeks a year; this is usually followed by a pain-free interval that averages a little less than 1 year. Cluster headache is characterized as chronic when there is less than 1 month of sustained remission without treatment. Patients are generally perfectly well between episodes. Onset is nocturnal in about 50% of patients, and men are affected three times more often than women. Patients with cluster headache tend to move about during attacks, pacing, rocking, or rubbing their head for relief; some may even become aggressive during attacks. This is in sharp contrast to patients with migraine, who prefer to remain motionless during attacks.
Cluster headache is associated with ipsilateral symptoms of cranial parasympathetic autonomic activation: conjunctival injection or lacrimation, rhinorrhea or nasal congestion, or cranial sympathetic dysfunction such as ptosis. The sympathetic deficit is peripheral and likely to be due to parasympathetic activation with injury to ascending sympathetic fibers surrounding a dilated carotid artery as it passes into the cranial cavity. When present, photophobia and phonophobia are far more likely to be unilateral and on the same side of the pain, rather than bilateral, as is seen in migraine. This phenomenon of unilateral photophobia/phonophobia is characteristic of TACs. Cluster headache is likely to be a disorder involving central pacemaker neurons in the posterior hypothalamic region (Fig. 447-3).
|
TREATMENT |
CLUSTER HEADACHE |
The most satisfactory treatment is the administration of drugs to prevent cluster attacks until the bout is over. However, treatment of acute attacks is required for all cluster headache patients at some time.
ACUTE ATTACK TREATMENT
Cluster headache attacks peak rapidly, and thus a treatment with quick onset is required. Many patients with acute cluster headache respond very well to oxygen inhalation. This should be given as 100% oxygen at 10–12 L/min for 15–20 min. It appears that high flow and high oxygen content are important. Sumatriptan 6 mg SC is rapid in onset and will usually shorten an attack to 10–15 min; there is no evidence of tachyphylaxis. Sumatriptan (20 mg) and zolmitriptan (5 mg) nasal sprays are both effective in acute cluster headache, offering a useful option for patients who may not wish to self-inject daily. Oral sumatriptan is not effective for prevention or for acute treatment of cluster headache.
PREVENTIVE TREATMENTS (TABLE 447-8)
|
PREVENTIVE MANAGEMENT OF CLUSTER HEADACHE |
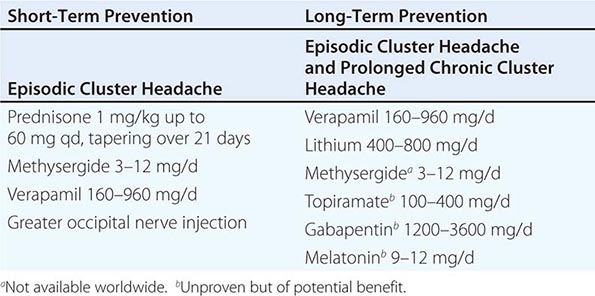
The choice of a preventive treatment in cluster headache depends in part on the length of the bout. Patients with long bouts or those with chronic cluster headache require medicines that are safe when taken for long periods. For patients with relatively short bouts, limited courses of oral glucocorticoids or methysergide (not available in the United States) can be very useful. A 10-day course of prednisone, beginning at 60 mg daily for 7 days and followed by a rapid taper, may interrupt the pain bout for many patients. Lithium (400–800 mg/d) appears to be particularly useful for the chronic form of the disorder.
Many experts favor verapamil as the first-line preventive treatment for patients with chronic cluster headache or prolonged bouts. While verapamil compares favorably with lithium in practice, some patients require verapamil doses far in excess of those administered for cardiac disorders. The initial dose range is 40–80 mg twice daily; effective doses may be as high as 960 mg/d. Side effects such as constipation and leg swelling can be problematic. Of paramount concern, however, is the cardiovascular safety of verapamil, particularly at high doses. Verapamil can cause heart block by slowing conduction in the atrioventricular node, a condition that can be monitored by following the PR interval on a standard electrocardiogram (ECG). Approximately 20% of patients treated with verapamil develop ECG abnormalities, which can be observed with doses as low as 240 mg/d; these abnormalities can worsen over time in patients on stable doses. A baseline ECG is recommended for all patients. The ECG is repeated 10 days after a dose change in patients whose dose is being increased above 240 mg daily. Dose increases are usually made in 80-mg increments. For patients on long-term verapamil, ECG monitoring every 6 months is advised.
NEUROSTIMULATION THERAPY
When medical therapies fail in chronic cluster headache, neurostimulation strategies can be used. Deep-brain stimulation of the region of the posterior hypothalamic gray matter has proven successful in a substantial proportion of patients, although its risk-benefit ratio makes it inappropriate with so many other options now available. Favorable results have also been reported with the less-invasive approach of occipital nerve stimulation, with sphenopalatine ganglion stimulation and with a noninvasive vagal nerve stimulator.
PAROXYSMAL HEMICRANIA
Paroxysmal hemicrania (PH) is characterized by frequent unilateral, severe, short-lasting episodes of headache. Like cluster headache, the pain tends to be retroorbital but may be experienced all over the head and is associated with autonomic phenomena such as lacrimation and nasal congestion. Patients with remissions are said to have episodic PH, whereas those with the nonremitting form are said to have chronic PH. The essential features of PH are unilateral, very severe pain; short-lasting attacks (2–45 min); very frequent attacks (usually more than five a day); marked autonomic features ipsilateral to the pain; rapid course (<72 h); and excellent response to indomethacin. In contrast to cluster headache, which predominantly affects males, the male-to-female ratio in PH is close to 1:1.
Indomethacin (25–75 mg tid), which can completely suppress attacks of PH, is the treatment of choice. Although therapy may be complicated by indomethacin-induced gastrointestinal side effects, currently there are no consistently effective alternatives. Topiramate is helpful in some cases. Piroxicam has been used, although it is not as effective as indomethacin. Verapamil, an effective treatment for cluster headache, does not appear to be useful for PH. In occasional patients, PH can coexist with trigeminal neuralgia (PH-tic syndrome); similar to cluster-tic syndrome, each component may require separate treatment.
Secondary PH has been reported with lesions in the region of the sella turcica, including arteriovenous malformation, cavernous sinus meningioma, pituitary pathology and epidermoid tumors. Secondary PH is more likely if the patient requires high doses (>200 mg/d) of indomethacin. In patients with apparent bilateral PH, raised cerebrospinal fluid (CSF) pressure should be suspected. It is important to note that indomethacin reduces CSF pressure. When a diagnosis of PH is considered, magnetic resonance imaging (MRI) is indicated to exclude a pituitary lesion.
SUNCT/SUNA
SUNCT (short-lasting unilateral neuralgiform headache attacks with conjunctival injection and tearing) is a rare primary headache syndrome characterized by severe, unilateral orbital or temporal pain that is stabbing or throbbing in quality. Diagnosis requires at least 20 attacks, lasting for 5–240 s; ipsilateral conjunctival injection and lacrimation should be present. In some patients, conjunctival injection or lacrimation is missing, and the diagnosis of SUNA (short-lasting unilateral neuralgiform headache attacks with cranial autonomic symptoms) can be made.
DIAGNOSIS The pain of SUNCT/SUNA is unilateral and may be located anywhere in the head. Three basic patterns can be seen: single stabs, which are usually short-lived; groups of stabs; or a longer attack comprising many stabs between which the pain does not completely resolve, thus giving a “saw-tooth” phenomenon with attacks lasting many minutes. Each pattern may be seen in the context of an underlying continuous head pain. Characteristics that lead to a suspected diagnosis of SUNCT are the cutaneous (or other) triggers of attacks, a lack of refractory period to triggering between attacks, and the lack of a response to indomethacin. Apart from trigeminal sensory disturbance, the neurologic examination is normal in primary SUNCT.
The diagnosis of SUNCT/SUNA is often confused with trigeminal neuralgia (TN) particularly in first-division TN (Chap. 455). Minimal or no cranial autonomic symptoms and a clear refractory period to triggering indicate a diagnosis of TN.
SECONDARY (SYMPTOMATIC) SUNCT SUNCT can be seen with posterior fossa or pituitary lesions. All patients with SUNCT/SUNA should be evaluated with pituitary function tests and a brain MRI with pituitary views.
Hemicrania Continua The essential features of hemicrania continua are moderate and continuous unilateral pain associated with fluctuations of severe pain; complete resolution of pain with indomethacin; and exacerbations that may be associated with autonomic features, including conjunctival injection, lacrimation, and photophobia on the affected side. The age of onset ranges from 11 to 58 years; women are affected twice as often as men. The cause is unknown.
OTHER PRIMARY HEADACHES
Primary Cough Headache Primary cough headache is a generalized headache that begins suddenly, lasts for several minutes, sometimes up to a few hours, and is precipitated by coughing; it is preventable by avoiding coughing or other precipitating events, which can include sneezing, straining, laughing, or stooping. In all patients with this syndrome, serious etiologies must be excluded before a diagnosis of “benign” primary cough headache can be established. A Chiari malformation or any lesion causing obstruction of CSF pathways or displacing cerebral structures can be the cause of the head pain. Other conditions that can present with cough or exertional headache as the initial symptom include cerebral aneurysm, carotid stenosis, and vertebrobasilar disease. Benign cough headache can resemble benign exertional headache (below), but patients with the former condition are typically older.
Primary Exercise Headache Primary exertional headache has features resembling both cough headache and migraine. It may be precipitated by any form of exercise; it often has the pulsatile quality of migraine. The pain, which can last from 5 min to 24 h, is bilateral and throbbing at onset; migrainous features may develop in patients susceptible to migraine. The duration tends to be shorter in adolescents than in older adults. Primary exertional headache can be prevented by avoiding excessive exertion, particularly in hot weather or at high altitude.
The mechanism of primary exertional headache is unclear. Acute venous distension likely explains one syndrome—the acute onset of headache with straining and breath holding, as in weightlifter’s headache. Because exertion can result in headache in a number of serious underlying conditions, these must be considered in patients with exertional headache. Pain from angina may be referred to the head, probably by central connections of vagal afferents, and may present as exertional headache (cardiac cephalgia). The link to exercise is the main clinical clue that headache is of cardiac origin. Pheochromocytoma may occasionally cause exertional headache. Intracranial lesions and stenosis of the carotid arteries are other possible etiologies.
Primary Headache Associated with Sexual Activity Three types of sex headache are reported: a dull bilateral ache in the head and neck that intensifies as sexual excitement increases; a sudden, severe, explosive headache occurring at orgasm; and a postural headache developing after coitus that resembles the headache of low CSF pressure. The last arises from vigorous sexual activity and is a form of low CSF pressure headache (Chap. 21). Headaches developing at the time of orgasm are not always benign; 5–12% of cases of subarachnoid hemorrhage are precipitated by sexual intercourse. Sex headache is reported by men more often than women and may occur at any time during the years of sexual activity. It may develop on several occasions in succession and then not trouble the patient again, even without an obvious change in sexual activity. In patients who stop sexual activity when headache is first noticed, the pain may subside within a period of 5 min to 2 h. In about half of patients, sex headache will subside within 6 months. About half of patients with sex headache have a history of exertional headaches, but there is no excess of cough headache. Migraine is probably more common in patients with sex headache.
Primary Thunderclap Headache Sudden onset of severe headache may occur in the absence of any known provocation. The differential diagnosis includes the sentinel bleed of an intracranial aneurysm, cervicocephalic arterial dissection, and cerebral venous thrombosis. Headaches of explosive onset may also be caused by the ingestion of sympathomimetic drugs or of tyramine-containing foods in a patient who is taking MAOIs, or they may be a symptom of pheochromocytoma. Whether thunderclap headache can be the presentation of an unruptured cerebral aneurysm is uncertain. When neuroimaging studies and lumbar puncture exclude subarachnoid hemorrhage, patients with thunderclap headache usually do very well over the long term. In one study of patients whose computed tomography (CT) scans and CSF findings were negative, ~15% had recurrent episodes of thunderclap headache, and nearly half subsequently developed migraine or TTH.
The first presentation of any sudden-onset severe headache should be diligently investigated with neuroimaging (CT or, when possible, MRI with MR angiography) and CSF examination. Formal cerebral angiography should be reserved for those cases in which no primary diagnosis is forthcoming and for clinical situations that are particularly suggestive of intracranial aneurysm. Reversible segmental cerebral vasoconstriction may be seen in primary thunderclap headache without an intracranial aneurysm. In the presence of posterior leukoencephalopathy, the differential diagnosis includes cerebral angiitis, drug toxicity (cyclosporine, intrathecal methotrexate/cytarabine, pseudoephedrine, or cocaine), posttransfusion effects, and postpartum angiopathy. Treatment with nimodipine may be helpful, although by definition, the vasoconstriction of primary thunderclap headache resolves spontaneously.
Cold-Stimulus Headache This refers to head pain triggered by application or ingestion/inhalation of something cold. It is bought on quickly and typically resolves within 10–30 min of the stimulus being removed. It is best recognized as “brain-freeze” headache or ice-cream headache when due to ingestion. Although cold may be uncomfortable at some level for many people, it is the reliable, severe, and somewhat prolonged nature of these pains that set them apart. The transient receptor potential cation subfamily M member 8 (TRPM8) channel, a known cold temperature sensor, may be a mediator of this syndrome.
External Pressure Headache External pressure from compression or traction on the head can produce a pain that may have some generalized component, although the pain is largely focused around the site of the pressure. It typically resolves within an hour of the stimulus being removed. Examples of stimuli include helmets, swimming goggles, or very long ponytails. Treatment is to recognize the problem and remove the stimulus.
Primary Stabbing Headache The essential features of primary stabbing headache are stabbing pain confined to the head or, rarely, the face, lasting from 1 to many seconds or minutes and occurring as a single stab or a series of stabs; absence of associated cranial autonomic features; absence of cutaneous triggering of attacks; and a pattern of recurrence at irregular intervals (hours to days). The pains have been variously described as “ice-pick pains” or “jabs and jolts.” They are more common in patients with other primary headaches, such as migraine, the TACs, and hemicrania continua.
Nummular Headache Nummular headache is felt as a round or elliptical discomfort that is fixed in place, ranges in size from 1–6 cm, and may be continuous or intermittent. Uncommonly it may be multifocal. It may be episodic but is more often continuous during exacerbations. Accompanying the pain there may be a local sensory disturbance, such as allodynia or hypesthesia. Local dermatologic or bony lesions need to be excluded by examination and investigation. This condition can be difficult to treat; tricyclics, such as amitriptyline, or anticonvulsants, such as topiramate or valproate, are most often tried.
Hypnic Headache This headache syndrome typically begins a few hours after sleep onset. The headaches last from 15 to 30 min and are typically moderately severe and generalized, although they may be unilateral and can be throbbing. Patients may report falling back to sleep only to be awakened by a further attack a few hours later; up to three repetitions of this pattern occur through the night. Daytime naps can also precipitate head pain. Most patients are female, and the onset is usually after age 60 years. Headaches are bilateral in most, but may be unilateral. Photophobia, phonophobia, and nausea are usually absent. The major secondary consideration in this headache type is poorly controlled hypertension; 24-h blood pressure monitoring is recommended to detect this treatable condition.
New Daily Persistent Headache Primary new daily persistent headache (NDPH) occurs in both males and females. It can be of the migrainous type, with features of migraine, or it can be featureless, appearing as new-onset TTH. Migrainous features are common and include unilateral headache and throbbing pain; each feature is present in about one-third of patients. Nausea, photophobia, and/or phonophobia occur in about half of patients. Some patients have a previous history of migraine; however, the proportion of NDPH sufferers with preexisting migraine is no greater than the frequency of migraine in the general population. At 24 months, ~86% of patients are headache-free. Treatment of migrainous-type primary NDPH consists of using the preventive therapies effective in migraine (see above). Featureless NDPH is one of the primary headache forms most refractory to treatment. Standard preventive therapies can be offered but are often ineffective. The secondary NDPHs are discussed elsewhere (Chap. 21).
448 |
Alzheimer’s Disease and Other Dementias |
ALZHEIMER’S DISEASE
Approximately 10% of all persons over the age of 70 years have significant memory loss, and in more than half, the cause is Alzheimer’s disease (AD). It is estimated that the median annual total cost of caring for a single patient with advanced AD is >$50,000, while the emotional toll for family members and caregivers is immeasurable. AD can manifest as young as the third decade, but it is the most common cause of dementia in the elderly. Patients most often present with an insidious loss of episodic memory followed by a slowly progressive dementia that evolves over years. In typical amnestic AD, brain imaging reveals atrophy that begins in the medial temporal lobes before spreading to lateral and medial parietal and temporal lobes and lateral frontal cortex. Microscopically, there are neuritic plaques containing amyloid beta (Aβ), neurofibrillary tangles (NFTs) composed of hyperphosphorylated tau filaments, and Aβ accumulation of in blood vessel walls in cortex and leptomeninges (see “Pathology,” below). The identification of causative mutations and susceptibility genes for AD has provided a foundation for rapid progress in understanding the biological basis of the disorder. The major genetic risk for AD is apolipoprotein ε4 (Apo ε4). Carrying one E4 allele increases the risk for AD by 2- to 3-fold, whereas two alleles increase the risk 16-fold.
CLINICAL MANIFESTATIONS
The cognitive changes of AD tend to follow a characteristic pattern, beginning with memory impairment and progressing to language and visuospatial deficits. Yet, approximately 20% of patients with AD present with nonmemory complaints such as word-finding, organizational, or navigational difficulty. In other patients, upstream visual processing dysfunction (referred to as posterior cortical atrophy syndrome) or a progressive “logopenic” aphasia are the primary manifestations of AD for years before progressing to involve memory and other cognitive domains. Still other patients may present with an asymmetric akinetic-rigid-dystonic (“corticobasal”) syndrome or a dysexecutive “frontal variant” of AD.
In the early stages of typical amnestic AD, the memory loss may go unrecognized or be ascribed to benign forgetfulness of aging. Once the memory loss becomes noticeable to the patient and spouse and falls 1.5 standard deviations below normal on standardized memory tests, the term mild cognitive impairment (MCI) is applied. This construct provides useful prognostic information, because approximately 50% of patients with MCI (roughly 12% per year) will progress to AD over 4 years. Increasingly, the MCI construct is being replaced by the notion of “early symptomatic AD” to signify that AD is considered the underlying disease (based on clinical or biomarker evidence) in a patient who remains functionally compensated. Even earlier in the course, “prodromal AD” refers to a person with biomarker evidence of AD (amyloid imaging positive with positron emission tomography or low cerebrospinal Aβ42 and mildly elevated tau) in the absence of symptoms. These refinements have been developed in anticipation of early-stage treatment and prevention trials that have already begun in humans. New evidence suggests that partial and sometimes generalized seizures herald AD and can occur even prior to dementia onset.
Eventually, with AD, the cognitive problems begin to interfere with daily activities, such as keeping track of finances, following instructions on the job, driving, shopping, and housekeeping. Some patients are unaware of these difficulties (anosognosia), but most remain acutely attuned to their deficits. Changes in environment (travel, relocation, hospitalization) tend to destabilize the patient. Over time patients become lost on walks or while driving. Social graces, routine behavior, and superficial conversation may be surprisingly intact, even into the later stages of the illness.
In the middle stages of AD, the patient is unable to work, is easily lost and confused, and requires daily supervision. Language becomes impaired—first naming, then comprehension, and finally fluency. Word-finding difficulties and circumlocution can be evident in the early stages, even when formal testing demonstrates intact naming and fluency. Apraxia emerges, and patients have trouble performing learned sequential motor tasks. Visuospatial deficits begin to interfere with dressing, eating, or even walking, and patients fail to solve simple puzzles or copy geometric figures. Simple calculations and clock reading become difficult in parallel.
In the late stages, some persons remain ambulatory, wandering aimlessly. Loss of judgment and reasoning is inevitable. Delusions are common, usually simple, with common themes of theft, infidelity, or misidentification. Approximately 10% of AD patients develop Capgras’ syndrome, believing that a caregiver has been replaced by an impostor. In contrast to dementia with Lewy bodies (DLB), where Capgras’ syndrome is an early feature, in AD this syndrome emerges late. Disinhibition and uncharacteristic belligerence may occur and alternate with passivity and withdrawal. Sleep-wake patterns are disrupted, and nighttime wandering becomes disturbing to the household. Some patients develop a shuffling gait with generalized muscle rigidity associated with slowness and awkwardness of movement. Patients often look parkinsonian (Chap. 449) but rarely have a high-amplitude, low-frequency tremor at rest. There is a strong overlap between Parkinson’s disease (PD) and AD, and some AD patients develop more classical PD features.
In the end stages, AD patients become rigid, mute, incontinent, and bedridden, and help is needed with eating, dressing, and toileting. Hyperactive tendon reflexes and myoclonic jerks (sudden brief contractions of various muscles or the whole body) may occur spontaneously or in response to physical or auditory stimulation. Often death results from malnutrition, secondary infections, pulmonary emboli, heart disease, or, most commonly, aspiration. The typical duration of AD is 8–10 years, but the course ranges from 1 to 25 years. For unknown reasons, some patients with AD show a steady decline in function while others have prolonged plateaus without major deterioration.
DIFFERENTIAL DIAGNOSIS
Early in the disease course, other etiologies of dementia should be excluded (see Tables 35-1, 35-3, and 35-4). Neuroimaging studies (computed tomography [CT] and magnetic resonance imaging [MRI]) do not show a single specific pattern with AD and may be normal early in the disease. As AD progresses, more distributed but usually posterior-predominant cortical atrophy becomes apparent, along with atrophy of the medial temporal memory structures (see Chap. 35, Fig. 35-1). The main purpose of imaging is to exclude other disorders, such as primary and secondary neoplasms, vascular dementia, diffuse white matter disease, and normal-pressure hydrocephalus (NPH). Imaging also helps to distinguish AD from other degenerative disorders, such as frontotemporal dementia (FTD) or Creutzfeldt-Jacob disease (CJD), which feature distinctive imaging patterns. Functional imaging studies, such as positron emission tomography (PET), reveal hypometabolism in the posterior temporal-parietal cortex in AD (see Fig. 35-1). PET can also be used to detect the presence of fibrillar amyloid in the brain (see Fig. 35-4), and amyloid PET positivity is becoming required for entry into treatment trials for AD. Barriers to interpretation continue, however, to limit the use of amyloid PET in routine clinical evaluation. Although amyloid binding with PET is typical for AD, many asymptomatic healthy older individuals show amyloid uptake, and the likelihood that these individuals will convert to clinical AD is still under study. Similarly, dementia due to a non-AD disorder can be the underlying etiology in a patient who is amyloid positive on imaging. Electroencephalogram (EEG) is normal or shows nonspecific slowing; prolonged EEG can be used to seek out intermittent nonconvulsive seizures. Routine spinal fluid examination is also normal. Cerebrospinal fluid (CSF) Aβ42 level is reduced, whereas the tau protein is elevated, but the test characteristics of these assays still make interpretation challenging in individual patients. Slowly progressive decline in memory and orientation, normal results on laboratory tests, and an MRI or CT scan showing only distributed or posteriorly predominant cortical and hippocampal atrophy are highly suggestive of AD. A clinical diagnosis of AD reached after careful evaluation is confirmed at autopsy about 90% of the time, with misdiagnosed cases usually representing one of the other dementing disorders described later in this chapter, a mixture of AD with vascular pathology, or DLB.
Simple clinical clues are useful in the differential diagnosis. Early prominent gait disturbance with only mild memory loss suggests vascular dementia or, rarely, NPH (see below). Resting tremor with stooped posture, bradykinesia, and masked facies suggest PD (Chap. 449). When dementia occurs after a well-established diagnosis of PD, PD dementia (PDD) is usually the correct diagnosis, but many patients with this diagnosis will show a mixture of AD and Lewy body disease at autopsy. The early appearance of parkinsonian features in association with fluctuating alertness, visual hallucinations, or delusional misidentification suggests DLB. Chronic alcoholism should prompt the search for vitamin deficiency. Loss of joint position and vibration sensibility accompanied by Babinski signs suggests vitamin B12 deficiency (Chap. 456). Early onset of a focal seizure suggests a metastatic or primary brain neoplasm (Chap. 118). Previous or ongoing depression raises suspicion for depression-related cognitive impairment, although AD can feature a depressive prodrome. A history of treatment for insomnia, anxiety, psychiatric disturbance, or epilepsy suggests chronic drug intoxication. Rapid progression over a few weeks or months associated with rigidity and myoclonus suggests CJD (Chap. 453e). Prominent behavioral changes with intact navigation and focal anterior-predominant atrophy on brain imaging are typical of FTD. A positive family history of dementia suggests either one of the familial forms of AD or one of the other genetic disorders associated with dementia, such as FTD (see below), HD (see below), prion disease (Chap. 453e), or rare hereditary ataxias (Chap. 450).
EPIDEMIOLOGY
The most important risk factors for AD are old age and a positive family history. The prevalence of AD increases with each decade of adult life, reaching 20–40% of the population over the age of 85. A positive family history of dementia suggests a genetic contribution to AD, although autosomal dominant inheritance occurs in only 2% of patients. Female sex is a risk factor independent of the greater longevity of women, and women who carry an Apo ε4 allele are more susceptible than are male ε4 carriers. A history of head trauma with concussion increases the risk for AD. AD is more common in groups with low educational attainment, but education influences test-taking ability, and it is clear that AD can affect persons of all intellectual levels. One study found that the capacity to express complex written language in early adulthood correlated with a decreased risk for AD. Numerous environmental factors, including aluminum, mercury, and viruses, have been proposed as causes of AD, but rigorous studies have failed to demonstrate to a significant role for any of these exposures. Similarly, several studies suggest that the use of nonsteroidal anti-inflammatory agents is associated with a decreased risk of AD, but this risk has not been confirmed in large prospective studies. Vascular disease, and stroke in particular, seems to lower the threshold for the clinical expression of AD. Also, in many patients with AD, amyloid angiopathy can lead to microhemorrhages, large lobar hemorrhages, ischemic infarctions most often in the subcortical white matter, or in rare cases an inflammatory leukoencephalopathy. Diabetes increases the risk of AD threefold. Elevated homocysteine and cholesterol levels; hypertension; diminished serum levels of folic acid; low dietary intake of fruits, vegetables, and red wine; and low levels of exercise are all being explored as potential risk factors for AD.
PATHOLOGY
At autopsy, the earliest and most severe degeneration is usually found in the medial temporal lobe (entorhinal/perirhinal cortex and hippocampus), lateral temporal cortex, and nucleus basalis of Meynert. The characteristic microscopic findings are neuritic plaques and NFTs (Fig. 448-1). These lesions may accumulate in small numbers during normal brain aging but dominate the picture in AD. Increasing evidence suggests that soluble amyloid species called oligomers may cause cellular dysfunction and represent the early toxic molecule in AD. Eventually, further amyloid polymerization and fibril formation lead to neuritic plaques, which contain a central core of amyloid, proteoglycans, Apo ε4, α-antichymotrypsin, and other proteins. Aβ is a protein of 39–42 amino acids that is derived proteolytically from a larger transmembrane protein, amyloid precursor protein (APP), when APP is cleaved by β and γ secretases (Fig. 448-2). The normal function of the Aβ peptides remains uncertain. APP has neurotrophic and neuroprotective properties. The plaque core is surrounded by a halo, which contains dystrophic, tau-immunoreactive neurites and activated microglia. The accumulation of Aβ in cerebral arterioles is termed amyloid angiopathy. NFTs are composed of silver-staining neuronal cytoplasmic fibrils composed of abnormally phosphorylated tau protein; they appear as paired helical filaments by electron microscopy. Tau binds to and stabilizes microtubules, supporting axonal transport of organelles, glycoproteins, neurotransmitters, and other important cargoes throughout the neuron. Once hyperphosphorylated, tau can no longer bind properly to microtubules and redistributes from the axon to throughout the neuronal cytoplasm and distal dendrites, compromising function. Finally, patients with AD often show comorbid DLB or vascular pathology. In animal models of AD, diminishing neuronal tau ameliorates the cognitive deficits and seizures, even though Aβ42 continues to accumulate, raising hope for tau-lowering therapies in humans. Biochemically, AD is associated with a decrease in the cortical levels of several proteins and neurotransmitters, especially acetylcholine, its synthetic enzyme choline acetyltransferase, and nicotinic cholinergic receptors. Reduction of acetylcholine reflects degeneration of cholinergic neurons in the nucleus basalis of Meynert that project throughout the cortex. There is also noradrenergic and serotonergic depletion due to degeneration of brainstem nuclei such as the locus coeruleus and dorsal raphe, where tau-immunoreactive neuronal cytoplasmic inclusions can be identified even in individuals lacking entorhinal cortex NFTs.
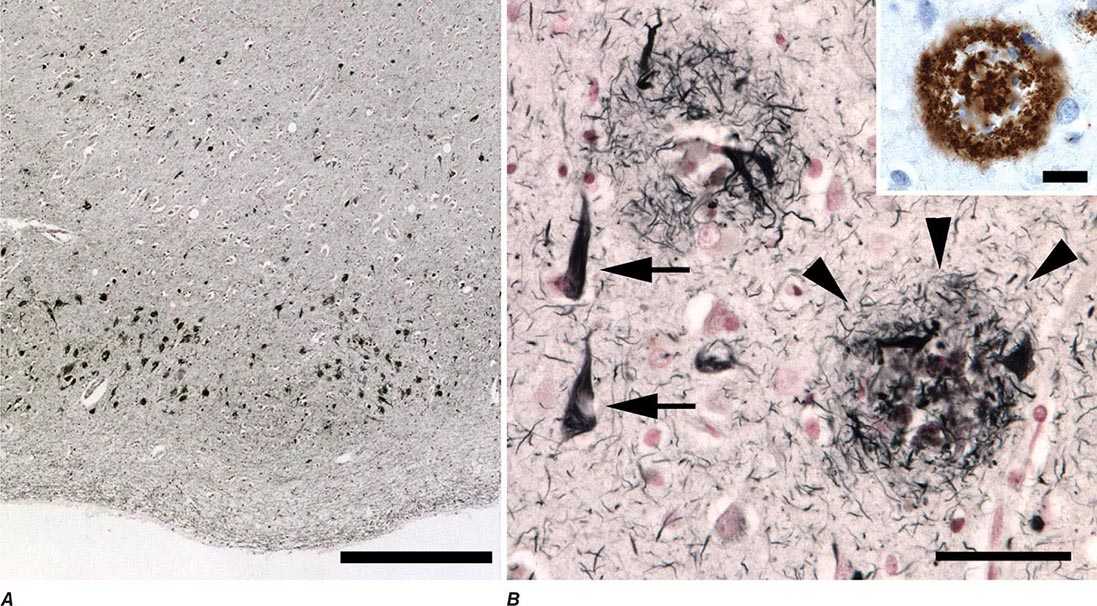
FIGURE 448-1 Neuropathology of Alzheimer’s disease. A. Early neurofibrillary degeneration, consisting of neurofibrillary tangles and neuropil threads, preferentially affects the medial temporal lobes, especially the stellate pyramidal neurons that compose the layer 2 islands of entorhinal cortex, as shown. B. Higher magnification view reveals the fibrillary nature of tangles (arrows) and the complex structure of neuritic plaques (arrowheads), whose major component is Aβ (inset shows immunohistochemistry for Aβ). Scale bars are 500 μM in A, 50 μM in B, and 20 μM in B inset.
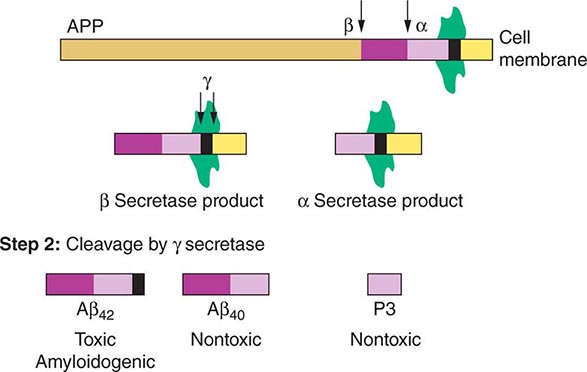
FIGURE 448-2 Amyloid precursor protein (APP) is catabolized by α, β, and γ secretases. A key initial step is the digestion by either β secretase (BASE) or α secretase (ADAM10 or ADAM17 [TACE]), producing smaller nontoxic products. Cleavage of the β secretase product by γ secretase (Step 2) results in either the toxic Aβ42 or the nontoxic Aβ40 peptide; cleavage of the α secretase product by γ secretase produces the nontoxic P3 peptide. Excess production of Aβ42 is a key initiator of cellular damage in Alzheimer’s disease (AD). Therapeutics for AD have focused on attempts to reduce accumulation of Aβ42 by antagonizing β or γ secretases, promoting α secretase, or clearing Aβ42 that has already formed by use of specific antibodies.
GENETIC CONSIDERATIONS
![]() Several genes play an important role in the pathogenesis of AD. One is the APP gene on chromosome 21. Adults with trisomy 21 (Down’s syndrome) consistently develop the typical neuropathologic hallmarks of AD if they survive beyond age 40 years, and many develop a progressive dementia superimposed on their baseline mental retardation. The extra dose of the APP gene on chromosome 21 is the initiating cause of AD in adult Down’s syndrome and results in excess cerebral amyloid production. Supporting this hypothesis, some families with early age-of-onset familial AD (FAD) have point mutations in APP. Although very rare, these families were the first examples of single-gene autosomal dominant transmission of AD.
Several genes play an important role in the pathogenesis of AD. One is the APP gene on chromosome 21. Adults with trisomy 21 (Down’s syndrome) consistently develop the typical neuropathologic hallmarks of AD if they survive beyond age 40 years, and many develop a progressive dementia superimposed on their baseline mental retardation. The extra dose of the APP gene on chromosome 21 is the initiating cause of AD in adult Down’s syndrome and results in excess cerebral amyloid production. Supporting this hypothesis, some families with early age-of-onset familial AD (FAD) have point mutations in APP. Although very rare, these families were the first examples of single-gene autosomal dominant transmission of AD.
Investigation of large families with multigenerational FAD led to the discovery of two additional AD-causing genes, the presenilins. Presenilin-1 (PS-1) is on chromosome 14 and encodes a protein called S182. Mutations in this gene cause an early-age-of-onset AD, with onset before the age of 60 and often before age 50, transmitted in an autosomal dominant, highly penetrant fashion. More than 100 different mutations have been found in the PS-1 gene in families from a wide range of ethnic backgrounds. Presenilin-2 (PS-2) is on chromosome 1 and encodes a protein called STM2. A mutation in the PS-2 gene was first found in a group of American families with Volga German ethnic background. Mutations in PS-1 are much more common than those in PS-2. The presenilins are highly homologous and encode similar proteins that at first appeared to have seven transmembrane domains (hence the designation STM), but subsequent studies have suggested eight such domains, with a ninth submembrane region. Both S182 and STM2 are cytoplasmic neuronal proteins that are widely expressed throughout the nervous system. They are homologous to a cell-trafficking protein, sel 12, found in the nematode Caenorhabditis elegans. Patients with mutations in the presenilin genes have elevated plasma levels of Aβ42, and PS-1 mutations produce increased Aβ42 in the media in cell culture. There is evidence that PS-1 is involved in the cleavage of APP at the γ secretase site and mutations in either gene (PS-1 or APP) may disturb γ secretase cleavage. Mutations in PS-1 are the most common cause of early-age-of-onset FAD, representing perhaps 40–70% of all cases. Mutations in PS-1 tend to produce AD with an earlier age of onset (mean onset 45 years) and a shorter, more rapidly progressive course (mean duration 6–7 years) than the disease caused by mutations in PS-2 (mean onset 53 years; duration 11 years). Although some carriers of PS-2 mutations have had onset of dementia after the age of 70, mutations in the presenilins rarely lead to late-age-of-onset AD. Clinical genetic testing for these uncommon mutations is available but likely to be revealing only in early-age-of-onset FAD and should be performed in association with formal genetic counseling.
The Apo ε gene on chromosome 19 is involved in the pathogenesis of AD. The protein, apolipoprotein E, participates in cholesterol transport (Chap. 421), and the gene has three alleles: ε2, ε3, and ε4. The Apo ε4 allele confers increased risk of AD in the general population, including sporadic and late-age-of-onset familial forms. Approximately 24–30% of the nondemented white population has at least one ε4 allele (12–15% allele frequency), and about 2% are ε4/ε4 homozygotes. Among patients with AD, 40–65% have at least one ε4 allele, a highly significant elevation compared with controls. Conversely, many AD patients have no ε4 allele, and ε4 carriers may never develop AD. Therefore, ε4 is neither necessary nor sufficient to cause AD. Nevertheless, the Apo ε4 allele represents the most important genetic risk factor for sporadic AD and acts as a dose-dependent disease modifier, with the earliest age of onset associated with the ε4 homozygosity. Precise mechanisms through which Apo ε4 confers AD risk or hastens onset remain unclear, but ε4 leads to less efficient amyloid clearance and to the production of toxic fragments from cleavage of the molecule. Apo ε can be identified in neuritic plaques and may also be involved in neurofibrillary tangle formation, because it binds to tau protein. Apo ε4 decreases neurite outgrowth in dorsal root ganglion neuronal cultures, perhaps indicating a deleterious role in the brain’s response to injury. Some evidence suggests that the ε2 allele may reduce AD risk. Use of Apo ε testing in AD diagnosis remains controversial. It is not indicated as a predictive test in normal persons because its precise predictive value is unclear, and many individuals with the ε4 allele never develop dementia. Many cognitively normal ε4 heterozygotes and homozygotes show decreased cerebral cortical metabolic function with PET, suggesting presymptomatic abnormalities due to AD or an inherited vulnerability of the AD-targeted network. In demented persons who meet clinical criteria for AD, finding an ε4 allele increases the reliability of diagnosis; however, the absence of an ε4 allele cannot be considered evidence against AD. Furthermore, all patients with dementia, including those with an ε4 allele, require a search for reversible causes of their cognitive impairment. Nevertheless, Apo ε4 remains the single most important biologic marker associated with AD risk, and studies of ε4’s functional role and diagnostic utility are progressing rapidly. The ε4 allele is not associated with risk for FTD, DLB, or CJD, although some evidence suggests that ε4 may exacerbate the phenotype of non-AD degenerative disorders, head trauma, and other brain injuries. Additional genes are also likely to be involved in AD, especially as minor risk alleles for sporadic forms of the disease. Genome-wide association studies have implicated the clusterin (CLU), phosphatidylinositol-binding clathrin assembly protein (PICALM), and complement component (3b/4b) receptor 1 (CR1) genes. CLU may play a role in synapse turnover, PICALM participates in clathrin-mediated endocytosis, and CR1 may be involved in amyloid clearance through the complement pathway. TREM2 is a gene involved with inflammation that increases the likelihood of dementia. Homozygous mutation carriers develop a frontal dementia with bone cysts (Nasu-Hakola disease), whereas heterozygotes are predisposed to the development of AD.
VASCULAR DEMENTIA
Dementia associated with cerebrovascular disease can be divided into two general categories: multi-infarct dementia and diffuse white matter disease (also called leukoaraiosis, subcortical arteriosclerotic leukoencephalopathy, or Binswanger’s disease). Cerebrovascular disease appears to be a more common cause of dementia in Asia than in Europe and North America, perhaps due to the increased prevalence of intracranial atherosclerosis. Individuals who have had strokes may develop chronic cognitive deficits, commonly called multi-infarct dementia. The strokes may be large or small (sometimes lacunar) and usually involve several different brain regions. The occurrence of dementia depends partly on the total volume of damaged cortex. Patients typically report previous discrete episodes of sudden neurologic deterioration. Many patients with multi-infarct dementia have a history of hypertension, diabetes, coronary artery disease, or other manifestations of widespread atherosclerosis. Physical examination may show focal neurologic deficits such as hemiparesis, a unilateral Babinski sign, a visual field defect, or pseudobulbar palsy. Recurrent strokes result in a stepwise disease progression. Neuroimaging reveals multiple areas of infarction. Thus, the history and neuroimaging findings differentiate this condition from AD; however, both AD and multiple infarctions are common and sometimes co-occur. With normal aging, there is also an accumulation of amyloid in cerebral blood vessels, leading to a condition called cerebral amyloid angiopathy (without dementia), which predisposes older persons to lobar hemorrhage and brain microhemorrhages. AD patients appear to be at increased risk for amyloid angiopathy, and this association may explain some of the observed links between AD and stroke.
Some individuals with dementia are discovered on MRI to have bilateral T2 signal hyperintensities in the subcortical white matter, termed diffuse white matter disease, often occurring in association with lacunar infarctions (see Fig. 35-2). The dementia may be insidious in onset and progress slowly, features that distinguish it from multi-infarct dementia, but other patients show a stepwise deterioration more typical of multi-infarct dementia. Early symptoms include mild confusion, apathy, anxiety, psychosis, and memory, spatial, or executive deficits. Marked difficulties in judgment and orientation and dependence on others for daily activities develop later. Euphoria, elation, depression, or aggressive behaviors are common as the disease progresses. Pyramidal and cerebellar signs may be present, and a gait disorder is seen in at least half of these patients. With advanced disease, urinary incontinence and dysarthria with or without other pseudobulbar features (e.g., dysphagia, emotional lability) are frequent. Seizures and myoclonic jerks appear in a minority of patients. Often, this disorder results from chronic ischemia due to occlusive disease of small, penetrating cerebral arteries and arterioles (microangiopathy). Any disease-causing stenosis of small cerebral vessels may be the critical underlying factor, although hypertension is the major cause. The term Binswanger’s disease should be used with caution, because it does not clearly identify a single entity.
Other rare causes of white matter disease also present with dementia, such as adult metachromatic leukodystrophy (arylsulfatase A deficiency) and progressive multifocal leukoencephalopathy (Chap. 164). A dominantly inherited form of white matter disease is known as cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL), discussed later in “Other Causes of Dementia.”
Mitochondrial disorders can present with stroke-like episodes and can selectively injure basal ganglia or cortex. Many such patients show other findings suggestive of a neurologic or systemic disorder such as ophthalmoplegia, retinal degeneration, deafness, myopathy, neuropathy, or diabetes. Diagnosis is difficult, but serum or (especially) CSF levels of lactate and pyruvate may be abnormal, and biopsy of affected tissue, preferably muscle, may be diagnostic.
Treatment of vascular dementia must be focused on preventing new ischemic injury by stabilizing or removing the underlying causes, such as hypertension, diabetes, smoking, or lack of exercise. Recovery of lost cognitive function is not likely, although fluctuations with periods of improvement are common.
FRONTOTEMPORAL LOBAR DEGENERATION SPECTRUM
Frontotemporal dementia (FTD) refers to a group of clinical syndromes united by underlying frontotemporal lobar degeneration (FTLD) pathology. FTD most often begins in the fifth to seventh decades and is nearly as prevalent as AD in this age group. Early studies suggested that FTD may be more common in men than women, although more recent reports cast doubt on this finding. Although a family history of dementia is common, autosomal dominant inheritance is seen in only 10–20% of all FTD cases.
The clinical heterogeneity seen in familial and sporadic FTD is remarkable. Three core clinical syndromes have been described (Fig. 448-3). In the behavioral variant (bvFTD), the most common FTD syndrome, social and emotional systems dysfunction manifests as apathy, disinhibition, compulsivity, loss of empathy, and overeating, often but not always accompanied by deficits in executive control. Two forms of primary progressive aphasia (PPA), the semantic and nonfluent/agrammatic variants, are commonly due to FTLD and included under the FTD umbrella. In the semantic variant, patients slowly lose the ability to decode word, object, person-specific, and emotion meaning, whereas patients with the nonfluent/agrammatic variant develop profound inability to produce words, often with prominent motor speech impairment. Any of these three clinical syndromes, but most often bvFTD, may be accompanied by motor neuron disease (MND), in which case the term FTD-MND is applied. In addition, the corticobasal syndrome (CBS) and progressive supranuclear palsy syndrome (PSP-S) can be considered part of the FTLD clinical spectrum. Furthermore, patients may evolve from any of the major syndromes described above to have prominent features of another syndrome.
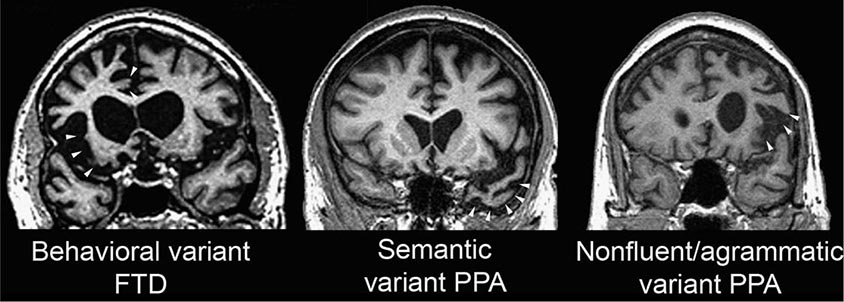
FIGURE 448-3 Three major frontotemporal dementia (FTD) clinical syndromes. Coronal magnetic resonance imaging sections from representative patients with behavioral variant FTD (left), semantic dementia (center), and progressive nonfluent aphasia (right). Areas of early and severe atrophy in each syndrome are highlighted (white arrowheads). The behavioral variant features anterior cingulate and frontoinsular atrophy, spreading to orbital and dorsolateral prefrontal cortex. Semantic variant primary progressive aphasia (PPA) shows prominent temporopolar atrophy, more often on the left. Nonfluent/agrammatic variant PPA is associated with dominant frontal opercular and dorsal insula degeneration.
Findings at the bedside are dictated by the anatomic localization of the disorder. Right hemisphere-predominant or symmetric anterior cingulate/medial prefrontal, orbital, and anterior insular degeneration predicts bvFTD. Patients with nonfluent/agrammatic PPA show left (dominant) frontal opercular and precentral gyrus degeneration, whereas left anterior temporal atrophy presents with semantic variant PPA. Visuoconstructive ability, arithmetic calculations, and navigation may remain normal late into any FTD syndrome. Many patients with nonfluent aphasia or bvFTD later develop PSP-S, as disease spreads into diencephalic and brainstem structures, or CBS-like features, as disease moves into dorsal and lateral perirolandic cortices.
The most common autosomal dominantly inherited mutations causing FTD involve the C9ORF72 (chromosome 9), GRN (chromosome 17), and MAPT (chromosome 17) genes. Hexanucleotide (GGGGCC) expansions in the noncoding portion of C9ORF72 are the most recently identified and represent the most common genetic cause of familial or sporadic FTD (usually presenting as bvFTD with or without MND) and amyotrophic lateral sclerosis (ALS). The expansion is associated with reduced C9ORF72 mRNA expression, nuclear mRNA foci containing transcribed portions of the expansion and other mRNAs, neuronal cytoplasmic inclusions containing dipeptide repeat proteins translated from the repeat mRNA, and transactive response DNA-binding protein of 43 kDa (TDP-43) neuronal cytoplasmic and glial inclusions. The pathogenic significance of these various features is a topic of vigorous investigation. MAPT mutations lead to a change in the alternate splicing of tau or cause loss of function in the tau molecule, thereby altering microtubule binding. With GRN, mutations in the coding sequence of the gene encoding progranulin protein result in mRNA degradation due to nonsense-mediated decay, providing a rare example of an autosomal dominant mutation that leads to haploinsufficiency and leads to a ~50% reduction in circulating progranulin protein levels. Intriguingly, a patient with GRN mutations on both chromosomes was recently reported to develop neuronal ceroid lipofuscinosis, focusing investigators on the lysosome as a site of molecular dysfunction in GRN-related FTD. Progranulin is a growth factor that binds to tumor necrosis factor (TNF) receptors and participates in tissue repair and tumor growth. How progranulin mutations lead to FTD remains unknown, but the most likely mechanisms include lysosomal dysfunction and enhanced neuroinflammation. Both MAPT and GRN mutations are associated with parkinsonian features, whereas ALS is rare. Infrequently, mutations in the valosin-containing protein (VCP, chromosome 9) and charged multivesicular body protein 2b (CHMP2b, chromosome 3) genes also lead to autosomal dominant familial FTD. Mutations in the TARDBP (encoding TDP-43) and FUS (encoding fused in sarcoma [FUS]) genes (see below) cause familial ALS, sometimes in association with an FTD syndrome, although a few patients presenting with FTD alone have been reported.
The gross pathologic hallmark of FTLD is a focal atrophy of frontal, insular, and/or temporal cortex, which can be visualized with neuroimaging studies (Fig. 448-3) and is often profound at autopsy. Despite the appearance of advanced disease, however, imaging studies suggest that atrophy often begins focally in one hemisphere before spreading to anatomically interconnected regions, including basal ganglia. Loss of cortical serotonergic innervation is seen in many patients. In contrast to AD, the cholinergic system is relatively spared in FTD, which accounts for the poor efficacy of acetylcholinesterase inhibitors in this group.
Although early studies suggested that 15–30% of patients with FTD showed underlying AD at autopsy, progressive refinement in clinical diagnosis has improved pathologic prediction accuracy, and most patients diagnosed with FTD at a dementia clinic with expertise in FTD will show underlying FTLD pathology. Microscopic findings seen across all patients with FTLD include gliosis, microvacuolation, and neuronal loss, but the disease is subtyped according to the protein composition of neuronal and glial inclusions, which contain either tau or TDP-43 in ~90% of patients, with the remaining ~10% showing inclusions containing FUS (Fig. 448-4).
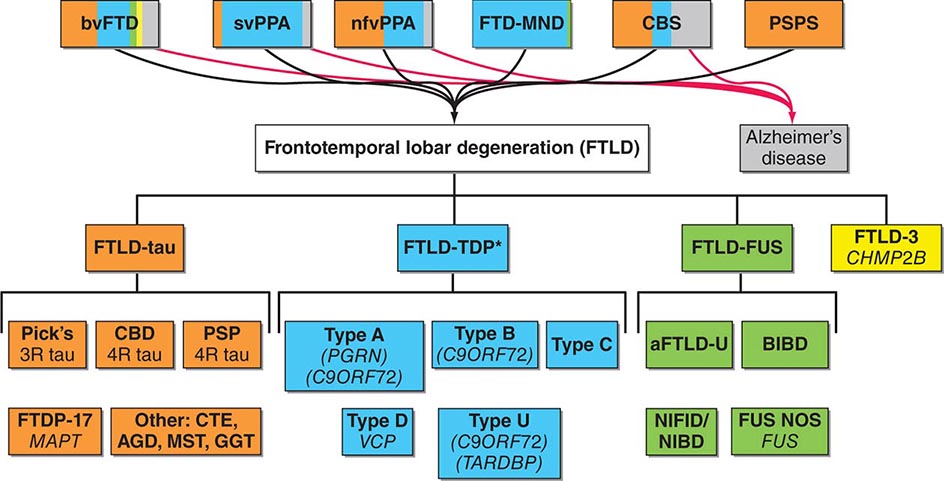
FIGURE 448-4 Frontotemporal dementia syndromes are united by underlying frontotemporal lobar degeneration pathology, which can be divided according to the presence of tau, TPD-43, or FUS-containing inclusions in neurons and glia. Correlations between clinical syndromes and major molecular classes are shown with colored shading. Despite improvements in clinical syndromic diagnosis, a small percentage of patients with some frontotemporal dementia syndromes will show Alzheimer’s disease neuropathology at autopsy (gray shading). aFTLD-U, atypical frontotemporal lobar degeneration with ubiquitin-positive inclusions; AGD, argyrophilic grain disease; BIBD, basophilic inclusion body disease; bvFTD, behavioral variant frontotemporal dementia; CBD, corticobasal degeneration; CBS, corticobasal syndrome; CTE, chronic traumatic encephalopathy; FTD-MND, frontotemporal dementia with motor neuron disease; FTDP-17, frontotemporal dementia with parkinsonism linked to chromosome 17; FUS, fused in sarcoma; GGT, globular glial tauopathy; MST, multisystem tauopathy; nfvPPA, nonfluent/agrammatic variant primary progressive aphasia; NIBD, neurofilament inclusion body disease; NIFID, neuronal intermediate filament inclusion disease; PSP, progressive supranuclear palsy; PSPS, progressive supranuclear palsy syndrome; svPPA, semantic variant primary progressive aphasia; Type U, unclassifiable type.
The toxicity and spreading capacity of tau aggregates underlies the pathogenesis of many familial cases and is emerging as a key factor in sporadic tauopathies, although loss of tau microtubule stabilizing function may also play a role. TDP-43 and FUS, in contrast, are RNA/DNA binding proteins whose roles in neuronal function are still being actively investigated, but one key role may be the chaperoning of mRNAs to the distal neuron for activity-dependent translation within dendritic spines. Because these proteins also form intracellular aggregates and produce similar anatomic progression, protein toxicity and spreading may also factor heavily in the pathogenesis of these FTLD-TDP and FTLD-FUS.
Increasingly, misfolded proteins in neurodegenerative disease are being recognized as having “prion-like” properties in that they can template the misfolding of their natively folded protein counterparts, a process that creates exponential amplification of protein misfolding within a cell and may promote transcellular and even transsynaptic protein propagation between cells. This hypothesis could provide a unifying explanation for the stereotypical patterns of disease spread observed in each syndrome (Chap. 444e).
Although the term Pick’s disease was once used to describe a progressive degenerative disorder characterized by selective involvement of the anterior frontal and temporal neocortex and pathologically by intraneuronal cytoplasmic inclusions (Pick bodies), it is now used only in reference to a specific FTLD-tau histopathologic entity. Classical Pick bodies are argyrophilic, staining positively with the Bielschowsky silver method (but not with the Gallyas method) and also with immunostaining for hyperphosphorylated tau. Recognition of the three FTLD major molecular classes has allowed delineation of distinct FTLD subtypes within each class. These subtypes, based on the morphology and distribution of the neuronal and glial inclusions (Fig. 448-5), account for the vast majority of patients, and some subtypes show strong clinical or genetic associations (Fig. 448-4). Despite this progress, available data do not allow reliable prediction of the underlying FTLD subtype, or even the major molecular class, based on clinical features alone. Molecular PET imaging with ligands chosen to bind misfolded tau protein shows great promise and is already being applied to the study of patients with AD and FTD. Because FTLD-tau and FTLD-TDP account for 90% of FTLD patients, the ability to detect pathologic tau protein deposition in vivo will greatly improve prediction accuracy, especially when amyloid PET imaging is negative.
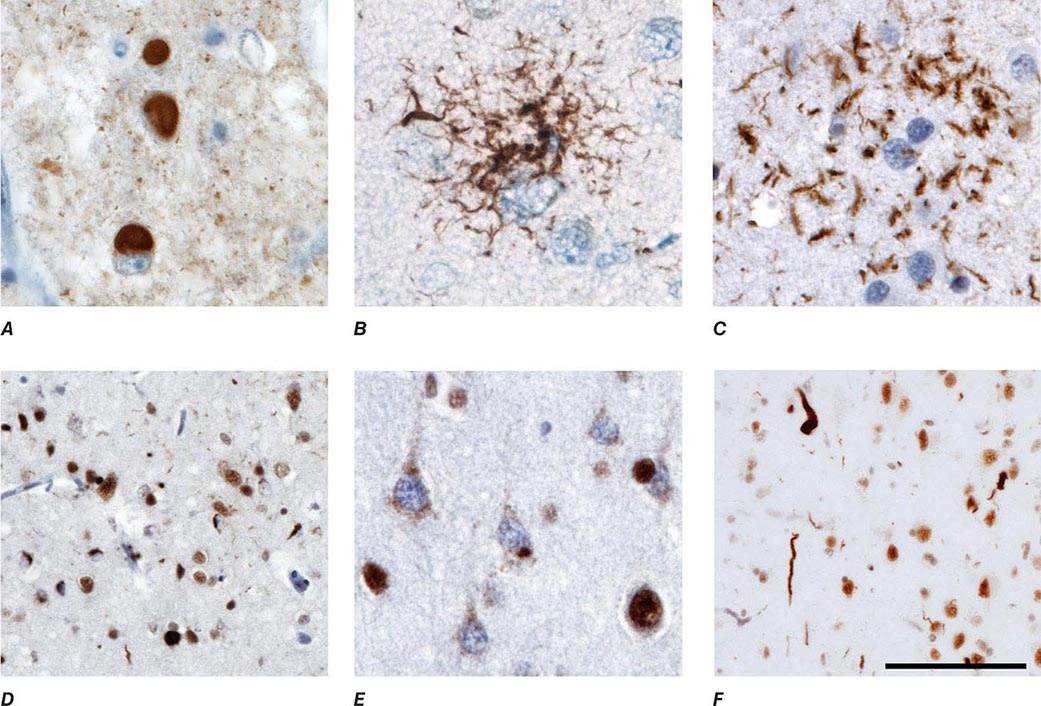
FIGURE 448-5 Neuropathology in frontotemporal lobar degeneration (FTLD). FTLD-tau (A–C) and FTLD-TDP (D–F) account for over 90% of patients with FTLD, and immunohistochemistry reveals characteristic lesions in each of the major histopathologic subtypes within each class: (A) Pick bodies in Pick’s disease; (B) a tufted astrocyte in progressive supranuclear palsy; (C) an astrocytic plaque in corticobasal degeneration; (D) small compact or crescentic neuronal cytoplasmic inclusions and short, then neuropil threads in FTLD-TDP, type A; (E) diffuse/granular neuronal cytoplasmic inclusions (with a relative paucity of neuropil threads) in FTLD-TDP, type B; and (F) long, tortuous dystrophic neurites in FTLD-TDP, type C. TDP can be seen within the nucleus in neurons lacking inclusions but mislocalizes to the cytoplasm and forms inclusions in FTLD-TDP. Immunostains are 3-repeat tau (A), phospho-tau (B and C), and TDP-43 (D–F). Sections are counterstained with hematoxylin. Scale bar applies to all panels and represents 50 μm in A, B, C, and E and 100 μm in D and F.
The burden on caregivers of patients with FTD is extremely high, especially when the illness disrupts core emotional and personality functions of the loved one. Treatment is symptomatic, and there are currently no therapies known to slow progression or improve symptoms. Many of the behaviors that may accompany FTD, such as depression, hyperorality, compulsions, and irritability, can be ameliorated with antidepressants, especially SSRIs. The co-association with motor disorders such as parkinsonism necessitates the careful use of antipsychotics, which can exacerbate this problem.
Progressive supranuclear palsy syndrome (PSP-S; also known as Steele-Richardson-Olszewski syndrome) is a degenerative disorder that involves the brainstem, basal ganglia, limbic structures, and selected areas of cortex. Clinically, PSP-S begins with falls and executive or subtle personality changes (such as mental rigidity, impulsivity, or apathy). Shortly thereafter, a progressive oculomotor syndrome ensues that begins with square wave jerks, followed by slowed saccades (vertical worse than horizontal) before resulting in progressive supranuclear ophthalmoparesis. Dysarthria, dysphagia, and symmetric axial rigidity can be prominent features that emerge at any point in the illness. A stiff, unstable posture with hyperextension of the neck and a slow, jerky, toppling gait are characteristic. Frequent unexplained and sometimes spectacular falls are common secondary to a combination of axial rigidity, inability to look down, and poor judgment. Even once patients have severely limited voluntary eye movements, they retain oculocephalic reflexes (demonstrated using a vertical doll’s head maneuver); thus, the oculomotor disorder is supranuclear. The dementia overlaps with bvFTD, featuring apathy, frontal-executive dysfunction, poor judgment, slowed thought processes, impaired verbal fluency, and difficulty with sequential actions and with shifting from one task to another. These features are common at presentation and often precede the motor syndrome. Some patients with a pathologic diagnosis of PSP begin with a nonfluent aphasia or motor speech disorder and progress to classical PSP-S. Response to L-dopa is limited or absent; no other treatments exist. Death occurs within 5–10 years of onset. Like Pick’s disease, increasingly the term PSP is used to refer to a specific histopathologic entity within the FTLD-tau class. In PSP, accumulation of hyperphosphorylated 4-repeat tau is seen within neurons and glia. Neuronal inclusions often take the form of NFTs, which may be large, spherical (“globose”), and coarse in brainstem, cerebellar dentate, and diencephalic neurons. Tau deposition is most prominent in subcortical structures (including the subthalamic nucleus, globus pallidus, substantia nigra, locus coeruleus, periaqueductal gray, tectum, oculomotor nuclei, and dentate nucleus of cerebellum). Neocortical NFTs, like those in AD, often take on a more flame-shaped morphology, but on electron microscopy, PSP tangles can be shown to consist of straight tubules rather than the paired helical filaments found in AD. Furthermore, PSP is associated with prominent tau-positive glial pathologies, such as tufted astrocytes (Fig. 448-5), thorny astrocytes, and coiled oligodendroglial inclusions (“coiled bodies”). Most patients with PSP-S show PSP at autopsy, although small numbers will show another tauopathy (corticobasal degeneration [CBD] or Pick’s disease; Fig. 448-4).
In addition to its overlap with FTD and CBS (see below), PSP is often confused with idiopathic Parkinson’s disease (PD). Although elderly patients with PD may have restricted upgaze, they do not develop downgaze paresis or other abnormalities of voluntary eye movements typical of PSP. Dementia occurs in ~20% of patients with PD, often due to the emergence of a full-blown DLB-like syndrome. Furthermore, the behavioral syndromes seen with DLB differ from PSP (see below). Dementia in PD becomes more likely with increasing age, increasing severity of extrapyramidal signs, long disease duration, and the presence of depression. Patients with PD who develop dementia also show cortical atrophy on brain imaging. Neuropathologically, there may be AD-related changes in the cortex, LBD-related α-synuclein inclusions in both the limbic system and cortex, or no specific microscopic changes other than gliosis and neuronal loss. PD is discussed in detail in Chap. 449.
Corticobasal syndrome (CBS) is a slowly progressive dementia-movement disorder associated with severe atrophy in perirolandic cortex and basal ganglia (substantia nigra and striatopallidum). Patients typically present with asymmetric onset of rigidity, dystonia, myoclonus, and apraxia of one limb, at times associated with alien limb phenomena in which the limb exhibits unintended motor actions such as grasping, groping, drifting, or undoing. Eventually CBS becomes bilateral and leads to dysarthria, slow gait, action tremor, and typically a frontal-predominant dementia. Whereas CBS refers to the clinical syndrome, CBD refers to a specific histopathologic FTLD-tau entity (Fig. 448-4). Although CBS was once thought to be pathognomonic for CBD, increasingly it has been recognize that CBS can be due to CBD, PSP, FTLD-TDP, or even AD. In CBD, the microscopic features include ballooned, achromatic, tau-positive neurons; astrocytic plaques (Fig. 448-5); and other dystrophic glial tau pathomorphologies that overlap with those seen in PSP. Most specifically, CBD features a severe tauopathy burden in the subcortical white matter, consisting of threads and oligodendroglial coiled bodies. As shown in Fig. 448-4, patients with bvFTD, nonfluent/agrammatic PPA, and PSP-S may also show CBD at autopsy, emphasizing the importance of distinguishing clinical and pathologic constructs and terminology. Treatment of CBS remains symptomatic; no disease-modifying therapies are available.
PARKINSON’S DISEASE DEMENTIA AND DEMENTIA WITH LEWY BODIES
The parkinsonian dementia syndromes are under increasing study, with many cases unified by Lewy body and Lewy neurite pathology that ascends from the low brainstem up through the substantia nigra, limbic system, and cortex. The DLB clinical syndrome is characterized by visual hallucinations, parkinsonism, fluctuating alertness, falls, and often rapid eye movement (REM) sleep behavior disorder (RBD). Dementia can precede or follow the appearance of parkinsonism. Hence, one pathway occurs in patients with long-standing PD without cognitive impairment, who slowly develop a dementia that is associated with visual hallucinations and fluctuating alertness. When this occurs after an established diagnosis of PD, many use the term Parkinson’s disease dementia (PDD). In others, the dementia and neuropsychiatric syndrome precede or co-emerge with the parkinsonism, and this constellation is referred to as DLB. Both PDD and DLB may be accompanied or preceded by symptoms referable to brainstem pathology below the substantia nigra including constipation, orthostatic lightheadedness, or RBD, and many researchers conceptualize these disorders as points on a spectrum of α-synuclein pathology.
Patients with PDD and DLB are highly sensitive to metabolic perturbations, and in some patients, the first manifestation of illness is a delirium, often precipitated by an infection, new medicine, or other systemic disturbance. A hallucinatory delirium induced by L-dopa, prescribed for parkinsonian symptoms attributed to PD, may likewise provide the initial clue to a PDD or DLB diagnosis. Conversely, patients with mild cognitive deficits and hallucinations may receive typical or atypical antipsychotic medications, which induce profound parkinsonism at low doses due to a subclinical DLB-related nigral dopaminergic neuron loss. Even without an underlying precipitant, fluctuations can be marked in DLB, with episodic confusion or even stupor admixed with lucid intervals. Despite the fluctuating pattern, however, the core clinical features persist, unlike delirium, which resolves following correction of the inciting factor. Cognitively, DLB features relative preservation of memory but more severe visuospatial and executive deficits than seen in patients with early AD.
The key neuropathologic feature in DLB is the presence of Lewy bodies and Lewy neurites throughout specific brainstem nuclei, substantia nigra, amygdala, cingulate gyrus, and, ultimately, the neocortex. Lewy bodies are intraneuronal cytoplasmic inclusions that stain with periodic acid–Schiff (PAS) and ubiquitin but are now identified with antibodies to the presynaptic protein, α-synuclein. Lewy bodies are composed of straight neurofilaments 7–20 nm long with surrounding amorphous material and contain epitopes recognized by antibodies against phosphorylated and nonphosphorylated neurofilament proteins, ubiquitin, and α-synuclein. Lewy bodies are typically found in the substantia nigra of patients with idiopathic PD, where they can be readily seen with hematoxylin-and-eosin staining. A profound cholinergic deficit, owing to basal forebrain and pedunculopontine nucleus involvement, is present in many patients with DLB and may be a factor responsible for the fluctuations, inattention, and visual hallucinations.
Due to the frequent comorbidity with AD and the cholinergic deficit in DLB, cholinesterase inhibitors often provide significant benefit, reducing hallucinosis, stabilizing delusional symptoms, and even helping with RBD in some patients. Exercise programs maximize motor function and protect against fall-related injury. Antidepressants are often necessary. Atypical antipsychotics may be required for psychosis but can worsen extrapyramidal syndromes, even at low doses, and increase risk of death. Patients with DLB are extremely sensitive to dopaminergic medications, which must be carefully titrated; tolerability may be improved by concomitant use of a cholinesterase inhibitor.
OTHER CAUSES OF DEMENTIA
Prion diseases such as Creutzfeldt-Jakob disease (CJD) are rare neurodegenerative conditions (prevalence ~1 per million) that produce dementia. CJD is a rapidly progressive disorder associated with dementia, focal cortical signs, rigidity, and myoclonus, causing death <1 year after first symptoms appear. The rapidity of progression seen with CJD is uncommon in AD so that the distinction between the two disorders is usually straightforward. CBD and DLB, more rapid degenerative dementias with prominent movement abnormalities, are more likely to be mistaken for CJD. The differential diagnosis for CJD includes other rapidly progressive dementing conditions such as viral or bacterial encephalitides, Hashimoto’s encephalopathy, central nervous system (CNS) vasculitis, lymphoma, or paraneoplastic/autoimmune syndromes. The markedly abnormal periodic complexes on EEG and cortical ribboning and basal ganglia hyperintensities on fluid-attenuate inversion recovery MRI are diagnostic features of CJD, although rarely, prolonged focal or generalized seizures can produce a similar imaging appearance. Prion diseases are discussed in detail in Chap. 453e.
Huntington’s disease (HD) (Chap. 449) is an autosomal dominant degenerative brain disorder. HD clinical hallmarks include chorea, behavioral disturbance, and executive impairment. Symptoms typically begin in the fourth or fifth decade, but there is a wide range, from childhood to >70 years. Memory is frequently not impaired until late in the disease, but attention, judgment, self-awareness, and executive functions are often deficient at an early stage. Depression, apathy, social withdrawal, irritability, and intermittent disinhibition are common. Delusions and obsessive-compulsive behavior may occur. Disease duration is variable but typically lasts approximately 15 years.
Normal-pressure hydrocephalus (NPH) is a relatively uncommon but treatable syndrome. The clinical, physiologic, and neuroimaging characteristics of NPH must be carefully distinguished from those of other dementias associated with gait impairment. Historically, many patients treated for NPH have suffered from other dementias, particularly AD, vascular dementia, DLB, and PSP. For NPH, the clinical triad includes an abnormal gait (ataxic or apractic), dementia (usually mild to moderate, with an emphasis on executive impairment), and urinary urgency or incontinence. Neuroimaging reveals enlarged lateral ventricles (hydrocephalus) with little or no cortical atrophy, although the sylvian fissures may appear propped open (so-called “boxcarring”), which can be mistaken for perisylvian atrophy. This syndrome is a communicating hydrocephalus with a patent aqueduct of Sylvius (see Fig. 35-3), in contrast to aqueductal stenosis, in which the aqueduct is small. Lumbar puncture opening pressure falls in the high-normal range, and the CSF protein, glucose, and cell counts are normal. NPH may be caused by obstruction to normal CSF flow over the cerebral convexities and delayed resorption into the venous system. The indolent nature of the process results in enlarged lateral ventricles with relatively little increase in CSF pressure. Presumed edema, stretching, and distortion of subfrontal white matter tracts may lead to clinical symptoms, but the precise underlying pathophysiology remains unclear. Some patients provide a history of conditions that produce meningeal scarring (blocking CSF resorption) such as previous meningitis, subarachnoid hemorrhage, or head trauma. Others with long-standing but asymptomatic congenital hydrocephalus may have adult-onset deterioration in gait or memory that is confused with NPH. In contrast to AD, the patient with NPH complains of an early and prominent gait disturbance without cortical atrophy on CT or MRI.
Numerous attempts to improve NPH diagnosis with various special studies and predict the success of ventricular shunting have been undertaken. These tests include radionuclide cisternography (showing a delay in CSF absorption over the convexity) and various efforts to monitor and alter CSF flow dynamics, including a constant-pressure infusion test. None has proven to be specific or consistently useful. A transient improvement in gait or cognition may follow lumbar puncture (or serial punctures) with removal of 30–50 mL of CSF, but this finding has also not proved to be consistently predictive of postshunt improvement. Perhaps the most reliable strategy is a period of close inpatient evaluation before, during, and after lumbar CSF drainage. Occasionally, when a patient with AD presents with gait impairment (at times due to comorbid subfrontal vascular injury) and absent or only mild cortical atrophy on CT or MRI, distinguishing NPH from AD can be challenging. Hippocampal atrophy on MRI favors AD, whereas a characteristic “magnetic” gait with external hip rotation, low foot clearance, and short strides, along with prominent truncal sway or instability, favors NPH. The diagnosis of NPH should be avoided when hydrocephalus is not detected on imaging studies, even if the symptoms otherwise fit. Thirty to fifty percent of patients identified by careful diagnosis as having NPH will improve with ventricular shunting. Gait may improve more than cognition, but many reported failures to improve cognitively may have resulted from comorbid AD. Short-lasting improvement is common. Patients should be carefully selected for shunting, because subdural hematoma, infection, and shunt failure are known complications and can be a cause for early nursing home placement in an elderly patient with previously mild dementia.
Intracranial hypotension, sometimes called sagging brain syndrome, is a disorder caused by low CSF pressure, leading to downward pressure on the subcortical structures and disruption of cerebral function. It presents in a variable manner with headache, often exacerbated by coughing or a Valsalva maneuver or by moving from lying to standing. Other common symptoms include dizziness, vomiting, disruption of sleep-wake cycles, and sometimes a progressive bvFTD-like syndrome. Although sometimes idiopathic, this syndrome can be caused by CSF leaks secondary to lumbar puncture, head trauma, or spinal cord arachnoid cysts. Treatment consists of finding and patching CSF leaks.
Dementia can accompany chronic alcoholism (Chap. 467) and may result from associated malnutrition, especially of B vitamins, particularly thiamine. Other poorly defined aspects of chronic alcoholism may, however, also produce cerebral damage. A rare idiopathic syndrome of dementia and seizures with degeneration of the corpus callosum has been reported primarily in male Italian red wine drinkers (Marchiafava-Bignami disease).
Thiamine (vitamin B1) deficiency causes Wernicke’s encephalopathy (Chap. 330). The clinical presentation features a malnourished patient (frequently but not necessarily alcoholic) with confusion, ataxia, and diplopia resulting from inflammation and necrosis of periventricular midline structures, including dorsomedial thalamus, mammillary bodies, midline cerebellum, periaqueductal gray matter, and trochlear and abducens nuclei. Damage to the dorsomedial thalamus correlates most closely with the memory loss. Prompt administration of parenteral thiamine (100 mg intravenously for 3 days followed by daily oral dosage) may reverse the disease if given in the first days of symptom onset. Prolonged untreated thiamine deficiency can result in an irreversible and profound amnestic syndrome (Korsakoff’s syndrome) or even death.
In Korsakoff’s syndrome, the patient is unable to recall new information despite normal immediate memory, attention span, and level of consciousness. Memory for new events is seriously impaired, whereas knowledge acquired prior to the illness remains relatively intact. Patients are easily confused, disoriented, and cannot store information for more than a few minutes. Superficially, they may be conversant, engaging, and able to perform simple tasks and follow immediate commands. Confabulation is common, although not always present. There is no specific treatment because the previous thiamine deficiency has produced irreversible damage to the medial thalamic nuclei and mammillary bodies. Mammillary body atrophy may be visible on MRI in the chronic phase (see Fig. 330-6).
Vitamin B12 deficiency, as can occur in pernicious anemia, causes a megaloblastic anemia and may also damage the nervous system (Chaps. 128 and 456). Neurologically, it most commonly produces a spinal cord syndrome (myelopathy) affecting the posterior columns (loss of vibration and position sense) and corticospinal tracts (hyperactive tendon reflexes with Babinski signs); it also damages peripheral nerves (neuropathy), resulting in sensory loss with depressed tendon reflexes. Damage to myelinated axons may also cause dementia. The mechanism of neurologic damage is unclear but may be related to a deficiency of S-adenosyl methionine (required for methylation of myelin phospholipids) due to reduced methionine synthase activity or accumulation of methylmalonate, homocysteine, and propionate, providing abnormal substrates for fatty acid synthesis in myelin. Use of histamine blockers or metformin, vegan diets, autoimmunity against gastric parietal cells, and various causes of malabsorption are the typical causes for vitamin B12 deficiency. The neurologic sequelae of vitamin B12 deficiency may occur in the absence of hematologic manifestations, making it critical to avoid using the complete blood count (CBC) and blood smear as a substitute for measuring B12 blood levels. Treatment with parenteral vitamin B12 (1000 μg intramuscularly daily for a week, weekly for a month, and monthly for life for pernicious anemia) stops progression of the disease if instituted promptly, but complete reversal of advanced nervous system damage will not occur.
Deficiency of nicotinic acid (pellagra) is associated with skin rash over sun-exposed areas, glossitis, and angular stomatitis (Chap. 96e). Severe dietary deficiency of nicotinic acid along with other B vitamins such as pyridoxine may result in spastic paraparesis, peripheral neuropathy, fatigue, irritability, and dementia. This syndrome has been seen in prisoners of war and in concentration camps but should be considered in any malnourished individual. Low serum folate levels appear to be a rough index of malnutrition, but isolated folate deficiency has not been proved as a specific cause of dementia.
CNS infections usually cause delirium and other acute neurologic syndromes. However, some chronic CNS infections, particularly those associated with chronic meningitis (Chap. 165), may produce a dementing illness. The possibility of chronic infectious meningitis should be suspected in patients presenting with a dementia or behavioral syndrome, who also have headache, meningismus, cranial neuropathy, and/or radiculopathy. Between 20 and 30% of patients in the advanced stages of HIV infection become demented (Chap. 226). Cardinal features include psychomotor retardation, apathy, and impaired memory. This syndrome may result from secondary opportunistic infections but can also be caused by direct infection of CNS neurons with HIV. Neurosyphilis (Chap. 206) was a common cause of dementia in the preantibiotic era; it is now uncommon but can still be encountered in patients with multiple sex partners, particularly among patients with HIV. Characteristic CSF changes consist of pleocytosis, increased protein, and a positive Venereal Disease Research Laboratory (VDRL) test.
Primary and metastatic neoplasms of the CNS (Chap. 118) usually produce focal neurologic findings and seizures rather than dementia, but if tumor growth begins in the frontal or temporal lobes, the initial manifestations may be memory loss or behavioral changes. A paraneoplastic syndrome of dementia associated with occult carcinoma (often small-cell lung cancer) is termed limbic encephalitis. In this syndrome, confusion, agitation, seizures, poor memory, emotional changes, and frank dementia may occur. Paraneoplastic encephalitis associated with NMDA receptor antibodies presents as a progressive psychiatric disorder with memory loss and seizures; affected patients are often young women with ovarian teratoma (Chap. 122).
A nonconvulsive seizure disorder (Chap. 445) may underlie a syndrome of confusion, clouding of consciousness, and garbled speech. Often, psychiatric disease is suspected, but an EEG demonstrates the epileptic nature of the illness. If recurrent or persistent, the condition may be termed complex partial status epilepticus. The cognitive disturbance often responds to anticonvulsant therapy. The etiology may be previous small strokes or head trauma; some cases are idiopathic.
It is important to recognize systemic diseases that indirectly affect the brain and produce chronic confusion or dementia. Such conditions include hypothyroidism; vasculitis; and hepatic, renal, or pulmonary disease. Hepatic encephalopathy may begin with irritability and confusion and slowly progress to agitation, lethargy, and coma.
Isolated vasculitis of the CNS (CNS granulomatous angiitis) (Chaps. 385 and 446) occasionally causes a chronic encephalopathy associated with confusion, disorientation, and clouding of consciousness. Headache is common, and strokes and cranial neuropathies may occur. Brain imaging studies may be normal or nonspecifically abnormal. CSF analysis reveals a mild pleocytosis or protein elevation. Cerebral angiography can show multifocal stenoses involving medium-caliber vessels, but some patients have only small-vessel disease that is not revealed on angiography. The angiographic appearance is not specific and may be mimicked by atherosclerosis, infection, or other causes of vascular disease. Brain or meningeal biopsy demonstrates endothelial cell proliferation and mononuclear infiltrates within blood vessel walls. The prognosis is often poor, although the disorder may remit spontaneously. Some patients respond to glucocorticoids or chemotherapy.
Chronic metal exposure represents a rare cause of dementia. The key to diagnosis is to elicit a history of exposure at work or home. Chronic lead poisoning from inadequately fire-glazed pottery has been reported. Fatigue, depression, and confusion may be associated with episodic abdominal pain and peripheral neuropathy. Gray lead lines appear in the gums, usually accompanied by an anemia with basophilic stippling of red blood cells. The clinical presentation can resemble that of acute intermittent porphyria, including elevated levels of urine porphyrins as a result of the inhibition of δ-aminolevulinic acid dehydrase. The treatment is chelation therapy with agents such as ethylenediamine tetraacetic acid (EDTA). Chronic mercury poisoning produces dementia, peripheral neuropathy, ataxia, and tremulousness that may progress to a cerebellar intention tremor or choreoathetosis. The confusion and memory loss of chronic arsenic intoxication is also associated with nausea, weight loss, peripheral neuropathy, pigmentation and scaling of the skin, and transverse white lines of the fingernails (Mees’ lines). Treatment is chelation therapy with dimercaprol (BAL). Aluminum poisoning is rare but was documented with the dialysis dementia syndrome, in which water used during renal dialysis was contaminated with excessive amounts of aluminum. This poisoning resulted in a progressive encephalopathy associated with confusion, nonfluent aphasia, memory loss, agitation, and, later, lethargy and stupor. Speech arrest and myoclonic jerks were common and associated with severe and generalized EEG changes. The condition has been eliminated by the use of deionized water for dialysis.
Recurrent head trauma in professional athletes may lead to a dementia previously referred to as “punch-drunk” syndrome or dementia pugilistica but now known as chronic traumatic encephalopathy (CTE) to signify its relevance to contact sport athletes other than boxers. The symptoms can be progressive, beginning late in an athlete’s career or, more often, after retirement. Early in the course, a personality change associated with social instability and sometimes paranoia and delusions occurs. Later, memory loss progresses to full-blown dementia, often associated with parkinsonian signs and ataxia or intention tremor. At autopsy, the cerebral cortex shows changes tau-immunoreactive NFTs that are more prominent than amyloid plaques (which are usually diffuse or absent rather than neuritic). NFTs and tau-positive reactive astrocytes are often clustered in the depths of cortical sulci, and TDP-43 inclusions have also been reported, highlighting the overlap with the FTLD spectrum. Loss of neurons in the substantia nigra is a variable feature.
Chronic subdural hematoma (Chap. 457e) is also occasionally associated with dementia, often in the context of underlying cortical atrophy from conditions such as AD or HD.
Transient global amnesia (TGA) is characterized by the sudden onset of a severe episodic memory deficit, usually occurring in persons over the age of 50 years. Often the amnesia occurs in the setting of an emotional stimulus or physical exertion. During the attack, the individual is alert and communicative, general cognition seems intact, and there are no other neurologic signs or symptoms. The patient may seem confused and repeatedly ask about his or her location in place and time. The ability to form new memories returns after a period of hours, and the individual returns to normal with no recall for the period of the attack. Frequently no cause is determined, but cerebrovascular disease, epilepsy (7% in one study), migraine, or cardiac arrhythmias have all been implicated. Approximately one-quarter of patients experience recurrent attacks. Rare instances of permanent memory loss have been reported in patients with TGA-like spells, usually representing ischemic infarction of the hippocampus or dorsomedial thalamic nucleus bilaterally. Seizure activity due to AD should always be suspected in this syndrome.
The ALS/parkinsonian/dementia complex of Guam is a rare degenerative disease that has occurred in the Chamorro natives on the island of Guam. Individuals may have any combination of parkinsonian features, dementia, and MND. The most characteristic pathologic features are the presence of NFTs in degenerating neurons of the cortex and substantia nigra and loss of motor neurons in the spinal cord, although recent reanalysis has shown that some patients with this illness also show coexisting TDP-43 pathology. Epidemiologic evidence supports a possible environmental cause, such as exposure to a neurotoxin or an infectious agent with a long latency period. One interesting but unproven candidate neurotoxin occurs in the seed of the false palm tree, which Guamanians traditionally used to make flour. The ALS syndrome is no longer present in Guam, but a dementing illness with rigidity continues to be seen.
Rarely, adult-onset leukodystrophies, lysosomal storage diseases, and other genetic disorders can present as a dementia in middle to late life. Metachromatic leukodystrophy (MLD) causes a progressive psychiatric or dementia syndrome associated with extensive, confluent frontal white matter abnormality. MLD is diagnosed by measuring arylsulfatase A enzyme activity in white blood cells. Adult-onset presentations of adrenoleukodystrophy have been reported in female carriers, and these patients often feature spinal cord and posterior white matter involvement. Adrenoleukodystrophy is diagnosed with measurement of plasma very-long-chain fatty acids. CADASIL is another genetic syndrome associated with white matter disease, often frontally and temporally predominant. Diagnosis is made with skin biopsy, which shows osmophilic granules in arterioles, or, increasingly, through genetic testing for mutations in Notch 3. The neuronal ceroid lipofuscinoses are a genetically heterogeneous group of disorders associated with myoclonus, seizures, vision loss, and progressive dementia. Diagnosis is made by finding eosinophilic curvilinear inclusions within white blood cells or neuronal tissue.
Psychogenic amnesia for personally important memories can be seen. Whether this results from deliberate avoidance of unpleasant memories, outright malingering, or unconscious repression remains unknown and probably depends on the patient. Event-specific amnesia is more likely to occur after violent crimes such as homicide of a close relative or friend or sexual abuse. It may develop in association with severe drug or alcohol intoxication and sometimes with schizophrenia. More prolonged psychogenic amnesia occurs in fugue states that also commonly follow severe emotional stress. The patient with a fugue state suffers from a sudden loss of personal identity and may be found wandering far from home. In contrast to neurologic amnesia, fugue states are associated with amnesia for personal identity and events closely associated with the personal past. At the same time, memory for other recent events and the ability to learn and use new information are preserved. The episodes usually last hours or days and occasionally weeks or months while the patient takes on a new identity. On recovery, there is a residual amnesia gap for the period of the fugue. Very rarely does selective loss of autobiographic information reflect a focal injury to the brain areas involved with these functions.
Psychiatric diseases may mimic dementia. Severely depressed or anxious individuals may appear demented, a phenomenon sometimes called pseudodementia. Memory and language are usually intact when carefully tested, and a significant memory disturbance usually suggests an underlying dementia, even if the patient is depressed. Patients in this condition may feel confused and unable to accomplish routine tasks. Vegetative symptoms, such as insomnia, lack of energy, poor appetite, and concern with bowel function, are common. Onset is often more abrupt, and the psychosocial milieu may suggest prominent reasons for depression. Such patients respond to treatment of the underlying psychiatric illness. Schizophrenia is usually not difficult to distinguish from dementia, but occasionally the distinction can be problematic. Schizophrenia generally has a much earlier age of onset (second and third decades) than most dementing illnesses and is associated with intact memory. The delusions and hallucinations of schizophrenia are usually more complex, bizarre, and threatening than those of dementia. Some chronic schizophrenics develop an unexplained progressive dementia late in life that is not related to AD. Conversely, FTD, HD, vascular dementia, DLB, AD, or leukoencephalopathy can begin with schizophrenia-like features, leading to the misdiagnosis of a psychiatric condition. Later age of onset, significant deficits on cognitive testing, or the presence of abnormal neuroimaging suggest a degenerative condition. Memory loss may also be part of a conversion disorder. In this situation, patients commonly complain bitterly of memory loss, but careful cognitive testing either does not confirm the deficits or demonstrates inconsistent or unusual patterns of cognitive problems. The patient’s behavior and “wrong” answers to questions often indicate that he or she understands the question and knows the correct answer.
Clouding of cognition by chronic drug or medication use, often prescribed by physicians, is an important cause of dementia. Sedatives, tranquilizers, and analgesics used to treat insomnia, pain, anxiety, or agitation may cause confusion, memory loss, and lethargy, especially in the elderly. Discontinuation of the offending medication often improves mentation.




