CHAPTER 65 ALZHEIMER’S DISEASE
DEFINITION
Two excellent sets of clinical criteria with good clinicopathological correlation are given in Tables 65-1 and 65-2.
|
Memory impairment: impaired ability to learn new information or to recall previously learned information
|
DSM-IV, Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition (American Psychiatric Association, 1994); HIV, human immunodeficiency virus.
TABLE 65-2 NINCDS-ARDA Criteria for Diagnosis of Probable Alzheimer’s Disease
CT, computed tomographic; EEG, electroencephalographic; NINCDS-ADRDA, National Institute of Neurological Disorders and Stroke–Alzheimer’s Disease Related Disorders Association.
Adapted from McKhann, G, Drachman D, Folstein M, et al: Clinical diagnosis of Alzheimer’s disease: report of the NINCDS/ADRDA Work Group under the auspices of Department of Health Services Task Force on Alzheimer’s Disease. Neurology 1984; 34:939-944.
EPIDEMIOLOGY
Alzheimer’s disease causes about 70% of cases of dementia.1 In the Framingham study, Bachman and colleagues reported that the incidence of Alzheimer’s disease increases from 3.5 per 1000 per year between the ages of 65 and 69 to 72.8 per 1000 per year at ages 85 to 89 years.2 Bachman and colleagues found that the incidence of Alzheimer’s disease doubled with every 5 years of age. Alzheimer’s disease prevalence is low among persons younger than 65 years, but it increases to 10% to 30% among persons older than 85.3
In 1907, when Alois Alzheimer described the disease named for him, mean life expectancy was 42 years; since then, in developed countries, life expectancy has greatly increased.4 For example, in the United States, 50% of persons are expected to live past 75 and 25% past 85. With age being such an important risk factor and so many people living into the high-risk ages, the prevalence of Alzheimer’s disease has greatly increased; it is 4.5 million in the United States, according to the 2000 census.5 Because of the staggering cost of caring for patients with Alzheimer’s disease (estimated at $100 billion annually in the United States6) and because “baby boomers” are reaching the high-risk period, it can be argued strongly that the world should invest heavily in finding ways to prevent this disease. In fact, Brookmeyer and associates7 predicted that if the onset of the disease is delayed by an average of 5 years, the present-day U.S. prevalence of Alzheimer’s disease would remain constant (at 4.5 million) instead of increasing to an anticipated 13 million in 2047.
RISK AND PUTATIVE PROTECTIVE FACTORS FOR ALZHEIMER’S DISEASE
Table 65-3 lists risk factors and Table 65-4 the putative protective factors for Alzheimer’s disease.
TABLE 65-4 Putative Protective Factors for Alzheimer’s Disease
Genetics
Researchers have discovered three genes that, if they have undergone mutation, cause early-onset Alzheimer’s disease. Although they account for fewer than 1% of cases of Alzheimer’s disease, the knowledge of how these genes probably cause Alzheimer’s disease has been helpful for understanding the pathogenesis of this disease. Table 65-5 lists the genes from the Alzheimer Disease and Frontotemporal Dementia Mutation Database and tabulates for each gene the number of mutations and families reported. By January 2005, investigators had reported a total 404 families with 189 different mutations in these three genes.
| Gene | Number of Mutations | Total Number of Families Published |
|---|---|---|
| APP | 18 | 48 |
| PSEN1 | 141 | 280 |
| PSEN2 | 10 | 16 |
Based on Alzheimer Disease and Frontotemporal Dementia Mutation Database, maintained by Marc Cruts and Roos Rademakers (www.molgen.ua.ac.be/ADMutations/, accessed June 8, 2006).
APP, amyloid precursor protein; PSEN, presenilin.
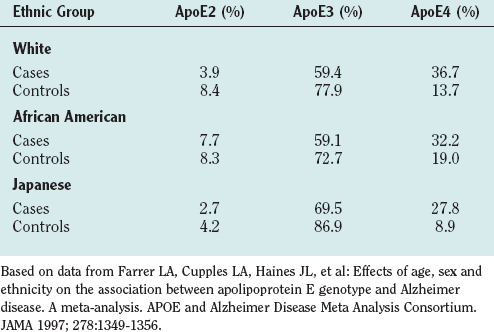
Many studies (summarized in a meta-analysis by Farrer and colleagues8) have established apolipoprotein E4 (ApoE4) as an important risk factor for Alzheimer’s disease. ApoE is a lipoprotein that carries cholesterol and has three forms: ApoE2, ApoE3, and ApoE4. The allelic prevalence varies in different ethnic groups. Table 65-6 reveals that persons with Alzheimer’s disease have a higher prevalence of ApoE4. Although community-based studies do not show ApoE4 as a risk factor for Alzheimer’s disease in African American patients, a large clinically based series reported that it is.9 One of Graff-Radford and colleagues’ findings in that report was that ApoE4 is associated with early-onset risk. In the community-based studies, there were very few early-onset cases.
TABLE 65-6 Apolipoprotein (Apo) Genotypes Related to Alzheimer’s Disease in Different Population Groups, Divided into Cases and Controls
There is strong evidence that there are undiscovered genetic factors related to late-onset Alzheimer’s disease.10 Although many candidate genes have been proposed, associations have often not been reproduced by different investigators. Therefore, researchers are working on refining their techniques and working out the pitfalls in case-control methods. Difficulties in the studies so far include heterogeneity of populations, the possible complexity of the factors causing the disease, the fact that many of the controls are probably susceptible themselves to the disease (as they age), and the fact that the effect size is too small to detect with the size of the samples used.
Amyloid Hypothesis
When Alzheimer described the disease histologically, he noted the deposition of a substance in the form of plaques that was later identified as the β amyloid protein (Aβ) by Glenner and Wong.11 Subsequently, many lines of evidence have led to the hypothesis that brain deposition of this protein causes Alzheimer’s disease.
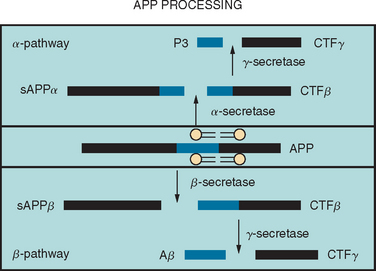
Aβ is produced by the sequential proteolytic cleavage of the amyloid precursor protein (APP) (coded for on chromosome 21) by enzymes known as secretases (Fig. 65-1). There are two pathways, one of which leads to the production of the Aβ peptides and the other does not. In the amyloidogenic pathway, the first step involves cleavage of APP at the amino terminus by an aspartyl protease (β-secretase), resulting in the formation of soluble APPβ and a membrane-bound APP carboxy terminal fragment (CTF-β). Cleavage of this CTF-β by γ-secretase results in the formation of Aβ peptides of variable length, including a 40–amino acid Aβ peptide (Aβ40) and a 42–amino acid peptide (Aβ42). The γ-secretase cleavage requires the presence of two membrane proteases, presenilin-1 and presenilin-2; it is thought that these presenilins may, in fact, be part of the γ-secretase complex. Other parts of this complex include nicastrin, a type I membrane protein that was originally purified, and two other membrane proteins, APH-1 and PEN-2. Together with presenilin, these three components appear to constitute a minimal functional γ-secretase complex. It is also possible that these “accessory” proteins are involved in substrate presentation. The non-amyloidogenic pathway is carried out by α-secretase, which cleaves the APP protein in the middle into soluble APPα and CTF-β, which, in turn, is cleaved by γ-secretase into P3 and CTF-γ (see Fig. 65-1).
Aβ42 constitutes less than 10% of the total secreted amyloid protein and is more likely to form fibrils than is Aβ40.12 Aβ42 is found in the brains of all patients with Alzheimer’s disease, and Aβ40 is found in two thirds.13 Both accumulate as extracellular plaques consisting of β-pleated sheets, but before plaque formation, a combination of fewer Aβ molecules form oligomers, which may mediate the neurotoxic effects seen in Alzheimer’s disease. Understanding of the generation of Aβ has proceeded at a faster rate than understanding of the pathways that lead to neuronal dysfunction and death. Aβ may be neurotoxic directly, may initiate inflammation, may cause oxidative stress, or may affect calcium homeostasis, or it may lead to a combination of all of these. Table 65-7 lists some of these factors supporting the amyloid hypothesis.
TABLE 65-7 Factors Supporting the Amyloid Hypothesis as a Cause of Alzheimer’s Disease
Aβ, β amyloid protein; CSF, cerebrospinal fluid.
Arguments against the Amyloid Hypothesis
The anatomical distribution of the tangle (tau) pathology is correlated better with the clinical picture than is distribution of the Aβ plaques.14 Because of this, some authors have proposed that tau, rather than Aβ, is the cause of the disease. Furthermore, there are many patients with extensive Aβ plaques who are not demented. Their condition is referred to as pathological aging in the literature.15 Because tau pathology is accelerated in the double-transgenic mouse16 by Aβ deposition, it is possible that Aβ deposition may be partially responsible for the tau pathology. Also, in the cases of early-onset Alzheimer’s disease, such as those caused by the APP mutations, there is typical tau pathology. Thus, attributing the disease exclusively to either the Aβ or tau pathology is incorrect; rather, it is much more likely that both pathological processes are involved. In further studies, investigators should work out how they interact, rather than ruling out one. Other important factors found in brains of patients with Alzheimer’s disease, including oxidative stress, inflammation, cell loss, apoptosis, and synapse loss, are probably crucial in the pathogenesis. Many researchers are studying the relationship of these factors to the amyloid and tau pathology.
PATHOLOGY
A definite diagnosis of Alzheimer’s dementia requires histopathological confirmation.
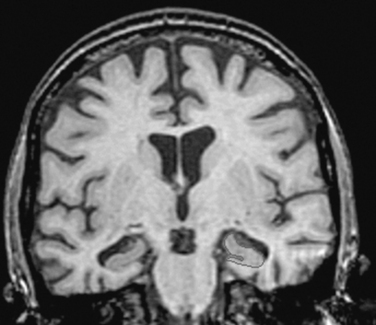
On gross examination (Fig. 65-2), the brain weight varies between 900 and 1200 g. The loss of brain weight is more marked in the early-onset form of Alzheimer’s disease, whereas in the late-onset form, there is substantial overlap with findings in age-matched controls. The temporal and parietal lobes are more affected than the frontal lobes. The occipital lobe may be involved but is relatively spared in the majority of cases. On examination, the diseased brain may appear normal in the early stages, but closer inspection usually reveals temporal lobe atrophy of its medial aspect with thinning of the cortical mantle. Other features include enlargement of the third and lateral ventricles, normal pigmentation of the substantia nigra, and pallor of the locus ceruleus in advanced cases. Alteration of pigmentation in the brainstem raises the possibility of other neurodegenerative disease (for example, Lewy body disease). In Alzheimer’s disease, the olfactory bulb is consistently smaller than expected. On occasion, atrophy may be asymmetrical or focal, and cases of Alzheimer’s disease with focal atrophy may be associated with focal cognitive deficits such as progressive aphasia, Balint’s syndrome, or frontal dementia.
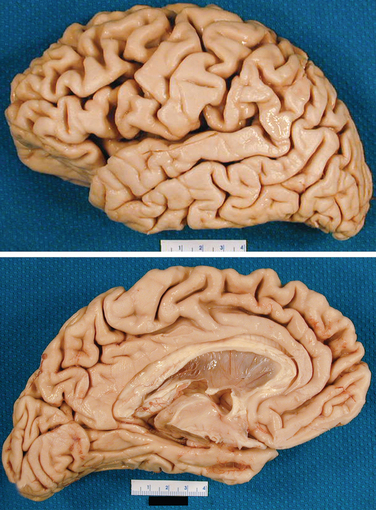
Figure 65-2 Gross appearance of the brain of a patient with Alzheimer’s disease.
(Courtesy of Dennis Dickson, MD.)
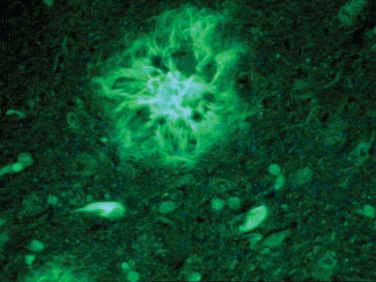
The cardinal histopathological lesions of Alzheimer’s disease are senile plaques composed of extracellular amyloid deposits (Fig. 65-3) and neurofibrillary tangles composed of intraneuronal tau protein aggregates (Fig. 65-4). The plaques are detected by silver staining (Bielschowsky stain) or by specialized amyloid stains (thioflavin S, Congo red). The β-pleated sheet structure gives rise to the staining characteristics: that is, a birefringent apple-green appearance under polarized light. Some plaques are “diffuse,” and these may be present in nondemented individuals. These plaques do not have dystrophic neurites (nerve endings containing tau protein), unlike the “cored” or neuritic plaques, which have deposition of extracellular amyloid surrounded by tau-containing neurites. Neurofibrillary tangles are filamentous inclusions that are intraneuronal and are composed of hyperphosphorylated aggregates of the microtubule-associated protein tau. Electron microscopy reveals that tau consists of a paired helical filament structure. Initially, tau collects in the nucleus and becomes aggregated into paired helical filaments, after which it may be attached to the protein ubiquitin. These tau tangles affect the microtubules, which, in turn, interfere with neuronal and dendrite/axonal transport. The neurons die and are surrounded by phagocytes removing the debris. Eventually, the tangles become extracellular structures.
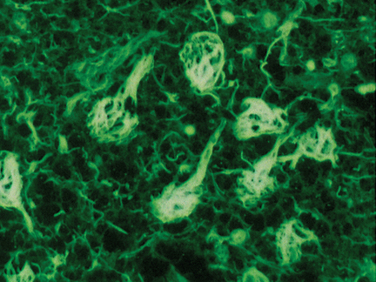
Nonamyloid Components of Plaques
Other Alzheimer’s disease–associated pathological changes include the following:
PATHOLOGICAL DIAGNOSTIC CRITERIA
Histopathological changes seen in patients with Alzheimer’s disease may overlap with changes seen in a nondemented elderly persons and may occur in patients with other neurodegenerative disease also (e.g., vascular dementia, Lewy body dementia). Different criteria are used in pathological assessment of Alzheimer’s disease cases. They include the Khachaturian criteria, the Consortium to Establish a Registry for Alzheimer’s Disease (CERAD) Guidelines for the Diagnosis of Alzheimer’s Disease, and the National Institute on Aging–Ronald and Nancy Reagan Institute criteria.17–19
The CERAD criteria, which are widely used and reproducible between different laboratories,20 involve the following:
 Macroscopic features, such as brain weight, regional atrophy, atrophy of specific regions (hippocampus, entorhinal cortex), the presence of lacunar infarcts, and the color of the substantia nigra and locus ceruleus.
Macroscopic features, such as brain weight, regional atrophy, atrophy of specific regions (hippocampus, entorhinal cortex), the presence of lacunar infarcts, and the color of the substantia nigra and locus ceruleus.
The National Institute on Aging–Ronald and Nancy Reagan Institute criteria19 constitute the most recent attempt to solve the disadvantages of the first two classifications. They include an age-related plaque score and a staging of neurofibrillary tangle pathology (Braak’s staging system); combination of the two yields an estimate of the likelihood that a patient has Alzheimer’s disease, which is classified as high, intermediate, or low probability.
DIAGNOSIS
The diagnosis of Alzheimer’s disease in the clinic comprises a complete history, a complete physical examination, laboratory studies, special tests, and a neuropsychological evaluation. There is a continuum from normal aging to dementia, including an entity called mild cognitive impairment. Published criteria for this entity are listed in Table 65-8.
Adapted from Petersen RC, Smith GE, Waring SC, et al: Mild cognitive impairment: clinical characterization and outcome. Arch Neurol 1999; 56:303-308.
History
With regard to patients with dementia, physicians should obtain the history from both the patient and an informant. In fact, patients with memory difficulty may be unaware and even deny they have a memory problem (this is a form of anosognosia). While documenting the history, physicians should keep the differential diagnosis in mind (see Table 65-9).
HIV, human immunodeficiency virus.
Medical Conditions That May Contribute to Dementia
Medication
The patient and the informant should provide a list of all prescription and nonprescription medications and food supplements. Ideally, they should bring the containers of all the medications and supplements the patient takes. Table 65-10 contains a list of medications that may adversely affect cognition.
| Antibiotics | Metronidazole |
| Anticholinergic agents |
Examination
The patient should be given a standardized short mental status test such as the Kokmen Short Test of Mental Status21 (Table 65-11) or the Mini-Mental State Examination. The patient’s anterograde memory should be tested with questions about current events or famous persons, such as the current president’s and spouse’s names and previous presidents’ and their spouses’ names. (The clinician should be sure that the patient has been exposed to this information.) Cardiovascular risk factors such as hypertension, arterial bruits, arrhythmias, and heart murmurs should be documented. A full neurological examination should be completed. The clinician should pay attention to focal deficits such as visual field cuts, pareses, sensory loss, and ataxia. The patient should be evaluated for extrapyramidal problems such as hypokinesia, rigidity, masklike facies, micrographia, and glabellar reflex. The clinician should examine the patient’s gait, looking at step size, speed of walking, arm swing, ability to turn, and how far apart the legs are. The presence of palmomental and snout reflexes is not particularly helpful because these are common in normal individuals. The grasp reflex occurs late in the course of the disease.
| Subtests | Testing | Maximum Score |
|---|---|---|
| Orientation | Name, address, current location (building), city, state, date (day), month, year | 8 |
| Attention | Digit span (present at 1 per second; record longest correct span) | 7 |
| 2-9-6-8-3 | ||
| 5-7-1-9-4-6 | ||
| 2-1-5-9-3-6-2 | ||
| Learning & Immediate Recall | Learn four unrelated words: apple, Mr. Johnson, charity, tunnel | 4 |
| Record the number of trials for acquisition (maximum of 4 trials) | ||
| Calculation | 5 × 13 = | 4 |
| 65 − 7 = | ||
| 58/2 = | ||
| 29 + 11 = | ||
| Abstraction/similarities | Similarities: orange/banana, dog/horse, table/bookcase | 3 |
| Information | President; first president; number of weeks per year; define an island | 4 |
| Construction | Copy the Necker cube | 4 |
| Draw a clock face showing 11:10 | ||
| Recall | The four words apple, Mr. Johnson, charity, tunnel | 4 |
| Total score* | 38 |
* Total score = sum of the subtest scores − (number of trials for acquisition − 1). For example, if a patient learned all 4 words on the first trial, nothing is subtracted from the sum of the subtest scores. If a patient required 4 trials to learn the 4 words, then 3 was subtracted from the sum of the subtest score.
From Kokmen E, Naessens JM, Offord KP: A short test of mental status: description and preliminary results. Mayo Clin Proc 1987; 62:281-288.
Special Tests
The following tests are indicated in the routine workup of a patient with dementia: laboratory studies, including complete blood cell count and measurements of thyroid-stimulating hormone; measurement of vitamin B12 levels and rapid plasma reagin; and neuroimaging (computed tomography or magnetic resonance imaging). Other tests may be ordered as the clinical situation dictates. It is not necessary to perform a lumbar puncture in the routine assessment of a patient with Alzheimer’s disease,22,23 but it is indicated when the following diagnoses are suspected: central nervous system infection, prion disease, hydrocephalus in the presence of normal cerebrospinal fluid pressure, and nonvasculitic autoimmune inflammatory meningoencephalitis. Lumbar puncture should also be considered when the manifestation is atypical: for example, a rapid course or an early onset of disease. Cerebrospinal fluid can be analyzed for markers of prion disease (14-3-3 protein) and Alzheimer’s disease (tau protein and Aβ42 level). Neuropsychological test results characterize the pattern of cognitive strengths and weaknesses and, in this way, are helpful in diagnosis. Also, in mild cases, they may be helpful in determining whether there is a deficit. Furthermore, these tests may be used in monitoring patients over time.
Markers for Alzheimer’s Disease
“The Consensus Report of the Working Group on: ‘Molecular and Biochemical Markers of Alzheimer’s Disease’”24 proposed that a diagnostic marker for Alzheimer’s disease should have as many of the following features as possible:







To date, the best markers are a combination of the cerebrospinal fluid tau and Aβ42 proteins. In an excellent review, Blennow and Hampel25 noted that the sensitivity and specificity for cerebrospinal fluid tau protein are 81% and 90%, respectively, and for cerebrospinal fluid Aβ42 protein, they are 86% and 90%, respectively. Because the clinical diagnosis without these markers is close to these measurements, most physicians do not use these markers routinely.
Neuroimaging: Distinguishing Cognitively Normal Individuals from Patients with Alzheimer’s Disease
Using structural magnetic resonance imaging, investigators have reported that with regard to visual inspection of anteromedial temporal lobe atrophy, the sensitivity is 83% to 85% and the specificity is 96% to 98% in distinguishing clinically diagnosed Alzheimer’s disease cases from controls.26 It is possible to outline the areas of interest in Alzheimer’s disease, such as the hippocampus, and to use this information for diagnosis and longitudinal change over time. An example is shown in Figure 65-5. Temporal and parietal hypometabolism characterize the typical positron emission tomography (PET) findings in Alzheimer’s disease. Published estimates of PET sensitivity and specificity, in which pathologically verified cases of Alzheimer’s disease were distinguished from controls by functional imaging, have ranged from 63% to 82%.27 It is now possible to image Alzheimer’s disease plaques and tangles directly with carbon 11 or fluorine 18 isotopes. Using the Pittsburgh B compound (Fig. 65-6), researchers imaged amyloid plaques and were able to distinguish patients with Alzheimer’s disease from controls in most cases.28
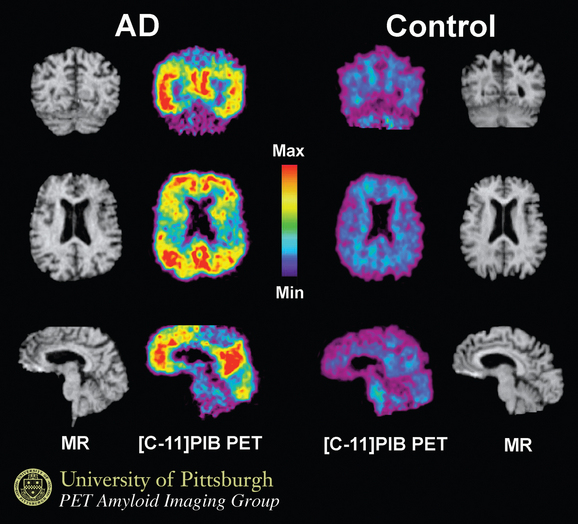
MANAGEMENT
Pharmacological Treatment
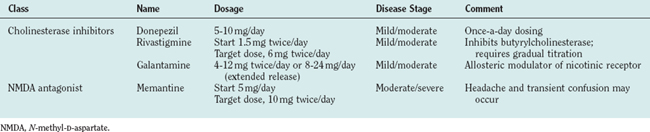
Current pharmacological treatment involves use of a cholinesterase inhibitor, which increases brain acetylcholine concentrations. In 2003, the U.S. Food and Drug Administration approved the use of memantine, a glutamate receptor antagonist, for treatment of moderate to severe Alzheimer’s disease. These two classes of agents represent the mainstay of current treatment and are listed in Table 65-12. Memantine may be used with any of the cholinesterase inhibitors or on its own; if the patient cannot tolerate cholinesterase inhibitors it may be used alone.
Therapies in Development
One of the targets identified is the Aβ protein. Academic institutions and pharmaceutical companies are investigating a number of strategies. These include drugs that inhibit the enzymes that form Aβ: that is, γ-secretase and β-secretase inhibitors. Experimentally, immunizing transgenic mice may prevent and remove amyloid from their brains; therefore, active and passive immunization are being investigated. One group has shown that heavy metal modulators, such as clioquinol, remove amyloid from the brain; this medication and newer generations of it are being investigated. Another strategy is to prevent amyloid from forming fibrils, and at present, such a medication has entered trials. Knowledge that some nonsteroidal anti-inflammatory agents decrease amyloid in the brains of transgenic mice has led to investigations of these agents. Some researchers are using antioxidant strategies.
Safety Issues
Driving
Patients with memory impairment are at increased risk for automobile accidents and therefore should be made aware of this.29 The American Academy of Neurology guidelines on driving and Alzheimer’s disease indicate that persons with a Clinical Dementia Rating Scale (CDR) score of 0.5 are at increased risk for accidents, but the risk is no greater than that for other groups at high risk, such as men aged 18 to 25 years or those with blood alcohol levels exceeding 0.08%. Patients can be offered a road test to evaluate their driving ability. Patients with a score of 1 or greater (equivalent to a Mini-Mental State Examination score of less than 25) are at great risk and should be advised not to drive.
Living Situation
The health provider should assess the patient’s living situation to make sure the patient is safe. Advice should be tailored for each situation. Factors to consider include severity of the problem, amount of family support available, patients’ behav iors, finances, and community options. Weapons should be removed or disabled. At the beginning, patients often live with a spouse or independently with regular monitoring by family members. Later, options include in-home respite care, day care, and assisted living facilities. In the later stages, the patient may need to be placed in a nursing home. Determining factors in making the decision to place a patient in a nursing home include severity of the disease, behavioral symptoms, incontinence, caregiver stress or ill health, and the family’s financial situation.
Management of Agitation in Dementia
Establishing a Cause
Medical
Infection (especially urinary infection), medication (particularly anticholinergics, analgesics, sedatives, and antipsychotics) (see Table 65-10), dehydration, electrolyte imbalance, constipation, pain, system dysfunction (biliary, cardiac, pulmonary, renal, and endocrine), and neurological dysfunction (cerebrovascular disease, seizure, and subdural hemorrhage) can affect the degree of agitation.
Intervention
Management of mild agitation may require only an environmental intervention or a redirection by a caregiver, depending on the cause. Environmental interventions include the provision of a predictable routine for the patient, a noise-free environment with control of access to prevent wandering, pictures of family members in a patient’s room, use of a night light in the room, reassurance of the patient by a family member or caregiver with whom the patient is familiar, and encouragement of the patient to participate in recreational activities available in the facility. If agitation is either severe or prolonged, then medication is necessary. One review showed evidence that olanzapine and risperidone are efficacious, but the effects are modest, and there is an increased risk for stroke.30 Another report indicates that quetiapine in doses of 25 to 50 mg twice per day was not useful in institutionalized patients with agitation.31 At the time of writing, quetiapine has not been found to be associated with an increased risk of stroke and cardiovascular disease. The chapter authors often prescribe quetiapine as a first choice; higher doses are more frequently needed than those tested in the cited study.
Doody RS, Stevens JC, Beck C, et al. Practice parameter: management of dementia (an evidence-based review). Report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology. 2001;56:1154-1156.
Golde TE. Alzheimer disease therapy: can the amyloid cascade be halted? J Clin Invest. 2003;111:11-18.
Hardy J. Toward Alzheimer therapies based on genetic knowledge. Annu Rev Med. 2004;55:15-25.
Schenk D, Hagen M, Seubert P. Current progress in beta-amyloid immunotherapy. Curr Opin Immunol. 2004;16:599-606.
1 Fratiglioni L, Launer LJ, Andersen K, et al. Incidence of dementia and major subtypes in Europe: a collaborative study of population-based cohorts. Neurologic Diseases in the Elderly Research Group. Neurology. 2000;54:S10-S15.
2 Bachman DL, Wolf PA, Linn RT, et al. Incidence of dementia and probable Alzheimer’s disease in a general population: the Framingham Study. Neurology. 1993;43:515-519.
3 Mayeux R. Epidemiology of neurodegeneration. Annu Rev Neurosci. 2003;26:81-104.
4 Alzheimer A. Uber eine eigenartige Erkrankung der Hirnrinde. Allgemeine Zeitschrift Fur Pschiatrie und phychish-Gerichtliche Medizin (Berlin). 1907;64:146-148.
5 Hebert LE, Scherr PA, Bienias JL, et al. Alzheimer disease in the US population: prevalence estimates using the 2000 census. Arch Neurol. 2003;60:1119-1122.
6 Small G, Rabins P, Barry P, et al. Diagnosis and treatment of Alzheimer’s disease and related disorders. Consensus Statement of the American Association for Geriatric Psychiatry, the Alzheimer’s Association, and the American Geriatrics Society. JAMA. 1997;278:1363-1371.
7 Brookmeyer R, Gray S, Kawas C. Projections of Alzheimer’s disease in the United States and the public health impact of delaying disease onset. Am J Public Health. 1998;88:1337-1342.
8 Farrer LA, Cupples LA, Haines JL, et al. Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta-analysis. APOE and Alzheimer Disease Meta Analysis Consortium [Comment]. JAMA. 1997;278:1349-1356.
9 Graff-Radford NR, Green RC, Go RC, et al. Association between apolipoprotein E genotype and Alzheimer disease in African American subjects. Arch Neurol. 2002;59:594-600.
10 Ertekin-Taner N, Graff-Radford N, Younkin LH, et al. Linkage of plasma Aβ42 to a quantitative locus on chromosome 10 in late-onset Alzheimer’s disease pedigrees. Science. 2000;290:2303-2304.
11 Glenner GG, Wong CW. Alzheimer’s disease and Down’s syndrome: sharing of a unique cerebrovascular amyloid fibril protein. Biochem Biophys Res Commun. 1984;122:1131-1135.
12 Kanai M, Matsubara E, Isoe KEA. Longitudinal study of cerebrospinal fluid levels of tau, Ab1–40, and Ab1–42(3) in Alzheimer’s disease: a study in Japan. Ann Neurol. 1998;44:17-26.
13 Gravina SA, Ho L, Eckman CB, et al. Amyloid beta protein (A beta) in Alzheimer’s disease brain. Biochemical and immunocytochemical analysis with antibodies specific for forms ending at A beta 40 or A beta 42(43). J Biol Chem. 1995;270:7013-7016.
14 Arnold SE, Hyman BT, Flory J, et al. The topographical and neuroanatomical distribution of neurofibrillary tangles and neuritic plaques in the cerebral cortex of patients with Alzheimer’s disease. Cereb Cortex. 1991;1:103-116.
15 Crystal H, Dickson D, Fuld P, et al. Clinico-pathologic studies in dementia: nondemented subjects with pathologically confirmed Alzheimer’s disease. Neurology. 1988;38:1682-1687.
16 Lewis J, Dickson DW, Lin WL, et al. Enhanced neurofibrillary degeneration in transgenic mice expressing mutant tau and APP. Science. 2001;293:1487-1491.
17 Fillenbaum GG, Huber MS, Beekly D, et al. The Consortium to Establish a Registry for Alzheimer’s Disease (CERAD). Part XIII. Obtaining autopsy in Alzheimer’s disease. Neurology. 1996;46:142-145.
18 Khachaturian Z. Diagnosis of Alzheimer’s disease. Arch Neurol. 1985;42:1097-1105.
19 Newell KL, Hyman BT, Growdon JH, et al. Application of the National Institute on Aging (NIA)–Reagan Institute criteria for the neuropathological diagnosis of Alzheimer disease. J Neuropathol Exp Neurol. 1999;58:1147-1155.
20 Mirra S, Gearling M, McKeel D, et al. Interlaboratory comparison of neuropathology assessments in Alzheimer’s disease: a study of the Consortium to Establish a Registry for Alzheimer’s Disease (CERAD). J Neuropathol Exp Neurol. 1994;53:303-315.
21 Kokmen E, Naessens J, Offord K. A Short Test of Mental Status: description and preliminary results. Mayo Clin Proc. 1987;62:281-288.
22 Becker P, Feussner J, Mulrow C, et al. The role of lumbar puncture in the evaluation of dementia: the Durhar Veterans Administration/Duke University Study. J Am Geriatr Soc. 1985;33:392-396.
23 Hammestrom D, Zimmer B. The role of lumbar puncture in the evaluation of dementia: the University of Pittsburgh Study. J Am Geriatr Soc. 1985;33:397-400.
24 Consensus Report of the Working Group on. “Molecular and Biochemical Markers of Alzheimer’s disease.” The Ronald and Nancy Reagan Research Institute of the Alzheimer’s Association and the National Institute on Aging Working Group. Neurobiol Aging. 1998;19:109-116. [Erratum in Neurobiol Aging 1998; 19:285].
25 Blennow K, Hampel H. CSF markers for incipient Alzheimer’s disease [Review]. Lancet Neurol. 2003;2:605-613.
26 Wahlund LO, Julin P, Johansson SE, et al. Visual rating and volumetry of the medial temporal lobe on magnetic resonance imaging in dementia: a comparative study. J Neurol Neurosurg Psychiatry. 2000;69:630-635.
27 Hoffman JM, Welsh-Bohmer KA, Hanson M, et al. FDG PET imaging in patients with pathologically verified dementia. J Nucl Med. 2000;41:1920-1928.
28 Klunk WE, Engler H, Nordberg A, et al. Imaging brain amyloid in Alzheimer’s disease with Pittsburgh Compound-B. Ann Neurol. 2004;55:306-319.
29 Dubinsky RM, Stein AC, Lyons K. Practice parameter: risk of driving and Alzheimer’s disease (an evidence-based review): report of the quality standards subcommittee of the American Academy of Neurology [Review]. Neurology. 2000;54:2205-2211.
30 Sink KM, Holden KF, Yaffe K. Pharmacological treatment of neuropsychiatric symptoms of dementia: a review of the evidence [Review]. JAMA. 2005;293:596-608.
31 Ballard C, Margallo-Lana M, Juszczak E, et al. Quetiapine and rivastigmine and cognitive decline in Alzheimer’s disease: randomised double blind placebo controlled trial. BMJ. 2005;330:874.
32 Mayeux R. Apolipoprotein E, Alzheimer disease, and African Americans [Comment]. Arch Neurol. 2003;60:161-163.
33 Green RC, Cupples LA, Go R, et al. Risk of dementia among white and African American relatives of patients with Alzheimer disease. JAMA. 2002;287:329-336.
34 Goate A, Chartier-Harlin M, Mullan M, et al. Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer’s diseases. Nature. 1991;349:704-706.
35 Levy-Lehad E, Wijsman E, Nemens EEA. A familial Alzheimer’s disease locus on chromosome 1. Science. 1995;269:970-973.
36 St. George-Hyslop P, Haines J, Rogaev E, et al. Genetic evidence for a novel familial Alzheimer’s disease locus on chromosome 14. Nat Genet. 1992;2:330-334.
37 Pericak-Vance M, Bebout J, Gaskell P, et al. Linkage studies in familial Alzheimer’s disease: evidence for chromosome 19 linkage. Am J Hum Genet. 1991;48:1034-1050.
38 Kamboh MI. Molecular genetics of late-onset Alzheimer’s disease. Ann Hum Genet. 2004;68:381-404.
39 Schupf N, Kapell D, Nightingale B, et al. Specificity of the five-fold increase in AD in mothers of adults with Down syndrome. Neurology. 2001;57:979-984.
40 Andersen K, Launer LJ, Dewey ME, et al. Gender differences in the incidence of AD and vascular dementia: The EURODEM Studies. EURODEM Incidence Research Group. Neurology. 1999;53:1992-1997.
41 Guo Z, Cupples LA, Kurz A, et al. Head injury and the risk of AD in the MIRAGE study. Neurology. 2000;54:1316-1323.
42 Plassman BL, Havlik RJ, Steffens DC, et al. Documented head injury in early adulthood and risk of Alzheimer’s disease and other dementias. Neurology. 2000;55:1158-1166.
43 Casserly I, Topol E. Convergence of atherosclerosis and Alzheimer’s disease: inflammation, cholesterol, and misfolded proteins. Lancet. 2004;363:1139-1146.
44 Snowdon DA, Kemper SJ, Mortimer JA, et al. Linguistic ability in early life and cognitive function and Alzheimer’s disease in late life. Findings from the Nun Study. JAMA. 1996;275:528-532.
45 Verghese J, Lipton RB, Katz MJ, et al. Leisure activities and the risk of dementia in the elderly. N Engl J Med. 2003;348:2508-2516.
46 Szekely C, Thorne J, Zandi PP, et al. Nonsteroidal anti-inflammatory drugs for the prevention of Alzheimer’s disease: a systematic review. Neuroepidemiology. 2004;23:159-169.
47 Wolozin B, Kellman W, Ruosseau P, et al. Decreased prevalence of Alzheimer disease associated with 3-hydroxy-3-methyglutaryl coenzyme A reductase inhibitors. Arch Neurol. 2000;57:1439-1443.
48 Shumaker SA, Legault C, Rapp SR, et al. Estrogen plus progestin and the incidence of dementia and mild cognitive impairment in postmenopausal women: the Women’s Health Initiative Memory Study: a randomized controlled trial. JAMA. 2003;289:2651-2662.
49 Yaffe K, Sawaya G, Lieberburg I, et al. Estrogen therapy in postmenopausal women: effects on cognitive function and dementia. JAMA. 1998;279:688-695.
50 Seshadri S, Beiser A, Selhub J, et al. Plasma homocysteine as a risk factor for dementia and Alzheimer’s disease. N Engl J Med. 2002;346:476-483.
51 Morris MC, Evans DA, Bienias JL, et al. Dietary niacin and the risk of incident Alzheimer’s disease and of cognitive decline. J Neurol Neurosurg Psychiatry. 2004;75:1093-1099.
52 Yaffe K, Barnes D, Nevitt M, et al. A prospective study of physical activity and cognitive decline in elderly women: women who walk [Comment]. Arch Intern Med. 2001;161:1703-1708.
53 Orgogozo JM, Dartigues JF, Lafont S, et al. Wine consumption and dementia in the elderly: a prospective community study in the Bordeaux area. Rev Neurol. 1997;153:185-192.
54 Morris MC, Evans DA, Bienias JL, et al. Consumption of fish and n-3 fatty acids and risk of incident Alzheimer disease [Comment]. Arch Neurol. 2003;60:940-946.
55 Morris MC, Evans DA, Bienias JL, et al. Dietary intake of antioxidant nutrients and the risk of incident Alzheimer disease in a biracial community study. JAMA. 2002;287:3230-3237.
56 Scheuner D, Eckman C, Jensen M, et al. Secreted amyloid beta-protein similar to that in the senile plaques of Alzheimer’s disease is increased in vivo by the presenilin 1 and 2 and APP mutations linked to familial Alzheimer’s disease. Nat Med. 1996;2:864-870.
57 Graff-Radford N, Eckman C, O’Brien P, et al. Plasma amyloid β protein (Aβ) in Down’s syndrome (DS): implications for Alzheimer’s disease. Neurology. 1997;47:A378.
58 Schupf N, Patel B, Silverman W, et al. Elevated plasma amyloid beta-peptide 1–42 and onset of dementia in adults with Down syndrome. Neurosci Lett. 2001;301:199-203.
59 Tokuda T, Fukushima T, Ikeda S, et al. Plasma levels of amyloid β proteins Ab1–40 and Ab1–42(43) are elevated in Down’s syndrome. Ann Neurol. 1997;41:271-273.
60 Gearing M, Mori H, Mirra S. Aβ-peptide length and apolipoprotein E genotype in Alzheimer’s disease. Ann Neurol. 1996;39:395-399.
61 Graff-Radford N, Eckman C, Hutton M, et al. Plasma amyloid (Aβ) levels in relatives of Alzheimer’s disease patients. Neurology. 1998;48:A314.






